Пиримидинилсульфонамидные соединения (варианты), способ получения пиримидинилсульфонамидных соединений (варианты), фармацевтическая композиция, способ лечения заболевания, опосредованного интегрином α4, способ снижения и/или предупреждения воспалительного компонента заболевания или аутоиммунного ответа
Номер патента: 17110
Опубликовано: 28.09.2012
Авторы: Конрэди Андрей В., Сэмкоу Кристофер, Смит Дженифер Л., Сюй Ин-Цзы





Формула / Реферат
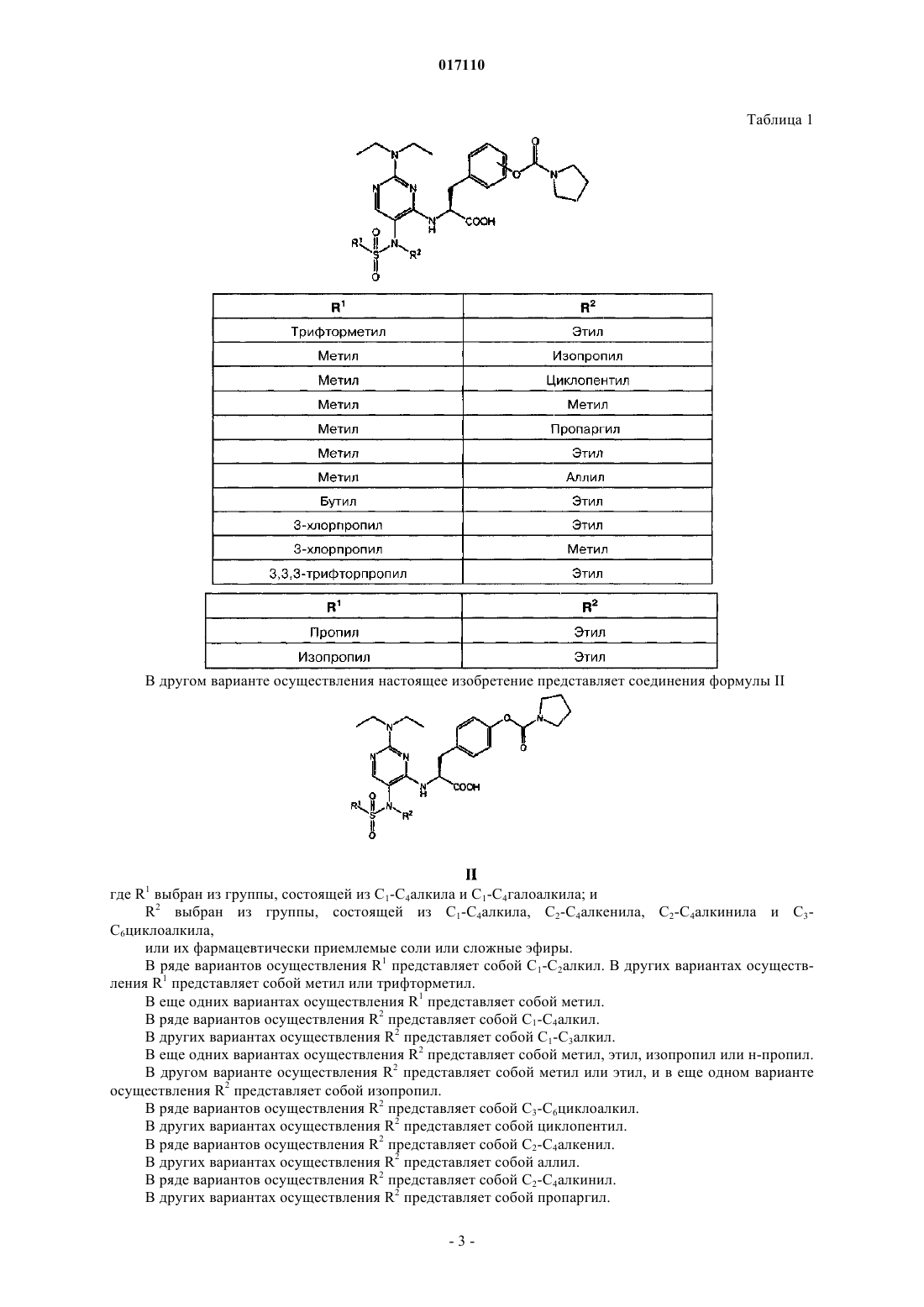
1. Пиримидинилсульфонамидные соединения, охватываемые общей общей формулой I

в которой R1 выбран из группы, включающей С1-С4алкил и С1-С4галоалкил; а
R2 выбран из группы, включающей С1-С4алкил, С2-С4алкенил, С2-С4алкинил и С3-С6циклоалкил;
или их фармацевтически приемлемые соли или сложные эфиры.
2. Соединения по п.1, в которых R1 представляет собой С1-С2алкил.
3. Соединения по п.1, в которых R1 представляет собой метил или трифторметил.
4. Соединения по п.1, в которых R1 представляет собой метил.
5. Соединения по п.1, в которых R2 представляет собой С1-С4алкил.
6. Соединения по п.1, в которых R2 представляет собой С1-С3алкил.
7. Соединения по п.6, в которых R2 представляет собой метил или этил.
8. Соединения по п.6, в которых R2 представляет собой изопропил.
9. Соединения по п.1, в которых R2 представляет собой С3-С6циклоалкил.
10. Соединения по п.9, в которых R2 представляет собой циклопентил.
11. Соединения по п.1, в которых R2 представляет собой С2-С4алкенил.
12. Соединения по п.11, в которых R2 представляет собой аллил.
13. Соединения по п.1, в которых R2 представляет собой С2-С4алкинил.
14. Соединения по п.13, в которых R2 представляет собой пропаргил.
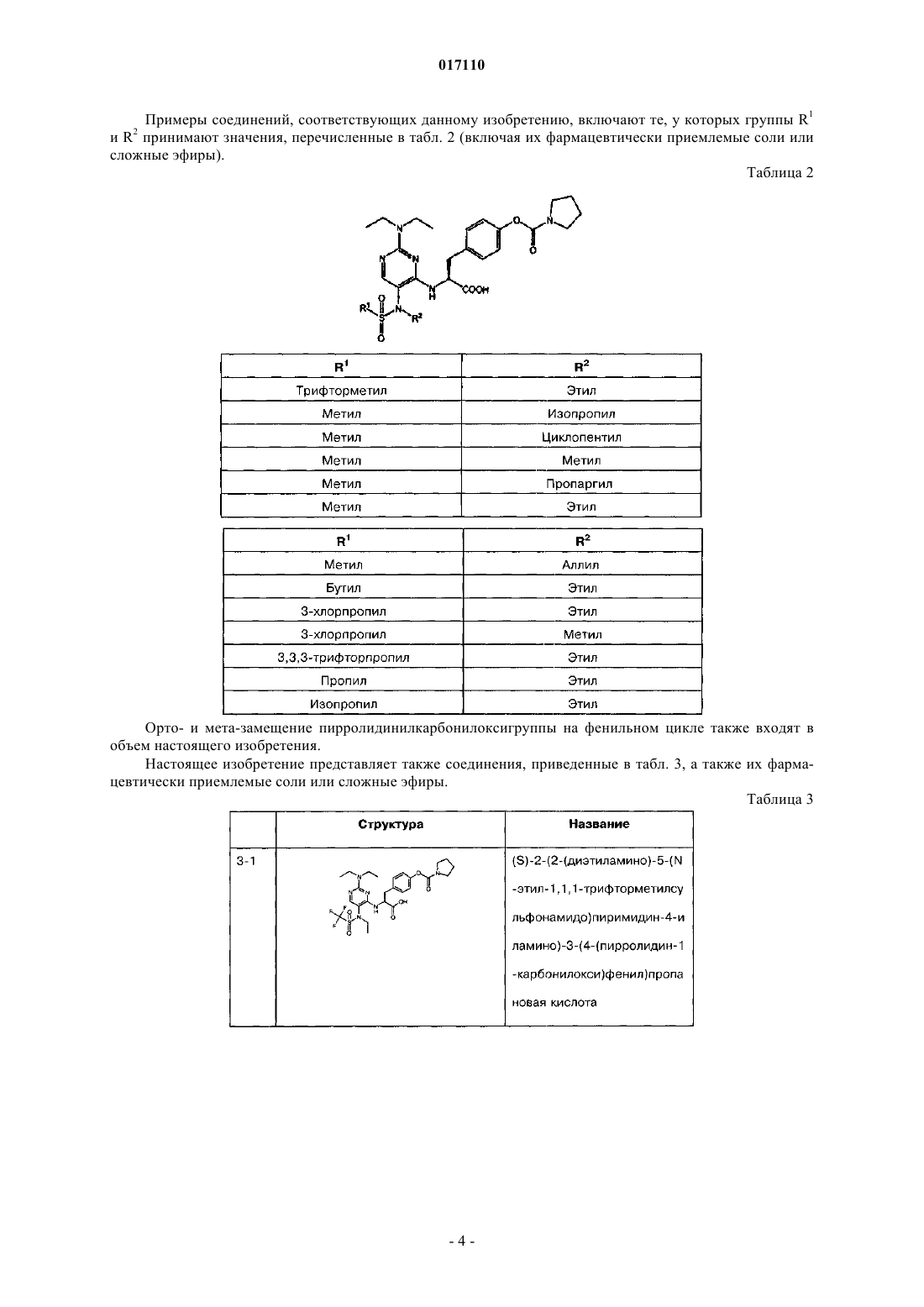
15. Пиримидинилсульфонамидные соединения, охватываемые общей общей формулой II

в которой R1 выбран из группы, включающей С1-С4алкил и С1-С4галоалкил; а
R2 выбран из группы, включающей С1-С4алкил, С2-С4алкенил, С2-С4алкинил и С3-С6циклоалкил,
или их фармацевтически приемлемые соли или сложные эфиры.
16. Соединения по п.15, в которых R1 представляет собой С1-С2алкил.
17. Соединения по п.15, в которых R1 представляет собой метил или трифторметил.
18. Соединения по п.15, в которых R1 представляет собой метил.
19. Соединения по п.15, в которых R2 представляет собой С1-С4алкил.
20. Соединения по п.15, в которых R2 представляет собой С1-С3алкил.
21. Соединения по п.20, в которых R2 представляет собой метил или этил.
22. Соединения по п.20, в которых R2 представляет собой изопропил.
23. Соединения по п.16, в которых R2 представляет собой С3-С6циклоалкил.
24. Соединения по п.23, в которых R2 представляет собой циклопентил.
25. Соединения по п.15, в которых R2 представляет собой С2-С4алкенил.
26. Соединения по п.25, в которых R2 представляет собой аллил.
27. Соединения по п.15, в которых R2 представляет собой С2-С4алкинил.
28. Соединения по п.27, в которых R2 представляет собой пропаргил.
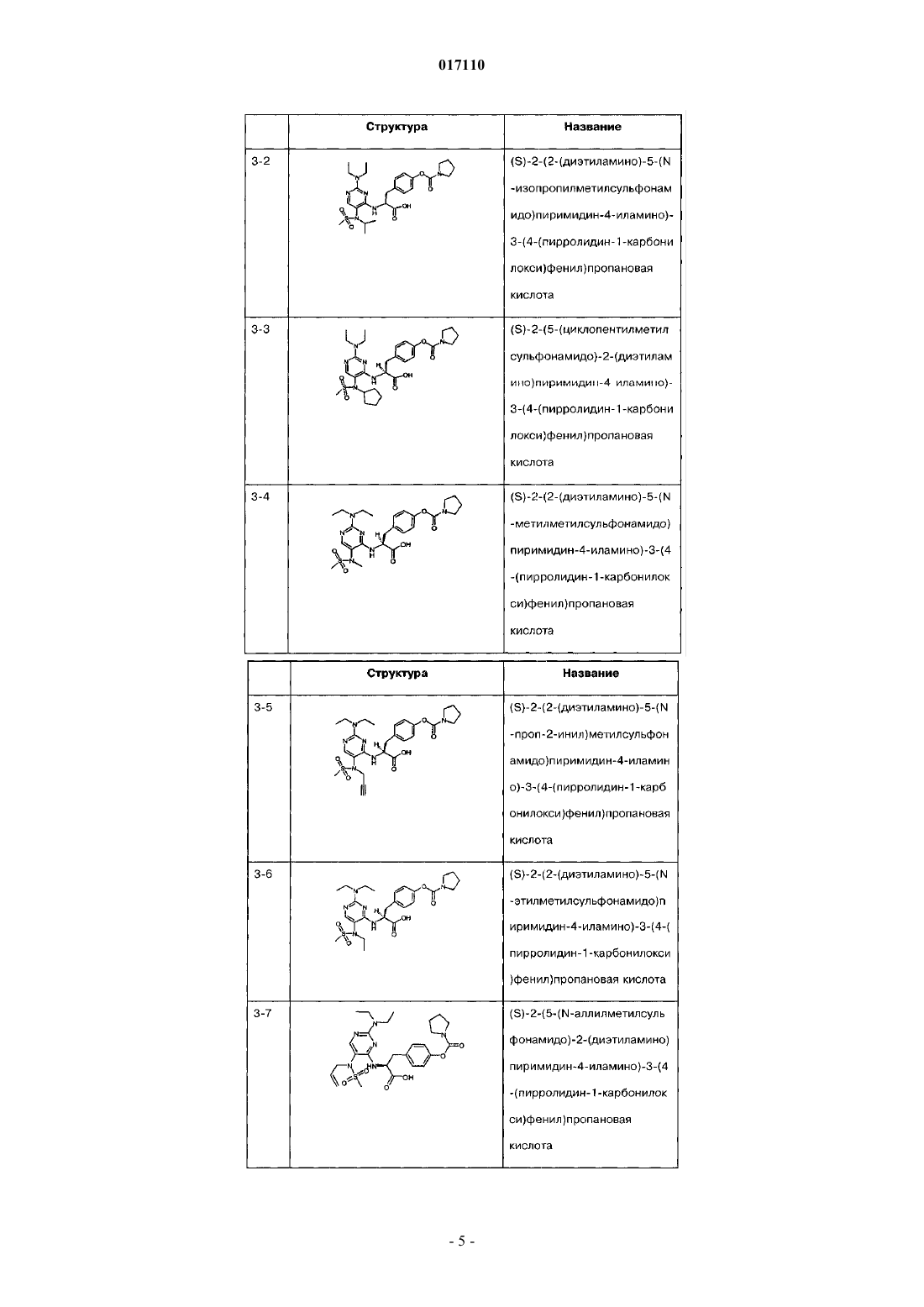
29. Пиримидинилсульфонамидные соединения, выбранные из группы, включающей
(S)-2-(2-(диэтиламино)-5-(N-этил-1,1,1-трифторметилсульфонамидо)пиримидин-4-иламино)-3-(4-(пирролидин-1- карбонилокси)фенил)пропановую кислоту;
(S)-2-(2-(диэтиламино)-5-(N-изопропилметилсульфонамидо)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту;
(S)-2-(5-(N-циклопентилметилсульфонамидо)-2-(диэтиламино)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту;
(S)-2-(2-(диэтиламино)-5-(N-метилметилсульфонамидо)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту;
(S)-2-(2-(диэтиламино)-5-(N-(проп-2-инил)метилсульфонамидо)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту;
(S)-2-(2-(диэтиламино)-5-(N-этилметилсульфонамидо)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту;
(S)-2-(5-(N-аллилметилсульфонамидо)-2-(диэтиламино)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту;
(S)-2-(2-(диэтиламино)-5-(N-этилбутилсульфонамидо)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту;
(S)-2-(5-(3-хлор-N-этилпропилсульфонамидо)-2-(диэтиламино)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту;
(S)-2-(5-(3-хлор-N-метилпропилсульфонамидо)-2-(диэтиламино)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту;
(S)-2-(2-(диэтиламино)-5-(N-этил-3,3,3-трифторпропилсульфонамидо)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту;
(S)-2-(2-(диэтиламино)-5-(N-этилпропилсульфонамидо)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту и
(S)-2-(2-(диэтиламино)-5-(N-этил-2-метилпропилсульфонамидо)пиримидин-4-иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановую кислоту;
а также их фармацевтически приемлемые соли или сложные эфиры.
30. Фармацевтическая композиция, включающая терапевтически эффективное количество по меньшей мере одного соединения по п.1 и фармацевтически приемлемый носитель.
31. Способ лечения заболевания, опосредованного, по меньшей мере частично, интегрином α4 у больного, заключающийся в том, что указанному больному вводят фармацевтическую композицию, включающую фармацевтически приемлемый носитель и терапевтически эффективное количество по меньшей мере одного из соединений, охватываемых общей формулой

