Способ лечения инфекционного заболевания
Номер патента: 17702
Опубликовано: 28.02.2013
Авторы: Сун Эркуан, Кинг Чи-Син Ричард, Ли Лих-Хуей, Ву По-Ли








Формула / Реферат
1. Способ лечения инфекционного заболевания, включающий введение нуждающемуся в этом субъекту путем парентеральной инъекции или инфузии эффективного количества состава, содержащего
D,L-малат соединения формулы I

воду и обеспечивающее изотоничность средство в концентрации от 0,2 до 13% мас./об., при этом соединение и обеспечивающее изотоничность средство растворены в воде.
2. Способ по п.1, где инфекционное заболевание представляет собой инфекцию мочевыводящих путей, простатит, респираторную инфекцию, остеомиелит, гонорею, Mycobacterium tuberculosis, комплекс Mycobacterium avium, острые приступы хронического бронхита, пневмонию, синусит, инфекционную диарею, Helicobacter pylori, кожную инфекцию, гинекологическую инфекцию или внутрибрюшную инфекцию.
3. Способ по п.1, где инфекционное заболевание вызвано грамотрицательными или грамположительными бактериями.
4. Способ по п.1, где инфекционное заболевание вызвано анаэробными бактериями.
5. Способ по п.1, где инфекционное заболевание вызвано устойчивым к метициллину S. aureus или мультирезистентным S. pneumoniae.
6. Способ по п.1, где обеспечивающее изотоничность средство представляет собой хлорид натрия.
7. Способ по п.6, где концентрация соединения в составе составляет от 0,2 до 45 мМ, а концентрация хлорида натрия в составе составляет от 0,2 до 1,3% мас./об.
8. Способ по п.7, где состав вводят посредством внутривенной инъекции или инфузии.
9. Способ по п.7, где концентрация хлорида натрия в составе составляет 0,9% мас./об.
10. Способ по п.7, где инфекционное заболевание представляет собой инфекцию мочевыводящих путей, простатит, респираторную инфекцию, остеомиелит, гонорею, Mycobacterium tuberculosis, комплекс Mycobacterium avium, острые приступы хронического бронхита, пневмонию, синусит, инфекционную диарею, Helicobacter pylori, кожную инфекцию, гинекологическую инфекцию или внутрибрюшную инфекцию.
11. Способ по п.7, где инфекционное заболевание вызвано грамотрицательными или грамположительными бактериями.
12. Способ по п.7, где инфекционное заболевание вызвано анаэробными бактериями.
13. Способ по п.7, где инфекционное заболевание вызвано устойчивым к метициллину S. aureus или мультирезистентным S. pneumoniae.
14. Способ по п.7, где состав дополнительно содержит стабилизирующее средство, где стабилизирующее средство выбрано из группы, состоящей из гистидина, лизина, глицина, сахарозы, фруктозы, трегалозы и их смеси.
15. Способ по п.7, где состав дополнительно содержит буфер, где буфер выбран из группы, состоящей из ацетата, цитрата, тартрата, лактата, сукцината, малата или фосфата.
16. Способ по п.7, где состав дополнительно содержит антиоксидант, где антиоксидант выбран из группы, состоящей из дисульфита натрия, бутилированного гидроксианизола, цистеина, гентизиновой кислоты, моноглутамата натрия, тиогликолята натрия и аскорбиновой кислоты.
17. Способ по п.7, где состав дополнительно содержит стабилизирующее средство в концентрации 0,1-1,0% мас./об. и буфер в концентрации 0,01-5% мас./об.
18. Способ по п.1, где обеспечивающее изотоничность средство выбрано из группы, состоящей из глицерина, лактозы, маннита, декстрозы, сульфата натрия и сорбита.
19. Способ по п.18, где обеспечивающее изотоничность средство представляет собой декстрозу и его концентрация в составе составляет 1-7% мас./об.
20. Способ по п.19, где состав дополнительно содержит стабилизирующее средство в концентрации 0,1-1,0% мас./об. и буфер в концентрации 0,01-5% мас./об.
21. Способ по п.19, где состав вводят посредством внутривенной инъекции или инфузии.
Текст
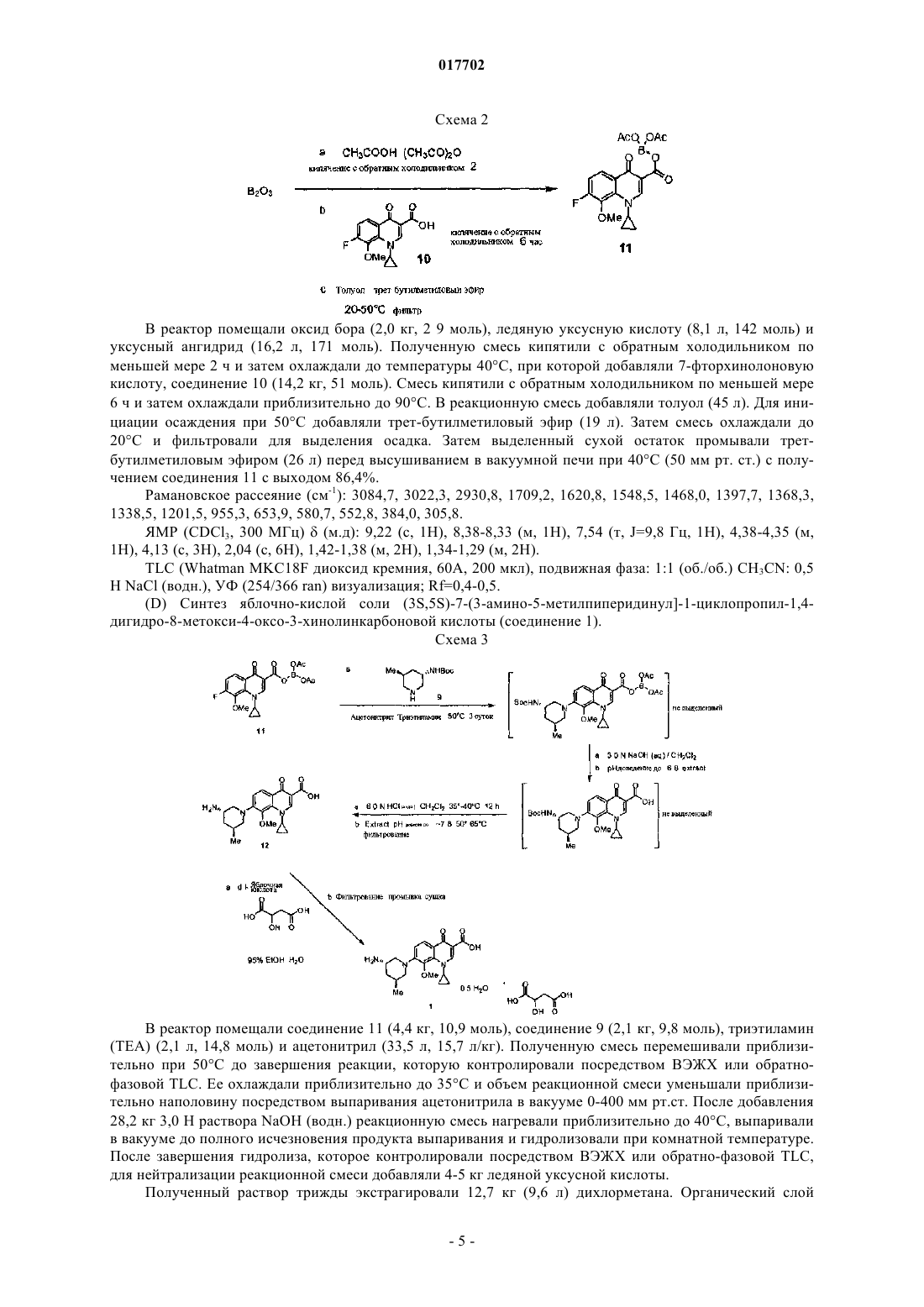
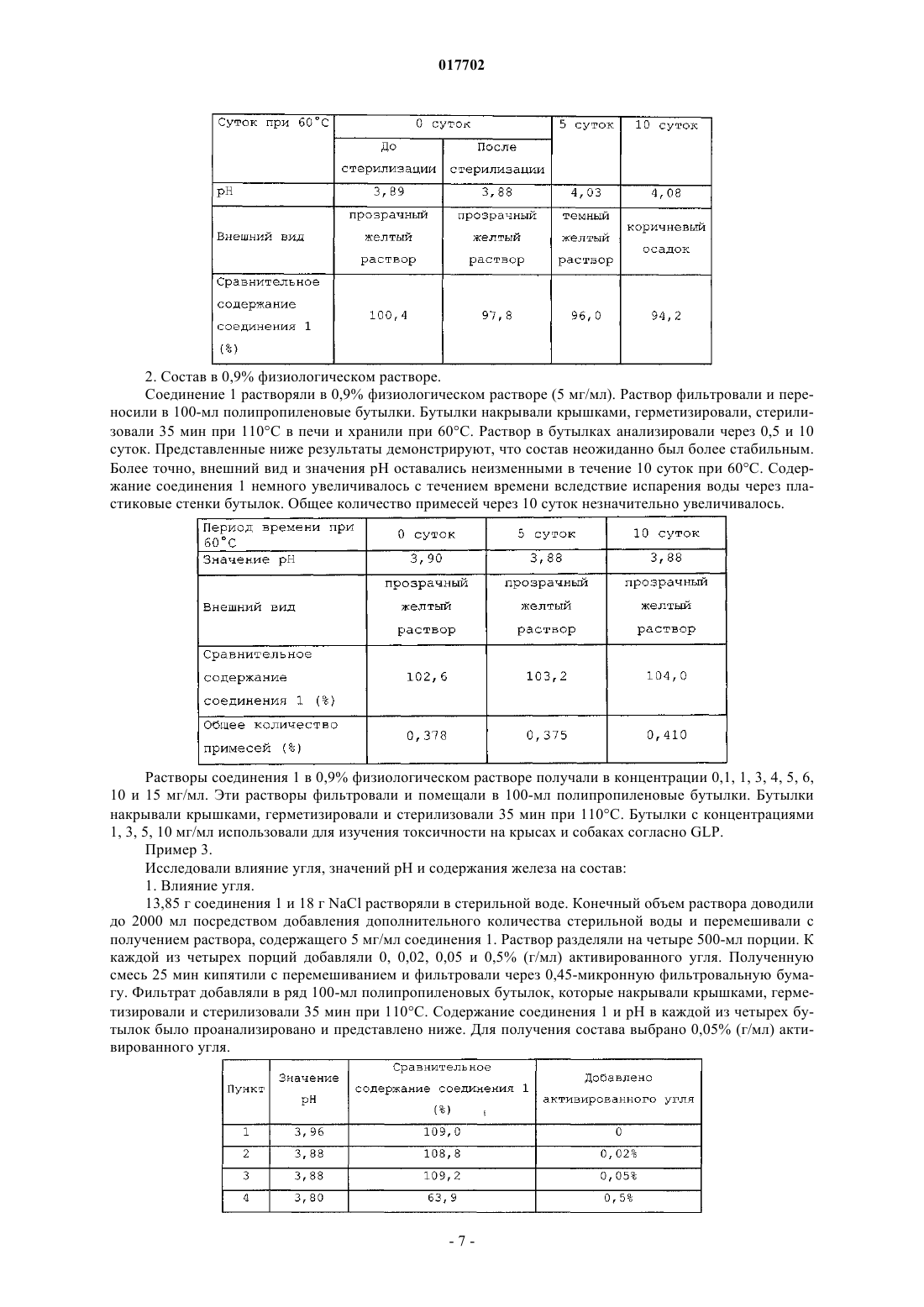
(56) RU-C2-2193558 Министерство здравоохранения Украины. Национальная академия наук Украины. Государственный научный центр лекарственных средств. Технология и стандартизация лекарств, т. Данное изобретение относится к способу лечения инфекционного заболевания, включающему введение нуждающемуся в этом субъекту путем парентеральной инъекции или инфузии эффективного количества состава, содержащего D,L-малат соединения формулы I Формула 1 воду и обеспечивающее изотоничность средство в концентрации от 0,2 до 13% мас./об., при этом соединение и обеспечивающее изотоничность средство растворены в воде. Изобретение обеспечивает эффективное лечение инфекционного заболевания введением данного состава индивидууму посредством парентеральной инъекции или инфузии. 017702 Предпосылки изобретения Парентеральная инъекция противомикробного лекарственного средства представляет собой один из наиболее эффективных способов лечения инфекций, особенно инфекций, устойчивых к метициллинуStaphylococcus aureus и устойчивых к метициллину Streptococcus pneumoniae. Для них необходимо применение водного состава, который представляет собой стабильный раствор. Сущность изобретения В одном из аспектов данное изобретение относится к противомикробному парентеральному составу(например, внутривенному составу), который содержит соединение формулы I, представленной ниже: Формула 1 воду и обеспечивающее изотоничность средство. Соединение и обеспечивающее изотоничность средство растворены в воде с образованием парентерального состава. Соединение включает его соли и пролекарственные средства. Например, соли могут образовываться между положительно заряженными аминогруппами соединения и анионом. Подходящие анионы включают, но не ограничиваются ими, хлорид, бромид, иодид, сульфат, нитрат, фосфат, D- или L-малат,D,L-малат, цитрат, тозилат, мезилат, D- или L-соль винной кислоты, D,L-тартрат, фумарат, трифторацетат, L-глутамат, D-глюкуронат, малеат, лактат