Производные пирроло [3.4-с] пиразола, обладающие активностью как ингибиторы киназы
Номер патента: 9462
Опубликовано: 28.12.2007
Авторы: Молль Юрген, Форте Барбара, Варази Марио, Вианелло Паола, Фанчелли Даниеле







Формула / Реферат
1. Способ лечения клеточных пролиферативных нарушений, вызванных и/или связанных с измененной активностью протеинкиназы, включающий введение млекопитающему, нуждающемуся в этом, эффективного количества соединения формулы (I)

в которой R представляет собой водород или метил;
R1 представляет собой гидрокси или линейный или разветвленный C1-C3алкил или
C1-C3алкоксигруппу;
R2 представляет собой водород или атом галогена;
X представляет собой двухвалентную группу, выбранную из метилена (-CH2-) или фторметилена
(-CHF-), или X означает гетероатом или гетероатомную группу, выбранную из кислорода (-O-) или азота (-NR'-), где R' представляет собой атом водорода, линейную или разветвленную C1-C4aлкильную группу или C3-С6циклоалкильную группу;
или его фармацевтически приемлемой соли.
2. Способ по п.1 лечения клеточных пролиферативных нарушений, вызванных и/или связанных с измененной активностью Авроракиназ.
3. Способ по п.2, где Авроракиназа представляет собой Аврору-2.
4. Способ по п.1, где клеточные пролиферативные нарушения выбраны из группы, включающей рак, болезнь Альцгеймера, вирусные инфекции, аутоиммунные заболевания и нейродегенеративные расстройства.
5. Способ по п.4, где рак выбран из группы, включающей карциному, плоскоклеточную карциному, гемопоэтические опухоли миелоидного или лимфоидного происхождения, опухоли мезенхимного происхождения, опухоли центральной и периферической нервной системы, меланому, семиному, тератокарциному, остеосаркому, пигментозную ксеродерму, кератоксантому, фолликулярный рак щитовидной железы и саркому Капоши.
6. Способ по п.1, где клеточное пролиферативное нарушение выбрано из группы, включающей доброкачественную гиперплазию простаты, семейный аденоматозный полипоз, нейрофиброматоз, псориаз, пролиферацию клеток гладких мышц сосудов, связанную с атеросклерозом, фиброз легких, артрит, гломерулонефрит и послеоперационный стеноз и рестеноз.
7. Способ по п.1, дополнительно включающий воздействие на млекопитающего, нуждающегося в этом, лучевой терапии или химиотерапевтической схемы в комбинации по меньшей мере с одним цитостатическим или цитотоксическим агентом.
8. Способ по п.1, где млекопитающее, нуждающееся в нем, представляет собой человека.
9. Способ по п.2, где происходит ингибирование Аврора-2-киназы.
10. Соединение формулы (I)

в которой R представляет собой водород или метил;
R1 представляет собой гидрокси или линейный или разветвленный C1-C3алкил или
C1-C3алкоксигруппу;
R2 представляет собой водород или атом галогена;
X представляет собой двухвалентную группу, выбранную из метилена (-CH2-) или фторметилена
(-CHF-), или X означает гетероатом или гетероатомную группу, выбранную из кислорода (-O-) или азота (-NR'-), где R' представляет собой атом водорода, линейную или разветвленную C1-C4алкильную группу или C3-C6циклоалкильную группу;
или его фармацевтически приемлемая соль.
11. Соединение формулы (I) по п.10, где R означает водород или метил; R1 выбран из гидрокси, метила или метокси; R2 означает водород или атом фтора; X выбран из метилена, фторметилена, -О- или -NR', где R' принимает значения, определенные в п.10.
12. Соединение формулы (I) по п.10, необязательно в виде его фармацевтически приемлемой соли, выбранное из группы, включающей:
(1) N-{5-[(2R)-2-метокси-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-(4-метилпиперазин-1-ил)бензамид;
(2) N-{5-[(2R)-2-метокси-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-(4-этилпиперазин-1-ил)бензамид;
(3) N-{5-[(2R)-2-метокси-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-(4-изопропилпиперазин-1-ил)бензамид;
(4) N-{5-[(2R)-2-метокси-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-(4-циклопропилпиперазин-1-ил)бензамид;
(5) 4-(3,4-диметилпиперазин-1-ил)-N-{5-[(2R)-2-метокси-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}бензамид;
(6) N-{5-[(2R)-2-метокси-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-пиперидин-1-илбензамид;
(7) 4-(4-фторпиперидин-1-ил)-N-{5-[(2R)-2-метокси-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}бензамид;
(8) N-{5-[(2R)-2-метокси-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-морфолин-4-илбензамид;
(9) 4-(4-трет-бутилпиперазин-1-ил)-N-{5-[(2R)-2-метокси-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}бензамид;
(10) N-{5-[(2R)-2-гидрокси-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-(4-метилпиперазин-1-ил)бензамид;
(11) N-{5-[(2R)-2-метил-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-(4-метилпиперазин-1-ил)бензамид;
(12) N-{5-[(2R)-2-метил-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-(4-этилпиперазин-1-ил)бензамид;
(13) N-{5-[(2R)-2-метил-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-(4-изопропилпиперазин-1-ил)бензамид;
(14) N-{5-[(2R)-2-метил-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-(4-циклопропилпиперазин-1-ил)бензамид;
(15) 4-(3,4-диметилпиперазин-1-ил)-N-{5-[(2R)-2-фенилпропаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}бензамид;
(16) N-{5-[(2R)-2-фенилпропаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-пиперидин-1-илбензамид;
(17) 4-(4-фторпиперидин-1-ил)-N-{5-[(2R)-2-метил-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}бензамид;
(18) N-{5-[(2R)-2-метил-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}-4-морфолин-4-илбензамид;
(19) 4-(4-трет-бутилпиперазин-1-ил)-N-{5-[(2R)-2-фенилпропаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил}бензамид.
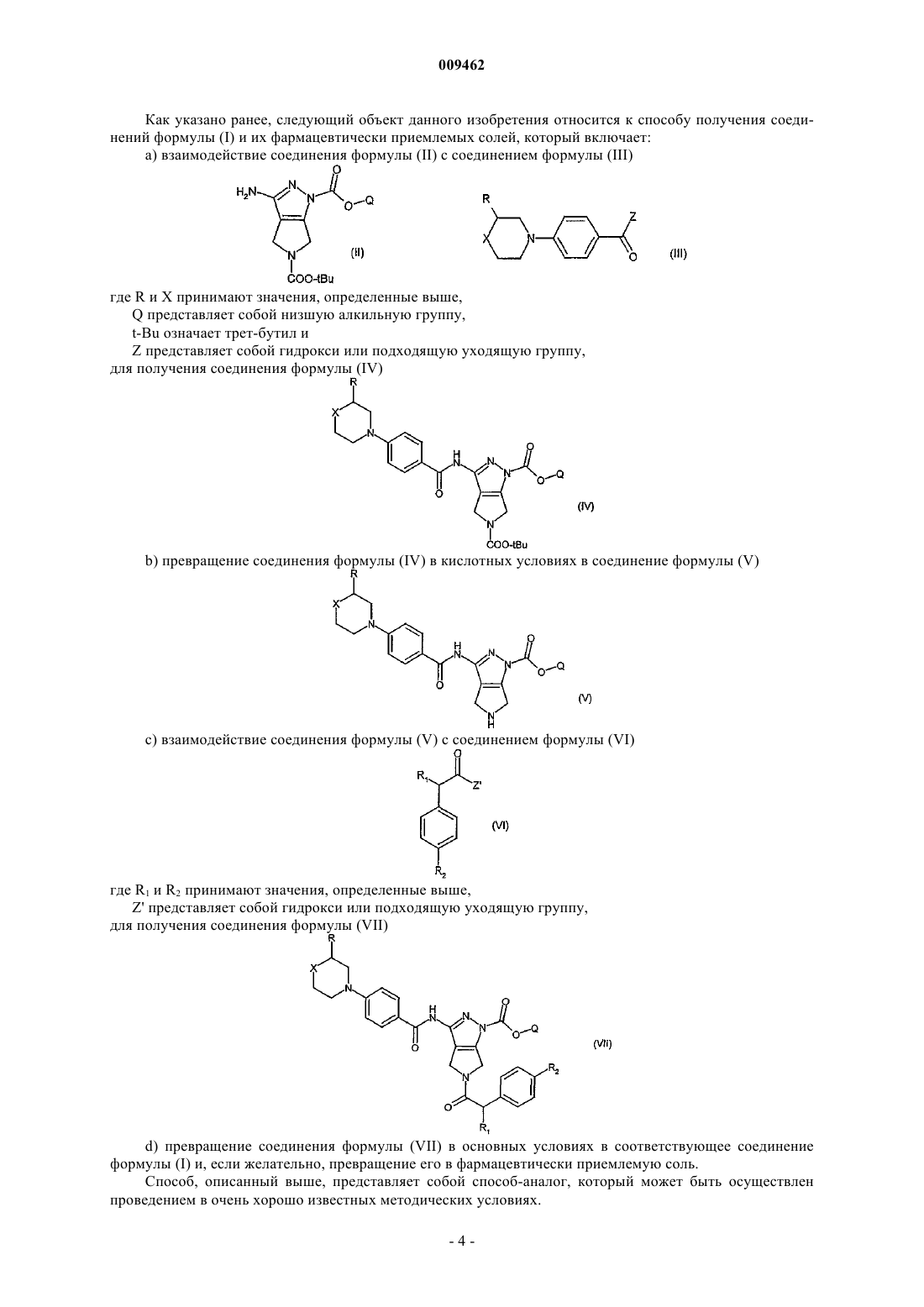
13. Способ получения соединений формулы (I) или их фармацевтически приемлемых солей по п.10, включающий:
а) взаимодействие соединения формулы (II) с соединением формулы (III)

где R и X принимают значения, определенные в п.10,
Q представляет собой низшую алкильную группу,
t-Bu означает трет-бутил и
Z представляет собой гидрокси или подходящую уходящую группу,
для получения соединения формулы (IV)

b) превращение соединения формулы (IV) в кислотных условиях в соединение формулы (V)

с) взаимодействие соединения формулы (V) с соединением формулы (VI)

где R1 и R2 принимают значения, определенные в п.10, и Z' представляет собой гидрокси или подходящую уходящую группу,
для получения соединения формулы (VII)

d) превращение соединения формулы (VII) в основных условиях в соответствующее соединение формулы (I) и, если желательно, превращение его в фармацевтически приемлемую соль.