в которой R1 выбран из группы, включающей С1-С4алкил и С1-С4галоалкил; а
R2 выбран из группы, включающей С1-С4алкил, С2-С4алкенил и С2-С4алкинил;
или их фармацевтически приемлемых солей или сложных эфиров.
32. Способ по п.31, в котором указанное заболевание выбрано из группы, включающей астму, рассеянный склероз и воспалительное заболевание кишечника.
33. Способ по п.31, в котором указанное заболевание представляет собой болезнь Крона.
34. Способ по п.31, в котором указанное заболевание представляет собой ревматоидный артрит.
35. Способ снижения и/или предупреждения воспалительного компонента заболевания у больного млекопитающего, который заключается в том, что указанному млекопитающему вводят фармацевтическую композицию, включающую фармацевтически приемлемый носитель и терапевтически эффективное количество по меньшей мере одного из соединений, охватываемых общей формулой

в которой R1 выбран из группы, включающей С1-С4алкил и С1-С4галоалкил; а
R2 выбран из группы, включающей С1-С4алкил, С2-С4алкенил и С2-С4алкинил;
или их фармацевтически приемлемых солей или сложных эфиров.
36. Способ снижения и/или предупреждения аутоиммунного ответа у млекопитающего больного, который заключается в том, что указанному млекопитающему вводят фармацевтическую композицию, включающую фармацевтически приемлемый носитель и терапевтически эффективное количество по меньшей мере одного из соединений, охватываемых общей формулой

в которой R1 выбран из группы, включающей С1-С4алкил и С1-С4галоалкил; а
R2 выбран из группы, включающей С1-С4алкил, С2-С4алкенил и С2-С4алкинил;
или их фармацевтически приемлемых солей или сложных эфиров.
37. Способ получения пиримидинилсульфонамидных соединений, охватываемых общей формулой I

в которой R1 выбран из группы, включающей С1-С4алкил и С1-С4галоалкил; а
R2 выбран из группы, включающей С1-С4алкил, С2-С4алкенил, С2-С4алкинил и С3-С6циклоалкил,
или их фармацевтически приемлемых солей или сложных эфиров,
в котором осуществляют взаимодействие соединения формулы III

где Pg представляет собой защитную группу карбоксила, с С1-С4альдегидом или кетоном, С2-С4алкенилальдегидом или кетоном, С2-С4алкинилальдегидом или кетоном, С3-С6циклоалкилкетоном и бензальдегидом в условиях восстановительного аминирования после восстановления нитрогруппы в формуле III до первичного амина с образованием соединения формулы IV

затем полученное соединение формулы IV вводят во взаимодействие с сульфонилгалогенидом формулы R1SO2Z, где Z представляет собой галоген, с образованием соединения формулы V

после чего из полученного соединения формулы (V) удаляют защитную группу карбоксила с образованием соединения формулы I.
38. Способ получения пиримидинилсульфонамидных соединений, охватываемых общей формулой I

в которой R1 выбран из группы, включающей С1-С4алкил и С1-С4галоалкил; а
R2 выбран из группы, включающей С1-С4алкил, С2-С4алкенил, С2-С4алкинил и С3-С6циклоалкил,
или их фармацевтически приемлемых солей или сложных эфиров,
в котором осуществляют взаимодействие соединения формулы VI

где Pg представляет собой защитную группу карбоксила, с избытком R1SO2X с образованием соединения формулы VII

затем из полученного соединения формулы VII избирательно удаляют одну группу -SO2R1 с образованием соединения формулы VIII

после чего полученное соединение формулы VIII вводят во взаимодействие с алкилирующим агентом формулы R2-X, где X представляет собой галоген, или с диметилсульфатом, где R2 представляет собой метил, с образованием соединения формулы IX

