Бета-аминотетрагидроимидазо – (1, 2 – а) – пиразины и тетрагидротриазоло – (4, 3 – а ) – пиразины как ингибиторы дипептидилпептидазы для лечения или предотвращения диабета
Номер патента: 6845
Опубликовано: 28.04.2006
Авторы: Ким Доосеоп, Эдмондсон Скотт Д., Фишер Майкл Г., Ксу Дзинйоу, Маккосс Малкольм, Вебер Энн Э., Парми Эмма Р.












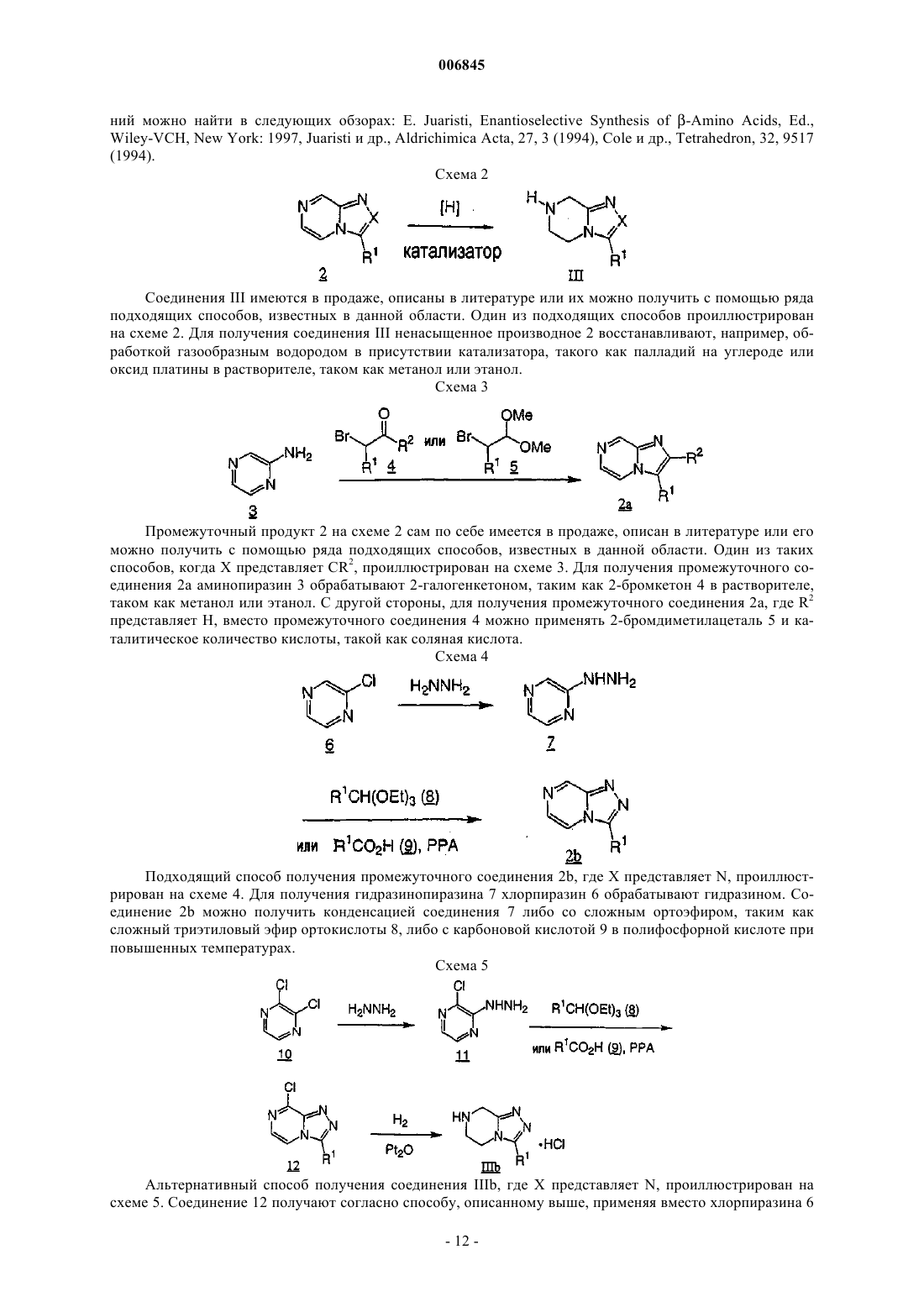
