Антагонисты ccr1 для лечения воспалительных артритов, демиелинизационных воспалительных заболеваний
Номер патента: 7748
Опубликовано: 29.12.2006
Авторы: Харриман Джеральдин Си.Би., Гош Шомир, Карсон Кеннет Дж.














Формула / Реферат
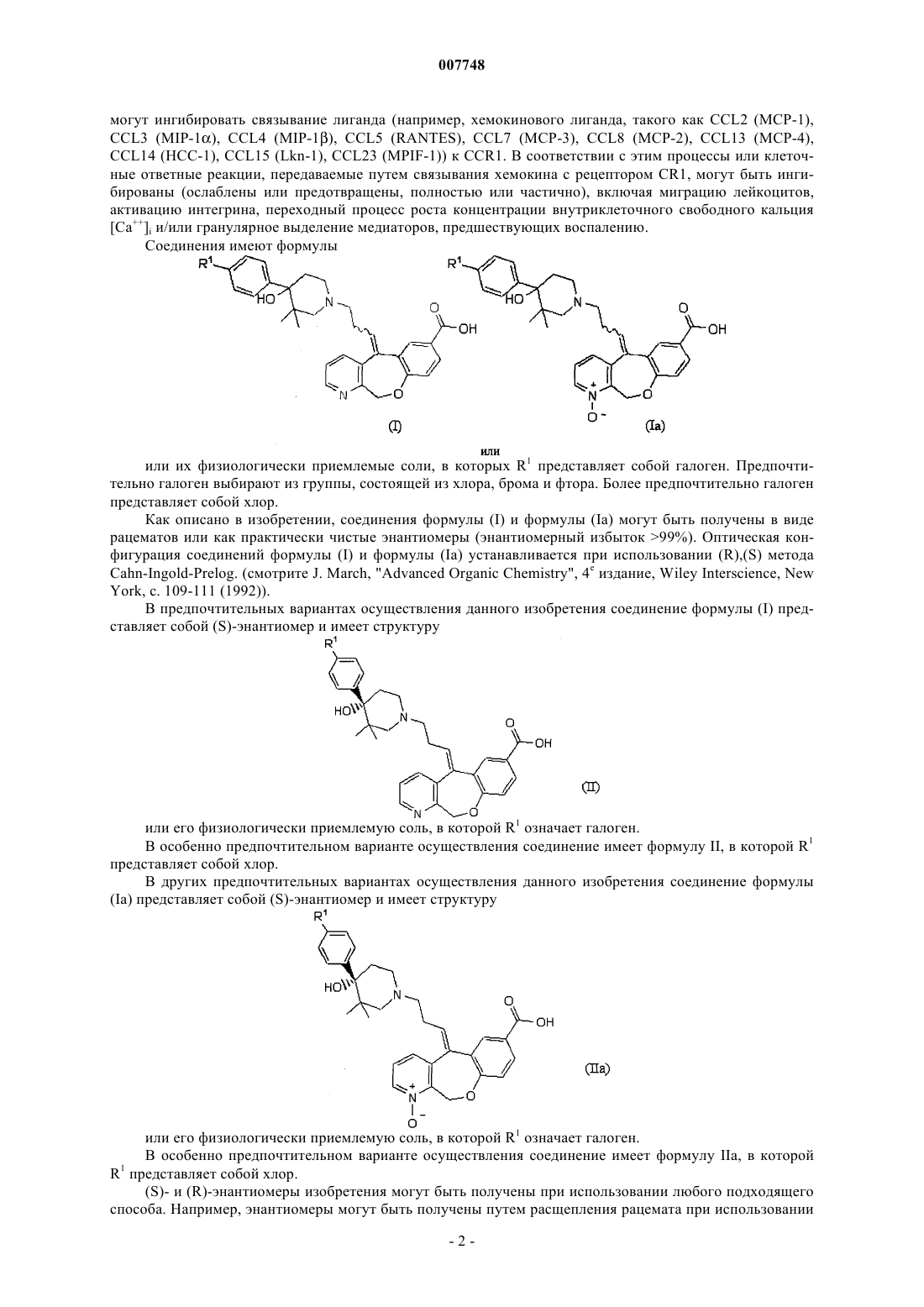
1. Соединение, имеющее формулу

или его физиологически приемлемая соль, в котором R1 означает галоген.
2. Соединение по п.1, в котором R1 выбирают из группы, состоящей из хлора, фтора и брома.
3. Соединение по п.2, в котором R1 представляет собой хлор.
4. Фармацевтическая композиция, содержащая соединение по п.1 и физиологически приемлемый носитель или наполнитель.
5. Способ лечения заболевания, характеризующегося патогенным восстановлением лейкоцитов, патогенной активацией лейкоцитов или патогенным восстановлением и активацией лейкоцитов, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения по п.1.
6. Соединение, имеющее структуру

или его физиологически приемлемая соль, в котором R1 означает галоген.
7. Соединение по п.6, в котором R1 выбирают из группы, состоящей из хлора, фтора и брома.
8. Соединение по п.7, в котором R1 представляет собой хлор.
9. Фармацевтическая композиция, содержащая соединение по п.6 и физиологически приемлемый носитель или наполнитель.
10. Способ лечения заболевания, характеризующегося патогенным восстановлением лейкоцитов, патогенной активацией лейкоцитов или патогенным восстановлением и активацией лейкоцитов, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения по п.6.
11. Способ по п.10, в котором R1 представляет собой хлор.
12. Способ по п.10, в котором указанное заболевание является острым или хроническим воспалительным заболеванием.
13. Способ по п.10, в котором указанное заболевание выбирают из группы, состоящей из воспалительного артрита, воспалительного демиелинизационного заболевания, атеросклероза, артериосклероза, рестеноза, ишемического/реперфузионного повреждения, сахарного диабета, псориаза, воспалительных заболеваний кишечника, отторжения трансплантированных органов, реакции трансплантата против хозяина, аллергии и астмы.
14. Способ по п.13, в котором указанное заболевание является воспалительным артритом.
15. Способ по п.14, в котором указанный воспалительный артрит является ревматоидным артритом.
16. Способ по п.13, в котором указанное заболевание является воспалительным демиелинизационным заболеванием.
17. Способ по п.16, в котором указанное воспалительное демиелинизационное заболевание является рассеянным склерозом.
18. Способ антагонизации рецептора 1 С-С-хемокина у субъекта, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения по п.1.
19. Соединение, имеющее формулу

или его физиологически приемлемая соль, в котором R1 представляет собой галоген.
20. Фармацевтическая композиция, содержащая соединение по п.19 и физиологически приемлемый носитель или наполнитель.
21. Способ лечения заболевания, характеризующегося патогенным восстановлением лейкоцитов, патогенной активацией лейкоцитов или патогенным восстановлением и активацией лейкоцитов, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения по п.19.
22. Способ по п.21, в котором указанное заболевание выбирают из группы, состоящей из воспалительного артрита, воспалительного демиелинизационного заболевания, атеросклероза, артериосклероза, рестеноза, ишемического/реперфузионного повреждения, сахарного диабета, псориаза, воспалительных заболеваний кишечника, отторжения трансплантированных органов, реакции трансплантата против хозяина, аллергии и астмы.
23. Способ лечения острого или хронического воспалительного заболевания, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения по п.19.
24. Способ по п.23, в котором указанное заболевание является воспалительным артритом.
25. Способ по п.24, в котором указанный воспалительный артрит является ревматоидным артритом.
26. Способ по п.23, в котором указанное заболевание является воспалительным демиелинизационным заболеванием.
27. Способ по п.26, в котором указанное воспалительное демиелинизационное заболевание является рассеянным склерозом.
28. Способ антагонизации рецептора 1 С-С-хемокина у субъекта, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения по п.19.
29. Соединение по любому из пп.1-3 или пп.6-8, применяемое в терапии или диагностике.
30. Соединение по п.19, применяемое в терапии или диагностике.