а затем из полученного соединения формулы IX удаляют защитную группу карбоксила с образованием соединения формулы I.
Текст
ПИРИМИДИНИЛСУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ (ВАРИАНТЫ), СПОСОБ ПОЛУЧЕНИЯ ПИРИМИДИНИЛСУЛЬФОНАМИДНЫХ СОЕДИНЕНИЙ (ВАРИАНТЫ),ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ,ОПОСРЕДОВАННОГО ИНТЕГРИНОМ 4, СПОСОБ СНИЖЕНИЯ И/ИЛИ ПРЕДУПРЕЖДЕНИЯ ВОСПАЛИТЕЛЬНОГО КОМПОНЕНТА ЗАБОЛЕВАНИЯ ИЛИ АУТОИММУННОГО ОТВЕТА Изобретатель: В настоящем изобретении предлагаются пиримидинилсульфонамидные соединения, охватываемые общей формулой I связывающие VLA-4. Некоторые из указанных соединений ингибируют также адгезию лейкоцитов и, в частности, адгезию лейкоцитов, опосредованную VLA-4. Предложенные соединения являются эффективными при лечении воспалительных заболеваний у человека или животных, таких как астма, болезнь Альцгеймера, атеросклероз, деменция при СПИДе, диабет, воспалительное заболевание кишечника, болезнь Крона, ревматоидный артрит, трансплантация ткани, метастазы опухоли и ишемия миокарда. Соединения по настоящему изобретению можно также применять для лечения воспалительных заболеваний головного мозга, таких как рассеянный склероз. 017110 Область техники, к которой относится изобретение Настоящее изобретение относится к пиримидинилсульфонамидным соединениям, которые ингибируют адгезию лейкоцитов, в частности адгезию лейкоцитов, опосредованную интегринами 4, где интегрин 4 предпочтительно представляет собой VLA-4. Настоящее изобретение относится также к фармацевтическим композициям, включающим вышеуказанные соединения, а также способам лечения, например, воспаления с использованием либо соединений, либо фармацевтических композиций, соответствующих данному изобретению. Сведения о предшествующем уровне техники Библиографический список. Указанные ниже публикации приведены в данной заявке в виде надстрочных цифр. 1. Hemler and Takada, Европейская публикация патентной заявки 330506, опубликована 30 августа 1989 г. 2. Elices et al., Cell., 60:577 584 (1990) 3. Springer, Nature, 346:425 434 (1990) 4. Osborn, Cell., 62:3 6 (1990) 5. Vedder et al., Surgery, 106:509 (1989) 6. Pretolani et al., J. Exp. Med., 180:795 (1994) 7. Abraham et al., J. Clin. Invest., 93:776 (1994) 8. Mulligan et al., J. Immunology, 150:2407 (1993) 9. Cybulsky et al., Science, 251:788 (1991) 10. Li et al., Arterioscier. Thromb., 13:197 (1993) 11. Sasseville et al., Am. J. Path., 144:27 (1994) 12. Yang et al., Proc. Nat. Acad. Science (USA), 90:10494 (1993) 13. Burkly et al., Diabetes, 43:529 (1994) 14. Baron et al., J. Clin. Invest., 93:1700 (1994) 15. Hamann et al., J. Immunology, 152:3238 (1994) 16. Yednock et al., Nature, 356:63 (1992) 17. Baron et al., J. Exp. Med., 177:57 (1993) 18. van Dinther-Janssen et al., J. Immunology, 147:4207 (1991) 19. van Dinther-Janssen et al., Annals. Rheumatic Dis., 52:672 (1993) 20. Elices et al., J. Clin. Invest., 93:405 (1994) 21. Postigo et al., J. Clin. Invest., 89:1445 (1991) 22. Paul et al., Transpl. Proceed., 25:813 (1993) 23. Okarhara et al., Can. Res., 54:3233 (1994) 24. Paavonen et al., Int. J. Can., 58:298 (1994) 25. Schadendorf et al., J. Path., 170:429 (1993) 26. Bao et al., Diff., 52:239 (1993) 27. Lauri et al., British J. Cancer, 68:862 (1993) 28. Kawaguchi et al., Japanese J. Cancer Res., 83:1304 (1992) 29. Konradi et al., PCT/USOO/01686, filed, January 21, 2000 Все вышеуказанные публикации включены в данном контексте в виде ссылки во всей своей полноте.and Takada1, представляет собой член семейства интегрина 1 клеточных поверхностных рецепторов,каждый из которых включает две субъединицы, -цепь и -цепь. VLA-4 содержит 4-цепь и 1-цепь. Существует по меньшей мере девять интегринов 1, все имеющие одинаковую 1-цепь и каждый имеющий отличную от других -цепь. Все указанные девять рецепторов связывают различный комплемент разных молекул клеточного матрикса, таких как фибронектин, ламинин и коллаген. VLA-4, например,связывается с фибронектином. VLA-4 также связывает нематриксные молекулы, которые экспрессируются эндотелиальными и другими клетками. Данные нематриксные молекулы включают VCAM-I, который экспрессируется на цитокин-активированных человеческих эндотелиальных клетках пупочной вены в культуре. Различные эпитопы VLA-4 отвечают за активности связывания фибронектина и VCAM-I, и показано, что каждая активность ингибируется независимо.2 Межклеточная адгезия, опосредованная VLA-4 и другими клеточными поверхностными рецепторами, ассоциирована с рядом воспалительных реакций. В области повреждения или другого воспалительного фактора активированные клетки сосудистого эндотелия экспрессируют молекулы, которые являются адгезивными в отношении лейкоцитов. Механизм адгезии лейкоцитов к эндотелиальным клеткам включает, отчасти, распознавание и связывание клеточных поверхностных рецепторов на лейкоцитах с соответствующими молекулами клеточной поверхности на эндотелиальных клетках. Будучи связанными,лейкоциты мигрируют через стенку кровеносного сосуда, чтобы проникнуть в область повреждения и высвободить химические медиаторы для борьбы с инфекцией. В плане обзоров по адгезивным рецепторам иммунной системы см., например, работы Springer3 и Osborn4.-1 017110 Воспалительные нарушения головного мозга, такие как экспериментальный аутоиммунный энцефаломиелит (EAE), рассеянный склероз (MS) и менингит, представляют собой примеры нарушений центральной нервной системы, в которых механизм адгезии эндотелия/лейкоцитов приводит в результате к разрушению здоровой ткани головного мозга в других отношениях. Большие количества лейкоцитов мигрируют через гематоэнцефалический барьер (BBB) у пациентов с данными воспалительными заболеваниями. Лейкоциты высвобождают токсичные медиаторы, которые вызывают обширное повреждение ткани, приводящее в результате к поврежденной проводимости нервов и параличу. В других системах органов повреждение ткани происходит также через механизм адгезии, приводя в результате к миграции или активации лейкоцитов. Например, показано, что начальное повреждение сердечной ткани после ишемии миокарда может быть в последствии осложнено проникновением лейкоцитов в поврежденную ткань, вызывая еще одно повреждение (Vedder et al.)5. Другие воспалительные или медицинские состояния, опосредованные механизмом адгезии, включают, например, астму 6-8, болезнь Альцгеймера, атеросклероз 9-10, деменцию при СПИДе 11, диабет 12-14 (включая острый ювенильный диабет), воспалительное заболевание кишечника 15 (включая язвенный колит и болезнь Крона), рассеянный склероз 16-17, ревматоидный артрит 18-21, трансплантацию тканей 22, метастазы опухоли 23-28, менингит,энцефалит, инсульт и другие церебральные травмы, нефрит, ретинит, атопический дерматит, псориаз,ишемию миокарда и острое опосредованное лейкоцитами повреждение легкого, такое как происходит при респираторном дистресс-синдроме взрослых. Замещенные аминопиримидины описаны как класс соединений, ингибирующих связывание VLA-4 с VCAM-I, и, соответственно, проявляют противовоспалительные свойства 29. Хотя данные соединения обладают антагонистическими свойствами в отношении данного связывания, повышенная биодоступность данных соединений повышала бы их эффективность. Сущность изобретения Настоящее изобретение описывает соединения, их фармацевтически приемлемые соли и сложные эфиры, их композиции, синтез и способы лечения VLA-4-опосредованных заболеваний. Основываясь на данных in vivo, полученных для тех соединений, соответствующих настоящему изобретению, которые были оценены таким образом, данные соединения, как предполагают, проявляют повышенную биодоступность при пероральной доставке, которую измеряют принятым анализом площади под кривой (AUC). В одном варианте осуществления настоящее изобретение представляет соединения формулы IR2 выбран из группы, состоящей из С 1-С 4 алкила, С 2-С 4 алкенила, С 2-С 4 алкинила и С 3 С 6 циклоалкила,или их фармацевтически приемлемые соли или сложные эфиры. В ряде вариантов осуществления R1 представляет собой С 1-С 2 алкил. В других вариантах осуществления R1 представляет собой метил или трифторметил. В еще одних вариантах осуществления R1 представляет собой метил. В ряде вариантов осуществления R2 представляет собой С 1-С 4 алкил. В других вариантах осуществления R2 представляет собой С 1-С 3 алкил. В еще одних вариантах осуществления R2 представляет собой метил, этил, изопропил или н-пропил. В другом варианте осуществления R2 представляет собой метил или этил, и в еще одном варианте осуществления R2 представляет собой изопропил. В ряде вариантов осуществления R2 представляет собой С 3-С 6 циклоалкил. В других вариантах осуществления R2 представляет собой циклопентил. В ряде вариантов осуществления R2 представляет собой С 2-С 4 алкенил. В других вариантах осуществления R2 представляет собой аллил. В ряде вариантов осуществления R2 представляет собой С 2-С 4 алкинил. В других вариантах осуществления R2 представляет собой пропаргил. Примеры соединений, соответствующих настоящему изобретению, включают те, у которых группыR1 и R2 принимают значения, перечисленные в табл. 1 (включая их фармацевтически приемлемые соли или сложные эфиры). В другом варианте осуществления настоящее изобретение представляет соединения формулы IIR2 выбран из группы, состоящей из С 1-С 4 алкила, С 2-С 4 алкенила, С 2-С 4 алкинила и С 3 С 6 циклоалкила,или их фармацевтически приемлемые соли или сложные эфиры. В ряде вариантов осуществления R1 представляет собой С 1-С 2 алкил. В других вариантах осуществления R1 представляет собой метил или трифторметил. В еще одних вариантах осуществления R1 представляет собой метил. В ряде вариантов осуществления R2 представляет собой С 1-С 4 алкил. В других вариантах осуществления R2 представляет собой С 1-С 3 алкил. В еще одних вариантах осуществления R2 представляет собой метил, этил, изопропил или н-пропил. В другом варианте осуществления R2 представляет собой метил или этил, и в еще одном варианте осуществления R2 представляет собой изопропил. В ряде вариантов осуществления R2 представляет собой С 3-С 6 циклоалкил. В других вариантах осуществления R2 представляет собой циклопентил. В ряде вариантов осуществления R2 представляет собой С 2-С 4 алкенил. В других вариантах осуществления R2 представляет собой аллил. В ряде вариантов осуществления R2 представляет собой С 2-С 4 алкинил. В других вариантах осуществления R2 представляет собой пропаргил.-3 017110 Примеры соединений, соответствующих данному изобретению, включают те, у которых группы R1 и R принимают значения, перечисленные в табл. 2 (включая их фармацевтически приемлемые соли или сложные эфиры). Таблица 2 2 Орто- и мета-замещение пирролидинилкарбонилоксигруппы на фенильном цикле также входят в объем настоящего изобретения. Настоящее изобретение представляет также соединения, приведенные в табл. 3, а также их фармацевтически приемлемые соли или сложные эфиры. Таблица 3 В другом аспекте данное изобретение представляет фармацевтические композиции, включающие фармацевтически приемлемый носитель и терапевтически эффективное количество по меньшей мере одного соединения, определенного в данном контексте.-6 017110 В одном из аспектов, относящихся к способам, данное изобретение направлено на способ лечения заболевания, опосредованного, по меньшей мере частично, интегрином 4, предпочтительно VLA-4, у пациента, причем способ включает введение фармацевтической композиции, включающей фармацевтически приемлемый носитель и терапевтически эффективное количество по меньшей мере одного соединения, соответствующего изобретению. Соединения и фармацевтические композиции по настоящему изобретению используют для лечения патологических состояний, опосредованных, по меньшей мере, отчасти интегринами 4, где интегрин 4 предпочтительно представляет собой VLA-4, или адгезией лейкоцитов. Указанные патологические состояния включают, например, астму, болезнь Альцгеймера, атеросклероз, деменцию при СПИДе, диабет(включая острый ювенильный диабет), воспалительное заболевание кишечника (включая язвенный колит и болезнь Крона), рассеянный склероз, ревматоидный артрит, трансплантацию тканей, метастазы опухоли, менингит, энцефалит, инсульт и другие церебральные травмы, нефрит, ретинит, атонический дерматит, псориаз, ишемию миокарда и острое опосредованное лейкоцитами повреждение легкого, такое как происходит при респираторном дистресс-синдроме взрослых. Другие патологические состояния включают, но без ограничения перечисленным, воспалительные состояния, такие как узелковая эритема, аллергический конъюнктивит, ретробульбарный неврит, увеит,аллергический ринит, анкилозирующий спондилит, псориатический артрит, васкулит, синдром Рейтера,системная красная волчанка, прогрессирующий системный склероз, полимиозит, дерматомиозит, грануломатоз Вегнера, аортит, саркоидоз, лимфоцитопению, темпоральный артериит, перикардит, миокардит,застойная сердечная недостаточность, узелковый полиартериит, синдромы гиперчувствительности, аллергия, гиперэозинофильные синдромы, синдром Чарга-Штраусса, хроническое обструктивное легочное заболевание, гиперчувствительный пневмонит, хронический активный гепатит, интерстициальный цистит, аутоиммунная эндокринная недостаточность, первичный билиарный цирроз, аутоиммунная апластичная анемия, хронический персистирующий гепатит и тиреоидит. В одном варианте осуществления патологическое состояние, опосредованное интегрином 4, представляет собой воспалительное заболевание. В другом варианте осуществления патологическое состояние представляет собой аутоиммунное заболевание. В ряде вариантов осуществления вышеуказанное заболевание выбрано из группы, включающей астму, рассеянный склероз и воспалительное заболевание кишечника. В других вариантах осуществления указанное заболевание представляет собой болезнь Крона. В еще одних вариантах осуществления указанное заболевание представляет собой ревматоидный артрит. В другом аспекте настоящее изобретение представляет способ получения соединения формулы IR2 выбран из группы, состоящей из С 1-С 4 алкила, С 2-С 4 алкенила, С 2-С 4 алкинила и С 3 С 6 циклоалкила; или их фармацевтически приемлемых солей или сложных эфиров, причем данный способ включает:a) взаимодействие соединения формулы III где Pg представляет собой защитную группу карбоксила, с С 1-С 4 альдегидом или кетоном, C2 С 4 алкенилальдегидом или кетоном, С 2-С 4 алкинилальдегидом или кетоном, С 3-С 6 циклоалкилкетоном и-7 017110 бензальдегидом в условиях восстановительного аминирования после восстановления нитрогруппы в формуле III до первичного амина для получения соединения формулы IVb) взаимодействие соединения IV с сульфонилгалогенидом формулы R1SO2Z, где Z представляет собой гало, при котором образуется соединение формулы Vc) удаление защитной группы карбоксила для получения соединения формулы I. В другом аспекте настоящее изобретение представляет способ получения соединений формулы IR2 выбран из группы, состоящей из С 1-С 4 алкила, С 2-С 4 алкенила, С 2-С 4 алкинила и С 3 С 6 циклоалкила,или их фармацевтически приемлемых солей или сложных эфиров, причем данный способ включаетa) взаимодействие соединения формулы VI где Pg представляет защитную группу карбоксила, с избытком R'SO2X для получения соединения формулы VIIb) избирательное удаление одной группы -SO2R' из соединения формулы VII для получения соединения формулы VIIIc) взаимодействие соединения VIII с алкилирующим агентом формулы R2-X, где X представляет собой гало, или диметилсульфатом, где R2 представляет собой метил, с образованием соединения формулы IXd) удаление защитной группы карбоксила для получения соединения формулы I. Сведения, подтверждающие возможность осуществления изобретения Как утверждается выше, настоящее изобретение относится к соединениям, которые ингибируют адгезию лейкоцитов и, в частности, адгезию лейкоцитов, опосредованную, по меньшей мере частично, интегринами 4, предпочтительно VLA4. Тем не менее, для описания настоящего изобретения в дальнейших деталях сначала будут даны определения используемых терминов. Определения. Если не указано иначе, следующие ниже термины, используемые в описании и формуле изобретения, имеют значения, приведенные ниже. Как используют в данном контексте и пока не указано иначе, термин "алкил" относится к одновалентным неразветвленным и разветвленным углеводородным группам, имеющим от 1 до 4 атомов углерода и предпочтительно 1-3 атома углерода. Данный термин иллюстрирует такие группы, как метил,этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил. Термин "алкенил" относится к неразветвленным или разветвленным одновалентным углеводородным группам, состоящим из 2-4 атомов углерода и предпочтительно из 2-3 атомов углерода и имеющим по меньшей мере 1 и предпочтительно 1 центр виниловой (C=C) ненасыщенности. Примеры данных алкенильных групп включают винил (-CH=CH2), аллил (-CH2CH=CH2), н-пропен-1-ил (-CH=CHCH3), нбутен-2-ил (-CH2CH=CHCH3) и т.п. В данный термин включают цис- и транс-изомеры и смеси данных изомеров. Термин "алкинил" относится к неразветвленным или разветвленным одновалентным углеводородным группам, содержащим от 2 до 4 атомов углерода и предпочтительно 2-3 атома углерода и имеющим по меньшей мере 1 и предпочтительно 1 центр ацетиленовой -CC- ненасыщенности. Примеры данных алкинильных групп включают ацетиленил (-CCH), пропаргил (-CH2CCH), н-пропин-1-ил-9 017110 Термин "гало" или "галоген" относится к фтору, хлору, брому и йоду и предпочтительно представляет собой фтор или хлор. Термин "галоалкил" относится к алкильным группам, содержащим от 1 до 5 галогрупп. Предпочтительно, когда данные группы содержат от 1 до 3 галогрупп и 1-2 атома углерода. Примеры галоалкильных групп включают галометил (например, фторметил), дигалометил (например, дифторметил), тригалометил (например, трифторметил), галоэтил (например, 2-хлорэт-1-ил), тригалоэтил (например, 2,2,2 трифторэт-1-ил), галопропил (например, 3-хлорпроп-1-ил) и тригалопропил (например, 3,3,3 трифторпроп-1-ил)."Фармацевтически приемлемый носитель" означает носитель, который используют при получении фармацевтической композиции, которая в основном безопасна, нетоксична и не является ни биологически, ни иным образом нежелательной и включает носитель, который приемлем для ветеринарного применения, а также в качестве фармацевтического применения для человека. "Фармацевтически приемлемый носитель", как используют в описании и формуле изобретения, включает как один, так и более чем один такой носитель. Термин "фармацевтически приемлемая соль" относится к солям, которые сохраняют биологическую эффективность и свойства соединений, соответствующих данному изобретению, и которые не являются биологически или иным образом нежелательными. Во многих случаях соединения, соответствующие данному изобретению, способны к формированию солей кислот и/или оснований в силу присутствия амино- и/или карбоксильных групп или подобных им групп. Фармацевтически приемлемые основно-аддитивные соли можно получить из неорганических и органических оснований. Соли, полученные из неорганических оснований, включают, например, соли натрия, калия, лития, аммония, кальция и магния. Соли, полученные из органических оснований, включают, но без ограничения перечисленным, соли первичных, вторичных и третичных аминов, таких как алкиламины, диалкиламины, триалкиламины, замещенные алкиламины, ди(замещенный алкил)амины,три(замещенный алкил)амины, алкениламины, диалкениламины, триалкенил амины, замещенные алкениламины, ди(замещенный алкенил)амины, три(замещенный алкенил)амины, циклоалкиламины,ди(циклоалкил)амины, три(циклоалкил)амины, замещенные циклоалкиламины, дизамещенные циклоалкиламины,тризамещенные циклоалкиламины,циклоалкениламины,ди(циклоалкенил)амины,три(циклоалкенил)амины, замещенные циклоалкениламины, дизамещенный циклоалкениламин, тризамещенные циклоалкениламины, ариламины, диариламины, триариламины, гетероариламины, дигетероариламины, тригетероариламины, гетероциклические амины, дигетероциклические амины, тригетероциклические амины, смешанные ди- и триамины, в которых по меньшей мере два из заместителей на амине различны и выбраны из группы, состоящей из алкила, замещенного алкила, алкенила, замещенного алкенила, циклоалкила, замещенного циклоалкила, циклоалкенила, замещенного циклоалкенила, арила, гетероарила, гетероцикла и т.п. Включены также амины, в которых два или три заместителя совместно с атомом азота амина формируют гетероциклическую или гетероарильную группу. Примеры подходящих аминов включают, только в качестве иллюстрации, изопропиламин, триметиламин, диэтиламин, три(изопропил)амин, три(н-пропил)амин, этаноламин, 2-диметиламиноэтанол,трометамин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, N-алкилглюкамины, теобромин, пурины, пиперазин, пиперидин, морфолин, N-этилпиперидин и т.п. Следует также иметь в виду, что другие производные карбоновых кислот можно также использовать в практической реализации данного изобретения, например амиды карбоновых кислот, включая карбоксамиды, низший алкилкарбоксамиды, диалкилкарбоксамиды и т.п. Фармацевтически приемлемые кислотно-аддитивные соли можно получить из неорганических и органических кислот. Соли, полученные из неорганических кислот, включают соли хлористоводородной кислоты, бромисто-водородной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты и т.п. Соли, полученные из органических кислот, включают соли уксусной кислоты, пропионовой кислоты, гликолевой кислоты, пировиноградной кислоты, щавелевой кислоты, яблочной кислоты, малоновой кислоты, янтарной кислоты, малеиновой кислоты, фумаровой кислоты, винной кислоты, лимонной кислоты, бензойной кислоты, коричной кислоты, миндальной кислоты, метансульфоновой кислоты,этансульфоновой кислоты, п-толуолсульфоновой кислоты, салициловой кислоты и т.п. Термин "фармацевтически приемлемый катион" относится к катиону фармацевтически приемлемой соли. Следует иметь в виду, что не предусмотрено включение в данном контексте всех замещенных групп, определенных в данном контексте, полимеров, полученных с помощью определенных заместителей с дальнейшими заместителями на них самих (например, замещенного арила, имеющего замещенную арильную группу в качестве заместителя, который сам является замещенным замещенной арильной группой и т.п.). В данных случаях максимальное число таких заместителей составляет три. Иначе говоря,каждое из вышеприведенных определений ограничено пределом, так что, например, замещенные арильные группы ограничены группой -замещенный арил-(замещенный арил)-(замещенный арил). Термин(1) предупреждение заболевания, т.е. создание условий, при которых симптомы заболевания не раз- 10017110 виваются у млекопитающего, которое может подвергнуться воздействию или предрасположено к заболеванию, но еще не испытывает или не проявляет симптомов заболевания,(2) подавление заболевания, т.е. остановку или снижение уровня развития заболевания или его клинических симптомов или(3) облегчение заболевания, т.е. создание условий для обратного развития заболевания или его клинических симптомов. Термин "терапевтически эффективное количество" означает количество соединения, которое при введении млекопитающему для лечения заболевания достаточно для осуществления данного лечения заболевания."Терапевтически эффективное количество" будет варьировать в зависимости от соединения, заболевания и его тяжести и возраста, массы тела и т.п. млекопитающего, лечение которого предусматривают. Интегрины представляют собой большое семейство гомологичных трансмембранных линкерных белков, которые являются основными рецепторами на клетках животных для связывания большинства белков внеклеточного матрикса, таких как коллаген, фибронектин и ламинин. Интегрины представляют собой гетеродимеры, состоящие из -цепи и -цепи. В настоящее время идентифицировано двадцать различных гетеродимеров интегрина, включающих 9 различных -субъединиц и 14 различных -субъединиц. Термин "интегрины 4" относится к классу гетеродимерных связанных с ферментом клеточных поверхностных рецепторов, которые содержат субъединицу 4, спаренную с любой из -субъединиц. VLA-4 является примером интегрина 4 и представляет собой гетеродимер субъединиц 4 и 1, и его также называют интегрин 41. Получение соединений Соединения, соответствующие настоящему изобретению, можно получить из легкодоступных исходных материалов, используя общие способы и процедуры. Следует иметь в виду, что когда приводят типичные или предпочтительные условия процесса (т.е. температуры реакции, время, молярные соотношения реагентов, растворители, давления и т.д.), могут быть также использованы другие условия, если не указано иначе, реакции могут варьировать в зависимости от конкретных используемых реагентов и растворителей, но данные условия может определить компетентный специалист в области техники путем рутинных методик оптимизации. Кроме того, как будет очевидно компетентным специалистам в области техники, могут быть необходимы принятые защитные группы, чтобы предупредить вступление ряда функциональных групп в нежелательные реакции. Подходящие защитные группы для различных функциональных групп, а также подходящие условия для защиты и снятия защиты с конкретных функциональных групп хорошо известны в области техники. Например, многие защитные группы описаны в монографии T.W. Greene и G.M.Wuts, Protecting Groups in Organic Synthesis (Защитные группы в органическом синтезе), 2 изд., Wiley,New York, 1991, и ссылках, приведенных в данном контексте. Более того, соединения, соответствующие настоящему изобретению, будут, как правило, содержать один или более хиральных центров. Соответственно при необходимости данные соединения можно получить или выделить в виде чистых стереоизомеров, т.е. в виде индивидуальных энантиомеров или диастереомеров или в виде смесей, обогащенных стереоизомерами. Все указанные стереоизомеры (и обогащенные смеси) включены в объем настоящего изобретения, если не указано иначе. Чистые стереоизомеры (или обогащенные смеси) можно получить, используя, например, оптически активные исходные материалы или стереоселективные реагенты, хорошо известные в области техники. Альтернативно можно разделить рацемические смеси данных соединений, используя, например, хиральную колоночную хроматографию, хиральные разделяющие агенты и т.п. Большинству соединений, соответствующих настоящему изобретению, присваивают названия, используя ChemDraw v. 10.0 (имеется в фирме Cambridgesoft, 100 Cambridge Park Drive, Cambridge, MA 02140). В одном варианте осуществления соединения, соответствующие настоящему изобретению, можно получить, как показано ниже на схеме 1, на которой только с целью иллюстрации R1 представляет собой метил и R2 представляет собой изопропил. где Pg представляет собой защитную группу карбоксила, такую как бензил, трет-бутил и т.п. Схема 1 особенно эффективна при получении соединений, в которых R2 представляет собой алкил или циклоалкил. На схеме 1 исходные 5-аминопиримидиновые промежуточные продукты, соединение 1.1, описаны в деталях в патенте СШАUS 7026328 В 1 и только с целью иллюстрации показаны на данной схеме как 4-замещенные фенилаланиновые производные. Конечно, следует иметь в виду, что 2- и 3-замещенные фенилаланиновые производные получали бы с помощью аналогичной последовательности реакций. В частности, на схеме 1 проводят реакцию 5-амино-2-диэтиламино-4-замещенного пиримидина, соединение 1.1 (полученного из соответствующего 5-нитропиримидина путем восстановления 5% Pd/C или 5% PtO2 по массе), в принятых условиях восстановительного аминирования с небольшим избытком C1 С 4 льдегида или кетона, который на схеме 1 иллюстрируют ацетоном. На схеме 1 5-аминогруппа соединения 1.1 образует промежуточный имин (не показан), который in situ восстанавливают до соответствующего амина, соединения 1.2, принятыми восстановителями, такими как цианоборогидрид натрия,борогидрид натрия, водород над подходящим катализатором, таким как PtO2, и т.п. Реакцию проводят в подходящем инертном носителе, таком как тетрагидрофуран, метиленхлорид и т.п. Реакцию поддерживают при температуре от приблизительно 0 до приблизительно 30C, пока реакция практически не завершится, что, как правило, происходит в течение приблизительно 0,5-16 ч. После завершения реакции соединение 1.2 выделяют принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение, хроматографию, фильтрование и т.п. или, альтернативно, используют на следующей стадии без очистки и/или выделения. Превращение аминогруппы в соединении 1.2 в соответствующую алкилсульфониламидогруппу соединения 1.3 осуществляют принятыми способами. Например, в одном способе соединение 1.2 взаимодействует с небольшим избытком алкансульфонилгалогенида, такого как метансульфонилхлорид в присутствии подходящего основания, такого как триэтиламин, диизопропилэтиламин и т.п., с целью связывания образованной кислоты. Предпочтительно, когда реакцию проводят в подходящем инертном растворителе, таком как тетрагидрофуран, диоксан, хлороформ и т.п. Предпочтительно, когда реакцию проводят при температуре от приблизительно -5 до 30C и продолжают, пока реакция практически не завершится, что, как правило, происходит в течение 0,5-16 ч. После завершения реакции соединение 1.3 можно выделить принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение, хроматографию, фильтрование и т.п., или, альтернативно, использовать на следующей стадии без очистки и/или выделения. Алкилсульфонилгалогениды представляют собой либо известные соединения, либо соединения, которые можно получить принятыми способами синтеза. Данные соединения, как правило, получают из соответствующей сульфоновой кислоты, т.е. из соединений формулы R1-SO3H, где R1 такое, как определено выше, при использовании трихлорида фосфора и пентахлорида фосфора. Реакцию, как правило,проводят путем взаимодействия сульфоновой кислоты с приблизительно 2-5 мол. экв. трихлорида фосфора или пентахлорида фосфора, либо в неразбавленном виде, либо в инертном растворителе, таком как дихлорметан, при температуре в интервале от 0 до приблизительно 80C в течение от приблизительно 1 до приблизительно 48 ч с получением сульфонилхлорида. Альтернативно, сульфонилхлорид можно получить из соответствующего тиолового соединения, т.е. из соединения формулы R1-SH, где R1 такое, как определено выше, путем обработки тиола хлором (Cl2) и водой в принятых условиях реакции. Примеры сульфонилхлоридов для применения в настоящем изобретении включают, но без ограничения перечисленным, метансульфонилхлорид, этансульфонилхлорид, 2-пропансульфонилхлорид, 1 бутансульфонилхлорид, трифторметансульфонилхлорид, 2,2,2-трифторэтансульфонилхлорид и т.п. Затем защитную группу карбоксила соединения 1.3 удаляют в принятых условиях, получая соединение 1.4, соединение формулы I. В одном варианте осуществления защитную группу трет-бутила мож- 12017110 но удалить путем взаимодействия с муравьиной кислотой. В другом варианте осуществления защитную группу бензила можно удалить путем взаимодействия с водородом в присутствии катализатора палладий/уголь, как правило, в протонном растворителе, таком как метанол, при повышенном давлении водорода. После завершения реакции соединение 1.4 можно выделить принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение, хроматографию, фильтрование и т.п. В другом варианте осуществления соединения, соответствующие настоящему изобретению, можно получить, как показано на схеме 2. Схема 2 где R1 и R2 такие, как определено в данном контексте; Pg представляет собой защитную группу карбоксила, а X означает гало. На схеме 2 исходные 5-аминопиримидиновые промежуточные продукты, соединение 1.1, описаны в деталях в патенте СШАUS 7026328 B1 и только с целью иллюстрации показаны на данной схеме как 4-замещенные фенилаланиновые производные. Конечно, следует иметь в виду, что 2- и 3-замещенные фенилаланиновые производные получали бы с помощью аналогичной последовательности реакций. В частности, на схеме 2 проводят реакцию 5-амино-2-диэтиламино-4-замещенного пиримидина, соединение 1.1 (полученного из соответствующего 5-нитропиримидина путем восстановления 5% Pd/C или 5% PtO2 по массе), с небольшим избытком R1-сульфонилгалогенида, такого как метансульфонилхлорид,в присутствии подходящего основания, такого как триэтиламин, диизопропилэтиламин и т.