Производные пиридина, пригодные для ингибирования системы обмена натрия/кальция
Номер патента: 8539
Опубликовано: 29.06.2007
Авторы: Карьялайнен Арто, Левийоки Йоуко, Коскелайнен Туула, Раску Сирпа, Отсомаа Леена, Поллеселло Пьеро




Формула / Реферат
1. Производные пиридина формулы (I)

где
X представляет -О-;
Z представляет -CHR12-;
Y представляет -СН2-, -С(О)-, CH(OR13)-;
пунктирная линия представляет необязательную двойную связь, в этом случае Z является -CR12-, a Y представляет -С(О)-;
R2 и R3 независимо представляют Н, галоген, -ОН, бензилокси или группу формулы (IIIа)

R1 представляет Н, CN, галоген, -CONH2, -COOR15, -CH2NR15R18, NHC(O)R5, NHCH2R5, NHR20, NR21R22, -NHC(NH)NHCH3, или в случае, когда существует необязательная двойная связь, или в случае, когда R2 или R3 является бензилокси или группой формулы (IIIа), или в случае, когда пиридиновое кольцо связано с атомом кислорода в 3-, 4- или 5-положении, R1 может также представлять собой -NO2 или NR16R17;
R4 представляет Н, -NO2, CN, галоген, -CONH2, -NR16R17;
R5 представляет С1-7алкил, замещенный 1-2 заместителями, выбранными из группы, состоящей из галогена, амино и гидрокси, или карбоксиС1-7алкил, -CHR6NR7R8 или одну из следующих групп:

W представляет N;
Q представляет CHR14, NR9, S или О;
R6 представляет Н;
R7 и R8 независимо представляют Н, C1-7ацил, C1-7алкил или гидрокси С1-7алкил;
R9 представляет Н, C1-7 алкил или фенил;
R10 и R11 независимо представляют Н или C1-7алкил;
R12 представляет Н;
R13 представляет Н;
R14 представляет Н, -ОН, -COOR15;
R15 представляет Н или C1-7алкил;
R16 и R17 независимо представляют Н, C1-7ацил, C1-7алкилсульфонил или -C(S)NHR18;
R18 представляет Н или С1-7алкил;
R19 представляет Н или -ОН;
R20 представляет пиридинильную группу, необязательно замещенную группой -NO2;
R21 и R22 представляют C1-7алкил;
и их фармацевтически приемлемые соли и сложные эфиры.
2. Соединение по п.1, где R1 представляет -NHC(O)R5, X представляет О, Y представляет CH2 и Z представляет CHR12.
3. Соединение по п.2, где Z представляет СН2 и R5 представляет С1-7алкил, замещенный 1-2 заместителями, выбранными из группы, состоящей из галогена, амино и гидрокси, или карбокси С1-7алкил, HR6NR7R8 или одну из следующих групп:

4. Соединение по п.1, где R2 или R3 является бензилокси или группой формулы (IIIа)

5. Соединение по п.4, где R4 является NO2.
6. Соединение по п.4 или 5, где R1 является NO2.
Текст
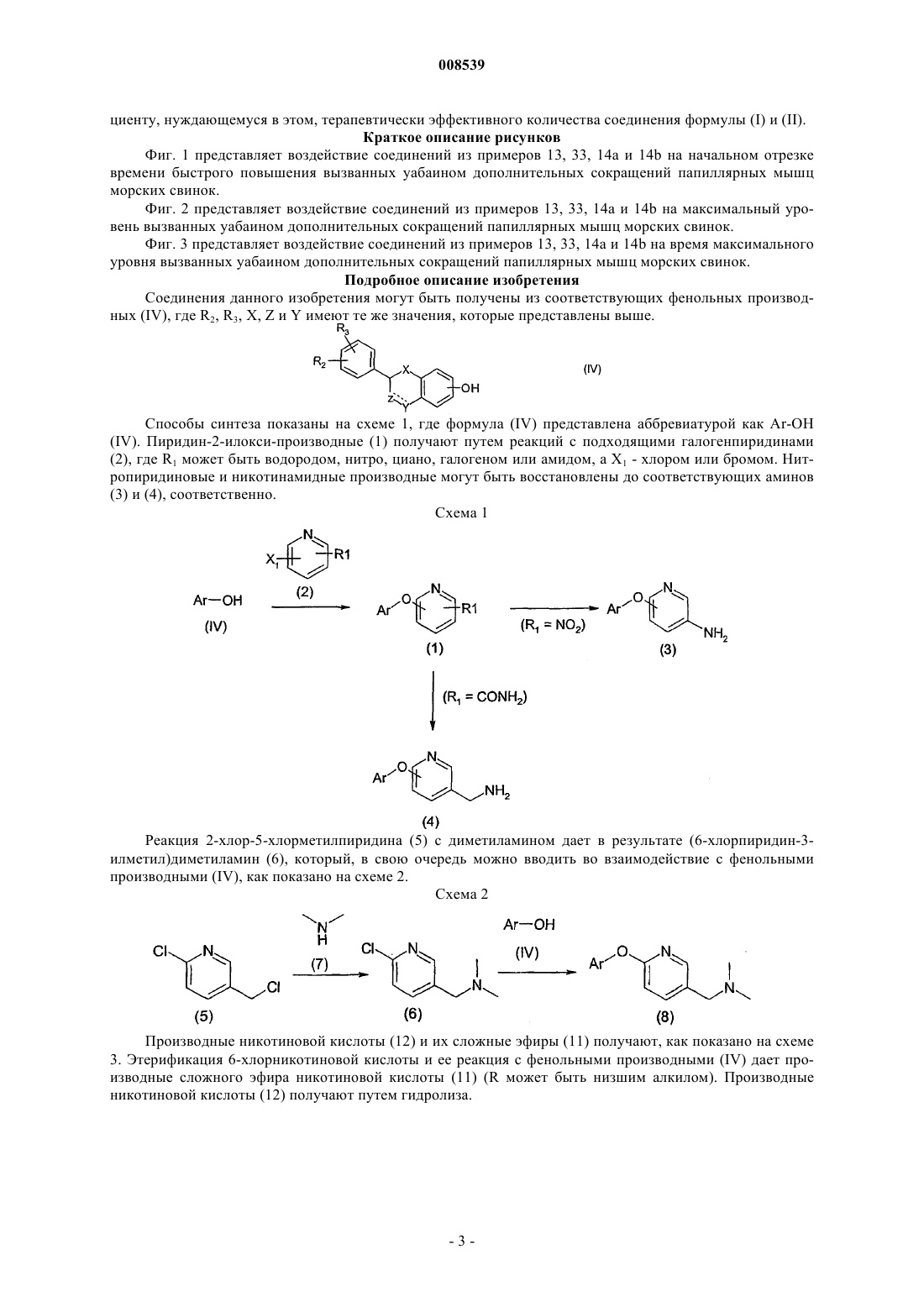
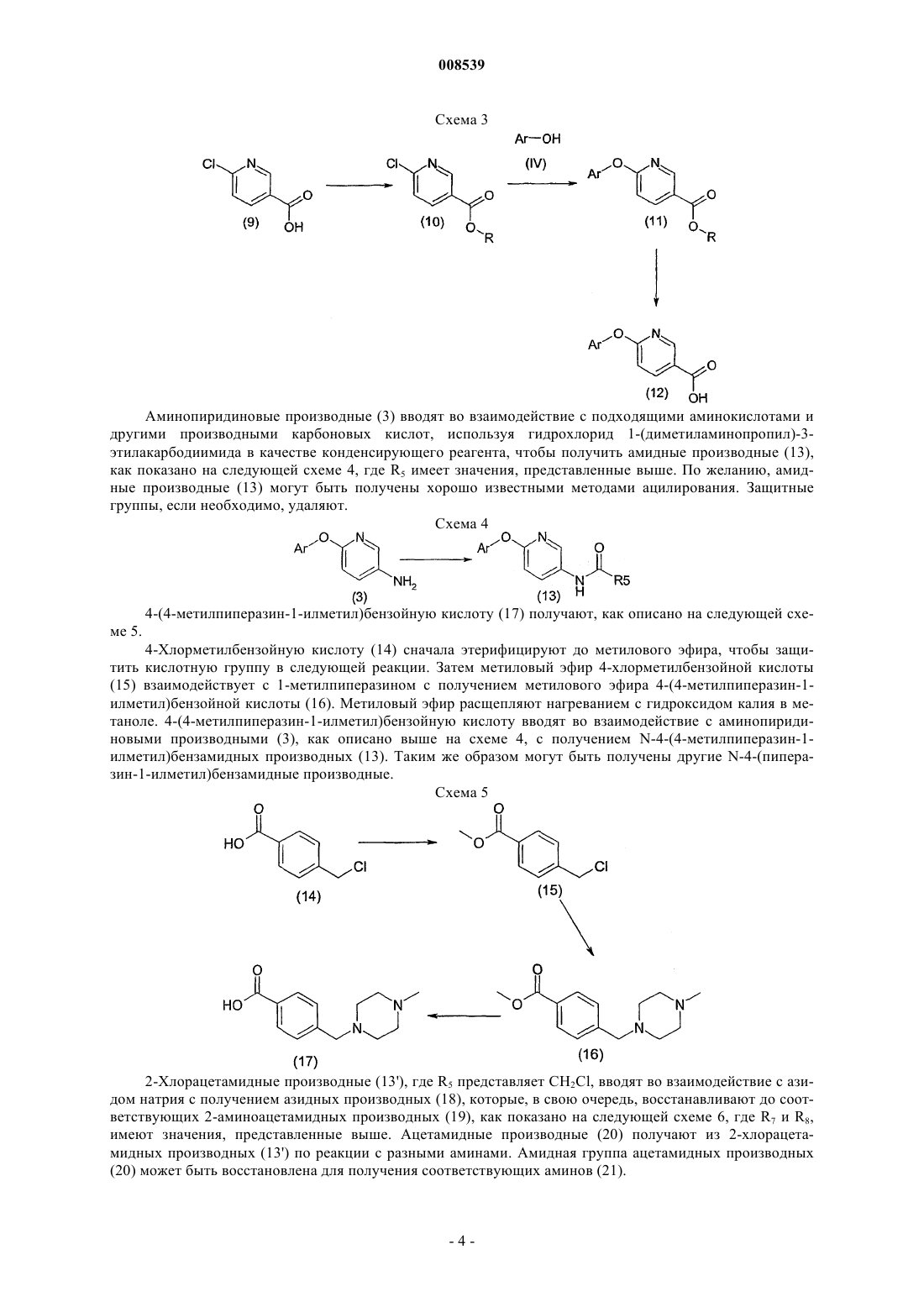
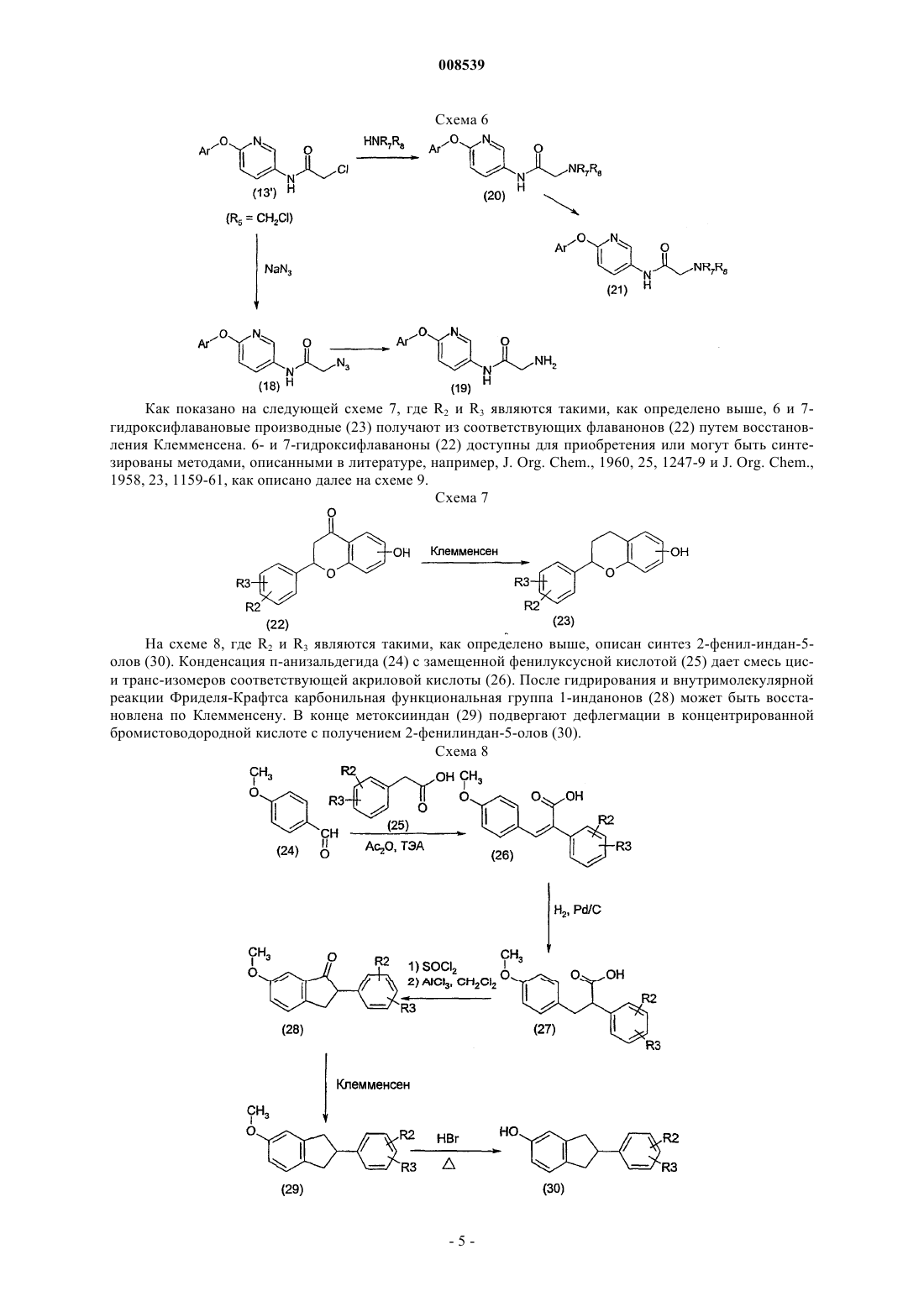
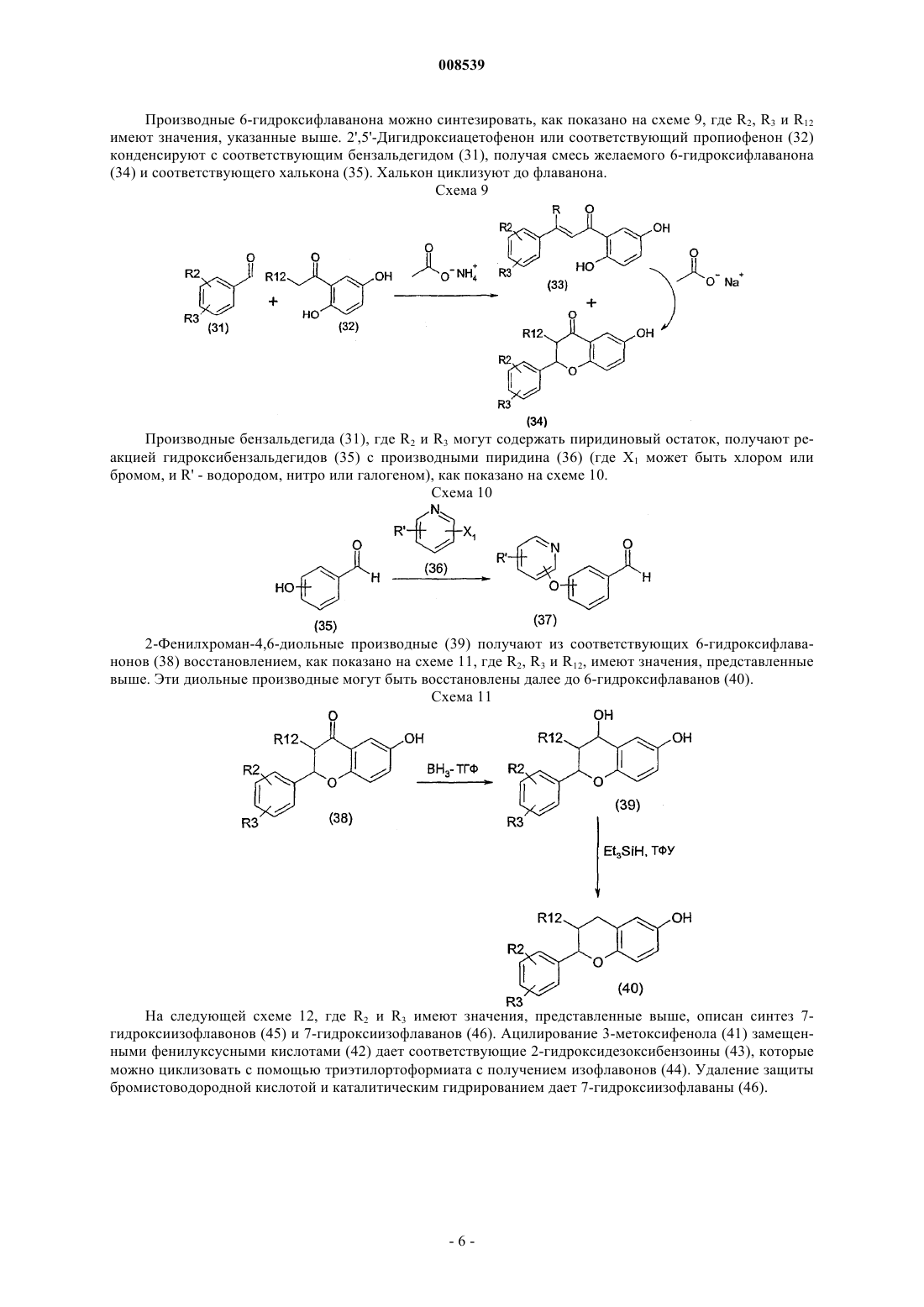
008539 Область техники Данное изобретение относится к новым терапевтически активным соединениям и их фармацевтически приемлемым солям и сложным эфирам. Данное изобретение относится также к фармацевтическим композициям, содержащим данные соединения в качестве активных ингредиентов. Соединения данного изобретения являются сильными ингибиторами механизма обмена Na+/Ca2+. Уровень техники Механизм обмена Na+/Ca2+ является одним из механизмов транспорта ионов, который регулирует концентрацию ионов натрия и кальция в клетках. Соединения, которые селективно ингибируют механизм обмена Na+/Ca2+ и, тем самым, предотвращают избыточное накопление Са 2+ в клетках, считаются полезными для предотвращения запуска механизма повреждения клетки сердечной мышцы и т.п. после ишемии и реперфузии. Такие соединения пригодны, например, для лечения ишемических заболеваний,таких как болезни сердца, ишемические церебральные заболевания, ишемические почечные болезни, и для защиты клеток во время тромболитической терапии, ангиопластики, операции шунтирования коронарной артерии или трансплантации органов и при аритмиях. Соединения, способные к ингибированию системы обмена Na+/Ca2+, были описаны ранее, например, в патентных публикациях WO 97/09306, ЕР 0978506, ЕР 1031556, JP 11049752 и JP 11302235. Сущность изобретения Обнаружено, что соединения формулы (I) или (II) являются особенно сильными ингибиторами механизма обмена Na+/Ca2+ и особенно подходят для лечения аритмий. Соединения данного изобретения имеют структуру, представленную формулой (I) или (II)Y представляет -СН 2-, -С(О)-, CH(OR13)-, -O-, -S-; при условии, что в случае, когда Z является валентной связью, Y не является С(О); пунктирная линия представляет необязательную двойную связь, в этом случае Z является -CR12-, aR2 и R3, независимо, представляют Н, низший алкил, низший алкокси, -NO2, галоген, -CF3, -ОН,бензилокси или группу формулы (IIIа)R1 представляет Н, CN, галоген, -CONH2, -COOR15, -CH2NR15R18, NHC(O)R5, NHCH2R5, NHR20,NR21R22, NHC(NH)NHCH3 или, в случае, когда соединение представлено формулой (II), где существует необязательная двойная связь, или в случае, когда R2 или R3 является бензилокси или группой формулы(IIIа), или в случае, когда пиридиновое кольцо в формуле (I) или (II) связано с атомом кислорода в 3-, 4 или 5-положении, R1 может также представлять собой -NO2 или NR16R17;R5 представляет алкил, замещенный 1-3 заместителями, выбранными из группы, состоящей из галогена, амино и гидрокси, или карбоксиалкил, в котором алкильная часть является необязательно замещенной 1-3 заместителями, выбранными из группы, состоящей из галогена, амино и гидроксила,-CHR6NR7R8 или одну из следующих групп:R7 и R8 независимо представляют Н, ацил, низший алкил или низший гидроксиалкил;R9 представляет низший алкил или фенил;R20 представляет пиридинильную группу, необязательно замещенную группой -NO22;R21 и R22 представляют низший алкил; и их фармацевтически приемлемые соли и сложные эфиры. К одной группе предпочтительных соединений и их фармацевтически приемлемых солей и сложных эфиров относятся соединения формулы (Ib) или (IIb), где значения R1, R2, R3, X, Y и Z представлены выше. К подгруппе предпочтительных соединений и их фармацевтически приемлемых солей и сложных эфиров относятся соединения формулы (Iс) или (IIс), где R1, R2, R3, X, Y и Z имеют значения, представленные выше. К другой группе предпочтительных соединений их фармацевтически приемлемых солей и сложных эфиров относятся соединения формулы (I) или (II), где R1 представляет -NHC(O)R5, X является О, Y является CH2 и Z является CHR12. К одной из подгрупп предпочтительных соединений и их фармацевтически приемлемых солей и сложных эфиров относятся соединения формулы (I) или (II), где R1 представляет -NHC(O)R5, X является O, Y является СН 2 и Z является СН 2 и R5 представляет алкил, замещенный 1-3 заместителями, выбранными из группы, состоящей из галогена, амино и гидрокси, или карбоксиалкил, в котором алкильная часть является необязательно замещенной 1-3 заместителями, выбранными из группы, состоящей из галогена, амино и гидроксила, -CHR6NR7R8 или одну из следующих групп: К другой группе предпочтительных соединений и их фармацевтически приемлемых солей и сложных эфиров относятся соединения, у которых R2 или R3 является бензилокси или группой формулы (IIIа) К одной из подгрупп предпочтительных соединений их фармацевтически приемлемых солей и сложных эфиров относятся соединения, где R4 и R1 являются NO2. Данное изобретение представляет также фармацевтическую композицию, содержащую соединение формулы (I) или (II) вместе с фармацевтически приемлемым носителем. Данное изобретение, кроме того, представляет способ ингибирования механизма обмена Na+/Ca2+ в клетке, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) и (II). Данное изобретение дополнительно включает способ предотвращения избыточного накопления ионов Са 2+ в клетках, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или (II). Данное изобретение, кроме того, представляет способ лечения аритмий, включающий введение па-2 008539 циенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) и (II). Краткое описание рисунков Фиг. 1 представляет воздействие соединений из примеров 13, 33, 14 а и 14b на начальном отрезке времени быстрого повышения вызванных уабаином дополнительных сокращений папиллярных мышц морских свинок. Фиг. 2 представляет воздействие соединений из примеров 13, 33, 14 а и 14b на максимальный уровень вызванных уабаином дополнительных сокращений папиллярных мышц морских свинок. Фиг. 3 представляет воздействие соединений из примеров 13, 33, 14 а и 14b на время максимального уровня вызванных уабаином дополнительных сокращений папиллярных мышц морских свинок. Подробное