Аралкилтетрагидропиридины, их получение и содержащие их фармацевтические композиции
Номер патента: 7324
Опубликовано: 25.08.2006
Авторы: Бурри Бернар, Кардамон Розанна, Барони Марко, Казелла Пьер






Формула / Реферат
1. Соединение формулы (I)

где X представляет собой N или СН;
R1 представляет собой атом водорода или галогена или группу CF3;
n означает целое число от 1 до 5;
А представляет собой группу формулы (а)-(d):

где m означает 1 или 2;
и его соли или сольваты и его N-оксиды.
2. Соединение по п.1, где n означает 1.
3. Соединение по п.1 или 2, где R1 представляет собой группу CF3.
4. Соединение по пп.1-3, где X представляет собой СН и R1 находится в 3-положении бензола.
5. Соединение по пп.1-3, где X представляет собой N и пиридин замещен в 2,6-положениях.
6. Соединение по п.1, выбранное из следующих соединений:
1-[2-(2,3-дигидро-1Н-циклопента[b]нафталин-6-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,
1-[2-(2,3-дигидро-1Н-инден-5-ил)этил]-4-[3-трифторметилфенил]-1,2,3,6-тетрагидропиридин,
1-[2-(5,6,7,8-тетрагидронафталин-2-ил)этил]-4-[3-трифторметилфенил]-1,2,3,6-тетрагидропиридин,
1-[2-(2,3-дигидро-1Н-циклопента[b]нафталин-6-ил)этил]-4-(6-хлорпирид-2-ил)-1,2,3,6-тетрагидро-пиридин,
1-[2-(2,3-дигидро-1Н-циклопента[b]нафталин-6-ил)этил]-4-(6-трифторметилпирид-2-ил)-1,2,3,6-тет-рагидропиридин,
1-[3-(2,3-дигидро-1Н-инден-5-ил)пропил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,
1-[4-(2,3-дигидро-1Н-инден-5-ил)бутил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,
1-[2-(флуорен-2-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,
1-[2-(9,10-дигидрофенантрен-2-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,
1-[2-(2,3-дигидро-1Н-циклопента[b]нафталин-6-ил)этил]-(4-(3-трифторметилфенил)-1,2,3,6-тетра-гидропиридин,
1-[2-(флуорен-3-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,
1-[2-(2,3-дигидро-1Н-циклопента[а]нафталин-8-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетра-гидропиридин,
1-[2-(5,6,7,8-тетрагидрофенантрен-3-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,
1-[2-(5,6,7,8-тетрагидроантрацен-2-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,
их солей и сольватов и их N-оксидов.
7. Способ получения соединения (I) по п.1, отличающийся тем, что
(а) соединение формулы (II)

где R1 такой, как определено в п.1, подвергают взаимодействию с кислотой формулы (III) или одним из ее функциональных производных

где n и А такие, как определено в п.1,
(б) восстанавливают карбонильную группу полученного таким образом соединения формулы (IV)

(в) дегидратируют промежуточный пиперидинол формулы (V)

(г) полученное таким образом соединение формулы (I) выделяют и, возможно, превращают в одну (один) из его солей или сольватов или N-оксидов.
8. Соединение формулы (IIIш)

где Аш представляет собой группу формулы (а), (с) или (d), как определено в п.1, n означает целое число от 1 до 5 и m означает 1 или 2, и его соли или сольваты.
9. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы (I) или одну (один) из его фармацевтически приемлемых солей, сольватов или N-оксидов по любому из пп.1-6.
10. Композиция по п.9, отличающаяся тем, что она содержит от 0,001 до 100 мг активного ингредиента.
11. Применение соединения формулы (I) или одной (одного) из его фармацевтически приемлемых солей, сольватов или N-оксидов по любому из пп.1-6 для приготовления аналгезирующих лекарственных средств и/или лекарственных средств, предназначенных для лечения заболеваний, связанных с иммунными и воспалительными нарушениями.
12. Лекарственное средство для лечения заболеваний, опосредованных цитокином TNF-альфа, отличающееся тем, что оно состоит из одного из соединений формулы (I) или одной (одного) из его солей, сольватов или N-оксидов по любому из пп.1-6.
Текст
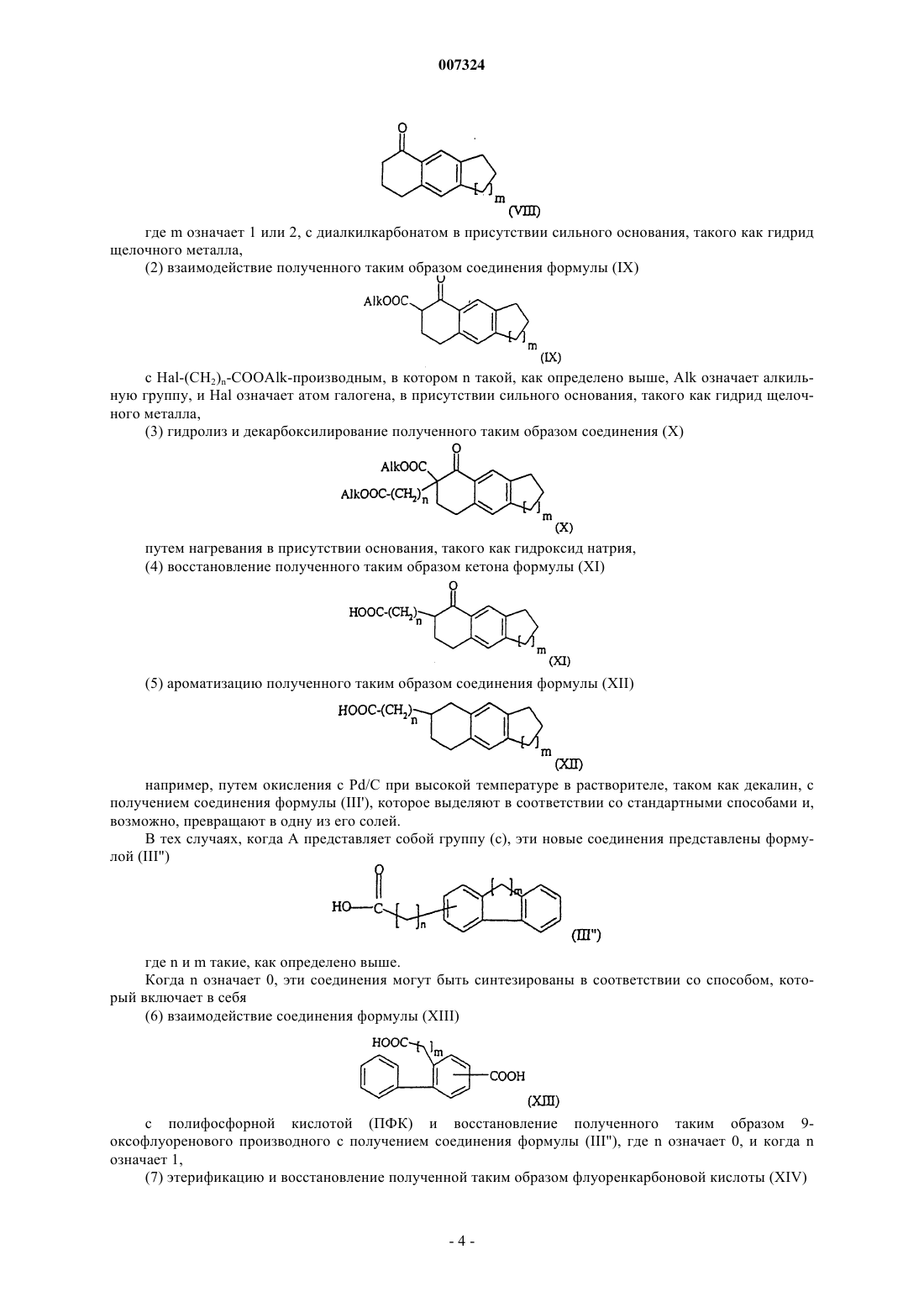
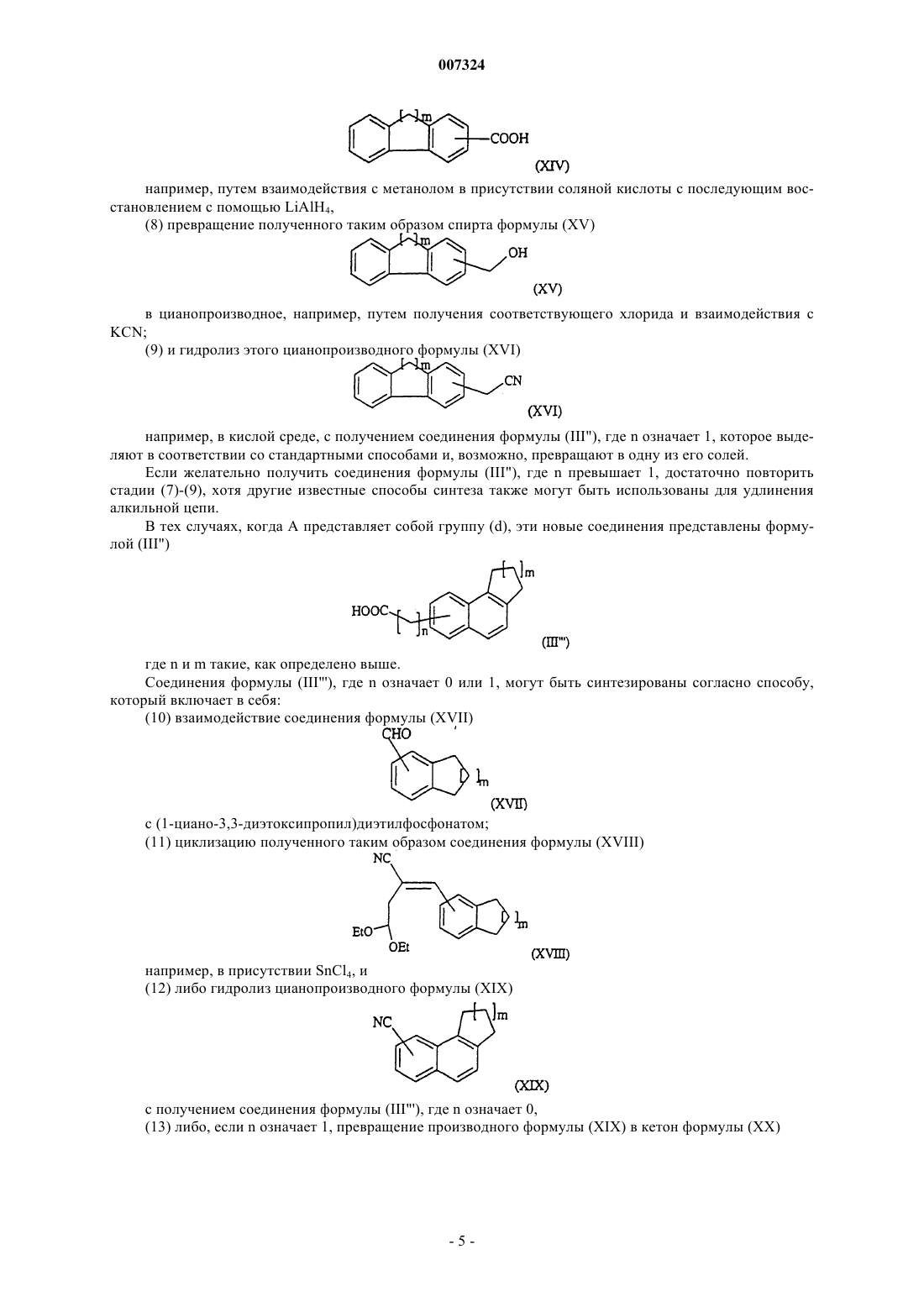
007324 Настоящее изобретение касается новых аралкилтетрагидропиридинов, фармацевтических композиций, содержащих их, способа их получения и промежуточных соединений, синтезируемых в этом способе. ЕР 0458697 описывает производные нафтилалкилтетрагидропиридина, обладающие модулирующей перистальтику кишечника активностью.Bourri с соавт. (Proc. Natl. Acad. Sci. 1999, 96 (22):12855-12859) описали активность соединения,названного SR 57746 (1-(2-нафт-2-илэтил)-4-(3-трифторметил)-1,2,3,6-тетрагидропиридин), в модели экспериментального аутоиммунного энцефаломиелита (experimental autoimmune encephalomyelitis, ЕАЕ) и в модуляции TNF-альфа (Tumor Necrosis Factor, фактор некроза опухолей). Было обнаружено, что некоторые тетрагидропиридины, замещенные частично насыщенным аралкильным радикалом, обладают мощной активностью в плане модуляции TNF-альфа.TNF-альфа представляет собой цитокин, который недавно вызвал интерес как медиатор иммунитета, воспаления, клеточной пролиферации, фиброза и т.д. Этот медиатор присутствует в избытке в воспаленной синовиальной ткани и играет важную роль в патогенезе аутоиммунитета (Annu. Rep. Med. Chem.,1997, 32:241-250). Таким образом, настоящее изобретение, согласно одному из его аспектов, касается аралкилтетрагидропиридинов формулы (I)R1 представляет собой атом водорода или галогена или группу CF3;n означает целое число от 1 до 5; А представляет собой группу формулы (а)-(d): где m означает 1 или 2; и их солей или сольватов и их N-оксидов. В настоящем описании термин "галоген" обозначает атом, выбранный из хлора, брома, йода и фтора. Предпочтительными являются соединения, где n означает 1. Другими предпочтительными соединениями являются соединения, где R1 представляет собой группу CF3. Другими предпочтительными соединениями являются соединения, где X представляет собой СН, иR1 находится в 3-положении бензола. Другими предпочтительными соединениями являются соединения, где X представляет собой СН, иR1 представляет собой группу CF3. Другими предпочтительными соединениями являются соединения, где X представляет собой N, и пиридин замещен в 2,6-положениях. Соли соединений формулы (I) по настоящему изобретению включают в себя как соли присоединения фармацевтически приемлемых неорганических или органических кислот, такие как гидрохлорид,гидробромид, сульфат, гидросульфат, дигидрофосфат, цитрат, малеат, тартрат, фумарат, глюконат, метансульфонат, 2-нафталинсульфонат и т.