п. с целью связывания образующейся кислоты. Предпочтительно, когда реакцию проводят в подходящем инертном растворителе, таком как тетрагидрофуран, диоксан, хлороформ и т.п. Предпочтительно, когда реакцию проводят при температуре от приблизительно -5 до 30C и продолжают, пока реакция практически не завершится, что, как правило, происходит в течение 0,5-16 ч. После завершения реакции соединение 1.5 можно выделить принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение,хроматографию, фильтрование и т.п., или, альтернативно, использовать на следующей стадии без очистки и/или выделения. Избирательное удаление одной группы R1SO2- из соединения 1.5 осуществляют в принятых условиях. Например, реакция соединения 1.5 с основанием в протонном растворителе, таком как метанол, этанол или вода, необязательно в присутствии ТГФ и т.п., например в смеси 1:1 метанол/тетрагидрофуран или смеси 1:1 вода/тетрагидрофуран, дает соединение 1.6. Реакционная смесь включает избыток подходящего основания, такого как карбонат калия, карбонат натрия и т.п., и предпочтительно, когда реакцию поддерживают при повышенных температурах, таких как 20-60C. Реакцию продолжают практически до завершения, которое, как правило, происходит в течение 24-144 ч. После завершения реакции соединение 1.6 можно выделить принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение, хроматографию, фильтрование и т.п., или, альтернативно, используют на следующей стадии без очистки и/или выделения. Реакция соединения 1.6 с избытком алкилгалогенида, диалкилсульфата, алкенилгалогенида, алкинилгалогенида или циклоалкилгалогенида (т.е. Х-R2-"соединение галогенида") протекает в принятых условиях с получением соединения 1.7. Реакцию, как правило, проводят путем взаимодействия соединения 1.6 с приблизительно от 1,1 до 20 экв. соединения галогенида в инертном растворителе, таком как ацетон, хлороформ, метиленхлорид и т.п., в присутствии основания, такого как карбонат калия, триэти- 13017110 ламин и т.п., для связывания кислоты, образующейся во время реакции. Предпочтительно, когда реакцию проводят при от приблизительно 20 до 60C и продолжают практически до завершения реакции,которое, как правило, происходит в течение 0,1-16 ч. После завершения реакции соединение 1.6 можно выделить принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение, хроматографию, фильтрование и т.п., или, альтернативно, используют на следующей стадии без очистки и/или выделения. Затем защитую группу карбоксила соединения 1.7 удаляют в принятых условиях, получая соединение 1.8, соединение формулы I. В одном варианте осуществления защитную группу трет-бутила можно удалить путем взаимодействия с муравьиной кислотой. В другом варианте осуществления защитную группу бензила можно удалить путем взаимодействия с водородом в присутствии катализатора палладий/уголь, как правило, в протонном растворителе, таком как метанол при повышенном давлении водорода. После завершения реакции соединение 1.8 можно выделить принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение, хроматографию, фильтрование и т.п. В еще одном варианте осуществления соединения, соответствующие данному изобретению, получают, как показано ниже на схеме 3. Схема 3 где R1 такое, как определено выше, Pg представляет собой защитную группу карбоксила, такую как бензил, трет-бутил и т.п., и R2 представляет собой алкильную, алкенильную, алкинильную или фенилалкиленовую группу, имеющую структуру CH2, присоединенную к группе йода. На вышеприведенной схеме исходные 5-аминопиримидиновые промежуточные продукты, соединение 1.1, описаны в деталях в патенте СШАUS 7026328 В 1 и только с целью иллюстрации показаны на данной схеме как 4-замещенные фенилаланиновые производные. Конечно, следует иметь в виду, что 2- и 3-замещенные фенилаланиновые производные получали бы с помощью аналогичной последовательности реакций. В частности, на схеме 1 5-амино-2-диэтиламино-4-замещенный пиримидин, соединение 1.1 (полученный из соответствующего 5-нитропиримидина путем восстановления 5% Pd/C или 5% PtO2 по массе),превращают в соответствующий трифторацетамид, соединение 1.8 принятыми способами. Например,небольшой избыток трифторуксусного ангидрида смешивают с соединением 1.1 в подходящем инертном разбавителе, таком как тетрагидрофуран, метиленхлорид, пиридин и т.п. Реакцию поддерживают при температуре от приблизительно 0C до приблизительно 30C, пока реакция практически не завершится,что, как правило, происходит в течение приблизительно 0,5-16 ч. После завершения реакции соединение 1.8 выделяют принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение, хроматографию, фильтрование и т.п., или, альтернативно, используют на следующей стадии без очистки и/или выделения. Превращение соединения 1.8 в соответствующий N(R2'), N-трифторацетамидопиримидин, соединение 1.9, снова осуществляют принятыми способами. Например, избыток галогенида R2'-I смешивают с соединением 1.8 в подходящем инертном носителе, таком как ДМФ, в присутствии избытка подходящего- 14017110 основания, такого как карбонат калия. В одном варианте осуществления используют приблизительно 2 экв. R2'-I и карбоната калия. Реакцию поддерживают в условиях окружающей среды в запечатанном контейнере и продолжают, пока реакция практически не завершится, что, как правило, происходит в течение 20-72 ч. После завершения реакции соединение 1.9 выделяют принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение, хроматографию, фильтрование и т.п., или, альтернативно,используют на следующей стадии без очистки и/или выделения. Затем трифторацетильную группу удаляют с получением соответствующего амина, соединения 1.10. В данном варианте осуществления трифторацетильная группа действует как защитная группа амина. Как указано выше, данная реакция обычно протекает, например, путем контактирования соединения 1.9 с большим избытком подходящего основания, такого как карбонат калия, в смеси воды и протонного растворителя, такого как метанол. Реакцию проводят при повышенных температурах, таких как 40-60C,и продолжают, пока реакция практически не завершится. После завершения реакции соединение 1.10 выделяют принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение, хроматографию, фильтрование и т.п., или, альтернативно, используют на следующей стадии без очистки и/или выделения. Далее превращение аминной группы в соединении 1.10 в соответствующую алкилсульфониламидогруппу, соединение 1.11, осуществляют принятыми способами. Например, в одном способе соединение 1.10 контактирует с небольшим избытком алкилсульфонилгалогенида в присутствии подходящего основания, такого как триэтиламин, дизопропилэтиламин и т.п., с целью связывания образующейся кислоты. Предпочтительно, когда реакцию проводят в подходящем инертном растворителе, таком как тетрагидрофуран, диоксан, хлороформ и т.п. Предпочтительно, когда реакцию проводят при температуре от приблизительно 0 до 30C и продолжают, пока реакция практически не завершится, что, как правило, происходит в течение 2-48 ч. После завершения реакции соединение 1.11 выделяют принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение, хроматографию, фильтрование и т.п. или, альтернативно, используют на следующей стадии без очистки и/или выделения. Затем защитную группу карбоксила соединения 1.11 можно удалить в принятых условиях, получая соединение 1.12, соединение формулы I. В одном варианте осуществления защитную группу трет-бутила можно удалить путем взаимодействия с муравьиной кислотой. В другом варианте осуществления защитную группу бензила можно удалить путем взаимодействия с водородом в присутствии катализатора палладий/уголь, как правило, в протонном растворителе, таком как метанол, при повышенном давлении водорода. После завершения реакции соединение 1.12 можно выделить принятыми способами, включая нейтрализацию, упаривание, экстракцию, осаждение, хроматографию, фильтрование и т.п. Изобретение также включает сложные эфиры соединений, соответствующих настоящему изобретению. Получение сложных эфиров иллюстрируют на различных схемах, описанных выше, таких как схема 1 (соединение 1.3), на схеме 2 (соединение 1.7) и на схеме 3 (соединение 1.11). Более того, пример 1 описывает получение (S)-4-(3-трет-бутокси-2-(2-(диэтиламино)-5-(N-изопропилметилсульфонамидо) пиримидин-4-иламино)-3-оксопропил)фенилпирролидин-1-карбоксилата, и пример 4 описывает получение (S)-4-(3-трет-бутокси-2-(2-(диэтиламино)-5-(N-(проп-2-инил)метилсульфонамидо)пиримидин-4-иламино)-3-оксопропил)фенилпирролидин-1-карбоксилата. Сложные эфиры кислот, соответствующих настоящему изобретению, можно также получить из кислот, как правило, хорошо известных в области техники. Например, сложные метиловые эфиры аминокислот можно получить, используя способ, предложенный в статье Brenner и Huber, Helv. Chim. Acta 1953, 36, 1109. Фармацевтические препараты. При использовании в качестве фармацевтических агентов соединения, соответствующие настоящему изобретению, обычно вводят в форме фармацевтических композиций. Данные соединения можно вводить множеством путей, включая пероральный, ректальный, чрескожный, подкожный, внутривенный,внутримышечный и интраназальный. Данные соединения эффективны как в инъекционной, так и в пероральной композициях. Такие композиции получают способом, хорошо известным в области фармацевтики, и они включают по меньшей мере одно активное соединение. Настоящее изобретение также включает фармацевтические композиции, которые содержат в качестве активного ингредиента по меньшей мере одно соединение из вышеприведенных формул I-II, ассоциированных с фармацевтически приемлемыми носителями. При получении композиций, соответствующих настоящему изобретению, активный ингредиент обычно смешивают с наполнителем, разбавляют наполнителем или заключают в носитель, который может быть в форме капсулы, пакетиков, бумажного или иного контейнера. Используемый наполнитель, как правило, представляет собой наполнитель,подходящий для введения человеку или животным. Когда наполнитель служит в качестве разбавителя,он может представлять собой твердый, полутвердый или жидкий материал, который действует как транспортный агент, носитель или среда для активного ингредиента. Таким образом, композиции могут находиться в форме таблеток, пилюль, порошков, лепешек, пакетиков, облаток, эликсиров, суспензий,эмульсий, растворов, сиропов, аэрозолей (как твердых, так и в жидкой среде), мазей, содержащих, на- 15017110 пример, до 10 мас.% активного соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных инъекционных растворов и стерильных упакованных порошков. При приготовлении препарата может потребоваться измельчение активного соединения для получения частиц подходящего размера перед смешиванием с другими ингредиентами. Если активное соединение в существенной степени нерастворимо, его обычно измельчают для получения частиц размером меньше чем 200 меш. Если активное соединение в существенной степени водорастворимо, размер частиц обычно доводят измельчением до получения практически однородного распределения в препарате, например, приблизительно 40 меш. Некоторые примеры подходящих наполнителей включают лактозу, декстрозу, сахарозу, сорбит,манит, крахмалы, акациевую камедь, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция,микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу. Препараты могут дополнительно включать скользящие агенты, такие как тальк, стеарат магния и минеральное масло; смачивающие агенты, эмульгаторы и суспендирующие агенты; консерванты, такие как метил- и пропилгидроксибензоаты; подсластители и вкусовые добавки. Композиции, соответствующие изобретению, могут быть составлены так, чтобы обеспечивать быстрое, замедленное или задержанное высвобождение активного ингредиента после введения пациенту при использовании способов, известных в уровне техники. Введение терапевтических агентов с помощью внутривенного препарата хорошо известно в фармацевтической промышленности. Внутривенный препарат должен обладать рядом качеств помимо того,что он представляет собой только композицию, в которой растворен лечебный фактор. Например, препарат должен способствовать общей стабильности активного ингредиента(ов), кроме того, производство препарата должно быть экономически эффективным. Все из данных факторов в конечном счете определяют общий успех и применимость внутривенного препарата. Другие дополнительные добавки, которые могут быть включены в фармацевтические препараты соединений, соответствующих настоящему изобретению, представлены следующими: растворители этанол, глицерин, пропиленгликоль; стабилизаторы - этилендиаминтетрауксусная кислота (ЭДТА), лимонная кислота; антимикробные консерванты - бензиловый спирт, метилпарабен, пропилпарабен; забуферивающие агенты - лимонная кислота/цитрат натрия, гидротартрат калия, гидротартрат натрия, уксусная кислота/ацетат натрия, малеиновая кислота/малеат натрия, гидрофталат натрия, фосфорная кислота/дигидрофосфат калия, фосфорная кислота/гидрофосфат динатрия и модификаторы тоничности - хлорид натрия, маннит, декстроза. Присутствие буфера может быть необходимо для поддержания водного pH в интервале от приблизительно 4 до приблизительно 8 и более предпочтительно в интервале от приблизительно 4 до приблизительно 6. Буферная система представляет собой, как правило, смесь слабой кислоты и ее растворимой соли, например цитрата натрия/лимонной кислоты, или однокатионную либо двукатионную соли двухосновной кислоты, например гидротартрат калия, гидротартрат натрия, фосфорная кислота/дигидрофосфат калия, фосфорная кислота/гидрофосфат динатрия. Используемое количество буферной системы зависит от (1) требуемого pH и (2) количества лекарственного препарата. Как правило, используемое количество буфера составляет 0,5:1 - 50:1 в мольном соотношении состава буфер:лекарственный препарат (где моли буфера принимают как объединенные моли ингредиентов буфера, например цитрата натрия и лимонной кислоты) для поддержания pH в интервале 4-8 и, как правило, используют мольное соотношение 1:1 - 10:1 буфера (объединенного) к присутствующему лекарственному препарату. Одним из используемых в изобретении буферов является цитрат натрия/лимонная кислота в интервале 5-50 мг/мл цитрата натрия к 1-15 мг/мл лимонной кислоты, что достаточно для поддержания водного pH 4-6 композиции. Буферный агент может также способствовать предотвращению осаждения лекарственного препарата при образовании растворимого комплекса металла с растворенными ионами металла, например Ca,Mg, Fe, Al, Ba, которые могут выделяться из стеклянных контейнеров или резиновых пробок либо присутствовать в обычной водопроводной воде. Агент может действовать как конкурентный комплексирующий агент с лекарственным препаратом и образовывать растворимый комплекс металла, приводящий к присутствию нежелательных частиц. Кроме того, присутствие агента, например хлорида натрия, в количестве приблизительно 1-8 мг/мл,для подведения тоничности до одинакового значения с кровью человека может потребоваться, чтобы избежать набухания или сморщивания эритроцитов при введении внутривенного препарата, приводящего к нежелательным побочным эффектам, таким как тошнота или диарея, и, возможно, к ассоциированным нарушениям крови. Как правило, тоничность препарата соответствует тоничности крови человека,которая лежит в интервале 282-288 мОсм/кг и, как правило, составляет 285 мОсм/кг, что эквивалентно осмотическому давлению, соответствующему 0,9% раствору хлорида натрия. Внутривенный препарат можно ввести путем прямой внутривенной инъекции, внутривенного болюса или можно ввести путем инфузии посредством добавления к соответствующему инфузионному раствору, такому как инъекционный 0,9% хлорид натрия или другому совместимому инфузионному рас- 16017110 твору. Предпочтительно, когда композиции готовят в виде единичной пероральной лекарственной формы,где каждая доза содержит от приблизительно 1 до приблизительно 250 мг, чаще от приблизительно 5 до приблизительно 100 мг, например от приблизительно 10 до приблизительно 30 мг активного ингредиента. Термин "единичная лекарственная форма" относится к физически раздельным единицам, подходящим в качестве единичных доз для человека и животных, причем каждая единица включает предварительно заданное количество активного начала, рассчитанное так, чтобы получить требуемый терапевтический эффект, в сочетании с подходящим фармацевтическим наполнителем. Активное соединение эффективно в широком диапазоне доз и, как правило, вводится в терапевтически эффективном количестве. Однако следует иметь в виду, что количество соединения, вводимое фактически, будет определять лечащий врач в свете соответствующих обстоятельств, включая состояние,лечение которого предусматривают, выбранный путь введения, действительное соединение, которое вводят, возраст, массу тела и ответ конкретного больного, тяжесть симптомов у больного и т.п. Для получения композиций в твердой форме, таких как таблетки, основной активный ингредиент смешивают с фармацевтическим наполнителем с формированием твердой предварительной перед получением препарата композицией, содержащей однородную смесь соединения, соответствующего настоящему изобретению. Называя данные предварительные перед получением препарата композиции однородными, имеют в виду, что активный ингредиент распределен равномерно по всей композиции, поэтому композицию можно легко разделить на одинаково эффективные единичные лекарственные формы,такие как таблетки, пилюли и капсулы. Данный твердый предварительный препарат затем разделяют на унифицированные лекарственные формы вышеописанного типа, содержащие, например, от 0,1 до приблизительно 500 мг активного ингредиента, соответствующего настоящему изобретению. Таблетки или пилюли, соответствующие настоящему изобретению, могут быть покрытыми и иным образом составлены, чтобы представить лекарственную форму, дающую преимущество пролонгированного действия. Например, таблетка или пилюля могут включать внутренний и наружный лекарственные компоненты, причем последний может находиться в форме покрытия на первом. Два компонента могут быть разделены энтеросолюбильным слоем, который служит для придания устойчивости к распаду в желудке и позволяет внутреннему компоненту проходить интактным в двенадцатиперстную кишку или задерживать его высвобождение. Множество материалов может быть использовано для данных энтеросолюбильных слоев или покрытий, причем данные материалы включают ряд полимерных кислот и смесей полимерных кислот с такими материалами, как шеллак, цетиловый спирт и ацетат целлюлозы. Жидкие формы, в которые могут быть включены новые композиции, соответствующие настоящему изобретению, для введения перорально или путем инъекции включают водные растворы, соответственно, сиропы с вкусовыми добавками, водные или масляные суспензии и эмульсии с вкусовыми добавками со съедобными маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и аналогичные фармацевтические носители. Композиции для ингаляции или инсуффляции включают растворы и суспензии в фармацевтически приемлемых водных или органических растворителях или их смесях и порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые наполнители, как описано выше. Предпочтительно, когда композиции вводят пероральным или назальным респираторным путем для получения местного или системного эффекта. Композиции в предпочтительных фармацевтически приемлемых растворителях можно разбрызгивать при использовании инертных газов. Разбрызгиваемые растворы можно вдыхать непосредственно из распылителя, или распылитель может быть присоединен к маскам для лица, тенту или дыхательному аппарату с переменным положительным давлением. Раствор,суспензию или порошковые композиции можно вводить предпочтительно перорально или назально из устройств, которые доставляют препарат соответствующим образом. Следующие примеры препаратов иллюстрируют фармацевтические композиции, соответствующие настоящему изобретению. Пример препарата 1. Получают твердые желатиновые капсулы, содержащие следующие ингредиенты: Вышеуказанные ингредиенты смешивают и наполняют ими твердые желатиновые капсулы в количестве 340 мг. Пример препарата 2. Состав для таблеток получают при использовании нижеуказанных ингредиентов: Компоненты измельчают и прессуют с формированием таблеток, массой 240 мг каждая. Пример препарата 3. Получают ингаляционный препарат в виде сухого порошка, содержащий следующие компоненты: Активный ингредиент смешивают с лактозой и добавляют смесь в ингаляционное устройство для сухого порошка. Пример препарата 4. Таблетки, каждая из которых содержит 30 мг активного ингредиента, получают следующим образом: Активный ингредиент, крахмал и целлюлозу пропускают через сито 20 меш США и тщательно перемешивают. Раствор поливинилпирролидона смешивают с полученными в результате порошками, которые затем пропускают через сито 16 меш США. Полученные таким образом гранулы сушат при 5060C и пропускают через сито 16 меш США. Карбоксиметилкрахмал натрий, стеарат магния и тальк предварительно пропускают через сито 30 меш США и затем добавляют к гранулам, которые после перемешивания прессуют на таблетировочной машине, получая таблетки массой 120 мг каждая. Пример препарата 5. Капсулы, каждая из которых содержит 40 мг лекарственного препарата, изготавливают следующим образом: Активный ингредиент, крахмал и стеарат магния смешивают, пропускают через сито 20 меш США и наполняют полученной смесью твердые желатиновые капсулы в количестве 150 мг. Пример препарата 6. Суппозитории, содержащие по 25 мг активного ингредиента каждый, получают следующим образом: Активный ингредиент пропускают через сито 60 меш США и суспендируют в глицеридах насыщенных жирных кислот, предварительно расплавленных с использованием минимального необходимого нагревания. Затем смесь выливают в форму для суппозиториев номинальной емкостью 2,0 г и позволяют охладиться. Пример препарата 7. Суспензии, каждая из которых содержат 50 мг лекарственного препарата/дозу 5,0 мл, получают следующим образом: Активный ингредиент, сахарозу и ксанановую камедь смешивают, пропускают через сито 10 меш США и затем смешивают с предварительно приготовленным раствором микрокристаллической целлюлозы и карбоксиметилцеллюлозы натрия в воде. Бензоат натрия, вкусовые добавки и краситель разводят в некотором количестве воды и добавляют при перемешивании. Затем добавляют воду в достаточном количестве для получения требуемого объема. Активный ингредиент, крахмал и стеарат магния смешивают, пропускают через сито 20 меш США и полученной смесью наполняют твердые желатиновые капсулы в количестве 425,0 мг. Пример препарата 9. Подкожный препарат можно получить следующим образом: Пример препарата 10. Внутривенный препарат можно получить следующим образом: В другом препарате, используемом в способах, соответствующих настоящему изобретению, применяют устройства для чрескожной доставки ("пластыри"). Данные чрескожные пластыри можно использовать для получения непрерывной или прерывающейся инфузии соединений, соответствующих настоящему изобретению, в контролируемых количествах. Конструкция и применение чрескожных пластырей для доставки фармацевтических агентов хорошо известны в области техники. См., например, патент США 5023252, выданный 11 июня 1991 г., в данном контексте включенный в виде ссылки. Данные пластыри можно сконструировать для непрерывной, пульсирующей доставки или доставки фармацевтических агентов по требованию. Часто бывает желательно или необходимо ввести фармацевтическую композицию в головной мозг либо прямо, либо непрямым образом. Методы прямого введения обычно включают размещение катетера для доставки лекарственного препарата в желудочковую систему хозяина, чтобы обойти гематоэнцефалический барьер. Одна из данных имплантируемых систем доставки, используемая для доставки биологических факторов в специфические анатомические области тела, описана в патенте США 5011472, который в данном контексте включен в виде ссылки. Непрямые методы обычно включают приготовление композиций, чтобы получить скрытую форму лекарственного препарата путем превращения гидрофильных лекарственных препаратов в лекарственные препараты, растворимые в жирах. Получение скрытой формы, как правило, осуществляют блокированием гидроксильных, карбонильных, сульфатных и первичных аминогрупп, присутствующих в лекарственном препарате, чтобы сделать лекарственный препарат более растворимым в липидах и способным к транспорту через гематоэнцефалический барьер. Альтернативно доставку гидрофильных лекарственных препаратов можно усилить внутриартериальной инфузией гипертонических растворов, которые могут временно открыть гематоэнцефалический барьер. Другие подходящие препараты, предназначенные для использования в настоящем изобретении,можно найти в справочнике Remington's Pharmaceutical Sciences (Справочник Ремингтона по фармацевтическим наукам), Mace Publishing Company, Philadelphia, PA, 17 изд. (1985). Как отмечено выше, соединения, описанные в данном контексте, подходят для использования в- 20017110 множестве вышеописанных систем доставки лекарственных препаратов. Кроме того, для увеличения полупериода существования введенного соединения в сыворотке in vivo соединения могут быть инкапсулированы, введены в полость липосом, получены в виде коллоида или могут быть использованы другие известные методы, которые обеспечивают увеличенный полупериод существования соединения в сыворотке. Существует множество способов получения липосом, как описано, например, в патентах США 4235871, 4501728 и 4837028, выданных Szoka et al., каждый из которых включен в данном контексте в виде ссылки. Соединения, обладающие желательной биологической активностью, можно модифицировать необходимым образом, чтобы получить требуемые свойства, такие как улучшенные фармакологические свойства (например, стабильность in vivo, биодоступность) или способность к определению в диагностических областях применения. Стабильность можно оценить множеством способов, например измерением полупериода существования белков при инкубировании с пептидазами или человеческой плазмой или сывороткой. Описан ряд таких анализов стабильности белка (см., например, статью Verhoef et al., Eur. J.Drug Metab. Pharmacokinet., 1990. 15(2):83-93). Конъюгаты, соответствующие настоящему изобретению, представляют собой антагонисты VLA-4 и предусмотрены для получения улучшенного удерживания in vivo по сравнению с неконъюгированными соединениями. Данное усовершенствованное удерживание конъюгата в организме приводило бы к более низким требуемым дозам лекарственного препарата, что, в свою очередь, давало бы в результате меньше побочных эффектов и более низкую вероятность токсичности. Кроме того, лекарственный препарат можно вводить пациенту менее часто, достигая при этом аналогичного или улучшенного терапевтического эффекта. Ожидают, что конъюгаты, соответствующие данному изобретению, будут проявлять ингибирование in vivo адгезии лейкоцитов к эндотелиальным клеткам, опосредованной VLA-4, посредством конкурентного связывания с VLA-4. Предпочтительно, когда соединения, соответствующие настоящему изобретению, можно использовать во внутривенных препаратах для лечения заболеваний, опосредованныхVLA-4 или адгезией лейкоцитов. Такие заболевания включают воспалительные заболевания у млекопитающих пациентов, такие как астма, болезнь Альцгеймера, атеросклероз, деменция при СПИДе, диабет(включая острый ювенильный диабет), воспалительное заболевание кишечника (включая язвенный колит и болезнь Крона), рассеянный склероз, ревматоидный артрит, трансплантацию тканей, метастазы опухоли, менингит, энцефалит, инсульт и другие церебральные травмы, нефрит, ретинит, атопический дерматит, псориаз, ишемию миокарда и острое опосредованное лейкоцитами повреждение легкого, такое как происходит при респираторном дистресс-синдроме взрослых. Препараты, соответствующие настоящему изобретению, особенно эффективны при лечении воспалительного заболевания кишечника, рассеянного склероза Крона и ревматоидного артрита. Соответствующие модели in vivo для демонстрации эффективности при лечении воспалительных состояний включают EAE (экспериментальный аутоиммунный энцефаломиелит) у мышей, крыс, морских свинок и приматов, а также другие модели воспалений, зависимые от интегринов 4. Воспалительное заболевание кишечника или "IBD" относится к группе нарушений, которые являются причиной того, что кишка воспаляется, как правило, проявляющиеся симптомами, включающими абдоминальные спазмы и боль, диарею, потерю массы тела и кишечное кровотечение. IBD является общим термином для двух близких заболеваний - язвенного колита ("UC") и болезни Крона ("CD"). Болезнь Крона ("CD") представляет собой хроническое аутоиммунное нарушение, которое приводит в результате к воспалению желудочно-кишечного (ЖК) тракта. Хотя может быть включена любая область ЖК тракта, CD наиболее часто поражает тонкую кишку и/или толстую кишку. При болезни Крона могут принимать участие все слои кишки, и нормальный здоровый участок кишки может находиться между очагами кишки, пораженной болезнью. CD ассоциируется с фиброзом, стенозом и образованием трещин, фистул между больным трактом и прилежащими органами (т.