Кристаллические оксалатные соли диамидного соединения


















Формула / Реферат
1. Кристаллическая оксалатная соль 1-(2-{[4-(4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}фенилкарбамоил)бутил]метилкарбамоил}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты, где кристаллическую оксалатную соль выбирают из:
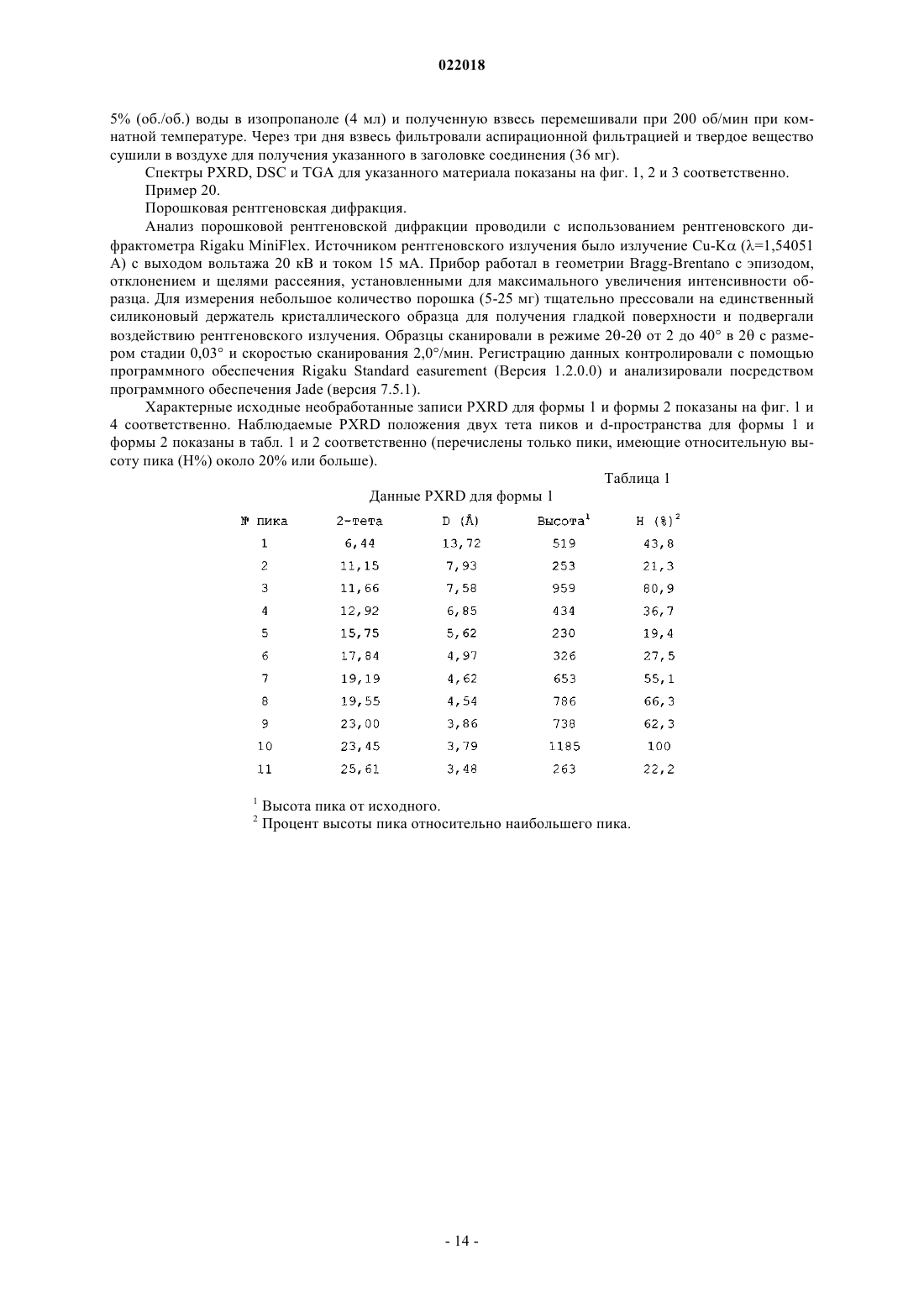
(a) сольвата изопропанола, характеризующегося порошковой рентгенограммой, включающей пики дифракции на 2θ значениях 11,66±0,20, 15,75±0,20, 19,55±0,20, 23,00±0,20 и 23,45±0,20;
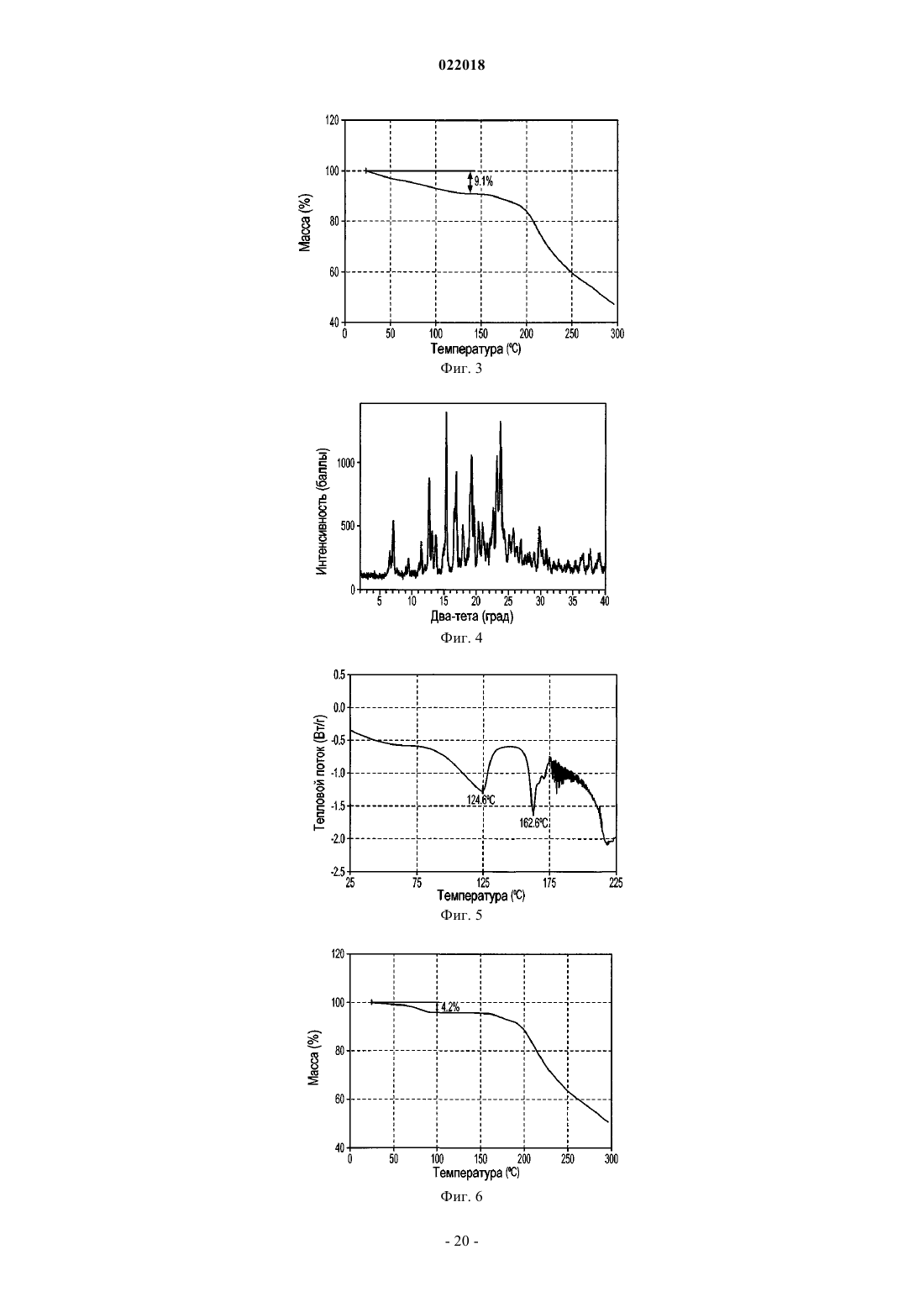
(b) гидрата, характеризуемого порошковой рентгенограммой, включающей пики дифракции на 2θ значениях 15,32±0,20, 16,90±0,20, 19,25±0,20 и 23,73±0,20.
2. Кристаллическая оксалатная соль по п.1, где кристаллической оксалатной солью является сольват изопропанола, характеризуемый порошковой рентгенограммой, включающей пики дифракции на 2θ значениях 11,66±0,20, 15,75±0,20, 19,55±0,20, 23,00±0,20 и 23,45±0,20.
3. Кристаллическая оксалатная соль по п.2, где кристаллическая оксалатная соль характеризуется порошковой рентгенограммой, в которой пиковые положения, по существу, соответствуют пиковым положениям графика, показанного на фиг. 1.
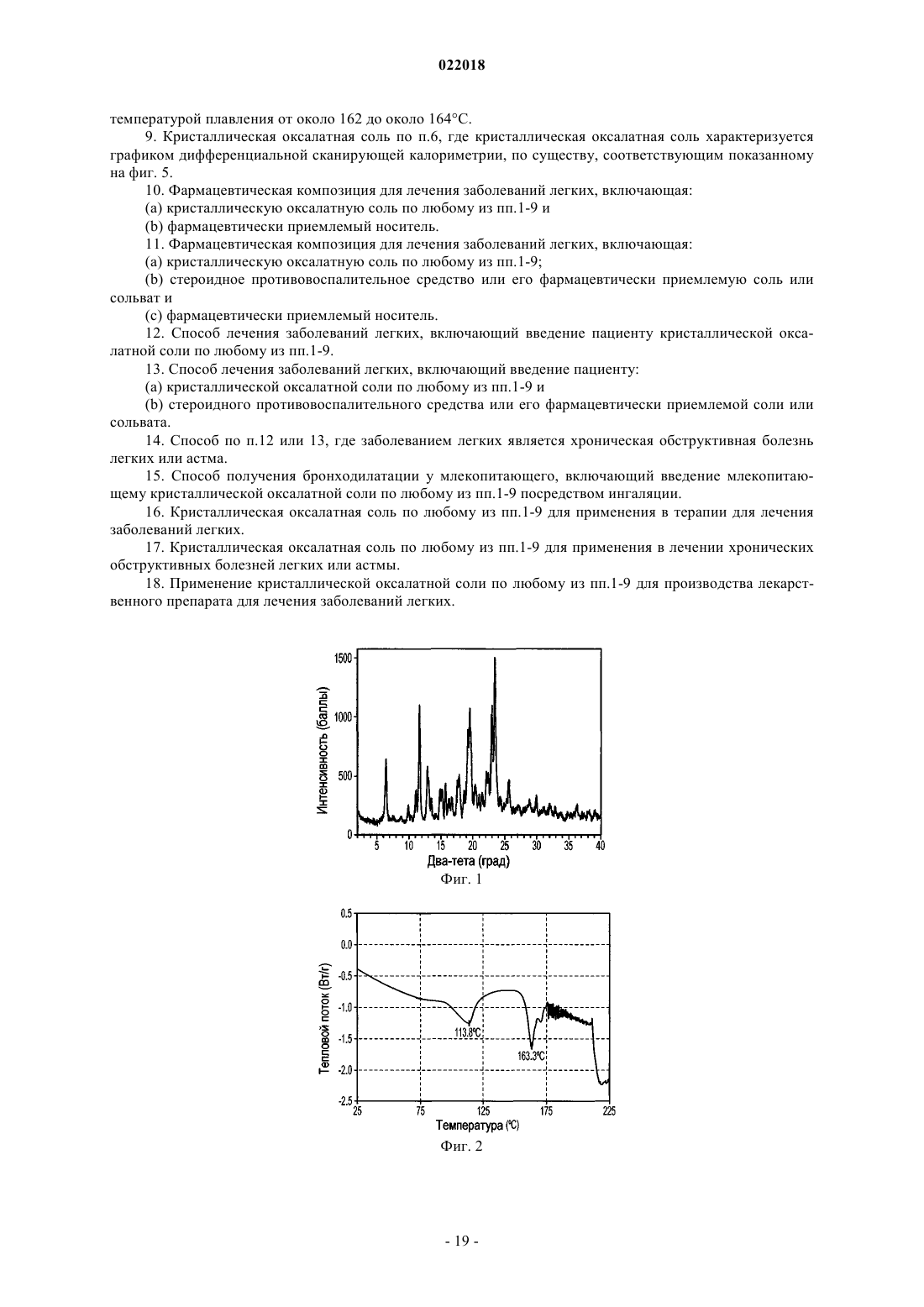
4. Кристаллическая оксалатная соль по п.2, где кристаллическая оксалатная соль характеризуется температурой плавления от около 162 до около 164°С.