д., так и соли присоединения, которые дают возможность осуществить соответствующее разделение или кристаллизацию соединений формулы (I), такие как пикрат или оксалат, или соли присоединения оптически активных кислот, например камфорсульфоновой кислоты и миндальной кислоты или замещенной миндальной кислоты. Согласно настоящему изобретению соединения формулы (I) могут существовать в виде Nоксидных производных, в частности они могут нести N-оксидную группу на тетрагидропиридине. Соединения формулы (I) могут быть синтезированы способом, который включает в себя(а) взаимодействие соединения формулы (II) где R1 такой, как определено выше, с кислотой формулы (III) или одним из ее функциональных производных где n и А такие, как определено выше,(б) восстановление карбонильной группы полученного таким образом соединения формулы (IV)(в) дегидратацию полученного таким образом промежуточного пиперидинола формулы (V)(г) выделение полученного таким образом соединения формулы (I) и, возможно, превращение его в одну (один) из его солей или сольватов или в его N-оксидные производные. Реакция стадии (а) может быть соответствующим образом осуществлена в органическом растворителе при температуре, варьирующей от -10 С до температуры флегмы реакционной смеси. Может быть предпочтительным осуществлять реакцию при низкой температуре, если реакция экзотермическая, как в том случае, когда хлорид используют в качестве функционального производного кислоты формулы (III). В качестве соединения формулы (III) можно использовать либо свободную кислоту, возможно активированную (например с помощью ВОР, а именно три(диметиламино)бензотриазол-1-илоксифосфония гексафторфосфата), либо одно из ее функциональных производных, такое как, например, ангидрид, смешанный ангидрид, активный эфир или галогенангдрид, предпочтительно бромид. Среди активных эфиров особенно предпочтительным является п-нитрофениловый эфир, но метоксифениловый, тритиловый и бензгидриловый эфиры и тому подобное также подходят. В качестве растворителя реакции предпочтительно использовать галогенированный растворитель,такой как метиленхлорид, дихлорэтан, 1,1,1-трихлорэтан, хлороформ или тому подобное, но другие органические растворители, совместимые с использующимися реагентами, например диоксан, тетрагидрофуран или углеводород, такой как гексан, также могут быть использованы. Реакция может быть соответствующим образом осуществлена в присутствии акцептора протонов,например карбоната щелочного металла или третичного амина, такого как триэтиламин. Стадия восстановления (б) может быть соответствующим образом осуществлена посредством подходящих восстанавливающих агентов, таких как борановые комплексы, например борандиметилсульфид ([СН 3]2S-ВН 3), гидрид алюминия или комплекс литий-алюминий гидрид, в инертном органическом растворителе при температуре от 0 С до температуры флегмы реакционной смеси в соответствии со стандартными способами. Выражение "инертный органический растворитель" подразумевает растворитель, который не препятствует реакции. Такими растворителями являются, например, простые эфиры, такие как диэтиловый эфир, тетрагидрофуран (ТГФ), диоксан или 1,2-диметоксиэтан. Согласно предпочтительному режиму, процесс осуществляют с боран-диметилсульфидом, использующимся в избытке относительно исходного соединения (II), при температуре флегмы, возможно в инертной атмосфере. Восстановление завершается обычно через несколько часов.-2 007324 Дегидратацию на стадии (в) легко выполнить, например, используя смесь уксусной и серной кислот при температуре, варьирующей от комнатной температуры до температуры флегмы использующегося растворителя. Согласно предпочтительному способу, реакцию стадии (в) осуществляют в смеси уксусная кислота/серная кислота в соотношении 3/1 по объему, путем нагревания при температуре примерно 80-100 С в течение 1-3 ч. Желаемое соединение выделяют в соответствии со стандартными способами в виде свободного основания или одной из его солей. Свободное основание может быть превращено в одну из его солей путем простого солеобразования в органическом растворителе, таком как спирт, предпочтительно этанол или 2 пропанол, простой эфир, такой как 1,2-диметоксиэтан, этилацетат, ацетон или углеводород, такой как гексан. Полученное соединение формулы (I) возможно превращают в одно из его N-оксидных производных. Соединения формулы (I), несущие N-оксидную группу на атоме азота тетрагидропиридина, могут быть получены путем окисления соответствующего соединения формулы (I). В этом случае соединение формулы (I) подвергают реакции окисления в соответствии со стандартными способами, например взаимодействию с м-хлорнадбензойной кислотой в подходящем растворителе, и выделяют согласно обычным способам, хорошо известным специалистам в данной области. Соединения формулы (I) также могут быть получены из тетрагидропиридина формулы (VI) в которой X и R1 такие, как определено выше, посредством реакции конденсации с соединением формулы (VII) в которой n и А такие, как определено выше, и L представляет собой уходящую группу, выделения полученного таким образом соединения формулы (I) и возможного его превращения в одну из его солей или сольватов. В качестве уходящей группы "L" можно использовать, например, галогеногруппу или любую другую группу, подходящую для конденсации с соединением формулы (VI). Реакцию конденсации осуществляют путем смешивания исходных соединений (VI) и (VII) в органическом растворителе, таком как спирт, например метанол или бутанол, в присутствии основания, такого как, например, карбонат щелочного металла, при температуре, варьирующей от комнатной температуры до температуры флегмы выбранного растворителя, в соответствии со стандартными способами. Исходные соединения формулы (II) известны или могут быть получены аналогично известным соединениям. Соединения формулы (III) в которой А представляет собой группу формулы (а), (с) или (d), как определено выше, n означает целое число от 1 до 5, и m означает 1 или 2, и их соли или сольваты являются новыми соединениями и представляют собой следующий аспект настоящего изобретения. В тех случаях, когда А представляет собой группу (а), эти новые соединения представлены формулой (III') где n и m такие, как определено выше, и могут быть синтезированы способом, который включает в себя: где m означает 1 или 2, с диалкилкарбонатом в присутствии сильного основания, такого как гидрид щелочного металла,(2) взаимодействие полученного таким образом соединения формулы (IX) с Наl-(СН 2)n-COOAlk-производным, в котором n такой, как определено выше, Alk означает алкильную группу, и Hal означает атом галогена, в присутствии сильного основания, такого как гидрид щелочного металла,(3) гидролиз и декарбоксилирование полученного таким образом соединения (X) путем нагревания в присутствии основания, такого как гидроксид натрия,(4) восстановление полученного таким образом кетона формулы (XI)(5) ароматизацию полученного таким образом соединения формулы (XII) например, путем окисления с Pd/C при высокой температуре в растворителе, таком как декалин, с получением соединения формулы (III'), которое выделяют в соответствии со стандартными способами и,возможно, превращают в одну из его солей. В тех случаях, когда А представляет собой группу (с), эти новые соединения представлены формулой (III") где n и m такие, как определено выше. Когда n означает 0, эти соединения могут быть синтезированы в соответствии со способом, который включает в себя(6) взаимодействие соединения формулы (XIII) с полифосфорной кислотой (ПФК) и восстановление полученного таким образом 9 оксофлуоренового производного с получением соединения формулы (III"), где n означает 0, и когда n означает 1,(7) этерификацию и восстановление полученной таким образом флуоренкарбоновой кислоты (XIV) например, путем взаимодействия с метанолом в присутствии соляной кислоты с последующим восстановлением с помощью LiAlH4,(8) превращение полученного таким образом спирта формулы (XV) в цианопроизводное, например, путем получения соответствующего хлорида и взаимодействия с(9) и гидролиз этого цианопроизводного формулы (XVI) например, в кислой среде, с получением соединения формулы (III"), где n означает 1, которое выделяют в соответствии со стандартными способами и, возможно, превращают в одну из его солей. Если желательно получить соединения формулы (III"), где n превышает 1, достаточно повторить стадии (7)-(9), хотя другие известные способы синтеза также могут быть использованы для удлинения алкильной цепи. В тех случаях, когда А представляет собой группу (d), эти новые соединения представлены формулой (III") где n и m такие, как определено выше. Соединения формулы (III"'), где n означает 0 или 1, могут быть синтезированы согласно способу,который включает в себя:(10) взаимодействие соединения формулы (XVII)(11) циклизацию полученного таким образом соединения формулы (XVIII)(12) либо гидролиз цианопроизводного формулы (XIX) с получением соединения формулы (III"'), где n означает 0,(13) либо, если n означает 1, превращение производного формулы (XIX) в кетон формулы (XX) например, посредством реакции Гриньяра с CH3Mgl; и(14) превращение кетона (XX) в соответствующую кислоту формулы (III"'), где n означает 1. Если желательно получить соединения формулы (III"'), где n превышает 1, то достаточно повторить стадии (7)-(9), описанные выше, начиная с кислоты формулы (III"'), полученной на стадии (14), или использовать другие способы синтеза, известные для удлинения алкильной цепи. При осуществлении циклизации стадии (11), начиная с бета-альдегида, можно получить два производных формулы (XIX), так как образование ароматического кольца может осуществляться путем циклизации в одном из двух соседних положений. В этом случае образованные таким способом изомеры следует разделить, например, посредством хроматографии на колонке с силикагелем, и продолжать с использованием нужного изомера. Реакции, описанные выше, в основном хорошо известны специалистам в данной области. Подробные примеры этих способов в любом случае описаны в экспериментальной части. Соединения по настоящему изобретению обладают полезными свойствами в отношении ингибирования TNF-. Эти свойства были продемонстрированы с помощью теста, направленного на измерение влияния молекул на синтез TNF-, индуцированный у мышей Balb/c с помощью липополисахарида (ЛПС) изEscherichia coli (055:B5, Sigma, St. Louis, Mo). Исследуемые продукты вводят перорально группам из 5 самок 7-8-недельных мышей Balb/c(Charles River, Франция). Через один час внутривенно вводят ЛПС (10 мкг/мышь). Кровь от каждого животного берут через 1,5 ч после введения ЛПС. Образцы центрифугируют и получают плазму, которую замораживают при -80 С. TNF- измеряют, используя коммерческие наборы (R и D, Abingdon, UK). В этом тесте обнаружили, что представленные соединения по настоящему изобретению являются очень активными в ингибировании синтеза TNF- даже в очень низких дозах. Благодаря этой активности и их низкой токсичности, соединения формулы (I) и их соли или сольваты могут быть использованы для лечения заболеваний, связанных с иммунными и воспалительными нарушениями, или в качестве анальгетиков. В частности, соединения формулы (I) могут быть использованы для лечения атеросклероза, аутоиммунных заболеваний, заболеваний, вызывающих демиелинизацию нейронов (таких как рассеянный склероз), астмы, ревматоидного артрита, фиброзных заболеваний,идиопатического легочного фиброза, муковисцидоза, гломерулонефрита, ревматоидного спондилита,остеоартрита, подагры, резорбции кости и хряща, остеопороза, болезни Педжета, множественной миеломы, увеоретинита, септического шока, сепсиса, эндотоксического шока, реакции трансплантат-противхозяина, отторжения трансплантата, респираторного дистресс-синдрома взрослых, силикоза, асбестоза,легочного саркоидоза, болезни Крона, язвенного колита, бокового амиотрофического склероза, болезни Альцгеймера, болезни Паркинсона, диссеминированной красной волчанки, гемодинамического шока,ишемических патологий (инфаркта миокарда, ишемии миокарда, коронаспазма, стенокардии, сердечной недостаточности, сердечного приступа), постишемических реинфузных приступов, малярии, микобактериальных инфекций, менингита, лепры, вирусных инфекций (ВИЧ, цитомегаловирус, вирус герпеса),оппортунистических инфекций, ассоциированных со СПИДом, туберкулеза, псориаза, атопического дерматита и контактного дерматита, диабета, кахексии, рака и повреждений, опосредованных радиацией. Соединения формулы (I) и их фармацевтически приемлемые соли и сольваты предпочтительно вводят перорально. В фармацевтических композициях по настоящему изобретению для перорального применения активное начало можно вводить животным и человеку для лечения вышеупомянутых болезней в лекарственных формах в виде смеси со стандартными фармацевтическими носителями. Подходящие лекарственные формы включают, например, таблетки, которые могут распадаться, желатиновые капсулы, порошки, гранулы и пероральные растворы или суспензии. В тех случаях, когда твердую композицию приготавливают в виде таблеток, основной активный ингредиент смешивают с фармацевтическим носителем, таким как желатин, крахмал, лактоза, стеарат магния, тальк, аравийская камедь или тому подобное. Таблетки могут быть покрыты сахарозой или другими подходящими веществами, или, альтернативно, они могут быть обработаны так, чтобы обладать отсроченной или пролонгированной активностью, и так, чтобы они непрерывно высвобождали предопределенное количество активного агента. Препарат в форме желатиновых капсул получают путем смешивания активного ингредиента с разбавителем и заполнения полученной смесью мягких или твердых желатиновых капсул.