Новый способ синтеза ивабрадина и его аддитивных солей с фармацевтически приемлемой кислотой





Формула / Реферат
1. Способ синтеза ивабрадина формулы (I)
отличающийся тем, что соединение формулы (VI)
взаимодействует с тиолом в органическом растворителе с получением гемитиоацеталя формулы (VII)
где R представляет собой замещенную или незамещенную, линейную или разветвленную алкильную группу, замещенную или незамещенную арильную группу, замещенную или незамещенную бензильную группу или группу CH2CO2Et,
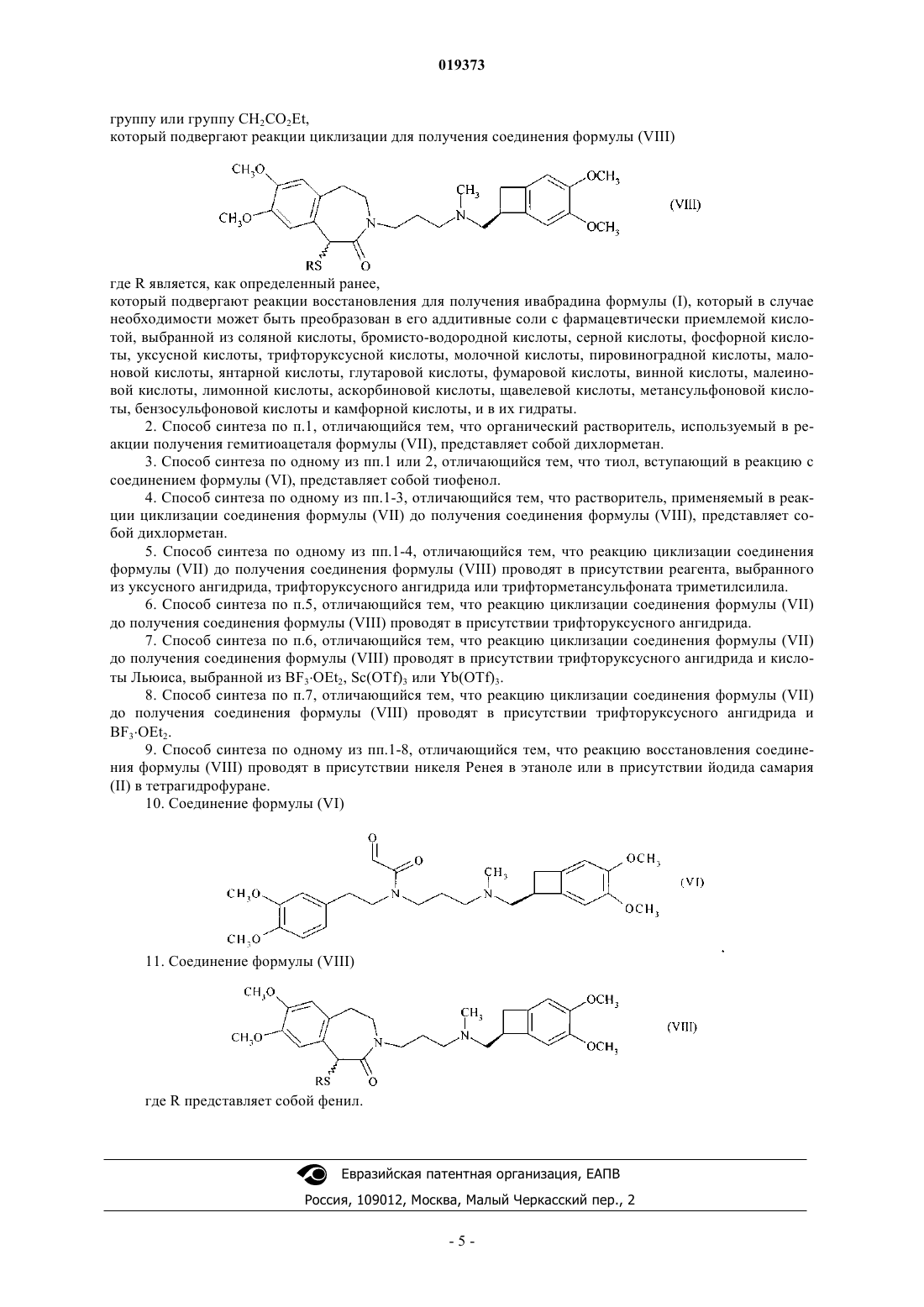
который подвергают реакции циклизации для получения соединения формулы (VIII)
где R является, как определенный ранее,
который подвергают реакции восстановления для получения ивабрадина формулы (I), который в случае необходимости может быть преобразован в его аддитивные соли с фармацевтически приемлемой кислотой, выбранной из соляной кислоты, бромисто-водородной кислоты, серной кислоты, фосфорной кислоты, уксусной кислоты, трифторуксусной кислоты, молочной кислоты, пировиноградной кислоты, малоновой кислоты, янтарной кислоты, глутаровой кислоты, фумаровой кислоты, винной кислоты, малеиновой кислоты, лимонной кислоты, аскорбиновой кислоты, щавелевой кислоты, метансульфоновой кислоты, бензосульфоновой кислоты и камфорной кислоты, и в их гидраты.
2. Способ синтеза по п.1, отличающийся тем, что органический растворитель, используемый в реакции получения гемитиоацеталя формулы (VII), представляет собой дихлорметан.
