Производные триазола, обладающие противогрибковой активностью, предназначенные для человека и животных
Номер патента: 3189
Опубликовано: 27.02.2003
Авторы: Скиоппакасси Джованна, Альбини Энрико, Наполетано Маоро, Фраире Кристина








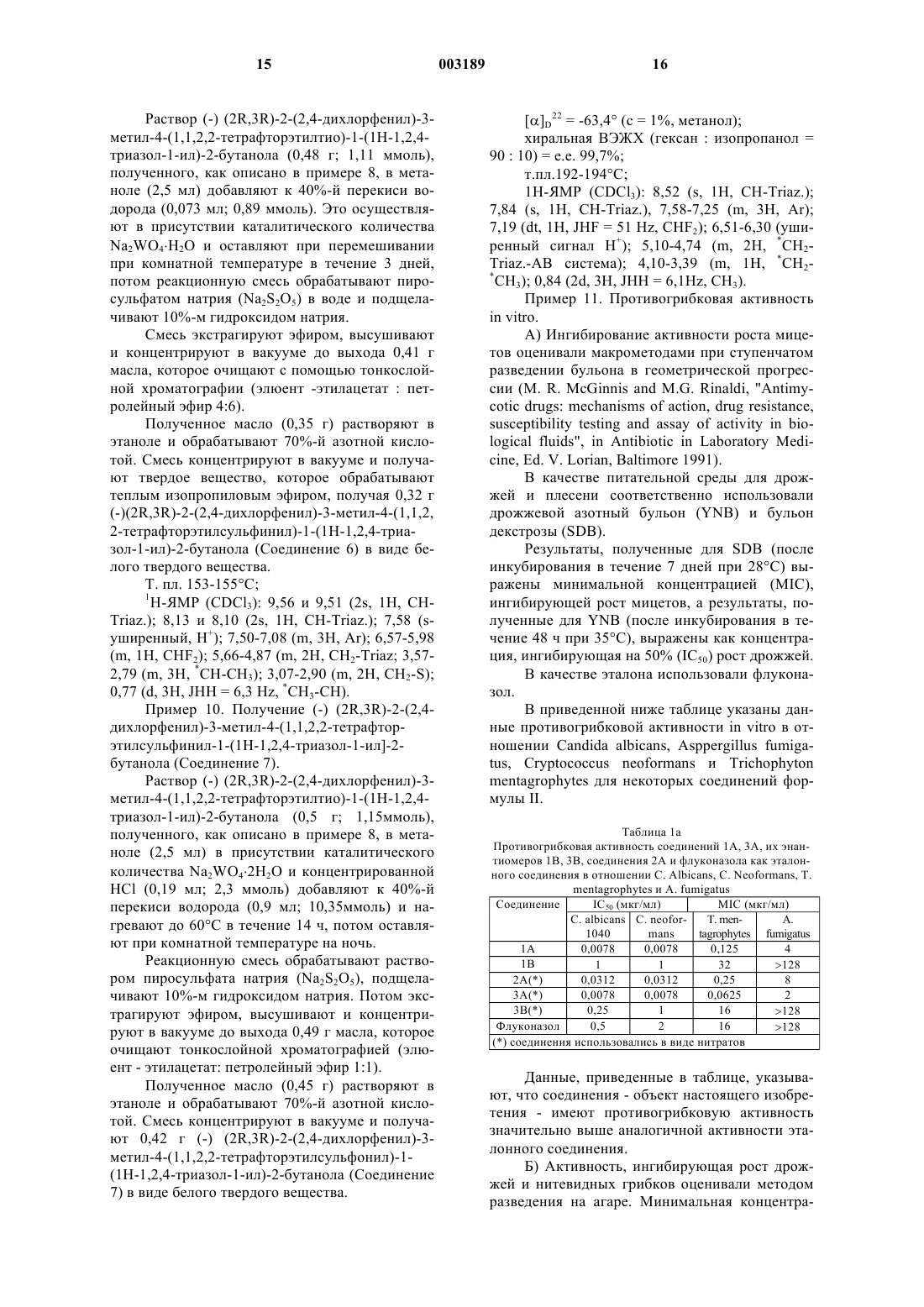
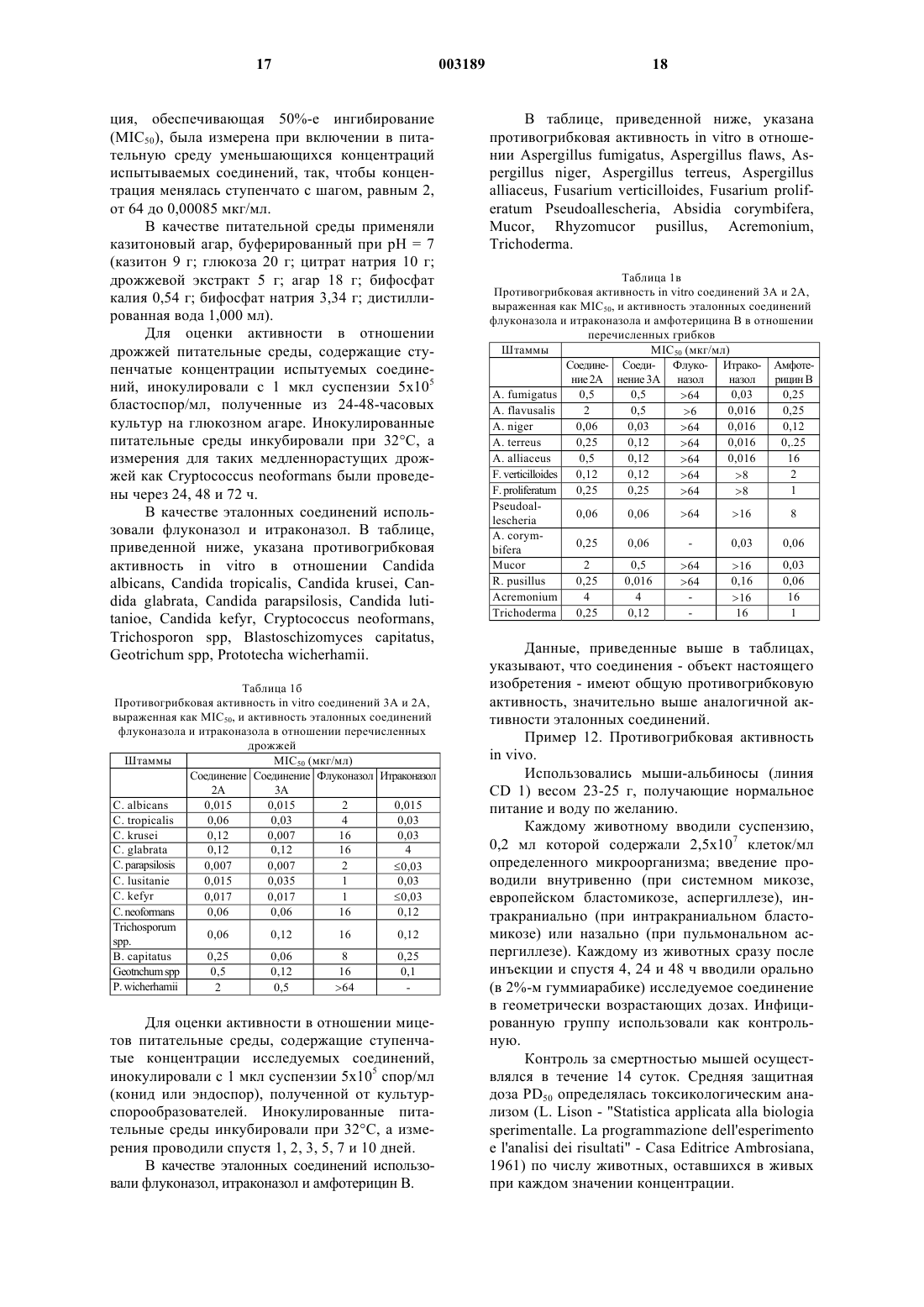
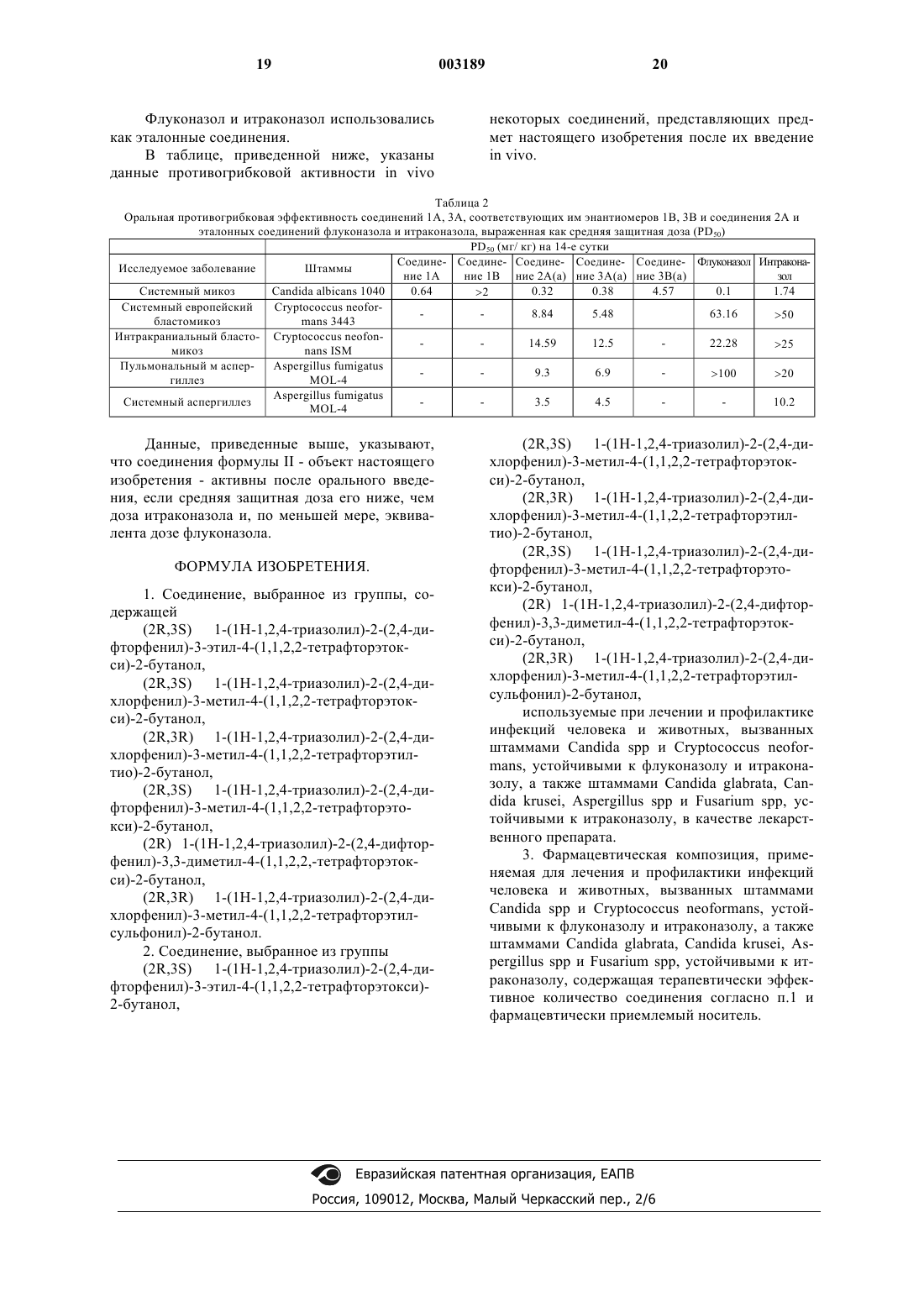
Формула / Реферат
1. Соединение, выбранное из группы, содержащей
(2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дифторфенил)-3-этил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол,
(2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол,
(2R,3R) 1-(1H-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилтио)-2-бутанол,
(2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дифторфенил)-3-метил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол,
(2R) 1-(1H-1,2,4-триазолил)-2-(2,4-дифторфенил)-3,3-диметил-4-(1,1,2,2,-тетрафторэтокси)-2-бутанол,
(2R,3R) 1-(1H-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилсульфонил)-2-бутанол.
2. Соединение, выбранное из группы
(2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дифторфенил)-3-этил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол,
(2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол,
(2R,3R) 1-(1H-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилтио)-2-бутанол,
(2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дифторфенил)-3-метил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол,
(2R) 1-(1H-1,2,4-триазолил)-2-(2,4-дифторфенил)-3,3-диметил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол,
(2R,3R) 1-(1H-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилсульфонил)-2-бутанол,
используемые при лечении и профилактике инфекций человека и животных, вызванных штаммами Candida spp и Cryptococcus neoformans, устойчивыми к флуконазолу и итраконазолу, а также штаммами Candida glabrata, Candida krusei, Aspergillus spp и Fusarium spp, устойчивыми к итраконазолу, в качестве лекарственного препарата.
3. Фармацевтическая композиция, применяемая для лечения и профилактики инфекций человека и животных, вызванных штаммами Candida spp и Cryptococcus neoformans, устойчивыми к флуконазолу и итраконазолу, а также штаммами Candida glabrata, Candida krusei, Aspergillus spp и Fusarium spp, устойчивыми к итраконазолу, содержащая терапевтически эффективное количество соединения согласно п.1 и фармацевтически приемлемый носитель.