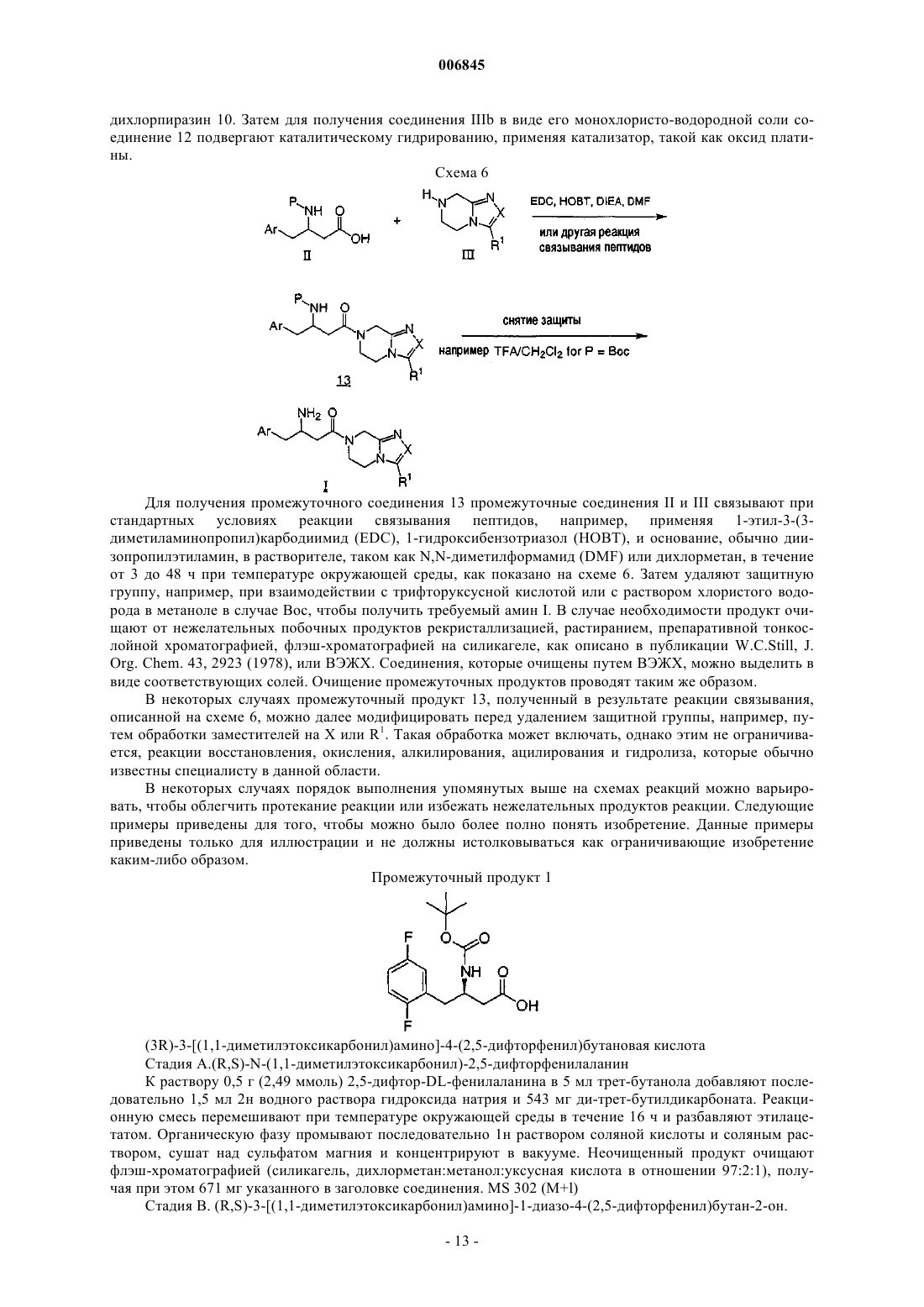








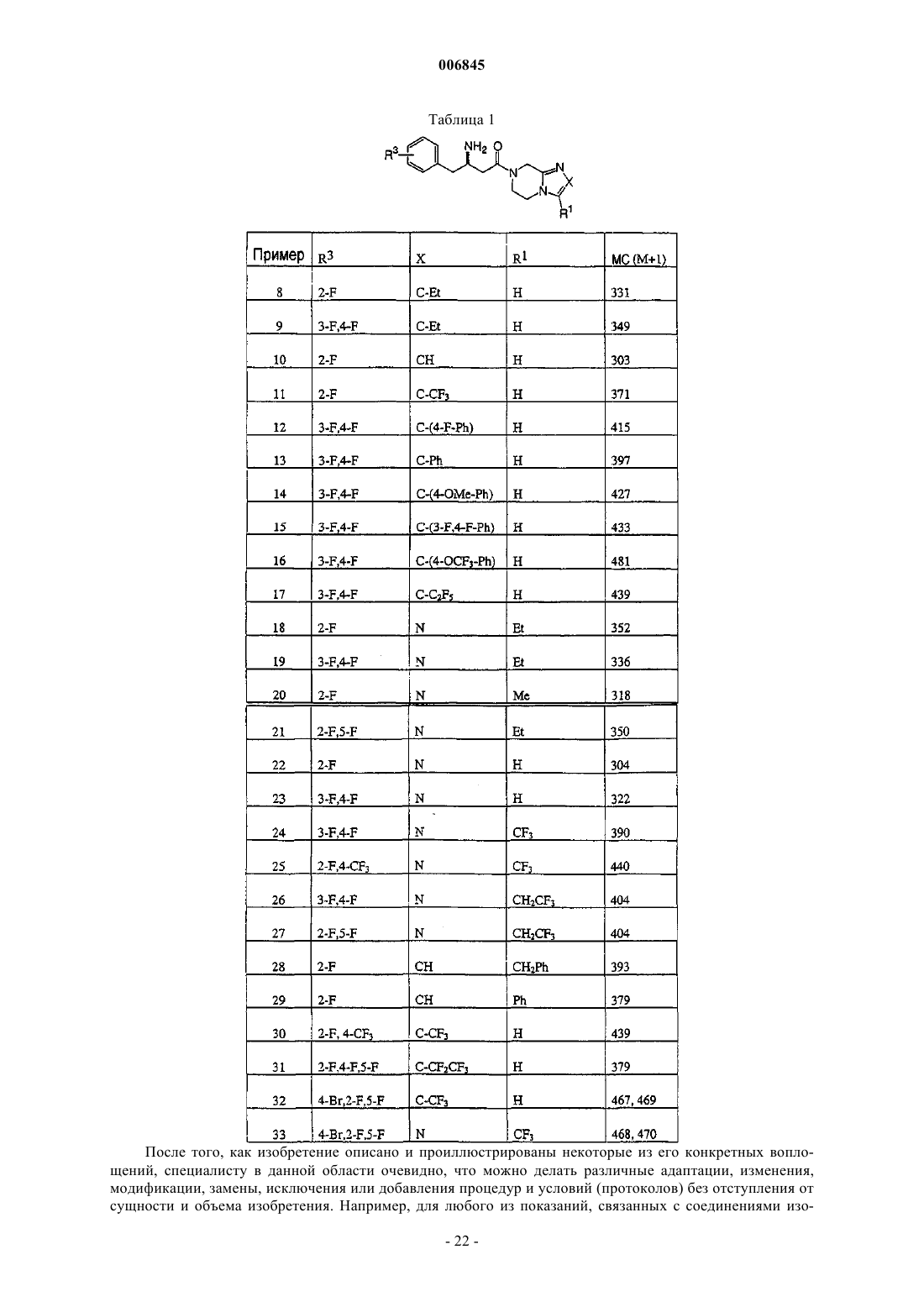

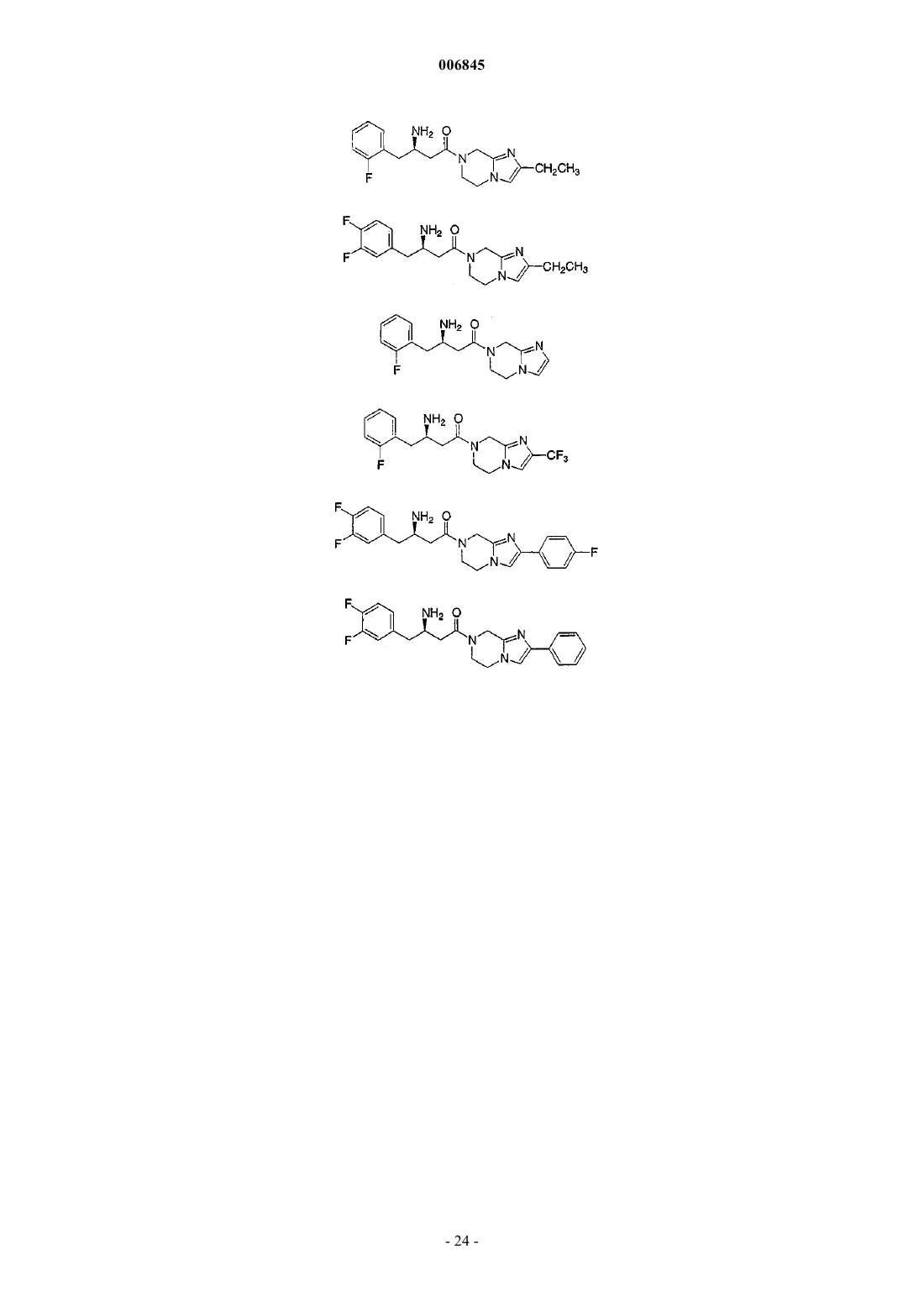

Формула / Реферат
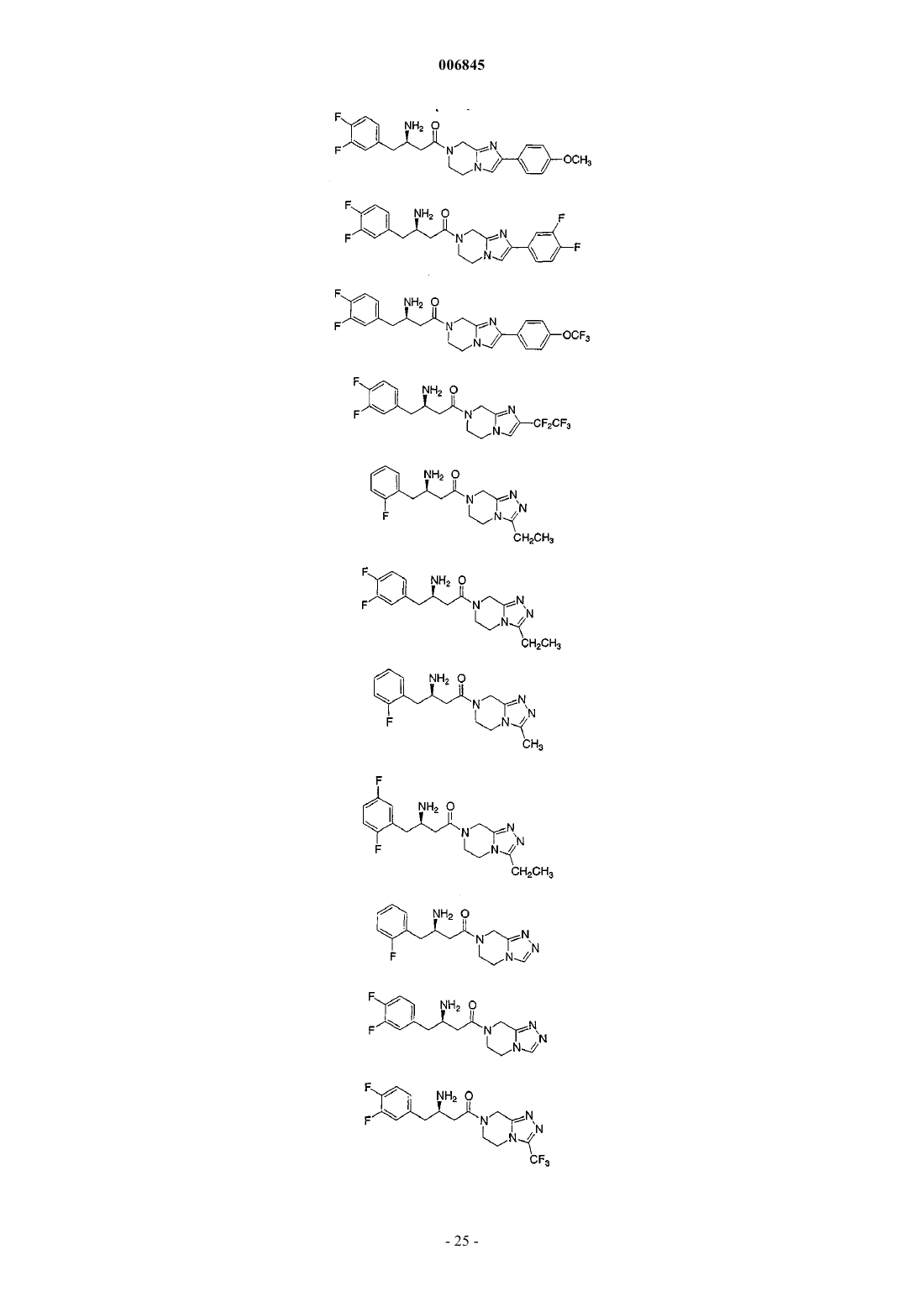
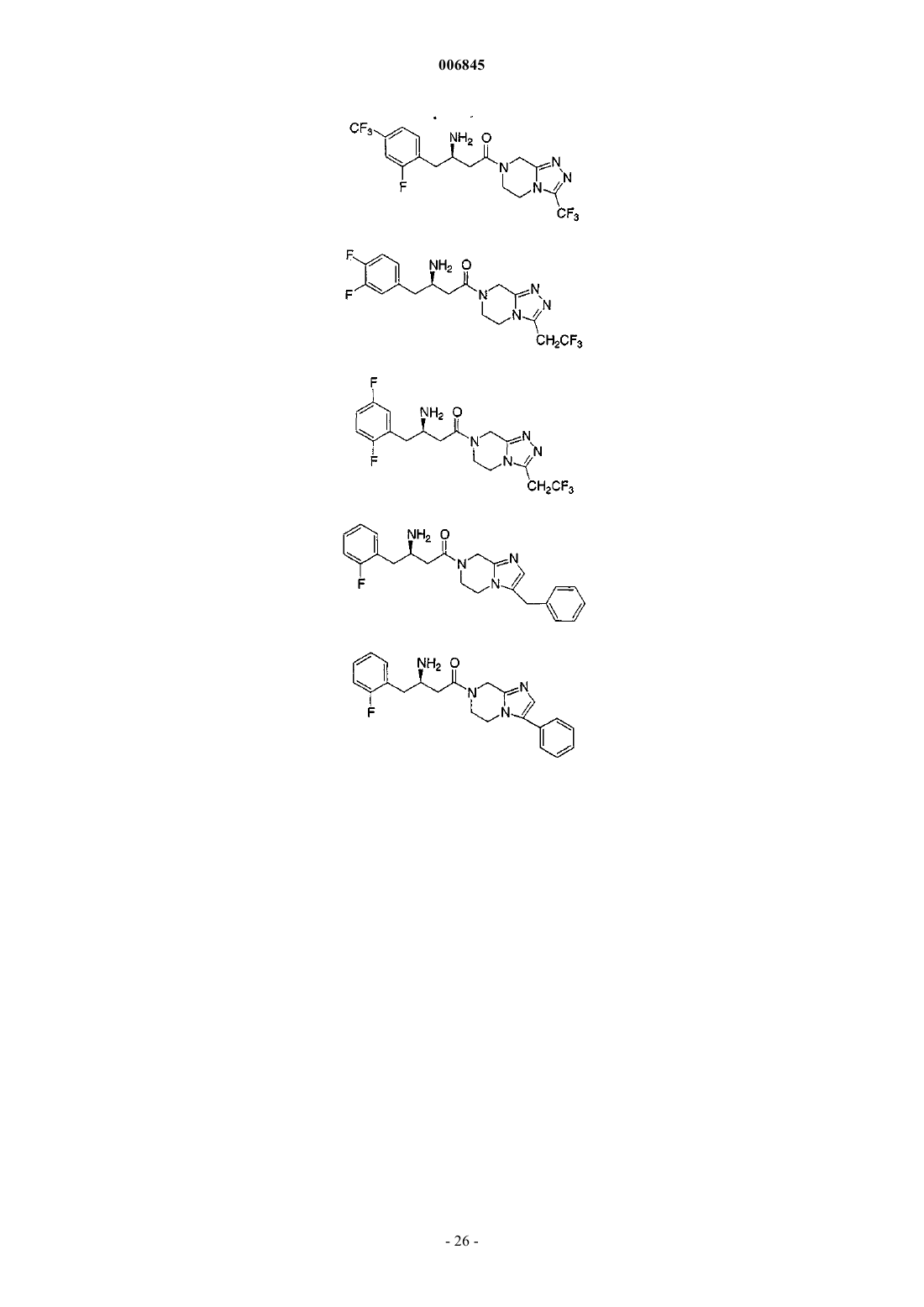
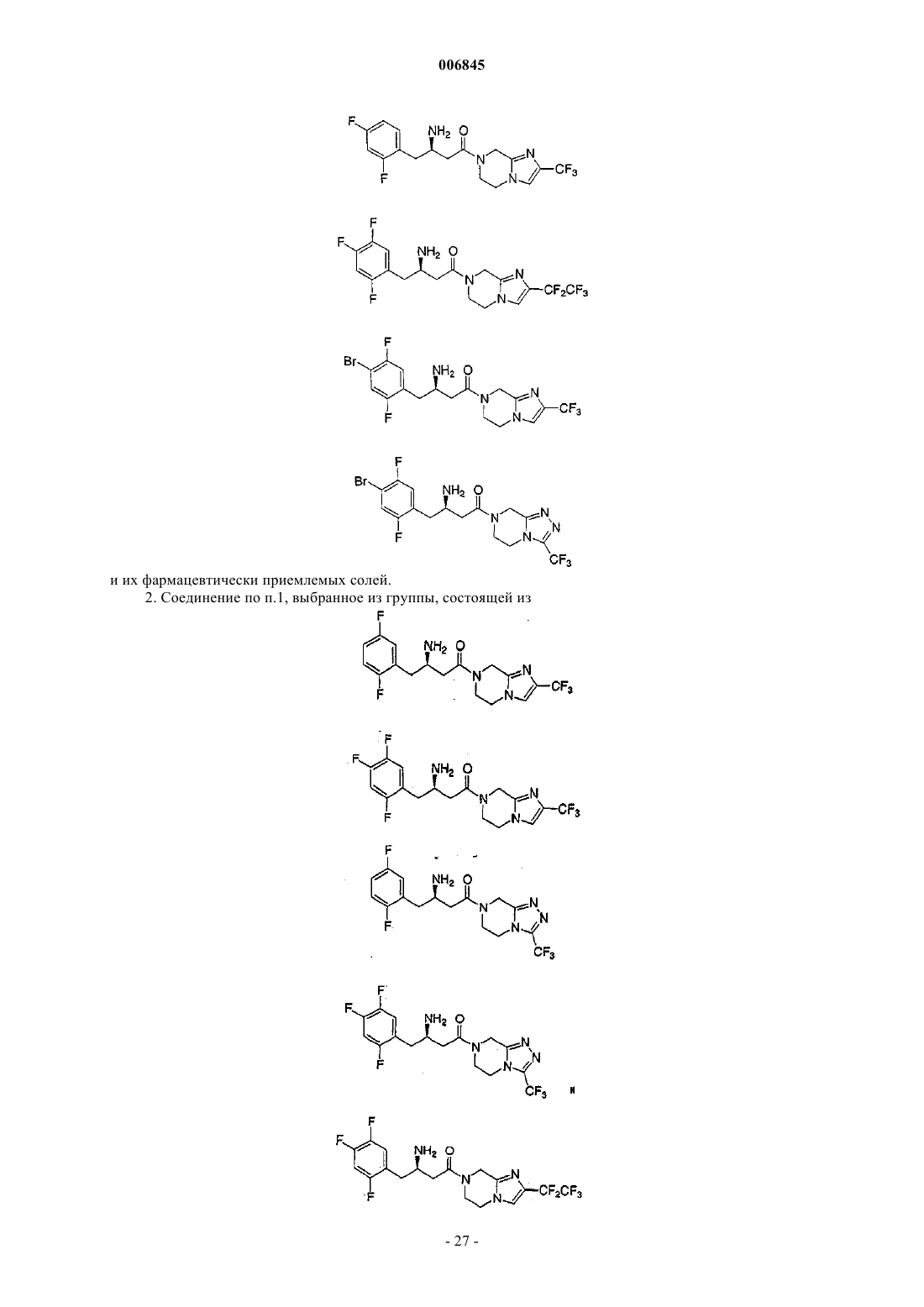
1. Соединение, выбранное из группы, состоящей из







и их фармацевтически приемлемых солей.
