Способ стереоселективного синтеза бициклических гетероциклов
Номер патента: 23562
Опубликовано: 30.06.2016
Авторы: Остермайер Маркус, Сантагостино Марко, Дойблер Юрген, Хухлер Гюнтер, Клинг Штефан




Формула / Реферат
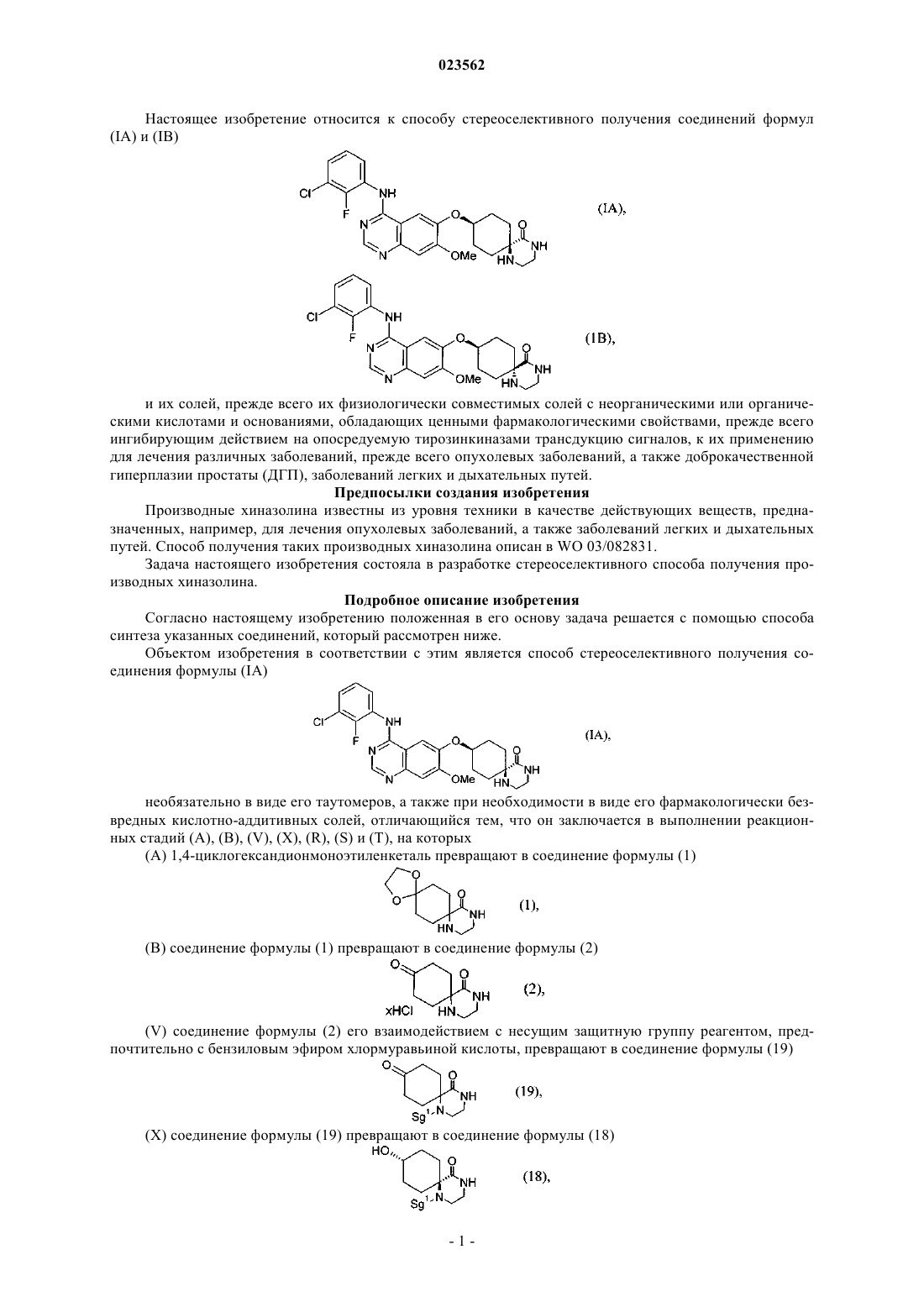
1. Способ стереоселективного получения соединения формулы (IA)

необязательно в виде его таутомеров, а также при необходимости в виде его фармакологически безвредных кислотно-аддитивных солей, отличающийся тем, что он заключается в выполнении реакционных стадий (А), (В), (V), (X), (R), (S) и (Т), на которых
(А) 1,4-циклогександионмоноэтиленкеталь превращают в соединение формулы (1)

(В) соединение формулы (1) превращают в соединение формулы (2)

(V) соединение формулы (2) его взаимодействием с несущим защитную группу реагентом превращают в соединение формулы (19)

(X) соединение формулы (19) превращают в соединение формулы (18)

(R) соединение формулы (18) его взаимодействием с соединением формулы (23)

превращают в соединение формулы (21)

(S) в соединении формулы (21) отщепляют защитные группы с получением соединения формулы (22)

и
(Т) соединение формулы (22) хлорируют и затем подвергают взаимодействию с 3-хлор-2-фторанилином,
при этом стадии (А)-(Т) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, а защитная группа Sg2 может представлять собой необязательно замещенный бензил.
2. Способ стереоселективного получения соединений формулы (1А), отличающийся тем, что он заключается в выполнении стадий (R), (S) и (Т), на которых
(R) соединение формулы (18) путем его взаимодействия с соединением формулы (23)

превращают в соединение формулы (21)

(S) в соединении формулы (21) отщепляют защитные группы с получением соединения формулы (22)

и
(Т) соединение формулы (22) хлорируют и затем подвергают взаимодействию с 3-хлор-2-фторанилином,
при этом стадии (R)-(T) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, а защитная группа Sg2 может представлять собой необязательно замещенный бензил.
3. Способ стереоселективного получения соединения формулы (18), отличающийся тем, что он заключается в выполнении стадий (А), (В), (V) и (X), на которых
(А) 1,4-циклогександионмоноэтиленкеталь превращают в соединение формулы (1)

(В) соединение формулы (1) превращают в соединение формулы (2)

(V) соединение формулы (2) путем его взаимодействия с несущим защитную группу реагентом превращают в соединение формулы (19)

(X) соединение формулы (19) превращают в соединение формулы (18)

при этом стадии (А), (В), (V) и (X) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил.
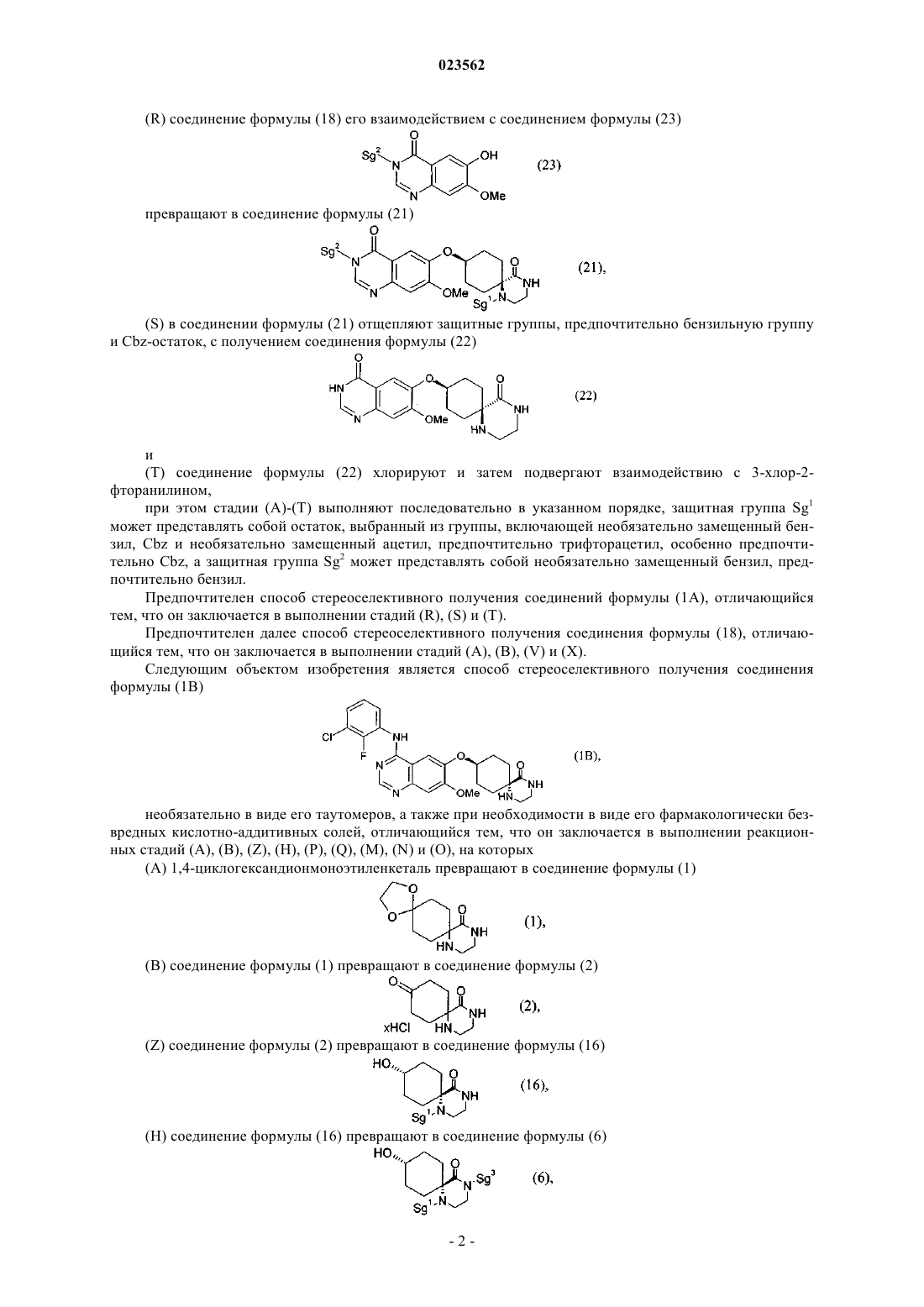
4. Способ стереоселективного получения соединения формулы (1В)