Текст
1 Настоящее изобретение касается противогрибковых соединений, предназначенных для человека и животных, в частности, азольных соединений, имеющих противогрибковую активность при лечении и профилактике инфекций человека и животных, вызванных грибками и дрожжами. Среди известных в литературе противогрибковых соединений важный класс составляют так называемые азольные производные, которые в класс включают некоторые используемые в терапии соединения, такие как флуконазол (The Merck Index, XI ed No. 4054, page 645),итраконазол (The Merck Index, XI ed No. 5131,page. 825), и кетоконазол (The Merck Index, XIed No. 5181, page 835). Однако, насколько известно, ни одно из этих соединений не отличается противогрибковой активностью в отношении некоторых условно-патогенных штаммов грибков, вызывающих даже смертельно опасные инфекции у больных с иммунно-депрессивным состоянием. Среди азольных производных, известных как противогрибковые для человека и животных, используется ряд соединений, характеризующихся наличием фрагмента четвертичного спирта в их формуле в которой Az представляет собой триазольную или имидазольную группу, X' предпочтительно представляет собой хлор, фтор, или трифторметил, R является предпочтительно водородом,фтором или бромом; R' и R", которые могут быть разными или одинаковыми, представлены водородом или группой алкилов; Y является S,SO, SO2 или О; А - алкил; в дальнейшем описываются указанные азолы четвертичного спирта. Среди этих азолов четвертичного спирта отметим, например, соединения, описанные в заявках на Европейский патент 54974 (Sumitomo Chemical Company Limited),61835 (Imperial Chemical Industries PLC),107392 (PfiserLimited),140154 (Sumitomo Chemical Company Limited),178533 (Sumitomo Pharmaceutical Company Limited),435081 (SS Pharmaceutical Co. Ltd.),473387 (Sankyo Company Limited). Было обнаружено, что некоторые из этих соединений отличаются общей противогрибковой активностью, а также местной и системной активностью. Однако, насколько нам известно,единственным разрабатываемым соединением является одно, известное как генаконазол,(2R,3R)(2,4-дихлорфенил)[1-(метилсульфонил)этил]-1 Н-1,2,4-триазол-1-этанол и раскрытое в заявке на Европейский патент 178533. Недавно в заявке на Европейский патент 679647 (Nihon Nohyaku Co. Ltd.) были рас 003189 2 крыты высшие гомологи азола четвертичного спирта формулы I, обладающие высокой противогрибковой активностью в отношении Candidaalbicans и характеризующиеся тем, что А представляет собой замещенный по выбору фенил или гетероцикл. Было обнаружено, что отдельные энантиомеры азольных производных формулыZ представляет собой СН или N;R3, R4, R5, которые могут быть одинаковыми или разными, представляют собой водород или C1-C4 алкил при условии, что R4 отличен отR6 представляет собой C1-C5 полифторалкильную группу, содержащую, по меньшей мере, два атома фтора, и по выбору другие атомы галогенов, выбранные из группы, состоящей из хлора и брома; отличаются широким диапазоном противогрибковой активности в отношении патогенных грибков, поражающих человека и животных, в частности, также штаммов грибков, устойчивых к противогрибковым препаратам, используемым в терапии, и против условно-патогенных штаммов грибков, вызывающих инфекции у субъектов с иммунно-депрессивным состоянием, и активных как местно, так и системно. Некоторые соединения формулы II, в рацемической смеси, особенно те, в которых Х представляет собой О или S, описаны в заявке на Европейский патент 315946 (Presidenza delCoordinamento delle Iniziative per la Ricerca Scientifica e Technologica) и, как отмечено, они полезны в сельском хозяйстве как иммунные агенты против патогенных грибков и как регуляторы роста полезной растительности. Таким образом, объектом настоящего изобретения являются соединения формулыR3, R4, R5, которые могут быть одинаковыми или разными, представляют собой водород или C1-C2 алкил при условии, что R4 отличен отR6 представляет собой C1-C5 полифторалкильную группу, содержащую, по меньшей мере, два атома фтора и их фармацевтически приемлемые кислые соли. Указанное условие к формуле II-A сделано для исключения соединений, запатентованных в заявке на Европейский патент 272679. Другой объект настоящего изобретения составляют соединения формулы II-A и их фармацевтически приемлемые кислые соли, используемые в качестве лекарственного средства. Атом углерода, отмеченный звездочкой,имеет абсолютную конфигурацию, определенную в этой формуле. Поэтому, согласно Cahn,Ingold и Prelog, этот атом имеет R- или Sконфигурацию, в зависимости от приоритетного порядка его заместителей. Соединения формулы II-A могут также содержать второй хиральный центр (если R3 и R4 отличны друг от друга) и, таким образом, они являются диастереоизомерами. Диастереоизомеры формулы II-A, представляющие собой объект настоящего изобретения, являются треодиастереоизомерами. Соединения формулы II-A имеют мощный и широкий диапазон противогрибковой активности, в частности, в отношении штаммов Candida spp и Cryptococcus neoformans, устойчивых к флуконазолу и итраконазолу, и в отношенииFusarium spp, устойчивых к итраконазолу, и, как предыдущие соединения, в отношении патогенных штаммов, чувствительных к грибковым инфекциям пациентов с иммуно-депрессивным состоянием, они полезны для лечения и профилактики грибковых и дрожжевых инфекций человека и животных. Обозначением C1-C2 алкил для R3, R4 и R5 указывают метил, этил, причем предпочтительны метил и этил. Обозначением C1-C5 полифторалкильная группа, которая содержит, по меньшей мере, два атома фтора, предпочтительно указывают 1,1,2,2-тетрафторэтил. Соли соединений формулы II-A представляют собой соли фармацевтически приемлемых органических и неорганических кислот, таких как соляная, бромисто-водородная, иодистоводородная, азотная, фосфорная, уксусная, щавелевая, яблочная, бензойная, бензолсульфоновая, метансульфоновая, 4-метилбензолсульфоновая, фумаровая, молочная, винная, лимонная и глюконовая. Предпочтительными соединениями формулы II-A являются такие, в которых R1 представляет собой хлор или фтор, R2 является во 003189 4 дородом, хлором или фтором, R3 - этил или метил, R4 и R5, которые могут быть одинаковыми или разными, представляют собой водород, метил или этил, Z представляет собой N, а R6 представляет собой 1,1,2,2-тетрафторэтил группу. Более предпочтительными соединениями формулы II-A являются такие, в которых R1 представляет собой хлор или фтор, R2 является водородом, хлором или фтором, R3 -этил или метил, R4 и R5, которые могут быть одинаковыми или разными, являются водородом, метилом или этилом, Z представляет собой N, R6 представляет собой 1,1,2,2-тетрафторэтилгруппу, а Х представляет собой О или SO2. Ниже приведены характерные примеры предпочтительных соединений формулы II-А:(2R,3R) 1-(1 Н-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилсульфонил)-2-бутанол Получение соединений формулы II-А, в которой Х представляет собой О или S, может быть проведено по схеме синтеза, уже описанной в указанной заявке на Европейский патент 315946, используя подходящие промежуточные соединения в оптически активной