Кристаллический 2, 5-дион-3-(1-метил-1н-индол-3-ил) -4-[1-(пиридин -2- илметил)пиперидин-4-ил] -1н-индол-3-ил-1н-пирролмоногидрохлорид
Номер патента: 7463
Опубликовано: 27.10.2006
Авторы: Фол Маргарет Мэри, Буш Джули Кей, Рутзел-Иденс Сузен Мари



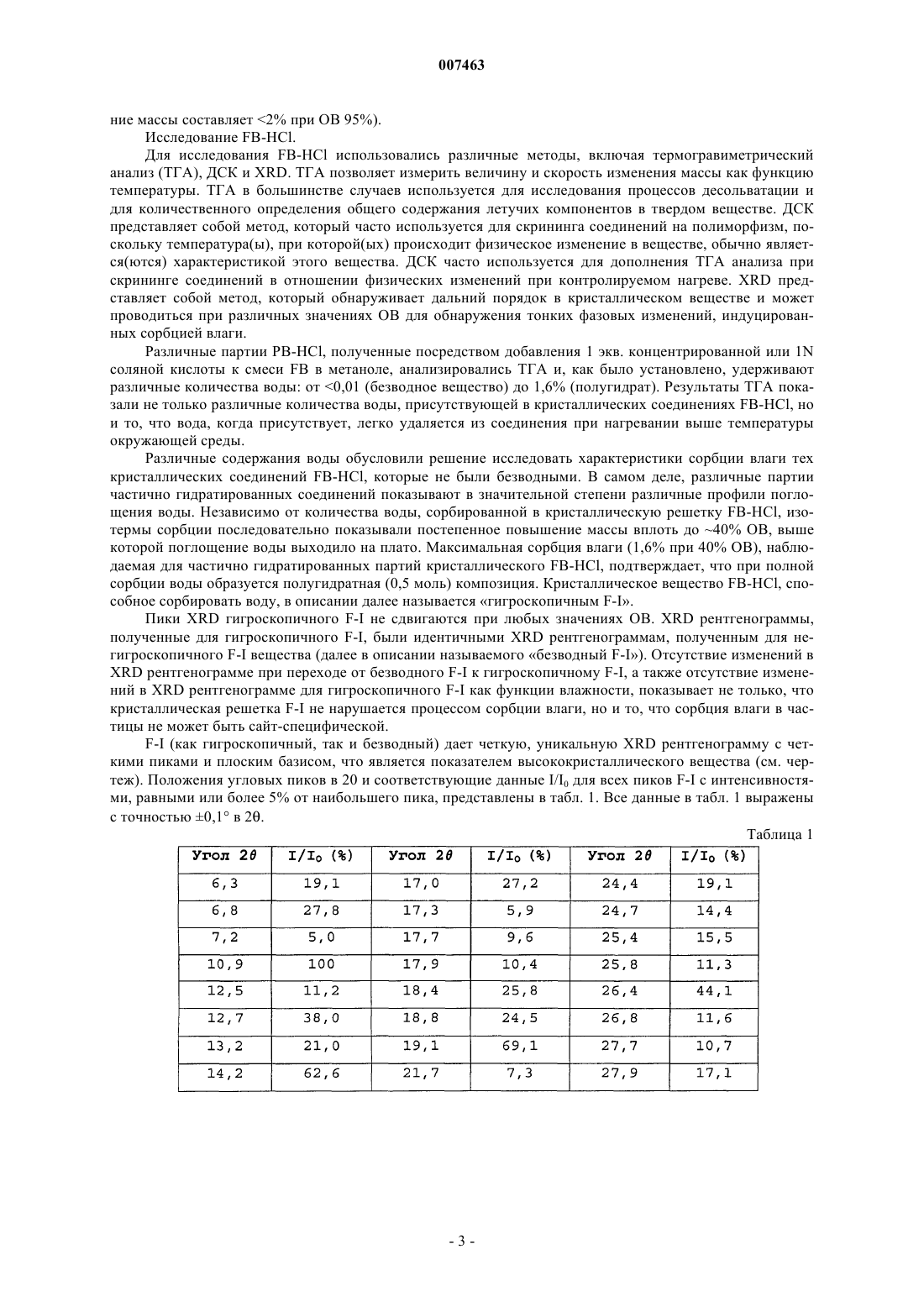





Формула / Реферат
1. Кристаллический 2,5-дион-3-(1-метил-1Н-индол-3-ил)-4-[1-(пиридин-2-илметил)пиперидин-4-ил]-1Н-индол-3-ил-1Н-пирролмоногидрохлорид, его гидрат или их смесь.
2. Кристаллический 2,5-дион-3-(1-метил-1Н-индол-3-ил)-4-[1-(пиридин-2-илметил)пиперидин-4-ил]-1Н-индол-3-ил-1Н-пирролмоногидрохлорид, его гидрат или их смесь, имеющие рентгенограмму, которая содержит следующие пики: 6,8+0,1, 10,9+0,1, 14,2+0,1 и 16,6+0,1ш в 2q; когда рентгенограмма получена при использовании медного источника излучения (CuKa; l=1,54056 Е).
3. Кристаллический моногидрохлорид по п.2, имеющий рентгенограмму, которая дополнительно содержит следующие пики: 6,3+0,1, 7,2+0,1, 12,5+0,1 и 17,0+0,1ш в 2q.
4. Фармацевтическая композиция, содержащая соль по любому из пп.1-3 и фармацевтический носитель.
5. Применение соединения по любому из пп.1-3 в качестве активного вещества в способе лечения человека терапией.
6. Применение по п.5 при лечении неходжкинской лимфомы.
7. Применение по п.5 при лечении глиобластомы.
8. Применение по п.5 при лечении немелкоклеточного рака легких.
9. Применение соединения по любому из пп.1-3 в качестве активного вещества для производства лекарственного средства для лечения неходжкинской лимфомы.
10. Применение соединения по любому из пп.1-3 в качестве активного вещества для производства лекарственного средства для лечения глиобластомы.
11. Применение соединения по любому из пп.1-3 в качестве активного вещества для производства лекарственного средства для лечения немелкоклеточного рака легких.
