(e)-n-{3-[1-(8-фтор-11н-10-окса-1-азадибензо[a,d]циклогептен-5-илиден)пропил]фенил}метансульфонамид в качестве модулятора рецепторов глюкокортикоидов для лечения ревматизма












Формула / Реферат
1. Соединение, представляющее собой (Е)-N-{3-[1-(8-фтор-11Н-10-окса-1-азадибензо[a,d]циклогептен-5-илиден)пропил]фенил}метансульфонамид формулы

или фармацевтически приемлемая соль данного соединения.
2. Соединение по п.1, представляющее собой (Е)-N-{3-[1-(8-фтор-11H-10-окса-1-азадибензо[a,d]циклогептен-5-илиден)пропил]фенил}метансульфонамид.
3. Применение соединения или его соли по п.1 или 2 в качестве лекарственного средства для лечения ревматоидного артрита, остеоартрита, ревматизма, астмы, аллергического ринита, системной красной волчанки, хронической обструктивной болезни легких, болезни Крона, воспалительного заболевания кишечника и язвенного колита.
4. Применение по п.3 в качестве лекарственного средства для лечения ревматоидного артрита.
5. Применение соединения или его соли по п.1 или 2 для лечения ревматоидного артрита, остеоартрита, ревматизма, астмы, аллергического ринита, системной красной волчанки, хронической обструктивной болезни легких, болезни Крона, воспалительных заболеваний кишечника или язвенного колита.
6. Применение по п.5 для лечения ревматоидного артрита.
7. Фармацевтическая композиция для лечения ревматоидного артрита, остеоартрита, ревматизма, астмы, аллергического ринита, системной красной волчанки, хронической обструктивной болезни легких, болезни Крона, воспалительного заболевания кишечника и язвенного колита, содержащая соединение или соль по любому из пп.1 или 2 в сочетании с одним или более фармацевтически приемлемыми носителями, эксципиентами или разбавителями.
8. Фармацевтическая композиция для лечения ревматоидного артрита, содержащая соединение или соль по любому из пп.1 или 2 в сочетании с одним или более фармацевтически приемлемыми носителями, эксципиентами или разбавителями.
Текст
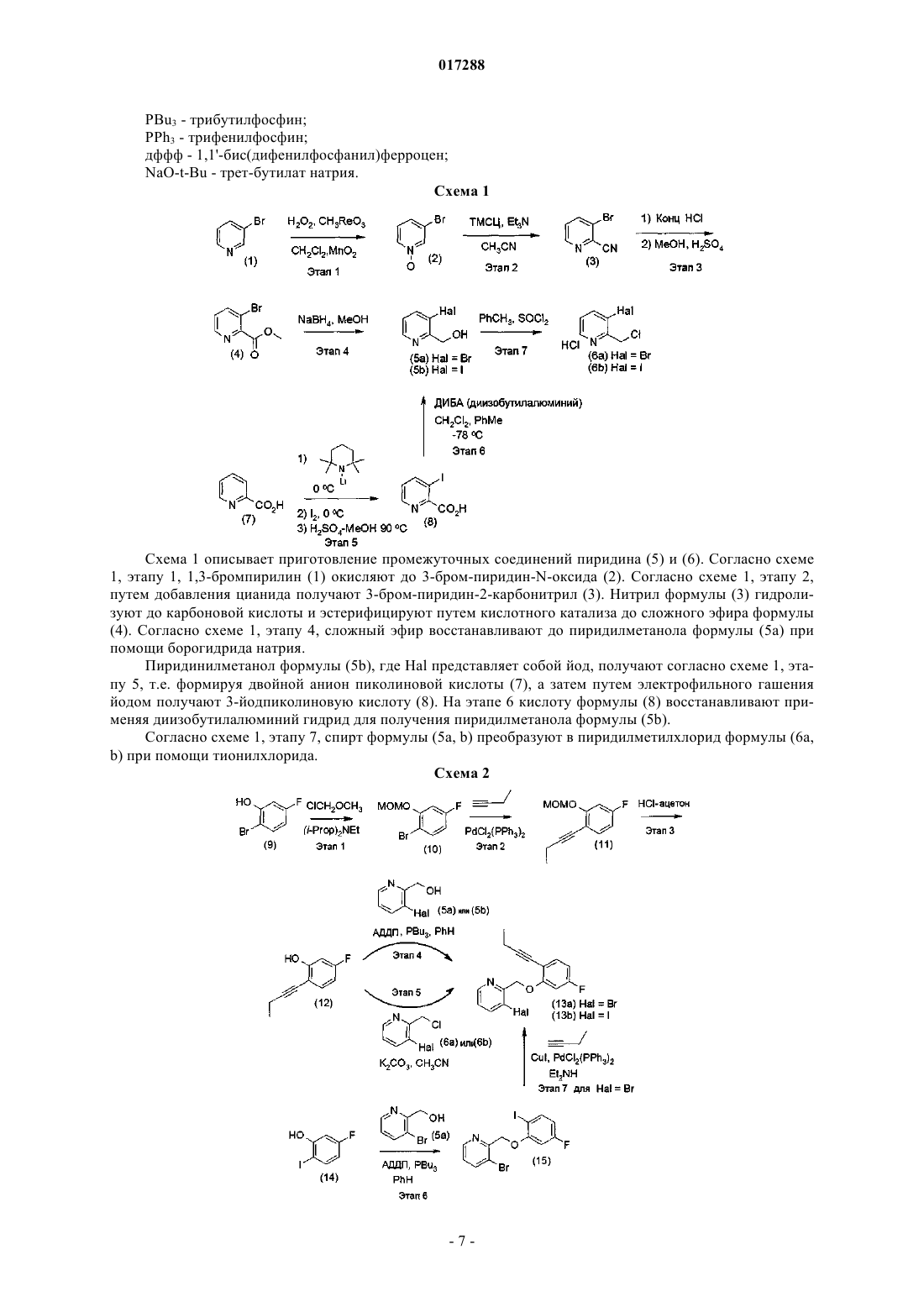
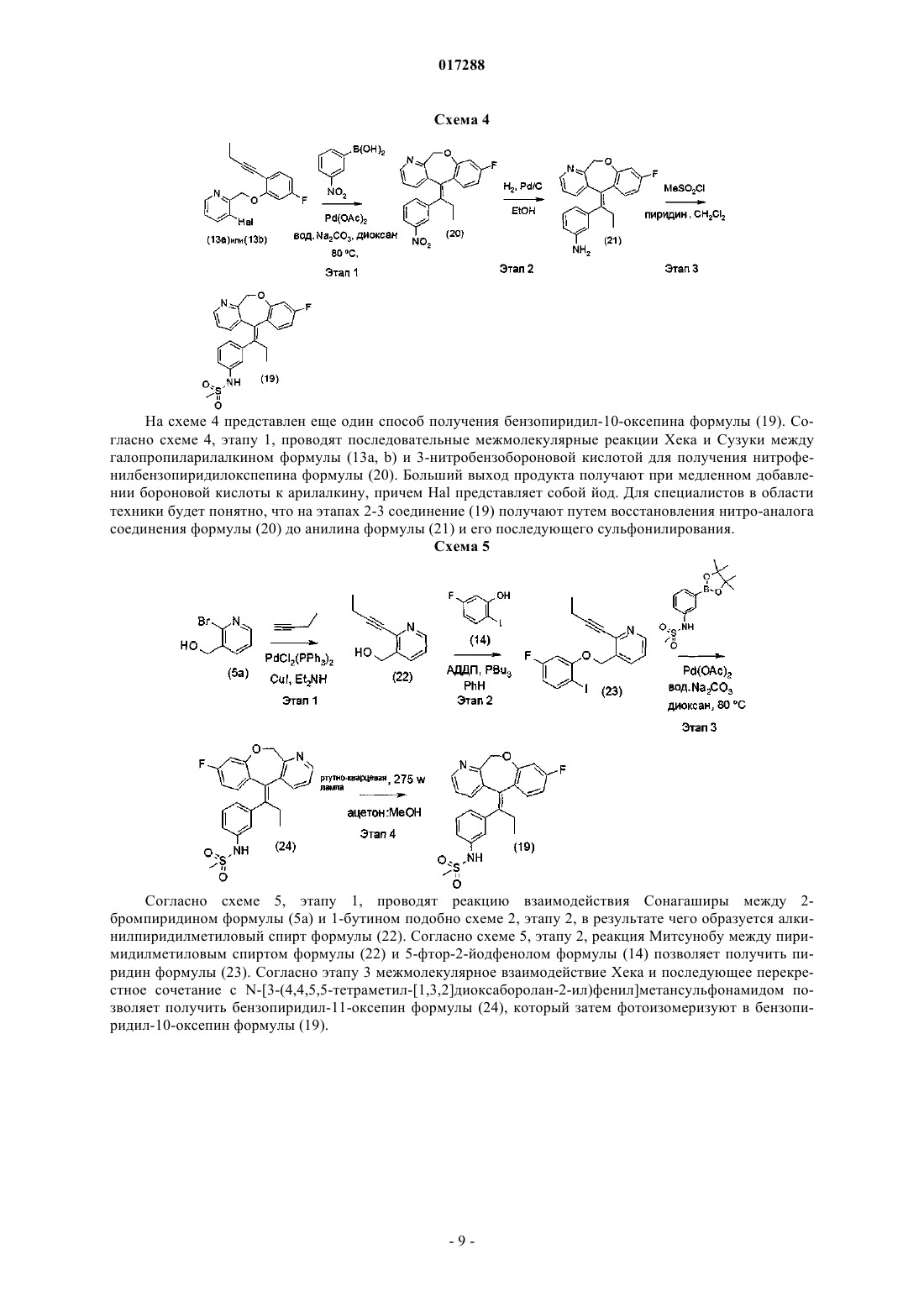
(E)-N-3-[1-(8-ФТОР-11 Н-10-ОКСА-1-АЗАДИБЕНЗО[a,d]ЦИКЛОГЕПТЕН-5 ИЛИДЕН)ПРОПИЛ]ФЕНИЛМЕТАНСУЛЬФОНАМИД В КАЧЕСТВЕ МОДУЛЯТОРА РЕЦЕПТОРОВ ГЛЮКОКОРТИКОИДОВ ДЛЯ ЛЕЧЕНИЯ РЕВМАТИЗМА Согласно настоящему изобретению предложено соединение (I) Карсон Мэттью Уилльям, Коулан Майкл Джозеф (US) Медведев В.Н. (RU) или фармацевтически приемлемая соль данного соединения; фармацевтическая композиция,предназначенная для лечения ревматоидного артрита, остеоартрита, ревматизма, астмы,аллергического ринита, системной красной волчанки, хронической обструктивной болезни легких, болезни Крона, воспалительного заболевания кишечника и язвенного колита, содержащая соединение (I) или его фармацевтически приемлемую соль в сочетании с одним или более фармацевтически приемлемыми носителями, эксципиентами или разбавителями; и применение соединения (I) или его фармацевтически приемлемой соли в качестве лекарственного средства для лечения ревматоидного артрита, остеоартрита, ревматизма, астмы, аллергического ринита,системной красной волчанки, хронической обструктивной болезни легких, болезни Крона,воспалительного заболевания кишечника и язвенного колита.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) 017288 Настоящее изобретение относится к группе терапевтических средств, применяемых для лечения или предотвращения воспалительных и иммунных нарушений, восприимчивых к стероидным глюкокортикоидам, фармацевтическим композициям, содержащим указанные средства, способам лечения или предотвращения воспалительных или иммунных заболеваний пациентов, а также к промежуточным соединениям и способам, используемым при синтезе терапевтических средств. Как встречающиеся в природе, так и синтетические стероидные глюкокортикоиды применяются на протяжении уже более пятидесяти лет для лечения острых и хронических воспалительных и иммунных заболеваний, таких как ревматоидный артрит, остеоартрит, ревматизм, астма, аллергический ринит, системная красная волчанка, хроническая обструктивная болезнь легких, болезнь Крона, воспалительные заболевания кишечника и язвенный колит. Тем не менее, применение глюкокортикоидов часто связано с тяжелыми и иногда необратимыми побочными действиями, такими как потеря костной массы/остеопороз, гипергликемия, сахарный диабет, повышенное давление, глаукома, атрофия мышц, синдром Иценко-Кушинга и психоз. Таким образом, сохраняется потребность в альтернативных способах лечения, которые бы использовали положительное действие стероидных глюкокортикоидов, но при этом имели пониженную вероятность или частоту возникновения сопутствующих побочных эффектов. Глюкокортикоиды регулируют транскрипцию генов после формирования комплекса с рецептором глюкокортикоидов (РГ). После связывания глюкортикоидов комплекс РГ-глюкокортикоид перемещается в клеточное ядро, где связывается с глюкокортикоидными элементами гормонального ответа (ГЭГО) в промоутерных участках определенных генов. Затем комплекс РГ-глюкокортикоид/ГЭГО, в свою очередь, активирует (трансактивация) или ингибирует транскрипцию проксимально расположенных генов. В другом случае комплекс РГ-глюкокортикоид может осуществлять негативную регуляцию транскрипции генов при помощи процесса, не включающего связывание ДНК. В этом процессе, называемом трансрепрессией, комплекс РГ-глюкокортикоид попадает в ядро и непосредственно взаимодействует (при помощи белок-белкового взаимодействия) с другими транскрипционными факторами, подавляя их способность стимулировать транскрипцию генов и, следовательно, экспрессию белков. Поиск РГ-лигандов, являющихся подходящей заменой стероидных глюкокортикоидов, затрудняется тем, что рецепторы других стероидных гормонов, например рецептор андрогена (РА), рецептор минералокортикоидов (РМ) и рецептор прогестерона (РП), опосредующие другие физиологические процессы,имеют участки связывания лигандов, гомологичные РГ. В результате есть вероятность того, что РГлиганды проявят перекрестную активность с данными рецепторами. Таким образом, желаемым свойством заместителя стероидных глюкокортикоидов является то, что он обладает большим сродством к РГ по отношению к другим рецепторам гормонов. Последние исследования показывают, что противовоспалительное действие глюкокортикоидов может обеспечиваться и без связывания РГ с ДНК. Вследствие этого считается, что механизмы действия глюкокортикоидов, опосредованные белок-белковыми взаимодействиями РГ (например, трансрепрессией), достаточны для того, чтобы вызвать противовоспалительный ответ. К тому же, в настоящее время считается, что многие побочные эффекты глюкокортикоидной терапии (например, гипергликемия, сахарный диабет, глаукома и мышечная атрофия) в основном опосредованы трансактивационными механизмами, следующими за связыванием РГ с ДНК. Таким образом, существует необходимость в получении средства, способного дифференцировать РГ-опосредованную трансрепрессию и РГ-опосредованную трансактивацию. Более того, также особенно желательным является получение средства, обладающего сниженной способностью модулировать (т.