е. мочевым пузырем, другими сегментами кишки, кожей) и абсцессом. У больных CD, как правило, присутствует диарея, абдоминальная боль и потеря массы тела. Абдоминальная боль обычно является бессимптомной и может быть связана с болезненной воспаленной массой. Часто наблюдают повышение температуры, потерю массы тела, стоматит, перианальную фистулу и/или трещину, артрит и узелковую эритему. С CD связана значительная заболеваемость, особенно у пациентов с заболеванием, не контролируемым имеющимися в настоящее время лекарственными препаратами. До 75% пациентов с умеренным-тяжелым заболеванием требуют хирургического вмешательства и до 75% данных пациентов будут после хирургического вмешательства подвержены рецидивам болезни в течение 10 лет и до 50% будут подвергнуты повторному хирургическому вмешательству в течение 20 лет. Данный высокий уровень рецидивов указывает на необходимость новых эффективных лекарственных препаратов как для применения при активной форме заболевания,так и для поддержания ремиссии при заболевании. Язвенный колит или "UC" представляет собой хроническое эпизодическое воспалительное заболевание толстой кишки и прямой кишки, характеризующееся кровавой диареей. Язвенный колит является воспалительным ответом, ограниченным в значительной степени слизистой и подслизистой кишечника. Язвенный колит можно классифицировать по его местонахождению: "проктит" включает только прямую- 21017110 кишку, "проктосигмоидит" поражает прямую кишку и сигмовидную кишку, "левосторонний колит" охватывает всю левую сторону толстой кишки, "панколит" охватывает воспалением всю кишку. Лимфоциты и макрофаги многочисленны в повреждениях, вызываемых воспалительной болезнью кишки, и могут вносить вклад в воспалительные повреждения. Иллюстративную модель воспалительного заболевания кишечника на животных (IBD) используют с участием трансгенных крыс HLA-B27. Данные крысы способны к сверхэкспрессии молекулы HLA-B27 человека (ген тяжелой цепи и -глобулина), которая ассоциирована со спондилоартропатиями, группой воспалительных состояний, поражающих скелет. Перед началом воспалительных изменений скелета у данных животных развивается негрануломатозное воспаление в тонкой кишке и диффузные абсцессы крипт на толстой кишке, патология, которая близка к патологии болезни Крона у человека. Исследования эффективности проводят модели IBD на трансгенных мышах HLA-B27 c использованием соединений, соответствующих настоящему изобретению, как описано в примере J ниже. Астма представляет собой заболевание, характеризующееся повышенной чувствительностью трахеобронхиального дерева к различным стимулам, потенцирующим пароксизмальное сокращение бронхиальных дыхательных путей. Стимулы вызывают высвобождение различных медиаторов воспаления изIgE-покрытых мастоцитов, включая гистамин, хемотаксические факторы эозинофилов и нейтрофилов,лейкотриены, простагландин и фактор активации тромбоцитов. Высвобождение данных факторов рекрутирует базофилы, эозинофилы и нейтрофилы, которые вызывают воспалительное повреждение. Некоторые подходящие модели на животных для изучения астмы in vivo могут включать модель астмы на крысах и модель на овцах, как описано в примере E. Атеросклероз является заболеванием артерий (например, коронарной, сонной, аорты и подвздошной). Основное повреждение, атерома, состоит из приподнятой локальной бляшки в интиме, имеющей липидное ядро и покрывающую фиброзную оболочку. Атеромы нарушают артериальный кровоток и ослабляют пораженные артерии. Инфаркты миокарда и головного мозга представляют собой основное последствие данного заболевания. Макрофаги и лейкоциты рекрутируют в атеромы и вносят вклад в воспалительное повреждение. Ревматоидный артрит представляет собой хроническое рецидивирующее воспалительное заболевание, которое в основном вызывает повреждение и разрушение суставов. Ревматоидный артрит обычно сначала поражает мелкие суставы рук и ног, но затем может включать кисти, локти, лодыжки и колени. Артрит возникает от взаимодействия синовиальных клеток с лейкоцитами, которые путем инфильтрации проходят из кровотока в синовиальную выстилку суставов. См., например, монографию Paul, Immunology (Иммунология) (3 изд., Raven Press, 1993). Со временем может произойти эрозия костей, разрушение хряща и полная потеря целостности сустава. В конечном счете могут быть поражены многие системы органов. Повреждение сустава при ревматоидном артрите начинается с пролиферации синовиальных макрофагов и фибробластов после запускающего события, возможно, аутоиммунного или инфекционного. Происходит инфильтрация лимфоцитов в периваскулярные области и пролиферация эндотелиальных клеток. Затем имеет место неоваскуляризация. Кровеносные сосуды в пораженном суставе закупориваются маленькими сгустками воспалительных клеток. С течением времени воспаленная синовиальная ткань начинает вести себя неорганизованно, образуя ткань инвазивного паннуса. Паннус проникает и разрушает хрящ и кость. Высвобождается множество цитокинов, интерлейкинов, протеиназ и факторов роста, вызывая дальнейшее разрушение сустава и развитие системных осложнений. См. раздел Firestein(Учебник по ревматологии Келли), 7 изд., Philadelphia: W.B. Saunders, 2005:996-1042. Подходящие модели на животных для изучения ревматоидного артрита могут включать артрит, вызываемый адъювантами ("AIA"), и артрит, вызываемый коллагеном ("CIA"), как описано в примерах G и Н в данном контексте. Другим показателем для соединений, соответствующих настоящему изобретению, является лечение отторжения органа или трансплантата, опосредованного VLA-4. В последние годы достигнуто значительное повышение эффективности хирургических способов трансплантации тканей и органов, таких как кожа, почка, сердце, легкое, поджелудочная железа и костный мозг. Возможно, основной значительной проблемой является отсутствие удовлетворительных агентов для индукции иммунотолерантности у реципиента к трансплантированному аллотрансплантату или органу. Когда аллогенные клетки или органы трансплантируют хозяину (т.е. донор и реципиент являются разными субъектами одного вида), иммунная система хозяина, вероятно, усиливает иммунный ответ на чужеродные антигены трансплантата (болезнь хозяин против трансплантата), приводя к разрушению трансплантированной ткани. Клетки CD8+,клетки CD4 и моноциты, все они участвуют в отторжении трансплантированных тканей. Соединения,соответствующие настоящему изобретению, которые связываются с интегрином 4, используют, среди прочего, для блокирования иммунных ответов, индуцированных аллоантигеном, у реципиента, препятствуя, таким образом, участию данных клеток в разрушении трансплантированной ткани или органа. См.,- 22017110 например, статьи Paul et al., Transplant International 9, 420-425 (1996); Georczynski et al., Immunology 87,573-580 (1996); Georcyznski et al., Transplant. Immunol. 3, 55-61 (1995); Yang et al., Transplantation 60, 7176 (1995); Anderson et al., APMIS 102, 23-27 (1994). Похожим является способ применения соединений, соответствующих настоящему изобретению,которые связываются с VLA-4, при модуляции иммунного ответа, участвующего в болезни "трансплантат против хозяина" ("GVHD"). См., например, статью Schlegel et al., J. Immunol. 155, 3856-3865 (1995).GVHD представляет собой потенциально летальное заболевание, которое происходит, когда иммунологически компетентные клетки переносятся аллогенному реципиенту. В данной ситуации иммунокомпетентные клетки донора могут атаковать ткани реципиента. Ткани кожи, кишечного эпителия и печени являются частыми мишенями и могут быть разрушены во время протекания GVHD. Заболевание представляет особенно тяжелую проблему, когда трансплантируют иммунную ткань, как при трансплантации костного мозга, но менее тяжелая GVHD, кроме того, также описана в других случаях, включая трансплантаты сердца и печени. Лечебные факторы, соответствующие настоящему изобретению, используют,среди прочего, для блокирования активации донорных T-клеток, нарушая, таким образом, их способность лизировать клетки-мишеии хозяина. Дополнительное применение соединений, соответствующих настоящему изобретению, представляет собой ингибирование метастазов опухолей. Показано, что ряд опухолевых клеток экспрессирует VLA4, и соединения, которые связывают VLA-4, блокируют адгезию данных клеток к эндотелиальным клеткам. См. статьи Steinback et al., Urol. Res. 23, 175-83 (1995); Orosz et al., Int. J. Cancer 60, 867-71 (1995);Freedman et al., Leuk. Lymphoma 13, 47-52 (1994); Okahara et al., Cancer Res. 54, 3233-6 (1994). Дополнительное применение соединений, соответствующих настоящему изобретению, представляет собой лечение рассеянного склероза. Рассеянный склероз является прогрессирующим неврологическим аутоиммунным заболеванием, которое поражает по приблизительной оценке 250000-350000 человек в Соединенных Штатах Америки. Считают, что рассеянный склероз является результатом специфической аутоиммунной реакции, в которой некоторые лейкоциты атакуют и инициируют разрушение миелина, отделяющего покрытия, закрывающего нервные волокна. На модели рассеянного склероза на животных показано, что мышиные моноклональные антитела, направленные против VLA-4, блокируют адгезию лейкоцитов к эндотелию и, таким образом, препятствуют воспалению центральной нервной системы и последующему параличу у животных 16. Фармацевтические композиции, соответствующие изобретению, пригодны для применения в ряде систем доставки лекарственных препаратов. Подходящие для применения в настоящем изобретении препараты можно найти в справочнике Remington's Pharmaceutical Sciences (Справочник Ремингтона по фармацевтическим наукам), Mace Publishing Company, Philadelphia, PA, 17 изд. (1985). Количество, вводимое пациенту, будет варьировать в зависимости от того, что вводят, с какой целью, такой как профилактики или лечение, состояния пациента, способа введения и т.п. При терапевтическом применении композиции вводят пациенту, уже страдающему от заболевания, в количестве, достаточном для излечения или по меньшей мере частичного снятия симптомов заболевания и его осложнений. Количество, адекватное для осуществления этого, определяют как "терапевтически эффективная доза". Количества, эффективные для данного применения, будут зависеть от болезненного состояния,которое лечат, а также от мнения лечащего врача, зависящего от таких факторов, как тяжесть воспаления, возраст, масса тела и общее состояние пациента и т.п., с учетом данных, полученных на соответствующей модели на животном, такие как представлены в данном контексте. Способы оценки подходящих для человека доз, основанные на данных результатах, известны в области техники. (См, например, монографию Wagner, J.G. Pharmacokinetics for the Pharmaceutical Scientist. (Фармакокинатика для фармацевтов) Technomic, Inc., Lancaster, PA 1993). Композиции, вводимые пациенту, находятся в форме вышеописанных фармацевтических композиций. Данные композиции можно стерилизовать с помощью принятых способов стерилизации или можно стерилизовать фильтрованием. Полученные в результате водные растворы можно упаковать для применения в том виде, в котором они находятся, или лиофилизировать, причем перед использованием лиофилизированный препарат смешивают со стерильным водным носителем. Соединения, обладающие требуемой биологической активностью, можно модифицировать по необходимости, получая требуемые свойства, такие как улучшенные фармакологические признаки (например, стабильность in vivo, биодоступность) или способность определяться в диагностических областях применения. Стабильность можно оценить рядом способов, например измерением полупериода существования белков при инкубировании с пептидазами или человеческой плазмой или сывороткой. Описан ряд данных анализов стабильности белков (см., например, статью Verhoef et al., Eur. J. Drug Metab. Pharmacokinet., 1990, 15(2):83-93). Терапевтическая доза соединений, соответствующих настоящему изобретению, будет варьировать в соответствии, например, с конкретным применением, для которого проводят лечение, способом введения соединения, состоянием здоровья и общим состоянием больного и мнением лечащего врача. Например,для внутривенного введения доза, как правило, будет находитьтся в интервале от приблизительно 20 до приблизительно 2000 мкг/кг массы тела, предпочтительно от приблизительно 20 до приблизительно 500- 23017110 мкг, более предпочтительно от приблизительно 100 до приблизительно 300 мкг/кг массы тела. Подходящие интервалы доз для интраназального введения в основном составляют от приблизительно 0,1 пг до 1 мг/кг массы тела. Эффективные дозы можно экстраполировать из кривых зависимости доза-ответ, полученных на основании тест-систем in vitro или на моделях на животных. Соединения, соответствующие данному изобретению, также способны к связыванию или антагонистическому действию в отношении активности интегринов 41 и 47. Согласно этому, соединения,соответствующие настоящему изобретению, используют также для предупреждения или реверсии симптомов, нарушений или заболеваний, вызываемых связыванием данных интегринов с их соответствующими лигандами. В другом аспекте изобретения соединения и композиции, описанные в данном контексте, могут быть использованы для ингибирования миграции иммунных клеток из кровотока в центральную нервную систему в случае, например, рассеянного склероза, или в области, которые приводят в результате к вызываемой воспалением деструкции миелина. Предпочтительно, когда реагенты ингибируют миграцию иммунных клеток таким образом, что это ингибирует демиелинизцию и далее может способствовать ремиелинизации. Реагенты могут также препятствовать демиелинизации и стимулировать ремиелинизацию центральной нервной системы при родственных метаболических нарушениях, при которых инфильтрация иммунных клеток действует на развитие миелинового покрытия в основном в ЦНС. Предпочтительно, когда реагенты также снижают паралич при введении пациенту с параличом, вызванным демиелинизирующим заболеванием или состоянием. Воспалительные заболевания, лечение которых предусмотрено композициями, соединениями и способами, описанными в данном контексте, включают, в основном, состояния, связанные с демиелинизацией. Гистологически миелиновые патологии представляют собой либо демиелинизацию, либо дисмиелинизацию. Демиелинизация подразумевает разрушение миелина. Дисмиелинизация относится к нарушенному образованию или поддержанию миелина в результате дисфункции олигодендроцитов. Предпочтительно, когда композиции и способы, описанные в данном контексте, предусматривают лечение заболеваний и состояний, связанных с демиелинизацией, и помощь при ремиелинизации. Дополнительные заболевания или состояния, лечение которых предусматривают, включают менингит, энцефалит и повреждения спинного мозга и состояния, которые, как правило, вызывают демиелинизацию в результате воспалительной реакции. Композиции, соединения и коктейли, описанные в данном контексте, предусматривают для применения при лечении состояний и заболеваний, ассоциированных с демиелинизацией. Заболевания и состояния, включающие демиелинизацию, содержат, но без ограничения перечисленным, рассеянный склероз, врожденные нарушения обмена веществ (например, фенилкетонурию (PKU), болезнь Тея-Сакса,болезнь Ниманна-Пика, болезнь Гоше, синдром Гурлера, болезнь Краббе и другие формы лейкодистрофии, которые поражают развивающуюся оболочку), невропатии с патологической миелинизацией (например, синдром Джиллиана-Барра, хроническая иммунная демиелинизирующая полиневропатия(CIDP), многоочаговая CIDP, многоочаговая двигательная невропатия (MMN), синдром анти-MAG (миелин-ассоциированного гликопротеина), синдром GALOP (аббревиатура от слов Gait disorder, Autoantibody, Late-age, Onset, Polyneuropathy (полиневропатия, развивающаяся в старческом возрасте, сопровождающаяся появлением антител и нарушением походки), синдром антител к сульфатидам, синдром антител к GM2, синдром POEMS (аббревиатура от слов Polyneuropathy, Organomegaly, Endocrinopathy, MProtein и Skin changes (полиневропатия, органомегалия, эндокринопатия, изменения М-белка и кожи),известный также как синдром Кроу-Фуказе и болезнь Такацуки, периневрит, синдром igM к антителамGDIb), демиелинизацию, связанную с лекарственными препаратами (например, вызываемую введением хлороквина, FK506, перексилина, прокаинамида и цимельдина), другие врожденные демиелинизирующие состояния (например, углевод-дефицитный гликопротеин, синдром Кокейна, врожденное гипомиелинирование, врожденную мышечную дистрофию, болезнь Фарбера, синдром Маринеско-Шегрена, метахроматическую лейкодистрофию, болезнь Пелицеуса-Мерцбахера, болезнь Рефсума, прионовые состояния и болезнь Салла) и другие демиелинизирующие состояния (например, менингит, энцефалит (известный также как диссеминированный энцефаломиелит, ADEM) или повреждение спинного мозга) или заболевания. Имеются различные модели заболеваний, которые могут быть использованы для изучения данных заболеваний in vivo. Например, модели на животных включают, но без ограничения перечисленным Наиболее распространенным демиелинизирующим заболеванием является рассеянный склероз("MS"), но многие другие метаболические и воспалительные нарушения приводят в результате к недостаточной или патологической миелинизации. MS представляет собой хроническое неврологическое заболевание, которое в большинстве случаев возникает в молодости. Только в Соединенных Штатах Америки насчитывают приблизительно 350000 случаев MS. За исключением травмы MS является наиболее частой причиной нетрудоспособности вследствие неврологического заболевания в молодости и в зрелые годы. Причину MS еще предстоит определить. MS характеризуется хроническим воспалением, демиелинизацией и глиозом (рубцеванием). Демиелинизация может привести в результате как к отрицательным,так и к положительным эффектам на аксональную проводимость. Патологии, связанные с положительной проводимостью, включают замедленную аксональную проводимость, различное блокирование проводимости, которое происходит в присутствии высоко-, но не низкочастотных серий импульсов, или полное блокирование проводимости. Патологии положительной проводимости включают генерацию эктопических импульсов, одновременный или последующий механический стресс и патологическое "перекрестное связывание" демиелинизированных аксонов. Т-клетки, реактивные в отношении миелиновых белков, либо миелинового основного белка (МВР),либо миелинового протеолипидного белка (PLP), как показано, опосредуют воспаление ЦНС при экспериментальном аллергическом энцефаломиелите. Наблюдают также пациентов, имеющих повышенные уровни иммуноглобулина (Ig) ЦНС. Кроме того, возможно, что некоторые из повреждений тканей, наблюдаемые при MS, опосредованы продуктами цитокинов активированных T-клеток, макрофагов или астроцитов. В настоящее время 80% пациентов, у которых диагностирован MS, живут 20 лет после начала развития болезни. Варианты лечения для ведения MS включают: (1) лечение, направленное на модификацию течения заболевания, включая лечение острых ухудшений, и направленное на длительное подавление заболевания; (2) лечение симптомов MS; (3) предупреждение и лечение медицинских осложнений и(4) управление вторичными личностными и социальными проблемами. Начало MS может быть резким или таким мягким, что не заставляет больного обратиться за медицинской помощью. Наиболее распространенные симптомы включают слабость в одной или нескольких конечностях, нечеткость зрения вследствие ретробульбарного неврита, сенсорные нарушения, двоение в глазах и атаксия. Течение болезни можно разделить на три основные категории: (1) рецидивирующийMS, (2) хронический прогрессирующий MS и (3) неактивный MS. Рецидивирующий MS характеризуется повторяющимися приступами неврологической дисфункции. Приступы MS, как правило, включают от нескольких дней до нескольких недель и за ними может последовать полное, частичное выздоровление или не последовать выздоровление. Выздоровление после приступов, как правило, происходит в течение нескольких недель-нескольких месяцев после максимального проявления симптомов, хотя редко некоторое выздоровление может продолжаться в течение 2 или более лет. Хронический прогрессирующий MS приводит к постепенному ухудшению без периодов стабилизации или ремиссии. Данная форма развивается у больных с рецидивирующим MS в предшествующей истории болезни, хотя у 20% больных не возвращаются никакие рецидивы. Хотя при прогрессирующем течении могут иметь место острые рецидивы. Третья форма представляет собой неактивный MS. Неактивный MS характеризуется фиксированными неврологическими недостаточностями вариабельной величины. Большинство пациентов с неактивным MS имеют предшествующую историю болезни рецидивирующим MS. Течение болезни зависит также от возраста пациента. Например, благоприятные прогностические- 25017110 факторы включают раннее начало (исключая детство), рецидивирующее течение и незначительную остаточную нетрудоспособность в течение 5 лет после начала. Напротив неблагоприятный прогноз ассоциирован с началом в позднем возрасте (т.е. в возрасте 40 лет и старше) и прогрессирующим течением. Данные варианты взаимозависимы, поскольку хронический прогрессирующий MS имеет тенденцию к началу в более позднем возрасте, чем рецидивирующий MS. Нетрудоспособность при хроническом прогрессирующем MS обычно обусловлена прогрессирующей параплегией или квадриплегией (параличом) у пациентов. В одном аспекте изобретения предпочтительно, когда пациентов будут лечить, когда пациент находится в состоянии ремиссии, а не в стадии рецидива заболевания. Кратковременное применение либо адренокортикотропного гормона, либо пероральных кортикостероидов (например, перорального преднизона или внутривенного метилпреднизолона) является единственной специфической лечебной мерой для лечении пациентов с острым ухудшением MS. Более новые способы лечения MS включают лечение пациента интерфероном -1b, интерфероном-1a и Copaxone (ранее известным как сополимер 1). Показано, что данные три лекарственных препарата существенно снижают уровень рецидивов заболевания. Данные лекарственные препараты вводятся самим больным внутримышечно или подкожно. Однако ни один из современных способов лечения не подавляет демиелинизацию, не говоря о том,что он способствует или дает возможность спонтанной ремиелинизации или уменьшает паралич. Один аспект изобретения предусматривает лечение MS агентами, описанными в данном контексте, либо в виде монотерапии, либо в комбинации с другими стандартными способами лечения. Облучение может вызывать демиелинизацию. Считают, что токсичность в отношении центральной нервной системы (ЦНС), обусловленная облучением, является причиной (1) повреждения сосудистых структур, (2) уничтожения предшественников олигодендроцитов-2 астроцитов и зрелых олигодендроцитов, (3) уничтожения популяций нервных стволовых клеток в гиппокампе, мозжечке и коре головного мозга и генерализованных изменений экспрессии цитокинов. Наибольшее радиационное повреждение обусловлено радиотерапией, применяемой при лечении некоторых форм рака. В качестве обзора см. статью Belka et al., 2001 Br. J. Cancer 85: 1233-9. Однако воздействие излучения может быть также причиной у космонавтов (см. статью Hopewell, 1994 Adv. Space Res. 14: 433-42), а также событием, обусловленным воздействием радиоактивных веществ. Предусматривают также паллиативное или облегчающее лечение данных состояний и заболеваний. Примеры Следующие примеры синтеза и биологические примеры предлагают для иллюстрации данного изобретения и никоим образом не должны быть истолкованы как ограничивающие объем настоящего изобретения. Если не указано иначе, все температуры даны в градусах Цельсия. В нижеприведенных примерах следующие сокращения имеют следующие значения. Если сокращение не определено, оно имеет свое общепринятое значение.= ангстремы,br s = широкий синглет,BSA = бычий сывороточный альбумин,d = дублет,dd = дублет дублетов,dq = дублет квартетов,dсекстет = дублет секстетов,ДМФ = диметилформамид,EC50 = доза, при которой желательный ответ присутствует у 50% популяции,ЭДТА = этилендиаминтетрауксусная кислота,EtOAc = этилацетат,EtOH = этанол,Et3N = триэтиламин,ЕМ = длина волны эмиссии (в нм),ЕХ = длина волны возбуждения (в нм),г = грамм,HBSS = сбалансированный солевой раствор Хенка,HEPES = 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота,HPLC = высокоэффективная жидкостная хроматография,час или ч = часы,IC50 = концентрация ингибитора, которая требуется для ингибирования фермента in vitro,in. = дюйм,i.p. = внутрибрюшинно,i-PrOH = изопропанол,кг = килограмм,л = литр,- 26017110 ЖХ/МС = жидкостная хроматография/масс-спектрометрия,гл = мультиплет,м 2 = квадратные метры,М = молярный,мбар = миллибар,мг = миллиграмм,МГц = мегагерц,мин = минуты,мл = миллилитры,мм = миллиметры,мМ = миллимолярный,ммоль = миллимоли,мОсм = миллиосмоль МТВЕ = метил-трет-бутиловый эфир,m/z или M/Z = соотношение мас./заряд,Н = нормальный,нг = нанограммы,нм = нанометры,ЯМР = ядерно-магнитный резонанс,PBS = забуференный фосфатом солевой раствор,PBS = PBS с добавление кальция и магния,Промиль = частиц/миллион,psi = фунты/квадратный дюйм,р.о. = per os (перорально), дословно "через рот", включает пероральный зонд,q = квартет,q.s. = в достаточном количестве,Rf = фактор удерживания (соотношение расстояние, пп пройденное субстанцией/расстояние, пройденное фронтом растворителя),об./мин = количество оборотов в минуту,кт или КТ = комнатная температура,Rt = время удерживания,s = синглет,t = триплет,ТФА = трифторуксусная кислота,ТГФ = тетрагидрофуран,ТСХ = тонкослойная хроматография,УФ = ультрафиолет,мас./мас. = соотношение массы к массе,мас./об. = соотношение массы к объему,мкг = микрограммы,мкм = микроны,мкМ = микромолярный. Пример 1. Получение (S)-2-(2-(диэтиламино)-5-(N-изопропилметилсульфонамидо)пиримидин-4 иламино)-3-(4-(пирролидин-1-карбонилокси)фенил)пропановой кислоты. Протокол синтеза, используемый в примере 1, кратко показан на схеме 4, иллюстрируемой ниже. На схеме 4 соединение 4 получают последовательно в трех емкостях из 5-нитропиримидинового соединения 1. Протокол синтеза согласно схеме 4 существенно упрощает получение данного соединения благодаря по меньшей мере одному фактору из нижеследующих: 1) существенно ускоренная стадия восстановления нитрогруппы; 2) рационализированная последовательность восстановления/восстановительного аминирования,которую проводят в той же колбе с тем же растворителем и тем же катализатором, вследствие чего манипуляции сокращаются и воздействие воздуха на чувствительные к кислороду продукты сводится к минимуму; 3) условия стадии восстановительного аминирования сводят к минимуму генерацию побочного продукта бис-изопропиламинопиримидина, устраняя, таким образом, необходимость хроматографической очистки соединения 3; 4) описаны условия, посредством которых возможно очистить промежуточный продукт моноизопропиламинопиримидин, соединение 2, тритурацией соответствующей соли L-винной кислоты (поэтому необходимость отдельной очистки соединения 2 также становится необязательной вследствие усовершенствований стадии восстановительного аминирования), и 5) определены условия отдельной очистки соединения 3 посредством кристаллизации из МТВЕгексана или МТВЕ-циклогексана. В стадиях реакции схемы 4 экспресс-хроматографию проводят с помощью Biotage Flash 75L при использовании силикагелевых картриждей KP-Sil 800 г (32-63 мкМ, 60 , 500-550 м 2/г). Описывают Rf для аналитической тонкослойной хроматографии с использованием тонких пластинок 250 мкМ для нормальной фазы ЕМ Sciences Silica Gel 60 F(254). ЯМР-спектры получают на спетрометре Varian Gemini 300 МГц (300 МГц для 1 Н-спектров и 75 МГц для 13 С-спектров). Аналитическую ВЭЖХ проводят на приборе Agilent 1100 Series HPLC с колонкой Phenomenex Luna, 3 мкм, С-18, 304,6 мм. Детектором является УФ при 210 нм. Используют растворители 0,1% ТФА в воде и 0,1% ТФА в ацетонитриле. Стандартная скорость потока составляет 1,5 мл/мин, и стандартный способ называют M1 с использованием градиента растворителя, изменяющегося от 20 до 70% CH3CN в течение 2,33 мин. Альтернативный способ называют М 2 со скоростью потока 2 мл/мин и изменением градиента от 20 до 70% CH3CN в течение 1,75 мин. Способ М 15 имеет скорость потока 1,5 мл/мин. При составе растворителей, изменяющемся от 20 до 70% CH3CN в течение 10 мин, поддерживая 70% в течение 2 мин с последующим линейным возрастанием до 95% в течение 1 мин и поддержанием на уровне 95% в течение 2 мин. ЖХ/МС проводят на приборе Agilent 1100 Series HPLC с Series 1100 MSD с электроспрей ионизацией (пока не указано иначе,как с химической ионизацией). Колонка и условия соответствуют свободно протекающей ВЭЖХ. Стадия 1. Получение (S)-4-(3-трет-бутокси-2-(2-(диэтиламино)-5-(изопропиламино)пиримидин-4 иламино)-3-оксопропил)фенилпирролидин-1-карбоксилата (2). Нитропиримидинкарбамат 1 (100 г, 189 ммоль) и PtO2 (6,33 г, 27,85 ммоль) суспендируют в 360 мл влажного ТГФ (5% Н 2 О). Смесь перемешивают при комнатной температуре под водородом (60 psi (4200 г/см 2. Через 3 ч ТСХ (50% EtOAc/гексаны на силикагеле) показывает полное восстановление нитрогруппы (анализ ТСХ на диоксиде кремния с EtOAc показывает Rf=0,2 (полоса) для аминопиримидина иRf=0,86 для исходного нитропиримидинкарбамата). В этом плане использование PtO2 для обеих стадий в данном двухстадийном процессе дает возможность проведения реакции в одной емкости с введением такой характеристики, как резкое повышение скорости восстановления нитрогруппы. В любом случае предпринимают меры для сведения к минимуму воздействия воздуха/кислорода, поскольку аминопиримидиновый продукт чувствителен к окислению. Этанол (200 мл), ацетон (21 мл, 1,5 экв.) и ледяную уксусную кислоту (3,0 мл, 0,28 экв.) добавляют к раствору аминопиримидина в колбу для гидрогенизации. После откачивания воздуха и продувания в колбу под давлением накачивают Н 2 (60 psi (4200 г/см 2. Восстановительному аминированию позволяют протекать в течение ночи. ТСХ на силикагеле с использованием EtOAc в качестве элюента дает Rf=0,41(полоса) для изопропиламинопиримидина и Rf=0,11 для исходного аминопиримидинкарбамата. Как ТСХ, так и ЖХ/МС подтверждают завершенную реакцию при практическом отсутствии образованного бис-изопропиламинопиримидина. При необходимости ВЭЖХ можно использовать как альтернативное средство для мониторирования протекания реакции. Неочищенный реакционный раствор разбавляютEtOAc (1 л) и фильтруют через слой основного оксида алюминия (400 мл). Оксид алюминия промываютEtOAc (200 мл) и EtOH (200 мл) и объединенные органические растворы концентрируют в вакууме. Из колбы удаляют газ под N2. Вязкое масло перерастворяют в безводном толуоле (700 мл) и концентрируют. После удаления газа из колбы под азотом продукт сушат снова путем азеотропного удаления других 400 мл толуола. Получают вязкое масло красновато-коричневого цвета. Как доказано ЖХ/МС, при данном способе образуется очень мало примеси бисизопропиламинопиримидинкарбамата по сравнению с предшествующими способами, при которых требуется удаление примеси бис-изопропиламинопиримидинкарбамата с помощью хроматографии. Если требуется формальная очистка моноизопропиламинопиримидина на стадии 2, его можно осадить из ТГФ/эфира в виде соли (L)-винной кислоты и тритурировать. Приведем лабораторный пример:(5,09 г, выход 99,6%) L-винную кислоту (1,42 г) растворяют в горячем ТГФ (45 мл). Горячий раствор винной кислоты добавляют к смоле изопропиламинопиримидина 2 (5,1 г). Смесь взбалтывают и нагревают, пока не станет однородной. Раствор изменяет цвет с розово-фиолетового на коричнево-желтый. Раствор концентрируют в вакууме, получая коричнево-желтую смолу. Добавляют эфир (150 мл), после чего наблюдают замасливание. Эфирную смесь концентрируют в вакууме. Добавляют ацетон (20 мл) и затем эфир (200 мл) и снова наблюдают образование вязкого масла. Смесь концентрируют в третий раз. Добавляют метиленхлорид (5-10 мл), а затем эфир ( 80 мл). Наблюдают образование осадка коричневожелтого цвета под супернатантом яркого оранжево-розового цвета. Смесь фильтруют. Осадок промывают эфиром (50 мл) и затем снова смесью (60 мл) ацетона и эфира (1:1). Осадок сушат под вакуумом в течение ночи, получая твердое вещество кремового цвета (4,9 г, выход 76%). Маленькую аликвоту твердой соли винной кислоты растворяют в изо-PrOH и EtOH и пропускают через маленький слой основного оксида алюминия, получая свободное основание. Аликвоту свободного основания анализируют ТСХ и ЖХ/МС. Оставшуюся соль суспендируют в смеси CH2Cl2 (250 мл) и 1 Н MaHCO3 (150 мл). При перемешивании и небольшом барботировании твердое вещество растворяется и амин свободного основания экстрагируют в органический слой. Водный слой экстрагируют еще раз EtOAc (150 мл) и органические экстракты объединяют и сушат над MgSO4 (прибл.150 г). Высушенный органический раствор пропускают через слой основного оксида алюминия (прибл.100 г), получая раствор светло-розового цвета, который концентрируют в вакууме, получая смолу коричневожелтого/розового цвета (3,28 г, выход 64% от исходного нитрокарбамата). Исследуют ряд других кислот в попытке образования солей с моноизопропиламинопиримидинкарбаматом 2. п-Толуолеульфоновая кислота и метансульфоновая кислота дают масло. Твердые соли могли бы образовываться с HCl и H3PO4, но винная кислота, по-видимому, дает наиболее благоприятные характеристики растворимости. Соли HCl и фосфорной кислоты, по-видимому, легко растворяются в CH2Cl2,изо-PrOH и ацетоне, тогда как соль винной кислоты, по-видимому, наиболее не растворима в CH2Cl2 и только частично растворима в других растворителях. Стадия 2. Получение (S)-4-(3-трет-бутокси-2-(2-(диэтиламино)-5-(N-изопропилметилсульфон- 29
МПК / Метки
МПК: A61P 37/00, A61P 11/06, A61P 29/00, C07D 239/50, A61K 31/506
Метки: варианты, снижения, ответа, аутоиммунного, соединений, пиримидинилсульфонамидных, интегрином, предупреждения, воспалительного, композиция, alpha;4, фармацевтическая, соединения, опосредованного, получения, заболевания, способ, компонента, пиримидинилсульфонамидные, лечения
Код ссылки
<a href="https://eas.patents.su/30-17110-pirimidinilsulfonamidnye-soedineniya-varianty-sposob-polucheniya-pirimidinilsulfonamidnyh-soedinenijj-varianty-farmacevticheskaya-kompoziciya-sposob-lecheniya-zabolevaniya-oposredo.html" rel="bookmark" title="База патентов Евразийского Союза">Пиримидинилсульфонамидные соединения (варианты), способ получения пиримидинилсульфонамидных соединений (варианты), фармацевтическая композиция, способ лечения заболевания, опосредованного интегрином α4, способ снижения и/или предупреждения воспалительного компонента заболевания или аутоиммунного ответа</a>
Пиримидиниламидные соединения (варианты), включающая их фармацевтическая композиция и способ лечения заболевания, опосредованного a4-интегринами