14. Способ по п.13, где в соединениях формулы (II) Q представляет собой линейную или разветвленную C1-C4алкильную группу.
15. Способ по п.14, где Q представляет собой метил или этил.
16. Способ по п.13, где в соединениях формул (III) и (VI) Z и Z' представляют собой, каждый независимо, гидрокси или подходящую уходящую группу.
17. Способ по п.16, где уходящая группа представляет собой атом хлора.
18. Способ по п.13, где стадию (b) проводят в кислотных условиях в присутствии хлористо-водородной, трифторуксусной или метансульфоновой кислоты.
19. Способ по п.13, где стадию (d) проводят в основных условиях в присутствии гидроксида натрия, калия или лития или третичного амина, такого как триэтиламин.
20. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, определенных в п.10, и по меньшей мере один фармацевтически приемлемый наполнитель, носитель и/или разбавитель.
21. Фармацевтическая композиция по п.20, содержащая дополнительно один или несколько химиотерапевтических агентов.
22. Набор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, определенные в п.10, или их фармацевтические композиции, определенные в п.20, и один или несколько химиотерапевтических агентов в качестве комбинированного препарата для одновременного, раздельного или последовательного применения в противораковой терапии.
23. Применение соединения формулы (I) или его фармацевтически приемлемой соли, определенных в п.10, для получения лекарственного средства, обладающего противоопухолевой активностью.
Текст
009462 Область техники, к которой относится изобретение Данное изобретение относится к производным пирролопиразола, к способу их получения, к фармацевтическим композициям, содержащим эти соединения, и к применению их в качестве терапевтических агентов, особенно в лечении рака и нарушений пролиферации клеток. Уровень техники Нарушение функции протеинкиназ (PK) является признаком многочисленных заболеваний. Большая часть онкогенов и протоонкогенов, участвующих в развитии злокачественных опухолей человека,кодируют PK. PK причастны также ко многим доброкачественным заболеваниям, таким как доброкачественная гиперплазия простаты, семейный аденоматоз, полипоз, нейрофиброматоз, псориаз, пролиферация клеток гладких мышц сосудов, связанная с атеросклерозом, фиброз легких, артрит, гломерулонефрит, а также послеоперационный стеноз и рестеноз.PK причастны также к воспалительным состояниям и к размножению вирусов и паразитов. PKs также играют главную роль в патогенезе и развитии нейродегенеративных расстройств. В качестве основной ссылки о снижении активности PK или дисрегуляции см., например, CurrentOpinion in Chemical Biology, 1999, 3, 459-465. Среди некоторых известных в данной области протеинкиназ, участвующих в развитии раковых клеток, имеются Авроракиназы, в частности Аврора-2. Было установлено, что Аврора-2 избыточно экспрессируется в ряде различных типов опухолей. Ее генный локус картируется как 20q13, хромосомный регион, часто сверхэкспрессированный при многих видах рака, включая рак молочной железы [Cancer Res., 1999, 59(9), 2041-4] и толстой кишки. 20q13 амплификация коррелирует с неблагоприятным прогнозом у пациентов с узелковым негативным раком молочной железы, и повышенная экспрессия Авроры-2 является указанием на неблагоприятный прогноз и пониженное время жизни у пациентов с раком мочевого пузыря [J. Natl. Cancer Inst., 2002,94(17), 1320-9]. В качестве основной ссылки о роли Авроры-2 в анормальной функции центросом при раке см. также Molecular Cancer Therapeutics, 2003, 2, 589-595. Сущность изобретения Объект данного изобретения относится к соединениям, которые применимы в терапии в качестве агентов против носителя заболеваний, вызываемых и/или связанных с дисрегуляцией активности протеинкиназы и, более конкретно, активности Авроракиназ. Другой аспект относится к соединениям, которым свойственна ингибирующая активность в отношении протеинкиназы и, более конкретно, ингибирующая активность в отношении Авроракиназ. Авторы в настоящей работе открыли, что некоторым пирролопиразолам и их производным свойственна ингибирующая активность в отношении протеинкиназы, например ингибирующая активность в отношении Авроракиназ. Более конкретно, соединения данного изобретения применимы в лечении различных типов рака,включающих, но без ограничения только ими карциному, такую как карцинома мочевого пузыря, молочной железы, толстой кишки, почки, печени, легкого, включая мелкоклеточный рак легкого, пищевода, желчного пузыря, яичника, поджелудочной железы, желудка, шеи, щитовидной железы, простаты и кожи, включая плоскоклеточную карциному; гемопоэтические опухоли лимфоидного происхождения, включающие лейкоз, острый лимфоцитарный лейкоз, острый лимфобластомный лейкоз, лимфому В-клеток, лимфому Т-клеток, лимфому Ходжкина (Hodgkin's), лимфому не-Ходжкина, клеточную лимфому волос и лимфому Беркитта (Burkett's); гемопоэтические опухоли миелоидного происхождения, включающие острые и хронические миелогенные лейкозы, миелодиспастический синдром и промиелоцитарный лейкоз; опухоли мезенхимного происхождения, включающие фибросаркому и рабдомиосаркому; опухоли центральной и периферической нервной системы, включающие астроцитому, нейробластому, глиому и шванному; другие опухоли, включающие меланому, семиному, тератокарциному, остеосаркому, пигментозную ксеродерму, кератоксантому, фолликулярный рак щитовидной железы и саркому Капоши (Kaposi's). Из-за ключевой роли PKs и Авроракиназ в регулировании пролиферации клеток данные пирролопиразолы также применимы в лечении множества клеточных пролиферативных нарушений, таких как,например, доброкачественная гиперплазия простаты, семейный аденоматоз, полипоз, нейрофиброматоз,псориаз, пролиферация клеток гладких мышц сосудов, связанная с атеросклерозом, фиброз легких, артрит, гломерулонефрит и послеоперационный стеноз и рестеноз. Таким образом, в первом варианте осуществления данное изобретение относится к способу лечения клеточных пролиферативных нарушений, вызванных и/или связанных с измененной активностью протеинкиназы, который включает введение млекопитающему, нуждающемуся в этом, эффективного количества соединения формулы (I) в которой R представляет собой водород или метил;R1 представляет собой гидрокси либо линейный или разветвленный C1-C3 алкил или алкоксигруппу;R2 представляет собой водород или атом галогена;X представляет собой двухвалентную группу, выбранную из метилена (-CH2-) или фторметилена(-CHF-), либо X означает гетероатом или гетероатомную группу, выбранную из кислорода (-O-) или азота (-NR'-), где R' представляет собой атом водорода, линейную или разветвленную C1-C4 алкильную группу или C3-C6 циклоалкильную группу,или его фармацевтически приемлемой соли. Вышеприведенный способ позволяет лечить клеточные пролиферативные нарушения, вызванные и/или связанные с измененной активностью Авроракиназ. В предпочтительном варианте осуществления способа, описанного выше, клеточное пролиферативное нарушение представляет собой рак. Специфические типы рака, которые могут быть подвергнуты лечению, включают карциному, спиналиому, гемопоэтические опухоли миелоидного или лимфоидного происхождения, опухоли мезенхимного происхождения, опухоли центральной и периферической нервной системы, меланому, семиному,тератокарциному, остеосаркому, пигментозную ксеродерму, кератоксантому, фолликулярный рак щитовидной железы и саркому Капоши. Данное изобретение также относится к соединению формулы (I) в которой R представляет собой водород или метил;R1 представляет собой гидрокси или линейный или разветвленный C1-C3 алкил или алкоксигруппу;R2 представляет собой водород или атом галогена;X представляет собой двухвалентную группу, выбранную из метилена (-CH2-) или фторметилена(-CHF-), или X означает гетероатом или гетероатомную группу, выбранную из кислорода (-О-) или азота(-NR'-), где R' представляет собой атом водорода, линейную или разветвленную C1-C4 алкильную группу или C3-C6 циклоалкильную группу,или к его фармацевтически приемлемой соли. Данное изобретение также включает способы синтеза пирролопиразолов формулы (I) и фармацевтически