е. усиливать, частично усиливать, частично ослаблять или ослаблять) транскрипционную активность других рецепторов стероидных гормонов. Задачей настоящего изобретения является обеспечение средства, связывающегося с РГ с большим сродством, чем с рецепторами других стероидных гормонов. Более конкретно, задачей является обеспечение средства, обладающего в 10 раз большим сродством к РГ, чем к РА, РМ И РП. Еще одной задачей данного изобретения является обеспечение средства, обладающего выраженными противовоспалительными свойствами в сравнении со свойством вызывать побочные эффекты, связанные с терапией глюкокортикоидами. Более конкретно, задачей изобретения является обеспечение средства, обладающего выраженными противовоспалительными свойствами по сравнению со способностью вызывать потерю костной массы или остеопороз. Еще одной задачей данного изобретения является обеспечение средства,обладающего ограниченной способностью к модулированию активности других рецепторов стероидных гормонов, РА, РМ и РП. Модуляторы РГ широко известны в данной области техники. Например, международная публикация WO 04/052847 описывает класс модуляторов рецепторов трициклических стероидных гормонов, которые могут применяться при лечении заболеваний, восприимчивость к модуляции рецепторов минералокортикоидов и рецепторов глюкокортикоидов. Неожиданно было обнаружено, что при выборе соединения из ряда, предложенного в документе WO 04/052847 (соединение представлено ниже в данном описании как соединение (1, было выявлено новое терапевтическое средство, обладающее неожиданным профилем активности, позволяющим предположить, что данное средство может оказаться весьма эффективным при лечении воспалительных и иммунных нарушений, восприимчивых к стероидным глюкокортикоидам.-1 017288 Соответственно настоящее изобретение относится к соединению (1)(Е)-N-3-[1-(8-фтор-11H-10-окса-1-азадибензо[a,d]циклогептен-5 илиден)пропил]фенилметансульфонамид или фармацевтически приемлемой соли данного соединения. В предпочтительном варианте настоящее изобретение относится к соединению, представляющему собой(Е)-N-3-[1-(8-фтор-11H-10-окса-1-азадибензо[a,d]циклогептен-5-илиден)пропил]фенилметансульфонамид. В другом варианте настоящее изобретение относится к применению указанного выше соединения или его соли в качестве лекарственного средства для лечения ревматоидного артрита, остеоартрита, ревматизма, астмы, аллергического ринита, системной красной волчанки, хронической обструктивной болезни легких, болезни Крона, воспалительного заболевания кишечника и язвенного колита. Наиболее предпочтительным применением является применение в качестве лекарственного средства для лечения ревматоидного артрита. В еще одном варианте настоящее изобретение относится к применению указанного выше соединения или его соли для лечения ревматоидного артрита, остеоартрита, ревматизма, астмы, аллергического ринита, системной красной волчанки, хронической обструктивной болезни легких, болезни Крона, воспалительных заболеваний кишечника или язвенного колита. Наиболее предпочтительным применением является применение для лечения ревматоидного артрита. В другом варианте настоящее изобретении относится к фармацевтической композиции для лечения ревматоидного артрита, остеоартрита, ревматизма, астмы, аллергического ринита, системной красной волчанки, хронической обструктивной болезни легких, болезни Крона, воспалительного заболевания кишечника и язвенного колита,содержащей(E)-N-3-[1-(8-фтор-11H-10-окса-1 азадибензо[a,d]циклогептен-5-илиден)пропил]фенилметансульфонамид или его фармацевтически приемлемую соль в сочетании с одним или более фармацевтически приемлемыми носителями, эксципиентами или разбавителями. Наиболее предпочтительна фармацевтическая композиция для лечения ревматоидного артрита, содержащая указанное выше соединение или его соль в сочетании с одним или более фармацевтически приемлемыми носителями, эксципиентами или разбавителями. Ревматоидный артрит (РА) - хроническое заболевание, характеризующееся постоянным воспалением синовиальной ткани суставов, чаще всего поражающее возрастную группу от 30 до 50 лет. Ревматоидный артрит является наиболее распространенной формой воспалительного артрита, причем женщины подвержены указанному заболеванию в два раза чаще, чем мужчины. Считается также, что применение соединения (1) для лечения или предупреждения воспалительных или иммунных заболеваний связано с уменьшенной вероятностью или частотой случаев возникновения побочных эффектов, обычно проявляющихся при глюкокортикоидной терапии. Одним из таких побочных эффектов глюкокортикоидной терапии является остеопороз или остеопороз, вызванный глюкокортикоидами (ОВГ). ОВГ - наиболее распространенный случай остеопороза, вызванного лекарственными средствами. Известно, что он возникает почти у 50% пациентов, проходящих длительную (т.е. более 6 месяцев) глюкокортикоидную терапию. В частности, считается, что применение соединения (1) связано с уменьшенной вероятностью или частотой случаев возникновения остеопороза. Если не указано иное, данное изобретение включает фармацевтически приемлемые соли соединения (1), а также сольваты свободного основания соединения (1) и их фармацевтически приемлемые соли. Тем не менее, наиболее предпочтительной является свободное основание соединения (1). Термин "фармацевтически приемлемые соли", использованный в настоящем изобретении, относится к солям соединения (1), являющимся практически безвредными для живых организмов. Примеры фармацевтически приемлемых солей и способов их изготовления широко представлены в данной области техники. См. например Stahl et al., "Handbook of Pharmaceutical Salts: Properties, Selection and Use", VCHA/Wiley-VCH,(2002); Gould, P.L., "Salt selection for basic drugs", International Journal of Pharmaceutics, 33: 201-217 (1986); а также Bastin et al. "Salt Selection and Optimization Procedures for Pharmaceutical New Chemical Entities",Organic Process Research and Development, 4: 427-435 (2000). Используемый в настоящем изобретении термин "пациент" относится к человеку или другому млекопитающему, например собаке, кошке, корове, обезьяне, лошади или овце. В частности, термин "паци-2 017288 ент" относится к человеку. Термин "лечение", используемый в настоящем изобретении, включает предотвращение, ограничение, замедление, остановку или реверсию развития или степени тяжести наличествующего симптома или заболевания. Термин "профилактика", используемый в настоящем изобретении, обозначает предотвращение или подавление коэффициента заболеваемости или возникновения симптома или заболевания. Соединение (1) или фармацевтически приемлемая соль данного соединения могут быть приготовлены в виде части фармацевтической композиции для введения. Фармацевтические композиции, как таковые, содержащие соединение (1) или его фармацевтически приемлемую соль в сочетании с одним или более фармацевтически приемлемым носителем, эксципиентом или разбавителем, являются важным вариантом осуществления изобретения. Примеры фармацевтических композиций и способов их изготовления широко известны в данной области техники. См., например, REMINGTON: THE SCIENCE AND(1) в суспензии с 0,5% метилцеллюлозой, 1% лаурилсульфатом натрия и 0,1% пеногасителем 1510 в 0,01 н. HCl. Предпочтительная композиция согласно настоящему изобретению содержит соединение (1) или его фармацевтически приемлемую соль, приготовленное в виде капсулы или таблетки. Соединение (1) или композиции, содержащие соединение (1), могут быть введены в организм любым путем, сохраняющим биологическую доступность соединения, в том числе оральным и парентеральным путем. Например, соединение (1) или композиции, содержащие соединение (1), могут быть введены орально, подкожно, внутримышечно, внутривенно, трансдермально, интраназально, ректально,трансбуккально и т.