Имидазопиридины в качестве противовирусных средств против респираторно-синцитиального вируса
Номер патента: 24681
Опубликовано: 31.10.2016
Авторы: Вендевилль Сандрин Мари Элен, Ху Лили, Ван Хоф Стивен Морис Паула, Йонкерс Тим Хьюго Мария, Коиманс Людвиг Поль, Тахри Абделлах, Рабуассон Пьер Жан-Мари Бернар















Формула / Реферат
1. Соединение формулы I или его аддитивная соль

где X представляет собой С или N, когда X находится в положении 5, и X представляет собой С, когда X находится в любом другом положении, отличном от положения 5;
Y представляет собой С или N, когда Y находится в положении 7', и Y представляет собой С, когда Y находится в любом другом положении, отличном от положения 7';
R1 присутствует, когда X=С, и R1 выбран из Н или галогена;
R1 отсутствует, когда X=N;
R2 выбран из группы, состоящей из Н, -(CR7R8)n-R9, -CºC-CH2-R9 и -C=C-R9;
R3 выбран из группы, состоящей из C1-C10-алкила, C3-C7-циклоалкила;
R4 присутствует и представляет собой Н, когда Y представляет собой С;
каждый R7 и R8 независимо выбран из Н, C1-C10-алкила, или R7 и R8, взятые вместе, образуют 6-членное гетероциклическое кольцо с атомами N и О;
R9 выбран из группы, состоящей из C1-C6-алкила, C1-C6-алкокси, C3-C7-циклоалкила, ОН, CN, F, CONR7R8, COOR7;
n представляет собой целое число от 2 до 6.
2. Соединение по п.1, где R2 выбран из группы, состоящей из Н, -(CR7R8)n-R9 и -CºC-CH2-R9.
3. Соединение по п.1 или 2, где все атомы X представляют собой С.
4. Соединение по пп.1, 2 или 3, где X-R1 в пара-положении к N, образующему мостиковую связь имидазольного и пиридинового кольца, выбран из группы, состоящей из С-Н, C-Cl и С-Br, и все другие R1 представляют собой Н.
5. Соединение по любому из предшествующих пунктов, где R7 и R8 представляют собой Н и n равно 2-4.
6. Соединение по любому из предшествующих пунктов, где R9 выбран из группы, состоящей из ОН, F и вторичного C1-C6-алкила.
7. Соединение по любому из предшествующих пунктов, где R2 представляет собой -CºC-CH2-R9, где R9 выбран из группы, состоящей из C1-C6-алкокси, C1-C6-алкила с неразветвленной цепью и C1-C6-алкила с разветвленной цепью.
8. Соединение по любому из предшествующих пунктов, где R3 представляет собой C3-C7-циклоалкил.
9. Соединение по любому из предшествующих пунктов, где R3 представляет собой циклопропил.
10. Соединение по любому из предшествующих пунктов, где R3 представляет собой изопропил.
11. Соединение по любому из предшествующих пунктов, где Y в пара-положении к N-R3 представляет собой С.
12. Применение соединения по любому из пп.1-11 в качестве лекарственного средства для лечения респираторно-синцитиальных вирусных инфекций.
13. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения по любому из пп.1-11.
14. Способ получения фармацевтической композиции по п.13, где указанный способ включает тщательное смешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения по любому из пп.1-11.
15. Применение соединения по любому из пп.1-11 в качестве лекарственного средства для ингибирования репликации респираторно-синцитиального вируса.
16. Применение соединения по любому из пп.1-11 для получения лекарственного средства для ингибирования репликации респираторно-синцитиального вируса.
Текст
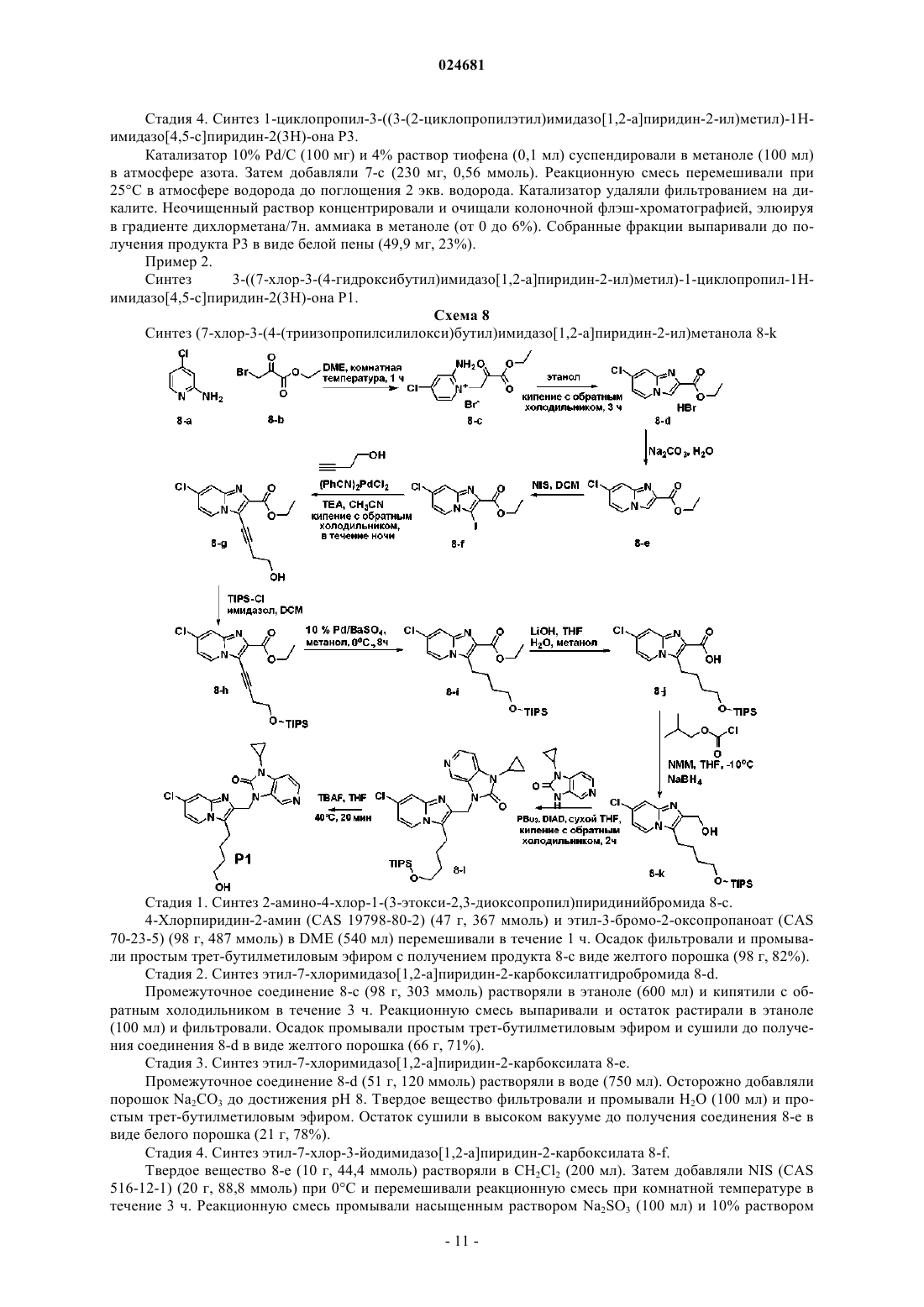
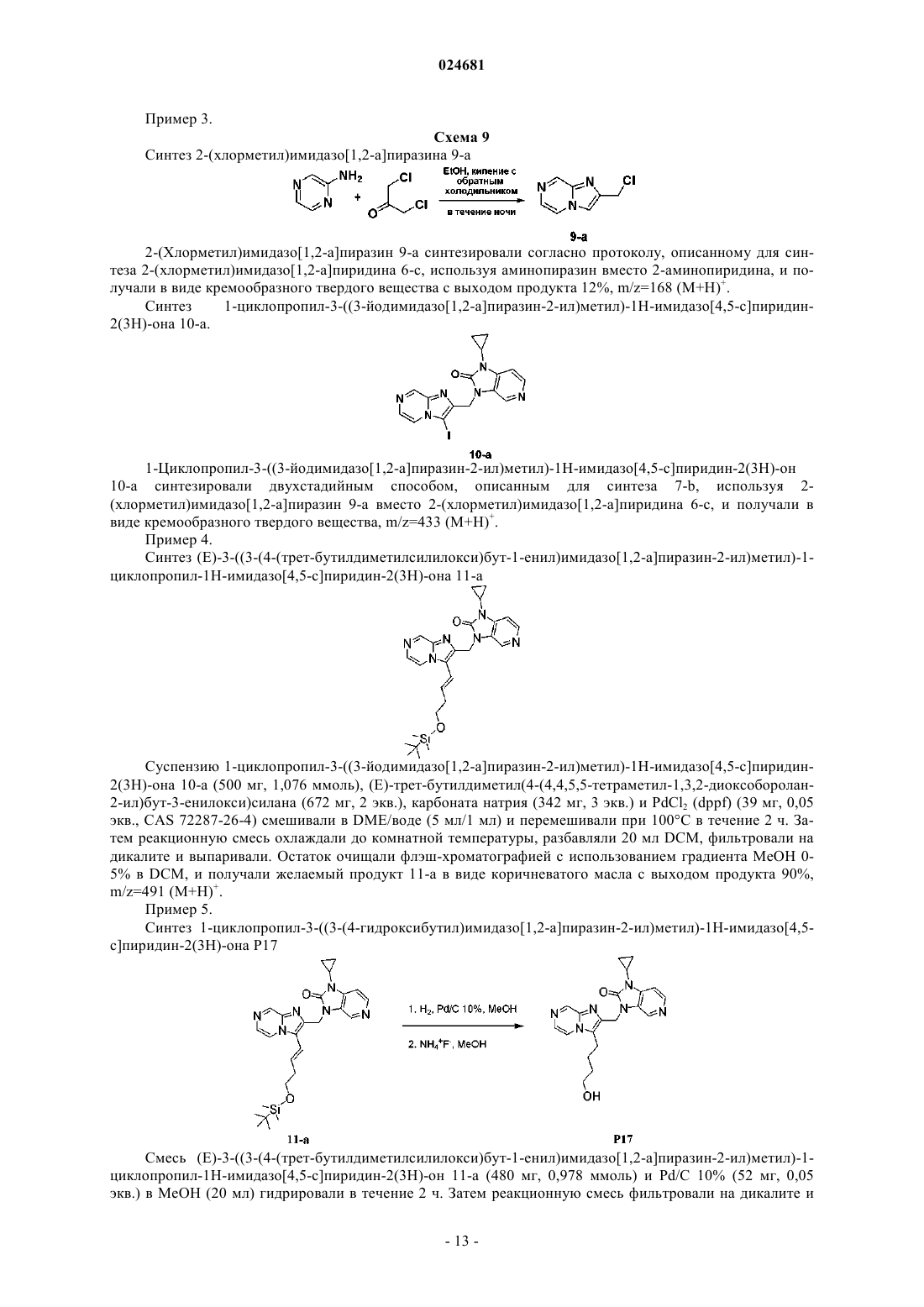
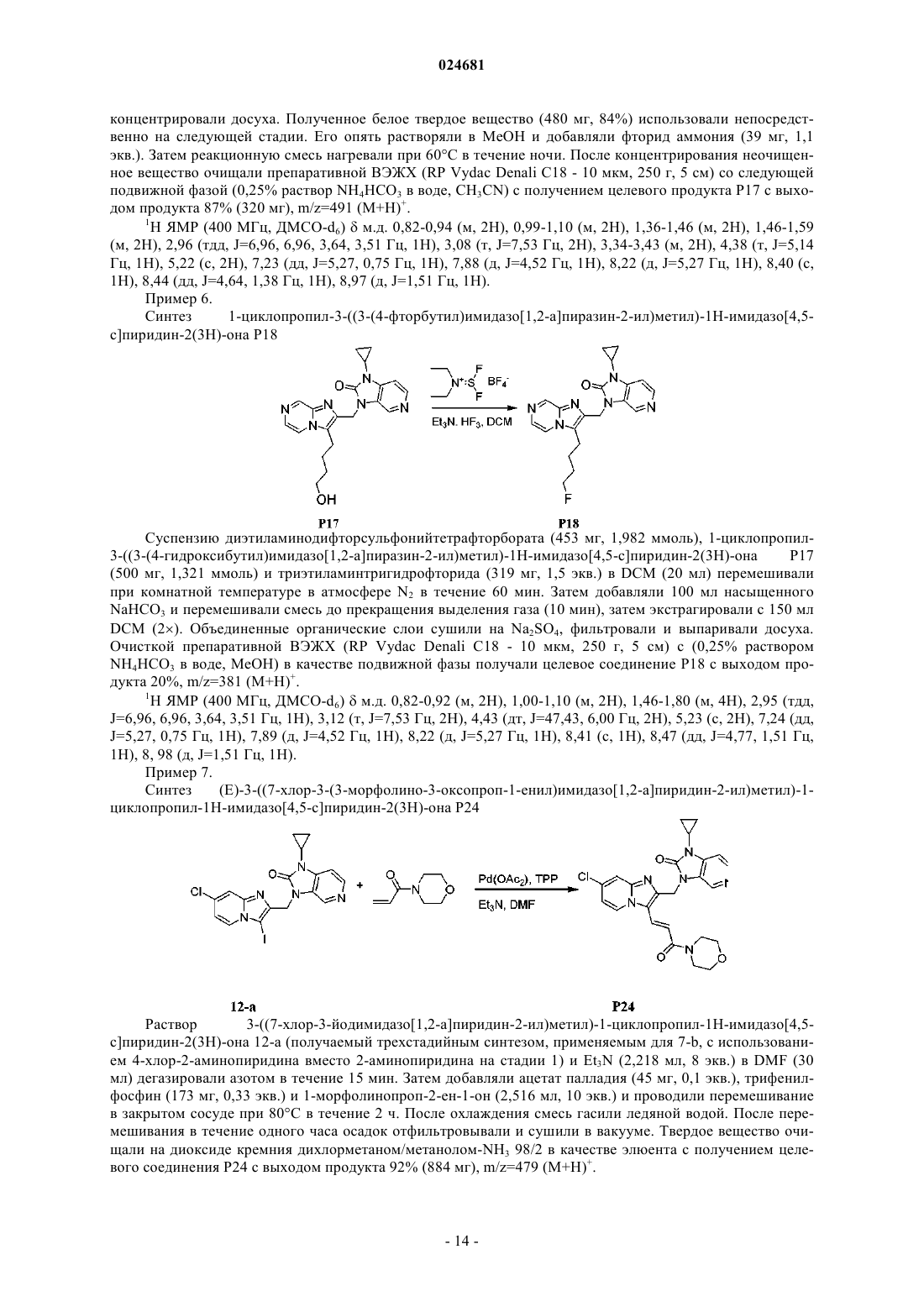
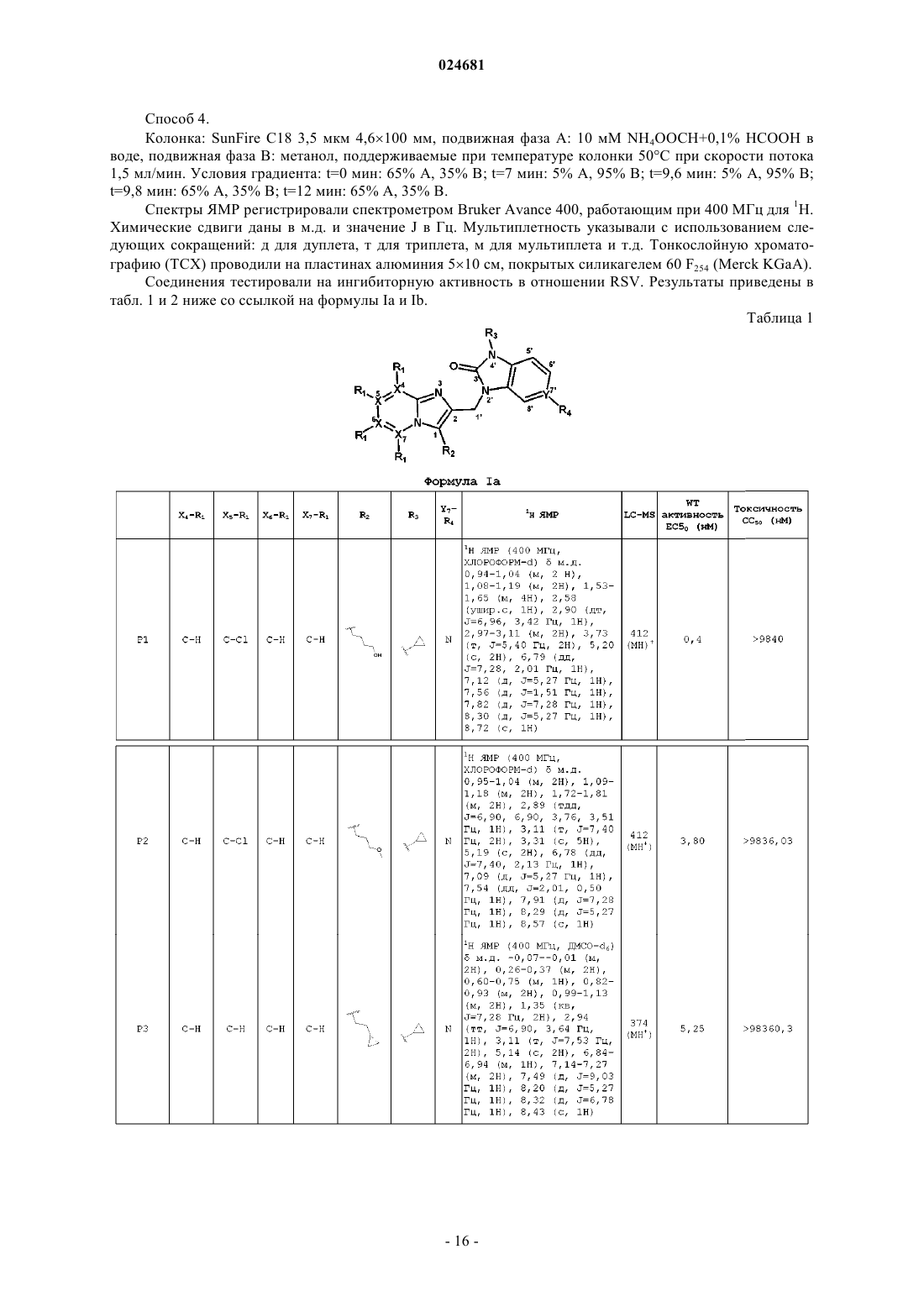
ИМИДАЗОПИРИДИНЫ В КАЧЕСТВЕ ПРОТИВОВИРУСНЫХ СРЕДСТВ ПРОТИВ РЕСПИРАТОРНО-СИНЦИТИАЛЬНОГО ВИРУСА Раскрыты соединение, удовлетворяющее формуле I, обладающее ингибирующей активностью в отношении репликации респираторно-синцитиального вируса, и его аддитивная соль,фармацевтические композиции, содержащие это соединение в качестве активного ингредиента,способ получения таких композиций и применение соединений формулы I для лечения респираторно-синцитиальной вирусной инфекции. Коиманс Людвиг Поль, Ху Лили,Йонкерс Тим Хьюго Мария,Рабуассон Пьер Жан-Мари Бернар,Тахри Абделлах, Ван Хоф Стивен Морис Паула, Вендевилль Сандрин Мари Элен (BE) Медведев В.Н. (RU)(71)(73) Заявитель и патентовладелец: ЯНССЕН САЙЕНСИЗ АЙРЛЭНД ЮCИ (IE) Область техники, к которой относится изобретение Изобретение относится к имидазопиридинам, обладающим противовирусной активностью, в частности обладающим ингибирующей активностью в отношении репликации респираторно-синцитиального вируса (RSV). Изобретение дополнительно относится к фармацевтическим композициям, содержащим эти соединения, и применению этих соединений для лечения респираторно-синцитиальной вирусной инфекции. Уровень техники, предшествующий изобретениюRSV человека или респираторно-синцитиальный вирус наряду с вирусом бычьего RSV представляет собой крупный РНК-вирус, член семейства Paramyxoviridae, подсемейства pneumoviridae. RSV человека вызывает спектр заболеваний дыхательных путей у людей любого возраста во всем мире. Он является основной причиной заболеваний нижних дыхательных путей в грудном и детском возрасте. Более половины всех детей сталкиваются с RSV в свои первые годы жизни, и почти все в свои первые два года. Инфекция у маленьких детей может вызывать поражение легких, которое персистирует годами и может способствовать хроническому заболеванию легких в более позднем возрасте (хроническому свистящему дыханию, астме). Дети более старшего возраста и взрослые при инфекции RSV часто страдают (тяжелой) вирусной инфекцией верхних дыхательных путей. В престарелом возрасте восприимчивость опять увеличивается, и RSV вызывает ряд вспышек пневмонии у пожилых людей, приводящих к значительной смертности. Инфекция вирусом из данной подгруппы не защищает против последующей инфекции изолятомRSV из той же самой подгруппы в следующий зимний сезон. Таким образом, повторное инфицирование является распространенным, несмотря на то, что существует только два подтипа А и В. В настоящее время одобрено только три лекарственных средства для использования против инфекции RSV. Первым является рибавирин, аналог нуклеозида, который обеспечивает аэрозольное лечение тяжелой инфекции RSV у госпитализированных детей. Аэрозольный путь введения, токсичность (риск тератогенного действия), стоимость и высокая вариабельность эффективности ограничивают его применение. Другие два лекарственных средства RespiGam (RSV-IG) и Synagis (паливизумаб), иммуностимуляторы на основе поликлональных и моноклональных антител, предназначены для профилактического применения. Оба являются дорогостоящими, и их необходимо вводить парентерально. Все другие попытки до настоящего времени получить безопасную и эффективную вакцину противRSV оказались безуспешными. Инактивированные вакцины не защищают от заболевания, и фактически в некоторых случаях усиливают заболевание при последующей инфекции. Живые аттенуированные вакцины прошли испытания с ограниченным успехом. Очевидно, что существует необходимость в эффективном нетоксичном и простом для введения лекарственном средстве против репликации RSV. Особенно предпочтительным является предоставление лекарственных средств против репликации RSV, которые можно вводить перорально. Ссылочный документ с заглавием "противовирусные средства на основе имидазопиридина и имидазопиримидина" представляет собой WO 01/95910, который фактически относится к противовирусным средствам на основе бензимидазола. Указанный документ относится к соединениям, которые обладают противовирусной активностью, даже со значениями ЕС 50 в широком диапазоне от 0,001 вплоть до 50 мкМ (которые обычно не проявляют желаемую биологическую активность). Другая ссылка, относящаяся к противовирусным средствам против RSV на основе замещенного 2-метилбензимидазола в аналогичном диапазоне активностей, представляет собой WO 03/053344. Другая ссылка, относящаяся к предшествующему уровню техники, на соединения аналогичного диапазона активностей в отношении противовирусных средств на основе бензимидазолона представляет собой WO 02/26228. Ссылка по связи структура-активность в отношении ингибирования RSV для соединений 5-замещенного бензимидазола представляет собой работу Х.А. Wang et al., Bioorganic and Medicinal Chemistry Letters 17 (2007), 45924598. Желательно предоставить новые лекарственные средства, которые обладают противовирусной активностью. В частности, было бы желательно предоставить новые лекарственные средства, которые обладают ингибирующей активностью в отношении репликации RSV. Кроме того, было бы желательно выявить структуры соединений, которые позволяют получить противовирусные виды биологической активности порядка величины более сильных областей известного уровня техники (т.