приемлемых солей, а также фармацевтические композиции, содержащие их. Более полная оценка данного изобретения и его многие сопутствующие преимущества могут быть восприняты по мере лучшего понимания их с помощью отсылки к следующему подробному описанию. Подробное описание изобретения Некоторые гетероциклические соединения известны в данной области как ингибиторы протеинкиназы. Среди них 3-карбоксамидопиразолы и 3-уреидопиразолы и их производные раскрыты как ингибиторы протеинкиназы в международных патентных заявках WO 01/12189, WO 01/12188, WO 02/48114 иWO 02/70515, все они составлены от имени заявителя заявки на данное изобретение. Конденсированные бициклические соединения, включающие остаток пиразола и обладающие ингибирующей киназу активностью, также раскрыты в WO 00/69846, WO 02/12242 и в WO 03/028720, еще-2 009462 не опубликованной заявке PCT/ЕР 03/04862 (имеющей приоритет по дате подачи заявки на патент США 60/381092 от 17 мая 2002 г.), принадлежащей заявителю заявки на данное изобретение. В дополнение к вышеизложенному производные аминофенилпиперазина или аминофенилпиперидина, обладающие ингибирующей активностью в отношении пренилтрансферазных протеинов, раскрыты в WO 02/30927, принадлежащей Pierre Fabre Medicament. Соединения данного изобретения попадают в объем общей формулы вышеупомянутой заявкиWO 02/12242, введенной в данное описание в виде ссылки, но не представлены в ней конкретными примерами. Соединения формулы (I) данного изобретения имеют асимметричные атомы углерода и поэтому могут существовать как индивидуальные оптические изомеры, как рацемические смеси или в виде любой другой смеси, содержащей преобладающее количество одного из двух оптических изомеров, которые все подразумеваются в рамках данного изобретения. Кроме того, применение в качестве противоопухолевого агента всех возможных изомеров и их смесей, метаболитов и фармацевтически приемлемых биопредшественников (иначе называемых как пролекарства) соединений формулы (I) также входит в объем данного изобретения. Пролекарства представляют собой любые ковалентносвязанные соединения, которые высвобождают активное исходное лекарственное соединение формулы (I) in vivo. В случаях, когда соединения могут существовать в таутомерных формах, каждая форма предполагается включенной в данное изобретение независимо от того, существует ли в равновесии или в преобладающей одной форме. Таким образом, если не оговорено особо, в случаях, когда указана только одна из следующих таутомерных форм формулы (Ia) или (Ib), оставшаяся другая форма предполагается включенной в объем данного изобретения В данном описании, если не оговорено особо, под термином "линейный или разветвленный C1-C3 или C1-C4 алкил" авторы имеют в виду любую из групп, такую как, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и втор-бутил. Под термином "линейный или разветвленный C1-C3 алкокси" авторы имеют в виду любую из групп,такую как, например, метокси, этокси, н-пропокси и изопропокси. Под термином "атом галогена" авторы имеют в виду атом фтора, хлора, брома или йода. Под термином "C3-C6 циклоалкил" авторы имеют в виду любую из групп, такую как циклопропил,циклобутил, циклопентил или циклогексил. Очевидно, что в зависимости от природы группы X этот же гетероцикл, будучи присоединенным к фениленовому фрагменту соединений формулы (I), может представлять собой пиперидино,4-фторпиперидино, пиперазино, 4-алкилпиперазино, 4-циклоалкилпиперазино или морфолиноцикл. Фармацевтически приемлемые соли соединений формулы (I) включают кислотно-аддитивные соли с неорганическими или органическими кислотами, такими как, например, азотная, хлористо-водородная,бромисто-водородная, серная, перхлорная, фосфорная, уксусная, трифторуксусная, пропионовая, гликолевая, молочная, щавелевая, малоновая, яблочная, малеиновая, винная, лимонная, бензойная, коричная,миндальная, метансульфоновая, изэтионовая и салициловая кислоты. Предпочтительный класс соединений данного изобретения представлен производными формулы(I), в которой R означает водород или метил; R1 выбран из гидрокси, метила или метокси; R2 означает водород или атом фтора; X выбран из метилена, фторметилена, -О- или -NR', где R' принимает значения,данные выше. В качестве ссылки на любое определенное соединение формулы (I), необязательно в виде фармацевтически приемлемой соли, см. экспериментальную часть и формулу изобретения.-3 009462 Как указано ранее, следующий объект данного изобретения относится к способу получения соединений формулы (I) и их фармацевтически приемлемых солей, который включает: а) взаимодействие соединения формулы (II) с соединением формулы (III) где R и X принимают значения, определенные выше,Q представляет собой низшую алкильную группу,t-Bu означает трет-бутил иZ представляет собой гидрокси или подходящую уходящую группу,для получения соединения формулы (IV)b) превращение соединения формулы (IV) в кислотных условиях в соединение формулы (V) с) взаимодействие соединения формулы (V) с соединением формулы (VI) где R1 и R2 принимают значения, определенные выше,Z' представляет собой гидрокси или подходящую уходящую группу,для получения соединения формулы (VII)d) превращение соединения формулы (VII) в основных условиях в соответствующее соединение формулы (I) и, если желательно, превращение его в фармацевтически приемлемую соль. Способ, описанный выше, представляет собой способ-аналог, который может быть осуществлен проведением в очень хорошо известных методических условиях.-4 009462 В соответствии со стадией (а) способа взаимодействие между соединениями формул (II) и (III) может быть проведено различными путями согласно обычным способам ацилирования аминопроизводных. Как пример, соединение формулы (II) может быть подвергнуто взаимодействию с ацилхлоридом формулы (III), где Z означает атом хлора в качестве подходящей уходящей группы. Предпочтительно данную реакцию проводят при температуре, изменяющейся от комнатной температуры до примерно 60C, в подходящем растворителе, таком как, например, тетрагидрофуран или дихлорметан, и в присутствии акцептора протона, такого как триэтиламин или диизопропилэтиламин. В соединениях формулы (II) Q представляет собой низшую алкильную группу, напримерC1-C4 алкильную группу, более предпочтительно метил или этил. В соответствии со стадией (b) способа из соединения формулы (IV) может быть легко удалена защита у атома азота пирролидина при обработке кислотой. Данная реакция может быть удобно проведена в присутствии минеральной или органической кислоты, такой как, например, хлористо-водородная, трихлоруксусная или метансульфоновая кислота, в подходящем растворителе, таком как дихлорметан, 1,4-диоксан, низший спирт (например, метанол или этанол), при температуре, находящейся в интервале от комнатной температуры до примерно 40C, и в течение периода времени, изменяющегося примерно от 1 до примерно 48 ч. Соединение формулы (V), полученное таким образом, затем подвергается взаимодействию, согласно стадии (с) способа, с соединением формулы (VI). Из вышесказанного квалифицированному специалисту ясно, что данная реакция ацилирования может быть выполнена различными путями и в различных методических условиях, которые широко известны в данной области для приготовления карбоксамидов. Реакция между соединением формулы (V) и карбоновой кислотой формулы (VI), где Z' означает гидрокси, может быть проведена в присутствии конденсирующего агента, такого как, например,карбодиимид, т.е. 1,3-дициклогексилкарбодиимид, 1,3-диизопропилкарбодиимид, 1-(3-диметиламинопропил)-3-этилкарбодиимид,N-циклогексилкарбодиимид-N'-пропилоксиметилполистирол илиN,N-диметилформамид, при температуре, находящейся в интервале примерно от -10C до кипения с обратным холодильником, в течение подходящего периода времени, т.