5. Кристаллическая оксалатная соль по п.2, где кристаллическая оксалатная соль характеризуется графиком дифференциальной сканирующей калориметрии, по существу, соответствующим показанному на фиг. 2.
6. Кристаллическая оксалатная соль по п.1, где кристаллическая оксалатная соль представляет собой гидрат, характеризуемый порошковой рентгенограммой, включающей пики дифракции на 2θ значениях 15,32±0,20, 16,90±0,20, 19,25±0,20 и 23,73±0,20.
7. Кристаллическая оксалатная соль по п.6, где кристаллическая оксалатная соль характеризуется порошковой рентгенограммой, в которой пиковые положения, по существу, соответствуют пиковым положениям графика, показанного на фиг. 4.
8. Кристаллическая оксалатная соль по п.6, где кристаллическая оксалатная соль характеризуется температурой плавления от около 162 до около 164°С.
9. Кристаллическая оксалатная соль по п.6, где кристаллическая оксалатная соль характеризуется графиком дифференциальной сканирующей калориметрии, по существу, соответствующим показанному на фиг. 5.
10. Фармацевтическая композиция для лечения заболеваний легких, включающая:
(a) кристаллическую оксалатную соль по любому из пп.1-9 и
(b) фармацевтически приемлемый носитель.
11. Фармацевтическая композиция для лечения заболеваний легких, включающая:
(a) кристаллическую оксалатную соль по любому из пп.1-9;
(b) стероидное противовоспалительное средство или его фармацевтически приемлемую соль или сольват и
(c) фармацевтически приемлемый носитель.
12. Способ лечения заболеваний легких, включающий введение пациенту кристаллической оксалатной соли по любому из пп.1-9.
13. Способ лечения заболеваний легких, включающий введение пациенту:
(а) кристаллической оксалатной соли по любому из пп.1-9 и
(b) стероидного противовоспалительного средства или его фармацевтически приемлемой соли или сольвата.
14. Способ по п.12 или 13, где заболеванием легких является хроническая обструктивная болезнь легких или астма.
15. Способ получения бронходилатации у млекопитающего, включающий введение млекопитающему кристаллической оксалатной соли по любому из пп.1-9 посредством ингаляции.
16. Кристаллическая оксалатная соль по любому из пп.1-9 для применения в терапии для лечения заболеваний легких.
17. Кристаллическая оксалатная соль по любому из пп.1-9 для применения в лечении хронических обструктивных болезней легких или астмы.
18. Применение кристаллической оксалатной соли по любому из пп.1-9 для производства лекарственного препарата для лечения заболеваний легких.
Текст
КРИСТАЛЛИЧЕСКИЕ ОКСАЛАТНЫЕ СОЛИ ДИАМИДНОГО СОЕДИНЕНИЯ Изобретение относится к кристаллическим оксалатным солям 1-(2-[4-(4-[(R)-2-гидрокси-2-(8 гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты. Настоящее изобретение также относится к композициям, содержащим такую кристаллическую оксалатную соль, способам применения такой кристаллической оксалатной соли для, например, лечения заболеваний легких и к процессам получения такой кристаллической оксалатной соли.(71)(73) Заявитель и патентовладелец: ТЕРЕВАНС РЭСПИРЭЙТОРИ КАМПАНИ ЭЛ ЭЛ СИ (US) Предшествующий уровень техники изобретения Область изобретения Настоящее изобретение относится к новым кристаллическим солям диамидного соединения. Настоящее изобретение также относится к фармацевтическим композициям, содержащим такие кристаллические соли, процессам и промежуточным продуктам, применимым для получения таких кристаллических солей, и способам применения таких кристаллических солей для, например, лечения заболеваний легких. Состояние области техники В WO 2010/123766 А 1, опубликованном 28 октября 2010 г., описаны диамидные соединения, которые, вероятно, могут быть применимыми в качестве терапевтических средств для лечения заболеваний легких, таких как хронические обструктивные болезни легких (ХОБЛ (COPD и астма. Одним соединением, описанным в указанной заявке, является 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2 дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 иловый эфир бифенил-2-илкарбаминовой кислоты, чья химическая структура представлена формулой I Терапевтические средства, применимые для лечения легочных заболеваний, обычно вводят непосредственно в дыхательный тракт с использованием ингаляционного устройства, такого как ингалятор сухого порошка (DPI), ингалятор с дозирующим клапаном (MDI) или небулайзерный ингалятор. При рецептировании терапевтического средства для применения в таких устройствах целесообразно иметь стабильную кристаллическую форму терапевтического средства, которое может быть микронизировано или иным образом рецептировано и хранится без существенного разложения или потери кристалличности. В частности, кристаллическая форма не должна быть избыточно гигроскопичной или легко впитывающей влагу. Кроме того, кристаллическая форма должна иметь относительно высокую температуру плавления (т.е. более чем около 130 С) для минимизации термического разложения во время обработки и хранения. Однако образование кристаллических форм органических соединений является крайне непредсказуемым. Не существует надежного метода для предсказания того, какая, если есть, форма органического соединения будет кристаллической. Более того, не существует способов для предсказания, какая, если есть, кристаллическая форма будет иметь физические свойства, желаемые для применения, например, в фармацевтических ингаляционных устройствах. В настоящее время не описано кристаллической формы 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси 2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбамовой кислоты. Соответственно, существует необходимость в стабильной кристаллической форме такого соединения. В частности, существует необходимость в стабильной, негигроскопичной кристаллической форме, которая имеет приемлемый уровень гигроскопичности и относительно высокую температуру плавления. Сущность изобретения Настоящее изобретение относится к аддитивным солям щавелевой кислоты 1-(2-[4-(4-[(R)-2 гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты в форме кристаллического твердого вещества. Такие кристаллические оксалатные соли включают кристаллические оксалатные соли сольвата изопропанола и кристаллические оксалатные гидратные соли. Определенные кристаллические оксалатные соли по изобретению были охарактеризованы как негигроскопичные и имеющие приемлемые свойства гигроскопичности при воздействии атмосферной влаги. Более того, кристаллические оксалатные соли по настоящему изобретению имеют относительно высокую температуру плавления по меньшей мере около 160 С. Соответственно, в одном аспекте настоящее изобретение обеспечивает кристаллическую оксалатную соль 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2 илкарбаминовой кислоты. В одном варианте осуществления изобретения кристаллической оксалатной солью по изобретению является сольват изопропанола, характеризуемый порошковой рентгенограммой с пиками в 2 значениях 11,660,20, 15,750,20, 19,550,20, 23,000,20 и 23,450,20. Такая кристаллическая форма или полиморф обозначена как форма 1. В другом варианте кристаллическая оксалатная соль по изобретению представляет собой гидрат,характеризующийся порошковой рентгенограммой с пиками на 2 значениях 15,320,20, 16,900,20,19,250,20 и 23,730,20. Такая кристаллическая форма или полиморф обозначена как форма 2. В одном варианте осуществления изобретения кристаллическая оксалатная соль по изобретению представляет собой гидрат, полученный при воздействии на кристаллическую оксалатную соль 1-(2-[4(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты от около 80 до около 100% относительной влажности в течение периода от около 1 до около 5 дней. В другом аспекте настоящее изобретение обеспечивает композицию, включающую кристаллическую оксалатную соль по изобретению. В одном варианте осуществления изобретения композицией по изобретению является фармацевтическая композиция, включающая (а) кристаллическую оксалатную соль по изобретению и (b) фармацевтически приемлемый носитель. В другом варианте осуществления изобретения композицией является фармацевтическая композиция, включающая (а) кристаллическую оксалатную соль по изобретению; (b) стероидное противовоспалительное средство или его фармацевтически приемлемую соль или сольват; (с) фармацевтически приемлемый носитель. В другом аспекте настоящее изобретение обеспечивает кристаллическую оксалатную соль по изобретению для применения в лечении. В одном варианте осуществления изобретения кристаллическую оксалатную соль по изобретению используют в лечении заболеваний легких. В определенном варианте осуществления изобретения заболеванием легких является хроническая обструктивная болезнь легких. В другом определенном варианте осуществления изобретения заболеванием легких является астма. В одном варианте осуществления настоящее изобретение обеспечивает способ лечения болезни легких, способ включает введение пациенту кристаллической оксалатной соли по изобретению. В другом варианте осуществления изобретения, настоящее изобретение обеспечивает способ лечения заболевания легких, способ включает введение пациенту (а) кристаллической оксалатной соли по изобретению; и (b) стероидного противовоспалительного средства или его фармацевтически приемлемой соли или сольвата. В еще одном варианте осуществления изобретения настоящее изобретение обеспечивает способ получения у млекопитающего бронходилатации, способ включает введение млекопитающему кристаллической оксалатной соли по изобретению посредством ингаляции. В другом варианте осуществления настоящее изобретение обеспечивает кристаллическую оксалатную соль по изобретению для применения в производстве лекарственного средства. Другие аспекты и варианты осуществления настоящего изобретения описаны в настоящем документе. Краткое описание чертежей Различные аспекты настоящего изобретения проиллюстрированы со ссылками на сопутствующие чертежи. На фиг. 1 показана порошковая рентгенограмма (PXRD) кристаллической оксалатной соли сольвата изопропанола 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2 илкарбаминовой кислоты (форма 1). На фиг. 2 показана запись дифференциальной сканирующей калориметрии (DSC) кристаллической оксалатной соли сольвата изопропанола 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2 дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 илового эфира бифенил-2-илкарбаминовой кислоты (форма 1). На фиг. 3 показана запись термического гравиметрического анализа (TGA) кристаллической оксалатной соли сольвата изопропанола 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2 дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 илового эфира бифенил-2-илкарбаминовой кислоты (форма 1). На фиг. 4 показана порошковая рентгенограмма кристаллической оксалатной соли гидрата изопропанола 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (форма 2). На фиг. 5 показана запись дифференцирующей сканирующей калориметрии (DSC) кристаллической оксалатной соли гидрата изопропанола 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2 дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 илового эфира бифенил-2-илкарбаминовой кислоты (форма 2). На фиг. 6 показана запись термического гравиметрического анализа (TGA) кристаллической окса-2 022018 латной соли гидратаизопропанола 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2 дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 илового эфира бифенил-2-илкарбаминовой кислоты (форма 2). На фиг. 7 показана запись динамического поглощения влаги (DMS) кристаллической оксалатной соли гидрата изопропанола 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5 ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (форма 2). Подробное описание изобретения Настоящее изобретение относится к аддитивным солям щавелевой кислоты 1-(2-[4-(4-[(R)-2 гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты в кристаллическом твердом состоянии, включая сольваты изопропанола и гидраты. Кристаллические оксалатные соли по изобретению могут присутствовать в виде одной или более отдельных кристаллических твердых форм, и настоящее изобретение охватывает все такие формы, включая смеси форм, если не указано иначе. В одном варианте осуществления настоящее изобретение обеспечивает первую кристаллическую оксалатную твердую форму аддитивной соли щавелевой кислоты соединения по формуле I, называемую здесь форма 1. Форма 1 представляет собой форму сольвата изопропанола. В другом варианте осуществления настоящее изобретение обеспечивает вторую кристаллическую оксалатную твердую форму аддитивной соли щавелевой кислоты соединения по формуле I, называемую в настоящем описании форма 2. Форма 2 представляет собой гидрат. Соединение по формуле I содержит один хиральный центр, имеющий (R) конфигурацию. Однако специалист в области техники понимает, что минимальные количества (S) стереоизомера могут присутствовать в композициях по настоящему изобретению, если не указано иначе, с условием, что какие-либо свойства композиции в целом не нарушаются в присутствии такого изомера. Соединение по формуле I и его промежуточные продукты были названы с использованием характеристики AutoNom коммерчески доступного программного обеспечения MDL ISIS/Draw (Symyx, SantaClara, California). Определения. При описании соединений, композиций, методов и процессов настоящего изобретения следующие термины имеют следующие значения, если не указано иначе. Термин "температура плавления" обозначает температуру, при которой наблюдают максимальный эндотермический тепловой ток при дифференциальной сканирующей калориметрии. Термин "микронизированный" или "в микронизированной форме" обозначает частицы, в которых по меньшей мере около 90% частиц имеют диаметр менее чем около 10 мкм. Термин "сольват" обозначает ассоциацию, такую как комплекс или агрегат, одной или более молекул растворенного вещества, т.