Производные карбоксамидотиазолов, их получение, содержащие их фармацевтические композиции
Номер патента: 3093
Опубликовано: 26.12.2002
Авторы: Молимар Жан-Шарль, Олльеро Доминик, Буажегрэн Робер, Броден Роже, Биньон Эрик







Формула / Реферат
1. Соединение формулы

в которой
R6a означает хлор или метил;
R10, R11, R13 означают, каждый независимо друг от друга, водород, метил, этил, гидроксил, ацетилоксигруппу, метоксигруппу, этоксигруппу, метилтиогруппу, трифторметил, аминогруппу или галоген; при условии, что один или два из заместителей R10, R11, R13 отличаются от водорода;
или его соли или сольваты.
2. Соединение по п.1 формулы

в которой один или два из заместителей R10a, R11a, R13a означают метил, метоксигруппу, хлор, фтор, трифторметил, причем другие или другой из заместителей означает водород, или его соли, или сольваты.
3. Соединение по п.1, которое выбирают из группы, состоящей из
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5-метилиндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5,7-диметилиндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-4-метоксииндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-4-метилиндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-4,5-диметилиндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5-метоксииндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5-хлориндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-4,5-дихлориндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-4,7-диметилиндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-4,5-диметоксииндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-7-метоксииндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-7-метилиндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5,7-дихлориндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-4,7-диметоксииндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5-метокси-7-метилиндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5-метил-7-хлориндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5-хлор-7-метилиндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5-хлор-7-фториндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-4-метил-7-хлориндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-4-метил-5-хлориндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5-хлор-7-трифторметилиндол-1-уксусной кислоты;
2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-4-метокси-7-метилиндол-1-уксусной кислоты;
или его соли или сольваты.
4. Соединение по п.3, представляющее собой 2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5-метилиндол-1-уксусную кислоту, или ее соли или сольваты.
5. Соединение по п.3, представляющее собой 2-(4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5,7-диметилиндол-1-уксусную кислоту, или ее соли или сольваты.
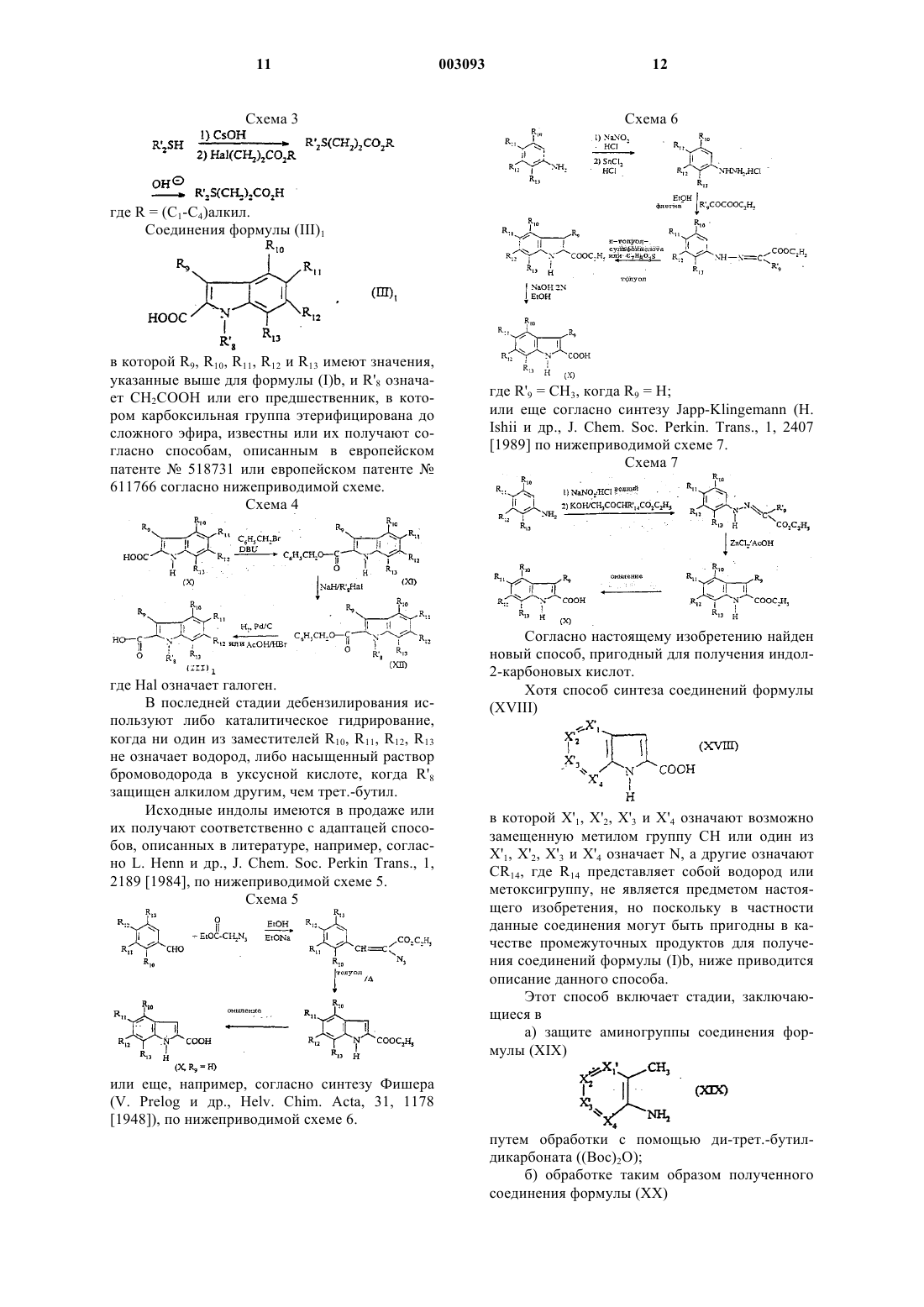
6. Способ получения соединения формулы (Ib) по п.1, или его солей или сольватов, характеризующийся тем, что он включает следующие стадии:
а) конденсация 2-аминотиазола формулы

в которой
R1 означает замещенный фенил формулы

где R6 означает хлор или метил; и
R2 означает

с кислотой формулы
R'3COOH,
в которой R'3 означает функциональное производное кислоты

в котором кислотная группа защищена;
причем R10, R11 и R13 имеют указанные в п.1 значения;
б) превращение полученного таким образом соединения в соединение формулы (Ib) путем удаления защитной группы от защищенной кислотной группы; и
в) выделение таким образом полученного соединения формулы (Ib) таким, какое есть, или в форме одной из его солей, или одного из его сольватов.
7. Фармацевтическая композиция, содержащая эффективную дозу, по крайней мере, одного соединения формулы (Ib) по любому из пп.1-5 или одной (одного) из его фармацевтически приемлемых солей, сольватов или гидратов.
8. Применение соединения по любому из пп.1-5 для получения лекарственных средств, предназначенных для борьбы с заболеваниями, лечение которых требует стимуляции рецепторов ССК-А холецистокинина.
9. Применение по п.8 для получения лекарственных средств, предназначенных для лечения нарушений желудочно-кишечной сферы.
10. Применение по п.8 для получения лекарственных средств, предназначенных для лечения нарушений центральной нервной системы.
11. Применение соединения по любому из пп.1-5 для получения лекарственных средств, пригодных при лечении ожирения.
12. Применение соединения по любому из пп.1-5 для получения лекарственных средств, пригодных при лечении синдрома раздражения толстой кишки.
13. Применение соединения по любому из пп.1-5 для получения лекарственных средств, пригодных при лечении дискинезии.
Текст
1 Область техники, к которой относится изобретение Предметом настоящего изобретения являются производные тиазола - агонисты холецистокинина (ССК) и в особенности агонисты рецепторов типа А холецистокинина (ССК-А),способ их получения и содержащие их лекарственные средства. Предшествующий уровень техники ССК представляет собой пептид, который в ответ на прием пищи выделяется на периферическом уровне и способствует регуляции многочисленных пищеварительных процессов[1994]). ССК идентифицирован также в головном мозге и может быть нейропептидом, в высшей степени эффективным в качестве нейромодулятора церебральных функций за счет стимуляции рецепторов типа ССК-В (Crawley J.N. и др.,Peptides, 15 (4), 731-735 [1994]). В центральной нервной системе ССК взаимодействует с нейронной трансмиссией, опосредуемой допамином (Crawley J.N. и др., ISIS Atlas of Sci., Pharmac, 84-90 [1988]). Он принимает участие также в механизмах, включающих ацетилхолин,GABA (g-аминобутановая кислота), серотонин,подобные опиатам синтетические наркотические препараты, соматостатин, вещество Р, и в ионных каналах. Его введение вызывает физиологические модификации: пальпебральное опущение, гипотермия, гипогликемия, каталепсия, и изменения в поведении: гиполокомоторика, уменьшение эксплорации, аналгезия, изменение способности к обучению, изменение полового поведения и состояния насыщения. ССК проявляет свою биологическую активность через посредство, по крайней мере,двух типов рецепторов: рецепторы ССК-А, локализованные главным образом на периферии, и рецепторы ССК-В, присутствующие в основном в коре головного мозга. Рецепторы ССК-А периферического типа также имеются в некоторых областях центральной нервной системы, включающих посттремальную область, ядро одиночного пути и межножковое ядро (Моran Т.Н. и др., Brain Research, 362, 175-179 [1986]; Hill D.R. и др., J. Neurosci., 10, 1070-1081 [1990]), однако,с видовыми различиями (Hill D.R. и др., J.Moran T.N. и др., Trends in Pharmacol. Sci., 12,232-236 [1991]). На периферии, через посредство рецепторов ССК-А (Moran T.N. и др Brain Research,362, 175-179 [1986]), ССК замедляет опорожнение желудка, модулирует интестинальную моторику, стимулирует везикулярное сокращение,повышает желчеотделение и контролирует пан 003093[1987]). Роль ССК в сигнале насыщения заключается фактически в том, что плазматические концентрации ССК, зависящие от состава еды (высокие концентрации протеинов или липидов),после приема пищи превосходят таковые, наблюдаемые до приема пищи (Izzo R.S. и др.,Regul. Pept., 9, 21-34 [1984]; Pfeiffer А. и др.,Eur. J. Clin. Invest., 23, 57-62 [1993]; LieverseR.J., Gut, 35, 501 [1994]). У пациентов с булимией происходит уменьшение выделения ССК,индуцированное приемом пищи (Geraciotti T.D.[1997]), и снижение концентраций ССК в спинно-мозговой жидкости (Lydiard R.B. и др., Am.J. Psychiatry, 150, 1099-1101 [1993]). В Тлимфоцитах, причем клеточный компартмент может отражать центральные нейронные секреции, базальные концентрации ССК значительно ниже у пациентов, больных Bulimia nervosa(Brambilla F. и др., Psychiatry Research., 37, 5156 [1995]). Обработки (например, L-фенилаланином или ингибиторами трипсина), которые повышают секрецию эндогенного ССК, провоцируют уменьшение приема пищи у некоторых видов, в том числе у человека (Hill A.J. и др.,Physiol. Behav., 48, 241-246 [1990]; Ballinger А.В. и др., Metabolism, 43, 735-738 [1994]). Точно также, введение экзогенного ССК способствует уменьшению приема пищи у многочисленных видов, в том числе у человека (Crawley J.N. и др., Peptides, 15, 731-755 [1994]). Ингибирование в отношении приема пищи за счет ССК опосредуется рецептором ССК-А. В самом деле, девазепид, селективный антагонист рецепторов ССК-А, ингибирует вызывающее потерю аппетита действие ССК, тогда как селективные агонисты этих рецепторов ингибируют прием пищи (Asin К.Е. и др., Pharmacol.R.L. и др., J. Med. Chem., 37, 1562-1568 [1994]). Кроме того, крысы OLEFT, не выражающие рецептор ССК-А, не чувствительны к вызывающему потерю аппетита действию ССК (Miyasaka К. и др., 180, 143-146 [1994]). Базирующаяся на этой очевидности ключевой роли ССК в сигнале периферического насыщения является бесспорной пригодность агонистов и антагонистов ССК в качестве лекарственных средств при лечении некоторых нарушений отношения к пище, ожирения и диабета. Агонист рецепторов ССК также может быть использован в терапии при лечении нарушений эмоционального, полового и относящегося к памяти поведения (Itoh S. и др., Drug.(Drugs of Future, 17 (3), 197-206 [1992]). Агонисты рецепторов ССК-А холецистокинина описаны в литературе. Например, некоторые продукты, обладающие такими свойствами, описаны в европейском патенте 383690 и международных заявках 90/06937, 95/28419,96/11701 или еще в международной заявке 96/11940. Большинство агонистов рецепторов ССКА, описанных на сегодняшний день, имеют пептидную природу. Так, FPL 14294, происходящий от ССК-7, представляет собой эффективный агонист рецепторов ССК-А, неселективный по отношению к рецепторам ССК-В. Он обладает сильной ингибирующей активностью в отношении приема пищи в случае крысы и собаки после введения через нос (Simmons R.D. и др.,Pharmacol. Biochem. Behav., 47 (3), 701-708[1994]; Kaiser E.F. и др., Faseb., 5, A864 [1991]). Точно также показано, что А-71623, тетрапептидный селективный агонист рецепторов ССКА, является эффективным в моделях анорексии в течение периода 11 дней и вызывает значительное снижение прибавки в весе по отношению к контролю у грызунов и собакоподобных обезьян (Asin K.E. и др., Pharmacol. Biochem.Behav., 42, 699-704 [1992]). Таким же образом структурные аналоги А-71623, обладающие хорошей эффективностью и селективностью в отношении рецепторов ССК-А, проявляют сильную, вызывающую потерю аппетита активность у крысы (Elliott R.L. и др., J. Med. Chem., 37,309-313 [1994]; Elliott R.L. и др., J. Med. Chem.,37, 1562-1568 [1994]). GW 5823 (Henke B.R. и др., J. Med. Chem., 39, 2655-2658 [1996]; HenkeB.R. и др., J. Med. Chem., 40, 2706-2725 [1997]); бензо-1,5-диазепин является агонистом рецепторов ССК-А in vitro. Эта молекула также активна при введении перорально в отношении сокращения желчного пузыря у мыши и в отношении приема пищи в случае крысы. Производные тиазола - антагонисты холецистокинина описываются в европейском патенте 432040 и заявке на европейский патент 518731. В заявке на европейский патент 518731 описываются соединения, которые взаимодействуют с рецепторами гастрина и холецистокинина формулы 1Z1 особенно может означать индолильную группу формулы в которой Xi имеет различные значения и Rz может означать водород; (C1-C4)алкил; возможно этерифицированную карбоксиалкиленовую группу формулы Z2COOR, в которой Z2 означает (C1-C4)алкилен и R означает водород, бензил,(С 1-С 6)-алкил; карбамоилалкиленовую группу формулы Z2CONRIVRV, в которой RIV и RV, каждый независимо, означают водород, (C1-C6) алкил или с азотом образуют насыщенный гетероциклил; ацильную группу формулы CORVI, в которой RVI означает (C1-C4)алкил или фенил; алкоксикарбонильную группу формулыCOORVII, в которой RVII означает трет.-бутил или бензил. Однако среди соединений формулы 1, описанных в этой заявке, ни одно не включает индолильной группы, содержащей одновременно группы Xi и Rz, отличные от водорода. Соединения формулы 1 и их соли описываются в европейском патенте 518731 как ингибирующие фиксацию холецистокинина с его рецепторами. Они являются более или менее селективными в отношении рецепторов типа А и В и более или менее эффективными антагонистами гастрина. В качестве характерного из этих соединений можно особенно назвать калиевую соль 2[4-(2-хлорфенил)тиаз-2-илкарбамоил]индол-1 уксусной кислоты или SR 27897 В, известную в качестве эффективного антагониста рецепторов ССК-А (Eur. J. Pharmacol., 1, 13-19 [1993]). В заявке на европейский патент 611766 описываются агонисты рецепторов холецистокинина в тесте панкреатической амилазы. Эти соединения отвечают формуле 2 в которой R означает водород, ацетил или группу CH2COOR', причем R' представляет собой водород или (C1-C4)алкил; также как их фармацевтически приемлемые соли. Сущность изобретения Предметом настоящего изобретения конкретно является получение нового семейства производных тиазола, особенно представляющих интерес из-за их агонистической активности в отношении рецепторов ССК-А. Соединения согласно изобретению составили объект систематических исследований в целях охарактеризовывания их возможности вытеснять [125I]-CCK из его сайтов связывания, имеющихся в панкреатических мембранах крысы (рецепторы ССК-А),или клеток ЗТЗ, выражающих человеческий рекомбинантный рецептор ССК-А; их агонистического свойства в отношении рецепторов ССК-А по их способности индуцировать in vitro мобилизацию внутриклеточного кальция в клетках ЗТЗ, выражающих человеческий рецептор ССК-А. Предпочтительно производные тиазола согласно настоящему изобретению обладают способностью связываться с рецепторами ССК-А и стимулировать как ССК мобилизацию внутриклеточного кальция в клеточной линии, выражающей человеческий рекомбинантный рецептор ССК-А. Оказывается, что они являются агонистами рецепторов ССК-А. Соединения согласно настоящему изобретению, кроме того, были исследованы in vivo,оценивая их способность блокировать опорожнение желудка у мыши. Как и ССК, эти соединения блокируют у мыши опорожнение желудка и ведут себя, следовательно, in vivo как агонисты рецепторов ССК-А. Неожиданно они оказываются более эффективными агонистами, чем производные тиазола, описанные в заявке на европейский патент 611766. Эти лучшие характеристики определены, с одной стороны, in vitro, по мобилизации внутриклеточного кальция и, с другой стороны,in vivo, при их введении интраперитонеально,по их способности блокировать опорожнение желудка у мыши. 6 Следовательно, одним из объектов настоящего изобретения являются соединения формулы (I)bR6a означает хлор или метил;R10, R11, R13 означают, каждый независимо друг от друга, водород, метил, этил, гидроксил,ацетилоксигруппу, трифторметил, аминогруппу или галоген;R9 и R12 означают водород; при условии,что один или два из заместителей R10, R11, R13 отличаются от водорода; или их соли или сольваты. Солями присоединения этих соединений являются таковые, получаемые с неорганическими или органическими основаниями; предпочтительными являются нетоксичные фармацевтически приемлемые соли, однако, предметом изобретения являются также другие соли,используемые для выделения или очистки соединений формулы (I)b. Соли соединений формулы (I)b включают соли с органическими или неорганическими основаниями, например, соли щелочных или щелочно-земельных металлов, как соли натрия,калия, кальция, причем предпочтительными являются соли натрия и калия, или с амином,таким как трометамол, или еще соли аргинина,лизина, N-метил-D-глюкамина или любого физиологически приемлемого амина. Настоящее изобретение относится также к сольватам, образуемым соединениями формулы(I)b с водой или неорганическими или органическими кислотами, такими как соляная кислота,бромоводородная кислота, трифторуксусная кислота, серная кислота, сульфокислота, фосфорная кислота, нафталин-2-сульфокислота, птолуолсульфокислота. Наиболее предпочтительную группу соединений согласно изобретению составляют соединения формулы (I)с в которой один или два из заместителей R10a,R11a, R13a означают метил, метоксигруппу, хлор,фтор или трифторметил, причем другие или другой из заместителей означает водород,или их соли или сольваты. Особенно предпочтительны следующие соединения: 8 2-(4-(4-хлор-2,5-диметоксифенил)-5-(2 циклогексилэтил)тиазол-2-илкарбамоил)-5 хлор-7-трифторметилиндол-1-уксусная кислота; 2-(4-(4-хлор-2,5-диметоксифенил)-5-(2 циклогексилэтил)тиазол-2-илкарбамоил)-4 метокси-7-метилиндол-1-уксусная