3. Способ синтеза по одному из пп.1 или 2, отличающийся тем, что тиол, вступающий в реакцию с соединением формулы (VI), представляет собой тиофенол.
4. Способ синтеза по одному из пп.1-3, отличающийся тем, что растворитель, применяемый в реакции циклизации соединения формулы (VII) до получения соединения формулы (VIII), представляет собой дихлорметан.
5. Способ синтеза по одному из пп.1-4, отличающийся тем, что реакцию циклизации соединения формулы (VII) до получения соединения формулы (VIII) проводят в присутствии реагента, выбранного из уксусного ангидрида, трифторуксусного ангидрида или трифторметансульфоната триметилсилила.
6. Способ синтеза по п.5, отличающийся тем, что реакцию циклизации соединения формулы (VII) до получения соединения формулы (VIII) проводят в присутствии трифторуксусного ангидрида.
7. Способ синтеза по п.6, отличающийся тем, что реакцию циклизации соединения формулы (VII) до получения соединения формулы (VIII) проводят в присутствии трифторуксусного ангидрида и кислоты Льюиса, выбранной из BF3×OEt2, Sc(OTf)3 или Yb(OTf)3.
8. Способ синтеза по п.7, отличающийся тем, что реакцию циклизации соединения формулы (VII) до получения соединения формулы (VIII) проводят в присутствии трифторуксусного ангидрида и BF3×OEt2.
9. Способ синтеза по одному из пп.1-8, отличающийся тем, что реакцию восстановления соединения формулы (VIII) проводят в присутствии никеля Ренея в этаноле или в присутствии йодида самария (II) в тетрагидрофуране.
10. Соединение формулы (VI)
11. Соединение формулы (VIII)
где R представляет собой фенил.