форме. Соединения формулы II-A, в которой Х представляет собой SO2, могут быть получены из соответствующих соединений формулы II-A,в которой Х представляет собой S при использовании подходящей технологии окисления. В качестве окислительных агентов предпочтительно использование перекиси водорода, гипогалогенидов или надкислот, возможно в присутствии катализаторов. Предпочтительно получение соединений формулы II-A, в которой Х представляет собой О или S, проводить добавлением полифторолефина формулы в которой X1 и Х 2, которые могут быть разными или одинаковыми, представляют собой F, Сl или СF3; или взаимодействием полифторированного спирта формулы 5 в которой R7 является C1-C4 полифторалкильной группой, содержащей, по меньшей мере, два атома фтора; с оптически активным промежуточным соединением формулы где R1, R2, R3, R4, R5 и Z были определены выше,а Х представляет собой О или S; или с их реакционноспособным производным, таким как сложный эфир, например, мезилат, тозилат или трифторметансульфонат. Оптически активные промежуточные соединения формулы V могут быть получены известными методами. Например, соединения формулы V можно получить разделением соответствующих рацемических смесей, таких, как описаны выше в заявке на Европейский патент 315946 или в патенте США 5134152. С другой стороны, соединения формулы V,в которой Х представляет собой S, могут быть получены известными методами из соответствующих соединений формулы V, где Х представляет собой О. Более того, было обнаружено, что ферментное расщепление при селективном ацилировании с последующим гидролизом имеет ряд преимуществ для получения соединений формулы V, в которой Х представляет собой О, а R3 и R4 - метил. Хотя известно ферментное расщепление при селективном ацилировании для получения низших гомологов третичного спирта соединений формулы V (см., например, заявку на Международный патент WO 94/24305 - Zeneca Limited), удивителен тот факт, что оно может применяться также для получения соединений формулы V, в которой Х представляет собой О, a R3 и R4 - метил. Действительно, в этих соединениях два атома углерода не только находятся между тем фрагментом, который будет ацилирован, и асимметричным атомом углерода, но, кроме того, один из этих атомов углерода не замещен,что вызывает заметное пространственное затруднение. Поэтому следующий объект настоящего изобретения представляет собой процесс ферментного расщепления соединений формулы V,в которой Х представляет собой О, а R3 и R4 представляют собой метил; этот процесс включает их стереоселективное ацилирование с последующим гидролизом. С другой стороны, соединения формулы V,в которой Х представляет собой О, a R5 - атом водорода, можно получить восстановлением соответствующих кислот формулы в которой R1, R2, R3, R4, и Z были определены выше. Соединение формулы VI известны, или их можно легко получить известными методами(Bartroli J. et al., J. Org. Chem., 1995, 60, 30003012). Получение солей соединений формулы IIA можно осуществить по общепринятой технологии, например, смешиванием в растворе эквимолярных количеств соединений формулы IIA и выбранной кислоты, и отделением полученных солей осаждением и фильтрацией или выпариванием растворителя. Соединения формулы II-A и их соли являются противогрибковыми соединениями, полезными для лечения и профилактики грибковых и дрожжевых инфекций человека и животных. Действительно, соединения II-А, являющиеся объектом настоящего изобретения, характеризуются противогрибковой активностью в отношении дрожжей, нитеобразных грибков,дерматофитов и диморфных грибков. Противогрибковую активность оценивалиin vitro как IC50 и как MIC (минимальная ингибиторная концентрация) на ряде штаммов, таких, например, как Candida albicans, Cryptococcus neoformans, Trichophyton mentagrophytes,Aspergillus fumigatus, Candida parapsilosis, Candida lusitaniae, Candida kefyr, Candida tropicalis,Candida krusei, Candida glabrata, Aspergillus niger и Fusarium spp. Важно подчеркнуть, что соединения формулы II-A - объекта настоящего изобретения эффективны в отношении всех рассматриваемых штаммов Candida spp. и Cryptococcus neoformans, содержащих штаммы, устойчивые к флуконазолу, итраконазолу и генакозолу. Особенно отмеченная противогрибковая активность была обнаружена в отношении штаммов Candida glabrata и Fusarium spp., устойчивых к флуконазолу и итраконазолу, и штаммов Candida krusei и Aspergillus fumigatus,устойчивых к флуконазолу и генакозолу, причем все они являются патогенным агентами,вызывающими инфекции субъектов с иммуннодепрессивным состоянием. Противогрибковую активность соединений формулы II-A - объекта настоящего изобретения- также оценивали в сравнении с активностью соответствующих энантиомеров. В некоторых случаях активность этих энантиомеров была сравнима с активностью указанных соединений, тем не менее в целом она значительно ниже, чем энантиомер соединений формулы II-A - объекта настоящего изобретения. 7 Противогрибковую активность in vivo оценивали при интраперитонеальном и пероральном введении образца Candida штамма Candida albicans, чувствительного к флуконазолу и итраконазолу, мышам. 50%-я защитная доза (PD50) соединений формулы II-A была установлена в тестах in vivo,и они показали, по меньшей мере, сравнимость с одним из указанных соединений. Поэтому объект настоящего изобретения соединения формулы II-A - активны в широком диапазоне глубоких микозов, но особенно против условно-патогенных агентов, вызывающих инфекции у субъектов с иммунно-депрессивным состоянием; они могут быть вводимы местно, перорально и парентерально и имеют надежный терапевтический показатель. Соединения формулы II-A полезны при лечении и профилактике человека и животных от системных инфекций и инфекций слизистых оболочек, вызванных грибками и дрожжами. Как было подчеркнуто, исследование фармакологической активности соединений формулы II-A показало также активность в отношении штаммов, устойчивых к противогрибковым препаратам, используемым в терапии и против недавно выделенных штаммов, вызывающих инфекции субъектов с иммунно-депрессивным состоянием. Это особенно удивительно, принимая во внимание, что соединения, раскрытые в заявке на Европейский патент 315946, были названы иммуннизирующими агентами при грибковых патологиях полезной растительности при грибковых патологиях и регуляторами роста растительного происхождения, причем их активность в этих качествах ограничивалась использованием в сельском хозяйстве. Фармакологическая активность соединений формулы II-A еще более удивительна, если принять во внимание то, что эти соединения имеют некоторые структурные фрагменты, общие для азольных четвертичных спиртов, описанных в литературе, в то же время характеризуются наличием двух атомов углерода в цепи между атомом углерода, связанным с гидроксигруппой, и кислородом или атомом серы, и простым полифторированным алкилом в эфире или фрагментом, содержащим серу. Насколько известно, комбинация этих структурных характеристик никогда не была описана в литературе для представителей класса соединений азольных четвертичных спиртов с противогрибковой активностью, предназначенных