е. оксалатной соли соединения по формуле I, и одной или более молекул растворителя. Характерные растворители включают, в качестве примера, воду и изопропанол. Когда растворителем является вода, образуемым сольватом является гидрат. Термин "терапевтически эффективное количество" обозначает количество, достаточное для эффективного лечения при введении пациенту, нуждающемуся в лечении. Термин "процесс лечения" или "лечение" обозначает:(a) предотвращение возникновения заболевания или медицинского состояния, т.е. профилактическое лечение пациента или субъекта;(b) облегчение заболевания или медицинского состояния, т.е. устранение или регресс заболевания или медицинского состояния у пациента;(c) подавление заболевания или медицинского состояния, т.е. замедление или остановка развития заболевания и или медицинского состояния у пациента; или(d) облегчение симптомов заболевания или медицинского состояния у пациента. Термин "стандартная лекарственная форма" обозначает физически дискретную единицу, подходящую для введения пациенту, т.е. каждая единица содержит заранее определенное количество терапевтического средства, рассчитанное для получения терапевтического эффекта или отдельно или в комбинации с одной или более дополнительными единицами. Например, такие стандартные лекарственные формы могут быть капсулами с сухим порошком для ингалятора, отмеренными дозами из ингалятора с дозирующим клапаном, капсулами, таблетками, пилюлями и подобными. Характерные кристаллические оксалатные соли по изобретению. Были обнаружены кристаллические оксалатные соли 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2 оксо-1,2-дигидрохинолин-5-ил)этиламино]метил-фенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты, существующие по меньшей мере в двух различных кристаллических формах. Для целей настоящего изобретения такие формы называют в настоящем описании форма 1 и форма 2. Форма 1 представляет собой сольват изопропанола, отличающийся порошковой рентгенограммой(PXRD), имеющей достоверные пики дифракции, среди других пиков в 2 значениях около 11,660,20,15,750,20, 19,550,20, 23,000,20 и 23,450,20. Форма 1 имеет график дифференциальной сканирующей калориметрии (DSC), которая имеет пик эндотермического теплового тока от около 162 до около 164 С,например около 163,3 С. Форма 1 включает от около 1 до около 2 мол.экв. изопропанола на 1 мол.экв. оксалатной соли соединения по формуле I, включая от около 1,25 до около 1,75 мол.экв., например как 1,5 мол.экв. Форма 2 представляет собой гидрат, отличающийся порошковой рентгенограммой (PXRD), имеющей достоверные пики дифракции, среди других пиков, на 2 значениях около 15,320,20, 16,900,20,19,250,20 и 23,730,20. Форма 2 имеет график дифференцирующей сканирующей калориметрии (DSC),которая проявляет пик эндотермического теплового тока от около 162 до около 164 С, например около 162,6 С. Форма 2 включает от около 1 до около 3 мол.экв. воды на 1 мол.экв. оксалатной соли соединения по формуле I, включая от около 1,5 до около 2 мол.экв. Кристаллические оксалатные соли по настоящему изобретению обычно содержат от около 0,90 до около 1,10 мол.экв. щавелевой кислоты на 1 мол.экв. соединения по формуле I, включая от около 0,95 до около 1,05 мол.экв. щавелевой кислоты на 1 мол.экв. соединения по формуле I. В определенном варианте осуществления изобретения соль щавелевой кислоты по настоящему изобретению содержит около 1 мол.экв. щавелевой кислоты на 1 мол.экв. соединения по формуле I. Молярное соотношение определяют с использованием обычных способов, таких как 13 С ЯМР, элементный анализ, ионный анализ или ВЭЖХ. Кристаллические оксалатные соли по настоящему изобретению обычно получают с использованием щавелевой кислоты или дигидрата щавелевой кислоты и 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2 оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты. Щавелевая кислота и дигидрат щавелевой кислоты являются коммерчески доступными, например, от Sigma-Aldrich, St. Louis, МО. 1-(2-[4(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты, используемый в настоящем изобретении, может быть легко получен из коммерчески доступных исходных материалов и реагентов с использованием методик, описанных в примерах ниже, или с использованием методик, описанных в принадлежащей тому же правообладателю США, описанной в разделе предшествующей области техники настоящего изобретения. Способы или процессы для получения кристаллических оксалатных солей по настоящему изобретению представлены в примерах. Обычно 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2 дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 иловый эфир бифенил-2-илкарбаминовой кислоты контактирует с от около 0,90 до около 1,1 мол.экв.,например, 1,0 мол.экв., щавелевой кислоты или дигидрата щавелевой кислоты в разбавителе. Обычно такую реакцию проводят при температуре, варьирующейся от около 0 до около 60 ЕС, включая от около 20 до около 35 ЕС, например, от около 25 до около 30 ЕС. Подходящие инертные разбавители для указанной реакции включают, но не ограничивается, метанол, изопропанол и подобные, необязательно содержащие воду. Необязательно реакционная смесь может быть обработана ультразвуком с использованием обычного оборудования для облегчения кристаллизации. Способ получения кристаллической оксалатной соли по настоящему изобретению может необязательно включать применение затравочного кристалла для получения преимущественно определенной кристаллической формы. Например, с использованием затравочного кристалла формы 2 может быть получена кристаллическая оксалатная гидратная соль, имеющая, по существу, такую же форму, как затравочный кристалл, т.е. форму 2. Такие затравочные кристаллы могут быть использованы при исходном образовании кристаллической оксалатной соли по изобретению или они могут быть использованы для рекристаллизации кристаллической или частично кристаллической формы. Обычно затравочные кристаллы получают посредством медленной кристаллизации без перемешивания и без применения охлаждения. В качестве иллюстрации для получения затравочных кристаллов соединение по формуле I обычно растворяют в растворителе при температуре, достаточной для обеспечения растворения. Например, соединение по формуле I растворяют в горячей 10% (об./об.) воде в метаноле и затем позволяют остыть до комнатной температуры. Полученному раствору позволяют медленно испаряться для получения кристаллической оксалатной соли по изобретению. Полученные кристаллы выделяют фильтрацией или другими обычными средствами. Альтернативно затравочные кристаллы могут быть получены из предшествующего препарата кристаллического материала. Было обнаружено, что кристалличность формы 2 улучшается, когда кристаллический материал хранится при комнатной температуре при высокой влажности. Например, когда образец формы 2 хранили при комнатной температуре при 84% относительной влажности, кристалличность улучшалась в течение периода около 5 дней, что оценивали по рентгенограмме PXRD образца. Композиции, комбинации и рецептуры. Кристаллические оксалатные соли по настоящему изобретению для получения композиций могут быть смешаны с другими материалами, такими как терапевтические средства, носители, вспомогательные вещества, разбавители, растворители и подобные. В одном варианте осуществления изобретения кристаллическую оксалатную соль по изобретению рецептируют с носителем или вспомогательным веществом для получения фармацевтической композиции или рецептуры. Такие фармацевтические композиции обычно содержат терапевтически эффективное количество кристаллической оксалатной соли по изобретению. В некоторых случаях, однако, фармацевтическая композиция может содержать более чем терапевтически эффективное количество, например,концентрированную партию композиции, или менее чем терапевтически эффективное количество, например, стандартную лекарственную форму, предназначенную для введения нескольких доз. Фармацевтическая композиция обычно содержит от около 0,01 до около 95% по массе кристаллической оксалатной соли по изобретению, включая, например, от около 0,05 до около 30% по массе или от около 0,1 до около 10% по массе. Такие фармацевтические композиции могут быть получены с использованием обычных носителей или вспомогательных веществ. Выбор определенного носителя или вспомогательного вещества или комбинаций носителей или вспомогательных веществ будет зависеть от различных факторов, таких как тип введения или заболевание или медицинское состояние, которое лечат. Множество подходящих носителей и вспомогательных веществ для получения фармацевтических композиций являются коммерчески доступными. Например, материалы могут быть закуплены у Sigma (St. Louis, МО). Методики и материалы для получения фармацевтических композиций, подходящих для определенного типа введения, описаны в фармацевтической области техники, включая, например, Remington: The Science and Practice ofPharmacy, 20th Edition, Lippincott WilliamsWhite, Baltimore, Maryland (2000); и H.C. Ansel et al, Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott WilliamsWhite, Baltimore, Maryland (1999). Характерные примеры фармацевтически приемлемых носителей включают, но не ограничиваются:(1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; (4) порошковая трагакантовая камедь; (5) солод; (6) желатин; (7) тальк; (8) отвержденные масла и воск, такие как масло какао и воска для суппозиториев; (9) жидкие масла (при комнатной температуре), такие как арахисовое масло, хлопковое масло, саффлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные вещества, такие как гидроксид магния и гидроксид алюминия; (15) альгиновая кислота; (16) апирогенная вода; (17) изотонический солевой раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатный буферный раствор; (21) прессованные газы-пропелленты,такие как хлорфторуглероды и гидрофторуглероды; и (22) другие нетоксичные совместимые вещества,используемые в фармацевтических композициях. Фармацевтическую композицию обычно получают путем тщательного и равномерного перемешивания или измельчения кристаллической оксалатной соли по изобретению с фармацевтически приемлемым носителем и любыми другими необязательными ингредиентами. Если необходимо или желательно,полученная, однородно перемешанная смесь затем может быть формована или загружена в таблетки,капсулы, пилюли, канистры, картриджи, дисперсеры и подобные с использованием обычных методик и оборудования. В одном варианте осуществления изобретения фармацевтическая композиция является подходящей для ингаляционного введения. Фармацевтические композиции для ингаляционного введения обычно находятся в форме аэрозоля или порошка. Такие композиции обычно вводят с использованием ингаляционных устройств, таких как ингалятор с дозирующим клапаном (MDI), ингалятор сухого порошка (DPI),небулайзерный ингалятор или подобные устройства доставки. В определенном варианте осуществления изобретения фармацевтическую композицию вводят путем ингаляции с использованием ингалятора сухого порошка. Такие ингаляторы сухого порошка обычно вводят фармацевтическую композицию в виде свободнотекущего порошка, который диспергируется в потоке воздуха пациента во время вдоха. С целью получения свободно текущей порошковой композиции терапевтическое средство обычно рецептируют