и ацетат. Подобным образом, отрицательно заряженные карбоксилаты на соединении могут образовывать соли с катионами. Подходящие катионы включают, но не ограничиваются ими, ион натрия, ион калия, ион магния, ион кальция и катион аммония, такой как ион тетраметиламмония. Примеры пролекарственных средств включают сложные эфиры и другие фармацевтически приемлемые производные, которые после введения индивидууму способны образовывать соединения, описанные выше (см. Goodman and Gilman's, Pharmacological basis of Therapeutics, 8th ed.,McGraw-Hill, Int. Ed. 1992, "Biotransformation of Drugs"). Кроме того, соединение, содержащее ассиметрические центры, может существовать в виде рацематов, рацемических смесей, индивидуальных энантиомеров, индивидуальных диастереомеров и диастереометрических смесей. Обеспечивающее изотоничность средство, такое как неэлектролиты и электролиты, регулирует уровень осмотического давления. См. патент США 6015810. Примеры включают, но не ограничиваются ими, глицерин, лактозу, маннит, декстрозу, хлорид натрия, сульфат натрия и сорбит. В составе по данному изобретению концентрация соединения может составлять от 0,2 до 45 мМ, а концентрация обеспечивающего изотоничность средства может составлять от 0,2 до 13% мас./об., в частности - от 0,2 до 1,3% мас./об. Концентрацию обеспечивающего изотоничность средства ("% мас./об.") рассчитывают как отношение массы (г) обеспечивающего изотоничность средства к объему (литр) состава. Состав по данному изобретению также может содержать буфер, стабилизирующее средство или антиоксидант. Примером состава по данному изобретению является состав, содержащий яблочно-кислую соль соединения в концентрации от 0,2 до 45 мМ, хлорид натрия в концентрации 0,9% мас./об., стабилизирующее средство в концентрации 0,1-1,0% мас./об. и буфер в концентрации 0,01-5% мас./об. В качестве другого примера состав содержит от 0,2 до 45 мМ яблочно-кислой соли соединения, декстрозу в концентрации 1-7% мас./об., стабилизирующее средство в концентрации 0,1-1,0% мас./об. и буфер в концентрации 0,01-5% мас./об. Подобно обеспечивающему изотоничность средству концентрации стабилизирующего средства,буфера и антиоксиданта также рассчитывают как отношение массы вещества и объема состава. В другом аспекте данное изобретение описывает способ лечения инфекционного заболевания посредством введения индивидууму эффективного количества описанного выше состава посредством парентеральной инъекции. Инфекционное заболевание может вызывать инфекция грамположительными бактериями, грамотрицательными бактериями, анаэробными бактериями, устойчивыми к метициллину S.aureus или мультирезистентным S. pneumoniae. Примеры инфекционного заболевания включают, но не ограничиваются ими, инфекцию мочевыводящих путей, простатит, респираторную инфекцию, остеомиелит, гонорею, Mycobacterium tuberculosis, комплекс Mycobacterium avium, острые приступы хронического бронхита, пневмонию, синусит, инфекционную диарею, Helicobacter pylori, кожную инфекцию, гинекологическую инфекцию и внутрибрюшную инфекцию. Также в объем данного изобретения входит применение описанного выше состава в качестве парентеральной инъекции для лечения инфекционного заболевания. Подробности одного или нескольких вариантов осуществления изобретения указаны в описании,приведенном ниже. Другие особенности, назначения и преимущества изобретения будут ясны из описания и из формулы изобретения.-1 017702 Подробное описание изобретения Соединение формулы I, используемое для практического осуществления данного изобретения,можно синтезировать стандартными способами. См. пример 1 ниже. Синтезированное таким образом соединение можно дополнительно очищать посредством хроматографии на испарительной колонке, высокоэффективной жидкостной хроматографии, кристаллизации или любых других подходящих способов. Для получения парентерального состава по данному изобретению можно просто смешивать в необходимых пропорциях и в любой последовательности соединение формулы I, обеспечивающее изотоничность средство и воду. Например, можно смешивать определенное количество соединения в определенной концентрации с физиологическим раствором (водным раствором, содержащим хлорид натрия, обеспечивающее изотоничность средство) Смешивание можно обеспечить встряхиванием, взбалтыванием или вихревым перемешиванием и его контролируют с целью обеспечения отсутствия сильного пенообразования при растворении твердых ингредиентов в воде. На любой стадии получения можно применять стерилизацию, например автоклавирование. Также состав по данному изобретению может содержать одно или несколько добавочных средств,таких как буфер, стабилизирующее средство и антиоксидант. Примеры буфера включают, но не ограничиваются ими, ацетат, цитрат, тартрат, лактат, сукцинат, малат и фосфат. Примеры стабилизирующего средства включают, но не ограничиваются ими, гистидин, лизин, глицин, сахарозу, фруктозу, трегалозу и их смесь. Примеры антиоксиданта включают, но не ограничиваются ими, бисульфит натрия, бутилированный гидроксианизол, цистеин, гентизиновую кислоту, глутамат мононатрия, тиогликолят натрия и аскорбиновую кислоту. Добавочные средства можно включать в состав на любой стадии его получения. Как известно специалистам в данной области, подходящую для достижения надлежащего эффекта концентрацию добавочного средства в составе можно определить стандартными способами. Состав по данному изобретению можно применять сразу после получения или можно сохранять для дальнейшего применения. Для немедленного применения можно предоставлять набор, состоящий из флакона с соединением формулы I и другого флакона с обеспечивающим изотоничность средством или с водным раствором, содержащим обеспечивающее изотоничность средство. Альтернативно можно