кислота; или их соли или сольваты. В высшей степени предпочтительными являются 2-(4-(4-хлор-2,5-диметоксифенил)-5-(2 циклогексилэтил)тиазол-2-илкарбамоил)-5 метилиндол-1-уксусная кислота; 2-(4-(4-хлор-2,5-диметоксифенил)-5-(2 циклогексилэтил)тиазол-2-илкарбамоил)-5,7 диметилиндол-1-уксусная кислота; или их соли или сольваты, особенно соли натрия и калия, или их сольваты. Вторым объектом настоящего изобретения является способ получения соединений формулы (I)b согласно изобретению. Этот способ характеризуется тем, что он включает следующие стадии: а) конденсация 2-аминотиазольного производного формулы (II) в которой R1 означает замещенный фенил формулы где R6 означает хлор или метил; и с кислотой формулы R'3 СООН (III),в которой R'3 означает функциональное производное кислоты в котором кислотная группа защищена;R10, R11 и R13 указанные выше для формулы (I)b; б) при необходимости, превращение полученного соединения в соединение формулы (I)b путем удаления защитной группы от защищенной кислотной функциональной группы заместителя R'3; и в) таким образом, полученное соединение формулы (I)b выделяют таким, какое есть, или в форме одной из его солей или одного из его сольватов. Стадию а) способа обычно осуществляют в основной среде. В качестве функционального производного кислоты формулы (III) можно использовать реакционноспособную кислоту,ангидрид, смешанный ангидрид или реакционноспособный эфир вышеуказанной карбоновой кислоты. В качестве защитной для кислотной функции группы используют сложный эфир, такой как, например, (C1-C4)алкиловый сложный эфир. Смешанные ангидриды могут быть получены путем воздействия алкилхлорформиата на кислоту в присутствии основания, обычно третичного амина, такого как триэтиламин, причем эту реакцию чаще всего осуществляют в растворителе, таком как дихлорметан, дихлорэтан или хлороформ. Конденсацию аминотиазола формулы (II) с кислотой формулы (III) в форме активированного сложного эфира, получаемого, например,путем воздействия 1-гидроксибензотриазола на кислоту в присутствии дициклогексилкарбодиимида согласно методике, описанной в J. Am.Chem. Soc., 93, 6318-6319 [1971], или путем воздействия 1-бензотриазолилокситрис(диметиламино)фосфонийгексафторфосфата (ВОР) согласно методике, описанной в Synthesis, 751-752[1976], можно осуществлять в растворителе,природу которого выбирают в зависимости от растворимости соединений и типа активации кислотной функциональной группы, предпочтительно в присутствии основания, например, третичного амина, такого как триэтиламин; реакцию обычно проводят при температуре от 0 до 30 С. Соединения формулы (I)b, в которых одинR10, R11, R13 означает гидроксил, получают из соединений формулы (I)b, в которых один заместитель (или заместители) означает ацетилоксигруппу, причем другие заместители являются идентичными, получают путем гидролиза в основной среде. Аминотиазолы формулы (II) получают известными способами, такими как способы, описанные в европейском патенте 518731 и заявке на европейский патент 611766. В общем, тиомочевину вводят во взаимодействие с галогенированным кетоном формулы(IV) согласно следующей реакционной схеме: Схема 1 быть получены путем воздействия брома в кислой среде, бромида двухвалентной меди или фенилтриметиламмонийтрибромида (РТТ) на соединение формулы (V)R2CH2COR1 в которой R1, R2 имеют значения, указанные выше для формулы (II), в органическом растворителе, таком как этилацетат, хлорсодержащий растворитель или их смесь. Кетоны формулы (V) обычно получают по реакции Фриделя-Крафтса в присутствии кислоты Льюиса, такой как, например, АlСl3 илиTiCl4. Галогенкетоны формулы (IV) также можно получать по реакции Фриделя-Крафтса при использовании соответствующего галоидангидрида кислоты HalCOCHHalR2 (VI) и надлежащим образом замещенного бензола (R1H=C6H3Bull., 39 (9), 2400-2407 [1991]. Аминотиазол формулы (II) также может быть получен в одну стадию, исходя из замещенного ацетофенона формулы (V) путем воздействия на него последовательно брома или фенилтриметиламмонийтрибромида в растворителе, таком как дихлорметан или тетрахлорид углерода, затем тиомочевины в спирте, таком как, например, этанол или метанол. Аминотиазолы формулы (II) также можно получать при использовании реакции Hoesch[1945], или согласно Satchell и др., The Chemistry of the Carbonyl Group, изд. S. Patai, Interscience, 1 (5), 233-302 [1966]) с последующей конденсацией с тиомочевиной. Галоидангидриды кислоты формулы (VI) получают из соответствующей кислоты формулы R2CH2COOH (VII) обычными способами,например, путем воздействия тионилхлорида или оксалилхлорида. Кислоты формулы (VII) известны или их получают известными способами. Особенно можно использовать триэтилфосфонокротонат и поступать согласно нижеприводимой реакционной схеме для получения соединений формулы R'2-(СН 2)3-СO2 Н, в которой R'2 представляет собой (C5-C7)циклоалкил. Схема 2(IV) имеют такие же значения, что и указанные для соединения формулы (II). Hal представляет собой галоген, предпочтительно бром или хлор. Галогенированные кетоны формулы (IV) могут быть получены общеизвестными способами, принципы которых описываются в общих руководствах. Например, бромкетоны могут где р = 1, 2 или 3. Кислоту формулы R'2-S-(СН 2)2-СООН, в которой R'2 представляет собой (С 5-С 7)циклоалкил, можно получать из соединения формулыR'2SH путем воздействия на него гидроксидом цезия, затем эфиром галогеналкановой кислоты,согласно нижеприводимой реакционной схеме. в которой R9, R10, R11, R12 и R13 имеют значения,указанные выше для формулы (I)b, и R'8 означает СН 2 СООН или его предшественник, в котором карбоксильная группа этерифицирована до сложного эфира, известны или их получают согласно способам, описанным в европейском патенте 518731 или европейском патенте 611766 согласно нижеприводимой схеме. Схема 4 где Hal означает галоген. В последней стадии дебензилирования используют либо каталитическое гидрирование,когда ни один из заместителей R10, R11, R12, R13 не означает водород, либо насыщенный раствор бромоводорода в уксусной кислоте, когда R'8 защищен алкилом другим, чем трет.-бутил. Исходные индолы имеются в продаже или их получают соответственно с адаптацей способов, описанных в литературе, например, согласно L. Неnn и др., J. Chem. Soc. Perkin Trans., 1,2189 [1984], по нижеприводимой схеме 5. Схема 5 где R'9 = СН 3, когда R9 = Н; или еще согласно синтезу Japp-Klingemann (H.[1989] по нижеприводимой схеме 7. Схема 7 Согласно настоящему изобретению найден новый способ, пригодный для получения индол 2-карбоновых кислот. Хотя способ синтеза соединений формулы в которой X'1, Х'2, Х'3 и Х'4 означают возможно замещенную метилом группу СН или один изCR14, где R14 представляет собой водород или метоксигруппу, не является предметом настоящего изобретения, но поскольку в частности данные соединения могут быть пригодны в качестве промежуточных продуктов для получения соединений формулы (I)b, ниже приводится описание данного способа. Этот способ включает стадии, заключающиеся в а) защите аминогруппы соединения формулы (XIX)[1948]), по нижеприводимой схеме 6. путем обработки с помощью ди-трет.-бутилдикарбоната Вос)2O); б) обработке таким образом полученного соединения формулы (XX) алкиллитием, таким как н-BuLi или втор.-BuLi; в) конденсации таким образом полученного литийсодержащего производного с эфиром щавелевой кислоты, таким как этилоксалат или бензилоксалат; г) циклизации в кислой среде; д) омылении таким образом полученного сложного эфира формулы (XXI) в которой А означает этил, или гидрогенолизе таким образом полученного сложного эфира формулы (XXI), в которой А означает бензил. В стадии б) введение лития осуществляют согласно D. Hands и др Synthesis, 877-882[1991]. В варианте, заключающемся в использовании на стадии в) бензилоксалата вместо этилоксалата, избегают промежуточных стадий омыления и этерификации до образования сложного эфира. Стадия г) может быть осуществлена в присутствии трифторуксусной кислоты или путем нагревания в присутствии 6 н. НСl. Указанный выше способ может быть эффективно использован для получения индол-2 карбоновых кислот формулы (Х)b в которой R10b, R11b, R12b, R13b, каждый независимо друг от друга, означают водород или метил, из ортометиланилина. В этом частном случае способ включает стадии, заключающиеся в а) защите аминогруппы ортометиланилина формулы (XI)b путем обработки с помощью ди-трет.бутилдикарбоната (Вос)2O; б) обработке таким образом полученного соединения формулы (ХII)b 14 алкиллитием, таким как н-BuLi или втор.-BuLi; в) конденсации таким образом полученного литийсодержащего производного с эфиром щавелевой кислоты, таким как этилоксалат или бензилоксалат; г) циклизации в кислой среде; д) омылении таким образом полученного сложного эфира формулы (XIII)b в которой А означает этил, или гидрогенолизе таким образом полученного сложного эфира формулы (ХIII)b, в которой А означает бензил. В этих частных случаях, где либо R13b = R12b = СН 3 и R10b = R11b = Н,либо R13b = R10b = СН 3 и R11b = R12b = Н,на стадии д) получают 2 сложных эфира формул Эти соединения могут быть разделены известными из органической химии методами,например, путем хроматографии. Производное 1H-пирроло[3,2-b]пиридин-2 карбоновой кислоты можно получать, например, согласно нижеприводимой реакционной схеме. Схема 8 Когда исходные пиридины имеют один или несколько метокси-заместителей, описанных в схемах 8 и 9 реакции позволяют получать соединения формулы (III)3, замещенные в пиридиновом кольце одной или несколькими метоксигруппами. Производные пирролопиридинкарбоновых кислот формулы (III)3 в которой один из X1, Х 2, Х 3, Х 4 означает N,другие означают CR14, где R14 означает Н или метоксигруппу и R'8 означает СН 2 СООН или его предшественник, в котором карбоксильная группа этерифицирована, также могут быть получены известными способами. Производные 1H-пирроло[2,3-с]пиридин 2-карбоновых кислот и 1H-пирроло-[3,2-с] пиридин-2-карбоновых кислот могут быть получены из соответствующих метилпиридинов,нитрованных в орто-положении к метилу, согласно модификации способа Reissert, как описывается, например, у В. Frydman и др., J. Org.Het. Chem., 775-776 [1969]. Производное 1H-пирроло[3,2-b]пиридин-2 карбоновой кислоты может быть получено, например, следуя нижеприводимой реакционной схеме 10. Схема 10 Согласно изобретению соединения формулы (I)b включают также такие, в которых один или несколько атомов водорода, углерода или галогена, особенно хлора или фтора, заменены их радиоактивным изотопом, например, тритием или углеродом-14. Такие меченые соединения пригодны для поисковых работ, в области метаболизма или фармакокинетики, в биохимических опытах в качестве меченых лигандов рецепторов. Соединения формулы (I)b являлись предметом исследований в отношении связывания invitro рецепторов ССК-А и ССК-В при использовании метода, описанного в Eur. J. Pharmacol.,232, 13-19 [1993]. Они обладают сильным сродством к рецепторам ССК-А (ингибирующая концентрация ИК 50 порядка 10-9 моль) и отчетливо более слабым сродством к рецепторам ССК-В, иногда с соотношением, по крайней мере, равным 100 между этими двумя сродствами. В качестве примера, соединение примера 5 связывается с человеческим рецептором ССК-А с сильным сродством (ИК 50 = 0,56 нмоль), выше такового ССК (ИК 50 = 1,17 нмоль), и сродство этого со 003093 16 единения к человеческому рецептору ССК-В является слабым (ИК 50 = 162 нмоль). Агонистическую активность соединений по отношению к рецепторам ССК-А оценивалиin vitro в ЗТЗ-клетках, выражающих человеческий рецептор ССК-А, путем определения мобилизации внутриклеточного кальция ([Са]i),по методике, представленной Lignon M.F. и др.,Eur. J. Pharmacol., 245, 241-245 [1993]. Концентрацию кальция [Са]i оценивали с помощьюFura-2 в качестве флуоресцентного зонда по методу двойной длины волны возбуждения. Соотношение излучаемой флуоресценции при двух длинах волн после калибровки дает концентрацию [Са]i (G. Grynkiewicz и др., J. Biol. Chem.,260, 3440-3450 [1985]). Как и ССК, соединения согласно изобретению повышают концентрацию внутриклеточного кальция ([Са]i) при ЭК 50 (эффективная концентрация, индуцирующая 50% эффекта ССК), ниже или равной 100 нмоль. Следовательно, они действуют как агонисты рецепторов ССК-А. На этом основании они более эффективны, чем соединения, описанные в европейском патенте 611766, которые не проявляют никакого агонистического свойства по отношению к [Са]i при концентрации 100 нмоль. В качестве примера соединение примера 5 стимулирует повышение концентрации внутриклеточного кальция до того же самого уровня, как и сам ССК, следовательно, оно действует как тотальный агонист. Его действие проявляется при очень слабой дозе (ЭК 50 = 1,27 нмоль), как в случае самого ССК (ЭК 50 = 1,28 нмоль). Изучение агонистического действия соединений на опорожнение желудка осуществляли следующим образом. Самок мышей Swissalbino CDI (20-25 г) доводили до состояния сильного голода в течение 18 ч. В день эксперимента, интраперитонеально вводили продукты (в виде суспензии в 1%-м растворе карбоксиметилцеллюлозы или 0,6%-м растворе метилцеллюлозы) или соответствующий эксципиент,за 30 мин до введения содержащей уголь еды(0,3 мл на мышь суспензии в воде 10% порошкообразного угля, 5% гуммиарабика и 1% карбоксиметилцеллюлозы или 0,6% метилцеллюлозы). Мышей умерщвляли спустя 5 мин путем свертывания шеи и опорожнение желудка оценивали по наличию угля в кишечнике за сфинктором привратника (Eur. J. Pharmacol., 232, 1319 [1993]). Соединения формулы (I)b блокируют опорожнение желудка как и сам ССК и, следовательно, действуют как агонисты рецепторов ССК-А. Соединения согласно изобретению имеют ЭД 50 (эффективная доза, индуцирующая 50% эффекта ССК), ниже или равные 0,1 мг/кг при введении интраперитонеально. В тех же самых условиях соединения, описанные в европейском патенте 611766, не проявляют агонистического свойства по отно 17 шению к опорожнению желудка в дозе 0,1 мг/кг и имеют ЭД 50 выше 1 мг/кг при введении интраперитонеально. В качестве примера соединение примера 5 очень активно in vitro, и оно полностью ингибирует опорожнение желудка с ЭД 50, составляющей 1,9 мкг/кг, при введении интраперитонеально. Следовательно, соединения формулы (I)b особенно эффективны, в качестве агонистов рецепторов ССК-А и ССК, для получения лекарственных средств, предназначенных для борьбы с заболеваниями, лечение которых требует стимуляции рецепторов ССК-А холецистокинина. В особенности соединения формулы (I)b используются для получения лекарственных средств, предназначенных для лечения некоторых нарушений желудочно-кишечной сферы(профилактика желчных конкрементов, синдрома раздражения толстой кишки), отношения к пище, ожирения или ассоциированных патологий, таких как диабет и гипертония. Соединения формулы (I)b индуцируют состояние насыщения и, таким образом, могут быть использованы для уменьшения приема пищи, лечения булимии, ожирения и вызывать потерю в весе. Соединения формулы (I)b также пригодны для получения лекарственных средств, предназначенных для лечения нарушений центральной нервной системы, особенно нарушений эмоционального, полового и относящегося к памяти поведения, психозов и, в частности, шизофрении, болезни Паркинсона, дискинезии, как замедленная дискинезия или лицевая дискинезия,после длительного лечения нейролептическими средствами. Они могут быть также использованы для лечения нарушений инстинктивной потребности, т.