Текст
НОВЫЙ СПОСОБ СИНТЕЗА ИВАБРАДИНА И ЕГО АДДИТИВНЫХ СОЛЕЙ С ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ КИСЛОТОЙ Способ синтеза ивабрадина формулы (I) и его аддитивных солей с фармацевтически приемлемой кислотой.(71)(73) Заявитель и патентовладелец: ЛЕ ЛАБОРАТУАР СЕРВЬЕ (FR) Настоящее изобретение относится к способу синтеза ивабрадина формулы (I) или 3-3-(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8 диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-она, к его аддитивным солям с фармацевтически приемлемой кислотой и к их гидратам. Ивабрадин, как и его аддитивные соли с фармацевтически приемлемой кислотой, и в особенности его гидрохлорид, обладает очень ценными фармакологическими и терапевтическими свойствами, в частности брадикардическими свойствами, которые делают эти соединения полезными при лечении или профилактике различных клинических случаев миокардической ишемии, таких как стенокардия, инфаркт миокарда и сопутствующие нарушения ритма, а также при различных патологиях, включающих нарушения ритма, а именно наджелудочковые нарушения ритма, и при сердечной недостаточности. Получение и использование в терапии ивабрадина и его аддитивных солей с фармацевтически приемлемой кислотой, и в особенности его гидрохлорида, было описано в европейском патенте ЕР 0534859. Этот патент описывает синтез гидрохлорида ивабрадина из соединения формулы (II) которое расщепляется для получения соединения формулы (III) которое подвергают взаимодействию с соединением формулы (IV) чтобы получить соединение формулы (V) каталитическое гидрирование которого дает ивабрадин, который в таком случае превращается в свой гидрохлорид. Недостаток этого пути синтеза состоит в том, что ивабрадин получают только с выходом 1%. Принимая во внимание фармацевтическую ценность этого соединения, было важным получить его эффективным способом синтеза, который обеспечивает ивабрадин с хорошим выходом. Настоящее изобретение относится к способу синтеза ивабрадина формулы (I) который характеризуется тем, что соединение формулы (VI) подвергают взаимодействию с тиолом в органическом растворителе для получения гемитиоацеталя формулы (VII) где R представляет собой замещенную или незамещенную, при необходимости перфторированную, линейную или разветвленную алкильную группу, замещенную или незамещенную арильную группу, замещенную или незамещенную бензильную группу или группу CH2CO2Et,которую подвергают реакции циклизации, чтобы получить соединение формулы (VIII) где R является, как определенный ранее,который подвергают реакции восстановления для получения ивабрадина формулы (I), который в случае необходимости может быть преобразован в его аддитивные соли с фармацевтически приемлемой кислотой, выбранной из соляной кислоты, бромисто-водородной кислоты, серной кислоты, фосфорной кислоты, уксусной кислоты, трифторуксусной кислоты, молочной кислоты, пировиноградной кислоты, малоновой кислоты, янтарной кислоты, глутаровой кислоты, фумаровой кислоты, винной кислоты, малеиновой кислоты, лимонной кислоты, аскорбиновой кислоты, щавелевой кислоты, метансульфоновой кислоты, бензосульфоновой кислоты и камфорной кислоты, и в их гидраты. Предпочтительным растворителем, используемым в реакции образования гемитиоацеталя формулы(VII), является дихлорметан. Предпочтительным тиолом, выбранным для вступления в реакцию с соединением формулы (VI),является тиофенол. Растворитель, предпочтительно применяемый для реакции циклизации соединения формулы (VII) до получения соединения формулы (VIII), представляет собой дихлорметан. В предпочтительной форме осуществления изобретения реакцию циклизации соединения формулы(VII) до получения соединения формулы (VIII) проводят в присутствии реагента, выбранного из уксусного ангидрида, трифторуксусного ангидрида или трифторметансульфоната триметилсилила. Более предпочтительно реакцию циклизации соединения формулы (VII) до