предоставлять набор, состоящий из флакона с соединением формулы I и другого флакона с водным раствором обеспечивающего изотоничность средства (например, физиологическим раствором). Набор также может содержать одно или несколько добавочных средств, таких как стабилизирующее средство, буфер или антиоксидант. Непосредственно перед введением для получения состава можно смешивать содержащиеся в наборе вещества. Состав по данному изобретению можно применять для лечения инфекционного заболевания посредством введения нуждающемуся в лечении индивидууму эффективного количества состава посредством парентеральной инъекции или инфузии. Как применяют в настоящем документе, термин "лечение" определен как введение эффективного количества состава индивидууму с инфекционным заболеванием, симптомом инфекции, заболеванием или нарушением, вторичным относительно инфекции, или с предрасположенностью к инфекции с целью лечения, ослабления, облегчения, устранения или улучшения инфекционного заболевания, симптома инфекции или нарушения, вторичного относительно инфекции, или предрасположенности к инфекции. Термин "эффективное количество" относится к количеству состава, обеспечивающему терапевтический эффект у получающего лечение индивидуума. Как применяют в настоящем документе термин "парентеральный" включает подкожный, внутрикожный, внутривенный, внутримышечный, внутрисуставной, внутриартериальный, внутрисиновиальный, внутригрудинный, внутрипозвоночный, внутрирубцовый и внутричерепной инъекционные или инфузионные способы введения. Среди них предпочтительными являются внутривенная инъекция или инфузия. Без дополнительного уточнения полагают, что приведенное выше описание адекватно представляет настоящее изобретение. Таким образом, следующие далее примеры, следует истолковывать исключительно как иллюстративные и ни в коем случае не ограничивающие оставшуюся часть описания. Таким образом, все процитированные в настоящем документе публикации включены в качестве ссылки в полном объеме. Пример 1. Соль яблочной кислоты (3S,5S)-7-[3-амино-5-метилпиперидинил]-1-циклопропил-1,4-дигидро-8 метокси-4-оксо-3-хинолинкарбоновой кислоты (соединение 1) синтезируют, как указано ниже: В 50-л реактор помещали соединение 2 (5,50 кг, 42,60 моль), метанол (27 л) и охлаждали до 1015C. В пределах периода 65 мин через дополнительную воронку добавляли тионилхлорид (10,11 кг, 2,0 экв.) с внешним охлаждением для поддержания температуры ниже 30. Получаемый раствор перемешивали при 25C в течение 1,0 ч, после чего метанол удаляли при пониженном давлении. Маслянистый остаток переводили в азеотропную смесь этилацетатом (32,5 л) для удаления метанола, растворяли в этилацетате (27,4 л), помещали в 50-л реактор и нейтрализовали медленным добавлением триэтиламина(3,6 кг) ниже 30C. Получаемую суспензию фильтровали с удалением триэтиламингидрохлорида. Фильтрат помещали в 50-л реактор вместе с DMAP (0,53 кг). В пределах периода 30 мин через нагретую горячей водой дополнительную воронку добавляли ди-трет-бутилдикарбонат (8,43 кг) при температуре 20-30C. По данным анализа TLC реакция завершалась через 1 ч. Органическую фазу промывали ледяным 1 Н HCl (27,5 л), насыщали раствором бикарбоната натрия (17,5 л), высушивали над сульфатом магния и фильтровали. После удаления этилацетата при пониженном давлении получали кристаллическую взвесь, растирали ее с МТВЕ (10,0 л) и фильтровали с получением соединения 3 в виде белого твердого вещества (5,45 кг, 52,4%). Аналитически рассчитано для C11H17NO5: C, 54,3; H, 7,04; N, 5,76. Выявлено: C, 54,5; Н, 6,96; N,5,80. HRMS (ESI+) Ожидалось для CuH18NO5, [M+H] 244,1185. Установлено 244,1174; 1H ЯМР (CDCl3 500 МГц): =4, 54 (дд, J=3,1, 9,5 Гц, 1H), 3,7 (с, 3H), 2,58-2,50 (м, 1H), 2,41 (ддд, 1H,J=17,6, 9,5, 3,7), 2,30-2,23 (м, 1H), 1,98-1,93 (м, 1H), 1,40 (с, 9 Н); 13 С ЯМР (CDCl3, 125,70 МГц)173,3, 171,9, 149,2, 83,5, 58,8, 52,5, 31,1, 27,9, 21,5; Мр 70,2C. В 50-л реактор помещали соединение 3 (7,25 кг, 28,8 моль), DME (6,31 кг) и реагент Bredereck (7,7 кг, 44,2 моль). Раствор перемешивали и нагревали до 75C5C в течение 3 ч. Реакционную смесь охлаждали до 0C на протяжении 1 ч, в течение которого образовывался преципитат. Смесь выдерживали в течение часа при 0, фильтровали и высушивали в вакууме по меньшей мере 30 ч при 30C5C с выходом соединения 4 (6,93 кг, 77,9%) в виде белого твердого кристаллического вещества. Аналитически рассчитано для C14H22N2O5: C, 56,4; H, 7,43; N, 9,39. Выявлено C, 56,4; H, 7,32; N,9,48; HRMS (ESI+) Ожидалось для C14H22N2O5, [M+H] 299,1607. Аналитически рассчитано 299,1613; 1H ЯМР (CDCl3, 499,8 МГц)7,11 (с, 1H), 4,54 (дд, 1H, J=10,8, 3,6), 3,74 (с, 3H), 3,28-3,19 (м, 1H),3,00 (с, 6H), 2,97-2,85 (м, 1H), 1,48 (с, 9 Н); 13 С ЯМР (CDCl3, 125,7 МГц)172,6, 169,5, 150,5, 146,5, 90,8, 82,2, 56,0, 52,3, 42,0, 28,1, 26,3. МР 127,9C. В 45,5-л реактор Pfaudler помещали 5% порошок палладия с углеродом (50% влажность, 0,58 кг влажн. вес.) ESCAT 142 (Engelhard Corp. N.J, US), соединение 4 (1,89 кг, 6,33 моль) и изопропанол (22,4 кг). После перемешивания в атмосфере водорода под давлением 704 кг/м 2 в течение 18 ч при 45C реакционную смесь охлаждали до комнатной температуры и фильтровали через слой целита (0,51 кг). Фильтрат выпаривали при пониженном давлении с получением густого масла, которое затвердевало после отстаивания с получением соединения 5 (1,69 кг, 100%) в виде 93:7 диастереоизомерной смеси. Образец смеси продукта очищали при помощи препаративной ВЭЖХ с получением вещества для получения аналитических данных. Аналитически рассчитано для C12H19NO5: C, 56,0; H, 7,44; N, 5,44. Выявлено C, 55,8; H, 7,31; N,5,44; MS (ESI+) Ожидалось для C12H19NO5, [M+H] 258,1342. Аналитически рассчитано 258,1321; 1-3 017702 В 50-л реактор помещали соединение 5 (3,02 