Текст
007463 Предпосылки изобретения Соединения формулы I и их фармацевтически приемлемые соли, полезные в качестве ингибиторов протеинкиназы С, описаны в публикации европейского патента 817627 (Heath et al.). В примере 49 данной публикации описано свободное основание формулы FB Хотя FB, несомненно, является очень эффективным фармацевтическим средством, неожиданные трудности возникли при его крупномасштабном производстве. Так, непредсказуемое образование сольватов усложнило коммерческий синтез до такой степени, что возникла необходимость в разработке альтернативной формы для крупномасштабного коммерческого производства. В данном контексте в WO 02/02094 и WO 02/02116 конкретно описано применение дигидрохлоридной соли FB (FB-2HCl) для лечения рака и для ингибирования роста опухоли в качестве единственного терапевтического средства или в сочетании с противоопухолевым средством или лучевой терапией. К сожалению, в настоящее время установлено, что FB-2HCl является гигроскопичным. Кроме того, хотя в соответствии с оптической микроскопией кажется, что FB-2HCl является кристаллическим, более детальное исследование порошковой рентгенографией (XRD) показало, что данное вещество фактически является лишь слабо кристаллическим. Неожиданно в соответствии с данным изобретением было установлено, что моногидрохлоридная соль FB может быть воспроизводимо получена в промышленном масштабе, обладает незначительной гигроскопичностью, является достаточно стабильной для применения в препаратах для перорального введения и может быть получена в высококристаллическом состоянии. Краткое описание изобретения Данное изобретение относится к кристаллическому 2,5-дион-3-(1-метил-1 Н-индол-3-ил)-4-[1(пиридин-2-илметил)пиперидин-4-ил]-1H-индол-3-ил-1 Н-пирролмоногидрохлориду, его гидрату или его смесям. Данное изобретение относится также к кристаллическому 2,5-дион-3-(1-метил-1 Н-индол-3-ил)-4-[1(пиридин-2-илметил)пиперидин-4-ил]-1 Н-индол-3-ил-1 Н-пирролмоногидрохлориду, его гидрату или его смесям, имеющему рентгенограмму которая включает следующие пики: 6,80,1, 10,90,1, 14,20,1 и 16,60,1 в 2; когда рентгенограмма получена с использованием медного источника излучения (CuK;=1,54056 ). Данное кристаллическое вещество далее в описании называется F-I. Данное изобретение относится также к фармацевтической композиции, содержащей F-I и фармацевтический носитель. В другом воплощении фармацевтический препарат данного изобретения может приспосабливаться для применения при лечении рака и для применения при ингибировании роста опухоли. Кроме того, данное изобретение относится к способам лечения рака и к способам ингибирования роста опухоли, которые включают введение млекопитающему, нуждающемуся в этом, эффективного количества F-I. Изобретение относится также к F-I для лечения рака и для ингибирования роста опухоли. Другое воплощение данного изобретения предлагает применение F-I для изготовления лекарственного средства для лечения рака и для изготовления лекарственного средства для ингибирования роста опухоли. Краткое описание чертежа Чертеж представляет собой XRD рентгенограмму F-I.-1 007463 Подробное описание изобретения Перед раскрытием проблем, связанных с возможностью крупномасштабного производства FB вследствие того, что FB может не обладать оптимальными свойствами биологической доступности, был проведен скрининг in situ солей для идентификации солей FB, обладающих улучшенными свойствами. Таким скринингом оценивали растворимость солей, образованных in situ в водной среде. Растворимость,полученная in situ для данной соли, не является непосредственно предсказанием равновесной растворимости кристаллической(их) формы(форм) такой соли. Однако данный in situ скрининг может использоваться для определения приоритетных солей для синтеза и исследований в процессе выбора соли. На основании этих данных для синтеза и исследования были выбраны пять из семнадцати солей монокислоты. Такими солями являлись цитрат, метансульфонат (мезилат), фосфат, тартрат и моногидрохлорид (FBHCl). Кроме того, был также синтезирован, охарактеризован и проанализирован FB-2HCl. Некоторые свойства указанных солей, а также FB обсуждены ниже. Цитрат, мезилат, фосфат и тартрат. Цитратная соль, полученная из метанола, является нерастворимой в воде. Мезилатная соль является гигроскопичной и показывает повышение массы до 2% при относительной влажности (ОВ) 70% и повышение массы выше 15% при ОВ 95%. Хотя фосфатная соль демонстрирует быстрое растворение и высокую растворимость сразу после получения, растворимость фосфата падает до 71 мкг/мл при длительном хранении. Фосфатная соль также в некоторой степени гигроскопична и дает гистерезис при десорбции воды, что говорит о возможности образования гидрата. Тартрат является лишь слабо гигроскопичным, демонстрируя 1% повышение массы при ОВ до 70%. Исходя из этого и других интересных исходных данных, тартрат был подвернут краткому полиморф/сольват-скринингу для определения его приемлемости для крупномасштабного производства и применения в качестве фармацевтического средства. Тартратная соль была сначала выделена (титрованием свободного основания с винной кислотой) в виде кристаллического гидрата. Гидрат затем подвергался перекристаллизации для определения, могут ли быть получены другие фармацевтически релевантные кристаллические формы тартратной соли. Число растворителей, приемлемых для применения при перекристаллизации, было ограничено относительно слабой растворимостью данной соли во многих растворителях, включая полярные, протонные растворители (Н 2O, метанол, этанол и изопропиловый спирт) и многие апротонные растворители (ацетон, этилацетат, метилэтилкетон и тетрагидрофуран). Достаточная растворимость наблюдалась только в диметилформамиде, диметилсульфоксиде и органических (органических/водных) смесях. Для достижения растворения зачастую требовались повышенные температуры. Тартратная соль обычно не получалась в проведенных экспериментах перекристаллизации. Вместо этого чаще образовывалась кристаллическая форма FB. Несольватированная форма тартрата не была получена. Данные результаты подтверждают, что выделение тартратной соли FB может быть затруднено,возможно, вследствие меньшей растворимости различных кристаллических форм FB по сравнению с тартратной солью и относительно небольшого отличия в рKа FB и винной кислоты.FB-2HCl. Исследовалась растворимость FB-2HCl в воде в различных условиях, и в концентрациях до 10 мг/мл растворы FB-2HCl стабильны в течение периода до 10 дней при температуре окружающей среды. Однако растворы, выдерживаемые при 50 С, показывали интенсивное осаждение до первого контрольного срока (6 дней). При концентрациях 40 мг/мл было отмечено быстрое осаждение в пределах нескольких минут при комнатной температуре. XRD