Фармацевтическая композиция ингибитора протеазы вируса гепатита c











Формула / Реферат
1. Жидкая фармацевтическая композиция для лечения инфекции вируса гепатита C, содержащая:
(а) 5-30 мас.% соединения формулы (1)

или его фармацевтически приемлемой соли;
(б) 30-60 мас.% фармацевтически приемлемого липида, выбранного из группы, включающей жирные кислоты, моно-, ди- или триглицериды со средней или длинной цепью, эфиры пропиленгликоля и жирных кислот, эфиры сорбита и жирных кислот, неводорастворимые витамины и их смеси; и
(в) 20-50 мас.% фармацевтически приемлемого гидрофильного поверхностно-активного вещества, выбранного из группы, включающей полиэтоксилированные растительные масла, полиэтоксилированные токоферолы, полиэтоксилированные эфиры сорбита и жирных кислот, соли желчных кислот, лецитины и их смеси.
2. Фармацевтическая композиция по п.1, в которой соединение формулы (1) присутствует в виде натриевой соли.
3. Фармацевтическая композиция по п.1 или 2, дополнительно содержащая 5-15% фармацевтически приемлемого гидрофильного растворителя, выбранного из группы, включающей пропиленгликоль, полипропиленгликоль, полиэтиленгликоль, глицерин, этанол, диметилизосорбид, гликофурол, пропиленкарбонат, диметилацетамид, воду и их смеси.
4. Фармацевтическая композиция по одному из предыдущих пунктов, где композиция при разведении водным раствором с использованием массового соотношения водного раствора и композиции 100:1 образует водную дисперсию, абсорбция которой превышает примерно 1,0 при длине волны примерно 400 нм.
5. Фармацевтическая композиция по одному из предыдущих пунктов, содержащая:
(а) 10-20 мас.% соединения формулы (1) в виде его натриевой соли;
(б) 40-50 мас.% фармацевтически приемлемого липида, выбранного из моноглицеридов каприловых и каприновых жирных кислот, диглицеридов каприловых и каприновых жирных кислот и их смесей;
(в) 25-35 мас.% фармацевтически приемлемого гидрофильного поверхностно-активного вещества, выбранного из токоферилполиэтиленгликольсукцината, полиоксила 40 гидрогенизированного касторового масла и полиоксила 35 касторового масла и их смесей;
(г) 5-10 мас.% фармацевтически приемлемого гидрофильного растворителя, выбранного из пропиленгликоля, полиэтиленгликоля, этанола, воды и их смесей.
Текст
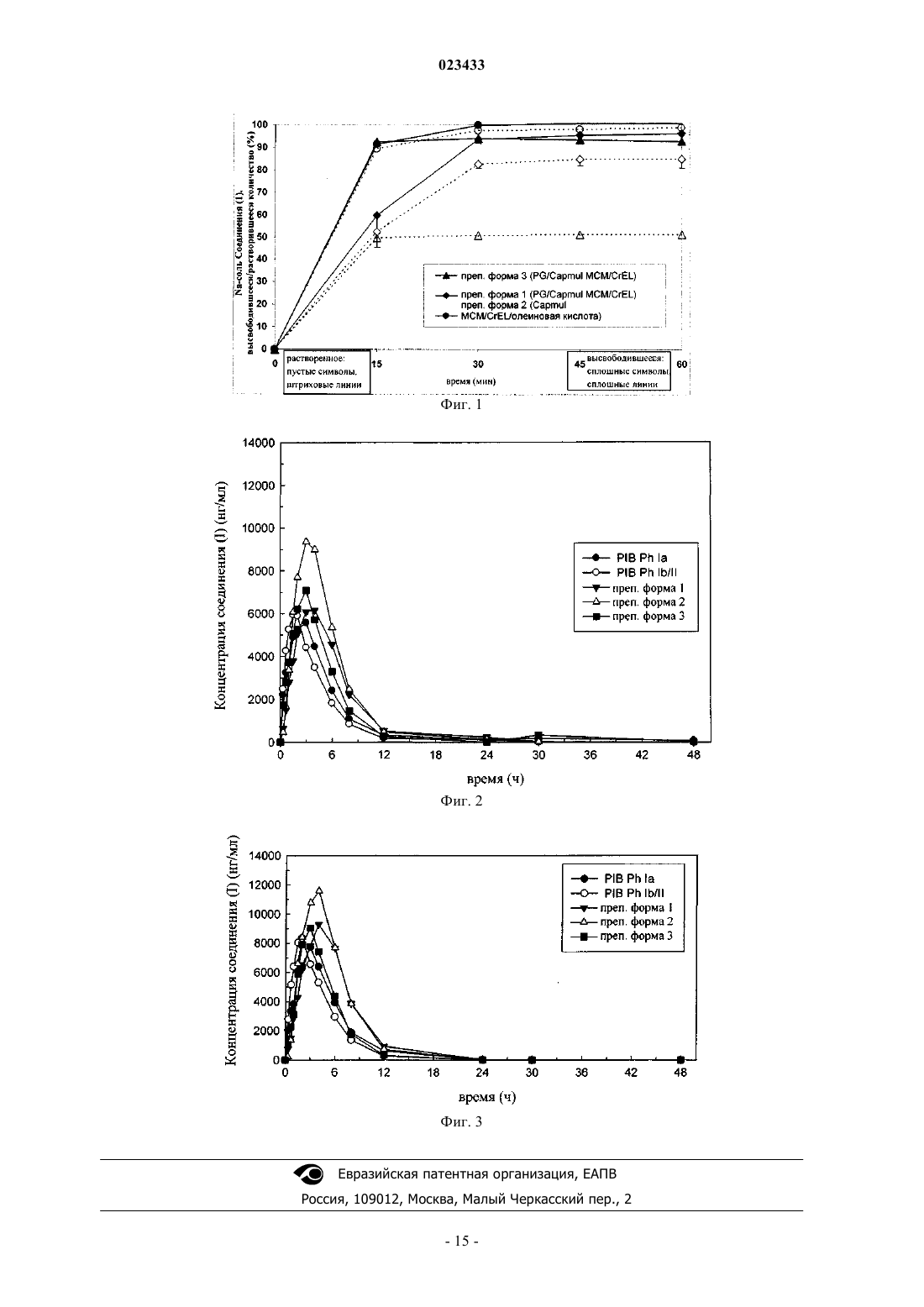
ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ИНГИБИТОРА ПРОТЕАЗЫ ВИРУСА ГЕПАТИТА C В изобретении приведены описание фармацевтической композиции ингибитора протеазы вируса гепатита С, которую можно использовать для орального введения в виде капсулы,заполненной жидкостью или полутвердым веществом, и способы применения этой композиции для ингибирования репликации вируса гепатита С (HCV) и лечения инфекции, вызываемойHCV. Жидкая фармацевтическая композиция, предлагаемая в настоящем изобретении,содержит 5-30 мас.% соединения формулы I или его фармацевтически приемлемой соли в сочетании с 30-60 мас.% фармацевтически приемлемыми липидами, выбранными из группы, включающей жирные кислоты, моно-, ди- или триглицериды со средней или длинной цепью, эфиры пропиленгликоля и жирных кислот, эфиры сорбита и жирных кислот,неводорастворимые витамины и их смеси, и 20-50 мас.% гидрофильными поверхностноактивными веществами, выбранными из группы, включающей полиэтоксилированные растительные масла, полиэтоксилированные токоферолы, полиэтоксилированные эфиры сорбита и жирных кислот, соли желчных кислот, лецитины и их смеси.(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) Область техники, к которой относится изобретение Настоящее изобретение в целом относится к фармацевтической композиции ингибитора протеазы вируса гепатита C, способам применения этой композиции для ингибирования репликации вируса гепатита C (HCV) и лечения инфекции, вызываемой HCV. Предпосылки создания изобретения Следующее соединение (1):(обозначено далее в настоящем описании как "соединение (1)") является известным в качестве селективного и эффективного ингибитора сериновой протеазы NS3 HCV. Соединение (1) относится к группе ациклических пептидов, являющихся ингибиторами HCV, которые описаны в US 6323180 и US 7514557 и US 7585845. Соединение (1) описано конкретно в качестве соединения 1055 в US 7585845 и соединения 1008 в US 7514557. Соединение (1) можно получать согласно процессам, описанным в вышеуказанных документах, которые полностью включены в настоящее описание в качестве ссылок. Предпочтительными формами соединения (1) являются кристаллические формы, прежде всего кристаллическая форма натриевой соли, которую можно получать с помощью методов, представленных в настоящем описании в разделе "Примеры". Химическую структуру соединение (1) можно представить также с помощью другого изображения,представленного ниже, которое эквивалентно вышеуказанной структуре:. Общая проблема, связанная с ингибиторами протеаз, заключается в том, что эти соединения являются липофильными и обладают слабой растворимостью в воде. Вследствие слабой растворимости в воде в организме пациента не может происходить удовлетворительная абсорбция стандартных твердых и жидких фармацевтических композиций, содержащих такие ингибиторы. Установлено, что среди различных факторов, которые могут оказывать влияние на биологическую доступность вводимого оральным путем лекарственного средства (включая растворимость в воде, абсорбцию лекарственного средства в желудочно-кишечном тракте, величину дозы и эффект первого прохождения), растворимость в воде часто является одним из наиболее важных факторов. Слабая растворимость соединений в воде часто приво-1 023433 дит либо к нарушению абсорбции, либо к недостаточной абсорбции в пищеварительном тракте и тем самым вызывает более низкий, чем это требуется, ответ. Соединение (1) является цвиттерионом и обладает способностью образовывать соли с сильными кислотами и основаниями. Попытки получить соли этого соединения в твердой форме, что позволило бы существенно повышать растворимость в воде и, как следствие, биологической доступность, оказались безуспешными. Таким образом, в данной области существует необходимость в создании фармацевтических композиций соединения (1), обладающих повышенной биологической доступностью. Ранее были описаны методы получения фармацевтических препаративных форм, содержащих определенные липофильные макроциклические соединения. Например, у Cavanak, в US 4388307 описано получение препаративных форм в виде эмульсии, содержащих поступающие в продажу циклоспорины, а у Hauer с соавторами в US 5342625 и у Meizner с соавторами в WO 93/20833 описано получение микроэмульсий и преконцентратов (предварительных концентратов) микроэмульсий циклоспорина. Кроме того, у Komiya с соавторами в US 5504068 описано получение улучшенных препаративных форм циклоспорина, предназначенных для местного применения. Примеры "самоэмульгирующихся" препаративных форм липофильных соединений приведены уLipari с соавторами в WO 96/36316, в которой описан самоэмульгирующийся преконцентрат, содержащий липофильное соединение, d-альфа-токоферилполиэтиленгликоль(1000)сукцинат (ТПГС) и липофильную фазу. У Gao с соавторами в US 6121313 описана самоэмульгирующаяся препаративная форма ингибитора пираноновой протеазы, содержащая производное пиранона, смесь моно- и диглицеридов,один или несколько растворителей и одно или несколько поверхностно-активных веществ; а у Gao с соавторами в US 6231887 В 1 описана самоэмульгирующаяся препаративная форма ингибитора пираноновой протеазы, содержащая производное пиранона, амин, один или несколько растворителей и одно или несколько поверхностно-активных веществ. У Crison с соавторами