описание изобретения Соединения данного изобретения могут быть получены из соответствующих фенольных производных (IV), где R2, R3, X, Z и Y имеют те же значения, которые представлены выше. Способы синтеза показаны на схеме 1, где формула (IV) представлена аббревиатурой как Ar-ОН(IV). Пиридин-2-илокси-производные (1) получают путем реакций с подходящими галогенпиридинами(2), где R1 может быть водородом, нитро, циано, галогеном или амидом, а X1 - хлором или бромом. Нитропиридиновые и никотинамидные производные могут быть восстановлены до соответствующих аминов Реакция 2-хлор-5-хлорметилпиридина (5) с диметиламином дает в результате (6-хлорпиридин-3 илметил)диметиламин (6), который, в свою очередь можно вводить во взаимодействие с фенольными производными (IV), как показано на схеме 2. Схема 2 Производные никотиновой кислоты (12) и их сложные эфиры (11) получают, как показано на схеме 3. Этерификация 6-хлорникотиновой кислоты и ее реакция с фенольными производными (IV) дает производные сложного эфира никотиновой кислоты (11) (R может быть низшим алкилом). Производные никотиновой кислоты (12) получают путем гидролиза. Аминопиридиновые производные (3) вводят во взаимодействие с подходящими аминокислотами и другими производными карбоновых кислот, используя гидрохлорид 1-(диметиламинопропил)-3 этилакарбодиимида в качестве конденсирующего реагента, чтобы получить амидные производные (13),как показано на следующей схеме 4, где R5 имеет значения, представленные выше. По желанию, амидные производные (13) могут быть получены хорошо известными методами ацилирования. Защитные группы, если необходимо, удаляют. Схема 4 4-(4-метилпиперазин-1-илметил)бензойную кислоту (17) получают, как описано на следующей схеме 5. 4-Хлорметилбензойную кислоту (14) сначала этерифицируют до метилового эфира, чтобы защитить кислотную группу в следующей реакции. Затем метиловый эфир 4-хлорметилбензойной кислоты(15) взаимодействует с 1-метилпиперазином с получением метилового эфира 4-(4-метилпиперазин-1 илметил)бензойной кислоты (16). Метиловый эфир расщепляют нагреванием с гидроксидом калия в метаноле. 4-(4-метилпиперазин-1-илметил)бензойную кислоту вводят во взаимодействие с аминопиридиновыми производными (3), как описано выше на схеме 4, с получением N-4-(4-метилпиперазин-1 илметил)бензамидных производных (13). Таким же образом могут быть получены другие N-4-(пиперазин-1-илметил)бензамидные производные. Схема 5 2-Хлорацетамидные производные (13'), где R5 представляет CH2Cl, вводят во взаимодействие с азидом натрия с получением азидных производных (18), которые, в свою очередь, восстанавливают до соответствующих 2-аминоацетамидных производных (19), как показано на следующей схеме 6, где R7 и R8,имеют значения, представленные выше. Ацетамидные производные (20) получают из 2-хлорацетамидных производных (13') по реакции с разными аминами. Амидная группа ацетамидных производных(20) может быть восстановлена для получения соответствующих аминов (21). Как показано на следующей схеме 7, где R2 и R3 являются такими, как определено выше, 6 и 7 гидроксифлавановые производные (23) получают из соответствующих флаванонов (22) путем восстановления Клемменсена. 6- и 7-гидроксифлаваноны (22) доступны для приобретения или могут быть синтезированы методами, описанными в литературе, например, J. Org. Chem., 1960, 25, 1247-9 и J. Org. Chem.,1958, 23, 1159-61, как описано далее на схеме 9. Схема 7 На схеме 8, где R2 и R3 являются такими, как определено выше, описан синтез 2-фенил-индан-5 олов (30). Конденсация п-анизальдегида (24) с замещенной фенилуксусной кислотой (25) дает смесь циси транс-изомеров соответствующей акриловой кислоты (26). После гидрирования и внутримолекулярной реакции Фриделя-Крафтса карбонильная функциональная группа 1-инданонов (28) может быть восстановлена по Клемменсену. В конце метоксииндан (29) подвергают дефлегмации в концентрированной бромистоводородной кислоте с получением 2-фенилиндан-5-олов (30). Схема 8-5 008539 Производные 6-гидроксифлаванона можно синтезировать, как показано на схеме 9, где R2, R3 и R12 имеют значения, указанные выше. 2',5'-Дигидроксиацетофенон или соответствующий пропиофенон (32) конденсируют с соответствующим бензальдегидом (31), получая смесь желаемого 6-гидроксифлаванона(34) и соответствующего халькона (35). Халькон циклизуют до флаванона. Схема 9 Производные бензальдегида (31), где R2 и R3 могут содержать пиридиновый остаток, получают реакцией гидроксибензальдегидов (35) с производными пиридина (36) (где X1 может быть хлором или бромом, и R' - водородом, нитро или галогеном), как показано на схеме 10. Схема 10 2-Фенилхроман-4,6-диольные производные (39) получают из соответствующих 6-гидроксифлаванонов (38) восстановлением, как показано на схеме 11, где R2, R3 и R12, имеют значения, представленные выше. Эти диольные производные могут быть восстановлены далее до 6-гидроксифлаванов (40). Схема 11 На следующей схеме 12, где R2 и R3 имеют значения, представленные выше, описан синтез 7 гидроксиизофлавонов (45) и 7-гидроксиизофлаванов (46). Ацилирование 3-метоксифенола (41) замещенными фенилуксусными кислотами (42) дает соответствующие 2-гидроксидезоксибензоины (43), которые можно циклизовать с помощью триэтилортоформиата с получением изофлавонов (44). Удаление защиты бромистоводородной кислотой и каталитическим гидрированием дает 7-гидроксиизофлаваны (46). На следующей схеме 13 описан синтез 2-фенил-2,3-дигидробензо[1,4]оксатиин-6-ола (50). Реакция 2-меркаптобензол-1,4-диола (47) с эпоксидом стирола (48) в присутствии основания дает сульфид (49). Циклизация с помощью кислотной ионообменной смолы дает 2-фенил-2,3-дигидробензо[1,4]оксатиин-6 ол (50). Схема 13 На следующей ниже схеме 14 описан синтез 6-фенил-5,6,7,8-тетрагидронафталин-2-ола (55) и 6 гидрокси-2-фенил-3,4-дигидро-2 Н-нафталин-1-она (54). Катализируемое палладием -арилирование 6 метокси-1-тетралона (51) дает 6-метокси-2-фенил-3,4-дигидро-2 Н-нафталин-1-он (53), что, после деметилирования, приводит к фенольному соединению (54). Восстановление триэтилсиланом дает 6-фенил 5,6,7,8-тетрагидронафталин-2-ол (55). Схема 14-7 008539 На следующей схеме 15, где R2 и R3 имеют значения, представленные выше, и R является подходящей защитной группой, описан синтез 2,3-дигидро-2-фенилбензо[1,4]диоксин-6 олов (60). После того,как защитные группы 2,5-дигидроксиацетофенона удаляют, этот кетон подвергают перегруппировке с перкислотами и после гидролиза это дает фенол (56). Фенол (56) конденсируют с галогенкетоном, и после восстановления и удаления защитных групп гидроксифенол (59) циклизуют до 2,3-дигидро-2 фенилбензо[1,4]-диоксин-6-ола (60). Схема 15 Дигидроксифлавановые производные (61) можно вводить во взаимодействие с пиридиновыми производными таким же образом, как описано для соединения (1) на схеме 1. 4-Хроманольное производное(62), где R является ОН, может быть восстановлено до соответствующего флавана триэтилсиланом в кислой среде. 5-нитропиридиновые производные (62) восстанавливают до соответствующих 2 аминопроизводных (63), которые, в свою очередь, можно ацилировать, или мезилировать, или вводить во взаимодействие с разными производными аминокислот или карбоновых кислот, как описано на схеме 4 для соединения (13). Когда нитрогруппу в бензилоксипроизводных (65) восстанавливают гидрированием с использованием палладия в качестве катализатора, получают [6-(5-аминопиридин-2-илокси)хроман-2-ил]фенольные производные (68), которые, в свою очередь, могут быть ацилированы или мезилированы. Эти фенольные производные (69) могут быть затем введены во взаимодействие с пиридиновыми производными (2) с получением производных, подобных (70), как показано на следующей схеме 17. Восстановление цинком приводит к аминам, подобным (66), которые, в свою очередь, ацилируют, мезилируют или вводят во взаимодействие с разными производными аминокислот и карбоновых кислот, как описано на схеме 4 для соединения (13). Схема 17-9 008539 Алкильные производные аминопиридинов (3) могут быть получены восстановительным аминированием, как показано на схеме 18 для диметиламиновых производных (71). В ходе реакции аминогруппа частично мигрирует из 5 в 4 положение. Схема 18 На следующей схеме 19 описан синтез 1-метил-3-(пиридинил)тиомочевины (73) и производных Nметил-N'-(пиридинил)гуанидина (74). Аминопиридиновые производные (3) вводят во взаимодействие с метилизотиоцианатом с получением производных тиомочевины (73), которые затем обрабатывают сначала метилйодидом, а затем метанольным раствором аммиака, чтобы получить гуанидиновые производные (74). Схема 19 Соли и сложные эфиры данных соединений, когда это применимо, можно получить известными методами. В качестве активных лекарственных средств пригодны физиологически приемлемые соли. Примерами являются соли с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота или азотная кислота, и соли с органическими кислотами, такими как метансульфоновая кислота,лимонная кислота или винная кислота. Физиологически приемлемые сложные эфиры также пригодны в качестве активных лекарственных средств. Примерами являются сложные эфиры с алифатическими или ароматическими кислотами, такими как уксусная кислота, или с алифатическими или ароматическими спиртами, такими как этанол. Термин алкил в соответствии с данным описанием, отдельно или в составе другой группы, включает радикалы с линейной, разветвленной и циклизованной цепью до 18 атомов углерода, предпочтительно от 1 до 7 атомов углерода. Термин низший алкил в соответствии с данным описанием, отдельно или в составе другой группы, включает радикалы с линейной, разветвленной и циклизованной цепью от 1 до 7 атомов углерода. Конкретными примерами алкильного и низшего алкильного остатков, соответственно, являются метил, этил, пропил, изопропил, бутил, трет-бутил, пентил, циклопентил, гексил, циклогексил, октил, децил и додецил, включая их изомеры с разветвленной цепью. Термин алкокси в соответствии с данным описанием, отдельно или в составе другой группы,включает алкильную группу, которой дано определение выше, связанную с атомом кислорода. Термин ацил в соответствии с данным описанием, отдельно или в составе другой группы, относится к алкилкарбонильной или алкенилкарбонильной группе, причем алкильной и алкенильной группам дано определение выше. Соединения данного изобретения можно вводить пациенту в терапевтически эффективных количествах, которые обычно находятся в интервале от примерно 0,05 до 200 мг, предпочтительно от 0,1 до 100 мг, более предпочтительно от 0,5 до 50 мг в сутки в зависимости от возраста, веса, состояния пациента,пути введения и используемого ингибитора обмена Na+/Ca2+. Соединения данного изобретения могут быть изготовлены в виде дозированных лекарственных форм с применением методик, известных специалистам. Их можно давать пациенту как таковые или в комбинации с подходящими фармацевтическими наполнителями в виде таблеток, гранул, капсул, суппозиториев, эмульсий, суспензий или растворов. Выбор подходящих ингредиентов для композиции является обычным для специалистов в данной области. Очевидно, что также можно использовать подходящие носители, растворители, гелеобразующие ингредиенты, формирующие дисперсию ингредиенты, антиоксиданты, красители, подсластители, повышающие смачивание соединения и другие ингредиенты, обычно используемые в данной области техники. Композиции, содержащие данное активное соединение могут назначаться энтерально или парентерально,причем пероральный путь является предпочтительным. Содержание активного соединения в композиции составляет от примерно 0,5 до 100%, предпочтительно от примерно 0,5 до примерно 20% от общего веса композиции. Эксперименты Действие соединений данного изобретения испытывали при аритмиях, вызванных уабаином на папиллярных мышцах морских свинок. Методы Папиллярные мышцы морских свинок помещали в горизонтальную кювету для мышц. Крючок, соединенный с датчиком усилия соединяли с другим концом мышцы. Мышечным препаратам задавали ритм в 1 Гц электрической стимуляцией через платиновые электроды. Модифицированный раствор Ти- 10008539 роде использовали для увлажнения мышечных препаратов. Состав раствора Тироде был следующим(мМ): NaCl 135, MgCl26H2O 1, KCl 5, CaCl22H2O 2, NaHCO3 15, Na2HPO42H2O 1 и глюкоза 10. Раствор Тироде насыщали газом карбогеном (95% O2, 5% СO2) до получения рН 7,4. Эксперименты проводили при 37 С. Обнаружение и анализ напряженности сокращений производили с помощью системы измерения силы и потенциала действия (ACFO v. 1.0, Fision Ltd, Finland). Ингибирование вызванных уабаином аритмий Уабаин, путем блокирования натрий-калиевой АТФ-азы, повышает внутриклеточное содержание натрия, который заменяется на кальций путем NCX. Повышенная внутриклеточная концентрация кальция приводит к перегрузке саркоплазматического ретикулума (СР) и спонтанному выделению кальция из СР, вызывающему задержку последующих поляризаций (ЗПП). Эквивалентами для ЗПП в сигнале усилия являются дополнительные сокращения (ДС), которые наблюдаются как спонтанные сокращения после прохождения регулируемого сокращения. Оценивали антиаритмическое воздействие соединений из примеров 13, 33, 14 а и 14b. Результаты показаны на фиг. 1-3. На фиг. 1 показано воздействие данных соединений на время начала быстрого повышения вызванных уабаином дополнительных сокращений. На фиг. 2 показано воздействие данных соединений на максимальную силу вызванных уабаином дополнительных сокращений папиллярных мышц морских свинок. На фиг. 3 показано воздействие данных соединений на время максимальной силы вызванных уабаином дополнительных сокращений у папиллярных мышц морских свинок. В основном соединения данного изобретения замедляют появление и снижают амплитуду дополнительных сокращений. Соединение из примера 33 при концентрации 10 мкМ способно полностью подавлять появление вызванного уабаином второго дополнительного сокращения. Примеры В примерах 1-11 в основном описано получение промежуточных продуктов для соединений данного изобретения. Получение соединений данного изобретения в основном описано в примере 12 далее. Пример 1. Промежуточные соединения.a) 2-Фенилхроманольные промежуточные соединения. 2-Фенилхроман-6-ол. Цинк (5,4 г, 83,2 ммоль), хлорид ртути (II) (340 мг), концентрированную соляную кислоту (0,2 мл) и воду смешивали при комнатной температуре в течение 15 мин, и смесь декантировали. 6-Гидроксифлаванон (1,0 г) добавляли в виде суспензии в смеси уксусной кислоты (25 мл), концентрированной соляной кислоты (5,2 мл) и воды (2 мл). Реакционную смесь нагревали с обратным холодильником в течение 1,5 ч. После охлаждения до комнатной температуры реакционную смесь фильтровали, и фильтрат экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным растворомNaHCO3, затем водой и сушили Na2SO4. 2-Фенилхроман-6-ол очищали колоночной хроматографией с использованием гептана-этилацетата (2:1) в качестве элюента. 1(ддд, 1H, J= -16,7, 5,5, 3,3 Гц), 2,10 (м, 1H), 1,94 (м, 1H). Используя ту же методику, которая описана выше для 2-фенилхроман-6-ола, но заменяя 6-гидроксифлаванон 7-гидроксифлаваноном, получали 2-фенилхроман-7-ол,1b) 5-Нитро-2-(2-(незамещенный)фенилхроманилокси)пиридиновые промежуточные соединения. 5-Нитро-2- (2-фенилхроман-6-илокси)пиридин. Фторид калия (225 мг) добавляли в раствор 2-фенилхроман-6-ола (300 мг) в безводном ДМФ (3 мл). После перемешивания полученной смеси при 120 С в течение 30 мин добавляли 2-хлор-5-нитропиридин(195 мг). Реакционную смесь перемешивали в течение еще 6,5 ч при 120 С. После охлаждения до комнатной температуры добавляли 1 М раствор HCl, и смесь экстрагировали этилацетатом. Объединенные органические слои промывали водой, затем насыщенным раствором NaCl и сушили Na2SO4. 5-Нитро-2(2-фенилхроман-6-илокси)пиридин перекристаллизовывали из ацетон-2-пропанола (1:5). 1H ЯМР (400 МГц, d6-ДMCO) : 9,00 (д, 1H, J=2,9 Гц), 8,60 (дд, 1H, J=9,2, 2,9 Гц), 7,47-7,32 (м, 5 Н),7,20 (д, 1H, J=9,2 Гц), 7,00-6,89 (м, 3 Н), 5,15 (дд, 1H, J=10,1, 2,2 Гц), 2,99 (ддд, 1H, J=-16,8, 11,3, 6,2 Гц),2,75 (ддд, 1H, J=-16,8, 5,4, 3,3 Гц), 2,18 (м, 1H), 2,02 (м, 1H). Используя ту же методику, которая описана выше для 5-нитро-2-(2-фенилхроман-6-илокси)пиридина, но заменяя 2-фенилхроман-6-ол на 2-фенилхроман-7-ол, получали 5-нитро-2-(2-фенилхроман-7-илокси)пиридин. 1- 11008539 Пример 2. Промежуточные соединения. а) Хроман-4-оновые промежуточные соединения. 