с подходящим вспомогательным веществом, таким как лактоза, крахмал, маннит, декстроза, полимолочная кислота (PLA), полилактид-со-гликолид (PLGA) или их комбинации. Обычно терапевтическое средство является микронизированным и комбинированным с подходящим носителем для получения композиции, подходящей для ингаляции. Соответственно в одном варианте осуществления изобретения кристаллическая оксалатная соль по изобретению находится в микронизированной форме. Характерная фармацевтическая композиция для применения в ингаляторе сухого порошка включает лактозу и кристаллическую оксалатную соль по изобретению в микронизированной форме. Такая композиция сухого порошка может быть получена, например, путем смешивания сухой измельченной лактозы с терапевтическим средством и последующей сушки и измельчения компонентов. Затем композицию обычно загружают в диспенсер сухого порошка или в картриджи для ингаляции или капсулы для применения с устройством доставки сухого порошка. Ингаляционные устройства доставки сухого порошка, подходящие для введения терапевтических средств посредством ингаляции, описаны в области техники, и примеры таких устройств являются коммерчески доступными. Например, характерные ингаляционные устройства или продукты доставки сухого порошка включают Aeolizer (Novartis); Airmax (IV АХ); ClickHaler (Innovata Biomed); Diskhaler (GlaxoSmithKline); Diskus/Accuhaler (GlaxoSmithKline); Easyhaler (Orion Pharma); Eclipse (Aventis); FlowCaps(Hovione); Handihaler (Boehringer Ingelheim); Pulvinal (Chiesi); Rotahaler (GlaxoSmithKline); SkyeHaler/Certihaler (SkyePharma); Twisthaler (Schering-Plough); Turbuhaler (AstraZeneca); Ultrahaler (Aventis) и подобные. В другом определенном варианте осуществления изобретения фармацевтическую композицию вводят посредством ингаляции с использованием ингалятора с дозирующим клапаном. Такие ингаляторы с дозирующим клапаном обычно вводят отмеренное количество терапевтического средства с использованием прессованного газа-пропеллента. Соответственно фармацевтические композиции, вводимые с использованием ингалятора с дозирующим клапаном, обычно включают раствор или суспензию терапевтического средства в сжиженном пропелленте. Может быть использован любой подходящий сжиженный пропеллент, включая гидрофторалканы (HFA), такие как 1,1,1,2-тетрафторэтан (HFA 134 а) и 1,1,1,2,3,3,3 гептафтор-н-пропан, (HFA 227), и хлорфторуглероды, такие как CCI3F. В определенном варианте осуществления изобретения пропеллентами являются гидрофторалканы. В некоторых вариантах осуществления изобретения композиция гидрофторалкана содержит со-растворитель, такой как этанол или пентан,и/или поверхностно-активное вещество, такое как триолеат сорбита, олеиновая кислота, лецитин и глицерин. Характерная фармацевтическая композиция для применения в ингаляторе с дозирующим клапаном содержит от около 0,01% до около 5% по массе кристаллической оксалатной соли по изобретению, от около 0% до около 20% по массе этанола и от около 0% до около 5% по массе поверхностно-активного вещества с оставшимся пропеллентом HFA. Такие композиции обычно получают путем добавления охлажденного или прессованного гидрофторалкана в подходящий контейнер, содержащий терапевтическое средство, этанола (если есть) и поверхностно-активного вещества (если есть). Для получения суспензии терапевтическое средство микронизируют и затем смешивают с пропеллентом. Затем композицию загружают в аэрозольный контейнер, который обычно образует часть ингаляционного устройства с дозирующим клапаном. Ингаляционные устройства с дозирующим клапаном, подходящие для введения терапевтических средств путем ингаляции, описаны в области техники, и примеры таких устройств являются коммерчески доступными. Например, характерные ингаляционные устройства или продукты с дозирующим клапаном включают ингаляционную систему AeroBid (Forest Pharmaceuticals), ингаляционный аэрозоль(Schering), ингаляционный аэрозоль Serevent (GlaxoSmithKline) и подобные. В другом определенном варианте осуществления изобретения фармацевтическую композицию вводят путем ингаляции с использованием небулайзерного ингалятора. Такое небулайзерное устройство обычно дает поток воздуха высокой скорости, который вызывает распыление фармацевтической композиции в виде тумана, который попадает в дыхательный тракт пациента. Соответственно, при рецептировании для применения в небулайзерном ингаляторе терапевтическое средство может быть растворено в подходящем носителе для получения раствора. Альтернативно терапевтическое средство может быть микронизировано и смешано с подходящим носителем для получения суспензии микронизированных частиц. Характерная фармацевтическая композиция для применения в небулайзерном ингаляторе включает изотоническую водную суспензию, включающую от около 0,05 мкг/мл до около 10 мг/мл кристаллической оксалатной соли по изобретению в микронизированной форме. В одном варианте осуществления изобретения раствор имеет рН от около 4 до около 6. Небулайзерные устройства, подходящие для введения терапевтических средств путем ингаляции,описаны в области техники, и примеры таких устройств являются коммерчески доступными. Например,характерные небулайзерные устройства или продукты включают ингалятор Respimat Softmist (BoehringerIngelheim), легочную систему доставки AERx (Aradigm Corp.), небулайзер многократного использованияPARI LC Plus (Pari GmbH) и подобные. Если желательно кристаллическую оксалатную соль по изобретению можно вводить в комбинации с одним или более другими терапевтическими средствами. В таком аспекте изобретения кристаллическую оксалатную соль по изобретению или физически перемешивают с другим терапевтическим средством для получения композиции, содержащей оба средства, или каждое средство отдельно вводят пациенту или одновременно, или последовательно. Например, кристаллическую оксалатную соль по изобретению можно комбинировать со вторым терапевтическим средством с использованием обычных методик и оборудования для получения компози-6 022018 ции, включающей кристаллическую оксалатную соль по изобретению и второе терапевтическое средство. Кроме того, терапевтическое средство может быть скомбинировано с фармацевтически приемлемым носителем для получения фармацевтической композиции, включающей кристаллическую оксалатную соль по изобретению, второе терапевтическое средство и фармацевтически приемлемый носитель. В указанном варианте осуществления изобретения компоненты композиции обычно смешивают или измельчают для получения физической смеси. Физическую смесь затем вводят пациенту с использованием любого пути, описанного в настоящем описании. Альтернативно терапевтические средства могут оставаться разделенными и отдельными перед введением пациенту. В указанном варианте осуществления изобретения терапевтические средства физически не смешивают вместе до введения, но вводят одновременно или последовательно в качестве отдельных средств или композиций. Например, кристаллическую оксалатную соль по изобретению можно вводить путем ингаляции одновременно или последовательно с другим терапевтическим средством и использованием ингаляционного устройства доставки, в котором используют отдельные контейнеры (например, блистерные упаковки) для каждого терапевтического средства. Альтернативно комбинацию можно вводить с использованием отдельных устройств доставки, т.е. одного устройства доставки для каждого терапевтического средства. Кроме того, терапевтические средства можно вводить различными путями введения, т.е. один путем ингаляции и другой путем перорального введения. Любое терапевтическое средство, совместимое с кристаллической оксалатной солью по изобретению, может быть использовано в комбинации с такими солями. В определенном варианте осуществления изобретения вторым терапевтическим средством является такое, которое эффективно вводят путем ингаляции. В качестве иллюстрации характерные типы терапевтических средств, которые могут быть использованы, включают, но не ограничиваются, противовоспалительные средства (включая кортикостероиды и глюкокортикоиды), нестероидные противовоспалительные средства (НПВС) и ингибиторы PDE4; бронходилататоры, такие как ингибиторы PDE3, модуляторы аденозина 2b и агонисты 2 адренергических рецепторов; противоинфекционные средства, такие как грамположительные антибиотики, грамотрицательные антибиотики и противовирусные средства; антигистаминные средства; ингибиторы протеаз; афферентные блокаторы, такие как D2 агонисты и модуляторы нейрокинина; и антагонисты мускариновых рецепторов (антихолинергические средства). Множество примеров таких терапевтических средств известно в области техники. Подходящие дозы для таких других терапевтических средств также известны в области техники и обычно варьируются от около 0,05 мкг/сутки до около 500 мг/сутки. В одном варианте осуществления изобретения второе терапевтическое средство является применимым для лечения заболевания легких. В определенном варианте осуществления изобретения кристаллическую оксалатную соль по изобретению вводят в комбинации со стероидным противовоспалительным средством. Характерные стероидные противовоспалительные средства включают, но не ограничиваются, беклометазона дипропионат; будесонид; бутиксокорт пропионат; 20R-16,17-[бутилиденбис(окси)]-6,9-дифтор-11-гидрокси 17-(метилтио)андроста-4-ен-3-он (RPR-106541); циклезонид; дексаметазон; S-фторметиловый эфир 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17 карботиоевой кислоты; S-фторметиловый эфир 6,9-дифтор-11-гидрокси-16-метил-17-[(4-метил 1,3-тиазол-5-карбонил)окси]-3-оксоандроста-1,4-диен-17-карботиоевой кислоты;(S)-(2 оксотетрагидрофуран-3S-иловый) эфир 6,9-дифтор-11-гидрокси-16-метил-3-оксо-17 пропионилоксиандроста-1,4-диен-17-карботиоевой кислоты; флунизолид; флутиказона фуроат; флутиказона пропионат; метилпреднизолон; мометазона фуроат; преднизолон; преднизон; рофлепонид; ST126; триамцинолона ацетонид и подобные и их фармацевтически приемлемые соли и сольваты. Такие стероидные противовоспалительные средства являются коммерчески доступными или могут быть получены с использованием обычных методик и реагентов. Например, получение и применение стероидных противовоспалительных средств описано в патенте США 6537983, поданном 25 марта 2003 г., патенте США 6750210 В 2, поданном 15 июня 2004 г., патенте США 6759398 В 2, поданном 6 июля 2004 г.,патенте США 6858596 В 2, поданном 22 февраля 2005 г., патенте США 7101866 В 2, поданном 5 сентября 2006 г., и ссылках, цитированных в них. При использовании стероидное противовоспалительное средство обычно вводят в количестве, которое дает терапевтически полезный эффект при совместном введении с кристаллической оксалатной солью по изобретению. Обычно стероидное противовоспалительное средство вводят в количестве, достаточном для обеспечения от около 0,05 до около 500 мкг средства на дозу. Характерные фармацевтические композиции включают, но не ограничиваются, следующие примеры.A. Композиция сухого порошка. Микронизированную кристаллическую оксалатную соль по изобретению (100 мг) перемешивали с сухой измельченной лактозой (25 г). Такую измельченную смесь затем загружали в отдельные блистеры снимающейся блистерной упаковки в количестве, достаточном для обеспечения от около 10 мкг до около 500 мкг кристаллической оксалатной соли по изобретению на дозу. Содержимое блистеров вводили с использованием ингалятора сухого порошка.B. Композиция сухого порошка. Микронизированную кристаллическую оксалатную соль по изобретению (1 г) перемешивали с сухой измельченной лактозой (200 г) для получения партии композиции, имеющей массовое соотношение кристаллической оксалатной соли к лактозе 1:200. Измельченную композицию упаковывали в ингаляционное устройство для сухого порошка, способное вводить от около 10 до около 500 мкг кристаллической оксалатной соли по изобретению на дозу.C. Композиция сухого порошка. Микронизированную кристаллическую оксалатную соль по изобретению (100 мг) и микронизированное стероидное противовоспалительное средство (500 мг) перемешивали с сухой измельченной лактозой (30 г). Измельченную смесь затем загружали в отдельные блистеры снимающейся блистерной упаковки в количестве, достаточном для обеспечения от около 10 мкг до около 500 мкг кристаллической оксалатной соли по изобретению на дозу. Содержимое блистеров вводили с использованием ингалятора сухого порошка.D. Композиция ингалятора с дозирующим клапаном. Микронизированную кристаллическую оксалатную соль по изобретению (10 г) диспергировали в растворе, полученном путем растворения лецитина (0,2 г) в деминерализованной воде (200 мл). Полученную суспензию лиофилизировали и затем микронизировали для получения микронизированной композиции, включающей частицы, имеющие средний диаметр менее чем около 1,5 мкм. Микронизированную композицию затем загружали в картриджи для ингалятора с дозирующим клапаном, содержащие прессованный 1,1,1,2-тетрафторэтан в количестве, достаточном для обеспечения от около 10 мкг до около 500 мкг кристаллической оксалатной соли по изобретению на дозу при введении посредством ингалятора с дозирующим клапаном. Применение. 