1,4-двузамещенные производные пиперазина в качестве уроселективных блокаторов альфа1-адренорецепторов
Номер патента: 6941
Опубликовано: 30.06.2006
Авторы: Гупта Джанг Бахадур, Чугн Анита, Хегде Лаксминарайан Г., Ананд Нитья, Синха Неелима, Джайн Санджай







Формула / Реферат
1. Соединения структурной формулы I

а также их фармацевтически приемлемые соли, амиды, энантиомеры, диастереомеры, N-оксиды, пролекарства, метаболиты или их полиморфы, где А - это прямая или разветвленная С1-С4-алкильная цепь; R - циннамил, бензил, замещенный бензил, моно- или двузамещенная фенильная группа с заместителями, независимо выбранными из группы, состоящей из гидрокси, нитро, трифторалкоксигруппы и (дигалодифенил) метила.
2. Соединения, выбранные из группы, состоящей из
2-[3-{4-(2-метоксифенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 01);
2-[3-{4-(4-фторфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1H-изоиндол-1,3(2Н)-диона (соединение 04);
2-[3-{4-(3-трифторметилфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 05);
2-[3-{4-(2-фторфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 06);
2-[3-{4-(3,4-диметилфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 07);
2-[3-{4-(2-метокси-5-фторфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 08);
2-[3-{4-(2-этилфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 09);
2-[3-{4-(2,4-дифторфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1H-изоиндол-1,3(2Н)-дион (соединение 10);
2-[3-{4-(2-этоксифенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1H-изоиндол-1,3(2Н)-диона (соединение 11);
2-[3-{4-(2-метил-5-хлорфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 12);
2-[3-{4-(бензил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2H)-диона (соединение 14);
2-[3-{4-(циннамил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 15);
2-[3-{4-(4-нитрофенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 16);
2-[3-{4-(3-хлор-4-метилфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 17);
2-[3-{4-(4-фтор-2-метоксифенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 18);
2-[3-{4-(бис-4-фторфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 19);
2-[3-{4-(2,4-дихлорфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 20);
2-[3-{4-(2,4-диметоксифенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона(соединение 21);
2-[3-{4-(2,6-диметилфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2H)-диона (соединение 22);
2-[3-{4-(2-изопропоксифенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 23);
2-[3-{4-(2-пропоксифенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1H-изоиндол-1,3(2H)-диона (соединение 24);
2-[3-{4-(2-n-гексилоксифенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 25);
2-[3-{4-(2,5-диметоксифенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 26);
2-[3-{4-(4-трет-бутилфенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1H-изоиндол-1,3(2Н)-диона (соединение 27);
2-[3-{4-(2-метокси-6-гидроксифенил)пиперазин-1-ил}пропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 28);
2-[3-{4-(2-метоксифенил)пиперазин-1-ил}-3-метилпропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 29);
2-[3-{4-(2-метоксифенил)пиперазин-1-ил}-2-метилпропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 30);
2-[3-{4-(2-этоксифенил)пиперазин-1-ил}-3-метилпропил]-3а,4,7,7а-тетрагидро-1Н-изоиндол-1,3(2Н)-диона (соединение 31).
3. Способ оказания селективного противодействия a 1-адренергическим рецепторам у млекопитающего, включающий введение указанному млекопитающему соединения структурной формулы I

его фармацевтически приемлемых солей, амидов энантиомеров, диастереомеров, N-оксидов, пролекарств, метаболитов или их полиморфов, где А - это прямая или разветвленная С1-С4алкильная цепь; R - циннамил, бензил, замещенный бензил, фенил, моно- или двузамещенная фенильная группа с заместителями, независимо выбранными из группы, состоящей из галогена, гидрокси, C1-С6алкила, C1-С6алкокси, трифторметила, нитро и трифторалкоксигруппы и (дигалодифенил) метила.
4. Способ лечения доброкачественной гиперплазии простаты у млекопитающего, включающий введение указанному млекопитающему соединения структурной формулы I

его фармацевтически приемлемых солей, амидов энантиомеров, диастереомеров, N-оксидов, пролекарств, метаболитов или их полиморфов, где А - это прямая или разветвленная С1-С4-алкильная цепь; R - циннамил, бензил, замещенный бензил, фенил, моно- или двузамещенная фенильная группа с заместителями, независимо выбранными из группы, состоящей из галогена, гидрокси, C1-С6алкила, C1-С6алкокси, трифторметила, нитро и трифторалкоксигруппы и (дигалодифенил) метила.
5. Фармацевтическая композиция, содержащая соединение по п.1 или 3 и фармацевтически приемлемый носитель.
6. Способ селективного противодействия a 1-адренергическим рецепторам у млекопитающего, включающий введение указанному млекопитающему фармацевтической композиции по п.5.
7. Способ лечения доброкачественной гиперплазии простаты у млекопитающего, включающий введение указанному млекопитающему фармацевтической композиции по п.6.
8. Способ получения соединений формулы I

или их фармацевтически приемлемых солей, амидов энантиомеров, диастереомеров, N-оксидов, пролекарств, метаболитов или их полиморфов, где А- это прямая или разветвленная С1-С4-алкильная цепь; R - циннамил, бензил, замещенный бензил, моно- или двузамещенная фенильная группа с заместителями, независимо выбранными из группы, состоящей из гидрокси, нитро, трифторалкоксигруппы и (дигалодифенил) метила, включающий взаимодействие цис-1,2,3,6-тетрагидрофталевого ангидрида формулы II