е. примерно от 30 мин до примерно 96 ч. Указанная реакция необязательно проводится в присутствии подходящего катализатора, например 4-диметиламинопиридина, или в присутствии дополнительного конденсирующего реагента, такого какN-гидроксибензотриазол. Реакция между соединением формулы (V) и соединением формулы (VI) может быть также проведена, например, методом смешанных ангидридов с применением алкилхлорформиата, такого как этил,изобутил или изопропилхлорформиат, в присутствии третичного основания, такого как триэтиламин,N,N-диизопропилэтиламин или пиридин, в подходящем растворителе, таком как, например, толуол,дихлорметан, хлороформ, тетрагидрофуран, ацетонитрил, диэтиловый эфир, 1,4-диоксан илиN,N-диметилформамид, при температуре, находящейся в интервале примерно от -30C до комнатной температуры. Реакция между соединением формулы (V) и карбоксильным производным формулы (VI), где Z' означает подходящую уходящую группу, может быть проведена в присутствии третичного основания, такого как триэтиламин, N,N-диизопропилэтиламин или пиридин, в подходящем растворителе, таком как толуол, дихлорметан, хлороформ, диэтиловый эфир, тетрагидрофуран, ацетонитрил или N,Nдиметилформамид, при температуре, находящейся в интервале примерно от -10C до кипения с обратным холодильником. Соединения формулы (V) характеризуются присутствием асимметричного атома углерода с присоединенным к нему R1, который ниже отмечен звездочкой Следовательно, соединения формулы (VI) могут быть либо в форме одного из энантиомеров, либо в виде их смесей, также многочисленных рацемических смесей. Ясно, что в зависимости от природы соединения формулы (VI), применяемого по способу данного изобретения, могли бы быть получены соответствующие соединения формулы (VII), имеющие надлежащую определенную стереохимию при том же самом атоме углерода. Согласно предпочтительному варианту осуществления данного изобретения стадию (с) проводят взаимодействием подходящего соединения формулы (VI), которое находится в заданной энантиомерной форме.-5 009462 Кроме того, если применяются рацемические смеси соединения формулы (VI), и всякий раз, когда желательно получить конечные соединения формулы (I) в оптически чистой форме, потребуется оптическое разделение промежуточного соединения формулы (VII) или, в альтернативном случае, конечного соединения формулы (I) с помощью обработки обычными способами. Как пример, обычные методики для разделения рацематов включают, например, раздельную кристаллизацию диастереомерных солей или препаративную хиральную ВЭЖХ (HPLC). Наконец, в соответствии со стадией (d) способа с соединения формулы (VII) удаляется защита при атоме азота пиразола обработкой обычными способами, приводящими, например, к селективному гидролизу карбаматной группы. Как пример, данная реакция может быть проведена в основных условиях, например в присутствии гидроксида натрия, калия или лития или третичного амина, такого как триэтиламин, и в подходящем растворителе, таком как N,N-диметилформамид, метанол, этанол, тетрагидрофуран, вода или их смеси. Обычно реакция проводится при температуре, находящейся в интервале от комнатной температуры до примерно 60C, в течение периода времени примерно от 30 мин до примерно 96 ч. Наконец, фармацевтически приемлемые соли соединений формулы (I) или, в альтернативном случае, свободные соединения из их солей могут быть получены традиционными способами. Исходные продукты способа данного изобретения известны или легко получаются известными способами. Как пример, получение соединения формулы (II), где Q представляет собой этил, раскрыто в вышеупомянутой международной патентной заявке WO 02/12242 (см., в частности, пример 26 на с. 249; то же самое соединение названо здесь как сложный 5-трет-бутиловый эфир, сложный 1-этиловый эфир 3 амино-4,6-дигидропирроло[3.4-с]пиразол-1,5-дикарбоновой кислоты). Аналогичным путем с таким же успехом могут быть приготовлены дополнительные соединения формулы (II), где Q представляет собой низшую алкильную группу, отличную от этила. Соединения формул (III) и (VI), например те, в которых Z и Z' представляют собой атом галогена,например атом хлора, являются либо известными, либо могут быть легко получены из соответствующих известных карбоновых кислот по методикам традиционных способов. Кроме того, квалифицированному специалисту в данной области должно быть ясно, что если соединение формулы (I), приготовленное по вышеприведенному способу с охватом любых его вариантов,получено в виде смеси изомеров, то их разделение на индивидуальные изомеры формулы (I), выполняемое по традиционным методикам, находится все же в объеме данного изобретения. Фармакология Соединения формулы (I) активны как ингибиторы протеинкиназы, более конкретно, как ингибиторы Авроракиназ и поэтому применимы, например, для ограничения неконтролируемой пролиферации опухолевых клеток. В терапии они могут быть использованы в лечении различных опухолей, таких как названные ранее, а также в лечении других клеточных пролиферативных нарушений, таких как псориаз, пролиферация клеток гладких мышц сосудов, связанная с атеросклерозом, и послеоперационный стеноз и рестеноз. Ингибирующая активность и эффективность выбранных соединений определены по способу анализа, основанному на применении SPA технологии (Amersham Pharmacia Biotech). Анализ включает перенос радиоактивно меченого фосфатного фрагмента киназой на биотинилированный субстрат. Образующемуся 33P-меченому биотинилированному продукту позволяют связаться со стрептавидин-покрытымиSPA гранулами (нагрузка биотина 130 пмоль/мг) и измеряют световое излучение сцинтилляционным счетчиком. Определение ингибирования активности Авроры-2. Киназная реакция: 8 мкМ биотинилированного пептида (4 повтора LRRWSLG), 10 мкМ АТФ(0,5 мкКи Р 33 у-АТР), 7,5 нг Авроры-2, ингибитор в конечном объеме, равном 30 мкл буфера (HEPES 50 мМ, рН 7,0, MgCl2 10 мМ, 1 мМ DTT, 0,2 мг/мл BSA, 3 мкМ ортованадат) добавляли в каждую ячейку 96-луночного планшета с U-образным дном. После 60-минутной инкубации при комнатной температуре реакцию прекращали и биотинилированный пептид поглощали добавлением 100 мкл суспензии гранул. Стратификация: 100 мкл CsCl2 5 М добавляли в каждую лунку и оставляли на 4 ч перед определением радиоактивности с помощью счетчика Top-Count. Определение IC50: ингибиторы испытывали при различных концентрациях, находящихся в интервале от 0,0015 до 10 мкМ. Экспериментальные данные анализировали с помощью компьютерной программы GraphPad Prizm с использованием четырехпараметрового логарифмического уравнения у = нижнее значение + (верхнее - нижнее)/(1 + 10 logIC50 - х)наклон,где х представляет собой логарифм концентрации ингибитора,у представляет собой ответ; у принимает значения от нижнего до верхнего по сигмоидному типу. Вычисление Ki: Экспериментальный способ: реакцию проводили в буфере (10 мМ Tris, рН 7,5, 10 мМ MgCl2,0,2 мг/мл BSA, 7,5 мМ DTT), содержащем 3,7 нМ фермента, гистон и АТФ (постоянное отношение не содержащий радиоактивности/меченый АТФ 1/3000). Реакцию прекращали с помощью ЭДТА и субстрат-6 009462 располагали на фосфомембране (96-луночные планшеты Multiscreen от Millipore). После интенсивного промывания планшеты оценивали на счетчике. Измеряли контроль для каждого АТФ (время нуль) и концентрации гистона. Моделирование опыта: скорости реакции измеряли при четырех концентрациях АТФ, субстрата(гистон) и ингибитора. Матрицу 80-точечной концентрации строили на основе соответствующих величин АТФ и Km величин субстрата и IC50 величин ингибитора (0,3, 1, 3, 9 указывали на Km или IC50 величины). Предварительный опыт по оценке временного режима в отсутствие ингибитора и при различных концентрациях АТФ и субстрата позволяет выбрать одно окончательное время (10 мин) в линейном диапазоне реакции для эксперимента по определению Ki. Оценка кинетических параметров: кинетические параметры оценивали совместной нелинейной регрессией методом наименьших квадратов, используя уравнение 1 (конкурентный ингибитор относительно АТФ, произвольный механизм) и полный набор данных (80 точек) где А = АТФ,В = субстрат,I = ингибитор,Vm = максимальная скорость,Ka, Kb, Ki - константы диссоциации АТФ, субстрата и ингибитора соответственно, и- кооперативный фактор связывания между субстратом и АТФ и связывания между субстратом и ингибитором соответственно. Соединения данного изобретения, кроме того, были испытаны in vitro для оценки антипролиферативного эффекта на культурах