Текст
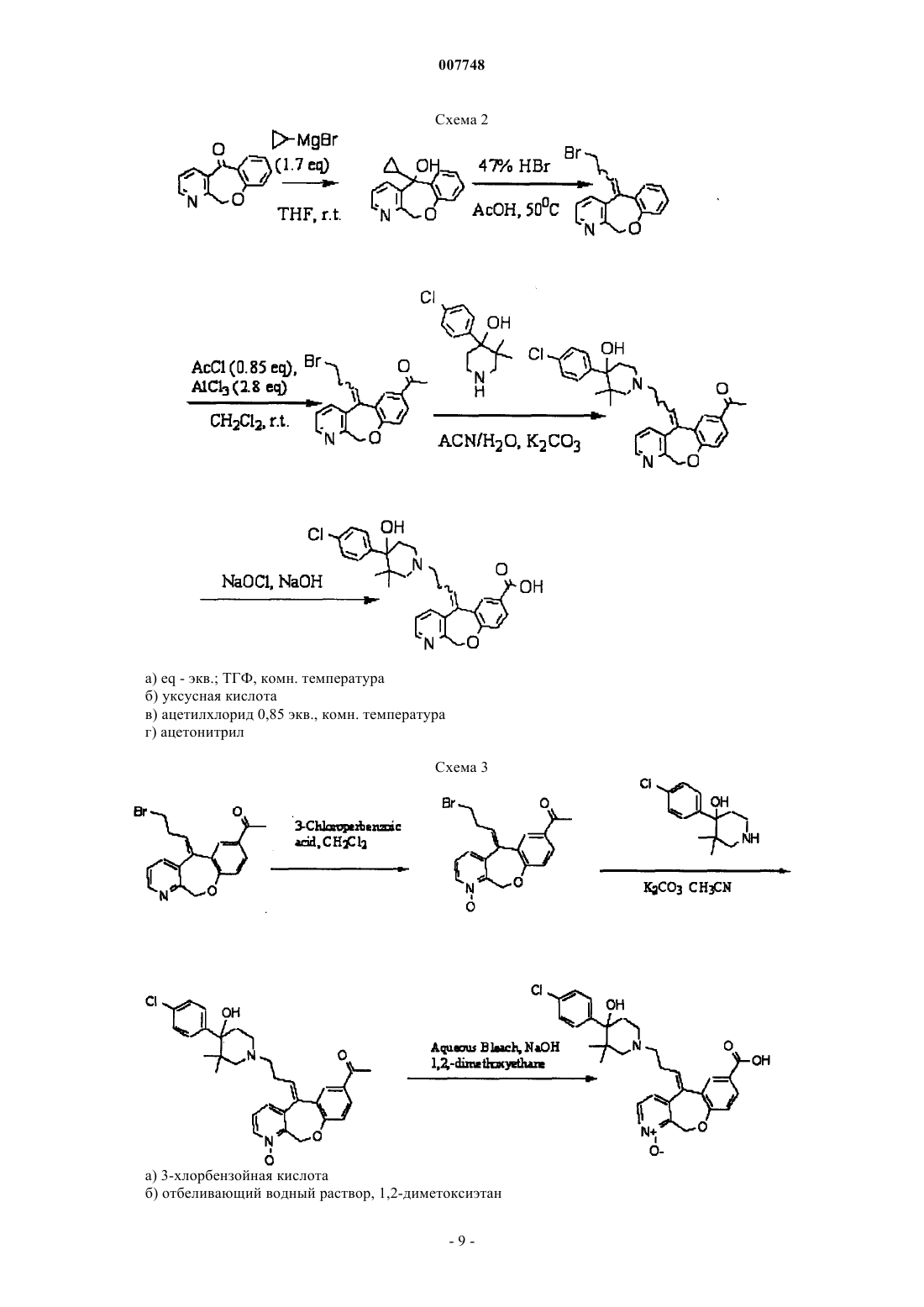
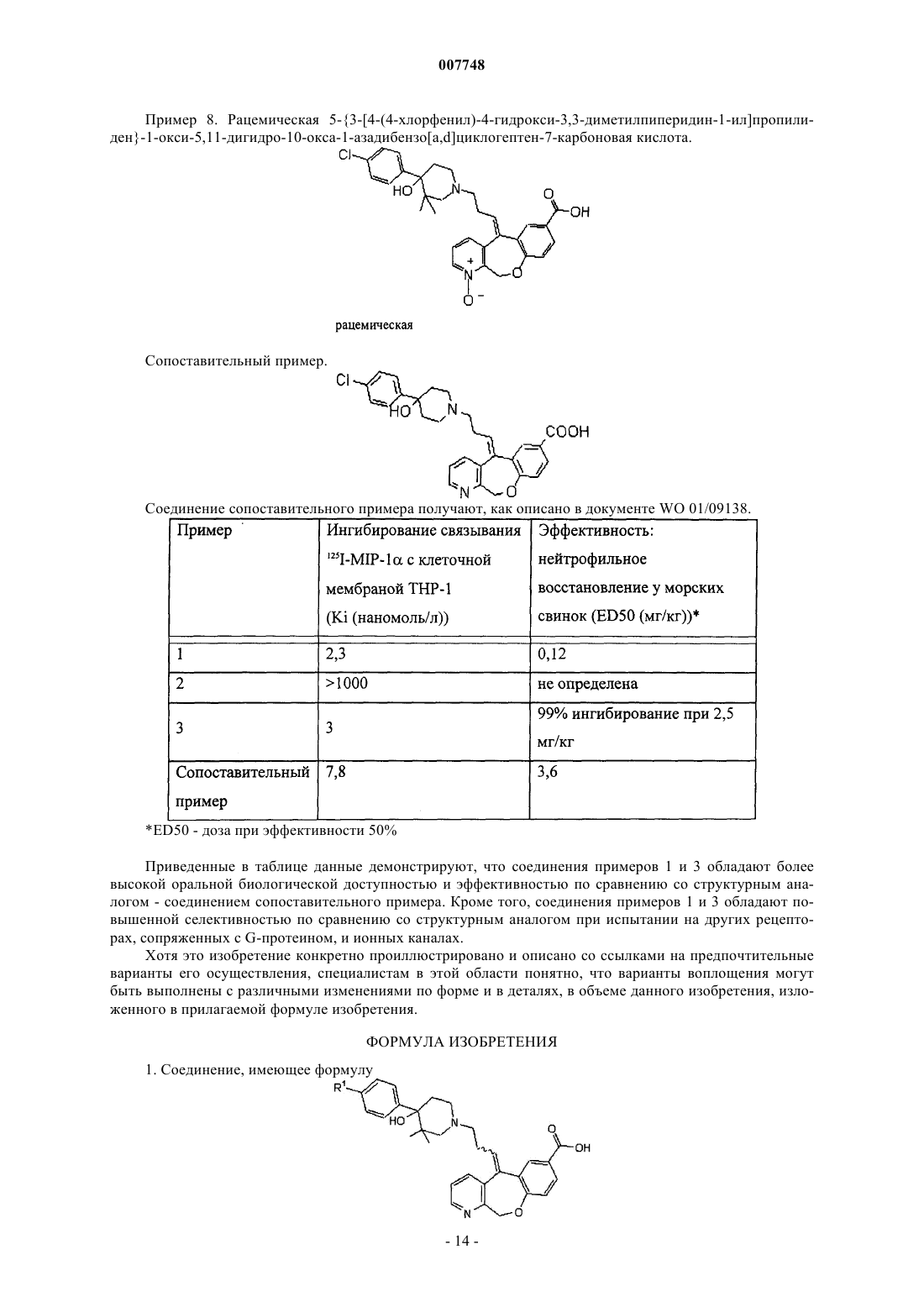
007748 Предпосылки создания изобретения Цитокины-хемоатрактанты или хемокины представляют собой семейство медиаторов, предшествующих воспалению, которые способствуют восстановлению и активации множества поколений лейкоцитов, таких как Т-лимфоциты. Хемокины могут выделяться во многих видах тканевых клеток после активации. Выделение хемокинов в местах воспаления является промежуточным звеном развивающейся миграции клеток эффектора при хроническом воспалении. Хемокины связаны в первичную структуру и содержат четыре сохраненных цистеина, которые образуют дисульфидные связи. Семейство хемокинов включает С-Х-С-хемокины (-хемокины) и С-С-хемокины (-хемокины), в которых первые два сохраненных цистеина разделяются промежуточным остатком или являются смежными, соответственно(Baggiolini M., Dahinden C.A., Immunology Today, т. 15, с. 127-133 (1994. Рецепторы хемокина являются членами надсемейства рецепторов со спаренным G-протеином(GPCR), в которых распределены структурные признаки, отражающие общий механизм действия сигнальной трансдукции (Gerard С., Gerard N.P., Аnnu. Rev. Immunol, т. 12, с. 775-808 (1994); Gerard С.,Gerard N.P., Curr. Opin. Immunol., т. 6, с. 140-145 (1994. Сохраненные признаки включают семь гидрофобных областей, охватывающих плазменные мембраны, которые соединяются посредством гидрофильных внеклеточных и внутриклеточных петель. Большая часть первичной гомологической последовательности встречается в гидрофобных межмембранных областях, причем гидрофильные области являются более разнообразными. Первый рецептор для С-С-хемокинов, который был подвергнут клонированию и экспрессии, связывает МIР-1 и RANTES. Соответственно, этот рецептор MIP-1/RANTES был обозначен как С-С-хемокиновый рецептор 1 (также называется CCR-1 или CKR-1; Neote, К., и др., Cell, т. 72, с. 415425 (1993); Horuk и др., WO 94/11504, 26.05.94; Gao J. и др., J. Exp. Med., т. 177, с. 1421-1427 (1993. Рецептор CCR1 также связывает хемокины CCL2 (МСР-1), CCL4 (MIP-1), CCL7 (МСР-3), CCL8 (МСР-2),CCL13 (МСР-4), CCL14 (НСС-1), CCL15 (Lkn-1), CCL23 (MPIF-1). (Murphy P.M. и др., International Unionof Pharmacology. XXII. Nomenclature for Chemokine Receptors, Pharmacol. Reviews, т. 52, c.145-176 (2000. Способность хемокинов, таких как RANTES и MIP-1, вызывать направленную миграцию моноцитов и запоминающую популяцию циркулирующих Т-клеток указывает, что хемокины и рецепторы хемокинов могут играть решающую роль в хронических воспалительных заболеваниях, так как эти заболевания характеризуются деструктивными инфильтратами Т-клеток и моноцитов (например, смотрите Schall, Т. и др., Nature, т. 347, с. 669-71 (1990. Небольшие молекулы антагонистов взаимодействия между рецепторами С-С-хемокина (например,CCR1) и их лигандами, включающими RANTES и MIP-1, могли бы обеспечить соединения, пригодные для торможения патогенных процессов, "запускаемых" посредством взаимодействия лигандов рецептора. Краткое описание изобретения Изобретение относится к соединениям, имеющим формулы или их физиологически приемлемым солям, в которых R1 представляет собой атом галогена. Кроме того, изобретение относится к способу лечения заболевания, характеризующегося патогенным восстановлением лейкоцитов, или патогенной активацией лейкоцитов, или патогенным восстановлением и активацией лейкоцитов. Этот способ включает в себя введение субъекту, нуждающемуся в лечении, эффективного количества описанного здесь соединения. Изобретение также относится к композициям, содержащим описанное здесь соединение и фармацевтически или физиологически приемлемый носитель или наполнитель. Кроме того, изобретение относится к применению описанных здесь соединений в терапии (включая паллиативную, лечебную и профилактическую терапию) или диагностике и к применению таких соединений при производстве лекарственных препаратов для лечения конкретных заболеваний или состояний,которые описаны здесь (например, воспалительный артрит (например, ревматоидный артрит), воспалительное демиелинизационное заболевание (например, рассеянный склероз. Подробное описание изобретения Изобретение относится к соединениям, которые являются антагонистами С-С хемокинового рецептора 1 (CCR1), композициям, содержащим эти соединения, и к способу лечения заболеваний или нарушений, который включает в себя введение одного или нескольких соединений. Соединения-антагонисты-1 007748 могут ингибировать связывание лиганда (например, хемокинового лиганда, такого как CCL2 (МСР-1),CCL3 (MIP-1), CCL4 (MIP-1), CCL5 (RANTES), CCL7 (МСР-3), CCL8 (МСР-2), CCL13 (МСР-4),CCL14 (НСС-1), CCL15 (Lkn-1), CCL23 (MPIF-1 к CCR1. В соответствии с этим процессы или клеточные ответные реакции, передаваемые путем связывания хемокина с рецептором CR1, могут быть ингибированы (ослаблены или предотвращены, полностью или частично), включая миграцию лейкоцитов,активацию интегрина, переходный процесс роста концентрации внутриклеточного свободного кальция или их физиологически приемлемые соли, в которых R1 представляет собой галоген. Предпочтительно галоген выбирают из группы, состоящей из хлора, брома и фтора. Более предпочтительно галоген представляет собой хлор. Как описано в изобретении, соединения формулы (I) и формулы (Iа) могут быть получены в виде рацематов или как практически чистые энантиомеры (энантиомерный избыток 99%). Оптическая конфигурация соединений формулы (I) и формулы (Iа) устанавливается при использовании (R),(S) методаYork, с. 109-111 (1992. В предпочтительных вариантах осуществления данного изобретения соединение формулы (I) представляет собой (S)-энантиомер и имеет структуру или его физиологически приемлемую соль, в которой R1 означает галоген. В особенно предпочтительном варианте осуществления соединение имеет формулу II, в которой R1 представляет собой хлор. В других предпочтительных вариантах осуществления данного изобретения соединение формулы или его физиологически приемлемую соль, в которой R1 означает галоген. В особенно предпочтительном варианте осуществления соединение имеет формулу IIа, в которой(S)- и (R)-энантиомеры изобретения могут быть получены при использовании любого подходящего способа. Например, энантиомеры могут быть получены путем расщепления рацемата при использовании-2 007748 хиральной хроматографии или перекристаллизации. Предпочтительно (S)- и/или (R)-энантиомеры получают путем стереоспецифического синтеза, как описано в изобретении. В соответствии с традиционными способами изображения структурных формул соединений терминальная метильная группа в описанном здесь соединении может быть изображена в виде прямой линии с группой СН 3 или без нее на конце: Описанные в изобретении соединения могут быть получены в виде Е- и Z-конфигурационных изомеров. Специально указывается, что это изобретение включает соединения с Е-конфигурацией и Z-конфигурацией вокруг двойной связи, соединяющей трициклическую функциональную группу с остатком молекулы, и способ лечения субъекта соединениями Е-конфигурации, Z-конфигурации и их смесями. Соответственно, в структурных формулах, приведенных в изобретении, символ означает как Е-конфигурацию, так и Z-конфигурацию. Одна конфигурация может обладать большей активностью, чем другая. Предпочтительно пиридильное кольцо и пиперидинильное кольцо имеют цис-конфигурацию, как показано в формуле (II) и формуле (IIа). Изобретение включает все изомерные формы и рацемические смеси раскрытых соединений и способ лечения субъекта как чистыми изомерами, так и их смесями, в том числе рацемическими смесями. Описанные здесь соединения могут быть получены и прописаны в виде нейтральных соединений,солей, сложных эфиров, амидов и/или пролекарств. Используемые здесь термины "фармацевтически или физиологически приемлемые соли, сложные эфиры, амиды и пролекарства" означают те соли (например,карбоксилатные соли, аддитивные соли аминокислот), сложные эфиры, амиды и пролекарства соединений настоящего изобретения, которые подходят для использования в контакте с тканями субъекта без несоответствующей токсичности, раздражения, аллергической реакции, и т.п., в соответствии с разумным соотношением преимущества/риска, и являются эффективными для намеченного применения соединений, а также амфотерные формы соединений изобретения, если это возможно. Фармацевтически или физиологически приемлемые кислотные аддитивные соли описанных здесь соединений включают соли, произведенные из нетоксичных неорганических кислот, таких как хлористоводородная, азотная, фосфорная, серная, бромисто-водородная, иодисто-водородная, фтористоводородная, фосфористая и т.п., и соли, произведенные из нетоксичных органических кислот, таких как алифатические моно- и дикарбоновые кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановые кислоты, алкандикарбоновые кислоты, ароматические кислоты, алифатические и ароматические сульфоновые кислоты и т.