6-Гидрокси-2-(4-фторфенил)хроман-4-он. 2',5'-Дигидроксиацетофенон (3,0 г) растворяли в теплой ледяной уксусной кислоте (40 мл). Добавляли 4-фторбензальдегид (2,4 мл) и ацетат аммония (1,97 г). Реакционную смесь нагревали с обратным холодильником в течение 8 ч. Ей давали остыть до комнатной температуры и выливали на лед. Образовавшийся осадок отфильтровывали, получая в результате 4,23 г смеси 2-(4-фторфенил)-6-гидроксихроман-4-она и 1-(2,5-дигидроксифенил)-3-(4-фторфенил)пропенона. Полученную смесь растворяли в этаноле (75 мл) и добавляли ацетат натрия (3,4 г). Реакционную смесь нагревали с обратным холодильником в течение 5 ч. Затем ей давали остыть до комнатной температуры и разбавляли водой и фильтровали. 2-(4-Фторфенил)-6-гидроксихроман-4-он перекристаллизовывали из уксусной кислоты. 1-16,9, 2,8 Гц). Используя ту же методику, которая описана выше для 6-гидрокси-2-(4-фторфенил)хроман-4-она, но заменяя 4-фторбензальдегид соответствующим бензальдегидом, получали 2-(3-фторфенил)-6-гидроксихроман-4-он. 1b) Хроман-4,6-диольные промежуточные соединения. 2-(4-Фторфенил)хроман-4,6-диол. В суспензию 2-(4-фторфенил)-6-гидроксихроман-4-она (3,4 г) в безводном ТГФ (34 мл) каплями добавляли раствор боран-ТГФ комплекса (20 мл, 1,0 М в ТГФ) в атмосфере азота. Реакционную смесь нагревали с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры ее выливали в ледяной 2 М раствор HCl. 2-(4-Фторфенил)хроман-4, 6-диол отфильтровывали. 1H ЯМР (400 МГц, d6-ДМСО) : 8,84 (с, 1H), 7,48 (м, 2 Н), 7,21 (м, 2 Н), 6,89 (д, 1H, J=2,7 Гц), 6,59 (д, 1H, J=8,7 Гц), 6,54(дд, 1H, J=8,7, 2,7 Гц), 5,42 (ушир.с, 1H), 5,12 (д, 1H, J=10,7 Гц), 4,87 (м, 1H), 2,25 (м, 1H), 1,89 (м, 1H). Используя ту же методику, которая описана выше для 2-(4-фторфенил)хроман-4,6-диола, но заменяя 2-(4-фторфенил)-6-гидроксихроман-4-он соответствующим 2-фенил-6-гидроксихроман-4-оном, получали 2-(3-фторфенил)хроман-4,6-диол. 1(д, 1H, J=8,0 Гц), 4,70 (д, 1H, J=10,6 Гц), 1,94 (м, 1H), 0,73 (д, 3H, J=6,7 Гц). с) Хроман-6-ольные промежуточные соединения. 2-(4-Фторфенил)хроман-6-ол. Триэтилсилан (14 мл) медленно добавляли в раствор 2-(4-фторфенил)хроман-4,6-диола (2,9 г) в дихлорметане (58 мл). Затем в реакционную смесь по каплям добавляли трифторуксусную кислоту (27 мл),и перемешивали ее при комнатной температуре в течение 1 ч. Реакционную смесь выливали в ледяную воду и экстрагировали дихлорметаном. Остаток выпаривали при пониженном давлении с толуолом с получением 2-(4-фторфенил)хроман-6-ола. 1H ЯМР (400 МГц, CDCl3) : 7,38 (м, 2 Н), 7,06 (м, 2 Н), 6,77 (д, 1H, J=8,6 Гц), 6,61 (дд, 1H, J=8,6, 2,9 Гц) 6,57 (д, 1H, 8,6 Гц), 4,97 (дд, 1H, J=10,2, 2,4 Гц), 2,95 (ддд, 1H, J=-16,8, 11,4, 6,2 Гц), 2,74 (ддд, 1H, J=16,8, 5,3, 3,1 Гц), 2,15 (м, 1H), 2,05 (м, 1H). Используя ту же методику, которая описана выше для 2-(4-фторфенил)хроман-6-ола, но заменяя 2(4-фторфенил)хроман-4,6-диол соответствующим 2-фенилхроман-4,6-диолом, получали 2-(3-фторфенил)хроман-6-ол. 1d) 2-[2-Фенилхроман-6-илокси]-5-нитропиридиновые промежуточные соединения. 2-[2-(4-Фторфенил)хроман-6-илокси]-5-нитропиридин. 2-[2-(4-Фторфенил)хроман-6-илокси]-5-нитропиридин получали, как описано для 5-нитро-2-(2 фенилхроман-6-илокси)пиридина в примере 1(b), исходя из 160 мг 2-(4-фторфенил)хроман-6-ола. 1H ЯМР (400 МГц, d6-ДМСО) : 9,04 (дд, 1H, J=2,9, 0,4 Гц), 8,60 (дд, 1H, J=9,1, 2,9 Гц), 7,51 (м, 2 Н),7,24 (м, 1H), 7,20 (дд, 1H, J=9,1, 0,4 Гц), 7,01 (д, 1H, J=2,8 Гц), 6,96 (дд, 1H, J=8,7, 2,8 Гц), 6,91 (д, 1H, 8,7 Гц), 5,15 (дд, 1H, J=10,3, 2,2 Гц), 2,94 (м, 1H), 2,76 (м, 1H) 2,17 (м, 1H), 2,01 (м, 1H). Используя ту же методику, которая описана выше для 2-[2-(4-фторфенил)хроман-6-илокси]-5 нитропиридина, но заменяя 2-(4-фторфенил)хроман-6-он соответствующим 2-фенилхроман-6-олом, получали 2-[2-(3-фторфенил)хроман-6-илокси]-5-нитропиридин. 1(5-Нитропиридин-2-илокси)-2-фенилхроман-4-оновые промежуточные соединения. 6-(5-Нитропиридин-2-илокси)-2-фенилхроман-4-он. 6-(5-Нитропиридин-2-илокси)-2-фенилхроман-4-он получают, как описано для 5-нитро-2-(2 фенилхроман-6-илокси)пиридина в примере 1(b) с использованием 200 мг 6-гидроксифлаванона. 1H ЯМР (400 МГц, d6-ДМСО) : 9,03 (ушир.с, 1H), 8,64 (д, 1H, J=9,0 Гц), 7,59-7,41 (м, 7 Н), 7,31 (д,1H, J=9,0 Гц), 7,23 (д, 1H, 8,8 Гц), 5,75 (дд, 1H, J=12,3, 2,9 Гц), 3,30 (дд, 1H, J=-16,3, 12,3 Гц), 2,87 (дд, 1H,J=-16,3, 2,9 Гц). Используя ту же методику, которая описана выше для 6-(5-нитропиридин-2-илокси)-2 фенилхроман-4-она, но заменяя 6-гидроксифлаванон соответствующим 2-фенилхроманоновым производным, получали 7-(5-нитропиридин-2-илокси)-2-фенилхроман-4-он. 1H ЯМР (400 МГц, d6-ДМСО) : 9,03 (д, 1H, J=2,9 Гц), 8,64 (дд, 1H, J=9,1, 2,9 Гц), 7,59-7,56 (м, 3H),7,50-7,32 (м, 4 Н) 7,30 (д, 1H, J=9,1 Гц), 7,18 (д, 1H, J=8,9 Гц), 5,38 (д, 1H, J=12,5 Гц), 3,36 (дд, 1H, J=12,5,6,9 Гц), 0,86 (д, 3H, J=6,9 Гц). Пример 4. Промежуточное соединение. 2-(2,3-Дигидро-2-фенилбензо[1,4]диоксин-6-илокси)-5-нитропиридин. а) 1-[2,5-бис(Бензилокси)фенил]этанон. Смесь 1-(2,5-дигидроксифенил)этанона (3,16 г), бензилхлорида (7,04 г), карбоната калия (12,4 г) и 18-краун-6 (30 мг) в 2-бутаноне (50 мл) нагревали с обратным холодильником в течение 5 ч. После охлаждения осадок отфильтровывали. Фильтрат выпаривали досуха при пониженном давлении и к остатку добавляли эфир (50 мл). Раствор промывали дважды разбавленным раствором гидроксида натрия, дважды разбавленной соляной кислотой, сушили над сульфатом натрия и, по существу, выпаривали досуха при пониженном давлении. Остаток растирали с холодным н-гептаном (30 мл), и осадок отфильтровывали вакуумфильтрованием с получением после сушки 2,85 г 1-[2,5-бис (бензилокси) фенил] этанона. 1b) 2,5-бис(Бензилокси)фениловый эфир уксусной кислоты. Раствор 1-[2,5-бис(бензилокси)фенил]этанона (2,25 г) и 40% перуксусной кислоты (1,63 мл) в уксусной кислоте (5,4 мл) перемешивали при 60 С в течение 1 ч. После охлаждения до комнатной температуры осажденный продукт собирали фильтрованием, промывали холодным эфиром и сушили при пониженном давлении. 2,5-бис(Бензилокси)фениловый эфир уксусной кислоты перекристаллизовывали из 2-пропанола. Выход составляет 1,87 г. 1c) 2,5-бис(Бензилокси)фенол. Раствор 2,5-бис(бензилокси)фенилового эфира уксусной кислоты (1,85 г) и 5 М раствор гидроксида натрия (10,6 мл) в этаноле (11 мл) нагревали с обратным холодильником в течение 6,5 ч. После того, как этанол выпаривали при пониженном давлении, прозрачный раствор подкисляли разбавленной соляной кислотой. Осажденный продукт собирали фильтрованием, промывали холодной водой и сушили при пониженном давлении. Выход составляет 0,56 г. 1d) 2-[2,5-бис(Бензилокси)фенокси]-1-фенилэтанон. Смесь 2,5-бис(бензилокси)фенола (0,28 г), 2-бромацетофенона (0,22 г), гидрокарбоната калия (0,25 г) и 18-краун-6 (3 мг) в ацетонитриле (4,2 мл) перемешивали при 22 С в течение одной недели. Смесь фильтровали и выпаривали досуха при пониженном давлении. Остаток растирали со смесью эфира (8,2 мл) и воды (1,4 мл) при температуре ледяной бани. Продукт собирали фильтрованием, промывали холодным эфиром и сушили при пониженном давлении, выход составляет 0,14 г. 1e) 2-[2,5-Бис(бензилокси)фенокси]-1-фенилэтанол. К раствору 2-[2,5-бис(бензилокси)фенокси]-1-фенилэтанона (0,14 г) в метаноле (0,5 мл) и тетрагидрофуране (1,9 мл) добавляли при температуре 0 С борогидрид натрия (6,5 мг). Реакционную смесь перемешивали 15 мин при температуре 0 С и 2 ч при температуре 22 С. После того, как остаток перемешивали при 22 С 0,5 ч, продукт фильтровали, промывали холодной водой и сушили при пониженном давле- 18008539 нии. Выход составляет 0,09 г. 1f) 2-(2-Гидрокси-2-фенилэтокси)бензол-1,4-диол. Раствор 2-[2,5-бис(бензилокси)фенокси]-1-фенилэтанола (3,9 г) в этаноле (175 мл) гидрировали в присутствии 10% палладия на угле (100 мг) при 30 фунт/дюйм 2. Катализатор удаляли фильтрованием и растворитель выпаривали при пониженном давлении. Остаток перекристаллизовывали из смеси толуолаэтилацетата 8:1 (15 мл). Выход 2-(2-гидрокси-2-фенилэтокси)бензол-1,4-диола составляет 1,2 г. 1g) 2,3-Дигидро-2-фенилбензо[1,4]диоксин-6-ол. Раствор 2-(2-гидрокси-2-фенилэтокси)бензол-1,4-диола (1,2 г) в толуоле (75 мл) нагревали с катализатором амберлист 15 (Amberlyst 15) (0,5 г) при нагревании с обратным холодильником в течение 7 ч. После фильтрования растворитель выпаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (толуол/этилацетат/уксусная кислота, 8:1:1). Выход 2,3-дигидро-2 фенилбензо[1,4]диоксин-6-ола составляет 0,5 г. 1h) 2-(2,3-Дигидро-2-фенилбензо[1,4]диоксин-6-илокси)-5-нитропиридин. Раствор 2,3-дигидро-2-фенилбензо[1,4]диоксин-6-ола (80 мг), 2-хлор-5-нитропиридина (56 мг) и карбоната калия (52 мг) в диметилформамиде (1,0 мл) перемешивали при 120 С в течение 2 ч. После охлаждения смеси добавляли воду (10 мл) и осажденный продукт отфильтровывали, промывали водой и 2 пропанолом и сушили при пониженном давлении. Выход составляет 60 мг, и т.