клеток и ингибирующего эффекта на клеточный цикл. Определение клеточной пролиферации in vitro. Клеточную линию HCT-116 рака толстой кишки человека высевали при 5000 кл./см 2 в 24-луночный планшет (Costar), используя среду F12 (Gibco), содержащую 10% FCS (EuroClone, Italy), 2 мМL-глутамин и 1% пенициллин/стрептомицин, и поддерживали при 37C, в 5% CO2 и при 96% относительной влажности. На следующий день планшеты обрабатывали дважды 5 мкл раствора соединений в соответствующем разбавлении, начиная от 10 мМ исходного образца в ДМСО. Две необработанные контрольные ячейки включали в каждый планшет. Через 72 ч после обработки среду удаляли и клетки извлекали из каждой ячейки, применяя 0,5 мл 0,05% (мас./об.) трипсина, 0,02% (мас./об.) ЭДТА (Gibco). Образцы разбавляли 9,5 мл изотона (Coulter) и снимали данные с помощью счетчика Multisizer 3 cell(Beckman Coulter). Данные представляли в виде процента от контрольных ячеек% CTR = (обработанный - чистый)/(контроль - чистый). Величины IC50 рассчитывали по методу LSW с применением Microsoft Excel аппроксимации сигмоидальной кривой. Определение in vitro клеточного цикла. Клеточную линию HCT-116 рака толстой кишки человека высевали при 5000 кл./см 2 в 24-луночный планшет (Costar) и культивировали, как отмечено выше. Клетки в их экспоненциальной фазе роста обрабатывали в течение 24 ч различными концентрациями соединения. Также собирали супернатант культуральной среды для того, чтобы избежать потери отделившихся апоптических или митотических клеток. После этого клетки промывали, применяя PBS, и отделяли 0,05% (мас./об.) трипсином, 0,02% (мас./об.) ЭДТА (Gibco). Активность трипсина прерывали использованием культуральной среды. Слитные и неслитные фракции клеток объединяли и центрифугировали при 2000 об/мин в течение 10 мин. Клетки ресуспендировали в PBS и снимали данные с помощью счетчика Multisizer 3 cell (Beckman Coulter). Для фиксации добавляли этанол (70%, об./об.) и клетки выдерживали в течение ночи при -20C. 1 млн фиксированных клеток центрифугировали при 2000 об/мин в течение 5 мин и промывали с помощью PBS и затем окрашивали в течение 1 ч при комнатной температуре в темноте добавлением 200 мкл: 25 мкг/мл пропидий йодид (Sigma) и 15 мкг/мл рибонуклеазы A (RNAse A, Sigma),0,001% (об./об.) Nonidet P40 (Sigma) в цитрате натрия (0,1% мас./об., рН 7,5). Образцы анализировали проточной цитометрией при 488 нм возбуждении (FACSCalibur, Beckton Dickinson) с программным обеспечением Cell Quest 3,0 (Beckton Dickinson). Обычно отбирали 10000 событий (активация двойных различных модулей DDM и образование только единичных клеток) и профили клеточного цикла фиксировали с помощью CellQuest (Verity Software). Распределение клеточного цикла популяции рассчитывали с помощью модифицированной модели в программном обеспечении Modfit 3.1 (Verity Software) и выражали в % G0/G1, S, G2/M и полиплоидии. По данным вышеприведенных анализов соединения формулы (I) изобретения обладали значительной ингибирующей активностью в отношении протеинкиназы, например ингибирующей активностью в отношении Авроры-2. В качестве примера в таблице представлены экспериментальные данные четырех типичных соединений изобретения, испытанных в качестве ингибиторов киназы Аврора-2 (IC50 нМ), по-7 009462 их клеточному антипролиферативному эффекту (IC50 нМ) и по их способности осуществлять блокирование клеточного цикла и вызывать полиплоидию (% G2/M + полиплоидия при 200 нМ). Интересно, что те же самые соединения были испытаны в сравнении со структурно очень близким соединением предшествующего уровня, определенным здесь как ссылочное соединение, которое, в частности, раскрыто в вышеупомянутой заявке WO 02/12242 - см. с. 160, строки 5-7; ссылочное соединение там названо как N-5-фенилацетил-4,6-дигидропирроло[3.4-с]пиразол-3-ил-4-(4-метилпиперазино)бензамид Неожиданно обнаружено, что полученная ингибирующая активность соединений данного изобретения в отношении Авроры-2 является постоянной и значительно превышающей подобную активность ссылочного соединения. Кроме того, те же самые соединения обладали клеточным антипролиферативным эффектом вместе со способностью блокировать клеточный цикл и вызывать полиплоидию, значительно превышая подобные эффекты ссылочного соединения, испытанного в тех же условиях. Из изложенного очевидно, что новые соединения формулы (I) данного изобретения обладают биологическим профилем, рассматриваемым как целое, который неожиданно превышает активность ближайшего предшествующего уровня по заявке WO 02/12242, и, следовательно, являются особенно предпочтительными в терапии против пролиферативных нарушений, связанных с измененной активностью киназы Аврора-2. Соединения данного изобретения могут быть введены как индивидуальные агенты или, альтернативно, в комбинации с известными противораковыми терапиями, такими как лучевая терапия, или как режим химиотерапии в комбинации с цитостатическими или цитотоксическими агентами, антибиотика-8 009462 ми, алкилирующими агентами, антиметаболитами, гормональными агентами, иммунологическими агентами, агентами типа интерферона, ингибиторами циклооксигеназы (например, ингибиторы COX-2),ингибиторами матриксметаллопротеазы, ингибиторами теломеразы, ингибиторами тирозинкиназы, агентами против рецептора фактора роста, анти-HER агентами, анти-EGFR агентами, агентами против развития кровеносных сосудов (например, ингибиторы ангиогенеза), ингибиторами фарнезилтрансферазы,ингибиторами ras-raf сигнал-трансдукторной системы клетки, ингибиторами клеточного цикла, другимиcdks ингибиторами, агентами, связывающими тубулин, ингибиторами топоизомеразы I, ингибиторами топоизомеразы II и т.п. В случае приготовления фиксированной дозы в таких комбинированных продуктах соединения данного изобретения используются в интервале дозировок, описанных ниже, и другое фармацевтически активное средство в интервале утвержденных дозировок. Соединения формулы (I) могут быть применены последовательно с известными противораковыми агентами, когда комбинированный препарат противопоказан. Соединения формулы (I) данного изобретения, подходящие для введения млекопитающим, например людям, могут быть введены обычными путями и уровень доз зависит от возраста, массы, состояний пациента и пути введения. Например, подходящая дозировка, принятая для перорального введения соединения формулы (I),может находиться в интервале примерно от 10 до примерно 500 мг на дозу, от 1 до 5 приемов в сутки. Соединения данного изобретения могут быть введены в различных лекарственных формах, например перорально в форме таблеток, капсул, сахарных таблеток или таблеток с пленочным покрытием, жидких растворов или суспензий; ректально в форме суппозиториев; парентерально, например внутримышечно,или путем внутривенной, и/или внутриоболочечной, и/или внутриспинальной инъекции или инфузии. Данное изобретение также включает фармацевтические композиции, содержащие соединение формулы (I) или его фармацевтически приемлемую соль в сочетании с фармацевтически приемлемым наполнителем, который может быть носителем или разбавителем. Фармацевтические композиции, содержащие соединения данного изобретения, обычно приготавливают следующими традиционными способами и вводят в подходящей фармацевтической форме. Например, твердые формы для перорального введения могут содержать вместе с активным соединением разбавители, например лактозу, декстрозосахарозу, сахарозу, целлюлозу, кукурузный или картофельный крахмал; смазывающие агенты, например двуокись кремния, тальк, стеариновую кислоту, стеарат магния или кальция и/или полиэтиленгликоли; связующие вещества, например крахмалы, аравийскую камедь, желатинированную метилцеллюлозу, карбоксиметилцеллюлозу или поливинилпирролидон; дезинтегрирующие агенты, например крахмал, альгиновую кислоту, альгинаты или натрийгликолат крахмала; вспенивающиеся смеси; красители; подсластители; смачивающие вещества, такие как лецитин, полисорбаты, лаурилсульфаты; в целом, нетоксичные и фармакологически неактивные вещества, используемые в фармацевтических препаратах. Данные фармацевтические препараты могут быть произведены известным