Диариловые эфиры в качестве антагониста опиоидного рецептора
Номер патента: 8861
Опубликовано: 31.08.2007
Авторы: Такеути Кумико, Фритц Джеймс Эрвин, Матт Джеймс Эдвард Мл., Томас Элизабет Мария, Куимби Стивен Джеймс, Холловэй Уилльям Глен, Смит Дана Рей, Диас Буэсо Нурия, Педрегаль-Терсеро Консепсион, Уолф Чад Нолан, Митч Чарльз Ховард, Бланко-Пилладо Мария-Хесус, Сиджел Майлз Гудман, Де Ла Торре Марта Гарсия, Стаки Расселл Дин, Чаппелл Марк Дональд





Формула / Реферат
1. Соединение, выбранное из группы, состоящей из
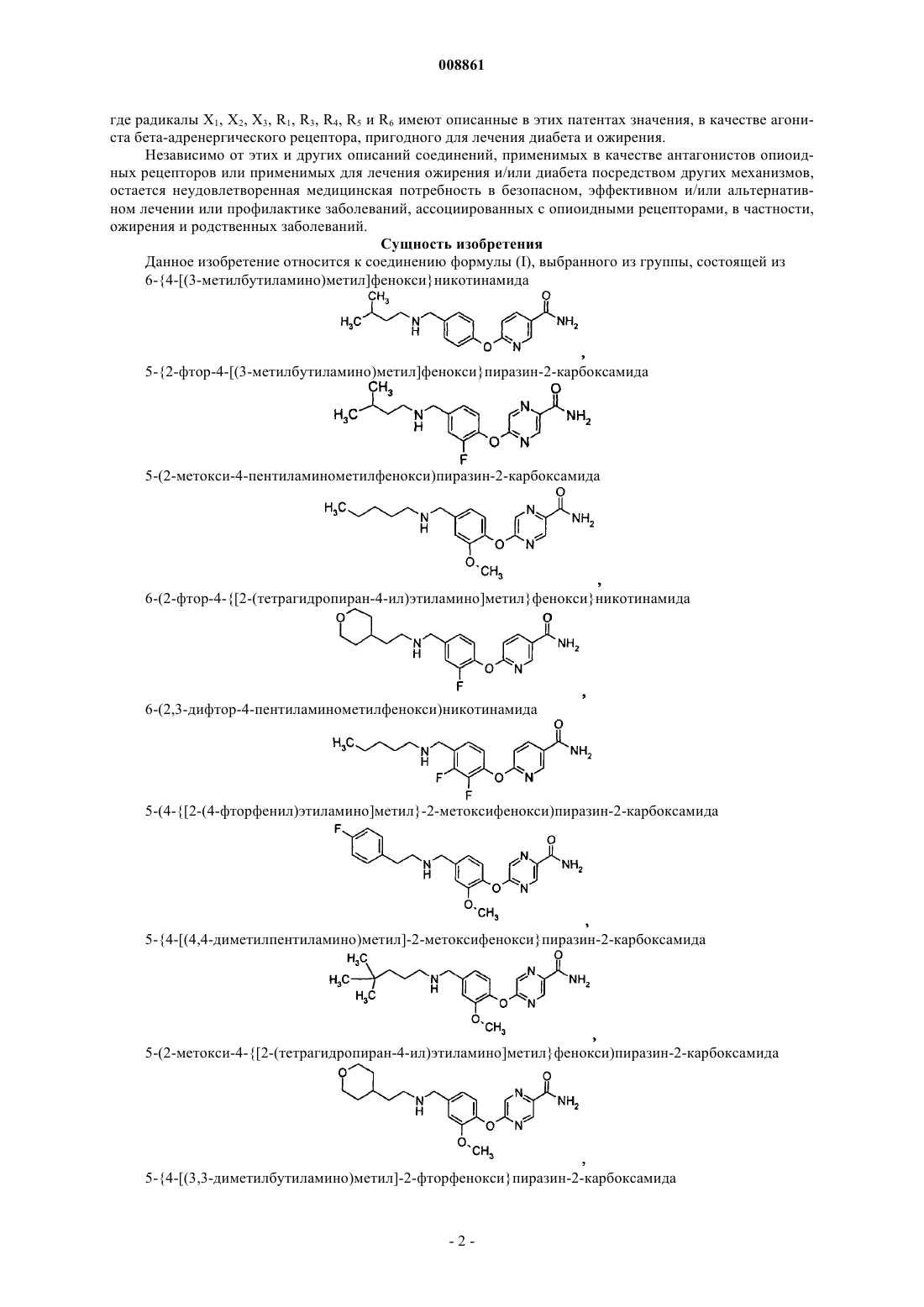
6-{4-[(3-метилбутиламино)метил]фенокси}никотинамида

5-{2-фтор-4-[(3-метилбутиламино)метил]фенокси}пиразин-2-карбоксамида

5-(2-метокси-4-пентиламинометилфенокси)пиразин-2-карбоксамида

6-(2-фтор-4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси}никотинамида

6-(2,3-дифтор-4-пентиламинометилфенокси)никотинамида

5-(4-{[2-(4-фторфенил)этиламино]метил}-2-метоксифенокси)пиразин-2-карбоксамида

5-{4-[(4,4-диметилпентиламино)метил]-2-метоксифенокси}пиразин-2-карбоксамида

5-(2-метокси-4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси)пиразин-2-карбоксамида

5-{4-[(3,3-диметилбутиламино)метил]-2-фторфенокси}пиразин-2-карбоксамида

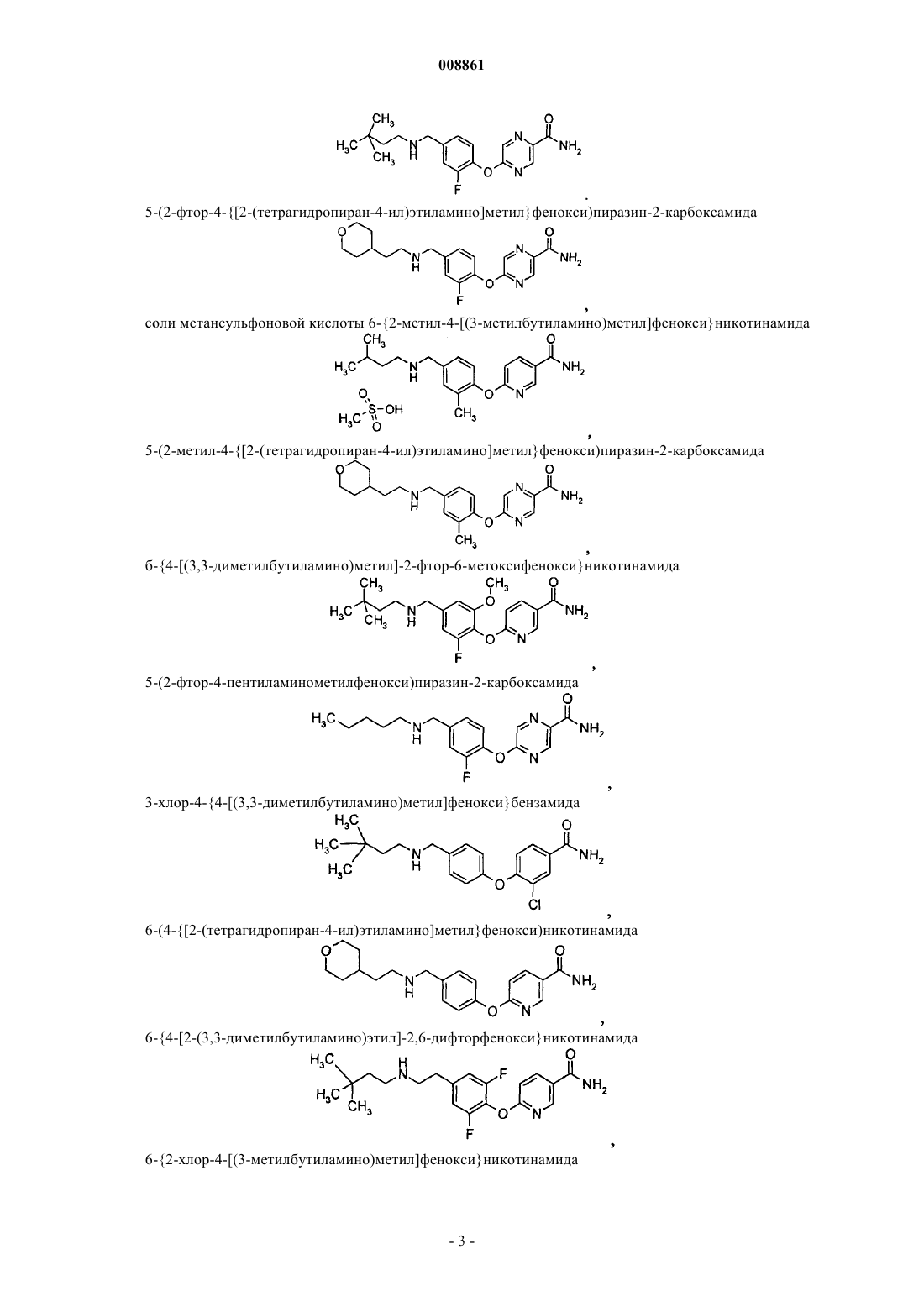
5-(2-фтор-4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси)пиразин-2-карбоксамида

соли метансульфоновой кислоты 6-{2-метил-4-[(3-метилбутиламино)метил]фенокси}никотинамида

5-(2-метил-4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси)пиразин-2-карбоксамида