1-(2-[4-(4-[(R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5 ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-иловый эфир бифенил 2-илкарбаминовой кислоты обладает активностью агониста и 2 адренергического рецептора и мускаринового рецептора, и, следовательно, ожидают, что оксалатная соль указанного соединения будет применимой в качестве терапевтического средства для лечения медицинских состояний, опосредованных 2 адренергическими рецепторами или мускариновыми рецепторами, т.е. медицинских состояний, которые облегчаются лечением агонистами 2 адренергических рецепторов или агонистами мускариновых рецепторов. Такие медицинские состояния известны в области техники, как проиллюстрировано, например, руководствами Eglen et al., Muscarinic Receptor Subtypes: Pharmacology and Therapuetic Potential, DNP 10(8), 462-469 (1997); Emilien et al., Current Therapeutic Uses and Potential of beta-Adrenoceptor Agonistsand Antagonists, European J. Clinical Pharm., 53(6), 389-404 (1998) и ссылками, цитированными в них. Например, такие медицинские состояния включают, в качестве иллюстрации, заболевания легких или заболевания, ассоциированные с обратимой обструкцией дыхательных путей, такие как хроническая обструктивная болезнь легких, астма, бронхит, эмфизема, пневмосклероз и подобные. Хронические обструктивные болезни легких или ХОБЛ известны в области техники как включающие множество респираторных состояний, включая хронический обструктивный бронхит и эмфизему, что проиллюстрировано руководствами Barnes, Chronic Obstructive Pulmonary Disease, N. Engl. J. Med., 2000: 343:269-78 и ссылками, цитированными в них. Другие состояния включают преждевременные роды, депрессию, застойную сердечную недостаточность, заболевания кожи (например, воспалительные, аллергические, псориатические и пролиферативные заболевания кожи), состояния, при которых желательно снижение пептической активности (например, пептические язвы или язвы желудка), и заболевания, поражающие мышцы. В одном аспекте настоящее изобретение обеспечивает кристаллическую оксалатную соль по изобретению для применения в терапии. Например, кристаллическая оксалатная соль по изобретению может быть использована для лечения заболеваний легких, таких как хроническая обструктивная болезнь легких или астма. При использовании для лечения заболеваний легких кристаллическую оксалатную соль по настоящему изобретению вводят пациенту, нуждающемуся в лечении. В одном варианте осуществления изобретения кристаллическую оксалатную соль вводят посредством ингаляции. Кристаллическую оксалатную соль по изобретению можно вводить в количестве нескольких доз в сутки (например, два раза в сутки или три раза в сутки), в одной дозе в сутки (например, один раз в день), одну дозу в неделю или по необходимости. Обычно доза для лечения заболеваний легких будет варьироваться от около 10 мкг/сутки до около 1000 мкг/сутки. При введении млекопитающему путем ингаляции кристаллическая оксалатная соль по настоящему изобретению обычно вызывает расширение бронхов, и, следовательно, кристаллическая оксалатная соль по настоящему изобретению является применимой в качестве средства для получения бронходилатации у млекопитающего, такого как человек, собака, морская свинка, крыса и подобные. В указанном вариан-8 022018 те осуществления изобретения кристаллическую оксалатную соль по изобретению обычно вводят млекопитающему путем ингаляции в количестве, дающем бронходилатацию. Обычно доза для получения бронходилатации варьируется от около 0,25 до около 20 мкг/кг. При использовании в качестве терапевтического средства кристаллические оксалатные соли по настоящему изобретению необязательно вводят в комбинации с одним или более другими терапевтическими средствами. В частности, путем введения кристаллической оксалатной соли по настоящему изобретению со стероидным противовоспалительным средством, может быть получена тройная легочная терапия,т.е. активность агониста 2 адренергического агониста, активность антагониста мускариновых рецепторов и противовоспалительная активность с использованием только двух терапевтических средств. Так как фармацевтические композиции (и комбинации), содержащие два терапевтических средства, обычно легче рецептировать и/или вводить по сравнению с композициями, содержащими три терапевтических средства, такие двухкомпонентные композиции обеспечивают существенное преимущество относительно композиций, содержащих три терапевтических средства. Соответственно, в определенном варианте осуществления изобретения фармацевтические композиции, комбинации и способы по настоящему изобретению дополнительно включают стероидное противовоспалительное средство или его фармацевтически приемлемую соль или сольват. Свойства и применение кристаллических оксалатных солей по настоящему изобретению могут быть продемонстрированы с использованием различных исследований in vitro и in vivo, известных специалисту в области техники. Например, характерные исследования описаны в следующих примерах. Примеры Следующие примеры представлены для иллюстрации различных характерных вариантов осуществления и аспектов настоящего изобретения и не предназначены для ограничения рамок настоящего изобретения какие-либо образом, если не указано иначе. Все реагенты, исходные материалы и растворители, используемые в следующих примерах, покупали у коммерческих поставщиков (таких как Sigma-Aldrich Chemical Company, St. Louis, МО) и использовали без дополнительной очистки, если не указано иначе. Следующие сокращения использовали для разбавителей:EtOAc=этилацетат; МеОН=метанол и THF=тетрагидрофуран. Спектры 1 Н-ЯМР записывали на спектрометре 400 МГц Varian AS400, если не указано иначе. Химические сдвиги представлены в виде значенийв ч./млн относительно тетраметилсилана (TMS) в качестве внутреннего стандарта. Константы связывания (J значения) даны в герц (Гц) и множества представлены с использованием следующих сокращений: s=синглет, d=дуплет, t=триплет, q=квартет,m=мультиплет, br=широкий, nd=не определяли. Условия жидкостной хроматографии масс-спектроскопии (ЖХ-MC). Данные ЖХ-МС получали с использованием системы для жидкостной хроматографии Agilent 1100Quadrupole LC/MS (Perkin-Elmer Sciex Instruments). Используемыми системами растворителей были: Растворитель А: 98% вода и 2% ацетонитрил (об/об) + 1 мл/л TFA Растворитель В: 90% ацетонитрил и 10% вода (об/об) + 1 мл/л TFA Скорость тока: 500 мкл/мин Градиент: (метод 10-90): от 10% В до 90% В в течение 3 мин(Метод 10-70): от 10% В до 70% В в течение 3 мин. Условия ВЭЖХ. ВЭЖХ проводили с использованием системы ВЭЖХ HP 1100 Series (Agilent Technologies) и колонки ZORBAX Rapid Resolution 3,5 мкм Rx, Bonus-RP (размер частиц 3,5 мкм; 2,1 мм 50 мм) (AgilentTechnologies) или колонки ВЭЖХ Ascentis Express C18 (размер частиц 2,7 мкм, 3,0 мм 3 см). Используемыми системами растворителей были: Растворитель А: 98% вода и 2% ацетонитрил (об/об) + 1 мл/л TFA Растворитель В: 90% ацетонитрил и 10% вода (об/об) + 1 мл/л TFA Скорость тока: 500 мкл/мин Градиент: (Метод 10-50): от 10% В до 50% В в течение 6 мин(Метод 2-90): от 2% В до 90% В в течение 6 мин. Пример 1. Получение 3-[4-(бифенил-2-карбамоилокси)пиперидин-1-ил]пропионовой кислоты. Перемешиваемый раствор пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (50,0 г,168,7 ммоль) (см., например, патентную публикацию США 2006/0035931 А 1, опубликованную 16 февраля 2006 г.) и акриловой кислоты (15,1 мл, 219,3 ммоль) в DCM (500 мл) нагревали при 50 С в течение ночи. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в МеОН(600 мл). Полученный раствор нагревали при 75 С в течение 2 ч и затем позволяли стоять при комнатной температуре в течение около 48 ч. Полученное твердое вещество собирали фильтрацией, промывали МеОН и сушили для получения указанного в заголовке соединения (61,5 г, 99% выход). Пример 2. Получение гидрохлорида метил 5-метиламинопентаноата. Перемешиваемый раствор 1-метил-2-пиперидинона (4,40 мл, 40,0 ммоль) в водном гидроксиде натрия (4 М, 11,0 мл, 44,0 ммоль) нагревали при 100 С в течение 15 ч. Реакционную смесь охлаждали до комнатной температуры и затем подкисляли до рН 2 с помощью концентрированной соляной кислоты. Затем смесь концентрировали при пониженном давлении для получения сырой 5-метиламинопентановой кислоты в виде розовато-белого твердого вещества. К сырой 5-метиламинопентановой кислоте добавляли МеОН (40,0 мл, 987 ммоль) и концентрированную соляную кислоту (0,33 мл, 4,0 ммоль). Полученный мутный раствор нагревали при 60 С в течение 39 ч, когда ЖХ-МС показывала оставшийся исходный материал. Добавляли дополнительную концентрированную соляную кислоту (0,33 мл, 4,0 ммоль) и полученную смесь нагревали при 60 С в течение 33 ч и затем при 65 С в течение дополнительных 24 ч. ЖХ-МС показывала оставшийся исходный материал. Реакционную смесь концентрировали при пониженном давлении и к остатку добавляли раствор хлороводорода в МеОН (1,25 М). Полученную смесь нагревали при 60 С в течение 72 ч, когда по ЖХ-МС не наблюдали остатков исходного материала. Реакционную смесь частично концентрировали при пониженном давлении и образующееся твердое вещество удаляли фильтрацией, промывая МеОН. Затем фильтрат концентрировали при пониженном давлении для получения гидрохлорида метил 5-метиламинопентаноата (7,57 г, 100% выход) в виде светло-желтого твердого вещества. ЖХ-МС (Метод 2-90): Rt 1,10 мин; m/z 146,4 [М+Н]+. 1H ЯМР (CD3OD)4,86 (с), 3,66 (с), 3,30 (т), 3,00 (т), 2,69 (с), 2,41 (т), 1,71 (м). Пример 3. Получение метилового эфира 5-(3-[4-(бифенил-2-илкарбамоилокси)пиперидин-1 ил]пропионилметиламино)пентановой кислоты. Смесь гидрохлорида метил 5-метиламинопентаноата (7,27 г, 40,0 ммоль), 3-[4-(бифенил-2 илкарбамоилокси)пиперидин-1-ил]пропионовой кислоты (13,3 г, 36,0 ммоль) и 1-гидрокси-7 азабензотриазола (5,14 г, 37,8 ммоль) в DCM (160 мл) и 2,6-лутидина (12,5 мл, 108 ммоль) перемешивали при комнатной температуре в течение 3 ч. Добавляли гидрохлорид N-(3-диметиламинопропил)-N'этилкарбодиимида (10,4 г, 54,0 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли насыщенный водный раствор бикарбоната натрия (100 мл) и слои разделяли. Водный слой экстрагировали DCM (50 мл) и органические слои комбинировали, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией с испарительной колонкой на силикагеле (30-100% EtOAc в гексанах; затем 2-10% МеОН в DCM) для получения указанного в заголовке соединения (12,38 г, 69% выход) в виде светло-желтого густого масла/белого твердого вещества. ЖХ-МС (Метод 2-90): Rt 2,43 мин; m/z 496,6 [М+Н]+. 1(т, 1 Н), 3,29 (м, 1 Н), 2,97 (с, 2 Н), 2,91 (с, 1 Н), 2,70 (м, 4 Н), 2,49 (м, 2 Н), 2,34 (м, 4 Н), 1,92 (м, 2 Н), 1,60 (м,5 Н). Пример 4. Получение 5-(3-[4-(бифенил-2-илкарбамоилокси)пиперидин-1-ил]пропионилметиламино)пентановой кислоты. К смеси метилового эфира 5-(3-[4-(бифенил-2-илкарбамоилокси)пиперидин-1-ил]пропионилметиламино)пентановой кислоты (10,21 г, 20,60 ммоль), трет-бутилового спирта (20 мл) и воды (20 мл) добавляли 1:1 смесь LiOH:вода (1,97 г, 41,2 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 4 ч и затем рН смеси доводили до около рН 2 с использованием соляной кислоты(1 Н). Водный слой экстрагировали DCM (280 мл) и органические слои комбинировали, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении для получения указанного в заголовке соединения (12,23 г, количественно) в виде не совсем белого пенного твердого вещества(содержащего остаточный трет-бутиловый спирт). ЖХ-МС (Метод 2-90): Rt 2,32 мин; m/z 482,4 [М+Н]+. Пример 5. Получение 2-(4-нитрофенил)-1,3-диоксолана. Перемешиваемый раствор п-нитробензальдегида (101,5 г, 672 ммоль), этиленгликоля (112 мл) и птолуолсульфоновой кислоты (12,8 г, 67,2 ммоль) в толуоле (800 мл) нагревали в колбе, снабженной ловушкой Dean-Stark, при 120 С в течение 4 ч. После охлаждения до комнатной температуры реакционную смесь концентрировали при пониженном давлении. К остатку добавляли насыщенный водный раствор бикарбоната натрия (800 мл) и полученную смесь перемешивали при комнатной температуре в течение 15 мин. Полученное твердое вещество выделяли фильтрацией и сушили в вакууме для получения ука- 10022018 занного в заголовке соединения (121,8 г, 92% выход) в виде желтого твердого вещества. 