способом, например посредством смешивания, гранулирования, таблетирования и способами покрытия сахарами и оболочкой. Жидкие дисперсии для перорального введения могут представлять собой, например, сиропы,эмульсии и суспензии. Как пример, сиропы могут содержать в качестве носителя сахарозу или сахарозу с глицерином и/или маннит и сорбит. Суспензии и эмульсии могут содержать в качестве примеров носителей природную смолу, агар,альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензии или растворы для внутримышечных инъекций могут содержать, наряду с активным соединением,фармацевтически приемлемый носитель, например стерильную воду, оливковое масло, этилолеат, гликоли, например пропиленгликоль, если желательно, подходящее количество гидрохлорида лидокаина. Растворы для внутривенных инъекций или инфузий могут содержать в качестве носителя стерильную воду или предпочтительно они могут находиться в виде стерильных, водных, изотонических, физиологических растворов или могут содержать пропиленгликоль как носитель. Суппозитории могут содержать, наряду с активным соединением, фармацевтически приемлемый носитель, например масло какао, полиэтиленгликоль, сурфактант, такой как сложный эфир жирной кислоты и полиоксиэтиленсорбитана, или лецитин. Ниже приведены следующие примеры для лучшей иллюстрации данного изобретения, но без ограничения только ими.-9 009462 Примеры Общие методы. Флэш-хроматографию выполняли на силикагеле (Merck grade 9385, 60 А). ВЭЖХ/МС (HPLC/MS) выполняли на колонке Waters X Terra RP 18 (4,650 мм, 3,5 мкм), используя систему ВЭЖХ Waters 2790,снабженную 996 Waters PDA детектором, и на Micromass mod. ZQ ординарном квадрупольном массспектрометре, снабженном электрораспылительным ионным источником (ESI). Подвижная фаза А представляла собой 5 мМ ацетатаммонийный буфер (рН 5,5, уксусная кислота/ацетонитрил 95:5),подвижная фаза В представляла собой H2O/ацетонитрил (5:95). Градиент от 10 до 90% В выдерживали 8 мин, 90% В - 2 мин. УФ-определение при 220 и 254 нм. Скорость потока 1 мл/мин. Объем пробы 10 мкл. Полное сканирование, интервал масс от 100 до 800 amu (атомные единицы массы). Капиллярное напряжение составляло 2,5 KB; источник температуры был при 120C; конус/пучок имел 10 В. Время удерживания (ВЭЖХ, комнатная температура) дано в минутах при 220 или 254 нм. Массы даны как отношение m/z. 1 Н-ЯМР спектроскопия выполнена на Mercury VX 400 при 400,45 МГц, снабженном 5 мм двойной резонансной пробой (1 Н 15N-31P ID PFG Varian). Пример 1. Получение 5-трет-бутил 1-этил 3-амино-4,6-дигидропирроло[3.4-с]пиразол-1,5 дикарбоксилата. Раствор этилхлоркарбоната (8,9 мл, 93 ммоль) в тетрагидрофуране (ТГФ, 250 мл) медленно добавляли к смеси трет-бутил 3-амино-4,6-дигидропирроло[3.4-с]пиразол-5(1 Н)-карбоксилата (20 г, 89 ммоль) и диизопропилэтиламина (DIEA, 92 мл, 528 ммоль) в ТГФ (500 мл) при 0-5C. Реакцию поддерживали при той же температуре в течение 2 ч, затем смеси позволяли достигнуть комнатной температуры и перемешивали в течение ночи. Полученную смесь упаривали в вакууме досуха и образовавшийся остаток экстрагировали этилацетатом (AcOEt) и водой. Органический слой отделяли, сушили над сульфатом натрия и упаривали досуха. Смесь очищали флэш-хроматографией (элюент: этилацетат/циклогексан от 4/6 до 7/3), получая 19 г указанного в заголовке соединения в виде белого твердого вещества [М+Н]+ 297. Пример 2. Получение 5-трет-бутил 1-этил 3-[4-(4-метилпиперазин-1-ил)бензоил]амино-4,6 дигидропирроло[3.4-с]пиразол-1,5-дикарбоксилата. Оксалилхлорид (23,2 мл, 265 ммоль) добавляли к суспензии 4-(4-метил-1-пиперазинил)бензойной кислоты (11,7 г, 53 ммоль) в дихлорметане (DCM, 320 мл) и диметилформамиде (ДМФА, 0,52 мл). После кипячения смеси с обратным холодильником в течение 6,5 ч летучие компоненты тщательно удаляли при пониженном давлении (при этом остаток три раза переводили в толуол). Образовавшийся дигидрохлорид 4-метилпиперазинобензоилхлорида добавляли порциями к раствору 5-трет-бутил 1-этил 3-амино-4,6-дигидропирроло[3.4-с]пиразол-1,5-дикарбоксилата (13,1 г,44,3 ммоль) в сухом ТГФ (620 мл) и DIEA (54,4 мл, 0,32 моль) при перемешивании при комнатной температуре. Полученную суспензию перемешивали 16 ч при комнатной температуре и 1 ч при 40C. После удаления растворителя при пониженном давлении остаток обрабатывали AcOEt (600 мл) и органический слой промывали водным карбонатом натрия (200 мл), насыщенным солевым раствором(200 мл) и сушили над сульфатом натрия. Растворитель выпаривали, остаток растирали в смеси диэтилового эфира (Et2O, 135 мл) и AcOEt(15 мл), фильтровали, сушили в вакууме при 40C, получая 20 г указанного в заголовке соединения в виде белого порошка [М+Н]+ 499. Аналогичным способом и при взаимодействии 5-трет-бутил 1-этил 3-амино-4,6-дигидропирроло[3.4-с]пиразол-1,5-дикарбоксилата с соответствующим ацилхлоридным производным получали следующие соединения: 5-трет-бутил 1-этил 3-[4-(4-этилпиперазин-1-ил)бензоил]амино-4,6-дигидропирроло[3.4-с]пиразол-1,5-дикарбоксилат, [М+Н]+ 513,5-трет-бутил 1-этил 3-[4-(4-изопропилпиперазин-1-ил)бензоил]амино-4,6-дигидропирроло[3.4(5)с]пиразол-1,5-дикарбоксилат, [М+Н]+ 527,5-трет-бутил 1-этил 3-[4-(4-циклопропилпиперазин-1-ил)бензоил]амино-4,6-дигидропирроло[3.4 с]пиразол-1,5-дикарбоксилат, [М+Н]+ 525,5-трет-бутил 1-этил 3-[4-(3,4-диметилпиперазин-1-ил)бензоил]амино-4,6-дигидропирроло[3.4-с]пиразол-1,5-дикарбоксилат, [М+Н]+ 513,5-трет-бутил 1-этил 3-[(4-пиперидин-1-илбензоил)амино]-4,6-дигидропирроло[3.4-с]пиразол-1,5 дикарбоксилат, [М+Н]+ 484,5-трет-бутил 1-этил 3-[4-(4-фторпиперидин-1-ил)бензоил]амино-4,6-дигидропирроло[3.4-с]пиразол-1,5-дикарбоксилат, [М+Н]+ 502,5-трет-бутил 1-этил 3-[(4-морфолин-4-илбензоил)амино]-4,6-дигидропирроло [3.4-с]пиразол-1,5 дикарбоксилат, [М+Н]+ 486,- 10009462 5-трет-бутил 1-этил 3-[4-(4-трет-бутилпиперазин-1-ил)бензоил]амино-4,6-дигидропирроло[3.4(5)с]пиразол-1,5-дикарбоксилат, [М+Н]+ 541. Пример 3. Получение тригидрохлорида этил 3-[4-(4-метилпиперазин-1-ил)бензоил]амино-5,6 дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилата. 4 н. раствор хлористо-водородной кислоты в диоксане (122 мл, 488 ммоль) добавляли по каплям к перемешиваемому раствору 5-трет-бутил 1-этил 3-[4-(4-метилпиперазин-1-ил)бензоил]амино-4,6 дигидропирроло[3.4-с]пиразол-1,5-дикарбоксилата (19,5 г, 39,2 ммоль), полученному в примере 2, в сухом DCM (240 мл); при этом почти сразу происходило осаждение белого твердого вещества. Полученную смесь перемешивали при комнатной температуре в течение 24 ч; после разбавления эфиром Et2O(100 мл) твердое вещество фильтровали, интенсивно промывали Et2O и сушили в вакууме при 50C, получая 20,1 г указанного в заголовке соединения, использованного на следующей стадии без дальнейшей очистки, [М+Н]+ 399. 1H-ЯМР (ДМСО-d6)м.д.: 1,4 (т, 3H); 2,8 (д, 3H); 3,2 (м, 4 Н); 3,5 (м, 2 Н); 4,1 (м, 2 Н); 4,4 (кв., 2 Н); 4,6 (м, 4 Н); 7,1-8,0 (м, 4 Н); 10,3 (ушир.с, 2 Н); 10,7 (ушир.