необязательно в виде его таутомеров, а также при необходимости в виде его фармакологически безвредных кислотно-аддитивных солей, отличающийся тем, что он заключается в выполнении реакционных стадий (А), (В), (Z), (Н), (Р), (Q), (M), (N) и (О), на которых
(А) 1,4-циклогександионмоноэтиленкеталь превращают в соединение формулы (1)

(В) соединение формулы (1) превращают в соединение формулы (2)

(Z) соединение формулы (2) превращают в соединение формулы (16)

(Н) соединение формулы (16) превращают в соединение формулы (6)

(Р) соединение формулы (6) его взаимодействием с соединением формулы (23)

превращают в соединение формулы (7)

(Q+M) в соединении формулы (7) отщепляют защитные группы с получением соединения формулы (12)

и
(N+О) хлорируют соединение формулы (12), превращая его в соединение формулы (13), и затем подвергают взаимодействию с 3-хлор-2-фторанилином,

при этом стадии (А)-(О) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, защитная группа Sg2 может представлять собой необязательно замещенный бензил, а защитная группа Sg3 может быть выбрана из группы, включающей Boc и аллилоксикарбонил.
5. Способ стереоселективного получения соединения формулы (1В), отличающийся тем, что он заключается в выполнении стадий (А), (В), (С), (D), (Е) или (F), (G), (P), (Q+M) и (N+О), на которых
(А) 1,4-циклогександионмоноэтиленкеталь превращают в соединение формулы (1)

(В) соединение формулы (1) превращают в соединение формулы (2)

(С) соединение формулы (2) превращают в соединение формулы (3)

(D) соединение формулы (3) превращают в соединение формулы (4)

(Е) или (F) соединение формулы (4) превращают в соединение формулы (5)

без выделения при этом соединения формулы (5) на стадии (F) и
(G) соединение формулы (5) превращают в соединение формулы (6)

(Р) соединение формулы (6) путем его взаимодействия с соединением формулы (23)

превращают в соединение формулы (7)

(Q+M) в соединении формулы (7) отщепляют защитные группы с получением соединения формулы (12)

и
(N+О) хлорируют соединение формулы (12), превращая его в соединение формулы (13), и затем подвергают взаимодействию с 3-хлор-2-фторанилином

при этом стадии (А)-(О) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, а защитная группа Sg3 может представлять собой остаток, выбранный из группы, включающей Boc и аллилоксикарбонил.
6. Способ стереоселективного получения соединения формулы (1В), отличающийся тем, что он заключается в выполнении стадий (А), (В), (С), (D), (Е) или (F), (G), (I), (J), (K), (L) и (N+О), на которых
(А) 1,4-циклогександионмоноэтиленкеталь превращают в соединение формулы (1)

(В) соединение формулы (1) превращают в соединение формулы (2)

(С) соединение формулы (2) превращают в соединение формулы (3)

(D) соединение формулы (3) превращают в соединение формулы (4)

(Е) или (F) соединение формулы (4) превращают в соединение формулы (5)

без выделения при этом соединения формулы (5) на стадии (F) и
(G) соединение формулы (5) превращают в соединение формулы (6)

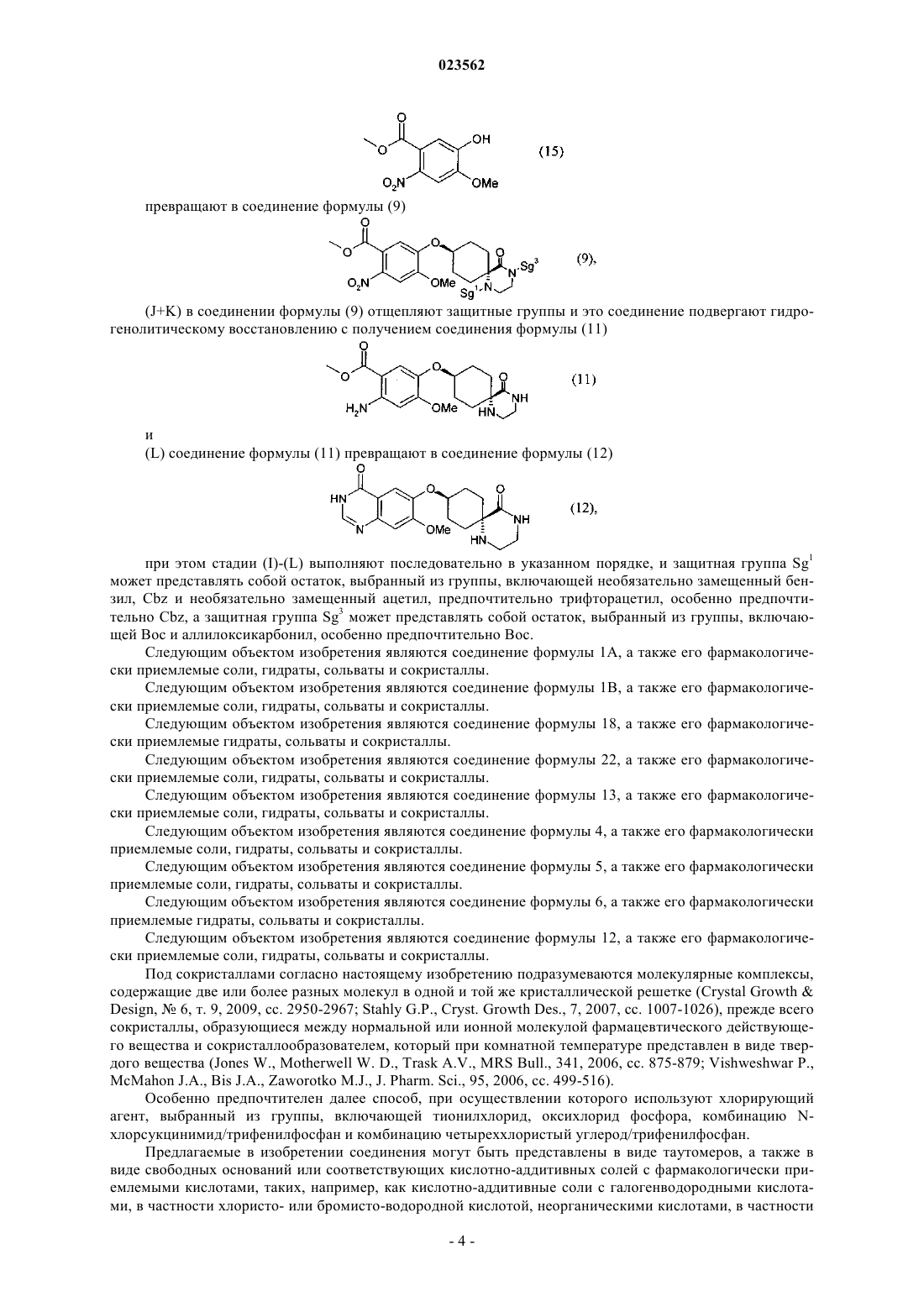
(I) соединение формулы (6) его взаимодействием с соединением формулы (15)

превращают в соединение формулы (9)

(J+K) в соединении формулы (9) отщепляют защитные группы и это соединение подвергают гидрогенолитическому восстановлению с получением соединения формулы (11)

и
(L) соединение формулы (11) превращают в соединение формулы (12)

и (N+О) хлорируют соединение формулы (12), превращая его в соединение формулы (13), и затем подвергают взаимодействию с 3-хлор-2-фторанилином,

при этом стадии (А)-(О) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, а защитная группа Sg3 может представлять собой остаток, выбранный из группы, включающей Boc и аллилоксикарбонил.
7. Соединение формулы (18)

в которой защитная группа Sg1 представляет собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил.
8. Соединение формулы (22)

9. Соединение формулы (13)

10. Соединение формулы (4)

в которой защитная группа Sg3 представляет собой остаток, выбранный из группы, включающей Boc и аллилоксикарбонил.
11. Соединение формулы (5)

в которой защитная группа Sg3 представляет собой остаток, выбранный из группы, включающей Boc и аллилоксикарбонил.
12. Соединение формулы (6)