д. В качестве альтернативы соединение может быть введено посредством непрерывной инфузии. Тем не менее, следует понимать, что оральное введение является наиболее предпочтительным. Термин "эффективное количество", используемый в настоящем изобретении, относится к количеству или дозе соединения (1), которое после однократного или многократного введения пациенту обеспечивает желаемый эффект у пациента в процессе установления диагноза или лечения. Эффективное количество может легко определить лечащий врач как специалист в данной области с учетом ряда показателей, таких как вид млекопитающего, его размер, возраст, общее состояние здоровья, наличествующее заболевание, степень тяжести заболевания, реакция индивидуального пациента, конкретное вводимое соединение, способ введения, биологическая доступность вводимого препарата, выбранный режим дозирования и применение сопутствующих лекарственных средств. Биологическая активность. В настоящем изобретении "Kd" обозначает константу равновесия диссоциации комплекса лигандрецептор; "Ki" обозначает константу равновесия диссоциации комплекса лекарственное средстворецептор и является показателем концентрации лекарственного средства, которое свяжется с половиной сайтов связывания в состоянии равновесии; "IC50" обозначает концентрацию средства, обеспечивающую 50% от максимального ингибирования, возможного для данного средства, или, в другом случае, концентрацию средства, обеспечивающую вытеснение 50% лиганда, связывающегося с рецептором; "ЕС 50" обозначает концентрацию средства, обеспечивающую 50% максимального возможного ответа на данное средство; "ED50" обозначает дозу вводимого терапевтического средства, обеспечивающую 50% максимального ответа организма на данное средство. Анализ ядерного связывания рецепторов гормонов. Величину Ki определяют с помощью анализа конкурентного связывания рецепторов и лигандов,применяя клеточные лизаты из зародышевых клеток почки человека HEK293, экспрессирующие повышенные количества РГ человека (рецепторы глюкокортикоидов), РА (рецептора андрогена), РМ (рецепторы минералокортикоидов) или РП (рецепторы прогестерона). Вкратце, конкурентно-связывающий анализ стероидных рецепторов проводят в буфере, содержащем 20 ммоль буфера Гепеса (pH 7,6), 0,2 ммоль ЭДТА, 75 ммоль NaCl, 1,5 ммоль MgCl2, 20% глицерин,20 ммоль молибдата натрия, 0,2 ммоль ДТТ (дитиотреитол), 20 мкг/мл апротинина и 20 мкг/мл леупептина. Обычно конкурентно-связывающий анализ стероидных рецепторов включает меченые лиганды,такие как 0,5 нмоль [3H]-дексаметазона для связывания РГ, 0,36 нмоль [3H]-метилтриэнолона для связывания РА, 0,25 нмоль [3H]-альдостерона для связывания РМ, 0,29 нмоль [3H]-метилтриэнолона для связывания РП, и либо 20 мкг 293-РГ лизата, 22 мкг 293-РА лизата, 20 мкг 293-РМ лизата, либо 40 мкг 293 РП лизата на лунку. Чаще всего анализ проводят с использованием 96-луночных планшетов. Конкурентные исследуемые соединения добавляют в разных концентрациях, варьирующих от 0,01 нмоль до 10 мкмоль. Неспецифическое связывание определяется в присутствии 500 нмоль дексаметазона для связывания с РГ, 500 нмоль альдостерона для связывания с РМ или 500 нмоль метилтриэнолона для связывания с РА и РП. Реакции связывания (140 мкл) выдерживают при температуре 4C в течение ночи, затем в каждую реакцию добавляют по 70 мкл буфера из активированного угля и декстрана (на 50 мкл контрольного буфера приходится по 0,75 г активированного угля и 0,25 г декстрана). Планшеты помещают в-3 017288 орбитальный встряхиватель при 4C для перемешивания в течение 8 мин. Затем их центрифугируют при 4C на 3000 оборотов в течение 10 мин. После этого аликвоту 120 мкл смеси реакции связывания переносят на другой 96-луночный планшет, и в каждую лунку добавляют 175 мкл сцинтилляционной жидкости Wallac Optiphase Hisafe 3. Планшеты запечатывают и энергично встряхивают в орбитальном встряхивателе. После 2-часовой инкубации планшеты просматривают в счетчике Wallac Microbeta. Данные используют для подсчета предполагаемого IC50 и процента ингибирования в 10 мкмоль. Kd для [3H]-дексаметазона для связывания с РГ, [3H]-альдостерона для связывания с МР или [3H]метилтриэнолона для связывания с РП определяют насыщением связывания. По значениям IC50 для соединений определяют значения Ki при использовании формулы Ченга-Прусова. Протоколы анализа связывания, подобные описанным выше, могут быть без труда составлены специалистом в данной области. После проведения вышеуказанных процедур соединение (1) показало значение Ki приблизительно 0,2 нмоль в результате анализа связывания с ГР, Ki примерно 6,7 нмоль в результате анализа связывания с АР, Ki приблизительно 9,2 нмоль в результате анализа связывания с РМ иKi примерно 32 нмоль в анализе связывания с РП (величины, получившиеся в ходе 7 экспериментов). Таким образом, соединение (1) является сильным лигандом РГ человека и, к тому же, обладает в 15 и выше раз большим сродством к РГ, по отношению к каждому из РМ, РА и РП человека. Для того чтобы продемонстрировать способность соединений, предложенных в данном изобретении, модулировать активность рецепторов стероидных гормонов (например, агонизировать, частично агонизировать, частично антагонизировать или антагонизировать), проводили биологические исследования, в которых определяли функциональную модуляцию экспрессии целевых генов в клетках, временно трансфицированных конструктом гена белка ядерных рецепторов и гена-репортера гормонального ответа. Растворители, реагенты и лиганды, задействованные в функциональном анализе, коммерчески легко доступны или могут быть изготовлены специалистом в данной области. Исследования функциональной модуляции ядерных рецепторов гормонов. Клетки почки эмбриона человека HEK293, трансфицируют плазмидами рецептора стероидных гормонов и гена-репортера при помощи реагента трансфекции Fugene. Вкратце, плазмиду репортера, содержащую две копии андроген-чувствительного элемента (ARE) пробазина и промотор ТК (тимидинкиназы), расположенные