п. Такие кислотные аддитивные соли включают, например, сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, ацетат, трифторацетат, пропионат, каприлат, изобутират, оксалат, малонат, сукцинат, соль пробковой кислоты, соль себациновой кислоты, фумарат, малеат, манделат,бензоат, хлорбензоат, метилбензоат, динитробензоат, фталат, бензолсульфонат, толуолсульфонат, фенилацетат, цитрат, лактат, малеат, тартрат и метансульфонат. Кроме того, рассматриваются соли аминокислот, такие как аргинат, глюконат, галактуронат и т.п. (например, смотрите Berge S.M. и др., "Pharmaceutical Salts", J. Pharma. Sci., т. 66, c. 1 (1977. Кислотные аддитивные соли соединений, которые содержат основную группу (например, аминную), могут быть получены с использованием подходящих способов. Например, кислотные аддитивные соли могут быть получены путем контактирования соединения в форме свободного основания с достаточным количеством желаемой кислоты, для того чтобы получить соль традиционным способом. Форма свободного основания может быть регенерирована путем контактирования солевой формы с основанием и выделения свободного основания традиционным образом. Форма свободного основания соединения может несколько отличаться от солевых форм по определенным физическим свойствам, таким как растворимость в полярных растворителях. Фармацевтически или физиологически приемлемые основные аддитивны соли могут образоваться с подходящими металлами или аминами, такими как щелочные и щелочно-земельные металлы или органические амины. Примеры металлов, которые подходят для использования в качестве катионов в основных аддитивных солях, включают натрий, калий, магний, кальций и т.п. Амины, которые подходят для использования в качестве катионов в основных аддитивных солях, включают N,N'-дибензилэтилендиамин, хлорпрокаин, холин, дианоламин, дициклогексиламин, этилендиамин, N-метилглюкамин и прокаин (например, смотрите Berge S.M. и др., "Pharmaceutical Salts", J. Pharma. Sci., т. 66, c. 1 (1977. Основные аддитивные соли соединений, которые содержат кислотную группу (например, карбоновую кислоту), могут быть получены при использовании подходящих способов. Например, соединение в форме свободной кислоты может контактировать с достаточным количеством желаемого основания, для того чтобы получить соль традиционным образом. Форма свободной кислоты может быть регенерирована путем контактирования солевой формы с подходящей кислотой и выделения свободной кислоты тра-3 007748 диционным образом. Форма свободной кислоты соединения может несколько отличаться от формы аддитивной соли основания по определенным физическим свойствам, таким как растворимость в полярных растворителях. Термин "пролекарство" относится к соединениям, которые могут быть превращены in vivo (например, после введения животному), за счет процессов обмена веществ или других процессов, с образованием соединения указанной выше формулы, например путем гидролиза в крови. Обширное обсуждение проблемы приведено в работах Т. Higuchi и V. Stella, "Пролекарства как новые системы подачи", Vol. 14Pharmaceutical Association and Pergamon Press, 1987, которые обе введены в это изобретение как ссылки. Подходящие пролекарства включают фармацевтически или физиологически приемлемые сложные эфиры и амиды описанных здесь соединений. Примеры фармацевтически или физиологически приемлемых сложных эфиров соединений этого изобретения включают C1-С 6-алкиловые сложные эфиры. В определенных вариантах алкильная группа алкилового сложного эфира представляет собой C1-С 6-алкильную группу с прямой или разветвленной цепочкой. Кроме того, приемлемые алкиловые сложные эфиры включают C5-С 7-циклоалкиловые сложные эфиры, а также арилалкиловые сложные эфиры, такие как бензил (но не ограниченные им). Предпочтительными являются сложные эфиры C1-C4. Сложные эфиры соединений настоящего изобретения могут быть получены с использованием любого подходящего способа. Примеры фармацевтически или физиологически приемлемых амидов соединений этого изобретения включают амиды, произведенные из аммиака, первичных C1-С 6-алкиламинов и вторичных C1-С 6-диалкиламинов, в которых алкильные группы имеют прямую или разветвленную цепочку. В случае вторичных аминов, этот амин также может находиться в форме 5- или 6-членного гетероцикла, содержащего один атом азота. Предпочтительными являются амиды, произведенные из аммиака, первичных C1-С 3-алкиламинов и вторичных C1-С 2-диалкиламинов. Амиды соединений изобретения могут быть получены с использованием любого подходящего способа. Композиции Изобретение также относится к фармацевтическим и/или физиологическим композициям, которые содержат одно или несколько описанных здесь соединений. Такие композиции могут быть составлены для назначения любым желаемым путем, таким как перорально, местным путeм, ингаляцией (например,интрабронхиальной, интраназальной, оральной ингаляцией или интраназальными каплями), ректально,трансдермально или парэнтерально. Обычно композиции содержат соединение изобретения (то есть одно или несколько соединений) в виде активного компонента и (одного или нескольких) подходящего носителя, разбавителя, наполнителя, вспомогательного средства и/или предохранителя. Рецептура назначаемого соединения будет изменяться в соответствии с выбранным способом назначения (например, раствор, эмульсия, капсула). Могут быть использованы традиционные методики получения фармацевтических рецептур. (Более подробно см. "Remington's Pharmaceutical Science", 18th Edition, Mack Publishing.(1990); Baker, et al., "Controlled Release of Biological Active Agents", John WileySons (1986), вышеуказанные рекомендации включены в это изобретение как ссылки.) Микроорганизмы, присутствующие в композициях, могут быть подавлены с помощью различных антибактериальных и/или противогрибковых агентов, например парабензоатов, хлорбутанола, спиртов(например, фенол, бензиловый спирт), сорбиновой кислоты и т.п. Кроме того, может быть целесообразным включение изотонических агентов, например cахаров, хлорида натрия и т.п. Композиции, подходящие для парэнтеральной инъекции, могут включать в себя физиологически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки для перевода их в стерильные растворы или дисперсии для инъекции. Примеры подходящих водных и неводных носителей, разбавителей, растворителей, наполнителей или носителей включают физиологический раствор, фосфатный буферный раствор, Hank's раствор, Ringer's-лактат и т.п., этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и т.п.), растительные масла (такие как оливковое масло) и органические сложные эфиры для инъекций, такие как этилолеат, или их любые подходящие смеси. Текучесть можно регулировать, например, путем использования покрытия, такого как лецитин, за счет поддержания необходимого размера частиц, в случае дисперсии, и за счет использования поверхностно-активных веществ. Когда желательна продолжительная абсорбция инъецируемой фармацевтической композиции, могут быть добавлены агенты, которые задерживают абсорбцию, например моностеарат алюминия и желатин. Твердые формы дозировки для орального назначения включают, например, капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых формах дозировки активный компонент (например, одно или несколько соединений изобретения) может смешиваться с одним или несколькими носителями или наполнителями, такими как цитрат натрия или дикальций фосфат; (а) наполнителями или разбавителями,например крахмалом, лактозой, сахарозой, глюкозой, маннитом, кремниевой кислотой, полиэтиленгликолями и т.п.; (б) связующими агентами, например карбоксиметилцеллюлозой, альгинатами, желатином,поливинилпирролидоном, сахарозой и смолой акации; (в) увлажнителями, например глицерином; (г) разрушающими агентами, например агар-агаром, карбонатом кальция, картофельным или тапиоковым крахмалом, альгиновой кислотой, некоторыми комплексными силикатами и карбонатом натрия; (д) рас-4 007748 твором добавок-замедлителей, например парафина; (е) ускорителями абсорбции, например соединениями четвертичного аммония; (ж) смачивающими агентами, например цетиловым спиртом и моностеаратом глицерина; (з) адсорбентами, например каолином и бентонитом; и (и) смазочными материалами, например тальком, стеаратом кальция, стеаратом магния, твердыми полиэтиленгликолями, лаурилсульфатом натрия или их смесями. Твердые композиции, такие как композиции для орального назначения, также могут содержать буферные агенты. Такие твердые композиции или твердые композиции, которые аналогичны описанным, могут быть предоставлены, по желанию, в заполненных мягких или твердых желатиновых капсулах. Твердые формы дозировки, такие как таблетки, драже, капсулы, пилюли и гранулы, могут быть получены с покрытиями и оболочками, такими как кишечно-растворимые покрытия или другие подходящие покрытия или оболочки. Некоторые такие покрытия и/или оболочки хорошо известны из уровня техники, и могут содержать непрозрачные агенты, и кроме того, могут иметь такой состав, что активное соединение или соединения выделяются в определенной части кишечного тракта в замедленном режиме. Примеры внедряемых композиций, которые могут быть использованы, представляют собой полимерные вещества и воски. Кроме того, активные соединения могут быть использованы в микроинкапсулированной форме, если это целесообразно, например, с одним или несколькими из вышеупомянутых носителей или наполнителей. Жидкие формы дозировки для орального назначения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Кроме активных соединений, жидкие формы дозировки могут содержать подходящий носитель или наполнитель, такой как вода или другие растворители,солюбилизирующие агенты и эмульгаторы, как, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла, в том числе хлопковое масло, арахисовое масло, кукурузное масло, оливковое масло, касторовое масло и кунжутное масло, глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и эфиры жирных кислот сорбитана или смеси этих веществ и т.