в которой защитная группа Sg3 представляет собой остаток, выбранный из группы, включающей Boc и аллилоксикарбонил.
13. Соединение формулы (12)

Текст
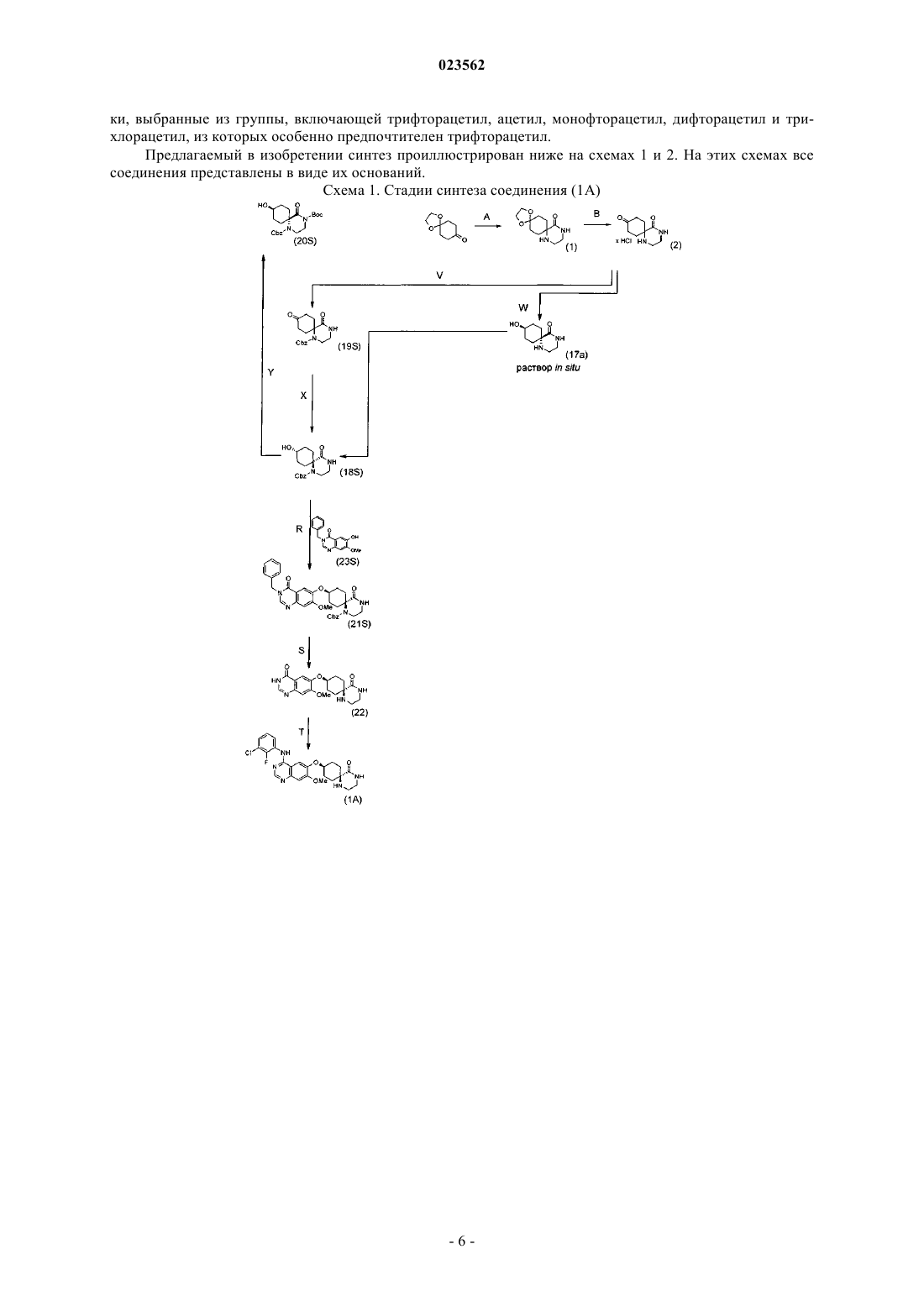
СПОСОБ СТЕРЕОСЕЛЕКТИВНОГО СИНТЕЗА БИЦИКЛИЧЕСКИХ ГЕТЕРОЦИКЛОВ(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) В изобретении описан способ стереоселективного получения соединений формул (1 А) и (1 В) и их солей, прежде всего их физиологически совместимых солей с неорганическими или органическими кислотами и основаниями, обладающих ценными фармакологическими свойствами, прежде всего ингибирующим действием на опосредуемую тирозинкиназами трансдукцию сигналов, а также описано их применение для лечения различных заболеваний,прежде всего опухолевых заболеваний, а также доброкачественной гиперплазии простаты,заболеваний легких и дыхательных путей. Настоящее изобретение относится к способу стереоселективного получения соединений формул и их солей, прежде всего их физиологически совместимых солей с неорганическими или органическими кислотами и основаниями, обладающих ценными фармакологическими свойствами, прежде всего ингибирующим действием на опосредуемую тирозинкиназами трансдукцию сигналов, к их применению для лечения различных заболеваний, прежде всего опухолевых заболеваний, а также доброкачественной гиперплазии простаты (ДГП), заболеваний легких и дыхательных путей. Предпосылки создания изобретения Производные хиназолина известны из уровня техники в качестве действующих веществ, предназначенных, например, для лечения опухолевых заболеваний, а также заболеваний легких и дыхательных путей. Способ получения таких производных хиназолина описан в WO 03/082831. Задача настоящего изобретения состояла в разработке стереоселективного способа получения производных хиназолина. Подробное описание изобретения Согласно настоящему изобретению положенная в его основу задача решается с помощью способа синтеза указанных соединений, который рассмотрен ниже. Объектом изобретения в соответствии с этим является способ стереоселективного получения соединения формулы (IA) необязательно в виде его таутомеров, а также при необходимости в виде его фармакологически безвредных кислотно-аддитивных солей, отличающийся тем, что он заключается в выполнении реакционных стадий (А), (В), (V), (X), (R), (S) и (Т), на которых(V) соединение формулы (2) его взаимодействием с несущим защитную группу реагентом, предпочтительно с бензиловым эфиром хлормуравьиной кислоты, превращают в соединение формулы (19)(S) в соединении формулы (21) отщепляют защитные группы, предпочтительно бензильную группу и Cbz-остаток, с получением соединения формулы (22)(Т) соединение формулы (22) хлорируют и затем подвергают взаимодействию с 3-хлор-2 фторанилином,при этом стадии (А)-(Т) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, предпочтительно трифторацетил, особенно предпочтительно Cbz, а защитная группа Sg2 может представлять собой необязательно замещенный бензил, предпочтительно бензил. Предпочтителен способ стереоселективного получения соединений формулы (1 А), отличающийся тем, что он заключается в выполнении стадий (R), (S) и (Т). Предпочтителен далее способ стереоселективного получения соединения формулы (18), отличающийся тем, что он заключается в выполнении стадий (А), (В), (V) и (X). Следующим объектом изобретения является способ стереоселективного получения соединения формулы (1 В) необязательно в виде его таутомеров, а также при необходимости в виде его фармакологически безвредных кислотно-аддитивных солей, отличающийся тем, что он заключается в выполнении реакционных стадий (А), (В), (Z), (H), (P), (Q), (М), (N) и (О), на которых(Q+M) в соединении формулы (7) отщепляют защитные группы с получением соединения формулы(N+О) соединение формулы (12) хлорируют и затем подвергают взаимодействию с 3-хлор-2 фторанилином,при этом стадии (А)-(О) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, предпочтительно трифторацетил, особенно предпочтительно Cbz, а защитная группа Sg2 может представлять собой необязательно замещенный бензил, предпочтительно бензил. Следующим объектом изобретения является способ стереоселективного получения соединения формулы (1 В), отличающийся тем, что вместо стадий [(Z), (Н)] выполняют стадии [(С), (D), (Е) или (F) и без выделения при этом соединения формулы (5) на стадии (F) и при этом стадии (C)-(G) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, предпочтительно трифторацетил, особенно предпочтительно Cbz, а защитная группа Sg3 может представлять собой остаток, выбранный из группы, включающей Boc и аллилоксикарбонил, особенно предпочтительно Boc. Следующим объектом изобретения является способ стереоселективного получения соединения формулы (1 В), отличающийся тем, что вместо стадий [(Р), (Q), (М)] выполняют стадии [(I), (J), (K), (L)],на которых(J+K) в соединении формулы (9) отщепляют защитные группы и это соединение подвергают гидрогенолитическому восстановлению с получением соединения формулы (11) при этом стадии (I)-(L) выполняют последовательно в указанном порядке, и защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, предпочтительно трифторацетил, особенно предпочтительно Cbz, а защитная группа Sg3 может представлять собой остаток, выбранный из группы, включающей Boc и аллилоксикарбонил, особенно предпочтительно Boc. Следующим объектом изобретения являются соединение формулы 1 А, а также его фармакологически приемлемые соли, гидраты, сольваты и сокристаллы. Следующим объектом изобретения являются соединение формулы 1B, a также его фармакологически приемлемые соли, гидраты, сольваты и сокристаллы. Следующим объектом изобретения являются соединение формулы 18, а также его фармакологически приемлемые гидраты, сольваты и сокристаллы. Следующим объектом изобретения являются соединение формулы 22, а также его фармакологически приемлемые соли, гидраты, сольваты и сокристаллы. Следующим объектом изобретения являются соединение формулы 13, а также его фармакологически приемлемые соли, гидраты, сольваты и сокристаллы. Следующим объектом изобретения являются соединение формулы 4, а также его фармакологически приемлемые соли, гидраты, сольваты и сокристаллы. Следующим объектом изобретения являются соединение формулы 5, а также его фармакологически приемлемые соли, гидраты, сольваты и сокристаллы. Следующим объектом изобретения являются соединение формулы 6, а также его фармакологически приемлемые гидраты, сольваты и сокристаллы. Следующим объектом изобретения являются соединение формулы 12, а также его фармакологически приемлемые соли, гидраты, сольваты и сокристаллы. Под сокристаллами согласно настоящему изобретению подразумеваются молекулярные комплексы,содержащие две или более разных молекул в одной и той же кристаллической решетке (Crystal GrowthDesign,6, т. 9, 2009, cc. 2950-2967; Stahly G.P., Cryst. Growth Des., 7, 2007, cc. 1007-1026), прежде всего сокристаллы, образующиеся между нормальной или ионной молекулой фармацевтического действующего вещества и сокристаллообразователем, который при комнатной температуре представлен в виде твердого вещества (Jones W., Motherwell W. D., Trask A.V., MRS Bull., 341, 2006, cc. 875-879; Vishweshwar P.,McMahon J.A., Bis J.A., Zaworotko M.J., J. Pharm. Sci., 95, 2006, cc. 499-516). Особенно предпочтителен далее способ, при осуществлении которого используют хлорирующий агент, выбранный из группы, включающей тионилхлорид, оксихлорид фосфора, комбинацию Nхлорсукцинимид/трифенилфосфан и комбинацию четыреххлористый углерод/трифенилфосфан. Предлагаемые в изобретении соединения могут быть представлены в виде таутомеров, а также в виде свободных оснований или соответствующих кислотно-аддитивных солей с фармакологически приемлемыми кислотами, таких, например, как кислотно-аддитивные соли с галогенводородными кислотами, в частности хлористо- или бромисто-водородной кислотой, неорганическими кислотами, в частности фосфорной кислотой или серной кислотой, или органическими кислотами, в частности щавелевой, фумаровой, дигликолевой, толуолсульфоновой, бензойной, янтарной, малеиновой, салициловой, яблочной или метансульфоновой кислотами. На описанных выше отдельных стадиях способа предпочтительно применение нижеуказанных растворителей, выбираемых из соответствующей группы. На стадии:A: CH2Cl2, CHCl3, тетрагидрофуран (ТГФ) и диоксан; В: НОАс, H2O, водные растворы следующих растворителей: EtOH, ТГФ, изо-PrOH, МеОН, Nметил-2-пирролидон (N-МП) и диметилформамид (ДМФ);Z: водный NaOH и водный KOH, а также дополнительно к тому или другому EtOH, MeOH, ТГФ; Р: N-МП, диоксан, ТГФ и CH2Cl2;L: н-PrOH, EtOH, MeOH, N-МП и АЦН. Описанные выше отдельные стадии способа предпочтительно проводить при температуре, лежащей в указанных ниже пределах. На стадии: А: предпочтительно от -15 до 40 С, особенно предпочтительно от -10 до 10 С,В: предпочтительно от 20 до 75 С, особенно предпочтительно от 35 до 55 С,V: предпочтительно от 0 до 50 С, особенно предпочтительно от 10 до 35 С,X: предпочтительно от 0 до 60 С, особенно предпочтительно от 5 до 35 С,R: предпочтительно от 5 до 100 С, особенно предпочтительно от 15 до 40 С,S: предпочтительно от 50 до 80 С, особенно предпочтительно от 65 до 80 С,Т: предпочтительно от 10 до 80 С, особенно предпочтительно от 15 до 50 С,Z: предпочтительно от 0 до 60 С, особенно предпочтительно от 10 до 35 С,Н: предпочтительно от 15 до 60 С, особенно предпочтительно от 15 до 30 С,Р: предпочтительно от 10 до 80 С, особенно предпочтительно от 15 до 35 С,Q: предпочтительно от 0 до 80 С, особенно предпочтительно от 50 до 70 С,М: предпочтительно от 20 до 90 С, особенно предпочтительно от 60 до 80 С,N: предпочтительно