Номер патента: 15388
Опубликовано: 31.08.2011
Авторы: Смит Дженифер Ли, Конрэди Андрей В., Сюй Ин-Цзы, Стэпэнбэк Фрэнк, Сэмкоу Кристофер Майкл, Фукуда Джури И., Росситер Кассандра Инес
МПК: A61K 31/506, C07D 239/50, A61P 29/00...
Метки: способ, включающая, фармацевтическая, варианты, соединения, a4-интегринами, лечения, композиция, опосредованного, пиримидиниламидные, заболевания
Формула / Реферат:
1. Пиримидиниламидные соединения общей формулы Iгде R1выбран из группы, включающей C1-C4-алкил, C1-C4-галогеналкил, гетероарил- и -NR5R6, где R5и R6 независимо выбраны из группы, включающей водород и C1-C4-алкил, или R5 и R6совместно с атомом азота, с которым они связаны, формируют гетероцикл;R2 представляет собой C1-C4-алкил;R3 и R4совместно с атомом азота, с которым они связаны, образуют гетероцикл;или их фармацевтически приемлемые соли или...
Способ лечения натализумабом воспалительного и/или аутоиммунного заболевания (варианты)

Номер патента: 16626
Опубликовано: 30.06.2012
Автор: Йеднок Теодор А.
МПК: A61K 39/395
Метки: аутоиммунного, заболевания, варианты, натализумабом, воспалительного, лечения, способ
Формула / Реферат:
1. Способ лечения натализумабом воспалительного и/или аутоиммунного заболевания у больного, в котором указанному больному вводят первую дозу натализумаба в течение первого периода приема натализумаба, измеряют в течение указанного периода содержание двухвалентного натализумаба в плазме или сыворотке больного, затем на основании измеренного содержания двухвалентного натализумаба рассчитывают вторую дозу натализумаба, которая повышает безопасность...
Соединения-агонисты гетероциклических рецепторов (варианты), фармацевтическая композиция на их основе, способ лечения диабета и метаболических расстройств, способ стимулирования выработки инсулина (варианты) и способы снижения уровня глюкозы и триглицеридов в крови посредством указанных соединений

