Дипиридодиазепиноны в качестве ингибиторов обратной транскриптазы
Номер патента: 7920
Опубликовано: 27.02.2007
Авторы: Дезиель Робер, О'мира Джеффри, Йоаким Кристиан, Симоно Брюно, Оджилви Уилльям У.

















Формула / Реферат
1. Соединение формулы

где R2 обозначает Н, галоген, C1-C4алкил, OC1-C4алкил, NHCl-С4алкил или N(C1-С4алкил)2; R4 обозначает Н или CH3; R5 обозначает Н или CH3 при условии, что и R4, и R5 не имеют одинаковое значение; R12 обозначает Н, галоген, С1-С4алкил, CF3 или NO2; R13 обозначает Н, С1-С4алкил, галоген, ОН или NH2 при условии, что R12 и R13 оба не обозначают Н; и R14 обозначает COOR14a, где R14a обозначает Н или C1-C6алкил; или R14 обозначает C2-C4алкенил-COOR14a, где R14a имеет указанные выше значения; или R14 обозначает C1-C4алкил-COOR14a, где R14a имеет указанные выше значения;
или его соль, или пролекарство.
2. Соединение по п.1, где R2 обозначает Н, галоген, C1-C4алкил, ОС1-С4алкил или N(C1-C4алкил)2 и R4 и R5 не имеют одинаковых значений.
3. Соединение по п.2, где R2 обозначает Н, Cl, F, С1-С4алкил, ОС1-С4алкил или N(С1-С4алкил)2.
4. Соединение по п.3, где R2 обозначает Н, Cl, F, CH3, ОМе или OEt.
5. Соединение по п.4, где R2 обозначает Н.
6. Соединение по п.1, где R4 обозначает Н.
7. Соединение по п.1, где R5 обозначает CH3.
8. Соединение по п.1, где R12 обозначает галоген, С1-С4алкил, CF3 или NO2.
9. Соединение по п.8, где R12 обозначает Вr, Cl, CH3 или СН3СН2.
10. Соединение по п.9, где R12 обозначает CH3 или СН3СН2.
11. Соединение по п.1, где R13 обозначает Н, CH3 галоген, ОН или NH2.
12. Соединение по п.11, где R13 обозначает Н, CH3 или ОН.
13. Соединение по п.12, где R13 обозначает Н.
14. Соединение по п.1, где R14 обозначает СООН, СООМе, С2-С4алкенил-СООН или С1-С4алкил-СООН.
15. Соединение по п.14, где R14 обозначает СООН, СН=СН-СООН, СН2СООН или СН2СН2СООН.
16. Соединение по п.15, где R14 обозначает СООН.
17. Соединение формулы (I) по п.1

где R2 обозначает Н, Cl, F, CH3, ОМе или OEt; R4 обозначает Н; R5 обозначает CH3; R12 обозначает Br, Сl, CH3 или СН2СН3; R13 обозначает Н, CH3 или ОН и R14 обозначает СООН, СН=СН-СООН, СН2СООН или СН2СН2СООН; или его соль, или пролекарство.
18. Соединение по п.17, где R2 обозначает Н; R4 обозначает Н; R5 обозначает CH3; R12 обозначает CH3 или СН3СН2; R13 обозначает Н и R14 обозначает СООН; или его соль, или пролекарство.
19. Фармацевтическая композиция, предназначенная для лечения или предупреждения ВИЧ-инфекции, содержащая соединение формулы I по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
20. Способ лечения или предупреждения ВИЧ-инфекции, заключающийся в том, что пациенту вводят ингибирующее ВИЧ количество соединения формулы I по п.1 или его фармацевтически приемлемой соли.
21. Способ лечения или предупреждения ВИЧ-инфекции, заключающийся в том, что пациенту вводят ингибирующее ВИЧ количество фармацевтической композиции по п.19, содержащей соединение формулы I по п.1 или его фармацевтически приемлемую соль.
22. Способ лечения или предупреждения ВИЧ-инфекции, заключающийся в том, что вводят соединение формулы I по п.1 в сочетании с антиретровирусным лекарственным средством.
23. Способ предупреждения перинатальной передачи ВИЧ-1 от матери к ребенку, заключающийся в том, что матери перед родами вводят соединение формулы I по п.1.
24. Применение соединения формулы I по п.1 или его фармацевтически приемлемой соли для приготовления лекарственного средства, предназначенного для лечения или предупреждения ВИЧ-инфекции.
25. Применение соединения формулы I по п.1 или его фармацевтически приемлемой соли для приготовления лекарственного средства, предназначенного для предупреждения перинатальной передачи ВИЧ-1 от матери к ребенку перед родами.
Текст
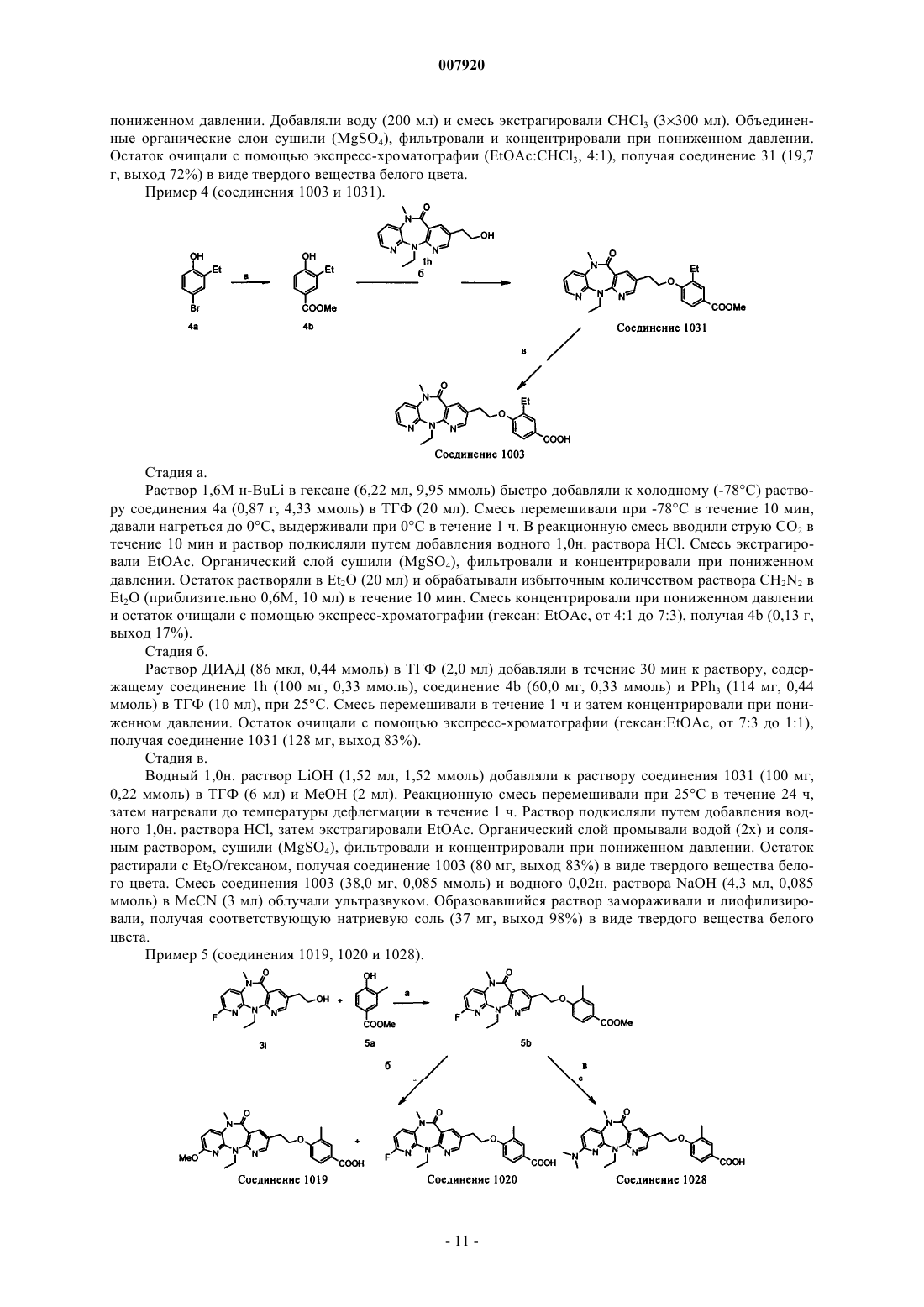
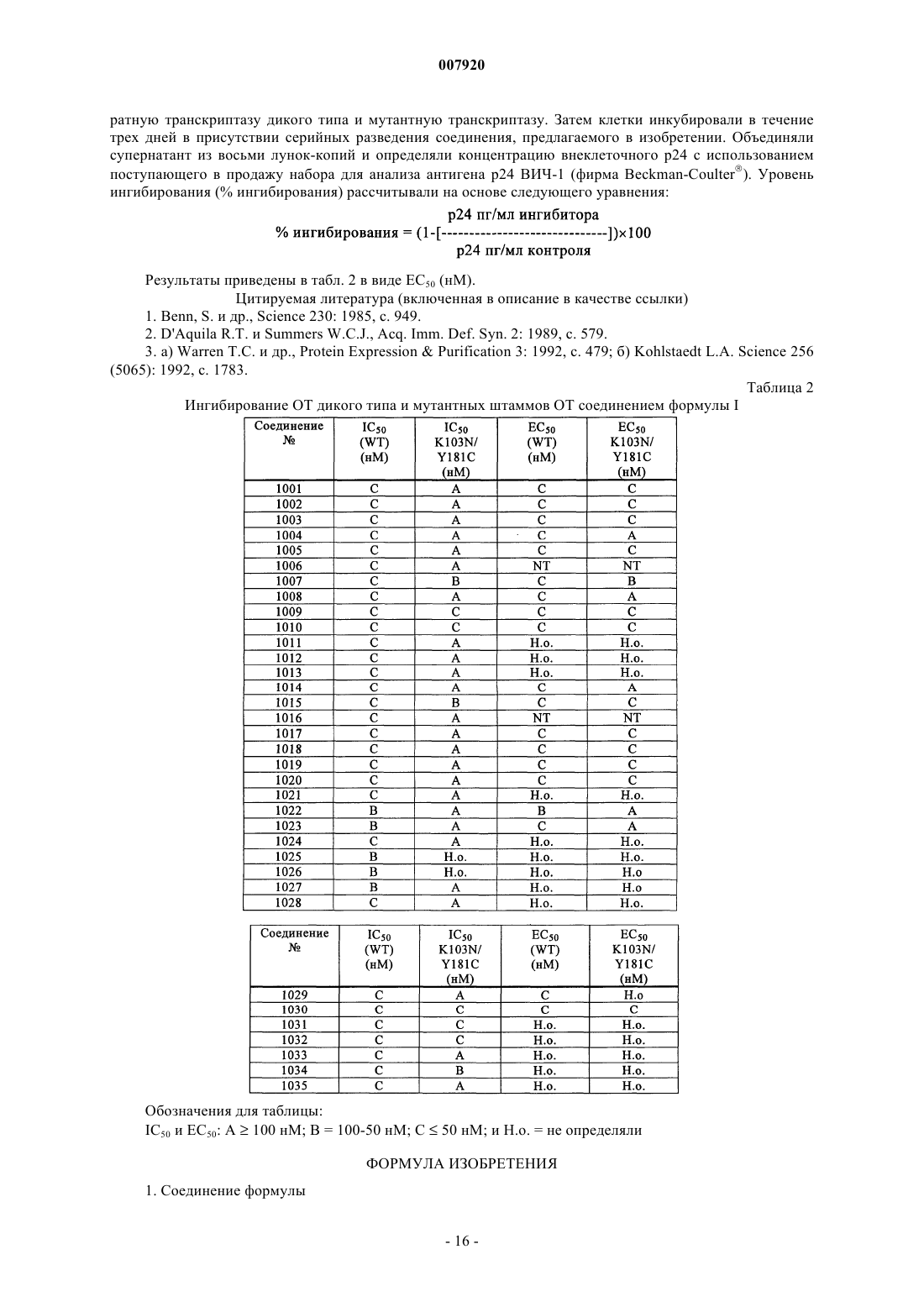
007920 Область техники, к которой относится изобретение Изобретение относится к новым соединениям и их фармацевтически приемлемым солям, их применению индивидуально или в сочетании с другими терапевтическими агентами для лечения или профилактики ВИЧ-инфекции и к фармацевтическим композициям, содержащим такие соединения. Предпосылки создания изобретения Заболевание, известное как синдром приобретенного иммунодефицита (СПИД), вызывается вирусом иммунодефицита человека (ВИЧ), прежде всего штаммом, известным как ВИЧ-1. Для репликации ВИЧ в клетке-хозяине информация, содержащаяся в вирусном геноме, должна быть интегрирована в ДНК клетки-хозяина. Однако ВИЧ представляет собой ретровирус, т.е. вирус, у которого генетическая информация считывается с РНК. Таким образом, цикл репликации ВИЧ включает стадию транскрипции вирусного генома (РНК) с образованием ДНК, этот процесс является обратным по отношению к нормальному ходу событий. Транскрипция вирусной РНК с образованием ДНК происходит с помощью фермента, называемого поэтому обратной транскриптазой (ОТ). Вирион ВИЧ содержит копию ОТ вместе с вирусной РНК. Известно, что обратная транскриптаза обладает тремя ферментативными функциями: она действует в качестве РНК-зависимой ДНК-полимеразы, в качестве рибонуклеазы и ДНК-зависимой ДНКполимеразы. В качестве РНК-зависимой ДНК-полимеразы, ОТ осуществляет транскрипцию копии одноцепочечной ДНК вирусной РНК. В качестве рибонуклеазы ОТ расщепляет исходную вирусную РНК и высвобождает ДНК, полученную непосредственно на матрице исходной РНК. Наконец, в качестве ДНКзависимой ДНК-полимеразы ОТ создает вторую комплементарную цепь ДНК, используя первую цепь ДНК в качестве матрицы. Две цепи образуют двухцепочечную ДНК, которая интегрируется в геном клеток-хозяев с помощью другого фермента, называемого интегразой. Соединения, которые обладают способностью ингибировать ферментативные функции обратной транскриптазы ВИЧ-1, могут ингибировать репликацию ВИЧ-1 в инфицированных клетках. Такие соединения можно применять для предупреждения или лечения вызванной ВИЧ-1 инфекции у больных людей, как это установлено при использовании известных ингибиторов ОТ, таких как 3'-азидо-3'дезокситимидин (AZT), 2',3'-дидезоксиинозин (ddI), 2',3'-дидезоксицитидин (ddC), d4T, 3TC, невирапин,делавирдин, эфавиренц и абакавир, которые являются основными лекарственными средствами, разрешенными для применения при лечении СПИД. Как и при любом антивирусной терапии применение ингибиторов ОТ для лечения СПИД в конечном итоге может приводить к снижению чувствительности вируса к конкретному лекарственному средству. Устойчивость (пониженная чувствительность) к таким лекарственным средствам является результатом мутаций, затрагивающих сегмент обратной транскриптазы гена pol. К настоящему времени определены характеристики нескольких мутантных штаммов ВИЧ и установлено, что устойчивость к известным терапевтическим агентам обусловлена мутациями в гене ОТ. Одними из наиболее часто встречающихся в клинических условиях мутантами являются: мутант Y181C, в котором остаток тирозина (Y) в кодоне 181 заменен на остаток цистеина (С), и K103N, в котором остаток лизина (K) в положении 103 заменен на остаток аспарагина (N). Другие мутанты, которые возникают с увеличивающейся частотой при лечении с использованием известных антивирусных лекарственных средств, представляют собой мутанты с одной мутацией V106A, G190A, Y188C и P236L; и мутанты с двумя мутациями K103N/Y181C,K103N/P225H, K103N/V108I и K103N/L100I. Продолжающееся применение антивирусных соединений для предупреждения ВИЧ-инфекции несомненно будет приводить к увеличению случаев возникновения новых устойчивых штаммов ВИЧ. Таким образом, существует насущная потребность в создании новых ингибиторов ОТ, которые обладают различными параметрами эффективности в отношении различных мутантов. Соединения, имеющие трициклические структуры, которые являются ингибиторами