получения соединения формулы (VIII) проводят в присутствии трифторуксусного ангидрида. Еще более предпочтительно реакцию циклизации соединения формулы (VII) до получения соединения формулы (VIII) проводят в присутствии трифторуксусного ангидрида и кислоты Льюиса, выбранной из BF3OEt2, Sc(OTf)3 или Yb(OTf)3. Еще более предпочтительно реакцию циклизации соединения формулы (VII) до получения соединения формулы (VIII) проводят в присутствии трифторуксусного ангидрида и BF3OEt2. Реакцию соединения формулы (VIII) предпочтительно проводят в присутствии никеля Ренея в этаноле или в присутствии йодида самария (II) в тетрагидрофуране. Соединения формул (VI), (VII) и (VIII) являются новыми продуктами, пригодными в качестве промежуточных соединений синтеза в химической или фармацевтической промышленности, в частности при синтезе ивабрадина, его аддитивных солей с фармацевтически приемлемой кислотой и их гидратов,и на основании этого являются неотъемлемой частью настоящего изобретения. Список используемых сокращений: ДМФ: N,N-диметилформамид; ДМСО: диметилсульфоксид; ТГФ: тетрагидрофуран; ИК: инфракрасный. Нижеследующие примеры поясняют изобретение. Точки плавления (ТК) были измерены с помощью столика Кфлера (Kfler) (BK). Инфракрасные спектры были зарегистрированы на аппарате Bruker, tensor 27, вспомогательное устройство ATR GoldenGate. Продукты помещали на пластину чистыми. Пример 1. трет-Бутил [2-(3,4-диметоксифенил)этил]карбамат. К раствору 2-(3,4-диметоксифенил)этиламина (10 г; 55,2 ммоль) в дихлорметане (200 мл) добавляют трет-бутил дикарбамат (12 г; 55,2 ммоль). После 1 ч контактирования при температуре окружающей среды реакционную смесь концентрируют под сниженным давлением. Остаток ресуспендируют в пентане (100 мл) и после 1 ч контактирования при температуре окружающей среды суспензию фильтруют через фритту. Получают 13,2 г продукта, указанного в заголовке, в виде твердого вещества. Выход = 85%. ТП = 652C. Пример 2. трет-Бутил 3-(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7 ил]метил(метил)амино]пропил[2-(3,4-диметоксифенил)этил]карбамат. К раствору соединения, полученного в предыдущей стадии (7,6 г; 27 ммоль), в 40 мл ДМФ при температуре окружающей среды маленькими фракциями добавляют NaH (60% в масле) (1,14 г; 28,5 ммоль). После 1 ч контактирования при температуре окружающей среды добавляют раствор 3-хлоро-N-[(7S)3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метил-1-пропанамина (7,68 г; 27 ммоль) в 16 мл ДМФ, затем реакционную смесь нагревают до 80C в течение 3 ч. После охлаждения до температуры окружающей среды реакционную смесь выливают в смесь дистиллированной воды и льда. Водную фазу экстрагируют этилацетатом. Объединенные органические фазы высушивают над MgSO4, затем концентрируют под сниженным давлением. Остаток очищают хроматографией на силикагелеN-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N'-[2-(3,4 диметоксифенил)этил]-N-метил-1,3-пропандиамин. 9 г (17 ммоль) соединения, полученного в предыдущей стадии, растворяют в 2,8 N этанольном растворе соляной кислоты. После 2 ч контактирования при температуре окружающей среды реакционную смесь концентрируют под сниженным давлением. Остаток ресуспендируют в 1N растворе гидроксида натрия, затем водную фазу экстрагируют этилацетатом. После высушивания органической фазы надMgSO4 и концентрирования под сниженным давлением получают 6,8 г продукта, указанного в заголовке,в виде масла. Выход = 93%. ИК:= 3304, 2793, 1261, 1236, 1205, 1153 см-1. Пример 4. N-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-N-[2-(3,4-диметоксифенил)этил]-2-гидроксиацетамид. Стадия 1. 