от 15 до 85 С, особенно предпочтительно от 70 до 85 С,О: предпочтительно от 0 до 80 С, особенно предпочтительно от 10 до 50 С,С: предпочтительно от 0 до 65 С, особенно предпочтительно от 15 до 30 С,D: предпочтительно от 10 до 80 С, особенно предпочтительно от 20 до 40 С,Е: предпочтительно от 0 до 40 С, особенно предпочтительно от 0 до 15 С,F: предпочтительно от 0 до 45 С, особенно предпочтительно от 10 до 25 С,G: предпочтительно от 0 до 45 С, особенно предпочтительно от 10 до 25 С,I: предпочтительно от 0 до 50 С, особенно предпочтительно от 15 до 30 С,J: предпочтительно от 0 до 85 С, особенно предпочтительно от 40 до 70 С,K: предпочтительно от 10 до 60 С, особенно предпочтительно от 15 до 35 С,L: предпочтительно от 60 до 97 С, особенно предпочтительно от 85 до 97 С. На стадиях K, М и S предпочтительно использовать катализаторы, выбираемые из группы, включающей Pd/C и Pd(OH)2, из которых предпочтителен Pd/C. В качестве защитных групп предпочтительно использовать таковые, выбираемые из группы, включающей бензил, Cbz, трифторацетил и Boc. Используемые в приведенных выше формулах и в приведенном выше описании сокращение "Boc" означает трет-бутилкарбамат, а сокращение "Cbz" означает бензилоксикарбонил. Под выражением "необязательно замещенный бензил" подразумеваются, например, остатки, выбранные из группы, включающей бензил, пара-метоксибензил, пара-метилбензил и 1-фенилэтил, из которых особенно предпочтителен бензил. Под выражением "необязательно замещенный ацетил" подразумеваются, например, остат-5 023562 ки, выбранные из группы, включающей трифторацетил, ацетил, монофторацетил, дифторацетил и трихлорацетил, из которых особенно предпочтителен трифторацетил. Предлагаемый в изобретении синтез проиллюстрирован ниже на схемах 1 и 2. На этих схемах все соединения представлены в виде их оснований. Схема 1. Стадии синтеза соединения (1A) Схема 2. Стадии синтеза соединения (1 В) В последующих примерах проиллюстрированы способы получения соединений формул (1 А) и (1 В). Эти примеры должны рассматриваться как поясняющие настоящее изобретение и не ограничивают его объем. Пример 1. 1,4-Диокса-9,12-диазадиспиро[4.2.5.2]пентадекан-13-он. Стадия А. К охлажденной до -5 С смеси из 250 г 1,4-циклогександионмоноэтилен-кеталя, 18,2 г бензилтриэтиламмонийхлорида и 1,57 г цианида натрия в 1 л дихлорметана по каплям добавляют 127,5 мл этилендиамина в 194 мл хлороформа. Затем при температуре в пределах примерно от -10 до 0 С в течение последующих 9 ч по каплям добавляют 407,5 мл 50%-ного раствора едкого натра. Через 14,5 ч при температуре в пределах от -5 до 25 С по каплям добавляют 500 мл концентрированной соляной кислоты. Осадок отфильтровывают и дважды промывают дихлорметаном порциями по 500 мл. Фильтрат разделяют на фазы. Водную фазу дважды экстрагируют дихлорметаном порциями по 1 л и однократно дихлорметаном порцией 500 мл. Объединенные органические фазы сушат над сульфатом натрия и концентрируют в вакууме. Далее добавляют 500 мл н-бутилацетата и продолжают концентрировать до тех пор, пока не останется 820 г суспензии. Затем при 50 С в течение 20 мин добавляют 3 л метил-трет-бутилового эфира. Осадок отделяют вакуум-фильтрацией и дважды промывают метил-трет-бутиловым эфиром порциями по 200 мл. После сушки получают 247 г продукта. Масс-спектр (ESI+): m/z = 227 [М+Н]+. Стадия В. К 500 г 1,4-диокса-9,12-диазадиспиро[4.2.5.2]пентадекан-13-она в 2,5 л уксусной кислоты в течение 45 мин по каплям добавляют 310 мл 10-молярной HCl в этаноле. Через 3 ч при 35-45 С в течение 20 мин по каплям добавляют 10 л изопропанола. Суспензию охлаждают до 15 С и фильтруют. Осадок промывают дважды изопропанолом порциями по 1 л и дважды метил-трет-бутиловым эфиром порциями по 1 л. После сушки твердого вещества получают 386 г продукта в виде гидрохлорида. Масс-спектр (ESI+): m/z = 183 [М+Н]+. Стадия С. 380 г гидрохлорида 1,4-диазаспиро[5.5]ундекан-5,9-диона в 3,8 л ацетонитрила в течение 1 ч смешивают с 320 мл 30%-ного раствора метилата натрия в метаноле. Далее добавляют 18 г карбоната натрия и смесь перемешивают в течение 18 ч. После этого отгоняют 2 л растворителя и остаток фильтруют. Осадок на фильтре дважды промывают ацетонитрилом порциями по 100 мл и фильрат, содержащий продукт, непосредственно используют далее в реакции на следующей стадии. Пример 3. трет-Бутиловый эфир 5,9-диоксо-1,4-диазаспиро[5.5]ундекан-4-карбоновой кислоты. Стадия D. К раствору полученной на предыдущей стадии смеси, содержащей 1,4-диазаспиро[5.5]ундекан-5,9 дион, добавляют 480 г карбоната калия и 10 г 4-(диметиламино)пиридина. Далее в течение 200 мин по каплям добавляют 415 г ди-трет-бутилдикарбоната в 415 мл ацетонитрила. Через 18,5 ч добавляют 10 г 4(диметиламино)пиридина и 100 г ди-трет-бутилдикарбоната в 100 мл ацетонитрила. Через 200 мин добавляют 100 г ди-трет-бутилдикарбоната в 100 мл ацетонитрила. Через 90 мин добавляют 50 г ди-третбутилдикарбоната в 50 мл ацетонитрила. Через 1 ч добавляют 2 л воды. После разделения фаз водную фазу промывают 1 л метил-трет-бутилового эфира. Объединенные органические фазы промывают 1 л 10%-ного раствора карбоната калия и 500 мл насыщенного раствора поваренной соли. Органическую фазу концентрируют в вакууме. К суспензии добавляют 1,5 л н-бутилацетата и вновь концентрируют. Далее добавляют еще 2 л н-бутилацетата и вновь концентрируют. Оставшуюся суспензию нагревают до 55 С и медленно смешивают с 1 л метил-трет-бутилового эфира. Суспензию охлаждают до 22 С. Осадок отфильтровывают и промывают 500 мл н-бутилацетата и 500 мл метил-трет-бутилового эфира. После сушки твердого вещества получают 296 г продукта. Масс-спектр (ESI+): m/z = 283 [М+Н]+. Пример 4. трет-Бутиловый эфир (цис)-9-гидрокси-5-оксо-1,4-диазаспиро[5.5]ундекан-4-карбоновой кислоты. Стадия Е. К смеси из 159 терт-бутилового эфира 5,9-диоксо-1,4-диазаспиро[5.5]ундекан-4-карбоновой кислоты в 1140 мл воды при 1 С в течение 17 мин по каплям добавляют 6,4 г борогидрида натрия в 100 мл воды. После этого капельную воронку промывают 30 мл воды. Через 50 мин добавляют 318 мл насыщенного раствора карбоната калия и после перемешивания в течение 1 ч при 10 С осадок отделяют вакуумфильтрацией и дважды промывают 10%-ным раствором карбоната калия порциями по 200 мл. После сушки осадок в течение 4,5 ч перемешивают в 1,6 л воды. Далее добавляют 350 мл насыщенного раствора карбоната калия, после перемешивания в течение 15 мин осадок отделяют вакуум-фильтрацией и промывают 200 мл 10%-ного раствора карбоната калия. После сушки осадок в течение 20 мин перемешивают в 500 мл тетрагидрофурана. После фильтрации, промывки 200 мл тетрагидрофурана и упаривания фильтрата получают 65,5 г продукта. Масс-спектр (ESI+): m/z = 285 [М+Н]+. Стадия F. К раствору 113 г трет-бутилового эфира 5,9-диоксо-1,4-диазаспиро[5.5]ундекан-4-карбоновой кислоты в 1150 мл ТГФ и 25 мл воды при 16 С в течение 20 мин по каплям добавляют 3,8 г борогидрида натрия в 30 мл воды. Через 45 мин добавляют 0,42 г борогидрида натрия. Через 35 мин добавляют 0,42 г борогидрида натрия. Еще через 35 мин добавляют 0,1 г борогидрида натрия. Через 15 мин добавляют 10 мл ацетона и реакционную смесь дважды промывают насыщенным раствором поваренной соли порциями по 500 мл. Органическую фазу используют непосредственно в реакции на следующей стадии. Масс-спектр (ESI+): m/z = 285 [М+Н]+. Пример 5. 1-Бензиловый и 4-трет-бутиловый эфиры (цис)-9-гидрокси-5-оксо-1,4-диазаспиро[5.5]ундекан-1,4-дикарбоновой кислоты. Стадия G. К органической фазе из полученной на предыдущей стадии смеси добавляют 112 мл насыщенного раствора карбоната калия, а затем в течение 20 мин по каплям добавляют 59 мл бензилового эфира хлормуравьиной кислоты. Через 16 ч добавляют 400 мл воды и фазы разделяют. Органическую фазу промывают 900 мл насыщенного раствора карбоната калия и дважды насыщенным раствором поваренной соли порциями по 450 мл. Далее органическую фазу сушат над сульфатом магния и затем концентрируют. После отгонки 1 л добавляют 450 мл метилциклогексана и смесь концентрируют далее. После этого еще дважды добавляют по 100 мл метилциклогексана и смесь продолжают концентрировать до тех пор, пока не останется 168 г сырого продукта. Этот сырой продукт трижды перекристаллизовывают из смеси метанол/вода в соотношении 1:1. После сушки получают 86 г продукта. Масс-спектр (ESI+): m/z = 419 [М+Н]+. Стадия Н. Смесь из 500 мг бензилового эфира (цис)-9-гидрокси-5-оксо-1,4-диазаспиро[5.5]ундекан-1-карбоновой кислоты, 217 мг карбоната калия, 686 мг ди-трет-бутилдикарбоната и 192 мг 4-(диметиламино)пиридина в 10 мл ацетонитрила в течение 4 ч перемешивают при комнатной температуре. Затем смесь очищают путем двукратной хроматографии на силикагеле, получая 420 мг продукта. Масс-спектр (ESI+): m/z = 419 [М+Н]+. Пример 6. Метиловый эфир 5-гидрокси-4-метокси-2-нитробензойной кислоты. Смесь из 500 г метилового эфира 4,5-диметокси-2-нитробензойной кислоты и 625 г гидроксида калия в 2300 мл воды нагревают до 95 С с выдержкой при этой температуре в течение 18,5 ч. После охлаждения смесь фильтруют до получения прозрачного фильтрата, который разбавляют 3 л воды. Полученный раствор смешивают с 950 мл уксусной кислоты и через 1 ч отфильтровывают осадок. Этот осадок суспендируют в 3250 мл этилацетата и затем добавляют 100 мл воды и 200 мл 12 н. соляной кислоты. Через 1,5 ч фазы разделяют и водную фазу экстрагируют 700 мл этилацетата. Объединенные органические фазы сушат над сульфатом магния и после фильтрации концентрируют. Затем упаривают совместно с 200 мл метилциклогексана. Остаток совместно с 1600 мл метанола и 100 мл концентрированной серной кислоты в течение 16,5 ч нагревают с обратным холодильником. Далее смесь концентрируют до начала кристаллизации. Затем добавляют 1000 мл воды и смесь перемешивают до образования гомогенной суспензии. Осадок отфильтровывают, промывают 500 мл воды и суспендируют в 1000 мл воды. После перемешивания в течение 1,5 ч осадок отфильтровывают и промывают 500 мл воды. После сушки осадка на фильтре получают 364 г продукта. Масс-спектр (ESI-): m/z = 226 [М-Н]+. Пример 7. 