ближе к 5'-концу относительно кДНК репортера люцеферазы, трансфицируют в клетки HEK293 с плазмидой, конститутивно экспрессирующей рецептор андрогена (РА) человека под контролем вирусного ЦМВ (цитомегаловируса). Репортерную плазмиду, содержащую две копии GRE пробазина и промотор ТК (тимидинкиназы), расположенные ближе к 5'-концу относительно кДНК люциферазы, трансфицируют с плазмидой, конститутивно экспрессирующей либо рецептор глюкокортикоида человека (ГР), либо рецептор минералокортикоида человека (МР), либо рецептор прогестерона человека (РП) под контролем вирусного промотора ЦМВ (цитомегаловирус) промотора. Клетки трансфицируют в Т 150 см колбах в модифицированной по способу Дульбекко среде Игла (DMEM) с 5% эмбриональной бычьей сывороткой (ЭБС), очищенной активированным углем. После инкубации в течение ночи трансфицированные клетки трипсинизируют, помещают d 9-луночные планшеты на среду DMEM,содержащую 5% очищенную активированным углем ЭБС, выдерживают 4 ч, а затем добавляют исследуемые соединения в различной концентрации, варьирующей от приблизительно 0,01 нмоль до 10 мкмоль. В случае исследования воздействия антагонистов в среду добавляют низкие концентрации агониста для каждого соответствующего рецептора (0,25 нмоль дексаметазона для РГ, 0,3 нмоль метилтриэнолона для РА, 0,05 нмоль промегестона для РП и 0,05 нмоль альдостерона для РМ). После выдерживания с исследуемыми соединениями в течение 24 ч клетки лизируют и измеряют активность люциферазы при помощи стандартных методик. Данные используют для построения логистической кривой с четырьмя параметрами, с помощью которой можно определить величины ЕС 50. Процент эффективности (соединений с интенсивным максимальным ответом) или процент максимальной стимуляции (соединения с неинтенсивным максимальным ответом) определяют по отношению к максимальной стимуляции, полученной при воздействии контрольных агонистов: 100 нмоль метилтриэнолона для анализа РА, 30 нмоль промегестона для анализа РП, 30 нмоль альдостерона для анализа РМ и 100 нмоль дексаметазона для анализа РГ. Сходным же образом можно определить величины IC50 - при использовании данных исследования воздействия антагонистов. Процент ингибирования также можно определить по отношению к ответу, полученному в присутствии только агониста, как описано выше. В результате исследований, проведенных по выше описанным методикам, соединение (1) показало следующий профиль активации транскрипции: для РГ примерно 44% эффективности со значением показателя ЕС 50 примерно 1,8 нмоль; для РА примерно 4,4% эффективности со значением показателя ЕС 50 выше 10 мкмоль; для РМ примерно 1% эффективности со значением показателя ЕС 50 больше 10 мкмоль; для РП примерно 54% эффективности со значением показателя ЕС 50 выше 10 мкмоль (значения, полученные в ходе 4-5 экспериментов).-4 017288 Исследование трансрепрессии, опосредованной рецепторами глюкокортикоидов. 1. IL-ip-стимулируемая выработка интерлейкинов IL-6 в фибробластах CCD-39SK кожи человека. Вкратце, фибробласты CCD-39SK кожи человека (20 тысяч клеток на лунку), полученные из Американской коллекции типовых культур (АТСС), помещают в 96-луночные планшеты в бессывороточную среду с добавлением 10% эмбриональной бычьей сыворотки (ЭБС), 100 ед./мл пенициллина, 100 мкг/мл стрептомицина и 2 ммоль/л L-глютамина. Клетки выдерживают во влажной камере с 5% содержаниемCO2 при температуре 37C. В ячейки добавляют исследуемые соединения в разных концентрациях, варьирующих от финальной концентрации 4,65 пмоль до 4,64 мкмоль. Для положительного контроля применяют 0,1 мкмоль дексаметазона. После 1 ч дополнительной обработки исследуемым соединением добавляют IL-1 в конечной концентрации 1 нг/мл, а затем оставляют реакционную смесь на ночь. Из каждой ячейки удаляют 10 мкл надосадочной жидкости и определяют концентрацию интерлейкина IL-6 с помощью набора ELISA, причем концентрации подсчитываются при определении коэффициента поглощения при длине волны 450 нм. 2. LPS-стимулированная выработка TNF-g в РМА-дифференцированных клетках U937 (LPSлипополисахарид; TNF-g - ФНО-альфа). Клетки-предшественники моноцитов человека U937, полученные из АТСС, выращивают в полнойRPMI 1640 среде с 10% содержанием ЭБС. Для обеспечения возможности дифференцировки моноцитов в адгезивные макрофаги, клетки U937 промывают в бескальциевой безмагниевой среде, и ресуспендируют в свежей среде RPMI, содержащей 20 нмоль форболмиристатацетата (ФМА), в течение ночи. После дифференциации к клеткам, находящимся в 96-луночных планшетах, добавляют исследуемые соединения в разных концентрациях, варьирующих от 4,65 пмоль до 4,64 мкмоль. После 1 ч дополнительной обработки исследуемым соединением добавляют липополисахарид в конечной концентрации 100 нг/мл и оставляют реакционную смесь на ночь. Из каждой ячейки на другой 96-луночный планшет переносят по 25 мкл надосадочной жидкости без клеток, после чего измеряют производительность по TNF- с помощью набора ELISA, причем концентрации подсчитываются при определении коэффициента поглощения при длине волны 450 нм. В результате исследований, проведенных по выше описанным методикам, соединение (1) вызывает примерно 90% или больше максимального ингибирования эндогенной экспрессии IL-6 и TNF- со значением показателя IC50 примерно 8,5 и 21 нмоль соответственно (сообщаются усредненные данные, полученные в ходе 15 экспериментов (исследование IL-6) и 4 экспериментов (исследование TNF-. Таким образом, соединение (1) является эффективным и полным трансрепрессором (вызывает примерно 90% и выше максимального ингибрования) эндогенной продукции провоспалительных белков IL6 и TNF-. К тому же, соединение (1) показывает лишь частичную агонистическую активность (примерно 50% и ниже максимального действия) в стимуляции РГ/ГЭГО-опосредованной транскрипции. Таким образом, соединение (1) проявляет дифференцированный профиль активности, вызывая полную РГопосредованную трансрепрессию, но при этом только частично вызывая РГ/ГЭГО-опосредованную трансактивацию. Более того, в исследованиях влияния на функциональные модуляции других рецепторов стероидов соединение (1) проявляет лишь частичную активность в стимулировании экспрессии генов, опосредованной РА, РМ и РП. Экспериментальные модели на животных. Модель каррагинанового отека стопы (КОС). Каррагинаны представляют собой группу полисахаридов, которые способны вызывать острый воспалительный ответ у животных. Главные симптомы воспаления, такие как отечность, повышенная болевая чувствительность и покраснение кожи, проявляются на месте укола сразу после введения вещества. Модель КОС является общепризнанной моделью воспаления. Она может применяться для оценки противовоспалительного действия лигандов рецепторов глюкокортикоидов. Для того чтобы оценить противовоспалительное действие соединения (1), последнее приготавливают в смеси с носителем, содержащим 0,5% карбоксиметилцеллюлозы и 0,25% полисорбата Твин-80, а затем вводят орально при помощи зонда