Двойные ингибиторы pi3 киназы/mtor




















Формула / Реферат
1. Соединение, представляющее собой 8-[5-(1-гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2Н-имидазо[4,5-с]хинолин-2-он, или его фармацевтически приемлемая соль.
2. Соединение по п.1, отличающееся тем, что представляет собой 8-[5-(1-гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2Н-имидазо[4,5-с]хинолин-2-он.
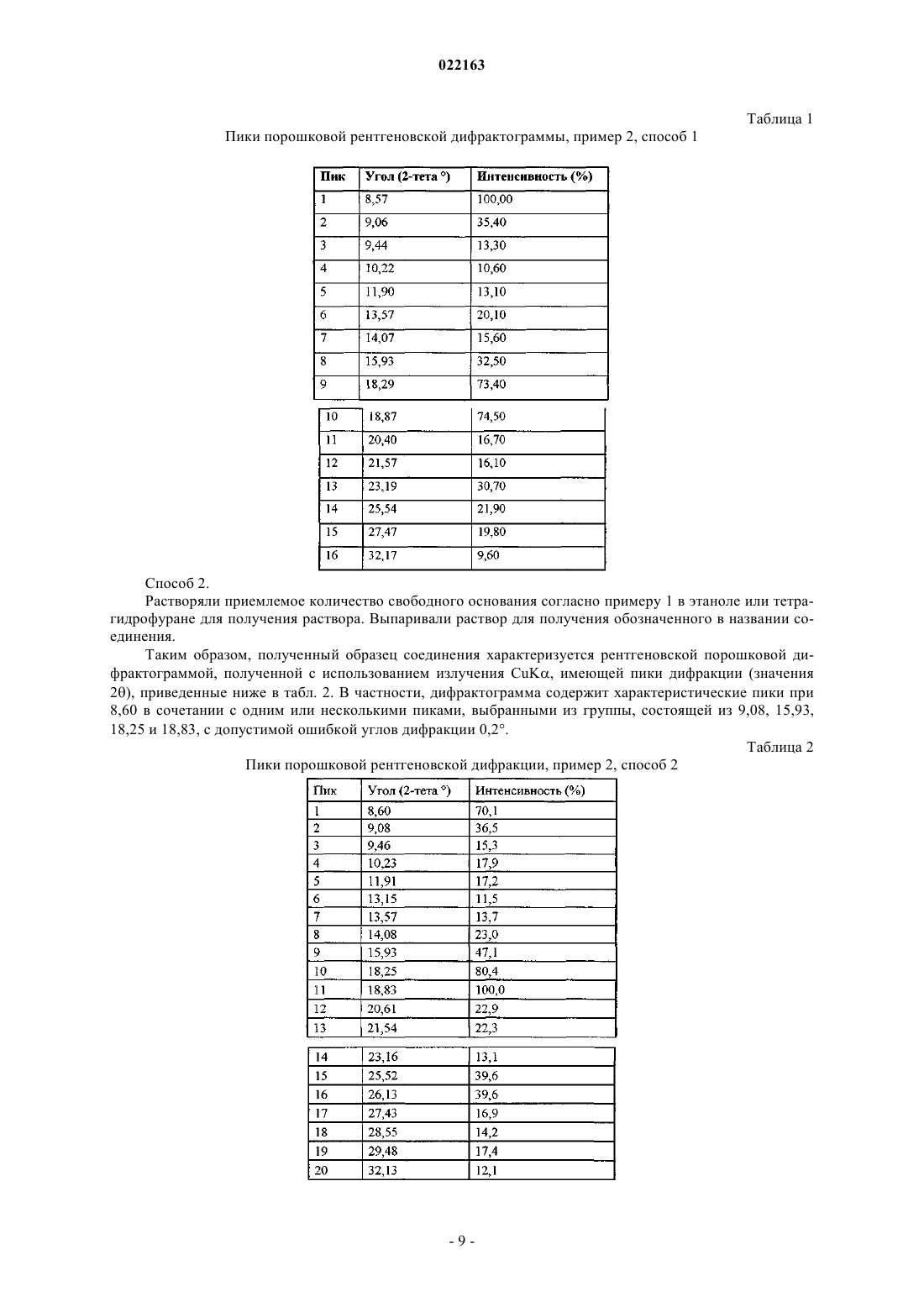
3. Соединение по п.2, отличающееся тем, что представляет собой 8-[5-(1-гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2Н-имидазо[4,5-с]хинолин-2-он в кристаллической форме, характеризующейся рентгеновской порошковой дифрактограммой, содержащей характеристические пики в 2θ±0,2, при 8,57 и одном или нескольких из 9,06, 15,93, 18,29 и 18,87.
4. Фармацевтическая композиция для лечения рака, содержащая соединение или соль по любому из пп.1-3 и фармацевтически приемлемый носитель, растворитель или наполнитель.
5. Применение соединения или соли по любому из пп.1-3 для лечения рака.
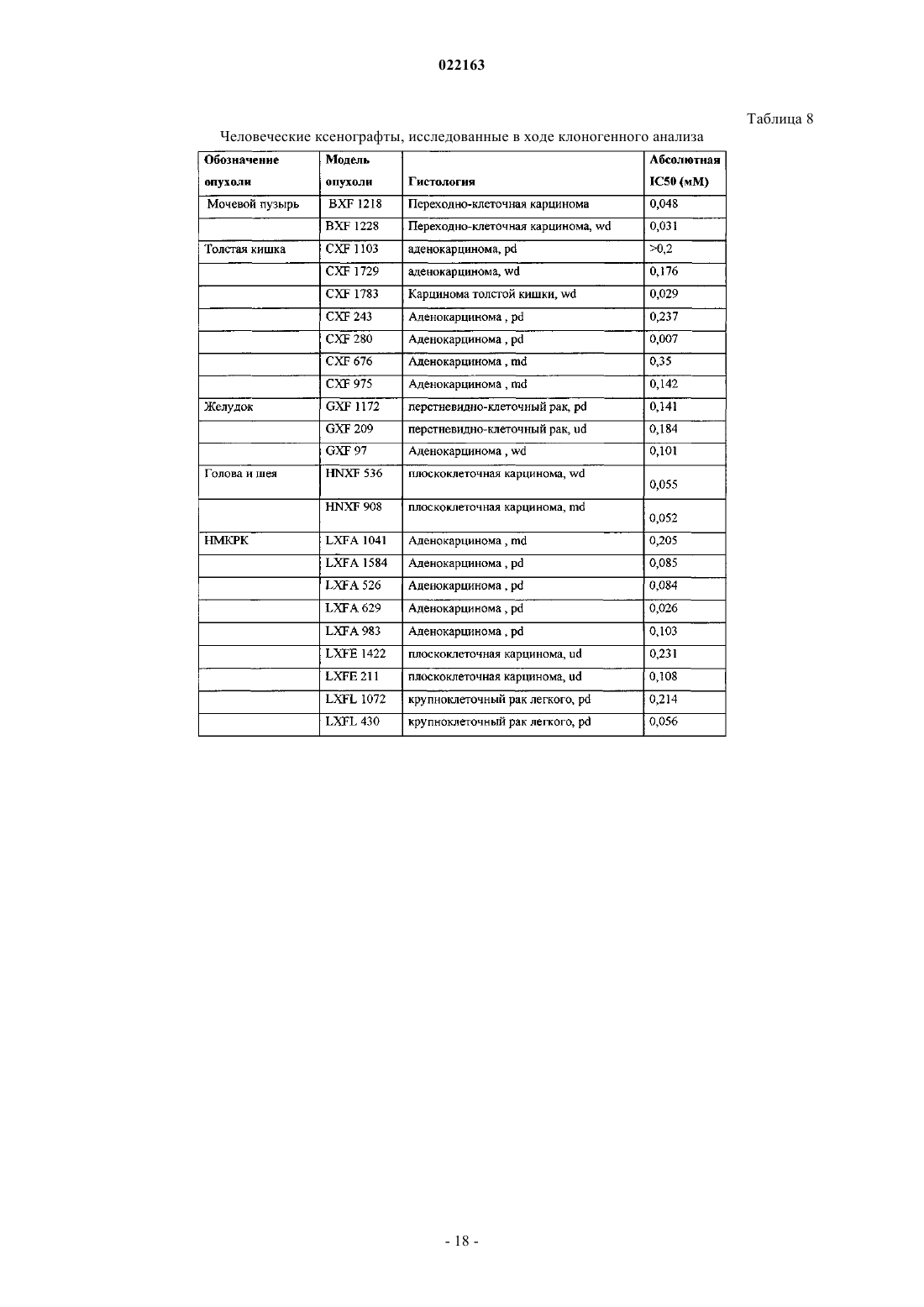
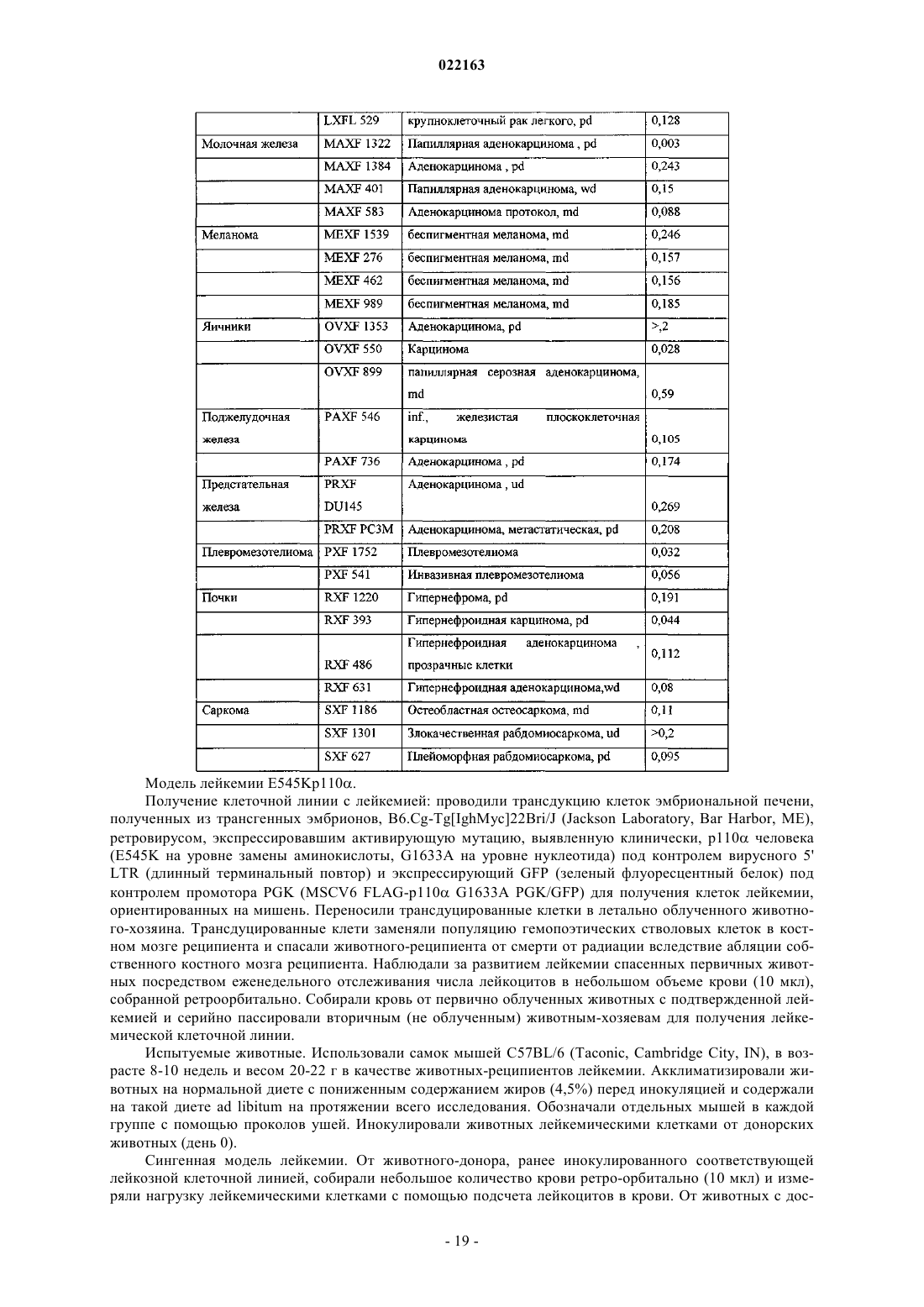
6. Применение по п.5, при этом рак выбран из группы, состоящей из рака мочевого пузыря, рака толстой кишки, рака желудка, рака головы и шеи, немелкоклеточного рака легкого (НМРЛ), рака молочной железы, меланомы, рака яичников, рака поджелудочной железы, рака предстательной железы, глиобластомы, рака легкого, рака почки, саркомы, рака кроветворной и лимфоидной тканей, рака центральной нервной системы, рака шейки матки, рака эндометрия, рака печени, рака кожи, рака желудка, рака щитовидной железы, рака верхних отделов желудочно-кишечного тракта и дыхательных путей и рака мочеполовой системы.