6-{4-[(3,3-диметилбутиламино)метил]-2-фтор-6-метоксифенокси}никотинамида

5-(2-фтор-4-пентиламинометилфенокси)пиразин-2-карбоксамида

3-хлор-4-{4-[(3,3-диметилбутиламино)метил]фенокси}бензамида

6-(4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси)никотинамида

6-{4-[2-(3,3-диметилбутиламино)этил]-2,6-дифторфенокси}никотинамида

6-{2-хлор-4-[(3-метилбутиламино)метил]фенокси}никотинамида

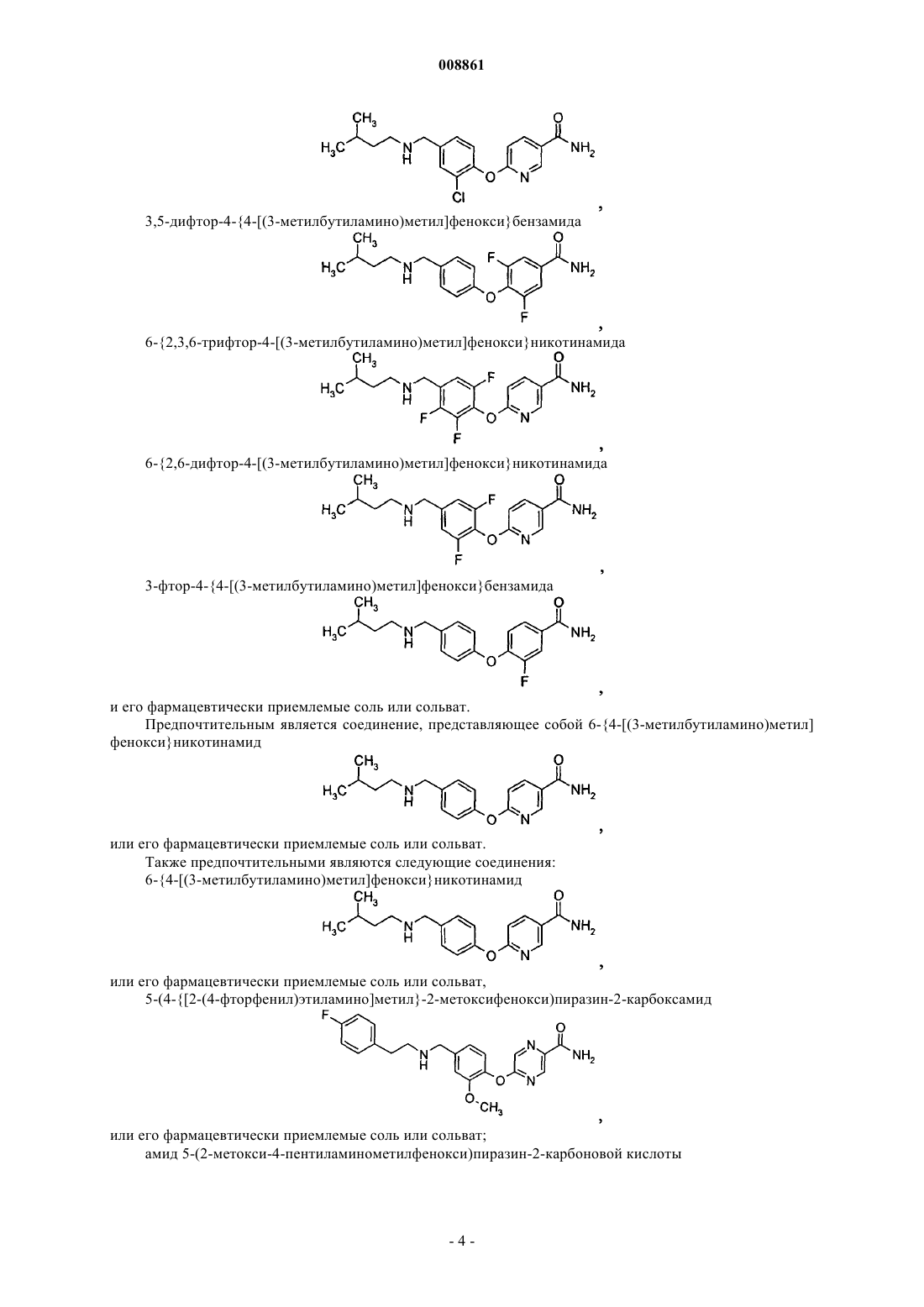
3,5-дифтор-4-{4-[(3-метилбутиламино)метил]фенокси}бензамида

6-{2,3,6-трифтор-4-[(3-метилбутиламино)метил]фенокси}никотинамида

6-{2,6-дифтор-4-[(3-метилбутиламино)метил]фенокси}никотинамида

3-фтор-4-{4-[(3-метилбутиламино)метил]фенокси}бензамида

и его фармацевтически приемлемые соль или сольват.
2. Соединение 6-{4-[(3-метилбутиламино)метил]фенокси}никотинамид

или его фармацевтически приемлемые соль или сольват.
3. Соединение 5-(4-{[2-(4-фторфенил)этиламино]метил}-2-метоксифенокси)пиразин-2-карбоксамид

или его фармацевтически приемлемые соль или сольват.
4. Соединение амид 5-(2-метокси-4-пентиламинометилфенокси)пиразин-2-карбоновой кислоты

или его фармацевтически приемлемые соль или сольват.
5. 6-(2-Фтор-4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси)никотинамид

или его фармацевтически приемлемые соль или сольват.
6. Соединение 5-(2-метокси-4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси)пиразин-2-карбоксамид

или его фармацевтически приемлемые соль или сольват.
7. Соединение по любому из пп.1-6, где фармацевтически приемлемой солью является соль хлористо-водородной кислоты, соль метансульфоновой кислоты, гидробромидная соль, бисульфатная соль или соль винной кислоты.
8. Соль хлористо-водородной кислоты соединения 6-{4-[(3-метилбутиламино)метил]фенокси}никотинамида

9. Соединение соль метансульфоновой кислоты 6-(2-фтор-4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси)никотинамида

10. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по любому из пп.1-9 и носитель, разбавитель и/или эксципиент.
11. Применение соединения по любому из пп.1-9 для приготовления лекарственного средства для лечения или предупреждения ожирения и родственных заболеваний.
12. Применение соединения по любому из пп.1-9 для приготовления лекарственного средства для лечения и/или ослабления симптомов, ассоциированных с ожирением и родственными заболеваниями.
13. Применение соединения по п.11 или 12, где родственное заболевание выбрано из группы, состоящей из диабета, диабетических осложнений, диабетической ретинопатии, атеросклероза, гиперлипидемии, гипертриглицеринемии, гипергликемии и гиперлипопротеинемии.
14. Применение соединения по п.11, где лекарственное средство предназначено для лечения или предупреждения ожирения.
15. Применение соединения по любому из пп.1-9 для приготовления лекарственного средства для супрессии (подавления) аппетита.
16. Применение соединения по любому из пп.1-9 для приготовления лекарственного средства для снижения веса.
Текст
008861 Данное изобретение относится к области медицинской химии. Данное изобретение относится конкретно к соединениям, применимым в качестве опиоидных антагонистов, способам лечения, способам применения и их фармацевтическим композициям. Обычно сообщаются три типа опиоидных рецепторов, опиоидные рецепторы мю, каппа и дельта. Недавнее доказательство указывает на взаимодействия между рецепторными димерными комбинациями рецепторов ,и/или(называемыми гетеродимерами), которые также способствуют опиоидной активности. Предполагалось, что опиоидные рецепторы и их нормальная регуляция или ее отсутствие участвуют в патологических состояниях, в том числе синдроме раздраженного кишечника, тошноте, рвоте, зудящих дерматозах, депрессии, привыкании к курению и алкоголю, половой дисфункции, апоплексическом ударе и травме у животных. Таким образом, неудивительно, что было показано, что способность антагонистически связывать опиоидные рецепторы вызывает ослабляющий, превентивный и/или лечебный эффекты у животных, в том числе людей, пораженных одним или несколькими из этих патологических состояний. Недавно, было обнаружено, что некоторые антагонисты опиоидных рецепторов увеличивают метаболическое потребление энергии и уменьшают массу у крыс, страдающих ожирением, при сохранении мышечной массы. Эти открытия указывают на то, что эффективный опиоидный антагонист может быть пригоден для предупреждения, лечения и/или ослаблении действия ожирения. С учетом процента населения, которое страдает ожирением в Западных сообществах, и опосредованных расходов, связанных с лечением эффектов и симптомов ожирения и родственных заболеваний, важность этих открытий не может быть преувеличенной. Хотя были описаны многие опиоидные антагонисты, продолжается поиск альтернативных, и/или улучшенных, или более эффективных антагонистов, имеющих общее преимущество для пациента с малыми побочными действиями или с отсутствием побочных действий. Патент США 4891379 описывает фенилпиперидиновые опиоидные антагонисты, используемые для лечения диабета и ожирения. В частности, в патенте США 4891379 описано соединение LY 255582, представленное структурой В патенте США 4191771 также описаны соединения, используемые в качестве опиоидных антагонистов. Были также получены бициклические аналоги фенилпиперидина, используемые в качестве опиоидных антагонистов в Wentland, et al., Bioorganic and Medicinal Chemistry Letters 11 (2001) 623-626; см. также Wentland, et al., Bioorganic and Medicinal Chemistry Letters 11 (2001) 1717-1721. Наконец, заявка на европейский патент с номером ЕР 1072592 А 2, поданная 18 мая 2000 г., описывает соединения фенилпиперидина формулы 1 где A, D, R1, R2, R3, X и n имеют значения, приведенные в описании, которые пригодны в профилактике и в лечении заболеваний, опосредованных опиоидными рецепторами, таких как зуд. В патенте США 6140352 и родственных патентах описано соединение формулы 1-1 008861 где радикалы X1, Х 2, Х 3, R1, R3, R4, R5 и R6 имеют описанные в этих патентах значения, в качестве агониста бета-адренергического рецептора, пригодного для лечения диабета и ожирения. Независимо от этих и других описаний соединений, применимых в качестве антагонистов опиоидных рецепторов или применимых для лечения ожирения и/или диабета посредством других механизмов,остается неудовлетворенная медицинская потребность в безопасном, эффективном и/или альтернативном лечении или профилактике заболеваний, ассоциированных с опиоидными рецепторами, в частности,ожирения и родственных заболеваний. Сущность изобретения Данное изобретение относится к соединению формулы (I), выбранного из группы, состоящей из 6-4-[(3-метилбутиламино)метил]феноксиникотинамида соли метансульфоновой кислоты 6-2-метил-4-[(3-метилбутиламино)метил]феноксиникотинамида и его фармацевтически приемлемые соль или сольват. Предпочтительным является соединение, представляющее собой 6-4-[(3-метилбутиламино)метил] феноксиникотинамид или его фармацевтически приемлемые соль или сольват. Также предпочтительными являются следующие соединения: 6-4-[(3-метилбутиламино)метил]феноксиникотинамид или его фармацевтически приемлемые соль или сольват,5-(4-[2-(4-фторфенил)этиламино]метил-2-метоксифенокси)пиразин-2-карбоксамид или его фармацевтически приемлемые соль или сольват; амид 5-(2-метокси-4-пентиламинометилфенокси)пиразин-2-карбоновой кислоты или его фармацевтически приемлемые соль или сольват; 6-(2-фтор-4-[2-(тетрагидропиран-4-ил)этиламино]метилфенокси)никотинамид или его фармацевтически приемлемые соль или сольват; 5-(2-метокси-4-[2-(тетрагидропиран-4-ил)этиламино]метилфенокси)пиразин-2-карбоксамид или его фармацевтически приемлемые соль или сольват. Наиболее предпочтительны соли хлористо-водородной кислоты, соли метансульфоновой кислоты,гидробромидная соль, бисульфатная соль или соль винной кислоты соединений, указанных выше. В частности, предпочтительны следующие соли: соль хлористо-водородной кислоты соединения 6-4-[(3 метилбутиламино)метил]феноксиникотинамида а также соль метансульфоновой метилфенокси)никотинамида Данное изобретение относится также к фармацевтической композиции, содержащей любое из выше названных соединений и носитель, разбавитель и/или эксципиент. Соединения настоящего изобретения пригодны для приготовления лекарственного средства для лечения и/или профилактики ожирения и родственных заболеваний, в том числе нарушений питания (булимии, нервной анорексии и т.д.), диабета, диабетических осложнений, депрессии, связанной с ожирением, тревожности, связанной с ожирением, эпилептического припадка, гипертензии, внутримозгового кровоизлияния, застойной сердечной недостаточности, нарушений сна, атеросклероза, апоплексического удара, гиперлипидемии, гипертриглицеридемии, гипергликемии, гиперлипопротеинемии, злоупотребления веществами, передозировки лекарственных средств, компульсивных поведенческих нарушений (таких как облизывание лапы у собаки) и пагубных поведенческих привычек, таких как страсть к игре и алкоголизм. Данное изобретение относится к применению соединения в качестве супрессоров аппетита. В другом варианте осуществления данное изобретение относится к применению любого из указанных соединений для снижения веса при сохранении или минимальной потери мышечной массы. Термины уменьшение интенсивности симптомов, предупреждение, профилактика, профилактический и предупреждают, используемые в описании, взаимозаменяемы и обозначают уменьшение тяжести симптомов, ассоциированных с ожирением и родственными заболеваниями у пациента, пораженного ими, или уменьшение вероятности того, что реципиент подвергнется описанным в описании патологическим состояниям или их осложнениям или разовьет эти состояния и их осложнения. В контексте данного изобретения термин эффективное количество является синонимом эффек-5 008861 тивной дозы и обозначает количество соединения, представленного выше, которое является достаточным в описании, или нескольких введениях для предупреждения, уменьшения симптомов или лечения состояния или его пагубных действий, описанных в описании, или количество соединения по изобретению, которое является достаточным для противодействия опиоидным рецепторам для достижения этих целей данного изобретения. Термин фармацевтически приемлемое в данном контексте является определением и означает по существу безвредное для пациента-реципиента. Термин активный ингредиент обозначает в данном контексте соединение по изобретению или комбинацию их. Термин "фармацевтическая композиция" включает в себя продукт, содержащий активный ингредиент (определенный выше) и инертный ингредиент (ингредиенты), которые составляют носитель, или другие компоненты вводимого лекарственного средства, а также любой продукт, который получают,прямо или опосредованно, из комбинации, комплексообразования или агрегации любых двух или более ингредиентов или при диссоциации одного или нескольких ингредиентов, или из других типов реакций или путем взаимодействия одного или нескольких из этих ингредиентов. Таким образом, фармацевтическая композиция данного изобретения включает в себя любую эффективную композицию, приготовленную смешиванием соединения данного изобретения и фармацевтически приемлемого носителя. Фармацевтические композиции данного изобретения применимы для лечения и/или предупреждения ожирения или родственных заболеваний. Термин родственные заболевания обозначает в данном контексте такие симптомы, заболевания или состояния, которые вызываются, обостряются, индуцируются или относятся к состоянию ожирения. Такие заболевания, состояния и/или симптомы включают в себя, но не ограничиваются ими, нарушения питания (булимию, нервную анорексию и т.д.), диабет, диабетические осложнения, диабетическую ретинопатию, половые/репродуктивные нарушения, связанную с ожирением депрессию, связанную с ожирением тревожность, эпилептический припадок, гипертензию, внутримозговое кровоизлияние, застойную сердечную недостаточность, нарушения сна, атеросклероз, ревматоидный артрит, апоплексический удар,гиперлипидемию, гипертриглицеридемию, гипергликемию и гиперлипопротеинемию. В данном контексте термины связанная с ожирением депрессия и связанная с ожирением тревожность обозначают состояния депрессии и тревожности соответственно, которые являются симптоматическими определенных страдающих ожирением пациентов и, возможно, вызываются знанием или самосознанием того, что они имеют ожирение, и, возможно, связаны с реальной или сознаваемой реакцией восприятия или неодобрения определенным индивидуумом, индивидуумами или обществом в целом. Связанная с ожирением депрессия или тревожность может обычно ослабляться или лечиться, как лечится и/или предупреждается состояние ожирения, введением соединения по изобретению. Термин подходящий растворитель относится к любому растворителю или смеси растворителей,инертных в отношении протекающей реакции, которые солюбилизируют реагирующие вещества с образованием среды, в которой будет происходить желаемая реакция. Термин общий (совместный) растворитель обозначает растворитель, который используется для достаточного растворения двух или более компонентов реакции или смеси по отдельности перед реакцией или смешиванием, т.е. растворитель, общий для более чем одного из реагентов или компонентов смеси. В данном контексте термин сольват является формой соединения данного изобретения, в которой кристалл или кристаллы соединения данного изобретения образованы из стехиометрического или нестехиометрического количества соединения по изобретению и растворителя. Типичные сольватирующие растворители включают в себя, например, воду, метанол, этанол, ацетон и диметилформамид. В тех случаях, когда соединение данного изобретения имеет кислотные или основные функциональные группы, могут быть образованы различные соли, которые являются более растворимыми в воде и/или более физиологически пригодными, чем исходное соединение. Примерами фармацевтически приемлемых солей являются, но не ограничиваются ими, соли щелочных и щелочно-земельных металлов,таких как литий, натрий, калий, кальций, магний, алюминий и т.п. Соли готовят обычно из свободной кислоты обработкой этой кислоты в растворе основанием или обработкой этой кислоты ионообменной смолой. В это определение фармацевтически приемлемых солей включены относительно нетоксичные, неорганические и органические основно-аддитивные соли соединений данного изобретения, например, с катионами аммония, четвертичного аммония и амина, полученные из азотистых оснований достаточной основности для образования солей с соединениями данного изобретения (см., например, S.M. Berge, etal., "Pharmaceutical Salts", J. Phar. Sci., 66: 1-19 (1977. Кроме того, основная группа (основные группы) соединения данного изобретения могут взаимодействовать с подходящими органическими или неорганическими кислотами с образованием солей, таких как ацетат, бензолсульфонат, бензоат, бикарбонат,бисульфат, битартрат, борат, гидробромид, камзилат, карбонат, клавуланат, цитрат, хлорид, эдетат, эдизилат, эстолат, эзилат, фторид, фумарат, глюцептат, глюканат, глутамат, гликолиларсанилат, гексилрезорцинат, гидрохлорид, гидроксинафтоат, гидроиодид, изотионат, лактат, лактобионат, лаурат, малеат,-6 008861 манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напзилат, нитрат, олеат, оксалат,пальмитат, пантотенат, фосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, таннат, тартрат, тозилат, трифторацетат, трифторметансульфонат и валерат. Предпочтительные соли в соответствии с данным изобретением включают в себя гидрохлоридную соль, гидробромидную соль, бисульфатную соль, соль метансульфоновой кислоты, соль п-толуолсульфоновой кислоты, битартратную, ацетатную и цитратную соль. Соединение данного изобретения может находиться в виде любого из его изомеров, стереохимических изомеров или региоизомеров, и все входят в объем данного изобретения. Некоторые соединения данного изобретения могут иметь один или более хиральных центров и, следовательно, могут существовать в оптически активных формах. Подобным образом, когда эти соединения содержат алкенильную или алкениленовую группу, может существовать возможность цис- и транс-изомерных форм этих соединений. Данным изобретением рассматриваются R- и S-изомеры и их смеси, в том числе рацемические смеси, а также смеси энантиомеров или цис- и транс-изомеров. Дополнительные асимметричные атомы углерода могут быть представлены в группе заместителя, такой как алкильная группа. Предполагается,что все такие изомеры, а также их смеси включены в объем данного изобретения. Если желательным является конкретный изомер, он может быть получен способами, хорошо известными в данной области, с использованием стереоспецифических реакций с исходными материалами, которые содержат асимметричные центры и уже являются разделенными, или, альтернативно, способами, которые приводят к смесям стереоизомеров, с последующим разделением известными способами. Например, рацемическая смесь может взаимодействовать с единственным энантиомером некоторого другого соединения, т.е. хирально разделяющим агентом. Это изменяет рацемическую форму на смесь стереоизомеров и диастереомеров, так как они имеют различные точки плавления, различные точки кипения и различные растворимости и могут быть разделены общепринятыми способами, такими как кристаллизация. Международная заявка РСТ WO 02/078693 А 2, опубликованная 10 октября 2002 г., описывает соединения формулы где R1, R2, R3, R4 и X имеют описанные в описании значения, в качестве антагонистов 5-НТ 6-рецептора для лечения нарушений, включающих в себя когнитивные нарушения, связанные со старением нарушения, нарушения настроения, психоз и т.