плавл. равна 163-170 С. 1H ЯМР (ДМСО-d6) : 4,16 (дд, J=8,5, 11,6 Гц, 1H), 4,47 (дд, J=11,6, 2,6 Гц, 1H), 5,28 (дд, J=2,6, 8,5 Гц, 1H), 6,75 (дд, J=2,6, 8,8 Гц, 1H), 6,88 (д, J=2,6 Гц, 1H), 7,05 (д, J=8,8 Гц, 1H), 7,21 (д, J=9,1 Гц, 1H),7,39-7,52 (м, 5 Н), 8,60 (дд, J=2,8, 9,1 Гц, 1H), 9,05 (д, J=2,8 Гц, 1H). Пример 5. Промежуточное соединение. 5-Нитро-2-(6-фенил-5,6,7,8-тетрагидронафталин-2-илокси)пиридин. а) 6-Метокси-2-фенил-3,4-дигидро-2 Н-нафталин-1-он. Смесь ацетата палладия (II) (0,57 г), рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (1,91 г) и третбутоксида калия (4,15 г) в безводном толуоле перемешивали в атмосфере аргона в течение 10 мин. Добавляли бромбензол (5,34 г) и 6-метокси-1-тетралон (3,0 г), сольватированный в безводном толуоле, и смесь перемешивали при 100 С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в насыщенный водный раствор хлорида аммония и экстрагировали этиловым эфиром. Органический экстракт промывали насыщенным раствором соли, сушили и выпаривали. Сырой продукт очищали флэш-хроматографией на силикагеле с использованием толуола и толуол-этилацетата (9:1) в качестве элюента. 1b) 6-Гидрокси-2-фенил-3,4-дигидро-2 Н-нафталин-1-он. 6-Метокси-2-фенил-3,4-дигидро-2 Н-нафталин-1-он (1,0 г) нагревали с обратным холодильником с 47% HBr (20 мл) до исчезновения исходного вещества. Смесь выливали в воду и экстрагировали этилацетатом. Этилацетат сушили и выпаривали. Данный продукт перекристаллизовывали из толуола. 1c) 6-Фенил-5,6,7,8-тетрагидронафталин-2-ол. К раствору 6-гидрокси-2-фенил-3,4-дигидро-2 Н-нафталин-1-она (50 мг) в трифторуксусной кислоте добавляли триэтилсилан (98 мг). Смесь нагревали и выдерживали при 60 С в течение 3 ч. Растворитель выпаривали, к остатку добавляли воду и смесь экстрагировали этилацетатом. Органический экстракт сушили и выпаривали. 1H ЯМР (400 МГц, d6-ДМСО) : 9,02 (с, 1H), 7,18-7,32 (м, 5 Н), 6,87 (д, 1H, J=7,9),6,50-6,53 (м, 2 Н), 2,68-2,92 (м, 5 Н), 1,94-1,99 (м, 1H), 1,81-1,89 (м, 1H).(23 мг) в безводном диметилформамиде нагревали и выдерживали при 120 с до исчезновения исходного вещества. Добавляли воду и 1 н HCl, и смесь экстрагировали этилацетатом. Этилацетат промывали насыщенным раствором соли и водой, сушили и выпаривали. Продукт перекристаллизовывали из толуола. 1- 19008539 Пример 6. Промежуточное соединение. 6-(5-Нитропиридин-2-илокси)-2-фенил-3,4-дигидро-2 Н-нафталин-1-он. 6-(5-Нитропиридин-2-илокси)-2-фенил-3,4-дигидро-2 Н-нафталин-1-он получали, как описано для 5 нитро-2-(6-фенил-5,6,7,8-тетрагидронафталин-2-илокси)пиридина в примере 5(d) с использованием 50 мг 6-гидрокси-2-фенил-3,4-дигидро-2 Н-нафталин-1-она, 33 мг 2-хлор-5-нитропиридина и 37 мг фторида калия. 1(3-Фторфенил)уксусную кислоту (3,7 г) и 3-метоксифенол (3,0 г) растворяли в BF3EtO (60 мл, 20 экв) в атмосфере аргона. Смесь перемешивали при 60-70 С до исчезновения исходных веществ (9 ч) и выливали в большой объем ледяной воды. После экстрагирования этилацетатом объединенные органические слои промывали водой, сушили и выпаривали. Сырой продукт очищали колоночной хроматографией с использованием CH2Cl2 в качестве элюента. 1b) 3-(3-Фторфенил)-7-метоксихромен-4-он. 2-(3-Фторфенил)-1-(2-гидрокси-4-метоксифенил)этанон (1, 76 г) растворяли в пиридине (88 мл). Добавляли пиперидин (8,8 мл) и триэтилортоформиат (88 мл), и смесь перемешивали при 120 с в течение 3,5 ч. После того, как смесь выливали в воду и подкисляли конц. HCl, сырой продукт отфильтровывали. Очистка колоночной хроматографией с использованием гептана-этилацетата (7:3) в качестве элюента давала 3-(3-фторфенил)-7-метоксихромен-4-он. 1(18 мл) до исчезновения исходного вещества. Смесь выливали в воду и остаток отфильтровывали и сушили, получая 3-(3-фторфенил)-7-гидроксихромен-4-он. 1d) 3-(3-Фторфенил)хроман-7-ол. 3-(3-Фторфенил)-7-гидроксихромен-4-он (160 мг) растворяли в этаноле (40 мл) и добавляли 10% палладия на угле (400 мг). Реакционную смесь гидрировали в течение 6 ч при нормальном давлении и комнатной температуре. Затем ее фильтровали через целит и промывали этанолом. Растворитель выпаривали при пониженном давлении с получением 3-(3-фторфенил)хроман-7-ола. 1e) 2-[3-(3-Фторфенил)хроман-7-илокси]-5-нитропиридин. 2-[3-(3-Фторфенил)хроман-7-илокси]-5-нитропиридин получали, как описано для 5-нитро-2-(2 фенилхроман-6-илокси)пиридина в примере 1(b), используя 125 мг 3-(3-фторфенил)хроман-7-ола. Продукт перекристаллизовывали из этанола. 1(м, 1H), 3,06 (м, 2 Н). Используя ту же методику, которая описана выше для 3-(3-фторфенил)хроман-7-ола, но заменяя 3(3-фторфенил)-7-гидроксихромен-4-он 7-гидрокси-3-фенилхромен-4-оном, получали 3-фенилхроман-7-ол. 1H ЯМР (400 МГц, d6-ДМСО) : 8,18 (ушир.с, 1H), 7,31-7,34 (м, 4 Н), 7,25-7,27 (м, 1H), 6,88 (д, 1H,J=8,2 Гц), 6,30 (дд, 1H, J=8,2, 2,4 Гц), 6,20 (д, 1H, J=2,4 Гц), 4,21 (дд, 1H, J=10,3, 3,6 Гц), 4,00 (т, 1H, 10,3 Гц), 3,13 (м, 1H), 2,84-2,87 (м, 2 Н). Используя ту же методику, которая описана выше для 2-[3-(3-фторфенил)хроман-7-илокси]-5 нитропиридина, но заменяя 3-(3-фторфенил)хроман-7-ол 3-фенилхроман-7-олом, получали 5-нитро-2-(3-фенилхроман-7-илокси)пиридин. 1H ЯМР (400 МГц, d6-ДМСО) : 9,05 (д, 1H, J=2,9 Гц), 8,61 (дд, 1H, J=9,1, 2,9 Гц), 7,34-7,38 (м, 4 Н),7,27-7,30 (м, 1H), 7,22 (м, 2 Н), 6,70-6,74 (м, 2 Н), 4,31 (дд, 1H, J=10,4, 3,5 Гц), 4,12 (т, 1H, 10,4 Гц), 3,24 (м,1H), 3,01-3,11 (м, 2 Н). 7-Гидрокси-3-фенилхромен-4-он доступен для приобретения или может быть синтезирован спосо- 20008539 бами, описанными для 3-(3-фторфенил)-7-гидроксихромен-4-она. Пример 8. Промежуточное соединение. 5-Нитро-2-(2-фенил-2,3-дигидробензо[1,4]оксатиин-6-илокси)пиридин. а) 2-(2-Гидрокси-1-фенилэтилсульфанил)бензол-1,4-диол. К перемешиваемому раствору 2-меркаптобензол-1,4-диола (0,5 г) и карбоната калия (0,49 г) в воде(5 мл) добавляли 2-фенилоксиран (0,40 мл) в атмосфере аргона. Смесь перемешивали при комнатной температуре в течение 2,5 ч и затем обрабатывали 2 М HCl и экстрагировали этилацетатом. Объединенные органические слои промывали водой и насыщенным раствором соли, сушили и выпаривали. Сырой продукт очищали колоночной хроматографией, используя гептан-этилацетат (1:1) в качестве элюента. 1b) 2-Фенил-2,3-дигидробензол [1,4]оксатиин-6-ол. Раствор 2-(2-гидрокси-1-фенилэтилсульфанил) бензол-1,4-диола (0,83 г) в безводном толуоле (60 мл) перемешивали с амберлистом 15 (0,5 г) при 60 С до исчезновения исходного вещества. После того,как смесь фильтровали и растворитель выпаривали, сырой продукт очищали колоночной хроматографией, используя гептан-этилацетат (1:1) в качестве элюента. 1H ЯМР (400 МГц, CDCl3) : 7,41 (м, 4 Н), 7,33-7,40 (м, 1H), 6,81 (д, 1H, J=8,7 Гц), 6,61 (д, 1H, J=3,0 Гц), 6,51 (дд, 1H, J=8,7, 3,0 Гц), 5,10 (дд, 1H, J=9,6, 1,9 Гц), 3,28 (дд, 1H, J=13,0, 9,6 Гц), 3,06 (дд, 1H,J=13,0, 1,9 Гц). с) 5-Нитро-2-(2-фенил-2,3-дигидробензо[1,4] оксатиин-6-илокси)пиридин. 5-Нитро-2-(2-фенил-2,3-дигидробензо[1,4]оксатиин-6-илокси)пиридин получали, как описано для 5 нитро-2-(2-фенилхроман-6-илокси)пиридина в примере 1(b), используя 269 мг 2-фенил-2,3 дигидробензо[1,4]оксатиин-6-ола. Продукт перекристаллизовывали из этанола. 1(м, 1H), 7,02 (д, 1H, J=9,1 Гц), 6,99 (д, 1H, J=8,9 Гц), 6,95 (д, 1H, J=2,8 Гц), 6,82 (дд, 1H, J=8,9, 2,8 Гц),5,21 (дд, 1H, J=9,7, 1,9 Гц), 3,31 (дд, 1H, 13,2, 9,7 Гц), 3,11 (дд, 1H, 13,2, 1,9 Гц). Пример 9. Промежуточное соединение 5-Нитро-2-(2-фенилиндан-5-илокси)пиридин. а) 3-(4-Метоксифенил)-2-фенилакриловая кислота. Триэтиламин добавляли к раствору п-анизальдегида (10 г) и фенилуксусной кислоты (10 г) в уксусном ангидриде (25 мл). Реакционную смесь перемешивали при 90 С в течение 8 ч. Реакционную смесь охлаждали и добавляли водный (600 мл) раствор карбоната калия (81 г). После добавления реакционную смесь нагревали и выдерживали при 60 С в течение часа. Перед нейтрализацией концентрированной соляной кислотой реакционную смесь охлаждали ниже 10 С. Осадок отфильтровывали и промывали водой. 1b) 3-(4-Метоксифенил)-2-фенилпропионовая кислота. 