Текст
Настоящее изобретение обеспечивает соединение имидазо[4,5-с]хинолин-2-она или его фармацевтически приемлемую соль, которое ингибирует как PI3K, так и mTOR и поэтому подходит для лечения рака. Барда Дэвид Энтони, Мадер Мэри Маргарет (US) Лыу Т.Н. (RU)(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Фосфоинозитид-3-киназы (PI3Ks) представляют собой семейство липидных киназ, которые запускают внутриклеточные сигнальные каскады, регулирующие широкий спектр клеточных процессов. Например, активация PI3K инициирует каскад сигнальной трансдукции, который способствует росту, выживаемости и метаболизму раковых клеток. Мишень рапамицина у млекопитающих (mTOR) является ключевым узлом сигналлинга, координирующим прохождение по клеточному циклу и рост клеток в ответ на генетические, эпигенетические условия и условия окружающей среды. Регуляция путей, участвующих в передаче сигналов mTOR, нарушена в предраковых тканях человека. С учетом роли, которую играют PI3K и mTOR в путях клеточного цикла, ингибирование и PI3K, и mTOR может быть полезным для лечения некоторых заболеваний человека, таких как рак. Ингибиторы PI3K и двойные ингибиторы PI3K/mTOR широко известны в данной области техники. В публикации WO 2010/038165 описаны некоторые соединения имидазол[1,5]нафтиридина, которые относят к модуляторам или ингибиторам фермента PI3-K и/или двойным ингибиторам PI3-K/mTOR. В публикациях WO 2010/139731 и WO 2010/139747 описаны некоторые соединения имидазолхинолина,предложенные для применения для лечения заболеваний, опосредованных протеинкиназами или липидными киназами, в частности заболеваний, опосредованных PI3K. Сохраняется необходимость в создании альтернативных ингибиторов PI3K/mTOR, особенно в мощных ингибиторах PI3K/mTOR с полезными физическими свойствами, такими как улучшенная растворимость, и/или желательными клиническими свойствами, такими как улучшенная активность in vivo,и/или фармакокинетическими свойствами, которые могут быть использованы при лечении заболеваний,обусловленных пролиферацией клеток, таких как рак. Настоящее изобретение обеспечивает мощные ингибиторы PI3K/mTOR. Более конкретно, настоящее изобретение обеспечивает мощные ингибиторыPI3K/mTOR с полезными физическими свойствами и/или желательными клиническими свойствами, которые являются полезными противораковыми агентами. Настоящее изобретение обеспечивает соединение, которое представляет собой 8-[5-(1-гидрокси-1 метилэтил)пиридин-3-ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2 он или его фармацевтически приемлемую соль. Согласно конкретному варианту реализации настоящее изобретение обеспечивает соединение, которое представляет собой 8-[5-(1-гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2-метоксипропил]-3 метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2-он. В другом варианте реализации оно представляет собой 8-[5-(1-гидрокси-1-метилэтил)пиридин-3 ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2-он в кристаллической форме. Настоящее изобретение также обеспечивает 8-[5-(1-гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2 метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2-он в кристаллической форме, которая характеризуется рентгеновской порошковой дифрактограммой, содержащей характеристические пики, в 20,2 при 8,57 и один или несколько из 9,06, 15,93, 18,29 и 18,87. Настоящее изобретение обеспечивает фармацевтическую композицию, содержащую 8-[5-(1 гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5 с]хинолин-2-он или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель,растворитель или наполнитель. Настоящее изобретение обеспечивает фармацевтическую композицию, содержащую 8-[5-(1 гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5 с]хинолин-2-он или его фармацевтически приемлемую соль, дополнительно содержащую один или несколько лекарственных ингредиентов. Настоящее изобретение обеспечивает способ лечения рака, включающий введение пациенту, который в этом нуждается, эффективного количества 8-[5-(1-гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2 метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2-она или его фармацевтически приемлемой соли. Настоящее изобретение обеспечивает использование 8-[5-(1-гидрокси-1-метилэтил)пиридин-3-ил]1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2-она или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения рака. Настоящее изобретение обеспечивает 8-[5-(1-гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2 метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2-он или его фармацевтически приемлемую соль для применения для лечения. Настоящее изобретение обеспечивает 8-[5-(1-гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2 метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2-он или его фармацевтически приемлемую соль для применения для лечения рака. Более того, настоящее изобретение обеспечивает предпочтительные варианты реализации способов и применений согласно настоящему описанию, в которых рак выбран из группы, состоящей из рака мочевого пузыря, рака толстой кишки, рака желудка, рака головы и шеи, НМРЛ, рака молочной железы,меланомы, рака яичников, рака поджелудочной железы, рака предстательной железы, глиобластомы,-1 022163 рака легкого, рака почки, саркомы, рака кроветворной и лимфоидной тканей, рака центральной нервной системы, рака шейки матки, рака эндометрия, рака печени, рака кожи, рака желудка, рака щитовидной железы, рака верхних отделов желудочно-кишечного тракта и дыхательных путей и рака мочеполовой системы. Следует понимать, что используемые выше и во всем описании изобретения следующие термины,если не указано иное, имеют следующие значения. Термин "фармацевтически приемлемая соль" или "фармацевтически приемлемые соли" обозначает относительно нетоксичные, неорганические и органические соли соединений согласно настоящему изобретению. Термины "лечение", "лечить" и т.п. включают замедление или реверсию прогрессирования заболевания. Эти термины также включают облегчение, уменьшение интенсивности симптомов, ослабление,устранение или уменьшение одного или нескольких симптомов расстройства или состояния, даже если расстройство или состояние в действительности не устранено и даже если прогрессирование заболевания или состояния само по себе не замедлилось или направление хода заболевания не изменено."Терапевтически эффективное количество" или "эффективное количество" означает количество соединения или его фармацевтически приемлемой соли согласно настоящему изобретению или фармацевтической композиции, содержащей соединение или его фармацевтически приемлемую соль согласно настоящему изобретению, которое будет вызывать биологический или медицинский ответ или оказывать желаемый терапевтический эффект на ткань, систему, животное, млекопитающее или человека, которого добивается исследователь, ветеринар, врач или другой доктор-клиницист. Соединения по настоящему изобретению способны вступать в реакцию, например, с рядом неорганических и органических кислот с образованием фармацевтически приемлемых солей. Такие фармацевтически приемлемые соли и обычная методика их получения хорошо известны в данной области. См.,например, P. Stahl, et al., Handbook of Pharmaceutical Salts: Properties, Selection, and Use, (VCHA/WileyVCH, 2002); S.M. Berge, et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, Vol. 66, No. 1,January 1977. Соединения согласно настоящему изобретению предпочтительно изготавливают в виде фармацевтических композиций с использованием одного или нескольких фармацевтически приемлемых носителей, разбавителей или наполнителей и вводят различными путями. Предпочтительно такие композиции предназначены для перорального, подкожного или внутривенного введения. Такие фармацевтические композиции и способы их приготовления хорошо известны в данной области техники. См., например,Remington: The Science and Practice of Pharmacy (A. Gennaro, et al., eds., 21st ed., Mack Publishing Co.,2005). Количество соединения согласно настоящему изобретению, которое будут фактически вводить, определит лечащий врач при соответствующих обстоятельствах, включая состояние, подлежащее лечению,выбранный путь введения, вводимое конкретное соединение согласно настоящему изобретению, возраст,вес и реакцию отдельного пациента, а также тяжесть симптомов пациента. Дневная доза обычно находится в пределах от примерно 1 до примерно 2000 мг. В некоторых случаях уровни доз меньше нижнего предела вышеуказанного диапазона могут быть более чем достаточными, тогда как в других случаях могут быть использованы еще большие дозы. Соединения согласно настоящему изобретению могут быть получены различными методами, известными в данной области техники, а также согласно описанию в разделах "Приготовления" и "Примеры" ниже. Специфические этапы синтеза для каждого из описанных путей могут быть объединены различными способами для получения соединений согласно настоящему изобретению. Реагенты и исходные материалы обычно доступны в готовом виде для специалиста в данной области. Другие могут быть получены стандартными методами органической химии и химии гетероциклических соединений и методами аналогичных синтезу известных структурно подобных соединений, и процедурам, описанным в разделах "Приготовления" и "Примеры" ниже, включая любые новые процедуры. Следующие разделы "Приготовления" и "Примеры" приведены с целью более подробной иллюстрации изобретения и представляют собой типичный синтез соединений. Названия соединений согласно настоящему изобретению обычно присваиваются с использованием SymyxDraw 3.2. Согласно настоящему описанию следующие термины имеют указанные значения: АТФ означает аденозинтрифосфат; AUC означает площадь под кривой; ЦНС означает центральную нервную систему;DMEM означает среду Игла в модификации Дульбекко; ДМСО означает диметилсульфоксид; ДТТ означает дитиотреитол; 4 Е-ВР 1 означает белок 1, связывающий 4 Е; ФБС означает фетальную бычью сыворотку; ЭДТА означает этилендиаминтетрауксусную кислоту; EGTA обозначает этиленгликоль-бис-(аминоэтиловый эфир)-N,N,N',N'-тетрауксусную кислоту; GFP означает зеленый флуоресцентный белок;GST означает глутатион-S-трансферазу; ГЭЦ означает гидроксиэтилцеллюлозу; HEPES обозначает N-2 гидроксиэтилпиперазин-N'-2-этансульфоновую кислоту; ВЭЖХ обозначает жидкостную хроматографию при высоких давлениях; IC50 обозначает ингибирующую концентрацию, равную половине максимальной; IMDM обозначает среду Дульбекко в модификации Исков; MEM обозначает минимально обогащенную среду; МС обозначает масс-спектроскопию; ЯМР обозначает ядерный магнитный резонанс; НМКРЛ обозначает немелкоклеточный рак легкого; PBS обозначает фосфатно-солевой буфер; PGK обозначает фосфоглицерат киназу; PIP2 обозначает фосфатидилинозитол-(4,5)-бисфосфат; РО обозначает перорально; POPS обозначает пальмитил-олеил фосфатидилсерин; PPI обозначает ингибитор протонной помпы; RPMI обозначает мемориальный институт Roswell Park; RT обозначает комнатную температуру;TFA обозначает трифторуксусную кислоту; TMED50 обозначает порог минимальной эффективной дозы;TR-FRET обозначает резонансный перенос энергии возбуждения с временным разрешением; Tris обозначает трис(гидроксиметил)аминометан; Triton-X обозначает 4-(1,1,3,3-тетраметилбутил)фенилполиэтиленгликоль t-октилфеноксиполиэтоксиэтанол полиэтиленгликоль трет-октилфенил эфир, Tween20 обозначает полисорбат 20; XRD обозначает рентгеновскую дифракцию порошка. Пример получения 1. (2S)-2-Метоксипропан-1-амин гидрохлорид Охладить раствор трет-бутил [(2S)-2-гидроксипропил]карбамата (870 г, 4,96 моль) в тетрагидрофуране (10 л) до 5C. Добавить гидрид натрия (60% дисперсия, 248 г, 6,21 моль, 1,25 экв.) порциями в течение 8 мин. Перемешивать реакционную смесь при 5C в течение 30 мин. Добавить метилйодид (387 мл,6,21 моль, 1,25 экв.) по каплям в течение 5 мин и дать реакции продолжаться при 5-10C в течение 45 мин. Остановить реакцию добавлением воды (1 л) и провести экстракцию этилацетатом (4 л). Получить органический слой и сконцентрировать его в вакууме для получения остатка. Провести одновременное испарение с толуолом (21 л), разбавить дихлорметаном (2 л) и отфильтровать. Промыть остаток дополнительным дихлорметаном (500 мл). Концентрировать объединенный жидкий фильтрат дихлорметана в вакууме для получения неочищенного промежуточного соединения трет-бутил N-[(2S)-2 метоксипропил]карбамата (880 г, 94%). Суспендировать неочищенное промежуточное соединение трет-бутилN-[(2S)-2 метоксипропил]карбамат (806 г, 4,26 ммоль) в дихлорметане (3,22 л) и добавить 4 М HCl в диоксане (2,66 л) по каплям в течение 30 мин при 15C. Перемешивать реакционную смесь при комнатной температуре в течение 2 ч. Отфильтровать и промыть остаток дополнительным дихлорметаном (2250 мл) и удалить летучие вещества в вакууме для получения сухого остатка. Добавить трет-бутил эфир (2 л) к осадку и отфильтровать; промыть осадок дополнительным метил трет-бутил эфиром (2500 мл), высушить и дальше промыть осадок ацетоном (4500 мл) для получения белого порошка соединения, обозначенного в названии (171 г, 32%). 