ВИЧ-1, описаны в US 5366972. Другие ингибиторы обратной транскриптазы ВИЧ-1 описаны у Hargrave и др., J. Med.Chem., 34, (1991), с. 2231. В US 5705499 описаны 8-арилалкил- и 8-арилгетероалкил-5,11-дигидро-6 Н-дипиридо[3,2-В:2',3'Е][1,4]диазепины в качестве ингибиторов ОТ. Установлено, что указанные в качестве примеров соединения обладают определенной активностью в отношении ОТ дикого типа и мутантного ВИЧ-1, прежде всего Y181C, но при этом они менее эффективны в отношении других мутантов, несущих одну мутацию,таких как K103N. В WO 01/96338A1 и US 6420359 описаны диазепиновые структуры, содержащие хинолиновые и хинолин-N-оксидные заместители, в качестве ингибиторов ОТ. Приведенные в качестве примеров соединения обладают активностью в отношении штаммов ВИЧ дикого типа (WT), мутантов с одной и двумя мутациями. Краткое изложение сущности изобретения В изобретении предложены замещенные производные бензойной кислоты, которые являются эффективными ингибиторами ОТ ВИЧ-1 дикого типа (WT) и мутантных штаммов с двумя мутациями, прежде всего, мутантного штамма с двумя мутациями K103N/Y181C. Первым объектом изобретения является соединение формулы IR13 обозначает Н, С 1-С 4 алкил, галоген, ОН или NH2, при условии, что R12 и R13 оба не обозначают Н; иR14 обозначает COOR14a, где R14a обозначает Н или C1-C6 алкил; или R14 обозначает С 2-С 4 алкенилCOOR14a, где R14a имеет указанные выше значения; или R14 обозначает С 1-С 4 алкил-COOR14a, где R14a имеет указанные выше значения; или его соль или пролекарство. Вторым объектом изобретения является фармацевтическая композиция, предназначенная для лечения или предупреждения ВИЧ-инфекции, которая содержит представленное выше в настоящем описании соединение формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Третьим объектом изобретения является способ лечения или предупреждения ВИЧ-инфекции, заключающийся в том, что пациенту вводят ингибирующее ВИЧ количество соединения формулы I, или его фармацевтически приемлемой соли, или фармацевтической композиции, которые указаны в настоящем описании. Четвертым объектом изобретения является способ лечения или предупреждения ВИЧ-инфекции,заключающийся в том, что вводят фармацевтическую композицию, содержащую описанное выше соединение формулы I в сочетании с антиретровирусным лекарственным средством. Пятым объектом изобретения является способ лечения или предупреждения перинатальной передачи ВИЧ от матери к ребенку, заключающийся в том, что матери перед родами вводят указанное в настоящем описании соединение формулы I или фармацевтическую композицию, как они описаны выше. Подробное описание изобретения Определения. Если не указано иное, то в описании используются следующие определения. В контексте настоящего описания понятия "C1-С 2 алкил", "С 1-С 4 алкил" и "C1-C6 алкил" индивидуально или в сочетании с другим радикалом обозначают ациклические алкильные радикалы, содержащие вплоть до двух, четырех или шести атомов углерода соответственно. Примерами таких радикалов являются метил, этил, пропил, бутил, 1-метилэтил, 1-метилпропил, 2-метилпропил и 1,1-диметилэтил. В контексте настоящего описания понятие "C2-C4 алкенил" индивидуально или в сочетании с другим радикалом обозначает ненасыщенный ациклический радикал, содержащий от двух до четырех атомов углерода. В контексте настоящего описания понятие "галоген" обозначает атом галогена и включает фтор,хлор, бром и йод. В контексте настоящего описания понятие "фармацевтически приемлемая соль" обозначает соли,которые получены из фармацевтически приемлемых оснований и являются нетоксичными. Примерами пригодных оснований могут служить холин, этаноламин и этилендиамин. Подразумевается, что под объем изобретения подпадают также Na+-, K+- и Са-соли (см. также Birge S.M. и др., Pharmaceutical salts, J.Pharm. Sci., 66, (1977), с. 1-19, включено в настоящее описание в качестве ссылки). В контексте настоящего описания понятие "пролекарство" обозначает фармакологически приемлемые производные, из которых в результате биотрансформации образуется действующее лекарственное средство, относящееся к соединениям формулы I. Примерами таких производных являются (но не ограничиваясь ими) сложные эфиры и амиды (см. Goodman и Gilman, The Pharmacological Basis of Therapeutics, 9-е изд., изд-во McGraw-Hill, Int. Ed., Biotransformation of Drugs, 1995, с. 11-16, включено в настоящее описание в качестве ссылки). Подробное описание предпочтительных вариантов осуществления изобретения Предпочтительно R2 обозначает Н, галоген, С 1-С-4 алкил, О-С 1-С-4 алкил или N(C1-C-4)2. Предпочтительно R2 обозначает Н, Cl, F, С 1-С 4 алкил, О-С 1-C-4 алкил или N-(С 1-С-4 алкил)2. Более предпочтительно R2 обозначает Н, Cl, F, CH3, ОМе или OEt. Наиболее предпочтительно R2 обозначает Н. Предпочтительно R4 и R5 оба имеют одинаковое значение. Более предпочтительно R4 обозначает Н. Более предпочтительно R5 обозначает CH3. Предпочтительно R12 обозначает галоген, C1-С-4 алкил, CF3 или NO2. Более предпочтительно R12-2 007920 обозначает Вr, Сl, CH3 или СН 3 СН 2. Наиболее предпочтительно R12 обозначает CH3 или СН 3 СН 2. Предпочтительно R13 обозначает Н, CH3, галоген, ОН или NH2. Более предпочтительно R13 обозначает Н, CH3 или ОН. Наиболее предпочтительно R13 обозначает Н. Предпочтительно R14 обозначает СООН, СООМе, С 2-С 4 алкенилСООН или С 1-С 4 алкилСООН. Более предпочтительно R14 обозначает СООН, СН=СН-СООН, СН 2 СООН или СН 2 СН 2 СООН. Наиболее предпочтительно R14 обозначает СООН. Соединения формулы I являются эффективными ингибиторами ВИЧ дикого типа, а также обладают способностью ингибировать мутантный фермент, несущий две мутации K103N/Y181C. Соединения формулы I обладают ингибирующей активностью в отношении обратной транскриптазы ВИЧ-1. При введении в виде пригодных лекарственных средств их можно применять для лечения СПИД, состояния, предшествующего синдрому приобретенного иммунодефицита (ARC) и родственных заболеваний, связанных с ВИЧ-1-инфекцией. Таким образом, следующим объектом изобретения является способ лечения вызываемой ВИЧ-1 инфекции, который заключается в том, что человеку, инфицированному ВИЧ-1, вводят терапевтически эффективное количество описанного выше нового соединения формулы I. Как для лечения, так и для профилактики соединение можно применять также для предупреждения перинатальной передачи ВИЧ-1 от матери к ребенку путем введения соединения матери перед родами. Соединения формулы I можно вводить в виде однократной дозы или в виде разделенных доз оральным или парентеральным путем. Пригодная оральная доза соединения формулы I может составлять от примерно 0,5 мг до 3 г в день. Предпочтительно оральная доза соединения формулы I составляет от примерно 100 до 800 мг в день для пациента весом 70 кг. В случае композиций для парентерального введения пригодная стандартная доза может составлять от 0,1 до 250 мг соединения, предпочтительно от 1 до 200 мг. Однако следует иметь в виду, что вводимая доза может варьироваться от пациента к пациенту и доза для каждого конкретного пациента зависит от решения лечащего врача, который в качестве критерия при назначении соответствующей дозы должен учитывать размер и состояние пациента, а также реакцию пациента на лекарственное средство. Если соединения, предлагаемые в настоящем изобретении, вводят оральным путем, то их можно вводить в форме фармацевтических композиций, которые содержат лекарственное средство в сочетании с совместимым фармацевтическим носителем. Такой носитель может представлять собой инертный органический или неорганический носитель, пригодный для орального введения. Примерами таких носителей являются вода, желатин, тальк, крахмал, стеарат магния, аравийская камедь, растительные масла,полиалкиленгликоли, вазелин и т.