кг, 11,7 моль), абсолютный этанол (8,22 кг) и МТВЕ(14,81 кг). При 0C5C малыми порциями добавляли борогидрат натрия (1,36 кг, 35,9 моль). Наблюдали небольшое выделение пузырьков газа. Реакционную смесь нагревали до 10C5C и в течение часа порциями добавляли дигидрат хлорида кальция (2,65 кг) при 10C5C. Реакционной смеси позволяли нагреваться до 20C5C в течение одного часа, а затем перемешивали дополнительные 12 ч при 20C5C. После охлаждения реакционной смеси до -5C5C медленно добавляли ледяной 2 Н HCl(26,9 кг) при 0C5C. Перемешивание останавливали. Удаляли нижнюю водную фазу. В течение 5 мин реактор заполняли водным насыщенным раствором бикарбоната натрия (15,6 кг) при перемешивании. Снова останавливали перемешивание и удаляли нижнюю водную фазу. В реактор помещали сульфат магния (2,5 кг) и перемешивали по меньшей мере 10 мин. Смесь фильтровали через нутч-фильтр и концентрировали при пониженном давлении с получением соединения 6 (1,80 кг, 66%). Аналитически рассчитано для C11H23NO4: C, 56,6 H, 9,94; N, 6,00. Выявлено C, 56,0; H, 9,68; N, 5,96;HRMS (ESI+) Ожидалось для C11H24NO4, [M+H] 234,1705. Аналитически рассчитано 234,1703; 1 Н ЯМР (CDCl3, 500 МГц)6,'34 (d, J=8,9 Гц, 1H, NH), 4,51 (т, J=5,8, 5,3 Гц, 1H, NHCHCH2OH),4,34 (т, J=5,3, 5,3 Гц, 1H, CH3CHCH2OH), 3,46-3,45, (м, 1H, NHCH), 3,28 (дд, J=10,6, 5,3 Гц,NHCHCHHOH), 3,21 (дд, J=10,2, 5,8 Гц, 1H, CH3CHCHHOH), 3,16 (дд, J=10,2, 6,2 Гц, 1H,NHCHCHHOH), 3,12 (дд, J=10,6, 7,1 Гц, 1H, CH3CHCHHOH), 1,53-1,50 (м, 1H, CH3CHCHHOH), 1,35 (с,9 Н, O(CH3)3, 1,30 (ддд, J=13,9, 10,2, 3,7 Гц, 1H, NHCHCHHCH), 1,14 (ддд, J=13,6, 10,2, 3,4 Гц, 1H,NHCHCHHCH), 0,80 (д, J=6,6 Гц, 3H, СН 3); 13 С ЯМР (CDCl3, 125,7 МГц) : 156,1, 77,9, 50,8, 65,1, 67,6,65,1, 35,6, 32,8, 29,0, 17,1. Мр 92,1C. В 50-л реактор помещали раствор соединения 6 (5,1 кг) в изопропилацетате (19,7 кг). Реакционную смесь охлаждали до 15C5C и при этой температуре добавляли триэтиламин (7,8 кг). Затем реактор охлаждали до 0C5C и добавляли метансульфонилхлорид (MsCl) (6,6 кг). Реакционную смесь перемешивали в течение нескольких часов и завершение реакции определяли посредством ВЭЖХ или TLC. Реакцию подавляли насыщенным водным раствором бикарбоната. Органическую фазу выделяли и успешно промывали охлажденным 10% водным раствором триэтиламина, охлажденным водным раствором HCl,охлажденным насыщенным водным раствором бикарбоната и, наконец, насыщенным водным солевым раствором. Органическую фазу высушивали, фильтровали и концентрировали в вакууме при температуре ниже 55C5C с получением соединения 7 в виде твердой/жидкой кристаллической взвеси, которую использовали в последующей реакции без дополнительного очищения. 50-л реактор с помещенными 9,1 кг очищенного бензиламина нагревали до температуры 55C, при которой добавляли раствор соединения 7 (8,2 кг) в 1,2-диметоксиэтане (14,1 кг). После добавления реакционную смесь перемешивали при 60C5C в течение нескольких часов и завершение реакции определяли посредством ВЭЖХ или TLC. Реакцию охлаждали до температуры окружающей среды и растворитель удаляли в вакууме. Остаток растворяли в 11,7 кг 15% (об./об.,), раствора этилацетата/гексана и обрабатывали при перемешивании 18,7 кг 20% (вес) раствора водного карбоната калия. После отстаивания получали трехфазную смесь. Собирали верхний органический слой. Выделенный средний слой дважды экстрагировали 11,7 кг порциями 15% (об./об.) раствора этилацетата/гексана. Смешанные органические слои концентрировали в вакууме с получением масляного остатка. Затем остаток очищали при помощи хроматографии с получением соединения 8 в виде масла. В 40-л сосуд высокого давления в потоке азота помещали 0,6 кг твердого палладия на углероде(E101, 10 мас.%) 50% влажности. Затем в реактор в атмосфере азота добавляли раствор соединения 8 (3,2 кг) в 13,7 кг абсолютного этанола. Реактор прочищали азотом, а затем накачивали водород до 704 кг/м 2. Затем реакционную смесь нагревали до 45C. Реакцию контролировали посредством TLC или LC. После завершения реакционную смесь охлаждали до температуры окружающей среды, удаляли газ и прочищали азотом. Смесь фильтровали через слой целита и твердый сухой остаток промывали 2,8 кг абсолютного этанола. Фильтрат концентрировали в вакууме с получением соединения 9 в виде воскового сухого остатка. Коэффициент удерживания (Rf) TLC (диоксид кремния F254, 70:30 об./об. этилацетат-гексан, пятно(C) Синтез хелата сложного боронового эфира 1-циклопропил-7-фтор-8-метокси-4-оксо-1,4 дигидрохинолин-3-карбоновой кислоты (соединение 11) В реактор помещали оксид бора (2,0 кг, 2 9 моль), ледяную уксусную кислоту (8,1 л, 142 моль) и уксусный ангидрид (16,2 л, 171 моль). Полученную смесь кипятили с обратным холодильником по меньшей мере 2 ч и затем охлаждали до температуры 40C, при которой добавляли 7-фторхинолоновую кислоту, соединение 10 (14,2 кг, 51 моль). Смесь кипятили с обратным холодильником по меньшей мере 6 ч и затем охлаждали приблизительно до 90C. В реакционную смесь добавляли толуол (45 л). Для инициации осаждения при 50C добавляли трет-бутилметиловый эфир (19 л). Затем смесь охлаждали до 20C и фильтровали для выделения осадка. Затем выделенный сухой остаток промывали третбутилметиловым эфиром (26 л) перед высушиванием в вакуумной печи при 40C (50 мм рт. ст.) с получением соединения 11 с выходом 86,4%. Рамановское рассеяние (см-1): 3084,7, 3022,3, 2930,8, 1709,2, 1620,8, 1548,5, 1468,0, 1397,7, 1368,3,1338,5, 1201,5, 955,3, 653,9, 580,7, 552,8, 384,0, 305,8. ЯМР (CDCl3, 300 МГц)(м.