1 Суспендируют этиловый эфир 6-бром-4-гидроксихинолин-3-карбоксикислоты (11 г, 37 ммоль) в безводном диметилформамиде (148,6 мл) в атмосфере азота. Добавляют фосфорилхлорид (20,7 мл, 222 ммоль, 6 экв.) с помощью шприца в течение 5 мин и интенсивно перемешивают при комнатной температуре в течение 3 ч. Останавливают реакцию посредством выливания смеси в ледяную воду (1,5 л) и продолжают перемешивать до тех пор, пока лед не растает. Образовавшийся осадок получают фильтрованием, промывают водой и оставляют до полного высыхания для получения обозначенного в названии соединения (11,4 г, 94%). MS (ESI) m/z (М+Н)+ 314,0, 316,0. В другом случае готовят этиловый эфир 6-бром-4-гидроксихинолин-3-карбоксикислоты согласно описанию ниже и используют для приготовления обозначенного в названии соединения. Растворяют диэтил 2-4-бромфенил)амино)метилен)малонат (25,6 г, 74,8 ммоль) в 2-метилтетрагидрофуране (107 мл) и переносят в насос. Затем насос подает от 19 до 21 мл данного раствора в реактор 25 мл при 240-260 С при давлении азота 575-700 psi для того, чтобы оставаться выше упругости пара раствора реагентов. Через приблизительно 60-180 мин нахождения при данной температуре полученная суспензия выходит из реактора через клапан и затем через погружную трубу в зону разгерметизации и охлаждения объемом 10 мл на дне реактора. Затем второй по очереди клапан отправляет суспензию в находящийся на одной оси нагнетательный фильтр. Эту последовательность опустошения реактора дополнительно повторяют два раза перед повторным заполнением реактора согласно описанию выше для гарантии того, что остатки суспензии удаляются до минимального объема и для обеспечения давления азота на каждом листовом фильтре. Автоматический цикл повторяют несколько раз, и осадок накапливается со временем на одном и том же листовом фильтре. Если выработку проводят в течение нескольких дней, будут параллельно использоваться по меньшей мере 2 фильтра для того, чтобы можно было промыть отключенный фильтр и удалить осадок без остановки реактора с прерывистым потоком. После проведения 5 дополнительных циклов подобным образом в реактор направляют 2-метилтетрагидрофуран (20 мл) и переносят в фильтр. Этот режим можно назвать режимом с прерывистым потоком, полунепрерывным или последовательным автоматизированным. В любом случае, аспекты этого режима работы, которые делают его похожим на непрерывную реакцию, заключаются в том, что температура и давление в реакторе не меняются со временем, а всегда остаются в условиях реакции, нагревание и охлаждение происходит очень быстро для реагентов, поступающих в реактор, и продуктов, вытекающих из реактора, период нагревания и охлаждения являются масштабируемыми, и время нахождения реагентов и продуктов в реакторе одинаковы в любом масштабе благодаря контролю входящих и исходящих потоков и возможности внешнего нагрева/охлаждения по отношению к реактору. После высушивания собирают этиловый эфир 6-бром-4 гидроксихинолин-3-карбоксикислоты (4,66 г, 21%). Полученные фильтраты, обогащенные этил 2-4 бромфенил)амино)метилен)малонатом, концентрируют для удаления этанола, достигающего 7,8 г, и перерастворяют в 2-метилтетрагидрофуране. Часть данного материала (4,8 г) повторно подвергают воздействию условий реакции и собирают дополнительный этиловый эфир 6-бром-4-гидроксихинолин-3 карбоксикислоты (0,56 г) для получения общей доли выхода 25%. 1 Способ 1. Добавляют раствор этил 5-бромпиридин-3-карбоксилата (22,5 г, 98 ммоль) в тетрагидрофуране (350 мл) в раствор 3M бромида метилмагния в диэтиловом эфире (98 мл, 293 ммоль, 3 экв.) при внутренней температуре ниже 30C. После завершения реакции охлаждают смесь на ледяной бане и останавливают реакцию насыщенным водным хлоридом аммония и продолжают перемешивать до растворения большей части осадка. Добавляют воду (500 мл) и проводят экстракцию с использованием этилацетата (3500 мл). Высушивают органический слой над сульфатом натрия, фильтруют и концентрируют органический слой до осадка. Очищают осадок с помощью колоночной хроматографии на силикагеле с элюцией этилацетатом:гексаном (1:1) для получения обозначенного в названии продукта в виде масла (19,4 г, 92%). MS(ESI) m/z (M+Н)+ 214,9, 216,9. Способ 2. Добавляют раствор метил 5-бромпиридин-3-карбоксилата (173 г, 800 ммоль) в тетрагидрофуране(2,6 л) по каплям в 3 М раствор метилмагнийбромида в диэтиловом эфире (800 мл, 3,0 экв.) в течение 30 мин в охлаждающей бане при внутренней температуре ниже 18C. Перемешивают смесь при комнатной температуре в течение 1 ч, затем охлаждают ее до 0C и останавливают реакцию насыщенным водным раствором хлорида аммония (500 мл). Добавляют насыщенный водный раствор бикарбоната натрия (1 л) и оставляют разделяться на слои. Получают органический слой и концентрируют его толуолом (1 л) для азеотропного связывания воды для получения обозначенного в названии соединения (168 г, 97%) в виде оранжевого масла. 1 Тщательно продували азотом смесь 2-(5-бром-3-пиридил)пропан-2-ола (18,5 г, 85,7 ммоль),бис(пинаколат)диборона (44 г, 171 ммоль, 2 экв.) и ацетата калия (25,2 г, 257 ммоль, 3 экв.) в 1,4 диоксане (428 мл). Добавляют (1,1'-бис(дифенилфосфин)ферроцен)палладий(II) хлорид (3,5 г, 4,3 ммоль),удаляют смесь и продувают через реакцию дважды азот. Нагревают смесь при 90C в течение ночи. Охлаждают и разбавляют этилацетатом (1 л) и обрабатывают ультразвуком в течение 30 мин. Фильтруют через подложку из Celite и высушивают жидкий фильтрат над сульфатом натрия. После фильтрации концентрируют органическую жидкость и повторно растворяют в этилацетате (1 л) и снова фильтруют через подложку из Celite. Концентрируют фильтрат и суспендируют осадок в диэтиловом эфире (100 мл), а затем в гексане (700 мл). В течение короткого времени обрабатывают ультразвуком и фильтруют для получения сухого остатка для получения обозначенного в названии соединения в виде неочищенного Суспендировали этил-6-бром-4-хлорхинолин-3-карбоксилат (389 г, 1,24 моль) и (2S)-2 метоксипропан-1-амин гидрохлорид (171 г, 1,36 моль, 1,1 экв.) в этаноле (5,84 л). Добавляют диизопропилэтиламин (474 мл) и нагревают смесь при 50 С в течение ночи. Через 16 ч охлаждают реакцию до комнатной температуры и концентрируют в вакууме. Добавляют метил-трет-бутиловый эфир (2 л) к осадку и перемешивают в течение 20 мин. Фильтруют концентрат и промывают его метил-третбутиловым эфиром (2250 мл). Концентрируют и фильтруют в вакууме для получения обозначенного в названии соединения, по существу, с количественным выходом. Это соединение будет использовано на следующем этапе без дальнейшей очистки. MS (ESI) m/z (M+Н)+ 367,0, 369,0. Пример получения 6. 6-Бром-4-(2S)-2-метоксипропил]амино]хинолин-3-карбоксикислота Добавляют раствор гидроксида натрия (296,7 г, 7,42 моль, 6 экв.) в воде (454 мл) в раствор этил-6 бром-4-(2S)-2-метоксипропил]амино]хинолин-3-карбоксилата (454 г, 1,24 моль) в тетрагидрофуране(4,54 л) при комнатной температуре. Нагревают смесь при 50 С в течение ночи. Через 18 ч охлаждают реакцию до 0 С, добавляют 37% водный HCl по каплям в течение 30 мин до рН 6 (приблизительно 450 мл) при температуре ниже 23 С. Фильтруют образовавшийся осадок через фильтровальную бумагу и промывают его последовательно в воде (2 л), ацетоне (2 л) и метил-трет-бутиловом эфире (2 л). Высушивают белый остаток для получения обозначенного в названии соединения (359 г, 86%). MS (ESI) m/z Суспендируют 6-бром-4-(2S)-2-метоксипропил]амино]хинолин-3-карбоксикислоту (510 г, 1,5 моль) в диметилформамиде (7,65 л) и добавляют триэтиламин (419 мл, 3 моль, 2 экв.) при 70 С. Добавляют дифенилфосфоновый азид (390 мл, 1,8 моль, 1,2 экв.) по каплям в течение 30 мин. Нагревают смесь до 70 С (внутренняя температура) в течение 1 ч. Охлаждают до 10 С и разбавляют водой (5 мл). Перемешивают смесь в течение 1 ч, фильтруют осадок, промывают его водой (21 л) и метил-трет-бутиловым эфиром (21 л) и затем оставляют его сушиться для получения обозначенного в названии соединения в виде белого порошка (445 г, 88%). MS (ESI) m/z (M+Н)+ 335,9, 337,9. Пример получения 8. 8-Бром-1-[(2S)-2-метоксипропил]-3-метилимидазол[4,5-с]хинолин-2-он и тетра-N-бутиламмоний бромид (3 г, 9,3 ммоль) в дихлорметане (150 мл). Добавляют 2 М водного гидроксида натрия (75 мл, 150 ммоль) при комнатной температуре. Добавляют йодометан (7,5 мл, 120 ммоль) и интенсивно перемешивают смесь в течение ночи при 28 С. Дают произойти разделению фаз. Концентрируют органические соединения в вакууме. Промывают осадок в ацетоне (50 мл) для удаления тетра-N-бутиламмоний бромида. Фильтруют смесь для получения обозначенного в названии соединения в виде сухого порошка (5 г, 48%). MS (ESI) m/z (M+Н)+ 350,0, 352,0. Способ 2. Суспендируют 8-бром-1-[(2S)-2-метоксипропил]-3H-имидазол[4,5-с]хинолин-2-он (285 г, 847,7 ммоль) и тетра-N-бутиламмоний бромид (82 г, 254 ммоль) в дихлорметане (2,85 л). Добавляют 2 М водного гидроксида натрия (1,7 л, 3,4 моль) при комнатной температуре. Добавляют диметилсульфат (160,8 мл, 1,7 моль) и интенсивно перемешивают смесь в течение 3 ч. Дают произойти разделению фаз и получают органический слой. Концентрируют органический слой в вакууме и суспендируют в воде (2,4 л) в течение 30 мин. Фильтруют образовавшийся твердый осадок и промывают его в воде (2500 мл), гексане(2500 мл) и высушивают. Обозначенное соединение получают в виде белого порошка (207 г, 70%). MS(ESI) m/z (М+H)+ 350,0, 352,0. Способ 3. Суспендируют 8-бром-1-[(2S)-2-метоксипропил]-3H-имидазол[4,5-с]хинолин-2-он (50 г, 149 ммоль) и тетра-N-бутиламмоний бромид (14,4 г, 44,7 ммоль) в дихлорметане (500 мл). Добавляют 8% раствор гидроксида натрия (600 ммоль). Добавляют йодметан (23,2 г, 163,4 ммоль) и перемешивают при комнатной температуре в течение 22 ч. Отделяют органическую фазу и промывают водой (250 мл). Затем органическую фазу концентрируют и перекристаллизуют из дихлорметана, потом высушивают при температуре ниже 65 С для получения обозначенного в названии соединения (42,7 г, 82%). 1 Н ЯМР (CDCl3, 400 МГц)8,70 (s, 1H), 8,50 (d, 1H, J = 2,4 Гц), 7,97 (d, 1H, J = 9,2 Гц), 7,65 (d, 1H, J = 9,2, 2 Гц), 4,32 (m, 2 Н), 3,82 (m, 1 Н), 3,79 (s, 3 Н), 3,28 (s, 3 Н), 1,32 (d, 3 Н, J = 6,4 Гц). Пример получения 9. Этил 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-карбоксилат(2,44 г, 8,7 ммоль) и ацетат калия (42,65 г, 435 ммоль) в раствор бис(пинаколат)диборона (35,88 г, 141,3 ммоль) в диметилформамиде (175 мл). Продувают азот через смесь в течение 15 мин и затем нагревают до 80-85 С. Затем медленно добавляют в смесь этил 5-бромникотинат (25,0 г, 108,8 ммоль) в диметилформамиде (75 мл) при 80-85 С и перемешивают образовавшуюся смесь при 80-85 С в течение 4-5 ч. Охлаждают реакционную смесь до 15-35 С и затем добавляют метил-трет-бутиловый эфир (250 мл) и воду (250 мл). Фильтруют смесь через диатомит и разделяют органический и водный слои. Проводят обратную экстракцию водного слоя метил-трет-бутиловым эфиром (250 мл). Промывают объединенный органический слой солевым раствором (150 мл) и водой (150 мл). Концентрируют органические соединения в вакууме, проводят перекристаллизацию сырого продукта метил-трет-бутиловым эфиром/гептаном (1:6) и затем высушивают его при температуре ниже 55 С для получения обозначенного в названии соединения в виде серого твердого вещества (18,67 г, 62%). 