п. Соединения формулы I можно применять в сочетании с антиретровирусным лекарственным средством, известным специалисту в данной области, в качестве комбинированного лекарственного средства для одновременного, раздельного или последовательного введения для лечения или предупреждения ВИЧ-инфекции у индивидуума. Примеры антиретровирусных лекарственных средств, которые можно применять при комбинированном лечении с использованием соединений формулы I, включают (но не ограничиваясь ими) нуклеозидные/нуклеотидные ингибиторы обратной транскриптазы (такие как АЗТ и тенофовир), ненуклеозидные ингибиторы обратной транскриптазы (такие как невирапин), ингибиторы протеазы (такие как ритановир), ингибиторы вирусного слияния (такие как Т-20), антагонисты CCR5L-870,810), ингибиторы тирозинаминотрансферазы (ТАТ), другие находящиеся на стадии исследования лекарственные средства (такие как PRO-542, BMS-806, ТМС-114 или AI-183), противогрибковые или антибактериальные агенты (такие как флуконазол), и иммуномодуляторы (такие как левамизол). Кроме того, соединение формулы I можно применять в сочетании с другим соединением формулы I. Фармацевтические композиции можно приготавливать стандартным образом и конечные лекарственные формы могут представлять собой твердые лекарственные формы, например таблетки, драже,капсулы и т.п., или жидкие лекарственные формы, например растворы, суспензии, эмульсии и т.п. Фармацевтические композиции можно подвергать стандартной фармацевтической обработке, такой как стерилизация. Кроме того, фармацевтические композиции могут содержать стандартные вспомогательные вещества, такие как консерванты, стабилизаторы, эмульгаторы, корригенты, смачивающие агенты, буферы, соли для изменения осмотического давления и т.п. Твердые носители, которые можно применять,включают, например крахмал, лактозу, маннит, метилцеллюлозу, микрокристаллическую целлюлозу,тальк, вторичный кислый фосфат кальция и высокомолекулярные полимеры (такие как полиэтиленгликоль). Для парентерального применения соединение формулы I можно вводить в виде водного или неводного раствора, суспензии или эмульсии в фармацевтически приемлемом масле или смеси жидкостей,которые могут содержать бактериостатические агенты, антиоксиданты, консерванты, буферы или другие растворенные вещества, придающие раствору изотоничность с кровью, загустители, суспендирующие агенты или иные фармацевтически приемлемые добавки. Такие добавки включают, например, тартратный, цитратный и ацетатный буферы, этанол, пропиленгликоль, полиэтиленгликоль, комплексообразующие агенты (такие как ЭДТК), антиоксиданты (такие как бисульфит натрия, метабисульфит натрия и-3 007920 аскорбиновая кислота), высокомолекулярные полимеры (такие как жидкие полиэтиленоксиды) для регулирования вязкости и полиэтиленовые производные ангидридов сорбита. При необходимости можно добавлять также консерванты, такие как бензойная кислота, метил- или пропилпарабен, бензалконийхлорид и другие производные четвертичного аммония. Соединения, предлагаемые в изобретении, можно вводить также в виде растворов для назального введения и они могут содержать помимо соединений, предлагаемых в настоящем изобретении, пригодные буферы, агенты для регулирования тоничности, противомикробные консерванты, антиоксиданты и повышающие вязкость агенты в водном носителе. Примерами повышающих вязкость агентов являются поливиниловый спирт, производные целлюлозы, поливинилпирролидон, полисорбаты или глицерин. Противомикробные консерванты, которые можно добавлять, включают бензалконийхлорид, тимерозал,хлорбутанол или фенилэтиловый спирт. Кроме того, соединения, предлагаемые в изобретении, можно вводить в виде суппозитория. Метод и синтез. Специалист в области синтетической органической химии на основе своих знаний может получать соединения, предлагаемые в изобретении. Примеры реакционных схем приведены ниже на схемах 1-4. Заместители R2, R4, R5, R12, R13 и R14 имеют значения, указанные в описании. Схема 1 Получение промежуточных продуктов, где R4 обозначает Me В целом метод состоит в том, что путем ароматического замещения (SNAR) соединения 1(i) Et-NH2 получают промежуточное соединение 1(ii). Затем путем галогенирования положения 5 с помощью бромирующего агента (например, NBS или брома) получают 1(iii). В результате замыкания кольца соединения 1(iii) с помощью проводимой в присутствии основания реакции SnAR образуется трициклический промежуточный продукт 1(iv). Путем введения заместителя R2 посредством ароматического замещения хлора в положении С-2 соединения 1(iv) получают промежуточное соединение 1(v). Схема 2 Получение промежуточных продуктов, где R5 обозначает Me Последовательность реакций на схеме 2 аналогична последовательности, описанной у J.M. Klunder и др.; J. Med. Chem. 41, 1998, с. 2960-2971 и у C.L. Cywin и др.; J. Med. Chem., 41, 1998, с. 2972-2984. В-4 007920 целом метод заключается в том, что путем ароматического замещения (SNAR) соединения 2(i) Et-NH2 получают промежуточный продукт 2(ii). Путем восстановления нитрогруппы (например, с помощью каталитического гидрирования) получают соединение 2(iii). В результате реакции конденсации, проводимой в присутствии основания, соединения 2(iii), например с 5-бром-2-хлор-3-пиридинкарбонилхлоридом, получают соединение 2(iv). После замыкания кольца соединения 2(iv), которое осуществляют путем реакции SnAR в присутствии основания, получают трициклический промежуточный продукт 2(v). Метильную группу R5 в соединении 2(vi) можно вводить с помощью известного в данной области алкилирования с использованием, например метилйодида. Схема 3 Получение промежуточных продуктов, где R2 обозначает С 1-С 4 алкил В целом метод заключается в том, что путем реакции конденсации соединений 3(i) и 3(ii), которую проводят в присутствии основания, получают промежуточный продукт 3(iii). Путем ароматического замещения (SNAR) соединения 3(iii) Et-NH2 получают промежуточный продукт 3(iv). Замыкание кольца соединения 3(iv) проводят с помощью SnAR-реакции в присутствии основания, получая трициклический промежуточный продукт, из которого путем алкилирования получают промежуточный продукт 3(v). Схема 4 Альтернативный вариант получения соединения, где R4 обозначает Me В целом метод заключается в том, что с помощью реакции конденсации соединений 4(i) и 3(ii) в присутствии основания получают промежуточный продукт 4(ii). Путем ароматического замещения(SNAR) соединения 4(ii) Et-NH2 получают промежуточный продукт 4(iii). Путем замыкания кольца 4(iii) с помощью реакции SnAR в присутствии основания получают трициклический промежуточный продукт,представляющий собой соединение 1(v).-5 007920 Схема 5 Введение производных бензойной кислоты В целом метод заключается в том, что бромсодержащее производное 5(i), синтезированное согласно методу, представленному в описании, подвергают реакции сочетания с реагентом, представляющим собой аллилолово, в апротонном растворителе (например, ДМФ) в присутствии катализатора, в результате чего образуются С-8-заместители 5(ii). После окисления двойной связи (например, путем озонолиза с получением озонида), и последующего восстановления получают С-8-гидроксиэтильный заместитель 5(iii). С помощью реакции типа реакции Мицунобу, нафтильные производные 5(iv), 5(v) или 5(vi), где Y имеет значения, указанные для R14, за исключением СООН, конденсируют с соединением 5(iii), получая соединение формулы I. В альтернативном варианте, если Y обозначает предшественник группы R14, например СООCH3, можно применять реакцию типа реакции Мицунобу для конденсации соединения 5(iv) или 5(v) с соединением 5(iii), и после этого Y можно химическим путем превращать в заместители R14,например путем омыления СООCH3, с образованием СООН, в результате чего получают соединение формулы I. Как указано выше, соединение, предлагаемое в изобретении, ингибирует ферментативную активность ОТ ВИЧ-1. Путем тестирования этих соединений согласно описанному ниже методу установлено,что они ингибируют РНК-зависимую ДНК-полимеразную активность ОТ ВИЧ-1. Установлено (данные не представлены), что они ингибируют также ДНК-зависимую ДНК-полимеразную активность ОТ ВИЧ 1. С помощью описанного ниже анализа на основе обратной транскриптазы (ОТ) соединения можно тестировать в отношении их способности ингибировать РНК-зависимую ДНК-полимеразную активность ОТ ВИЧ-1. Таким путем было проведено тестирование некоторых конкретных соединений, которые описаны в приведенных ниже примерах. Результаты тестирования приведены в табл. 1 в виде значений IC50 иEC50. Примеры Ниже изобретение более подробно проиллюстрировано на примерах, не ограничивающих его объем. Все реакции проводили в атмосфере азота или аргона, если не указано иное. Температуры даны в градусах Цельсия. Проценты или соотношения для растворов представляют собой об./об.