п. По желанию, композиция также может включать смачивающие агенты, эмульгаторы, суспендирующие агенты, подслащивающие вещества, вкусовые вещества и/или отдушки. Суспензии могут содержать суспендирующие агенты, такие как этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар, трагакант и т.п. По желанию,могут быть использованы смеси суспендирующих агентов. Свечи (например, для ректального или вагинального введения) могут быть получены путем смешивания одного или нескольких соединений изобретения с подходящими нераздажающими наполнителями или носителями, такими как масло какао, полиэтиленгликоль или суппозиторный воск, который является твердым при комнатной температуре, но жидким при температуре тела и плавится в прямой кишке или влагалище, и при этом выделяется активный компонент. Формы дозировки для местного назначения включают мази, порошки, спреи и средства для ингаляции. Активный компонент может смешиваться в подходящих условиях (например, в стерильных условиях) с физиологически приемлемым носителем и любыми предохранителями, буферами или газамивытеснителями, которые могут потребоваться. Офтальмологические рецептуры, глазные мази, порошки и растворы также могут быть получены, например, с использованием подходящих носителей или наполнителей. Для ингаляций соединения могут быть солюбилизированы и залиты в подходящее дозирующее устройство для введения (например, распылитель, пульверизатор или аэрозольный распределитель под давлением). Количество активного компонента (одно или несколько соединений изобретения) в композиции может изменяться приблизительно от 0,1 до 99,9 мас.%. Предпочтительно количество активного компонента составляет приблизительно от 10 до 90% или приблизительно от 20 до 80 мас.%. Унифицированная доза препарата может содержать приблизительно от 1 до 1000 мг активного компонента, предпочтительно приблизительно от 10 до 100 мг активного компонента. По желанию, композиция также может содержать другие совместимые терапевтические агенты, такие как теофиллин, -адренергические бронхолитические средства, кортикостероиды, антигистамины, противоаллергические агенты, иммунодепрессивные агенты (например, циклоспорин A, FK-506, преднизон, метилпреднизолон), гормоны (например,адренокортикотропный гормон (АСТН, цитокины (например, интерфероны (например, IFN-1a, IFN1b и т.п. В одном варианте осуществления данного изобретения композиция содержит (S)-5-3-[4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден-5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-карбоновую кислоту и физиологически приемлемый носитель или наполнитель. В другом варианте осуществления композиция практически не содержит (R)-5-3-[4-(4-хлорфенил)-4-гидрокси-3,3 диметилпиперидин-1-ил]пропилиден-5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-карбоновой кислоты (содержит, по меньшей мере, приблизительно 98% или приблизительно, по меньшей мере, 99% энантиомерного избытка (S)-5-3-[4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден-5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-карбоновой кислоты).-5 007748 В другом варианте осуществления композиция содержит (S)-5-3-[4-(4-хлорфенил)-4-гидрокси-3,3 диметилпиперидин-1-ил]пропилиден-5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-карбоновую кислоту, (R)-5-3-[4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден-5,11-дигидро 10-окса-1-азадибензо[а,d]циклогептен-7-карбоновую кислоту и физиологически приемлемый носитель или наполнитель. В одном варианте осуществления композиция содержит рацемическую 5-3-[4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден-5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-карбоновую кислоту. В других вариантах отношение (S)-энантиомера к (R)-энантиомеру (по массе) составляет, по меньшей мере, около 2:1, или приблизительно 5:1, или около 10:1, или приблизительно 20:1, или около 50:1. В одном варианте осуществления композиция содержит (S)-5-3-[4-(4-хлорфенил)-4-гидрокси-3,3 диметилпиперидин-1-ил]пропилиден-1-окси-5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-карбоновую кислоту и физиологически приемлемый носитель или наполнитель. В другом варианте осуществления композиция практически не содержит (R)-5-3-[4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден-1-окси-5,11-дигидро-10-окса-1-азадибензо[a,d]циклогептен-7-карбоновую кислоту(содержит, по меньшей мере, приблизительно 98% или, по меньшей мере, около 99% энантиомерного избытка (S)-5-3-[4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден-1-окси-5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-карбоновой кислоты). В другом варианте осуществления композиция содержит (S)-5-3-[4-(4-хлорфенил)-4-гидрокси-3,3 диметилпиперидин-1-ил]пропилиден-1-окси-5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-карбоновую кислоту, (R)-5-3-[4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден-1-окси 5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-карбоновую кислоту и физиологически приемлемый носитель или наполнитель. В одном варианте осуществления композиция содержит рацемическую 5-3-[4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден-1-окси-5,11-дигидро-10-окса 1-азадибензо[a,d]циклогептен-7-карбоновую кислоту. В других вариантах осуществления отношение (S)энантиомера к (R)-энантиомеру (по массе) составляет, по меньшей мере, около 2:1, или приблизительно 5:1, или около 10:1, или приблизительно 20:1, или приблизительно 50:1. Терапевтические методы Изобретение также относится к способу лечения (например, паллиативному, лечебному, профилактическому) заболевания или нарушения, связанного с патогенным восстановлением, активацией или восстановлением и активацией лейкоцитов, передаваемого хемокинами или действием рецептора хемокина,в том числе хронических и острых воспалительных нарушений. Используемый здесь термин "патогенное восстановление, активация или восстановление и активация лейкоцитов" относится к восстановлению лейкоцитов (например, накоплению лейкоцитов в поле зрения воспаления или повреждения) и/или активации (например, физиологическое состояние, в котором лейкоциты осуществляют эффекторный механизм), что влияет на состояния, процессы или результаты заболевания или нарушения, которые подлежат лечению. Например, для субъекта, пораженного рассеянным склерозом, восстановление и/или активация Т-клеток в центральной нервной системе рассматривается как "патогенное восстановление лейкоцитов, патогенная активация лейкоцитов или патогенное восстановление и активация лейкоцитов", поскольку восстановленные и активированные Т-клетки дают вклад в демиелинизационную характеристику этого заболевания. Аналогично, для субъекта, пораженного ревматоидным артритом, восстановление и/или активация Т-клеток в сочленениях (например, в синовиальной ткани или жидкости) рассматривается как "патогенное восстановление лейкоцитов, патогенная активация лейкоцитов или патогенное восстановление и активация лейкоцитов", поскольку восстановленные и активированные Т-клетки влияют на характеристику повреждения тканей при ревматоидном артрите. Заболевания и нарушения, характеризующиеся патогенным восстановлением лейкоцитов, патогенной активацией лейкоцитов или патогенным восстановлением и активацией лейкоцитов, которые могут быть излечены в соответствии со способами, описанными в изобретении, включают, например, острые и хронические воспалительные нарушения, характеризующиеся наличием CCL2 (МСР-1), CCL3 (MIP-1),CCL4 (MIP-1), CCL5 (RANTES), CCL7 (МСР-3), CCL8 (МСР-2), CCL13 (МСР-4), CCL14 (НСС-1),CCL15 (Lkn-1) и/или CCL23 (MPIF-1) чувствительных клеток, таких как Т-клетки, моноциты или эозинофильные клетки. Такие заболевания или нарушения включают (но не ограничиваются перечисленными) воспалительный артрит (например, ревматоидный артрит), воспалительные демиелинизационные заболевания (например, рассеянный склероз), атеросклероз, артериосклероз, рестеноз, ишемическое/реперфузионное повреждение, сахарный диабет (например, сахарный диабет 1-го типа), псориаз, воспалительные заболевания кишечника, такие как язвенный колит и болезнь Крона, отторжение (острое или хроническое) трансплантированных органов и тканей (например, острое отторжение аллотрансплантата, хроническое отторжение аллотрансплантата), реакцию "трансплантата против хозяина", а также аллергические болезни и астму. Другие заболевания, связанные с аберрантным восстановлением и/или активацией лейкоцитов, которые могут излечиваться (в том числе профилактическое лечение) с применением способов,раскрытых в данном изобретении, представляют собой воспалительные заболевания, связанные с вирус-6 007748 ной (например, вирус иммунодефицита человека (ВИЧ, бактериальной или грибковой инфекцией, такие как связанный со СПИДом энцефалит, связанный со СПИДом пятнисто-папулезный дерматит, связанная со СПИДом интерстициальная пневмония, связанная со СПИДом энтеропатия, связанное со СПИДом перипортальное печеночное воспаление и связанный со СПИДом гломерулонефрит. Этот способ включает в себя введение субъекту, нуждающемуся в лечении, эффективного количества соединения(т.