1 Н ЯМР (ацетон-d6, 400 МГц)1,40-1,43 (m, 15 Н), 4,44 (q, J=7,1 Гц, 2 Н), 8,57 (t, J=1,9 Гц, 1H), 9,02 В трехгорлую колбу, содержащую 8-бром-1-[(2S)-2-метоксипропил]-3-метилимидазо[4,5 с]хинолин-2-он (35 г, 100 ммоль), этил 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3 карбоксилат (29,1 г, 105 ммоль), этилацетат натрия (28,7 г, 350 ммоль), 1,1'-бис(дифенилфосфин)ферроцен-палладия(II) дихлорид дихлорметан (0,817 г, 1,0 ммоль), добавляют 1,4-диоксан (200 мл) и воду (200 мл) в атмосфере N2. Затем нагревают реакционную смесь до 85 С и продолжают перемешивать в течение 10 ч. Охлаждают реакционную смесь до комнатной температуры. Фильтруют смесь через KieselguhrSilica-Thiol для удаления катализатора. Добавляют воду (400 мл) по капле и осаждают осадок. Фильтруют смесь и промывают твердый осадок водой (400 мл). Перемешивают твердый осадок в этилацетате (70 мл) при комнатной температуре в течение 1 ч; фильтруют, промывают этилацетатом (70 мл) и высушивают в вакууме при температуре ниже 65 С для получения обозначенного в названии соединения (33,6 г,80%). 1 Способ 1. Растворяли 8-бром-1-[(2S)-2-метоксипропил]-3-метилимидазол[4,5-с]хинолин-2-он (0,600 г, 1,7 ммоль) и 2-[5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3-пиридил]пропан-2-ол (0,9 г, 3,43 ммоль) в тетрагидрофуране (75 мл) и воде (7,5 мл) в закрытой пробирке. Продували через смесь азот. Добавляли фторид калия (400 мг, 6,89 ммоль), трис(дибензилиденацетон)дипалладий(0) (200 мг, 0,22 ммоль) и тритрет-бутилфосфония тетрафторборат (200 мг, 0,68 ммоль). Закрывали реакцию в азоте и нагревали при 65-70 С в течение ночи. Охлаждали смесь до комнатной температуры, фильтровали для удаления неорганического остатка. Концентрировали фильтрат и разбавляли его дихлорметаном (120 мл) и водой (30 мл). Отделяли органический слой и высушивали его над порошком сульфата магния. Концентрировали его в вакууме до коричневого масла. Очищали осадок с использованием хроматографии на колонке с силикагелем с элюирующим растворителем, состоящим на 30-65% из этилацетата в гексане, затем с 0-7% метанолом в дихлорметане. Концентрировали фракции, содержащие продукт, и одновременно выпаривали по очереди с диэтиловым эфиром (210 мл), ацетоном (10 мл), ацетоном и диэтиловым эфиром (по 10 мл). Высушивали твердый остаток для получения обозначенного в названии соединения в виде оранжевого порошка (0,30 г, 44%). MS (ESI) m/z (M+Н)+ 407,0. Способ 2. Продували азот через суспензию 2-(5-бром-3-пиридил)пропан-2-ола (100 г, 464 ммоль, 1,25 экв.),4,4,5,5-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолана (141,4 г, 557 ммоль, 1,5 экв.) и ацетата калия (127,5 г, 1,3 моль) в 1,4-диоксане (2,6 л) в течение 30 мин при комнатной температуре. Добавляли аддукт дихлор-бис-дифенилфосфин)ферроцетенил)палладий(II) дихлорметана(9 г, 11,14 ммоль) в атмосфере азота и нагревали смесь до 90 С. Перемешивали смесь в течение 3 ч. Охлаждали реакционную смесь до 80 С и затем добавляли 8-бром-1-[(2S)-2-метоксипропил]-3-метилимидазо[4,5-с]хинолин-2-он (130 г, 371 ммоль), раствор карбоната натрия (118 г, 1,1 моль) в воде (910 мл) и аддукт дихлор-бис-фенилфосфин)ферроценил)палладий(II) дихлорметана (9 г, 11,14 ммоль). Перемешивали смесь в течение 1,5 ч при той же температуре. Давали разделиться на фазы для получения органического слоя и охлаждали до 40 С перед концентрированием в вакууме. Очищали осадок (350 г) с помощью колоночной хроматографии на силикагеле с градиентом смеси элюирующего растворителя из дихлорметана/этилацетата/метанола от 1:1:1 до 1:1:20. Получали фракции, содержащие продукт. Концентрировали и суспендировали в этилацетате (10 л/кг) при 40 С в течение 15 мин, фильтровали и промывали осадок в этилацетате (21 л/кг) и метил-трет-бутиловом эфире (22 л/кг). Растворяли промытый осадок в метаноле (10 л/кг), обрабабывали SiliaBond Thiol (0,4 г/г) для удаления остатков металлов. Перемешивали суспензию при 23 С в течение 4 ч и фильтровали. Промывали осадок метанолом (1 л/кг). Смешивали все фильтраты и то, что осталось после промывки метанолом, и концентрировали в вакууме. Оставляли осадок в растворителе (приблизительно 100 мл). Материал кристаллизовался из растворителя. Фильтровали твердый материал и высушивали его при 1 мбар/40 С в течение ночи для получения обозначенного в названии соединения в виде белого порошка (77 г, 51%). MS (ESI) m/z (M+Н)+ 407,1. 1(dd, J = 8,3, 15,2 Гц, 1H), 4,37 (dd, J = 4,2, 15,2 Гц, 1H), 3,85-3,80 (m, 1H), 3,57 (s, 3H), 3,12 (s, 3H), 1,56 (d,J = 1,2 Гц, 6 Н), 1,26 (d, J = 6,1 Гц, 3H). Способ 3. В раствор этил 5-[1-[(2S)-2-метоксипропил]-3-метил-2-оксоимидазо[4,5-с]хинолин-8-ил]пиридин-3 карбоксилата (32,0 г, 76,1 ммоль) в тетрагидрофуране (320 мл) медленно добавляли раствор MeMrBr(1,4 М в тетрагидрофуране/толуоле, 221,4 г, 6,92 Х) под защитой N2 при температуре ниже 0 С. Перемешивали смесь при -5-0 С в течение 1 ч под защитой N2. Медленно добавляли раствор NH4Cl (15%, 320 мл) для остановки реакции при поддержании температуры ниже 25 С. Затем добавляли этилацетат (320 мл). Нагревали смесь до 25-30 С и перемешивали ее в течение получаса. После разделения на два слоя проводили обратную экстракцию водного слоя тетрагидрофураном (160 мл). Промывали объединенный органический слой солевым раствором (192 мл). Добавляли активированный уголь (1,6 г) в органический слой и перемешивали при 65-75 С в течение 4-5 ч. Фильтровали смесь с использованием KieselguhrSilica-Thiol (1,6 г). Перемешивали органический слой при 65-75 С в течение 2-3 ч; затем фильтровали смесь через Kieselguhr Silica-Thiol. Органический слой удаляли в вакууме и затем проводили перекристаллизацию этилацетат/тетрагидрофураном для получения 8-[5-(1-гидрокси-1-метилэтил)пиридин-3 ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2-она в виде светложелтого твердого вещества (22,34 г, 72,2%). В трехгорлую колбу добавляли 8-[5-(1-гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2 метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2-он, полученный согласно описанию в данном способе 3 (50,0 г, 123 ммоль), и тетрагидрофуран (1500 мл). Перемешивали смесь и нагревали ее до 45-55 С для получения абсолютного раствора. Фильтровали, затем концентрировали фильтрат в вакууме ниже 45 С до 2,0-3,0 V и добавляли этилацетат (500 мл), затем концентрировали в вакууме ниже 45 С до 7-8 V. Перемешивали смесь при 70-80 С в течение 6-10 ч, затем охлаждали ее до 20-25 С и фильтровали. Добавляли этилацетат (135-140 г) и этанол (13,5-14,0 г) к осадку. Перемешивали смесь при 70-80 С в течение 6-10 ч, затем охлаждали ее до 20-25 С и фильтровали. Концентрировали в вакууме для получения обозначенного в названии соединения в виде бледно-желтого твердого вещества (37,9 г,75,8%). 1H ЯМР (CDCl3, 400 МГц)8,86 (d, 1H, J = 2 Гц), 8,76 (d, 1H, J = 2,4 Гц), 8,71 (s, 1H), 8,64 (d, 1H, J = 1,6 Гц), 8,21 (t, 2 Н), 7,85 (d, 1 Н, J = 6,8 Гц), 4,36 (q, 2 Н), 3,88 (m, 1 Н), 3,62 (s, 3 Н), 3,23 (s, 3 Н), 2,79 (s, 1 Н),1,95 (s, 1 Н), 1,71 (d, 6 Н, J = 0,8 Гц), 1,33 (d, 3 Н, J = 6,4 Гц). Пример 2. 8-[5-(1-Гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3 дигидро-2 Н-имидазо[4,5-с]хинолин-2-он, форма I. Способ 1. Неочищенное свободное основание (пример 1) смешивали с этилацетатом, при этом из коричневатого раствора начинал выпадать белый осадок. Осадок фильтровали в герметизированном рабочем боксе с вмонтированными перчатками, размещенном внутри вытяжки, и оставляли сушиться в вакууме в герметизированном рабочем боксе в течение ночи. Продукт оставляли в вакууме в течение ночи (11 г, выход 63,18%). Порошковые рентгеновские дифрактограммы (XRD) кристаллического осадка получали с использованием порошкового дифрактометра Bruker D4 Endeavor, оснащенного источником излучения CuK ( = 1,54060 ) и детектором Vantec, работающим при 35 кВ и 50 мА. Образец исследовали в интервале 2 углов от 4 до 40 с шагом 0,0087 и экспозицией 0,5 с/шаг, с дивергенцией 0,6 мм, 5,28 мм фиксированным антирассеивающим растром и щелью детектора 9,5 мм. Сухой порошок упаковывали в кварцевый держатель образца и гладкую поверхность получали с использованием предметного стекла. Хорошо известно в области кристаллографии, что для любой кристаллической формы расположения угловых пиков могут слегка отличаться. Например, положения пиков могут смещаться из-за различий в температуре или влажности, при которых анализируют образец, смещения образца или от присутствия или отсутствия внутреннего стандарта. В данном случае вариабельность положений пиков 0,2 2-углов учитывает такие возможные вариации, не препятствуя однозначной идентификации указанной кристаллической формы. Определение кристаллической формы может быть сделано на основе любого уникального сочетания отличительных пиков (в единицах 2), обычно наиболее выдающихся пиков. Дифрактограммы кристаллической формы, полученные при температуре и влажности окружающей среды, корректируются с учетом стандартных пиков NIST 675 при 2 8,85 и 26,77. Таким образом, полученный образец соединения характеризуется рентгеновской порошковой дифрактограммой, полученной с использованием излучения CuK, имеющей пики дифракции (значения 2), приведенные ниже в табл. 1. В частности, дифрактограмма содержит характеристические пики при 8,57 и один или несколько из 9,06, 15,93, 18,29 и 18,87, с допустимой ошибкой углов дифракции 0,2. Таблица 1 Пики порошковой рентгеновской дифрактограммы, пример 2, способ 1 Способ 2. Растворяли приемлемое количество свободного основания согласно примеру 1 в этаноле или тетрагидрофуране для получения раствора. Выпаривали раствор для получения обозначенного в названии соединения. Таким образом, полученный образец соединения характеризуется рентгеновской порошковой дифрактограммой, полученной с использованием излучения CuK, имеющей пики дифракции (значения 2), приведенные ниже в табл. 2. В частности, дифрактограмма содержит характеристические пики при 8,60 в сочетании с одним или несколькими пиками, выбранными из группы, состоящей из 9,08, 15,93,18,25 и 18,83, с допустимой ошибкой углов дифракции 0,2. Таблица 2 Пики порошковой рентгеновской дифракции, пример 2, способ 2 Пример 3 может быть получен посредством перемешивания смеси метанолатной формы свободного основания (метанолат представляет собой кристаллическую форму, получаемую из раствора свободного основания в метаноле) вместе с безводной формой свободного основания (см. пример 2) в приемлемом количестве воды в течение 24 ч. В другом случае суспендировали безводную форму свободного основания (см. пример 2) в растворе ацетон/вода (соотношение 95:5, aw = 0,57), и затравливание моногидратной формой приводило к полному превращению безводной формы I в желаемый моногидрат в течение 24 ч. Условия получения порошковой рентгеновской дифрактограммы (XRD) в примере 3 точно такие же, как и условия, описанные в примере 2. Таким образом, полученный образец обозначенного в названии соединения характеризуется порошковой рентгеновской дифрактограммой, полученной с использованием излучения CuK, имеющей пики дифракции (величины 2), приведенные ниже в табл. 3. В частности, дифрактограмма содержит пик при 13,57 в сочетании с одним или несколькими пиками, выбранными из группы, состоящей из 6,75,9,71, 16,35, 16,98 и 19,54, с допустимой ошибкой углов дифракции 0,2. Таблица 3 Пики порошковой рентгеновской дифрактограммы, пример 3 Пример 4 можно получить посредством суспендирования свободного основания (53,5 мг) в ацетоне(2 мл) и последующего встраивания L-малеиновой кислоты (22 мг). Твердые вещества растворяли до прозрачного раствора. Белый кристаллический осадок осаждали из раствора. Осадок фильтровали в вакууме и высушивали. Высушивали соль малата в вакуумной печи (65 С) в течение ночи для получения обозначенного в названии соединения. Условия получения порошковой рентгеновской дифрактограммы (XRD) в примере 4 точно такие же, как и условия, описанные в примере 2. Таким образом, полученный образец обозначенного в названии соединения характеризуется порошковой рентгеновской дифрактограммой, полученной с использованием излучения CuK, имеющей пики дифракции (величины 2), приведенные ниже в табл. 4. В частности, дифрактограмма содержит пик