2-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил[2-(3,4-диметоксифенил)этил]амино-2-оксоэтил ацетат. К раствору соединения, полученного в предыдущей стадии (6,8 г; 15,8 ммоль), в 200 мл дихлорметана при 0C добавляют триэтиламин (3,1 г; 22 ммоль), затем по каплям ацетоксиацетилхлорид (2,1 мл; 19 ммоль). Через 0,5 ч контактирования при температуре окружающей среды реакционную смесь промывают дистиллированной водой, затем органическую фазу высушивают над MgSO4. После концентрирования органической фазы под сниженным давлением получают 8 г продукта, указанного в заголовке, в виде масла и без очистки используют в следующей стадии. Стадия 2. N-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-N-[2-(3,4-диметоксифенил)этил]-2-гидроксиацетамид. К раствору соединения, полученного в предыдущей стадии (8 г), в 80 мл смеси из вода/метанол(2/1) добавляют K2CO3 (8,3 г; 60,4 ммоль). После 1 ч контактирования при температуре окружающей среды реакционную смесь концентрируют под сниженным давлением, затем остаток ресуспендируют в дистиллированной воде. После экстрагирования этилацетатом объединенные органические фазы высушивают над MgSO4, затем концентрируют под сниженным давлением. Получают 6,2 г продукта, указанного в заголовке, в виде масла. Выход = 81% (2 этапа). ИК:= 3406, 2794, 1641, 1261, 1236, 1205, 1153 см-1. Пример 5. N-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-N-[2-(3,4-диметоксифенил)этил]-2-оксоацетамид. К раствору оксалилхлорида (0,6 мл; 6,77 ммоль) в 25 мл дихлорметана при -78C добавляют раствор ДМСО (0,9 мл; 12,32 ммоль) в 5 мл дихлорметана. После 1 ч контактирования при -78C в течение 0,5 ч добавляют раствор соединения, полученного в предыдущей стадии (3 г; 6,16 ммоль), в 25 мл дихлорметана. После 1 ч контактирования при -78C добавляют триэтиламин (4,3 мл; 30,8 ммоль) затем реакционную смесь взбалтывают в течение 3 ч при температуре окружающей среды и затем выливают в насыщенный водный раствор NaHCO3. Органическую фазу высушивают над MgSO4, затем концентрируют под сниженным давлением. Получают 2,7 г продукта, указанного в заголовке, в виде масла. Выход = 89%. ИК:= 2788, 1645, 1589, 1261, 1236, 1207, 1151 см-1. Пример 6. 3-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8-диметокси-1-(фенилсульфанил)-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он. К раствору соединения, полученного в предыдущей стадии (2,6 г; 5,17 ммоль), в 60 мл дихлорметана добавляют тиофенол (0,53 мл; 5,17 ммоль). После 1 ночи контактирования при температуре окружающей среды постепенно добавляют трифторуксусный ангидрид (6,5 мл; 47 ммоль), затем BF3OEt2 (6,5 мл; 26 ммоль). Реакционную смесь взбалтывают в течение 3 ч при температуре окружающей среды, затем выливают в насыщенный водный раствор NaHCO3. Остаток, полученный после высушивания органической фазы над MgSO4, затем концентрирования под сниженным давлением очищают хроматографией на силикагеле (CH2Cl2/EtOH/NH4OH 28%: 97/3/0,3). Получают 1,15 г продукта, указанного в заголовке, в виде масла. Выход = 38%. ИК:= 2790, 1641, 1245, 1205, 1174 см-1. Пример 7. 3-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он. К раствору соединения, полученного в предыдущей стадии (0,8 г; 1,39 ммоль), в этаноле добавляют никель Ренея (2,5 г) (50% в H2O). После 1 ч контактирования при кипячении в колбе с обратным холодильником суспензию охлаждают, затем фильтруют через целит. Получают 600 мг продукта, указанного в заголовке, в виде масла. Выход = 94%. ИК:= 2788, 1646, 1519, 1461, 1245, 1105 см-1. Пример 8. Гидрохлорид 3-3-(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7 ил]метил(метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-она. Продукт, указанный в заголовке, получают исходя из продукта, полученного в предыдущей стадии,следуя порядку выполнения, описанному в патенте ЕР 0534859 (пример 2, стадия Е). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ синтеза ивабрадина формулы (I) отличающийся тем, что соединение формулы (VI) взаимодействует с тиолом в органическом растворителе с получением гемитиоацеталя формулы (VII) где R представляет собой замещенную или незамещенную, линейную или разветвленную алкильную группу, замещенную или незамещенную арильную группу, замещенную или незамещенную бензильную группу или группу CH2CO2Et,который подвергают реакции циклизации для получения соединения формулы (VIII) где R является, как определенный ранее,который подвергают реакции восстановления для получения ивабрадина формулы (I), который в случае необходимости может быть преобразован в его аддитивные соли с фармацевтически приемлемой кислотой, выбранной из соляной кислоты, бромисто-водородной кислоты, серной кислоты, фосфорной кислоты, уксусной кислоты, трифторуксусной кислоты, молочной кислоты, пировиноградной кислоты, малоновой кислоты, янтарной кислоты, глутаровой кислоты, фумаровой кислоты, винной кислоты, малеиновой кислоты, лимонной кислоты, аскорбиновой кислоты, щавелевой кислоты, метансульфоновой кислоты, бензосульфоновой кислоты и камфорной кислоты, и в их гидраты. 2. Способ синтеза по п.1, отличающийся тем, что органический растворитель, используемый в реакции получения гемитиоацеталя формулы (VII), представляет собой дихлорметан. 3. Способ синтеза по одному из пп.1 или 2, отличающийся тем, что тиол, вступающий в реакцию с соединением формулы (VI), представляет собой тиофенол. 4. Способ синтеза по одному из пп.1-3, отличающийся тем, что растворитель, применяемый в реакции циклизации соединения формулы (VII) до получения соединения формулы (VIII), представляет собой дихлорметан. 5. Способ синтеза по одному из пп.1-4, отличающийся тем, что реакцию циклизации соединения формулы (VII) до получения соединения формулы (VIII) проводят в присутствии реагента, выбранного из уксусного ангидрида, трифторуксусного ангидрида или трифторметансульфоната триметилсилила. 6. Способ синтеза по п.5, отличающийся тем, что реакцию циклизации соединения формулы (VII) до получения соединения формулы (VIII) проводят в присутствии трифторуксусного ангидрида. 7. Способ синтеза по п.6, отличающийся тем, что реакцию циклизации соединения формулы (VII) до получения соединения формулы (VIII) проводят в присутствии трифторуксусного ангидрида и кислоты Льюиса, выбранной из BF3OEt2, Sc(OTf)3 или Yb(OTf)3. 8. Способ синтеза по п.7, отличающийся тем, что реакцию циклизации соединения формулы (VII) до получения соединения формулы (VIII) проводят в присутствии трифторуксусного ангидрида иBF3OEt2. 9. Способ синтеза по одному из пп.1-8, отличающийся тем, что реакцию восстановления соединения формулы (VIII) проводят в присутствии никеля Ренея в этаноле или в присутствии йодида самария
МПК / Метки
МПК: C07D 223/16, A61K 31/55, A61P 9/00
Метки: аддитивных, кислотой, способ, фармацевтически, новый, приемлемой, солей, синтеза, ивабрадина
Код ссылки
<a href="https://eas.patents.su/6-19373-novyjj-sposob-sinteza-ivabradina-i-ego-additivnyh-solejj-s-farmacevticheski-priemlemojj-kislotojj.html" rel="bookmark" title="База патентов Евразийского Союза">Новый способ синтеза ивабрадина и его аддитивных солей с фармацевтически приемлемой кислотой</a>
Новый способ получения функционализированных бензоциклобутенов и их применение в синтезе ивабрадина и его аддитивных солей с фармацевтически приемлемой кислотой

Номер патента: 17503
Опубликовано: 30.01.2013
Авторы: Бодуан Оливье, Пиккарди Рикардо, Пегльон Жан-Луи, Шомонте Манон, Одик Никола
МПК: C07C 213/02, C07C 213/10, C07C 213/08...
Метки: функционализированных, кислотой, фармацевтически, бензоциклобутенов, солей, приемлемой, применение, новый, получения, ивабрадина, аддитивных, способ, синтезе
Формула / Реферат:
1. Способ получения соединений формулы (IV)где R1, R2, R3 и R4, которые могут быть идентичными или различными, каждый, представляют собой атом водорода, линейную или разветвленную (C1-C6)алкильную группу, линейную или разветвленную (C1-C6)алкоксигруппу, атом фтора, атом хлора, защищенную аминогруппу, защищенную гидроксильную группу, алкоксикарбонильную группу, в которой алкоксильная группа является линейной или разветвленной (C1-C6), или...