е. в начале указанного выше диапазона до 50 мкМ), и предпочтительно на уровне приблизительно наиболее активном, более предпочтительно даже более высокой активности, чем соединения, описанные в данной области. Дополнительно желательно найти соединения, обладающие пероральной противовирусной активностью. Сущность изобретения Для более эффективного решения одной или более из указанных выше потребностей в одном из аспектов изобретение относится к противовирусным соединениям имидазопиридина, представленным формулой I, или их аддитивной соли где X представляет собой С или N, когда X находится в положении 5, и X представляет собой С, когда X находится в любом другом положении, отличном от положения 5;Y находится в любом другом положении, отличном от положения 7';R4 присутствует и представляет собой Н, когда Y представляет собой С; каждый R7 и R8 независимо выбран из Н, C1-C10-алкила, или R7 и R8, взятые вместе, образуют 6 членное гетероциклическое кольцо с атомами N и О;n представляет собой целое число от 2 до 6. В другом аспекте изобретение относится к применению указанных выше соединений в качестве лекарственного средства для лечении инфекций RSV у теплокровных животных, предпочтительно людей. В еще одном аспекте изобретение относится к применению указанных выше соединений в качестве лекарственного средства для ингибирования репликации респираторно-синцитиального вируса. В другом аспекте изобретение относится к применению соединения, как определено выше, для получения лекарственного средства для ингибирования репликации респираторно-синцитиального вируса. В дополнительном аспекте изобретение относится к фармацевтической композиции, содержащей соединение, как определено выше, и фармацевтически приемлемый эксципиент. Еще в одном дополнительном аспекте изобретение относится к способу получения указанной фармацевтической композиции, включающему тщательное смешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы I. Подробное описание изобретения Молекулы формулы I, в отличие от известного уровня техники, содержат с одной стороны (левая сторона молекулы, как обозначено) ароматическое шестичленное кольцо, конденсированное с имидазольным кольцом, где шестичленное кольцо включает по меньшей мере один атом азота, являющийся общим с имидазольным кольцом. Кратко, его обозначают как замещенную группу имидазопиридина. Следует понимать, что термин "пиридин" применяют в случае, когда все атомы X представляют собой С,но настоящий краткий термин "имидазопиридин" включает все варианты, представленные в формуле I для шестичленного кольца, т.е. независимо от того, один или более атомов X представляют собой С илиN, например имидазопиразины. Изобретение в широком смысле основано на разумном признании того, что такие соединения "имидазопиридина", как правило, обладают представляющей интерес ингибирующей активностью в отношении RSV. Кроме того, эти соединения обеспечивают виды активности против RSV в высоких областях(нижняя граница значений ЕС 50) диапазона, доступного в указанных выше ссылках. В частности, можно обнаружить, что молекулярные структуры на основе таких соединений даже превосходят описанные в ссылках соединения в отношении видов биологической активности. Настоящее изобретение дополнительно описано в отношении конкретных вариантов осуществления и в отношении определенных примеров, но изобретение не ограничено ими, а только формулой изобретения. Когда в настоящем описании и формуле изобретения используют термин "содержащий", он не исключает другие элементы или стадии. Когда используют существительное в единственном числе, оно также включают множественное число этого существительного, если конкретно не указано что-либо другое. Термин "пролекарство", как используется на всем протяжении данного описания, означает фармакологически приемлемые производные, т.е. сложные эфиры и амиды, такие, что получаемый продукт биотрансформации производного представляет собой активное лекарственное средство. Таким образом,-2 024681 включена ссылка Goodman and Gilman (The Pharmacological Basis of Therapeutics, 8th ed.; McGraw-Hill, Int.Ed. 1992, "Biotransformation of Drugs", p. 13-15), в которой описаны пролекарства в общем смысле. Пролекарства характеризуются хорошей водорастворимостью и биодоступностью и легко метаболизируютсяin vivo в активные ингибиторы. Как используется в настоящем описании, C1-C6-алкил в качестве группы или части группы обозначает насыщенные углеводородные радикалы с неразветвленной или разветвленной цепью, содержащие от 1 до 6 атомов углерода, такие как метил, этил, пропил, 1-метилэтил, бутил, пентил, гексил, 2 метилбутил и т.п.C1-C10-алкил в качестве группы или части группы обозначает насыщенные углеводородные радикалы с неразветвленной или разветвленной цепью, содержащие от 1 до 10 атомов углерода, такие как группы, обозначаемые как C1-C6-алкил, и гептил, октил, нонил, 2-метилгексил, 2-метилгептил, децил, 2 метилнонил и т.п.C1-C6-алкокси в качестве группы или части группы обозначает О-C1-C6-алкильный радикал, где C1C6-алкил имеет, независимо, приведенное выше значение.C3-C7-циклоалкил представляет собой общее обозначение циклопропила, циклобутила, циклопентила, циклогексила или циклогептила. Термин "-(CR7R8)n-", используемый в настоящем описании, обозначает n повторений подгруппыCR7R8, где каждая из этих подгрупп определена независимо. Термин "галоген" представляет собой общее обозначение фтора, хлора, брома и йода. Следует отметить, что положения радикалов в любой молекулярной группе, используемой в определениях, могут находиться в любом месте такой группы при условии, что она является химически стабильной. Радикалы, используемые в определениях переменных, включают все возможные изомеры, если не указано иное. Например, пентил включает 1-пентил, 2-пентил и 3-пентил. Когда любая переменная встречается более одного раза в любом компоненте, каждое определение является независимым. Используемый далее в настоящем описании термин "соединения формулы I", или "соединения по настоящему изобретению", или аналогичный термин включает соединения общей формулы I и их аддитивные соли. Следует понимать, что некоторые соединений формулы I могут содержать один или более центров хиральности и существовать в виде стереохимически изомерных форм. Термин "стереохимически изомерные формы", как используется далее в настоящем описании, определяет все возможные соединения, состоящие из аналогичных атомов, связанных связями в той же самой последовательности, но имеющих различные трехмерные структуры, которые не являются равнозначными тем, которыми могут обладать соединения формулы I. Если не упомянуто или указано иное, химическое обозначение соединения включает смесь всех возможных стереохимически изомерных форм, которыми может обладать указанное соединение. Указанная смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Для терапевтического применения соли соединений формулы I представляют собой такие, где противоион является фармацевтически приемлемым. Однако соли кислот и оснований, которые не являются фармацевтически приемлемыми, также могут находить применение, например, при получении или очистке фармацевтически приемлемого соединения. Подразумевают, что фармацевтически приемлемые аддитивные соли кислот и аддитивные соли оснований, как указано в настоящем описании, включают терапевтически активные нетоксичные формы аддитивных солей кислот и оснований, которые могут образовывать соединения формулы I. Фармацевтически приемлемые аддитивные соли кислот можно получать подходящим способом посредством обработки формы основания такой соответствующей кислотой. Соответствующие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например хлористо-водородную или бромисто-водородную кислоту, серная, азотная, фосфорная и т.п. кислоты, или органические кислоты, такие как, например, уксусная, пропионовая, гидроксиуксусная, молочная, пировиноградная, щавелевая (т.е. этандиовая), малоновая, янтарная (т.е. бутандиовая кислота), малеиновая, фумаровая, яблочная (т.