-соотношения,если не указано иное. Сокращения или символы, используемые в описании, имеют следующие значения: ДЭАД: диэтилазодикарбоксилат; ДИАД: диизопропилазодикарбоксилат; ДИЭА: диизопропилэтиламин; ДМАП: 4-(диметиламино)пиридин; ДМСО: диметилсульфоксид; ДМФ: диметилформамид; ДЦК: дициклогексилкарбодиимид;Et2O: диэтиловый эфир; ЖХВР: жидкостная хроматография высокого разрешения;Ph: фенил; ТБЭ: Трис-борат-ЭДТК; ТБТУ: тетрафторборат 2-(1H-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония; ТФК: трифторуксусная кислота; ТГФ: тетрагидрофуран; БОЕ: бляшкообразующие единицы; ДЭПК: диэтилпирокарбонат; ДТТ: дитиотреитол; ЭДТА: этилендиаминотетраацетат; УМФ: уридин-5'-монофосфат; УТФ: уридин-5'-трифосфат; МЭС: 2-(н-морфолино)этансульфоновая кислота; ДСН-ПААГ: гель-электрофорез в полиакриламидном геле в присутствии додецилсульфата натрия; ММСО: предельное значение пропускаемой молекулярной массы; бис-Трис-пропан: 1,3-бистрис(гидроксиметил)метиламинопропан;GSH: восстановленный глутатион; ОБГ: н-октилD-глюкозид. Синтез В следующих примерах проиллюстрированы способы получения соединений, предлагаемых в изобретении. Пример 1. Стадия а. К раствору 2-хлор-3-нитропиридина 1 а (51 г, 325 ммолей) в ТГФ (650 мл) добавляли 2 М раствор этиламина в ТГФ (365 мл, 731 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь сливали в воду (1,5 л) и образовавшийся твердый продукт фильтровали и сушили при пониженном давлении, получая соединение 1b (52 г). Стадия б. Раствор 2-(этиламино)-3-нитропиридина 1b (52 г) в МеОН (600 мл) перемешивали в течение ночи при комнатной температуре в атмосфере водорода (1 атм) в присутствии 20% Рd(ОН)2/С (10,4 г). Катализатор удаляли путем фильтрации через диатомовую землю. Фильтрат концентрировали при пониженном давлении, получая соединение 1 с в виде твердого вещества черного цвета (39 г, выход 88% после стадий а и б). Стадия в. К охлажденному раствору 3-амино-2-(этиламино)пиридина 1 с (30,6 г, 223 ммоль) в MeCN (740 мл) добавляли твердый NaHCO3 (56,3 г, 669 ммоль). Через 5 мин добавляли неочищенный 5-бром-2-хлор-3 пиридинкарбонилхлорид (полученный из 5-бром-2-гидрокси-3-пиридинкарбоновой кислоты и SOCl2 (1 экв., 223 ммоль) [как описано у Т.W. Gero и др. в Synth. Commun. 19, 1989, с. 553-559 (документ включен в настоящее описание в качестве ссылки) за исключением водной обработки]. Через 2 ч реакционную смесь сливали на лед/H2O (1,5 л) и образовавшийся твердый продукт фильтровали, промывали H2O и-7 007920 затем гексаном. После сушки при пониженном давлении в течение ночи получали соединение 1d в виде твердого вещества черного цвета (54,9 г, выход 69%). Стадия г. К раствору 2-хлор-N-2-(этиламино)-3-пиридинил-5-бром-3-пиридинкарбоксамида 1d (54,9 г,154,4 ммоль) в пиридине (308 мл) при 50 С добавляли по каплям 1 М раствор NaHMDS в ТГФ (355 мл,355 ммоль). Через 10 мин реакционной смеси давали охладиться до комнатной температуры и затем сливали на ледяную воду (2 л). Образовавшийся твердый продукт фильтровали, промывали водой и затем гексаном. После сушки при пониженном давлении в течение ночи получали соединение 1 е (36 г, выход 75%) в виде твердого вещества темно-зеленого цвета. Стадия д. К раствору 8-бром-5,11-дигидро-11-этил-6 Н-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она 1 е (36,7 г,115 ммоль) в ДМФ (380 мл) добавляли NaH (3,5 г, 138 ммоль) и смесь нагревали до 50 С в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры и обрабатывали MeI (14,3 мл, 230 ммоль). Через 1,5 ч реакционную смесь сливали на ледяную воду. Твердый продукт фильтровали, промывали водой и затем гексаном, получая после сушки соединение 1f (37,9 г, выход 99%) в виде твердого вещества темно-серого цвета. Стадия е. Аллилтрибутилолово (30,7 мл, 99,0 ммоль) и Pd(Ph3P)4 (5,20 г, 4,50 ммоль) добавляли к дегазированному (путем пропускания N2 через раствор в течение 30 мин) раствору 8-бром-5,11-дигидро-11-этил 5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она 1f (30,0 г, 90,0 ммоль) в ДМФ (450 мл) при комнатной температуре. Смесь перемешивали при 90 С в течение 1,5 ч, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали с помощью экспрессхроматографии (гексан: EtOAc, от 8:2 до 7:3), получая соединение 1g (22,19 г, выход 84%). Стадия ж. Струей озонированного кислорода барботировали холодный (-78 С) раствор 5,11-дигидро-11-этил 5-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она 1g (22,19 г, 75,4 ммоль) в СН 2 Сl2(150 мл) и МеОН (150 мл) в течение 2,5 ч. Затем раствор барботировали струей N2 в течение 15 мин и после этого к раствору добавляли твердый NaBH4 (4,99 г, 132 ммоль). Реакционной смеси давали нагреться до комнатной температуры. Через 1 ч добавляли водный насыщенный NH4Cl (200 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Органические растворители удаляли при пониженном давлении. К остатку добавляли воду (300 мл) и СНСl3 (300 мл). Фазы разделяли и водный слой экстрагировали СНСl3 (3300 мл). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (EtOAc:CHCl3,4:1) получая соединение 1h (16,1 г, выход 72%) в виде твердого вещества белого цвета. Пример 2. Стадия а. Охлажденный на льду раствор EtNH2 (49,8 г, 1,10 моль) в толуоле (200 мл) добавляли в течение 15 мин к охлажденному на льду раствору 2,6-дихлор-3-нитропиридина 2 а (100,0 г, 0,52 моль) в толуоле (225 мл). Смесь перемешивали при 0 С в течение 45 мин. Добавляли воду (500 мл) и EtOAc (500 мл) и разделяли фазы. Органический слой последовательно промывали водой (200 мл) и соляным раствором (200 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Оставшийся твердый-8 007920 продукт перекристаллизовывали из МеОН, получая соединение 2b (83,7 г, выход 80%) в виде игл желтого цвета. Стадия б. Соединение 2 с получали методом, аналогичным методу, описанному в примере I, стадия б. Стадия в. Раствор 5-бром-2-хлор-3-пиридинкарбонилхлорида (30,0 г, 97,0 ммоль) в MeCN (100 мл) добавляли через канюлю при комнатной температуре к раствору 3-амино-6-хлор-2-(этиламино)пиридина 2 с (16,6 г,97,0 ммоль) в MeCN (180 мл), содержащем твердый NaHCO3 (14,2 г, 169 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли воду (200 мл) и смесь перемешивали в течение 10 мин. Образовавшуюся суспензию фильтровали. Твердый продукт промывали водой, затем гексаном и сушили при пониженном давлении, получая соединение 2d (28,4 г, выход 75%). Стадия г. 1 М раствор NaHMDS в ТГФ (167,5 мл, 167,5 ммоль) медленно добавляли к раствору 5-бром-2-хлорN-2-(этиламино)-6-хлор-3-пиридинил-3-пиридинкарбоксамида 2d (28,4 г, 72,8 ммоль) в пиридине (146 мл), нагретом до 50 С. Реакционную смесь перемешивали при 50 С в течение 1,5 ч. Затем смесь сливали на смесь воды и льда (1 