с 1-амино-4-замещенным пиперазинил алканом формулы III

где А и R - те же, что указано выше.
9. Способ получения соединений формулы I

или их фармацевтически приемлемых солей, амидов энантиомеров, диастереомеров, N-оксидов, пролекарств, метаболитов или их полиморфов, где А - это прямая или разветвленная С1-С4-алкильная цепь; R - циннамил, бензил, замещенный бензил, фенил, моно- или двузамещенная фенильная группа с заместителями, независимо выбранными из группы, состоящей из галогена, гидрокси, C1-С6 алкила, C1-С6 алкокси, трифторметила, нитро, трифторалкоксигруппы и (дигалодифенил) метила, включающий взаимодействие 1-(w-галоалкил)цис-3а,4,7,7а-тетрагидрофталамида формулы IV

где А - то же, что указано выше, с 1-замещенным пиперазином формулы V

где R - тo же, что указано выше.
Текст
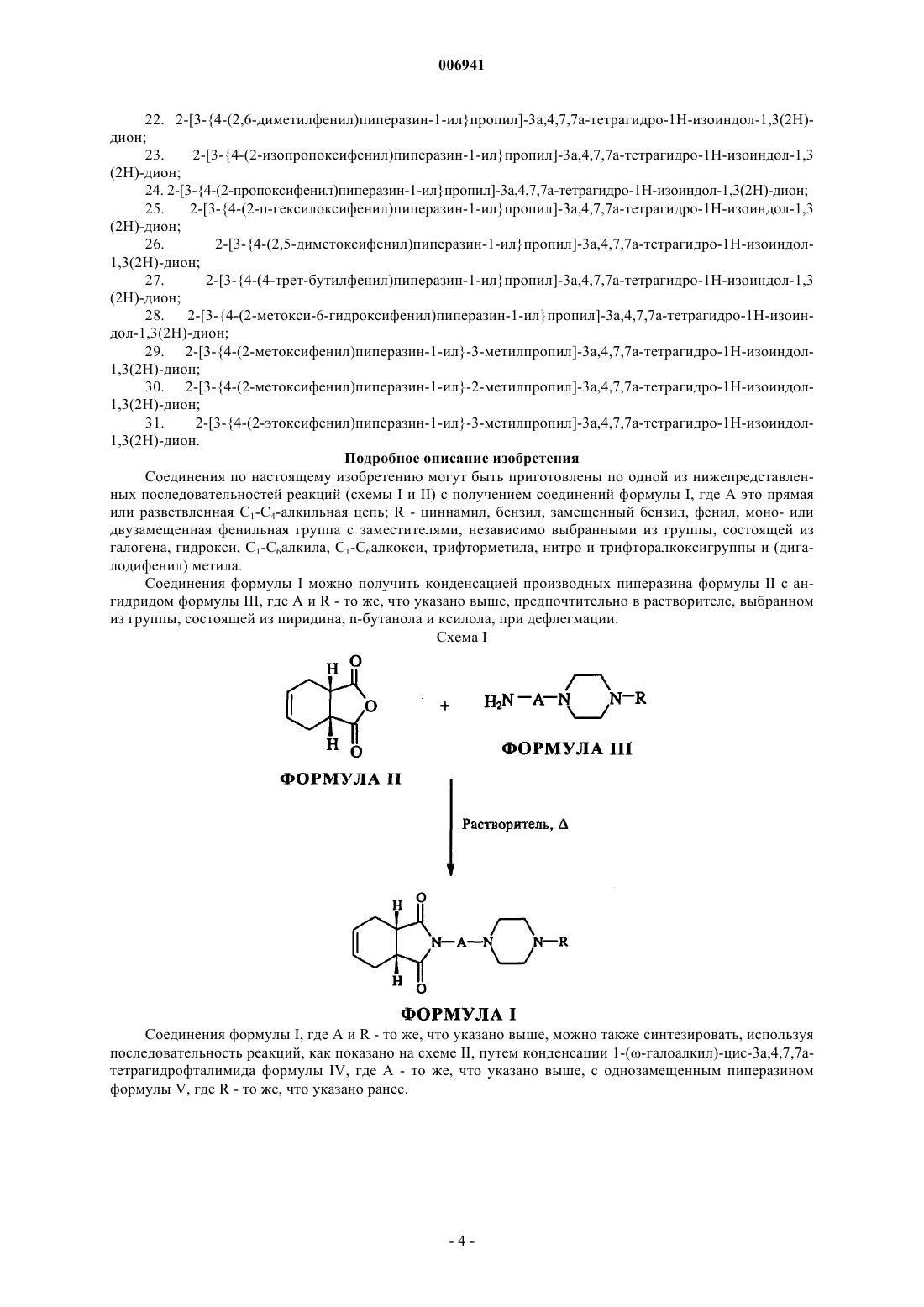
006941 Область изобретения Настоящее изобретение относится к некоторым новым 1,4-двузамещенным производным пиперазина формулы I и их фармацевтически приемлемым кислотно-аддитивным солям, обладающим превосходной уроселективной антагонистической активностью в отношении 1-адренорецепторов, превышающей активность ранее описанных соединений. Соединения по настоящему изобретению являются перспективными для лечения симптомов доброкачественной гиперплазии простаты (ДГП). Изобретение относится также к способам получения новых соединений, к фармацевтическим композициям, содержащим эти соединения, и способу лечения симптомов доброкачественной гиперплазии простаты с применением этих соединений. Область техники Доброкачественная гиперплазия простаты является обычным заболеванием у пожилых лиц мужского пола, и у значительного процента мужчин с ДГП развивается обструкция мочевого пузыря. Считается,что обструкции, вызванной ДГП, свойственны два основных компонента, а именно статический компонент, связанный с увеличенной массой ткани простаты, и динамический компонент, связанным с чрезмерным сокращением простаты и уретры. Наиболее успешное лечение основано на использовании антагонистов -адренергических рецепторов и модуляции уровней андрогенов ингибиторами 5-редуктазы. Ингибиторы 5-редуктазы имеют ограниченную эффективность в отношении незамедлительного симптоматического и уродинамического облегчения. Антагонисты 1-адренергических рецепторов проявляют намного большую эффективность и обеспечивают немедленные субъективные симптоматические улучшения и поэтому являются предпочтительными в лечении для ослабления симптомов доброкачественной гиперплазии простаты. 1-Адренорецепторы присутствуют также в кровеносных сосудах и играют важную роль в регулировании кровяного давления. Таким образом, антагонисты 1-адренорецепторов имеют особую важность, так как первоначально они разрабатывались в качестве противогипертонических агентов и вероятно обладают также полезным воздействием на дисфункцию липидов и резистентность к инсулину, которая обычно связана с эссенцйальной гипертензией. Наиболее часто применяемыми для лечения ДГП лекарствами являются длительно действующие антагонисты 1-адренорецепторов, теразозин, доксазозин и тамсулозин, представленные ниже. Однако с этими лекарствами связаны действующие на сосуды побочные эффекты (например, постуральная гипертензия, обморок, головокружение, головная боль и т. д.) из-за отсутствия селективности действия на простатические и сосудистые 1-адренорецепторы.-1 006941 На протяжении последнего десятилетия проводился интенсивный поиск уроселективных антагонистов 1-адренорецепторов для лечения ДГП, которые позволили бы избежать сердечно-сосудистых побочных эффектов, связанных с применяемыми в настоящее время лекарствами. Несомненно, что антагонисты 1-адренорецепторов, которым присуща более высокая селективность в отношении простатических 1-адренорецепторов, предоставляют возможность получить улучшенные уродинамические показатели. Это подчеркивает важность отыскания антагонистов, которые дадут улучшенные уродинамические показатели без побочных эффектов, связанных с существующими лекарствами. В последнее время идентифицировано три подтипа 1-рецепторов, а именно 1A, 1B и 1D, что может обеспечить значительное улучшение фармакологической селективности блокаторов 1. Эти подтипы имеют разное распределение в тканях, при этом 1A-рецепторы преобладают в нижнем мочевом пути и менее распространены в сосудистой системе. Это делает возможным получение агентов селективного действия в отношении к патологическим уродинамическим состояниям. Уроселективный антагонист 1 мог бы иметь более высокую эффективность, и была бы достигнута более полная блокада простатических 1-адренорецепторов, если увеличение дозы его приема не было бы ограничено сердечнососудистыми побочными эффектами. Была оценена действенность веществ в отношении к агонисту, т. е. стимулированное повышение уретрального давления относительно снижения кровяного давления или блокада вызванного агонистом кровяного давления. Многие селективные антагонисты описаны в Hieble и др. Ехр opin Invest Drugs; 6, 367-387 (1997) и в Kenny и др. J.Med. Chem.;40, 1293-1315 (1997). Во многих из этих структурных последовательностях были подробно изучены зависимости активности структур, и многочисленные высокоселективные соединения были идентифицированы. Настоящее изобретение направлено на получение новых антагонистов 1, а именно 1,4 двузамещенных соединений пиперазина, обладающих большей селективностью в отношении к 1A- адренорецепторам и тем самым снимающих симптомы ДГП. В литературе имеются многочисленные описания фармакологической активности фенилпиперазинов. Так, в Eur. J. Med. Chem. - Chimica Therapeutica, 12, 173-176 (1977) описаны замещенные трифторметилфенилпиперазины, имеющие