е. гидроксибутандиовая кислота), винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламиновая, салициловая, п-аминосалициловая, памовая и подобные им кислоты. И наоборот, формы указанных кислот можно преобразовывать в форму свободного основания обработкой соответствующим основанием. Соединения формулы I, содержащие кислый протон, также можно преобразовывать в их нетоксичные формы солей металлов или аддитивных солей аминов обработкой соответствующими органическими и неорганическими основаниями. Соответствующие формы основных солей включают, например,аммонийные соли, соли щелочных металлов и щелочно-земельных металлов, например литиевые, натриевые, калиевые, магниевые, кальциевые соли и т.п., соли с органическими основаниями, например,бензатиновые, N-метил-D-глюкаминовые, гидрабаминовые соли, и соли с аминокислотами, такими как,-3 024681 например, аргинин, лизин и т.п. Следует понимать, что соединения по изобретению по отношению к указанным выше находящимся слева и справа частям формулы I предоставляют широкий спектр модификаций. Не выходя за рамки объема изобретения, ниже более подробно описаны определенные варианты осуществления. В предпочтительном варианте осуществления R2 выбирают из группы, состоящей из Н,-(CR7R8)n-R9 и -CC-CH2-R9. В другом предпочтительном варианте все атомы X представляют собой С. В предпочтительном варианте осуществления X-R1 в пара-положении к N, образующему мостиковую связь имидазольного и пиридинового кольца, выбирают из группы, состоящей из С-Н, C-Cl и С-Br, и все другие R1 представляют собой Н. Предпочтительно R7 и R8 представляют собой Н, n равно 2-4. В другом предпочтительном варианте R9 выбирают из группы, состоящей из ОН, F и вторичногоC1-C6-алкила. В еще одном предпочтительном варианте R2 представляет собой -CC-CH2-R9, где R9 выбран из группы, состоящей из C1-C6-алкокси, C1-C6-алкила с неразветвленной цепью и C1-C6-алкила с разветвленной цепью. Предпочтительно R3 представляет собой C3-C7-циклоалкил. Еще более предпочтительно, когда R3 представляет собой циклопропил. В другом предпочтительном варианте R3 представляет собой изопропил. Предпочтительно Y в пара-положении к N-R3 представляет собой С. Предпочтительные соединения представляют собой соединения, перечисленные в табл. 1 ниже. Более предпочтительные представляют собой соединения под номерами Р 1, Р 2, Р 3, Р 4, Р 5, Р 6, Р 7, Р 8 и Р 9. Наиболее предпочтительные представляют собой соединения Р 1, Р 2, Р 3 и Р 4. Соединение формулы I можно получать описанными ниже способами, известными в данной области способами синтеза органической химии или модификаций и получения производных, которые хорошо известны специалистам в данной области. Используемые в настоящем описании исходные вещества являются коммерчески доступными, или их можно получать общепринятыми, известными в данной области способами, такими как способы, описанные в обычных справочниках. Предпочтительные способы включают, но не ограничиваются этим, описанные ниже способы. Во время любой из следующих ниже последовательностей синтеза может быть необходимо и/или желательно защищать чувствительные или реакционноспособные группы на любой из представляющих интерес молекул. Это можно проводить посредством общепринятых защитных групп, таких как описанные в T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Chemistry, John WileySons, 1999, которая таким образом полностью включена посредством ссылки. Соединения формулы I или их фармацевтически приемлемые соли можно получать в соответствии со схемами реакций, описанными ниже в настоящем описании. Если не указано иное, заместители на схемах представляют собой такие, как определено выше. Выделение и очистку продуктов проводят общепринятыми способами, которые известны специалисту-химику. Следующие ниже схемы являются примерами способов получения соединений формулы I. На приведенных ниже схемах используемые числа, включая числа от I до XVIII, используют для удобства обозначения формул на схемах. Подразумевают, что использование чисел от I до XVIII на приведенных ниже схемах не означает, что обозначаемые такими числами соединения соответствуют соединениям формул от I до XVIII, которые описаны в настоящем описании выше и перечислены в прилагаемой формуле изобретения. Схема 1 Общий синтез соединений типа формулы I На схеме 1 проиллюстрирован способ получения соединений формулы I, где R1-R9, X и Y являются такими, как определено выше. Соединение типа IV можно получать сочетанием 2-гидроксиметиленимидазопиридина II-а и N3 замещенного бензимидазолона III известным в данной области способом, таким как реакция Мицунобу,в которой используют азадиизопропилдикарбоксилат и трифенилфосфин в подходящем растворителе,таком как, но не ограничиваясь им, DMF или THF. Альтернативно, соединения формулы I можно получать замещением W, который представляет собой галогенид, II-b, предпочтительно хлор или сульфонат,II-с, такой как мезилат или тозилат, в присутствии основания, такого как, но не ограничиваясь им, гидрида натрия, карбоната калия или карбоната цезия, в подходящем растворителе, таком как DMF или THF. Для преобразования соединения типа IV в соединение типа V можно использовать галогенирующие реагенты, такие как, но не ограничиваясь ими, N-йодсукцинимид, и CH3CN может представлять собой подходящий растворитель для этой реакции. Можно получать соединение типа VI сочетанием алкила с соединением типа V известным в данной области способом, таким как реакция сочетания Соногашира. Восстановление тройной связи можно проводить каталитическим путем с использованием водорода в присутствии катализатора, такого как палладий или платина, в подходящем растворителе, таком как метанол, или стехиометрическим путем с использованием железа в присутствии хлорида аммония или хлорида олова, в присутствии концентрированной хлористо-водородной кислоты с получением соединения формулы I. Схема 2 Общий синтез соединений типа II-а Синтез соединений типа II-а, как правило, можно проводить, как представлено на схеме 2. Соединение типа IX можно синтезировать сочетанием коммерчески доступного соединения типа VII с коммерчески доступным соединением типа VIII, в котором галоген предпочтительно представляет собой бром,посредством реакции сочетания опосредованной основанием. Возможные основания касательно этой реакции представляют собой, но не ограничиваются ими, K2CO3, Cs2CO3, триэтиламин и гидрид натрия. Подходящий растворитель для этого типа опосредованного основанием сочетания представляет собойDME. Соединение X можно получать после внутримолекулярного замыкания кольца посредством нагревания. Преобразование сложного алкилового эфира соединения X в спирт II-а проводили с гидридом ме-5 024681 талла, таким как алюмогидрид лития или борогидрид натрия, в подходящем растворителе, таком как THF или метанол. Схема 3 Общий синтез соединений типа II-b и II-с На схеме 3 показаны варианты синтеза соединений типа II-b и II-с. Обработкой спирта II-а реагентами, такими как, но не ограничиваясь ими, SOCl2, PBr3, p-TsCl,MsCl, получают 2-хлорметилиндол II-b и промежуточное соединение II-с в присутствии органического основания, такого как триэтиламин или диизопропилэтиламин в подходящем растворителе, таком как дихлорметан. Это проиллюстрировано способом 1. Альтернативно, соединение типа II-b также можно получать посредством внутримолекулярного замыкания кольца коммерчески доступного соединения типа XI и также коммерчески доступного соединения типа XII. Подходящий растворитель для такой реакции может представлять собой этанол. Это проиллюстрировано способом 2. Схема 4 Общий синтез соединений типа III Соединения III можно синтезировать способом, проиллюстрированным на схеме 4. СоединениеXIV получают реакцией замещения Z, который представляет собой галогенид, предпочтительно фтор или хлор, или алкоксигруппу, предпочтительно метокси, соединения XIII амином в подходящем растворителе, таком как THF или DMF, в присутствии органического основания, такого как триэтиламин или диизопропилэтиламин. Восстановление нитрогруппы до амина XV можно проводить каталитическим путем с использованием водорода в присутствии катализатора, такого как палладий или платина, в подходящем растворителе, таком как метанол, или стехиометрическим путем с использованием железа в присутствии хлорида аммония или хлорида олова, в присутствии