в US 5993858 описана самоэмульгирующаяся препаративная форма эксципиентов, которая представляет собой эмульсию, содержащую масло или другой липидный продукт, поверхностно-активное вещество и гидрофильное вспомогательное поверхностно-активное вещество. У Patel с соавторами в US 6294192 и 6451339 описаны композиции, предназначенные для введения гидрофобного терапевтического средства, которые содержат носитель, состоящий из комбинации гидрофильного поверхностно-активного вещества и гидрофобного поверхностно-активного вещества. УAylwin с соавторами в US 6652880 описана жидкая фармацевтическая композиция, в которой действующее вещество растворено в жидком наполнителе, включающем глицерид жирной кислоты с длинной цепью и липофильное поверхностно-активное вещество. Для определенных соединений, обладающих активностью в отношении HCV, разработана также самоэмульгирующаяся система для введения лекарственных средств (СЭСВЛ), описанная в US 6828301 и 7157424 и публикации заявки на патент США 2004/0033959. Однако в данной области сохраняется необходимость в фармацевтической препаративной форме соединения (1), которая является в достаточной степени оптимизированной, стабильной и обладает биологической доступностью. Краткое изложение сущности изобретения Настоящее изобретение позволяет решать вышеуказанные проблемы с помощью имеющей липидную основу фармацевтической композиции соединения (1), которую можно применять для орального введения с использованием капсулы, заполненной жидким или полутвердым веществом. Имеющие липидную основу фармацевтические композиции, предлагаемые в настоящем изобретении, относятся к типу самоэмульгирующейся системы для введения лекарственных средств (далее в контексте настоящего описания "СЭСВЛ"), и они обладают приемлемой стабильностью и биологической доступностью и поэтому являются наиболее пригодными для терапевтического введения соединения (1). Все фармацевтические композиции, предлагаемые в настоящем изобретении, содержат 5-30 мас.% соединения (1) или его фармацевтически приемлемой соли в сочетании с 30-60 мас.% фармацевтически приемлемыми липидами, выбранными из группы, включающей жирные кислоты, моно-, ди- или триглицериды со средней или длинной цепью, эфиры пропиленгликоля и жирных кислот, эфиры сорбита и жирных кислот, неводорастворимые витамины и их смеси, и 20-50 мас.% гидрофильными поверхностно-активными веществами, выбранными из группы, включающей полиэтоксилированные растительные масла, полиэтоксилированные токоферолы, полиэтоксилированные эфиры сорбита и жирных кислот,соли желчных кислот, лецитины и их смеси. Композиции, предлагаемые в настоящем изобретении, необязательно могут содержать один или несколько дополнительных ингредиентов, таких, например, как фармацевтически приемлемые гидрофильные растворители, отверждающие агенты, антиоксиданты и т.д., как это будет описано более подробно ниже. Фармацевтические композиции являются жидкими или полутвердыми и их предпочтительно заключают в капсулы, предназначенные для орального введения. Предлагаемые в изобретении композиции можно использовать в способе лечения вызываемой вирусом гепатита C инфекции у млекопитающего, заключающимся в том, что вводят млекопитающему в терапевтически эффективном количестве фармацевтическую композицию, предлагаемую в настоящем изобретении. Краткое описание чертежей На чертежах показано: на фиг. 1 - результаты оценки растворимости in vitro трех препаративных форм, предлагаемых в настоящем изобретении, которые представлены в виде высвободившегося/растворившегося количества (в процентах) Na-соли соединения (1) в зависимости от времени; на фиг. 2 - средние концентрации соединения (1) в плазме шести собак после введения пяти различных препаративных форм Na-соли соединения (1); на фиг. 3 - средние концентрации соединения (1) в плазме трех собак после введения пяти различных препаративных форм Na-соли соединения (1). Подробное описание изобретения Определение применяемых понятий и условных обозначений. Понятия, определения которых не приведены конкретно в настоящем описании, имеют значения,которые специалист в данной области должен придавать им на основе содержания и контекста описания. Однако если не указано иное, то перечисленные ниже понятия и обозначения, используемые в описании,имеют следующие значения. Понятие "примерно" означает, что рассматриваемая величина находится в пределах 20%, предпочтительно в пределах 10% и наиболее предпочтительно в пределах 5% от данной величины или диапазона. Например, "примерно 10%" означает от 8 до 12%, предпочтительно от 9 до 11% и наиболее предпочтительно от 9.5 до 10,5%. Если понятие "примерно" употребляется применительно к диапазону значений,например "примерно от X до Y%", то подразумевается, что понятие "примерно" касается вариации как нижней (X), так и верхней (Y) границы указанного диапазона. Например, понятие "примерно от 0,1 до 10%" эквивалентно понятию "примерно от 0,1 до примерно 10%". Все проценты, указанные для количеств ингредиентов в композициях, представляют собой мас.% в пересчете на массу всей композиции. В контексте настоящего описания понятие "фармацевтически приемлемая" по отношению к субстанции обозначает субстанцию, которую, по мнению специалистов в области медицины, можно использовать для контакта с тканями человека и низших животных, не вызывая при этом излишнего токсического действия, раздражения, аллергической реакции и т.п., и которая характеризуется приемлемым соотношением польза/риск и обладает эффективностью при требуемом применении при ее использовании в составе фармацевтической композиции. Понятие "полутвердый" обозначает материал, который не является ни твердым (материал, который обладает упругими свойствами), ни жидким (материал, который обладает вязкими свойствами) и обладает как характеристиками вязкости, так и характеристиками упругости. Примерами полутвердых материалов являются гели, мази, кремы и жидкости, обладающие большой вязкостью. Понятия "лечить" или "лечение" обозначают лечение инфекции, вызываемой вирусом гепатита C, у пациента и включают:(I) предупреждение возникновения инфекции, вызываемой вирусом гепатита C, у пациента, прежде всего, в том случае, когда пациент предрасположен к такому болезненному состоянию, но оно еще не диагностировано у него;(II) ингибирование или облегчение инфекции, вызываемой вирусом гепатита C, т.е. прекращение или замедление ее развития; или(III) ослабление инфекции, вызываемой вирусом гепатита C, т.е. достижение ее регресса или излечения болезненного состояния. Понятие "терапевтически эффективное количество" обозначает количество соединения, предлагаемого в изобретении, которое при введении пациенту, нуждающемуся в этом, является достаточным для лечения инфекции, вызываемой вирусом гепатита C. Специалист в данной области может определить такое терапевтически эффективное количество стандартными методами на основе своих знаний, существующего уровня техники и настоящего описания. Предпочтительные варианты осуществления изобретения Основным вариантом осуществления настоящего изобретения является фармацевтическая композиция, содержащая 5-30 мас.% соединения формулы (1) или его фармацевтически приемлемой соли, 3060 мас.% фармацевтически приемлемых липидов, выбранных из группы, включающей: жирные кислоты,моно-. ди- или триглицериды со средней или длинной цепью, эфиры пропиленгликоля и жирных кислот,эфиры сорбита и жирных кислот, неводорастворимые витамины и их смеси, и 20-50 мас.