анализ и ионная хроматография (для определения содержания хлорида) осажденных кристаллов подтвердили, что данный осадок представлял собой FB-HCl.FB. Продукт синтеза, описанного ниже в получении 1, обычно представляет собой несольватированную кристаллическую форму FB. Данная несольватированная форма (называемая далее в описании форма IFB) является предпочтительной, поскольку она хорошо кристаллизуется в процессе получения, быстро фильтруется и дает продукт высокой чистоты (общее содержание родственных веществ (ОСРВ) 0,77%). Однако в очень похожих условиях реакции иногда также выделяется сольват, содержащий тетрагидрофуран (ТГФ) (частота проявления 10-20%). Данный кристаллический сольват фильтруется очень медленно и захватывает некоторые примеси, что приводит к более высокому ОСРВ в продукте (2,42-4,78%). Высокое значение ОСРВ, связанное с данным сольватом, требует, когда присутствует, повторной обработки выделенного сольвата. Несмотря на значительные исследования, причина образования сольвата,содержащего ТГФ, неизвестна. Потеря контроля при получении формы I FB ограничивает его потенциал для разработки в качестве конечного активного фармацевтического ингредиента (АФИ, API).FB-HCl, полученный посредством добавления 1 экв. концентрированной или 1N соляной кислоты к смеси FB в низшем спирте, например метаноле, изопропаноле или 2-бутаноле, или в смеси низшего спирта и воды, является кристаллическим и имеет начальную температуру плавления примерно 256 С,что определено дифференциальной сканирующей калориметрией (ДСК). FB-HCl, полученный способом,описанным в примере 1, является относительно негигроскопичным при ОВ в интервале 0-70% (повыше-2 007463 ние массы составляет 2% при ОВ 95%). Исследование FB-HCl. Для исследования FB-HCl использовались различные методы, включая термогравиметрический анализ (ТГА), ДСК и XRD. ТГА позволяет измерить величину и скорость изменения массы как функцию температуры. ТГА в большинстве случаев используется для исследования процессов десольватации и для количественного определения общего содержания летучих компонентов в твердом веществе. ДСК представляет собой метод, который часто используется для скрининга соединений на полиморфизм, поскольку температура(ы), при которой(ых) происходит физическое изменение в веществе, обычно является(ются) характеристикой этого вещества. ДСК часто используется для дополнения ТГА анализа при скрининге соединений в отношении физических изменений при контролируемом нагреве. XRD представляет собой метод, который обнаруживает дальний порядок в кристаллическом веществе и может проводиться при различных значениях ОВ для обнаружения тонких фазовых изменений, индуцированных сорбцией влаги. Различные партии РВ-НСl, полученные посредством добавления 1 экв. концентрированной или 1N соляной кислоты к смеси FB в метаноле, анализировались ТГА и, как было установлено, удерживают различные количества воды: от 0,01 (безводное вещество) до 1,6% (полугидрат). Результаты ТГА показали не только различные количества воды, присутствующей в кристаллических соединениях FB-HCl, но и то, что вода, когда присутствует, легко удаляется из соединения при нагревании выше температуры окружающей среды. Различные содержания воды обусловили решение исследовать характеристики сорбции влаги тех кристаллических соединений FB-НСl, которые не были безводными. В самом деле, различные партии частично гидратированных соединений показывают в значительной степени различные профили поглощения воды. Независимо от количества воды, сорбированной в кристаллическую решетку FB-НСl, изотермы сорбции последовательно показывали постепенное повышение массы вплоть до 40% ОВ, выше которой поглощение воды выходило на плато. Максимальная сорбция влаги (1,6% при 40% ОВ), наблюдаемая для частично гидратированных партий кристаллического FB-HCl, подтверждает, что при полной сорбции воды образуется полугидратная (0,5 моль) композиция. Кристаллическое вещество FB-HCl, способное сорбировать воду, в описании далее называется гигроскопичным F-I. Пики XRD гигроскопичного F-I не сдвигаются при любых значениях OB. XRD рентгенограммы,полученные для гигроскопичного F-I, были идентичными XRD рентгенограммам, полученным для негигроскопичного F-I вещества (далее в описании называемого безводный F-I). Отсутствие изменений вXRD рентгенограмме при переходе от безводного F-I к гигроскопичному F-I, а также отсутствие изменений в XRD рентгенограмме для гигроскопичного F-I как функции влажности, показывает не только, что кристаллическая решетка F-I не нарушается процессом сорбции влаги, но и то, что сорбция влаги в частицы не может быть сайт-специфической.F-I (как гигроскопичный, так и безводный) дает четкую, уникальную XRD рентгенограмму с четкими пиками и плоским базисом, что является показателем высококристаллического вещества (см. чертеж). Положения угловых пиков в 20 и соответствующие данные I/I0 для всех пиков F-I с интенсивностями, равными или более 5% от наибольшего пика, представлены в табл. 1. Все данные в табл. 1 выражены с точностью 0,1 в 2. Таблица 1 В области кристаллографии хорошо известно, что для любой данной кристаллической формы относительные интенсивности дифракционных пиков могут меняться вследствие предпочтительной ориентации, которая является результатом таких факторов, как кристаллическая морфология. Когда имеют место влияния предпочтительной ориентации, интенсивности пиков изменяются, но положения характеристических пиков полиморфа являются неизменными. См., например, The United States Pharmacopeia 23,National Formulary 18, pages 1843-1844, 1995. Кроме того, в области кристаллографии также хорошо известно, что для любой данной кристаллической формы положения угловых пиков могут незначительно изменяться. Например, положения пиков могут сдвигаться вследствие изменения температуры, при которой анализировался образец, при замене образца или при наличии или отсутствии внутреннего стандарта. В данном случае изменчивость положения пика на 0,1 в 2 будет учитывать эти возможные изменения, не мешая точной идентификации кристаллической соли данного изобретения. Хорошо известным и широко применяемым методом исследования кристаллических форм является метод, называемый в литературе методом Финка ("Fink" method). В методе Финка для начального исследования используются четыре наиболее интенсивные линии и затем следующие четыре наиболее интенсивные линии. Обычно, согласно методу Финка, исходя из интенсивностей пиков, а также из положения пика, F-I может идентифицироваться наличием пиков при 6,80,1, 10,90,1, 14,20,1 и 16,60,1 в 2; когда картина получена с использованием медного источника излучения (=1,54056). Наличие F-I может быть дополнительно подтверждено пиками при 6,30,1, 7,20,1, 12,50,1 и 17,00,1 в 2; когда