е. для регуляции желаний потребления, в особенности, потребления сахаров, жира, алкоголя или лекарств и обычно аппетитных ингредиентов. Никакого симптома токсичности не наблюдается при использовании этих соединений в фармакологически активных дозах, и их безвредность, следовательно, совместима с их использованием в качестве лекарственных средств для лечения вышеуказанных нарушений и заболеваний. Предметом настоящего изобретения также являются фармацевтические композиции, содержащие эффективную дозу, по крайней мере,одного соединения согласно изобретению или его фармацевтически приемлемой соли или сольвата, и, в желательном случае, в смеси с приемлемыми эксципиентами. Вышеуказанные эксципиенты выбирают в зависимости от фармацевтической формы и желательного способа введения. В фармацевтических композициях настоящего изобретения для перорального, подъязычного, подкожного, внутримышечного, внутривенного, локального, внутритрахеального, внут 003093 18 риносового, чрескожного, ректального или внутриглазного введения действующие начала вышеприведенной формулы (I)b, или их возможные соли, могут быть введены, в унитарных формах введения, в смеси с обычными фармацевтическими носителями, животным и людям для профилактики или лечения вышеуказанных нарушений или заболеваний. Соответствующие унитарные формы введения включают формы для перорального приема, такие как таблетки,желатиновые капсулы, порошки, гранулы и пероральные растворы или суспензии, формы для подъязычного, трансбуккального, внутритрахеального, внутриносового введения, формы для подкожного, внутримышечного или внутривенного введения и формы для ректального введения. Для локального применения можно использовать соединения согласно изобретению в кремах, помадах, лосьонах или примочках для глаз. Для того чтобы достичь желательного профилактического или терапевтического эффекта, доза действующего начала может изменяться от 0,01 до 50 мг на кг массы тела в день. Каждая разовая доза может содержать 0,51000 мг, предпочтительно 1-500 мг активных ингредиентов в комбинации с фармацевтическим носителем. Эту разовую дозу можно вводить 1-5 раз в день для введения суточной дозировки 0,5-5000 мг, предпочтительно 1-2500 мг. Когда приготовляют твердую композицию в форме таблеток, основной активный ингредиент смешивают с фармацевтическим эксципиентом, таким как желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналогичные вещества. Таблетки можно покрывать сахарозой, производным целлюлозы или другими соответствующими веществами или еще их можно обрабатывать таким образом, чтобы они обладали пролонгированной или замедленной активностью и чтобы они высвобождали непрерывно предопределенные количества действующего начала. Лекарственную форму в виде желатиновых капсул получают путем смешения активного ингредиента с разбавителем и выливания полученной смеси в мягкие или твердые желатиновые капсулы. Композиция в форме сиропа или эликсира или для введения в форме капель может содержать активный ингредиент вместе с подсластителем, предпочтительно некалорийным, метилпарабеном и пропилпарабеном в качестве антисептика, также как с вкусовой добавкой и соответствующим красителем. Порошки или гранулы, диспергирующиеся в воде, могут содержать активный ингредиент в смеси с диспергаторами или смачивателями,или суспендирующими компонентами, как поливинилпирролидон, также как с подсластителями или вкусовыми добавками. Для ректального введения используют суппозитории, которые получают со связующи 19 ми, плавящимися при ректальной температуре,как, например, масло какао или полиэтиленгликоли. Для парентерального введения используют водные суспензии, солевые изотонические растворы или стерильные и вводимые путем инъекции растворы, которые содержат фармакологически приемлемые диспергаторы и/или смачиватели, как, например, пропиленгликоль или бутиленгликоль. Действующее начало может быть также использовано для получения лекарственной формы в виде микрокапсул, в случае необходимости вместе с одним или несколькими носителями или добавками, или же с матрицами, такими как полимер или циклодекстрин (лоскут,формы с пролонгированным высвобождением). Композиции согласно изобретению могут быть использованы при лечении или профилактике различных болезненных состояний, где ССК представляет терапевтический интерес. Композиции согласно настоящему изобретению могут содержать, кроме продуктов вышеприведенной формулы (I)b или их фармацевтически приемлемых солей, другие действующие начала, которые могут быть пригодны для лечения вышеуказанных нарушений или заболеваний. Предпочтительно, фармацевтические композиции согласно настоящему изобретению содержат в качестве действующего начала, по крайней мере, одно соединение вышеприведенной формулы (I)b или одну (один) из его фармацевтически приемлемых солей, сольватов или гидратов. Сведения, подтверждающие возможность осуществления изобретения Нижеследующие примеры даны в качестве иллюстрации и не ограничивают объема охраны настоящего изобретения. А. Получение производных 2-aминoтиазолов Препаративный пример 1.1. 2-Амино-5 циклогексилэтил-4-(2,5-диметокси-4-метилфенил)тиазол. А). 4-Циклогексилбутирилхлорид. В течение 4 ч кипятят с обратным холодильником 50 г 4-циклогексанбутановой кислоты в 160 мл тионилхлорида. После выпаривания избыточного количества тионилхлорида, целевой продукт перегоняют. Т кип. = 70-80 С при давлении 400 Па. Б). 4-Циклогексил-1-(2,5-диметокси-4 метилфенил)бутан-1-он. Смешивают 9,4 г полученного на предыдущей стадии соединения с 6,66 г АlCl3 в 150 мл ССl4 и при температуре 4 С добавляют по каплям 7,6 г 2,5-диметокси-4-метилбензола, затем температуру поддерживают в диапазоне 5-10 С в течение 3 ч. Реакционную смесь гидролизуют с помощью разбавленного и охлажденного раствора НСl, затем органическую фазу декантируют и сушат над сульфатом магния. Раствори 003093 20 тель выпаривают, получая целевой продукт. Т пл. = 53,5-54,5 С. В). 2-Амино-5-циклогексилэтил-4-(2,5 диметокси-4-метилфенил)тиазол. Все количество полученного на предыдущей стадии соединения бромируют с помощью 2,5 мл брома в ССl4 при комнатной температуре. Промывают водой, сушат над сульфатом магния, растворитель выпаривают, затем обрабатывают с помощью 100 мл этанола и 8 г тиомочевины. После кипячения с обратным холодильником в течение 3 ч, выпаривают, затем реакционную смесь обрабатывают насыщенным раствором карбоната натрия. Экстрагируют этилацетатом, сушат над сульфатом магния, растворитель выпаривают, затем остаток хроматографируют на колонке с диоксидом кремния,элюируя смесью дихлорметана с этилацетатом(объемное соотношение 70/30); получают 9,3 г целевого соединения. Т пл. = 112 С. Препаративный пример 1.2. 2-Амино-4-(5 хлор-2,4-диметоксифенил)-5-циклогексилэтилтиазол. А). 4-Циклогексил-1-(5-хлор-2,4-диметоксифенил)бутан-1-он. Смешивают 10 г 2,4-диметоксихлорбензола и 10,93 г полученного на стадии А препаративного примера 1.1 хлорангидрида 4 циклогексанбутановой кислоты в 100 мл ССl4 при температуре 0 С и добавляют 6,35 мл TiCl4. После перемешивания в течение 2 ч при температуре 0 С, выливают в охлажденный 1 н. раствор НСl, затем органическую фазу отделяют путем декантации и промывают 0,5 н. растворомNaOH. После высушивания над сульфатом магния и выпаривания растворителя получают 20 г целевого продукта. Б). 2-Амино-4-(5-хлор-2,4-диметоксифенил)-5-циклогексилэтилтиазол. С помощью 3,15 г брома в 20 мл ССl4 обрабатывают 20 г полученного на предыдущей стадии соединения в 200 мл ССl4, промывают водой и отделяют органическую фазу, которую сушат над сульфатом магния, затем выпаривают. Обрабатывают этанолом, затем в течение 3 ч кипятят с обратным холодильником в присутствии 7 г тиомочевины. После выпаривания растворителя обрабатывают этилацетатом, затем промывают насыщенным раствором карбоната натрия и сушат над сульфатом магния. После выпаривания растворителя и перевода в порошкообразное состояние в смеси гептана с диэтиловым эфиром (объемное соотношение 50/50) получают 12,55 г целевого продукта. Т пл. = 113 С. Препаративный пример 1.3. 2-Амино-5 циклогексилэтил-4-(2,4-диметоксифенил)тиазол. А). 4-Циклогексил-1-(2,4-диметоксифенил) бутан-1-он. Смешивают 3,53 г АlСl3 и 5 г хлорангидрида 4-циклогексанбутановой кислоты в 100 мл 21 ССl4 и добавляют по каплям при температуре 0 С 5 г 1,3-диметоксибензола. После перемешивания в течение 3 ч при температуре 0 С всю смесь выливают в охлажденный разбавленный раствор НСl. Органическую фазу отделяют путем декантации, затем промывают с помощью 0,5 н. раствора NaOH. После высушивания над сульфатом магния и выпаривания растворителя хроматографируют на колонке с силикагелем 60 Н (выпускаемой в продажу фирмой Е. Merck,Дармштадт), элюируя толуолом, с получением 4,5 г целевого соединения в виде масла. Б). 2-Амино-5-циклогексилэтил-4-(2,4 диметоксифенил)тиазол. В 50 мл тетрагидрофурана вносят 4,44 г полученного на предыдущей стадии продукта и добавляют по каплям раствор 5,75 г фенилтриметиламмонийтрибромида (РТТ) в 50 мл тетрагидрофурана. После перемешивания в течение 15 мин при температуре 0 С выливают в смесь воды с дихлорметаном, затем экстрагируют дихлорметаном. После высушивания над сульфатом магния органическую фазу выпаривают,остаток обрабатывают с помощью 100 мл этанола, затем кипятят с обратным холодильником в течение 4 ч в присутствии 6 г тиомочевины. После выпаривания растворителя обрабатывают этилацетатом и промывают последовательно раствором карбоната натрия, затем водой. Органическую фазу сушат над сульфатом магния и выпаривают. Остаток переводят в порошкообразное состояние с помощью смеси диэтилового эфира с гептаном (объемное соотношение 50/50), получая 4,18 г целевого соединения. Т пл. = 122 С. Препаративный пример 1.4. 2-Амино-4-(4 хлор-2,5-диметоксифенил)-5-циклогексилэтилтиазол. А). 4-Циклогексил-1-(2,5-диметокси-4 хлорфенил)бутан-1-он. К суспензии 2,8 г АlСl3 в 20 мл ССl4 при температуре -4 С и в атмосфере азота добавляют раствор 3,77 г хлорангидрида 4-циклогексанбутановой кислоты в 10 мл CCl4. Добавляют по каплям 5,2 г 2,5-диметоксихлорбензола в 10 млCCl4, затем перемешивают в течение 3 ч при комнатной температуре. Реакционную смесь гидролизуют с помощью разбавленного раствора НСl, затем декантируют и экстрагируют органическую фазу дихлорметаном. После высушивания над сульфатом магния выпаривают,затем очищают путем хроматографии на диоксиде кремния, элюируя смесью гептана с дихлорметаном (объемное соотношение 60/40),затем чистым дихлорметаном. Получают 3,37 г целевого соединения. Т пл. = 80-81 С. Б). 2-Амино-4-(4-хлор-2,5-диметоксифенил)-5-циклогексилэтилтиазол. Все количество полученного на предыдущей стадии продукта растворяют в 50 мл дихлорметана. При комнатной температуре добавляют раствор 1,66 г брома в 10 мл дихлормета 003093 22 на, затем органическую фазу промывают водой и сушат над сульфатом магния. После выпаривания остаток обрабатывают с помощью 30 мл этанола и добавляют 1,6 г тиомочевины, затем в течение ночи кипятят с обратным холодильником. Этанол выпаривают, затем остаток обрабатывают 50%-м водным раствором карбоната натрия и дихлорметаном. После перемешивания в течение 1 ч декантируют, экстрагируют дихлорметаном, затем объединенные органические фазы сушат над сульфатом магния и растворитель выпаривают. Добавляют гептан, продукт кристаллизуют путем порошкования. Отфильтровывают и высушивают, получая 3,42 г целевого соединения. Т пл. = 110-111 С. Препаративный пример 1.5. 2-Амино-5 циклогексилэтил-4-(5-этокси-2-метокси-4-метилфенил)тиазол. А). 4-Циклогексил-1-(2,5-диэтокси-4-метилфенил)бутан-1-он. К суспензии 2,9 г АlСl3 в 40 мл ССl4 при температуре -4 С добавляют раствор 4 г хлорангидрида 4-циклогексилбутановой кислоты в 10 мл ССl4. Добавляют по каплям 4,2 г 2,5 диэтокситолуола в 20 мл ССl4. После выдерживания в течение 4 ч при температуре +4 С, реакционную смесь выливают в разбавленный охлажденный раствор НСl. Добавляют дихлорметан, декантируют, затем органическую фазу сушат над сульфатом магния. После выпаривания растворителя продукт очищают путем хроматографии на силикагеле 60 Н, элюируя толуолом. Получают 5,33 г целевого соединения. Т пл. = 49-50 С. Б). 4-Циклогексил-1-(5-этокси-2-гидрокси 4-метилфенил)бутан-1-он. Раствор 5,33 г полученного на предыдущей стадии продукта в 80 мл безводного дихлорметана, охлажденный до температуры -5 С, обрабатывают с помощью 16 мл 1 М раствора трихлорида бора. Перемешивают в течение 5 мин при температуре -5 С и реакционную среду выливают в охлажденный разбавленный раствор НСl. После перемешивания в течение 30 мин декантируют. Органическую фазу сушат над сульфатом магния и выпаривают. Продукт очищают путем хроматографии на диоксиде кремния, элюируя смесью дихлорметана с гептаном(объемное соотношение 50/50). Получают 3,88 г целевого соединения в виде твердого вещества бледно-желтого цвета. Т пл. = 48-49 С. В). 4-Циклогексил-1-(5-этокси-2-метокси 4-метилфенил)бутан-1-он. Смешивают 2 г полученного на предыдущей стадии продукта и 2 г 50%-го водного раствора гидроксида цезия в 50 мл метанола. После выпаривания остаток обрабатывают изопропанолом и выпаривают досуха. Полученное твердое вещество желтого цвета растворяют в 10 мл диметилформамида и добавляют 5 мл метилиодида. После нагревания в течение 2 ч при температуре 80 С дихлорметан выпаривают и оста 23 ток обрабатывают водой. Соединение экстрагируют дихлорметаном. Органическую фазу сушат над сульфатом магния и выпаривают. Остаток очищают путем хроматографии на диоксиде кремния, элюируя дихлорметаном; получают 2,12 г целевого продукта. Т пл. = 37-38 С. Г). 2-Амино-5-циклогексилэтил-4-(5 этокси-2-метокси-4-метилфенил)тиазол. К охлажденному до температуры +4 С раствору 2,12 г полученного на предыдущей стадии продукта в 30 мл тетрагидрофурана добавляют по каплям раствор 2,5 г фенилтриметиламмонийтрибромида в 20 мл тетрагидрофурана. Перемешивают в течение четверти часа при температуре +4 С, затем реакционную массу выливают в смесь воды со льдом. Водную фазу экстрагируют 3 раза по 100 мл дихлорметаном. Органические фазы объединяют и сушат над сульфатом магния, затем выпаривают. Остаток обрабатывают с помощью 30 мл этанола и добавляют 1,1 г тиомочевины. После кипячения с обратным холодильником в течение 3 ч и охлаждения, этанол выпаривают и полученное масло обрабатывают с помощью 5%-го раствора карбоната натрия. Экстрагируют два раза дихлорметаном. Дихлорметановые фазы сушат над сульфатом магния и выпаривают. Остаток кристаллизуют путем перевода в порошкообразное состояние в гептане. После отфильтровывания и высушивания получают 2,19 г целевого соединения. Т пл. = 96-97 С. Препаративный пример 1.6. 2-Амино-5 циклогексилтиометил-4-(2,5-диметокси-4 метилфенил)тиазол. А). Этиловый эфир 3-циклогексилтиопропионовой кислоты. В 200 мл метанола растворяют 25 г циклогексилмеркаптана и 64,66 г 50%-го водного раствора гидроксида цезия. После выпаривания досуха, осуществляют две азеотропные перегонки с изопропанолом, затем сухой продукт растворяют в 100 мл диметилформамида, добавляют 40 г этилового эфира 3-бромпропионовой кислоты и нагревают в течение 2 ч при температуре 80 С. После охлаждения, образовавшееся твердое вещество отфильтровывают,затем промывают минимальным количеством диметилформамида. Диметилформамид выпаривают, остаток обрабатывают диэтиловым эфиром, промывают водой, 5%-м раствором карбоната натрия, затем водой. Сушат над сульфатом натрия, затем выпаривают. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с гептаном (объемное соотношение 50/50), с получением 38,86 г целевого соединения в виде жидкости. Б). 3-Циклогексилтиопропионовая кислота. Все количество продукта предыдущей стадии вносят в 200 мл метанола и добавляют раствор 17 г NaOH в 50 мл воды. После выдерживания в течение ночи при комнатной температу 003093 24 ре метанол выпаривают, остаток обрабатывают водой, затем водную фазу экстрагируют диэтиловым эфиром. Эфирные фазы удаляют, водную фазу подкисляют до рН 2 путем добавления концентрированной НСl, затем экстрагируют 3 раза дихлорметаном, сушат над сульфатом магния и выпаривают, получая 31,1 г целевого продукта в маслянистой форме. В). Хлорангидрид 3-циклогексилтиопропионовой кислоты. В 40 мл дихлорметана растворяют 4 г полученного на предыдущей стадии соединения и обрабатывают с помощью 3,5 г оксалилхлорида,добавляют 3 капли диметилформамида, затем перемешивают в течение 30 мин и выпаривают досуха. Продукт используют таким, какой есть,на следующей стадии. Г). 2-Амино-5-циклогексилтиометил-4(2,5-диметокси-4-метилфенил)тиазол. Следуя вышеописанным методикам, обрабатывают 2,5-диметокситолуолом в присутствии TiCl4, затем осуществляют бромирование кетона путем воздействия фенилтриметиламмонийтрибромида и вводят во взаимодействие с тиомочевиной, получая 4,13 г целевого продукта. Т пл. = 143-144 С. Препаративный пример 1.7. 2-Амино-5 циклогексилметилтио-4-(2,5-диметокси-4-метилфенил)тиазол. А). 1-(2,5-Диметокси-4-метилфенил)этан 1-он. Суспензию 17,52 г хлорида алюминия в смеси 100 мл ССl4 и 100 мл дихлорметана охлаждают до температуры +4 С в атмосфере азота,добавляют по каплям 10,4 г ацетилхлорида, затем раствор 20 г 2,5-диметокситолуола в 20 мл дихлорметана. После перемешивания в течение 4 ч при температуре +4 С, реакционную среду выливают на лед, к которому добавлено несколько мл концентрированной НСl, и перемешивают в течение 30 мин. Декантируют, экстрагируют дихлорметаном, объединенные органические фазы промывают водным 5%-м раствором карбоната натрия, сушат над сульфатом натрия и выпаривают. Продукт кристаллизуют,переводят в порошкообразное состояние в 150 мл гептана, отфильтровывают, затем промывают гептаном, получая 21,03 г целевого соединения. Т пл. = 75-77 С. Б). 