л) и через 1 ч фильтровали образовавшуюся суспензию. Твердый продукт промывали водой и сушили при пониженном давлении, получая соединение 2 е (23,4 г, выход 91%). Стадия д. Твердый NaH (60% масляная дисперсия, 3,46 г, 86,1 ммоль) добавляли в течение 30 мин к раствору 8-бром-2-хлор-5,11-дигидро-11-этил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она 2 е (23,4 г, 66,3 ммоль) в ДМФ (220 мл) при 50 С. Смесь перемешивали при 50 С в течение 1 ч, затем давали нагреться до комнатной температуры. Смесь сливали на воду (1 л) и образовавшуюся суспензию фильтровали. Твердый продукт последовательно промывали водой и гексаном, затем сушили при пониженном давлении, получая соединение 2f (23,0 г, выход 94%). Стадия е. Аллилтрибутилолово (21,3 мл, 68,7 ммоль) и Pd(Ph3P)4 (3,61 г, 3,12 ммоль) добавляли к дегазированному (путем пропускания N2 через раствор в течение 30 мин) раствору 8-бром-2-хлор-5,11-дигидро 11-этил-5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она 2f (23,0 г, 62,5 ммоль) в ДМФ (312 мл). Смесь нагревали до 90 С в течение 2 ч. Смесь концентрировали при пониженном давлении. Остаток очищали с помощью экспресс-хроматографии (гексан:EtOAc, 7:3) получая соединение 2g (13,4 г, выход 65%). Стадия ж. Озонированный кислород вводили в холодный (-78 С) раствор 2-хлор-5,11-дигидро-11-этил-5 метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она 2g (13,4 г, 40,7 ммоль) в МеОН (102 мл) и СН 2 Сl2 (102 мл) до полного поглощения алкена. Раствор барботировали азотом для удаления избытка O3. Затем небольшими порциями добавляли твердый NaBH4 (2,69 г, 71,1 ммоль) и смеси давали медленно нагреться до комнатной температуре. Через 1 ч добавляли водный насыщенный NH4Cl (150 мл) и смесь перемешивали в течение 20 мин. Органические растворители удаляли при пониженном давлении. К водному раствору добавляли воду (100 мл). Раствор экстрагировали СНСl3 (3200 мл). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью экспресс-хроматографии (EtOAc:CHCl3, 4:1), получая соединение 2h-9 007920 Стадия а. К смеси концентрированной серной кислоты (600 мл) и дымящей азотной кислоты (90%, 400 мл) в ледяной бане (внутреннюю температуру поддерживали на уровне 5-10 С) добавляли по каплям 2,6 дифторпиридин 3 а (200 г, 1,74 моль). Образовавшуюся смесь перемешивали в течение ночи при комнатной температуре. Смесь медленно сливали на 3 кг льда и экстрагировали Et2O (22 л). Объединенные органические слои промывали водным 1,5 н. NaOH (21 л), затем водным насыщенным NaHCO3 (400 мл) или до достижения значения рН примерно 8-9. Органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении до постоянной массы (для удаления непрореагировавшего 2,6-дифторпиридина: 10-12%). В результате получали соединение 3b в виде жидкости желтого цвета (207,3 г, выход 74%). Стадия б. К раствору 2,6-дифтор-3-нитропиридина 3b (45,7 г, 285 ммоль) в ТГФ (500 мл) при -40 С добавляли по каплям раствор этиламина (25,7 г, 570 ммоль) в ТГФ (250 мл). Через 30 мин реакционную смесь концентрировали при пониженном давлении и остаток растворяли в EtOAc. Органическую фазу промывали соляным раствором, сушили (MgSO4), фильтровали и концентрировали. Образовавшийся твердый продукт очищали с помощью экспресс-хроматографии (15% EtOAc в гексане), получая соединение 3 с (43,2 г, выход 82%) в виде твердого вещества желтого цвета. Стадия в. Раствор 2-этиламино-6-фтор-3-нитропиридина 3 с (43,2 г, 230 ммоль) в ТГФ (1 л) перемешивали в течение ночи при комнатной температуре в атмосфере водорода (1 атм) в присутствии 20% Pd(OH)2/C(4,35 г). Катализатор удаляли фильтрацией через диатомовую землю. Фильтрат концентрировали при пониженном давлении, получая соединение 3d (36,3 г, выход 95%) в виде твердого вещества черного цвета. Стадия г. К охлажденному раствору (4 С) 3-амино-2-этиламино-6-фторпиридина 3d (31,0 г, 200 ммоль) вMeCN (160 мл) добавляли твердый NaHCO3 (50,4 г, 600 ммоль). Через 15 мин добавляли раствор 5-бром 2-хлор-3-пиридинкарбонилхлорида (1 экв., 200 ммоль) в MeCN (155 мл). После выдерживания в течение 60 мин при комнатной температуре реакционную смесь сливали на воду (1,2 л) и перемешивали в течение 30 мин. Образовавшийся твердый продукт фильтровали, сушили в течение ночи при пониженном давлении при 50 С. В результате получали соединение 3 е (73,7 г, выход 99%) в виде твердого вещества черного цвета. Стадия д. К раствору 2-хлор-N-2-(этиламино)-6-фтор-3-пиридинил-5-бром-3-пиридинкарбоксамида 3 е (73,5 г, 216 ммоль) в пиридине (435 мл) при 50 С добавляли по каплям 1 М раствор NaHMDS в ТГФ (520 мл,520 ммоль). Через 10 мин реакционной смеси давали нагреться до комнатной температуры, после чего сливали на ледяную воду (2 л). Образовавшийся твердый продукт фильтровали, промывали водой и затем гексаном. Твердый продукт сушили при пониженном давлении, получая соединение 3f (50,6 г, выход 69%) в виде твердого вещества темно-зеленого цвета. Стадия е. К раствору 8-бром-5,11-дигидро-11-этил-2-фтор-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она 3f(44 г, 130,5 ммоль) в ДМФ (520 мл) добавляли NaH (4,28 г, 178 ммоль) и смесь нагревали до 50 С в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры и обрабатывали MeI (24,4 мл, 522 ммоль). Через 1,5 ч реакционную смесь сливали на ледяную воду. Твердый продукт фильтровали, промывали водой и затем гексаном, сушили при пониженном давлении, получая соединение 3g (43,2 г, выход 94%) в виде твердого вещества темно-серого цвета. Стадия ж. Аллилтрибутилолово (32,0 мл, 103,4 ммоль) и Pd(Ph3P)4 (5,43 г, 4,70 ммоль) добавляли к дегазированному (путем пропускания N2 через раствор в течение 45 мин) раствор 8-бром-5,11-дигидро-11-этил-2 фтор-5-метил-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она 3g (33,0 г, 94,0 ммоль) в ДМФ (470 мл). Через 1, 2, 3, 4 и 5 ч добавляли дополнительные количества Pd(Ph3)4 (1,09 г, 0,94 ммоль) для завершения реакции. Смесь нагревали до 90 С в течение 6 ч. Смесь концентрировали при пониженном давлении. Остаток очищали с помощью экспресс-хроматографии (гексан:EtOAc, от 8:2 до 7:3), получая соединение 3h (22,4 г, выход 76%). Стадия з. Струей озонированного кислорода барботировали холодный (-78 С) раствор 5,11-дигидро-11-этил 2-фтор-5-метил-8-(2-пропенил)-6H-дипиридо[3,2-b:2',3'-е][1,4]диазепин-6-она 3h (22,38 г, 71,6 ммоль) в СН 2 Сl2 (100 мл) и МеОН (100 мл) в течение 3 ч. После этого раствор барботировали в течение 15 мин струей N2 и затем к раствору добавляли твердый NaBH4 (5,05 г, 133 ммоль). Реакционной смеси давали нагреться до комнатной температуры. Через 1 ч к реакционной смеси добавляли дополнительную порцию NaBH4 (1,62 г, 43,0 ммоль). Еще через 1 ч добавляли водный насыщенный NH4Cl (150 мл) и смесь перемешивали при комнатной температуре в течение 30 мин. Органические растворители удаляли при- 10007920 пониженном давлении. Добавляли воду (200 мл) и смесь экстрагировали CHCl3 (3300 мл). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью экспресс-хроматографии (EtOAc:CHCl3, 4:1), получая соединение 31 (19,7 г, выход 72%) в виде твердого вещества белого цвета. Пример 4 (соединения 1003 и 1031). Стадия а. Раствор 1,6 М н-BuLi в гексане (6,22 мл, 9,95 ммоль) быстро добавляли к холодному (-78 С) раствору соединения 4 а (0,87 г, 4,33 ммоль) в ТГФ (20 мл). Смесь перемешивали при -78 С в течение 10 мин,давали нагреться до 0 С, выдерживали при 0 С в течение 1 ч. В реакционную смесь вводили струю СO2 в течение 10 мин и раствор подкисляли путем добавления водного 1,0 н. раствора НСl. Смесь экстрагировали EtOAc. Органический слой сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток растворяли в Et2O (20 мл) и обрабатывали избыточным количеством раствора CH2N2 вEt2O (приблизительно 0,6 М, 10 мл) в течение 10 мин. Смесь концентрировали при пониженном давлении и остаток очищали с помощью экспресс-хроматографии (гексан: EtOAc, от 4:1 до 7:3), получая 4b (0,13 г,выход 17%). Стадия б. Раствор ДИАД (86 мкл, 0,44 ммоль) в ТГФ (2,0 мл) добавляли в течение 30 мин к раствору, содержащему соединение 1h (100 мг, 0,33 ммоль), соединение 4b (60,0 мг, 0,33 ммоль) и PPh3 (114 мг, 0,44 ммоль) в ТГФ (10 мл), при 25 С. Смесь перемешивали в течение 1 ч и затем концентрировали при пониженном давлении. Остаток очищали с помощью экспресс-хроматографии (гексан:EtOAc, от 7:3 до 1:1),получая соединение 1031 (128 мг, выход 83%). Стадия в. Водный 1,0 н. раствор LiOH (1,52 мл, 1,52 ммоль) добавляли к раствору соединения 1031 (100 мг,0,22 ммоль) в ТГФ (6 мл) и МеОН (2 мл). Реакционную смесь перемешивали при 25 С в течение 24 ч,затем нагревали до температуры дефлегмации в течение 1 ч. Раствор подкисляли путем добавления водного 1,0 н. раствора HCl, затем экстрагировали EtOAc. Органический слой промывали водой (2 х) и соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток растирали с Еt2O/гексаном, получая соединение 1003 (80 мг, выход 83%) в виде твердого вещества белого цвета. Смесь соединения 1003 (38,0 мг, 0,085 ммоль) и водного 0,02 н. раствора NaOH (4,3 мл, 0,085 ммоль) в MeCN (3 мл) облучали ультразвуком. Образовавшийся раствор замораживали и лиофилизировали, получая соответствующую натриевую соль (37 мг, выход 98%) в виде твердого вещества белого цвета. Пример 5 (соединения 1019, 1020 и 1028).- 11007920 Стадия а. Раствор ДИАД (86 мкл, 0,44 ммоль) в ТГФ (2,0 мл) добавляли в течение 2 ч к раствору, содержащему соединение 3i (106 мг, 0,34 ммоль), соединение 5 а (56,0 мг, 0,34 ммоль) и PPh3 (114 мг, 0,44 ммоль) в ТГФ (7 мл), при 25 С. Смесь перемешивали в течение 1 ч и затем концентрировали при пониженном давлении. Остаток очищали с помощью экспресс-хроматографии (гексан:EtOAc, от 7:3 до 1:1), получая соединение 5b (115 мг, выход 73%) в виде твердого вещества белого цвета. Стадия б. Водный 1,0 н. раствор LiOH (1,0 мл, 1,0 ммоль) добавляли к раствору соединения 5b (100 мг, 0,21 ммоль) в МеОН (6 мл). Реакционную смесь перемешивали при 25 С в течение 24 ч. Раствор подкисляли путем добавления водного 1,0 н. раствора HCl и затем экстрагировали EtOAc. Органический слой промывали водой и соляным раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью экспресс-хроматографии (гексан:EtOAc:АсОН, 50:50:1), получая сначала соединение 1019 (25 мг, выход 25%) в виде твердого вещества белого цвета, а затем соединение 1020 (48 мг, выход 50%) в виде твердого вещества белого цвета. Соответствующие натриевые соли получали путем обработки водным 0,02 н. раствором NaOH. Стадия в. 1,0 М раствор диметиламина в ТГФ (5,0 мл, 5,0 ммоль) и водный 1,0 н. раствор LiOH (1,0 мл, 1,0 ммоль) добавляли к раствору соединения 5b (50,0 мг, 0,11 ммоль) в изо-PrOH (3 мл). Реакционную смесь нагревали до температуры дефлегмации в течение 48 ч. Добавляли водный 1,0 н. раствор HCl (2 мл) и смесь экстрагировали EtOAc. Органический слой промывали водой и соляным раствором, сушили(MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью экспресс-хроматографии (гексан:EtOAc:АсОН, 50:50:1), получая соединение 1028 (18 мг, выход 35%) в виде твердого вещества белого цвета. Соответствующую натриевую соль получали путем обработки водным 0,02 н. раствором NaOH. Пример 6 (соединение 1014). Стадия а. К раствору кислоты 6 а (1,00 г, 5,55 ммоль) в CH2Cl2 (50 мл) добавляли оксалилхлорид (0,73 мл, 8,3 ммоль) и ДМФ (100 мкл). Реакционную смесь перемешивали в течение 90 мин, затем добавляли EtOH(15 мл) и реакционную смесь перемешивали еще в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении, остаток разбавляли EtOAc и промывали последовательно водой, соляным раствором, сушили (MgSO4), фильтровали и концентрировали, получая сложный эфир 6b, который использовали без дополнительной очистки. Стадия б. К раствору сложного эфира 6b в CH2Cl2 (50 мл) добавляли 1 М раствор BBr3 в CH2Cl2 (7,2 мл, 7,20 ммоль). После выдерживания в течение 3 ч при комнатной температуре реакционную смесь охлаждали до 0 С и добавляли EtOH (5 мл). Реакционную смесь перемешивали в течение 30 мин при комнатной температуре, затем концентрировали при пониженном давлении. Остаток разбавляли EtOAc и последовательно промывали насыщенным водным раствором NaHCO3, водой и соляным раствором, сушили(7,0 мл), при комнатной температуре. Смесь перемешивали в течение 4 ч и затем концентрировали при пониженном давлении. Остаток очищали с помощью экспресс-хроматографии (гексан:EtOAc; от 30:70 до 50:50), получая соединение 6d (46 мг, выход 29%) в виде пены белого цвета. Стадия г. К раствору сложного эфира 6d (44 мг, 0,09 ммоль) в смеси ТГФ (3 мл) и МеОН (1 мл) добавляли водный 1 н. раствор LiOH (1,0 мл, 1,0 ммоль). После выдерживания в течение 4 ч при комнатной температуре добавляли 1 н. HCl (2 мл). Смесь экстрагировали EtOAc. Органический слой промывали водой и соляным раствором, сушили (MgSO4), фильтровали и концентрировали досуха, получая соединение 1014(39 мг, выход 93%) в виде твердого вещества белого цвета. Соответствующую натриевую соль получали путем обработки 1 экв. водного гидроксида натрия и образовавшийся раствор лиофилизировали, получая пушистое твердое вещество белого цвета. Таблица 1 Анализ обратной транскриптазы (ОТ) Теоретические основы анализа. В число ферментов, которые кодируются вирусом иммунодефицита человека (ВИЧ-1), входит обратная транскриптаза (1), получившая такое название потому, что она транскрибирует копию ДНК на матрице РНК. Эту активность можно количественно измерять с помощью бесклеточного ферментативного анализа, который основан на установленном факте, что обратная транскриптаза обладает способностью использовать синтетическую матричную poly r(C), примированную биотинилированным олигонуклеотидом d(G), для транскрипции радиоактивномеченной цепи ДНК с использованием в качестве субстрата 3 Н-дГТФ. В описанном ниже методе анализа используется фермент дикого типа (который является доминирующей формой фермента, обнаруженной у пациентов, инфицированных ВИЧ-1), однако, в методе можно применять также мутантные ОТ-ферменты (например, Y181C, полученный путем сайтнаправленного мутагенеза, в котором остаток тирозина в кодоне 181 заменен на остаток цистеина) с использованием аналогичных условия проведения анализа. Этот анализ позволяет оценивать эффективность соединений в отношении их способности ингибировать мутантные ферменты. Материалы. а). Получение фермента. Некоторые мутанты ОТ ВИЧ-1 клона ВН 10 IIIВ были получены от Dr. С.-K. Shih (фирма BoehringerpKRT2, несущий только ген p66 ОТ, находящийся под контролем оперона lac/промотора trc, получали отDr. W. Summers (Йельский университет) (2). Путем сайтнаправленного мутагенеза в ген ОТ дикого типа интродуцировали различные аминокислотные замены. Клоны ОТ субклонировали в бактериальном экспрессионном векторе pKK233-2. Полученные клоны включали клон дикого типа, Val106Ala, Tyr181Cys,Tyr188Cys, Tyr188Leu, Gly190Ala и Pro236Leu. Другие клоны были получены заявителями путем сайтнаправленного мутагенеза ОТ pKK233-2 и включали Lys103Asn, Lys103Asn/Tyr181Cys, Lys103Asn/Leu100Ile, Lys103Asn/Pro225His и Lys103Asn/Val108Ile. б). Очистка фермента. Очистку рекомбинантной обратной транскриптазы осуществляли с помощью комбинации описанных ранее методов (3). Для инициации роста предварительной культуры, которую выращивали при 37 С,использовали индивидуальную колонию из планшета, содержащего свежие трансформированные клетки линии JM109. Этой предварительной культурой инокулировали два литра среды для роста. После достижения ОП 6001,5 (5-6 ч при 37 С), экспрессию гена ОТ индуцировали с помощью ИПТГ (конечная концентрация 1 мМ) и ферментацию осуществляли еще в течение нескольких часов при 37 С. После центрифугирования отбрасывали супернатанты, пеллетированные клетки пулировали и помещали на хранение при -80 С до осуществления очистки. Клеткам давали оттаять при 4 С в течение ночи и суспендировали в буфере для лизиза (MES 50 мМ рН 6, ЭДТА 1 мМ, 10 об.