с, 1 Н); 11,4 (с, 1 Н). Следуя вышеописанной методике и исходя из соответствующего промежуточного продукта, аналогично получали следующие соединения: тригидрохлорид этил 3-[4-(4-этилпиперазин-1-ил)бензоил]амино-5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилата, [М+Н]+ 413,тригидрохлорид этил 3-[4-(4-изопропилпиперазин-1-ил)бензоил]амино-5,6-дигидропирроло[3.4 с]пиразол-1(4 Н)-карбоксилата, [М+Н]+ 427,тригидрохлорид этил 3-[4-(4-циклопропилпиперазин-1-ил)бензоил]амино-5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилата, [М+Н]+ 425,тригидрохлорид этил 3-[4-(3,4-диметилпиперазин-1-ил)бензоил]амино-5,6-дигидропирроло[3.4 с]пиразол-1(4 Н)-карбоксилата, [М+Н]+ 413,этил 3-[(4-пиперидин-1-илбензоил)амино]-5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат,[М+Н]+ 384,тригидрохлорид этил 3-[4-(4-фторпиперидин-1-ил)бензоил]амино-5,6-дигидропирроло[3.4 с]пиразол-1(4 Н)-карбоксилата, [М+Н]+ 402,тригидрохлорид этил 3-[4-(4-морфолин-4-ил)бензоил]амино-5,6-дигидропирроло[3.4-с]пиразол 1(4 Н)-карбоксилата, [М+Н]+ 386,тригидрохлорид этил 3-[4-(4-трет-бутилпиперазин-1-ил)бензоил]амино-5,6-дигидропирроло[3.4 с]пиразол-1(4 Н)-карбоксилата, [М+Н]+ 441. Пример 4. Получение этил 5-[(2R)-2-метокси-2-фенилэатаноил]-3-[4-(4-метилпиперазин-1 ил)бензоил]амино-5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилата. Оксалилхлорид (10,12 мл, 115 ммоль) добавляли к раствору R-(-)метоксифенилуксусной кислоты (1,91 г, 11,5 ммоль) в DCM (90 мл) и ДМФА (0,50 мл). После перемешивания смеси при комнатной температуре в течение 16 ч летучие компоненты тщательно отгоняли при пониженном давлении. Раствор полученного R-(-)метоксифенилацетилхлорида в DCM (20 мл) добавляли по каплям к раствору тригидрохлорида этил 3-[4-(4-метилпиперазин-1-ил)бензоил]амино-5,6-дигидропирроло[3.4 с]пиразол-1(4 Н)-карбоксилата (4,5 г, 8,9 ммоль) в DCM (400 мл) и DIEA (11,8 мл, 69 ммоль) при перемешивании при комнатной температуре. Образовавшийся раствор перемешивали в течение 20 ч при комнатной температуре. Реакционную смесь затем промывали водным карбонатом натрия (200 мл), насыщенным солевым раствором (200 мл) и сушили над сульфатом натрия. Растворитель выпаривали и остаток растирали в смеси Et2O (100 мл) и AcOEt (10 мл), фильтровали и сушили в вакууме при 40C, получая 3,94 г указанного в заголовке соединения в виде белого порошка, использованного на следующей стадии без дополнительной очистки, [М+Н]+ 547. 1H-ЯМР (ДМСО-d6)м.д.: 1,3 (т, 3H); 2,3 (д, 3H); 2,6 (м, 4 Н); 3,3-3,4 (м, 7 Н); 4,3 (кв., 2 Н); 4,6-4,9 (м,4 Н); 5,1 (д, 1 Н); 7,0-8,0 (м, 9 Н); 11,1 (д, 1 Н). Следуя вышеописанной методике и исходя из соответствующего ацилхлоридного производного,аналогично получали следующие соединения: этил 5-[(2R)-2-метокси-2-фенилэтаноил]-3-[4-(4-этилпиперазин-1-ил)бензоил]амино-5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 561,этил 5-[(2R)-2-метокси-2-фенилэтаноил]-3-[4-(4-изопропилпиперазин-1-ил)бензоил]амино-5,6 дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 575,этил 5-[(2R)-2-метокси-2-фенилэтаноил]-3-[4-(4-циклопропилпиперазин-1-ил)бензоил]амино-5,6 дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 573,этил 3-[4-(3,4-диметилпиперазин-1-ил)бензоил]амино-5-[(2R)-2-метокси-2-фенилэтаноил]-5,6 дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 561,этил 5-[(2R)-2-метокси-2-фенилэтаноил]-3-[(4-пиперидин-1-илбензоил)амино]-5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 532,- 11009462 этил 3-[4-(4-фторпиперидин-1-ил)бензоил]амино-5-[(2R)-2-метокси-2-фенилэтаноил]-5,6 дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 550,этил 5-[(2R)-2-метокси-2-фенилэтаноил]-3-[(4-морфолин-4-илбензоил)амино]-5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 534,этил 3-[4-(4-трет-бутилпиперазин-1-ил)бензоил]амино-5-[(2R)-2-метокси-2-фенилэтаноил]-5,6 дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 589,этил 5-[(2R)-2-метил-2-фенилэтаноил]-3-[4-(4-этилпиперазин-1-ил)бензоил]амино-5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 545,этил 5-[(2R)-2-метил-2-фенилэтаноил]-3-[4-(4-изопропилпиперазин-1-ил)бензоил]амино-5,6 дигидропирроло [3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 559,этил 5-[(2R)-2-метил-2-фенилэтаноил]-3-[4-(4-циклопропилпиперазин-1-ил)бензоил]амино-5,6 дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 557,этил 3-[4-(3,4-диметилпиперазин-1-ил)бензоил]амино-5-[(2R)-2-метил-2-фенилэтаноил]-5,6 дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 545,этил 5-[(2R)-2-метил-2-фенилэтаноил]-3-[(4-пиперидин-1-илбензоил)амино]-5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 516,этил 3-[4-(4-фторпиперидин-1-ил)бензоил]амино-5-[(2R)-2-метил-2-фенилэтаноил]-5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 534,этил 5-[(2R)-2-метил-2-фенилэтаноил]-3-[(4-морфолин-4-илбензоил)амино]-5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 518,этил 3-[4-(4-трет-бутилпиперазин-1-ил)бензоил]амино-5-[(2R)-2-метил-2-фенилэтаноил]-5,6 дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 573,этил 5-[(2R)-2-гидрокси-2-фенилэтаноил]-3-[4-(4-метилпиперазин-1-ил)бензоил]амино-5,6 дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилат, [М+Н]+ 533. Пример 5. Получение N-5-[(2R)-2-метокси-2-фенилэтаноил]-1,4,5,6-тетрагидропирроло[3.4-с]пиразол-3-ил-4-(4-метилпиперазин-1-ил)бензамида (1). Раствор этил 5-[(2R)-2-метокси-2-фенилэтаноил]-3-[4-(4-метилпиперазин-1-ил)бензоил]амино 5,6-дигидропирроло[3.4-с]пиразол-1(4 Н)-карбоксилата (3,94 г, 7,2 ммоль) в метаноле (МеОН, 130 мл) и триэтиламине (Et3N, 13 мл) перемешивали при комнатной температуре в течение 16 ч (выделялось некоторое количество осадка). Твердое вещество отделяли и промывали эфиром Et2O, получая 1,6 г указанного в заголовке соединения. Раствор упаривали до нескольких миллилитров и отделяли вторую порцию твердого продукта (1,62 г). Обе порции объединяли и анализировали с помощью ЖХ-МС (LC-MS) (чистота около 90% при 254 и 220 нМ). После хроматографической очистки (короткая колонка с силикагелем, DCM/MeOH 45:5) получали 2,83 г (83%) указанного в заголовке соединения в виде белого твердого вещества. Т.пл. 289C; [М+Н]+ 475. 1H-ЯМР (ДМСО-d6)м.д.: 2,21 (с, 3H); 2,43 (м, 4 Н); 3,29 (м, 7 Н); 4,20-4,90 (м, 4 Н); 5,09 (с, 1 Н); 6,80-8,00 (м, 9 Н); 10,6 (ушир., 1 Н); 12,09 (ушир., 1 Н). Аналогичным способом через гидролиз соединений примера 4 в присутствии основания получены следующие соединения:(19) 4-(4-трет-бутилпиперазин-1-ил)-N-5-[(2R)-2-фенилпропаноил]-1,4,5,6-тетрагидропирроло[3.4 с]пиразол-3-илбензамид, [М+Н]+ 501. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ лечения клеточных пролиферативных нарушений, вызванных и/или связанных с измененной активностью протеинкиназы, включающий введение млекопитающему, нуждающемуся в этом,эффективного количества соединения формулы (I) в которой R представляет собой водород или метил;R1 представляет собой гидрокси или линейный или разветвленный C1-C3 алкил илиR2 представляет собой водород или атом галогена;X представляет собой двухвалентную группу, выбранную из метилена (-CH2-) или фторметилена(-CHF-), или X означает гетероатом или гетероатомную группу, выбранную из кислорода (-O-) или азота(-NR'-), где R' представляет собой атом водорода, линейную или разветвленную C1-C4aлкильную группу или C3-С 6 циклоалкильную группу; или его фармацевтически приемлемой соли. 2. Способ по п.1 лечения клеточных пролиферативных нарушений, вызванных и/или связанных с измененной активностью Авроракиназ. 3. Способ по п.2, где Авроракиназа представляет собой Аврору-2. 4. Способ по п.1, где клеточные пролиферативные нарушения выбраны из группы, включающей рак, болезнь Альцгеймера, вирусные инфекции, аутоиммунные заболевания и нейродегенеративные расстройства. 5. Способ по п.4, где рак выбран из группы, включающей карциному, плоскоклеточную карциному,гемопоэтические опухоли миелоидного или лимфоидного происхождения, опухоли мезенхимного происхождения, опухоли центральной и периферической нервной системы, меланому, семиному, тератокарциному, остеосаркому, пигментозную ксеродерму, кератоксантому, фолликулярный рак щитовидной железы и саркому Капоши. 6. Способ по п.1, где клеточное пролиферативное нарушение выбрано из группы, включающей доброкачественную гиперплазию простаты, семейный аденоматозный полипоз, нейрофиброматоз, псориаз,пролиферацию клеток гладких мышц сосудов, связанную с атеросклерозом, фиброз легких, артрит, гломерулонефрит и послеоперационный стеноз и рестеноз. 7. Способ по п.1, дополнительно включающий воздействие на млекопитающего, нуждающегося в этом, лучевой терапии или химиотерапевтической схемы в комбинации по меньшей мере с одним цитостатическим или цитотоксическим агентом. 8. Способ по п.1, где млекопитающее, нуждающееся в нем, представляет собой человека. 9. Способ по п.2, где происходит ингибирование Аврора-2-киназы. 10. Соединение формулы (I) в которой R представляет собой водород или метил;R1 представляет собой гидрокси или линейный или разветвленный C1-C3 алкил илиR2 представляет собой водород или атом галогена;X представляет собой двухвалентную группу, выбранную из метилена (-CH2-) или фторметилена(-CHF-), или X означает гетероатом или гетероатомную группу, выбранную из кислорода (-O-) или азота(-NR'-), где R' представляет собой атом водорода, линейную или разветвленную C1-C4 алкильную группу или C3-C6 циклоалкильную группу; или его фармацевтически приемлемая соль. 11. Соединение формулы (I) по п.10, где R означает водород или метил; R1 выбран из гидрокси, метила или метокси; R2 означает водород или атом фтора; X выбран из метилена, фторметилена, -О- или NR', где R' принимает значения, определенные в п.10. 