1-Бензиловый и 4-трет-бутиловый эфиры (транс)-9-(2-метокси-5-метоксикарбонил-4 нитрофенокси)-5-оксо-1,4-диазаспиро[5.5]ундекан-1,4-дикарбоновой кислоты. диазаспиро[5.5]ундекан-1,4-дикарбоновой кислоты, 53,74 г метилового эфира 5-гидрокси-4-метокси-2 нитробензойной кислоты (15) и 74,34 г трифенилфосфина в 764 мл диоксана при комнатной температуре в течение часа по каплям добавляют 58,75 мл диизопропилового эфира азодикарбоновой кислоты. Через 17 ч добавляют 5 мл диизопропилового эфира азодикарбоновой кислоты и смесь перемешивают еще в течение 1,5 ч. Смесь, содержащую продукт, без очистки непосредственно используют в реакции на следующей стадии. Масс-спектр (ESI+): m/z = 645 [M+NH4]+. Пример 8. Бензиловый эфир (транс)-9-(2-метокси-5-метоксикарбонил-4-нитрофенокси)-5-оксо-1,4 диазаспиро[5.5]ундекан-1-карбоновой кислоты. Стадия J. К полученной на предыдущей стадии смеси, содержащей 1-бензиловый и 4-трет-бутиловый эфиры(транс)-9-(2-метокси-5-метоксикарбонил-4-нитрофенокси)-5-оксо-1,4-диазаспиро[5.5]ундекан-1,4 дикарбоновой кислоты, добавляют 130 мл 4-молярной HCl в диоксане. Реакционную смесь нагревают до 60 С. Через 2 ч добавляют еще 13 мл 4-молярной HCl в диоксане. Затем реакционный раствор охлаждают до комнатной температуры и смешивают с 500 мл насыщенного раствора карбоната калия. Органическую фазу промывают 500 мл насыщенного раствора карбоната калия и 200 мл насыщенного раствора поваренной соли. Органическую фазу, содержащую продукт, без очистки непосредственно используют в реакции на следующей стадии. Масс-спектр (ESI+): m/z = 528 [М+Н]+. Пример 9. Метиловый эфир (транс)-2-амино-4-метокси-5-(5-оксо-1,4-диазаспиро[5.5]ундец-9 илокси)бензойной кислоты Стадия K. К полученной на предыдущей стадии смеси, содержащей бензиловый эфир (транс)-9-(2-метокси-5 метоксикарбонил-4-нитрофенокси)-5-оксо-1,4-диазаспиро[5.5]ундекан-1-карбоновой кислоты, добавляют 12,4 г Pd (10%-ного) на угле и 500 мл метанола. После гидрирования водородом в течение 1,5 ч при 3 барах смесь концентрируют до остаточного объема 600 мл. Затем смесь разбавляют 1,8 л диоксана и фильтруют до прозрачности. Далее в течение 45 мин по каплям добавляют 59 мл 4-молярной HCl в диоксане, по истечении последующих 30 мин осадок отделяют вакуум-фильтрацией и дважды промывают диоксаном порциями по 200 мл. После сушки твердого вещества получают 98,6 г продукта в виде гидрохлорида. Масс-спектр (ESI+): m/z = 364 [М+Н]+. Пример 10. (транс)-9-(4-Гидрокси-7-метоксихиназолин-6-илокси)-1,4-диазаспиро[5.5]ундекан-5-он. Стадия L. 88 г гидрохлорида метилового эфира (транс)-2-амино-4-метокси-5-(5-оксо-1,4-диазаспиро[5.5]ундец-9-илокси)бензойной кислоты и 25 г формамидинацетата в 1,8 л н-пропанола в течение 17 ч нагревают с обратным холодильником. Затем смесь охлаждают до 28 С и перемешивают в течение 4 ч при этой температуре. После охлаждения до 14 С осадок отфильтровывают и промывают 200 мл холодного н-пропанола. После сушки твердого вещества получают 44 г продукта в виде гидрохлорида. Масс-спектр (ESI+): m/z = 359 [М+Н]+. Стадия М. К смеси из 1,7 г бензилового эфира (транс)-9-(3-бензил-7-метокси-4-оксо-3,4-дигидрохиназолин-6 илокси)-5-оксо-1,4-диазаспиро[5.5]ундекан-1-карбоновой кислоты в 30 мл уксусной кислоты и 3 мл воды добавляют 300 мг палладия (10%-ного) на угле. После гидрирования в течение 22 ч при 70 С смесь Стадия N. 10 г гидрохлорида (транс)-9-(4-гидрокси-7-метоксихиназолин-6-илокси)-1,4-диазаспиро[5.5]ундекан-5-она и 12 г трифенилфосфина суспендируют в 450 мл диоксана. Затем отгоняют 250 мл растворителя и при 41 С по каплям добавляют 6,45 г N-хлорсукцинимида в 100 мл ацетонитрила. После этого реакционную смесь нагревают с обратным холодильником. Через 100 мин смесь охлаждают до 29 С и добавляют 150 мл метилтетрагидрофурана. Осадок отфильтровывают и трижды промывают метилтетрагидрофураном порциями по 50 мл. После сушки при 30 С получают 12 г темного твердого вещества, которое содержит продукт в виде гидрохлорида и которое без очистки используют в реакции на следующей стадии. Масс-спектр (ESI+): m/z = 377 [M+H]+. Пример 12. (транс)-9-[4-(3-Хлор-2-фторфениламино)-7-метоксихиназолин-6-илокси]-1,4-диазаспиро[5.5]ундекан-5-он. Стадия О. 12 г содержащего примеси гидрохлорида (транс)-9-(4-хлор-7-метоксихиназолин-6-илокси)-1,4 диазаспиро[5.5]ундекан-5-она с предыдущей стадии при комнатной температуре в течение 90 мин порциями добавляют к раствору 3,9 г 3-хлор-2-фторанилина в 60 мл 2 н. соляной кислоты. Суспензию нагревают до 40 С с выдержкой при этой температуре в течение 60 мин. Затем добавляют 60 мл толуола и смесь охлаждают до комнатной температуры. Через 50 мин фильтруют и осадок промывают 50 мл толуола и 50 мл насыщенного раствора NaCl. После сушки при 40 С получают 10 г твердого вещества, содержащего продукт. Этот продукт очищают путем основной хроматографии на силикагеле. Масс-спектр (ESI+): m/z = 486 [М+Н]+. 1 Н-ЯМР (400 МГц, ДМСО): 9,60 (1H, s); 8,37 (1H, s); 7,82 (1 Н, s); 7,45-7,54 (2 Н, m), 7,36 (1 Н, s); 7,28 Стадия Р. К суспензии 1,3 г 3-бензил-6-гидрокси-7-метокси-3H-хиназолин-4-она, 2 г 1-бензилового и 4-третбутилового эфиров (цис)-9-гидрокси-5-оксо-1,4-диазаспиро[5.5]ундекан-1,4-дикарбоновой кислоты и 1,8 г трифенилфосфина в 10 мл N-метил-2-пирролидона в течение 90 мин по каплям добавляют 1,36 мл диизопропилового эфира азодикарбоновой кислоты. Затем смесь перемешивают в течение 4 ч. Смесь, содержащую продукт, используют непосредственно на следующей стадии. Масс-спектр (ESI+): m/z = 683 [М+Н]+. Пример 14. Бензиловый эфир (транс)-9-(3-бензил-7-метокси-4-оксо-3,4-дигидрохиназолин-6 илокси)-5-оксо-1,4-диазаспиро[5.5]ундекан-1-карбоновой кислоты. Стадия О. К полученной на предыдущей стадии смеси, содержащей 1-бензиловый и 4-трет-бутиловый эфиры(транс)-9-(3-бензил-7-метокси-4-оксо-3,4-дигидрохиназолин-6-илокси)-5-оксо-1,4-диазаспиро[5.5]ундекан-1,4-дикарбоновой кислоты, добавляют 2,5 мл 4-молярной HCl в диоксане. Через 19 ч добавляют 2 мл 4-молярной HCl в диоксане и смесь нагревают до 40 С. Через 3 ч температура повышается до 60 С,смесь разбавляют 60 мл диоксана и добавляют 10 мл 4-молярной HCl в диоксане. Через 16 ч смесь концентрируют в вакууме и остаток растворяют в 50 мл дихлорметана. После трехкратной промывки водой порциями по 50 мл органическую фазу концентрируют. Остаток очищают хроматографией на силикагеле. Соответствующие фракции концентрируют и остаток вываривают совместно с 150 мл этилацетата. После выделения и сушки осадка получают 2,1 г продукта. Масс-спектр (ESI+): m/z = 583 [М+Н]+. Пример 15. Бензиловый эфир (цис)-9-(3-бензил-7-метокси-4-оксо-3,4-дигидрохиназолин-6-илокси)5-оксо-1,4-диазаспиро[5.5]ундекан-1-карбоновой кислоты.(транс)-9-гидрокси-5-оксо-1,4-диазаспиро[5.5]ундекан-1-карбоновой кислоты и 2,79 г трифенилфосфина в 20 мл N-метил-2-пирролидона при охлаждении по каплям добавляют 2,1 мл диизопропилового эфира азодикарбоновой кислоты. Через 20 мин добавляют 20 мл N-метил-2-пирролидона и смесь перемешивают в течение 4 ч. Осадок отделяют вакуум-фильтрацией при 0 С и промывают 50 мл метил-третбутилового эфира. После сушки получают 3,3 г продукта, который все еще содержит N-метил-2 пирролидон. Масс-спектр (ESI+): m/z = 583 [М+Н]+. Пример 16. (цис)-9-(4-Гидрокси-7-метоксихиназолин-6-илокси)-1,4-диазаспиро[5.5]ундекан-5-он Стадия S. К смеси из 1,7 г бензилового эфира (цис)-9-(3-бензил-7-метокси-4-оксо-3,4-дигидрохиназолин-6 илокси)-5-оксо-1,4-диазаспиро[5.5]ундекан-1-карбоновой кислоты в 30 мл этанола и 10 мл 1-молярной соляной кислоты добавляют 300 мг палладия (10%-ного) на угле. После гидрирования в течение 25 ч при 80 С смесь фильтруют и раствор упаривают досуха, получая 1,4 г сырого продукта. Этот сырой продукт вываривают совместно с 100 мл этанола и после фильтрации фильрат концентрируют. Остаток суспендируют в 50 мл ацетонитрила и после добавления 1 г карбоната калия перемешивают в течение 23 ч. Затем смесь концентрируют и после добавления 20 мл дихлорметана и 4 мл метанола очищают хроматографией на силикагеле. Таким путем получают 500 мг продукта. Масс-спектр (ESI+): m/z = 359 [М+Н]+. Пример 17. (цис)-9-[4-(3-Хлор-2-фторфениламино)-7-метоксихиназолин-6-илокси]-1,4-диазаспиро[5.5]ундекан-5-он. Стадия Т. К смеси из 100 мг 7-метокси-6-(5-оксо-1,4-диазаспиро[5.5]ундец-9-илокси)-3H-хиназолин-4-она и 0,23 мл триэтиламина в 5 мл ацетонитрила добавляют 0,13 мл окситрихлорида фосфора. Через 1 ч добавляют 0,04 мл 3-хлор-2-фторанилина. Через 18 ч добавляют 1 мл воды и смесь концентрируют до объема 2 мл. После очистки препаративной ЖХВД получают 95 мг продукта. Масс-спектр (ESI+): m/z = 486 [М+Н]+. 