2. Соединение по п.1, выбранное из группы, состоящей из


или его фармацевтически приемлемая соль.
3. Соединение по п.2, которое представляет

или его фармацевтически приемлемая соль.
4. Соединение по п.2, которое представляет

или его фармацевтически приемлемая соль.
5. Соединение по п.2, которое представляет

или его фармацевтически приемлемая соль.
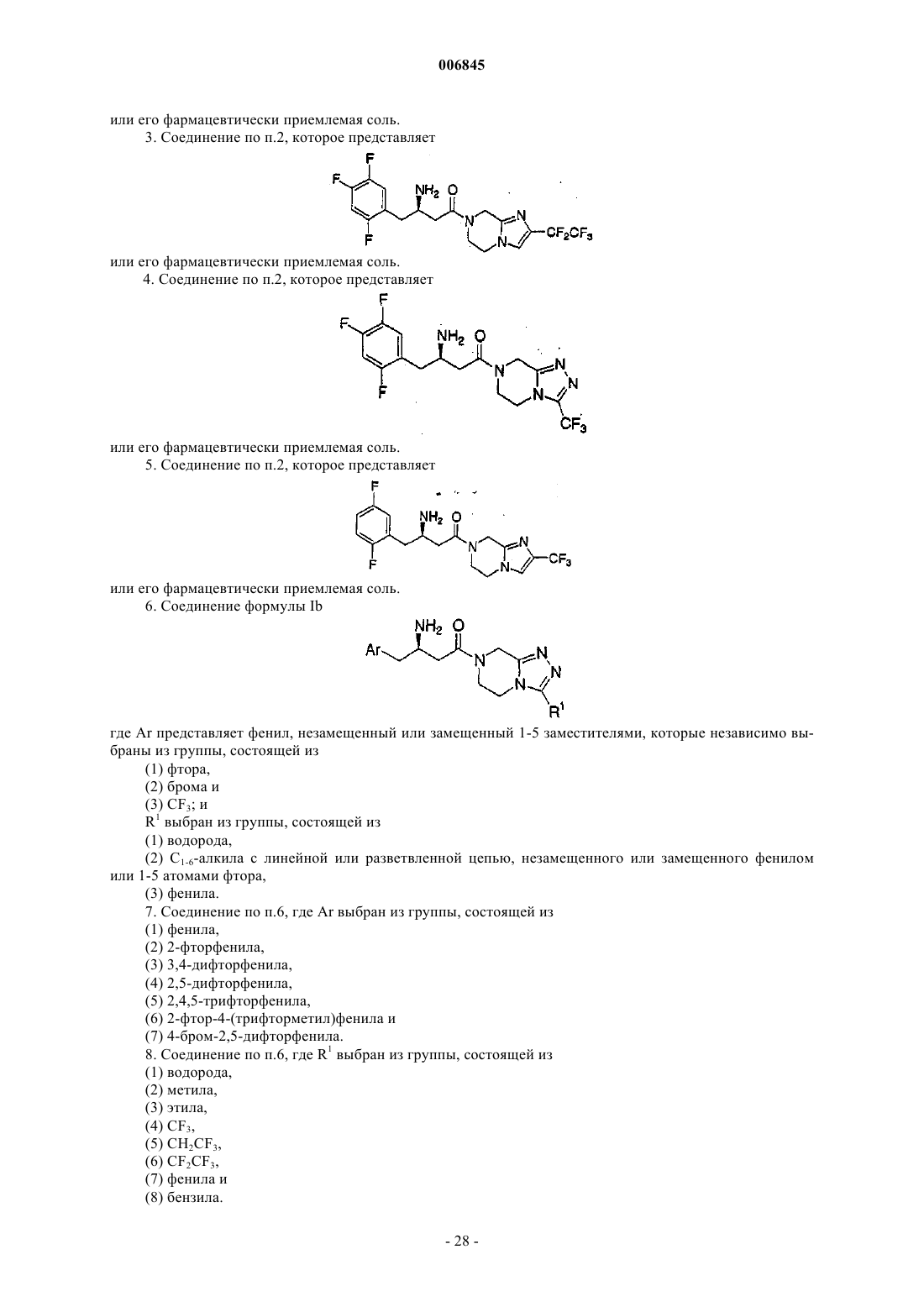
6. Соединение формулы Ib

где Аr представляет фенил, незамещенный или замещенный 1-5 заместителями, которые независимо выбраны из группы, состоящей из
(1) фтора,
(2) брома и
(3) CF3; и
R1 выбран из группы, состоящей из
(1) водорода,
(2) C1-6-алкила с линейной или разветвленной цепью, незамещенного или замещенного фенилом или 1-5 атомами фтора,
(3) фенила.