циклоимидоалкильные боковые цепи, показанные ниже: Эти соединения являются потенциальными анорексическими агентами, не оказывающими побочные действия на центральную нервную систему (ЦНС). Другие родственные соединения, которые были получены в качестве анксиолитических, нейролептических, антидиабетических и антиаллергических агентов, описаны в следующих источниках:Soc, Vol. LV, 819-821 (1978 и N-(N4-арилпиперазинилалкил)фталимидов (J. Indian. Chem. Soc, Vol. LVI,1002-1005 (1979. Было показано, что эти соединения проявляют антигипертензивную активность и успокаивающее действие на ЦНС у экспериментальных животных. Однако ни в одном из вышеприведенных источниках не раскрыта и не упоминается селективная,блокирующая 1-адренорецепторы активность описанных в них соединений, поэтому они не пригодны для лечения симптомов доброкачественной гиперплазии простаты. Синтез 1-(4-арилпиперазин-1-ил)[N-(,-дикарбоксимидо)]алканов, пригодных в качестве уроселективных блокаторов 1-адренорецепторов, описан в патентах США 6083950 и 6090809. Данные соединения обладают хорошей блокирующей 1-адренорецепторы активностью и селективностью, а одно их этих соединений проходит II-ю фазу клинических испытаний. В настоящее время установлено, что структурная модификация этих соединений от глутаримида до тетрагидрофталимида повышает блокирующую адренорецепторы активность, а также сильно увеличивает селективность в отношении 1A-адренорецепторов по сравнению с активностью, блокирующей 1B-2 006941 адренорецепторы, что является существенным требованием к соединениям, пригодным быть кандидатами для лечения ДГП. Цели изобретения Целью настоящего изобретения является создание новых производных арилпиперазина, которые проявляют более высокую 1A-адренергическую блокирующую активность, более селективны, чем имеющиеся известные соединения, и пригодны для лечения доброкачественной гиперплазии простаты. Целью изобретения является также создание способа синтеза этих новых соединений. Дополнительная цель настоящего изобретения - создание содержащих новые соединения композиций, пригодных для лечения доброкачественной гиперплазии простаты. Сущность изобретения Отмеченные выше цели достигаются при помощи нового класса производных пиперазина общей формулы I, представленной ниже, их фармацевтически приемлемых солей, амидов, энантиомеров, диастереомеров, N-оксидов, пролекарств, метаболитов или полиморфов, где А - это прямая или разветвленная С 1-С 4-алкильная цепь; R циннамил, бензил, замещенный бензил, фенил, моно- или двузамещенная фенильная группа с заместителями, независимо выбранными из группы, состоящей из галогена, гидрокси, C1-С 6 алкила, C1-С 6 алкокси,трифторметила, нитро- и трифторалкокси-группы и (дигалодифенил) метила. Галоген в формуле I может быть выбран из группы, состоящей из хлора, фтора, йода; C1-С 6 алкил может быть выбран из метила, этила, n-пропила, изопропила, бутила, трет-бутила, а C1-С 6 алкокси может быть выбран из метокси, этокси, n-пропокси, изо-пропокси или гексилокси. Настоящее изобретение предлагает также фармацевтические композиции для лечения доброкачественной гиперплазии простаты. Эти композиции содержат эффективное количество по меньшей мере одного из соединений формулы I или эффективное количество по меньшей мере одной физиологически приемлемой кислотно-аддитивной соли такого соединения с фармацевтически приемлемым носителем. Пояснительный перечень конкретных соединений по изобретению представлен ниже: 1. 2-[3-4-(2-метоксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1H-изоиндол-1,3(2 Н)дион; 2. 2-[3-4-(3-хлорфенил)пиперазин-1-ил пропил]-3 а,4,7,7 а-тетрагидро-1H-изоиндол-1,3(2 Н)-дион; 3. 2-[3-4-(2-метилфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)-дион; 4. 2-[3-4-(4-фторфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)-дион; 5. 2-[3-4-(3-трифторметилфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1H-изоиндол 1,3(2 Н)-дион; 6. 2-[3-4-(2-фторфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1H-изоиндол-1,3(2 Н)-дион; 7. 2-[3-4-(3,4-диметилфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)дион; 8. 2-[3-4-(2-метокси-5-фторфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)-дион; 28. 2-[3-4-(2-метокси-6-гидроксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)-дион; 29. 2-[3-4-(2-метоксифенил)пиперазин-1-ил-3-метилпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол 1,3(2 Н)-дион; 30. 2-[3-4-(2-метоксифенил)пиперазин-1-ил-2-метилпропил]-3 а,4,7,7 а-тетрагидро-1H-изоиндол 1,3(2 Н)-дион; 31. 2-[3-4-(2-этоксифенил)пиперазин-1-ил-3-метилпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол 1,3(2 Н)-дион. Подробное описание изобретения Соединения по настоящему изобретению могут быть приготовлены по одной из нижепредставленных последовательностей реакций (схемы I и II) с получением соединений формулы I, где А это прямая или разветвленная С 1-С 4-алкильная цепь; R - циннамил, бензил, замещенный бензил, фенил, моно- или двузамещенная фенильная группа с заместителями, независимо выбранными из группы, состоящей из галогена, гидрокси, C1-С 6 алкила, C1-C6 алкокси, трифторметила, нитро и трифторалкоксигруппы и (дигалодифенил) метила. Соединения формулы I можно получить конденсацией производных пиперазина формулы II с ангидридом формулы III, где А и R - то же, что указано выше, предпочтительно в растворителе, выбранном из группы, состоящей из пиридина, n-бутанола и ксилола, при дефлегмации. Схема I Соединения формулы I, где А и R - то же, что указано выше, можно также синтезировать, используя последовательность реакций, как показано на схеме II, путем конденсации 1-(-галоалкил)-цис-3 а,4,7,7 атетрагидрофталимида