% фармацевтически приемлемых гидрофильных поверхностно-активных веществ, выбранных из группы, включающей: полиэтоксилированные растительные масла, полиэтоксилированные токоферолы, полиэтоксилированные эфиры сорбита и жирных кислот, соли желчных кислот, лецитины и их смеси. В более конкретном варианте осуществления изобретения фармацевтическая композиция состоит практически из 5-30 мас.% соединения формулы (1) или его фармацевтически приемлемой соли, 30-60 мас.% фармацевтически приемлемых липидов, выбранного из группы включающей: жирные кислоты, моно-, ди- или триглицериды со средней или длинной цепью, эфиры пропиленгликоля и жирных кислот, эфиры сорбита и жирных кислот, неводорастворимые витамины и их смеси, и 20-50 мас.% фармацевтически приемлемых гидро-3 023433 фильных поверхностно-активных веществ, выбранных из группы, включающей: полиэтоксилированные растительные масла, полиэтоксилированные токоферолы, полиэтоксилированные эфиры сорбита и жирных кислот, соли желчных кислот, лецитины и их смеси. Соединение (1). Соединение (1) можно применять в свободной форме или в форме его фармацевтически приемлемой соли. Понятие "фармацевтически приемлемая соль" относится к соли соединения (1), которую, по мнению специалистов в области медицины, можно использовать для контакта с тканями человека и низших животных, не вызывая при этом излишнего токсического действия, раздражения, аллергической реакции и т.п., и которая характеризуется приемлемым соотношением польза/риск, как правило, является растворимой или диспергируемой в воде или масле и обладает эффективностью при требуемом применении. Понятие относится к фармацевтически приемлемым кислотно-аддитивным солям и фармацевтически приемлемым солям присоединения оснований. Перечень приемлемых солей представлен, например,у S.M. Birge et al., J. Pharm. Sci., 66, 1977, p. 1-19 и в US 7585845. Различные соли, перечисленные вUS 7585845, включены в настоящее описание в качестве ссылки. Наиболее предпочтительной формой соединения (1), которую можно применять в композиции,предлагаемой в настоящем изобретении, является натриевая соль соединения (1). Методы получения кристаллической формы натриевой соли описаны ниже в разделе "Примеры". Натриевая соль соединения(1) может находиться в кристаллическом, аморфном состоянии или представлять собой смесь указанных форм. Могут иметь место также различные виды полиморфизма известной кристаллической лекарственной субстанции, которые представлены в настоящем описании. Лекарственную субстанцию, представляющую собой соединение (1), можно применять непосредственно в нативном состоянии или ее можно подвергать соответствующей обработке для (1) снижения степени агломерации частиц лекарственной субстанции и/или (2) снижения распределения по размерам первичных частиц лекарственной субстанции. Этот процесс может представлять собой просеивание, деагломерацию, размалывание с помощью ударной мельницы, размалывание с помощью струйной мельницы или их комбинации с целью снижения продолжительности смешения при изготовлении нерасфасованного содержимого, предназначенного для капсулирования. Количество, в котором соединение (1), представляющее собой действующее вещество, может присутствовать в составе системы на основе липидов, может варьироваться в широких пределах или его можно регулировать в широких пределах в зависимости от предполагаемого пути введения, эффективности конкретного применяемого действующего вещества, серьезности инфекции, вызываемой вирусом гепатита C, и требуемой концентрации. В конкретном варианте осуществления изобретения соединение формулы (1) присутствует в системе на основе липидов в количестве примерно от 5 до 30 мас.%, предпочтительно примерно от 10 до 20 мас.%. Липиды. Как хорошо известно в данной области, эмпирическим параметром, который, как правило, используют для характеризации относительной гидрофильности и гидрофобности соединений, является гидрофильно-липофильный баланс (значение "ГЛБ"). Поверхностно-активные вещества с более низкими значениями ГЛБ являются более гидрофобными и характеризуются более высокой растворимостью в маслах, а поверхностно-активные вещества с более высокими значениями ГЛБ являются более гидрофильными и характеризуются более высокой растворимостью в водных растворах. При применении значений ГЛБ в качестве грубого ориентирующего критерия, как правило, считается, что к гидрофильным поверхностно-активным веществам относятся соединения, значение ГЛБ для которых выше примерно 10, а также анионогенные, катионогенные и цвиттерионные соединения, для которых шкала на основе ГЛБ,как правило, не применима. Аналогично этому, к гидрофобным поверхностно-активным веществам относятся соединения, значение ГЛБ для которых ниже примерно 10. Фармацевтически приемлемые липиды, которые можно применять в композиции, предлагаемой в настоящем изобретении, представляют собой любой липидный продукт, который имеет значение гидрофильно-липофильного баланса (значение "ГЛБ") более низкое или равное 10 (ГЛБ 10), отличающийся ограниченной растворимостью в воде. Примерами фармацевтически приемлемых липидов, которые можно применять, являются многочисленные смешивающиеся с водой продукты, такие, например, как жирные кислоты, моно-, ди- или триглицериды со средней или длинной цепью, эфиры пропиленгликоля и жирных кислот, эфиры сорбитана и жирных кислот, неводорастворимые витамины и их смеси. В предпочтительном варианте осуществления изобретения фармацевтически приемлемый липид выбран из группы, включающей моноглицериды каприловых и каприновых жирных кислот, диглицериды каприловых и каприновых жирных кислот и их смеси (например, CAPMUL МСМ фирмы Abitech Corp.). Количество липида в композиции может варьироваться в широких пределах, и количество, оптимальное для конкретной композиции, должно зависеть от типа и количества других ингредиентов в композиции, что может определять специалист в области фармацевтики. Однако, как правило, фармацевтически приемлемый липид присутствует в количестве примерно от 30 до 60 мас.% или в количестве при-4 023433 мерно от 40 до 50 мас.%. Гидрофильное поверхностно-активное вещество. Для облегчения самоэмульгирования в композицию, предлагаемую в настоящем изобретении,включают фармацевтически приемлемое гидрофильное поверхностно-активное вещество, которое имеет значение ГЛБ, превышающее или равное 10 (ГЛБ 10), и отличается способностью хорошо смешиваться с водой. Примерами фармацевтически приемлемых гидрофильных поверхностно-активных веществ, которые можно применять, являются полиэтоксилированные растительные масла, полиэтоксилированные токоферолы, полиэтоксилированные эфиры сорбита и жирных кислот (например, Твин 80), соли желчных кислот, лецитины и их смеси. Предпочтительными поверхностно-активными веществами являются токоферилполиэтиленгликольсукцинат (ТПГС) (витамин Е ТПГС), полиоксил 40 гидрогенизированного касторового масла (Cremophor RH40) и полиоксил 35 касторового масла (Cremophor EL) и их смеси. Количество гидрофильных поверхностно-активных веществ в композиции может также варьироваться в широких пределах, и оптимальное количество для конкретной композиции должно зависеть от типа и количества других ингредиентов в композиции, что может определять специалист в области фармацевтики. Фармацевтически приемлемое гидрофильное поверхностно-активное вещество предпочтительно присутствует в количестве, составляющем вплоть до примерно от 20 до 50 мас.%, предпочтительно от 25 до 35 мас.