7. Соединение по п.6, где Аr выбран из группы, состоящей из
(1) фенила,
(2) 2-фторфенила,
(3) 3,4-дифторфенила,
(4) 2,5-дифторфенила,
(5) 2,4,5-трифторфенила,
(6) 2-фтор-4-(трифторметил)фенила и
(7) 4-бром-2,5-дифторфенила.
8. Соединение по п.6, где R1 выбран из группы, состоящей из
(1) водорода,
(2) метила,
(3) этила,
(4) CF3,
(5) CH2CF3,
(6) CF2CF3,
(7) фенила и
(8) бензила.
9. Соединение по п.8, где R1 выбран из группы, состоящей и:
(1) водорода,
(2) метила,
(3) этила,
(4) CF3 и
(5) CH2CF3.
10. Соединение по п.9, где R1 водород или CF3.
11. Соединение формулы Iс

где Аr представляет фенил, незамещенный или замещенный 1-5 заместителями, которые независимо выбраны из группы, состоящей из
(1) фтора,
(2) брома, и
(3) CF3; и
R1 выбран из группы, состоящей из:
(1) водорода,
(2) C1-6-алкила с линейной или разветвленной цепью, незамещенного или замещенного фенилом или 1-5 атомами фтора и
R2 выбран из
(1) водорода,
(2) C1-6-алкила с линейной или разветвленной цепью, незамещенного или замещенного 1-5 атомами фтора,
(3) фенила, незамещенного или замещенного 1-3 заместителями, независимо выбранными из фтора, ОСН3 и OCF3.
12. Соединение по п.11, где R2 выбран из группы, состоящей из
(1) водорода,
(2) метила,
(3) этила,
(4) CF3,
(5) CH2CF3,
(6) CF2CF3,
(7) фенила,
(8) (4-метокси)фенила,
(9) (4-трифторметокси)фенила,
(10) 4-фторфенила и
(11) 3,4-дифторфенила.
13. Соединение по п.12, где R2 представляет CF3 или CF2CF3.
14. Фармацевтическая композиция, содержащая инертный носитель и соединение по п.6 или 11.
15. Применение соединения по п.6 или 11 для изготовления лекарственного средства для применения для лечения состояния, выбранного из группы, выбранной из гипергликемии, диабета типа 2, ожирения, и липидных расстройств у млекопитающих.
Текст
006845 Предшествующий уровень техники Диабет относится к заболеванию, вызываемому большим количеством факторов, и характеризуется повышенными уровнями глюкозы в плазме или гипергликемией, наблюдаемой в состоянии натощак или после введения глюкозы во время проведения пробы на толерантность к глюкозе. Устойчивая или неконтролируемая гипергликемия связана с повышенной и преждевременной заболеваемостью и смертностью. Часто аномальный гомеостаз глюкозы связан как прямо, так и косвенно с альтерациями липидного, липопротеинового и аполипопротеинового обмена и другим метаболическим и гемодинамическим заболеванием. Следовательно, пациенты с сахарным диабетом типа 2 подвержены особенно высокому риску осложнений, связанных с крупными и мелкими кровеносными сосудами, включая коронарную болезнь сердца, удар, заболевание периферийных сосудов, гипертонию, нефропатию, невропатию и ретинопатию. Поэтому для клинической терапии и лечения сахарного диабета крайне необходим терапевтический контроль гомеостаза глюкозы, липидного обмена и гипертонии. Существует две общепризнанные формы диабета. При диабете типа 1, или инсулинзависимом сахарном диабете (IDDM), у пациентов вырабатывается мало инсулина или в общем случае инсулин, гормон, регулирующий утилизацию глюкозы, не вырабатывается. При диабете типа 2, или инсулиннезависимом сахарном диабете (NIDDM), уровни инсулина в плазме пациентов по сравнению с субъектами, не страдающими диабетом, часто такие же или даже выше; однако у таких пациентов развивается резистентность к инсулину, обладающему стимулирующим влиянием на метаболизм глюкозы и липидов в основных, восприимчивых к инсулину тканях, которые представляют мышечную, печеночную и жировую ткани, а уровни инсулина в плазме, несмотря на то, что они повышены, недостаточны для преодоления явно выраженной резистентности к инсулину. Резистентность к инсулину обусловлена, в первую очередь, не уменьшением числа инсулиновых рецепторов, а дефектом, связанным с инсулиновым рецептором после его связывания с инсулином, который еще не объяснен. Такая резистентность к восприимчивости инсулина приводит к недостаточной инсулиновой активации поглощения, окисления и отложения глюкозы в мышцах и неадекватному инсулиновому подавлению липолиза в жировой ткани и выработки глюкозы, а также секреции в печени. Применяемые в настоящее время способы лечения диабета типа 2, которые за много лет существенно не изменились, имеют известные ограничения. Несмотря на то, что физическая нагрузка и уменьшение потребляемых с пищей калорий разительно улучшают состояние больных диабетом, мало кто согласен с таким лечением из-за хорошо укоренившегося сидячего образа жизни и избыточного потребления пищевых продуктов, особенно продуктов, содержащих высокие количества насыщенных жиров. Повышение уровня инсулина в плазме путем введения производных сульфонилмочевины (например, толбутамида и глипизида) или меглитинида, которые стимулируют -клетки поджелудочной железы к выделению большего количества инсулина, и/или путем инъекции инсулина, когда производные сульфонилмочевины или меглитинид становятся неэффективными, может приводить к концентрациям инсулина, достаточно высоким для стимулирования тех самых резистентных к инсулину тканей. Однако в результате введения инсулина или стимуляторов секреции инсулина (производные сульфонилмочевины или меглитинид) уровни глюкозы в плазме могут стать опасно низкими, а из-за равномерного повышения уровней инсулина в плазме уровень резистентности к инсулину может повышаться. Бигуаниды повышают восприимчивость к инсулину, приводя к некоторой коррекции гипергликемии. Однако два бигуанида, фенформин и метформин, могут вызывать молочный ацидоз и тошноту/диарею. Метформин имеет меньше побочных эффектов, чем фенформин, и часто прописывается для лечения диабета типа 2. Глитазоны (например, 5-бензилтиазолидин-2,4-дионы) относятся к ранее описанному классу соединений, потенциально способных улучшать многие симптомы диабета типа 2. В некоторых моделях диабета типа 2 у животных такие агенты существенно повышают восприимчивость мышечной, печеночной и жировой ткани к инсулину, приводя к частичной или полной коррекции повышенных уровней глюкозы в плазме без появления гипогликемии. Имеющиеся в настоящее время в продаже глитазоны являются агонистами рецептора, активированного пероксисомным пролифератором (PPAR), прежде всего подтипа гамма-PPAR. Как полагают, гамма-агонизм PPAR отвечает за улучшенную восприимчивость к инсулину,которая наблюдается с глитазонами. Более новые агонисты PPAR, которые испытываются для лечения диабета типа 2, являются агонистами подтипа альфа, гамма или дельта или их комбинацией, и во многих случаях химически отличаются от глитазонов (то есть, они не являются тиазолидиндионами). Некоторые из глитазонов, такие как троглитазон, обладают серьезными побочными эффектами (например, вызывают интоксикацию печени). В настоящее время изучаются дополнительные способы лечения заболевания. Введенные недавно или все еще развивающиеся новые биохимические подходы включают лечение ингибиторами альфаглюкозидазы (например, акарбозой) и ингибиторами (протеин-тирозин)фосфатазы-1 В (РТР-1 В). В качестве лекарственных средств исследуются соединения, являющиеся ингибиторами фермента дипептидилпептидазы-IV (DP-IV или DPP-IV), которые также можно применять для лечения диабета, и в частности, диабета типа 2. См., например, следующие публикации: заявки WO 97/40832, WO 98/19998, патент США 5939560; Bioorg. Med. Chem. Lett., 6(10), 1163-1166 (1996); и Bioorg. Med.Chem. Lett., 6(22), 2745-2748 (1996). Применение ингибиторов DP-IV для лечения диабета типа 2 основа-1 006845 но на том факте, что DP-IV in vivo легко инактивирует глюкагон, подобный пептиду-1 (GLP-1) и полипептиду, угнетающему секрецию желудка (GIP). GLP-1 и GIP являются инкретинами и вырабатываются после потребления пищи. Инкретины стимулируют выработку инсулина. Ингибирование DP-IV приводит к уменьшению инактивации инкретинов, а это, в свою очередь, приводит к повышению эффективности инкретинов при стимулировании продуцирования инсулина поджелудочной железой. Поэтому ингибирование DP-IV приводит к повышению уровня сывороточного инсулина. Так как инкретины вырабатываются в организме преимущественно только после употребления пищи, ингибирование DP-IV, как ожидается, в промежутки времени между принятием пищи не будет увеличивать уровень инсулина, который может приводить к чрезмерно низкому сахару в крови (гипогликемии). Ингибирование DP-IV, как ожидается, будет повышать инсулин без увеличения риска гипогликемии, которая является опасным побочным эффектом, связанным с применением стимуляторов секреции инсулина. У ингибиторов DP-IV также есть другие обсуждаемые здесь терапевтические применения. До настоящего времени ингибиторы DP-IV широко не изучались, в частности, в отношении других заболеваний, отличных от диабета. Для того, чтобы найти улучшенные ингибиторы DP-IV для лечения диабета и потенциально других заболеваний и состояний, необходимы новые соединения. Краткое описание изобретения Настоящее изобретение относится к соединениям, являющимся ингибиторами фермента дипептидилпептидазы-IV (ингибиторы DP-IV) и применимым для лечения или профилактики заболеваний, в которые вовлечен фермент дипептидилпептидаза-IV, таких как диабет и, в частности, диабет типа 2. Изобретение также относится к фармацевтическим композициям, содержащим такие соединения, и применению таких соединений и композиций для профилактики или лечения таких заболеваний, в которые вовлечен фермент дипептидилпептидаза-IV. Подробное описание изобретения Настоящее изобретение относится к соединениям формулы Ib и Iс и фармацевтически приемлемым солям указанных соединений. Воплощение настоящего изобретения включает соединения формулы IbIb где Аr и R1 определены здесь; их фармацевтически приемлемые соли. Другое воплощение настоящего изобретения включает соединения формулы Iс где Ar, R1 и R2 определены здесь; их фармацевтически приемлемые соли и индивидуальные диастереомеры. В настоящем изобретении Аr предпочтительно представляет фенил, незамещенный или замещенный 1-5 заместителями, которые независимо выбраны из группы, состоящей из(3) CF3. В настоящем изобретении Аr более предпочтительно выбирают из группы, состоящей из(7) 4-бром-2,5-дифторфенила. В настоящем изобретении R1 предпочтительно выбирают из группы, состоящей из(2) C1-6-алкила с линейной или разветвленной цепью, незамещенного или замещенного фенилом-2 006845 или 1-5 атомами фтора,(3) фенила. В настоящем изобретении R1 более предпочтительно выбирают из группы, состоящей из:(7) бензила. В настоящем изобретении R1 более предпочтительно выбирают из группы, состоящей из(5) CH2CF3. В настоящем изобретении R1 наиболее предпочтительно представляет водород или CF3. В настоящем изобретении R2 предпочтительно выбирают из(1) водорода,(2) C1-6-алкила с линейной или разветвленной цепью, незамещенного или замещенного 1-5 атомами фтора,(3) фенила, незамещенного или замещенного 1-3 заместителями, независимо выбранными из фтора,ОСН 3 и OCF3. В настоящем изобретении R2 более предпочтительно выбирают из группы, состоящей из(10) 3,4-дифторфенила. В настоящем изобретении R2 наиболее предпочтительно представляет CF3 или CF2CF3. В настоящем изобретении R3 предпочтительно представляет F, Вr или CF3. Соединения настоящего изобретения могут содержать один или более асимметрических центров и,следовательно, могут существовать в виде рацематов и рацемических смесей, отдельных энантиомеров,смесей диастереомеров и индивидуальных диастереомеров. Соединения настоящего изобретения имеют один асимметрический центр на атоме углерода в бета-положении. В зависимости от природы различных заместителей в молекуле могут присутствовать дополнительные асимметрические центры. Каждый такой асимметрический центр независимо продуцирует два оптических изомера, при этом подразумевается, что все возможные оптические изомеры и диастереомеры в смесях, в виде чистых или частично очищенных соединений включены в объем настоящего изобретения. Подразумевается, что настоящее изобретение включает в себя все изомерные формы таких соединений. Некоторые из описанных здесь соединений содержат олефиновые двойные связи, и если не определено иначе, подразумевается, что в объем изобретения включены как Е, так и Z геометрические изомеры. Некоторые из описанных здесь соединений могут существовать в виде таутомеров, отличающихся присоединением водорода к различным атомам, которое сопровождается одним или более сдвигом двойных связей. Например, кетон и его енольная форма являются кето-енольными таутомерами. Индивидуальные таутомеры также как их смеси включены в объем настоящего изобретения. Формула I иллюстрирует структуру класса соединений без учета предпочтительной стереохимии. Формула Iа иллюстрирует предпочтительную стереохимию на атоме углерода, связанном с аминогруппой бета-аминокислоты, из которой получают данные соединения. Как известно в данной области, можно провести независимый синтез таких диастереомеров или можно достичь их разделения хроматографически при соответствующей модификации раскрываемой здесь методики. Их абсолютную стереохимию можно определить путем рентгенокристаллографического исследования кристаллических продуктов или кристаллических промежуточных продуктов, которые получают в случае необходимости с реактивом, содержащим асимметрический центр с известной абсолютной конфигурацией.