-6 007324 Препарат в форме сиропа или эликсира может содержать активный ингредиент в сочетании с подсластителем, предпочтительно бескалорийным подсластителем, метилпарабеном и пропилпарабеном в качестве антисептических агентов, а также корригентом и подходящим красителем. Водно-диспергируемые порошки или гранулы могут содержать активный ингредиент в виде смеси с диспергирующими или увлажняющими агентами, или суспендирующими агентами, такими как поливинилпирролидон, а также с подсластителями или улучшителями вкуса. Активное начало может быть также приготовлено в форме микрокапсул, возможно с одним или более чем одним носителем или добавкой. В фармацевтических композициях по настоящему изобретению активное начало может находиться также в виде комплекса включения в циклодекстринах или их простых эфирах или сложных эфирах. Количество активного начала, подлежащего введению, зависит, как всегда, от степени развития заболевания, а также от возраста и массы пациента. Тем не менее, стандартные дозы обычно содержат от 0,001 до 100 мг, предпочтительно от 0,01 до 50 мг, более предпочтительно от 0,1 до 20 мг активного начала и еще более предпочтительно от 0,5 до 10 мг. Согласно другому аспекту настоящее изобретение касается комбинации, включающей соединение формулы (I) или одну (один) из его фармацевтически приемлемых солей или сольватов и по меньшей мере одно соединение, выбранное из иммунодепрессантов, таких как интерферон бета-1b; адренокортикотропного гормона; глюкокортикоидов, таких как преднизон или метилпреднизолон; ингибиторов интерлейкина-1. Более подробно, настоящее изобретение касается комбинации, включающей соединение формулы(I), или одну из его фармацевтически приемлемых солей или сольватов и по меньшей мере одно соединение, выбранное из роквинимекса (roquinimex, 1,2-дигидро-4-гидрокси-N,1-диметил-2-оксо-3 хинолинкарбоксанилид), милорана (myloran, продукт от компании Autoimmune, содержащий бычий миелин), антегрена (моноклональное человеческое антитело от компаний Elan/Athena Neurosciences) и рекомбинантного интерферона бета-1b. Другими возможными комбинациями являются комбинации, включающие соединение формулы (I) или одну из его фармацевтически приемлемых солей или сольватов и блокатор калиевых каналов, такой как, например, фампридин (4-аминопиридин). Согласно другому аспекту настоящее изобретение касается способа лечения заболеваний, связанных с иммунными и воспалительными нарушениями, а также лечения болей, в частности атеросклероза,аутоиммунных заболеваний, заболеваний, вызывающих демиелинизацию нейронов (таких как рассеянный склероз), астмы, ревматоидного артрита, фиброзных заболеваний, идиопатического легочного фиброза, муковисцидоза, гломерулонефрита, ревматоидного спондилита, остеоартрита, подагры, резорбции кости и хряща, остеопороза, болезни Педжета, множественной миеломы, увеоретинита, септического шока, сепсиса, эндотоксического шока, реакции трансплантат-против-хозяина, отторжения трансплантата, респираторного дистресс-синдрома взрослых, силикоза, асбестоза, легочного саркоидоза, болезни Крона, язвенного колита, бокового амиотрофического склероза, болезни Альцгеймера, болезни Паркинсона, диссеминированной красной волчанки, гемодинамического шока, ишемических патологий (инфаркта миокарда, ишемии миокарда, коронароспазма, стенокардии, сердечной недостаточности, сердечного приступа), постишемических реинфузных приступов, малярии, микобактериальных инфекций, менингита, лепры, вирусных инфекций (ВИЧ, цитомегаловируса, вируса герпеса), оппортунистических инфекций, ассоциированных со СПИДом, туберкулеза, псориаза, атопического дерматита и контактного дерматита, диабета, кахексии, рака и повреждений, опосредованных радиацией, при котором вводят соединение формулы (I) или его фармацевтически приемлемую соль или сольват одно или в комбинации с другими активными началами. Последующие примеры иллюстрируют настоящее изобретение. Подготовительный пример 1. 1,2,3,6,7,8-Гексагидро-5 Н-циклопента[b]нафталин-5-он. Суспензию 61 г (0,457 моль) безводного AlCl3 в 188 мл смеси дихлорметан/нитрометан (соотношение 8/1) охлаждают до 0-5 С и к этой смеси прибавляют 22,15 г индана и порциями 22,5 г (0,225 моль) янтарного ангидрида. После выдерживания полученной смеси в течение одного часа при 0 С ее вливают в смесь воды и льда, добавляют 37%-ную соляную кислоту до получения прозрачного раствора. Среду экстрагируют этилацетатом, промывают водой, органическую фазу высушивают и растворитель выпаривают при пониженном давлении. Получают белое твердое вещество, которое растворяют в 500 мл диоксана. К этой смеси добавляют 4,6 мл серной кислоты (98%) в 50 мл этанола и 3,6 г 10% Pd/C. После выдерживания полученной смеси в течение 5 ч в атмосфере водорода катализатор отфильтровывают, растворитель выпаривают, остаток растворяют в воде и среду экстрагируют этилацетатом. Полученный-7 007324 продукт растворяют в 20 мл метанола и к этой смеси прибавляют 8 мл 1 н. водного раствора гидроксида натрия, и среду перемешивают при комнатной температуре в течение 2 ч. Растворитель выпаривают, добавляют воду, среду доводят до кислого рН, экстрагируют и выделяют белое твердое вещество. 38,7 г этого продукта растворяют в 1000 мл безводного метиленхлорида в атмосфере азота; среду охлаждают до 0 С и к этой смеси осторожно прибавляют 47 г (0,225 моль) PCl3, среду перемешивают при 0 С в течение двух часов и к этой смеси медленно прибавляют 121,8 г (0,467 моль) SnCl4, и через 10 мин среду оставляют нагреваться до комнатной температуры. После выдерживания в течение двух часов при комнатной температуре среду вливают в смесь воды и льда, экстрагируют метиленхлоридом и выделяют масло, которое затем очищают посредством хроматографии на колонке с силикагелем, элюируя смесью циклогексан/этилацетат (соотношение 8/2). Получают 6,0 г соединения, указанного в заголовке. Подготовительный пример 2. 2,3-Дигидро-1 Н-циклопента[b]нафталин-6-илуксусная кислота. Процедуру проводят в токе азота, при этом 3 г гидрида натрия (80%, 0,075 моль) перемешивают в 845 мл тетрагидрофурана и к этой смеси прибавляют 0,7 г (0,0375 моль) продукта из подготовительного примера 1, растворенного в 45 мл тетрагидрофурана. После 1-часового нагревания с обратным холодильником добавляют 9 мл (0,075 моль) диэтилкарбоната в 45 мл тетрагидрофурана и среду продолжают перемешивать при нагревании с обратным холодильником в течение 5 ч. Смесь охлаждают и к ней прибавляют насыщенный водный раствор хлорида аммония и воду до полного растворения, и среду экстрагируют этилацетатом. Среду промывают водой, органическую фазу высушивают и растворитель выпаривают при пониженном давлении. Полученное масло растворяют в 60 мл тетрагидрофурана и этот раствор при комнатной температуре в атмосфере азота прибавляют к суспензии гидрида натрия (80%) (1,6 г, 0,41 моль) в 30 мл тетрагидрофурана. Среду перемешивают в течение 30 мин и прибавляют 8,8 мл этилбромацетата (0,08 моль), и среду перемешивают при комнатной температуре в течение 5 ч. Прибавляют хлорид аммония и среду экстрагируют метиленхлоридом. Среду промывают водой, органическую фазу высушивают и растворитель выпаривают при пониженном давлении. Полученное таким образом масло растворяют в 600 мл 0,5 н. водно-спиртового раствора гидроксида натрия (соотношение ЕtOН/Н 2O составляет 2/1). Среду нагревают примерно при 40 С до тех пор, пока продукт полностью не превратится в его натриевую соль (образование продукта с основанием при проведении тонкослойной хроматографии,элюция смесью этилацетат/гексан (соотношение 3/7. По прошествии примерно 8 ч половину растворителя выпаривают при пониженном