для человека и животных. Соединения формулы II-A, предназначенные для человека и животных, можно вводить в смеси с подходящим носителем, выбранным с учетом способа введения. Таким образом, еще один объект настоящего изобретения включает фармацевтическую композицию, содержащую терапевтически эффективное количество одного из соединений 8 формулы II-A в смеси с фармацевтически приемлемым носителем. Например, соединения формулы II-A или их соли можно вводить орально в виде таблеток,капсул, растворов или суспензий. Для парентерального введения (например,внутривенного, внутримышечного или подкожного введения) соединения формулы II-А или их соли используют в форме стерильного водного раствора. Или, соединения формулы II-A или их соли можно вводить как суппозитории и пессарии. При местном введении соединения формулы II-A или их соли используются предпочтительно в виде кремов и порошков. При оральном и парентеральном введении ежедневная доза соединений формулы II-A обычно колеблется от 0,1 до 50 мг/кг (предпочтительно от 1 до 20 мг/кг), разделенная на один или более приемов. Для лучшей иллюстрации настоящего изобретения предлагаются следующие примеры. Пример 1. Ферментативное кинетическое расщепление 3-(2,4-дифторфенил-2,2-диметил)-4-(1 Н-1,2,4-триазол-1-ил)-1,3-бутандиола. а) Изомер(+) Фермент (3 г; липаза из Candida cilindracea тип VII - Sigma) добавили при энергичном перемешивании при 30 С к раствору 3-(2,4 дифторфенил)-2,2-диметил-4-(1 Н-1,2,4-триазол 1-ил)-1,3-бутандиола (2,97 г; 10 ммоль) в хлороформе (60 мл) и винилацетате (1,9 мл; 20 ммоль). Спустя 72 ч полученную суспензию отфильтровали, а потом добавили 3 г фермента,причем перемешивание продолжали еще 24 ч при 30 С. Суспензию отфильтровали, твердое вещество промыли хлороформом (20 мл), а органическую фазу выпаривали при пониженном давлении. Полученный остаток хроматографировали на силикагеле (элюент-этилацетат : гексан = 60 : 40) для получения (-) 3-(2,4-дифторфенил)-3 гидрокси-2,2-диметил-4-(1 Н-1,2,4-триазол-1 ил)бутилацетата (1,63 г; е.е. 88% хиральная ВЭЖХ - колонка CHIRACEL-OD, элюент - гексан:изопропанол = 70:30) и (+) 3-(2,4 дифторфенил)-2,2-диметил]-4-(1 Н-1,2,4-триазол-1-ил)-1,3-бутандиола (1,3 г; е.е. 79% хиральная ВЭЖХ). Полученный изомер(+) (1,3 г) снова обрабатывали липазой при тех же условиях в течение 17 ч. После этой обработки и выделения с использованием тонкослойной хроматографии получили (+) 3-(2,4-дифторфенил)-2,2-диметил 4-(1 Н-1,2,4-триазол-1-ил)-1,3-бутандиол (0,92 г; выход 62%) в виде белого твердого вещества.H-ЯМР идентичен спектру рацемической смеси. б) изомер (-)(-) 3-(2,4-дифторфенил)-3-гидрокси-2,2 диметил-4-(1 Н-1,2,4-триазол-1-ил) бутилацетат,полученный на предыдущей стадии (а) (1,63 г) растворили в 0,05 М рН 7 фосфатного буфера(400 мл) и ацетоне (50 мл). Полученный раствор при перемешивании при 30 С добавили к ферменту (1,6 г; липаза изCandida cilindracea типа VII), а значение рН поддерживали последующим добавлением 0,1 М гидроксида натрия. Спустя 5 ч значение рН довели до 4 с помощью концентрированной соляной кислоты, а экстракцию осуществляли этилацетатом (3 х 100 мл). Органическую фазу обезвоживали над сульфатом натрия и выпаривали при пониженном давлении до получения маслянистого остатка. После тонкослойной хроматографии на силикагеле (элюент - этилацетат : гексан = 60:40) был выделен (-) 3-(2,4-дифторфенил)-2,2 диметил-4-(1 Н-1,2,4-триазол-1-ил)-1,3-бутандиол (1.20 г; выход 80%) в виде белого твердого вещества.[]D20 = -46,2 (с = 1%, метанол); хиральная ВЭЖХ = е.е. 98,4%; т.пл.117-118 С; 1 Н-ЯМР идентичен спектру рацемической смеси. Пример 2. Получение (-) (2S,3R)-3-(2,4 дифторфенил)-2-метил-4-(1 Н-1,2,4-триазол-1 ил)-1,3-бутандиола. Раствор (2S,3R)-3-(2,4-дифторфенил)-3-гидрокси-2-метил-4-(1 Н-1,2,4-триазол-1-ил)бутановой кислоты (0,88 г; 2,97 ммоль) в безводном тетрагидрофуране (6,5 мл) по каплям добавляли к трифторэфирату бора (0,365 мл; 2,97 ммоль) и смесь перегоняли 30 мин. Полученный раствор добавляли по каплям при 55 С к 10 М раствору диметилсульфата бора в тетрагидрофуране (0,356 мл; 3,56 ммоль), перегонку продолжали в течение 6 ч. После охлаждения до 4 С добавили раствор вода : тетрагидрофуран = 1:1 (3 мл), а затем 5 М соды (8 мл) и смесь перегоняли 12 ч. Тетрагидрофуран выпарили при пониженном давлении, полученную смесь экстрагировали хлороформом (510 мл), потом органическую фазу обезвожили и выпарили. Кристаллизацией твердого остатка из бензол : гексан =1:1 (20 мл) был получен (-) (2S,3R)-3-(2,4-дифторфенил)-2 метил-4-(1 Н-1,2,4-триазол-ил)-1,3-бутандиол(dd, 1H); 3,80 (dd, 1H); 3,10 (s-уширенный, 1H); 2,45-2,25 (m, 1H); 0,85 (d, 3 Н). Аналогично были получены следующие соединения:(m, 2H, CH-CH2-СН 3); 0,81 (t, 3H, СН-СН 2 СН 3). Пример 3. Получение (-)2-(2,4-дифторфенил)-4-(1,1,2,2-тетрафторэтокси)-3,3-диметил-1-(1 Н-1,2,4-триазол-1-ил)-2-бутанол (Соединение 1 А). Раствор (-)3-(2,4-дифторфенил)-2,2-диметил-4-(1 Н-1,2,4-триазол-1-ил)-1,3-бутандиола (5 г; 16,8 ммоль), полученный, как это описано в примере 1, и диметилсульфоксид (8 мл) в толуоле (60 мл) при перемешивании при -5 С добавили к порошку гидроксида калия (533 г; 9,5 ммоль). Реакционную среду заместили тетрафторэтиленом и полученную смесь перемешивали при -5 С в течение 90 мин. После добавления воды (120 мл) органическую фазу промыли 5%-й соляной кислотой (80 мл) и обработали безводным гидрокарбонатом натрия (6,5 г) при перемешивании в течение 30 мин. Жидкую фазу отфильтровали, а растворитель выпарили при пониженном давлении. Полученный маслянистый остаток очистили тонкослойной хроматографией (элюент-гексан : этилацетат = 6 : 4), в результате получили указанное выше соединение 1 А (5,2 г; выход 79%) в виде белого твердого вещества. []D20 = - 48,5Triaz.); система АВ: VA = 4,19, VB = 3,75, JAB = 9,8 Hz, CH2-0; 1,06 (d, 3H, JHF = 2,4 Hz, СН 3); 0,98 (s, 3H, СН 3). Аналогично были получены следующие соединения:H-ЯМР спектр идентичен спектру соединения 1 А.[]D20 = - 54,7 (с = 1%, метанол); Хиральная ВЭЖХ (гексан: изопропанол = 80 : 20) = е.е. 99% нитрат (кристаллизуется из изопропилового эфира); т. пл. 147.5-148.5 С; 1 Н-ЯМР (CDCl3): 9,77 и 8,09 (2s, 2H,Triaz.); 7,39-6,69 (m, 5H, ОН е HNO3); 5,76 (tt,1H, JHF = 53,1 Hz, CHF2); система АВ: Va = 5,12, Vb = 4,95, Jab = 14,4 Hz, CH2-Triaz.; часть АВ системы АВХ: Va = 4,37, Vb = 3,96, Jab = 10,5 Hz, Jax = 7,4 Hz, Jbx = 4,8 Hz; CH2-O; 2,722,55 (m, 1H, СН-СН 3); 0,86-0,81 (s, 3 Н, СН 3 СН).[]D20= -77.7 (с = 1%, метанол); хиральная ВЭЖХ (гексан : изопропанол = 90 : 10) = е. е.99,8% нитрат (кристаллизуется из изопропилового эфира); т.пл 120,3-121,3 С; 1 Н-ЯМР (CDCl3): 9,74 и 8,07 (2s, 2H,Triaz.); 7,44 (d, 1H, JHH = 8,6 Hz, C-CH-CH-CCl); 7,35 (d, 1H, JHH = 2,2 Hz, C-Cl-CH-C-Cl); 7,08 (dd, 1H, C-CH-CH-C-Cl); 5,82 (tt, 1H, JHF = 53,2 Hz, CHF2); система АВ: Va = 5,67, Vb = 4,89, Jab = 14,3 Hz, CH2-Triaz.; часть АВ системы АВХ: Va = 4,44, Vb = 4,09, Jab = 10,8 Hz, Jax[]D20 = 56,1 (с = 1%, метанол); хиральная ВЭЖХ (гексан: изопропанол = 90:10) = е.