-3 006845 В случае необходимости рацемические смеси соединений можно разделить, чтобы выделить индивидуальные энантиомеры. Разделение можно провести с помощью способов, известных в данной области, таких как сочетание соединений рацемической смеси с энантиомерно чистым соединением для образования смеси диастереомеров с последующим разделением индивидуальных диастереомеров стандартными способами, такими как фракционная кристаллизация или хроматография. Реакция сочетания часто представляет собой образование солей с применением энантиомерно чистой кислоты или основания. Производные диастереомеров можно затем преобразовать до чистых энантиомеров путем отщепления добавленного хирального остатка. Рацемическую смесь соединений можно также разделить прямыми хроматографическими способами с применением хиральных стационарных фаз, способами, которые известны в данной области. С другой стороны, любой энантиомер соединения можно получить путем стереоселективного синтеза, применяя оптически чистые исходные материалы или реактивы известной конфигурации с помощью способов, хорошо известных в данной области. Термин фармацевтически приемлемые соли относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли, получаемые из неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и т.п. Особенно предпочтительны соли аммония, кальция, магния, калия и натрия. В твердой форме соли могут существовать в виде более чем одной кристаллической структуры, а также могут быть в форме гидратов. Соли, полученные из фармацевтически приемлемых, органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая существующие в природе замещенные амины, циклические амины и основные ионообменные полимеры, такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы,прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Когда соединение настоящего изобретения является основным, соли можно получить из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромисто-водородную, соляную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфокислоту и т.п.Особенно предпочтительны лимонная, бромисто-водородная, соляная, малеиновая, фосфорная, серная, фумаровая и винная кислоты. Понятно, что применяемые здесь ссылки на соединения формулы I предполагают также включение фармацевтически приемлемых солей. Как очевидно специалисту, применяемый здесь термин галоген подразумевает включение фтора,хлора, брома и йода. Точно так же обозначение C1-8 в C1-8-алкиле обозначает группу, содержащую 1, 2, 3,4, 5, 6, 7 или 8 атомов углерода, с линейным или разветвленным расположением атомов, так что C1-8 алкил, в частности, включает метил, этил, н-пропил, изопропил, н-бутил, изобутил, грет-бутил, пентил,гексил, гептил и октил. Аналогично обозначение С 0 в С 0-алкиле обозначает присутствие прямой ковалентной связи. Группа, обозначаемая как независимо замещенная заместителями, может быть независимо замещена большим числом таких заместителей. Применяемый здесь термин гетероцикл включает системы из 5- или 6-членных колец, которые представлены в следующем списке: бензимидазолил, бензодиоксанил, бензофуранил, бензопиразолил, бензотиадиазолил, бензотриазолил, бензотиофенил, бензоксадиазолил, бензоксазолил, карбазолил, карболинил, хроманил, циннолинил, фуранил, имидазолил,индолинил, индолил, индолазинил, индазолил, изобензофуранил, изоиндолил, изохинолил, изотиазолил,изоксазолил, нафтиридинил, оксадиазолил, оксазолил, пиразинил, пиразолил, пиридопиридинил, пиридазинил, пиридил, пиримидил, пирролил, хиназолинил, хинолинил, хиноксалинил, тетразолил, тиадиазолил, тиазолидинил, тиазолил, тиенил, триазолил, азетидинил, 1,4-диоксанил, гексагидроазепинил, пиперазинил, пиперидинил, пирролидинил, морфолинил, тиоморфолинил, дигидробензимидазолил, дигидробензофуранил, дигидробензотиофенил, дигидробензоксазолил, дигидрофуранил, дигидроимидазолил,дигидроиндолил, дигидроизооксазолил, дигидроизотиазолил, дигидрооксадиазолил, дигидрооксазолил,дигидропиразинил, дигидропиразолил, дигидропиридинил, дигидропиримидинил, дигидропирролил,дигидрохинолинил, дигидротетразолил, дигидротиадиазолил, дигидротиазолил, дигидротиенил, дигидротриазолил, дигидроазетидинил, метилендиоксибензоил, тетрагидрофуранил, тетрагидроимидазолил,тетрагидроизохинолинил и тетрагидротиенил. Иллюстрацией изобретения является применение описанных здесь и в примерах соединений. Определенные соединения, соответствующие настоящему изобретению, включают соединение, выбранное из группы, состоящей из соединений, описанных в следующих примерах, их фармацевтически приемлемых солей и их индивидуальных диастереомеров.-4 006845 Представленные соединения применимы для способа ингибирования фермента дипептидилпептидазы-IV, включающего введение эффективного количества соединения пациенту, такому как млекопитающее, нуждающемуся в таком ингибировании. Настоящее изобретение направлено на применение соединений, описанных здесь как ингибиторы активности фермента дипептидилпептидазы-IV. Кроме приматов, таких как человек, способ, соответствующий настоящему изобретению, можно применять для лечения ряда других млекопитающих. Например, можно применять для лечения млекопитающих, включая, но этим не ограничиваясь, коров, овец, коз, лошадей, собак, кошек, морских свинок,крыс или других представителей этих видов. Способ можно также применять для представителей других видов, таких как виды птиц (например, цыплята). Настоящее изобретение также относится к способу изготовления лекарственного средства для ингибирования активности фермента дипептидилпептидазы-IV у человека и животных, включающему объединение соединения настоящего изобретения с фармацевтическим носителем или разбавителем. Настоящие способы лечения предназначены для млекопитающего, предпочтительно человека,мужчины или женщины, нуждающегося в ингибировании активности фермента дипептидилпептидазыIV. Термин терапевтически эффективное количество означает количество обсуждаемого соединения,при котором будет установлена биологическая или терапевтическая реакция ткани, системы, животного или человека, и которое определяется исследователем, ветеринаром, врачом или другим клиницистом. Применяемый термин композиция включает продукт, содержащий определенные ингредиенты в определенных количествах, также как любой продукт, который получается, прямо или косвенно, при сочетании определенных ингредиентов в определенных количествах. Такой термин, относящийся к фармацевтической композиции, включает продукт, содержащий активный(е) ингредиент(ы) и инертный ингредиент(ы), являющийся носителем, также как любой продукт, который получается, прямо или косвенно, при сочетании, образовании комплексов или агрегации любых двух или более ингредиентов, или при диссоциации одного или более ингредиентов, или при других видах реакций или взаимодействий одного или более ингредиентов. Соответственно фармацевтические композиции настоящего изобретения включают любую композицию, изготовленную путем смешения соединения настоящего изобретения и фармацевтически приемлемого носителя. Под фармацевтически приемлемым понимается носитель, разбавитель или наполнитель, который совместим с другими ингредиентами композиции и не наносит вреда реципиенту. Под терминами введение и/или прием соединения следует понимать обеспечение индивидуума в случае необходимости лечения соединением, соответствующим изобретению, или пролекарством, соответствующим соединению изобретения. Применимость соединений, соответствующих настоящему изобретению, в качестве ингибиторов активности