2-Амино-4-(2,5-диметокси-4-метилфенил)тиазол. Раствор 21,03 г полученного на предыдущей стадии соединения в 300 мл дихлорметана обрабатывают путем прикапывания раствором 17,35 г брома в 70 мл дихлорметана. После декантации органическую фазу промывают водой,сушат над сульфатом магния, затем выпаривают. Обрабатывают с помощью 200 мл абсолютного этанола, затем добавляют 15,2 г тиомочевины и в течение ночи кипятят с обратным холодильником. Охлаждают на бане со льдом,отфильтровывают образовавшиеся кристаллы, 25 затем кристаллы обрабатывают водным 5%-м раствором карбоната натрия, после чего экстрагируют этилацетатом. Декантируют, органическую фазу сушат над сульфатом натрия, затем выпаривают. Остаток переводят в порошкообразное состояние в гептане, затем отфильтровывают, получая 17,74 г целевого соединения в кристаллической форме. Т пл. = 191-192 С. В). 4-(2,5-Диметокси-4-метилфенил)-2(2,5-диметилпиррол-1-ил)тиазол. В 300 мл бензола растворяют 17,74 г полученного на предыдущей стадии продукта, 20,3 г 2,5-гександиона и 8,51 г уксусной кислоты. После 24 ч азеотропной перегонки, реакционную смесь выливают в воду, затем нейтрализуют водным 5%-м раствором карбоната натрия. Декантируют, затем экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом натрия, затем выпаривают. Остаток хроматографируют на диоксиде кремния, элюируя дихлорметаном, с получением 19,46 г целевого соединения, которое кристаллизуют из гептана. Т пл. = 92-93 С. Г). 5-(Циклогексилметилтио)-4-(2,5-диметокси-4-метилфенил)-2-(2,5-диметилпиррол-1 ил)тиазол. Раствор 3,28 г полученного на предыдущей стадии соединения в 80 мл безводного тетрагидрофурана охлаждают до температуры -30 С,затем обрабатывают с помощью 8 мл 1,6 М раствора н-бутиллития в гексане. После выдерживания в течение 30 мин при температуре -30 С добавляют 650 мг серного цвета. Оставляют стоять для повышения температуры до комнатной, затем добавляют 3,4 г циклогексилметанолтозилата в виде раствора в 10 мл безводного тетрагидрофурана, перемешивают в течение 2 ч при комнатной температуре. Реакционную среду выливают в воду, затем экстрагируют диэтиловым эфиром, экстракт сушат над сульфатом натрия и выпаривают. Остаток хроматографируют на силикагеле 60 Н, элюируя толуолом. Получают 1,1 г целевого соединения. Т пл. = 117-118 С. Д). 2-Амино-5-циклогексилметилтио-4(2,5-диметокси-4-метилфенил)тиазол. В течение 36 ч кипятят с обратным холодильником смесь, содержащую 1,1 г полученного на предыдущей стадии соединения, 30 мл этанола, 4,5 мл воды и 3,7 г гидроксиламингидрохлорида. После выпаривания массу обрабатывают водным 5%-м раствором карбоната натрия, затем экстрагируют дихлорметаном, экстракт сушат над сульфатом магния и выпаривают, остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с этилацетатом (объемное соотношение 70/30). Получают 0,78 г целевого соединения. Т пл. = 111112 С. Препаративный пример 1.8. 2-Амино-5 циклогептилэтил-4-(2,5-диметокси-4-метилфенил)тиазол. 26 А). Этиловый эфир 4-циклогептилиденбут 2-еновой кислоты. В 7 мл диметоксиэтана растворяют 6,7 мл этилового эфира 4-(диэтоксифосфорил)бут-2 еновой кислоты, затем выливают в 40 мл диметоксиэтана, содержащего 1,3 г 60%-го гидрида натрия. После перемешивания в течение 45 мин охлаждают до температуры 10 С, затем добавляют по каплям 3,2 мл циклогептанона и оставляют стоять для повышения температуры до комнатной. После перемешивания в течение 4 ч выливают в холодную воду, затем экстрагируют диэтиловым эфиром, сушат и выпаривают. Остаток хроматографируют на диоксиде кремния,элюируя дихлорметаном. Получают 1,9 г целевого соединения. Б). Этиловый эфир 4-циклогептилбутановой кислоты. Все количество полученного на предыдущей стадии соединения гидрируют при комнатной температуре и атмосферном давлении в 20 мл этанола в присутствии 190 мг 10%-го палладия-на-угле. Получают 1,9 г целевого соединения. В). 4-Циклогептилбутановая кислота. В 20 мл метанола и 5 мл воды вносят 1,9 г полученного на предыдущей стадии соединения и 755 мг гидроксида натрия и перемешивают при комнатной температуре в течение 24 ч. Высушивают в вакууме, получая 1,5 г целевого соединения. Г). 4-Циклогептил-(2,5-диметокси-4-метилфенил)бутан-1-он. Охлаждают до температуры 0 С 1,5 г полученного на предыдущей стадии соединения и одну каплю диметилформамида в 20 мл дихлорметана, добавляют 0,71 мл оксалилхлорида,затем оставляют стоять для повышения температуры до комнатной. После перемешивания в течение 3 ч добавляют 1,2 г охлажденного до температуры 4 С 2,5-диметокситолуола, после чего порциями добавляют 1,2 г АlСl3. Перемешивают в течение 1,5 ч при температуре +4 С,затем в течение 2 ч при комнатной температуре. Реакционную смесь выливают в разбавленный раствор НСl, затем декантируют и экстрагируют диэтиловым эфиром. Органические фазы объединяют, промывают 1 н. раствором NaOH, затем концентрируют. Очищают путем хроматографии на диоксиде кремния, элюируя смесью этилацетата с пентаном (объемное соотношение 30/70), и получают 2,19 г целевого соединения. Д). 2-Амино-5-циклогептилэтил-4-(2,5 диметокси-4-метилфенил)тиазол. В 25 мл тетрагидрофурана растворяют 2,1 г полученного на предыдущей стадии соединения и в течение 5 мин добавляют 2,47 г фенилтриметиламмонийтрибромида. После перемешивания в течение 5 ч осадок отфильтровывают, промывают тетрагидрофураном, затем органическую фазу концентрируют, остаток обрабатывают с помощью 30 мл этанола, затем добав 27 ляют 0,5 г тиомочевины и кипятят с обратным холодильником в течение 48 ч. Реакционную среду концентрируют, обрабатывают 10%-м раствором карбоната натрия, экстрагируют диэтиловым эфиром, затем органическую фазу промывают, сушат и концентрируют. Остаток хроматографируют на диоксиде кремния, элюируя смесью этилацетата с пентаном (объемное соотношение 70/30). Препаративный пример 1.9. 2-Амино-5[(4,4-диметилциклогексил)этил]-4-(2,5-диметокси-4-метилфенил)тиазол. А). 4,4-Диметилциклогексанон. В 65 мл этилацетата вносят 6,7 г 4,4 диметилциклогекс-2-ен-1-она и 2 г 10%-го палладия-на-угле и гидрируют при комнатной температуре и атмосферном давлении вплоть до поглощения теоретически рассчитанного объема водорода. Катализатор отфильтровывают,затем фильтрат концентрируют, получая 6,3 г целевого продукта. Б). 2-Амино-5-[(4,4-диметилциклогексил) этил]-4- (2,5-диметокси-4-метилфенил)тиазол. Следуют описанной в предыдущем препаративном примере методике, осуществляя последовательно стадии А), Б), В), Г) и Д), и получают целевой продукт. Т пл. = 136-138 С. Препаративный пример 1.10. 2-Амино-5 циклопентилэтил-4-(2,5-диметокси-4-метилфенил)тиазол. Следуют методике, описанной в двух предыдущих препаративных примерах, получая целевое соединение. Т пл. = 80 С. Препаративный пример 1.11. 2-Амино-5 циклогексилэтил-4-(2,5-диметокси-4-этилфенил)тиазол. А). 1-(2,5-Диметоксифенил)этан-1-он. При температуре +5 С и в атмосфере азота в 200 мл ССl4 вводят 19,3 г АlСl3 и добавляют 11,36 г ацетилхлорида, затем маленькими порциями 20 г 1,4-диметоксибензола. После перемешивания в течение 3 ч при температуре +5 С выливают в разбавленный охлажденный раствор НСl. Отделяют органическую фазу, которую сушат над сульфатом магния, выпаривают, затем хроматографируют на диоксиде кремния,элюируя смесью дихлорметана с гептаном (объемное соотношение 50/50), с получением 24,71 г целевого продукта. Б). 1,4-Диметокси-2-этилбензол. Готовят смесь из 300 г цинка в виде порошка и 40 г хлорида двухвалентной ртути, которую добавляют к раствору 24 г полученного на предыдущей стадии соединения в 400 мл бензола и 100 мл концентрированной НСl при температуре 80 С. После перемешивания в течение 2 ч при температуре 80 С отфильтровывают, затем отделяют органическую фазу, которую сушат над сульфатом магния и выпаривают, после чего хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с геп 003093 28 таном (объемное соотношение 50/50), с получением 8,6 г целевого продукта. В). 2-Амино-5-циклогексилэтил-4-(2,5 диметокси-4-этилфенил)тиазол. Следуют методике, описанной в препаративном примере 1.3, получая целевой продукт. Т пл. = 88 С. Препаративный пример 1.12. 2-Амино-5 циклопентилметилтио-4-(2,5-диметокси-4 метилфенил)тиазол. А). 5-(Циклопентилметилтио)-2-(2,5-диметилпиррол-1-ил)-4-(2,5-диметокси-4-метилфенил)тиазол. Приготовляют раствор 3,28 г 4-(2,5 диметокси-4-метилфенил)-2-(2,5-диметилпиррол-1-ил)тиазола такого, который получен на стадии В) препаративного примера 1.7, в 80 мл тетрагидрофурана и охлаждают этот раствор до температуры -30 С; добавляют по каплям 8 мл 1,6 М гексанового раствора н-бутиллития в 10 мл тетрагидрофурана и перемешивают в течение 30 мин до достижения температуры 0 С. Охлаждают до температуры -30 С и добавляют 0,65 г серого цвета. Оставляют стоять для повышения температуры до 0 С и добавляют 3,5 г циклопентилметил-п-толуолсульфоната в 3 мл тетрагидрофурана. После перемешивания в течение 2 ч при комнатной температуре, выливают в воду со льдом, экстрагируют диэтиловым эфиром, затем экстракт сушат над сульфатом натрия и выпаривают. Остаток хроматографируют на силикагеле 60 Н, элюируя толуолом, с получением 0,47 г целевого продукта. Т пл. = 107,5-108,5 С. Б). 2-Амино-5-циклопентилметилтио-4(2,5-диметокси-4-метилфенил)тиазол. Смешивают 0,47 г полученного на предыдущей стадии соединения и 2 г гидроксиламингидрохлорида в 20 мл этанола и 3 мл воды и в течение 48 ч кипятят с обратным холодильником. После выпаривания растворителя остаток обрабатывают 5%-м раствором карбоната натрия, затем экстрагируют дихлорметаном, сушат над сульфатом магния и выпаривают. Остаток хроматографируют на диоксиде кремния,элюируя смесью дихлорметана с этилацетатом(объемное соотношение 80/20). Получают 0,32 г целевого соединения. Т пл. = 88-89 С. Препаративный пример 1.13. 2-Амино-5 циклогексилэтил-4-(2,6-диметокси-4-метилфенил)тиазол. А). 5-Циклогексилэтил-2-(2,5-диметилпиррол-1-ил)-4-(2,6-диметокси-4-метилфенил)тиазол. К 4,27 г 2-(2,5-диметилпиррол-1-ил)-4-(2,6 диметокси-4-метилфенил)тиазола при температуре -30 С добавляют 10 мл 1,6 М раствора нбутиллития в гексане и перемешивают в течение 30 мин при температуре -30 С. При температуре-45 С добавляют 4,2 г циклогексилэтанолтрифторметансульфоната, затем температуру повышают до 0 С, добавляют воду, экстрагируют 29 диэтиловым эфиром, экстракт сушат и выпаривают. Образовавшуюся смолу хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (объемное соотношение 1/1). Получают 0,52 г целевого соединения в виде бесцветной смолы. Б). 2-Амино-5-циклогексилэтил-4-(2,6 диметокси-4-метилфенил)тиазол. В течение ночи кипятят с обратным холодильником 0,4 г полученного на предыдущей стадии соединения в 5 мл этанола и 2 мл воды в присутствии 0,93 г гидроксиламингидрохлорида. Выливают в водный насыщенный раствор карбоната натрия, экстрагируют этилацетатом, сушат, затем выпаривают. Образовавшуюся смолу хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом (объемное соотношение 100/3). Получают 0,25 г целевого соединения в виде светлокоричневого масла. Препаративный пример 1.14. 2-Амино-4(2,5-диметокси-4-метилфенил)-5-(5,5-диметилгексил)тиазол. А).(2,5-Диметокси-4-метилфенил)-7,7 диметилоктан-1-он. Растворяют 1 г 7,7-диметилоктановой кислоты в 20 мл дихлорметана, охлажденного до температуры 0 С, добавляют 0,57 мл оксалилхлорида, затем перемешивают в течение 1 ч при температуре 0 С и в течение 2 ч при комнатной температуре. Добавляют 0,93 мл 2,5 диметокситолуола и одну каплю диметилформамида, охлаждают до температуры 0 С, затем добавляют 940 мг АlСl3 и перемешивают в течение 1 ч при температуре 0 С и в течение ночи при комнатной температуре. Выливают в 10%ый раствор НСl, экстрагируют диэтиловым эфиром, затем объединенные органические фазы промывают 2 н. раствором гидроксида натрия,сушат и концентрируют. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с пентаном (объемное соотношение 85/15), с получением 1,05 г целевого соединения. Б). 2-Амино-4-(2,5-диметокси-4-метилфенил)-5-(5,5-диметилгексил)тиазол. В 15 мл тетрагидрофурана растворяют 1,05 г полученного на предыдущей стадии соединения, затем добавляют 1,29 г фенилтриметиламмонийтрибромида и перемешивают в течение 4 ч при комнатной температуре. Выпавший осадок удаляют и фильтрат концентрируют. Остаток обрабатывают с помощью 15 мл этанола,затем добавляют 260 мг тиомочевины и в течение ночи кипятят с обратным холодильником. На следующий день экстрагируют диэтиловым эфиром, промывают водой, 1 н. растворомNaOH, сушат над сульфатом натрия и концентрируют. Остаток хроматографируют на диоксиде кремния, элюируя смесью этилацетата с пентаном (объемное соотношение 75/25). Получают 1,12 г целевого соединения. 31 Препаративный пример 1.29. 2-Амино-5 циклогексилэтил-4-(2,6-диметокси-4-изопропилфенил)тиазол. А). 1-Изопропенил-2,4-диметоксибензол. Растворяют 10,57 г 1-(2,4-диметоксифенил)этанона в 100 мл диэтилового эфира и 50 мл тетрагидрофурана в атмосфере азота. При температуре -50 С добавляют 55 мл 1,6 М раствора метиллития в диэтиловом эфире и перемешивают в течение 2 ч при температуре от -60 до-40 С, затем в течение 30 мин при температуре от -40 до 0 С и в течение 3 ч при комнатной температуре. Охлаждают до температуры 0 С и добавляют 70 мл 2 н. НСl. После декантации экстрагируют диэтиловым эфиром, затем органическую фазу сушат над сульфатом натрия. Концентрируют, затем обрабатывают с помощью 150 мл тетрагидрофурана и добавляют 75 мл 2 н. НСl. После перемешивания в течение 4 ч при комнатной температуре добавляют 100 мл воды, затем экстрагируют диэтиловым эфиром. Органическую фазу промывают раствором карбоната натрия, водой, затем сушат над сульфатом натрия и концентрируют. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с пентаном (объемное соотношение 70/30). Получают 5 г целевого соединения в виде масла. Б). 1-Изопропил-2,4-диметоксибензол. Растворяют 4,96 г полученного на предыдущей стадии соединения в 100 мл метанола и добавляют 0,2 г 10%-го палладия-на-угле, затем гидрируют при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают, затем концентрируют в вакууме, получая 4,2 г целевого соединения в виде масла. В). 2-Амино-5-циклогексилэтил-4-(2,6 диметокси-4-изопропилфенил)тиазол. Следуют вышеописанным методикам. Обрабатывают 1-изопропил-2,4-диметоксибензол с помощью 4-циклогексилбутирилхлорида в присутствии АlСl3, затем осуществляют бромирование кетона путем воздействия фенилтриметиламмонийтрибромида и вводят во взаимодействие с тиомочевиной, получая целевой продукт. Т пл. = 143 С. Б. Получение производных индол-2-карбоновой кислоты Препаративный пример 2.1. 1-(Метоксикарбонилметил)индол-2-карбоновая кислота. А). Бензиловый эфир индол-2-карбоновой кислоты. В трехгорлую колбу, при комнатной температуре, вводят 10,32 г индол-2-карбоновой кислоты в 70 мл тетрагидрофурана и добавляют 10,38 г карбонилдиимидазола, затем, по окончании газовыделения, добавляют 7,62 г бензилового спирта. После кипячения с обратным холодильником в течение 5 ч выливают в воду, экстрагируют дихлорметаном, сушат и выпаривают. Образовавшиеся кристаллы промывают изопро 003093 32 панолом, получая 13,62 г целевого соединения. Т пл. = 136 С. Б). Бензиловый эфир 1-(метоксикарбонилметил)индол-2-карбоновой кислоты. В трехгорлую колбу вводят 2,85 г гидрида натрия в виде 50%-й дисперсии в масле и 10 мл диметилформамида и пропускают путем барботирования азот. Добавляют 13,58 г полученного на предыдущей стадии соединения, растворенного в 50 мл диметилформамида, затем медленно добавляют 9,09 г метилбромацетата. После перемешивания в течение ночи при комнатной температуре выливают на лед, экстрагируют этилацетатом, сушат над сульфатом натрия и выпаривают; остаток хроматографируют на диоксиде кремния, элюируя смесью толуола с этилацетатом (объемное соотношение 95/5). Получают 6,84 г целевого соединения, которое кристаллизуют из смеси этанола с пентаном. Т пл. = 94 С. В). 1-(Метоксикарбонилметил)индол-2 карбоновая кислота. Смешивают 6,84 г полученного на предыдущей стадии продукта с 80 мл диметилформамида, 80 мл этанола и 100 мг 5%-го палладияна-угле и гидрируют при интенсивном перемешивании в течение 8 ч. Катализатор отфильтровывают, фильтрат выпаривают досуха, затем продукт кристаллизуют из смеси этанола с петролейным эфиром. Таким образом, получают 4,30 г целевого соединения. Т пл. = 190 С. Препаративный пример 2.2. 