формулы IV, где А - то же, что указано выше, с однозамещенным пиперазином формулы V, где R - то же, что указано ранее. Фармацевтически приемлемые, нетоксичные, кислотно-аддитивные соли полученных согласно настоящему изобретению соединений, имеющие эффективность свободных оснований формулы I, могут быть образованы с помощью неорганических или органических кислот широко известными способами и применены вместо свободных оснований. Типичными примерами подходящих кислот для образования таких кислотно-аддитивных солей являются яблочная, фумаровая, бензойная, аскорбиновая, памовая,янтарная, бис-метиленовая, салициловая, метансульфокислота, этандисульфокислота, уксусная, пропионовая, винная, лимонная, глюконовая, аспаргиновая, стеариновая, пальмитиновая, итаконовая, гликолевая, р-аминобензойная, глутаминовая, бензосульфаминовая, фосфорная, бромисто-водородная, серная,соляная и азотная кислоты и т. п. В объем настоящего изобретения включены также пролекарства соединений формулы I. В целом,такие пролекарства будут являться функциональными производными этих соединений, которые легко превращаются in vivo в определенные соединения. Известны традиционные процедуры выбора и получения соответствующих пролекарств. В объем изобретения входят также энантиомеры, диастереоизомеры, N-оксиды, фармацевтически приемлемые соли, амиды и полиморфные формы заявленных соединений, а также метаболиты, обладающие такой же активностью. Кроме того, к изобретению относятся фармацевтические композиции,содержащие молекулы вещества формулы I или его пролекарства, метаболиты, энантиомеры, диастереоизомеры, N-оксиды, фармацевтически приемлемые соли в сочетании с фармацевтически приемлемым носителем и факультативно наполнителем. Еще одним аспектом изобретения являются способы селективного блокирования 1A-рецепторов путем доставки эффективного количества соединений по изобретению в среду указанных рецепторов,например, во внеклеточную среду (или путем введения в млекопитающее, имеющее указанные рецепторы). Изобретение описано со ссылками на его отдельные варианты осуществления лишь с иллюстративной целью. Для специалиста в данной области техники будут очевидны многочисленные альтернативные варианты, входящие в объем настоящего изобретения. Представленные ниже примеры демонстрируют общее, а также конкретное синтетическое получение предпочтительных соединений. Примеры даны для пояснения подробностей изобретения и не накладывают ограничения на объем настоящего изобретения. Пример. Получение 2-[3-4-(2-метоксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Низоиндол-1,3(2 Н)-диона. Схема I. Смесь 1-амино-3-[4-(2-метоксифенил)пиперазин-1-ил]пропана (0,498 г, 2,0 ммоль) и цис 1,2,3,6-тетрагидрофталевого ангидрида (0,273 г, 1,8 ммоль) дефлегмировали в пиридине в течение 5 ч. После завершения реакции растворитель удалили под вакуумом, а остаток растворили в хлороформе (25 г). Хлороформовую фазу промыли водой (2x15 мл), высушили безводным сульфатом натрия и концентрировали под вакуумом. Полученное таким образом сырое соединение очистили силикагелем в хроматографической колонке (100-200 меш) с использованием хлороформа в качестве элюента (выход 0,502 г,72%). Гидрохлоридную соль получили путем добавления мольного количества эфирного раствора хлористого водорода к эфирному раствору свободного основания и фильтрацией собрали осажденную твердую фазу (точка плавл. 184-185 С). Схема II. Смесь 1-(3-бромпропил)-цис-3 а,4,7,7 а-тетрагидрофталимида (7,04 г, 25,88 ммоль), 1-(2 метоксифенил)пиперазин гидрохлорида (5,32 г, 23,29 ммоль), карбоната калия (7,14 г, 51,76 ммоль) и йодида калия (0,026 г, 1,55 ммоль) в N,N-диметилформамиде (27 мл) грели при 75-80 С в течение около-5 006941 12 ч. После завершения реакции растворитель выпарили под вакуумом, остаток суспендировали в воде(130 мл) и экстрагировали соединение с помощью дихлорметана (2 х 65 мл). Объединенный дихлорметановый слой промыли водой (2 х 30 мл), высушили безводным сульфатом натрия, а растворитель концентрировали под вакуумом с выходом 8,308 г (93%) неочищенного основания. Полученное таким образом соединение превратили в его гидрохлоридную соль (точка плавл. 184-185 С). Далее представлен пояснительный перечень соединений по изобретению, которые были синтезированы одним или несколькими из вышеописанных методов. 1. 2-[3-4-(2-метоксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)дион гидрохлорид; точка плавл. 184-185 С. 2. 2-[3-4-(3-хлорфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1H-изоиндол-1,3(2 Н)-дион гидрохлорид; точка плавл. 221-223 С. 3. 2-[3-4-(2-метилфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1H-изоиндол-1,3(2 Н)-дион гидрохлорид; точка плавл. 186-187 С. 4. 2-[3-4-(4-фторфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1H-изоиндол-1,3(2 Н)-дион гидрохлорид; точка плавл. 228-230 С. 5. 2-[3-4-(3-трифторметилфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)-дион гидрохлорид; точка плавл. 193-195 С. 27. 2-[3-4-(4-трет-бутилфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)дион гидрохлорид; точка плавл. 264-265 С. 28. 2-[3-4-(2-метокси-6-гидроксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоин-6 006941 дол-1,3(2 Н)-дион гидрохлорид; точка плавл. 267-268 С. 29. 2-[3-4-(2-метоксифенил)пиперазин-1-ил-3-метилпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол 1,3(2 Н)-дион гидрохлорид; точка плавл. 219-220 С. 30. 2-[3-4-(2-метоксифенил)пиперазин-1-ил-2-метилпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол 1,3(2 Н)-дион гидрохлорид; точка плавл. 184-185 С. 31. 2-[3-4-(2-этоксифенил)пиперазин-1-ил-3-метилпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол 1,3(2 Н)-дион гидрохлорид; точка плавл. 