1 Н-ЯМР (400 МГц, ДМСО): 9,58 (1 Н, s); 8,36 (1 Н, s); 7,81 (1 Н, s); 7,54 (1 Н, t); 7,49 (1 Н, t); 7,42 (1H,s); 7,29 (1 Н, t), 7,20 (1H, s); 4,49-4,58 (1 Н, m); 3,93 (3H, s); 3,11-3,15 (2 Н, m); 2,80-2,85 (2 Н, m); 2,38 (1H,s); 1,88-2,02 (4 Н, m); 1,69-1,81 (4 Н, m). Пример 18. Гидрохлорид (транс)-9-гидрокси-1,4-диазаспиро[5.5]ундекан-5-она и гидрохлорид Стадия U. К смеси из 500 мг гидрохлорида 1,4-диазаспиро[5.5]ундекан-5,9-диона в 5 мл воды добавляют 50 мг диоксида платины. После гидрирования в течение 3 ч смесь фильтруют и раствор концентрируют досуха. Далее его упаривают дважды совместно с н-пропанолом, который используют порциями по 50 мл, в результате чего остается 500 мг транс/цис-смеси гидрохлоридов 9-гидрокси-1,4-диазаспиро[5.5]ундекан-5 онов. Масс-спектр (ESI+): m/z = 185 [М+Н]+. Пример 19. Бензиловый эфир 5,9-диоксо-1,4-диазаспиро[5.5]ундекан-1-карбоновой кислоты Стадия V. К смеси из 20 г гидрохлорида 1,4-диазаспиро[5.5]ундекан-5,9-диона в 100 мл тетрагидрофурана и 82 мл 50%-ного раствора карбоната калия при охлаждении добавляют 14,4 мл бензилового эфира хлормуравьиной кислоты. Через 2,5 ч добавляют 250 мл воды и осадок отфильтровывают. После промывки 200 мл воды и 200 мл метил-трет-бутилового эфира и сушки получают 24,3 г продукта. Масс-спектр (ESI+): m/z = 317 [М+Н]+. Пример 20. Бензиловый эфир (транс)-9-гидрокси-5-оксо-1,4-диазаспиро[5.5]ундекан-1-карбоновой кислоты. Стадия W. К смеси из 5 г гидрохлорида 1,4-диазаспиро[5.5]ундекан-5,9-диона в 20 мл воды добавляют 50 мг диоксида платины. После гидрирования в течение 22 ч добавляют 25 мг диоксида платины. После гидрирования в течение 26 ч смесь фильтруют и фильрат смешивают с 35 г карбоната калия и 25 мл тетрагидрофурана. Далее добавляют 3,43 мл бензилового эфира хлормуравьиной кислоты и смесь перемешивают в течение 6 дней. Затем добавляют 25 г карбоната калия и смесь перемешивают в течение 4 дней. После этого добавляют 3,5 мл бензилового эфира хлормуравьиной кислоты. Через 20 ч добавляют 200 мл воды,перемешивают в течение 1 ч, осадок отделяют вакуум-фильтрацией и промывают 100 мл метил-третбутилового эфира. Таким путем получают 3,4 г твердого вещества, которое преимущественно состоит из продукта. Масс-спектр (ESI+): m/z = 319 [М+Н]+. Стадия X. К 20 г бензилового эфира 5,9-диоксо-1,4-диазаспиро[5.5]ундекан-1-карбоновой кислоты в 100 мл тетрагидрофурана, 100 мл этанола, 80 мл воды и 20 мл 0,1 н. раствора едкого натра добавляют 7,2 г борогидрида натрия. После перемешивания в течение 16,5 ч при комнатной температуре и в течение 1 ч при 60 С затем при охлаждении льдом по каплям добавляют 80 мл 2-молярной соляной кислоты и 200 мл воды. Через 2 ч осадок отделяют вакуум-фильтрацией и промывают 200 мл воды. После сушки осадка и очистки хроматографией на силикагеле получают 8 г продукта. Масс-спектр (ESI+): m/z = 319 [М+Н]+.(транс)-9-гидрокси-5-оксо-1,4 Стадия Y. Смесь из 200 мг бензилового эфира (транс)-9-гидрокси-5-оксо-1,4-диазаспиро[5.5]ундекан-1 карбоновой кислоты, 87 мг карбоната калия, 274 мг ди-трет-бутилдикарбоната и 76 мг 4(диметиламино)пиридина в 5 мл ацетонитрила перемешивают в течение 2 ч при комнатной температуре. Затем смесь очищают препаративной ЖХВД, получая 100 мг продукта. Масс-спектр (ESI+): m/z = 419 [М+Н]+. Пример 22. Бензиловый эфир (цис)-9-гидрокси-5-оксо-1,4-диазаспиро[5.5]ундекан-1-карбоновой кислоты. Стадия Z К раствору 75 г гидрохлорида 1,4-диазаспиро[5.5]ундекан-5,9-диона в 350 мл 1-молярного раствора едкого натра при комнатной температуре порциями добавляют 14,3 г борогидрида натрия. По истечении 35 мин затем при охлаждении в течение 30 мин по каплям добавляют 60 мл концентрированной соляной кислоты. Далее добавляют 390 г карбоната калия. После добавления 300 мл тетрагидрофурана и 67 мл бензилового эфира хлормуравьиной кислоты смесь нагревают до 48 С с выдержкой при этой температуре в течение 1,5 ч. Затем добавляют 900 мл метил-трет-бутилового эфира и после охлаждения до 22 С добавляют 1,6 л воды. После часового перемешивания суспензию подвергают вакуум-фильтрации и осадок на фильтре промывают 500 мл воды и 1 л метил-трет-бутилового эфира. После сушки осадка на фильтре получают 77 г продукта, который преимущественно состоит из цис-изомера. Масс-спектр (ESI+): m/z = 319 [М+Н]+. Пример 23. Метиловый эфир (цис)-1-(2-трет-бутоксикарбониламиноэтиламино)-4-гидроксициклогексанкарбоновой кислоты. Стадия ZZ. К раствору 500 мг трет-бутилового эфира 5,9-диоксо-1,4-диазаспиро[5.5]ундекан-4-карбоновой кислоты в 5 мл метанола добавляют 16,7 мг борогидрида натрия. Через 4 ч смесь концентрируют и упаривают совместно с тетрагидрофураном. Полученный остаток содержит продукт. Масс-спектр (ESI+): m/z = 317 [М+Н]+. Пример 24. mpem-Бутиловый эфир (цис)-[2-(4-гидрокси-1-гидроксиметилциклогексил-амино)этил]карбаминовой кислоты. Стадия ZZZ. К смеси 1 г трет-бутилового эфира 5,9-диоксо-1,4-диазаспиро[5.5]ундекан-4-карбоновой кислоты в 10 мл 1-молярного раствора карбоната калия при охлаждении добавляют 161 мг борогидрида натрия. После выдержки в течение 14,5 ч при 50 С добавляют 10 мл этилацетата и после разделения фаз органическую фазу концентрируют. После хроматографической очистки остатка на силикагеле выделяют 580 мг смеси, содержащей продукт. Масс-спектр (ESI+): m/z = 289 [М+Н]+. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ стереоселективного получения соединения формулы (IA) необязательно в виде его таутомеров, а также при необходимости в виде его фармакологически безвредных кислотно-аддитивных солей, отличающийся тем, что он заключается в выполнении реакционных стадий (А), (В), (V), (X), (R), (S) и (Т), на которых(V) соединение формулы (2) его взаимодействием с несущим защитную группу реагентом превращают в соединение формулы (19)(S) в соединении формулы (21) отщепляют защитные группы с получением соединения формулы(Т) соединение формулы (22) хлорируют и затем подвергают взаимодействию с 3-хлор-2 фторанилином,при этом стадии (А)-(Т) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, а защитная группа Sg2 может представлять собой необяза- 15023562 тельно замещенный бензил. 2. Способ стереоселективного получения соединений формулы (1 А), отличающийся тем, что он заключается в выполнении стадий (R), (S) и (Т), на которых(R) соединение формулы (18) путем его взаимодействия с соединением формулы (23)(S) в соединении формулы (21) отщепляют защитные группы с получением соединения формулы(Т) соединение формулы (22) хлорируют и затем подвергают взаимодействию с 3-хлор-2 фторанилином,при этом стадии (R)-(T) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, а защитная группа Sg2 может представлять собой необязательно замещенный бензил. 3. Способ стереоселективного получения соединения формулы (18), отличающийся тем, что он заключается в выполнении стадий (А), (В), (V) и (X), на которых(V) соединение формулы (2) путем его взаимодействия с несущим защитную группу реагентом превращают в соединение формулы (19) при этом стадии (А), (В), (V) и (X) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил. 4. Способ стереоселективного получения соединения формулы (1 В) необязательно в виде его таутомеров, а также при необходимости в виде его фармакологически безвредных кислотно-аддитивных солей, отличающийся тем, что он заключается в выполнении реакционных стадий (А), (В), (Z), (Н), (Р), (Q), (M), (N) и (О), на которых(Q+M) в соединении формулы (7) отщепляют защитные группы с получением соединения формулы(N+О) хлорируют соединение формулы (12), превращая его в соединение формулы (13), и затем подвергают взаимодействию с 3-хлор-2-фторанилином, при этом стадии (А)-(О) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, защитная группа Sg2 может представлять собой необяза- 17023562 тельно замещенный бензил, а защитная группа Sg3 может быть выбрана из группы, включающей Boc и аллилоксикарбонил. 5. Способ стереоселективного получения соединения формулы (1 В), отличающийся тем, что он заключается в выполнении стадий (А), (В), (С), (D), (Е) или (F), (G), (P), (Q+M) и (N+О), на которых без выделения при этом соединения формулы (5) на стадии (F) и(Р) соединение формулы (6) путем его взаимодействия с соединением формулы (23)(Q+M) в соединении формулы (7) отщепляют защитные группы с получением соединения формулы(N+О) хлорируют соединение формулы (12), превращая его в соединение формулы (13), и затем подвергают взаимодействию с 3-хлор-2-фторанилином при этом стадии (А)-(О) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, а защитная группа Sg3 может представлять собой остаток,выбранный из группы, включающей Boc и аллилоксикарбонил. 6. Способ стереоселективного получения соединения формулы (1 В), отличающийся тем, что он заключается в выполнении стадий (А), (В), (С), (D), (Е) или (F), (G), (I), (J), (K), (L) и (N+О), на которых без выделения при этом соединения формулы (5) на стадии (F) и(J+K) в соединении формулы (9) отщепляют защитные группы и это соединение подвергают гидрогенолитическому восстановлению с получением соединения формулы (11) и (N+О) хлорируют соединение формулы (12), превращая его в соединение формулы (13), и затем подвергают взаимодействию с 3-хлор-2-фторанилином, при этом стадии (А)-(О) выполняют последовательно в указанном порядке, защитная группа Sg1 может представлять собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил, а защитная группа Sg3 может представлять собой остаток,выбранный из группы, включающей Boc и аллилоксикарбонил. 7. Соединение формулы (18) в которой защитная группа Sg1 представляет собой остаток, выбранный из группы, включающей необязательно замещенный бензил, Cbz и необязательно замещенный ацетил. 8. Соединение формулы (22) в которой защитная группа Sg3 представляет собой остаток, выбранный из группы, включающей в которой защитная группа Sg3 представляет собой остаток, выбранный из группы, включающей в которой защитная группа Sg3 представляет собой остаток, выбранный из группы, включающей
МПК / Метки
МПК: C07D 239/70, C07D 403/12
Метки: гетероциклов, стереоселективного, бициклических, синтеза, способ
Код ссылки
<a href="https://eas.patents.su/22-23562-sposob-stereoselektivnogo-sinteza-biciklicheskih-geterociklov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ стереоселективного синтеза бициклических гетероциклов</a>
Способ стереоселективного синтеза циклических аминокислот