при 5,39 в сочетании с одним или несколькими пиками, выбранными из группы, состоящей из 10,33,12,16, 15,57 и 0,08, с допустимой ошибкой углов дифракции 0,2. Таблица 4 Пики порошковой рентгеновской дифракции, пример 4 Пример 5 можно получить посредством добавления свободного основания 8-[5-(1-гидрокси-1 метилэтил)пиридин-3-ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2 она (60,2 мг) к 1-бутанолу (0,5 мл) и последующего добавления 21,9 мг фумаровой кислоты. Добавление гептана (50,5 мл) дает густую белую суспензию, которую затем перемешивали при 90 С/500 об/мин. Пропускали через фильтр в вакууме и высушивали в атмосфере азота. Осадок терялся при восстановлении из фильтрата, но этого было достаточно для XRD. Дополнительно кристаллическую соль фумарата получали при добавлении свободного основания (101,0 мг) в 1-бутаноле (0,5 мл) и затем добавляли 34 мг фумаровой кислоты. Добавляли гептан (60,5 мл) и кристаллы-затравки соли фумарата из первого примера получения и перемешивали смесь при 90 С/500 об/мин в течение 1 ч. Получали осадок с помощью вакуумной фильтрации и высушивали осадок в атмосфере азота. Затем высушивали осадок в вакуумной печи (65 С) в течение ночи для получения обозначенного в названии соединения. Условия получения порошковой рентгеновской дифрактограммы (XRD) в примере 5 точно такие же, как и условия, описанные в примере 2. Таким образом, полученный образец обозначенного в названии соединения характеризуется порошковой рентгеновской дифрактограммой, полученной с использованием излучения CuK, имеющей пики дифракции (величины 2), приведенные ниже в табл. 5. В частности, дифрактограмма содержит пик при 5,10 в сочетании с одним или несколькими пиками, выбранными из группы, состоящей из 8,55,15,45, 15,78 и 22,50 с допустимой ошибкой углов дифракции 0,2. Таблица 5 Пики порошковой рентгеновской дифракции, пример 5 Ферментный анализ mTOR (FRAP1) in vitro. Использовали анализ киназы mTOR LanthaScreen (Invitrogen) для определения значений IC50 соединения по отношению к киназе mTOR. Он представляет собой формат анализа флуоресцентный резонансный перенос энергии с временным разрешением (Time Resolved Fluorescence Resonance EnergyTransfer, TR-FRET), при котором используется антитело, меченное тербием с длительным периодом полураспада в качестве донора, и 4 Е-ВР 1, меченный зеленым флуоресцентным белком (GFP) в качестве акцептора. Использовали соотношение TR-FRET для наблюдения за активностью mTOR, при этом усиление фосфорилирования белка приводит к повышению соотношения TR-FRET. Проводили киназную реакцию с использованием объема реакции 12,5 мкл в неглубоких черных 384-луночных планшетахProxiplate. Добавляли реагент для получения конечных условий реакции: 50-миллимолярная N-2 гидроксиэтилпиперазин-N'-2-этансульфоновая кислота (HEPES) рН 7,5, 1-миллимолярная этиленгликоль-бис-(-аминоэтиловый эфир)-N,N,N',N'-тетрауксусная кислота (EGTA), 0,01% Tween 20, 10 мМ хлорид марганца, 2 мМ DL-дитиотреит (DTT), 0,4-микромолярный GFP-4E-BP1 (физиологический субстрат mTOR, 4E-BP1, экспрессированный и очищенный в виде гибридного белка с зеленым флуоресцентным белком, Invitrogen), 70 нг/мл mTOR (рекомбинантный человеческий mTOR, остатки 1360-2549,меченный глутатион-S-трансферазой (GST), эксперссированный в клетках насекомых, Invitrogen), 4% диметилсульфоксид и серийные разведения соединения (разведенные 1:3 от 20,000 до 1 нМ). Добавляли фермент и субстрат соединения и затем добавляли аденозинтрифосфат (АТФ) до 10 мкМ для начала реакции. Инкубировали при комнатной температуре в течение 60 мин и затем добавили 12,5 мкл буфера для разведения антител, содержащего 4 нМ антитела против фосфотреонин-46 4 Е-ВР 1, меченного тербием, и 20 мМ этилендиаминотетрауксусной кислоты (EDTA), 0,67 мМ трис(гидроксиметил)аминоэтан гидрохлорида (Trizma) pH 7,5, 0,02% азида натрия и 0,01% нонилфенилполиэтилен гликоля (NonidetP40). Инкубировали при RT в течение 60 мин и считывали результат с использованием ридера планшетов EnVision с фильтром возбуждения с длиной волны 340 нм и фильтром испускания с длиной волны 495 нм и 520 нм. Сигнал, полученный с помощью фильтра на 520 нм (специфичного к GFP), делили на сигнал, полученный с помощью фильтра 495 нм (специфичного для тербия), для вычисления соотношения TR-FRET. Получали значение IC50 для каждого соединения с использованием данных относительного ингибирования, которые рассчитывали исходя из данных реакции по сравнению с контролями на планшетах (соотношение TR-FRET данных анализа по сравнению с контролями АТФ на планшетах). Использовали ACTIVITYBASE 4,0 для соответствия данных относительного ингибирования и данных десятибалльных концентраций соединений четырехпараметрическому логистическому уравнению. Соединение согласно настоящему изобретению исследуют с помощью данного анализа, проводимого согласно описанию выше. Например, соединение с примером 1 проанализировали и оказалось, что его абсолютное значение IC50 равно 0,165 мкМ (0,0925, n=5). Полученные результаты показывают, что соединения в рамках настоящего изобретения, являются сильными ингибиторами mTOR. Ферментный анализ фосфоинозитид-3-киназы(PI3K) in vitro. Использовали сцинтилляционный анализ сближения PI3K (PI3K SPA) для определения значенийIC50 соединений в отношении PI3K киназы. В этом анализе измеряется активность PI3K в присутствии ингибиторов соединений посредством измерения встраивания -Р 33-АТФ в фосфотидилинозитол (4,5) бисфосфат (PIP2) Проводили киназные реакции в объеме 40 мкл в 96-луночных белых плоскодонных полистирольных планшетах с половинным объемом лунок с прозрачным дном. Добавляли PI3K для начала реакции. Конечные условия реакции составляют 43,75 мМ 2,2-бис(гидроксиметил)-2,2',2"нитрилотриэтанола (бис-трис) рН 7,0, 306 мМ хлорида натрия (NaCl), 1,76 мМ октилфенилового эфира полиэтиленгликоля (Triton Х-100), 10 мкМ аденозинтрифосфата (АТФ), 2,9 мМ хлорида магния(MgCl2) и 1 Ci на лунку -Р 33-аденозинтрифосфата (-P33-АТР), 5,0 нМ рекомбинантного человеческого фермента PI3K, 0,2 мМ пальмитил-олеил-фосфатидилсерина (POPS), 0,04 мМ фосфотидилинозитол(4,5) бисфосфата (PIP2), 4% DMSO и серийные разведения соединения (разведенное 1:3 от 20,000 до 1 нМ). Инкубировали при RT в течение 90 мин после добавления PI3K. Реакцию останавливали добавлением 40 мкл остановочного буфера, содержащего 2,5 мг/мл бусин, связанных с неомицином (Amersham,Cat RPNQ0506) и 21 мМ этилендиаминтетрауксусной кислоты (EDTA). Центрифугировали планшеты в течение 30 мин при 1000 об/мин и измеряли радиоактивность с использованием Wallac Microbeta Trilux,нормализованного по Р 33. Получали значение IC50 для примера 1 посредством использования данных относительного ингибирования, которые рассчитывали исходя из данных реакции по сравнению с контролями на планшетах (активный фермент по сравнению с контролем фермента, ингибированным 62,5 ммоль ЭДТА). С использованием ACTIVITYBASE 4,0 проводили соответствие данных относительного ингибирования и данных десятибалльных концентраций соединений четырехпараметрическому логистическому уравнению. Соединение согласно настоящему изобретению исследовали с помощью данного анализа, проведенного в точности согласно описанию выше. Например, проанализировали соединение согласно примеру 1, и оказалось, что его абсолютное значение IC50 составляет 0,00607 мкМ (0,00338, n=2). Полученные результаты показывают, что соединения в рамках настоящего изобретения являются сильными ингибиторами PI3K. Ферментный анализ фосфоинозитид-3-киназы(PI3K) in vitro. Использовали сцинтилляционный анализ сближения PI3K (PI3K SPA) для определения значенийIC50 соединений в отношении PI3K киназы. В этом анализе измеряется активность PI3K в присутствии ингибиторов соединения посредством измерения встраивания -P33-АТФ в фосфотидилинозитол (4,5) бисфосфат (PIP2). Проводили киназные реакции в объеме 40 мкл в 96-луночных белых плоскодонных полистирольных планшетах с половинным объемом лунок с прозрачным дном. Добавляли PI3K для начала реакции. Конечные условия реакции составляли 43,75 мМ 2,2-бис(гидроксиметил)-2,2',2"нитрилотриэтанола (бис-трис) рН 7,0, 87,5 мМ хлорида натрия (NaCl), 1,76 мМ октилфенилового эфира полиэтиленгликоля (Triton Х-100), 40 мкМ аденозинтрифосфата (АТФ), 1 мМ хлорида магния (MgCl2) и 1 Ci на лунку -P33-аденозинтрифосфата (-P33-АТР), 6,0 нМ рекомбинантного человеческого фермента PI3K, 0,2 мМ пальмитил-олеил-фосфатидилсерина (POPS), 0,04 мМ фосфотидилинозитола (4,5) бисфосфата (PIP2), 4% DMSO и серийные разведения соединения (разведенное 1:3 от 20,000 до 1 нМ). Инкубировали при RT в течение 90 мин после добавления PI3K. Реакцию останавливали добавлением 40 мкл остановочного буфера, содержащего 2,5 мг/мл бусин, связанных с неомицином (Amersham, CatRPNQ0506) и 21 мМ этилендиаминтетрауксусной кислоты (EDTA). Центрифугировали планшеты в течение 30 мин при 1000 об/мин и измеряли радиоактивность с использованием Wallac Microbeta Trilux,нормализованного по Р 33. Получали значение IC50 для соединения посредством использования данных относительного ингибирования, которые рассчитывали исходя из данных реакции по сравнению с контролями на планшетах (активный фермент по сравнению с контролем фермента, ингибированным 62,5 ммоль ЭДТА). С использованием ACTIVITYBASE 4,0 проводили соответствие данных относительного ингибирования и данных десятибалльных концентраций соединений четырехпараметрическому логистическому уравнению. Соединение согласно настоящему изобретению исследовали с помощью данного анализа, проведенного в точности согласно описанию выше. Например, проанализировали соединение согласно примеру 1, и оказалось, что его абсолютное значение IC50 составляет 0,0776 мкМ (0,0401, n=2). Полученные результаты показывают, что соединения в рамках настоящего изобретения являются сильными ингибиторами PI3K. Ферментные анализы in vitro фосфоинозитид-3-киназы(PI3K) и фосфоинозитид-3-киназы(PI3K). Использовали анализ киназы Adapta для иммунологического определения АДФ на основе флуоресценции. Он представляет собой формат анализа флуоресцентный резонансный перенос энергии с временным разрешением (TR-FRET), в котором используется антитело к АДФ, меченное европием, и антитело к метке АДФ, меченное Alexa Fluor 647 (AF647), для наблюдения за выработкой АДФ кина- 13022163 зой. Использовали соотношение TR-FRET для наблюдения за активностью PI3K или PI3K, при этом увеличение фосфорилирования липидов и соответствующая повышенная продукция АДФ приводит к увеличению TR-FRET. Ферментативные реакции: проводили киназую реакцию для PI3K с использованием объема реакции 10 мкл в белых 384-луночных планшетах Corning для маленьких объемов (Corning3674). Добавляли реагенты для получения следующих конечных условий реакции: 0,47-2,6 нг PI3K (рекомбинантная полноразмерная человеческая PI3K, экспресированная и выделенная из клеток насекомых, Invitrogen) и 50-микромолярный PIP2: PS в 32,5-миллимолярном HEPES, рН 7,5, 50-миллимолярный хлорид натрия, 0,015% CHAPS, 1,5-миллимолярный хлорид магния, 0,5-миллимолярная EGTA, 25 миллимолярный АТФ, 1% DMSO и серийные разведения соединения (разведенного 1:3 от 20,000 до 1 наномолярного). Добавляли АТФ к соединению, затем добавляли смесь субстрата/киназы для начала реакции. Встряхивали планшеты в течение 30 с и затем инкубировали при комнатной температуре в течение 60 мин. Проводили киназную реакцию на PI3K с использованием 10 мкл объема реакции в белом 384-луночном планшете Corning для маленьких объемов (Corning3674). Добавляли реагенты для получения конечных условий реакции: 3,5-26 нг PI3K (рекомбинантная полноразмерная человеческаяPI3K, экспрессированная и выделенная из клеток насекомых, Invitrogen) и 50-микромолярный PIP2: PS в 32,5-миллимолярном HEPES, рН 7,5, 0,5-миллимолярном EGTA ,1,5-миллимолярный хлорид магния, 25 микромолярный АТФ, 1% DMSO и серийно разведенное соединение (разбавленное 1:3 от 20,000 до 1 наномолярного). Добавляли к соединению АТФ и затем добавляли смесь субстрата/киназы для начала реакции. Встряхивали планшеты в течение 30 с, центрифугировали в течение 2 мин при 1000g и затем инкубировали при комнатной температуре в течение 60 мин. Определение АДФ: добавляли 5 мкл