246-248 С. Все вышеуказанные точки плавления не корректировались и измерялись методом открытого капилляра с использованием прибора Buchi 535. Результаты фармакологических испытаний Оценка рецепторного связывания Оценку рецепторного связывания выполняли с использованием нативных -адренорецепторов. Сродство различных соединений к 1A- и 1B-подтипам адренорецепторов определяли исследованием их способности замещать специфическое связывание [3 Н]празозина в мембранах клеток крыс, в субмаксиллярной области и печени соответственно (Michel и др., Br J Pharmacol, 98, 883-889 (1989. Оценку связывания проводили согласно U'Prichard и др., (Eur J Pharmacol, 50: 87-89 (1978 с небольшими изменениями. Немедленно после умерщвления подопытных животных отделяли субмаксиллярные железы. Печень залили буферным раствором (трис-НСl 50 ммоль, NaCl 100 ммоль, ЭДТА 10 ммоль, рН 7,4). Ткани гомогенизировали в 10 объемах буферного раствора (трис-НСl 50 ммоль, NaCl 100 ммоль, ЭДТА 10 ммоль, рН 7,4). Гомогенат профильтровали через два слоя влажной марли, а фильтрат центрифугировали с силой 500 г в течение 10 мин. Затем надосадочную жидкость центрифугировали с силой 40000 г в течение 45 мин. Полученный таким образом осадок повторно суспендировали в равном объеме буферного раствора для анализа (трис-НСl 50 ммоль, ЭДТА 5 ммоль, рН 7,4) и хранили при -70 С до исследований. Мембранные гомогенаты (150-250 мкг протеина) инкубировали в 250 мкл буферного раствора для анализа (трис-НСl 50 ммоль, ЭДТА 5 ммоль, рН 7,4) при 24-25 С в течение 1 ч. Неспецифическое связывание определяли в присутствии 300 нмоль празозина. Инкубирование завершалось вакуумной фильтрацией в фильтрах из базальтового стекловолокна (GF/B волокно). Затем фильтры промывали ледяным буферным раствором (трис-НСl 50 ммоль, рН 7,4). Фильтрующие маты высушивали и определяли связанную радиоактивность, сохранившуюся на фильтрах. Путем нелинейной аппроксимации кривой с помощью программы G Pad Prism оценивали 50%-ную ингибиторную концентрацию (IC50) и равновесную постоянную диссоциации (Kd). Величину постоянной ингибирования Ki вычисляли на основе конкурентных исследований с использованием уравнения Ченга-Пруссофа (ChengPrusoff, Biochem Pharmacol, 1973, 22: 3099-3108): Ki = IC50/(l+L/Kd), где L - концентрация [3 Н]празозина в конкретном эксперименте (таблица I). Функциональные исследования in vitro Для исследования селективности действия предложенных соединений по отношению к разным подтипам -адренорецепторов изучалась способность этих соединений противодействовать сократительному ответу, вызванному агонистом 1-адренорецепторов в аорте (1D), простате (1A) и селезенке (1B). Ткани аорты и селезенки были выделены из анестезированных уретаном (1,5 г/кг) крыс-самцов Уистара. Выделенные ткани были установлены в ванночке для органов, содержащей буфер Кребса-Хенселейта следующего состава (ммоль): NaCl 118; KCl 4,7; CaCl2 2,5; MgSO47H2O 1,2; NaHCO3 25; KH2PO4 1,2; глюкоза 11,5. Буфер находился при 37oC и аэрировался смесью O2 (95%) и CO2 (5%). К тканям прикладывалось напряжение покоя 2 г (аорта) или 1 г (селезенка и простата). Сократительный ответ наблюдали с использованием силового датчика перемещений и регистрировали самописцем. Равновесное состояние в тканях достигалось в течение 2 часов. В конце периода установления равновесия концентрационные кривые отклика на норэпинефрин (аорта) и фенилэпинефрин (селезенка и простата) были получены в присутствии и отсутствии испытываемых соединений (при концентрациях 1,1 и 10 ммоль). Антагонистическое сродство вычислялось и выражалось как константа основности рКB (таблица II). Исследование уроселективности in vivo Чтобы оценить уроселективность in vivo, действия предложенных соединений изучались по среднему артериальному давлению (САД) и внутриуретральному давлению (ВУД) у находящихся в сознании гончих собак согласно методу Вrиnе и др. (Pharmacol 1996, 53: 356-368). Вкратце, за две недели до исследований кобелей оснастили приборами для постоянного непрерывного измерения кровяного артериального давления путем имплантации телеметрического передатчика (TL11M2-D70-PCT, Data Sci. International, St. Paul, MN. США) в бедренную артерию. В течение восстановительного периода животное привыкало находиться в ограничивающем его движения связывающем приспособлении. В день опыта голодающее с вечера животное помещали в связывающее приспособление. Имеющий наконечник баллонный катетер Свана-Ганза вводили в уретру до уровня простаты и надували баллон (Вrиnе и др., 1996). После фиксации начальной линии показаний регистрировали воздействие фенилэфрина (16 мкг/кг внутривенно) на САД и ВУД. Действие фенилэфрина на САД и ВУД регистрировали через 0,5, 1, 2, 3, 4, 6, 9-7 006941 и 24 часа после орального введения наполнителя или испытываемого лекарства. Изменения САД оперативно регистрировали с использованием программы Dataquest Software (Data Sci. International, Data Sci.International, St. Paul, MN. США), а ВУД регистрировали прибором Grass Polygraph (модель 7, Grass Instruments, США). Изменение действия фенилэфрина на САД и ВУД после введения испытываемого лекарства вычислялось как процентное изменение от контрольного значения. Подсчитывалась площадь под кривой, а отношение значений ВУД к САД использовалось для определения уроселективности (таблица Несмотря на то, что настоящее изобретение описано на основе его отдельных вариантов осуществления, для специалиста в данной области техники будут очевидны определенные модификации и эквиваленты, входящие в объем данного изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединения структурной формулы I а также их фармацевтически приемлемые соли, амиды, энантиомеры, диастереомеры, N-оксиды, пролекарства, метаболиты или их полиморфы, где А - это прямая или разветвленная С 1-С 4-алкильная цепь; R циннамил, бензил, замещенный бензил, моно- или двузамещенная фенильная группа с заместителями,независимо выбранными из группы, состоящей из гидрокси, нитро, трифторалкоксигруппы и (дигалодифенил) метила. 