концентрированной хлористоводородной кислоты. Циклизацией получаемого диамина XV с использованием CDI, фосгена или трифосгена в растворителе, таком как ацетонитрил или THF, получают соединение III. Альтернативно, соединения типа III можно получать, исходя из коммерчески доступных дианилинов XVI, которые можно подвергать циклизации замыканием кольца с использованием CDI, фосгена или трифосгена с образованием промежуточных соединений типа XVII. Алкилирование или сульфонилирование азота мочевины XVII можно проводить реакцией Мицунобу с использованием коммерчески дос-6 024681 тупных спиртов или замещением хлора в соединениях типа XVIII с образованием соединений формулыIII. Соединения формулы I можно преобразовывать в соответствующие формы N-оксида известными в данной области способами преобразования трехвалентного азота в его форму N-оксида. Указанную реакцию N-окисления, как правило, можно проводить посредством взаимодействия исходного вещества (I) с соответствующим органическим или неорганическим пероксидом. Соответствующие неорганические пероксиды включают, например, пероксид водорода, пероксиды щелочных и щелочно-земельных металлов, например пероксид натрия, пероксид калия, соответствующие органические пероксиды могут включать перкислоты, такие как, например, бензолкарбопероксовая кислота или галогензамещенная бензолкарбопероксовая кислота, например 3-хлорбензолкарбопероксовая кислота, пероксоалкановые кислоты,например пероксоуксусная кислота, алкилгидропероксиды, например трет-бутилгидропероксид. Подходящие растворители представляют собой, например, воду, низшие спирты, например этанол и т.п., углеводороды, например толуол, кетоны, например 2-бутанон, галогенированные углеводороды, например дихлорметан, и смеси таких растворителей. Чистые стереохимически изомерные формы соединений формулы I можно получать известными в данной области способами. Диастереомеры можно разделять физическими способами, такими как селективная кристаллизация, и хроматографическими способами, например, противоточной распределительной хроматографией, жидкостной хроматографией и т.п. Соединения формулы I, полученные описанными выше в настоящем описании способами, как правило, представляют собой рацемические смеси энантиомеров, которые можно разделять известными в данной области способами разделения. Рацемические соединения формулы I, которые являются в достаточной степени основными или кислотными, можно преобразовывать в соответствующие формы диастереомерных солей реакцией с подходящей хиральной кислотой, соответственно хиральным основанием. Затем указанные формы диастереомерных солей разделяют, например, селективной или фракционной кристаллизацией, и выделяют из них энантиомеры щелочью или кислотой. Альтернативный способ разделения энантиомерных форм соединений формулы I включает жидкостную хроматографию, в частности, жидкостную хроматографию с использованием хиральной неподвижной фазы. Указанные чистые стереохимически изомерные формы также можно получать из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ при условии, что реакция проходит стереоспецифически. Предпочтительно, если желательным является конкретный стереоизомер, указанное соединение синтезируют стереоспецифическими способами получения. В этих способах преимущественно применяют энантиомерно чистые исходные вещества. В дополнительном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы I, как указано в настоящем описании, или соединение любого из вариантов осуществления соединений формулы I, как указано в настоящем описании, и фармацевтически приемлемый носитель. Терапевтически эффективное количество в этом контексте представляет собой количество, достаточное для профилактического действия против, стабилизации или уменьшения вирусной инфекции, и в частности, вирусной инфекции RSV, у инфицированного индивидуума или индивидуума, подверженного риску инфицирования. В дополнительном аспекте настоящее изобретение относится к способу получения фармацевтической композиции, как указано в настоящем описании, который включает тщательное перемешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы I, как указано в настоящем описании, или соединения любого из вариантов осуществления соединений формулы I, как указано в настоящем описании. Таким образом, соединения по настоящему изобретению или их любого варианта осуществления можно формулировать в различных фармацевтических формах для введения. В качестве соответствующих композиций, можно перечислить все композиции, применяемые, как правило, для системного введения лекарственных средств. Для получения фармацевтических композиций по настоящему изобретению эффективное количество конкретного соединения необязательно в форме аддитивной соли или комплексного соединения с металлом в качестве активного ингредиента объединяют при тщательном перемешивании с фармацевтически приемлемым носителем, где носитель может находиться в различных формах в зависимости от формы желаемого препарата для введения. Такие фармацевтические композиции являются желательными в единичной дозированной форме, подходящей, в частности, для введения перорально, ректально, чрескожно или посредством парентеральной инъекции. Например, при получении композиций в пероральной дозированной форме можно применять любую из обычных фармацевтических сред, таких как, например, вода, гликоли, масла, спирты и т.п., в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы, или твердых носителей, таких как крахмалы, сахара, каолин, лубриканты, связывающие средства, дезинтегрирующие средства и т.п., в случае порошков, пилюлей, капсул и таблеток. Вследствие простоты введения таблетки и капсулы представляют собой наиболее предпочтительные пероральные единичные дозированные формы, в случае которых очевидно применяют твердые фармацевтические носители. Для парентеральных композиций носитель, как правило, включает стерильную воду, по меньшей мере, как большую часть, хотя может включать другие ингредиенты, например, для увеличения растворимости. Можно получать инъекционные растворы, например, в которых носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также можно получать инъекционные суспензии,в случае которых можно применять соответствующие жидкие носители, суспендирующие средства и т.п. Также включены твердые формы препаратов, которые необходимо преобразовывать непосредственно перед использованием в жидкие формы препаратов. В подходящих для чрескожного введения композициях носитель необязательно содержит средство для усиления проникновения и/или подходящее смачивающее средство, необязательно объединенное с подходящими добавками любой природы в малых пропорциях, где добавки не оказывают существенного вредного воздействия на кожу. Соединения по настоящему изобретению также можно вводить посредством пероральной ингаляции или инсуффляции способами и посредством составов, применяемыми в данной области для введения таким путем. Таким образом, в основном соединения по настоящему изобретению можно вводить в легкие в форме раствора, суспензии или сухого порошка, где раствор является предпочтительным. Любая система, предназначенная для доставки растворов, суспензий или сухих порошков посредством пероральной ингаляции или инсуффляции, является подходящей для введения соединений по настоящему изобретению. Таким образом, настоящее изобретение также относится к фармацевтической композиции, полученной с возможностью введения посредством ингаляции или инсуффляции через рот, содержащей соединение формулы I и фармацевтически приемлемый носитель. Предпочтительно соединения по настоящему изобретению вводят посредством ингаляции раствора в дозах в распыленном виде или в виде аэрозоля. Особенно предпочтительно формулировать указанные выше фармацевтические композиции в единичной дозированной форме для простоты введения и однородности дозирования. Единичная дозированная форма, как используется в настоящем описании, относится к физически дискретным единицам,подходящим в качестве стандартных доз, где каждая единица содержит предопределенное количество активного ингредиента, рассчитанное для оказания желаемого терапевтического действия, совместно с необходимым фармацевтическим носителем. Примеры таких единичных дозированных форм представляют собой таблетки (включая таблетки с насечкой или покрытые таблетки), капсулы, пилюли, суппозитории, пакетики с порошком, облатки, инъекционные растворы или суспензии и т.