смесь для детекции (30 мМ EDTA, 30 нМ антитела к АДФ, меченного европием, ЕС 60 концентрация метки АДФ для реакции с 5-100 мкМ АТФ, Invitrogen) в ферментные реакции PI3Kи . Встряхивали планшеты в течение 30 с, центрифугировали в течение 2 мин при 1000g и затем инкубировали при комнатной температуре в течение 60 мин. Считывали планшеты на планшетном ридере флуоресценции с использованием фильтра возбуждения на длину волны 340 нм и фильтров испускания на длину волны 665 нм и 615 нм. Сигнал, полученный с использованием фильтра на 665 нм (специфичный для эмиссии AF647 полиGT), делили на сигнал, полученный с использованием фильтра на 615 нм (специфичный для европия), для вычисления соотношения TR-FRET. Использовали соотношение TR-FRET для вычисления концентрации АДФ посредством обратного вычисления стандартной кривой АТФ/АДФ, которая соответствует сигмоидальной модели доза-отклик номер 205 (XLfit от IDBS). Получали значения IC50 для каждого соединения с использованием данных относительного ингибирования, которые вычисляли из данных реакции по сравнению с контролями на планшетах (концентрация АДФ экспериментальных точек по сравнению с контролями АДФ на планшетах). Использовали XLfit (IDBS) для соответствия данных относительного ингибирования и данных десятибалльных концентраций соединений сигмоидальной модели доза-отклик номер 205 (XLfit от IDBS). Соединение согласно настоящему изобретению исследовали в указанных анализах в точности согласно описанию выше. Например, проанализировали соединение согласно примеру 1, и оказалось, что его абсолютная величина IC50 составляет 0,0380 мкМ для PDK и абсолютная величина IC50 составляет 0,0238 мкМ для PI3K. Полученные результаты показывают, что соединения согласно настоящему изобретению являются сильными ингибиторами PI3K и PI3K. Ферментный анализ ДНК-зависимой протеинкиназы (DNA-PK) in vitro. Использовали формат Z'LYTE киназного анализа (Invitrogen) для определения значений IC50 против ДНК-ПК для соединения. Он представляет собой флуоресцентный сопряжнный ферментный формат анализа, основанный на чувствительности к протеолизу фосфорилированного дважды меченного субстрата пептида по сравнению с нефосфорилированным (Coumerin на аминоконце, флуоресцеин на карбоксиконце). Используют соотношение резонансного переноса энергии флуоресценции (FRET) для наблюдения за активностью ДНК-ПК, при этом фосфорилирование пептида защищает пептид от протеолитического расщепления и FRET субстрата сохраняется. Проводили реакцию с использованием объема реакции 10 мкл в 384-луночных черных планшетах Corning для малых объемов NBS (Corning 3676). Добавляли реагенты для получения конечных условий реакции: 50-миллимолярная N-2 гидроксиэтилпипепразин-N'-2-этансульфоновая кислота (HEPES) рН 7,5, 0,01% BRIJ-35 неионный сурфактант, 10-миллимолярный хлорид магния, 1-миллимолярная этиленгликоль-бис-(-аминоэтиловый эфир)-N,N,N',N'-тетрауксусная кислота (EGTA), 1 мМ DL-дитиотритол (DTT), 2,5 мкл/мл ДНК из тимуса теленка (СТ DNA), 3,88-27,3 нг ДНК-ПК, 2-микромолярного меченного Ser/Thr 26 пептида (Invitrogen),1% диметилсульфоксил и серийные разведения соединения (разведенного 1:3 от 20,000 до 1 нМ). Добавляли фермент и субстрат к соединению и затем добавляли 25,0-микромолярный аденозинтрифосфат(АТФ) для начала реакции. Встряхивали планшеты в течение 30 с и затем инкубировали при комнатной температуре в течение 60 мин. Добавляли 5 мкл разведения 1:16 раствора В Development Reagent (Invitrogen). Встряхивали планшеты в течение 30 с и затем инкубировали при комнатной температуре в тече- 14022163 ние 60 мин. Считывали планшеты на планшетном ридере флуоресценции с использованием фильтра возбуждения на длину волны 400 нм и фильтров испускания на длину волны 445 нм и 520 нм. Сигнал, полученный с использованием фильтра на 445 нм (специфичный для эмиссии Кумарина), делили на сигнал,полученный с использованием фильтра на 520 нм (специфичный для флуоресцеина), для вычисления соотношения TR-FRET. Получали значения IC50 для соединения с использованием данных относительного ингибирования, которые вычисляли из данных реакции по сравнению с контролями на планшетах(контроль DMSO для 0% ингибирования и отсутствие реакции АТФ для 100% ингибирования). Использовали XLfit (IDBS) для соответствия данных относительного ингибирования и данных десятибалльных концентраций соединений сигмоидальной модели доза-отклик (модель номер 205). Соединение согласно настоящему изобретению исследовали с помощью данного анализа, проведенного в точности согласно описанию выше. Например, проанализировали соединение согласно примеру 1, и оказалось, что его абсолютное значение IC50 составляет 0,00424 мкМ. Полученные результаты показывают, что соединения согласно настоящему изобретению являются сильными ингибиторами ДНКПК. Детекция AlphaScreen SureFire фосфорилированных киназ p70S6 (Thr389), AKT (Thr308) и AKT(Ser473) в клетках U87MG. Использовали AlphaScreen SureFire для киназы p-p70S6 (Thr389) (TGR Biosciences, TGRAS50K),p-AKT(Thr308) (TGR Biosciences, TGRA2S50K) и р-AKT(Ser473) (TGR Biosciences, TGRAS50K) для определения влияния примера 1 на образование эндогенной фосфорилированных киназ p70S6 (Thr389),AKT(Thr308) и AKT(Ser473) соответственно. В данном гомогенном анализе используется захватывание фосфорилированного аналита иммуно-сэндвичем и последующая детекция с помощью бусин Alphascreen с целью сгенерировать усиленный сигнал. Культивировали клетки U87MG в питательной среде U87MG, состоящей из DMEM (GIBCO 11965092) с добавлением 10% фетальной бычьей сыворотки (FBS, GIBCO, 10091-141), 1% заменимых аминокислот (GIBCO, 11140-050) и 1% пирувата натрия (GIBCO, 11360-070). Собирали клетки с использованием стандартных процедур культивирования и затем посчитали с использованием Vi-Cell. Посеяли 100 мкл клеток U87MG в питательную среду (50,000 клеток/лунка) в 96-луночные планшеты Costar 3596 и инкубировали в течение ночи при 37 С, 5% СО 2. В день анализа обрабатывали клетки примером 1 (20 мкл/лунка), разбавленным в среде, содержащей 6% DMSO. Инкубировали в течение часа при 37 С, затем удаляли среду и добавляли 50 мкл 1 лизирующего буфера SureFire (TGR Biosciences, компонент набора SureFire) в каждую лунку и инкубировали при комнатной температуре в течение 10 мин, осторожно встряхивая. Переносили 6 мкл лизата и 10 мкл реакционной смеси (60 частей реакционного буфера/10 частей активирующего буфера/1 часть донорских и 1 часть акцепторных бусин, Perkin Elmer, 6760617R) в 384-луночный проксипланшет (PerkinElmer, 6006280) для анализов киназы p-p70S6 kinase (Thr389) и p-AKT(Ser473). Закрывали планшет и инкубировали при RT в течение 4 ч. Переносили 4 мкл лизата и 5 мкл реакционной смеси (40 частей реакционного буфера/10 частей активирующего буфера/1 часть акцепторных бусин) в 384-луночный проксипланшет для анализа р-AKT (Thr308). Инкубировали при RT в течение 2 ч и затем добавляли 2 мкл смеси для разведения (20 частей буфера для разведения/1 часть донорских бусин) в каждую лунку. Закрывали планшет и инкубировали при RT еще 2 ч. Считывали планшеты с помощью Perkin Elmer EnVision, оснащенного TurboModule, с использованием стандартных настроек AlphaScreen (Ех 680 нм и Em520620 нм). Получали данные относительного ингибирования из данных реакции по сравнению с контролем на планшетах. Затем использовали ACTIVITYBASE 4,0 соответствия данных относительного ингибирования от данных десятибалльных концентраций соединения четырехпараметрическому логистическому уравнению для получения значения IC50 для примера 1. Соединение согласно настоящему изобретению исследовали с помощью данного анализа, проведенного в точности согласно описанию выше. Например, проанализировали соединение согласно примеру 1, и оказалось, что его абсолютные значения IC50 равны представленным в табл. 6. Полученные результаты показали, что соединения в рамках настоящего изобретения подавляют ферменты путей PI3K и Анализ пролиферации клеток. Использовали систему люминисцентного анализа жизнеспособности клеток CellTiter-Glo (коммер- 15022163 чески доступна у Promega) для измерения антипролиферативной активности примера 1 посредством определения количества жизнеспособных клеток в культуре на основе количественного определения присутствующего АТФ, который свидетельствует о наличии метаболически активных клеток. Посеяли клетки в 96-луночный планшет с плотностью 2000 клеток/лунка в 100 мкл специализированной для клеток среды (для U87MG использовали DMEM, 10% FBS, 25 мМ 4-(2-гидроксиэтил)-1 пиперазинэтансульфоновой кислоты (HEPES), 1,0 мМ пирувата натрия и 0,1 мМ заменимых аминокислот (АТСС Cat.30-2002); для НТ 1080 использовали среду MEM в модификации Игла, 10% FBS (ATCCCat.30-2003); для Н 1975, А 2780, SJSA-1 и 786-O использовали RPMI 1640, 10% FBS (АТСС Cat.302001); для А 204 использовали МакКоя 5 А, 10% FBS (ATCC Cat. 30-2007), в первую колонку добавляли только среду в качестве отрицательного контроля. Инкубировали планшеты в течение ночи при 37 С и 5% СО 2. На следующий день готовили стоковые растворы соединений в 1 мМ в DMSO и серийно разводили в DMSO в 96-луночном полипропиленовом планшете с круглым дном. Анализировали соединения в 10 концентрациях в двух повторностях, 4 соединения на планшет. Переносили 4 мкл серийных разведений в DMSO в 96-луночный планшет и добавляли 196 мкл питательной среды для получения 10 X стокового раствора для дозирования. Аккуратно переносили 11 мкл каждого стокового раствора для дозирования в соответствующую лунку планшета с клетками, получая концентрацию 0,2% в DMSO и конечный объем 111 мкл. Добавляли 11 мкл среды в контрольные колонки (колонка 12) и фоновой колонки (колонка 1). Инкубировали клетки с соединениями при 37 С, 5% СО 2 в течение 72 или 96 ч (для H1975, 786-O, НТ 1080, А 2780, А 204 и SJSA-1 использовали 72 ч и для U87MG использовали 96 ч). Готовили реагент CellTiter-Glo (Promega, Cat: G7571) и добавляли 100 л в каждую лунку после окончания инкубации, гомогенизировали клетки с помощью перемешивания на орбитальном шейкере в течение 2 мин и затем инкубировали при RT в течение 10 мин для стабилизации люминисцентного сигнала. Считывали необработанные данные люминисценции на ридере планшетов Wallac Victor V. Рассчитывали значения IC50 для примера 1 с использованием данных о проценте ингибирования. Четырехпараметрическую логистическую кривую подстраивали для ответа каждой дозы. Соединение согласно настоящему изобретению исследовали с помощью данного анализа, проведенного в точности согласно описанию выше. Например, исследовали соединение согласно примеру 1, и оказалось, что его абсолютные значения IC50 равны представленным в табл. 7. Полученные результаты показывают, что соединения согласно настоящему изобретению полезны для ингибирования пролиферации клеточных линий U87MG, H1975, 786-O, А 2780, НТ-1080, A204 и SJSA-1. Таблица 7 Клоногенный анализ опухоли Oncotest. Использовали коллекцию опухолевых ксенографтов человека, выращенных подкожно в иммунокомпромитированных голых мышах, Oncotest (GmbH of Freiburg, Germany) для измерения ответа примера 1 на различные типы опухолей. Ксенографты, непосредственно трансплантированные от пациентов и выращенные в голых мышах,сохраняли большую часть характеристик родительских опухолей пациента, включая гистологию и чувствительность к противораковым лекарственным средствам, что повторяло ответ пациента донора на стандартные противораковые средства в большой степени. Получали клетки опухоли непосредственно из человеческих донорских ксенографтов, выращенных в голых мышах. Измеряли ингибирование образования неприкрепленных колоний опухолевых клеток в мягком агаре. Тестировали пример 1 на моделях человеческих опухолевых ксенографтов, полученных от пациентов, представленных в табл. 10, которые включают 2-10 моделей 13 различных гистотипов опухолей человека, а именно рак мочевого пузыря, толстой кишки, желудка, головы и шеи, немелкоклеточный рак легкого (адено, плоскоклеточный рак и крупноклеточный), рак молочной железы, рак яичников, поджелудочной железы, предстательной железы и рак почек, а также меланома, плевромезотелиома и саркома,где md означает умеренно дифференцированная, pd - слабо