13 г 3-(4-метоксифенил)-2-фенилакриловую кислоту растворяли в 600 мл этилацетата и добавляли 2,6 г 10% палладия на угле в инертной атмосфере. Исходное вещество гидрировали при комнатной температуре с получением количественного выхода 3-(4-метоксифенил)-2-фенилпропионовой кислоты. 1c) 6-Метокси-2-фенилиндан-1-он. К раствору 3-(4-метоксифенил)-2-фенилпропионовой кислоты (4,6 г) в безводном дихлорметане (26 мл) добавляли две капли безводного ДМФ. Добавляли тионилхлорид (3 мл) и реакционную смесь перемешивали при 40 С в течение 4 ч. Растворитель выпаривали под вакуумом. Остаток растворяли в дихлорметане. Раствор охлаждали до 0-3 С. Этот раствор и хлорид алюминия (2,5 г) медленно смешивали в течение 4 ч, поддерживая температуру ниже 4 С. После смешивания реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакцию завершали выливанием в разбавленной ледяной раствор соляной кислоты. Слои разделяли и водный раствор экстрагировали дихлорметаном. Объединенные органические слои промывали водой, сушили и выпаривали. Сырой продукт растирали с получением 2,9 г 6-метокси-2-фенилиндан-1-она. 1d) 5-Метокси-2-фенилиндан. 5-Метокси-2-фенилиндан получали, как описано для 2-фенилхроман-6-ола в примере 1 (а), используя 600 мг 6-метокси-2-фенилиндан-1-она. 1- 21008539 холодильником в течение 5,5 ч. Реакционной смеси давали остыть до комнатной температуры и добавляли 20 мл ледяной воды, и ее экстрагировали дихлорметаном. Объединенные органические слои промывали насыщенным раствором соли и сушили с помощью Na2SO4. Растворители выпаривали с получением 2-фенилиндан-5-ола. 1f) 5-Нитро-2-(2-фенилиндан-5-илокси)пиридин. 5-Нитро-2-(2-фенилиндан-5-илокси)пиридин получали как описано для 2-фенилхроман-6 илокси)пиридина в примере 1(b), используя 107 мг 2-фенилиндан-5-ола. 1H-ЯMP (400 МГц, d6-ДМСО): 9,04 (д, 1H, J=2,9 Гц), 8,61 (дд, 1H, J=2,9, 9,1 Гц), 7,38-7,28 (м, 5 Н),7,24-7,20 (м, 2 Н), 7,11 (д, 1H, J=2,2 Гц), 7,00 (дд, 1H, J=2,2, 8,0 Гц), 3,72 (к, 1H, J=8,9 Гц), 3,36-3,28 (м,2 Н), 3,01 (дд, 2 Н, J=8,9, 15,3 Гц). Пример 10. Промежуточные соединения. 5-Аминопиридиновые промежуточные соединения. 5-Амино-2-(2-фенилхроман-6-илокси)пиридин. 5-Нитро-2-(2-фенилхроман-6-илокси)пиридин (2,26 г) растворяли в 350 мл ледяной уксусной кислоты. Несколькими порциями, из-за экзотермической реакции, добавляли порошок цинка (8,48 г). Смесь перемешивали при комнатной температуре в течение 2 ч и фильтровали. Цинк промывали ледяной уксусной кислотой. Кислоту выпаривали и добавляли толуол, и снова выпаривали. Смесь продукта растворяли в CH2Cl2 и промывали 1 М NaOH. Водную фазу дополнительно промывали CH2Cl2. Обе органические фракции объединяли и сушили над Na2SO4. Продукт очищали колоночной хроматографией. 1H ЯМР (400 МГц, d6-ДМСО) : 7,52 (д, 1H, J=2,8 Гц), 7,46-7,30 (м, 5 Н), 7,05 (дд, 1H, J=8,6, 3,0 Гц),6,82-6,72 (м, 3H), 6,69 (д, 1H, J=8,6 Гц), 5,08 (дд, 1H, J=10,0, 2,1 Гц), 5,00 (с, 2 Н), 3,00-2,87 (м, 1H), 2,742,64 (м, 1H), 2,19-2,10 (м, 1H), 2,05-1,91 (м, 1H). Используя ту же методику, которая описана выше для 5-амино-2-(2-фенилхроман-6-илокси)пиридина, но заменяя 5-нитро-2-(2-фенилхроман-6-илокси)пиридин соответствующим нитропиридиновым промежуточным соединением, получали 6-[2-(4-фторфенил)хроман-6-илокси]пиридин-3-иламин. 1(м, 1H), 2,17 (м, 1H), 1,97 (м, 1H). Используя ту же самую методику, которая описана для 5-амино-2-(2-фенилхроман-6-илокси)пиридина, но заменяя 5-нитро-2-(2-фенилхроман-6-илокси)пиридин на 2-[2-(3,4-дифторфенил)хроман-6-илокси]-5-нитропиридин,5-нитро-2-[2-(2-трифторметилфенил)хроман-6-илокси]пиридин,2-[2-(3-хлор-4-фторфенил)хроман-6-илокси]-5-нитропиридин,2-[2-(3-хлорфенил)хроман-6-илокси]-5-нитропиридин,2-[2-(2,4-дихлорфенил)хроман-6-илокси]-5-нитропиридин,2-[2-(3-бромфенил)хроман-6-илокси]-5-нитропиридин,2-[2-(4-этилфенил)хроман-6-илокси]-5-нитропиридин,2-(3-метил-2-фенилхроман-6-илокси)-5-нитропиридин,5-нитро-2-(2-фенилхроман-7-илокси)пиридин,7-(5-нитропиридин-2-илокси)-2-фенилхроман-4-он,3-метил-6-(5-нитропиридин-2-илокси)-2-фенилхроман-4-он,2-(2,3-дигидро-2-фенилбензо[1,4]диоксин-6-илокси)-5-нитропиридин,5-нитро-2-(6-фенил-5,6,7,8-тетрагидронафталин-2-илокси)пиридин,6-(5-нитропиридин-2-илокси)-2-фенил-3,4-дигидро-2 Н-нафталин-1-он,2-[3-(3-фторфенил)хроман-7-илокси]-5-нитропиридин,2-(3-фенилхроман-7-илокси)-5-нитропиридин,- 23008539 5-нитро-2-(2-фенилиндан-5-илокси)пиридин,5-нитро-2-(2-фенилиндан-5-илокси)пиридин,можно получить 6-[2-(3,4-дифторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(2-трифторметилфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(3-хлор-4-фторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(3-хлорфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(2,4-дихлорфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(3-бромфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(4-этилфенил)хроман-6-илокси]пиридин-3-иламин,6-(3-метил-2-фенилхроман-6-илокси)пиридин-3-иламин,5-амино-2-(2-фенилхроман-7-илокси)пиридин,7-(5-аминопиридин-2-илокси)-2-фенилхроман-4-он,6-(5-аминопиридин-2-илокси)-3-метил-2-фенилхроман-4-он,6-(2-фенил-2,3-дигидробензо[1,4]диоксин-6-илокси)пиридин-3-иламин,6-(6-фенил-5,6,7,8-тетрагидронафталин-2-илокси)пиридин-3-иламин,6-(5-аминопиридин-2-илокси)-2-фенил-3,4-дигидро-2 Н-нафталин-1-он,6-[3-(3-фторфенил)хроман-7-илокси]пиридин-3-иламин,6-(3-фенилхроман-7-илокси)пиридин-3-иламин,6-(2-фенилиндан-5-илокси)пиридин-3-иламин, соответственно. Пример 11. Промежуточные соединения. 3-Пиридинилоксибензальдегидные промежуточные соединения. 3-(5-Хлорпиридин-2-илокси)бензальдегид. 3-Гидроксибензальдегид (3,0 г) растворяли в безводном ДМФ (30 мл) в атмосфере азота. В раствор добавляли трет-бутоксид калия (3,0 г), и полученную смесь перемешивали в течение 30 мин. Добавляли 2,5-дихлорпиридин (3,6 г) и смесь перемешивали при 120 С в течение 1,5 ч. Реакционной смеси давали остыть до комнатной температуры и добавляли 1 М раствор HCl, и эту смесь экстрагировали этилацетатом. Объединенные органические фазы промывали водой и насыщенным раствором NaCl и сушили. Продукт очищали колоночной хроматографией, используя гептан-этилацетат (3:1) в качестве элюента. 1H ЯМР (400 МГц, d6-ДМСО) : 10,01 (с, 1H), 8,17 (дд, 1H, J=5,0, 1,7 Гц), 7,90 (ддд, 1H, J=8,5, 6,8,1,9 Гц), 7,77 (д, 1H, J=7,8 Гц), 7,66, (т, 1H, J=7,8 Гц), 7,63 (м, 1H), 7,50 (м, 1H), 7,18 (дд, 1H, J=6,9, 5,0 Гц),7,13 (д, 1H, 8,3 Гц). Пример 12. 6-(5-Нитропиридин-2-илокси)-2-фенилхромен-4-он. 6-(5-Нитропиридин-2-илокси)-2-фенилхромен-4-он получали, как описано для 5-нитро-2-(2-фенилхроман-6-илокси)пиридина в примере 1(b), исходя из 500 мг 6-гидроксифлавона. Продукт перекристаллизовывали из смеси 2-пропанола и ацетона. 1H ЯМР (300 МГц, d6-ДМСО) : 9,04 (д, 1H, J=2,9 Гц), 8,67(дд, 1H, J=9,0, 2,9 Гц), 8,16-8,13 (м, 2 Н), 7,95 (д, 1H, J=9,0 Гц), 7,82 (д, 1H, J=2,9 Гц), 7,63 (дд, 1H, J=9,1,2,9 Гц), 7,64-7,61 (м, 3H), 7,38 (д, 1H, J=9,1 Гц), 7,09 (с, 1H). Пример 13. 2-[2-(3-(5-Нитропиридин-2-илокси)фенил)хроман-6-илокси]-5-нитропиридин и его производные. а) 6-Гидрокси-2-(3-гидроксифенил)хроман-4-он (промежуточное соединение). 6-Гидрокси-2-(3-гидроксифенил)хроман-4-он получали, как описано для 6-гидрокси-2-(4-фторфенил)хроман-4-она в примере 2(a), но исходя из 3-гидроксибензальдегида. Продукт перекристаллизовывали из этанола. 1H ЯМР (400 МГц, d6-ДМСО) : 9,50 (ушир.с, 1H), 9,41 (ушир.с, 1H), 7,22-7,17 (м, 1H), 7,11 (д, 1H,J=3,0 Гц), 7,03 (дд, 1H J=3,0, 8,9 Гц), 6,64 (д, 1H, J=8,9 Гц), 6,92-6,90 (м, 2 Н), 6,76-6,73 (м, 1H), 5,46 (дд,1H J=2,9, 12,7 Гц), 3,09 (дд, 1H, J=12,7, 16,9 Гц), 2,75 (дд, 1H, J=2,9, 16,9 Гц). Подобным же образом получали 6-гидрокси-2-(4-гидроксифенил)хроман-4-он. 1b) 2-(3-Гидроксифенил)хроман-4,6-диол (промежуточное соединение). 2-(3-Гидроксифенил)хроман-4,6-диол получали, как описано для 2-(4-фторфенил)хроман-4,6-диола в примере 2(b), но исходя из 6-гидрокси-2-(3-гидроксифенил)хроман-4-она. 1H ЯМР (400 МГц, d6-ДМСО)(м, 1H), 6,58 (д, 1H, J=8,7 Гц), 6,53 (дд, 1H, J=2,7, 8,7), 5,01 (д, 1H, J=11,3 Гц), 4,86 (дд, 1H, J=6,2, 10,8 Гц),2,25-2,19 (м, 1H), 1,88-1,75 (м, 1H). Подобным же образом получали 2-(4-гидроксифенил)хроман-4,6-диол. 1H ЯМР (400 МГц, d6-ДМСО) : 8,82 (с, 1H), 8,17 (м, 1H), 7,86 (м, 1H), 7,43 (т, 1H, J=7,8 Гц), 7,29 (д,1H, J=7,8 Гц), 7,18 (с, 1H), 7,15-7,04 (м, 3H), 6,87 (д, 1H, J=2,7 Гц), 6,59 (д, 1H, J=8,7 Гц), 6,53 (дд, 1H,J=8,7, 2,7 Гц), 5,40 (д, 1H, J=7,0 Гц), 5,14 (д, 1H, J=11,6 Гц), 4,86 (м, 1H), 2,29 (м, 1H), 1,88 (м, 1H). с) 2-(3-Гидроксифенил)хроман-6-ол (промежуточное соединение). 