2. Соединения, выбранные из группы, состоящей из 2-[3-4-(2-метоксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)-диона(соединение 16); 2-[3-4-(3-хлор-4-метилфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)диона (соединение 17); 2-[3-4-(4-фтор-2-метоксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол 1,3(2 Н)-диона (соединение 18); 2-[3-4-(бис-4-фторфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)диона (соединение 19); 2-[3-4-(2,4-дихлорфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)диона (соединение 20); 2-[3-4-(2,4-диметоксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)диона(соединение 21); 2-[3-4-(2,6-диметилфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2H)диона (соединение 22); 2-[3-4-(2-изопропоксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)диона (соединение 23); 2-[3-4-(2-пропоксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1H-изоиндол-1,3(2H)диона (соединение 24); 2-[3-4-(2-n-гексилоксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)диона (соединение 25); 2-[3-4-(2,5-диметоксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол-1,3(2 Н)диона (соединение 26); 2-[3-4-(4-трет-бутилфенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1H-изоиндол-1,3(2 Н)диона (соединение 27); 2-[3-4-(2-метокси-6-гидроксифенил)пиперазин-1-илпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол 1,3(2 Н)-диона (соединение 28); 2-[3-4-(2-метоксифенил)пиперазин-1-ил-3-метилпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол 1,3(2 Н)-диона (соединение 29); 2-[3-4-(2-метоксифенил)пиперазин-1-ил-2-метилпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол 1,3(2 Н)-диона (соединение 30); 2-[3-4-(2-этоксифенил)пиперазин-1-ил-3-метилпропил]-3 а,4,7,7 а-тетрагидро-1 Н-изоиндол 1,3(2 Н)-диона (соединение 31). 3. Способ оказания селективного противодействия 1-адренергическим рецепторам у млекопитающего, включающий введение указанному млекопитающему соединения структурной формулы I его фармацевтически приемлемых солей, амидов энантиомеров, диастереомеров, N-оксидов, пролекарств, метаболитов или их полиморфов, где А - это прямая или разветвленная С 1-С 4 алкильная цепь; R циннамил, бензил, замещенный бензил, фенил, моно- или двузамещенная фенильная группа с заместителями, независимо выбранными из группы, состоящей из галогена, гидрокси, C1-С 6 алкила, C1-С 6 алкокси,трифторметила, нитро и трифторалкоксигруппы и (дигалодифенил) метила. 4. Способ лечения доброкачественной гиперплазии простаты у млекопитающего, включающий введение указанному млекопитающему соединения структурной формулы I его фармацевтически приемлемых солей, амидов энантиомеров, диастереомеров, N-оксидов, проле- 10006941 карств, метаболитов или их полиморфов, где А - это прямая или разветвленная С 1-С 4-алкильная цепь; R циннамил, бензил, замещенный бензил, фенил, моно- или двузамещенная фенильная группа с заместителями, независимо выбранными из группы, состоящей из галогена, гидрокси, C1-С 6 алкила, C1-С 6 алкокси,трифторметила, нитро и трифторалкоксигруппы и (дигалодифенил) метила. 5. Фармацевтическая композиция, содержащая соединение по п.1 или 3 и фармацевтически приемлемый носитель. 6. Способ селективного противодействия 1-адренергическим рецепторам у млекопитающего,включающий введение указанному млекопитающему фармацевтической композиции по п.5. 7. Способ лечения доброкачественной гиперплазии простаты у млекопитающего, включающий введение указанному млекопитающему фармацевтической композиции по п.6. 8. Способ получения соединений формулы I или их фармацевтически приемлемых солей, амидов энантиомеров, диастереомеров, N-оксидов, пролекарств, метаболитов или их полиморфов, где А- это прямая или разветвленная С 1-С 4-алкильная цепь; R циннамил, бензил, замещенный бензил, моно- или двузамещенная фенильная группа с заместителями,независимо выбранными из группы, состоящей из гидрокси, нитро, трифторалкоксигруппы и (дигалодифенил) метила, включающий взаимодействие цис-1,2,3,6-тетрагидрофталевого ангидрида формулы II с 1-амино-4-замещенным пиперазинил алканом формулы III где А и R - те же, что указано выше. 9. Способ получения соединений формулы I или их фармацевтически приемлемых солей, амидов энантиомеров, диастереомеров, N-оксидов, пролекарств, метаболитов или их полиморфов, где А - это прямая или разветвленная С 1-С 4-алкильная цепь; R циннамил, бензил, замещенный бензил, фенил, моно- или двузамещенная фенильная группа с заместителями, независимо выбранными из группы, состоящей из галогена, гидрокси, C1-С 6 алкила, C1-С 6 алкокси,трифторметила, нитро, трифторалкоксигруппы и (дигалодифенил) метила, включающий взаимодействие 1-(-галоалкил)цис-3 а,4,7,7 а-тетрагидрофталамида формулы IV
МПК / Метки
МПК: A61K 31/40, A61P 13/08, C07D 209/48
Метки: блокаторов, альфа1-адренорецепторов, 1,4-двузамещенные, пиперазина, качестве, уроселективных, производные
Код ссылки
<a href="https://eas.patents.su/12-6941-14-dvuzameshhennye-proizvodnye-piperazina-v-kachestve-uroselektivnyh-blokatorov-alfa1-adrenoreceptorov.html" rel="bookmark" title="База патентов Евразийского Союза">1,4-двузамещенные производные пиперазина в качестве уроселективных блокаторов альфа1-адренорецепторов</a>
Замещенные изохинолины в качестве нервно-мышечных блокаторов ультракраткосрочного действия