д. Однако соединения данного изобретения пригодны для лечения и/или предупреждения ожирения и родственных заболеваний. Соединения данного изобретения проявляют также ингибирование действия, возбуждающего аппетит, и, следовательно, применимы в качестве супрессоров аппетита либо в виде отдельной терапии, либо в виде комбинированной терапии вместе с физическими упражнениями и другими эффективными подавляющими аппетит или вызывающими потерю массы лекарственными средствами. Эффективность соединений данного изобретения продемонстрирована определением их активности на нескольких биологических моделях, включающих в себя сцинтилляционный анализ близости (SPA анализ связывания ГТФ-гамма), анализ связывания опиоидного рецептора ex vivo, анализ ожирения крысin vivo и непрямой калориметрический анализ, который измеряет энергетический баланс и дыхательный коэффициент. В этих моделях тестируемые соединения данного изобретения показывают лучшие или одинаковые результаты, чем сравнительные соединения. Основным ссылочным (сравнительным) соединением является высокоэффективный прежний кандидат для клинического испытания LY255582, описанный в патенте США 4891379, разработка которого была прекращена из-за отсутствия удовлетворительной пероральной биодоступности для человека. Было показано, что пероральное введение этого антагониста опиоидного рецептора LY255582 значительно сокращает прием пищи после острого и продолжительного лечения у крыс. Кроме того, продолжительное лечение LY255582 приводило к стойкому отрицательному энергетическому балансу, приводя к уменьшению общей массы тела у крыс с индуцированным диетой ожирением, которых кормили рационом с высоким содержанием жира. Интересно, что было обнаружено, что тест-соединения данного изобретения дают сходные или лучшие благотворные эффекты в сравнении с LY255582. Представляет также интерес вторичное наблюдение, что тестируемые соединения-пробы данного изобретения давали лучшие результаты в тестах авторов данного изобретения в сравнении с Naltrexone HCl. Соединение по изобретению существует в виде свободного основания или в виде фармацевтически приемлемой соли. Более предпочтительной является гидрохлоридная соль, бисульфатная соль, мезилатная соль или соль щавелевой кислоты. Получение соединений изобретения В типичном протоколе необязательно замещенный бензонитрил или пиридинкарбоксамид или их синтон, имеющие отщепляемую группу, такую как галоген, предпочтительно фтор, бром или хлор, или алкилсульфонил, или другую подходящую отщепляемую группу, взаимодействует с нуклеофильной группой, такой как, например, гидроксифенилкарбоксальдегид, или его синтон, или производное.-7 008861 Например, в соответствии со схемой 1, необязательно замещенный 4-фторбензонитрил взаимодействует с необязательно замещенным 4-гидроксибензальдегидом с образованием простого эфира, соединения 3, при основных условиях. Схема 1 Основные условия включают в себя применение оснований, выбранных из неорганических и органических оснований. Примеры используемых неорганических оснований включают в себя, но не ограничиваются ими, карбонат калия, гидрид натрия, карбонат натрия, гидроксид натрия, гидроксид калия, карбонат кальция и карбонат цезия. Примеры органических оснований включают в себя, но не ограничиваются ими, гексаметилдисилазид калия, н-бутиллитий, гексаметилфосфортриамид (НМРТ) и т.п. Эти основные условия дополняются присутствием растворителя, предпочтительно органического растворителя. Предпочтительные органические растворители включают в себя протонные растворители или полярные апротонные растворители. Наиболее предпочтительные растворители включают в себя диметилформамид, метанол, диметилацетамид (ДМА), диметилсульфоксид. Наиболее предпочтительные условия основной реакции включают в себя применение карбоната калия в диметилацетамиде при температурах приблизительно 60-100 С. Нитрильное соединение формулы 3 превращают в карбоксамид 4 посредством гидролиза, известного специалисту. Например, соединение формулы 3 взаимодействует с карбонатом калия или другим подходящим основанием в присутствии пероксида водорода в подходящем органическом растворителе, т.е. ДМСО или ДМФА. Полученное амидное соединение 4 подвергают восстановительному аминированию подходящим образом замещенным амином. Восстановительное аминирование может выполняться двухстадийно или одностадийно в зависимости от стабильности промежуточного имина. Соединение 4 взаимодействует с первичным или вторичным амином (предпочтителен первичный амин) в метаноле в качестве растворителя. Могут добавляться молекулярные сита для усиления эффективности образования имина. Во второй стадии к реакционной смеси добавляют восстанавливающий агент, обычно боргидрид натрия или другой гидридный восстанавливающий агент. Ход реакции может быть подвергнут мониторингу с использованием ТСХ, ВЭЖХ,ЖХ-МС или других аналитических способов, известных специалисту, для определения по существу завершения каждой стадии и выбора момента добавления следующего реагента. В результате восстановительного аминирования соединения 4 получают соединение формулы 5, которое является целевым соединением данного изобретения. Аналоги соединений 3 и 5, имеющие одну или несколько групп заместителя R, могут быть получены с использованием подходящим образом замещенных исходных материалов или взаимопревращением функциональности заместителя. Например, исходная группа-заместительR может быть защищена и освобождена от защитной группы подходящим образом для получения желаемого конечного заместителя R. Альтернативно, исходный заместитель R может быть превращен посредством известных реакций 1, 2 или 3 стадиями в другие желаемые заместители R. Другой протокол, иллюстрируемый в схеме 2, показывает использование карбоксамидного исходного материала для получения, например, соединений, имеющих пиридинильное В-кольцо. Применение карбоксамидного исходного материала является особенно предпочтительным для соединений данного изобретения, в которых В-кольцо является пиридинильной, пиридазинильной, пиразинильной или пиримидинильной группой. Карбоксамид может быть введен в виде части исходного материала, когда подходящий заменитель для В-кольца является коммерчески доступным, или может быть получен для определенных групп, как обсуждается в примерах. Например, использование пиридинкарбоксамида, никотинамида или их замещенных аналогов приводит к замещенным производным или аналогам соединений формулы 4 а или 5 а, которые являются также соединениями данного изобретения. Первичные и вторичные амины применимы для восстановительного аминирования для превращения соединения (4 а) в соединение (5 а). Примеры применимых аминов включают в себя, но не ограничиваются ими, фенетиламин, 3-метилбутиламин, пропиламин, изопропиламин, бензиламин и изопентиламин. Соединения, полученные посредством этой и других схем, описанных в описании или известных специалисту, могут быть дополнительно преобразованы в кислотно-аддитивную соль, как показано, например, в схеме 2 А. Схема 2 А Схема 2 А показывает получение гидрохлоридной соли (6 а') соединения 5 а (схема 2), где RNH2 обозначает 3-метилбутиламин или другую аминную группу и R4 и R5, оба, обозначают водород. Соединение 5 а' растворяют в этаноле и добавляют небольшой избыток (например, 1,0-1,5 мол. экв.) 1 н. хлористоводородной кислоты при температурах в диапазоне от 0 С до комнатной температуры. Этой смеси дают кристаллизоваться при охлаждении или без охлаждения или ее упаривают с получением гидрохлоридной соли, которая может быть дополнительно очищена растиранием с подходящим органическим растворителем, таким как толуол, гексаны, диэтиловый эфир или их смеси. В альтернативном случае безводный НСl может барботироваться в холодный раствор соединения 5 а' до завершения реакции или до насыщения раствора, и эту смесь обрабатывают должным образом. Специалисту известны детали и варьируемые способы получения, выделения и очистки кислотно-аддитивных солей, и специалист может получить сравнимые результаты с использованием способов, подходящих для конкретного субстрата, без дополнительного экспериментирования. Модифицированный протокол для получения соединений данного изобретения показан на схеме 3,в которой реакцию нуклеофильного замещения для образования простой эфирной связи выполняют в конце этого синтеза, а не в более ранней стадии. По этому протоколу подходящим образом замещенный аминофенол восстановительно аминируют бензальдегидом, который является необязательно замещенным при необходимости. Восстановительное аминирование выполняют в присутствии боргидрида натрия или другого восстанавливающего агента и подходящего основания. Альтернативно и предпочтительно используют ди-трет-бутилдикарбонат(Вос 2O) для защиты начального свободного амина в виде Воc-защищенного амина. Затем полученное феноксисоединение 7 взаимодействует с источником В-кольца, таким как, например, фенил- или пиридинкарбоксамид, бензонитрил или пиридинонитрил или их синтон. Связывание источников колец В и А выполняют при основных условиях с получением простого эфира 8 а и 8b для приведенного выше примера. Когда связанный продукт существует в виде смеси изомеров, как в случае 8 а и 8b, эти изомеры могут быть разделены или использованы непосредственно в следующей стадии. В следующей стадии нитрильную группу, если она присутствует, как в данном примере, гидролизуют до карбоксамида, как обсуждалось ранее. Защитная группа может быть удалена с использованием хлористо-водородной кислоты или трифторуксусной кислоты с помощью процедур, известных специалисту. Специалисту известно, что подходящим образом замещенные аналоги соединений формулы 10 а или 10b могут быть получены с использованием подходящим образом замещенных исходных материалов или их заменителей, которые могут быть превращены в желаемые заместители. Соединения формулы I, имеющие различные длины алкильных цепей в боковой цепи амино, могут быть получены в одном случае реакциями карбонильного удлинения. Примером является модифицированная реакция типа реакции Виттига, показанная в схеме 4. Протокол схемы 4 и известные варианты его обеспечивают возможность манипуляции длины цепи и/или заместителей боковой цепи аминогруппы. В соответствии с протоколом, необязательно замещенный 4-гидроксибензальдегид, т.е. соединение 11, взаимодействует с необязательно замещенным бензонитрилом, имеющим подходящую отщепляемую группу, например, галоген, алкилсульфонил и т.д. Затем никотинонитрил 12 или его аналог подвергают реакции карбонильного удлинения, такой как, например,реакция Виттига и ее вариации (см. Organophosphorous Agents in Organic Synthesis, J.I.G. Cadogan, Ed.,Academic Press London (1979); см. также J. March, Advanced Organic Chemistry, 3rd Edition, Wiley Interscience, New York New York, (1995). В приведенном примере альдегид 12 взаимодействует с метоксиметилтрифенилфосфином (доступным из Aldrich chemical Company, Milwaukee, USA) с использованием сильного основания, такого как, например, н-бутиллитий, втор-бутиллитий и т.п., с образованием начального карбаниона. Полученный винилметиловый простой эфир 13 гидролизуют с использованием сильной кислоты, такой как п-толуолсульфоновая кислота, НСl или серная кислота, с получением нового альдегида. Затем этот альдегид реагирует с подходящим амином с последующим восстановлением с образованием продукта 14 восстановительного аминирования. Детали каждой стадии в описанных в описании схемах приведены в экспериментальном разделе или могут быть найдены в приведенных в качестве ссылок руководствах по органическому синтезу или являются известными специалисту. Некоторые реакции, такие как образование разных илидов для реакции Виттига и родственные реакции, протекают лучше при пониженных температурах в диапазоне от приблизительно -10 до приблизительно -70 С. Другие реакции протекают лучше при повышенных температурах в диапазоне от приблизительно 30 до приблизительно 150 С и еще другие реакции протекают лучше при температуре окружающей среды в диапазоне от приблизительно 15 до приблизительно 30 С. Соединения данного изобретения, в которых группы R1 и R2 объединяются друг с другом и с атомом азота с образованием азотсодержащего гетероцикла, могут быть получены, например, в соответствии со схемой 5. Схема 5 В соответствии со схемой 5, восстановительное аминирование альдегида выполняют с использованием циклического амина, имеющего желаемый размер кольца и/или желаемые заместители. Например,- 11008861 взаимодействие соединения необязательно замещенного циклического амина, такого как, например, необязательно замещенный пирролидин (как показано), с альдегидом 4 приводит к образованию соединения 16, имеющего R1 и R2, соединенные вместе, с образованием азотсодержащего гетероциклического амина. Соединения формулы I, в которых R1 или R2 вместе с А-кольцом с образованием азотсодержащего гетероцикла, могут быть получены, как показано на следующей схеме 6. Схема 6 Приведенная выше схема показывает получение кольца бензо[d]азепина в качестве репрезентативного примера. Как показано, реакция 3-метоксифенацетилхлорида (17) с диметилацеталем метиламиноацетальдегида приводит к образованию соединения 18. Соединение (18) циклизуется с соединением 19 азепин-2-оном. Соединение 19 восстанавливается до соединения тетрагидробензо[d]азепин-2-она с использованием, например, литийалюминийгидрида в ТГФ или 5% палладия-на-угле в этилацетате. Это соединение дополнительно дезоксигенируют и восстанавливают до соединения 20 тетрагидробензо[d]азепина. Сначала соединение 20 защищают в виде трифторацетамида, деметилируют трибромидом бора в подходящем полярном апротонном растворителе и затем дают ему реагировать, например, с 6 хлорникотинамидом для образования соответствующего эфирного продукта. Трифторацетамидную защитную группу удаляют основным гидролизом, т.е. аммиаком в метаноле, и замещение на азоте азепина приводит к соединениям 22 данного изобретения. Такие замещения могут выполняться с использованием основания, такого как карбонат натрия или калия, в присутствии электрофила, т.е. алкил-, бензил- или арилгалогенида. Подробные процедуры для использования на практике приведенного выше протокола,как и других описанных выше протоколов, могут быть найдены в экспериментальном разделе. Подробности для отдельных стадий описанных в описании протоколов также могут быть найдены в литературе или могут быть известны специалистам. Соединения формулы I, в которых В-кольцо является позиционным изомером пиридина, может быть получено, как показано на схеме 7. Схема 7 Как показано выше, диазотирование с последующим бромированием 2-амино-5-фторпиридина (23) дает соединение 24 2-бром-5-фторпиридин. Соединение 2-бром-5-фторпиридин превращают в этоксикарбонильное производное посредством реакции гидроксикарбонилирования с последующей этерификацией начальной карбоксильной группы. Катализируемая палладием реакция гидроксикарбонилирования известна специалисту и также описана в традиционных справочных руководствах по органической химии. Для варианта реакции гидроксикарбонилирования с использованием трифлатной отщепляемой группы см. Sandro Sacchi and Alessandro Lupi, Palladium Catalyzed Hydroxycarbonylation of Vinyl and ArylTriflates: Synthesis of ,-Unsaturated and Aromatic Carboxylic Acids, Tetrahedron Letters, Vol. 33, No. 27,pp. 3939-3942 (1992). Полученный эфир может быть гидролизован до кислоты, которую затем превращают в карбоксамид реакцией сочетания в присутствии агента связывания, например, такого как EDCl. Альтернативно, соединение 2-бром-5-фторпиридин может быть превращено в нитрил реакцией с цианидом меди в полярном апротонном растворителе, таком как ДМФА. Затем этот нитрил гидролизуют, как обсуждалось ранее, с получением соответствующего карбоксамида 26. Специалисту известно, что катализируемые палладием реакции цианирования с использованием цианида меди, источника палладия и лиганда являются доступными для выполнения обсуждаемой выше реакции цианирования со сходными или, возможно, улучшенными выходами. Карбоксамидное соединение 26 взаимодействует с замещенным или незамещенным 4-гидроксибензальдегидом, защищенным в виде ацеталя 28. Затем полученный продукт этерификации подвергают восстановительному аминированию амином в присутствии боргидрида натрия или другого подходящего восстанавливающего агента с получением соединения 29 данного изобретения, как показано в схеме. Соединения формулы I, в которых кольцо В является пиразинилом, могут быть получены, например, в соответствии со схемой 8, приведенной ниже: Схема 8 Соединения, в которых R1 и/или R2 обозначает независимо циклическую группу, т.