рентгенограмма получена с использованием медного источника излучения (=1,54056). Форма I FB относительно гигроскопичного F-I и безводного F-I. Были выполнены экстенсивные определения равновесной растворимости гигроскопичного и безводного F-I в различных водных средах при температуре окружающей среды. Дополнительно была определена равновесная растворимость формы I FB при температуре окружающей среды. Образцы анализировались высокоэффективной жидкостной хроматографией (ВЭЖХ) после 24 ч равновесного состояния в соответствующих растворителях. Результаты представлены в табл. 2. Таблица 2 Данные равновесной растворимости показали, что хотя F-I (гигроскопичный и безводный) и формаI FB имеют близкие растворимости в буфере с рН 2,2, F-I значительно менее растворим, чем форма I FB,в 0,01N HCl и имитаторе кишечной жидкости (SIF) (питание). Значительных различий между гигроскопичным и безводным F-I в любой испытываемой среде не наблюдалось. Результаты исследования растворимости подтверждают, что контроль объемного состава (гигро-4 007463 скопичных относительно безводных частиц) F-I, как API, не является определяющим с точки зрения биодоступности. Для подтверждения того, что возможность изменения гигроскопичности в образцах F-I не должна оказывать отрицательного влияния на биодоступность, были определены также характеристические скорости растворения гигроскопичного и безводного F-I. Для сравнения также была определена характеристическая скорость растворения формы I FB. Поскольку форма I FB растворялась слишком быстро (10% 100 мг компакта растворялись в течение 10 мин) и гигроскопичный и безводный F-I растворялись слишком медленно (отсутствие заметного растворения в течение 10 мин), точные характеристические скорости растворения получить было невозможно. Результаты определения характеристических скоростей растворения представлены в табл. 3. Таблица 3 Процент 100 мг компакта, растворенный в течение 10 мин Данные растворения и растворимости in vitro, обсужденные выше, подтверждают, что форма I FB должна предоставлять преимущества биодоступности in vivo относительно F-I. Для подтверждения этого предположения были определены параметры фармакокинетики в плазме формы I FB у женской особи гончей собаки при ее кормлении после разового перорального введения с помощью зонда 5 мг/кг формыI FB или F-I в эксперименте перекрестного планирования, 6 собак произвольно подразделили на две группы для приема разовых доз формы I FB с последующим приемом двумя неделями позже разовой дозы F-I или наоборот. В 1-й и 14-й дни 3 собаки получали форму I FB и 3 другие собаки получали F-I и через 0,5, 1, 2, 3, 4, 8, 12 и 24 ч после введения отбирали образцы крови. Концентрации формы I FB определяли жидкостной хроматографией в сочетании с масс-спектрометрией. Полученные концентрации затем использовали для определения фармакокинетических параметров, представленных в табл. 4. Таблица 4FB, день 14 - прием F-I. Неожиданно на основании данных растворимости и растворения in vitro плазменная экспозиция дляF-I из расчета на площадь под кривой зависимости концентрации от времени (ППК) для периода времени от 0 до 24 ч и от 0 до бесконечности была значительно более высокой, чем экспозиция, полученная для формы I FB. Скорость абсорбции, как оказалось, не изменялась, когда время достижения Cmax(tmax) находилось в интервале от 1 до 2 ч как для формы I FB, так и для F-I. Повышенная экспозиция для F-I была наиболее вероятно обусловлена повышенной биодоступностью, поскольку клиренс, как оказалось, не изменял данных аналогий в значениях кажущегося периода полувыведения. Синтез Получение FB. Стадия 1 - смесь 2-пиколилхлорида гидрохлорида (7,0 г, 42,7 ммоль), 4-пиперидона моногидрата гидрохлорида (6,88 г, 44,8 ммоль), порошкообразного карбоната натрия (18,3 г, 173 ммоль) и ацетонитрила (70 мл) перемешивают в течение 45 мин при температуре окружающей среды, 45 мин при 40 С, 45 мин при 50 С, 45 мин при 60 С и затем нагревают до 70 С с энергичным перемешиванием. Реакцию контролируют ВЭЖХ (колонка Zorbax RX-C8 25 см, ацетонитрил/Н 3 РO4 буфер при рН 3,0, =250 нм) по исчезновению пиколилхлорида. По завершении реакции смеси дают охладиться до комнатной температуры, затем смесь фильтруют для удаления нерастворимых твердых веществ, затем осадок на фильтре промывают ацетонитрилом (225 мл). Фильтрат концентрируют до небольшого объема (30 мл) и растворитель заменяют 41 мл этилацетата. Раствор быстро перемешивают и нагревают до 55 С, затем обра-5 007463 батывают в течение 30 мин раствором камфорсульфоновой кислоты (9,91 г, 42,67 ммоль) в этилацетате(77 мл). Полученной суспензии дают охладиться до комнатной температуры и затем перемешивают в течение 3 ч. Осадок фильтруют, промывают этилацетатом (230 мл) и сушат в вакууме при 45 С, получая 15,6 г (87%) соли камфорсульфоновой кислоты. Стадия 2 - в 3-горлую колбу объемом 1 л, снабженную рубашкой, в атмосфере N2 добавляют продукт стадии 1 (1,0 экв., 33,3 г), 2-(2,2-диметоксиэтил)анилин (Fukuyama et al., Tet. Lett., 39 (l-2):71-74,1998; 1,0 экв., 14,3 г) и пропионовую кислоту (115 мл). Реакционную смесь перемешивают при 20-24 С до растворения (15-30 мин). Смесь охлаждают до температуры в интервале от -10 до -15 С, затем добавляют 1,0 М NaBH(OPr)3 в тетрагидрофуране (115 мл) в течение по меньшей мере 2 ч в атмосфере N2,поддерживая температуру внутри колбы -10 С. Завершение реакции восстановительного аминирования подтверждают ВЭЖХ (колонка Zorbax C-8, рН 3,0 (1,5 мл триэтиламина/1,5 мл Н 3 РO4/1 л Н 2O. Исходный градиент: 80% воды/20% ацетонитрила. Конечный градиент (45 мин): 20% воды/80% ацетонитрила). После подтверждения завершения реакции добавляют этилацетат (200 мл) и температуру реакции доводят до 0 С. Значение рН раствора доводят до 10,0 осторожным добавлением 25% NaOH (315 г) и реакционной смеси дают возможность нагреться до 47-52 С. Реакционную смесь перемешивают в течение 30-60 мин при 47-52 С. Перемешивание прекращают и реакционной смеси дают возможность расслаиваться в течение по меньшей мере 15 мин при 47-52 С. Нижний водный слой удаляют и органический слой промывают водным 20% раствором NaCl (150 мл). Смесь перемешивают в течение 30 мин при 47-52 С, затем перемешивание прекращают и смеси дают возможность расслаиваться в течение 15 мин. Нижний водный слой удаляют и с помощью вакуумной отгонки объем реакционной смеси снижают до 65-85 мл. К реакционной смеси снова добавляют этилацетат (100 мл) и смесь охлаждают до 23-25 С. К смеси добавляют трифторуксусную кислоту (30 мл) в течение по меньшей мере 30 мин. Смесь нагревают до 2931 С и реакцию проводят до тех пор, пока ВЭЖХ анализ не покажет, что исходный аддукт аминирования присутствует в количестве менее 1,0%. После подтверждения завершения реакции к смеси добавляют