самцам крыс линии Спраг-Доули (180-200 г). Для сравнения орально в комбинации с тем же носителем вводят преднизолон. Через 2 ч 1% каррагинан в 50 мкл 0,9% апирогенного солевого раствора вводят субплантарно в правую заднюю лапу. Через 3 ч после инъекции крыс умертвляют при помощи CO2. Лапы ампутируют и взвешивают на микровесах. Затем их анатомируют, выполняя несколько разрезов на поверхности лапы и сразу же погружают в жидкий азот до замораживания. Затем замороженные лапы центрифугируют для извлечения эксудата. В эксудате измеряют концентрацию IL-1, цитокина, вырабатывающегося во время воспалительной реакции, с помощью набора ELISA, следуя инструкциям производителя. Также определяют суммарный белок с помощью набора для анализа белка. Абсолютный уровень IL-1 нормализуют до концентрации нг IL-1/мг суммарного белка. Ингибированный соединением (1) каррагинан спровоцировал увеличение лапы в массе, причем значение показателя ED50 составило примерно 2,8 мг/кг. Также воздействие соединения (1) привело к снижению уровня IL-1 в эксудате лапы, при этом значение показателя ED50 составило примерно-5 017288 3,2 мг/кг. Напротив, лечение преднизолоном на данной модели подавляло увеличение массы лапы (значение показателя ED50 6,6 мг/кг) и сократило уровень содержания IL-1, при этом значение показателяED50 составило меньше 1 мг/кг (представлено среднее значение показателя ED50 из 5 независимых опытов). Анализ сывороточного остеокальцина. Потеря костной массы/остеопороз и обусловленный этим повышенный риск перелома являются частыми и серьезными неблагоприятными эффектами глюкокортикоидной терапии. Считается, что, по меньшей мере, отчасти остеопороз, вызванный воздействием глюкокортикоидов,является следствием угнетения костеобразования. Измерение сывороточного остеокальцина, биологического маркера костеобразования, является признанным способом оценки неблагоприятного влияния глюкокортикоидной терапии на кости. Для оценки влияния соединения (1) на костеобразование последнее приготавливают в смеси с носителем, содержащем 5% карбоксиметилцеллюлозы и 0,25% полисорбата Твин-80, а затем вводят орально при помощи зонда самцам мышей линии Свисс-Вебстер в возрасте 16 недель (Harlan Industries, Индианаполис) в течение 7 дней. Для сравнения орально в комбинации с тем же носителем вводят преднизолон. Сбор сыворотки производят 24 ч спустя последнего введения; уровень содержания остеокальцина измеряют методом конкурентного радиоиммуноанализа в 96-луночных планшетах. Вкратце, каждую лунку планшета Multiscreen, содержащую 2,5 мкл сыворотки мыши, 2,5 мкл козьего антимышиного остеокальцина, 0,625 мкл нормальной сыворотки козы и 119,375 мкл буфера для радиоиммуноанализа(0,1225 М NaCl, 0,01 М NaH2PO4, pH 7,4, 0,025 М тетрафосфата EDTA, 0,1% (вес./об.) бычьего сывороточного альбумина (БСА) и 0,1% (вес./об.) полисорбата Твин 20), выдерживают в орбитальном встряхивателе на скорости 80 об/мин, в течение 18 ч при 4C. После добавления 0,2 мкКи/мл [125I] остеокальцина мышей в 25 мкл буфера для радиоиммуноанализа в каждую лунку, планшеты выдерживают в орбитальном встряхивателе на скорости 80 об/мин в течение 24 ч при 25C. Комплекс осаждают в течение 2 ч при 25C путем добавления IgG осла (антикозий) в 0,2 М Na2HPO4, рН 7,4, 5% (вес./об.) полиэтиленгликоля, 125 мкл на лунку. Осадок собирают путем вакуумной фильтрации и один раз промывают дистиллированной водой (по 100 мкл на ячейку). Фильтры перфорируют, радиоактивность измеряют при помощи счетчика гамма-излучения. Радиоактивность, зафиксированная на фильтрах от исследуемых образцов, обратно пропорциональна концентрации остеокальцина. Калибровочная кривая очищенного остеокальцина мышей применяется для вычисления концентрации сывороточного остеокальцина в исследуемых образцах. Если сравнивать суточные дозы с аппроксимированием значений ED50 полученных для модели КОС крысы (3 мг/кг в день для соединения (1) и 10 мг/кг в день для преднизолона), соединение (1) вызвало меньшее снижение уровня сывороточного остеокальцина, чем преднизолон. Способы приготовления соединения (1) широко известны в данной области техники. Например,международная публикация WO 04/052847 описывает основные используемые методики. Кроме того,международная публикация WO 05/066161 описывает дополнительные основные используемые методики. Приведенные ниже схемы, промежуточные соединения и примеры дополнительно иллюстрируют изобретение и представляют стандартные пути синтеза соединения (1). Реагенты и исходные материалы легко доступны или могут быть без труда синтезированы специалистом в данной области. Следует понимать, что схемы, промежуточные соединения и примеры изложены здесь для иллюстрирования изобретения и не ограничивают его и что специалисты в данной области техники могут внести определенные изменения в варианты осуществления данного изобретения. Названия соединений согласно данному изобретению в основном получены из ChemDraw Ultra, версия 7.0.1. Используемые в настоящем изобретении сокращения обозначают: ДМСО - диметилсульфоксид; ДИАК - диизопропилазодикарбоксилат; АДДП - 1,1'-(азодикарбонил)дипиперидин; ТГФ - тетрагидрофуран; ДМФ - диметилформамид; ТМСЦ - триметилсилилцианид; ТЭА или Et3N - триэтиламин; ДМЭ - 1,2-диметоксиэтан; Схема 1 описывает приготовление промежуточных соединений пиридина (5) и (6). Согласно схеме 1, этапу 1, 1,3-бромпирилин (1) окисляют до 3-бром-пиридин-N-оксида (2). Согласно схеме 1, этапу 2,путем добавления цианида получают 3-бром-пиридин-2-карбонитрил (3). Нитрил формулы (3) гидролизуют до карбоновой кислоты и эстерифицируют путем кислотного катализа до сложного эфира формулы(4). Согласно схеме 1, этапу 4, сложный эфир восстанавливают до пиридилметанола формулы (5 а) при помощи борогидрида натрия. Пиридинилметанол формулы (5b), где Hal представляет собой йод, получают согласно схеме 1, этапу 5, т.е. формируя двойной анион пиколиновой кислоты (7), а затем путем электрофильного гашения йодом получают 3-йодпиколиновую кислоту (8). На этапе 6 кислоту формулы (8) восстанавливают применяя диизобутилалюминий гидрид для получения пиридилметанола формулы (5b). Согласно схеме 1, этапу 7, спирт формулы (5 а, b) преобразуют в пиридилметилхлорид формулы (6 а,b) при помощи тионилхлорида. Схема 2-7 017288 Схема 2 показывает разные способы получения основного промежуточного вещества (13 а, b). Согласно схеме 2, этапу 1, 5-фтор-3-бромфенол (9) защищают для получения метоксиметилового эфира формулы (10). Согласно схеме 2, этапу 2, проводят реакцию сочетания Сонагаширы между защищенным бромфенолом формулы (10) и 1-бутином для получения арилалкина формулы (11). Реакцию можно проводить либо с диэтиламином, либо с триэтиламином. Бромиды арила объединяют при повышенных температурах (70C), иодиды арила - при комнатной температуре. Снимают защиту фенола, как показано на схеме 1, этап 3, при помощи HCl-ацетона для получения фенола формулы (12). В качестве альтернативы фенол защищают как эфир ТГФ и снимают защиту