е. одного или нескольких соединений), которые описаны в изобретении. Используемый здесь термин "воспалительное демиелинизационное заболевание" относится к острым и хроническим воспалительным заболеваниям, характеризующимся демиелинизацией ткани центральной нервной системы. Воспалительное демиелинизационное заболевание может быть острым воспалительным демиелинизационным заболеванием, например острым рассеянным энцефаломиелитом,синдромом Guillain-Barre или острым геморрагическим лейкоэнцефалитом. В других вариантах осуществления воспалительное демиелинизационное заболевание может представлять собой хроническое воспалительное демиелинизационное заболевание, например рассеянный склероз, хроническую воспалительную демиелинизационную полирадикулоневропатию. Предпочтительный вариант осуществления данного изобретения обеспечивает способ лечения рассеянного склероза (PC), который включает в себя введение субъекту, нуждающемуся в лечении, эффективного количества соединения формулы (I), (Iа), (II) или (IIа). Рассеянный склероз имеет различные проявления, и клиническое течение PC может быть сгруппировано в четырех категориях: рецидивирующий-ремиттирующий, первичный прогрессирующий, вторичный прогрессирующий и прогрессирующийрецидивирующий. Способ согласно данному изобретению может быть использован для лечения PC, который находится в любом из четырех общепризнанных клинических состояний. В соответствии с этим соединение согласно данному изобретению может быть введено пациенту с прогрессирующим течениемPC, для того чтобы замедлять или предупреждать развитие неврологического повреждения. Кроме того,соединение данного изобретения может быть введено субъекту с рецидивирующим-ремиттирующим,вторичным прогрессирующим или прогрессирующим-рецидивирующим PC с целью подавления рецидива (например, острый приступ). Например, соединение согласно данному изобретению может быть введено субъекту с рецидивирующим-ремиттирующим PC в течение ремиттирующей фазы заболевания, для того чтобы предотвратить или задержать рецидив. Используемый здесь термин "воспалительный артрит" относится к тем заболеваниям сочленений, в которых иммунная система вызывает или обостряет воспаление в сочленениях, и включает ревматоидный артрит, ювенильный ревматоидный артрит и спондилоартропатические заболевания, такие как анкилозирующий спондилит, реактивный артрит, синдром Рейтера, псориазный артрит, псориазный спондилит, энтеропатический артрит, энтеропатический спондилит, начало ювенильной спондилоартропатии и недифференцированная спондилоартропатия. Воспалительный артрит обычно характеризуется инфильтрацией синовиальной ткани и/или синовиальной жидкости лейкоцитами. В другом предпочтительном варианте изобретение обеспечивает способ лечения ревматоидного артрита, который включает введение субъекту, нуждающемуся в лечении, эффективного количества соединения формулы (I), (Ia), (II) или (IIа). Предпочтительно термин "субъект" означает птицу или млекопитающее, такое как человек (Homosapiens), но также может означать животное, нуждающееся в ветеринарном лечении, например домашнее животное (например, собаки, кошки и т.п.), сельскохозяйственное животнoе (например, коровы, овцы,домашняя птица, свиньи, лошади и т.п.) и лабораторное животное (например, крысы, мыши, морские свинки и т.п.). Термин "эффективное количество" соединения означает количество, которое подавляет связывание хемокина с рецептором (например, CCR1) и тем самым ингибирует один или несколько процессов, опосредованных путем связывания в субъекте, с заболеванием, связанным с патогенным восстановлением лейкоцитов, патогенной активацией лейкоцитов или патогенным восстановлением и активацией лейкоцитов. Примеры таких процессов включают миграцию лейкоцитов, активацию интегрина, кратковременное увеличение концентрации внутриклеточного свободного кальция [Ca2+]i и/или гранулярное выделение медиаторов, предшествующих воспалению. "Эффективное количество" соединения означает количество, с которым может достигаться желаемый терапевтический и/или профилактический эффект, или такое количество, которое приводит к предупреждению или ослаблению симптомов, связанных с заболеванием, ассоциированным с патогенным восстановлением лейкоцитов, патогенной активацией лейкоцитов или патогенным восстановлением и активацией лейкоцитов. Количество соединения, назначаемого индивидууму, будет зависеть от типа и серьезности заболевания и от характеристик индивидуума, таких как общее состояние здоровья, возраст, пол, масса тела и переносимость лекарственных препаратов. Кроме того, это количество зависит от степени, серьезности и типа заболевания. Специалист в этой области сможет определить подходящие формы дозировки, зависящие от этих и других факторов. Кроме того, антагонист функции хемокинового рецептора может быть назначен в сочетании с одним или несколькими дополнительными терапевтическими агентами, такими как теофиллин, -адренергические бронхолитические средства, кортикостероиды, антигистамины, противоаллергические агенты, иммунодепрессивные агенты (например, циклоспорин A, FK-506, преднизон,-7 007748 метилпреднизолон), гормоны (например, адренокортикотропный гормон (АСТН, цитокины (например,интерфероны (например, IFN-1a, IFN-1b и т.п. Когда соединение данного изобретения назначается в сочетании с другим терапевтическим агентом, соединение и агент могут быть назначены таким образом,чтобы обеспечить перекрывание фармакологической активности, например, одновременно или последовательно. Соединение может быть введено любым подходящим способом, в том числе, например, орально в капсулах, суспензии или таблетках или путем парэнтерального назначения. Парэнтеральное введение может включать, например, системное введение, такое как путем внутримышечной, внутривенной, подкожной или внутрибрюшинной инъекции. Кроме того, соединение может быть введено орально (например, с пищей), трансдермально, локально, путем ингаляции (например, внутрибронхиально, интраназально, оральной ингаляцией или интраназальными каплями) или ректально, в зависимости от заболевания или состояния, подлежащего лечению. Предпочтительными способами назначения являются оральное или парэнтеральное введение. Соединение может назначаться индивидуально в виде части фармацевтической или физиологической композиции. Активность соединений настоящего изобретения можно оценить с использованием подходящих исследований, таких как анализ связывания рецептора или анализ хемотаксиса. Например, как описано в примерах, небольшие молекулы антагонистов связывания МIР-1 идентифицируются с использованием клеточных мембран ТНР-1. Конкретно, для идентификации небольших молекул антагонистов, которые блокируют связывание МIP-1, используется высокопроизводительный анализ связывания рецептора, в котором проводится наблюдение связывания меченого 125I-МIР-1 с клеточными мембранами ТНР-1. Кроме того, соединения настоящего изобретения могут быть идентифицированы в силу их способности ингибировать стадии активации, которые запускаются посредством связывания хемокина (например,CCL2 (МСР-1), CCL3 (MIP-1), CCL4 (MIP-1), CCL5 (RANTES), CCL7 (МСР-3), CCL8 (МСР-2), CCL13(МСР-4), CCL14 (НСС-1), CCL15 (Lkn-1), CCL23 (MPIF-1 с его рецептором (CCR-1), такие как хемотаксис, активация интегрина и гранулярное выделение медиатора. Они также могут быть идентифицированы благодаря их способности блокировать хемотаксис, индуцированный хемокином (например, CCL2(МСР-4), CCL14 (НСС-1), CCL15 (Lkn-1), CCL23 (MPIF-1, например, в клетках HL-60, Т-клетках, моноядерных клетках периферической крови или эозинофила. Примеры Схема 1 Стадия 1. трет-Бутиловый эфир 3,3-диметил-4-оксопиперидин-1-карбоновой кислоты. В сухую 2-литровую, двугорлую, круглодонную колбу, оборудованную магнитной мешалкой, холодильником и большой водяной баней при 10 С, добавляют трет-бутиловый эфир 4-оксопиперидин-1 карбоновой кислоты (125 г, 628 ммоль) и 1 л безводного тетрагидрофурана (ТГФ). В образовавшийся желтый раствор добавляют метилиодид (85 мл, 1365 ммоль). Затем по частям добавляют трет-бутилат натрия (150 г, 1560 ммоль) в течение 30 мин. Наблюдается выделение