е. насыщенный или ненасыщенный моноциклический карбоцикл, могут быть получены, как показано ниже в схеме 9. Схема 9 выполняется реакцией амина 33, включающего в себя А-кольцо, с галогенникотинамидом, например, 6-хлорникотинамидом или галогенникотинонитрилом, с образованием соединения 34 данного изобретения. Схема 9 При использовании галогенникотинонитрила гидролиз полученного нитрила с образованием амидного производного был описан ранее. Сам амид 33 получают восстановительным аминированием 4 гидроксифенацетальдегида и соответствующего амина. Сам фенацетальдегид является продажным продуктом или может быть получен из соответствующего бензальдегида реакциями карбонильного удлинения, т.е. реакцией Виттига или модифицированной реакцией Виттига, как обсуждалось ранее. Альтернативный протокол показан на схеме 10. Схема 10 Как показано на схеме 10, субстрат амина, имеющий А-кольцо, т.е. 4-гидроксифенетиламин, защищают при аминогруппе, например, Вос-защитной группой или другими типичными аминозащитными группами. Вос-защищенный амин 35 связывают с компонентом В-кольца, т.е. 6-хлорникотинамидом (как показано), или никотинонитрилом, или бензонитрилом, или их аналогом, или производным. Затем этот связанный продукт освобождают от защитных групп и подвергают восстановительному аминированию циклическим кетоном, имеющим желаемую группу R1 и/или R2 для этой структуры и в объеме формулыI. Для показанного примера защищенный трет-бутилдиметилсилильной (TBDMS) группой 3 гидроксициклогексанон 37 взаимодействует с амином 36, имеющим уже на месте кольца А и В, с обра- 13008861 зованием желаемого соединения 38 данного изобретения после десилилирования. Предпочтительные условия реакции для каждой стадии описанных в описании реакций или схем приведены в экспериментальном разделе или известны специалисту или предлагаются в литературе или могут быть определены минимальным рутинным экспериментированием специалистом в соответствии с некоторыми или всеми приведенными в описании или цитируемыми в ссылках описаниями. Заместители, такие как R и R-группы, используемые в этих схемах, приведены только для иллюстрации и не должны рассматриваться как ограничение количества и/или типа заместителей. Специалисту известны типы заместителей и их разнообразия, которые являются подходящими и/или возможными для конкретного положения. Обычно, хотя для целей иллюстрации используют конкретный субстрат или конкретное соединение, не предполагается ограничение пригодности конкретной схемы для других соединений в пределах границ данного изобретения, если нет других указаний. Специалисту известно, что соединения формулы II могут быть также получены с использованием приведенных выше схем и описанных в экспериментальном разделе процедур. Схема 11 Некоторые соединения данного изобретения могут быть также получены протоколами, приведенными в схеме 11. Например, соединения формулы I или II, имеющие группы у, другие, чем водород, могут быть легко получены присоединением Михаэля нитрометана к альдегиду, например, альдегиду 39,имеющему желаемые заместители А-кольца. Полученный продукт восстанавливают с получением насыщенного амина. Когда r является метилом, продукт 41 освобождают от защитных групп реакцией с ВВr3 с последующими процедурами, описанными в описании и/или известными специалисту. Полученный гидроксиамин необязательно защищают, например, с использованием Вос-группы, с получением соединения 42. Затем защищенное аминосоединение 42 взаимодействует с подходящим образом замещенным бензамидом, или никотинонитрилом, или никотинамидом с образованием соединения формулыI или II после дополнительной обработки, описанной ранее. Способ применения изобретения Как отмечалось выше, соединения данного изобретения применимы в блокировании действия агонистов при опиоидных рецепторах ,и/или . Таким образом, данное изобретение обеспечивает также способ блокирования рецептора , ,или комбинации этих рецепторов (гетеродимера) у млекопитающего, предусматривающий введение указанному млекопитающему блокирующей рецептор дозы соединения формулы I или II. Термин блокирующая рецептор доза в данном контексте обозначает количество соединения формулы I или II, необходимое для блокирования рецептора ,илиили комбинации этих рецепторов (гетеродимера) после введения млекопитающему, нуждающемуся в блокировании рецептора ,илиили комбинации этих рецепторов (гетеродимера). Соединения формулы I или II или их комбинации являются эффективными в широком диапазоне доз. Например, дневная доза обычно находится в диапазоне от приблизительно 0,05 до приблизительно 250 мг/кг массы тела. При лечении взрослых людей предпочтительным является диапазон от приблизительно 0,5 до приблизительно 100 мг/кг в одной дозе или в разделенных дозах. Однако будет понятно, что количество фактически вводимого соединения будет определяться врачом в свете релевантных обстоятельств, в том числе подлежащего лечению состояния, выбора вводимого- 14008861 соединения, возраста, массы и реакции индивидуального пациента, тяжести симптомов пациента и выбранного способа введения. Таким образом, приведенные выше диапазоны доз не предназначены для ограничения объема данного изобретения каким-либо обоазом. Эти соединения могут вводиться различными способами, такими как пероральный, трансдермальный, подкожный, интраназальный, внутримышечный и внутривенный способы. Было показано, что различные физиологические функции могут подвергаться действию или испытывать влияние рецепторов ,илиили комбинации рецепторов (гетеродимеров) в головном мозге. Считают, что соединения данного изобретения способны лечить нарушения, ассоциированные с этими рецепторами или их комбинациями, такие как нарушения питания, передозировка опиоидов, депрессия,курение, алкоголизм, половая дисфункция, шок, апоплексический удар, повреждение спинного мозга и травма головы. Как таковое, данное изобретение обеспечивает также способы лечения вышеупомянутых нарушений блокированием действия агонистов при рецепторах ,иили комбинациях рецепторов(гетеродимере). Было обнаружено, что соединения данного изобретения проявляют превосходную активность в анализе связывания опиоидных рецепторов, который измеряет способность этих соединений блокировать рецепторы , , иили комбинацию этих рецепторов (гетеродимер). Анализ связывания ГТФS Был разработан формат анализа на основе SPA ГТФS на основе прежних форматов анализов опиоидных (Emmerson et al., J. Pharm. Exp. Ther. 278, 1121, 1996; Horng et al., Society for Neuroscience Abstracts, 434,6, 2000) и мускариновых (DeLapp et al., JPET 289, 946, 1999) рецепторов. Мембраны ресуспендировали в 20 мМ HEPES, 100 мМ NaCl, 5 мМ МgСl2, 1 мМ ДТТ и 1 мМ ЭДТА. Пятьдесят (50) мл соединения ГТФ[35S], суспензии мембран (20 мкг на лунку) и покрытые агглютинином зародыши пшеницы SPA-гранулы (1 мг на лунку) добавляли в 96-луночные аналитические планшеты, где лунки имели прозрачное дно. ГДФ (200 мМ) добавляли к раствору мембран перед добавлением в аналитические планшеты. Планшеты герметизировали и инкубировали в течение 4 ч при комнатной температуре,затем помещали в холодильник на ночь, чтобы эти гранулы выпали в осадок. Было определено, что стабильность сигнала при 4 С сохраняется в течение 60 ч. Планшеты согревали до комнатной температуры и считали в сцинтиляционном счетчике Wallac Microbeta. Для анализов антагонистов добавляли специфические агонисты в следующих концентрациях: (MOR) DAMGO 1 мкМ, (DOR) DRDPE 30 нМ, (KOR)U69593 300 нМ. Константы связывания определяли по уравнению Cheng-Prusoff (см. Cheng and Prusoff,Biochem. Pharmacol. 22, 3099, 1973). Результаты, полученные для репрезентативной пробы соединений данного изобретения в анализе связывания ГТФS, показаны в табл. 1 ниже. Таблица 1 Связывание рецептора ex vivo Для того чтобы связать аффинность связывания и антагонистическую эффективность in vitro с активностью и эффективностью in vivo, авторы данного изобретения разработали анализ связывания рецептора ex vivo в головном мозге крысы. Этот анализ измеряет различие в ассоциации (связывании) неселективного радиолиганда (3 Н-дипренорфина) опиоидного рецептора высокой аффинности в ткани головного мозга, выделенной из животных, получающих носитель, против тест-соединения обработки(меньшее связывание 3 Н-дипренорфина соответствует большей ассоциации тест-соединения с опиоидными рецепторами). Исследования с использованием анализа связывания рецептора ex vivo продемонстрировали положительную корреляцию между активностью (силой и продолжительностью активности),которая также коррелирует с 24-часовой эффективностью в индуцированном пищевым рационом ожирении крыс.- 15008861 Способы. Анализ связывания опиоидного рецептора ex vivo измеряет связывание 3 Н-дипренорфина (радиолиганда для рецепторов ,ис аффинностью 0,1-0,4 нМ) в полосатом теле/прилежащем ядре; области головного мозга, которая содержит высокую плотность рецепторов ,и , после перорального введения соединений. Экспериментально дозу скрининга 7 мг/кг соединения или носителя перорально вводят крысам. Спустя 6 ч после введения соединения животных умерщвляют и полосатое тело/прилежащее ядро выделяют и гомогенизируют в 10 объемах (мас./об.) буфера для связывания. Затем этот гомогенат используют в анализе связывания гомогената с использованием насыщающей концентрации 3 Н-дипренорфина в течение 30 мин. Гомогенизацию и анализ выполняют при 4 С для минимизации перераспределения соединения в части связывания in vitro этого анализа. Результаты выражены (табл. 2) в виде % ингибирования связывания дипренорфина, основанного на различии специфического связывания между обработанными соединением животными и контрольными животными, обработанными одним носителем. Таблица 2 Анализ острого воздействия посредством кормления (анализ на ожирение крыс) Эффективность соединений данного изобретения дополнительно подтверждали результатами анализа ожирения крыс, показанными в табл. 3. Результаты этого анализа показывают, что соединения данного изобретения проявляют ингибирование опиоидных рецепторов на уровне, сравнимом с уровнем,получаемым с прежним клиническим кандидатным соединением LY255582, описанным и заявленным в патенте США 4891379, или на уровне, превосходящем уровень, получаемый с LY255582. Непрямой калориметрический анализ Двадцатичетырехчасовой расход энергии (ЕЕ) и дыхательный коэффициент (RQ) измеряли непрямой калориметрией с использованием калориметрической системы с незамкнутым контуром (Oxymax,Columbus Instrument Int. Corp., USA). RQ является отношением объема продуцируемого СO2 (VCO2) к объему потребляемого O2 (VO2). ЕЕ рассчитывали как произведение теплотворной способности кислорода (CV) и VO2 на килограмм массы тела, где CV = 3,815+1,232 (RQ). Общее количество расходуемых калорий рассчитывали для определения суточной утилизации горючего (энергии в виде пищи). Для расчета доли белков, жиров и углеводов, которая используется во время 24-часового периода, авторы использовали предложение Flatt (см. Flatt JP 1991 Assessment of daily and cumulative carbohydrate and fatbalances in mice. J. Nutr. Biochem. 2:193-202) и формулы, а также другие производные константы (см. EliaM., Livesey G. 1992 Energy expenditure and fuel selection in biological systems: the theory and practice of calculation based on indirect calorimetry and tracer methods. World Rev Nutr Diet 70:68-131). Измеряли также потребление на протяжении 24-часового периода. Минимальная эффективная доза (MED) для ингибирования потребления пищи выражена как самая низкая доза, которая вызывала уменьшение потребления пищи, которое было значимо отличающимся от контролей, обработанных носителем. Результаты, полученные для пробы соединений данного изобретения с использованием непрямого калориметрического анализа, показаны ниже в табл. 4. Таблица 4 Баланс энергии = поглощение калорий минус утилизация (ккал/кг/день)- 17008861 Непрямой калориметрический анализ, описанный выше, показывает, что минимальная эффективная доза для ингибирования потребления пищи на уровне, значимо отличающемся от уровня, достигаемого с контрольной дозой носителя, для соединений данного изобретения была сравнима с дозой ссылочного соединения или была лучшей в сравнении со ссылочным соединением. Композизия Соединение данного изобретения предпочтительно представлено в форме фармацевтической композиции, содержащей фармацевтически приемлемый носитель, разбавитель или эксципиент и соединение данного изобретения. Такие композиции будут содержать от приблизительно 0,1 до приблизительно 90,0 мас.% соединения данного изобретения (активного ингредиента). Как таковое, данное изобретение обеспечивает также фармацевтические композиции, содержащие соединение данного изобретения и фармацевтически приемлемый носитель, разбавитель или эксципиент. В приготовлении композиций данного изобретения активный ингредиент обычно смешивают с носителем или разбавляют носителем или заключают в носитель, который может быть в форме капсулы,подушечки, бумаги или другого контейнера. Когда этот носитель служит в качестве разбавителя, он может быть твердым, полутвердым или жидким материалом, который действует в качестве носителя, эксципиента или среды для активного ингредиента. Таким образом, эта композиция может быть в форме таблеток, пилюль, порошков, пастилок, подушечек, крахмальных облаток, эликсиров, эмульсий, растворов, сиропов, суспензий, аэрозолей (в виде твердого вещества или жидкой среды) и мягких и жестких желатиновых капсул. Примеры подходящих носителей, эксципиентов и разбавителей включают в себя лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, аравийскую камедь, фосфат кальция, альгинаты, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, трагакант, желатин, сироп,метилцеллюлозу, метил- и пропилгидроксибензоаты, тальк, стеарат магния, воду и минеральное масло. Эти композиции могут также включать в себя увлажняющие агенты, эмульгирующие и суспендирующие агенты или улучшающие вкус и запах агенты. Композиции данного изобретения могут быть приготовлены таким образом, чтобы обеспечивать быстрое, пролонгированное или задержанное высвобождение активного ингредиента после введения пациенту с использованием процедур, хорошо известных в данной области. Для перорального введения активный ингредиент, соединение данного изобретения, может быть смешан с носителями и разбавителями и сформован в таблетки или заключен в желатиновые капсулы. Эти композиции предпочтительно готовят в виде стандартной дозированной формы, причем каждая доза содержит от приблизительно 1 до приблизительно 500 мг, более часто приблизительно 5-300 мг активного ингредиента. Термин "стандартная дозированная форма" обозначает физически дискретные единицы, подходящие в качестве стандартных доз для субъектов-людей и других млекопитающих, причем каждая единица содержит заранее определенное количество активного материала, рассчитанное для получения терапевтического эффекта, вместе с подходящим фармацевтическим носителем. Для более полной иллюстрации этой операции данного изобретения обеспечены следующие примеры композиций. Эти примеры являются лишь иллюстративными и не предназначены для ограничения объема данного изобретения. Эти композиции могут использовать в качестве активного ингредиента любое из соединений данного изобретения. Композиция 1. Жесткие желатиновые капсулы готовят с использованием следующих ингредиентов. Указанные выше ингредиенты смешивают и наполняют ими жесткие желатиновые капсулы в количествах 460 мг. Композиция 2. Капсулы, каждая из которых содержит 20 мг лекарственного средства, готовят следующим образом. Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают, пропускают через сито 45 меш США и наполняют этой смесью жесткую желатиновую капсулу. Композиция 3. Капсулы, каждая из которых содержит 100 мг активного ингредиента, готовят следующим образом. Указанные выше ингредиенты тщательно смешивают и помещают в пустую желатиновую капсулу. Композиция 4. Таблетки, каждая из которых содержит 10 мг активного ингредиента, готовят следующим образом. Активный ингредиент, крахмал и целлюлозу пропускают через сито 45 меш США и тщательно смешивают. Раствор поливинилпирролидона смешивают с полученным порошком и затем смесь пропускают через сито 14 меш США. Полученные таким образом гранулы сушат при 50-60 С и пропускают через сито 18 меш США. Затем натрий-карбоксиметилкрахмал, стеарат магния и тальк, предварительно пропущенные через сито 60 меш США, добавляют к этим гранулам, которые после смешивания прессуют на таблетировочной машине с получением таблетки с массой 100 мг. Композиция 5. Смесь для таблеток может быть получена с использованием указанных ниже ингредиентов. Эти компоненты смешивают и прессуют с получением таблеток с массой 665 мг каждая. Композиция 6. Суспензии, каждая из которых содержит 5 мг лекарственного средства на дозу 5 мл, готовят следующим образом. Лекарственное средство пропускают через сито 45 меш США и смешивают с натрийкарбоксиметилцеллюлозой и сиропом для образования гладкой пасты. К этой пасте добавляют при перемешивании раствор бензойной кислоты, ароматизатор и краситель, разбавленные некоторым количеством воды. Затем добавляют достаточное количество воды для получения требуемого объема. Коипозиция 7. Готовят раствор для аэрозоля, содержащий следующие компоненты. Активное соединение смешивают с этанолом и эту смесь добавляют к части пропеллента 22, охлажденной до -30 С, и переносят в устройство для заполнения. Затем требуемое количество подают в контейнер из нержавеющей стали и разбавляют дополнительно остальным количеством пропеллента. Затем присоединяют к этому контейнеру клапанные элементы. Пример 1. 6-[4-(2-Бензиламиноэтил)фенокси]никотинамид.- 20008861 Бензальдегид (7,5 мл, 74 ммоль) добавляли к перемешиваемому раствору тирамина (10,00 г, 73 ммоль) и безводного метанола (90 мл). Реакционную смесь нагревали с обратным холодильником в течение 1 ч в атмосфере азота. Реакционную смесь охлаждали до 0 С и медленно добавляли боргидрид натрия (2,84 г, 75 ммоль). Перемешивали в течение 1 ч при комнатной температуре и затем концентрировали на роторном испарителе. Добавляли воду (100 мл) и перемешивали в течение 1,5 ч при комнатной температуре. Фильтровали и промывали водой с получением 10,11 г (61%) 4-(2-бензиламиноэтил) фенола: масс-спектр (распыление ионов): rn/z = 228,1 (М+1); 1(7,03 г, 44,90 ммоль). С использованием ловушки Дина-Старка реакционную смесь нагревали с обратным холодильником в атмосфере азота в течение 6 ч. Реакционные смеси охлаждали до комнатной температуры, отфильтровывали от твердых веществ и концентрировали большую часть растворителя на роторном испарителе. Остаток помещали в этилацетат (200 мл) и добавляли 1 н. хлористо-водородную кислоту(200 мл). Смесь перемешивали в течение 15 мин и отфильтровывали осадок с промыванием этилацетатом. Твердое вещество растворяли в 400 мл кипящей смеси метанол/вода 1:1. Добавляли 5 н. гидроксид натрия (35 мл) и давали этому раствору остыть до комнатной температуры. Раствор фильтровали и промывали водой с получением 19,74 г (83%) 6-[4-(2-бензиламиноэтил)фенокси]никотинамида: масс-спектр Бикарбонат натрия (0,0823 г, 0,0980 ммоль) добавляли к перемешиваемому раствору 6-[4-(2 бензиламиноэтил)фенокси]никотинамида (0,3061 г, 0,0881 ммоль), (2-бромэтил)бензола (0,135 мл, 0,988 ммоль) и ДМФА (5 мл). Реакционную смесь нагревали с обратным холодильником в течение 3 ч в атмосфере азота и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду (50 мл) и экстрагировали диэтиловым эфиром (350 мл). Экстракты в диэтиловом эфире сушили над безводным сульфатом магния. Концентрировали на роторном испарителе и очищали этот неочищенный продукт флэш-хроматографией на силикагеле с элюцией смесью 90% этилацетат/гексаны с получением 0,1538 г(39%) 6-4-[2-(бензилфенетиламино)этил]феноксиникотинамида: масс-спектр (распыление ионов): m/z = 452,1 (М+1); 1 Н ЯМР (CDCl3): 8,55 (д, 1 Н), 8,13 (дд, 1 Н), 7,29-7,11 (м, 14 Н), 7,01 (д, 2 Н), 6,92 (д, 1 Н), 3,71 (с, 1 Н),2,94-2,77 (м, 9 Н). По способу примера 1 получали следующие соединения: По способу примера 2 получали следующие соединения: Соединение примера 14 получают по способу примера 2. Примеры 15 А-15 Е. Стадия 1. 1-(2-Бромэтил)-3-хлорбензол. Трифенилфосфин (3,90 г, 14,9 ммоль) добавляли к перемешиваемому раствору 3-хлорфенетилового спирта (2,0 мл, 14,8 ммоль), тетрабромида углерода (4,91 г, 14,8 ммоль) и безводного дихлорметана (100 мл). Перемешивали в течение 5 ч в атмосфере азота при комнатной температуре и затем промывали водой (100 мл) и солевым раствором (100 мл). Дихлорметановый слой сушили над сульфатом магния,фильтровали и концентрировали на роторном испарителе с получением неочищенного продукта. Этот неочищенный продукт очищали флэш-хроматографией на силикагеле с элюцией 100% гексанами с получением 2,30 г (71%) 1-(2-бромэтил)-3-хлорбензола: ТСХ: Rf в 100% гексанах: 0,27; 1 Н ЯМР (CDCl3): 7,26-7,11 (м, 3 Н), 7,09-7,07 (м, 1 Н), 3,54 (т, 2 Н), 3,12 (т, 2 Н). Стадия 2. К перемешиваемому раствору 6-[4-(2-бензиламиноэтил)фенокси]никотинамида (0,3058 г, 0,8802 ммоль), бензальдегида (0,092 мл, 0,905 ммоль), ледяной уксусной кислоты (0,052 мл, 0,908 ммоль) и 1,2 дихлорэтана (8 мл) добавляли триацетоксиборгидрид натрия (0,2600 г, 1,227 ммоль). Перемешивали в течение 18 ч при комнатной температуре в атмосфере азота. Реакционную смесь выливали в 1 н. гидроксид натрия (50 мл) и экстрагировали диэтиловым эфиром (350 мл). Экстракты в диэтиловом эфире- 24008861 промывали солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали на роторном испарителе с получением неочищенного продукта. Этот неочищенный продукт очищали флэшхроматографией на силикагеле с элюцией смесью 75% этилацетат/гексаны с получением 0,2501 г (65%) 6-[4-(2-дибензиламиноэтил)фенокси]никотинамида: ВЭЖХ (30/70-90/10 ACN/(0,1% ТФУ в воде), колонка Zorbax SB-Phenyl 4,6 мм 15 см 5 мкм. Время удерживания: 8,14 мин, чистота: 99,7%; масс-спектр(распыление ионов): m/z = 438,0 (М+1). Следующие соединения (примеры 15 А-15 Е) получали из соответствующих коммерчески доступных спиртов, за исключением примеров 1-(3-бромпропил)-2-метилбензола и 2-(3-бромпропил)тиофена, в случае которых исходные спирты синтезировали. Получение спиртового исходного материала для примера 15D 3-о-толилпропан-1-ол. К безводному тетрагидрофурану (100 мл) добавляли 2-метилгидрокоричную кислоту (18,4 ммоль) и охлаждали до 0 С. Медленно добавляли литийалюминийгидрид (2,20 г, 58,0 ммоль) и удаляли баню со льдом спустя 20 мин. Перемешивали при комнатной температуре в атмосфере азота в течение 18 ч. Охлаждали реакционную смесь до 0 С и реакцию гасили медленным добавлением воды (2,2 мл), 15% гидроксида натрия (2,2 мл) и воды (6,6 мл). Соли алюминия отфильтровывали. К фильтрату добавляли солевой раствор (100 мл) и 5 н. гидроксид натрия (30 мл) и экстрагировали этилацетатом (3100 мл). Этилацетатные экстракты сушили над сульфатом магния, фильтровали и концентрировали на роторном испарителе с получением 2,65 г (96%) 3-о-толилпропан-1-ола: 1 Н ЯМР (CDCl3): 7,18-7,10 (м, 4 Н), 3,72 (т, 2 Н) , 2,72-2,69 (м, 2 Н), 2,33 (с, 3H), 1,90-1,83 (м, 2 Н), 1,60(ушир.с, 1 Н). Получение спиртового исходного материала для примера 15 Е 3-тиофен-2-илпропан-1-ол. При применении способа, сходного с примером 15D, использование 3-(2-тиенил)пропановой кисло- 25008861 ты дает указанное в заголовке соединение: 1 Н ЯМР (CDCl3): 7,12 (дд, 1 Н), 6,92 (дд, 1 Н), 6,82-6,80 (м, 1 Н), 3,70 (т, 2 Н), 2,96-2,92 (м, 2 Н), 1,981,91 (м, 2 Н), 1,67 (ушир.с, 1 Н). Пример 16. 6-(4-2-[Бензил-(3-оксо-3-фенилпропил)амино]этилфенокси)никотинамид. К перемешиваемому раствору ацетофенона (5,0 мл, 43 ммоль), параформальдегида (2,15 г), гидрохлорида диметиламина (4,54 г, 56 ммоль) и этанола (15 мл) добавляли концентрированную хлористоводородную кислоту (0,090 мл, 1,1 ммоль). Эту реакционную смесь нагревали с обратным холодильником в течение 18 ч в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, выливали в 1 н. гидроксид натрия (150 мл) и экстрагировали диэтиловым эфиром (350 мл). Экстракты в диэтиловым эфире сушили над сульфатом магния, фильтровали и концентрировали на роторном испарителе с получением неочищенного продукта. Этот неочищенный продукт растворяли в этаноле (70 мл) и добавляли иодметан (3,2 мл, 51 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч в атмосфере азота. Фильтровали и промывали этанолом и затем диэтиловым эфиром с получением 12,56 г (92%) иодида 3-триметиламмоний-1-фенилпропан-1-она: масс-спектр (распыление ионов): m/z = 193,0 (М+1); 1 Н ЯМР (ДMCO-d6): 8,08-8,06 (м, 2 Н), 7,72-7,67 (м, 1 Н), 7,60-7,55 (м, 2 Н), 3,70 (с, 4 Н), 3,14 (с, 6 Н),3,11 (с, 3H). Стадия 2. К перемешиваемому раствору 6-[4-(2-бензиламиноэтил)фенокси]никотинамида (0,3041 г, 0,8753 ммоль), карбоната натрия (0,1862 г, 1,757 ммоль) и диметилформамида (5 мл) добавляли иодид 3 триметиламмоний-1-фенилпропан-1-она (0,3612 г, 1,132 ммоль). Через реакционную смесь барботировали азот в течение 18 ч при комнатной температуре. Реакционную смесь выливали в 1 н. гидроксид натрия(50 мл) и экстрагировали диэтиловым эфиром (350 мл). Экстракты в диэтиловым эфире сушили над сульфатом магния, фильтровали и концентрировали на роторном испарителе с получением неочищенного продукта. Этот неочищенный продукт очищали флэш-хроматографией на силикагеле с элюцией смесью 90% этилацетат/гексаны с получением 0,1910 г (46%) 6-(4-2-[бензил-(3-оксо-3 фенилпропил)амино]этилфенокси)никотинамида: масс-спектр (распыление ионов): m/z = 480,1 (М+1); 1 Н ЯМР (СDCl3): 8,57 (д, 1 Н), 8,15 (дд, 1 Н), 7,90-7,88 (м, 2 Н), 7,57-7,53 (м, 1 Н), 7,46-7,42 (м, 2 Н),7,28-7,15 (м, 9 Н), 7,04-7,00 (м, 2 Н), 6,93 (д, 1 Н), 3,71 (с, 2 Н), 3,13-3,01 (м, 4 Н), 2,78 (с, 4 Н). Пример 17. 6-(4-2-[Бензил-(3-оксо-3-тиофен-2-илпропил)амино]этилфенокси)никотинамид. При применении способа, сходного с примером 16, использование циклогексилметилкетона дает указанное в заголовке соединение: масс-спектр (распыление ионов): m/z = 198,2 (М+1); 1 Н ЯМР (ДМСО-d6): 3,51-3,47 (м, 4 Н), 3,11 (с, 6 Н), 3,05 (с, 3H), 2,49-2,42 (м, 1 Н), 1,87-1,84 (м, 2 Н),1,73-1,60 (м, 3H), 1,31-1,12 (м, 5 Н). Стадия 2. При применении способа, сходного с примером 16, использование иодида 1-циклогексил-3 триметиламмонийпропан-1-она дает указанное в заголовке соединение: масс-спектр (распыление ионов): К 6-(4-2-[бензил-(3-оксо-3-фенилпропил)амино]этилфенокси)никотинамиду (0,1871 г, 0,3901 ммоль) добавляли метанол (10 мл) и охлаждали до 0 С. Добавляли боргидрид натрия (0,0664 г, 1,756 ммоль) и перемешивали в течение 1,5 ч при 0 С в атмосфере азота. Реакционную смесь выливали в солевой раствор (50 мл) и экстрагировали дизтиловым эфиром (350 мл). Экстракты в диэтиловым эфире сушили над сульфатом магния, фильтровали и концентрировали на роторном испарителе с получением неочищенного продукта. Этот неочищенный продукт очищали флэш-хроматографией на силикагеле с элюцией 100% этилацетатом с получением 0,0239 г (13%) 6-(4-2-[бензил-(3-гидрокси-3 фенилпропил)амино]этилфенокси)никотинамида: ВЭЖХ (30/70-90/10 ACN/(0,1% ТФУ в воде), колонка(0,1211 г, 0,2603 ммоль) (пример 3) и 1,2-дихлорэтана (5 мл) добавляли 1-хлорэтилхлорформиат (0,056 мл, 0,52 ммоль). Реакционную смесь нагревали с обратным холодильником в атмосфере азота в течение 1,5 ч. Добавляли метанол (7 мл) и нагревали с обратным холодильником в атмосфере азота в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли 2 М аммиак в метаноле (5 мл). Концентрировали на роторном испарителе с получением неочищенного продукта. Этот неочищенный продукт очищали флэш-хроматографией на силикагеле с элюцией смесью 1% концентрированный гидроксид аммония/10% этанол/хлороформ с получением 0,0654 г (67%) 6-4-[2-(3-фенилпропиламино)этил]феноксиникотинамида: ВЭЖХ (30/70-90/10 ACN/(0,1% ТФУ в воде), колонка ZorbaxSB-Phenyl 4,6 мм 15 см 5 мкм. Время удерживания: 7,48 мин, чистота: 99,2%; масс-спектр (распыление ионов): m/z = 376,2 (М+1). По способу примера 22 получали следующие соединения из соответствующих соединений, полученных в примерах 2-14: К перемешиваемому раствору тирамина (5,00 г, 36,5 ммоль) и безводного тетрагидрофурана добавляли ди-трет-бутилдикарбонат (9,75 г, 44,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч в атмосфере азота. Реакционную смесь концентрировали с получением неочищенного продукта. Этот неочищенный продукт очищали флэш-хроматографией на силикагеле с элюцией смесью 35% этилацетат/гексаны с получением 7,56 г (87%) трет-бутилового эфира [2-(4 гидроксифенил)этил]карбаминовой кислоты: масс-спектр (распыление ионов): m/z = 236,1 (М-l); 1 Н ЯМР (CDCl3): 7,01 (д, 2 Н), 6,77 (д, 2 Н), 6,10 (ушир.с, 1 Н), 4,61 (ушир.с, 1 Н), 3,34-3,32 (м, 2 Н),2,72-2,68 (м, 2 Н), 1,44 (с, 9 Н). Стадия 2. Трет-бутиловый эфир 2-[4-(5-карбамоилпиридин-2-илокси)фенил]этилкарбаминовой кислоты. К перемешиваемому раствору трет-бутилового эфира [2-(4-гидроксифенил)этил]карбаминовой кислоты (6,40 г, 27,0 ммоль) и безводного тетрагидрофурана (120 мл) добавляли трет-бутоксид (4,28 г, 36,2 ммоль). Перемешивали в течение 30 мин в атмосфере азота при комнатной температуре. Добавляли 6 хлорникотинамид (4,27 г, 27,2 ммоль) и нагревали с обратным холодильником в течение 18 ч в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, выливали в солевой раствор (150 мл) и экстрагировали диэтиловым эфиром (350 мл). Экстракты в диэтиловым эфире сушили над суль- 29008861 фатом магния, фильтровали и концентрировали на роторном испарителе с получением неочищенного продукта. Этот неочищенный продукт очищали флэш-хроматографией на силикагеле с элюцией смесью 0,7% концентрированный гидроксид аммония/7% этанол/хлороформ с получением 4,46 г (46%) третбутилового эфира 2-[4-(5-карбамоилпиридин-2-илокси)фенил]этилкарбаминовой кислоты: масс-спектр(распыление ионов): m/z = 358,1 (М+1); 1 Н ЯМР (ДМСО-d6): 8,58 (д, 1 Н), 8,22 (дд, 1 Н), 8,02 (ушир.с, 1 Н), 7,46 (ушир.с, 1 Н), 7,23 (д, 2 Н),7,06-7,02 (м, 3H), 6,92-6,89 (м, 1 Н), 3,17-3,12 (м, 2 Н), 2,69 (т, 2 Н), 1,35 (с, 9 Н). Стадия 3. К соединению стадии 2 примера 33 (5,12 г, 14,3 ммоль) добавляли дихлорметан (60 мл). К этой суспензии добавляли трифторуксусную кислоту (32,0 мл, 415 ммоль) и перемешивали в атмосфере азота в течение 1,5 ч. Реакционную смесь делили на три равные аликвоты и наносили каждую аликвоту на предварительно упакованный 10 г SCX картридж (небольшую колонку). Промывали метанолом (200 мл) и элюировали продукт из картриджа 2 М аммиаком в метаноле (100 мл). Объединяли промывки 2 М аммиаком в метаноле из трех картриджей и концентрировали на роторном испарителе с получением 3,11 г (84%) 6-[4-(2-аминоэтил)фенокси]никотинамида: масс-спектр (распыление ионов): m/z = 258,1 (М+1); 1 Н ЯМР (ДМСО-d6): 8,61 (д, 1 Н), 8,25 (дд, 1 Н), 8,04 (с, 1 Н), 7,49 (с, 1 Н), 7,30-7,23 (м, 2 Н), 7,11-7,03(соединения примера 33), 2-метоксибензальдегида (0,047 мл, 0,39 ммоль) и метанола (5 мл) добавляли молекулярные сита 3 . Реакционную смесь перемешивали в течение 18 ч на качалке с платформой при комнатной температуре. Добавляли боргидрид натрия и перемешивали в течение 1 ч при комнатной температуре. Реакционную смесь фильтровали для удаления молекулярных сит и наносили непосредственно на предварительно упакованный 10 г SCX картридж (небольшую колонку). Промывали метанолом (150 мл) и элюировали продукт из SCX-картриджа 2 М аммиаком в метаноле (50 мл). Концентрировали на роторном испарителе с получением 0,1253 г (85%) 6-4-[2-(2-метоксибензиламино)этил]фенокси никотинамида: ВЭЖХ (30/70-90/10 ACN/(0,1% ТФУ в воде), колонка Zorbax SB-Phenyl 4,6 мм 15 см 5 мкм. Время удерживания: 4,14 мин, чистота: 97,9%; масс-спектр (распыление ионов): m/z = 378,1 (М+1). По способу примера 34 получали следующие соединения:
МПК / Метки
МПК: A61P 3/04, C07D 213/82, A61K 31/4427, C07D 333/20, C07D 401/06, C07D 401/12, C07D 241/24, C07C 43/20, A61K 31/4412
Метки: качестве, рецептора, опиоидного, антагониста, диариловые, эфиры
Код ссылки
<a href="https://eas.patents.su/30-8861-diarilovye-efiry-v-kachestve-antagonista-opioidnogo-receptora.html" rel="bookmark" title="База патентов Евразийского Союза">Диариловые эфиры в качестве антагониста опиоидного рецептора</a>
Производные 3-азабицикло (3.1.0) гексана в качестве антагонистов опиоидного рецептора