12. Соединение формулы (I) по п.10, необязательно в виде его фармацевтически приемлемой соли,выбранное из группы, включающей:(19) 4-(4-трет-бутилпиперазин-1-ил)-N-5-[(2R)-2-фенилпропаноил]-1,4,5,6-тетрагидропирроло[3.4 с]пиразол-3-илбензамид. 13. Способ получения соединений формулы (I) или их фармацевтически приемлемых солей по п.10,включающий: а) взаимодействие соединения формулы (II) с соединением формулы (III) где R и X принимают значения, определенные в п.10,Q представляет собой низшую алкильную группу,t-Bu означает трет-бутил иZ представляет собой гидрокси или подходящую уходящую группу,для получения соединения формулы (IV)b) превращение соединения формулы (IV) в кислотных условиях в соединение формулы (V) с) взаимодействие соединения формулы (V) с соединением формулы (VI) где R1 и R2 принимают значения, определенные в п.10, и Z' представляет собой гидрокси или подходящую уходящую группу,для получения соединения формулы (VII)d) превращение соединения формулы (VII) в основных условиях в соответствующее соединение формулы (I) и, если желательно, превращение его в фармацевтически приемлемую соль. 14. Способ по п.13, где в соединениях формулы (II) Q представляет собой линейную или разветвленную C1-C4 алкильную группу. 15. Способ по п.14, где Q представляет собой метил или этил. 16. Способ по п.13, где в соединениях формул (III) и (VI) Z и Z' представляют собой, каждый независимо, гидрокси или подходящую уходящую группу. 17. Способ по п.16, где уходящая группа представляет собой атом хлора. 18. Способ по п.13, где стадию (b) проводят в кислотных условиях в присутствии хлористоводородной, трифторуксусной или метансульфоновой кислоты. 19. Способ по п.13, где стадию (d) проводят в основных условиях в присутствии гидроксида натрия,калия или лития или третичного амина, такого как триэтиламин. 20. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, определенных в п.10, и по меньшей мере один фармацевтически приемлемый наполнитель, носитель и/или разбавитель. 21. Фармацевтическая композиция по п.20, содержащая дополнительно один или несколько химиотерапевтических агентов. 22. Набор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, определенные в п.10, или их фармацевтические композиции, определенные в п.20, и один или несколько химиотерапевтических агентов в качестве комбинированного препарата для одновременного, раздельного или последовательного применения в противораковой терапии. 23. Применение соединения формулы (I) или его фармацевтически приемлемой соли, определенных в п.10, для получения лекарственного средства, обладающего противоопухолевой активностью.
МПК / Метки
МПК: C07D 487/04, A61P 35/00, A61K 31/4162
Метки: активностью, пиразола, пирроло, 3.4-с, киназы, обладающие, производные, ингибиторы
Код ссылки
<a href="https://eas.patents.su/17-9462-proizvodnye-pirrolo-34-s-pirazola-obladayushhie-aktivnostyu-kak-ingibitory-kinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пирроло [3.4-с] пиразола, обладающие активностью как ингибиторы киназы</a>
Производные хиноксалин-2,3-диона и их фармацевтически приемлемые соли, фармацевтические композиции, обладающие антагонистической активностью в отношении рецептора глютамата и противосудорожной активностью, способы лечения пациентов, страдающих от удара или заболеваний, при помощи этих соединений.

Номер патента: 762
Опубликовано: 24.04.2000
Авторы: Корнберг Брайэн Эдвард, Никам Шам, Рафферти Майкл Фрэнсис
МПК: C07D 241/44
Метки: фармацевтически, пациентов, отношении, производные, приемлемые, этих, заболеваний, помощи, рецептора, обладающие, композиции, противосудорожной, способы, хиноксалин-2,3-диона, лечения, страдающих, антагонистической, соли, соединений, глютамата, фармацевтические, активностью, удара
Формула / Реферат:
1. Производные хиноксалин-2,3-диона общей формулы I где R - группа - NR4R5, где R4 и R5 независимо друг от друга означают водород, низший алкил, незамещенный или замещенный низшим алкилом, низший циклоалкил, который может содержать 1 или 2 атома кислорода в качестве гетероатома, R1 и R2 независимо друг от друга означают водород или нитрогруппу, R3 - низший алкил, при этом радикалы R3 и R-CH2- могут каждый находиться в положениях 5 или...
Производные камптотецина, обладающие противоопухолевой активностью

Номер патента: 3605
Опубликовано: 26.06.2003
Авторы: Пенко Серджо, Карминати Паоло, Цунино Франко, Мерлини Лючио
МПК: A61K 31/435, C07D 491/22
Метки: камптотецина, противоопухолевой, активностью, производные, обладающие
Формула / Реферат:
1. Соединения формулы I в которой R1 обозначает группу -C(R5)=N-O(n)R4, в которой R4 обозначает нормальный или разветвленный C1-C8алкил, нормальную или разветвленную C1-C8алкенильную группу, C3-C10циклоалкил, (C3-C10)циклоалкилнормальный или разветвленный (C1-C8)алкил, (C6-C14)арил, (C6-C14)арилнормальный или разветвленный (C1-C6)алкил, гетероциклическую группу или гетероциклонормальный или разветвленный-(C1-C8)алкил, где гетероциклическая...
Тиольные производные, обладающие ингибирующей активностью в отношении металлопептидаз.

Номер патента: 991
Опубликовано: 28.08.2000
Авторы: Семераро Клаудио, Романьяно Стефано, Норчини Габриеле, Фантуччи Марио, Пеллачини Франко, Сантанджело Франческо
МПК: C07D 277/30, A61K 31/425, A61P 9/00...
Метки: обладающие, активностью, отношении, ингибирующей, производные, тиольные, металлопептидаз
Формула / Реферат:
1. Соединение формулы где R обозначает меркаптогруппу или группу R4COS, которая превращается в организме в меркаптогруппу; R1 обозначает С2-С4алкильную группу с прямой или разветвленной цепью или арильную или арилалкильную группу, имеющую от 1 до 6 атомов углерода в алкильном фрагменте, где арил обозначает фенил или 5- или 6-членный ароматический гетероцикл с одним или двумя гетероатомами, выбранными из группы, включающей азот, кислород и...
Производные азобициклооктанов и нонанов обладающие активностью в ингибировании dpp – iv

Номер патента: 8166
Опубликовано: 27.04.2007
Авторы: Араньи Петер, Урбан-Сабо Каталин, Т.Надь Лайош, Варга Мартон, Бата Имре, Балаж Ласло, Капуи Зольтан, Боронкаи Эва, Шушан Эдит, Батори Шандор, Сабо Тибор
МПК: A61P 3/10, C07D 207/06, A61K 31/46...
Метки: обладающие, активностью, производные, азобициклооктанов, нонанов, ингибировании
Формула / Реферат:
1. Соединения общей формулы (I) в которой R означает азотсодержащий ароматический фрагмент с одним или двумя циклами, состоящий из одного или двух ароматических циклов, предпочтительно пиридила, пиридазинила, пиримидинила, пиразинила, имидазолила, пиразолила, тиазолила, изотиазолила, оксазолила, изоксазолила, оксадиазолила, хинолинила, изохинолинила, циннолинила, фталазинила, хиназолинила, хиноксалинила, бензимидазолила, индазолила,...
Производные фосфоновой кислоты, обладающие ингибирующей активностью в отношении металлопептидаз

Номер патента: 743
Опубликовано: 28.02.2000
Авторы: Сантанджело Франческо, Норчини Габриеле, Ботта Даниела
МПК: C07K 5/062, A61K 38/05
Метки: фосфоновой, обладающие, отношении, кислоты, активностью, ингибирующей, производные, металлопептидаз
Формула / Реферат:
1. Соединение формулы где R обозначает С1-С6алкильную группу с прямой или с разветвленной цепью, необязательно замещенную одним или несколькими атомами фтора, арильную или арилалкильную группу с 1-6 атомами углерода в алкильном фрагменте, где арил обозначает фенильную, 1-нафтильную или 2-нафтильную группу или 5- или 6-членный ароматический гетероцикл с 1 или 2 гетероатомами, выбранными из группы, включающей азот, кислород и серу,...
Предыдущий патент: Усовершенствованный способ приготовления и очистки винилароматических мономеров
Следующий патент: Применение эритропоэтина
Случайный патент: Усовершенствования в области многофункциональных систем отождествления