тепла, особенно в начале добавления. Реакционную смесь осторожно нагревают до кипения, при этом интенсивность кипения регулируется скоростью добавления основания. Смесь дополнительно перемешивают еще 30 мин. Растворитель удаляют в вакууме. Маслянистый остаток обрабатывают раствором NH4Cl в воде (500 мл) и экстрагируют эфиром (3 раза по 200 мл). Объединенные органические слои промывают рассолом, сушат над Nа 2SO4 и фильтруют через короткий слой силикагеля. Растворитель удаляют в вакууме, и образовавшееся желтое масло начинает кристаллизоваться. Масло выдерживают в высоком вакууме в течение ночи. Смесь суспендируют в гексане (50-100 мл) и обрабатывают ультразвуком в течение 1 мин. Желтое твердое вещество собирают посредством фильтрации и промывают гексаном (100 мл). Первая порция трет-бутилового эфира 3,3-диметил-4-оксопиперидин-1-карбоновой кислоты имеет вид твердого желтого вещества(смотрите препарат (37) в статье Vice, S. и дp., J. Org. Chem., т. 66, c. 2487-2492 (2001. Спектр 1 Н-ЯМР (CDCl3, 300 МГц) : 1,13 (с, 6 Н), 1,49 (с, 9 Н), 2,49 (т, 2 Н), 3,43 (шир. с, 2 Н), 3,73 (т, 2 Н). Стадия 2. трет-Бутиловый эфир 4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-карбоновой кислоты. В двугорлую, 2-литровую, круглодонную колбу вставляют две капельные воронки емкостью 125 мл и магнитную мешалку. После монтажа колбу сушат в пламени, продувая ее сухим азотом. В колбу заливают ТГФ (700 мл) и 4-бромхлорбензол (33,7 г, 176 ммоль, 2,5 экв.). Образовавшийся раствор охлаждают до -78 С в бане сухой лед/ацетон. В одну из капельных воронок добавляют через канюлю 2,5 М раствор бутиллития (в 70 мл гексана, 175 ммоль, 2,5 экв.). Раствор бутиллития медленно добавляют в холодный раствор ТГФ в течение 1 ч. Смесь перемешивают еще в течение 0,5 ч, получая белую суспензию. Готовят раствор трет-бутилового эфира 3,3-диметил-4-оксопиперидин-1-карбоновой кислоты (16,0 г, 70,5 ммоль,1 экв.) в ТГФ (100 мл) и добавляют в реакционную смесь из второй капельной воронки в течение 1,75 ч. Образовавшуюся смесь перемешивают при -78 С в течение 2 ч; оказалось, что за это время реакция практически завершается, по данным анализа тонкослойной хроматографии (ТСХ). Добавляют насыщенный водный раствор NH4Cl (150 мл) и реакционной смеси дают нагреться до комнатной температуры. Добавляют воду (150 мл) и смесь экстрагируют этилацетатом (2+1 л). Объединенные экстракты промывают водой и рассолом, сушат над сульфатом магния, фильтруют и концентрируют. Твердый остаток растирают с этилацетатом и фильтруют. Жидкость над осадком концентрируют и растирают с эфиром. Затем образовавшуюся жидкость над осадком растирают со смесью диэтиловый эфир/петролейный эфир. Образовавшиеся твердые вещества объединяют, получая трет-бутиловый эфир 4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-карбоновой кислоты в виде беловатого твердого вещества. 1 Н-ЯМР (CDCl3, 300 МГц) : 0,82 (с, 6 Н), 1,34-1,44 (м, 2 Н), 1,49 (с, 9 Н), 2,67 (ддд, 1 Н), 3,10-3,70 (м,3 Н), 4,00-4,30 (м, 1 Н), 7,31 (д, 2 Н), 7,39 (д, 2 Н). Стадия 3. 4-(4-Хлорфенил)-3,3-диметилпиперидин-4-ол. В охлажденный (0 С) раствор трет-бутилового эфира 4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-карбоновой кислоты (10,42 г, 30,7 ммоль) в метиленхлориде (300 мл) медленно добавляют трифторуксусную кислоту (60 мл) в течение 1,25 ч. Образовавшийся желтый раствор перемешивают при 0 С еще в течение 1,5 ч. Смесь концентрируют при пониженном давлении и остаток растворяют в этилацетате (1,2 л) и промывают водным раствором гидроксида натрия (1 н., 150 мл). Водный слой экстрагируют дополнительно этилацетатом (200 мл) и объединенные экстракты промывают рассолом, сушат над сульфатом натрия, фильтруют и концентрируют. Образовавшийся твердый остаток растирают с эфиром,чтобы получить 4-(4-хлорфенил)-3,3-диметилпиперидин-4-ол в виде беловатого твердого вещества. 1 Н-ЯМР (CD3OD, 300 МГц) : 0,73 (с, 3 Н), 0,85 (с, 3 Н), 1,42 (ддд, 1 Н), 2,36 (д, 1 Н), 2,61 (ддд, 1 Н),2,91 (шир. дд, 1 Н), 3,08-3,19 (м, 2 Н), 7,26-7,32 (м, 2 Н), 7,44-7,50 (м, 2 Н). Масс-спектр (МС) m/z: 240 (М+1).- 10007748 Стадия 4. (S)-4-(4-Хлорфенил)-3,3-диметилпиперидин-4-ол. Совершенно чистую трехгорлую, 5-литровую колбу, снабженную верхней мешалкой, продувают азотом в течение 20 мин. В колбу добавляют рацемический 4-(4-хлорфенил)-3,3-диметилпиперидин-4-ол (202 г,843 ммоль), L-(+)-винную кислоту (114 г, 759 ммоль) и 4040 мл смеси бутанон:вода (9:1). Смесь нагревают до кипения. По частям добавляют воду (202 мл) в течение 45 мин (отношение бутанона к воде 6:1), чтобы полностью растворить твердую смесь. Кипячение с обратным холодильником продолжают еще 45 мин, затем выключают нагревание и колбе дают охладиться до комнатной температуры в течение ночи. Твердое вещество отделяют путем фильтрации с отсасыванием и сушат его приблизительно 3 суток в вакууме, получая S-энантиомер в виде L-(+) соли винной кислоты, которую распределяют между 1 М раствором NaOH и метиленхлоридом (промывают рассолом и сушат сульфатом натрия), для того чтобы получить свободное основание. 1 Н-ЯМР (CD3OD, 300 МГц) : 0,73 (с, 3 Н), 0,85 (с, 3 Н), 1,42 (ддд, 1 Н), 2,36 (д, 1 Н), 2,61 (ддд, 1 Н),2,91 (шир. дд, 1 Н), 3,08-3,19 (м, 2 Н), 7,26-7,32 (м, 2 Н), 7,44-7,50 (м, 2 Н).MC m/z: 240 (M+1). Стадия 5. 5-Циклопропил-5,11-дигидро[1]бензоксепино[2,3-b]пиридин-5-ол. Сухую трехгорлую, 2-литровую, круглодонную колбу снабжают магнитной мешалкой, стеклянной пробкой, резиновой мембраной и вводом аргона. В атмосфере аргона в колбу добавляют 50,0 г 5,11-дигидро[1]бензоксепино[3,4-b]пиридин-5-она (получен по способу Inoue и др., Synthesis 1, с. 113-116 (1997),(0,24 моля и 500 мл осушенного тетрагидрофурана и колбу охлаждают в бане со льдом. Свежеприготовленный раствор циклопропилмагнийбромида в тетрагидрофуране (50,0 г циклопропилмагнийбромида получают из бромистого циклопропила (0,41 моль) и 12,0 г магниевых стружек (0,49 моль) в 400 мл осушенного тетрагидрофурана) вводят через иглу шприца в течение 5 мин. Удаляют баню со льдом и смесь перемешивают в течение 30 мин. Реакционную смесь медленно выливают в 500 мл насыщенного раствора хлорида аммония, смесь экстрагируют двумя порциями (по 300 мл) этилацетата и объединенные органические экстракты промывают насыщенным водным раствором (300 мл) хлорида натрия. Органический раствор сушат безводным сульфатом магния, фильтруют и выпаривают (в вакууме аспиратора, приблизительно при 30 С). К твердому остатку добавляют 150 мл смеси гексан/этилацетат=1:1 (по объему) и эту смесь обрабатывают ультразвуком 15 мин, фильтруют и промывают смесью гексан/этилацетат (1:1 по объему), чтобы получить указанное в заголовке соединение в виде бледно-желтого твердого вещества. Стадия 6. 5-(3-Бромпропилиден)-5,11-дигидро[1]бензоксепино[2,3-b]пиридин. В 2-литровую колбу (форма баклажана) с магнитной мешалкой добавляют 75,0 г 5-циклопропил 5,11-дигидро[1]бензоксепино[2,3-b]пиридин-5-ола (0,30 моль) и 75 мл уксусной кислоты. Раствор охлаждают водой (около 10 С) и добавляют 120 мл водной бромисто-водородной кислоты (47%) в течение 5 мин. Реакционную смесь нагревают до 60 С, перемешивают в течение 1 ч и выпаривают (в вакууме аспиратора, приблизительно при 50 С) до объема около 200 мл. Реакционную смесь выливают в 1500 мл насыщенного водного раствора бикарбоната натрия, смесь экстрагируют двумя порциями (по 800 мл) этилацетата и объединенные органические экстракты промывают насыщенным водным раствором хлорида натрия (500 мл). Органический раствор сушат безводным сульфатом магния, фильтруют и выпаривают (в вакууме аспиратора, приблизительно при 30 С). Маслянистый остаток очищают путем хроматографии на 500 г силикагеля 60, с элюированием смесью 5:1-4:1 (по объему) гексан/этилацетат. Продукт элюирования выпаривают, получая соединение, указанное в заголовке, в виде бледно-желтого масла. Стадия 7. 7-Ацетил-5-(3-бромпропилиден)-5,11-дигидро[1]бензоксепино[2,3-b]пиридин. Сухую трехгорлую, 3-литровую, круглодонную колбу снабжают магнитной мешалкой, стеклянной пробкой, резиновой мембраной и вводом аргона. В атмосфере аргона в колбу добавляют 94,0 г 5-(3-бромпропилиден)-5,11-дигидро[1]бензоксепино[2,3-b]пиридина (0,30 моль) и 900 мл сухого дихлорметана и колбу охлаждают в бане со льдом. В раствор медленно добавляют 78,5 г хлористого алюминия (0,83 моль) и затем 17,8 мл ацетилхлорида (0,25 моль) и смесь перемешивают при 0 С в течение часа. Реакционную смесь выливают в 1500 г льда и разделяют слои. Водный слой экстрагируют тремя порциями (400 мл) этилацетата. Дихлорметановый слой и органические экстракты объединяют и промывают последовательно насыщенным водным раствором бикарбоната и насыщенным водным раствором хлорида натрия(1 л). Органический раствор сушат безводным сульфатом магния, фильтруют и выпаривают (в вакууме аспиратора, приблизительно при 30 С). Маслянистый остаток очищают хроматографически на 800 г силикагеля 60, который элюируют смесью 5:1-1:1 (по объему) гексан/этилацетат. Элюат выпаривают, получая соединение, указанное в заголовке, в виде бледно-желтого твердого вещества. Стадия 8. (S)-1-(5-3-[4-(4-Хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден-5,11 дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-ил)этанон. В суспензию (S)-4-(4-хлорфенил)-3,3-диметилпиперидин-4-ола (5,50 г, 22,94 ммоль) в ацетонитриле(200 мл) и воде (50 мл) добавляют карбонат калия (7,17 г, 51,9 ммоль) и затем твердый 1-[5-(3-бромпропилиден)-5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-ил]этанон (6,30 г, 17,3 ммоль). Гетерогенную смесь перемешивают при комнатной температуре 4 ч, нагревают до 50 С и перемешивают 13 ч. Смесь охлаждают до комнатной температуры и ацетонитрил удаляют при пониженном давлении. Водный слой экстрагируют этилацетатом (750 мл) и экстракт промывают рассолом, сушат над сульфатом натрия,фильтруют и концентрируют. Сырой остаток очищают хроматографически на силикагеле (смесь 3:1- 11007748 этилацетат:гексан), получая (S)-1-(5-3-[4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден-5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-ил)этанон в виде беловатого твердого вещества. 