Новый способ синтеза ивабрадина и его солей присоединения с фармацевтически приемлемой кислотой

Номер патента: 16353
Опубликовано: 30.04.2012
Авторы: Дессинье Эмея, Пеглион Жан-Луи, Серки Бернар
МПК: C07D 223/16
Метки: способ, солей, новый, приемлемой, фармацевтически, синтеза, присоединения, кислотой, ивабрадина
Формула / Реферат:
1. Способ синтеза соединения формулы (VIII) в рацемической или оптически активной формеотличающийся тем, что соединение формулы (II) в рацемической или оптически активной формереагирует с соединением формулы (IX)в присутствии соли переходного металла или лантаноида, в растворителе, для получения соединения формулы (X) в рацемической или оптически активной формекоторое преобразуют в соединение формулы (VIII) при действии донора водорода.2. Способ...
Новый способ синтеза ивабрадина и его солей присоединения с фармацевтически приемлемой кислотой

Номер патента: 16335
Опубликовано: 30.04.2012
Авторы: Пеглион Жан-Луи, Дессинье Эмея, Серки Бернар
МПК: C07D 223/16
Метки: солей, кислотой, способ, присоединения, новый, ивабрадина, фармацевтически, синтеза, приемлемой
Формула / Реферат:
1. Способ синтеза соединения формулы (VII) в рацемической или оптически активной формев которой R представляет собой атом водорода или метильную группу,отличающийся тем, что соединение формулы (VIII)реагирует с соединением формулы (IX) в рацемической или оптически активной форме, в форме свободного основания или солив которой R является таким, как определено выше,в присутствии соли переходного металла или лантаноида, в растворителе, для...
Новый способ синтеза 7,8-диметокси-1,3-дигидро-2h-3-бензазепин-2-она и его применение при синтезе ивабрадина и его солей присоединения с фармацевтически приемлемой кислотой

Номер патента: 17333
Опубликовано: 30.11.2012
Авторы: Лекув Жан-Пьер, Лерестиф Жан-Мишель, Бриго Даниель
МПК: C07D 223/16
Метки: новый, ивабрадина, синтезе, синтеза, применение, фармацевтически, кислотой, солей, приемлемой, 7,8-диметокси-1,3-дигидро-2h-3-бензазепин-2-она, присоединения, способ
Формула / Реферат:
1. Способ синтеза соединения формулы (I)который характеризируется тем, что (3,4-диметоксифенил)уксусную кислоту формулы (IV)превращают в соединение формулы (V)где группы R1 и R2, которые могут быть одинаковыми или разными, представляют собой линейные или разветвленные (С1-С6)алкоксигруппы или вместе с атомом углерода, к которому они присоединены, образуют 1,3-диоксановое, 1,3-диоксолановое или 1,3-диоксепановое кольцо,либо с помощью...
Способ синтеза ивабрадина и его солей присоединения с фармацевтически приемлемой кислотой

Номер патента: 16405
Опубликовано: 30.04.2012
Авторы: Пеглион Жан-Луис, Лерестиф Жан-Мишель, Лекув Жан-Пьер, Дессинье Эмея, Серки Бернар
МПК: C07C 47/47, C07D 223/16
Метки: кислотой, ивабрадина, солей, синтеза, присоединения, способ, фармацевтически, приемлемой
Формула / Реферат:
1. Способ синтеза соединения формулы (VI) в рацемической или оптически активной формегде А представляет собой Н2С-СН2 или НС=СН,который отличается тем, что соединение формулы (VII) в рацемической или оптически активной формеподвергают реакции восстановительного аминирования с соединением формулы (VIII)где А имеет значение, указанное выше, в присутствии восстановителя,в органическом растворителе или смеси органических растворителей.2. Способ...
Предыдущий патент: Активные ингредиенты для повышения защиты растений от стресса, вызванного абиотическими факторами, и способ их обнаружения
Следующий патент: Композиция телмисартана и способ ее получения
Случайный патент: Способ получения симвастатина и его производных