2-(3-Гидроксифенил)хроман-6-ол получали, как описано для 2-(4-фторфенил)хроман-6-ола в примере 2(c), но исходя из 2-(3-гидроксифенил)хроман-4,6-диола. 1H ЯМР (400 МГц, d6-ДМСО) : 9,38 (с,1H), 8,77 (с, 1H), 7,17-7,13 (м, 1H), 6,82-6,79 (м, 2 Н), 6,70-6,67 (м, 1H), 6,62 (д, 1H, J=8,6 Гц), 6,52-6,47 (м,2 Н), 4,89 (дд, 1H, J=2,1, 9,9 Гц), 2,86-2,82 (м, 1H), 2,65-2,59 (м, 1H), 2,09-2,04 (м, 1H), 1,91-1,85 (м, 1H). Подобным же образом получали 2-(3-Бензилоксифенил)хроман-6-ол. 1d) 2-[2-(3-(5-Нитропиридин-2-илокси)фенил)хроман-6-илокси]-5-нитропиридин. 2-[2-(3-(5-Нитропиридин-2-илокси)фенил)хроман-6-илокси]-5-нитропиридин получали, как описано для 5-нитро-2-(2-фенилхроман-6-илокси)пиридина в примере 1(b), но исходя из 2-(3-гидроксифенил) хроман-6-ола и используя 210 моль% 2-хлор-5-нитропиридина. 1(м, 1H), 7,06 (8,6 Гц), 7,04 (2,5 Гц), 6,96 (дд, 1H, J=8,6, 2,5 Гц), 5,18 (д, 1H, J=8,8 Гц), 2,98 (м, 1H), 2,72 (м,1H), 2,21 (м, 1H), 2,02 (м, 1H). Пример 14. 6-(5-Нитропиридин-2-илокси)-2-[3-(5-нитропиридин-2-илокси)фенил]хроман-4-ол и его производные. а) 6-(5-Нитропиридин-2-илокси)-2-[3-(5-нитропиридин-2-илокси)фенил]хроман-4-ол. 6-(5-Нитропиридин-2-илокси)-2-[3-(5-нитропиридин-2-илокси)фенил]хроман-4-ол получали, как описано для 5-нитро-2-(2-фенилхроман-6-илокси)пиридина в примере 1(b), но исходя из 2-(3-гидроксифенилхроман-4,6-диола) и используя 210 мол.% 2-хлор-5-нитропиридина. 1H ЯМР (400 МГц, d6-ДМСО) : 9,06 (д, 1H, J=2,8 Гц), 9,03 (д, 1H, J=2,8 Гц), 8,64 (дд, 1H, J=2,8, 9,1 Гц), 8,61 (дд, 1H, J=2,8, 9,1 Гц), 7,54 (т, 1H, J=7,9 Гц), 7,43 (д, 1H, J=7,9 Гц), 7,36 (с, 1H), 7,30 (д, 1H, J=9,1 Гц), 7,25-7,21 (м, 3H) 7,01 (дд, 1H, J=2,9, 8,7 Гц), 6,89 (д, 1H, J=8,7 Гц), 5,67 (д, 1H, J=6,4 Гц), 5,36 (д, 1H,J=10,8 Гц), 5,01-4,95 (м, 1H), 2,41-2,36 (м, 1H), 2,02-1,92 (м, 1H). Подобным же образом получали(малый, 2,07-1,98 (м, 1H, основной (большой. Пример 15. 2-2-[4-(5-Нитропиридин-2-илокси)фенил]хроман-6-илокси-5-нитропиридин. 2-2-[4-(5-Нитропиридин-2-илокси)фенил]хроман-6-илокси-5-нитропиридин получали, как описано для 2-(4-фторфенил)хроман-6-ола в примере 2(c), но исходя из 6-(5-нитропиридин-2-илокси)-2-[4-(5 нитропиридин-2-илокси)фенил]-хроман-4-ола. 1(м, 1H), 2,26-2,21 (м, 1H), 2,11-2,02 (м, 1H). Пример 16. 6-[2-(3-(5-Аминопиридин-2-илокси)фенил)хроман-6-илокси]пиридин-3-иламин и его производные. 6-[2-(3-(5-Аминопиридин-2-илокси)фенил)хроман-6-илокси]-пиридин-3-иламин получали, как описано для 5-амино-2-(2-фенилхроман-6-илокси)пиридина в примере 10, но исходя из 2-[2-(3-(5 нитропиридин-2-илокси)фенил)хроман-6-илокси]-5-нитропиридина. Продукт выделяли в виде дигидрохлорида. 1H ЯМР (300 МГц, d6-ДМСО) : 8,12 (м, 2 Н), 7,78 (м, 1H), 7,45 (т, 1H, J=7,8 Гц), 7,31 (д, 1H, J=7,2 Гц), 7,21 (с, 1H), 7,13-7,04 (м, 3H), 6,91-6,87 (м, 3H), 5,15 (д, 1H, J=9,8 Гц), 3,02-2,91 (м, 1H), 2,76-2,70 (м,1H), 2,23-2,17 (м, 1H), 2,05-1,93 (м, 1H). Подобным же образом получали 6-[2-(3-бензилоксифенил)хроман-6-илокси]пиридин-3-иламина гидрохлорид. 1- 26008539 добавляли 430 мг 10% палладия на угле в инертной атмосфере. Исходное вещество гидрировали при комнатной температуре с получением количественного выхода 3-[6-(5-аминопиридин-2-илокси)хроман 2-ил]фенол. 1(м, 1H), 1,96-1,89 (м, 1H). Пример 18. 2-Ацетиламино-N-[6-(2-фенилхроман-6-илокси)пиридин-3-ил]ацетамид. 5-Амино-2-(2-фенилхроман-6-илокси)пиридин (500 мг) и N-ацетилглицин (275 мг) растворяли в 35 мл дихлорметана. Добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (450 мг). Смесь перемешивали при комнатной температуре в течение 6 ч. Реакцию гасили добавлением воды, и образованный осадок отфильтровывали. 1H ЯМР (400 МГц, d6-ДМСО): 10,1 (с, 1H), 8,30 (д, 1H, J=2,7 Гц), 8,21 (т, 1H, J=5,7 Гц), 8,00 (дд,1H, J=2,7, 8,9 Гц), 7,47-7,30 (м, 5 Н), 6,95 (д, 1H, J=8,9 Гц), 6,87-6,84 (м, 3H), 5,12 (дд, 1H, J=1,90, 10,0 Гц),3,86 (д, 2 Н, J=5,7 Гц), 3,00-2,92 (м, 1H), 2,75-2,70 (м, 1H), 2,19-2,14 (м, 1H), 2,03-1,97 (м, 1H). Используя ту же самую методику, которая описана для 2-ацетиламино-N-[6-(2-фенилхроман-6 илокси)пиридин-3-ил]ацетамида, выше, но заменяя 5-амино-2-(2-фенилхроман-6-илокси)пиридина на 6-[2-(4-фторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(3-фторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(2-фторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(2,3-дифторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(2,4-дифторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(2,5-дифторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(2,6-дифторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(3,4-дифторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(3,5-дифторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(2-трифторметилфенил)хроман-6-илокси]пиридин-3 иламин,6-[2-(4-трифторметилфенил)хроман-6-илокси]пиридин-3 иламин,6-[2-(3-хлор-4-фторфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(2-хлорфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(3-хлорфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(2,4-дихлорфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(3-бромфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(4-этилфенил)хроман-6-илокси]пиридин-3-иламин,6-[2-(3-метоксифенил)хроман-6-илокси]пиридин-3-иламин,6-(3-метил-2-фенилхроман-6-илокси)пиридин-3-иламин,5-амино-2-(2-фенилхроман-7-илокси)пиридин,6-(5-аминопиридин-2-илокси)-2-фенилхроман-4-он,7-(5-аминопиридин-2-илокси)-2-фенилхроман-4-он,6-(5-аминопиридин-2-илокси)-3-метил-2-фенилхроман-4-он,6-(2-фенил-2,3-дигидробензо[1,4]диоксин-6-илокси)пиридин-3-иламин,6-(6-фенил-5,6,7,8-тетрагидронафталин-2-илокси)пиридин-3-иламин,6-(5-аминопиридин-2-илокси)-2-фенил-3,4-дигидро-2 Н-нафталин-1-он,6-(2-фенил-2,3-дигидробензо[1,4]оксатиин-6-илокси)пиридин-3-иламин,6-[3-(3-фторфенил)хроман-7-илокси]пиридин-3-иламин,6-(3-фенилхроман-7-илокси)пиридин-3-иламин,6-(5-аминопиридин-2-илокси)-2-фенилхромен-4-он,6-(2-фенилиндан-5-илокси)пиридин-3-иламин, получали 2-ацетиламино-N-6-[2-(4-фторфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(3-фторфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(2-фторфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(2,3-дифторфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(2,4-дифторфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(2,5-дифторфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(2,6-дифторфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(3,4-дифторфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N6-[2-(3,5-дифторфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(2-трифторметилфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(4-трифторметилфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(3-хлор-4-фторфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(2-хлорфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(3-хлорфенил)хроман-6-илокси]пиридин-3-илацетамид,- 27008539 2-ацетиламино-N-6-[2-(2,4-дихлорфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(3-бромфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(4-этилфенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-[2-(3-метоксифенил)хроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-6-(3-метил-2-фенилхроман-6-илокси]пиридин-3-илацетамид,2-ацетиламино-N-[6-(2-фенилхроман-7-илокси]пиридин-3 ил]ацетамид,2-ацетиламино-N-[6-(4-оксо-2-фенилхроман-6-илокси]пиридин-3-ил]ацетамид,2-ацетиламино-N-[6-(4-оксо-2-фенилхроман-7-илокси]пиридин-3-ил]ацетамид,2-ацетиламино-N-[6-(3-метил-4-оксо-2-фенилхроман-6-илокси]пиридин-3-ил]ацетамид,2-ацетиламино-N-[6-(2-фенил-2,3-дигидробензо[1,4]диоксин-6-илокси]пиридин-3-ил]ацетамид,2-ацетиламино-N-[6-(6-фенил-5,6,7,8-тетрагидронафталин-2-илокси)пиридин-3-ил]ацетамид,2-ацетиламино-N-[6-(5-оксо-6-фенил-5,6,7,8-тетрагидронафталин-2-илокси)пиридин-3-ил]ацетамид,2-ацетиламино-N-[6-(2-фенил-2,3-дигидробензо[1,4]оксатиин-6-илокси)пиридин-3-ил]ацетамид,2-ацетиламино-N-6-[3-(3-фторфенил)хроман-7-илокси]пиридин-3-илацетамид,2-ацетиламино-N-[6-(3-фенилхроман-7-илокси)пиридин-3 ил]ацетамид,2-ацетиламино-N-[6-(4-оксо-2-фенил-4 Н-хромен-6-илокси)пиридин-3-ил]ацетамид,2-ацетиламино-N-[6-(2-фенилиндан-5-илокси)пиридин-3 ил]ацетамид,соответственно. Пример 19. [6-(2-Фенилхроман-6-илокси)пиридин-3-ил]амид пиперидин-4-карбоновой кислоты. а) трет-Бутиловый эфир 4-[6-(2-Фенилхроман-6-илокси)пиридин-3-илкарбамоил]пиперидин-1 карбоновой кислоты. 