%. Гидрофильный растворитель. Композиция, предлагаемая в настоящем изобретении, может необязательно содержать также фармацевтически приемлемый гидрофильный растворитель, предназначенный для (1) повышения растворимости представляющей собой действующее вещество лекарственной субстанции и предупреждения ее осаждения из препаративной формы, (2) снижения продолжительности смешения при изготовлении нерасфасованной жидкой композиции, предназначенной для включения в капсулы, и/или (3) улучшения диспергируемости в воде препаративиной формы. Примерами гидрофильных растворителей, которые можно применять, являются, например, пропиленгликоль, полипропиленгликоль, полиэтиленгликоль,глицерин, этанол, диметилизосорбид, гликофурол, пропиленкарбонат, диметилацетамид, вода или их смеси. Предпочтительными гидрофильными растворителями являются пропиленгликоль, полиэтиленгликоль (например, ПЭГ 400), этанол, вода и их смеси. Количество растворителя в композиции может также варьироваться в широких пределах, и оптимальное количество для конкретной композиции должно зависеть от типа и количества других ингредиентов в композиции, и оно может быть легко определено специалистом в данной области. Однако, как правило, растворитель(и) присутствует(ют) в количестве, составляющем вплоть до 15 мас.%. Отверждающий агент. Композиция, предлагаемая в настоящем изобретении, может необязательно содержать также отверждающий агент для превращения жидкой препаративной формы в полутвердую после капсулирования в состоящие из двух частей твердые капсулы (например, твердые желатиновые капсулы и капсулы из ГПМЦ (гидроксипропилметилцеллюлоза. Примерами отверждающих агентов, которые можно применять, являются полиэтиленгликоли, полоксамеры, поливинилпирролидоны, поливиниловые спирты, производные целлюлозы, полиакрилаты, полиметакрилаты, сахара, полиолы и их смеси. Конкретными предпочтительными примерами являются высокомолекулярные ПЭГ, включая ПЭГ 3350, ПЭГ 6000, ПЭГ 8000,полоксамеры или их смеси. Применение отверждающего агента является наиболее важным для композиций, содержащих гликоль или этанол, для снижения активности пластификатора для желатина в отношении его миграции из жидкого содержимого капсулы в оболочку капсулы, что в результате повышает физическую стабильность, предупреждая размягчение и деформацию лекарственной формы. С другой стороны, этот подход применим также для содержащих ПЭГ 400 композиций для снижения гигроскопичности содержимого и хрупкости капсулы. При применении в композиции отверждающий агент предпочтительно присутствует в количестве, составляющем вплоть до примерно 50 мас.%, предпочтительно примерно от 1 до 20 мас.%. Необязательные дополнительные ингредиенты. При необходимости композиции, предлагаемые в настоящем изобретении, могут дополнительно содержать общепринятые фармацевтические добавки, которые являются необходимыми или желательными для получения требуемой препаративной формы, такие как антиоксиданты, замасливатели, разрыхлители, консерванты, буферы, стабилизаторы, акцепторы, загустители, красители, подслащивающие вещества, корригенты, ароматизаторы и т.д. Другие добавки, которые можно применять в композициях,предлагаемых в изобретении, описаны у Llinas-Brunet с соавторами, US 6323180 В 1. В одном из предпочтительных вариантов осуществления изобретения композиции, предлагаемые в настоящем изобретении, дополнительно содержат один или несколько антиоксидантов. Предпочтительными антиоксидантами являются, например, аскорбиновая кислота, сульфатиды, лимонная кислота, пропилгаллат, dlтокоферол, аскорбилпальмитат, бутилированный гидрокситолуол (БГТ) или бутилированный гидроксианизол (БГА). Количество антиоксиданта, если он присутствует, как правило, составляет от примерно 0,01 до 1 мас.%. В другом предпочтительном варианте осуществления изобретения композиция, предлагаемая в настоящем изобретении, может содержать также активный карбонильный (например, альдегиды, кетоны) акцептор (например, амины, включая Трис и меглумин) для снижения структурирования желатиновых капсул, что может оказывать нежелательное воздействие на высвобождение композиции из лекарственной формы. Стабилизаторы, которые можно применять, представляют собой, например, определенные щелочные агенты, включая амины, которые повышают кажущееся значение рН применяемой для заполнения капсул препаративной формы. Дополнительные предпочтительные варианты осуществления изобретения. В дополнительных вариантах осуществления изобретения композиция, предлагаемая в настоящем изобретении, отличается отсутствием (или наличием лишь ограниченных количеств) одного или несколько классов продуктов, которые, как правило, входят в фармацевтические препаративные формы. В контексте представленного ниже описания понятие "практически свободна от (практически не содержит) конкретного продукта", как правило, обозначает, что в препаративной форме продукт содержится не более чем в следовых количествах, например в количестве не более 1 мас.%, предпочтительно не более 0,5 мас.%, еще более предпочтительно не более 0,1 мас.%. Композиция, предлагаемая в настоящем изобретении, может отличаться одним или несколькими из следующих признаков:(1) она либо практически свободна от какого-либо аминового производного, либо не содержит никакого аминового производного;(2) она либо практически свободна от какого-либо спиртового производного, либо не содержит никакого спиртового производного;(3) она либо практически свободна от какого-либо триглицеридного производного, либо не содержит никакого триглицерида;(4) она либо практически свободна от какого-либо глицерида жирной кислоты с длинной цепью,либо не содержит никакого глицерида;(5) она либо практически свободна от какого-либо дополнительного поверхностно-активного вещества, либо не содержит никакого дополнительного поверхностно-активного вещества. Конкретным вариантом осуществления настоящего изобретения является фармацевтическая композиция, содержащая (или практически состоящая из):(а) соединение формулы (1) или его фармацевтически приемлемую соль в количестве примерно от 10 до 20 мас.%;(6) фармацевтически приемлемый липид в количестве примерно от 40 до 50 мас.%;(в) фармацевтически приемлемое гидрофильное поверхностно-активное вещество в количестве примерно от 25 до 35 мас.%;(г) фармацевтически приемлемый гидрофильный растворитель в количестве примерно от 5 до 15 мас.%. Другим конкретным вариантом осуществления настоящего изобретения является фармацевтическая композиция, содержащая (или практически состоящая из):(а) соединение формулы (1) в виде его натриевой соли в количестве примерно от 10 до 20 мас.%;(б) фармацевтически приемлемый липид, выбранный из моноглицеридов каприловых и каприновых жирных кислот, диглицеридов каприловых и каприновых жирных кислот и их смесей, в количестве примерно от 40 до 50 мас.%:(в) фармацевтически приемлемое гидрофильное поверхностно-активное вещество, выбранное из токоферилполиэтиленгликольсукцината, полиоксила 40 гидрогенизированного касторового масла и полиоксила 35 касторового масла и их смесей, в количестве примерно от 25 до 35 мас.%;(г) фармацевтически приемлемый гидрофильный растворитель, выбранный из пропиленгликоля,полиэтиленгликоля, этанола, воды и их смесей, в количестве примерно от 5 до 10 мас.