1H ЯМР (ДМСО-д 6) : =8,12 (д, 2 Н), 7,59 (д, 2 Н), 5,78 (с, 1 Н), 3, 8-4,0 (м, 4 Н). Пример 6. Получение 4-(1,3-диоксолан-2-ил)фениламина. К смеси диоксида платины (227 мг, 1,00 ммоль) и бикарбоната натрия (420 мг, 5,00 ммоль) в сухом азоте добавляли раствор 2-(4-нитрофенил)-1,3-диоксолана (976 мг, 5,00 ммоль) в EtOH (30,0 мл). Реакционную смесь барботировали водородом в течение 15 мин и затем перемешивали в атмосфере водорода(баллон) в течение 2 ч. Затем реакционную смесь фильтровали через подушку Целита, промывая МеОН. Фильтрат концентрировали при пониженном давлении для получения указанного в заголовке соединения(800 мг, 5 ммоль) и N,N-диизопропилэтиламина (1,26 мл, 7,26 ммоль) в DCM (48,4 мл) добавляли 1 гидрокси-7-азабензотриазол (692 мг, 5,08 ммоль) и гидрохлорид N-(3-диметиламинопропил)-N'этилкарбодиимида (1,39 г, 7,26 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь промывали насыщенным водным раствором бикарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении для получения указанного в заголовке соединения (3,04 г, 100% выход) в виде желтого твердого вещества. ЖХ-МС (Метод 10-70): Rt 2,67 мин; m/z 629,6 [М+Н]+. Пример 8. Получение 1-(2-[4-(4-формилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты. Перемешиваемую смесь 1-(2-[4-(4-(1,3-диоксолан-2-ил)фенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (3,04 г, 4,84 ммоль) в водной соляной кислоте (1 М, 10 мл) и ацетонитриле (10 мл) нагревали при 50 С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и к остатку добавляли насыщенный водный раствор бикарбоната натрия и DCM. Слои разделяли и органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении для получения указанного в заголовке соединения (2,83 г,100% выход). ЖХ-МС (Метод 10-70): Rt 2,67 мин; m/z 585,4 [М+Н]+. Пример 9. Получение 1-(2-[4-(4-[(R)-2-(трет-бутилдиметилсиланилокси)-2-(8-гидрокси-2-оксо-1,2 дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 илового эфира бифенил-2-илкарбаминовой кислоты. К перемешиваемому раствору 1-(2-[4-(4-формилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (2,83 г, 4,84 ммоль) и соли уксусной кислоты 5-[(R)-2-амино-1-(трет-бутилдиметилсиланилокси)этил]-8-гидрокси-1 Н-хинолин-2-она (1,91 г, 4,84 ммоль) в 1:1 смеси MeOH:DCM (40,0 мл, 312 ммоль) добавляли триацетоксиборогидрид натрия(3,08 г, 14,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем слои разделяли. Органический слой промывали насыщенным водным раствором бикарбоната натрия,сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении для получения желтого твердого вещества. Твердое вещество очищали испарительной хроматографией на силикагеле (0-30% МеОН в DCM+0,5% NH4OH) для получения указанного в заголовке соединения (3,60 г, 82% выход) в виде желтого твердого вещества. ЖХ-МС (Метод 10-70): Rt 2,72 мин; m/z 903,8 [М+Н]+. Пример 10. Получение соли дитрифторуксусной кислоты 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо 1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 илового эфира бифенил-2-илкарбаминовой кислоты. К перемешиваемому раствору 1-(2-[4-(4-[(R)-2-(трет-бутилдиметилсиланилокси)-2-(8-гидрокси-2 оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (3,60 г, 3,98 ммоль) в 9:1 смесиDCM:DMF (32,9 мл) добавляли тригидрофторид триэтиламина (1,95 мл, 12,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи и затем концентрировали при пониженном давлении. Остаток очищали ВЭЖХ (метод 10-70) для получения указанного в заголовке соединения (1,90 г, 46% выход) в виде белого твердого вещества. ЖХ-МС (Метод 10-70): Rt 2,12 мин; m/z 789,6 [М+Н]+. Пример 11. Получение соли дигидрофторуксусной кислоты 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо 1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 илового эфира бифенил-2-илкарбаминовой кислоты. К перемешиваемому раствору 1-(2-[4-(4-[(R)-2-(трет-бутилдиметилсиланилокси)-2-(8-гидрокси-2 оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (3,87 г, 4,28 ммоль) в смеси DCM (20 мл) и DMF (2,5 мл) добавляли тригидрофторид триэтиламина (2,1 мл, 13 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. DCM (68 мл) добавляли к реакционной смеси и затем, при перемешивании, по каплям добавляли EtOAc (68 мл). Образующийся светлоокрашенный осадок давал взвесь. Взвесь фильтровали через пористый стеклянный фильтр средней пористости и фильтрационный осадок промывали EtOAc (210 мл) и затем сухо прессовали на азотном прессе для получения указанного в заголовке соединения (3,73 г, 99% выход). Пример 12. Получение 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2 илкарбаминовой кислоты. Раствор соли дигидрофтористой кислоты 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2 дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 илового эфира бифенил-2-илкарбаминовой кислоты (15,2 г, 16,7 ммоль) в растворе DCM, содержащем 10% 0,5 М гидроксида аммония в МеОН (150 мл общего раствора), загружали на силикагель (80 г) в грубо фильтрованной стеклянной воронке, которую предварительно увлажнили раствором DCM, содержащим 10% 0,5 М гидроксида аммония в МеОН (200 мл). Силикагель промывали раствором DCM, содержащим 20% 0,5 М гидроксида аммония в МеОН (11000 мл), и затем раствором DCM, содержащим 25% 0,5 М гидроксида аммония в МеОН (11000 мл). Фильтрат концентрировали при пониженном давлении для получения желтого стеклообразного твердого вещества (13,6 г). Полученное вещество растворяли вDCM (600 мл) и полученный раствор фильтровали через фильтр из фриттованного стекла и фильтр промывали DCM (3150 мл). Фильтрат концентрировали при пониженном давлении и остаток нагревали до 50 С с использованием масляной бани и держали в вакууме в течение ночи для получения указанного в заголовке соединения (12,7 г) (содержащего около 6 мас.% DCM). Пример 13. Получение кристаллического гидрата оксалатной соли 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси 2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (форма 2). К перемешиваемому раствору 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (0,50 г, 0,63 ммоль) в THF (5 мл, 60 ммоль) по каплям добавляли дигидрат щавелевой кислоты (80 мг, 0,63 ммоль) в МеОН (2 мл, 50 ммоль). Немедленно образовывался не совсем белый осадок, и в течение периода 5 мин твердое вещество агломерировало с образованием липкого шарика. Затем реакционную смесь нагревали при 60 С в течение 2 ч, когда смесь образовывала не совсем белую густую взвесь очень мелких частиц. Полученную смесь медленно охлаждали до комнатной температуры и затем перемешивали в течение около 72 ч. Осуществляли попытку фильтровать смесь, но большая часть осадка проходила через фильтр. Затем фильтрат концентрировали при пониженном давлении для получения осадка. Приблизительно одну четверть осадка удаляли и растворяли в горячем растворе 10% (мас./мас.) воды в МеОН. Полученный раствор охлаждали до комнатной температуры, но осадок не образовывался. Затем раствору позволяли стоять и медленно испаряться при комнатной температуре в течение периода 4 дней, что давало частицы, проявляющие некоторое двойное преломление. Перемешивание продолжали с периодической обработкой ультразвуком в течение периода 5 дней для получения белой густой суспензии материала, проявляющей лучшее двойное лучепреломление. Полученное вещество собирали фильтрацией и сушили при высоком вакууме при комнатной температуре в течение 2 ч для получения указанного в заголовке соединения. Запись PXRD показала, что вещество является кристаллическим или полукристаллическим. Пример 14. Получение кристаллической оксалатной гидратной соли 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси 2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (форма 2). К перемешиваемому раствору 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (0,65 г, 0,824 ммоль) в МеОН (12,4 мл) добавляли по каплям раствор дигидрата щавелевой кислоты (104 мг, 0,824 ммоль) в воде (0,6 мл). Сразу образовывался слегка липкий осадок и к нему добавляли затравочные кристаллы из примера 13. Полученную взвесь перемешивали при комнатной температуре в течение около 72 ч и затем обрабатывали ультразвуком при 30 С в течение ночи. Затем взвесь охлаждали до 0 С, перемешивали в течение 30 мин и фильтровали для получения осадка. Фильтрованный осадок сушили на воздухе в течение 1 ч и затем сушили в высоком вакууме в течение 2 ч при комнатной температуре для получения указанного в заголовке соединения (705 мг,96% выход, 99% чистота). Пример 15. Воздействие на форму 2 высокой относительной влажности. Гидрат оксалата 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5 ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (приблизительно 100 мг, из примера 14) распределяли на фильтровальной бумаге, помещенной в чашку Петри. Чашку Петри помещали в эксикатор на пористом планшете над насыщенным водным раствором хлорида калия (100 мл) и эксикатор тщательно герметизировали. Относительная влажность внутри камеры оставалась приблизительно 84% ОВ, что измеряли гигрометром. Через три дня твердое вещество удаляли из эксикатора. Графики PXRD, DSC, TGA и DMS для полученного вещества показаны на фиг. 4, 5, 6 и 7 соответственно. Пример 16. Получение кристаллического гидрата оксалатной соли 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси 2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (форма 2). К перемешиваемому раствору 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (1,0 г, 1,27 ммоль) в МеОН (19 мл) добавляли по каплям раствор дигидрата щавелевой кислоты (160 мг, 1,269 ммоль) в воде (1,0 мл). Липкий желтоватый осадок образовывался немедленно. Реакционную смесь нагревали при 40 С в течение ночи и затем обрабатывали ультразвуком в течение 5 мин. Затем реакционную смесь нагревали при 40 С в течение 18 ч, охлаждали до 0 С, осадок собирали фильтрацией. Осадок сушили для получения указанного в заголовке соединения (1,05 г, 93% выход, 99% чистота). Пример 17. Воздействие на форму 2 высокой относительной влажности. Супернасыщенный водный раствор хлорида калия получали на дне эксикатора (твердый KCl оставался после тщательного встряхивания). Гидрат оксалата 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2 оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (500 мг, 0,569 ммоль, из примера 16) распределяли на бумаге для взвешивания, и бумагу для взвешивания помещали на пористый пластиковый планшет над раствором KCl в эксикаторе и эксикатор закрывали. Относительную влажность внутри эксикатора поддерживали на уровне 84% ОВ из-за равновесия давления пара раствора KCl. Записи PXRD получали на частях оксалатной соли через 3, 5 и 10 дней. Записи PXRD показали, что кристалличность оксалатной соли улучшалась до 5 дней. Изменение кристалличности было незначительным между 5 и 10 днями. Пример 18. Получение кристаллической оксалатной гидратной соли 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси 2-оксо-1,2-дигидрохинолин-5 ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (форма 2). К перемешиваемому раствору 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (11,0 г, 13,9 ммоль) в МеОН (400 мл) добавляли по каплям раствор дигидрата щавелевой кислоты (1,758 г, 13,9 ммоль) в воде (22 мл). Липкий осадок образовывался немедленно. Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем обрабатывали ультразвуком в течение 5 мин. Затем реакционную смесь перемешивали при комнатной температуре в течение 48 ч и затем нагревали при 40 С в течение 8 ч. Реакционной смеси позволяли медленно остыть до комнатной температуры и затем нагревали при 30 С в течении около 72 ч. Осадок собирали фильтрацией, промывали МеОН (150 мл) и сушили в высоком вакууме для получения указанного в заголовке соединения (11,6 г, 94% выход, 99% чистота). Пример 19. Получение кристаллической соли изопропанола сольвата оксалата 1-(2-[4-(4-[(R)-2-гидрокси-2(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (форма 1). Кристаллический гидрат оксалата 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2 дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 илового эфира бифенил-2-илкарбаминовой кислоты (приблизительно 40 мг) суспендировали в растворе 5% (об./об.) воды в изопропаноле (4 мл) и полученную взвесь перемешивали при 200 об/мин при комнатной температуре. Через три дня взвесь фильтровали аспирационной фильтрацией и твердое вещество сушили в воздухе для получения указанного в заголовке соединения (36 мг). Спектры PXRD, DSC и TGA для указанного материала показаны на фиг. 1, 2 и 3 соответственно. Пример 20. Порошковая рентгеновская дифракция. Анализ порошковой рентгеновской дифракции проводили с использованием рентгеновского дифрактометра Rigaku MiniFlex. Источником рентгеновского излучения было излучение Cu-K (=1,54051 А) с выходом вольтажа 20 кВ и током 15 мА. Прибор работал в геометрии Bragg-Brentano с эпизодом,отклонением и щелями рассеяния, установленными для максимального увеличения интенсивности образца. Для измерения небольшое количество порошка (5-25 мг) тщательно прессовали на единственный силиконовый держатель кристаллического образца для получения гладкой поверхности и подвергали воздействию рентгеновского излучения. Образцы сканировали в режиме 2-2 от 2 до 40 в 2 с размером стадии 0,03 и скоростью сканирования 2,0/мин. Регистрацию данных контролировали с помощью программного обеспечения Rigaku Standard easurement (Версия 1.2.0.0) и анализировали посредством программного обеспечения Jade (версия 7.5.1). Характерные исходные необработанные записи PXRD для формы 1 и формы 2 показаны на фиг. 1 и 4 соответственно. Наблюдаемые PXRD положения двух тета пиков и d-пространства для формы 1 и формы 2 показаны в табл. 1 и 2 соответственно (перечислены только пики, имеющие относительную высоту пика (Н%) около 20% или больше). Таблица 1 Данные PXRD для формы 1 Высота пика от исходного. Процент высоты пика относительно наибольшего пика. Высота пика от исходного. Процент высоты пика относительно наибольшего пика. Пример 21. Дифференциальная сканирующая калориметрия. Дифференциальную сканирующую калориметрию (DSC) проводили с использованием модуля ТАInstruments Model Q-100 с контроллером Thermal Analyst. Данные собирали и анализировали с использованием программного обеспечения ТА Instruments Thermal Solutions. Образец около 2 мг аккуратно отвешивали в алюминиевую банку с крышкой. Образец оценивали с использованием линейного нагревательного возрастания 10 С/мин от комнатной температуры до приблизительно 300 С. Ячейку DSC продували сухим азотом во время использования. Характерные графики DSC для формы 1 и формы 2 показаны на фиг. 2 и 5 соответственно. Графики DSC демонстрируют, что форма 1 и форма 2 имеют превосходную термическую стабильность с температурой плавления от около 162 до 164 С. Пример 22. Термогравиметрический анализ. Термогравиметрический анализ (TGA) проводили с использованием модуля ТА Instruments ModelQ-500, снабженного способностью высокого разрешения. Данные собирали и анализировали с использованием программного обеспечения ТА Instruments Thermal Solutions. Образец весом около 10 мг помещали в платиновую банку и сканировали со скоростью нагревания высокого разрешения от комнатной температуры до 300 С. Балансирующую и термостатные камеры продували током азота во время применения. Характерные записи TGA для формы 1 и формы 2 показаны на фиг. 3 и 6 соответственно. Пример 23. Динамическая оценка поглощения влаги. Динамическую оценку поглощения влаги (DMS) (также известную как профиль сорбции-десорбции влаги) проводили для образца формы 2 с использованием атмосферного микробаланса VTI, системыSGA-100 (VTI Corp., Hialeah, FL 33016). Использовали образец весом приблизительно 10 мг и в начале анализа влажность устанавливали на значении окружающей среды. Типичный анализ DMS состоял из трех сканирований: от влажности окружающей среды до 5% относительной влажности (ОВ), от 5% ОВ до 90% ОВ, от 90% ОВ до 5% ОВ со скоростью сканирования 5% ОВ/стадия. Массу измеряли каждые 2 мин и ОВ изменяли до следующего значения (+/-5% OB), когда масса образца оставалась стабильной в рамках 0,01% для 5 последовательных точек. Характерный график DMS для формы 2 показан на фиг. 7. График DMS фиг. 7 демонстрирует, что форма 2 имела недостоверное увеличение массы на около 1,6 мас.% в диапазоне влажности от 5% ОВ до 90% ОВ. Обратимый профиль сорбции/десорбции влаги демонстрирует, что форма 2 обладает приемлемой гигроскопичностью и не является легковпитывающей влагу. Профили PXRD соли были сходными до и после эксперимента DMS, предполагая, что в физической форме во время процессов сорбции влаги и десорбции влаги не возникает изменений. Биологические исследования и препараты. Пример А. Культура клеток и препарат мембран из клеток, экспрессирукщих человеческие мускариновые рецепторы M1, M2, М 3 и M4. Клеточные линии СНО, стабильно экспрессирующие человеческие мускариновые рецепторы подтипов hM1, hM2, hM3 и hM4 соответственно, выращивали почти до слияния в среде Hams F-12, дополненной 10% FBS и 250 мкг/мл генетицина. Клетки выращивали в 5% СО 2, 37 С-ном инкубаторе и поднимали с помощью 2 мМ EDTA в dPBS. Клетки собирали путем 5-минутного центрифугирования при 650g,и осадки клеток или хранили замороженными при -80 С, или получали мембраны непосредственно для применения. Для получения мембран осадки клеток ресуспендировали в лизирующем буфере и гомогенизировали с помощью разрушителя ткани Polytron PT-2100 (Kinematica AG; 20 секунд 2 запуска). Сырые мембраны центрифугировали при 40000g в течение 15 мин при 4 С. Затем осадки мембран ресуспендировали с помощью ресуспезионного буфера и снова гомогенизировали с помощью разрушителя ткани Polytron. Концентрацию белка в суспензии мембран определяли по методу, описанному в Lowry et al, 1951,Journal of Biochemistry, 193, 265. Все мембраны хранили замороженными в аликвотах при -80 С или использовали немедленно. Аликвоты полученных hM5 рецепторов мембран покупали у PerkmElmer, Inc. (Wellesley, MA) и хранили при -80 С до применения. Пример В. Анализ связывания меченого лиганда для мускариновых рецепторов. Анализ связывания меченого лиганда для клонированных мускариновых рецепторов проводили в 96-луночных планшетах микротитратора в общем объеме анализа 100 мкл. Мембраны клеток СНО, стабильно экспрессирующих или hM1, hM2, hM3, hM4 или hM5 подтипы мускариновых рецепторов, разводили в буфере для анализа до следующих специфических целевых концентраций белка (г/ячейку): 10 мкг для hM1, 10-15 мкг для hM2, 10-20 мкг для hM3/10-20 мкг для hM4 и 10-12 мкг для hM5 для получения сходных сигналов (ц/мин). Мембраны коротко гомогенизировали с использованием разрушителя тканиPolytron (10 секунд) перед добавлением в планшет для анализа. Исследования насыщения связывания для определения значений KD меченого лиганда проводили с использованием метилхлорида L-[N-метил-3 Н]скополамина ([3H]-NMS) (TRK666, 84,0 Ки/ммоль, Amersham Pharmacia Biotech, Buckinghamshire, England) в концентрациях, варьирующихся от 0,001 нМ до 20 нМ. Анализы вытеснения для определения значений Ki тестируемых соединений проводили с [3H]-NMS в количестве 1 нМ и одиннадцатью различными концентрациями тестируемого соединения. Тестируемые соединения исходно растворяли в концентрации 400 мкМ в буфере для разведения и затем серийно разводили в 5 раз с помощью буфера для разведения до конечных концентраций, варьирующихся от 10 пМ до 100 мкМ. Порядок добавления и объемы, добавленные в планшеты для анализа, были следующими: 25 мкл меченного лиганда, 25 мкл разведенного тестируемого соединения и 50 мкл мембран. Анализируемые планшеты инкубировали в течение 6 ч при 37 С. Реакции связывания терминировали быстрой фильтрацией через фильтровальные планшеты из стеклянных волокон GF/B (PerkinElmer, Inc.), предварительно обработанные 1% BSA. Фильтровальные планшеты промывали три раза промывочным буфером (10 мМ HEPES) для удаления несвязанной радиоактивности. Затем планшеты сушили на воздухе и к каждой ячейке добавляли 50 мкл жидкой сцинтилляционной жидкости Microscint-20 (PerkinElmer, Inc.). Затем планшеты считывали в жидкостном сцинтилляционном счетчике PerkinElmer Topcount (PerkinElmer, Inc.). Данные связывания анализировали анализом нелинейной регрессии с помощью пакета программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) с использованием односторонней конкурентной модели. Значения Ki для тестируемых соединений рассчитывали из наблюдаемых значений IC50 и значения KD меченного лиганда с использованием уравнения Cheng-Prusoff (Cheng Y;Prusoff WH. (1973) Biochemical Pharmacology, 22(23):3099-108). Ki значения преобразовывали в pKi значения для определения геометрического среднего и 95% доверительного интервала. Суммарную статистику затем снова преобразовывали обратно в значения Ki для представления данных. В проведенном анализе более низкие значения Ki обозначали, что тестируемое соединение имеет более высокую аффинность связывания с рецептором. Соль дифторуксусной кислоты 1-(2-[4-(4-[(R)-2 гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты имела значениеKi к hM3 0,1 нМ (данные округлены до ближайших 0,1 нМ) в этом анализе. Пример С. Культуры клеток и препараты мембран из клеток, экспрессирующих человеческие адренергические рецепторы 1, 2 или 3. Человеческие клеточные линии эмбриональных почек (НЕК-293), стабильно экспрессирующие клонированные человеческие адренергические рецепторы 1 и 2, или клеточные линии яичников Китайского хомяка (СНО), стабильно экспрессирующие клонированные человеческие 3 адренергические рецепторы, выращивали почти до слияния в среде DMEM или F-12 с 10% FBS в присутствии 500 мкг/мл Генетицина. Монослой клеток поднимали с помощью 2 мМ EDTA в PBS. Клетки осаждали центрифугированием при 1000 об/мин, и осадки клеток или хранили замороженными при -80 С, или получали мембраны непосредственно для применения. Для получения мембран, экспрессирующих 1 и 2, осадки клеток ресуспендировали в лизатном буфере (10 мМ HEPES/HCl, 10 мМ EDTA, рН 7,4 при 4 С) и гомогенизировали с использованием плотно подогнанного стеклянного гомогенизатора Dounce (30 запусков) на льду. Для более протеазачувствительных мембран, экспрессирующих 3 рецепторы, осадки клеток гомогенизировали в лизатном буфере (10 мМ Tris/HCl, рН 7,4), дополненном одной таблеткой "Таблетки смеси полного ингибитора протеаз с 2 мМ EDTA" на 50 мл буфера (Roche Molecular Biochemicals, Indianapolis, IN). Гомогенат центрифугировали при 20000g, и полученный осадок промывали один раз лизатным буфером посредством ресуспензии и центрифугировали, как описано в настоящем описании. Конечный осадок затем ресуспендировали в ледяном буфере для анализа связывания (75 мМ Tris/HCl рН 7,4, 12,5 мМ MgCl2, 1 мМ EDTA). Концентрацию белка суспензии мембран определяли способами, описанными в Lowry et al, 1951,Journal of Biological Chemistry, 193, 265; и Bradford, Analytical Biochemistry, 1976, 72, 248-54. Все мембраны хранили замороженными в аликвотах при -80 С или использовали немедленно. Пример D. Анализ для определения эффективности агониста адренергических рецепторов. Проводили цАМФ анализы в формате радиоиммуноанализа с использованием системы анализа активации аденилил циклазы Flashplate с [125I]-цАМФ (NEN SMP004, PerkinElmer Life Sciences Inc., Boston,MA), в соответствии с инструкциями производителя. Для указанного анализа клеточные линии НЕК-293,стабильно экспрессирующие клонированные человеческие 1 или 2 рецепторы, выращивали почти до слияния в DMEM, дополненной 10% FBS и Генетицином (500 мкг/мл); или клеточные линии CHO-K1,стабильно экспрессирующие клонированные человеческие 3 адренергические рецепторы, выращивали почти до слияния в среде Hams F-12, дополненной 10% FBS и Генетицином (250 мкг/мл). Клетки промывали PBS и открепляли в dPBS (Dulbecco's фосфатный буферный раствор, без CaCl2 и MgC.), содержащий 2 мМ EDTA или раствор трипсина-EDTA (0,05% трипсин/0,53 мМ EDTA). После подсчета клеток в счетчике клеток Coulter клетки осаждали центрифугированием при 1000 об/мин и ресуспендировали в стимуляционном буфере, содержащем IBMX (PerkinElmer Kit), заранее подогретом до комнатной температуры в концентрации от 1,6106 до 2,8106 клеток/мл. От около 40000 до 80000 клеток на ячейку использовали в указанном анализе. Тестируемые соединения (10 мМ в DMSO) разводили в PBS, содержащем 0,1% BSA, в Beckman Biomek-2000 и тестировали в 11 различных концентрациях, варьирующихся от 100 мкМ до 1 пМ. Реакционные смеси инкубировали в течение 10 мин при 37 С и останавливали путем добавления 100 мкл холодного буфера для определения, содержащего [125I]-цАМФ (NEN SMP004,PerkinElmer Life Sciences, Boston, MA). Количество образующегося цАМФ (пмоль/ячейку) рассчитывали на основании подсчетов, наблюдаемых для образцов и стандартов цАМФ, как описано в руководстве для пользователя производителя. Данные анализировали посредством