5-Амино-2-(2-фенилхроман-6-илокси)пиридин (500 мг) и N-(трет-бутоксикарбонил) изонипекотиновую кислоту (541 мг) растворяли в 40 мл ТГФ. Добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (451 мг). Смесь нагревали с обратным холодильником в течение нескольких часов. Реакцию гасили добавлением воды и экстрагировали этилацетатом. Объединенные органические слои промывали водой, насыщенным раствором карбоната натрия, сушили Na2SO4 и выпаривали. 1b) [6-(2-Фенилхроман-6-илокси)пиридин-3-ил]амид пиперидин-4-карбоновой кислоты. Смесь трет-бутилового эфира 4-[6-(2-фенилхроман-6-илокси)пиридин-3-илкарбамоил]пиперидин-1 карбоновой кислоты (860 мг) и 1 М HCl в диэтиловом эфире (13 мл) перемешивали при комнатной температуре в течение 24 ч. Осадок отфильтровывали и промывали эфиром. 1(дд, 1H, J=2,8, 8,8 Гц), 7,39-7,46 (м, 4 Н), 7,34-7,35 (м, 1H), 7,00 (д, 1H, J=8,8 Гц), 6,86-6,88 (м, 3H), 5,12 (д,1H, J=10,5 Гц), 4,04 (м, 1H), 2,96 (м, 1H), 2,72 (м, 1H), 2,17 (м, 1H), 2,01 (м, 1H), 1,47 (д, 3H, J=6,9 Гц). Пример 21. 2-Амино-3-метил-N-[6-(2-фенилхроман-6-илокси)пиридин-3 ил]бутирамид и его производные. 2-Амино-3-метил-N-[6-(2-фенилхроман-6-илокси)пиридин-3 ил]бутирамид и его производные получали, как описано для [6-(2-фенилхроман-6-илокси)пиридин-3-ил]амина пиперидин-4-карбоновой кислоты в примере 19 а) и b), но заменяя N-(трет-бутоксикарбонил)изонипекотиновую кислоту (S), (R) илиa) Трет-бутиловый эфир S)-1-6-[2-(3-гидроксифенил)хроман-6-илокси]пиридин-3-илкарбамоил 2-метилпропил)карбаминовой кислоты. Трет-бутиловый эфир S)-1-6-[2-(3-гидроксифенил)хроман-6-илокси]пиридин-3-илкарбамоил-2 метилпропил)карбаминовой кислоты получали, используя ту же самую методику, которая описана в примере 21 а) для трет-бутилового эфира (S)-2-метил-1-[6-(2-фенилхроман-6-илокси)пиридин-3 илкарбамоил] пропилкарбаминовой кислоты, но заменяя 5-амино-2-(2-фенилхроман-6-илокси)пиридин 3-[6-(5-аминопиридин-2-илокси)хроман-2-ил]фенолом (описан в примере 17). 1b) трет-Бутиловый эфир [(S)-2-метил-1-(6-2-[3-(5-нитропиридин-2-илокси)фенил]хроман-6 илоксипиридин-3-илкарбамоил)пропил]карбаминовой кислоты. трет-Бутиловый эфир [(S)-2-метил-1-(6-2-[3-(5-нитропиридин-2-илокси)фенил]хроман-6-илокси пиридин-3-илкарбамоил)пропил]-карбаминовой кислоты как описано для 5-нитро-2-(2-фенилхроман-6 илокси)пиридина в примере 1b). 1(S)-2-Амино-3-метил-N-(6-2-[3-(5-нитропиридин-2-илокси)фенил]хроман-6-илоксипиридин-3 ил)бутирамида гидрохлорид получали таким же образом, как описано для (S)-2-амино-3-метил-N-[6-(2 фенилхроман-6-илокси)-пиридин-3 ил] бутирамида в примере 21 b). 1[6-(2-Фенилхроман-6-илокси)пиридин-3-ил]амид пирролидин-2-карбоновой кислоты и его производные получали, как описано для [6-(2-фенилхроман-6-илокси)пиридин-3-ил]амина пиперидин-4 карбоновой кислоты в примере 19 а) и b), но заменяя N-(трет-бутоксикарбонил)изонипекотиновую кислоту 1-трет-бутиловым эфиром (S), (R) или (R,S)-пирролидин-1,2-дикарбоновой кислоты и 5-амино-2(2-фенилхроман-6-илокси)пиридин соответствующими 5-аминопиридиновыми производными. а) трет-Бутиловый эфир 2-[6-(2-фенилхроман-6-илокси)пиридин-3-илкарбамоил]пирролидин-1 карбоновой кислоты и его производные. трет-Бутиловый эфир 2-[6-(2-фенилхроман-6-илокси)пиридин-3-илкарбамоил]-(S)-пирролидин-1 карбоновой кислоты. 1
МПК / Метки
МПК: C07D 405/14, C07D 405/12, A61K 31/4433, C07D 411/12, C07D 213/75, A61P 9/00
Метки: производные, пиридина, обмена, пригодные, ингибирования, системы
Код ссылки
<a href="https://eas.patents.su/30-8539-proizvodnye-piridina-prigodnye-dlya-ingibirovaniya-sistemy-obmena-natriya-kalciya.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиридина, пригодные для ингибирования системы обмена натрия/кальция</a>
Новые соединения, являющиеся сильнодействующими ингибиторами механизма обмена ионов na+/ca2+ при лечении аритмии

Номер патента: 6580
Опубликовано: 24.02.2006
Авторы: Котовуори Пекка, Коскелайнен Туула, Тенхунен Юкка, Термякангас Олли, Раску Сирпа, Норе Пентти, Отсомаа Леена, Тиайнен Эйя, Карьялайнен Арто
МПК: C07C 217/14, A61K 31/353, A61K 31/135...
Метки: являющиеся, новые, ингибиторами, сильнодействующими, механизма, обмена, соединения, аритмии, лечении, ионов
Формула / Реферат:
1. Соединения формулы (I) в которой X представляет собой -O-, -CH2- или -C(O)-; Z представляет собой -CHR9- или валентную связь; Y представляет собой -CH2-, -C(O)-, CH(OR10)-, -CH(NR11R12)-, -O-, -S-, -S(O)- или -S(O2)-, при условии, что в том случае, когда Z представляет собой валентную связь, Y не является C(O); пунктирная линия представляет собой необязательную двойную связь в том случае, когда Z представляет собой -CR9- и Y представляет...
Фармацевтические композиции с контролируемым высвобождением, содержащие альгинат натрия и альгинат натрия-кальция

Номер патента: 7488
Опубликовано: 27.10.2006
Авторы: Кинцл Майя, Крамар Андрейка, Брезник Марьянка, Врецер Франц, Писек Роберт
МПК: A61K 9/22, A61K 9/16, A61K 31/7048...
Метки: фармацевтические, контролируемым, содержащие, натрия, высвобождением, альгинат, натрия-кальция, композиции
Формула / Реферат:
1. Фармацевтическая композиция с контролируемым высвобождением, содержащая (а) по меньшей мере один активный ингредиент, (б) альгинат натрия и (в) альгинат натрия-кальция, где массовое соотношение альгината натрия (б) и альгината натрия-кальция (в) составляет 1 к по меньшей мере 1,1. 2. Фармацевтическая композиция по п.1, где массовое соотношение альгината натрия (б) и альгината натрия- кальция (в) составляет от 1:1,1 до 1:10. 3....
Производные пиридина, ингибирующие фосфодиэстеразу iv

Номер патента: 4207
Опубликовано: 26.02.2004
Авторы: Матесанс-Бальестерос Мария Энкарнасьон, Диас-Мартинес Адольфо, Фрейн Эдди Жан Эдгар, Дильс Гастон Станислас Марселла
МПК: A61K 31/4427, C07D 401/06, A61P 29/00...
Метки: пиридина, ингибирующие, фосфодиэстеразу, производные
Формула / Реферат:
1. Соединение, имеющее формулу где L обозначает водород; -A-B- обозначает двухвалентный радикал формулы -CR4=CR5- (a-1) или -CHR4-CHR5- (a-2); D обозначает O или NR6; Q обозначает радикал формулы R1 обозначает водород; R2 обозначает водород, C1-6алкил или гидрокси; R3 обозначает водород или C1-6алкил; R4 и R5, каждый независимо, выбраны из водорода или C1-4алкила; R6 обозначает водород, C1-4алкил или циано; R7 и R8, каждый независимо,...
Производные индола, пригодные для лечения расстройств цнс

Номер патента: 6315
Опубликовано: 27.10.2005
Авторы: Келер Ян, Феллинг Якоб, Андерсен Ким, Банг-Андерсен Бенни
МПК: A61P 25/06, C07D 401/14, A61K 31/4045...
Метки: производные, индола, пригодные, расстройств, цнс, лечения
Формула / Реферат:
1. Замещенное производное индола формулы I где (a) один из Y1 и Y2 представляет собой N, который связан с Y4, а другой из Y1 и Y2 представляет собой CO, CS, SO или SO2 и Y4 представляет собой CH2; (b) один из Y1 и Y2 представляет собой N, который связан с Y4, а другой из Y1 и Y2 представляет собой CH2 и Y4 представляет собой CO, CS, SO или SO2; или (c) один из Y1 и Y2 представляет собой N, который связан с Y4, а другой из Y1 и Y2 представляет...
Система связи низкой стоимости и большого радиуса действия для обмена данными с удаленными или мобильными полевыми модулями и способ работы такой системы связи

Номер патента: 414
Опубликовано: 24.06.1999
Авторы: Уайт Джеффри Р., Нельсон Родни, Фитцджеральд Брендан Т., Поушок Эндрю Т., Белчер Дональд К., Дарби Альберт Д.
МПК: H04B 7/204
Метки: системы, радиуса, данными, связи, работы, система, стоимости, модулями, действия, мобильными, удаленными, полевыми, обмена, низкой, способ, большого
Формула / Реферат:
1. Двухсторонняя беспроводная система передачи данных, содержащая подсистему выходных сообщений для отправки выходных сообщений; подсистему входных сообщений, содержащую сеть, по меньшей мере, из двух работающих на радиочастотах базовых станций для приема входных сообщений, причем указанные базовые станции также непрерывно производят замеры в своих местах расположения комплекта частотных каналов в полосе радиочастот передачи входных сообщений...
Предыдущий патент: Способы и композиции для лечения сердечно-сосудистого заболевания доставкой генов in vivo
Следующий патент: Производные хинолина и изохинолина, способ их получения и их применение в качестве ингибиторов воспаления
Случайный патент: Композиция фруктового льда