Номер патента: 6708
Опубликовано: 24.02.2006
Авторы: Лирас Спирос, Макхарди Стэнтон Ферст, Хек Стивен Дональд
МПК: A61K 31/4178, A61K 31/403, C07D 209/52...
Метки: производные, рецептора, антагонистов, 3.1.0, качестве, 3-азабицикло, опиоидного, гексана
Формула / Реферат:
1. Соединение, описываемое формулой I, где X представляет собой H, галоген, -OH, -CN, -C1-C4алкил, замещенный атомами галогена в количестве от одного до трех, либо -O(C1-C4алкил), где C1-C4алкил в -O(C1-C4алкиле) необязательно замещен атомами галогена в количестве от одного до трех; Q представляет собой галоген, -OH, -O(C1-C4алкил), -NH2, -N(C1-C4алкил)(C1-C4алкил), -C(=O)NH2, -C(=O)NH(C1-C4алкил), C(=O)N(C1-C4алкил) (C1-C4алкил) или...
Сложные эфиры (+)-альфа-(2,3-диметоксифенил)-1-[2-(4-фторфенил)этил]-4-пиперидинметанола и их использование в качестве пролекарств антагониста mdl 110907 5ht2a рецепторов

Номер патента: 3667
Опубликовано: 28.08.2003
Авторы: Косли Рэймонд В.Мл., Карр Альберт А., Ван Хейфте Люк Е.
МПК: C07D 211/22, A61K 31/445, A61P 25/18...
Метки: +)-альфа-(2,3-диметоксифенил)-1-[2-(4-фторфенил)этил]-4-пиперидинметанола, эфиры, пролекарств, использование, качестве, 5ht2a, антагониста, сложные, 110907, рецепторов
Формула / Реферат:
1. Соединение формулы I Формула I где R представляет собой C4-C20алкил, или его стереоизомер, или его фармацевтически приемлемая соль. 2. Соединение по п.1, в котором R представляет собой C5-C20алкил с неразветвленной цепью. 3. Соединение по п.1, выбранное из группы, включающей (+)-a-(2,3-диметоксифенил)-1-[2-(4-фторфенил)этил]-4-пиперидинметанолдеканоат; (+)-a-(2,3-диметоксифенил)-1-[2-(4-фторфенил)этил]-4-пиперидинметанолгексаноат;...
Полиморфная форма 2-(r) – (1- (r) – (3,5-бис (трифторметил) фенил) этокси) -3- (s) – (4-фтор) фенил – 4 – (3 – (5-оксо-1h, 4h-1, 2, 4 – триазоло) метил)морфолина в качестве антагониста рецептора тахикинина