% глицерина, 0,02% мас./об. ОБГ, 0,02% мас./об. азида натрия). Добавляли лизоцим и смесь инкубировали на льду в течение 40 мин. После гомогенизации с использованием Dounce в присутствии лизоцима и облучения ультразвуком клетки центрифугировали в течение 30 мин. Собирали супернатант (S1) и помещали на хранение при 4 С. Полученный центрифугированием пеллет ресуспендировали в буфере для экстракции (MES 50 мМ рН 6, KPO4 50 мМ рН 6, KCl 100 мМ, 10 об.% глицерина, 0,02% мас./об. ОБГ, 0,02% мас./об. азида натрия) и перемешивали в течение 30 мин при 4 С. Эту вторую смесь снова центрифугировали и собирали супернатант (S2). Описанную выше процедуру повторяли еще два раза, получая супернатанты S3 и S4, и в течение ночи проводили последнюю экстракцию (S5). К объединенным супернатантам добавляли полимин (конечная концентрация 0,005%) для удаления нуклеиновых кислот. Этот раствор перемешивали в течение 75 мин при 4 С и центрифугировали в течение 1 ч. Супернатант (SS1) осаждали на льду с помощью 20% мас./об. сульфата аммония и перемешивали в течение 1 ч при 4 С. Затем смесь центрифугировали и образовавшийся супернатант (SS2) осаждали путем добавления 40% мас./об. сульфата аммония (всего 60%), пере- 14007920 мешивали в течение 1 ч и снова центрифугировали. Конечный дебрис (Р 1) хранили в течение ночи при 4 С перед проведением очистки на следующий день. Все стадии очистки осуществляли при 4 С, если не указано иное. Дебрис (Р 1) ресуспендировали в буфере MES, содержащем 50 мМ рН 6, KPO4 10 мМ рН 6, KCl 100 мМ, 10 об.% глицерина, 0,02% мас./об. ОБГ, 0,02% мас./об. азид натрия. Суспензию в течение ночи подвергали диализу в противотоке того же самого буфера с использованием диализных трубок с ММСО 1214 кДа. Диализат центрифугировали и супернатант фильтровали через фильтры типа Millex-PF с размером отверстий 0,8 мкм. Профильтрованный образец загружали в гидроксиапатитную колонку (объем слоя 30-мл) и промывали тем же самым буфером. Связанный фермент элюировали с использованием линейного градиента от 10 до 300 мМ KPO4 (200 мл) в указанном выше буфере. Фракции, содержащие гетеродимер р 66/р 51 (по данным анализа с помощью ДСН-ПААГ 8% и вестерн-блоттинга) объединяли для внесения в следующую колонку. Фракции, содержащие ОТ, разбавляли в два раза бис-Трис-буфером,содержащим пропан 50 мМ рН 7,0, 0,02% мас./об. ОБГ, 10 об.% глицерина, 0,02% мас./об. азида натрия и загружали в Hi-Trap гепарин-сефарозную колонку (объем слоя 5 мл) и промывали тем же самым буфером. Затем связанную ОТ элюировали с использованием линейного градиента от 0 до 1 М сульфата аммония (75 мл) в том же самом буфере. Содержащие ОТ фракции объединяли на основе данных анализа,полученных с помощью ДСН-ПААГ и вестерн-блоттинга. Концентрацию протеина в таком пуле определяли с помощью метода Брадфорда, используя в качестве стандарта БСА. Конечный ферментный препарат подвергали диализу в противотоке буфера, содержащего MES 50 мМ рН 6, KPO4 300 мМ рН 6, KCl 175 мМ, 10 об.% глицерина, 0,02% мас./об. азида натрия, разделяли на аликвоты и хранили при -80 С. Процедура анализа. Радиометрический ферментативный анализ был адаптирован к формату 96-луночного титрационного микропланшета и основан на использовании загруженных стрептавидином сцинтилляционных гранул. Метод анализа вкратце описан ниже. Фермент ОТ ВИЧ-1 подвергали оттаиванию и разбавляли соответствующим образом Трис/HCl-буфером, 50 мМ рН 7,8, содержащим NaCl 60 мМ, гексагидрат MgCl2 2 мМ, ДТТ 6 мМ, GSH 2 мМ и 0,02% мас./об. Chaps, получая 3 нМ фермент. К 30 мкл раствора этого фермента добавляли 10 мкл раствора ингибитора (от 50 мкМ до 2,5 нМ ингибитора в этом же самом буфере для анализа, дополненном 15 об.% ДМСО). Перед осуществлением следующей стадии планшет подвергали предварительной инкубации в течение 15 мин при комнатной температуре. На этой стадии предварительной инкубации наибольшая и наименьшая концентрации ингибитора составляли 12,5 мкМ и 0,62 нМ соответственно, а концентрация ДМСО составляла 3,75 об.%. Затем инициировали ферментативную реакцию путем добавления 10 мкл раствора субстрата. Конечная реакционная смесь содержала Трис/HCl 50 мМ рН 7,8, NaCl 60 мМ, MgCl26H2O 2 мМ, ДТТ 6 мМ, GSH 2 мМ, Chaps 0,02% мас./об. ДМСО 3 об.%, Poly rС 179 нМ, биотин дГ 15 18 нМ, дГТФ 288 нМ, 3 Н-дГТФ 71 нМ и 1-2 нМ фермент. На этой стадии инкубации наибольшая и наименьшая концентрации ингибитора составляли 10 мкМ и 0,5 нМ соответственно. После добавления субстратов планшет заклеивали пластиковым покрытием и инкубировали в течение 1 ч при 37 С в сухой камере. Затем реакцию прекращали путем добавления 75 мкл 0,5 М раствора ЭДТК, содержащего 5 мг/мл загруженных стрептавидином сцинтилляционных гранул. Планшет встряхивали в течение 2 мин при средней скорости и инкубировали в течение 1 ч при комнатной температуре. Затем добавляли 75 мкл 7 М раствора хлорида цезия, планшет встряхивали в течение 2 мин при средней скорости и снова инкубировали в течение 1 ч при комнатной температуре. Затем планшет заклеивали пластиковым покрытием и производили количественную оценку с использованием сцинтилляционного и люминисцентного счетчика для микропланшетов типа TopCount-NXT(фирма Packard). Радиоактивность в каждой лунке оценивали в течение 60 с. В каждой серии для получения экстремальных значений использовали пустые и контрольные лунки. Процент ингибирования рассчитывали следующим образом: С помощью описанного выше анализа тестировали соединение, предлагаемое в изобретении, в отношении ингибирования ОТ дикого типа (WT) и мутантных ферментов. Результаты приведены в табл. 2 в виде значений IC50 (нМ). Для подтверждения способности соединения ингибировать репликацию ВИЧ его тестировали согласно описанному ниже методу анализа человеческой Т-клеточной культуры (фирма Syncytia). Твердофазный иммуноферментный анализ (ELISA) для оценки активности в клеточной культуре. Способность соединения, предлагаемого в изобретении, ингибировать репликацию ВИЧ в клеточной культуре тестировали в формате 96-луночного планшета. Для разбавления соединения, а также среды для роста клеток использовали полную среду RPMI 1640, содержащую RPMI 1640+10% фетальной телячьей сыворотки, 10 мкг/мл гентамицина и 10 мкМ -меркаптоэтанола. Клеточную линию Тлимфоцитов С 8166 инфицировали с множественностью заражения 0,001 вирусами, кодирующими об- 15007920 ратную транскриптазу дикого типа и мутантную транскриптазу. Затем клетки инкубировали в течение трех дней в присутствии серийных разведения соединения, предлагаемого в изобретении. Объединяли супернатант из восьми лунок-копий и определяли концентрацию внеклеточного р 24 с использованием поступающего в продажу набора для анализа антигена р 24 ВИЧ-1 (фирма Beckman-Coulter). Уровень ингибирования (% ингибирования) рассчитывали на основе следующего уравнения:(5065): 1992, с. 1783. Таблица 2 Ингибирование ОТ дикого типа и мутантных штаммов ОТ соединением формулы IIС 50 и ЕС 50: А 100 нМ; В = 100-50 нМ; С 50 нМ; и Н.о. = не определяли ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы где R2 обозначает Н, галоген, C1-C4 алкил, OC1-C4 алкил, NHCl-С 4 алкил или N(C1-С 4 алкил)2; R4 обозначает Н или CH3; R5 обозначает Н или CH3 при условии, что и R4, и R5 не имеют одинаковое значение; R12 обозначает Н, галоген, С 1-С 4 алкил, CF3 или NO2; R13 обозначает Н, С 1-С 4 алкил, галоген, ОН или NH2 при условии, что R12 и R13 оба не обозначают Н; и R14 обозначает COOR14a, где R14a обозначает Н или C1C6 алкил; или R14 обозначает C2-C4 алкенил-COOR14a, где R14a имеет указанные выше значения; или R14 обозначает C1-C4 алкил-COOR14a, где R14a имеет указанные выше значения; или его соль, или пролекарство. 