д): 9,22 (с, 1H), 8,38-8,33 (м, 1H), 7,54 (т, J=9,8 Гц, 1H), 4,38-4,35 (м,1H), 4,13 (с, 3H), 2,04 (с, 6H), 1,42-1,38 (м, 2H), 1,34-1,29 (м, 2H). В реактор помещали соединение 11 (4,4 кг, 10,9 моль), соединение 9 (2,1 кг, 9,8 моль), триэтиламин(TEA) (2,1 л, 14,8 моль) и ацетонитрил (33,5 л, 15,7 л/кг). Полученную смесь перемешивали приблизительно при 50C до завершения реакции, которую контролировали посредством ВЭЖХ или обратнофазовой TLC. Ее охлаждали приблизительно до 35C и объем реакционной смеси уменьшали приблизительно наполовину посредством выпаривания ацетонитрила в вакууме 0-400 мм рт.ст. После добавления 28,2 кг 3,0 Н раствора NaOH (водн.) реакционную смесь нагревали приблизительно до 40C, выпаривали в вакууме до полного исчезновения продукта выпаривания и гидролизовали при комнатной температуре. После завершения гидролиза, которое контролировали посредством ВЭЖХ или обратно-фазовой TLC,для нейтрализации реакционной смеси добавляли 4-5 кг ледяной уксусной кислоты. Полученный раствор трижды экстрагировали 12,7 кг (9,6 л) дихлорметана. Органический слой-5 017702 смешивали и переносили в другой реактор. Объем реакционной смеси уменьшали приблизительно наполовину посредством испарения при 40C. После добавления 20,2 кг раствора 6,0 Н HCl (водн.), реакционную смесь перемешивали по меньшей мере 12 ч при 35C. После завершения реакции по показаниям ВЭЖХ или обратно-фазовой TLC продолжали перемешивание для разделения фаз. Органическую фазу удаляли и водный слой экстрагировали с помощью 12,7 кг (9,6 л) дихлорметана. Водный слой растворяли в 18,3 кг дистиллированной воды и нагревали приблизительно до 50C. Затем удаляли дихлорметан посредством выпаривания в вакууме (100-400 мм рт.ст.). Затем pH водного раствора доводили до 7,8-8,1 посредством добавления приблизительно 9,42 кг 3,0 Н NaOH (водн.) ниже 65C. Реакционную смесь перемешивали по меньшей мере час при 50C и затем охлаждали до комнатной температуры. Осадок выделяли фильтрованием с отсасыванием, дважды промывали 5,2 кг дистиллированной воды, высушивали по меньшей мере 12 ч с отсасыванием и затем еще 12 ч в конвекционной печи при 55C. Получали соединение 12 (3,2 кг, 79%) в виде твердого вещества. В реактор помещали 3,2 кг соединения 12 и 25,6 кг 95% этанола. В реактор добавляли 1,1 кг твердой D,L-яблочной кислоты. Смесь кипятили с обратным холодильником при температуре (80C). Осадок растворяли в дистиллированной воде (5,7 л) и добавляли 0,2 кг актированного угля. Реакционную смесь пропускали, через фильтр. Чистый фильтрат охлаждали до 45C и осаждали по меньшей мере 2 ч с кристаллизацией. Затем реакционную смесь охлаждали до 5C, осадок выделяли фильтрованием с отсасыванием, промывали 6,6 кг 95% этанола и высушивали с отсасыванием по меньшей мере 4 ч. Затем твердый остаток высушивали в конвекционной печи при 45C по меньшей мере 12 ч с получением 3,1 кг соединения 1 (выход: 70%). ЯМР (D2O, 300 МГц)(м.д.): 8,54 (с, 1H), 7,37 (д, J=9,0 Гц, 1H), 7,05 (д, J=9,0 Гц, 1H), 4,23-4,18 (м,1H), 4,10-3,89 (м, 1H), 3,66 (шир. с, 1H), 3,58 (с, 3H), 3,45 (д, J=9,0 Гц, 1H), 3,34 (d, J=9,3 Гц, 1H), 3,16 (д,J=12,9 Гц, 1H), 2,65 (дд, J=16,1, 4,1 Гц, 1H), 2,64-2,53 (м, 1H), 2,46 (дд, J=16,1, 8,0 Гц, 1H), 2,06 (шир.: с,1H), 1,87 (д, J=14,4 Гц, 1H), 1,58-1,45 (м, 1H), 1,15-0,95 (м, 2H), 0,91 (д, J=6,3 Гц, 3H), 0,85-0,78 (м, 2H). Соединение 1 растворяли в ацетонитриле/воде/муравьиной кислоте (12:88:0,2). Полученный раствор анализировали посредством градиентной обратно-фазовой ВЭЖХ с детекцией в УФ при 292 нм. Разделение осуществляли с использованием градиентного элюирования (см. таблицу), на колонке C8(Waters Symmetry Shield RP 8,5 мкм, 4,6150 мм) с подвижной фазой, содержащей ацетонитрил, воду и муравьиную кислоту, скоростью потока 1,5 мл/мин и при 30C. Составы подвижной фазы в зависимости от времени показаны в таблице ниже: Пример 2. Получали и исследовали растворы состава с декстрозой и хлоридом натрия: 1. Состав в 5% растворе декстрозы. Соединение 1 растворяли в 5% водном стерильном растворе декстрозы (5 мг/мл). Раствор фильтровали и переносили в 100-мл полипропиленовые бутылки. Бутылки накрывали крышками, герметизировали, стерилизовали 35 мин при 110C в печи и хранили при 60C. Раствор в бутылках анализировали через 0,5 и 10 суток. Представленные ниже результаты демонстрируют, что у раствора соединения 1 в 5% растворе декстрозы снижалось содержание активного компонента, увеличивалось значение pH, изменялся цвет раствора и образовывался коричневый осадок. 2. Состав в 0,9% физиологическом растворе. Соединение 1 растворяли в 0,9% физиологическом растворе (5 мг/мл). Раствор фильтровали и переносили в 100-мл полипропиленовые бутылки. Бутылки накрывали крышками, герметизировали, стерилизовали 35 мин при 110C в печи и хранили при 60C. Раствор в бутылках анализировали через 0,5 и 10 суток. Представленные ниже результаты демонстрируют, что состав неожиданно был более стабильным. Более точно, внешний вид и значения pH оставались неизменными в течение 10 суток при 60C. Содержание соединения 1 немного увеличивалось с течением времени вследствие испарения воды через пластиковые стенки бутылок. Общее количество примесей через 10 суток незначительно увеличивалось. Растворы соединения 1 в 0,9% физиологическом растворе получали в концентрации 0,1, 1, 3, 4, 5, 6,10 и 15 мг/мл. Эти растворы фильтровали и помещали в 100-мл полипропиленовые бутылки. Бутылки накрывали крышками, герметизировали и стерилизовали 35 мин при 110C. Бутылки с концентрациями 1, 3, 5, 10 мг/мл использовали для изучения токсичности на крысах и собаках согласно GLP. Пример 3. Исследовали влияние угля, значений pH и содержания железа на состав: 1. Влияние угля. 13,85 г соединения 1 и 18 г NaCl растворяли в стерильной воде. Конечный объем раствора доводили до 2000 мл посредством добавления дополнительного количества стерильной воды и перемешивали с получением раствора, содержащего 5 мг/мл соединения 1. Раствор разделяли на четыре 500-мл порции. К каждой из четырех порций добавляли 0, 0,02, 0,05 и 0,5% (г/мл) активированного угля. Полученную смесь 25 мин кипятили с перемешиванием и фильтровали через 0,45-микронную фильтровальную бумагу. Фильтрат добавляли в ряд 100-мл полипропиленовых бутылок, которые накрывали крышками, герметизировали и стерилизовали 35 мин при 110C. Содержание соединения 1 и pH в каждой из четырех бутылок было проанализировано и представлено ниже. Для получения состава выбрано 0,05% (г/мл) активированного угля.