давлении, среду подкисляют 15 мл 1 н. соляной кислоты и нагревают при 50 С в течение примерно 45 мин. Среду экстрагируют этилацетатом, органическую фазу высушивают и растворитель выпаривают при пониженном давлении. Полученный продукт растворяют в 60 мл уксусной кислоты и 4,5 мл серной кислоты (98%), разведенных в 9 мл уксусной кислоты, и прибавляют 1 г 10% Pd/C; среду затем гидрируют в течение 8 ч. Половину растворителя выпаривают, катализатор отфильтровывают, прибавляют воду и среду экстрагируют этилацетатом, органическую фазу высушивают и растворитель выпаривают. Полученный таким образом продукт растворяют в 100 мл метанола, прибавляют 200 мг паратолуолсульфоновой кислоты и среду кипятят с обратным холодильником в течение 4 ч. Растворитель частично выпаривают и прибавляют 50 мл 5%-ного водного раствора бикарбоната натрия, и среду экстрагируют диэтиловым эфиром, промывают водой и рассолом. Органическую фазу высушивают и растворитель выпаривают при пониженном давлении. Полученный таким образом продукт растворяют в декалине (20 мл), прибавляют 1,4 г 10% Pd/C и среду нагревают при 200 С в течение 48 ч. Катализатор отфильтровывают, растворитель выпаривают и сырой реакционный продукт очищают посредством хроматографии на колонке с силикагелем, элюируя смесью циклогексан/этилацетат (соотношение 9/1). 750 мг полученного таким образом продукта растворяют в 30 мл этанола, прибавляют 20 мл 1 н NaOH и среду нагревают при 50 С в течение 4 ч. Этанол выпаривают и среду подкисляют 1 н. HCl. Получают белое твердое вещество, т.пл. 185-188 С. Подготовительный пример 3. 5-(2-Бромэтил)индан. 2 мл (0,016 моль) индана в 33 мл метиленхлорида и 1,6 мл (0,019 моль) бромацетилбромида смешивают при 0 С. Медленно добавляют 2,27 г (0,017 моль) AlCl3 и среду оставляют нагреваться до комнатной температуры и перемешивают в течение 2 ч. Эту смесь вливают в смесь воды и льда и экстрагируют метиленхлоридом. Органическую фазу высушивают, фильтруют и растворитель выпаривают при пониженном давлении. Сырой реакционный продукт кристаллизуют из гексана. Отделяют белое твердое вещество (т.пл. 57,8-58,1 С), соответствующее продукту ацилирования. 1,67 г (7 ммоль) этого продукта растворяют в 3,9 мл триэтилсилана и 3,9 мл трифторуксусной кислоты и среду нагревают при 80 С в течение двух часов. Среду вливают в смесь льда и NaOH и экстрагируют этилацетатом. Получают соединение, указанное в заголовке. Подготовительный пример 4. 2-(2-Бромэтил)-5,6,7,8-тетрагидронафталин. Соединение, указанное в заголовке, получают, выполняя процедуру, как описано в подготовительном примере 3, но с использованием тетралина вместо индана.-8 007324 Подготовительный пример 5. 5-(3-Бромпропил)индан. Соединение, указанное в заголовке, получают, выполняя процедуру, как описано в подготовительном примере 3, но с использованием бромпропионилбромида вместо бромацетилбромида. Подготовительный пример 6. 5-(3-Бромбутил)индан. Соединение, указанное в заголовке, получают, выполняя процедуру, как описано в подготовительном примере 3, но с использованием бромбутаноилбромида вместо бромацетилбромида. Подготовительный пример 7. 2-(2-Бромэтил)флуорен. Соединение, указанное в заголовке, получают, выполняя процедуру, как описано в подготовительном примере 3, но с использованием флуорена вместо индана. Подготовительный пример 8. 2-(2-Бромэтил)-9,10-дигидрофенантрен. Соединение, указанное в заголовке, получают, выполняя процедуру, как описано в подготовительном примере 3, но с использованием 9,10-дигидрофенантрена вместо индана. Подготовительный пример 9. 3-Флуоренилуксусная кислота. 9 а) 9-Оксофлуорен-3-илкарбоновая кислота. Смесь 0,45 г (0,00186 моль) 2,5-бифенилдикарбоновой кислоты и 13,9 г полифосфорной кислоты(ПФК) нагревают до 200 С. По прошествии одного часа среду охлаждают до 100 С и прибавляют лед до получения объема примерно 100 мл. Среду фильтруют и к осадку прибавляют раствор 0,5 г NaOH в 90 мл воды, среду перемешивают в течение одного часа при 60 С. Среду подкисляют соляной кислотой и экстрагируют этилацетатом. Органическую фазу высушивают, фильтруют и растворитель выпаривают при пониженном давлении. Получают 0,31 г продукта, указанного в заголовке, в виде светлокоричневого твердого вещества. 9 б) Флуорен-3-илкарбоновая кислота. 0,27 г (0,0012 моль) продукта, полученного на предыдущей стадии, растворяют в 2,5 мл этиленгликоля и прибавляют 0,1 г NaOH и 0,25 мл гидразина (98%, d=1,03). Среду кипятят с обратным холодильником в течение 1,5 ч, оставляют охлаждаться до комнатной температуры, прибавляют 70 мл воды и среду подкисляют до рН=6 соляной кислотой. Среду экстрагируют этилацетатом, органическую фазу высушивают, фильтруют и растворитель выпаривают при пониженном давлении. Таким образом получают 0,4 г продукта, указанного в заголовке, в виде желтого твердого вещества. 9 в) Метиловый эфир флуорен-3-илкарбоновой кислоты. Сложный эфир продукта, полученного на предыдущей стадии, получают с помощью метанола в соляной кислоте. Раствор доводят до щелочного рН и экстрагируют этилацетатом с получением продукта,указанного в заголовке, в виде масла. 9 г) 3-Гидроксиметилфлуорен. Смесь 0,68 г LiAlH4 в 7 мл безводного этилового эфира охлаждают до 0 С в токе азота и по каплям прибавляют раствор 3,4 г (0,0152) продукта, полученного на предыдущей стадии, в 27 мл безводного этилового эфира и среду перемешивают в течение ночи. Затем прибавляют смесь воды и льда, среду экстрагируют этилацетатом, органическую фазу высушивают, фильтруют и растворитель выпаривают при пониженном давлении. Таким образом получают 3 г сырого продукта, который очищают посредством хроматографии на колонке с силикагелем, элюируя смесью циклогексан/этилацетат (соотношение 7/3), с получением 1,33 г продукта, указанного в заголовке, в виде белого твердого вещества. 9 д) 3-Хлорметилфлуорен. Раствор 35 мг (0,178 ммоль) продукта, полученного на предыдущей стадии, в 0,5 мл метиленхлорида охлаждают до 0 С и к нему по каплям прибавляют 0,13 мл тионилхлорида (1,178 ммоль). Смесь перемешивают в течение трех часов при комнатной температуре и к ней добавляют смесь воды и бикарбоната натрия до рН 8. Среду экстрагируют метиленхлоридом, органическую фазу высушивают, фильтруют и растворитель выпаривают при пониженном давлении. Таким образом получают 20 мг продукта, указанного в заголовке, в виде желтого масла. 9 е) 3-Цианометилфлуорен. 1,3 г (0,00605 г) продукта, полученного на предыдущей стадии, растворяют в 32 мл ДМСО и прибавляют 0,44 г (0,00666 моль) KCN. Среду нагревают при 80 С в течение 3 ч, к ней прибавляют смесь воды и льда и среду экстрагируют этилацетатом. Органическую фазу высушивают, фильтруют и растворитель выпаривают при пониженном давлении. Сырой продукт очищают посредством флэшхроматографии, элюируя смесью циклогексан/этилацетат (соотношение 8/2). Таким образом получают 250 мг продукта, указанного в заголовке. 