п., и их отдельные компоненты. Соединения формулы I проявляют противовирусные свойства. Вирусные инфекции, которые лечат соединениями и способами по настоящему изобретению,включают такие инфекции, которые вызваны орто- и парамиксовирусами, и в частности, респираторносинцитиальным вирусом (RSV) человека и бычьим респираторно-синцитиальным вирусом. Кроме того,ряд соединений по настоящему изобретению является активным против мутировавших штаммов RSV. Дополнительно для многих соединений по настоящему изобретению показан благоприятный фармакокинетический профиль, и они обладают привлекательными свойствами в отношении биодоступности,включая приемлемое время полужизни, AUC и пиковые значения и отсутствие неблагоприятных явлений, таких как недостаточно быстрое начало действия и удерживание в тканях. Противовирусную активность in vivo против RSV соединений по настоящему изобретению исследовали в тесте, как описано в экспериментальной части описания, и ее также можно продемонстрировать в анализе уменьшения выхода вируса. Противовирусную активность in vivo против RSV соединений по настоящему изобретению можно продемонстрировать на тестовой модели с использованием хлопковых хомячков, как описано у Wyde et al. (Antiviral Research (1998), 38, 31-42). Вследствие своих противовирусных свойств, в частности, своих свойств против RSV, соединения формулы I или их любого варианта осуществления, их аддитивные соли являются пригодными при лечении индивидуумов, переносящих вирусную инфекцию, в частности инфекцию RSV, и для профилактики таких инфекций. В основном соединения по настоящему изобретению могут быть пригодными при лечении теплокровных животных, инфицированных вирусами, в частности, респираторно-синцитиальным вирусом. Таким образом, соединения по настоящему изобретению или его любому варианту осуществления можно применять в качестве лекарственных средств. Указанное применение в качестве лекарственного средства или способ лечения включает системное введение индивидуумам, инфицированным вирусом,или индивидуумам, подверженным вирусным инфекциям, количества, эффективного для борьбы с состояниями, ассоциированными с вирусной инфекцией, в частности инфекцией RSV. Настоящее изобретение также относится к применению соединений по настоящему изобретению или их любого варианта осуществления при получении лекарственного средства для лечения или предотвращения вирусные инфекций, в частности инфекции RSV. Кроме того, раскрывается способ лечения теплокровного животного, инфицированного вирусом или подверженного риску инфицирования вирусом, в частности, RSV, включающий введение противовирусно эффективного количества соединения формулы I, как указано в настоящем описании, или соединения любого из вариантов осуществления соединений формулы I, как указано в настоящем описа-8 024681 нии. В основном предполагают, что противовирусно эффективное суточное количество составляет от 0,01 до 500 мг/кг массы тела, более предпочтительно от 0,1 до 50 мг/кг массы тела. Может оказаться подходящим введение необходимой дозы в виде двух, трех, четырех или более частей дозы через соответствующие интервалы в течение суток. Указанные части дозы можно формулировать в виде единичных дозированных форм, например, содержащих от 1 до 1000 мг, и частности от 5 до 200 мг активного ингредиента на единичную дозированную форму. Точное дозирование и частота введения зависят от конкретного используемого соединения формулы I, конкретного состояния, которое подлежит лечению, тяжести состояния, которое подлежит лечению, возраста, массы, пола, степени расстройства и общего физического состояния конкретного пациента, а также другого лекарственного средства, которое может принимать индивидуум, как хорошо известно специалистам в данной области. Кроме того, очевидно, что указанное эффективное суточное количество можно уменьшать или увеличивать в зависимости от реакции индивидуума, получающего лечение,и/или в зависимости от оценки врача, предписывающего соединения по настоящему изобретению. Таким образом, диапазоны эффективного суточного количества, указанные выше в настоящем описании, являются только рекомендательными. Также в качестве лекарственного средства можно использовать комбинацию другого противовирусного средства и соединения формулы I. Таким образом, раскрывается также продукт, содержащий(а) соединение формулы I и (b) другое противовирусное соединение, в качестве объединенного препарата для совместного, раздельного или последовательного применения при противовирусном лечении. Можно комбинировать различные лекарственные средства в одном препарате совместно с фармацевтически приемлемым носителем. Например, соединения по настоящему изобретению можно комбинировать с интерфероном бета или фактором некроза опухоли альфа для лечения или предотвращения инфекций RSV. Далее в настоящем описании изобретение проиллюстрировано со ссылкой на следующие ниже неограничивающие примеры. Пример 1. Ниже приведено подробное описание синтеза репрезентативных примеров по изобретению. Схема 5 Синтез 1-циклопропил-1 Н-имидазо[4,5-с]пиридин-2(3 Н)-она 5-d Стадия 1. Синтез N-циклопропил-3-нитропиридин-4-амина 5-b. 4-Метокси-3-нитропиридин 5-а (CAS 31872-62-5) (200 г, 1300 ммоль), циклопропиламин (CAS 76530-0) (185,5 г, 3250 ммоль) и DIEA (CAS 7087-68-5) (336 г, 2600 ммоль) в сухом этаноле (800 мл) кипятили с обратным холодильником в течение 3 ч. Смесь охлаждали до 0 С. Твердое вещество собирали фильтрацией. Осадок на фильтре промывали холодным этанолом (150 мл). Твердое вещество сушили до получения соединения 5-b в виде белого порошка (167 г, 72%). Стадия 2. Синтез N4-циклопропилпиридин-3,4-диамина 5-с. 5-b (167 г, 932 ммоль) в этаноле (1400 мл) подвергали гидрированию (50 фунт/дюйм 2) при 20 С в присутствии влажного 10% Pd/C (34 г) в качестве катализатора в течение ночи. После поглощения Н 2 (3 экв.) катализатор отфильтровывали и фильтрат выпаривали. Остаток промывали МТВЕ до получения соединения 5-с в виде желтого порошка (133 г, 95%). Стадия 3. Синтез 1-циклопропил-1 Н-имидазо[4,5-с]пиридин-2(3 Н)-она 5-d.(1800 мл) при 0 С. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 1 ч. Твердое вещество собирали фильтрацией и промывали CH3CN (200 мл) до получения соединения 5-d в виде белого порошка (101 г, 65%). Схема 6 Синтез 2-(хлорметил)имидазо[1,2-а]пиридина 6-с Стадия 1. Синтез 2-(хлорметил)имидазо[1,2-а]пиридина 6-с. Раствор 1,3-дихлорацетона (CAS 534-07-6) (14,8 г, 116,9 ммоль) в абсолютном этаноле (210 мл) перемешивали в 500 мл колбе, оснащенной магнитной мешалкой, обратным холодильником и щелью для пропускания воздуха. К реакционной смеси добавляли 2-аминопиридин 6-a (CAS 504-29-0) (10 г, 106,3 ммоль) при комнатной температуре. Затем смесь нагревали до кипения с обратным холодильником в течение ночи. Реакционную смесь концентрировали, отбирали остаток в воде (300 мл) и подщелачивали до рН 9 насыщенным раствором Na2CO3. Раствор экстрагировали дихлорметаном (3250 мл) и отмывали объединенные органические слои насыщенным солевым раствором (300 мл), сушили над MgSO4, фильтровали и концентрировали. Продукт очищали колоночной флэш-хроматографией, элюируя в градиенте дихлорметана/метанола от 0,1 до 2,5%. Концентрированием фракций получали продукт 6-с в виде розоватого твердого вещества (4,7 г,27%). Стадия 2. Синтез 1-циклопропил-3-(3-(2-циклопропилэтил)имидазо[1,2-а]пиридин-2-ил)метил-1 Нимидазо[4,5-с]пиридин-2(3 Н)-она Р 3. Схема 7 Синтез 1-циклопропил-3-3-(2-циклопропилэтил)имидазо[1,2-а]пиридин-2-ил)метил)-1Hимидазо[4,5-с]пиридин-2(3H)-она Р 3 Стадия 1. Синтез 1-циклопропил-3-(имидазо[1,2-а]пиридин-2-илметил)-1 Н-имидазо[4,5-с]пиридин 2(3 Н)-она 7-а. К раствору 5-d (4,5 г, 25,6 ммоль) в сухом DMF (100 мл) добавляли 60% дисперсию NaH (CAS 764669-7) (1,1 г, 28,2 ммоль) при 0 С. Немедленно происходило выделение газа. Реакционную смесь перемешивали при 0 С под аргоном в течение 30 мин. Раствор 6-с (4,7 г, 28,2 ммоль) в сухом DMF (25 мл) добавляли к реакционной смеси. Смесь нагревали до комнатной температуры и перемешивали под аргоном в течение ночи. К остатку добавляли воду (250 мл). Смесь экстрагировали этилацетатом и органический слой сушили над Na2SO4, фильтровали и выпаривали. Продукт