этилацетат (175 мл) и воду (30 мл) и осторожно доводят значение рН до 9,0 добавлением 25% NaOH (74 г) с нагреванием до 40-45 С. Полученную двухфазную смесь перемешивают в течение по меньшей мере 1 ч при 45-50 С и дают возможность снизиться до 8,60. Перемешивание прекращают и в течение по меньшей мере 15 мин при 45-50 С смеси дают возможность расслаиваться. Нижний водный слой удаляют и органический слой промывают водным 20% NaCl (125 мл) с перемешиванием при 45-50 С. Перемешивание продолжают в течение 30 мин, затем в течение 15 мин смеси дают осаждаться при 45-50 С,водный слой удаляют и реакционную смесь концентрируют до объема 100-150 мл отгонкой в вакууме. Добавляют изопропанол (400 мл) и смесь снова концентрируют до 200 мл, затем добавляют дополнительную порцию изопропанола (200 мл). Смесь концентрируют до конечного объема 200 мл отгонкой в вакууме и полученную суспензию оставляют на 3 ч при 43-45 С, затем охлаждают в течение 3-4 ч до-5 С. Продукт отфильтровывают при -5 С и промывают предварительно охлажденным (0 С) изопропанолом (240 мл). Продукт восстановительного аминирования сушат при 50-60 С при пониженном давлении. Стадия 3 - продукт стадии 2 (5,00 г, 17,2 ммоль) суспендируют в сухом трет-бутилметилэфире (70 мл, 14 об.) в N2 при 23 С. В полученную смесь при температуре окружающей среды одной порцией добавляют сухой ацетонитрил (20 мл, 4 об.) и полученный мутный раствор нагревают до 40 С. В полученную смесь по каплям в течение 30 мин добавляют раствор 2,0 М НСl в ацетонитриле (8,5 мл, 17,0 ммоль,0,99 экв.), поддерживая предварительно установленную в рубашке температуру на уровне 40 С. Полученную суспензию нагревают до 50 С и перемешивают в течение 1 ч. Смесь охлаждают до -10 С в течение 2-3 ч. По каплям в течение 3-5 мин добавляют оксалилхлорид (2,30 мл, 26,4 ммоль, 1,50 экв.), поддерживая температуру -5 С. Суспензию нагревают до 0 С и перемешивают в течение 1-2 ч до завершения реакции, что устанавливают ВЭЖХ. По каплям в течение 3-5 мин добавляют метанол (10 мл, 2 об.),поддерживая температуру в реакторе 10 С. Полученной суспензии дают возможность медленно нагреться до 23 С в течение 15-30 мин и затем перемешивают ее в течение 1-2 ч до завершения реакции. Суспензию охлаждают до 0-5 С, затем по каплям для доведения рН смеси до 7,8 добавляют 2N КОН (38 мл, 76 ммоль, 4,4 экв.), поддерживая температуру в реакторе 10 С. После корректировки рН погашенную реакционную смесь перемешивают при 10 С в течение 15-20 мин, затем удаляют нижний водный слой. Нижний водный слой снова экстрагируют трет-бутилметилэфиром (20 мл). Объединенные органические слои (100 мл) промывают водным 20% раствором NaCl (50 мл) в течение 20-30 мин при 10 С. Смесь оставляют для расслаивания на 15 мин, затем удаляют слой раствора соли. В органический слой добавляют Na2SO4 (15 г, безводный), смесь нагревают до 23 С, затем перемешивают в течение 1-12 ч. Реакционную смесь фильтруют, затем фильтрат концентрируют в вакууме. Остаток снова растворяют в этилацетате (100 мл), затем снова концентрируют. Добавляют этилацетат (35 мл) и CH3CN (1 мл), смесь нагревают до 45-50 С для растворения, затем охлаждают до 40 С в течение 1 ч. В неочищенную смесь необязательно вносят затравку (30 мг), затем охлаждают смесь до 23 С в течение 2 ч с получением суспензионных форм. По каплям в течение 20-30 мин к суспензии добавляют гептан (80 мл), затем смесь охлаждают до 0 С в течение 1-2 ч. Суспензию перемешивают в течение дополнительных 1-2 ч при 0 С,-6 007463 затем фильтруют. Осадок на фильтре промывают охлажденной смесью 2:1/гептан:этилацетат (15 мл),затем гептаном комнатной температуры (15 мл). Осадок на фильтре сушат в вакуумной печи при 50 С до постоянной массы, получая 5,60 г 1-(1-[(пиридин-2-ил)метил]пиперидин-4-ил)-3-(метоксикарбонилкарбонил)индола (87%). Стадия 4 - в 3-горлую колбу, снабженную капельной воронкой и устройством ввода азота, загружают продукт со стадии 3 (10,0 г (1,0 экв., 26,5 ммоль) и 1-метил-3-(аминокарбонилметил)индол (Faul etFicher0,03%, 72 мл, 7,2 об.). Суспензию охлаждают до температуры от -5 до -10 С на бане со смесью лед/ацетон. Добавляют трет-бутоксид калия (20% в тетрагидрофуране, 1,6 М, 36,4 мл, 2,2 экв., 58,3 ммоль) в течение 10-30 мин, поддерживая температуру реакции на уровне от -10 до 5 С. Реакционную смесь нагревают до 40-45 С и перемешивают в течение 1 ч для получения суспензии. Смесь охлаждают до 0-10 С на бане со смесью лед/вода и затем быстро добавляют воду (74 мл, предварительно охлажденную до 0-10 С). Реакционную смесь, незначительно нагретую до 15 С за счет экзотермической реакции,снова охлаждают до 0-10 С и значение рН доводят до 12,7-12,9 смесью концентрированной НСl (5,2 мл) и воды (15 мл) (требуется примерно 2/3 этой смеси). Значение рН смеси доводят до 7,3-7,8 оставшейся смесью HCl/вода в течение 20 мин, затем полученную смесь перемешивают в течение 30 мин при 010 С. Медленно в течение 20-30 мин добавляют воду (60 мл) при 0-10 С и реакционную смесь перемешивают в течение 1-2 ч. Смесь фильтруют на напорном фильтре, осадок промывают предварительно охлажденной смесью тетрагидрофурана (20 мл) и воды (60 мл) и сушат в течение ночи при 50 С в вакууме,получая FB. Пример 1. В 3-горлую колбу, снабженную нагревательным кожухом, холодильником и приемником дистиллята, загружают FB (59,0 г, 114,4 моль), 2-бутанол (949 мл, 16,1 об.), деионизированную воду (621,4 мл,10,5 об.) и НСl (пищевой сорт: 12,24 мл, 14,3 г, 0,21 об., 1,05 экв.). Реакционную смесь нагревают до температуры образования флегмы и половину растворителя удаляют отгонкой. Медленно в течение 2 ч добавляют 2-бутанол (27 об.), поддерживая постоянный уровень растворителя в реакционной колбе. Реакционную смесь охлаждают до комнатной температуры в течение 60 мин, затем охлаждают до 0-5 С и перемешивают в течение 1-2 ч. Продукт фильтруют и осадок на фильтре промывают 2 об. 2-бутанола и сушат в течение ночи при 50 С в вакууме, получая F-I. Элементный анализ: Вычислено дляXRD рентгенограмму получают на рентгеновском порошковом дифрактометре Siemens D5000,снабженном CuK источником (=1,54056 ) и детектором твердого состояния Kevex, работающем при 50 кВ и 40 мА с 1 мм дивергенцией и щелью приема и 0,1 мм щелью детектора. Каждый образец сканируют в интервале 2 от 4 до 35 с размером шага 0,02 и максимальной скоростью сканирования 3 с/шаг.XRD рентгенограмма для соединения, полученного в примере 1, описана в табл. 1 и представлена на чертеже. Фармацевтический препарат. Перед введением соль данного изобретения предпочтительно представляют в виде единичной дозированной формы. Таким образом, еще одним воплощением данного изобретения является фармацевтическая композиция, содержащая соль данного изобретения и фармацевтический носитель. Используемый в описании термин