Номер патента: 2224
Опубликовано: 28.02.2002
Авторы: Своринджен Рой Арчибальд, Бигхэм Эрик Кливленд, Пэйтел Сэнджей Шешикент, Бозуэлл Грейди Ивэн, Сэйвэриз Джон Джозеф, Борос Эрик Юджин, Сэймэно Винсент
МПК: C07D 217/20, A61K 31/47, A61P 41/00...
Метки: качестве, ультракраткосрочного, замещенные, блокаторов, изохинолины, нервно-мышечных, действия
Формула / Реферат:
1. Соединение формулы (I) где Х представляет собой галоген, h равно числу от 1 до 2, Y представляет собой водород или метокси, Z1 и Z2 представляют собой метил, W1 и W2 представляют собой углерод, и А представляет собой фармацевтически приемлемый анион. 2. Соединение формулы (I), которое включает в себя дихлорид...
Производные пиперазина

Номер патента: 669
Опубликовано: 28.02.2000
Авторы: Кониси Нобукийо, Мурано Кендзи, Сигенага Синдзи, Мияке Хироси, Мацуда Масааки, Хагивара Дайдзиро, Мацуда Хироси, Манабе Такаси
МПК: A61K 31/50, C07D 403/06
Метки: производные, пиперазина
Формула / Реферат:
1. Соединение формулы 2. Способ получения соединения по п.1, включающий реакцию соединения формулы или его соли, отличной от соли фумаровой кислоты, с фумаровой кислотой с получением соединения формулы 3. Фармацевтическая композиция, включающая в качестве активного ингредиента соединение по п.1 в сочетании с фармацевтически приемлемым нетоксичным носителем или разбавителем. 4. Способ лечения или профилактики заболеваний,...
Производные пиперазина с антагонистической активностью к рецептору ccr1