при помощи р-толуолсульфоната пиринидина(ПТСП) в метаноле для получения фенола формулы (12). Согласно схеме 2, этапу 4, реакция Митсунобу между алкинилфенолом формулы (12) и пиридинметиловым спиртом формулы (5) или (5b) позволяет получить галопиридиларилалкин формулы (13 а, b). Также к подходящим реагентам относятся ДИАК и трифенилфосфинин в ТГФ. В качестве альтернативы,на этапе 5, галопиридиларилалкин формулы (13 а, b) получают путем алкилирования фенола формулы(12) и пиридинметилхлорида формулы (6 а) или (6b) карбонатом калия в ацетонитриле. Другой вариант получения бромпиридиларилалкина формулы (13 а) показан на этапах 6 и 7. Согласно схеме 2, этапу 6, 5-фтор-2-йодфенол (Morice, С., et. al. Tetrahedron Lett. 2001, 42, 64 99-6502) соединяют в ходе реакции Митсунобу с пиридилметанолом формулы (5 а) для получения йодарилового эфира формулы (15). Согласно схеме 2, этапу 7, йодариловый эфир подвергают реакции сочетания Сонагаширы с 1-бутином для получения галопиридиларилалкина формулы (13 а). Схема 3 Согласно схеме 3, этапы 1 и 2, катализируемая платиной диборонация галопиридиларилалкина с формулой (13 а, b) приводит к образованию дибороновой кислоты формулы (16 а, b), которая образует винилбороновую кислоту формулы (17) при проведении реакции межмолекулярного взаимодействия Сузуки в более мягких условиях (около 0,01 моль). Следует отметить, что диборонация наиболее эффективна для соединения (13 а), причем Hal представляет собой бром. Согласно схеме 3, этапу 3, реакция межмолекулярного взаимодействия Сузуки между винилбороновой кислотой (17) и 3-йоданилином позволяет получить анилинбензопиридил-10-оксепин формулы(18). На этапе 4 анилин формулы (18) сульфонилируют метансульфонилхлоридом для получения конечного бензопиридил-10-оксепина формулы (19). В качестве альтернативы, согласно схеме 3, этапу 5, бензопиридил-10-оксепин формулы (19) получают непосредственно путем проведения реакции межмолекулярного взаимодействия Сузуки с N-(3-йодфенил)метансульфонамидом. На схеме 4 представлен еще один способ получения бензопиридил-10-оксепина формулы (19). Согласно схеме 4, этапу 1, проводят последовательные межмолекулярные реакции Хека и Сузуки между галопропиларилалкином формулы (13 а, b) и 3-нитробензобороновой кислотой для получения нитрофенилбензопиридилокспепина формулы (20). Больший выход продукта получают при медленном добавлении бороновой кислоты к арилалкину, причем Hal представляет собой йод. Для специалистов в области техники будет понятно, что на этапах 2-3 соединение (19) получают путем восстановления нитро-аналога соединения формулы (20) до анилина формулы (21) и его последующего сульфонилирования. Схема 5 Согласно схеме 5, этапу 1, проводят реакцию взаимодействия Сонагаширы между 2 бромпиридином формулы (5 а) и 1-бутином подобно схеме 2, этапу 2, в результате чего образуется алкинилпиридилметиловый спирт формулы (22). Согласно схеме 5, этапу 2, реакция Митсунобу между пиримидилметиловым спиртом формулы (22) и 5-фтор-2-йодфенолом формулы (14) позволяет получить пиридин формулы (23). Согласно этапу 3 межмолекулярное взаимодействие Хека и последующее перекрестное сочетание с N-[3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]метансульфонамидом позволяет получить бензопиридил-11-оксепин формулы (24), который затем фотоизомеризуют в бензопиридил-10-оксепин формулы (19). Согласно схеме 6, этапу 1, лактон формулы (25) взаимодействует с натриевой солью галофенола формулы (26), в результате чего образуется кислота формулы (27). Согласно схеме 6, этапу 3, проводят реакцию Фриделя-Крафтса для бензойной кислоты формулы (27) для получения бензопиридилоксепинона формулы (28). Согласно схеме 6, этапу 3, кетон формулы (28) преобразуют в алкенилбензопиридинилоксепин формулы (29) как смесь геометрических изомеров либо путем олифенирования Виттига, либо путем последовательного добавления/дегидратации церистого алкила. Согласно этапу 4 проводят реакцию алкенилбензопиридилоксепина формулы (29) и его изомера либо с бромом, либо с трибромидом 4(диметиламино)пиридиния с целью получения геометрических изомеров бромидов формул (30) и (31). Винилбромиды разделяют и видоизменяют в соответствующие сульфонамиды (19) или (24), как показано на этапах 5 и 6, реакцией перекрестного сочетания Сузуки с N-[3-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фенил]метансульфонамидом при нормальных условиях. Согласно схеме 6,этапу 7, проводят фотоизомеризацию сульфонамида формулы (24) в бензопиридил-10-оксепин формулы 2-Бром-5-фторфенол (30 г, 0,16 моль) растворяли в дихлорметане (170 мл) и охлаждали до 0C. К этому раствору добавляли диизопропилэтиламин (36 мл, 0,20 моль) при помощи шприца, а затем вносили хлорметилметиловый эфир (16 мл, 0,20 моль) при помощи капельной воронки. Раствор перемешивали и нагревали до комнатной температуры. 2 ч спустя смесь промывали насыщенным водным растворомNH4Cl, водой и солевым раствором. Органическую фазу высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали методом флэш-хроматографии (Biotage Si65M, 20% этилацетат/гексан) для получения 26,1 г (71%) соединения, указанного в названии, в виде чистого бесцветного масла. 1 Н ЯМР 400 МГц (CDCl3)7,46-7,53 (м, 1 Н), 6,92-6,99 (м, 1 Н), 6,63-6,70 (м, 1 Н), 5,26 (с, 2 Н), 3,55 1-Бром-4-фтор-2-метоксиметоксибензин (7,6 г, 32 ммоль) помещали в колбу, находящуюся под давлением, растворяли в диэтиламине (65 мл) и дегазировали азотом в течение 15 мин. Избыток бутина выходил при бурлении, к смеси добавляли CuI (1,9 г, 10 ммоль) и PdCl2(PPh3)2 (2,3 г, 3,3 ммоль), а затем- 10017288 колбу нагревали до 70C в течение 44 ч. Смесь разбавляли сложным эфиром, после чего промывали насыщенным водным раствором NH4Cl и солевым раствором. Органическую фазу высушивали над MgSO4,фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали методом флэш-хроматографии (Biotage Si65M, 3% ТГФ/гексан) для получения 6,12 г (91%) соединения, указанного в названии, в виде сухого вещества оранжевого цвета. ГХМС мг/экв. 208 [М]+. Промежуточное соединение 3. 2-Бут-1-инил-5-фторфенол 1-Бут-1-инил-4-фтор-2-метоксиметоксибензол (6,1 г, 29 ммоль) растворяли в 250 мл 10% раствора концентрированной соляной кислоты в ацетоне. Смесь помешивали при комнатной температуре в течение 5 ч. Смесь разбавляли сложным эфиром и промывали насыщенным водным раствором NaHCO3 и солевым раствором. Органическую фазу высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали методом флэш-хроматографии (Biotage Si65M,5% AcOEt/гексан) для получения 3,52 г (73%) соединения, указанного в названии, в виде масла оранжевого цвета. ГХМС мг/экв. 164 [М]+. Промежуточное соединение 4. 3-Бромпиридин 1-оксид Метилтриоксорений (100 мг, 0,401 ммоль) растворяли в дихлорметане (40 мл) и добавляли к раствору 3-бромпиридин (15,8 г, 100 ммоль), а затем 30% водный раствор Н 2 О 2 (22,7 мл). Двухфазную смесь перемешивали при комнатной температуре. 