-7 017702 2. Влияние pH. 2000 мл раствора соединения 1 в 0,9% физиологическом растворе получали способом, аналогичным описанному выше. Раствор разделяли на 6 равных порций. Значения pH для 6 порций доводили до 2,43, 3,00, 3,76, 4,51,6,01 и 7,13 посредством добавления разведенной соляной кислоты или гидроксида натрия. Внешний вид и состав растворов были проанализированы и представлены ниже. Из результатов видно, что соединение 1 с концентрацией 5 мг/мл в физиологическом растворе при pH 6,6 выпадало в осадок. 3. Влияние содержания железа. 1000 мл раствора соединения 1 в 0,9% физиологическом растворе получали способом, аналогичным описанному выше. Раствор разделяли на 2 равные порции. К одной порции добавляли 0,25 г активированного угля, а к другой добавляли 0,25 г активированного угля и 0,1 г порошка железа. Каждую из полученных смесей перемешивали и фильтровали в 100-мл полипропиленовую бутылку. Заполненные бутылки накрывали крышками, герметизировали и стерилизовали 35 мин при 110C. Внешний вид и pH каждого раствора представлены ниже. Основываясь на результатах, при получении парентерального состава следует избегать контакта с железом. 4. Влияние длительности и температуры стерилизации. 3000 мл раствора соединения 1 в 0,9% физиологическом растворе получали способом, аналогичным описанному выше. В раствор добавляли 1,5 г активированного угля (0,05% г/мл). Смесь перемешивали 15 мин и фильтровали. Фильтрат добавляли в ряд 100-мл полипропиленовых бутылок. Заполненные бутылки накрывали крышками, герметизировали и разделяли на четыре группы (7 бутылок/группа). Стерилизацию образцов проводили при 115C/35 мин, 110C/35 мин, 105C/35 мин. Содержание и уровень загрязнения соединения 1, а также pH каждой группы (включая контрольную группу) были измерены и представлены ниже. Основываясь на исследовании, выбрали стерилизацию парентерального состава при 110C в течение 35 мин. 5. Влияние низкой температуры (-15C). Бутылки, содержащие 5 мг/мл соединения 1 в 0,9% физиологическом растворе, хранили в моро-8 017702 зильной камере при -15C. Образцы анализировали через 0, 5 и 10 суток. Представленные ниже результаты показывают, что внешний вид, содержание соединения, pH и общее количество примесей оставались неизменными при -15C в течение 10 суток. 6. Влияние света на состав. Бутылки, содержащие раствор соединения 1 (5 мг/мл исходя из свободных оснований) в 0,9% физиологическом растворе помещали в световой короб с 4500 лк 500 лк. Образцы анализировали через 0,5 и 10 суток. Представленные ниже результаты показывают отсутствие изменений под действием яркого света. Пример 4. Получали растворы соединения 1 (5 мг/мл на основе свободного основания) в 0,9% физиологическом растворе, фильтровали и переносили в 100-мл полипропиленовые бутылки. Бутылки накрывали крышками, герметизировали, стерилизовали 35 мин при 110C, хранили 6 месяцев при 40C и относительной влажности (OB) 20% или 12 месяцев при 25C и OB 60% и проверяли, как показано в таблице ниже. В каждой пробной точке (обозначено как "X" в приведенной выше таблице) оценивали внешний вид, цвет, прозрачность, pH, поглощение в УФ, пробу и загрязнение образцов. Значимых изменений не выявлено. Пример 5. Активность соединения 1 in vitro и in vivo изучали следующим образом: 1. Антибактериальная активность in vitro. Из клинических образцов выделяли различные бактериальные штаммы, такие как грамположительные бактерии (например, Clostridium, Staphylococcus и Streptococcus), грамотрицательные бактерии (например, Moraxella, Haemophilus, Pseudomonas, Proteus и Bacteriodes), анаэробные и атипичные патогенные микроорганизмы. Антибактериальную активность соединения формулы I в отношении этих бактериальных штаммов определяли способами разведения в агаре, описанными в US National Committee forClinical Laboratory, M7-A6, 2003 и M11-A5, 2001. Из результатов видно, что соединение 1 проявляло сильную активность против широкого спектра бактерий, включая активность против грамположительных, грамотрицательных, анаэробных и атипичных патогенных микроорганизмов. Соль была особенно активна против Staphylococci и Streptococci,включая устойчивый к метициллину S. aureus (MRSA) и мультирезистентный S. pneumoniae. Минимальная ингибирующая концентрация (МИК 90) в отношении 90% выделенных патогенов чувствительных к-9 017702 ципрофлоксацину восприимчивых к метициллину S. aureus (MSSA) и MRSA составляла 0,06 мкг/мл. МИК 90 составила 0,5 мкг/мл против устойчивого к ципрофлоксацину MSSA и 1 мкг/мл - против устойчивого к ципрофлоксацину MFSA. Против восприимчивого, устойчивого к пенициллину и устойчивого к макролидам 5. pneumoniae МИК 90 составляла 0,12 мкг /мл. В данном исследовании значения МИК для всех Staphylococci и Streptococci составили 2 мкг/мл и 1 мкг/мл, соответственно. Рассматривая все грамположительные организмы, соединение 1 являлось в 4-8 раз более сильным, чем левофлоксацин, и в 2-4 раза более сильным, чем гатифлоксацин. Среди грамотрицательных микроорганизмов соединение 1 активно против Moraxella catarrhalis(МИК 90=0,03 мкг/мл), Haemophilus influenzae (МИК 90=0,12 мкг/мл) и Neisseria gonorrhoeae (МИК 90=0,06 мкг/мл). Соединение активно против большинства кишечных микроорганизмов с МИК 90=0,12 мкг/мл дляE. coli, МИК 90=1 мкг/мл для Klebsiella pneumoniae и МИК 90=0,5 мкг/мл для Proteus mirabilis. Оно также активно против многих выделенных Pseudomonas aeruginosa, а также анаэробных патогенов Clostridiumdifficile и Bacteroides species. 