фармацевтический означает, по существу, безвредный для пациента, принимающего лекарственное средство. Фармацевтические композиции данного изобретения изготавливают известными способами с использованием хорошо известных и доступных ингредиентов. Для получения препаратов данного изобретения активный ингредиент (например, F-I) обычно будет смешиваться с носителем, или разбавляться носителем, или вводиться в носитель, который может иметь форму капсулы, саше, бумажного или другого контейнера. Когда носитель служит в качестве разбавителя, он может быть твердым, полутвердым или жидким веществом, которое выступает в качестве растворителя, эксципиента или среды для активного ингредиента. Таким образом, композиции могут быть в форме таблеток, пилюль, порошков, лепешек,саше, крахмальной облатки, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозоля (в виде твердого вещества или в жидкой среде), мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильных расфасованных порошков. Некоторые примеры подходящих носителей, эксципиентов и разбавителей включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, аравийскую камедь, фосфат кальция, альгинаты, трагакант,желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, водный сироп, метилцеллюлозу, метил- и пропилгидроксибензоаты, тальк, стеарат магния и минеральное масло. Препараты могут дополнительно включать смазывающие агенты, смачивающие агенты, эмульгаторы и суспендирующие агенты, консерванты, подсластители или флаворанты. Композиции данного изобретения могут быть изготовлены с тем, чтобы обеспечить быстрое, пролонгированное или замедленное высвобождение активного ингредиента после введения пациенту. Представленные выше капсулы получают способом водного гранулирования, как описано ниже. Лактозу, часть кросповидона и активный ингредиент (F-I) добавляют в гранулятор и смешивают сухим способом в течение подходящего периода времени для получения порошков с однородными по размеру частицами. Грануляционный раствор, состоящий из повидона и полисорбата 80 в очищенной воде, распыляют при постоянной скорости на порошки при перемешивании их в установленных условиях. При достижении подходящей конечной точки гранулирования гранулятор останавливают и гранулированный продукт выгружают. Гранулированный продукт просеивают влажным просеиванием через подходящее сито для разрушения крупных агломератов, распределяют на поддоны, выложенные бумагой, и сушат в конвекционной печи до тех пор, пока содержание влаги не снижается до подходящего уровня. Размер гранул гранулированного продукта снижается до нужного интервала значений пропусканием через соизмельчитель или другой подходящий аппарат. Полученные измельченные порошки собирают, переносят в аппарат смешения и смешивают с заданным количеством стеарата магния и дополнительным количеством кросповидона до получения однородного распределения. После этого конечным порошкообразным продуктом заполняют твердые желатиновые капсулы вручную или на подходящей модели автоматизированного аппарата для наполнения капсул. После операции наполнения конечные капсулы визуально проверяют на наличие внешних дефектов. Для улучшения внешнего вида фармацевтического продукта капсулы могут быть обработаны для физического устранения порошка и отполированы вручную или с помощью автоматизированных процессов. Демонстрация функции. Соль данного изобретения является ингибитором ангиогенеза, индуцированного васкулярным эндотелиальным фактором роста (VEGF). По меньшей мере две системы испытания демонстрируют эти фармакологические активности: 1) F-I является мощным ингибитором VEGF-стимулированной пролиферации HUVEC клеток в культуре при 72-часовой экспозиции соединением; 2) F-I является высокоэффективным ингибитором VEGF-индуцированного неоангиогенеза в корнеальном микроочаге у крыс при пероральном введении животным в течение 10 дней. Данные системы испытания более полно описаны вWO 02/02116. Таким образом, соль данного изобретения эффективна при лечении рака и ингибировании роста опухоли. Применения. В качестве ингибиторов роста опухолей соль данного изобретения полезна для лечения рака мочевого пузыря, головного мозга, молочной железы, шейки матки, толстой кишки, пищевода, почки, головы-8 007463 и шеи, печени, легкого, яичника, поджелудочной железы, простаты и желудка. Соль данного изобретения также полезна для лечения сарком мягких тканей и остеосарком, а также для лечения болезни Ходжкина и неходжкинской лимфомы или развития злокачественной опухоли кровеносной системы (лейкемия). Предпочтительные способы применения соли данного изобретения относятся к ее применению для лечения рака мочевого пузыря, почки, головного мозга, молочной железы, толстой кишки, печени, легкого (немелкоклеточного рака), яичника и желудка и к ее применению для лечения неходжкинской лимфомы (например, диффузной лимфомы крупных В клеток и лимфомы клеток коры головного мозга) или развития злокачественных опухолей кровеносной системы (лейкемии). Еще более предпочтительные способы применения соли данного изобретения относятся к ее применению для лечения раковых заболеваний мозга, толстой кишки, легкого (немелкоклеточного рака),также к ее применению для лечения неходжкинской лимфомы, (-клеточной лимфомы и лейкемии, связанной с -клетками. Доза. Специалисту в данной области будет понятно, что количество соли данного изобретения, подлежащее введению в соответствии с данным изобретением, т.е. терапевтически эффективное количество,представляет собой количество, достаточное для получения антинеопластического действия, для индуцирования апоптоза или гибели клеток, и/или поддержания антиангиогенного действия. Обычно, количество соли данного изобретения, предназначенное для введения, определяется в каждом конкретном случае лечащим врачом. В качестве определяющих среди прочих факторов при назначении подходящей дозы будут учитываться степень развития и тип неоплазии, время введения относительно других терапевтических средств (если они имеют место), а также масса тела и возраст пациента. Обычно эффективная минимальная суточная доза соли данного изобретения, например, F-I, будет превышать примерно 200 мг (обычно 400 мг, например, 500 мг). Обычно максимальная эффективная суточная доза F-I не будет превышать примерно 700 мг. Однако в случае глиобластом (опухоли мозга) максимальная суточная доза F-I могла бы составлять 1400 мг. Точная доза для лечения глиобластомы может определяться в соответствии со стандартной практикой в области медицины способом титрования дозы реципиентом, т.