Номер патента: 4984
Опубликовано: 28.10.2004
Авторы: Блейкмор Дейвид Клайв, Уильямс Софи Кэролайн, Брайанз Джастин Стивен
МПК: C07C 227/32
Метки: способ, синтеза, стереоселективного, аминокислот, циклических
Формула / Реферат:
1. Способ получения соединения формулы I где R - C1-C10алкил или C3-C10циклоалкил, и его фармацевтически приемлемых солей, который включает a) добавление цианацетата формулы (A) NCЪCO2R1, где R1 - алкил или бензил, к смеси хирального циклопентанона формулы (1) растворителя, карбоновой кислоты и катализатора реакции Кневенагеля и перемешивание смеси в присутствии средств удаления воды для получения алкена формулы (2) b) добавление...
Способ получения фенильных гетероциклов, пригодных в качестве ингибиторов цог-2

Номер патента: 1629
Опубликовано: 25.06.2001
Авторы: Тилльер Ричард Д., Доллинг Ульф Х., Десмонд Ричард, Фрей Лайза Ф., Тшаен Дэвид М.
МПК: C07D 307/38
Метки: ингибиторов, пригодных, качестве, цог-2, способ, гетероциклов, фенильных, получения
Формула / Реферат:
1. Способ получения соединений формулы 1 где R2 является моно- или дизaмещенным фенилом, где заместитель выбран из группы, включающей: (1) водород, (2) галоген, (3) C1-6-алкокси, (4) C1-6-алкилтио, (5) CN, (6) СF3 и (7) C1-6-алкил, R3 и R3' независимо выбраны из водорода и С1-4-алкила, который заключается в том, что осуществляют: (b4) взаимодействие в N,N-диметилформамиде соединения формулы 2 с фенилуксусной кислотой формулы в...
Способ стереоселективного получения (-)-галофената и его интермедиатов