9 ж) 3-Флуоренилуксусная кислота. Цианопроизводное, полученное на предыдущей стадии, гидролизуют с помощью 6 н. соляной кислоты при кипячении с обратным холодильником. Добавляют смесь воды и льда и среду экстрагируют этилацетатом. Органическую фазу высушивают, фильтруют и растворитель выпаривают при пониженном давлении. Таким образом получают 0,22 г продукта, указанного в заголовке, в виде твердого вещества. Т.пл. 166-168 С. Подготовительный пример 10. 2,3-Дигидро-1 Н-циклопента[а]нафталин-8-илуксусная кислота. 10 а) 4,4-Диэтокси-2-индан-4-илметиленбутиронитрил.-9 007324 0,23 г (0,0092 моль) NaH (60%) в 20 мл безводного ТГФ охлаждают при 0 С в токе азота и прибавляют раствор 2,26 г (0,0085 моль) диэтилового эфира (1-циано-3,3-диэтоксипропил)фосфоновой кислоты в 16 мл ТГФ. Через 30 мин смесь становится прозрачной и к ней затем по каплям прибавляют раствор 1,1 г (0,0075 моль) 2,3-дигидро-1 Н-инден-8-илбензальдегида в 16 мл ТГФ. Среду перемешивают в течение одного часа при 0 С и прибавляют смесь воды и льда. Среду экстрагируют метиленхлоридом, органическую фазу промывают водным раствором NaOH, органическую фазу высушивают, фильтруют и растворитель выпаривают при пониженном давлении. Таким образом получают 2,49 г сырого продукта в виде оранжевого масла, которое очищают посредством флэш-хроматографии, элюируя смесью циклогексан/этилацетат (соотношение 95/5), с получением продукта, указанного в заголовке. 10 б) 8-Циано-2,3-дигидро-1 Н-циклопента[а]нафталин. Смесь 0,56 г (0,0021 моль) продукта, полученного на предыдущей стадии, в 14 мл метиленхлорида охлаждают до 0 С и к ней по каплям прибавляют 152 мл (0,0013 моль) SnCl4 (d=2,226). Среду перемешивают при комнатной температуре в течение 4 ч, затем к ней прибавляют бикарбонат натрия (10%) и среду экстрагируют метиленхлоридом. Органическую фазу высушивают, фильтруют и растворитель выпаривают при пониженном давлении. Таким образом получают 0,43 г продукта, указанного в заголовке, в виде светло-желтого твердого вещества. 10 в) 8-Метилкарбонил-2,3-дигидро-1 Н-циклопента[а]нафталин. 0,5 г (0,0025 моль) продукта, полученного на предыдущей стадии, растворяют в 20 мл безводного толуола и по каплям в токе азота прибавляют 1,6 мл (0,005 моль) 3 М йодида метилмагния. Среду перемешивают в течение ночи при комнатной температуре и затем к ней добавляют воду и соляную кислоту. Среду перемешивают в течение 15 мин, доводят до щелочного рН с помощью 5 М раствора NaOH и экстрагируют этилацетатом. Органическую фазу промывают, высушивают, фильтруют и растворитель выпаривают при пониженном давлении. Таким образом получают 0,69 г сырого продукта, который очищают посредством флэш-хроматографии, элюируя смесью гексан/диэтиловый эфир (соотношение 95/5), с получением 0,35 г продукта, указанного в заголовке. 10 г) 8-(Морфолинотиокарбонил)-2,3-дигидро-1 Н-циклопента[а]нафталин. 0,35 г (0,0016 моль) продукта, полученного на предыдущей стадии, растворяют в 0,35 мл (0,0016 ммоль) морфолина (d=0,999) и прибавляют 0,54 г (0,0020) серы и кристалл паратолуолсульфоновой кислоты (ПТСК). Среду кипятят с обратным холодильником в течение 6 ч и затем прибавляют метанол. Среду перемешивают при комнатной температуре в течение ночи. Растворитель удаляют при пониженном давлении и таким образом получают 2,5 г сырого продукта, который очищают посредством флэшхроматографии, элюируя смесью циклогексан/этилацетат (соотношение 9/1), с получением 0,16 г продукта, указанного в заголовке. 10 д) 2,3-Дигидро-1 Н-циклопента[а]нафталин-8-илуксусная кислота. 0,16 г (0,0005 моль) продукта, полученного на предыдущей стадии, растворяют в 7,6 мл раствора метанол/вода (1:1) и прибавляют 0,02 г твердого NaH. Среду кипятят с обратным холодильником в течение 6 ч, затем растворитель удаляют при пониженном давлении, остаток переносят в смесь воды и этилацетата, органическую фазу удаляют, водную фазу подкисляют и экстрагируют метиленхлоридом. Органическую фазу высушивают, фильтруют и растворитель выпаривают при пониженном давлении. Таким образом получают 20 мг продукта, указанного в заголовке. Подготовительный пример 11. 5,6,7,8-Тетрагидрофенантрен-3-илуксусная кислота. Продукт, указанный в заголовке, получают, следуя методике, описанной в подготовительном примере 10, но используя 5,6,7,8-тетрагидронафталин-1-илбензальдегид вместо 2,3-дигидро-1 Н-инден-7 илбензальдегида. Подготовительный пример 12. 5,6,7,8-Тетрагидроантрен-2-илуксусная кислота. Продукт, указанный в заголовке, получают, следуя методике, описанной в подготовительном примере 10, но с использованием 5,6,7,8-тетрагидронафталин-2-илбензальдегида вместо 2,3-дигидро-1 Нинден-7-илбензальдегида. Пример 1. 1-[2-(2,3-Дигидро-1 Н-циклопента[b]нафталин-6-ил)этил]-4-[3-трифторметилфенил]1,2,3,6-тетрагидропиридин и его гидрохлорид. 1 а) 1-[4-Гидрокси-4-(3-трифторметилфенил)-1-пиперидинил]-2-(2,3-дигидро-1 Н-циклопента[b] нафталин-6-ил)-1-этанон. 400 мг (1,76 моль) продукта из подготовительного примера 2 растворяют в 10 мл безводного метиленхлорида в атмосфере азота и прибавляют 430 мг (1,76 моль) 4-гидрокси-4-(3-трифторметилфенил) пиперидина, 0,79 г (1,76 моль) ВОР и 0,73 мл триэтиламина, среду перемешивают при комнатной температуре в течение 3 ч. Прибавляют 40 мл этилацетата, среду промывают 1 н. раствором соляной кислоты,- 10007324 затем 1 н. гидроксидом натрия и затем водой. Органическую фазу высушивают над сульфатом натрия и растворитель выпаривают. Получают 0,8 г соединения, указанного в заголовке, в виде масла. 1 б) 1-[2-(2,3-Дигидро-1 Н-циклопента[b]нафталин-6-ил)этил]-4-гидрокси-4-(3-трифторметилфенил) пиперидин. Продукт, полученный в примере 1 а, растворяют в 7 мл безводного ТГФ, среду кипятят с обратным холодильником, прибавляют 0,5 мл боран-диметилсульфида и среду кипятят с обратным холодильником в течение 4 ч. Среду охлаждают до 0-5 С и осторожно прибавляют 7 мл метанола. По прошествии 5 мин среду кипятят с обратным холодильником еще в течение 30 мин, растворитель выпаривают, остаток переносят в смесь вода/аммиак (соотношение 1/1), среду экстрагируют этилацетатом, эти две фазы разделяют и органическую фазу промывают водой. Среду высушивают над сульфатом натрия и растворитель выпаривают при пониженном давлении. Сырой кристаллический продукт очищают в 2-пропиловом эфире. Получают 0,37 г продукта, указанного в заголовке. Т.пл. 154-156 С. 1 в) 1-[2-(2,3-Дигидро-1 Н-циклопента[b]нафталин-6-ил)этил]-(4-(3-трифторметилфенил)-1,2,3,6 тетрагидропиридин и его гидрохлорид. 350 мг (0,8 ммоль) продукта, полученного на предыдущей стадии, растворяют в 10 мл уксусной кислоты, прибавляют 1 мл серной кислоты (96%) и среду нагревают при 80 С в течение 2 ч. Среду охлаждают, прибавляют концентрированный NH4OH и эту среду экстрагируют этилацетатом. Среду промывают водой, высушивают и упаривают при пониженном давлении с получением соединения, указанного в заголовке. Гидрохлорид получают с помощью раствора 2-пропанола, насыщенного соляной кислотой; т.