%. Метод получения и капсулирования. Композицию, предлагаемую в настоящем изобретении, можно получать общепринятым методом,например, методом, заключающемся в том, что перемешивают жидкие компоненты, например, фармацевтически приемлемый(ые) липид(ы), поверхностно-активное(ые) вещество(а) и растворитель(и); необязательно нагревают образовавшуюся смесь, если это требуется для расплавления в достаточной степени одного или нескольких компонентов смеси; добавляют соединение формулы (1) к образовавшейся смеси и продолжают перемешивание вплоть до полной или практически полной солюбилизации соединения формулы (1), например, до тех пор, пока раствор не станет визуально прозрачным. Затем с помощью известных методов изготовления из образовавшегося предназначенного для заполнения раствора приготавливают требуемую лекарственную форму, например, капсулы, включая капсулы с твердой оболочкой или мягкие гелевые капсулы (например, твердые или мягкие желатиновые капсулы). Примеры мягких желатиновых капсул, которые можно применять, включают капсулы, описанные в ЕР 649651 В 1 и US 5985321. В наиболее предпочтительном варианте осуществления изобретения композицию, предлагаемую в настоящем изобретении, капсулируют в мягкие эластичные капсулы, например мягкую желатиновую капсулу и мягкие капсулы, основа которых не имеет животное происхождение. Поскольку композиция может быть практически свободна от полярного растворителя, то ее преимуществом может являться возможность применения готовой стандартной гелевой композиции для включения в оболочку капсулы или простая разработка нового геля для оболочки капсулы, что минимизирует затраты на разработку и стоимость. Поскольку мягкая капсула может иметь больший объем заполнения, то преимуществом композиции, предлагаемой в настоящем изобретении, является более высокая загрузка капсулы лекарственным средством. Следует отметить, что композиция, предлагаемая в настоящем изобретении, может содержать также воду, входящую в лекарственную субстанцию, в эксципиенты (в частности, выбранные из поверхностно-активных веществ и растворителей, которые являются гидрофильными по своей природе), и образовавшуюся в процессе капсулирования. В частности, в процессе капсулирования препаративной формы,предназначенной для заполнения мягких желатиновых капсул, большое количество влаги может мигрировать в предназначенную для заполнения препаративную форму из влажных желатиновых лент. Важно удалять любую избыточную влагу из предназначенной для заполнения композиции с помощью соответствующего процесса сушки для того, чтобы избегать какого-либо осаждения и гидролитического расщепления лекарственной субстанции и избыточного размягчения капсулы. Как правило, полученные после завершения процесса мягкие желатиновые капсулы, предлагаемые в настоящем изобретении, должны содержать в предназначенной для заполнения препаративной форме воду в количестве не более 5 мас.% и более предпочтительно не более 3 мас.%. Одной из важных характерных особенностей, обнаруженных при создании композиций, предлагаемых в настоящем изобретении, является то, что растворимость натриевой соли соединения (1) повышается по мере снижения температуры. Это неожиданно установленное при создании изобретения свойство обеспечивает уникальную благоприятную возможность повышать стабильность лекарственного продукта путем хранение на холоде (например, в холодильнике), не заботясь о возможном осаждении лекарственного средства при пониженной температуре. В результате композиции, предлагаемые в настоящем изобретении, обладают неожиданным преимуществом, связанным с упаковыванием, которое позволяет применять более широкий спектр упаковочных материалов. Методы терапевтического применения. Соединения формулы (1) являются эффективными ингибиторами протеазы HCV и, следовательно,эти соединения и фармацевтические композиции, содержащие эти соединения, можно применять для ингибирования репликации HCV и для лечения инфекций, вызываемых HCV, у млекопитающих. Таким образом, настоящее изобретение относится также к лечению инфекции, вызываемой вирусом гепатита C,у млекопитающих, заключающемуся в том, что млекопитающему вводят в терапевтически эффективном количестве фармацевтическую композицию, предлагаемую в настоящем изобретении. Уровни доз соединений формулы (1) и различные схемы лечения, применяемые в монотерапии для предупреждения и лечения инфекции, вызываемой HCV, описаны в US 758545. Однако специалисту в данной области должно быть очевидно, что при использовании композиций, предлагаемых в настоящем изобретении, можно применять меньшие дозы в зависимости от уровня повышения биологической доступности. Можно применять также комбинированную терапию с использованием одного или нескольких дополнительных терапевтических или профилактических средств, как это подробно описано в US 7585845. Дополнительное(ые) средство(а) можно объединять с соединениями, предлагаемыми в настоящем изобретении, с получением однократной стандартной дозы или в альтернативном варианте это(и) дополнительное(ые) средство(а) можно вводить млекопитающему раздельно в качестве составной части многокомпонентной лекарственной формы. Обычный специалист в данной области на основе собственных знаний, известного уровня техники и настоящего описания может определять с помощью общепринятых методов необходимое терапевтически эффективное количество, в котором требуется вводить фармацевтическую композицию. Ниже настоящее изобретение более подробно проиллюстрировано на примерах. Эти примеры приведены с целью иллюстрации вариантов осуществления настоящего изобретения и никоим образом не направлены на ограничение его объема. Характеристики лекарственного продукта. Имеющую липидную основу систему введения лекарственных средств, предлагаемую в настоящем изобретении, выбирали в связи с липофильной природой натриевой соли соединения (1). Препаративные формы, включающие имеющую липидную основу СЭСВЛ (самоэмульгирующаяся система для введения лекарственных средств), позволяют решать связанную с ограниченной растворимостью проблему абсорбции. Поскольку лекарственная субстанция в лекарственной форме присутствует в растворе и поддерживается в растворе при контакте с водными средами благодаря способности препаративной формы к самоэмульгированию, абсорбция не ограничивается степенью растворения. При создании настоящего изобретения была разработана препаративная форма в виде мягкой капсулы, заполненной жидкостью, для применения в клинических испытаниях. Присущими ей важными характеристиками являются следующие: биологическая доступность (быстрое высвобождение в процессе растворения in vitro; в сочетании с сопоставимой с обнаруженной в опытах на собаках абсорбцией ранее известных препаративных форм в виде порошка для приготовления раствора для орального введения); стабильность (доказанная химическая и физическая стабильность лекарственной формы при оценке в условиях длительного хранения, принятых ICH (Международная конференция по гармонизации руководств по надлежащей клинической практике) при 25C/60%RH); технологичность изготовления (для удовлетворения требованиям эксперимента достаточно размера партий вплоть до 25 кг). Представленные в разделе "Примеры" данные демонстрируют очень высокую стабильность и характеристики биологической доступности препаративной формы в виде капсулы, предлагаемой в настоящем изобретении. Оптическую прозрачность препаративных форм, предлагаемых в настоящем изобретении, оценивали визуально и количественно определяли спектроскопически. Предназначенные для заполнения препаративные формы получали согласно методу, описанному ниже в примере 1 (наряду с соответствующими препаративными формами наполнителей и плацебо), и разводили в 100 раз с получением растворов в водных средах. Абсорбцию для каждого раствора оценивали при длине волны 400 и 450 нм, используя соответствующую стандарту очищенную воду, подробные результаты представлены в настоящем описании в разделе "Примеры". Указанные результаты свидетельствуют о том, что абсорбция для дисперсий составляет от 2,36 до 2,99 при 400 нм и от 0,35 до 2,96 при 450 нм. Таким образом, еще одним вариантом осуществления изобретения является фармацевтическая композиция, предлагаемая в настоящем изобретении, где при разведении водным раствором с использованием массового соотношения водного раствора и композиции 100:1 композиция образует водную дисперсию, для которой абсорбция превышает примерно 1,0 при длине волны примерно 400 нм и предпочтительно превышает примерно 2,0 при длине волны примерно 400 нм. Примеры Приготавливали три различные жидкие предназначенные для заполнения препаративные формы,две из которых капсулировали в мягкие гелевые капсулы (SGC) и одну капсулировали в капсулу с твердой оболочкой (HSC). Пример 1. Препаративная форма 1 в виде мягкой гелевой капсулы. Состав жидкой предназначенной для заполнения препаративной формы На основе вышеуказанной общей препаративной формы 1 приготавливали две конкретные препаративные формы лекарственного продукта в виде мягкой гелевой капсулы, а именно 40-миллиграммовый продукт и 120-миллиграммовый продукт: 42,30 мг Na-соли соединения (1) эквивалентно 40,0 мг активного фрагмента. 