е. введением пациенту первоначально низкой дозы соединения, например 200 или 400 мг, и постепенным повышением дозы до получения желаемого терапевтического эффекта. Способ введения. Соль данного изобретения может вводиться различными способами, включая пероральный, ректальный, трансдермальный, подкожный, местный, внутривенный, внутримышечный или интраназальный способы. Предпочтительным является пероральный способ введения. Комбинационная терапия. Соль данного изобретения может использоваться в сочетании с традиционными противоопухолевыми терапевтическими средствами для лечения млекопитающих, в частности людей, страдающих неоплазией. Методики обычного противоопухолевого терапевтического лечения, включая химиотерапевтическое лечение с использованием противоопухолевых средств и терапевтического облучения, являются легкодоступными и широко используемыми в медицине, например, см. Harrison's PRINCIPLES OFINTERNAL MEDICINE 11th edition, McGraw-Hill Book Company. В частности, кристаллическая соль данного изобретения может применяться для повышения противоопухолевого действия противоопухолевого средства. Для комбинационной терапии согласно данному изобретению подходит широкий спектр доступных противоопухолевых средств. Противоопухолевые средства, предназначенные для комбинационной терапии согласно данному изобретению, включают, но без ограничения только ими, алкилирующие средства, включая бусульфан,хлорамбуцил, циклофосфамид, ифосфамид, мелфалан, азотистый иприт, стрептозоцин, тиотепу, урацилазотистый иприт и триэтиленмеламин, темозоломид; антибиотики и растительные алкалоиды, включая актиномицин-D, блеомицин, криптофицины, даунорубицин, доксорубицин, идарубицин, иринотекан, Lаспарагиназу, митомицин-С, митрамицин, навелбин, паклитаксел, доцетаксел, топотекан, винбластин,винкристин и VP-16; гормоны и стероиды, включая аминоглютетимид, анастрозол, бикалутамид, DES,эстрамустин, этинилэстрадиол, флутамид, флуоксиместерон, госерелин, гидроксипрогестерон, летрозол,леупролид, медроксипрогестерон ацетат, мегестрол ацетат, метилпреднизолон, метилтестостерон, митотан, нилутамид, преднизолон, тамоксифен, тестостерон и триамикнолон; синтетические лекарственные средства, включая полностью транс-ретиноевую кислоту, BCNU (кармустин), карбоплатин (параплатин),CCNU (ломустин), цис-диаминодихлорплатину (цисплатин), дакарбазин, гексаметилмеламин, гидроксимочевину, левамизол, митоксантрон, оксалиплатин, прокарбазин; антиметаболиты, включая хлордеоксиаденозин, цитозинарабинозид, 2'-деоксикоформицин, флударабин фосфат, 5-флуороурацил, 5-FUDR,гемцитабин, 6-меркаптопурин, метотрексат, пеметрексед и тиогуанин; моноклональные антитела, включая ритуксимаб и трастузумаб; десенсибилизирующие соединения, включая ISIS 3521; и биологические лекарственные средства, включая альфа-интерферон, BCG, G-CSF, GM-CSF и интерлейкин-2 и т.п. Данные противоопухолевые средства проявляют цитотоксическое или противоопухолевое действие в раз-9 007463 личных специфических неопластических условиях (см. WO 02/02094). В предпочтительном воплощении данного изобретения одно или несколько противоопухолевых средств выбраны из группы, включающей BCNU, циклофосфамид, доксорубицин, преднизон или дексаметазон, винкристин, гемцитабин, цисплатин, 5-фторурацил, капецитибин, СРТ-11, карбоплатин, паклитаксел, доцетаксел, ритуксимаб и трастузумаб. Кристаллическая соль данного изобретения также может использоваться в сочетании с лучевой терапией. Обычно облучение используется для лечения участка солидной опухоли непосредственно или при введении брахитерапевтических имплантантов. Терапевтическое облучение, предназначенное для комбинационной терапии в соответствии с данным изобретением, представляет собой виды облучения, которые используются при лечении рака и включают, но без ограничения, рентгеновское излучение, гамма-излучение, излучение электронов высокой энергии и излучение с высоким переносом энергии (LET), таких как протоны, нейтроны и альфачастицы. Ионизирующее излучение применяется с помощью методов, хорошо известных специалисту в данной области. Например, рентгеновские лучи и гамма-лучи применяются с помощью внешних и/или внутритканевых средств из линейных ускорителей или радиоактивных источников. Электроны высокой энергии могут быть получены с помощью линейных ускорителей. Излучение с высоким переносом энергии также может применяться из радиоактивных источников, имплантированных внутритканево. Фраза в сочетании с означает, что кристаллическая соль данного изобретения вводится незадолго до, сразу после, одновременно или в любом сочетании до, после или одновременно, с такими другими противоопухолевыми терапевтическими средствами. Соль данного изобретения может вводиться в сочетании с более чем одним противоопухолевым терапевтическим средством. В предпочтительном воплощении соль данного изобретения вводится в интервале от 2 недель до 1 дня перед любой химиотерапией или в интервале от 2 недель до 1 дня перед любой лучевой терапией. В другом предпочтительном воплощении соль данного изобретения может вводиться в процессе противоопухолевой химиотерапии и лучевой терапии. При введении после такой химиотерапии или лучевой терапии соль данного изобретения предпочтительно дается в интервале от 1 до 14 дней после первичного лечения. Соль данного изобретения также может вводиться постоянно или полупостоянным образом в течение периода от примерно 2 недель до примерно 5 лет. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Кристаллический 2,5-дион-3-(1-метил-1 Н-индол-3-ил)-4-[1-(пиридин-2-илметил)пиперидин-4 ил]-1 Н-индол-3-ил-1 Н-пирролмоногидрохлорид, его гидрат или их смесь. 2. Кристаллический 2,5-дион-3-(1-метил-1 Н-индол-3-ил)-4-[1-(пиридин-2-илметил)пиперидин-4 ил]-1 Н-индол-3-ил-1 Н-пирролмоногидрохлорид, его гидрат или их смесь, имеющие рентгенограмму,которая содержит следующие пики: 6,80,1, 10,90,1, 14,20,1 и 16,60,1 в 2; когда рентгенограмма получена при использовании медного источника излучения (CuK; =1,54056 ). 3. Кристаллический моногидрохлорид по п.2, имеющий рентгенограмму, которая дополнительно содержит следующие пики: 6,30,1, 7,20,1, 12,50,1 и 17,00,1 в 2. 4. Фармацевтическая композиция, содержащая соль по любому из пп.1-3 и фармацевтический носитель. 5. Применение соединения по любому из пп.1-3 в качестве активного вещества в способе лечения человека терапией. 6. Применение по п.5 при лечении неходжкинской лимфомы. 7. Применение по п.5 при лечении глиобластомы. 8. Применение по п.5 при лечении немелкоклеточного рака легких. 9. Применение соединения по любому из пп.1-3 в качестве активного вещества для производства лекарственного средства для лечения неходжкинской лимфомы. 10. Применение соединения по любому из пп.1-3 в качестве активного вещества для производства лекарственного средства для лечения глиобластомы. 11. Применение соединения по любому из пп.1-3 в качестве активного вещества для производства лекарственного средства для лечения немелкоклеточного рака легких.