перекристаллизовывали в ацетонитриле до получения продукта 7-а в виде розоватого твердого вещества (3,19 г, 40%). Стадия 2. Синтез 1-циклопропил-3-3-йодимидазо[1,2-а]пиридин-2-ил)метил)-1 Н-имидазо[4,5 с]пиридин-2(3 Н)-она 7-b. К раствору 7-а (2,5 г, 8,29 ммоль) в сухом CH3CN (16 мл) добавляли N-йодсукцинимид (CAS 51612-1) (2,1 г, 9,11 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. В ходе реакции образовывался осадок. Реакционную смесь охлаждали до 0 С для обеспечения полного осаждения. Затем полученное твердое вещество отфильтровывали и промывали охлажденным ацетонитрилом. Полученные твердые вещества собирали и тщательно сушили. Так получали соединение 7-b в виде белого твердого вещества (3,2 г, 91%). Стадия 3. Синтез 1-циклопропил-3-3-(циклопропилэтинил)имидазо[1,2-а]пиридин-2-ил)метил)1 Н-имидазо[4,5-с]пиридин-2(3 Н)-она 7-с. К суспензии 7-b (500 мг, 1,16 ммоль) в сухом DMF (11,6 мл) добавляли дихлорбис(трифенилфосфин)палладий (CAS 13965-03-2) (244,8 мг, 0,36 ммоль), триэтиламин (CAS 121-44-8)(0,80 мл, 5,80 ммоль и йодид меди(I) (CAS 7681-65-4) (66,3 мг, 0,36 ммоль). Затем очень медленно добавляли циклопропилацетилен (CAS 6746-94-7) (0,36 мл, 4,17 ммоль), и перемешивали реакционную смесь при комнатной температуре в атмосфере азота в течение 2 суток. Смесь концентрировали на силикагеле и очищали колоночной флэш-хроматографией, элюируя в градиенте дихлорметана/7 н. NH3 в метаноле, начиная от 1 до 7,5%. Собранные фракции объединяли и концентрировали с получением соединения 7-с в виде желтоватой пены (235 мг, 49%). Стадия 4. Синтез 1-циклопропил-3-3-(2-циклопропилэтил)имидазо[1,2-а]пиридин-2-ил)метил)-1 Нимидазо[4,5-с]пиридин-2(3 Н)-она Р 3. Катализатор 10% Pd/C (100 мг) и 4% раствор тиофена (0,1 мл) суспендировали в метаноле (100 мл) в атмосфере азота. Затем добавляли 7-с (230 мг, 0,56 ммоль). Реакционную смесь перемешивали при 25 С в атмосфере водорода до поглощения 2 экв. водорода. Катализатор удаляли фильтрованием на дикалите. Неочищенный раствор концентрировали и очищали колоночной флэш-хроматографией, элюируя в градиенте дихлорметана/7 н. аммиака в метаноле (от 0 до 6%). Собранные фракции выпаривали до получения продукта Р 3 в виде белой пены (49,9 мг, 23%). Пример 2. Синтез 3-7-хлор-3-(4-гидроксибутил)имидазо[1,2-а]пиридин-2-ил)метил)-1-циклопропил-1 Нимидазо[4,5-с]пиридин-2(3 Н)-она Р 1. Схема 8 Синтез (7-хлор-3-(4-(триизопропилсилилокси)бутил)имидазо[1,2-а]пиридин-2-ил)метанола 8-k Стадия 1. Синтез 2-амино-4-хлор-1-(3-этокси-2,3-диоксопропил)пиридинийбромида 8-с. 4-Хлорпиридин-2-амин (CAS 19798-80-2) (47 г, 367 ммоль) и этил-3-бромо-2-оксопропаноат (CAS 70-23-5) (98 г, 487 ммоль) в DME (540 мл) перемешивали в течение 1 ч. Осадок фильтровали и промывали простым трет-бутилметиловым эфиром с получением продукта 8-с виде желтого порошка (98 г, 82%). Стадия 2. Синтез этил-7-хлоримидазо[1,2-a]пиридин-2-карбоксилатгидробромида 8-d. Промежуточное соединение 8-с (98 г, 303 ммоль) растворяли в этаноле (600 мл) и кипятили с обратным холодильником в течение 3 ч. Реакционную смесь выпаривали и остаток растирали в этаноле(100 мл) и фильтровали. Осадок промывали простым трет-бутилметиловым эфиром и сушили до получения соединения 8-d в виде желтого порошка (66 г, 71%). Стадия 3. Синтез этил-7-хлоримидазо[1,2-а]пиридин-2-карбоксилата 8-е. Промежуточное соединение 8-d (51 г, 120 ммоль) растворяли в воде (750 мл). Осторожно добавляли порошок Na2CO3 до достижения рН 8. Твердое вещество фильтровали и промывали Н 2 О (100 мл) и простым трет-бутилметиловым эфиром. Остаток сушили в высоком вакууме до получения соединения 8-е в виде белого порошка (21 г, 78%). Стадия 4. Синтез этил-7-хлор-3-йодимидазо[1,2-а]пиридин-2-карбоксилата 8-f. Твердое вещество 8-е (10 г, 44,4 ммоль) растворяли в CH2Cl2 (200 мл). Затем добавляли NIS (CAS 516-12-1) (20 г, 88,8 ммоль) при 0 С и перемешивали реакционную смесь при комнатной температуре в течение 3 ч. Реакционную смесь промывали насыщенным раствором Na2SO3 (100 мл) и 10% растворомK2CO3 (100 мл). Органический слой сушили над Na2SO4, фильтровали и выпаривали в вакууме до получения соединения 8-f в виде белого порошка (13,2 г, 85%). Стадия 5. Синтез этил-7-хлор-3-(4-гидроксибут-1-инил)имидазо[1,2-а]пиридин-2-карбоксилата 8-g. Смесь из промежуточного соединения 8-f (8,75 г, 25 ммоль), 3-бутин-1-ола (CAS 927-74-2) (10,5 г,150 ммоль), (PhCN)2PdCl2 (0,95 г, 2,5 ммоль) и триэтиламина (CAS 121-44-8) (14,5 мл, 150 ммоль) дегазировали N2 и кипятили с обратным холодильником в течение 3 ч в атмосфере N2. Растворитель отгоняли в вакууме, и остаток очищали колоночной флэш-хроматографией, элюируя петролейным эфиром/этилацетатом (1:3). Растворитель выпаривали, полученное твердое вещество промывали простым трет-бутилметиловым эфиром и сушили в высоком вакууме до получения соединения 8-g в виде белого твердого вещества (4,75 г, 66%). Стадия 6. Синтез этил-7-хлор-3-(4-(триизопропилсилилокси)бут-1-инил)имидазо[1,2-а]пиридин-2 карбоксилата 8-h. Смесь из соединения 8-g (1,9 г, 6,5 ммоль) и имидазола (CAS 288-32-4) (1,37 г, 19,5 ммоль) в сухомCH2Cl2 (40 мл) охлаждали на ледяной водяной бане. Затем капельно добавляли TIPS-C1 (CAS 13154-240) (1,87 г, 9,8 ммоль) при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь промывали водой и насыщенным солевым раствором. Органический слой сушили и выпаривали. Остаток очищали колоночной флэш-хроматографией, элюируя в градиенте, начиная с чистого петролейного эфира, продолжая этилацетатом/петролейным эфиром (1:4). После выпаривания фракций получали продукт 8-h в виде белого твердого вещества (2,8 г, 96%). Стадия 7. Синтез этил-7-хлор-3-(4-(триизопропилсилилокси)бутил)имидазо[1,2-а]пиридин-2 карбоксилата 8-i. Соединение 8-h (2 г, 4,4 ммоль) в метаноле подвергали гидрированию (1 атмосфера) в присутствии 5% Pd на BaSO4 (2 г) в качестве катализатора при 0 С в течение 6 ч. После поглощения Н 2 (2 экв.) смесь фильтровали. Фильтрат концентрировали в вакууме и очищали остаток препаративной ВЭЖХ (колонка:Grace, 5 мкм, 25200 мм, элюирование в градиенте CH3CN/вода от 83 до 100%, в присутствии 0,5% TFA,скорость потока 25 мл/мин). Собранные фракции объединяли и нейтрализовали насыщенным NaHCO3. Органический растворитель отгоняли в вакууме. Полученную водную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель отгоняли в вакууме до получения соединения 8-i в виде белого твердого вещества (0,4 г, 20%). Стадия 8. Синтез 7-хлор-3-(4-(триизопропилсилилокси)бутил)имидазо[1,2-а]пиридин-2-карбоновой кислоты 8-j. Промежуточное соединение 8-i (0,4 г, 0,88 ммоль) и LiOHH2O (CAS 1310-66-3) (0,10 г, 2,4 ммоль) суспендировали в смеси THF (4 мл), метанола (4 мл) и воды (4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 8 ч. Затем добавляли 1 н. раствор HCl для подкисления смеси до рН 5. Смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель отгоняли в вакууме до получения соединения 8-j в виде белого твердого вещества (0,32 г, 86%). Стадия 9. Синтез(7-хлор-3-(4-(триизопропилсилилокси)бутил)имидазо[1,2-а]пиридин-2 ил)метанола 8-k. Продукт 8-j (0,32 г, 0,75 ммоль) в сухом THF (10 мл) охлаждали на ледяной водяной бане. К этой охлажденной смеси капельно добавляли NMM (CAS 109-02-4) (0,15 г, 1,5 ммоль) и изобутилхлорформиат (CAS 543-27-1) (0,15 г, 1,1 ммоль). Смесь перемешивали при -10 С в течение 30 мин. Затем добавлялиNaBH4 (CAS 16940-66-2) (0,08 г, 2,2 ммоль) и опять перемешивали при -10 С в течение 30 мин. Капельно добавляли воду для гашения реакции и перемешивали полученную смесь в течение 1 ч. Смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором и сушили надNa2SO4. Растворитель отгоняли в вакууме и очищали остаток препаративной ТСХ, элюируя петролейным эфиром/этилацетатом (1:1) с получением продукта 8-k в виде белого порошка (0,1 г, 30%). Стадия 10 и 11. Синтез 3-7-хлор-3-(4-гидроксибутил)имидазо[1,2-а]пиридин-2-ил)метил)-1 циклопропил-1 Н-имидазо[4,5-с]пиридин-2(3 Н)-она Р 1. Промежуточное соединение 8-k (0,1 г, 0,24 ммоль), промежуточное соединение 5-d (0,085 г, 0,48 ммоль) и PBU3 (CAS 998-40-3) (0,145 г, 0,72 ммоль) растворяли в сухом THF, охлаждали на ледяной метанольной бане и дегазировали N2. Капельно добавляли DIAD (CAS 2446-83-5) (0,145 г, 0,72 ммоль) и смесь кипятили с обратным холодильником под N2 в течение 2 ч. Растворитель отгоняли в вакууме, и остаток очищали колоночной флэш-хроматографией, элюируя этилацетатом. После выпаривания фракций получали 0,2 г продукта 8-I в виде белой пены, но которая была загрязнена 50% PBU3O. Промежуточное соединение 8-I и TBAF-3H2O (CAS 429-41-4) (0,15 г, 0,47 ммоль) в THF (2 мл) перемешивали при 40 С в течение 20 мин. Растворитель отгоняли в вакууме. Твердый остаток промывали водой, простым трет-бутилметиловым эфиром и CH3CN. После тщательной сушки получали продукт Р 1 в виде белого порошка (62,0 мг, 62% общего выхода). 