пл. (гидрохлорид) 264-266 С. Пример 2. 1-[2-(2,3-Дигидро-1 Н-инден-5-ил)этил]-4-[3-трифторметилфенил]-1,2,3,6-тетрагидропиридин и его гидрохлорид. Продукт, полученный в подготовительном примере 3, растворяют в 17 мл бутанола. Прибавляют 0,84 г (3,2 моль) 4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридина и 0,9 г (6,5 ммоль) карбоната калия и среду кипятят с обратным холодильником в течение 5 ч. Растворитель выпаривают и остаток промывают водой. Его экстрагируют метиленхлоридом, органическую фазу высушивают и растворитель выпаривают при пониженном давлении. Остаток очищают на колонке с силикагелем, элюируя смесью циклогексан/этилацетат (соотношение 8/2), и получают соединение, указанное в заголовке. Гидрохлорид получают с помощью 2-пропанола, насыщенного соляной кислотой. Т.пл. (гидрохлорид) 252-255 С. Пример 3. 1-[2-(5,6,7,8-Тетрагидронафталин-2-ил)этил]-4-[3-трифторметилфенил]-1,2,3,6-тетрагидропиридин и его гидрохлорид. Соединения, указанные в заголовке, получают путем выполнения методики, описанной в примере 2, но с использованием продукта из подготовительного примера 4 вместо продукта из подготовительного примера 3; т.пл. (гидрохлорид) 263-267 С. Пример 4. 1-[2-(2,3-Дигидро-1 Н-циклопента[b]нафталин-6-ил)этил]-4-(6-хлорпирид-2-ил)-1,2,3,6 тетрагидропиридин и его гидрохлорид. Соединения, указанные в заголовке, получают путем выполнения методики, описанной в примере 1, но с использованием 4-(6-хлорпирид-2-ил)-1,2,3,6-тетрагидропиридина вместо 4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридина; т.пл. (гидрохлорид) 254-258 С. Пример 5. 1-[2-(2,3-Дигидро-1 Н-циклопента[b]нафталин-6-ил)этил]-4-(6-трифторметилпирид-2-ил)1,2,3,6-тетрагидропиридин и его гидрохлорид. Соединения, указанные в заголовке, получают путем выполнения методики, описанной в примере 1, но с использованием 4-(6-трифторметилпирид-2-ил)-1,2,3,6-тетрагидропиридина вместо 4-(3 трифторметилфенил)-1,2,3,6-тетрагидропиридина; т.пл. (гидрохлорид). 264-267 С. Пример 6. 1-[3-(2,3-Дигидро-1 Н-инден-5-ил)пропил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин и его гидрохлорид.- 11007324 Соединения, указанные в заголовке, получают путем выполнения методики, описанной в примере 2, но с использованием продукта из подготовительного примера 5 вместо продукта из подготовительного примера 3; т.пл. (гидрохлорид) 192-195 С. Пример 7. 1-[4-(2,3-Дигидро-1 Н-инден-5-ил)бутил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин и его гидрохлорид. Соединения, указанные в заголовке, получают путем выполнения методики, описанной в примере 2, но с использованием продукта из подготовительного примера 6 вместо продукта из подготовительного примера 3, т.пл. (гидрохлорид) 198-200 С. Пример 8. 1-[2-(Флуорен-2-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин и его гидрохлорид. Соединения, указанные в заголовке, получают путем выполнения методики, описанной в примере 2, но с использованием продукта из подготовительного примера 7 вместо продукта из подготовительного примера 3; т.пл. (гидрохлорид) 285-287 С. Пример 9. 1-[2-(9,10-Дигидрофенантрен-2-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин и его гидрохлорид. Соединения, указанные в заголовке, получают путем выполнения методики, описанной в примере 2, но с использованием продукта из подготовительного примера 8 вместо продукта из подготовительного примера 3; т.пл. (гидрохлорид) 256-258 С. Пример 10. 1-[2-(2,3-Дигидро-1 Н-циклопента[b]нафталин-6-ил)этил]-(4-(3-трифторметилфенил)1,2,3,6-тетрагидропиридина 1-оксид. 0,27 г м-хлорнадбензойной кислоты прибавляют к раствору 0,47 г (1,1 ммоль) 1 -[2-(2,3-дигидро 1 Н-циклопента[b]нафталин-6-ил)этил]-4-(6-трифторметилпирид-2-ил)-1,2,3,6-тетрагидропиридина в 40 мл метиленхлорида при температуре 0-5 С. Среду продолжают перемешивать при 0-5 С в течение двух часов, промывают насыщенным водным раствором бикарбоната натрия и эти две фазы разделяют. Органическую фазу высушивают, фильтруют и упаривают при пониженном давлении. Эту среду очищают посредством хроматографии, элюируя смесью метанол/этилацетат (соотношение 1/1), и получают указанный в заголовке продукт; т.пл. 116-117 С. Пример 11. 1-[2-(Флуорен-3-ил)этил]-(4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин и его гидрохлорид. Соединения, указанные в заголовке, получают путем выполнения методики, описанной в примере 1, но с использованием продукта из подготовительного примера 9 вместо продукта из подготовительного примера 2; т.пл. (гидрохлорид): 238-240 С. Пример 12. 1-[2-(2,3-Дигидро-1 Н-циклопента[а]нафталин-8-ил)этил]-4-(3-трифторметилфенил)1,2,3,6-тетрагидропиридин и его гидрохлорид.- 12007324 Соединения, указанные в заголовке, получают путем выполнения методики, описанной в примере 1, но с использованием продукта из подготовительного примера 10 вместо продукта из подготовительного примера 2; т.пл. (гидрохлорид): 212-213 С. Пример 13. 1-[2-(5,6,7,8-Тетрагидрофенантрен-3-ил)этил]-(4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин и его гидрохлорид. Соединения, указанные в заголовке, получают путем выполнения методики, описанной в примере 1, но с использованием продукта из Подготовительного примера 11 вместо продукта из подготовительного примера 2; т.пл. (гидрохлорид) 222-224 С. Пример 14. 1-[2-(5,6,7,8-Тетрагидроантрацен-2-ил)этил]-(4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин и его гидрохлорид. Соединения, указанные в заголовке, получают путем выполнения методики, описанной в примере 1, но с использованием продукта из подготовительного примера 12 вместо продукта из подготовительного примера 2, т.пл. (гидрохлорид) 252-253 С. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I)R1 представляет собой атом водорода или галогена или группу CF3;n означает целое число от 1 до 5; А представляет собой группу формулы (а)-(d): где m означает 1 или 2; и его соли или сольваты и его N-оксиды. 2. Соединение по п.1, где n означает 1. 3. Соединение по п.1 или 2, где R1 представляет собой группу CF3. 4. Соединение по пп.1-3, где X представляет собой СН и R1 находится в 3-положении бензола. 5. Соединение по пп.1-3, где X представляет собой N и пиридин замещен в 2,6-положениях. 6. Соединение по п.1, выбранное из следующих соединений: 1-[2-(2,3-дигидро-1 Н-циклопента[b]нафталин-6-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6 тетрагидропиридин,1-[2-(2,3-дигидро-1 Н-инден-5-ил)этил]-4-[3-трифторметилфенил]-1,2,3,6-тетрагидропиридин,1-[2-(5,6,7,8-тетрагидронафталин-2-ил)этил]-4-[3-трифторметилфенил]-1,2,3,6-тетрагидропиридин,1-[2-(2,3-дигидро-1 Н-циклопента[b]нафталин-6-ил)этил]-4-(6-хлорпирид-2-ил)-1,2,3,6-тетрагидропиридин,1-[2-(2,3-дигидро-1 Н-циклопента[b]нафталин-6-ил)этил]-4-(6-трифторметилпирид-2-ил)-1,2,3,6-тетрагидропиридин,- 13007324 1-[3-(2,3-дигидро-1 Н-инден-5-ил)пропил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,1-[4-(2,3-дигидро-1 Н-инден-5-ил)бутил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,1-[2-(флуорен-2-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,1-[2-(9,10-дигидрофенантрен-2-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,1-[2-(2,3-дигидро-1 Н-циклопента[b]нафталин-6-ил)этил]-(4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,1-[2-(флуорен-3-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,1-[2-(2,3-дигидро-1 Н-циклопента[а]нафталин-8-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,1-[2-(5,6,7,8-тетрагидрофенантрен-3-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,1-[2-(5,6,7,8-тетрагидроантрацен-2-ил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин,их солей и сольватов и их N-оксидов. 