е. 99%; нитрат (кристаллизованный из изопропилового эфира или ацетонитрила); т. пл. 89-91 С; 1Va = 4,42, Vb=4,21, Jab = 11,1 Hz, Jax = 7,7 Hz,Jbx = 2,6 Hz: CH2-O; 2,41-2,30 (m. 1H, СН-СH2); 1,32-1,18 (m, 2H, CH-CH2); 0,83 (d, 3H, JHH = 7,1 Hz, СН 3). Пример 4. Получение (-) (2R,3S)-2-(2,4 дихлорфенил)-4-(1,1,2,2-тетрафторэтокси)-3-метил-1-(1 Н-1,2,4-триазол-1-ил)-2-бутанола (Соединение 3 А). Иным путем, чем в примере 3, технологический процесс очистки указанного в заглавии соединения проводят так: неочищенное соединение (86,2 г) растворяют в 173 мл этанола при 30-35 С, потом фильтруют до получения совершенно чистого раствора, который по каплям добавляют в смесь воды (260 мл) и этанола (87 мл) при 20 С, в течение 90 мин. Смесь нагревают до 35 С при перемешивании в течение 90 мин, охлаждают до 2 С и снова перемешивают в течение 60 мин. Нерастворимое твердое вещество отфильтровывают, промывают смесью этанола и воды(1:1) и просушивают в печи при 50 С и в вакууме до получения постоянного веса. В результате выход составил 74 г (-) (2R,3S)-2-(2,4 дихлорфенил)-4-(1,1,2,2-тетрафторэтокси)-3-метил-1-(1 Н-1,2,4-триазол-1-ил)-2-бутанола (Соединение 3 А), чистота 99%; 13 53,4 Hz, CHF2); система AB = VA = 5,48, VB = 4,54, JAB = 14,4 Hz, CH2-Triaz.); 5,09 (уширенный сигнал, 1H, ОН); 4,54-3,98 (m, 2H, CH2-O); 3,14-2,97 (m, 1H, СН-СН 3); 0,67 (d, 3H, JHH = 7,1 Hz, СН 3-СН). Пример 5. Получение бутилового эфира 3(2,4-дихлорфенил)-3-гидрокси-2-метил-4-(1 Н 1,2,4-триазол-1-ил) метансульфоновой кислоты. Раствор (+) (2R,3S)-3-(2,4-дихлорфенил)-2 метил-4-(1 Н-1,2,4-триазол-1-ил)-1,3-бутандиола(0,735 мл; 9,46 ммоль) при 0 С. Реакцию проводят в течение 20 мин, сохраняя температуру 0 С, потом реакционную смесь выливают в воду(60 мл). Фазы разделяют и водную фазу потом дважды экстрагируют метилхлоридом. Собранные органические фазы промывают 5%-м водным раствором гидрокарбоната натрия, потом - предельно насыщенным водным раствором хлорида натрия и обезвоживают над сульфатом натрия. После выпаривания растворителя было получено 3,64 г (100%) бутилового эфира 3-(2,4 дихлорфенил)-3-гидрокси-2-метил-4-(1 Н-1,2,4 триазол-1-ил) метансульфоновой кислоты в виде белого твердого вещества, которое используется на следующей стадии. 1 Н-ЯМР (CDCl3): 7,84 и 7,79 (2s, 2H, HTriaz.); 7,50-7,00 (m, 3H, H-phenyl), 5,56 и 4,56S-[3-(2,4-дихлорфенил)-3-гидрокси-2-метил-4(1 Н-1,2,4-триазол-1-ил)бутила]. Бутиловый эфир 3-(2,4-дихлорфенил)-3 гидрокси-2-метил-4-(1 Н-1,2,4-триазол-1-ил) метансульфоновой кислоты (9,3 ммоль), полученный, как описано в примере 5, растворяют в этаноле (95 мл) и добавляют к тиоацетату калия (2,12 г). Смесь перегоняют в течение 2 ч, потом охлаждают на льду и полученный осадок отфильтровывают и промывают ледяным метиленхлоридом. Фильтрат концентрируют при пониженном давлении, добавляют воду (60 мл) и экстрагируют несколько раз метиленхлоридом. Органические фазы собирают и обезвоживают над сульфатом натрия. Таким образом, выход красноватого сырого продукта составляет 3,5 г, его очищают с помощью тонкослойной хроматографии на силикагеле 14 2H, СН 2-SСОСН 3); 2,75 (m, 1H, СН); 2,39 (s, 3H,СН 3 thioacetate); 0,70 (d, 3H, СН 3). Пример 7. Получение (2R,3R)-3-(2,4-дихлорфенил)-3-гидрокси-2-метил-4-(1 Н-1,2,4 триазол-1-ил)-1-бутандиола. В атмосфере азота при комнатной температуре раствор гидроксида кальция (0,266 г; 4,74 ммоль) в метаноле (40 мл) добавляют по каплям в раствор тиоацетатного эфира S-[3-(2,4 дихлорфенил)-3-гидрокси-2-метил-4-(1 Н-1,2,4 триазол-1-ил)бутила], полученного, как описано в примере 6 (1,72 г; 4,6 ммоль) в метаноле (40 мл). После взаимодействия в течение 10 мин реакцию гасят 5%-й НСl (4 мл) разбавленной водой (20 мл). Реакционную смесь концентрируют при пониженном давлении, потом разбавляют водой и несколько раз экстрагируют метиленхлоридом. Обезвоживание и выпаривание собранных органических фаз дает 1,6 г сырого вещества, которое очищают тонкой хроматографией на силикагеле (элюент - этилацетат: гексан = 1:1). Таким образом получили 1,2 г(Соединение 5). Исходя из (2R,3R)-3-(2,4-дихлорфенил)-3 гидрокси-2-метил-4-(1 Н-1,2,4-триазол-1-ил)-1 бутандиола (1,85 г; 5,57 ммоль), полученного,как описано в примере 7, и следуя методике примера 3, было получено сырое вещество, которое очищают тонкослойной хроматографией(элюент - петролейный эфир : этилацетат 6 : 4) и получают 2,15 г (-) (2R,3R)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилтио)-1(1 Н-1,2,4-триазол-1-ил)-2-бутанола (Соединение 5) в виде твердого белого вещества.Na2WO4 Н 2O и оставляют при перемешивании при комнатной температуре в течение 3 дней,потом реакционную смесь обрабатывают пиросульфатом натрия (Na2S2O5) в воде и подщелачивают 10%-м гидроксидом натрия. Смесь экстрагируют эфиром, высушивают и концентрируют в вакууме до выхода 0,41 г масла, которое очищают с помощью тонкослойной хроматографии (элюент -этилацетат : петролейный эфир 4:6). Полученное масло (0,35 г) растворяют в этаноле и обрабатывают 70%-й азотной кислотой. Смесь концентрируют в вакууме и получают твердое вещество, которое обрабатывают теплым изопропиловым эфиром, получая 0,32 г(-)(2R,3R)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилсульфинил)-1-(1 Н-1,2,4-триазол-1-ил)-2-бутанола (Соединение 6) в виде белого твердого вещества. Т. пл. 153-155 С; 1(m, 1H, СHF2); 5,66-4,87 (m, 2H, CH2-Triaz; 3,572,79 (m, 3H, СН-СН 3); 3,07-2,90 (m, 2H, CH2-S); 0,77 (d, 3H, JHH = 6,3 Нz, СН 3-СН). Пример 10. Получение (-) (2R,3R)-2-(2,4 дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилсульфинил-1-(1 Н-1,2,4-триазол-1-ил]-2 бутанола (Соединение 7). Раствор (-) (2R,3R)-2-(2,4-дихлорфенил)-3 метил-4-(1,1,2,2-тетрафторэтилтио)-1-(1 Н-1,2,4 триазол-1-ил)-2-бутанола (0,5 г; 1,15 ммоль),полученного, как описано в примере 8, в метаноле (2,5 мл) в присутствии каталитического количества Na2WO42 Н 2O и концентрированной НСl (0,19 мл; 2,3 ммоль) добавляют к 40%-й перекиси водорода (0,9 мл; 10,35 ммоль) и нагревают до 60 С в течение 14 ч, потом оставляют при комнатной температуре на ночь. Реакционную смесь обрабатывают раствором пиросульфата натрия (Na2S2O5), подщелачивают 10%-м гидроксидом натрия. Потом экстрагируют эфиром, высушивают и концентрируют в вакууме до выхода 0,49 г масла, которое очищают тонкослойной хроматографией (элюент - этилацетат: петролейный эфир 1:1). Полученное масло (0,45 г) растворяют в этаноле и обрабатывают 70%-й азотной кислотой. Смесь концентрируют в вакууме и получают 0,42 г (-) (2R,3R)-2-(2,4-дихлорфенил)-3 метил-4-(1,1,2,2-тетрафторэтилсульфонил)-1(1 Н-1,2,4-триазол-1-ил)-2-бутанола (Соединение 7) в виде белого твердого вещества.in vitro. А) Ингибирование активности роста мицетов оценивали макрометодами при ступенчатом разведении бульона в геометрической прогрессии (M. R. McGinnis and M.G. Rinaldi, "Antimycotic drugs: mechanisms of action, drug resistance,susceptibility testing and assay of activity in biological fluids", in Antibiotic in Laboratory Medicine, Ed. V. Lorian, Baltimore 1991). В качестве питательной среды для дрожжей и плесени соответственно использовали дрожжевой азотный бульон (YNB) и бульон декстрозы (SDB). Результаты, полученные для SDB (после инкубирования в течение 7 дней при 28 С) выражены минимальной