126,90 мг Na-соли соединения (1) эквивалентно 120,0 мг активного фрагмента. 3 Азот применяют для обработки, и он отсутствует в конечном продукте. 4 Приблизительная масса оболочки капсулы до сушки и полировки составляет 280 мг. Приблизительная масса оболочки капсулы после сушки и полировки составляет 198 мг. 5 Приблизительная масса оболочки капсулы до сушки и полировки составляет 590 мг. Приблизительная масса оболочки капсулы после сушки и полировки составляет 404 мг. 2 Пример 2. Препаративная форма 2 в виде мягкой гелевой капсулы. Состав жидкой предназначенной для заполнения препаративной формы На основе вышеуказанного общего состава приготавливали конкретную препаративную форму лекарственного продукта в виде 150-миллиграммовой мягкой гелевой капсулы. Пример 3. Препаративная форма 3 в виде капсулы с твердой оболочкой. Состав жидкой предназначенной для заполнения препаративной формы На основе вышеуказанного общего состава приготавливали конкретную препаративную форму лекарственного продукта в виде 150-миллиграммовой капсулы с твердой оболочкой. Получение препаративных форм 1-3. Лекарственную субстанцию измельчали с помощью струйной мельницы с целью удаления крупных агрегатов для того, чтобы продолжительность смешения при изготовлении предназначенной для заполнения нерасфасованной массы было постоянной и в достаточной степени короткой. Требуемое распределение частиц лекарственной субстанции по размерам необходимо снижать до значения 90 (об./об.), составляющего не более 10 мкм, и значения 98 (об./об.), составляющего не более 20 мкм, при измерении с помощью устройства фирмы Sympatec. Все эксципиенты, входящие в предназначенную для заполнения препаративную форму, объединяли в сосуде для смешения и перемешивали до достижения однородного состояния до внесения лекарственной субстанции. После добавления лекарственной субстанции перемешивание продолжали до тех пор, пока при визуальной оценке раствор для заполнения не становился прозрачным. В процессе приготовления раствора для заполнения применяли насыщение поверхностного слоя азотом, что является стандартной практикой. Раствор для заполнения пропускали через фильтр для удаления любых несоответствующих частиц. Включение нерасфасованной массы профильтрованного материала, предназначенного для заполнения, в капсулы осуществляли с использованием стандартной технологии для мягких желатиновых или твердых желатиновых капсул с осуществлением контроля в ходе процесса. Заполненные капсулы сушили и затем промывали раствором для полировки/отмывки до упаковывания, получая блестящие высококачественные с позиций фармацевтики капсулы. Пример 4. Изучение химической стабильности. Стабильность оценивали с использованием применяемой в качестве прототипа препаративной формы в виде капсулы с более высоким содержанием действующего вещества (150 мг в пересчете на натриевую соль) с такими же относительными количествами применяемых для заполнения эксципиентов и составом геля, что и у препаративной формы 1 (пример 1), и которая по качеству соответствовала препаративной форме 1. Анализ не выявил никаких существенных изменений анализируемого состава или профиля примесей после хранения в течение 12 месяцев при 25C/60% RH или 30C/70% RH. Пример 5. Изучение растворимости и биологической доступности. Для оценки препаративных форм 1-3 применяли тест на растворимость. Результаты продемонстрировали высвобождение предназначенной для заполнения препаративной формы из капсулы и диспергирование лекарственной субстанции в имеющей липидную основу самоэмульгирующейся системе для введения лекарственных средств при контакте с водными средами (см. фиг. 1). Тест на растворимость осуществляли согласно следующему протоколу: использовали 2150-миллиграммовых капсул для каждой препаративной формы; тест осуществляли, используя на сосуд по 500 мл раствора ацетатного буфера рН 4,5; скорость вращения 100 об/мин; тест с использованием корзины, 37 С. Кроме того, изучение in vivo на собаках биологической доступности СЭСВЛ-композиции в виде капсул продемонстрировало, что имеет место абсорбция, сопоставимая с абсорбцией ранее полученных оральных препаративных форм в виде растворов типа "порошок-во-флаконе", для которых установлено,что они обеспечивают достаточную экспозицию в клинических испытаниях на человеке. Протокол и результаты указанных опытов in vivo описаны ниже. Перекрестное 5-компонентное испытание препаративных форм на собаках породы Бигль. Животные/план испытания: Для перекрестного исследования использовали 6 самцов собак породы Бигль. Схема испытания предусматривала наличие однонедельного периода выведения препарата из организма между "визитами" 1 и 2 и "визитами" 3 и 4. Был предусмотрен также двухнедельный период выведения препарата из организма между "визитами" 2 и 3 и "визитами" 4 и 5. Предварительная обработка: пентагастрин, 6 мкг/кг IM за 1 ч до введения препаративных форм. Особенности кормления: голодание в течение ночи (кормление после 4 ч утра). Препаративные формы: А - Фаза Ia: раствор для орального введения, форма типа "порошок-во-флаконе" (PIB), доза 150 мг,48 мг/мл; Б - Фаза Ib/II: раствор для орального введения, форма типа "порошок-во-флаконе" (PIB), доза 150 мг, 48 мг/мл; В - Препаративная форма 1 SGC-капсула 1, доза 150 мг; Г - Препаративная форма 2 SGC-капсула 2, доза 150 мг; Д - Препаративная форма 3 HGC- капсула, доза 150 мг. Дозирование: Применяемые на фазе Ia и Ib/II оральные PIB-растворы содержали Na-соль соединения (1) в количестве 48 мг/мл. Для обработки собак использовали объем, составляющий примерно 3,13 мл (доза 150 мг) с последующим введением 50 мл воды через желудочный зонд. Препаративные формы В, Г и Д представляли собой капсулы, каждая из которых содержала Na-соль соединения (1) в количестве 150 мг. Собаки получали по одной капсуле с последующим введением 50 мл воды через желудочный зонд. Отбор образцов крови: Образцы крови (2 мл) получали до обработки через 0,33, 0,67, 1, 1,5, 2, 3, 4,6, 8, 12, 24, 30 и 48 ч после введения дозы. Антикоагулянт: Li-гепарин. Таблица 1 Обобщение фармакокинетических (ФК) параметров соединения (1) в организме собак породы Бигль (n=6) после орального введения Na-соли соединения (1) в виде пяти различных препаративных форма Данные представляют собой средние значения (% RSD (относительное стандартное отклонение,за исключением tmax, которое представлено в виде медианного значения (диапазон). В таблицу включены данные, полученные для всех собак, вне зависимости от наличия у них рвоты. Данные о соответствующих средних концентрациях соединения (1) в плазме всех собак (n=6) при использовании пяти различных препаративных форм Na-соли соединения (1) представлены на фиг. 2. Таблица 2 Обобщение фармакокинетических параметров соединения (1) в организме собак породы Бигль (n=3) после орального введения Na-соли соединения (1) в виде пяти различных препаративных форма Данные представляют собой средние значения (% RSD), за исключением tmax, которое представлено в виде медианного значения (диапазон). Из таблицы исключены все данные для всех препаративных форм, полученные для всех собак, у которых имела место рвота; 1492, 1912, 1916. Данные о соответствующих средних концентрациях соединения (1) в плазме трех собак при использовании пяти различных препаративных форм Na-соли соединения (1) (n=3), представлены на фиг. 3. Пример 6. Опыты по определения оптической прозрачности. Оптическую прозрачность препаративных форм, предлагаемых в настоящем изобретении, оценивали визуально и определяли количественно спектроскопически. Предназначенные для заполнения препаративные формы получали согласно методу, описанному для препаративной формы 1 (пример 1), а также получали соответствующие препаративные формы наполнителя и плацебо, и каждую из их разводили в 100 раз тремя водными средами с различными значениями рН. Абсорбцию каждого раствора определяли как непосредственно после приготовления, так и через 30 мин при длине волны 400 и 450 нм,используя соответствующую стандарту очищенную воду, полученные результаты представлены ниже. Результаты свидетельствуют о том, что абсорбция образовавшихся дисперсий составляет от 2,36 до 2,99 при 400 нм и от 0,35 до 2,96 при 450 нм. Препаративные формы Общая процедура: добавляли образец препаративной формы в количестве 0,1 г в 20-миллилитровый флакон для сцинтилляционного счетчика; добавляли во флакон 9,9 мл водной среды; хорошо диспергировали путем перемешивания вручную; сразу осуществляли количественную оценку или выдерживали в течение 30 мин; диспергировали образец перед считыванием в УФ; переносили аликвоту в УФ-ячейку с длиной оптического пути 1 см; определяли абсорбцию путем однократного измерения или в диапазоне длин волн. Реактивы. Водные среды: смоделированный раствор желудочного сока (SGF); ацетатный буфер; раствор, представляющий собой модель кишечного сока (SIF). Оборудование: спектрофотометр Cary 50 U V-Vis. Программное обеспечение: Программное обеспечение фирмы Cary "simple reads". Результаты определения оптической прозрачности (нулевой момент времени) Результаты определения оптической прозрачности (через 30 мин) Примеры 7-12. Приготовление Na-соли соединения (1). Методы, которые можно применять для получения аморфного соединения (1), описаны, в частности, в US 6323180, US 7514557 и US 7585845, которые включены в настоящее описание в качестве ссылки. Методы, которые можно применять для получения натриевой соли соединения (1), описаны, в частности, в публикации заявки на патент США 2010/0093792, а также изложены ниже в примерах. Пример 7. Получение соединения (1) типа А. Аморфное соединение (1) (партия 7, 13,80 г) добавляли в 1000-миллилитровую трехгорлую колбу. Добавляли в колбу абсолютный этанол (248,9 г). При перемешивании нагревали содержимое колбы со скоростью 60 С/ч до 74 С. (Твердые частицы не растворялись при 74 С). Затем равномерно при перемешивании, поддерживая температуру на уровне 74 С, добавляли воду (257,4 г) в течение 4 ч до получения густой суспензии. После завершения добавления воды температуру равномерно снижали до температуры окружающей среды со скоростью 8 С/ч и затем выдерживали при перемешивании в течение 6 ч при температуре окружающей среды. Образовавшиеся твердые частицы собирали фильтрацией и промывали 50 мл смеси 1/1 (мас./мас.) EtOH/вода. Влажные твердые частицы сушили в течение 30 мин в воронке с отсасыванием, пропуская N2 через осадок на фильтре. (XRPD-анализ (диффракция рентгеновских лучей на порошке) указанного образца продемонстрировал, что структура образца соответствуетEtOH-сольвату). Затем твердые частицы сушили при 65-70 С в вакууме (Р=25 мм рт. столба) и выпускали азот в течение 1,5 ч. С помощью XRPD подтверждали, что образовавшиеся твердые частицы (12,6 г,скорректированный выход 95,5%) представляли собой соединение (1) типа А. Пример 8. Получение натриевой соли соединения (1). Метод 1 2,1 г аморфной натриевой соли соединения (1) и 8,90 г ацетона вносили во флакон и перемешивали при температуре окружающей среды в течение 3 ч. Густую суспензию отфильтровывали от маточного раствора и образовавшиеся твердые частицы сушили в течение 20 мин в потоке азота. Собирали 1,51 г кристаллической натриевой соли соединения (1) в виде твердых частиц. Пример 9. Получение натриевой соли соединения (1) - Метод 2. 15,6 г соединения (1) типа А, 175 мл ацетона и 3,6 мл воды вносили в 250-миллилитровый реактор и нагревали до 53 С до растворения твердых частиц. В реактор добавляли 900 мкл 10,0 н. NaOH и в раствор вносили в качестве затравки соединение типа А. Затравочный раствор перемешивали при 53 С в течение 10 мин. Добавляли вторую порцию (900 мкл) 10,0 н. NaOH и систему перемешивали при 53 С в течение 30 мин с получением густой суспензии. Густую суспензию охлаждали до 19 С со скоростью охлаждения 15 С/ч и выдерживали в течение ночи при 19 С. Конечную образовавшуюся густую суспензию фильтровали и влажные твердые частицы промывали 15 мл ацетона. Твердые частицы сушили в течение 1 ч при 52 С в вакууме в потоке азота и затем выдерживали твердые частицы в лабораторном воздухе в течение 1 ч. Собирали 12,1 г кристаллической натриевой соли соединения (1) в виде твердых частиц. Пример 10. Получение натриевой соли соединения (1) - Метод 3. 25,4 кг аморфного соединения (1), 228 л ТГФ и 11,1 кг 10 мас.% NaOH (водный раствор) вносили в реактор. Соединения перемешивали при 25 С до растворения всех твердых частиц. Образовавшийся раствор профильтровывали и реактор и фильтр промывали 23 л ТГФ. 180 л растворителя удаляли дистилляцией при атмосферном давлении при 65 С. Добавляли 195 л МИБК (метилизобутилкетон) и 166 л растворителя удаляли путем вакуумной дистилляции при 44 С. В реактор вновь добавляли 161 л МИБК и 0,41 кг воды и содержимое нагревали до 70 С. Добавляли 255 г натриевой соли соединения (1) при 70 С и добавляли в течение 1,5 ч 1,42 л воды. После добавления воды густую суспензию выдерживали при 70 С в течение 45 мин и затем охлаждали до 45 С в течение 1 ч. Образовавшуюся густую суспензию фильтровали и промывали 64 л МИБК, содержащего 0,8 мас.% воды. Влажный осадок на фильтре сушили при 55 С с получением 25 кг кристаллической натриевой соли соединения (1). Пример 11. Получение натриевой соли соединения (1) - Метод 4. 2,00 г аморфного соединения (1), 9,96 г ТГФ и 0,11 г воды вносили в реактор и перемешивали при температуре окружающей среды до растворения твердых частиц. 0,820 мл 21 мас.%-ного раствора NaOEt в этаноле добавляли по каплям при перемешивании с получением раствора А. 15,9 г н-BuAc и 160 мкл воды вносили во второй реактор и нагревали до 65 С (раствор Б). 2,56 г раствора А добавляли к раствору Б при 65 С и в образовавшуюся смесь вносили в качестве затравки 40 мг натриевой соли соединения (1). Затравочной смеси давали созревать при 65 С в течение 45 мин. 2,56 г раствора Б добавляли к раствору А с использованием 4 отдельных промежутков времени и давали созревать в течение 45 мин. После конечного добавления и созревания густую суспензию охлаждали до 50 С в течение 1 ч и фильтровали. Влажный осадок на фильтре промывали 6 мл н-BuAc, содержащего 0,5 мас.% воды. Полученные итоге твердые частицы сушили при 50 С в вакууме, используя продувку азотом. Собирали кристаллическую натриевую соль соединения (1) в виде твердых частиц. Пример 12. Получение натриевой соли соединения (1) - Метод 5. При комнатной температуре добавляли при перемешивании раствор этоксида натрия в этаноле(21 мас.