7. Способ получения соединения (I) по п.1, отличающийся тем, что где R1 такой, как определено в п.1, подвергают взаимодействию с кислотой формулы (III) или одним из ее функциональных производных где n и А такие, как определено в п.1,(б) восстанавливают карбонильную группу полученного таким образом соединения формулы (IV)(в) дегидратируют промежуточный пиперидинол формулы (V)(г) полученное таким образом соединение формулы (I) выделяют и, возможно, превращают в одну(один) из его солей или сольватов или N-оксидов. 8. Соединение формулы (III) где А представляет собой группу формулы (а), (с) или (d), как определено в п.1, n означает целое число от 1 до 5 и m означает 1 или 2, и его соли или сольваты. 9. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы (I) или одну (один) из его фармацевтически приемлемых солей, сольватов или N-оксидов по любому из пп.1-6. 10. Композиция по п.9, отличающаяся тем, что она содержит от 0,001 до 100 мг активного ингредиента. 11. Применение соединения формулы (I) или одной (одного) из его фармацевтически приемлемых солей, сольватов или N-оксидов по любому из пп.1-6 для приготовления аналгезирующих лекарственных- 14007324 средств и/или лекарственных средств, предназначенных для лечения заболеваний, связанных с иммунными и воспалительными нарушениями. 12. Лекарственное средство для лечения заболеваний, опосредованных цитокином TNF-альфа, отличающееся тем, что оно состоит из одного из соединений формулы (I) или одной (одного) из его солей,сольватов или N-оксидов по любому из пп.1-6.
МПК / Метки
МПК: A61K 31/444, A61P 29/00, C07D 401/04, C07D 211/70
Метки: фармацевтические, аралкилтетрагидропиридины, композиции, получение, содержащие
Код ссылки
<a href="https://eas.patents.su/16-7324-aralkiltetragidropiridiny-ih-poluchenie-i-soderzhashhie-ih-farmacevticheskie-kompozicii.html" rel="bookmark" title="База патентов Евразийского Союза">Аралкилтетрагидропиридины, их получение и содержащие их фармацевтические композиции</a>
Производные карбоксамидотиазолов, их получение, содержащие их фармацевтические композиции

Номер патента: 3093
Опубликовано: 26.12.2002
Авторы: Броден Роже, Биньон Эрик, Буажегрэн Робер, Олльеро Доминик, Молимар Жан-Шарль
МПК: A61K 31/427, A61P 1/06, A61K 31/405...
Метки: композиции, фармацевтические, производные, карбоксамидотиазолов, получение, содержащие
Формула / Реферат:
1. Соединение формулы в которой R6a означает хлор или метил; R10, R11, R13 означают, каждый независимо друг от друга, водород, метил, этил, гидроксил, ацетилоксигруппу, метоксигруппу, этоксигруппу, метилтиогруппу, трифторметил, аминогруппу или галоген; при условии, что один или два из заместителей R10, R11, R13 отличаются от водорода; или его соли или сольваты. 2. Соединение по п.1 формулы в которой один или два из заместителей R10a, R11a,...
Фармацевтические композиции, содержащие олигосахариды, и их получение

Номер патента: 4768
Опубликовано: 26.08.2004
Авторы: Мурье Пьер, Висков Кристиан, Перрэн Элизабет
МПК: A61P 29/00, C07H 15/04, A61K 31/70...
Метки: олигосахариды, получение, композиции, фармацевтические, содержащие
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве действующего начала олигосахарид формулы в которой n обозначает целое число от 0 до 25, R1, R3, R4, R5, R6 и R8, одинаковые или разные, обозначают атом водорода или радикал SO3M, R2 и R7, одинаковые или разные, обозначают атом водорода или радикал SO3M или COCH3 и M обозначает натрий, кальций, магний или калий, или смесь указанных олигосахаридов. 2. Фармацевтическая композиция по п.1,...
Новые таксоиды, их получение и фармацевтические композиции, их содержащие

Номер патента: 1516
Опубликовано: 23.04.2001
Авторы: Бушар Эрве, Коммерсон Ален, Бурза Жан-Доминик
МПК: C07D 305/14, A61K 31/335, A61P 35/00...
Метки: содержащие, фармацевтические, композиции, таксоиды, новые, получение
Формула / Реферат:
1. Новые таксоиды общей формулы (I) в которой Z означает радикал общей формулы (II) в которой R1 означает радикал бензоил или алкоксикарбонил; R3 означает фенил; R4 означает радикал гидрокси или радикал алкокси, содержащий от 1 до 4 атомов углерода с прямой или разветвленной цепью, алканоилокси, алканоильная часть которого содержит от 2 до 4 атомов углерода с прямой или разветвленной цепью; R5 означает радикал алкокси, содержащий от 1 до...
Новые таксоиды,их получение и содержащие их фармацевтические композиции

Номер патента: 1533
Опубликовано: 23.04.2001
Авторы: Бушар Эрве, Коммерсон Алан
МПК: A61K 31/335, A61P 35/00, C07D 305/14...
Метки: фармацевтические, композиции, новые, таксоиды,их, получение, содержащие
Формула / Реферат:
1. Новые таксоиды общей формулы в которой Z обозначает радикал общей формулы в которой R1 обозначает бензоильный радикал или радикал R2-О-СО-, в котором R2 представляет собой алкильный радикал с 1-8 атомами углерода и R3 обозначает фенил или a - или b -нафтил; и либо R4 обозначает атом водорода, R6 и R7 вместе образуют кетонную функцию и R и R5 вместе образуют связь, либо R4 обозначает атом водорода или радикал алкокси с 1-6 атомами...
Смеси полисахаридов на основе гепарина, их получение и фармацевтические композиции, их содержащие

Номер патента: 5995
Опубликовано: 25.08.2005
Авторы: Диаз Жак, Перрэн Элизабет, Висков Кристиан, Пеке Кристель
МПК: C08B 37/10, A61K 31/715
Метки: фармацевтические, основе, содержащие, полисахаридов, получение, композиции, гепарина, смеси
Формула / Реферат:
1. Смеси сульфатированных полисахаридов, имеющих общую структуру полисахаридов, входящих в состав гепарина и обладающих следующими характеристиками: они имеют среднюю молекулярную массу в диапазоне от 1500 до 3000 Да, активность анти-Xa в диапазоне от 100 до 150 МЕ/мг, активность анти-IIa в диапазоне от 0 до 10 МЕ/мг и отношение активность анти-Xa/активность анти-IIa больше 10, полисахариды, входящие в состав смесей, содержат от 2 до 26...
Предыдущий патент: Способ получения n-(1-оксопентил)-n-[[2'-(1h-тетразол-5-ил)[1,1'-дифенил] -4-ил] метил]-l-валина (валсартана)
Следующий патент: Применение специфической дозы фондапаринукса натрия для лечения острого коронарного синдрома (окс)
Случайный патент: Производные триазоло[4,5-d]пирамидина и их применение в качестве антагонистов пуринового рецептора