концентрацией (MIC),ингибирующей рост мицетов, а результаты, полученные для YNB (после инкубирования в течение 48 ч при 35 С), выражены как концентрация, ингибирующая на 50% (IС 50) рост дрожжей. В качестве эталона использовали флуконазол. В приведенной ниже таблице указаны данные противогрибковой активности in vitro в отношении Candida albicans, Asppergillus fumigatus, Cryptococcus neoformans и Trichophytonmentagrophytes для некоторых соединений формулы II. Таблица 1a Противогрибковая активность соединений 1 А, 3 А, их энантиомеров 1 В, 3 В, соединения 2 А и флуконазола как эталонного соединения в отношении С. Albicans, С. Neoformans, Т. Данные, приведенные в таблице, указывают, что соединения - объект настоящего изобретения - имеют противогрибковую активность значительно выше аналогичной активности эталонного соединения. Б) Активность, ингибирующая рост дрожжей и нитевидных грибков оценивали методом разведения на агаре. Минимальная концентра 17 ция, обеспечивающая 50%-е ингибирование(МIС 50), была измерена при включении в питательную среду уменьшающихся концентраций испытываемых соединений, так, чтобы концентрация менялась ступенчато с шагом, равным 2,от 64 до 0,00085 мкг/мл. В качестве питательной среды применяли казитоновый агар, буферированный при рН = 7(казитон 9 г; глюкоза 20 г; цитрат натрия 10 г; дрожжевой экстракт 5 г; агар 18 г; бифосфат калия 0,54 г; бифосфат натрия 3,34 г; дистиллированная вода 1,000 мл). Для оценки активности в отношении дрожжей питательные среды, содержащие ступенчатые концентрации испытуемых соединений, инокулировали с 1 мкл суспензии 5 х 105 бластоспор/мл, полученные из 24-48-часовых культур на глюкозном агаре. Инокулированные питательные среды инкубировали при 32 С, а измерения для таких медленнорастущих дрожжей как Cryptococcus neoformans были проведены через 24, 48 и 72 ч. В качестве эталонных соединений использовали флуконазол и итраконазол. В таблице,приведенной ниже, указана противогрибковая активность in vitro в отношении Candidaalbicans, Candida tropicalis, Candida krusei, Candida glabrata, Candida parapsilosis, Candida lutitanioe, Candida kefyr, Cryptococcus neoformans,Trichosporon spp, Blastoschizomyces capitatus,Geotrichum spp, Prototecha wicherhamii. Таблица 1 б Противогрибковая активность in vitro соединений 3 А и 2 А,выраженная как MIC50, и активность эталонных соединений флуконазола и итраконазола в отношении перечисленных дрожжей ШтаммыMIC50 (мкг/мл) Соединение Соединение Флуконазол Итраконазол 2 А 3 А С. albicans 0,015 0,015 2 0,015 С. tropicalis 0,06 0,03 4 0,03 С. krusei 0,12 0,007 16 0,03 С. glabrata 0,12 0,12 16 4 С. parapsilosis 0,007 0,007 2 0,03 С. lusitanie 0,015 0,035 1 0,03 С. kefyr 0,017 0,017 1 0,03 С. neoformans 0,06 0,06 16 0,12 Для оценки активности в отношении мицетов питательные среды, содержащие ступенчатые концентрации исследуемых соединений,инокулировали с 1 мкл суспензии 5 х 105 спор/мл(конид или эндоспор), полученной от культурспорообразователей. Инокулированные питательные среды инкубировали при 32 С, а измерения проводили спустя 1, 2, 3, 5, 7 и 10 дней. В качестве эталонных соединений использовали флуконазол, итраконазол и амфотерицин В. 18 В таблице, приведенной ниже, указана противогрибковая активность in vitro в отношении Aspergillus fumigatus, Aspergillus flaws, Aspergillus niger, Aspergillus terreus, Aspergillusalliaceus, Fusarium verticilloides, Fusarium proliferatum Pseudoallescheria, Absidia corymbifera,Mucor, Rhyzomucor pusillus, Acremonium,Trichoderma. Таблица 1 в Противогрибковая активность in vitro соединений 3 А и 2 А,выраженная как MIC50, и активность эталонных соединений флуконазола и итраконазола и амфотерицина В в отношении перечисленных грибков ШтаммыMIC50 (мкг/мл) Соедине- Соеди- Флуко- Итрако- Амфотение 2A нение 3 А назол назол рицин В Данные, приведенные выше в таблицах,указывают, что соединения - объект настоящего изобретения - имеют общую противогрибковую активность, значительно выше аналогичной активности эталонных соединений. Пример 12. Противогрибковая активностьCD 1) весом 23-25 г, получающие нормальное питание и воду по желанию. Каждому животному вводили суспензию,0,2 мл которой содержали 2,5 х 107 клеток/мл определенного микроорганизма; введение проводили внутривенно (при системном микозе,европейском бластомикозе, аспергиллезе), интракраниально (при интракраниальном бластомикозе) или назально (при пульмональном аспергиллезе). Каждому из животных сразу после инъекции и спустя 4, 24 и 48 ч вводили орально(в 2%-м гуммиарабике) исследуемое соединение в геометрически возрастающих дозах. Инфицированную группу использовали как контрольную. Контроль за смертностью мышей осуществлялся в течение 14 суток. Средняя защитная доза PD50 определялась токсикологическим анализом (L. Lison - "Statistica applicata alla biologiae l'analisi dei risultati" - Casa Editrice Ambrosiana,1961) по числу животных, оставшихся в живых при каждом значении концентрации. Флуконазол и итраконазол использовались как эталонные соединения. В таблице, приведенной ниже, указаны данные противогрибковой активности in vivo 20 некоторых соединений, представляющих предмет настоящего изобретения после их введение Таблица 2 Оральная противогрибковая эффективность соединений 1 А, 3 А, соответствующих им энантиомеров 1 В, 3 В и соединения 2 А и эталонных соединений флуконазола и итраконазола, выраженная как средняя защитная доза (PD50)PD50 (мг/ кг) на 14-е сутки Соедине- Соедине- Соедине- Соедине- Соедине- Флуконазол ИнтраконаИсследуемое заболевание Штаммы ние 1A ние 1 В ние 2 А(а) ние 3 А(а) ние 3 В(а) зол Данные, приведенные выше, указывают,что соединения формулы II - объект настоящего изобретения - активны после орального введения, если средняя защитная доза его ниже, чем доза итраконазола и, по меньшей мере, эквивалента дозе флуконазола. ФОРМУЛА ИЗОБРЕТЕНИЯ. 1. Соединение, выбранное из группы, содержащей(2R,3S) 1-(1 Н-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол,(2R,3R) 1-(1 Н-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилтио)-2-бутанол,(2R,3S) 1-(1 Н-1,2,4-триазолил)-2-(2,4-дифторфенил)-3-метил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол,(2R) 1-(1 Н-1,2,4-триазолил)-2-(2,4-дифторфенил)-3,3-диметил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол,(2R,3R) 1-(1 Н-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилсульфонил)-2-бутанол,используемые при лечении и профилактике инфекций человека и животных, вызванных штаммами Candida spp и Cryptococcus neoformans, устойчивыми к флуконазолу и итраконазолу, а также штаммами Candida glabrata, Candida krusei, Aspergillus spp и Fusarium spp, устойчивыми к итраконазолу, в качестве лекарственного препарата. 3. Фармацевтическая композиция, применяемая для лечения и профилактики инфекций человека и животных, вызванных штаммамиCandida spp и Cryptococcus neoformans, устойчивыми к флуконазолу и итраконазолу, а также штаммами Candida glabrata, Candida krusei, Aspergillus spp и Fusarium spp, устойчивыми к итраконазолу, содержащая терапевтически эффективное количество соединения согласно п.1 и фармацевтически приемлемый носитель.