2. Эффективность in vivo. Антибактериальную эффективность соединения 1 in vivo оценивали в модели на мышах. Мышей обезболивали и интраназально инфицировали смертельным количеством S. pneumoniae Stp 6301. Через двенадцать, восемнадцать и двадцать четыре часа после инфицирования, мышам подкожно вводили композицию, содержащую соединение 1 или моксифлоксацин (в качестве положительного контроля), 0,7% молочную кислоту и 3% декстрозы при общих дозах 50, 25, 12,5, или 6,25 мг/кг. Половину из обработанных мышей умерщвляли через четыре часа после последнего лечения соединением 1 или моксифлоксацином и собирали их кровь и легочную ткань. Затем определяли количество живых бактерий в крови и легочной ткани. Легочную ткань также подвергали гистопатологическому исследованию. Другую половину мышей наблюдали 6 суток и регистрировали количество выживших особей. Результаты демонстрируют, что соединение 1 при всех испытанных дозах значительно снижало жизнеспособность бактерий в легких и крови по сравнению с получавшим плацебо контролем. Кроме того, антибиотик при всех, испытанных дозах обеспечивал полную защиту от смертельной инфекции(100% выживаемость). В данной модели легочной инфекции при одинаковых дозах соединение 1 было более эффективно, чем моксифлоксацин. Другие варианты осуществления Все свойства, описанные в данном описании, можно сочетать в любой комбинации. Все альтернативные свойства, удовлетворяющие аналогичным, эквивалентным или похожим условиям могут замещать любое свойство, описанное в данном описании. Таким образом, если явно не указано иначе, каждое описанное свойство является только примером общего ряда эквивалентных или сходных свойств. Из приведенного выше описания специалист в данной области может легко понять основные характеристики настоящего изобретения, и, не отклоняясь от его сущности и объема, может произвести различные изменения и модификации изобретения для его адаптации к различным условиям и применениям. Таким образом, другие варианты осуществления также находятся в объеме приведенной ниже формулы изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ лечения инфекционного заболевания, включающий введение нуждающемуся в этом субъекту путем парентеральной инъекции или инфузии эффективного количества состава, содержащего воду и обеспечивающее изотоничность средство в концентрации от 0,2 до 13% мас./об., при этом соединение и обеспечивающее изотоничность средство растворены в воде. 2. Способ по п.1, где инфекционное заболевание представляет собой инфекцию мочевыводящих путей, простатит, респираторную инфекцию, остеомиелит, гонорею, Mycobacterium tuberculosis, комплексMycobacterium avium, острые приступы хронического бронхита, пневмонию, синусит, инфекционную диарею, Helicobacter pylori, кожную инфекцию, гинекологическую инфекцию или внутрибрюшную инфекцию. 3. Способ по п.1, где инфекционное заболевание вызвано грамотрицательными или грамположительными бактериями. 4. Способ по п.1, где инфекционное заболевание вызвано анаэробными бактериями. 5. Способ по п.1, где инфекционное заболевание вызвано устойчивым к метициллину S. aureus или мультирезистентным S. pneumoniae.- 10017702 6. Способ по п.1, где обеспечивающее изотоничность средство представляет собой хлорид натрия. 7. Способ по п.6, где концентрация соединения в составе составляет от 0,2 до 45 мМ, а концентрация хлорида натрия в составе составляет от 0,2 до 1,3% мас./об. 8. Способ по п.7, где состав вводят посредством внутривенной инъекции или инфузии. 9. Способ по п.7, где концентрация хлорида натрия в составе составляет 0,9% мас./об. 10. Способ по п.7, где инфекционное заболевание представляет собой инфекцию мочевыводящих путей, простатит, респираторную инфекцию, остеомиелит, гонорею, Mycobacterium tuberculosis, комплекс Mycobacterium avium, острые приступы хронического бронхита, пневмонию, синусит, инфекционную диарею, Helicobacter pylori, кожную инфекцию, гинекологическую инфекцию или внутрибрюшную инфекцию. 11. Способ по п.7, где инфекционное заболевание вызвано грамотрицательными или грамположительными бактериями. 12. Способ по п.7, где инфекционное заболевание вызвано анаэробными бактериями. 13. Способ по п.7, где инфекционное заболевание вызвано устойчивым к метициллину S. aureus или мультирезистентным S. pneumoniae. 14. Способ по п.7, где состав дополнительно содержит стабилизирующее средство, где стабилизирующее средство выбрано из группы, состоящей из гистидина, лизина, глицина, сахарозы, фруктозы,трегалозы и их смеси. 15. Способ по п.7, где состав дополнительно содержит буфер, где буфер выбран из группы, состоящей из ацетата, цитрата, тартрата, лактата, сукцината, малата или фосфата. 16. Способ по п.7, где состав дополнительно содержит антиоксидант, где антиоксидант выбран из группы, состоящей из дисульфита натрия, бутилированного гидроксианизола, цистеина, гентизиновой кислоты, моноглутамата натрия, тиогликолята натрия и аскорбиновой кислоты. 17. Способ по п.7, где состав дополнительно содержит стабилизирующее средство в концентрации 0,1-1,0% мас./об. и буфер в концентрации 0,01-5% мас./об. 18. Способ по п.1, где обеспечивающее изотоничность средство выбрано из группы, состоящей из глицерина, лактозы, маннита, декстрозы, сульфата натрия и сорбита. 19. Способ по п.18, где обеспечивающее изотоничность средство представляет собой декстрозу и его концентрация в составе составляет 1-7% мас./об. 20. Способ по п.19, где состав дополнительно содержит стабилизирующее средство в концентрации 0,1-1,0% мас./об. и буфер в концентрации 0,01-5% мас./об. 21. Способ по п.19, где состав вводят посредством внутривенной инъекции или инфузии.