%; 306 мл) к раствору соединения (1) (745 г) в ТГФ (2000 мл) и воде (76,5 мл). После перемешивания в течение 30 мин смесь фильтровали и фильтр промывали ТГФ (85 мл). Образовавшийся раствор нагревали до 65 С и обрабатывали профильтрованным бутилацетатом (6640 мл, необязательно предварительно нагретым до 65 С) в течение 30 мин. Добавляли затравочные кристаллы (0,50 г) и смесь перемешивали при 65 С в течение 2 ч, при этом кристаллизация начиналась примерно через 30 мин. Суспензию охлаждали до 50 С в течение 1 ч и перемешивали при указанной температуре в течение еще 1 ч. Указанное в заголовке соединение выделяли фильтрацией, промывали профильтрованным бутилацетатом (765 мл, необязательно предварительно нагретый до 50 С) и сушили при 65 С в течение примерно 16 ч с получением кристаллической натриевой соли соединения (1) (725 г). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Жидкая фармацевтическая композиция для лечения инфекции вируса гепатита C, содержащая: или его фармацевтически приемлемой соли;(б) 30-60 мас.% фармацевтически приемлемого липида, выбранного из группы, включающей жирные кислоты, моно-, ди- или триглицериды со средней или длинной цепью, эфиры пропиленгликоля и жирных кислот, эфиры сорбита и жирных кислот, неводорастворимые витамины и их смеси; и(в) 20-50 мас.% фармацевтически приемлемого гидрофильного поверхностно-активного вещества,выбранного из группы, включающей полиэтоксилированные растительные масла, полиэтоксилированные токоферолы, полиэтоксилированные эфиры сорбита и жирных кислот, соли желчных кислот, лецитины и их смеси. 2. Фармацевтическая композиция по п.1, в которой соединение формулы (1) присутствует в виде натриевой соли. 3. Фармацевтическая композиция по п.1 или 2, дополнительно содержащая 5-15% фармацевтически приемлемого гидрофильного растворителя, выбранного из группы, включающей пропиленгликоль, полипропиленгликоль, полиэтиленгликоль, глицерин, этанол, диметилизосорбид, гликофурол, пропиленкарбонат, диметилацетамид, воду и их смеси. 4. Фармацевтическая композиция по одному из предыдущих пунктов, где композиция при разведении водным раствором с использованием массового соотношения водного раствора и композиции 100:1 образует водную дисперсию, абсорбция которой превышает примерно 1,0 при длине волны примерно 400 нм. 5. Фармацевтическая композиция по одному из предыдущих пунктов, содержащая:(а) 10-20 мас.% соединения формулы (1) в виде его натриевой соли;(б) 40-50 мас.% фармацевтически приемлемого липида, выбранного из моноглицеридов каприловых и каприновых жирных кислот, диглицеридов каприловых и каприновых жирных кислот и их смесей;(в) 25-35 мас.% фармацевтически приемлемого гидрофильного поверхностно-активного вещества,выбранного из токоферилполиэтиленгликольсукцината, полиоксила 40 гидрогенизированного касторового масла и полиоксила 35 касторового масла и их смесей;(г) 5-10 мас.% фармацевтически приемлемого гидрофильного растворителя, выбранного из пропиленгликоля, полиэтиленгликоля, этанола, воды и их смесей.
МПК / Метки
МПК: A61K 9/107, A61K 47/44, A61K 47/14, A61K 31/427, A61K 47/12, A61K 47/10, A61K 9/48
Метки: ингибитора, гепатита, вируса, композиция, фармацевтическая, протеазы
Код ссылки
<a href="https://eas.patents.su/16-23433-farmacevticheskaya-kompoziciya-ingibitora-proteazy-virusa-gepatita-c.html" rel="bookmark" title="База патентов Евразийского Союза">Фармацевтическая композиция ингибитора протеазы вируса гепатита c</a>
Ингибиторы протеазы вируса гепатита с

Номер патента: 20235
Опубликовано: 30.09.2014
Авторы: Лю Йо-Чин, Ли Куан-Юань, Чэнь Жун-Цзиунн, Кинг Чи-Син Ричард, Линь Чу-Чун, Чэнь Чих-Мин, Ло Пинь, Лю Чэнь-Фу
МПК: C07D 207/16, C07D 207/08, A61P 31/12...
Метки: ингибиторы, протеазы, вируса, гепатита
Формула / Реферат:
1. Соединение формулы (I)гдеR1 является С3-10циклоалкилом;R2 является C1-6алкилом или С3-10циклоалкилом;U является -NHSO2-;W является -(СН2)m-, -О(СН2)n- или -SO(CH2)n-,m равно 2 иn равно 1;X представляет собой -О-;Y представляет собойгдекаждый из V и Т является -N-;R представляет собой арил или гетероарил икаждый из А1 и А2 независимо являются арилом или гетероарилом, каждый из которых необязательно замещен галогеном, С1-6алкилом или...
Ингибиторы ns3 протеазы вируса гепатита с

Номер патента: 13331
Опубликовано: 30.04.2010
Авторы: Холловэй М.Катарин, Людмерер Стивен У., Макинтайр Чарльз Дж., Олсен Дэвид Б., Вакка Джозеф П., Макколи Джон А., Ливертон Найджел Дж., Радд Майкл Т.
МПК: A61P 31/14, C07K 5/08, A61K 38/06...
Метки: протеазы, вируса, гепатита, ингибиторы
Формула / Реферат:
1. Соединение формулы Iили его фармацевтически приемлемая соль,где р и q оба равны 1;R1 является CONR10SO2R6;R2 является C1-С6алкилом или С2-С6алкенилом, где указанный алкил или алкенил необязательно замещен 1-3 атомами галогена;R3 является C1-С8алкилом или C3-С8циклоалкилом;R5 является Н;R6 является C3-С6циклоалкилом;Y является С(=O);Z является О;М является С1-С12алкиленом или С2-С12алкениленом икаждый R10 независимо является Н или...
Кристаллические соли эффективного ингибитора вируса гепатита с

Номер патента: 21805
Опубликовано: 30.09.2015
МПК: A61K 31/427, A61P 31/18, C07D 417/14...
Метки: ингибитора, гепатита, соли, кристаллические, вируса, эффективного
Формула / Реферат:
1. Кристаллическая соль соединения формулы (1) и трометамина:2. Кристаллическая соль по п.1, на порошковой рентгеновской дифрактограмме которой наблюдается пик при 5,9° 2θ (±0,2° 2θ) при измерении с использованием излучения CuKα.3. Кристаллическая соль по п.1, на спектре 13С твердотельного ЯМР которой наблюдаются пики с химическими сдвигами при 178,3, 138,1 и 27,1 м.д. (±0,2 м.д.).4. Кристаллическая соль по п.1, на порошковой...
Ингибиторы серин-протеаз, в частности ns3 протеазы вируса гепатита c (hvc)

Номер патента: 1915
Опубликовано: 22.10.2001
Авторы: Бхисетти Говинда Рао, Фармер Люк Дж., Танг Роджер Д., Дейнинджер Дэвид Д, Харбесон Скотт Л., Мурко Марк А.
МПК: A61K 38/55, A61P 1/16, C07K 5/10...
Метки: hvc, частности, серин-протеаз, ингибиторы, гепатита, вируса, протеазы
Формула / Реферат:
1. Соединение структурной формулы (II) где W является m равно 0 или 1; каждый R2 представляет собой независимо водород, алкил, алкенил, арил, аралкил, аралкенил, циклоалкил, циклоалкилалкил, циклоалкенил, циклоалкенилалкил, гетероциклил, гетероциклилалкил, гетероциклилалкенил, гетероарил или гетероаралкил, или две R2 группы, которые связаны с одним и тем же атомом азота, образуют вместе с этим атомом азота 5-7-членную моноциклическую...
Фармацевтическая композиция тгп и ингибитора апф

Номер патента: 3470
Опубликовано: 26.06.2003
Авторы: Калвиньш Иварс, Бундулис Юрис, Скарда Илзе, Веверис Марис
МПК: A61P 9/00, A61K 31/205
Метки: композиция, фармацевтическая, апф, ингибитора, тгп
Формула / Реферат:
1. Фармацевтическая композиция, содержащая дигидрат 3-(2,2,2-триметилгидразиний)пропионата (ТГП) и ингибитор ангиотензинпревращающего фермента (АПФ). 2. Фармацевтическая композиция по п.1, в которой указанный ингибитор АПФ является (S)-1-{N-[1-(этоксикарбонил)-3-фенилпропил]-L-аланил}-L-пролином, т.е. эналаприлом. 3. Фармацевтическая композиция по п.2, в которой соотношение указанных активных соединений дигидрата...
Предыдущий патент: Дистанционно управляемое настраиваемое устройство и способ регулирования потока
Следующий патент: Устройство для переработки отходов
Случайный патент: Окклюзионное приспособление, направляемое с помощью чрескожного катетера