Номер патента: 2405
Опубликовано: 25.04.2002
Авторы: Крокер Луис, Макколи Джеймс
МПК: C07D 265/32, A61K 31/5375, A61P 25/28...
Метки: 4-фтор, 4h-1, 5-оксо-1h, 2-(r, рецептора, форма, 3,5-бис, этокси, полиморфная, метил)морфолина, фенил, антагониста, качестве, тахикинина, трифторметил, триазоло
Формула / Реферат:
1. Полиморфная форма соединения 2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фтор)фенил-4-(3-(5-оксо-1Н,4Н-1,2,4-триазоло)метил)морфолина, обозначенная как форма I, по существу, отличающаяся рентгеновской порошковой дифрактограммой с характерными отражениями приблизительно 12,0, 15,3, 16,6, 17,0, 17,6, 19,4, 20,0, 21,9, 23,6, 23,8 и 24,8ш (2 тэта). 2. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и...
Комбинация ингибитора pde и антагониста лейкотриенового рецептора

Номер патента: 7736
Опубликовано: 29.12.2006
Авторы: Воллин Штефан-Лутц, Бойме Рольф, Ваймар Кристиан, Бундшу Даниела
МПК: A61K 31/44, A61K 31/47, A61K 45/06...
Метки: комбинация, лейкотриенового, рецептора, антагониста, ингибитора
Формула / Реферат:
1. Фармацевтическая композиция, включающая первое действующее вещество, представляющее собой ингибитор PDE4, выбранный из 3-циклопропилметокси-4-дифторметокси-N-(3,5-дихлорпирид-4-ил)бензамида [МНН рофлумиласт] и его фармацевтически приемлемых производных, в смеси со вторым действующим веществом, представляющим собой антагонист лейкотриенового рецептора, выбранный из (R)-N-[3-[5-(4-фторбензил)тиен-2-ил]-1-метил-2-пропинил]-N-гидроксимочевины...
Синергетические комбинации антагониста рецептора nk1 и структурного аналога гамк

Номер патента: 4266
Опубликовано: 26.02.2004
Авторы: Хьюз Джон, Сингх Лакхбир
МПК: A61K 31/195, A61K 31/40, A61P 25/02...
Метки: комбинации, антагониста, структурного, рецептора, аналога, синергетические, гамк
Формула / Реферат:
1. Способ предупреждения или лечения хронической боли, при котором пациенту, нуждающемуся в лечении, вводят эффективное количество синергетической комбинации антагониста рецептора NK1 и аналога ГАМК. 2. Способ по п.1, где соотношение аналога ГАМК и антагониста рецептора NK1, выраженное в массовых частях, составляет от 50:1 до 1:1. 3. Способ по п.1, где соотношение аналога ГАМК и антагониста рецептора NK1, выраженное в массовых частях, составляет...
Предыдущий патент: Способ получения катализатора полимеризации олефинов
Следующий патент: Оральная дозированная форма пропиверина
Случайный патент: Игровая система