1-(трет.Бутоксикарбонилметил)индол-2-карбоновая кислота. А). Бензиловый эфир 1-(трет.-бутоксикарбонилметил)индол-2-карбоновой кислоты. Растворяют 50,25 г бензилового эфира индол-2-карбоновой кислоты (препаративный пример 2.1, стадия А) в 140 мл безводного диметилформамида, затем в токе сухого азота в течение 30 мин добавляют раствор 6,6 г гидрида натрия, в виде 80%-й дисперсии в масле, в 100 мл диметилформамида. Реакционную смесь перемешивают в течение 90 мин при комнатной температуре, затем ее охлаждают на бане со льдом и добавляют по каплям 42,91 мл трет.бутилбромацетата. После перемешивания в течение ночи при комнатной температуре, диметилформамид выпаривают и остаток обрабатывают дихлорметаном, затем водой. После перемешивания декантируют, затем водную фазу экстрагируют 2 раза дихлорметаном, объединенные органические фазы промывают насыщенным раствором NaCl, после чего сушат над сульфатом магния. После отфильтровывания и выпаривания остаток обрабатывают смесью 100 мл диэтилового эфира и 100 мл гептана. После перемешивания в течение 2 ч образовавшиеся кристаллы отфильтровывают, затем промывают с помощью 50 мл смеси гептана с диэтиловым эфиром (объемное соотношение 70/30) и высу 33 шивают в сушильном шкафу. Получают 58 г целевого продукта. Т пл. = 95-96 С. Б). 1-(трет.-Бутоксикарбонилметил)индол 2-карбоновая кислота. В 150 мл этанола и 150 мл диметилформамида растворяют 58 г полученного на предыдущей стадии сложного бензилового эфира; в токе азота добавляют 3 г 5%-го палладия-на-угле и гидрируют реакционную массу при атмосферном давлении. Фильтруют через целит, затем растворители выпаривают и остаток обрабатывают водой. После перевода в порошкообразное состояние образовавшееся твердое вещество промывают водой, затем растворяют его в 1 л этилацетата. Эту органическую фазу промывают 2 раза водой, затем сушат над сульфатом натрия. Отфильтровывают и выпаривают, образовавшееся твердое вещество переводят в порошкообразное состояние в присутствии 100 мл смеси гептана с диэтиловым эфиром (объемное соотношение 50/50). Снова отфильтровывают и высушивают в сушильном шкафу, получая 39,6 г целевого соединения в виде твердого вещества белого цвета. Т пл. = 156-157 С. Препаративный пример 2.3. 5-Метил-1(трет.-бутилоксикарбонилметил)индол-2-карбоновая кислота. А). Бензиловый эфир 5-метилиндол-2 карбоновой кислоты. Растворяют 2,43 г 5-метилиндол-2-карбоновой кислоты в 15 мл диметилформамида, добавляют 2,11 г DBU (1,8-диазабицикло[5.4.0] ундец-7-ен), затем перемешивают в течение 1 ч при комнатной температуре. Добавляют по каплям 2,61 г бензилбромида, затем перемешивают в течение 6 ч при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают этилацетатом, промывают водой, насыщенным раствором карбоната натрия, сульфатным буфером, затем водным насыщенным растворомNaCl. Сушат над сульфатом натрия, затем концентрируют. Остаток кристаллизуют из пентана; получают 3,3 г целевого соединения. Т пл. = 150-152 С. Б). Бензиловый эфир 5-метил-1-(трет.бутилоксикарбонилметил)индол-2-карбоновой кислоты. Растворяют 3,26 г полученного на предыдущей стадии соединения в 30 мл диметилформамида и добавляют порциями в атмосфере азота 0,65 г гидрида натрия в виде 50%-й дисперсии в масле. После перемешивания в течение 2,5 ч при комнатной температуре добавляют по каплям при температуре 80 С 2,64 г трет.бутилбромацетата и перемешивают в течение 4 ч при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают с помощью 150 мл сульфатного буфера, затем экстрагируют этилацетатом, сушат над сульфатом натрия и концентрируют. Получают 3,53 г целевого со 003093 34 единения, которое кристаллизуют из пентана. Т пл. = 80-82 С. В). 5-Метил-1-(трет.-бутилоксикарбонилметил)индол-2-карбоновая кислота. В 30 мл абсолютного этанола и 15 мл диметилформамида растворяют 3,47 г полученного на предыдущей стадии соединения и в атмосфере азота добавляют 0,3 г 10%-го палладияна-угле, затем гидрируют при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают, затем концентрируют в вакууме, получая 2,86 г целевого соединения, которое кристаллизуют из пентана. Т пл. = 174176 С. Препаративный пример 2.4. 4-Метокси-1(трет.-бутилоксикарбонилметил)индол-2-карбоновая кислота. А). Бензиловый эфир 4-метоксииндол-2 карбоновой кислоты. Растворяют 2,65 г 4-метоксииндол-2 карбоновой кислоты в 15 мл диметилформамида и добавляют 2,11 г DBU, затем перемешивают в течение 1 ч при комнатной температуре. Добавляют по каплям, при комнатной температуре,2,61 г бензилбромида и перемешивают в течение 5 ч при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают этилацетатом, промывают водой, насыщенным раствором карбоната натрия, сульфатным буфером,затем водным насыщенным раствором NaCl. Сушат над сульфатом натрия и концентрируют. Получают 3,57 г целевого соединения, которое кристаллизуют из пентана. Т пл. = 162-164 С. Б). Бензиловый эфир 4-метокси-1-(трет.бутилоксикарбонилметил)индол-2-карбоновой кислоты. Растворяют 3,3 г полученного на предыдущей стадии соединения в 30 мл диметилформамида, добавляют порциями в атмосфере азота 0,62 г гидрида натрия в виде 50%-й дисперсии в масле и перемешивают в течение 2,5 ч при комнатной температуре. Добавляют по каплям при комнатной температуре 2,52 г трет.-бутилбромацетата и перемешивают в течение 5 ч при комнатной температуре. Концентрируют в вакууме,остаток обрабатывают с помощью 150 мл сульфатного буфера, затем экстрагируют этилацетатом, сушат над сульфатом натрия и концентрируют. Получают 4,2 г целевого соединения, которое кристаллизуют из пентана. Т пл. = 9698 С. В). 4-Метокси-1-(трет.-бутилоксикарбонилметил)индол-2-карбоновая кислота. В 30 мл абсолютного этанола и 30 мл диметилформамида растворяют 4,13 г полученного на предыдущей стадии соединения и добавляют 0,4 г 10%-го палладия-на-угле, затем гидрируют при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают и концентрируют, получая 2,36 г целевого соединения, которое кристаллизуют из смеси дихлорметана с пентаном. Т пл. = 222-224 С. 35 Препаративный пример 2.5. 5-Хлор-1(этоксикарбонилметил)индол-2-карбоновая кислота. А). трет.-Бутиловый эфир 5-хлориндол-2 карбоновой кислоты. Растворяют 5,52 г 5-хлориндол-2-карбоновой кислоты в 40 мл диметилформамида, при комнатной температуре и в атмосфере азота добавляют 4,57 г карбонилдиимидазола, затем нагревают до температуры 40 С и добавляют 4,3 г DBU и 4,17 г трет.-бутанола. Нагревают при температуре 40 С в течение 3 ч, затем выпавший осадок отфильтровывают и фильтрат концентрируют в вакууме. Остаток обрабатывают этилацетатом, промывают 10%-м раствором карбоната натрия, насыщенным водным раствором NaCl, сульфатным буфером, затем сушат над сульфатом натрия и концентрируют. Остаток хроматографируют на диоксиде кремния, элюируя дихлорметаном, с получением 0,45 г целевого соединения. Т пл. = 140-142 С. Б). трет.-Бутиловый эфир 5-хлор-1(этоксикарбонилметил)индол-2-карбоновой кислоты. Растворяют 0,45 г полученного на предыдущей стадии соединения в 15 мл диметилформамида, затем при комнатной температуре и в атмосфере азота порциями добавляют 94 мг гидрида натрия в виде 50%-й дисперсии в масле. После перемешивания в течение 4 ч при комнатной температуре добавляют по каплям при комнатной температуре 0,33 г этилбромацетата и перемешивают в течение 4 ч при комнатной температуре. Концентрируют в вакууме,остаток обрабатывают этилацетатом, промывают сульфатным буфером, сушат надсульфатом натрия и концентрируют, получая 0,6 г целевого соединения в виде масла. В). 5-Хлор-1-(этоксикарбонилметил)индол-2-карбоновая кислота. Растворяют 0,6 г полученного на предыдущей стадии соединения в 10 мл дихлорметана, помещают на баню со льдом и добавляют 10 мл трифторуксусной кислоты, затем перемешивают в течение 4 ч на бане со льдом. После выдерживания в течение ночи при температуре 4 С, концентрируют в вакууме. Остаток кристаллизуют из пентана. После высушивания получают 0,37 г целевого соединения. Т пл. = 198-200 С. Препаративный пример 2.6. 1-(2 Этоксикарбонилбензил)индол-2-карбоновая кислота. А). Бензиловый эфир индол-2-карбоновой кислоты. Этот сложный эфир может быть получен согласно способу, альтернативному таковому,описанному в препаративном примере 2.1. В 500 мл диметилформамида растворяют 100 г индол-2-карбоновой кислоты, добавляют по каплям 93 мл DBU, затем 89,7 г бензилбромида. После перемешивания в течение ночи при 36 комнатной температуре, диметилформамид выпаривают, затем остаток выливают в воду. Выпавший осадок отфильтровывают, промывают водой, обрабатывают этилацетатом. Органическую фазу промывают 5%-м раствором карбоната натрия, сульфатным буфером, затем сушат над сульфатом натрия. После отфильтровывания, растворители выпаривают, остаток переводят в порошкообразное состояние в диэтиловом эфире, отфильтровывают и высушивают. Получают 133 г целевого соединения. Т пл. = 136 С. Б). Этиловый эфир 2-бромметилбензойной кислоты. Смесь, содержащую 8,2 г этилового эфира 2-метилбензойной кислоты, 10,7 г Nбромсукцинимида и 0,2 г бензоилпероксида, в 50 мл ССl4 кипятят с обратным холодильником при облучении. Спустя 45 мин раствор охлаждают и образовавшийся сукцинимид отфильтровывают. Органическую фазу промывают 5%-м раствором гидрокарбоната натрия, затем сушат над сульфатом магния и выпаривают. Продукт используют таким, какой есть, на следующей стадии. В). Бензиловый эфир 1-(2-этоксикарбонилбензил)индол-2-карбоновой кислоты. В атмосфере азота растворяют 12,56 г полученного на стадии А соединения в 50 мл диметилформамида, маленькими порциями добавляют 1,81 г гидрида натрия в виде 80%-й дисперсии в масле, поддерживая температуру ниже 20 С с помощью бани со льдом. После перемешивания в течение 1 ч при комнатной температуре охлаждают до температуры +4 С с помощью бани со льдом и добавляют по каплям полученный на предыдущей стадии продукт в виде раствора в 20 мл диметилформамида, затем перемешивают в течение ночи при комнатной температуре. Диметилформамид выпаривают,остаток обрабатывают смесью воды со льдом,экстрагируют 3 раза диэтиловым эфиром, затем органические фазы объединяют, промывают насыщенным раствором NaCl, сушат над сульфатом натрия и выпаривают. Продукт растворяют в толуоле, отфильтровывают и фильтрат хроматографируют на силикагеле 60 Н, элюируя толуолом. Получают 9 г целевого соединения в виде масла. Г). 1-(2-Этоксикарбонилбензил)индол-2 карбоновая кислота. Все количество полученного на предыдущей стадии продукта гидрируют в присутствии палладия-на-угле согласно обычному способу. Получают 6,13 г целевого соединения. Т пл. = 191-192 С. Препаративный пример 2.7. 1-(Метоксикарбонилэтил)индол-2-карбоновая кислота. А). Бензиловый эфир 1-(2-цианоэтил)индол-2-карбоновой кислоты. В 40 мл диоксана смешивают 1,6 мл 40%го водного раствора N-бензилтриметиламмонийгидроксида и 4 мл акрилонитрила, затем при 37 перемешивании добавляют 9,44 г бензилового эфира индол-2-карбоновой кислоты. В течение 24 ч нагревают при температуре 80 С, затем выливают в 500 мл воды со льдом. Выпавший осадок отфильтровывают, обрабатывают этилацетатом, затем сушат над сульфатом натрия и концентрируют, получая 10,1 г целевого соединения, которое кристаллизуется. Т пл. = 98100 С. Б). Бензиловый эфир 1-(метоксикарбонилэтил)индол-2-карбоновой кислоты. Растворяют 10,1 г полученного на предыдущей стадии соединения в 60 мл дихлорметана и добавляют 12 мл метанола и 120 мл насыщенного газообразным хлороводородом диэтилового эфира. После выдерживания в течение 72 ч при температуре 0 С, отфильтровывают образовавшийся имидат, затем осадок обрабатывают с помощью 30 мл воды и уксусной кислоты. Полученный раствор перемешивают в течение 2 ч при комнатной температуре, затем добавляют 50 мл 1 н. НСl и экстрагируют этилацетатом,сушат над сульфатом натрия и концентрируют,получая 9 г целевого соединения в виде масла. В). 1-(Метоксикарбонилэтил)индол-2-карбоновая кислота. Гидрируют в этаноле в присутствии палладия-на-угле 9 г полученного на предыдущей стадии соединения, получая 3,95 г целевого соединения. Т пл. = 118-120 С. Препаративный пример 2.8. 1-(трет.Бутоксикарбонилметил)-5-этилиндол-2-карбоновая кислота. А). 2-[(4-Этилфенил)гидразоно]пропионовая кислота. Это соединение получают согласно V.Prelog и др., Helv. Chem. Acta, 31, 1178 [1948]. В 150 мл концентрированной НСl растворяют 13,2 г 4-этиланилина. Охлаждают до температуры 0 С, затем добавляют при температуре ниже или равной 5 С 10,6 г NaNO2 в виде раствора в 40 мл воды. Спустя 5 мин, при температуре 5 С добавляют раствор SnCl2(2H2O) в 75 мл концентрированной НСl, затем выдерживают в течение 2,5 ч при температуре 0 С. Отфильтровывают, образовавшийся осадок промывают минимальным количеством воды при температуре 5 С, затем растворяют в 500 мл воды при температуре 5 С. При температуре 10 С добавляют 9,5 мл пирувиновой кислоты в 50 мл воды. После выдерживания в течение ночи в холодильнике, выпавший осадок отфильтровывают, затем обрабатывают с помощью 120 мл бензола. Сушат над сульфатом натрия, затем концентрируют, получая 11 г целевого соединения. Б). Этиловый эфир 2-[(4-этилфенил)гидразоно]пропионовой кислоты. В течение 2 ч кипятят с обратным холодильником 11 г полученного на предыдущей стадии соединения в 100 мл абсолютного этанола и 6 мл серной кислоты. Концентрируют до 38 трети объема, затем выливают в воду со льдом,экстрагируют диэтиловым эфиром, промывают раствором карбоната натрия, сушат над сульфатом натрия и концентрируют. Образовавшийся осадок обрабатывают пентаном, затем отфильтровывают, получая 7,9 г целевого соединения. В). Этиловый эфир 5-этилиндол-2-карбоновой кислоты. Нагревают до температуры кипения с обратным холодильником 9,6 г п-толуолсульфокислоты в 100 мл бензола, осторожно добавляют 7,9 г полученного на предыдущей стадии соединения и кипятят с обратным холодильником в течение полутора часов. Отфильтровывают нерастворимую часть, затем бензольный раствор промывают насыщенным раствором гидрокарбоната натрия, сушат, затем концентрируют. Остаток осаждают из пентана. Получают 5,4 г целевого продукта. Г). 5-Этилиндол-2-карбоновая кислота. В течение 3 ч кипятят с обратным холодильником 5,4 г полученного на предыдущей стадии соединения в 50 мл этанола и 4 мл воды,содержащей 3,4 г КОН. Среду концентрируют,остаток обрабатывают водой, промывают диэтиловым эфиром, водную фазу подкисляют путем добавления концентрированной НСl, затем отфильтровывают выпавший осадок, получая 3,95 г целевого соединения. Д). Бензиловый эфир 5-этилиндол-2 карбоновой кислоты. В 30 мл диметилформамида смешивают 4 г полученного на предыдущей стадии соединения, 3,14 мл DBU и 2,75 мл бензилбромида и перемешивают в течение 48 ч при комнатной температуре. Реакционную массу выливают в 300 мл воды при температуре 5 С, отфильтровывают выпавший осадок, затем промывают его водой при температуре 5 С, затем пентаном. Этот осадок обрабатывают с помощью 300 мл этилацетата, сушат над сульфатом натрия и концентрируют, получая 4,93 г целевого соединения. Е). Бензиловый эфир 5-этил-1-(трет.бутоксикарбонилметил)индол-2-карбоновой кислоты. Растворяют 4,93 г полученного на предыдущей стадии соединения в 40 мл диметилформамида, порциями добавляют 0,78 г гидрида натрия в виде 60%-й дисперсии в масле, затем в течение 30 мин нагревают при температуре 60 С. Оставляют охлаждаться и добавляют по каплям 3,1 мл трет.-бутилового эфира бромуксусной кислоты. После выдерживания в течение ночи при комнатной температуре, диметилформамид выпаривают, экстрагируют диэтиловым эфиром, промывают водой, сушат над сульфатом натрия, затем остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с пентаном (объемное соотношение 60/40),с получением 5,2 г целевого соединения. 39 Ж). 5-Этил-1-(трет.-бутоксикарбонилметил)индол-2-карбоновая кислота. При комнатной температуре и атмосферном давлении гидрируют 5,2 г полученного на предыдущей стадии соединения в 100 мл этанола и 120 мл этилацетата в присутствии 520 мг 10%-го палладия-на-угле. Катализатор отфильтровывают, промывают этилацетатом, затем концентрируют досуха, получая 4 г целевого соединения. Препаративный пример 2.9. 1-(трет.