МПК / Метки
МПК: C07D 249/08, A61P 31/10, A61K 31/4196
Метки: производные, триазола, человека, противогрибковой, обладающие, предназначенные, животных, активностью
Код ссылки
<a href="https://eas.patents.su/11-3189-proizvodnye-triazola-obladayushhie-protivogribkovojj-aktivnostyu-prednaznachennye-dlya-cheloveka-i-zhivotnyh.html" rel="bookmark" title="База патентов Евразийского Союза">Производные триазола, обладающие противогрибковой активностью, предназначенные для человека и животных</a>
Триазольные соединения, обладающие противогрибковой активностью, предназначенные для применения в области медицины и ветеринарии

Номер патента: 2142
Опубликовано: 24.12.2001
Авторы: Скьоппакасси Джованна, Альбини Энрико
МПК: C07D 249/08, A61K 31/4196, A61P 31/10...
Метки: предназначенные, соединения, ветеринарии, обладающие, триазольные, противогрибковой, области, активностью, медицины, применения
Формула / Реферат:
1. Соединения формулы где R1 обозначает хлор, фтор или трифторметил; R2 обозначает водород, хлор, фтор или трифторметил; Z обозначает N; R3, R4 и R5 имеют одинаковые или различные значения и обозначают водород или (С1-С4)алкил, при условии, что R4 имеет значение, отличное от R5, когда R3 обозначает водород; Х обозначает О или S; R6 обозначает (С1-С5)полифторалкил, содержащий, по крайней мере, два атома фтора и необязательно атомы другого...
Производные хиноксалин-2,3-диона и их фармацевтически приемлемые соли, фармацевтические композиции, обладающие антагонистической активностью в отношении рецептора глютамата и противосудорожной активностью, способы лечения пациентов, страдающих от удара или заболеваний, при помощи этих соединений.

Номер патента: 762
Опубликовано: 24.04.2000
Авторы: Никам Шам, Рафферти Майкл Фрэнсис, Корнберг Брайэн Эдвард
МПК: C07D 241/44
Метки: пациентов, противосудорожной, фармацевтически, отношении, способы, рецептора, композиции, этих, лечения, соли, заболеваний, обладающие, активностью, приемлемые, хиноксалин-2,3-диона, соединений, помощи, страдающих, производные, глютамата, удара, антагонистической, фармацевтические
Формула / Реферат:
1. Производные хиноксалин-2,3-диона общей формулы I где R - группа - NR4R5, где R4 и R5 независимо друг от друга означают водород, низший алкил, незамещенный или замещенный низшим алкилом, низший циклоалкил, который может содержать 1 или 2 атома кислорода в качестве гетероатома, R1 и R2 независимо друг от друга означают водород или нитрогруппу, R3 - низший алкил, при этом радикалы R3 и R-CH2- могут каждый находиться в положениях 5 или...
Тиольные производные, обладающие ингибирующей активностью в отношении металлопептидаз.

Номер патента: 991
Опубликовано: 28.08.2000
Авторы: Сантанджело Франческо, Романьяно Стефано, Норчини Габриеле, Фантуччи Марио, Пеллачини Франко, Семераро Клаудио
МПК: A61K 31/425, C07D 277/30, A61P 9/00...
Метки: отношении, производные, обладающие, тиольные, ингибирующей, активностью, металлопептидаз
Формула / Реферат:
1. Соединение формулы где R обозначает меркаптогруппу или группу R4COS, которая превращается в организме в меркаптогруппу; R1 обозначает С2-С4алкильную группу с прямой или разветвленной цепью или арильную или арилалкильную группу, имеющую от 1 до 6 атомов углерода в алкильном фрагменте, где арил обозначает фенил или 5- или 6-членный ароматический гетероцикл с одним или двумя гетероатомами, выбранными из группы, включающей азот, кислород и...
Производные фосфоновой кислоты, обладающие ингибирующей активностью в отношении металлопептидаз

Номер патента: 743
Опубликовано: 28.02.2000
Авторы: Ботта Даниела, Норчини Габриеле, Сантанджело Франческо
МПК: C07K 5/062, A61K 38/05
Метки: отношении, производные, кислоты, активностью, ингибирующей, обладающие, металлопептидаз, фосфоновой
Формула / Реферат:
1. Соединение формулы где R обозначает С1-С6алкильную группу с прямой или с разветвленной цепью, необязательно замещенную одним или несколькими атомами фтора, арильную или арилалкильную группу с 1-6 атомами углерода в алкильном фрагменте, где арил обозначает фенильную, 1-нафтильную или 2-нафтильную группу или 5- или 6-членный ароматический гетероцикл с 1 или 2 гетероатомами, выбранными из группы, включающей азот, кислород и серу,...
Соединения из класса ацилмочевин, предназначенные для лечения кокцидиоидомикоза у теплокровных животных

Номер патента: 1623
Опубликовано: 25.06.2001
Авторы: Бартш Роберт Карл, Грин Расселл Томас
МПК: A61P 11/00, A61K 31/17
Метки: класса, теплокровных, соединения, кокцидиоидомикоза, предназначенные, лечения, ацилмочевин, животных
Формула / Реферат:
1. Способ лечения или профилактики вызываемых С. immitis инфекций у теплокровных животных, включающий введение животному, нуждающемуся в таком лечении, терапевтически эффективного количества ацилмочевины, представленной следующей формулой где R1 обозначает незамещенный или замещенный фенил, нафтил, пиридил, пиридазинил, пиримидинил или пиразинил, R2 обозначает водород или С1-С6алкил, R3 обозначает R4-R8, каждый независимо друг от друга,...
Предыдущий патент: Замещенные пиридо- или пиримидосодержащие 6,6- или 6,7-бициклические производные
Следующий патент: Азаполициклические соединения, конденсированные с арилом
Случайный патент: Применение антагонистов cgrp для устранения приливов в период менопаузы