2. Соединение по п.1, где R2 обозначает Н, галоген, C1-C4 алкил, ОС 1-С 4 алкил или N(C1-C4 алкил)2 и 4R и R5 не имеют одинаковых значений. 3. Соединение по п.2, где R2 обозначает Н, Cl, F, С 1-С 4 алкил, ОС 1-С 4 алкил или N(С 1-С 4 алкил)2. 4. Соединение по п.3, где R2 обозначает Н, Cl, F, CH3, ОМе или OEt. 5. Соединение по п.4, где R2 обозначает Н. 6. Соединение по п.1, где R4 обозначает Н. 7. Соединение по п.1, где R5 обозначает CH3. 8. Соединение по п.1, где R12 обозначает галоген, С 1-С 4 алкил, CF3 или NO2. 9. Соединение по п.8, где R12 обозначает Вr, Cl, CH3 или СН 3 СН 2. 10. Соединение по п.9, где R12 обозначает CH3 или СН 3 СН 2. 11. Соединение по п.1, где R13 обозначает Н, CH3 галоген, ОН или NH2. 12. Соединение по п.11, где R13 обозначает Н, CH3 или ОН. 13. Соединение по п.12, где R13 обозначает Н. 14. Соединение по п.1, где R14 обозначает СООН, СООМе, С 2-С 4 алкенил-СООН или С 1-С 4 алкилСООН. 15. Соединение по п.14, где R14 обозначает СООН, СН=СН-СООН, СН 2 СООН или СН 2 СН 2 СООН. 16. Соединение по п.15, где R14 обозначает СООН. 17. Соединение формулы (I) по п.1 где R2 обозначает Н, Cl, F, CH3, ОМе или OEt; R4 обозначает Н; R5 обозначает CH3; R12 обозначает Br, Сl,CH3 или СН 2 СН 3; R13 обозначает Н, CH3 или ОН и R14 обозначает СООН, СН=СН-СООН, СН 2 СООН или СН 2 СН 2 СООН; или его соль, или пролекарство. 18. Соединение по п.17, где R2 обозначает Н; R4 обозначает Н; R5 обозначает CH3; R12 обозначаетCH3 или СН 3 СН 2; R13 обозначает Н и R14 обозначает СООН; или его соль, или пролекарство. 19. Фармацевтическая композиция, предназначенная для лечения или предупреждения ВИЧинфекции, содержащая соединение формулы I по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. 20. Способ лечения или предупреждения ВИЧ-инфекции, заключающийся в том, что пациенту вводят ингибирующее ВИЧ количество соединения формулы I по п.1 или его фармацевтически приемлемой соли. 21. Способ лечения или предупреждения ВИЧ-инфекции, заключающийся в том, что пациенту вводят ингибирующее ВИЧ количество фармацевтической композиции по п.19, содержащей соединение формулы I по п.1 или его фармацевтически приемлемую соль. 22. Способ лечения или предупреждения ВИЧ-инфекции, заключающийся в том, что вводят соединение формулы I по п.1 в сочетании с антиретровирусным лекарственным средством. 23. Способ предупреждения перинатальной передачи ВИЧ-1 от матери к ребенку, заключающийся в том, что матери перед родами вводят соединение формулы I по п.1. 24. Применение соединения формулы I по п.1 или его фармацевтически приемлемой соли для приготовления лекарственного средства, предназначенного для лечения или предупреждения ВИЧинфекции.- 17007920 25. Применение соединения формулы I по п.1 или его фармацевтически приемлемой соли для приготовления лекарственного средства, предназначенного для предупреждения перинатальной передачи ВИЧ-1 от матери к ребенку перед родами.
МПК / Метки
МПК: A61K 31/55, A61P 31/18, C07D 221/00, C07D 243/00, C07D 471/14
Метки: ингибиторов, обратной, дипиридодиазепиноны, качестве, транскриптазы
Код ссылки
<a href="https://eas.patents.su/19-7920-dipiridodiazepinony-v-kachestve-ingibitorov-obratnojj-transkriptazy.html" rel="bookmark" title="База патентов Евразийского Союза">Дипиридодиазепиноны в качестве ингибиторов обратной транскриптазы</a>
4,4-дизамещенные 3,4-дигидро-2(1н)-хиназолиноны, полезные в качестве ингибиторов обратной транскриптазы вич

Номер патента: 1991
Опубликовано: 22.10.2001
Авторы: Ко Соо Сунг, Корбетт Джеффри В.
МПК: A61P 31/18, C07D 239/80, A61K 31/517...
Метки: качестве, 4,4-дизамещенные, транскриптазы, вич, полезные, 3,4-дигидро-2(1н)-хиназолиноны, ингибиторов, обратной
Формула / Реферат:
1. Соединение формулы (I) или его стереоизомер, или фармацевтически приемлемая соль, где R1 обозначает C1-3-алкил, замещенный 1-7 атомами галогена; R2 выбран из C1-5-алкила, замещенного 1-2 R4, С2-5-алкенила, замещенного 1-2 R4, и С2-5-алкинила, замещенного одним R4; R3 каждый независимо выбран из C1-4-алкила, ОН, C1-4-алкокси, F, Cl, Br, I, NR5R5a, NO2, CN, C(O)R6, NHC(O)R7 и NHC(О)NR5R5a; альтернативно, если присутствуют два заместителя R3...
Ненуклеозидные ингибиторы обратной транскриптазы

Номер патента: 5598
Опубликовано: 28.04.2005
Автор: Симоно Брюно
МПК: A61K 31/55, C07D 471/14
Метки: ингибиторы, транскриптазы, ненуклеозидные, обратной
Формула / Реферат:
1. Соединение общей формулы I где R2 выбирают из группы, включающей H, F, Cl, C1-C4алкил, C3-C4циклоалкил и CF3; R4 обозначает H или Me; R5 обозначает H, Me или Et при условии, что R4 и R5 оба не обозначают Me, и если R4 обозначает Me, то R5 не обозначает Et; R11 обозначает Me, Et, циклопропил, пропил, изопропил или циклобутил и Q выбирают из группы, включающей или его фармацевтически приемлемая соль. 2. Соединение по п.1, где R11 обозначает...
Набор для анализа обратной транскриптазы и его применение

Номер патента: 4880
Опубликовано: 26.08.2004
Авторы: Гроновитз Симон, Петтерссон Ингвар, Келландер Клас
МПК: C12Q 1/68
Метки: транскриптазы, применение, набор, анализа, обратной
Формула / Реферат:
1. Набор для анализа обратной транскриптазы (ОТ), включающий в себя одну или несколько упаковок, содержащих связанную(ые) с твердой фазой матрицу (матрицы), представляющую(ие) собой полирибоадениловую кислоту (prA) и/или полидезоксиадениловую кислоту (pdA), приготавливаемую(ые) путем приведения в контакт твердой фазы на основе полистирола со связывающим раствором, содержащим 1-метилимидазол и prA и/или pdA, с последующими инкубацией, промывкой...
Способ кристаллизации ингибитора обратной транскриптазы с применением противорастворителя

Номер патента: 1805
Опубликовано: 27.08.2001
Авторы: Крокер Луис С., Кларк Уилльям, Кукура Джозеф Л.
МПК: C07D 265/18
Метки: транскриптазы, кристаллизации, ингибитора, способ, применением, обратной, противорастворителя
Формула / Реферат:
1. Способ кристаллизации соединения структурной формулы включающий следующие стадии: (1) растворение соединения в растворителе при соотношении от около 3,0 мл до около 10,0 мл растворителя на 1 г соединения; (2) фильтрацию раствора соединения для удаления твердых частиц вещества; (3) добавление к перемешиваемому раствору противорастворителя в течение периода времени от около 30 мин до около 1 ч при комнатной температуре для достижения точки...
Ненуклеозидные ингибиторы обратной транскриптазы как антагонисты пролиферации и индукторы дифференцировки клеток

Номер патента: 7004
Опубликовано: 30.06.2006
Авторы: Маттеи Элизабетта, Спадафора Коррадо, Паломбини Гильельмо, Лорендзини Родольфо Нелло, Лавия Патриция, Нерви Клара
МПК: A61K 31/55, A61K 31/495, A61K 31/535...
Метки: пролиферации, обратной, антагонисты, транскриптазы, ингибиторы, индукторы, дифференцировки, ненуклеозидные, клеток
Формула / Реферат:
1. Применение по меньшей мере одного соединения-ингибитора обратной транскриптазы для получения фармацевтической композиции для противодействия утрате дифференцировки клеток и уменьшения скорости пролиферации клеток при раковых и не раковых патологиях, где вышеуказанное соединение выбрано из класса соединений 5,11-дигидро-6Н-дипиридо[3,2-b:2',3'-е][1,4]диазепина. 2. Применение по п.1, где соединение способно стимулировать дифференцировку клеток....
Предыдущий патент: Средство, обладающее противовирусной активностью, и способ его получения
Следующий патент: Лечение атрофии мышц с помощью селективных модуляторов андрогенового рецептора
Случайный патент: Узел скважинного насоса и способ извлечения скважинных текучих сред