Бутоксикарбонилметил)-5-трифторметилиндол 2-карбоновая кислота. А). Этиловый эфир 2-[(4 трифторметил)фенилгидразоно]пропионовой кислоты. К смеси 200 мл воды и 32 мл концентрированной НСl добавляют по каплям 19,3 г 4 трифторметиланилина, охлаждают до температуры -5 С и добавляют 8,3 г NaNO2 в 20 мл воды. Кроме того, приготовляют раствор 98 г гидратированного ацетата натрия СН 3 СО 2Nа (3H2O) и 29 мл этилового эфира 2-метил-3 оксобутановой кислоты в 125 мл этанола и 90 г измельченного льда; охлаждают до температуры-10 С, добавляют вышеполученную реакционную смесь и выдерживают в течение 5 мин при температуре -10 С, затем оставляют стоять для повышения температуры до комнатной. Отфильтровывают осадок, который промывают водой, затем пентаном, получая 22,5 г целевого соединения. Б). 1-(трет.-Бутоксикарбонилметил)-5-трифторметилиндол-2-карбоновая кислота. Затем следуют методике, описанной для стадий В-Ж предыдущего препаративного примера, с целью получения целевого продукта. Препаративный пример 2.10. 1-(трет.Бутоксикарбонилметил)-4-трифторметилиндол 2-карбоновая кислота. Это соединение получают из 3 трифторметиланилина, используя методику,описанную в предыдущем препаративном примере. Препаративный пример 2.11. 5-Метил-1(этоксикарбонилпропил)индол-2-карбоновая кислота. А). Этиловый эфир 5-метилиндол-2 карбоновой кислоты. При температуре 0 С к 23,57 г птолуидина в 50 мл НСl и 100 мл воды добавляют 15,4 г NaNO2 в 4 мл воды; после перемешивания в течение 20 мин добавляют 18,2 г ацетата натрия. Кроме того, приготовляют при температуре 0 С смесь 28,8 г этилового эфира 2-метил 3-оксобутановой кислоты в 100 мл этанола, которую обрабатывают с помощью 11,22 г гидроксида калия в 20 мл воды и 200 г измельченного льда. Добавляют вышеприготовленный раствор соли диазония и перемешивают в течение 3 ч при температуре 0 С. После выдерживания в холодильнике в течение ночи выливают в на 003093 40 сыщенный раствор NaCl, затем экстрагируют этилацетатом, сушат над сульфатом магния и концентрируют. Остаток обрабатывают толуолом, добавляют 16 г п-толуолсульфокислоты и кипятят с обратным холодильником в течение ночи, удаляя воду с помощью аппарата Дина Старка. Реакционную среду хроматографируют на диоксиде кремния, элюируя толуолом, с получением 12 г целевого продукта. Т пл. = 133 С. Б). 5-Метилиндол-2-карбоновая кислота. В 50 мл этанола вносят 12 г полученного на предыдущей стадии сложного эфира и добавляют раствор 3 г гидроксида натрия в 30 мл воды. После перемешивания в течение 30 мин растворитель выпаривают, остаток обрабатывают водой, промывают этилацетатом, затем водные фазы подкисляют до рН 2 путем добавления концентрированной НСl, экстрагируют этилацетатом, экстракт сушат над сульфатом магния и концентрируют, получая 8,83 г целевого соединения. Т пл. = 218 С. В). Бензиловый эфир 5-метилиндол-2 карбоновой кислоты. Этот сложный эфир получают путем воздействия бензилбромида в присутствии DBU согласно обычным методикам. Т пл. = 141 С. Г). Бензиловый эфир 5-метил-1-(этоксикарбонилпропил)индол-2-карбоновой кислоты. В атмосфере азота в 5 мл диметилформамида вносят 0,6 г гидрида натрия в виде 60%-й дисперсии в масле, добавляют 3 г полученного на предыдущей стадии соединения и перемешивают в течение 2 ч. В реакционную среду добавляют при температуре 0 С 3,8 г этилового эфира иодбутановой кислоты в 10 мл диметилформамида. После перемешивания в течение 30 мин растворитель выпаривают, затем остаток обрабатывают этилацетатом. Промывают раствором карбоната натрия, затем сушат над сульфатом магния и концентрируют, получая 2,75 г целевого соединения в виде масла. Д). 5-Метил-1-(этоксикарбонилпропил) индол-2-карбоновая кислота. Все количество полученного на предыдущей стадии соединения гидрируют в течение 2 ч при комнатной температуре и атмосферном давлении в присутствии 1 г 10%-го палладия-наугле в 80 мл метанола и 20 мл диметилформамида. Катализатор отфильтровывают на целите. После выпаривания растворителей полученный продукт переводят в порошкообразное состояние в гептане и получают 1,54 г целевого соединения. Т пл. = 142 С. Препаративный пример 2.12. 1-(Этоксикарбонилпентил)индол-2-карбоновая кислота. А). Бензиловый эфир 1-(Этоксикарбонилпентил)индол-2-карбоновой кислоты. Перемешивают в течение 1 ч в атмосфере азота смесь, содержащую 3 г бензилового эфира индол-2-карбоновой кислоты в 30 мл безводного диметилформамида и 500 мг гидрида натрия в виде 60%-й дисперсии в масле. При темпера 41 туре 0 С добавляют 3 г этилового эфира 6 бромгексановой кислоты в 10 мл диметилформамида и перемешивают в течение ночи при комнатной температуре. Растворители выпаривают, остаток обрабатывают этилацетатом,промывают водой, сушат над сульфатом магния и концентрируют. Остаток хроматографируют на колонке с силикагелем 60 Н, элюируя смесью гептана с толуолом (объемное соотношение 50/50); получают 4,17 г целевого соединения. Б). 1-(Этоксикарбонилпентил)индол-2 карбоновая кислота. Все количество полученного на предыдущей стадии соединения гидрируют в течение 2 ч при комнатной температуре и атмосферном давлении в присутствии 1 г 5%-го палладия-наугле. Катализатор отфильтровывают на целите,затем хроматографируют на силикагеле 60 Н,элюируя смесью дихлорметана с метанолом(объемное соотношение 100/3). Получают 2,14 г целевого соединения. Т пл. = 62 С. Препаративный пример 2.13. 5,7-Диметил 1-(трет.-бутоксикарбонилметил)индол-2-карбоновая кислота. А). Этиловый эфир 5,7-диметилиндол-2 карбоновой кислоты. Этот сложный эфир может быть получен тремя разными способами. Способ 1. а). Этиловый эфир 2-[(2,4-диметилфенил) гидразоно]пропионовой кислоты. Растворяют 17,11 г 2,4-диметиланилина в 36 мл концентрированной НСl, разбавленной с помощью 280 мл воды. При температуре 0 С добавляют раствор 10,13 г нитрита натрия в 30 мл воды. Перемешивают в течение 15 мин при температуре 0 С, затем при температуре 0 С полученный раствор выливают в раствор 20,5 г этилового эфира 2-метил-3-оксобутановой кислоты в 150 мл этанола. В то же самое время добавляют 31,7 г гидроксида калия в виде раствора в 32 мл воды, затем перемешивают в течение 15 мин при температуре 0 С. Среду нейтрализуют с помощью 70,6 мл 2 н. НСl. Выпавший осадок отфильтровывают, промывают его водой, затем осадок растворяют в этилацетате. Сушат над сульфатом натрия и концентрируют. Кристаллический остаток обрабатывают диизопропиловым эфиром, затем отфильтровывают,получая 25,49 г целевого соединения. Т пл. = 146 С. б). Этиловый эфир 5,7-диметилиндол-2 карбоновой кислоты. В течение 3 ч нагревают при температуре 75 С 19 г полученного на предыдущей стадии продукта в 190 мл муравьиной кислоты. Реакционную среду выливают в 2,5 л холодной воды, выпавший осадок отфильтровывают и промывают его водой. Осадок растворяют в этилацетате, сушат над сульфатом натрия и концентрируют. Кристаллический остаток промывают гептаном, затем полученный продукт перекри 003093 42 сталлизовывают из диизопропилового эфира,получая 8,9 г целевого соединения. Т пл. = 141143 С. Способ 2. а). Этиловый эфир 3-(3,5-диметилфенил)2-азидопроп-2-еновой кислоты. При температуре -10 С и в атмосфере сухого азота к смеси 25 мл этанола и 50 мл 21%-го раствора этилата натрия в этаноле добавляют смесь 5 г 3,5-диметилбензальдегида и 19,3 г этилазидоацетата. Перемешивают в течение 1 ч при температуре -10 С и в течение 14,5 ч при температуре +5 С. Выливают в 100 мл воды,выпавший осадок отфильтровывают и промывают его водой. Осадок растворяют в диэтиловом эфире, сушат над сульфатом магния, затем выпаривают, используя продукт таким, какой есть, на следующей стадии. б). Этиловый эфир 5,7-диметилиндол-2 карбоновой кислоты. К 100 мл ксилола при температуре кипения с обратным холодильником добавляют по каплям раствор полученного на предыдущей стадии продукта в 100 мл ксилола. Спустя 2 ч реакционную среду выпаривают и кристаллический остаток промывают пентаном, получая 4,2 г целевого соединения. Т пл. = 146 С. Способ 3.a). N-(Воc)-2,4,6-триметиланилин. Нагревают до температуры кипения с обратным холодильником раствор 36 г Вос 2 О в 60 мл гептана. Добавляют по каплям 20,28 г 2,4,6 триметиланилина и кипятят с обратным холодильником в течение 3 ч. Охлаждают и фильтруют через диоксид кремния, элюируя дихлорметаном. Выпаривают, получая 33,5 г целевого соединения в виде кристаллического продукта белого цвета. Т пл. = 73-73,5 С. б). Этиловый эфир 5,7-диметилиндол-2 карбоновой кислоты. В атмосфере сухого азота охлаждают до температуры -40 С раствор 4,7 г полученного на предыдущей стадии продукта в 70 мл безводного тетрагидрофурана. Добавляют по каплям 34 мл 1,3 М раствора втор.-BuLi в циклогексане. Оставляют стоять в течение 30 мин для повышения температуры до -20 С. Охлаждают до-40 С и быстро добавляют этот раствор желтого цвета к раствору 5,9 г этилоксалата в 70 мл безводного тетрагидрофурана, затем оставляют стоять в атмосфере азота для повышения температуры до комнатной. Спустя 2 ч охлаждают до температуры +4 С и медленно добавляют 200 мл сульфатного буфера. Экстрагируют два раза диэтиловым эфиром, экстракт сушат над сульфатом магния и выпаривают. Маслянистый остаток обрабатывают 100 мл тетрагидрофурана и 160 мл 6 н. НСl. Нагревают при температуре 60 С в течение 1,5 ч, затем охлаждают. Экстрагируют диэтиловым эфиром, сушат над сульфатом магния и выпаривают. Продукт фильтруют через диоксид кремния, элюируя толуолом, с 43 получением 1,6 г целевого соединения. Т пл. = 140-141 С. Б). 5,7-Диметилиндол-2-карбоновая кислота. В течение 6 дней перемешивают 8,7 г полученного на предыдущей стадии продукта в 100 мл абсолютного этанола вместе со 100 мл 2 н. раствора гидроксида натрия, затем в течение 1 ч кипятят с обратным холодильником. Охлаждают до комнатной температуры и добавляют 20 мл концентрированной НСl. Выпавший осадок отфильтровывают, промывают его водой. Осадок растворяют в этилацетате, сушат над сульфатом натрия и концентрируют, получая 7,15 г целевого соединения. Т пл. = 254-256 С. В). Бензиловый эфир 5,7-диметилиндол-2 карбоновой кислоты. Этот сложный эфир может быть получен двумя разными способами. Способ 1. Растворяют 8,3 г полученного на предыдущей стадии продукта в 60 мл диметилформамида. Добавляют 6,68 г DBU и перемешивают в течение 15 мин при комнатной температуре,затем добавляют 8,25 г бензилбромида и перемешивают в течение 48 ч при комнатной температуре. Концентрируют в вакууме, затем остаток обрабатывают с помощью 500 мл воды. Отфильтровывают выпавший осадок, растворяют его в этилацетате, затем органический раствор промывают насыщенным водным раствором карбоната натрия, сульфатным буфером, насыщенным водным раствором NaCl. Сушат над сульфатом натрия, концентрируют, затем промывают остаток, который кристаллизуют из гептана. Получают 11,15 г целевого соединения. Т пл. = 130-131 С. Способ 2. В атмосфере азота охлаждают до температуры -40 С раствор 4,7 г N-Boc-2,4,6-триметиланилина, полученного на стадии а) указанного выше способа 3, в 70 мл тетрагидрофурана. Добавляют по каплям 34 мл 1,3 М раствора втор.BuLi в циклогексане и оставляют стоять в течение 30 мин для повышения температуры до-20 С. Охлаждают до температуры -40 С и быстро добавляют этот раствор желтого цвета к раствору 9,53 г бензилоксалата в 70 мл безводного тетрагидрофурана. Оставляют стоять в атмосфере азота для повышения температуры до комнатной. Спустя 2 ч охлаждают до температуры +4 С и добавляют 200 мл сульфатного буфера. Экстрагируют 2 раза диэтиловым эфиром,экстракт сушат над сульфатом магния и выпаривают. Остаток охлаждают до температуры+4 С и добавляют смесь 5 мл анизола, 20 мл дихлорметана и 20 мл трифторуксусной кислоты. Оставляют стоять для повышения температуры до комнатной. Спустя 3 ч выпаривают и остаток обрабатывают водой. Экстрагируют этилацетатом, декантируют и органическую фазу промывают 5%-м раствором карбоната 44 натрия. Сушат над сульфатом магния, выпаривают, затем фильтруют через диоксид кремния,элюируя толуолом. Получают 1,24 г целевого соединения. Т пл. = 132,5-133,5 С. Г). Бензиловый эфир 5,7-диметил-1-(трет.бутоксикарбонилметил)индол-2-карбоновой кислоты. В атмосфере азота 167 мг гидрида натрия в виде 60%-й дисперсии в масле вносят в 10 мл диметилформамида и порциями добавляют 1 г полученного на предыдущей стадии соединения. После перемешивания в течение 3 ч при комнатной температуре, добавляют 0,7 мл трет.бутилбромацетата в виде раствора в 1 мл диметилформамида и перемешивают в течение 12 ч. Избыточное количество гидрида натрия гидролизуют, затем реакционную среду концентрируют, обрабатывают этилацетатом, промывают водой, затем раствором карбоната натрия. Выпаривают, затем сушат над сульфатом натрия и остаток хроматографируют на диоксиде кремния, элюируя смесью пентана с дихлорметаном(объемное соотношение 60/40). Получают 940 мг целевого соединения. Т пл. = 115 С. Д). 5,7-Диметил-1-(трет.-бутоксикарбонилметил)индол-2-карбоновая кислота. Все количество полученного на предыдущей стадии соединения гидрируют при комнатной температуре и атмосферном давлении в присутствии 5%-го палладия-на-угле в 5 мл этанола и 20 мл этилацетата. Катализатор отфильтровывают на целите, затем выпаривают, получая 596 мг целевого соединения. Т пл. = 210 С. Препаративный пример 2.13-бис. 5,7 Диметил-1-(метоксикарбонилметил)индол-2 карбоновая кислота. А). Бензиловый эфир 5,7-диметил-1(метоксикарбонилметил)индол-2-карбоновой кислоты. Растворяют 11,03 г продукта, полученного на стадии В препаративного примера 2.13, в 50 мл СН 3 СN. Добавляют 1,35 г бензилтриэтиламмонийхлорида, 12,39 г карбоната калия и 7,55 г метилбромацетата. Кипятят с обратным холодильником в течение 3 ч, затем добавляют еще 1,64 г карбоната калия и 1,81 г метилбромацетата. Снова кипятят с обратным холодильником в течение 3 ч, затем неорганические вещества отфильтровывают. Фильтрат концентрируют,затем остаток хроматографируют на силикагеле 60 Н, элюируя толуолом. Получают 9,7 г целевого соединения. Т пл. = 91-93 С. Б). 5,7-Диметил-1-(метоксикарбонилметил) индол-2-карбоновая кислота. Растворяют 9,6 г полученного на предыдущей стадии продукта в 100 мл диметилформамида и 100 мл абсолютного этанола. Добавляют 900 мг 10%-го палладия-на-угле и гидрируют при комнатной температуре и атмосферном давлении. Фильтруют через hyflo, концентрируют в вакууме, затем остаток обрабатывают с помощью 300 мл воды. Осадок отфильтровы 45 вают, затем его растворяют в этилацетате, раствор сушат над сульфатом натрия, концентрируют, затем промывают остаток, который кристаллизуют из диизопропилового эфира. Получают 6,11 г целевого соединения. Т пл. = 221223 С. Препаративный пример 2.14. 5,6-Диметил 1-(трет.-бутоксикарбонилметил)индол-2-карбоновая кислота. А). 2,4,5-Триметиланилин. Растворяют 19,70 г 2,4,5-триметилнитробензола в 500 мл этанола, затем гидрируют при комнатной температуре и атмосферном давлении в присутствии 1 г 5%-го палладия-на-угле. Катализатор отфильтровывают на целите, затем среду выпаривают, получая 15,67 г целевого соединения. Б). (N-Boc)-2,4,5-триметиланилин. Доводят до температуры кипения с обратным холодильником раствор 41 г (Вос)2 О в 60 мл гептана, затем добавляют по каплям раствор 15,67 г полученного на предыдущей стадии соединения в 20 мл этилацетата и кипятят с обратным холодильником в течение 3 ч. После охлаждения продукт кристаллизуется, образовавшиеся кристаллы отфильтровывают, получая 13,68 г целевого соединения. Фильтрат выпаривают, обрабатывают гептаном и перемешивают,снова отфильтровывают 8,35 г кристаллов целевого продукта. Таким образом получают 22,03 г. Т пл. = 109-110 С. В). Этиловый эфир 5,6-диметилиндол-2 карбоновой кислоты. В атмосфере азота охлаждают до температуры -40 С раствор 4,70 г полученного на предыдущей стадии соединения в 70 мл тетрагидрофурана и добавляют по каплям 34 мл 1,3 М раствора втор.-бутиллития в циклогексане, затем перемешивают в течение 30 мин при температуре -40 С. Реакционную среду выливают в перемешиваемый при температуре -40 С раствор 5,9 г этилоксалата в 70 мл тетрагидрофурана и оставляют стоять для повышения температуры до комнатной. Охлаждают до температуры 0 С, затем добавляют 100 мл воды. Экстрагируют 3 раза диэтиловым эфиром, сушат над сульфатом натрия и выпаривают, затем хроматографируют на диоксиде кремния, элюируя дихлорметаном. Полученное твердое вещество обрабатывают смесью 60 мл воды, 60 мл 12 н. НСl и 60 мл тетрагидрофурана, затем нагревают при температуре 60 С в течение 3 ч. После охлаждения экстрагируют 3 раза диэтиловым эфиром, сушат над сульфатом натрия и выпаривают, затем остаток хроматографируют на силикагеле 60 Н, элюируя толуолом. Получают 1,37 г целевого соединения. Т пл. = 163,5-164,5 С. Г). 