Номер патента: 6243
Опубликовано: 27.10.2005
Авторы: Хэйворд Мэттью Меррилл, Посс Кристофер Стенли, Браун Мэттью Фрэнк, Шавня Андре, Лундквист Грегори Дин Мл., Блюмберг Лаура Кук
МПК: C07D 241/04, A61K 31/495, A61P 29/00...
Метки: рецептору, активностью, пиперазина, производные, антагонистической
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая форма; где a равно 0, 1, 2, 3, 4 или 5; b равно 0, 1 или 2; c равно 0, 1 или 2; d равно 0, 1, 2, 3 или 4; X представляет собой -O-, -S-, -CH2- или -NR6-; Y представляет собой (C6-C10)арил или (C2-C9)гетероарил; каждый R1 независимо представляет собой H-, HO-, галоген-, (C1-C8)алкил-, (C1-C8)алкил-O-, HO-(C1-C8)алкил-, NC-, H2N-, H2N-(C1-C8)алкил-, HO-(C=O)-, (C1-C8)алкил-(C=O)-,...
Производные 1-(1,2-дизамещенный пиперидинил)-4-замещенного пиперазина.

Номер патента: 909
Опубликовано: 26.06.2000
Авторы: Ван Росбрук Ив Эмиль Мария, Жансенс Франс Эдуард, Сюрлеро Доминик Луи Нестор Гилейн, Ленартс Йозеф Элизабет, Соммен Франсуа Мария
МПК: A61P 29/00, A61P 1/08, A61P 23/00...
Метки: 1-(1,2-дизамещенный, пиперидинил)-4-замещенного, пиперазина, производные
Формула / Реферат:
1. Соединение формулы его N-оксидная форма, фармацевтически приемлемая аддитивная соль или стереохимически изомерная форма; где n равно 0, 1 или 2; m равно 1 или 2, при условии, что если m равно 2, то n равно 1; р равно 1 или 2; =Q представляет =O или =NR3; X представляет ковалентную связь или двухвалентный радикал формулы -О-, -S-, -NR3-; R1 представляет Аr1, Аr1С1-6алкил или ди(Ar1)C1-6алкил, где каждая C1-6алкильная группа...
Галогензамещённые производные 4-фенил-1-пиперазина, -пиперидина и -тетрагидропиридина

Номер патента: 5013
Опубликовано: 28.10.2004
Авторы: Келер Ян, Фелдинг Якоб, Банг-Андерсен Бенни
МПК: A61P 25/00, C07D 403/02, A61K 31/495...
Метки: пиперидина, производные, галогензамещённые, тетрагидропиридина, 4-фенил-1-пиперазина
Формула / Реферат:
1. Соединение формулы I где W является C, CH или N и пунктирная линия, исходящая от W, обозначает связь, если W является C; и отсутствие связи, если W является N или CH; R1 и R2 независимо выбраны из водорода и галогена, при условии, что по крайней мере один из R1 и R2 является атомом галогена; R3 выбран из водорода, галогена, C1-6-алкила, C2-6-алкенила, C2-6-алкинила, трифторметила, C1-6-алкокси, арилокси, аралкокси, гидрокси, амино,...