дифференцированная, ud - недифференцированная и wd - хорошо дифференцированная. Приготовление суспензий отдельных клеток из ксенографтов опухоли человека. Выращивали ксенографты солидных опухолей человека подкожно посредством серии пассажей в бестимусных голых мышах (ЯМР I штамм nu/nu) и в стерильных условиях удаляли опухоли, механически дезагрегировали и затем инкубировали с коктейлем ферментов, состоящим из коллагеназы IV типа(41 U/мл), ДНазы I (125 U/мл), гиалуронидазы (100 U/мл) и диспазы II (1,0 U/мл) в среде RPMI 1640 при 37 С в течение 45 мин. Пропускали клетки через фильтр с размером пор 200 мкм и 50 мкм и дважды промывали стерильным буфером PBS. Определяли процент жизнеспособных клеток в камере Нойбауэра с использованием исключения трипанового синего. Процедура клоногенного анализа с использованием клеток из ксенографтов опухолей человека. Проводили клоногенный анализ в формате 24-луночного планшета согласно модифицированному анализу на двухслойном агаре (Hamburger et al., Science 197:461-643, 1997). Нижний слой состоял из 0,2 мл/лунка IMDM (с добавлением 20% (v/v) фетальной бычьей сыворотки, 0,01% (w/v) гентамицина) и 0,75% (w/v) агара. Добавляли 0,8104 - 5 104 клеток в 0,2 мл такой же питательной среды с добавлением 0,4% (w/v) агара и сеяли в нижний слой в 24-луночные планшеты. Наносили исследуемое соединение посредством постоянного предъявления (наложение лекарственного средства) в 0,2 мл питательной среды. Добавляли наложение лекарственного средства 24 ч после посева клеток в виде трехкратного концентрированного раствора. Включали шесть необработанных контрольных лунок и шесть обработанных концентрацией лекарственного средства лунок в каждом планшете. Инкубировали культуры при 37 С и 7,5% СО 2 в увлажненной атмосфере в течение 20 дней и внимательно наблюдали за образованием колоний с использованием инвертированного микроскопа. В течение этого времени in vitro рост опухоли приводил к образованию колоний с диаметром 50 мкм. В момент наибольшего роста колоний считали колонии с использованием автоматической системы анализа изображений (OMNICON 3600, BiosysGmbH). Окрашивали жизнеспособные колонии за 24 ч перед оценкой с использованием стерильного водного раствора 2-(4-йодфенил)-3-(4-нитрофенил)-5-фенилтетразолия хлорида (1 мг/мл, 100 мкл/лунка). Выражали действие лекарства в виде процента образования колоний. Сравнивали среднее число колоний в обработанных лунках со средним числом колоний в необработанном контроле (выражали относительное количество колоний в виде значения исследуемая группа против контроля, Т/С-значение [%]): Строили зависимость концентрации соединения от относительного числа колоний и определяли абсолютные значения IC50 и IC70 или концентрации лекарственного средства, необходимые для ингибирования образования колоний на 50% (Т/С = 50%) и 70% (Т/С = 30%) соответственно, посредством подбора кривой по двум точкам. Соединение согласно настоящему изобретению исследовали с помощью данного анализа, проведенного в точности согласно описанию выше. Например, исследовали соединение согласно примеру 1, и оказалось, что его абсолютные величины IC50 равны указанным в табл. 8. Данные результаты показывают, что соединения согласно настоящему изобретению полезны для ингибирования пролиферации данных клеточных линий, полученных от пациентов. Модель лейкемии Е 545Kp110. Получение клеточной линии с лейкемией: проводили трансдукцию клеток эмбриональной печени,полученных из трансгенных эмбрионов, B6.Cg-Tg[IghMyc]22Bri/J (Jackson Laboratory, Bar Harbor, ME),ретровирусом, экспрессировавшим активирующую мутацию, выявленную клинически, p110 человека(Е 545K на уровне замены аминокислоты, G1633A на уровне нуклеотида) под контролем вирусного 5'LTR (длинный терминальный повтор) и экспрессирующий GFP (зеленый флуоресцентный белок) под контролем промотора PGK (MSCV6 FLAG-p110 G1633A PGK/GFP) для получения клеток лейкемии,ориентированных на мишень. Переносили трансдуцированные клетки в летально облученного животного-хозяина. Трансдуцированные клети заменяли популяцию гемопоэтических стволовых клеток в костном мозге реципиента и спасали животного-реципиента от смерти от радиации вследствие абляции собственного костного мозга реципиента. Наблюдали за развитием лейкемии спасенных первичных животных посредством еженедельного отслеживания числа лейкоцитов в небольшом объеме крови (10 мкл),собранной ретроорбитально. Собирали кровь от первично облученных животных с подтвержденной лейкемией и серийно пассировали вторичным (не облученным) животным-хозяевам для получения лейкемической клеточной линии. Испытуемые животные. Использовали самок мышей C57BL/6 (Taconic, Cambridge City, IN), в возрасте 8-10 недель и весом 20-22 г в качестве животных-реципиентов лейкемии. Акклиматизировали животных на нормальной диете с пониженным содержанием жиров (4,5%) перед инокуляцией и содержали на такой диете ad libitum на протяжении всего исследования. Обозначали отдельных мышей в каждой группе с помощью проколов ушей. Инокулировали животных лейкемическими клетками от донорских животных (день 0). Сингенная модель лейкемии. От животного-донора, ранее инокулированного соответствующей лейкозной клеточной линией, собирали небольшое количество крови ретро-орбитально (10 мкл) и измеряли нагрузку лейкемическими клетками с помощью подсчета лейкоцитов в крови. От животных с дос- 19022163 таточной лейкозной нагрузкой собирали донорскую кровь, разбавляли фосфатно-солевым буфером (PBS) до 500000 лейкоцитов в 200 мкл и вводили 200 мкл животному ретро-орбитально в день 0 для инициирования лейкемии. Распределяли мышей, инокулированных клетками p110 (Е 545K)/Мус, по группам из пяти для обработки примером 1 и контрольную группу из десяти для введения носителя. С 5 по 11 день после инокуляции вводили каждой группе ежедневно с помощью желудочного зонда только растворитель; пример 1 при 5, 10, 20 мг тестируемого QD на 1 кг массы тела (мг/кг). Собирали не менее 10 мкл крови ретро-орбитально от животных на 12 день для оценки прогрессирования лейкемии с помощью анализа лейкемических клеток. Анализ лейкемических клеток. Собирали десять (10) мкл цельной крови от каждого исследованного животного и обрабатывали на Coulter TQ-Prep так, что красные кровяные клетки лизировали и фиксировали оставшиеся ядерные лейкоциты для анализа. Анализировали фиксированные клетки непосредственно или хранили их в темноте при 4 С для последующего анализа. Анализировали клетки методом флуоресцентно-активированный клеточный сортинг (FACS) с использованием Cytomics FC 500 (Beckman Coulter). Подсчитывали лейкемические клетки в определенном регионе диаграммы прямого/бокового светорассеяния (FS/SS) в каждом образце (определен как регион, показывающий мало/отсутствие лейкемических клеток у нормальных животных, но достаточно лейкемических клеток у контрольных животных с лейкемией). Нормализовали данные в лейкемических клетках в единице объема крови с использованием фиксированного отсечения флуоросфер Beckman Coulter Flow-Count в образце (одинаковое количество флуоросфер изначально добавляли в каждый образец крови, при этом равные количества в образце были равны равному объему, подсчитанному в образце). Исследуемое вещество. Еженедельно смешивали пример 1 с 1% гидроксиэтилцеллюлозой(ГЭЦ)/0,25% полисорбатом 80/0,05% понегасителем/очищенной водой и обрабатывали ультразвуком с использованием probe sonicator для суспендирования. Помещали полученное исследуемое вещество при 4 С и хранили в темноте до применения (ресуспендировали перед каждым введением). Статистический анализ. Сводили в таблицу данные проточной цитометрии с использованием программного обеспечения Beckman Coulter's CXP. Определили статистическую значимость эффектов в примере 1 с помощью критерия Даннета, однопараметрического ANOVA с использованием группы носителя в качестве контроля (JMP Statistical Discovery Software, SAS Institute). Соединение согласно настоящему изобретению исследовали с помощью данного анализа, проведенного в точности согласно описанию выше. Например, проанализировали соединение согласно примеру 1, и оказалось, что его значения %TGI равны представленным в табл. 9. Полученные результаты показывают, что соединения согласно настоящему изобретению ингибируют рост опухолей, чей рост управляется Е 545K PI3K, одной из распространенных мутаций, обнаруживаемых при многих видах рака у человека. Таблица 9 Результаты E545Kp110 на модели лейкемии для примера 1%TGI представляет собой % ингибирования роста опухоли по сравнению с контрольной необработанной группой,%TGI SEM представляет собой стандартную ошибку среднего %TGI. Модели опухолевых ксенографтов. Культивировали клетки глиобластомы человека U87MG и клетки карциномы почек человека 786-O,собирали и подкожно вводили в заднюю часть тела атимических голых мышей. Культивировали клетки немелкоклеточного рака легкого человека NCI-H1975, собирали и подкожно вводили в заднюю часть тела мышей CD-1 nu/nu. Готовили исследуемые соединения в приемлемом носителе и вводили с помощью зонда, когда образовывались опухоли (7-21 дней после имплантации). Ответ опухоли определяли посредством измерения объемов опухоли, проводимого дважды в неделю во время лечения. Массу тела брали в качестве общего параметра токсичности. Соединение согласно настоящему изобретению исследовали с помощью данного анализа, проведенного в точности согласно описанию выше. Например, проанализировали соединение согласно примеру 1, и оказалось, что его значения %TGI равны представленным в табл. 10. Полученные результаты показывают, что соединения согласно настоящему изобретению являются полезными для проявления дозозависимой противоопухолевой активности на модели U87MG, 786-O и NCI-Н 1975.%TGI представляет собой % ингибирования роста опухоли по сравнению с контрольной необработанной группой,%TGI SEM представляет собой стандартную ошибку среднего %TGI, и подчеркнутые величины указывают значимость. Определение ингибирования мишени PI3K и mTOR in vivo. Имплантировали клетки глиобластомы человека U87MG (5106) подкожно в заднюю часть атомических голых мышей в 0,2 мл матригеля. Через 10 дней после имплантации вводили дозу мышам перорально по протоколу определения зависимости от времени, однократной дозе/отдельному моменту времени или дозозависимому протоколу для определения TMED50 (пороговая минимальная эффективная доза). Мгновенно замораживали ткани при сборе и собирали кровь для определения содержания родительского соединения в плазме и расчета TMEC50 (пороговая минимальная эффективная концентрация) в случае исследований влияния дозы на эффективность. Гомогенизировали опухоли в 500 мкл XY лизирующего буфера (10 мкг/мл лейпептина, 10 мкг/мл ингибитора трипсина-химотрипсина, 10 мкг/мл тозилфенил-аланил-хлорметил кетона, 10 мкг/мл Апротинина, 60 мМ -Глицеролфосфата, 1% Тритон Х 100,25 мМ Трис рН 7,5, 2,5 мМ пирофосфата, 150 мМ NaCl, 2 мМ р-тозил-L-аргининметил эфир, 15 мМ паранитрофенил фосфат, 5 мМ бензамидин, 1 мМ ваданат натрия, 10 мМ фторид натрия, 50 мкг/мл фенилметан сульфонил фторид, 1 мМ 1,4-дитиотритол (DTT), 15 мМ EDTA рН 8,0, 5 мМ EGTA рН 8,0, 1 мкМ микроцистина, 1 мкМ окадаевой кислоты и 1 минитаблетка полного ингибитора протеазы Roche на 10 мл) с использованием пестика, не содержащего РНКаз (Kimble-Kontes). Лизаты аликвотировали и анализировали немедленно или хранили при -80 С для дальнейшего анализа. Использовали мультиплексный формат ИФА Meso Scale Discovery (Gaithersburg, MD) и измеряли in vivo ингибирование мишени PI3K иmTOR для оценки влияния на фосфорилирование треонина в положении 308 AKT, даунстрим эффектораmTORC2. Добавляли 20 мкг лизата к карбоновому электроду, содержащему 96-луночные планшеты,предварительно меченые соответствующими связывающими антителами. Гибридизовали интересующий белок с использованием детектирующего антитела, меченного рутением. Пропускали ток по электроду в присутствии буфера для считывания, содержащего сореагент ТРА, и количественно определяли и записывали свет, генерируемый электрохемилюминесценцией с использованием MSD Sector 6000. Рассчитывали процент ингибирования относительно группы сравнения, обработанной носителем, и проводили дисперсионный анализ с использованием JMP пакета программного обеспечения для определения статистической значимости. Соединение согласно настоящему изобретению исследовали с помощью данного анализа, проведенного в точности согласно описанию выше. Например, проанализировали соединение согласно примеру 1, и оказалось, что активность равна представленной в табл. 11, при этом подчеркнутые значения указывают значимость. Полученные результаты показали, что соединения согласно настоящему изобретению проявляют способность ингибировать PI3K и mTOR in vivo. Определение растворимости. Готовили раствор 2 мг/мл примера 2 в каждой требуемой среде посредством взвешивания приблизительно 1 мг соединения в пробирке и добавляли требуемый объем (т.