18 ч спустя к ней добавляли MnO2 (25 мг, 0,29 ммоль) и перемешивали при комнатной температуре в течение 2 ч. Смесь экстрагировали дихлорметаном, смешанные экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении для получения 9,52 г (55%) соединения, указанного в названии, в виде масла оранжевого цвета. ГХМС мг/экв. 174 [М-H]-. Промежуточное соединение 5. 3-Бромпиридин-2-карбонитрил 3-Бромпиридин 1-оксид (9,4 г, 54 ммоль) растворяли в ацетонитриле (60 мл), затем к раствору последовательно добавляли триэтиламин (15 мл) и триметилсилилцианид (21,7 мл, 163 ммоль). Смесь нагревали до 100C и перемешивали в течение 16 ч. Затем смесь охлаждали до 0C, вливали в 250 мл 5 М водного раствора NaOH и экстрагировали дихлорметаном. Смешанные экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученное вещество очищали методом флэш-хроматографии (Biotage Si65M, 20% AcOEt/гексан), что позволяло получить 7,8 г (79%) соединения, указанного в названии, в виде сухого вещества желтого цвета. ГХМС мг/экв. 182 [М-H]-. Промежуточное соединение 6. Метиловый эфир 3-бромпиридин-2-карбоновой кислоты 3-Бромпиридин-2-карбонитрил (14,7 г, 80,3 ммоль) растворяли в концентрированной соляной кислоте (50 мл) и нагревали до 110C в течение 18 ч. Смесь охлаждали до 0C, фильтровали, промывали небольшим количеством эфира и высушивали в сушильном шкафу при пониженном давлении. Полученное сухое вещество коричневого цвета растворяли в метаноле (80 мл), затем капельным путем добавляли концентрированную серную кислоту (6,6 мл), после чего к смеси добавляли водный раствор бикарбоната калия для создания щелочного рН среды. Затем смесь экстрагировали AcOEt. Смешанные экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении для получения 12,8 г (74%) соединения, указанного в названии, в виде сухого вещества белого цвета. ГХМС мг/экв. 215 [М-H]-. Промежуточное соединение 7. (3-Бромпиридин-2-ил)метанол(150 мл) и охлаждали до 0C. К смеси добавляли NaBH4 (11,2 г, 296 ммоль) порциями в 1 г. Смесь нагревали до комнатной температуры и перемешивали в течение 3 ч. Метанол удаляли при пониженном дав- 11017288 лении, затем добавляли AcOEt; раствор промывали насыщенным водным раствором хлорида аммония и солевым раствором. Органическую фазу высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении для получения 6,8 г (62%) соединения, указанного в названии, в виде сухого вещества белого цвета. ГХМС мг/экв. 187 [М-H]-. Промежуточное соединение 8. 3-Бром-2-(2-бут-1-инил-5-фторфеноксиметил)пиридин Раствор (3-бромпиридин-2-ил)метанола (3,09 г, 16,43 ммоль), 2-бут-1-инил-2-фторфенола (2,70 г,16,43 ммоль) и трифенилфосфина (6,46 г, 24,64 ммоль) в дихлорметане (162 мл) охлаждали до -5-0C. Через 15 мин к раствору капельным путем добавляли азодикарбоксилат диизопропила (АДДИ) (4,85 мл,24,64 ммоль). Спустя 1 ч растворитель выпаривали, а неочищенный продукт сразу очищали методом флэш-хроматографии (SiO2; 4% гексан: AcOEt) для получения 3,5 г (64%) соединения, указанного в названии. ЖХМС мг/экв. 334 [М+H]+. Промежуточное соединение 9. 8-Фтор-5-[1-(3-нитрофенил)пропилиден]-5,11-дигидро-10-окса-1 азадибензо[a,d]циклогептенPd(OAc)2 (0,18 г, 5%) и о-толилфосфин (0,32 г, 1,05 ммоль) добавляли к раствору 3-бром-2-(2-бут-1 инил-5-фторфеноксиметил)пиридина (3,50 г, 10,5 ммоль), карбоната натрия (3,39 г, 31,5 ммоль) и 3 нитрофенилбороновой кислоты (2,27 г, 13,06 ммоль) в 4:1 диоксане:воде (101 мл). Азот пропускали через смесь в течение 15 мин, затем реакционную смесь оставляли на ночь для перемешивания при 75C. После этого смесь охлаждали до комнатной температуры и твердую фазу фильтровали через Celite, добавляли воду и AcOEt, затем фазы декантировали. Водный слой экстрагировали при помощи AcOEt,объединенные органические слои высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали методом флэш-хроматографии (SiO2; 4% AcOEt: гексаны) для получения 1,49 г (38%) соединения, указанного в названии. ЖХМС мг/экв. 377 [М+H]+. Промежуточное соединение 10. 3-[1-(8-Фтор-11H-10-окса-1-азадибензо[a,d]циклогептен-5 илиден)пропил]фениламин 8-Фтор-5-[1-(3-нитрофенил)пропилиден]-5,11-дигидро-10-окса-1-азадибензо[а,d]циклогептен (1,49 г,3,96 ммоль) растворяли в этаноле (20 мл) и продували азотом. Добавляли Pd/C (0,15 г, 10%), смесь гидрогенизовали под давлением в 1 атмосферу в течение 2 ч. Смесь продували азотом, фильтровали черезCelite, твердую фазу промывали этанолом. Фильтрат концентрировали при пониженном давлении для получения 1,37 г (99%) соединения, указанного в названии. ЖХМС мг/экв. 347 [М+H]+. Пример 1. N-3-[1-(8-Фтор-11H-10-окса-1-азадибензо[a,d]циклогептен-5-илиден)пропил]фенилметансульфонамид(1,37 г, 3,96 ммоль) и пиридина (0,35 мл, 4,36 ммоль) в дихлорметане (10 мл) капельным путем добавляли хлорид метансульфонила (0,34 мл, 4,36 ммоль) при 0C. Спустя 2 ч добавляли 7% водный раствор бикарбоната натрия (20 мл), смесь перемешивали в течение 30 мин, декантировали, слои разделяли. Водную фазу промывали дихлорметаном (220 мл). Органические фазы объединяли и промывали солевым- 12017288 раствором, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении для получения 1,98 г неочищенного продукта. Затем его очищали методом флэш-хроматографии(Biotage), извлекали при помощи смеси гексан:этанол (9:1) для получения 1,35 г (80%) соединения,указанного в названии. ЖХМС мг/экв. 425 [М+H]+. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение,представляющее собой или фармацевтически приемлемая соль данного соединения. 2. Соединение по п.1,представляющее собой(Е)-N-3-[1-(8-фтор-11H-10-окса-1 азадибензо[a,d]циклогептен-5-илиден)пропил]фенилметансульфонамид. 3. Применение соединения или его соли по п.1 или 2 в качестве лекарственного средства для лечения ревматоидного артрита, остеоартрита, ревматизма, астмы, аллергического ринита, системной красной волчанки, хронической обструктивной болезни легких, болезни Крона, воспалительного заболевания кишечника и язвенного колита. 4. Применение по п.3 в качестве лекарственного средства для лечения ревматоидного артрита. 5. Применение соединения или его соли по п.1 или 2 для лечения ревматоидного артрита, остеоартрита, ревматизма, астмы, аллергического ринита, системной красной волчанки, хронической обструктивной болезни легких, болезни Крона, воспалительных заболеваний кишечника или язвенного колита. 6. Применение по п.5 для лечения ревматоидного артрита. 7. Фармацевтическая композиция для лечения ревматоидного артрита, остеоартрита, ревматизма,астмы, аллергического ринита, системной красной волчанки, хронической обструктивной болезни легких, болезни Крона, воспалительного заболевания кишечника и язвенного колита, содержащая соединение или соль по любому из пп.1 или 2 в сочетании с одним или более фармацевтически приемлемыми носителями, эксципиентами или разбавителями. 8. Фармацевтическая композиция для лечения ревматоидного артрита, содержащая соединение или соль по любому из пп.1 или 2 в сочетании с одним или более фармацевтически приемлемыми носителями, эксципиентами или разбавителями.