МПК / Метки
МПК: A61K 33/14, A61P 31/04, A61K 31/4709
Метки: лечения, инфекционного, способ, заболевания
Код ссылки
<a href="https://eas.patents.su/12-17702-sposob-lecheniya-infekcionnogo-zabolevaniya.html" rel="bookmark" title="База патентов Евразийского Союза">Способ лечения инфекционного заболевания</a>
Пиримидинилсульфонамидные соединения (варианты), способ получения пиримидинилсульфонамидных соединений (варианты), фармацевтическая композиция, способ лечения заболевания, опосредованного интегрином α4, способ снижения и/или предупреждения воспалительного компонента заболевания или аутоиммунного ответа

Номер патента: 17110
Опубликовано: 28.09.2012
Авторы: Сэмкоу Кристофер, Сюй Ин-Цзы, Смит Дженифер Л., Конрэди Андрей В.
МПК: A61P 29/00, A61K 31/506, A61P 11/06...
Метки: ответа, снижения, соединений, опосредованного, соединения, аутоиммунного, интегрином, пиримидинилсульфонамидные, воспалительного, пиримидинилсульфонамидных, компонента, композиция, alpha;4, заболевания, фармацевтическая, лечения, получения, способ, варианты, предупреждения
Формула / Реферат:
1. Пиримидинилсульфонамидные соединения, охватываемые общей общей формулой Iв которой R1 выбран из группы, включающей С1-С4алкил и С1-С4галоалкил; аR2 выбран из группы, включающей С1-С4алкил, С2-С4алкенил, С2-С4алкинил и С3-С6циклоалкил;или их фармацевтически приемлемые соли или сложные эфиры.2. Соединения по п.1, в которых R1 представляет собой С1-С2алкил.3. Соединения по п.1, в которых R1 представляет собой метил или трифторметил.4. Соединения...
Композиции для лечения заболеваний , опосредованных циклооксигеназой-2, способ лечения воспалительного заболевания

Номер патента: 1596
Опубликовано: 25.06.2001
Авторы: Гертц Барри, Винтерс Конрад, Эхрич Эллиот, Хэнкок Бруно
МПК: A61P 19/00, A61K 31/366
Метки: заболевания, циклооксигеназой-2, лечения, опосредованных, композиции, воспалительного, заболеваний, способ
Формула / Реферат:
1. Фармацевтическая композиция для лечения заболеваний, опосредованных циклооксигеназой-2, приемлемая для перорального введения один раз в сутки, включающая 12,5 или 25 или 50 мг 3-фенил-4-(4-метилсульфонил)фенил)-2-(5Н)-фуранона. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что дополнительно включает (a) микрокристаллическую целлюлозу, (b) моногидрат лактозы, (c) гидроксипропилцеллюлозу, (d) натрий кроскармелозу, (е) окись...
Применение индазолметоксиалкановой кислоты для лечения заболевания, связанного с уровнями триглицерида, холестерина и/или глюкозы и способ лечения указанных заболеваний

Номер патента: 16885
Опубликовано: 30.08.2012
Авторы: Гульельмотти Анджело, Бьонди Джузеппе
МПК: A61P 3/04, A61P 3/06, A61K 31/416...
Метки: кислоты, указанных, уровнями, способ, применение, связанного, заболеваний, холестерина, индазолметоксиалкановой, триглицерида, заболевания, глюкозы, лечения
Формула / Реферат:
1. Применение соединения формулы (I)где R и R', которые могут быть одинаковыми или разными, представляют собой Н или С1-5алкил иR'' представляет собой Н или С1-4алкил,когда R'' означает Н, в виде его соли с фармацевтически приемлемым органическим или неорганическим основанием, для приготовления фармацевтической композиции, для лечения заболевания, связанного с повышенными уровнями триглицерида, холестерина и/или глюкозы в крови, выбранного из...
Способ лечения заболевания

Номер патента: 15272
Опубликовано: 30.06.2011
Автор: Макинтош Дейрдр
МПК: A61K 38/35
Метки: заболевания, лечения, способ
Формула / Реферат:
1. Способ лечения заболевания, выбранного из ревматоидного артрита; неврита зрительного нерва; аутоиммунных заболеваний, включающих волчанку, псориаз, экзему, тиреоидит и полимиозит; злокачественных новообразований, в частности миелом, меланом и лимфом; воспалительных состояний; ожирения и сексуальной дисфункции, в частности эректильной дисфункции; включающий введение CRF (кортикотропинрилизинг-фактора) пациенту, нуждающемуся в этом.2. Способ...
Применение бактериальной комбинированной композиции для получения средства для профилактики и/или лечения гипероксалурии у людей и животных и способ профилактики и/или лечения указанного заболевания

Номер патента: 4257
Опубликовано: 26.02.2004
Автор: Де Симоне Клаудио
МПК: A23L 1/03, A61P 13/04, A61K 39/02...
Метки: профилактики, средства, получения, способ, лечения, гипероксалурии, комбинированной, заболевания, указанного, животных, бактериальной, применение, людей, композиции
Формула / Реферат:
1. Применение бактериальной комбинированной композиции, включающей в качестве штаммов бактерий (a) Streptococcus thermophilus в смеси с (b) по меньшей мере одним штаммом бактерий, выбранным из группы, состоящей из Lactobacillus brevis, Lactobacillus acidophilus, Lactobacillus plantarum, Bifidobacterium infantis, Bifidobacterium longum и Bifidobacterium breve или их смесей, для получения диетического, фармацевтического или ветеринарного средства...
Предыдущий патент: Цепь, содержащая звенья
Следующий патент: Резьбовое соединение для труб
Случайный патент: Расчетно-платежная система