е. 0,5 мл) соответствующей среды в каждую пробирку. Помещали закрытые пробирки на вращающую смесь в течение ночи (16 ч) при комнатной температуре и затем пропускали через фильтр с использованием фильтров 0,22 мкм UltrafreeMC (Millipore) и измеряли рН фильтрата (Orion 720A рН метр). Готовили образец для анализа ВЭЖХ посредством переноса 100 мкл фильтрата в пробирку ВЭЖХ и добавляли 900 мкл раствора 50% ацетонитрила в воде. Определяли растворимость с использованием метода ВЭЖХ (мобильная фаза ВЭЖХ из 15% ацетонитрила с 0,1% TFA и 85% воды с 0,1% TFA; колонка Bonus RP, 4,675 мМ, 3,5 см; детекторы на 264 нм UV; температура колонки 40 С; скорость пропускания 1,5 мл/мин; вводимый объем 1 мкл). рН 2 = 50 мМ фосфатного буфера при рН 2 рН 4 = 50 мМ фосфатного буфера при рН 4 рН 6 = 50 мМ фосфатного буфера при рН 6 рН 8 = 50 мМ фосфатного буфера при рН 8SGF = имитация желудочного сока (Aburub et al., Int. J.of Pharmaceutics, 347:16-22, 2008) Сытое = имитация желудочного сока в сытом состоянии(Dressman J et al., Pharma. Res., 15(1):11-21, 1998) Натощак = имитация желудочного сока натощакDressman J. et al., Pharma. Res., 15(1):11-21, 1998) Соединение согласно настоящему изобретению исследовали с помощью данного анализа, проведенного в точности согласно описанию выше. Например, проанализировали соединение согласно примеру 2, и оказалось, что результаты его растворимости равны представленным в табл. 12. Полученные результаты показывают, что пример 2 проявляет желаемую растворимость выше физиологического рН желудочно-кишечного тракта (ЖКТ). Это физико-химическое свойство поможет избежать изменчивости воздействия у онкологических больных, которые, скорее всего, будут принимать несколько препаратов,таких как ингибиторы протонной помпы (ИПП), что может привести к лекарственному взаимодействию с лекарственным средством, которое имеет переменную растворимость при рН ЖКТ выше физиологического. Это происходит из-за того, что изменения рН желудка (т.е. пациенты, принимающие или не принимающие ИПП или пищевой эффект) может привести к изменчивости воздействия из-за различий в растворимости. Предотвращение потенциального лекарственного взаимодействия особенно важно в онкологии из-за многочисленных препаратов, которые пациенты обычно получают в одно и то же время,узкого терапевтического окна многих противораковых препаратов и больших меж- и внутрииндивидуальных отличий пациентов. Соединение, имеющее предпочтительную растворимость, также позволяет избежать необходимости в сложных и дорогих лекарственных формах, которые могут быть использованы для увеличения системного воздействия, необходимого для эффективности из-за низкой растворимости или для уменьшения изменчивости воздействия из-за пищевого эффекта и ИПП. Исследование фармакокинетических свойств на собаках. Собаки породы бигль обычно используются для определения in vivo воздействия и фармакокинетических параметров фармацевтических продуктов. Хотя физиология желудка собак в некоторых аспектах отличается от человеческой, это полезно для прогнозирования абсорбции лекарственного средства и идентификации потенциальных проблем при нелинейной фармакокинетике. Для определения фармакокинетических параметров примера 1 у собак сукам и кобелям (до 4 животных на дозу, в отдельных исследованиях) давали пример 1 через желудочный зонд в 1% гидроксиэтилцеллюлозе, 0,25% полисорбате 80, 0,05% пеногасителя в суспензии в очищенной воде ("суспензия ГЭЦ"). Пределы вводимых доз составляют от 1 до 12 мг/кг в суспензии ГЭЦ. Собирали образцы крови в пробирки, содержащие калиевую соль этилендиаминотетрауксусной кислоты, от каждой собаки в 0 (до дозы), 0,5, 1, 2, 4, 8 и через 24 ч после дозы. Некоторые исследования также включали образцы, собранные в момент времени 0,25 ч и 12 ч. Эти образцы центрифугировали для получения плазмы, которую затем замораживали перед анализом. В образцах проводили осаждение бел- 23022163 ка и экстракты анализировали на наличие примера 1 с помощью жидкой хроматографии/тандемной массспектрометрии с использованием масс-спектрометра PE-Sciex API4000. Стандартные кривые варьировали от 1 до 5000 нг/мл. Концентрации в плазме выше верхнего предела определения измеряли с помощью разведения. Измеренные концентрации примера 1 сохраняли в системе Watson v.7.4, валидизированной системе управления лабораторной информацией, используемой для хранения и управления электронными данными, и определяли фармакокинетические параметры с помощью некомпартментной модели с использованием программного обеспечения WATSON. Соединение согласно настоящему изобретению исследовали с помощью данного анализа, проведенного в точности согласно описанию выше. Например, проанализировали соединение согласно примеру 1, и оказалось, что средняя AUC соответствует представленным в табл. 13. Значения площади под кривой (AUC) для примера 1 повышается линейно при дозе в пределах от 1 до 12 мг/кг, как показано в таблице ниже. Анализ линейной регрессии отдельных значений AUC дает корреляцию определения R2 0,86 и линейное уравнение у = 1474,3 х + 44,311. Анализ линейной регрессии средних значений AUC для каждой дозы приводит к корреляции определения R2 0,96 и линейному уравнению у = 1544,7 х-735,34. Эти результаты показывают, что соединения в рамках настоящего изобретения имеют линейные фармакокинетические свойства у собак выше соответствующего фармакологического диапазона доз, без признаков насыщения поглощения. Это является благоприятным свойством для создания лекарственных средств и клинического применения, способствующим предсказуемому увеличению при системном воздействии при пероральном введении. Таблица 13 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, представляющее собой 8-[5-(1-гидрокси-1-метилэтил)пиридин-3-ил]-1-[(2S)-2 метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2-он, или его фармацевтически приемлемая соль. 2. Соединение по п.1, отличающееся тем, что представляет собой 8-[5-(1-гидрокси-1 метилэтил)пиридин-3-ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2-он. 3. Соединение по п.2, отличающееся тем, что представляет собой 8-[5-(1-гидрокси-1 метилэтил)пиридин-3-ил]-1-[(2S)-2-метоксипропил]-3-метил-1,3-дигидро-2 Н-имидазо[4,5-с]хинолин-2 он в кристаллической форме, характеризующейся рентгеновской порошковой дифрактограммой, содержащей характеристические пики в 20,2, при 8,57 и одном или нескольких из 9,06, 15,93, 18,29 и 18,87. 4. Фармацевтическая композиция для лечения рака, содержащая соединение или соль по любому из пп.1-3 и фармацевтически приемлемый носитель, растворитель или наполнитель. 5. Применение соединения или соли по любому из пп.1-3 для лечения рака. 6. Применение по п.5, при этом рак выбран из группы, состоящей из рака мочевого пузыря, рака толстой кишки, рака желудка, рака головы и шеи, немелкоклеточного рака легкого (НМРЛ), рака молочной железы, меланомы, рака яичников, рака поджелудочной железы, рака предстательной железы, глиобластомы, рака легкого, рака почки, саркомы, рака кроветворной и лимфоидной тканей, рака центральной нервной системы, рака шейки матки, рака эндометрия, рака печени, рака кожи, рака желудка, рака щитовидной железы, рака верхних отделов желудочно-кишечного тракта и дыхательных путей и рака мочеполовой системы. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2
МПК / Метки
МПК: A61P 35/00, A61K 31/4745, C07D 471/04
Метки: ингибиторы, двойные
Код ссылки
<a href="https://eas.patents.su/25-22163-dvojjnye-ingibitory-pi3-kinazy-mtor.html" rel="bookmark" title="База патентов Евразийского Союза">Двойные ингибиторы pi3 киназы/mtor</a>
Триазины как ингибиторы киназы pi3 и mtor

Номер патента: 20317
Опубликовано: 30.10.2014
Авторы: Аирал-Калоустиан Семирамис, Венкатесан Аранапакам М., Денхардт Кристоф М., Чэнь Чзэчэн, Дос Сантос Освальдо, Верхейен Ерун К., Ши Мэнсяо, Делос Сантос Эфрен Гийермо, Ричард Дэвид Дж., Каплан Джошуа Аарон, Гопалсами Ариамала, Мансур Тарек С., Заск Ари, Каррэн Кевин Дж.
МПК: C07D 498/08, A61K 31/53, A61P 35/00...
Метки: триазины, ингибиторы, киназы
Формула / Реферат:
1. Соединение формулы Iгде R1 представляет собойгде R6, R7, R8, R9, каждый независимо, представляют собой водород и C1-C6-алкил илиR6 и R9 или R7 и R8 вместе с атомами углерода, к которым они присоединены, образуют необязательно замещенный 5-6-членный насыщенный цикл, содержащий 0 или 1 атом О;R2 представляет собой необязательно замещенный C6-C10-арил-NH-CO-R3;R3 представляет собой OR5, NR5R5, NHR5;R5 независимо выбирают из C1-C6-алкила;...
Ингибиторы pi3 киназы или mtor

Номер патента: 19700
Опубликовано: 30.05.2014
Авторы: Нормэн Марк Х., Рид Энтони Б., Ян Кевин, Джексон Клер Л.М., Лэнмэн Брайан А., Нишимуа Нобуко, Ву Бинь, Петтас Липин Х., Лю Лонбинь, Тадесс Сейфу, Смит Эдриан Л., Ляо Хонъю, Бо Юньсинь И., Херберич Брэдли Дж., Хон Фан-Цао, Тамайо Нуриа А., Вурц Райан, Эндрюс Кристин, Си Виктор Дж., Букер Шон, Д'энжело Ноэль
МПК: A61K 31/52, A61P 35/00, C07D 401/14...
Метки: ингибиторы, киназы
Формула / Реферат:
1. Соединение или его фармацевтически приемлемая соль, выбранное...
Способ получения пиридо-пиримидиновых ингибиторов mtor киназы, их солевых форм

Номер патента: 19092
Опубликовано: 30.01.2014
Авторы: Пауэлл Лин, Ро Стивен Антони, Блейд Хелен, Чёрчилл Гуидион Нью, Карри Анджела Шарлотте, Кенуорти Мартин Нил, Хайнс Питер Самьюэл, Добсон Бенджамин Чарлз
МПК: A61K 31/519, A61P 35/00, C07D 471/04...
Метки: солевых, пиридо-пиримидиновых, способ, получения, ингибиторов, киназы, форм
Формула / Реферат:
1. Способ получения соединения формулы 1где R1 представляет собой водород или OR3;R2 представляет собой CH2OR4, CN, CO2R5 или CONR6R7;R3 представляет собой С1-4алкильную группу;R4 представляет собой -COR8 группу, где R8 представляет собой вторичную С3-6алкильную или третичную С4-6алкильную группу;R5 представляет собой С1-4алкильную группу;R6 представляет собой водород или С1-4алкильную группу иR7 представляет собой С1-4алкильную...
Комбинированная терапия ингибитором киназы p70 s6 и ингибитором mtor

Номер патента: 18824
Опубликовано: 30.10.2013
Авторы: Донохо Грегори Пол, Джиджанейдж Сандаруван
МПК: A61K 31/519, A61K 31/436, A61K 45/06...
Метки: ингибитором, терапия, комбинированная, киназы
Формула / Реферат:
1. Продукт, включающий соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор mTOR, представляющий собой рапамицин, в виде комбинированного препарата для одновременного, раздельного или последовательного применения в терапии.2. Продукт, включающий соединение...
Ингибиторы акт и p70 s6-киназы

Номер патента: 18947
Опубликовано: 29.11.2013
Авторы: Шеферд Тимоти Алан, Джозеф Саджан, Дэлли Роберт Дин
МПК: A61P 35/00, C07D 473/00, A61K 31/52...
Метки: ингибиторы, s6-киназы, акт
Формула / Реферат:
1. Соединение формулыгде X представляет собой F, Cl, CF3, CN или Н;Y представляет собой F, Н или Cl;R1 и R2 независимо представляют собой Н, С1-С4алкил или СН2СН2ОН; или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют пирролидиновое кольцо, необязательно замещенное гидроксиметилом в положении 2 или гидроксилом в положении 3, или азетидиновое кольцо, замещенное гидроксилом в положении 3;R3 представляет собой Н или ОН;R6...
Предыдущий патент: Брезентовое закрывающее устройство для накопительного бункера
Следующий патент: (пиридин-4-ил)бензиламиды как аллостерические модуляторы альфа 7 nachr
Случайный патент: Соединения диосметина, способ их получения и фармацевтические композиции, которые их содержат