МПК / Метки
МПК: C07D 401/14, A61K 31/4468
Метки: кристаллический, 5-дион-3-(1-метил-1н-индол-3-ил, илметил)пиперидин-4-ил, 4-[1-(пиридин, 1н-индол-3-ил-1н-пирролмоногидрохлорид
Код ссылки
<a href="https://eas.patents.su/12-7463-kristallicheskijj-2-5-dion-3-1-metil-1n-indol-3-il-4-1-piridin-2-ilmetilpiperidin-4-il-1n-indol-3-il-1n-pirrolmonogidrohlorid.html" rel="bookmark" title="База патентов Евразийского Союза">Кристаллический 2, 5-дион-3-(1-метил-1н-индол-3-ил) -4-[1-(пиридин -2- илметил)пиперидин-4-ил] -1н-индол-3-ил-1н-пирролмоногидрохлорид</a>
3 -{(3r,4r)-4-метил-3-[метил-(7h-пиpроло[2,3-d] пиримидин-4-ил)-амино] пиперидин-1-ил}-3-оксопропионитрил и его фармацевтически приемлемые соли

Номер патента: 7251
Опубликовано: 25.08.2006
Авторы: Флэнэган Марк Эдвард, Кехер Кристиан, Уилкокс Гленн Эрнест, Врис Тон, Манчхоф Майкл Джон
МПК: A61K 31/505, C07D 487/04, A61P 37/06...
Метки: пиперидин-1-ил}-3-оксопропионитрил, соли, пиримидин-4-ил)-амино, приемлемые, 3r,4r)-4-метил-3-[метил-(7h-пиpроло[2,3-d, фармацевтически
Формула / Реферат:
Соединение 3-{(3R,4R)-4-метил-3-[метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино] пиперидин-1-ил}-3-оксопропионитрил или его фармацевтически приемлемая соль.
Композиция, включающая 5-[4-[2-(n-метил-n-(2-пиридил)амино)этокси]бензил]тиазолидин-2,4-дион

Номер патента: 2384
Опубликовано: 25.04.2002
Авторы: Грэнетт Джеффри Роджер, Росс Хамиш, Прайс Робин, Пэйтел Джай
МПК: A61P 3/00, A61K 31/44
Метки: композиция, 5-[4-[2-(n-метил-n-(2-пиридил)амино)этокси]бензил]тиазолидин-2,4-дион, включающая
Формула / Реферат:
1. Фармацевтическая композиция, включающая 5-[4-[2-(N-метил-N-(2-пиридил)амино)этокси]бензил]тиазолидин-2,4-дион (обозначаемый далее как соединение (I)), отличающаяся тем, что композиция включает от 2 до 12 мг соединения (I) в фармацевтически приемлемой форме и, необязательно, фармацевтически приемлемый носитель для него. 2. Композиция по п.1, которая включает от 2 до 4 мг соединения (I) в фармацевтически приемлемой форме. 3. Композиция по п.1,...
Производные n-[фенил(пиперидин-2-ил)метил]бензамида и их применение в терапии

Номер патента: 7225
Опубликовано: 25.08.2006
Авторы: Эстенн-Буту Женевьев, Севрен Мирей, Мага Паскаль, Марабу Бенуа, Вероник Коринн, Роже Пьер, Медеско Флоранс, Даргазанли Жихад
МПК: A61P 25/00, A61K 31/445, C07D 211/26...
Метки: n-[фенил(пиперидин-2-ил)метил]бензамида, применение, производные, терапии
Формула / Реферат:
1. Соединение в форме чистого оптического изомера (1R, 2R) или (1S, 2S) либо в форме трео-диастереоизомера, соответствующее общей формуле (I) где А представляет собой или группу общей формулы N-R1, где R1 представляет собой или атом водорода, или линейную или разветвленную (С1-С7)алкильную группу, возможно замещенную одним или более чем одним атомом фтора, или (С4-С7)циклоалкильную группу, или (С3-С7)циклоалкил(С1-С3)алкильную группу, или...
Способ получения [is-[1a,2b,3b,4a(s*)]]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3н-имидазо[4,5-b]пиридин-3-ил]-n-этил-2,3-дигидроксициклопентанкарбоксамида.

Номер патента: 1989
Опубликовано: 22.10.2001
Авторы: Ванасс Бенуа Дж., Цуей Чинг Т., Шах Харшавадан К., Леон Патрик, О'брайен Майкл К, Томпсон Майкл Д., Паунер Тори Х., Гарсиа Эрве, Вальтер Фрэнсис Л., Рейлли Лоренс В.
МПК: C07D 409/12
Метки: is-[1a,2b,3b,4a(s*)]]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3н-имидазо[4,5-b]пиридин-3-ил]-n-этил-2,3-дигидроксициклопентанкарбоксамида, способ, получения
Формула / Реферат:
1. Способ получения [1S-[1a,2b,3b,4a(S*)]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3Н-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамида (соединение (I)), включающий взаимодействие [1S-[1a,2b,3b,4a(S*)]]-4-[[3-амино-4-[[1-[(3-хлор-2-тиенил)метил]пропил]амино]-2-пиридинил]амино]-N-этил-2,3-дигидроксициклопентанкарбоксамида (соединение (IX)) со сложным эфиром ортоформиата, ацетатом формамидина или диметилацеталем...
Кристаллы, содержащие соль n-[2-(диэтиламино)этил]-5-[(5-фторо-2-оксо- 1,2-дигидро-3h-индол-3-илиден)метил]-2,4 -диметил-1н-пиррол-3- карбоксамида с яблочной кислотой, способы их получения и их композиции

Номер патента: 6445
Опубликовано: 29.12.2005
Авторы: Малоуни Марк Т., Холи Майкл, Флек Томас Дж., Прескотт Стивен П.
МПК: A61P 35/00, C07D 403/06, A61K 31/404...
Метки: карбоксамида, кристаллы, композиции, способы, кислотой, содержащие, n-[2-(диэтиламино)этил]-5-[(5-фторо-2-оксо, соль, 1,2-дигидро-3h-индол-3-илиден)метил]-2,4, яблочной, диметил-1н-пиррол-3, получения
Формула / Реферат:
1. Безводный кристалл, содержащий соль N-[2-(диэтиламино)этил]-5-[(5-фторо-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоксамида, имеющего структуру с яблочной кислотой. 2. Кристалл по п.1, где яблочной кислотой является L-яблочная кислота. 3. Безводный кристалл, содержащий соль N-[2-(диэтиламино)этил]-5-[(5-фторо-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоксамида с яблочной кислотой,...
Предыдущий патент: Способ радикальной регулируемой полимеризации акриловой кислоты и ее солей, полученные полимеры и их применение
Следующий патент: Антагонисты метаботропных рецепторов глутамата
Случайный патент: Дизамещенные бициклические гетероциклы, их получение и применение в качестве лекарственных средств