МПК / Метки
МПК: A61P 19/02, A61P 11/06, C07D 491/04, A61K 31/4353
Метки: глюкокортикоидов, e)-n-{3-[1-(8-фтор-11н-10-окса-1-азадибензо[a,d]циклогептен-5-илиден)пропил]фенил}метансульфонамид, качестве, ревматизма, рецепторов, модулятора, лечения
Код ссылки
<a href="https://eas.patents.su/14-17288-e-n-3-1-8-ftor-11n-10-oksa-1-azadibenzoadciklogepten-5-ilidenpropilfenilmetansulfonamid-v-kachestve-modulyatora-receptorov-glyukokortikoidov-dlya-lecheniya-revmatizma.html" rel="bookmark" title="База патентов Евразийского Союза">(e)-n-{3-[1-(8-фтор-11н-10-окса-1-азадибензо[a,d]циклогептен-5-илиден)пропил]фенил}метансульфонамид в качестве модулятора рецепторов глюкокортикоидов для лечения ревматизма</a>
Применение модулятора рецепторов глюкокортикоидов и агониста рецепторов глюкокортикоидов при получении лекарственного средства

Номер патента: 7483
Опубликовано: 27.10.2006
Авторы: Свик Эндрю Гордон, Лиу Кевин Кун-Чин, Морган Брэдли Пол, Дау Роберт Ли
МПК: A61K 31/325, A61K 31/122, A61K 31/05...
Метки: агониста, лекарственного, рецепторов, получении, глюкокортикоидов, средства, применение, модулятора
Формула / Реферат:
1. Применение соединения формулы II его изомера, пролекарства указанного соединения или изомера, или фармацевтически приемлемой соли указанного соединения, изомера или пролекарства; где R1 представляет собой (C1-С6)алкил, (С2-С6)алкенил, (С2-С6)алкинил или фенил, незамещенный или замещенный одним из следующих заместителей: -ОН, -NR12R13, -NR12C(О)-(С1-С4)алкил, -CN, -(С0-С2)алкил-hеt, -О-(C1-C3)алкил-С(О)-NR12R13,...
3-(4-{[4-(4-{[3-(3,3-диметил-1-пиперидинил)пропил]окси}фенил)-1-пиперидинил]карбонил}-1-нафталинил)пропановая или пропеновая кислота в качестве антагонистов н1 – и н3 – рецепторов для лечения воспалительных и аллергических расстройств

Номер патента: 14354
Опубликовано: 29.10.2010
Авторы: Винадер Бругаролас Мария Виктория, Ходжсон Саймон Тинби, Прокопиоу Панайиотис Александроу
МПК: A61P 11/00, A61K 31/4545, A61P 37/08...
Метки: воспалительных, лечения, качестве, пропеновая, аллергических, 3-(4-{[4-(4-{[3-(3,3-диметил-1-пиперидинил)пропил]окси}фенил)-1-пиперидинил]карбонил}-1-нафталинил)пропановая, рецепторов, антагонистов, кислота, расстройств
Формула / Реферат:
1. Соединение формулы (I)где нафталиновое кольцо замещено по положению 2, 3, 4, 5, 6, 7 или 8 заместителем R1и R1 представляет собой -СН2СН2СООН или -СН=С(СН3)СООН; или его соль.2. Соединение по п.1, где нафталиновое кольцо замещено по положению 2, 3, 4, 5, 6, 7 или 8 заместителем R1 и R1представляет собой -СН2СН2СООН, или его соль.3. 3-(4-{[4-(4-{[3-(3,3-Диметил-1-пиперидинил)пропил]окси}фенил)-1-пиперидинил]карбонил}-1-нафталинил)пропановая...
Полиморфная форма 2-(r) – (1- (r) – (3,5-бис (трифторметил) фенил) этокси) -3- (s) – (4-фтор) фенил – 4 – (3 – (5-оксо-1h, 4h-1, 2, 4 – триазоло) метил)морфолина в качестве антагониста рецептора тахикинина

Номер патента: 2405
Опубликовано: 25.04.2002
Авторы: Макколи Джеймс, Крокер Луис
МПК: A61P 25/28, C07D 265/32, A61K 31/5375...
Метки: трифторметил, полиморфная, 4h-1, фенил, форма, 4-фтор, триазоло, 2-(r, рецептора, качестве, 5-оксо-1h, антагониста, этокси, метил)морфолина, 3,5-бис, тахикинина
Формула / Реферат:
1. Полиморфная форма соединения 2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фтор)фенил-4-(3-(5-оксо-1Н,4Н-1,2,4-триазоло)метил)морфолина, обозначенная как форма I, по существу, отличающаяся рентгеновской порошковой дифрактограммой с характерными отражениями приблизительно 12,0, 15,3, 16,6, 17,0, 17,6, 19,4, 20,0, 21,9, 23,6, 23,8 и 24,8ш (2 тэта). 2. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и...
Производные 5-[4-(азетидин-3-илокси)фенил]-2-фенил-5н-тиазоло[5,4-c]пиридин-4-она и их использование в качестве рецепторов mch

Номер патента: 15559
Опубликовано: 31.08.2011
Авторы: Гардинир Кевин Мэттью, Брунавс Майкл, Гармен Дэвид Джозеф, Хембр Эрик Джеймс, Секереш Хелен Джейн
МПК: A61K 31/437, C07D 513/04
Метки: производные, 5-[4-(азетидин-3-илокси)фенил]-2-фенил-5н-тиазоло[5,4-c]пиридин-4-она, использование, качестве, рецепторов
Формула / Реферат:
1. Соединение формулыгде ------ отсутствует или возможно представляет собой связь;q представляет собой 1 или 2;R1 независимо выбран из водорода, -C1-C2-алкила, галогена, гидрокси, -C1-C2-галогеналкила, -C1-C3-алкокси, циано, -O-C3-C4-циклоалкила и -OC1-C2-галогеналкила;R2 выбран из группы, состоящей из водорода, -C1-C3-алкила, гидрокси, -C1-C3-алкокси, циано, -C1-C2-галогеналкила, -OC1-C2-галогеналкила и галогена;R3 выбран из группы, состоящей...
Модуляторы рецепторов глюкокортикоидов

Номер патента: 4886
Опубликовано: 26.08.2004
Авторы: Лиу Кевин Кун-Чин, Свик Эндрю Гордон, Дау Роберт Ли, Морган Брэдли Пол
МПК: A61K 31/05, C07C 35/42, C07D 213/40...
Метки: глюкокортикоидов, рецепторов, модуляторы
Формула / Реферат:
1. Соединение формулы II его изомер, пролекарство указанного соединения или изомера или фармацевтически приемлемая соль указанного соединения, изомера и пролекарства; где R1 представляет собой фенил, незамещенный или замещенный одним из следующих заместителей: -OH, -NR12R13, -NR12-C(O)-(C1-C4)алкил, -CN, -(C0-C2)алкил-het, -O-(C1-C3)алкил-C(O)-NR12R13, -NR12-(C0-C2)алкил-C(O)-NR12R13, -(C0-C2)алкил-NR12-SO2-R13, -NR12-SO2-het,...
Предыдущий патент: Способ получения морфийных соединений
Следующий патент: Синергическая пребиотическая композиция и ее применение
Случайный патент: Погружной выпариватель