1 Н-ЯМР (CDCl3) : 0,6-0,9 (6 Н, д), 1,2-1,6 (4 Н, м), 2,2-2,4 (4 Н, м), 2,55 (3 Н, с), 2,8 (2 Н, д), 5,3 (2 Н,шир. с), 6,25 (1 Н, т), 6,85 (1H, д), 7,27-7,4 (6 Н, м), 7,6-7,8 (2 Н, м), 8,0 (1 Н, д), 8,5 (1 Н, д).MC m/z: 517 (M+1). Стадия 9. В круглодонную колбу емкостью 25 мл помещают продукт стадии 8 (500 мг, 0,969 ммоль), водный 2 М раствор NaOH (4,84 ммоль, 2,42 мл), гипохлорит натрия (4% доступного хлора, 3,6 ммоль) и диметоксиэтан (10 об., 5 мл) и смесь перемешивают при комнатной температуре в течение ночи. Через 12 ч добавляют насыщенный водный раствор бисульфита натрия (5 мл), реакционную смесь экстрагируют этилацетатом (4x5 мл); органические слои объединяют и сушат над сульфатом натрия, фильтруют и выпаривают при пониженном давлении, чтобы получить 500 мг (выход 96%) желтого твердого вещества. Это твердое вещество растворяют в воде (20 об., 10 мл) и подкисляют уксусной кислотой до рН 6,15. При подкислении осаждается твердое вещество кремового цвета, которое фильтруют и помещают в вакуумный шкаф приблизительно на 2 суток, чтобы получить соединение, указанное в заголовке. 1 Н-ЯМР (CD3OD) : 0,75 (с, 3 Н), 0,86 (с, 3 Н), 1,63 (д, 1 Н), 2,49-2,66 (м, 2 Н), 2,70-2,89 (м, 2 Н), 2,993,23 (м, 5 Н), 5,10-5,50 (шир. с, 2 Н), 6,15 (т, 1 Н), 6,75 (д, 2 Н), 7,25-7,31 (м, 2 Н), 7,39-7,47 (м, 2 Н), 7,71-7,81 Часть 1. (R)-4-(4-Хлорфенил)-3,3-диметилпиперидин-4-ол. Рацемический 4-(4-хлорфенил)-3,3-диметилпиперидин-4-ол (0,500 г, 2,086 ммоль) растворяют в минимальном количестве горячего изопропилового спирта (приблизительно 5 мл). Горячий раствор фильтруют через слой ваты и переносят в раствор (1S)-(+)-10-камфорсульфоновой кислоты (0,484 г,2,086 ммоль) в изопропиловом спирте (около 3 мл). Смесь интенсивно перемешивают в течение нескольких минут, в течение которых образуется густой осадок, и оставляют ее охлаждаться до комнатной температуры в течение 0,25 ч. Твердые вещества отделяют путем фильтрации с отсасыванием и сушат в вакууме. Высушенную соль растворяют в горячем изопропиловом спирте (приблизительно 50 мл), фильтруют через слой ваты и раствору дают медленно охладиться до комнатной температуры, без перемешивания, в течение ночи. Твердое вещество, образовавшееся при охлаждении (95 мг, 19% от теории), отделяют путем фильтрации с отсасыванием; причем аналитически показано, что оно является энантиомерно чистым (с помощью ЖХВР). Эту соль суспендируют в этилацетате и нейтрализуют гидроксидом натрия(1 н. раствор). Однородную органическую фазу промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и сушат, получая R-4-(4-хлорфенил)-3,3-диметилпиперидин-4-ол. 1 Н-ЯМР (CD3OD, 300 МГц):0,73 (с, 3 Н), 0,85 (с, 3 Н), 1,42 (ддд, 1 Н), 2,36 (д, 1 Н), 2,61 (ддд, 1 Н),2,91 (шир, дд, 1 Н), 3,08-3,19 (м, 2 Н), 7,26-7,32 (м, 2 Н), 7,44-7,50 (м, 2 Н).MC m/z: 240 (М+1). Часть 2. Соединение получают практически так же, как описано в стадиях 5-9 примера 1, но заменяя (S)-4(4-хлорфенил)-3,3-диметилпиперидин-4-ол на (R)-4-(4-хлорфенил)-3,3-диметилпиперидин-4-ол. Пример 3. Рацемическая-5-3-[4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден 5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-карбоновая кислота. Этот рацемический материал получают практически так же, как описано в стадиях 5-9 примера 1,- 12007748 но заменяя (S)-4-(4-хлорфенил)-3,3-диметилпиперидин-4-ол на рацемический 4-(4-хлорфенил)-3,3-диметилпиперидин-4-ол. Пример 4. Получение ТНР-1 клеточной мембраны и анализ связывания. Мембраны готовят из ТНР-1 клеток (АТСС ТIB202). Клетки собирают путем центрифугирования,промывают 2 раза фосфатно-солевым буфером (ФСБ) и лепешки клеток замораживают при температуре от -70 до -85 С. Замороженные лепешки оттаивают в ледяном лизисном буфере, содержащем 5 мМ HEPES(N-2-гидроксиэтилпиперазин-N'-2-этансульфокислота) рН 7,5, 2 мМ ЭДТУК (этилендиаминтетрауксусная кислота) и по 5 мкг/мл каждого ингибитора протеазы: апротинина, лейпептина и химостатина, и 100 мкг/мл фенилметансульфонилфторида (ФМСФ - также ингибитор протеазы), при концентрации от 1 до 5107 клеток/мл. Эта процедура приводит к лизису клеток. Суспензию хорошо перемешивают, для того чтобы снова суспендировать все замороженные лепешки клеток. Ядра и остатки клеток удаляют путем центрифугирования при ускорении 400 g в течение 10 мин при температуре 4 С. Жидкость над осадком переносят в чистую пробирку и фрагменты мембран собирают путем центрифугирования при 25000 g в течение 30 мин при 4 С. Жидкость над осадком отсасывают и лепешку снова суспендируют в замораживающем буфере,содержащем 10 мМ HEPES рН 7,5, 300 мМ сахарозы и по 1 мкг/мл каждого: апротинина, лейпептина и химостатина, и 10 мкг/мл ФМСФ (приблизительно 0,1 мл на каждые 108 клеток). Все скопления клеток пептизируют, используя мини-устройство для гомогенизации, и определяют общую концентрацию белка,используя набор для анализа белков (Bio-Rad, фирма Herculec, CA,по каталогу 500-0002). Затем отбирают аликвоты мембранного раствора и сохраняют их при температуре от -70 до -85 С до использования. В анализе связывания используются описанные выше мембраны. Белок мембраны (от 2 до 20 мкг общего мембранного белка) выдерживают с рецептором МIР-1, меченым изотопом 125I (от 0,1 до 0,2 наномоль/л), с немеченым конкурентом MIP-1, или без него, или с различными концентрациями соединений. Реакции связывания проводят в связывающем буфере (от 60 до 100 мкл), содержащем 10 мМ HEPES с рН,равным 7,2, 1 мМ СаСl2, 5 мМ MgCl2 и 0,5% альбумина бычьей сыворотки (BSA), в течение 60 мин, при комнатной температуре. Реакции связывания прерывают, убирая мембраны посредством быстрой фильтрации через фильтры из стекловолокна (марки GF/B или GF/C, фирма Packard), которые предварительно смачивают в 0,3% растворе полиэтиленимина. Фильтры промывают связывающим буфером (приблизительно 600 мкл), содержащим 0,5 М NaCl, сушат и количественно определяют связанную радиоактивность,используя счетчик сцинтилляций. Значения активности испытанных соединений приведены в таблице. Пример 5. Модель оценки эффективности in vivo. Для оценки биологической/фармакодинамической активности соединений используют животную модель нейтрофильного восстановления в ответ на МIР-1. Соединения вводят перорально самкам морских свинокHartley (дозы изменяются приблизительно от 0,5 до 5,0 мг/кг), за 30 мин до внутрикожной инъекции мышиного рецептора МIР-1 (100 пикомоль/центр) или фосфатно-солевого буфера (ФСБ). Спустя 5 ч берут образцы пункционной биопсии кожи и проводят исследования с миелопероксидазой (МПО). Активность МПО используют в качестве индикатора нейтрофильного восстановления в месте инъекции. Результаты приведены в таблице. Пример 6. (S)-5-3-[4-(4-Хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил]пропилиден-1-окси 5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен-7-карбоновая кислота. Соединение сопоставительного примера получают, как описано в документе WO 01/09138.ED50 - доза при эффективности 50% Приведенные в таблице данные демонстрируют, что соединения примеров 1 и 3 обладают более высокой оральной биологической доступностью и эффективностью по сравнению со структурным аналогом - соединением сопоставительного примера. Кроме того, соединения примеров 1 и 3 обладают повышенной селективностью по сравнению со структурным аналогом при испытании на других рецепторах, сопряженных с G-протеином, и ионных каналах. Хотя это изобретение конкретно проиллюстрировано и описано со ссылками на предпочтительные варианты его осуществления, специалистам в этой области понятно, что варианты воплощения могут быть выполнены с различными изменениями по форме и в деталях, в объеме данного изобретения, изложенного в прилагаемой формуле изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, имеющее формулу- 14007748 или его физиологически приемлемая соль, в котором R1 означает галоген. 2. Соединение по п.1, в котором R1 выбирают из группы, состоящей из хлора, фтора и брома. 