2-(Хлорметил)имидазо[1,2-а]пиразин 9-а синтезировали согласно протоколу, описанному для синтеза 2-(хлорметил)имидазо[1,2-а]пиридина 6-с, используя аминопиразин вместо 2-аминопиридина, и получали в виде кремообразного твердого вещества с выходом продукта 12%, m/z=168 (M+H)+. Синтез 1-циклопропил-3-3-йодимидазо[1,2-а]пиразин-2-ил)метил)-1 Н-имидазо[4,5-с]пиридин 2(3 Н)-она 10-а. 1-Циклопропил-3-3-йодимидазо[1,2-а]пиразин-2-ил)метил)-1 Н-имидазо[4,5-с]пиридин-2(3 Н)-он 10-а синтезировали двухстадийным способом, описанным для синтеза 7-b, используя 2(хлорметил)имидазо[1,2-а]пиразин 9-а вместо 2-(хлорметил)имидазо[1,2-а]пиридина 6-с, и получали в виде кремообразного твердого вещества, m/z=433 (M+H)+. Пример 4. Синтез (Е)-3-3-(4-(трет-бутилдиметилсилилокси)бут-1-енил)имидазо[1,2-а]пиразин-2-ил)метил)-1 циклопропил-1 Н-имидазо[4,5-с]пиридин-2(3 Н)-она 11-а Суспензию 1-циклопропил-3-3-йодимидазо[1,2-а]пиразин-2-ил)метил)-1 Н-имидазо[4,5-с]пиридин 2(3 Н)-она 10-а (500 мг, 1,076 ммоль), (Е)-трет-бутилдиметил(4-(4,4,5,5-тетраметил-1,3,2-диоксоборолан 2-ил)бут-3-енилокси)силана (672 мг, 2 экв.), карбоната натрия (342 мг, 3 экв.) и PdCl2 (dppf) (39 мг, 0,05 экв., CAS 72287-26-4) смешивали в DME/воде (5 мл/1 мл) и перемешивали при 100 С в течение 2 ч. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли 20 мл DCM, фильтровали на дикалите и выпаривали. Остаток очищали флэш-хроматографией с использованием градиента МеОН 05% в DCM, и получали желаемый продукт 11-а в виде коричневатого масла с выходом продукта 90%,m/z=491 (M+H)+. Пример 5. Синтез 1-циклопропил-3-3-(4-гидроксибутил)имидазо[1,2-а]пиразин-2-ил)метил)-1 Н-имидазо[4,5 с]пиридин-2(3 Н)-она Р 17 Смесь (Е)-3-3-(4-(трет-бутилдиметилсилилокси)бут-1-енил)имидазо[1,2-а]пиразин-2-ил)метил)-1 циклопропил-1 Н-имидазо[4,5-с]пиридин-2(3 Н)-он 11-а (480 мг, 0,978 ммоль) и Pd/C 10% (52 мг, 0,05 экв.) в МеОН (20 мл) гидрировали в течение 2 ч. Затем реакционную смесь фильтровали на дикалите и концентрировали досуха. Полученное белое твердое вещество (480 мг, 84%) использовали непосредственно на следующей стадии. Его опять растворяли в МеОН и добавляли фторид аммония (39 мг, 1,1 экв.). Затем реакционную смесь нагревали при 60 С в течение ночи. После концентрирования неочищенное вещество очищали препаративной ВЭЖХ (RP Vydac Denali C18 - 10 мкм, 250 г, 5 см) со следующей подвижной фазой (0,25% раствор NH4HCO3 в воде, CH3CN) с получением целевого продукта Р 17 с выходом продукта 87% (320 мг), m/z=491 (M+H)+. 1 Н ЯМР (400 МГц, ДМСО-d6)м.д. 0,82-0,94 (м, 2 Н), 0,99-1,10 (м, 2 Н), 1,36-1,46 (м, 2 Н), 1,46-1,59(500 мг, 1,321 ммоль) и триэтиламинтригидрофторида (319 мг, 1,5 экв.) в DCM (20 мл) перемешивали при комнатной температуре в атмосфере N2 в течение 60 мин. Затем добавляли 100 мл насыщенногоNaHCO3 и перемешивали смесь до прекращения выделения газа (10 мин), затем экстрагировали с 150 млDCM (2). Объединенные органические слои сушили на Na2SO4, фильтровали и выпаривали досуха. Очисткой препаративной ВЭЖХ (RP Vydac Denali C18 - 10 мкм, 250 г, 5 см) с (0,25% растворомNH4HCO3 в воде, МеОН) в качестве подвижной фазы получали целевое соединение Р 18 с выходом продукта 20%, m/z=381 (М+Н)+. 1 Раствор 3-7-хлор-3-йодимидазо[1,2-а]пиридин-2-ил)метил)-1-циклопропил-1 Н-имидазо[4,5 с]пиридин-2(3 Н)-она 12-а (получаемый трехстадийным синтезом, применяемым для 7-b, с использованием 4-хлор-2-аминопиридина вместо 2-аминопиридина на стадии 1) и Et3N (2,218 мл, 8 экв.) в DMF (30 мл) дегазировали азотом в течение 15 мин. Затем добавляли ацетат палладия (45 мг, 0,1 экв.), трифенилфосфин (173 мг, 0,33 экв.) и 1-морфолинопроп-2-ен-1-он (2,516 мл, 10 экв.) и проводили перемешивание в закрытом сосуде при 80 С в течение 2 ч. После охлаждения смесь гасили ледяной водой. После перемешивания в течение одного часа осадок отфильтровывали и сушили в вакууме. Твердое вещество очищали на диоксиде кремния дихлорметаном/метанолом-NH3 98/2 в качестве элюента с получением целевого соединения Р 24 с выходом продукта 92% (884 мг), m/z=479 (M+H)+. Суспензию (Е)-3-7-хлор-3-(3-морфолино-3-оксопроп-1-енил)имидазо[1,2-а]пиридин-2-ил)метил)1-циклопропил-1 Н-имидазо[4,5-с]пиридин-2(3 Н)-она Р 24 (880 мг, 1,837 ммоль), Pd/C 10% (195 мг, 0,1 экв.) и дифенилсульфида (0,03 мл, 0,1 экв.) в MeOH/THF (150 мл, 1/1 смесь) гидрировали при комнатной температуре в течение 4 ч. Затем отфильтровывали катализатор на дикалите в токе азота, и выпаривали фильтрат досуха. Остаток растирали в ацетонитриле/простом изопропиловом эфире 1/1. Осадок собирали фильтрованием, сушили в вакууме и очищали препаративной ВЭЖХ (RP Vydac Denali C18 - 10 мкм, 200 г, 5 см) (0,25% раствором NH4HCO3 в воде, MeOH+CH3CN) в качестве подвижной фазы с выходом продукта 94 мг (10%) Р 26 и 241 мг (29%) Р 27 в виде белых твердых веществ, m/z (Р 26)=481 (М+Н)+; m/z(P27)=447 (M+H)+. Пример 9. Характеристика соединений и тест на ингибирующую активность в отношении RSV. Анализ ВЭЖХ-МС проводили одним из следующих ниже способов. Способ 1. Измерения ВЭЖХ проводили с использованием модуля Agilent 1100, включающего насос, детектор на диодной матрице (DAD) (используемая длина волны 220 нм), нагреватель колонки и колонку, как указано ниже. Поток из колонки разделялся на Agilent MSD серий G1946C и G1956A. Детектор МС был оснащен API-ES (ионизация распылением в электрическом поле при атмосферном давлении). Массспектры получали сканированием от 100 до 1000. Напряжение иглы капилляра составляло 2500 В для положительного режима ионизации и 3000 В для отрицательного режима ионизации. Напряжение фрагментации составляло 50 В. Температуру сушильного газа поддерживали при 350 С при потоке 10 л/мин. ВЭЖХ с обращенными фазами проводили на устройстве YMC-Pack ODS-AQ, колонке 502,0 мм 5 мм со скоростью потока 0,8 мл/мин. Использовали две подвижные фазы (подвижная фаза А: вода с 0,1% TFA,подвижная фаза В: ацетонитрил с 0,05% TFA). Сначала удерживали 100% А в течение 1 мин. Затем применяли градиент к 40% А и 60% В в течение 4 мин и удерживали в течение 2,5 мин. Использовали характерные объемы впрыска 2 мл. Температура термостата составляла 50 С (МС полярность: положительная). Способ 2. Измерения ВЭЖХ проводили с использованием модуля Agilent 1100, включающего насос, детектор на диодной матрице (DAD) (используемая длина волны 220 нм), нагреватель колонки и колонку, как указано ниже. Поток из колонки разделялся на Agilent MSD серий G1946C и G1956A. Детектор МС был оснащен API-ES (ионизация распылением в электрическом поле при атмосферном давлении). Массспектры получали сканированием от 100 до 1000. Напряжение иглы капилляра составляло 2500 В для положительного режима ионизации и 3000 В для отрицательного режима ионизации. Напряжение фрагментации составляло 50 В. Температуру сушильного газа поддерживали при 350 С при потоке 10 л/мин. ВЭЖХ с обращенными фазами проводили на устройстве YMC-Pack ODSAQ, колонке 502,0 мм 5 мм со скоростью потока 0,8 мл/мин. Использовали две подвижные фазы (подвижная фаза А: вода с 0,1% TFA, подвижная фаза В: ацетонитрил с 0,05% TFA). Сначала удерживали 90% А и 10% В в течение 0,8 мин. Затем применяли градиент к 20% А и 80% В в течение 3,7 мин и удерживали в течение 3 мин. Использовали характерные объемы впрыска 2 мл. Температура термостата составляла 50 С. (МС полярность: положительная). Способ 3. Колонка: XTerra MS C18 2,5 мкм, 4,650 мм, подвижная фаза А: 10 мМ NH4OOCH+0,1% HCOOH в воде, подвижная фаза В: метанол, поддерживаемые при температуре колонки 50 С при скорости потока 1,5 мл/мин. Условия градиента: t=0 мин: 65% А, 35% В; t=3,5 мин: 5% А, 95% В; t=5,5 мин: 5% А, 95% В; Способ 4. Колонка: SunFire C18 3,5 мкм 4,6100 мм, подвижная фаза А: 10 мМ NH4OOCH+0,1% HCOOH в воде, подвижная фаза В: метанол, поддерживаемые при температуре колонки 50 С при скорости потока 1,5 мл/мин. Условия градиента: t=0 мин: 65% А, 35% В; t=7 мин: 5% А, 95% В; t=9,6 мин: 5% А, 95% В;t=9,8 мин: 65% А, 35% В; t=12 мин: 65% А, 35% В. Спектры ЯМР регистрировали спектрометром Bruker Avance 400, работающим при 400 МГц для 1 Н. Химические сдвиги даны в м.