Номер патента: 16720
Опубликовано: 30.07.2012
Авторы: Клеменс Л.Эдвард, Мерфи Элисон, Уилсон Мария Е., Чэнь Синь, Чжу Ян, Рэббэт Кристофер Дж., Чжао Чжухунь, Ма Цзянюань, Сон Цзянгао, Нашашиби Имад, Чэн Пэн, Джонсон Джефри Д.
МПК: A61K 31/445, A61K 31/497, A01N 43/40...
Метки: композиция, уровня, метаболических, глюкозы, лечения, триглицеридов, соединения-агонисты, основе, способы, диабета, гетероциклических, соединений, стимулирования, фармацевтическая, крови, указанных, рецепторов, расстройств, инсулина, посредством, способ, варианты, выработки, снижения
Формула / Реферат:
1. Соединения-агонисты гетероциклических рецепторов, характеризующиеся молекулярной массой менее 1200 и охватываемые общей формулой (I)где X, Y и Z независимо выбраны из группы, включающей О, N, N(R3), S и C(R3), и по меньшей мере один из X, Y и Z выбран из О, N, N(R3) и S;нижний индекс r представляет собой целое число от 0 до 3;нижний индекс s представляет собой целое число от 0 до 3, а сумма r+s≤4;нижний индекс q представляет собой целое...
Соединения пиразинона, фармацевтическая композиция (варианты) и лекарственная форма, способ ингибирования сериновой протеазы, способ снижения тромбообразующей способности поверхности, способ лечения нарушенного протеолиза и тромбоза, связанного с различными заболеваниями, способ снижения свертывания крови у млекопитающего