3. Соединение по п.2, в котором R1 представляет собой хлор. 4. Фармацевтическая композиция, содержащая соединение по п.1 и физиологически приемлемый носитель или наполнитель. 5. Способ лечения заболевания, характеризующегося патогенным восстановлением лейкоцитов, патогенной активацией лейкоцитов или патогенным восстановлением и активацией лейкоцитов, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения по п.1. 6. Соединение, имеющее структуру или его физиологически приемлемая соль, в котором R1 означает галоген. 7. Соединение по п.6, в котором R1 выбирают из группы, состоящей из хлора, фтора и брома. 8. Соединение по п.7, в котором R1 представляет собой хлор. 9. Фармацевтическая композиция, содержащая соединение по п.6 и физиологически приемлемый носитель или наполнитель. 10. Способ лечения заболевания, характеризующегося патогенным восстановлением лейкоцитов,патогенной активацией лейкоцитов или патогенным восстановлением и активацией лейкоцитов, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения по п.6. 11. Способ по п.10, в котором R1 представляет собой хлор. 12. Способ по п.10, в котором указанное заболевание является острым или хроническим воспалительным заболеванием. 13. Способ по п.10, в котором указанное заболевание выбирают из группы, состоящей из воспалительного артрита, воспалительного демиелинизационного заболевания, атеросклероза, артериосклероза,рестеноза, ишемического/реперфузионного повреждения, сахарного диабета, псориаза, воспалительных заболеваний кишечника, отторжения трансплантированных органов, реакции трансплантата против хозяина, аллергии и астмы. 14. Способ по п.13, в котором указанное заболевание является воспалительным артритом. 15. Способ по п.14, в котором указанный воспалительный артрит является ревматоидным артритом. 16. Способ по п.13, в котором указанное заболевание является воспалительным демиелинизационным заболеванием. 17. Способ по п.16, в котором указанное воспалительное демиелинизационное заболевание является рассеянным склерозом. 18. Способ антагонизации рецептора 1 С-С-хемокина у субъекта, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения по п.1. 19. Соединение, имеющее формулу или его физиологически приемлемая соль, в котором R1 представляет собой галоген. 20. Фармацевтическая композиция, содержащая соединение по п.19 и физиологически приемлемый носитель или наполнитель. 21. Способ лечения заболевания, характеризующегося патогенным восстановлением лейкоцитов,патогенной активацией лейкоцитов или патогенным восстановлением и активацией лейкоцитов, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения- 15007748 по п.19. 22. Способ по п.21, в котором указанное заболевание выбирают из группы, состоящей из воспалительного артрита, воспалительного демиелинизационного заболевания, атеросклероза, артериосклероза,рестеноза, ишемического/реперфузионного повреждения, сахарного диабета, псориаза, воспалительных заболеваний кишечника, отторжения трансплантированных органов, реакции трансплантата против хозяина, аллергии и астмы. 23. Способ лечения острого или хронического воспалительного заболевания, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения по п.19. 24. Способ по п.23, в котором указанное заболевание является воспалительным артритом. 25. Способ по п.24, в котором указанный воспалительный артрит является ревматоидным артритом. 26. Способ по п.23, в котором указанное заболевание является воспалительным демиелинизационным заболеванием. 27. Способ по п.26, в котором указанное воспалительное демиелинизационное заболевание является рассеянным склерозом. 28. Способ антагонизации рецептора 1 С-С-хемокина у субъекта, который заключается в том, что вводят субъекту, нуждающемуся в лечении, эффективное количество соединения по п.19. 29. Соединение по любому из пп.1-3 или пп.6-8, применяемое в терапии или диагностике. 30. Соединение по п.19, применяемое в терапии или диагностике.
МПК / Метки
МПК: C07D 221/00, C07D 491/04, C07D 313/00
Метки: антагонисты, лечения, заболеваний, демиелинизационных, воспалительных, артритов
Код ссылки
<a href="https://eas.patents.su/17-7748-antagonisty-ccr1-dlya-lecheniya-vospalitelnyh-artritov-demielinizacionnyh-vospalitelnyh-zabolevanijj.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты ccr1 для лечения воспалительных артритов, демиелинизационных воспалительных заболеваний</a>
Производные 2-(пурин-9-ил)-тетрагидрофуран-3,4-диола, способы их получения, фармацевтическая композиция и способ лечения или профилактики воспалительных заболеваний с их применением

Номер патента: 3194
Опубликовано: 27.02.2003
Авторы: Чен Чуэн, Кокс Брайан, Казинз Ричард Питер Чарлз
МПК: A61K 31/52, A61P 29/00, C07D 473/32...
Метки: применением, 2-(пурин-9-ил)-тетрагидрофуран-3,4-диола, композиция, производные, воспалительных, профилактики, получения, лечения, заболеваний, фармацевтическая, способы, способ
Формула / Реферат:
1. Производное 2-(пурин-9-ил)-тетрагидрофуран-3,4-диола формулы (I) где R1 и R2 независимо представляют собой группу, выбранную из (1) C3-8циклоалкил-; (2) водород; (3) арил2CHCH2-; (4) C3-8циклоалкилC1-6алкил-; (5) C1-8алкил-; (6) арилC1-6алкил-; (7) R4R5N-C1-6алкил-; (8) C1-6алкил-CH(CH2OH)-; (9) арилC1-5алкил-CH(CH2OH)-; (10) C3-8циклоалкил, независимо замещенный одной или более чем одной группой -(CH2)pR6; (11) H2NC(=NH)NHC1-6алкил-; (12)...
Непептидильные ингибиторы vla-4-зависимого связывания клеток, применяемые при лечении воспалительных, аутоимунных и респираторных заболеваний

Номер патента: 4673
Опубликовано: 24.06.2004
Авторы: Миличи Энтони Джон, Чупак Луис Стэнли, Лау Ван Фанг, Дюплантье Аллен Якоб
МПК: A61P 37/00, A61K 31/421, C07D 413/04...
Метки: респираторных, аутоимунных, ингибиторы, клеток, непептидильные, лечении, vla-4-зависимого, применяемые, заболеваний, воспалительных, связывания
Формула / Реферат:
1. Соединение формулы (1.0.0) и его фармацевтические соли и другие пролекарственные производные, где A обозначает (C1-C6)алкил, циклоалкил, арил, гетероарил или гетероциклил, определенные ниже, где указанные алкил, циклоалкил, арил, гетероарил или гетероциклил необязательно замещены от 0 до 3 R9; или A выбран из группы, включающей следующие радикалы: A1-NHC(=O)NH-A2-, A1-NHC(=O)O-A2-, A1-OC(=O)NH-A2-, A1-NHSO2 NH-A2-, A1-NHC(=O)-A2-,...
Непептидные ингибиторы vla-4-зависимого клеточного связывания, полезные в лечении воспалительных, аутоиммунных и респираторных заболеваний

Номер патента: 4792
Опубликовано: 26.08.2004
Авторы: Чупак Луи Стэнли, Майлиси Энтони Джон, Даплантир Аллен Джейкоб
МПК: A61P 29/00, A61K 31/42, C07D 241/08...
Метки: vla-4-зависимого, респираторных, клеточного, ингибиторы, аутоиммунных, воспалительных, связывания, заболеваний, полезные, лечении, непептидные
Формула / Реферат:
1. Соединение формулы (1.0.0) и его фармацевтически приемлемые соли и другие пролекарственные производные, где A1 представляет собой фенил или пиридил, каждый возможно замещенный R10 в количестве от 1 до 2; B независимо выбран из группы, состоящей из следующего: где символ * указывает место присоединения группировки, представленной каждой формулой с (1.1.0) по (1.1.14) к группировке в формуле (1.0.0), а символ R показывает место...
Замещенные гетероциклические соединения для лечения воспалительных процессов

Номер патента: 4499
Опубликовано: 29.04.2004
Авторы: Хартманн Сьюзен Дж., Браун Дейвид Л., Роджир Дональд Дж., Картер Джеффери С., Мец Сьюзен, Талли Джон Дж., Хано Кэтлин Э., Бертеншо Стивен Р., Людвиг Синди Л., Нагараян Шринивасан Р., Обуковиц Марк Г., Девадас Балекудру, Грането Меттью Дж.
МПК: A61K 31/33, C07D 311/58, A61P 1/04...
Метки: замещенные, гетероциклические, процессов, соединения, лечения, воспалительных
Формула / Реферат:
1. Соединение формулы I' где X выбран из группы, состоящей из O, S, CRcRb и NRa; Ra выбран из группы, включающей водород, C1-C3-алкил, фенил-C1-C3-алкил, (замещенный фенил)-C1-C3-алкил, где фенильная группа несет от 1 до 3 заместителей, выбранных из группы, включающей C1-C6-алкил, гидроксил, галоид, галоидный алкил, нитрогруппу, циан, алкоксигруппу и C1-C6-алкиламиногруппу, ацил и карбокси-C1-C6-алкил; каждый из Rb и Rc независимо от другого...
Применение липополисахаридов для лечения кишечных воспалительных процессов

Номер патента: 5012
Опубликовано: 28.10.2004
Авторы: Гриммеке Ханс-Дитер, Калоусек Маркус Бруно, Заиболд Франк
МПК: A61P 29/00, A61K 31/715
Метки: липополисахаридов, лечения, кишечных, применение, воспалительных, процессов
Формула / Реферат:
1. Применение ЛПС и/или их производных вариантов для перорального или ректального лечения кишечных воспалительных процессов. 2. Применение по п.1, отличающееся тем, что используют агонистический ЛПС. 3. Применение по п.1 или 2, отличающееся тем, что используют ЛПС из E.coli. 4. Применение по пп.1-3, отличающееся тем, что используют ЛПС из серотипа 02:K1:H6 E.coli. 5. Применение по пп.1-4, отличающееся тем, что используют ЛПС из серотипа 02:K1:H6...
Предыдущий патент: Замещённые амины активные в отношении рецептора каннабиноида-1
Случайный патент: Защитное средство