д. и значение J в Гц. Мультиплетность указывали с использованием следующих сокращений: д для дуплета, т для триплета, м для мультиплета и т.д. Тонкослойную хроматографию (ТСХ) проводили на пластинах алюминия 510 см, покрытых силикагелем 60 F254 (Merck KGaA). Соединения тестировали на ингибиторную активность в отношении RSV. Результаты приведены в табл. 1 и 2 ниже со ссылкой на формулы Ia и Ib. Таблица 1 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы I или его аддитивная соль где X представляет собой С или N, когда X находится в положении 5, и X представляет собой С, когда X находится в любом другом положении, отличном от положения 5;Y находится в любом другом положении, отличном от положения 7';R4 присутствует и представляет собой Н, когда Y представляет собой С; каждый R7 и R8 независимо выбран из Н, C1-C10-алкила, или R7 и R8, взятые вместе, образуют 6 членное гетероциклическое кольцо с атомами N и О;n представляет собой целое число от 2 до 6. 2. Соединение по п.1, где R2 выбран из группы, состоящей из Н, -(CR7R8)n-R9 и -CC-CH2-R9. 3. Соединение по п.1 или 2, где все атомы X представляют собой С. 4. Соединение по пп.1, 2 или 3, где X-R1 в пара-положении к N, образующему мостиковую связь имидазольного и пиридинового кольца, выбран из группы, состоящей из С-Н, C-Cl и С-Br, и все другиеR1 представляют собой Н. 5. Соединение по любому из предшествующих пунктов, где R7 и R8 представляют собой Н и n равно 2-4. 6. Соединение по любому из предшествующих пунктов, где R9 выбран из группы, состоящей из ОН,F и вторичного C1-C6-алкила. 7. Соединение по любому из предшествующих пунктов, где R2 представляет собой -CC-CH2-R9,где R9 выбран из группы, состоящей из C1-C6-алкокси, C1-C6-алкила с неразветвленной цепью и C1-C6 алкила с разветвленной цепью. 8. Соединение по любому из предшествующих пунктов, где R3 представляет собой C3-C7 циклоалкил. 9. Соединение по любому из предшествующих пунктов, где R3 представляет собой циклопропил. 10. Соединение по любому из предшествующих пунктов, где R3 представляет собой изопропил. 11. Соединение по любому из предшествующих пунктов, где Y в пара-положении к N-R3 представ- 21024681 ляет собой С. 12. Применение соединения по любому из пп.1-11 в качестве лекарственного средства для лечения респираторно-синцитиальных вирусных инфекций. 13. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения по любому из пп.1-11. 14. Способ получения фармацевтической композиции по п.13, где указанный способ включает тщательное смешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения по любому из пп.1-11. 15. Применение соединения по любому из пп.1-11 в качестве лекарственного средства для ингибирования репликации респираторно-синцитиального вируса. 16. Применение соединения по любому из пп.1-11 для получения лекарственного средства для ингибирования репликации респираторно-синцитиального вируса.
МПК / Метки
МПК: C07D 471/04, A61K 31/437, C07D 487/04, A61P 31/12
Метки: качестве, противовирусных, респираторно-синцитиального, имидазопиридины, средств, против, вируса
Код ссылки
<a href="https://eas.patents.su/23-24681-imidazopiridiny-v-kachestve-protivovirusnyh-sredstv-protiv-respiratorno-sincitialnogo-virusa.html" rel="bookmark" title="База патентов Евразийского Союза">Имидазопиридины в качестве противовирусных средств против респираторно-синцитиального вируса</a>
Азабензимидазолы в качестве противовирусных средств в отношении респираторного синцитиального вируса

Номер патента: 21613
Опубликовано: 30.07.2015
Авторы: Тахри Абделлах, Ху Лили, Вендевилль Сандрин Мари Элен, Коиманс Людвиг Поль, Демэн Самюэль Доминик, Йонкерс Тим Хьюго Мария, Рабуассон Пьер Жан-Мари Бернар
МПК: A61K 31/437, C07D 471/04, A61P 31/12...
Метки: вируса, синцитиального, респираторного, средств, отношении, азабензимидазолы, противовирусных, качестве
Формула / Реферат:
1. Соединение формулы I или его аддитивная сольгде каждый X независимо представляет собой С или N, по меньшей мере один X = N;каждый Y независимо представляет собой С или N;R1 присутствует в тех случаях, когда X = С, и R1 выбран из группы Н, галогена, N(R5)2, CO(R6), C(=NH)NH2, CF3 и B(OH)2;R1 отсутствует в тех случаях, когда X = N;R2 представляет собой -(CR7R8)n-R9;R3 выбран из группы, состоящей из Н, C1-C10-алкила, C3-C7-циклоалкила, CH2CF3...
Производные бензимидазола и имидазопиридина в качестве ингибиторов репликации респираторно-синцитиального вируса

Номер патента: 4746
Опубликовано: 26.08.2004
Авторы: Вене Марк Гастон, Гийемон Жером Эмиль Жорж, Лякрамп Жан Фернан Арман, Янссенс Франс Эдуард, Андрис Кунрад Йозеф Лодевейк Марсель
МПК: C07D 401/14, A61K 31/4709, A61P 11/00...
Метки: вируса, репликации, качестве, производные, бензимидазола, респираторно-синцитиального, ингибиторов, имидазопиридина
Формула / Реферат:
1. Соединение формулы его аддитивная соль или стереохимически изомерная форма, где в указанной формуле -a1=a2-a3=a4- представляет двухвалентный радикал формулы -CH=CH-CH=CH- (a-1); -N=CH-CH=CH- (a-2); -CH=N-CH=CH- (a-3); -CH=CH-N=CH- (a-4) или -CH=CH-CH=N- (a-5); где каждый атом водорода в...
Морфолинилсодержащие бензимидазолы в качестве ингибиторов репликации респираторно-синцитиального вируса

Номер патента: 9876
Опубликовано: 28.04.2008
Авторы: Мейер Кристоф, Мюллер Филипп, Тиммерман Филип Мария Марта Берн, Фортэн Жером Мишель Клод, Виллебрордс Руди Эдмонд, Дубле Фредерик Марк Морис, Андрис Кунрад Йозеф Лодевейк, Бонфанти Жан-Франсуа, Жевер Том Валериус Жозефа
МПК: C07D 401/06, C07D 413/14, A61K 31/4184...
Метки: ингибиторов, респираторно-синцитиального, качестве, репликации, морфолинилсодержащие, бензимидазолы, вируса
Формула / Реферат:
1. Соединение, имеющее формулу его N-оксиды, аддитивные соли, четвертичные амины, комплексы с металлами и стереохимически изомерные формы, где G представляет собой прямую связь или C1-10алкандиил, необязательно замещенный одним или несколькими заместителями, индивидуально выбранными из группы заместителей, состоящей из гидрокси, C1-6алкилокси, Ar1C1-6алкилокси, C1-6алкилтио, Ar1C1-6алкилтио, НО(-CH2-CH2-O)n-, C1-6алкилокси(-CH2-CH2-O)n- или...
Бeнзимидазоловые ингибиторы респираторно-синцитиального вируса

Номер патента: 22972
Опубликовано: 31.03.2016
Авторы: Тахри Абделлах, Йонкерс Тим Хьюго Мария, Коиманс Людвиг Поль, Вендевилль Сандрин Мари Элен, Рабуассон Пьер Жан-Мари Бернар, Демэн Самюэль Доминик, Ху Лили
МПК: A61K 31/4188, A61P 11/00, C07D 235/26...
Метки: бeнзимидазоловые, ингибиторы, респираторно-синцитиального, вируса
Формула / Реферат:
1. Соединение, представленное формулой (I), его фармацевтически приемлемая соль присоединения кислот и оснований или стереохимический изомергде X представляет собой С или N;R1 представляет собой Н;R2 выбран из группы, состоящей из Br и Cl;R3 представляет собой -(CR6R7)n-R8;R4 выбран из группы, состоящей из С3-С7циклоалкила, С2-С10алкенила, CH2CF3 или -SO2CH3;R5 присутствует в том случае, когда X представляет собой С, где каждый R5 выбран...
Ингибиторы репликации респираторно-синцитиального вируса

Номер патента: 4939
Опубликовано: 28.10.2004
Авторы: Мерсман Катлен Петрус Мари-Жозе, Лякрамп Жан Фернан Арман, Янссенс Франс Эдуард, Андрис Кунрад Йозеф Лодевейк Марсель, Соммен Франсуа Мария, Гийемон Жером Эмиль Жорж
МПК: A61P 31/14, A61K 31/501, C07D 401/06...
Метки: ингибиторы, репликации, вируса, респираторно-синцитиального
Формула / Реферат:
1. Применение соединения для получения лекарственного средства для лечения вирусных инфекций, где указанное соединение представляет собой соединение формулы его пролекарство, N-оксид, аддитивную соль, четвертичный амин, комплекс с металлом или стереохимически изомерную форму, где в указанной формуле -a1=a2-a3=a4- представляет двухвалентный радикал формулы -CH=CH-CH=CH- (a-1); -N=CH-CH=CH- ...
Предыдущий патент: Композиция и способ для снижения агломерации гидратов
Следующий патент: Доильное устройство
Случайный патент: Автомобильное бензиновое топливо для двигателя внутреннего сгорания, способ работы двигателя и способ снижения выбросов