5,6-Диметилиндол-2-карбоновая кислота. Полученный на предыдущей стадии сложный эфир гидролизуют путем воздействия гидроксида натрия в метаноле, затем подкисления с 46 помощью концентрированной НСl. Т пл. = 266266,5 С. Д). Бензиловый эфир 5,6-диметилиндол-2 карбоновой кислоты. Этот сложный эфир получают путем воздействия бензилбромида в присутствии DBU согласно обычным способам. Т пл. = 172,5173,5 С. Е). Бензиловый эфир 5,6-диметил-1-(трет.бутоксикарбонилметил)индол-2-карбоновой кислоты. В атмосфере азота охлаждают до температуры -4 С 1,67 г полученного на предыдущей стадии соединения и порциями добавляют 150 мг гидрида натрия в виде 60%-й дисперсии в масле. После перемешивания в течение 30 мин при температуре -4 С добавляют по каплям раствор 0,7 г трет.-бутилбромацетата в5 мл диметилформамида. Оставляют повышаться температуру до комнатной и, спустя ночь, диметилформамид выпаривают, остаток обрабатывают водой, отфильтровывают твердое вещество,промывают водой, затем обрабатывают этилацетатом. Органическую фазу промывают 2 раза водой, сушат над сульфатом натрия и выпаривают, затем переводят в порошкообразное состояние в 70 мл смеси гептана с диэтиловым эфиром (объемное соотношение 50/20), отфильтровывают и высушивают, получая 2 г целевого соединения в виде твердого вещества белого цвета. Т пл. = 133-134 С. Ж). 5,6-Диметил-1-(трет.-бутоксикарбонилметил)индол-2-карбоновая кислота. Целевой продукт получают путем каталитического гидрирования в присутствии 5%-го палладия-на-угле. Т пл. = 240-241 С. Препаративный пример 2.15. 4,5-Диметокси-1-(трет.-бутоксикарбонилметил)индол-2-карбоновая кислота. А). Этиловый эфир 2-азидо-3-(2,3-диметоксифенил)акриловой кислоты. В атмосфере азота охлаждают до температуры -30 С раствор этилата натрия, приготовленный из 80 мл абсолютного этанола и 2,76 г натрия. Добавляют 4,99 г 2,3-диметоксибензальдегида и 15,5 г этилазидоацетата, затем перемешивают при температуре от -20 до -10 С в течение 2 ч. Реакционную среду выливают в 250 мл воды, содержащей 25 мл концентрированной НСl. Выпавший осадок отфильтровывают, промывают его водой, затем растворяют этот осадок в диэтиловом эфире. Сушат над сульфатом натрия и концентрируют, получая 5,86 г целевого соединения. Т пл. 50 С. Б). Этиловый эфир 4,5-диметоксииндол-2 карбоновой кислоты. Растворяют 5,85 г полученного на предыдущей стадии соединения в 200 мл толуола и в течение 7 ч кипятят с обратным холодильником. Выдерживают в течение 2 дней при комнатной температуре, затем концентрируют в вакууме. Остаток хроматографируют на силикагеле 60 Н, 47 элюируя смесью дихлорметана с метанолом(объемное соотношение 100/0,6). После кристаллизации из пентана получают 2,32 г целевого соединения. Т пл. = 129-131 С. Затем осуществляют обычные стадии для получения следующих соединений. В). 4,5-Диметоксииндол-2-карбоновая кислота. Т пл.=258-260 С. Г). Бензиловый эфир 4,5-диметоксииндол 2-карбоновой кислоты. Т пл. = 109-111 С. Д). Бензиловый эфир 4,5-диметокси-1(трет.-бутоксикарбонилметил)индол-2-карбоновой кислоты. Т пл. = 70-72 С. Е). 4,5-диметокси-1-(трет.-бутоксикарбонилметил)индол-2-карбоновая кислота. Т пл. = 206-208 С. Препаративный пример 2.16. 4,5-Дихлор 1-(этоксикарбонилметил)индол-2-уксусная кислота. Это соединение получают из 2,3 дихлорбензальдегида, следуя методике вышеописанного препаративного примера. Т пл. = 205-207 С. Препаративный пример 2.17. 3,5-Диметил 1-(метоксикарбонилэтил)индол-2-карбоновая кислота. А). Этиловый эфир 3,5-диметилиндол-2 карбоновой кислоты. В смесь 120 мл воды и 50 мл концентрированной НСl вводят 23,57 г п-толуидина, затем добавляют по каплям, при температуре 0 С,раствор 15,4 г NaNO2 в 40 мл воды и перемешивают в течение 20 мин при температуре 0 С. К таким образом полученному раствору птолуолдиазонийхлорида при температуре -10 С добавляют раствор 32 г этилового эфира 2-этил 3-оксобутановой кислоты в 150 мл этанола и 150 мл 20%-го раствора гидроксида натрия. После перемешивания в течение 30 мин при температуре -5 С подкисляют до рН 4 путем добавления разбавленной НСl и 1 л воды. За счет порошкования образуется твердое вещество красного цвета, которое отфильтровывают, затем высушивают в сушильном шкафу при температуре 40 С. Это твердое вещество обрабатывают 200 мл абсолютного этанола и 20 мл концентрированной серной кислоты, затем в течение 45 мин кипятят с обратным холодильником. Реакционную среду выливают в смесь воды со льдом, отфильтровывают, затем осадок обрабатывают этилацетатом, промывают водой и сушат над сульфатом магния. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с гептаном. Т пл. = 118 С. Б). 3,5-Диметилиндол-2-карбоновая кислота. Это соединение получают путем гидролиза полученного на предыдущей стадии сложного эфира. Т пл. = 177 С. 48 В). Бензиловый эфир 3,5-диметилиндол-2 карбоновой кислоты. Это соединение получают из полученной на предыдущей стадии кислоты путем воздействия бензилбромида в присутствии DBU. Т пл. = 91 С. Г). Бензиловый эфир 3,5-диметил-1-(2 цианоэтил)индол-2-карбоновой кислоты. Реакцию осуществляют согласно методике, аналогичной таковой, описанной в J. Chem.Soc. (С), 2599-2601 [1967] для N-цианоэтилирования индолов. В 20 мл диоксана вводят 0,5 мл водного 40%-го раствора N-бензилтриметиламмонийгидроксида и 2,2 мл акрилонитрила, затем добавляют шпателем 3 г полученного на предыдущей стадии соединения и после полного растворения нагревают при температуре 80 С в течение 24 ч. Реакционную среду выпаривают,обрабатывают этилацетатом, затем промывают раствором карбоната натрия и сушат над сульфатом магния. Остаток хроматографируют на диоксиде кремния, элюируя смесью гептана с толуолом (объемное соотношение 50/50). Получают 3,2 г целевого соединения. Т пл. = 71 С. Д) Бензиловый эфир 3,5-диметил-1(метоксикарбонилэтил)индол-2-карбоновой кислоты. В 30 мл дихлорметана и 3,8 мл метанола вносят 3,2 г полученного на предыдущей стадии соединения и добавляют 45 мл насыщенного газообразным хлороводородом диэтилового эфира. После выдерживания в течение 2 дней при температуре -4 С, отфильтровывают образовавшееся твердое вещество белого цвета,промывают его смесью диэтилового эфира с гептаном (объемное соотношение 50/50), затем обрабатывают с помощью 15 мл воды и 15 мл уксусной кислоты и в течение 30 мин нагревают при температуре 100 С. Добавляют 50 мл воды,экстрагируют дихлорметаном, затем сушат над сульфатом магния, получая 1,72 г целевого соединения в виде масла. Е). 3,5-Диметил-1-(метоксикарбонилэтил) индол-2-карбоновая кислота. Все количество полученного на предыдущей стадии соединения гидрируют при комнатной температуре и атмосферном давлении в 200 мл 90%-го этанола в присутствии 300 мг 5%-го палладия-на-угле. Катализатор отфильтровывают на целите, затем фильтрат переводят в порошкообразное состояние с гептаном, содержащим небольшое количество диэтилового эфира. Получают 0,8 г целевого соединения. Т пл. = 169 С. Препаративный пример 2.18. 5-(N-трет.Бутоксикарбониламино)-1-(трет.-бутоксикарбонилметил)индол-2-карбоновая кислота.A). 5-Нитроиндол-2-карбоновая кислота. В 200 мл этанола вносят 13 г этилового эфира 5-нитроиндол-2-карбоновой кислоты и обрабатывают в течение 12 ч с помощью 15 г 49 30%-го раствора NaOH. После выпаривания растворителя подкисляют концентрированной НСl, затем отфильтровывают, получая 10,8 г целевого соединения. Б). Бензиловый эфир 5-нитроиндол-2 карбоновой кислоты. Это соединение получают путем воздействия бензилбромида в присутствии DBU. Т пл. = 192 С.B). Бензиловый эфир 5-нитро-1-(трет.бутоксикарбонилметил)индол-2-карбоновой кислоты. Это соединение получают путем воздействия гидрида натрия, затем трет.-бутилбромацетата. Т пл. = 112 С. Г). 5-Амино-1-(трет.-бутоксикарбонилметил)индол-2-карбоновая кислота. Гидрируют 16 г полученного на предыдущей стадии соединения в 200 мл диметилформамида при атмосферном давлении и комнатной температуре в присутствии 200 мг 5%-го палладия-на-угле. Катализатор отфильтровывают на целите, растворитель выпаривают, затем переводят в порошкообразное состояние в минимальном количестве дихлорметана, получая 10,1 г целевого соединения. Т пл. = 128 С. Д). 5-(N-трет.-Бутоксикарбониламино)-1(трет.-бутоксикарбонилметил)индол-2-карбоновая кислота. В раствор, содержащий 30 мл диоксана, 30 мл воды и 1 мл триэтиламина, вносят 2 г полученного на предыдущей стадии соединения и добавляют по каплям при температуре 80 С раствор 2 г Вос 2 О в 10 мл диоксана. По окончании выделения диоксида углерода растворитель выпаривают, затем остаток обрабатывают этилацетатом. Промывают сульфатным буфером,затем сушат над сульфатом магния. Получают 2,44 г целевого соединения. Т пл. 300 С. Следуя вышеописанным методикам, получают производные индол-2-карбоновой кислоты, указанные в нижеследующей таблице. Таблица 2 В. Получение производных пирролопиридинкарбоновой кислоты Препаративный пример 3.1. 1-трет.Бутоксикарбонилметил-1 Н-пирроло[3,2-с]пиридин-2-карбоновая кислота. А). Бензиловый эфир 1H-пирроло[3,2-с] пиридин-2-карбоновой кислоты. В атмосфере азота и при температуре-40 С к 5 г 4-(N-Boc-амино)-3-метилпиридина добавляют 40 мл 1,7 М раствора трет.бутиллития в пентане. После перемешивания в течение 1 ч при температуре -40 С таким образом полученное литийсодержащее производное добавляют к раствору 12,9 г бензилового эфира щавелевой кислоты в 100 мл тетрагидрофурана при температуре -40 С. Перемешивают в течение 2 ч при повышении температуры до 0 С,затем добавляют 25 мл 6 н. НСl и нагревают в течение 1,5 ч при температуре 50 С. Доводят до рН 9 путем добавления 1 н. раствора NaOH, затем экстрагируют этилацетатом, сушат и выпаривают. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом (объемное соотношение 100/3), получая 2,7 г целевого соединения. Б). Бензиловый эфир 1-трет.-бутоксикарбонилметил-1H-пирроло[3,2-с]пиридин-2-карбоновой кислоты. 52 В атмосфере азота в 20 мл диметилформамида вносят 0,29 г гидрида натрия в виде 60%-й дисперсии в масле, затем при температуре 10 С добавляют 1,7 г полученного на предыдущей стадии соединения. После перемешивания в течение 45 мин добавляют 1,43 г трет.бутилового эфира бромуксусной кислоты. Оставляют стоять для повышения температуры до комнатной, перемешивают в течение 5 ч, затем реакционную массу выливают в воду, экстрагируют диэтиловым эфиром, сушат, затем выпаривают. Полученное желтое масло хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом (объемное соотношение 100/3), затем содержащие продукт фракции объединяют и хроматографируют повторно на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (объемное соотношение 1/1). Получают 1 г кристаллов желтого цвета. Т пл. = 94 С. В). 1-трет.-Бутоксикарбонилметил-1 Нпирроло[3,2-с]пиридин-2-карбоновая кислота. Осуществляют каталитическое гидрирование 1 г полученного на предыдущей стадии соединения в присутствии 170 мг 10%-го палладия-на-угле при атмосферном давлении и комнатной температуре в течение 4 ч. Образовавшийся осадок растворяют в диметилформамиде,затем отфильтровывают палладий-на-угле на целите, фильтрат выпаривают досуха, затем кристаллы промывают диэтиловым эфиром,получая 0,39 г целевого соединения. Т пл. = 265 С. Препаративный пример 3.2. 1-трет.Бутоксикарбонилметил-5-метокси-1 Н-пирроло[2,3-с]пиридин-2-карбоновая кислота. А). 5-(N-Boc-амино)-2-метокси-4-метилпиридин. К 5 г 5-(N-Boc-амино)-2-метоксипиридина в атмосфере азота и при температуре -60 С добавляют 41,7 мл 1,6 М раствора н-бутиллития в гексане и 7,7 г тетраметилэтилендиамина. Среда окрашивается в желтый цвет. Перемешивают в течение 4 ч до достижения температуры -10 С; выпадает осадок светло-желтого цвета. Охлаждают до температуры -40 С, затем добавляют 4,7 г метилиодида. Оставляют стоять для повышения температуры до комнатной, затем реакционную массу выливают в воду, экстрагируют этилацетатом, сушат и выпаривают. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (объемное соотношение 85/15). Б). Бензиловый эфир 5-метокси-1 Нпирроло[2,3-с]пиридин-2-карбоновой кислоты. При температуре -60 С и в атмосфере азота, 13,8 мл 1 М раствора н-бутиллития в гексане добавляют к 2,4 г полученного на предыдущей стадии соединения. Оставляют температуру повышаться до -20 С и перемешивают в течение 30 мин. Таким образом полученное литийсодержащее производное добавляют к 5,4 г бензи 53 локсалата и перемешивают в течение 2 ч при комнатной температуре. Выливают в воду, экстрагируют этилацетатом, сушат, затем выпаривают. Обрабатывают с помощью 100 мл тетрагидрофурана, затем добавляют 30 мл 6 н. НСl и нагревают при температуре 50 С в течение 1,5 ч. Доводят до рН 6 путем добавления 1 н. НСl,экстрагируют дихлорметаном, сушат, затем выпаривают. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом (объемное соотношение 100/2,5). Получают 0,9 г целевого соединения. Т пл. = 182 С. В). Бензиловый эфир 1-трет.-бутоксикарбонилметил-5-метокси-1H-пирроло[2,3-с]пиридин-2-карбоновой кислоты. К 0,14 г гидрида натрия, в виде 60%-й дисперсии в масле, в 10 мл диметилформамида при температуре 5 С добавляют 0,9 г полученного на предыдущей стадии соединения, перемешивают в течение 15 мин, затем при комнатной температуре добавляют 0,68 г трет.-бутилового эфира бромуксусной кислоты. После перемешивания в течение 5 ч при комнатной температуре выливают в воду, экстрагируют диэтиловым эфиром, сушат и выпаривают. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с этилацетатом (объемное соотношение 100/1). Получают 1,2 г целевого соединения. Т пл. = 110 С. Г). 1-трет.-Бутоксикарбонилметил-5 метокси-1 Н-пирроло[2,3-с]пиридин-2-карбоновая кислота. В течение 3 ч при атмосферном давлении и комнатной температуре в присутствии 120 мг 10%-го палладия-на-угле гидрируют 1,2 г полученного на предыдущей стадии соединения,растворенного в смеси этанола с диметилформамидом. Палладий-на-угле отфильтровывают на целите, фильтрат выпаривают досуха, затем остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом (объемное соотношение 100/3). Получают 0,41 г целевого соединения в виде белой пены. Препаративный пример 3.3. 1-трет.Бутоксикарбонилметил-1 Н-пирроло[2,3-b]пиридин-2-карбоновая кислота. А). Этиловый эфир 1 Н-пирроло[2,3-b] пиридин-2-карбоновой кислоты. При температуре ниже 5 С и в атмосфере азота к 5 г 2-(N-Boc)амино-3-метилпиридина в 100 мл тетрагидрофурана добавляют 30 мл нбутиллития. После перемешивания в течение 1 ч при температуре 0 С таким образом полученное литийсодержащее производное при температуре-3 С добавляют к раствору 7 г этилоксалата в 50 мл тетрагидрофурана. Оставляют стоять для повышения температуры до комнатной, затем реакционную массу медленно выливают в 25 мл 6 н. НСl при температуре 0 С, поддерживая температуру ниже 10 С. Нагревают в течение 2 ч при температуре 50 С, затем доводят до рН 3 54 путем добавления 1 н. раствора NaOH. Экстрагируют диэтиловым эфиром, органическую фазу обрабатывают раствором карбоната калия, затем сушат и выпаривают, получая 1,8 г целевого соединения в виде кристаллов белого цвета. Т пл. = 162 С. Б). 1H-Пирроло[2,3-b]пиридин-2-карбоновая кислота. В течение 3 ч перемешивают смесь, содержащую 2,4 г полученного на предыдущей стадии соединения и 2,5 г гидроксида натрия в смеси этанола с водой (объемное соотношение 20/20). Выпавший осадок отфильтровывают,затем растворяют в воде. При подкислении до рН 4 с помощью уксусной кислоты выпадает осадок белого цвета, который промывают водой, затем сушат, получая 1,3 г целевого соединения. Т пл. 260 С. В). Бензиловый эфир 1H-пирроло[2,3-b] пиридин-2-карбоновой кислоты. При комнатной температуре в течение 2 ч перемешивают смесь, содержащую 1,3 г полученного на предыдущей стадии соединения,1,27 г DBU и 1,37 г бензилбромида в 30 мл диметилформамида. После выпаривания растворителя остаток обрабатывают дихлорметаном и водой, затем экстрагируют сульфатным буфером. Сушат, затем выпаривают, образовавшиеся кристаллы промывают гептаном, получая 1,5 г целевого соединения. Т пл. = 176 С. Г). Бензиловый эфир 1-трет.-бутоксикарбонилметил-1H-пирроло[2,3-b]пиридин-2-карбоновой кислоты. Это соединение получают по методике,описанной для стадии В предыдущего препаративного примера. Д). 1-трет.-Бутоксикарбонилметил-1 Нпирроло[2,3-b]пиридин-2-карбоновая кислота. Это соединение получают путем гидрирования в присутствии палладия-на-угле. Т пл. = 104 С. Пример 1. 2-(5-Циклогексилэтил-4-(2,5 диметокси-4-метилфенил)тиазол-2-илкарбамоил)-5-метилиндол-1-уксусная кислотаСF3 СООН. А). трет.