анализа нелинейной регрессии с помощью пакета программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) с сигмоидным уравнением. Уравнение Cheng-Prusoff (Cheng Y, и Prusoff WFL, Biochemical Pharmacology, 1973, 22, 23, 3099-108) использовали для расчета значений EC50. В проведенном анализе более низкие значения ЕС 50 обозначают, что тестируемое соединение имеет более высокую функциональную активность в отношении тестируемого рецептора. Соль дифторуксусной кислоты 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5 ил)этиламино]метилфенилкарбамоил)-бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты имела значение EC50 для h2 1 нМ (данные округлены до ближайших 1 нМ) в указанном анализе. Пример Е. Анализ Эйнтховена. В указанном анализе измеряли способность тестируемого соединения обеспечивать бронхопротекцию относительно бронхоконстрикции, индуцированной метахолином (MCh) у крыс. Для всех исследований использовали самцов крыс Sprague-Dawley (Harlan, Indianapolis, IN) весом между 200 г и 350 г. Тестируемое соединение или носитель (стерильная деионизированная вода) дозировали посредст- 17022018Carlos, CA) с использованием 5 мл дозирующего раствора. Крыс подвергали воздействию аэрозоля, который получали из небулайзера LC Star модели Model 22F51 (PARI Respiratory Equipment, Inc.Midlothian, VA), запускаемого Bioblend (5% СО 2/95% атмосферный воздух), при давлении 22 фунтов на кв. дюйм. Крысам вводили 100 мкг тестируемого соединения, если не указано иначе. В заранее определенные временные точки крыс вводили в наркоз с помощью внутрибрюшинной инъекции (ИП) 120 мг/кг инактина (тиобутабарбитал). Дополнительную дозу (40 мг/кг, ИП) вводили,если животное отвечало на физические стимулы (например, укол лапы). Область хирургического вмешательства брили и производили 1-2 см срединный разрез в вентральной области шеи. Яремную вену выделяли и катетеризировали с помощью заполненного солевым раствором полиэтиленового катетера (РЕ-50) для возможности в/в введения MCh. Трахею рассекали свободно и катетеризировали иглой 14G (NE014, Small Parts, Miami Lakes, FL). После размещения трахеальной трубки каждую крысу вентилировали с использованием респиратора (Model 683, Harvard Apparatus, Inc., MA), установленного на рабочий объем 1 мл/100 г массы тела (но не более 2,5 мл объема) и скорость 90 вдохов в минуту. Т-соединитель помещали вдоль выдыхательной трубки респиратора для возможности измерения изменений в вентиляционном давлении (VP) с использованием трансдуктора Biopac, который соединяли с преамплификатором(TSD 137C). Температуру тела поддерживали на уровне 37 С с использованием электрической грелки. Изменения VP записывали с использованием программного обеспечения для сбора данных Acknowledge (Santa Barbara, CA). Исходные значения получали в течение по меньшей мере 2,5 мин. Затем крысам проводили некумулятивные внутривенные (в/в) инфузии 40 и 80 мкг/кг MCh. MCh вводили внутривенно в течение 2,5 мин через шприцевую помпу (sp210iw, World Precision Instruments, Inc., Sarasota, FL) со скоростью 2 мл/кг/мин, с 2-минутным интервалом между двумя введениями MCh. Изменения вентиляционного давления (см Н 2 О) у животных, получавших лечение, выражали как % ингибирования ответа на MCh относительно контрольных животных. Другие бронхоконстрикторы, такие как гистамин и ацетилхолин, могут быть использованы вместоMCh в этом исследовании. Кроме того, вместо крыс могут быть использованы морские свинки. В указанном исследовании более высокий % ингибирования MCh ответа показывает, что тестируемое соединение обеспечивает больший бронхопротективный эффект. Ингибирование более чем или равное 30% через 24 ч является показателем длительной продолжительности действия. Соль дитрифторуксусной кислоты 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5 ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты обеспечивает по меньшей мере около 40%-ное ингибирование через 24 ч в настоящем исследовании. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Кристаллическая оксалатная соль 1-(2-[4-(4-[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2 дигидрохинолин-5-ил)этиламино]метилфенилкарбамоил)бутил]метилкарбамоилэтил)пиперидин-4 илового эфира бифенил-2-илкарбаминовой кислоты, где кристаллическую оксалатную соль выбирают из:(a) сольвата изопропанола, характеризующегося порошковой рентгенограммой, включающей пики дифракции на 2 значениях 11,660,20, 15,750,20, 19,550,20, 23,000,20 и 23,450,20;(b) гидрата, характеризуемого порошковой рентгенограммой, включающей пики дифракции на 2 значениях 15,320,20, 16,900,20, 19,250,20 и 23,730,20. 2. Кристаллическая оксалатная соль по п.1, где кристаллической оксалатной солью является сольват изопропанола, характеризуемый порошковой рентгенограммой, включающей пики дифракции на 2 значениях 11,660,20, 15,750,20, 19,550,20, 23,000,20 и 23,450,20. 3. Кристаллическая оксалатная соль по п.2, где кристаллическая оксалатная соль характеризуется порошковой рентгенограммой, в которой пиковые положения, по существу, соответствуют пиковым положениям графика, показанного на фиг. 1. 4. Кристаллическая оксалатная соль по п.2, где кристаллическая оксалатная соль характеризуется температурой плавления от около 162 до около 164 С. 5. Кристаллическая оксалатная соль по п.2, где кристаллическая оксалатная соль характеризуется графиком дифференциальной сканирующей калориметрии, по существу, соответствующим показанному на фиг. 2. 6. Кристаллическая оксалатная соль по п.1, где кристаллическая оксалатная соль представляет собой гидрат, характеризуемый порошковой рентгенограммой, включающей пики дифракции на 2 значениях 15,320,20, 16,900,20, 19,250,20 и 23,730,20. 7. Кристаллическая оксалатная соль по п.6, где кристаллическая оксалатная соль характеризуется порошковой рентгенограммой, в которой пиковые положения, по существу, соответствуют пиковым положениям графика, показанного на фиг. 4. 8. Кристаллическая оксалатная соль по п.6, где кристаллическая оксалатная соль характеризуется температурой плавления от около 162 до около 164 С. 9. Кристаллическая оксалатная соль по п.6, где кристаллическая оксалатная соль характеризуется графиком дифференциальной сканирующей калориметрии, по существу, соответствующим показанному на фиг. 5. 10. Фармацевтическая композиция для лечения заболеваний легких, включающая:(a) кристаллическую оксалатную соль по любому из пп.1-9 и(b) фармацевтически приемлемый носитель. 11. Фармацевтическая композиция для лечения заболеваний легких, включающая:(a) кристаллическую оксалатную соль по любому из пп.1-9;(b) стероидное противовоспалительное средство или его фармацевтически приемлемую соль или сольват и(c) фармацевтически приемлемый носитель. 12. Способ лечения заболеваний легких, включающий введение пациенту кристаллической оксалатной соли по любому из пп.1-9. 13. Способ лечения заболеваний легких, включающий введение пациенту:(а) кристаллической оксалатной соли по любому из пп.1-9 и(b) стероидного противовоспалительного средства или его фармацевтически приемлемой соли или сольвата. 14. Способ по п.12 или 13, где заболеванием легких является хроническая обструктивная болезнь легких или астма. 15. Способ получения бронходилатации у млекопитающего, включающий введение млекопитающему кристаллической оксалатной соли по любому из пп.1-9 посредством ингаляции. 16. Кристаллическая оксалатная соль по любому из пп.1-9 для применения в терапии для лечения заболеваний легких. 17. Кристаллическая оксалатная соль по любому из пп.1-9 для применения в лечении хронических обструктивных болезней легких или астмы. 18. Применение кристаллической оксалатной соли по любому из пп.1-9 для производства лекарственного препарата для лечения заболеваний легких.
МПК / Метки
МПК: A61P 11/08, A61K 31/444, C07D 401/12
Метки: кристаллические, соединения, соли, диамидного, оксалатные
Код ссылки
<a href="https://eas.patents.su/22-22018-kristallicheskie-oksalatnye-soli-diamidnogo-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Кристаллические оксалатные соли диамидного соединения</a>
Новые кристаллические соли эпалрестата

Номер патента: 18600
Опубликовано: 30.09.2013
Авторы: Сталтс Джефри С., Мартин-Дойл Вильям, Калофонос Изабель, Калофонос Димитрис, Стэнли Г.Патрик, Хьюстон Травис Л.
МПК: A61K 31/42, A61P 3/10, A61K 31/41...
Метки: эпалрестата, новые, соли, кристаллические
Формула / Реферат:
1. Кристаллическая соль калиевого ангидрата 5-[(1Z,2E)-2-метил-3-фенилпропенилиден]-4-оксо-2-тиоксо-3-тиазолидинуксусной кислоты.2. Кристаллическая соль натриевого ангидрата 5-[(1Z,2E)-2-метил-3-фенилпропенилиден]-4-оксо-2-тиоксо-3-тиазолидинуксусной кислоты.3. Кристаллическая соль 1-(2-гидроксиэтил)пирролидин ангидрата 5-[(1Z,2E)-2-метил-3-фенилпропенилиден]-4-оксо-2-тиоксо-3-тиазолидинуксусной кислоты.4. Кристаллическая соль калиевого...
Кристаллические соли эффективного ингибитора вируса гепатита с

Номер патента: 21805
Опубликовано: 30.09.2015
МПК: C07D 417/14, A61P 31/18, A61K 31/427...
Метки: соли, гепатита, ингибитора, эффективного, кристаллические, вируса
Формула / Реферат:
1. Кристаллическая соль соединения формулы (1) и трометамина:2. Кристаллическая соль по п.1, на порошковой рентгеновской дифрактограмме которой наблюдается пик при 5,9° 2θ (±0,2° 2θ) при измерении с использованием излучения CuKα.3. Кристаллическая соль по п.1, на спектре 13С твердотельного ЯМР которой наблюдаются пики с химическими сдвигами при 178,3, 138,1 и 27,1 м.д. (±0,2 м.д.).4. Кристаллическая соль по п.1, на порошковой...
Кристаллические соли металл-глюкозамин сульфата и способы их приготовления

Номер патента: 6632
Опубликовано: 24.02.2006
Авторы: Среекумар Е.С., Бхат Рави Гаджанан, Мухопадхйай Триптикумар
МПК: A61P 19/02, C07H 5/04, A61K 31/70...
Метки: металл-глюкозамин, способы, соли, кристаллические, приготовления, сульфата
Формула / Реферат:
1. Новые кристаллические соли металл-глюкозамин сульфата с низким содержанием металла, формула I которых имеет следующий вид: где M представляет металл, выбираемый из Na или K. 2. Соединение по формуле I в соответствии с п.1, отличающееся тем, что в качестве M применяется Na (натрий). 3. Соединение по формуле I в соответствии с п.1, отличающееся тем, что в качестве M применяется K (калий). 4. Способ приготовления соединения по формуле I в...
Кристаллические формы [ r-(r*,r*)]-2-(4-фторфенил)-бета, дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино) карбонил]-1h-пиррол-1-гептановой кислоты кальциевой соли (2:1) (аторвастатин)

Номер патента: 5317
Опубликовано: 30.12.2004
Авторы: Берн Стивен Роберт, Коутс Дейвид Эндрю, Моррисон Генри Грант Второй, Гасхёрст Карен Сью, Крзизэниэк Джозеф Фрэнсис, Ли Женг Джейн, Парк Ири, Влахова Петинка Иванова
МПК: C07D 207/34, A61K 31/40
Метки: соли, кислоты, кристаллические, карбонил]-1h-пиррол-1-гептановой, кальциевой, 2:1, формы, r-(r*,r*)]-2-(4-фторфенил)-бета, дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино, аторвастатин
Формула / Реферат:
1. Кристаллическая Форма V аторвастатина или его гидрата, дающая дифракцию рентгеновских лучей на порошке, содержащую следующие значения 2Q, измеренные с использованием CuKa-излучения: 4.9 (уширенный), 6.0, 7.0, 8.0 (уширенный), 8.6, 9.9, 16.6, 19.0 и 21.1. 2. Кристаллическая Форма VI аторвастатина или его гидрата, дающая дифракцию рентгеновских лучей на порошке, содержащую следующие значения 2Q, измеренные с использованием CuKa-излучения: 7.2,...
Соли и кристаллические формы 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрила

Номер патента: 15677
Опубликовано: 31.10.2011
Авторы: Бэнцигер Маркус, Гарад Судхакар Девидасрао, Штовассер Франк
МПК: A61P 35/00, A61K 31/4745, C07D 471/04...
Метки: формы, соли, кристаллические, 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрила
Формула / Реферат:
1. Кристаллическая форма соединения формулы Iили гидрата или сольвата соединения формулы I, или соли соединения формулы I, или гидрата или сольвата соли соединения формулы I.2. Соединение I по п.1 в кристаллической форме A.3. Соединение по п.2, на рентгенограмме которого содержится пик при угле дифракции 2-тэта, равном 8,4±0,3°.4. Соединение I по п.1 в кристаллической форме B.5. Соединение по п.3, на рентгенограмме которого содержится пик при...
Предыдущий патент: Дверца стиральной машины с фронтальной загрузкой и стиральная машина с такой дверцей
Следующий патент: Этинильные производные в качестве положительных аллостерических модуляторов mglur5
Случайный патент: Способ переработки пластикового мусора любого типа, в частности смеси пластмасс