Номер патента: 15673
Опубликовано: 31.10.2011
Авторы: Чжу Ян, Чжао Зючюн, Ченг Пенг, Чен Ксин, Ма Цзингуан
МПК: A61K 31/40, A61K 31/495, A61K 31/4025...
Метки: получения, стереоселективного, интермедиатов, галофената, способ
Формула / Реферат:
1. Способ получения соединения, имеющего формулу (I)в которой R1 выбирают из группы, состоящей изкаждый R2независимо выбирают из группы, состоящей из (C1-C4)алкила, галогена, (C1-C4)галогеноалкила, аминогруппы, (C1-C4)аминоалкила, амидогруппы, (C1-C4)амидоалкила, (C1-C4)сульфонилалкила, (C1-C4)сульфамилалкила, (C1-C4)алкоксигруппы, (C1-C4)гетероалкила, карбоксигруппы и нитрогруппы;нижний индекс n равен 1, если R1 имеет формулу (a) или (b), и n...
Новый способ синтеза соединений ( 2s, 3аs, 7as )-1-[ (s)-аланил]октагидро -1н-индол-2-карбоновой кислоты и их применение для синтеза периндоприла

Номер патента: 9062
Опубликовано: 26.10.2007
Авторы: Ланглуа Паскаль, Дюбюффе Тьерри
МПК: C07K 5/06, C07K 5/02, C07D 209/42...
Метки: 1, новый, синтеза, кислоты, периндоприла, 3аs, s)-аланил]октагидро, способ, соединений, 1н-индол-2-карбоновой, применение
Формула / Реферат:
1. Способ синтеза соединений (I) в которой R1 представляет собой атом водорода или бензильную группу, a R2 - это защитная группа для группы амина, представляющая собой трет-бутоксикарбонильную группу, характеризующийся тем, что 1-(1-циклогексен-1-ил)пирролидин формулы (III) подвергают реакции с соединением серина формулы (IV) в которой R1 является таким, как определено для формулы (I), a R3 - это защитная группа для группы амина,...
Способ стереоселективного ферментативного гидролиза эфира 5-метил-3-нитрометилгексановой кислоты

Номер патента: 19285
Опубликовано: 28.02.2014
Авторы: Осл Дорис, Ремлер Питер, Альберт Мартин, Лушниг Дэниел, Зерек Фердинанд, Бергер Андреас, Де Соуза Доминик, Риезорст Ваандер, Салченеггер Джоерг, Шваб Хельмут
МПК: C12P 13/00, C07C 229/08, C07C 205/51...
Метки: гидролиза, стереоселективного, ферментативного, эфира, способ, кислоты, 5-метил-3-нитрометилгексановой
Формула / Реферат:
1. Способ стереоселективного ферментативного гидролиза эфира 5-метил-3-нитрометилгексановой кислоты (VIII), в котором рацемический эфир 5-метил-3-нитрометилгексановой кислоты (VIII)контактирует с ферментом с получением (R)-энантиомера эфира 5-метил-3-нитрометилгексановой кислоты (VIII) и (S)-энантиомера соли 5-метил-3-нитрометилгексановой кислоты (IX), где R1 представляет собой C1-C8-алкильную группу, а фермент выбирают из группы, состоящей из...
Предыдущий патент: Производные пиридина и пиразина в качестве модуляторов протеинкиназы, лекарственное средство и композиция, содержащая их
Следующий патент: Способ и устройство для контроля процесса покрытия методом осаждения
Случайный патент: Гетероциклические азотсодержащие соединения, их применение и содержащая их фармацевтическая композиция