-Бутиловый эфир 2-(5-циклогексилэтил-4-(2,5-диметокси-4-метилфенил)тиазол-2-илкарбамоил)-5-метилиндол-1-уксусной кислоты. В 2 мл диметилформамида смешивают 0,505 г соединения препаративного примера 1.1,0,45 г соединения препаративного примера 2.3,0,75 г ВОР (бензотриазол-1-илокситрис(диметиламино)фосфонийгексафторфосфат) и 0,17 г триэтиламина. После перемешивания в течение 11 дней при комнатной температуре выливают в 150 мл сульфатного буфера. Выпавший осадок отфильтровывают, затем промывают водой, по 55 сле чего его растворяют в дихлорметане, раствор сушат над сульфатом натрия и концентрируют. Остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с этилацетатом (объемное соотношение 100/1), получая 0,41 г целевого соединения. Б). 2-(5-Циклогексилэтил-4-(2,5-диметокси-4-метилфенил)тиазол-2-илкарбамоил)-5 метилиндол-1-уксусная кислотаСF3 СООН. Растворяют 0,41 г полученного на предыдущей стадии соединения в 15 мл трифторуксусной кислоты. После перемешивания в течение 4 ч при температуре 10 С концентрируют в вакууме. Остаток обрабатывают с помощью 250 мл воды, затем перемешивают в течение 1 ч при комнатной температуре. Выпавший осадок белого цвета отфильтровывают, затем сушат, получая 0,37 г целевого продукта. Т пл. = 140 С. Пример 1-бис. Натриевая соль 2-(5 циклогексилэтил-4-(2,5-диметокси-4-метилфенил)тиазол-2-илкарбамоил)-5-метилиндол-1 уксусной кислотыCF3COONa. Суспензию 0,47 г полученного в примере 1 соединения в 200 мл абсолютного этанола нагревают до температуры кипения с обратным холодильником, добавляют 0,68 мл 2 н. раствораNaOH и перемешивают в течение 10 мин при кипячении с обратным холодильником. Концентрируют в вакууме, остаток обрабатывают диэтиловым эфиром, образовавшийся кристаллический продукт отфильтровывают под вакуумом и промывают его диэтиловым эфиром. Получают 0,36 г целевого продукта. Т пл. = 170 С. Пример 2. 2-(5-Циклогексилэтил-4-(2,5 диметокси-4-метилфенил)тиазол-2-илкарбамоил)-4-метоксииндол-1-уксусная кислотаНСl. А). трет.-Бутиловый эфир 2-(5-циклогексилэтил-4-(2,5-диметокси-4-метилфенил)тиазол-2-илкарбамоил)-4-метоксииндол-1-уксусной кислоты. В 2 мл диметилформамида смешивают 0,505 г соединения препаративного примера 1.1,0,47 г соединения препаративного примера 2.4,0,75 г ВОР и 0,17 г триэтиламина. После перемешивания в течение 5 дней при комнатной температуре добавляют 50 мл сульфатного буфера. Выпавший осадок растворяют в дихлорметане, затем раствор сушат над сульфатом натрия и концентрируют. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с этилацетатом (объемное соотношение 100/2); получают 0,8 г целевого соединения в виде масла. Б). 2-(5-Циклогексилэтил-4-(2,5-диметокси-4-метилфенил)тиазол-2-илкарбамоил)-4 метоксииндол-1-уксусная кислотаНСl. Растворяют 0,9 г полученного на предыдущей стадии соединения в 10 мл дихлормета 003093 56 на, затем при температуре 10 С добавляют 10 мл трифторуксусной кислоты и перемешивают в течение 4 ч при температуре 10 С. Концентрируют в вакууме, остаток обрабатывают с помощью 100 мл диэтилового эфира, затем экстрагируют 2 раза по 25 мл 2 н. раствором NaOH. Водную фазу подкисляют с помощью 55 мл 2 н. НСl,затем выпавший осадок отфильтровывают. Его промывают водой, затем сушат, получая 0,68 г целевого соединения. Т пл. = 150 С. Пример 3. Натриевая соль 2-5 циклогексилэтил)-4-(2,5-диметокси-4-метилфенил)тиазол-2-илкарбамоил)индол-1-уксусной кислоты. А). Метиловый эфир 2-5-циклогексилэтил)-4-(2,5-диметокси-4-метилфенил)тиазол-2-илкарбамоил)индол-1-уксусной кислоты. В 30 мл дихлорметана смешивают 0,976 г соединения препаративного примера 1.1, 0,7 г 1 метоксикарбонилметилиндол-2-карбоновой кислоты (препаративный пример 2.1), 1,44 г ВОР и 0,33 г триэтиламина. После перемешивания в течение 3 дней при комнатной температуре добавляют 30 мл сульфатного буфера, декантируют, органическую фазу сушат над сульфатом натрия, затем концентрируют. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом (объемное соотношение 100/0,2). Получают 1,52 г целевого соединения. Т пл. = 144-146 С. Б). Натриевая соль 2-5-циклогексилэтил)4-(2,5-диметокси-4-метилфенил)тиазол-2-илкарбамоил)индол-1-уксусной кислоты. Растворяют 0,5 г полученного на предыдущей стадии соединения в 30 мл диоксана и добавляют 10 мл пропан-2-ола и 0,9 г 30%-го раствора гидроксида натрия. После перемешивания в течение 15 ч при комнатной температуре растворитель выпаривают. Остаток обрабатывают изопропанолом, образовавшиеся кристаллы отфильтровывают, промывают изопропанолом, диэтиловым эфиром, затем высушивают в сушильном шкафу. Получают 0,46 г целевого соединения. Т пл. 350 С. Натриевая соль кристаллизуется с одной молекулой NaOH. Пример 4. 2-5-Циклогексилэтил)-4-(2,5 диметокси-4-метилфенил)тиазол-2-илкарбамоил)индол-1-уксусная кислота. В 3 мл диметилформамида смешивают 0,7 г соединения препаративного примера 1.1, 0,53 г 1-трет.-бутоксикарбонилметилиндол-2-карбоновой кислоты (препаративный пример 2.2), 0,85 г ВОР и 0,25 мл триэтиламина. После перемешивания в течение ночи при комнатной температу 57 ре добавляют сульфатный буфер. Образовавшиеся кристаллы отфильтровывают, обрабатывают дихлорметаном. Органическую фазу промывают сульфатным буфером, затем раствором карбоната натрия и высушивают над сульфатом магния. Хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с этилацетатом (объемное соотношение 100/1). Полученный чистый продукт обрабатывают с помощью 10 мл трифторуксусной кислоты и перемешивают в течение 3 ч. После выпаривания обрабатывают водой, раствором карбоната натрия, затем добавляют концентрированную соляную кислоту вплоть до рН 5. Экстрагируют дихлорметаном, затем сушат над сульфатом магния; получают 0,42 г целевого соединения. Т пл. = 198 С. Пример 5. 2-(4-(4-Хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-илкарбамоил)-5,7-диметилиндол-1-уксусная кислота СF3 СООН. А). трет.-Бутиловый эфир 2-(4- (4-хлор-2,5 диметоксифенил)-5-(2-циклогексилэтил)тиазол 2-илкарбамоил)-5,7-диметилиндол-1-уксусной кислоты. Смешивают 250 мг соединения препаративного примера 2.13, 314 мг соединения препаративного примера 1.4, 0,34 мг триэтиламина и 365 мг ВОР в 10 мл дихлорметана и перемешивают в течение 3 дней при комнатной температуре. Экстрагируют диэтиловым эфиром,промывают водой, насыщенным раствором гидросульфата калия, затем сушат над сульфатом натрия и растворители выпаривают. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с этилацетатом (объемное соотношение 30/20). Получают 218 мг целевого соединения. Б). 2-(4-(4-Хлор-2,5-диметоксифенил)-5-(2 циклогексилэтил)тиазол-2-илкарбамоил)-5,7 диметилиндол-1-уксусная кислотаСF3 СООН. Смешивают 218 мг полученного на предыдущей стадии соединения с 2,5 мл трифторуксусной кислоты в 6 мл дихлорметана и перемешивают в течение 3 ч при комнатной температуре. Реакционную смесь концентрируют, обрабатывают диэтиловым эфиром и выпавший осадок отфильтровывают. Получают 160 мг целевого соединения. Т пл. = 173 С. Пример 5-2. Калиевая соль 2-(4-(4-хлор 2,5-диметоксифенил)-5-циклогексилэтилтиазол 2-илкарбамоил)-5,7-диметилиндол-1-уксусной кислоты 3 Н 2O. 58 А). Метиловый эфир 2-(4-(4-хлор-2,5 диметоксифенил)-5-циклогексилэтилтиазол-2 илкарбамоил)-5,7-диметилиндол-1-уксусной кислоты. Смешивают 5,9 г соединения препаративного примера 2.13-бис, 7,82 г соединения препаративного примера 1.4, 2,5 г триэтиламина и 10,9 г ВОР в 35 мл диметилформамида и перемешивают в течение 48 ч при комнатной температуре. Выливают в 2 л сульфатного буфера,выпавший осадок отфильтровывают, промывают его водой. Осадок растворяют в 400 мл этилацетата, промывают последовательно 2 раза с помощью 250 мл насыщенного раствора карбоната натрия, 150 мл раствора сульфатного буфера и 150 мл насыщенного раствора NaCl. Органическую фазу сушат над сульфатом натрия и выпаривают. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с этилацетатом (объемное соотношение 98/2). Полученные кристаллы промывают диизопропиловым эфиром. Получают 11,6 г целевого продукта. Т пл. = 202 С. Б). Калиевая соль 2-(4-(4-хлор-2,5 диметоксифенил)-5-циклогексилэтилтиазол-2 илкарбамоил)-5,7-диметилиндол-1-уксусной кислоты 3 Н 2 О. Смесь 2 г полученного на стадии А продукта, 1,33 г карбоната калия в 4,6 мл воды и 8 мл н-бутанола нагревают при температуре 90 С в течение 9 ч. После охлаждения до комнатной температуры добавляют воду и выпавший осадок отфильтровывают, промывают 3 раза по 50 мл водой, затем 3 раза по 20 мл диэтиловым эфиром и высушивают в вакууме. Получают 1,2 г целевого продукта. Т пл. = 250 С. Пример 5-3. 2-(4-(4-Хлор-2,5-диметоксифенил)-5-циклогексилэтилтиазол-2-илкарбамоил)-5,7-диметилиндол-1-уксусная кислота. Смесь 4 г полученного на стадии А примера 5-2 соединения, 2,65 г карбоната калия в 9,2 мл воды и 16 мл н-бутанола нагревают при температуре 90 С в течение 12 ч. После охлаждения до комнатной температуры добавляют 19,2 мл 2 н. НСl. Выпавший осадок белого цвета промывают 3 раза 200 мл воды, затем 3 раза 200 мл диэтилового эфира. Получают 3,1 г целевого продукта. Т пл. = 241 С. Пример 5-4. Натриевая соль 2-(4-(4-хлор 2,5-диметоксифенил)-5-циклогексилэтилтиазол 2-илкарбамоил)-5,7-диметилиндол-1-уксусной кислоты 1,5 Н 2 О. Т пл. = 200 С. Примеры 6 и 7. Натриевая соль 2-(4-(4 хлор-2,5-диметоксифенил)-5-циклогексилэтилтиазол-2-илкарбамоил)индол-1-уксусной кислоты и 2-(4-(4-хлор-2,5-диметоксифенил)-5-циклогексилэтилтиазол-2-илкарбамоил)индол-1-уксусная кислота. Следуют методике, описанной в примере 3, стадия А, исходя из соединений препаративных примеров 1.4 и 2.1, с целью получения метилового эфира 2-(4-(4-хлор-2,5-диметоксифенил)-5-(циклогексилэтил)тиазол-2-илкарбамоил)индол-1-уксусной кислоты. Т пл. = 145 С(стадия А). Следуя методике, описанной для стадии Б, получают натриевую соль целевого соединения (пример 6), которая кристаллизуется с одной молекулой NaOH. Т пл. = 252 С. К суспензии 0,7 г полученного на стадии А сложного эфира в 30 мл метанола добавляют 2 мл 2 н. раствора NaOH. Полученный раствор оставляют стоять в течение 18 ч, затем метанол выпаривают. Остаток обрабатывают водой, затем подкисляют путем добавления концентрированной НСl до рН 2. После перемешивания в течение 1 ч выпавший осадок отфильтровывают, промывают водой и высушивают в сушильном шкафу. Получают 0,63 г целевой кислоты В течение ночи перемешивают 0,88 г соединения препаративного примера 1.4, 0,7 г соединения препаративного примера 2.12, 1,2 г ВОР и 0,32 мл триэтиламина в 3 мл диметилформамида. Добавляют сульфатный буфер, выпавший осадок отфильтровывают и обрабатывают этилацетатом. Промывают сульфатным буфером, раствором карбоната натрия, затем сушат над сульфатом магния. Хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с этилацетатом (объемное соотношение 100/2). Полученный продукт омыляют в 10 мл этанола и 2 мл 4 н. раствора NaOH. Растворитель выпаривают, остаток обрабатывают водой, затем подкисляют до рН 2 путем добавления концентрированной НСl. Выпавший осадок отфильтровывают; получают 1,21 г целевого соединения. Т пл. = 121 С. Пример 9. 2-(5-Циклогексилэтил-4-(2,5 диметокси-4-метилфенил)тиазол-2-илкарбамоил)индол-1-бутановая кислота. В течение 4 ч перемешивают смесь 0,6 г соединения препаративного примера 2.21, 0,75 г соединения препаративного примера 1.1, 1 г ВОР и 0,3 г триэтиламина в 3 мл диметилфор 003093 60 мамида. Добавляют сульфатный буфер и отфильтровывают выпавший осадок. Его обрабатывают этилацетатом, полученный раствор промывают сульфатным буфером, раствором карбоната натрия, затем сушат над сульфатом магния и концентрируют в вакууме. Хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с этилацетатом (объемное соотношение 100/1). Полученный продукт омыляют в 10 мл этанола и 2 мл 4 н. раствора NaOH. Растворитель выпаривают, затем остаток переводят в порошкообразное состояние в воде с несколькими каплями концентрированной НСl, полученный осадок отфильтровывают и высушивают. Получают 0,82 г целевого соединения. Т пл. = 223 С. Следуя методикам, описанным в вышеприведенных примерах, получают соединения согласно изобретению, и соединения сравнения,исходя из соединений, полученных в препаративных примерах. Сведения о полученных соединениях приведены в табл. 3. Таблица 3
МПК / Метки
МПК: C07D 209/42, A61P 1/06, A61P 25/00, A61P 3/04, C07D 417/12, C07D 277/46, A61K 31/405, A61K 31/427
Метки: содержащие, композиции, производные, фармацевтические, получение, карбоксамидотиазолов
Код ссылки
<a href="https://eas.patents.su/30-3093-proizvodnye-karboksamidotiazolov-ih-poluchenie-soderzhashhie-ih-farmacevticheskie-kompozicii.html" rel="bookmark" title="База патентов Евразийского Союза">Производные карбоксамидотиазолов, их получение, содержащие их фармацевтические композиции</a>
Новые таксоиды,их получение и содержащие их фармацевтические композиции

Номер патента: 1533
Опубликовано: 23.04.2001
Авторы: Коммерсон Алан, Бушар Эрве
МПК: A61K 31/335, C07D 305/14, A61P 35/00...
Метки: содержащие, получение, фармацевтические, композиции, новые, таксоиды,их
Формула / Реферат:
1. Новые таксоиды общей формулы в которой Z обозначает радикал общей формулы в которой R1 обозначает бензоильный радикал или радикал R2-О-СО-, в котором R2 представляет собой алкильный радикал с 1-8 атомами углерода и R3 обозначает фенил или a - или b -нафтил; и либо R4 обозначает атом водорода, R6 и R7 вместе образуют кетонную функцию и R и R5 вместе образуют связь, либо R4 обозначает атом водорода или радикал алкокси с 1-6 атомами...
Новые таксоиды, их получение и фармацевтические композиции, их содержащие

Номер патента: 1516
Опубликовано: 23.04.2001
Авторы: Бурза Жан-Доминик, Бушар Эрве, Коммерсон Ален
МПК: A61K 31/335, A61P 35/00, C07D 305/14...
Метки: новые, получение, фармацевтические, таксоиды, содержащие, композиции
Формула / Реферат:
1. Новые таксоиды общей формулы (I) в которой Z означает радикал общей формулы (II) в которой R1 означает радикал бензоил или алкоксикарбонил; R3 означает фенил; R4 означает радикал гидрокси или радикал алкокси, содержащий от 1 до 4 атомов углерода с прямой или разветвленной цепью, алканоилокси, алканоильная часть которого содержит от 2 до 4 атомов углерода с прямой или разветвленной цепью; R5 означает радикал алкокси, содержащий от 1 до...
Новые соединения 1 – аза – 2 – алкил – 6 – арилциклоалкана, способ их получения и содержащие их фармацевтические композиции

Номер патента: 2620
Опубликовано: 27.06.2002
Авторы: Ренар Пьер, Лестаж Пьер, Лебрэн Мари-Сесиль, Рено Оливье, Гийон Жан, Пфеффер Брюно, Ро Сильвен, Даллемань Патрик
МПК: C07D 211/38, A61K 31/44, A61P 25/04...
Метки: получения, фармацевтические, композиции, соединения, содержащие, новые, алкил, аза, арилциклоалкана, способ
Формула / Реферат:
1. Соединения формулы (I) в которой n равно 0 или 1, R1 представляет атом водорода или арил-(C1-С6)алкильную группу, в которой алкильная часть является линейной или разветвленной, линейную или разветвленую (C1-С6)алкильную группу, линейную или разветвленную (C1-C6)ацильную группу, линейную или разветвленную (C1-C6)алкоксикарбонильную группу, арил-(C1-С6)алкоксикарбонильную группу, в которой алкокси часть является линейной или разветвленной, или...
Антибактериальные соединения карбапенема, содержащие их фармацевтические композиции и способы лечения

Номер патента: 1296
Опубликовано: 25.12.2000
Авторы: Близзард Тимоти А., Вилкенинг Роберт Р., Рэтклифф Рональд В.
МПК: C07D 477/14, A61K 31/428
Метки: соединения, способы, лечения, карбапенема, фармацевтические, содержащие, композиции, антибактериальные
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемые соли, где R1 обозначает Н или метил; СО2М обозначает карбоновую кислоту, карбоксилатный анион, фармацевтически приемлемую сложноэфирную группу или карбоновую кислоту, защищенную защитной группой; Р обозначает водород, гидроксил, F или гидроксил, защищенный гидроксилзащитной группой; каждый R независимо выбирают из -R*; -Q; водорода; галогена; -CN; -NO2; -NRaRb; -ORc; -SRc; -C(O)NRaRb;...
Фармацевтические композиции, содержащие вориконазол

Номер патента: 1924
Опубликовано: 22.10.2001
Автор: Хардинг Валери Дениз
МПК: A61K 47/40, A61P 31/10
Метки: фармацевтические, вориконазол, содержащие, композиции
Формула / Реферат:
1. Фармацевтическая композиция, содержащая вориконазол или его фармацевтически приемлемое производное и производное циклодекстрина формулы I где R1a-g, R2a-g и R3a-g независимо представляют собой ОН либо O(СН2)4SO3Н; при условии, что, по меньшей мере, один из R1a-g представляет собой O(СН2)4SO3Н; или его фармацевтически приемлемую соль. 2. Композиция по п.1, где среднее количество групп O(СН2)4SO3Н на молекулу формулы I находится в пределах...