Номер патента: 4884
Опубликовано: 26.08.2004
Авторы: Маркоутэн Томас П., Лу Тианбао, Томкзук Брюс Э.
МПК: C07K 5/078, A61P 7/02, A61K 38/05...
Метки: протеазы, форма, сериновой, пиразинона, способ, снижения, крови, связанного, способности, лекарственная, соединения, заболеваниями, свертывания, варианты, фармацевтическая, нарушенного, тромбообразующей, лечения, поверхности, ингибирования, различными, млекопитающего, тромбоза, протеолиза, композиция
Формула / Реферат:
1. Соединение формулы I или его сольват, гидрат или фармацевтически приемлемая соль, где W означает R1; R1 означает R2, R2(CH2)tC(R12)2, где t равно 0-3 и оба R12 одинаковые или различные, (R2)(OR12)CH(CH2)p, где p равно 1-4, R2C(R12)2(CH2)t, где t равно 1-3 и оба R12 одинаковые или различные, причем (R12)2 может быть объединен с атомом углерода, к которому он присоединен, с образованием от 3- до 7-членного циклоалкильного кольца; ...
Производное n,n’ -дифенилмочевины, фармацевтическая композиция (варианты) и способ лечения и предупреждения заболеваний и состояний с его использованием (варианты)

Номер патента: 10485
Опубликовано: 30.10.2008
Авторы: Дюма Жак, Уилхелм Скотт, Бойер Стивен, Ридль Бернд
МПК: C07D 213/81, A61P 35/00, A61K 31/44...
Метки: лечения, способ, варианты, производное, предупреждения, использованием, дифенилмочевины, композиция, фармацевтическая, заболеваний, состояний
Формула / Реферат:
1. Производное N,N'-дифенилмочевины формулы (I) его соль, или 1-оксопиридинпроизводное, или стереоизомер. 2. Производное по п.1, отличающееся тем, что его соль представляет собой основную соль органической или неорганической кислоты, выбранной из группы: хлористо-водородная, бромисто-водородная, серная, фосфорная, метансульфоновая, трифторметансульфоновая, бензолсульфоновая, п-толуолсульфоновая, 1-нафталинсульфоновая, 2-нафталинсульфоновая,...
Предыдущий патент: Система и способ обработки данных на периферийном устройстве
Следующий патент: Монокристаллический пластинчатый сульфат бария в косметических композициях
Случайный патент: Ароматизированные композиции мыла