фермента дипептидилпептидазы-IV можно продемонстрировать с помощью известной в данной области методики. Константы ингибирования определяют следующим образом. Применяют непрерывный флуорометрический анализ, используя в качестве субстрата Gly-Pro-АМС, который расщепляется под действием DP-IV с высвобождением флуоресцентной уходящей группы АМС. Такая реакция описывается следующими кинетическими параметрами: Кm=50 мкМ; kкат=75c-1; kкат/Km=l,5 х 106 М-1 с-1. Типичная реакционная смесь содержит приблизительно 50 пМ фермента, 50 мкМ Gly-Pro-AMC и буфер(100 мМ HEPES, рН 7,5, 0,1 мг/мл BSA) в общем реакционном объеме 100 мкл. Высвобождение АМС непрерывно контролируется в 96-луночном планшете, помещенном в флуорометр, при длине волны возбуждения 360 нм и длине волны излучения 460 нм. При таких условиях в течение 30 мин при 25 С продуцируется приблизительно 0,8 мкМ АМС. Применяемый в таких исследованиях фермент является растворимым (с исключенными транс-мембранным доменом и цитоплазматической областью) человеческим протеином, продуцируемым в системе бакуловирусной экспрессии (Вас-То-Вас, Gibco BRL). Найденные кинетические константы гидролиза Gly-Pro-АМС и GLP-1 соответствуют литературным значениям для нативного фермента. При измерении констант диссоциации соединений растворы ингибитора в ДМСО добавляют к реакционной смеси, содержащей фермент и субстрат (конечная концентрация ДМСО равна 1%). Все эксперименты проводят при комнатной температуре при описанных выше стандартных условиях реакции. При определении констант диссоциации (Ki) скорости реакции соответствуют нелинейной регрессии для уравнения Михаэлиса-Ментена для конкурентного ингибирования. Погрешности при воспроизведении констант диссоциации обычно не превышают двух раз. В частности, соединения, рассматриваемые в следующих примерах, при упомянутых выше испытаниях обычно имеют ингибирующую активность, характеризуемую значением IC50, по отношению к ферменту дипептидилпептидазе-IV приблизительно меньше 1 мкМ. Такой результат указывает на присущую соединениям активность в случае применения их в качестве ингибиторов активности фермента дипептидилпептидазы-IV. Фермент дипептидилпептидаза-IV (DP-IV) является протеином клеточной поверхности, который вовлечен в биологические функции широкого диапазона. Он широко распределен в тканях (кишечник,почки, печень, поджелудочная железа, плацента, тимус, селезенка, эпителиоциты, васкулярный эндотелий, лимфоидные и миелоидные клетки, сыворотка) и имеет разные уровни экспрессии в тканях и клетках разных типов. DP-IV идентична маркеру CD26 активации Т-клеток и может расщеплять ряд иммуно-5 006845 регуляторных, эндокринных и неврологических пептидов in vitro. Можно предположить потенциальное участие такой пептидазы в протекании ряда заболеваний человека или других видов. Соответственно, обсуждаемые соединения применимы для способа профилактики или лечения следующих заболеваний, расстройств и состояний. Диабет типа 2 и родственные расстройства: Хорошо известно, что инкретины GLP-1 и GIP быстро инактивируются DP-IV in vivo. Изучение мышей, дефицитных по DP-IV(-/-), и предварительные клинические испытания указывают, что ингибирование DP-IV повышает устойчивые концентрации GLP-1 и GIP,приводя к улучшению переносимости глюкозы. По аналогии с GLP-1 и GIP, вероятно, что другие пептиды семейства глюкагона, включенные в регуляцию глюкозы, также инактивируются DP-IV (например,РАСАР, глюкагон). Инактивация таких пептидов DP-IV может также играть роль в гомеостазе глюкозы. Поэтому ингибиторы DP-IV настоящего изобретения применяются для лечения диабета типа 2 и лечения и предотвращения многочисленных состояний, которыми диабет типа 2 часто сопровождается,включая метаболический синдром X, реактивную гипогликемию и диабетическую дислипидемию. Ожирение, обсуждаемое ниже, является другим состоянием, часто обнаруживаемым при диабете типа 2, которое может давать реакцию на лечение соединениями данного изобретения. Следующие заболевания, расстройства и состояния связаны с диабетом типа 2 и, следовательно,при лечении с помощью соединений данного изобретения их можно лечить, контролировать или в некоторых случаях предупреждать: (1) гипергликемия, (2) низкая переносимость глюкозы, (3) резистентность к инсулину, (4) ожирение, (5) расстройства липидного обмена, (6) дислипидемия, (7) гиперлипидемия, (8) гипертриглицеридемия, (9) гиперхолестеринемия, (10) низкие уровни HDL, (11) высокие уровни LDL,(12) атеросклероз и его последствия, (13) васкулярный рестеноз, (14) синдром раздраженной толстой кишки, (15) воспалительные заболевания кишечника, включая болезнь Крона и неспецифический язвенный колит, (16) другие воспалительные состояния, (17) панкреатит, (18) ожирение брюшной полости,(19) нейродегенеративное заболевание, (20) ретинопатия, (21) нефропатия, (22) невропатия, (23) синдромX, (24) овариальный гиперандрогенизм (синдром поликистоза яичников) и другие расстройства, где составляющим компонентом является резистентность к инсулину. Ожирение: ингибиторы DP-IV можно применять для лечения ожирения. Такое лечение основано на наблюдаемом ингибирующем влиянии GLP-1 и GLP-2 на всасывание пищи и опорожнение желудка. Экзогенное введение GLP-1 существенно уменьшает всасывание пищи и замедляет опорожнение желудка у человека (Am.J.Physiol. 277, R910-R916, 1999). ICV-введение GLP-1 крысам и мышам также значительно влияет на всасывание пищи (Nature Medicine 2, 1254-1258 (1996. У мышей GLP-lR(-/-) такого ингибирования всасывания съеденной пищи не наблюдается, что указывает на то, что данные эффекты опосредованы рецепторами GLP-1 мозга. По аналогии с GLP-1 вероятно, что GLP-2 также регулируется DP-IV.ICV-введение GLP-2 также ингибирует всасывание пищи, аналогично эффектам, наблюдаемым для GLP1 (Nature Medicine 6, 802-807, 2000). Дефицит гормона роста: ингибирование DP-IV можно применять для лечения дефицита гормона роста, исходя из гипотезы, что рилизинг-фактор гормона роста (GRF), пептид, стимулирующий высвобождение гормона роста из передней доли гипофиза, расщепляется ферментом DP-IV in vivo (WO 00/56297). Следующие данные свидетельствуют, что GRF является эндогенным субстратом: (1) GRF эффективно расщепляется in vitro с образованием неактивного продукта GRF[3-44] (BBA 1122, 147-153(1992; (2) GRF быстро разлагается в плазме до GRF[3-44]; указанные явления можно предотвращать воздействием ингибитора DP-IV дипротина А; и (3) GRF[3-44] обнаруживают в плазме трансгенных свиней с человеческим GRF (J. Clin. Invest. 83, 1533-1540 (1989. Следовательно, ингибиторы DP-IV можно применять для того же спектра симптомов, которые обсуждаются для стимуляторов секреции гормона роста. Повреждение кишечника: потенциальное применение ингибиторов DP-IV для лечения повреждения кишечника подсказано результатами исследований, указывающих, что глюкагоноподобный пептид-2(GLP-2), вероятный эндогенный субстрат для DP-IV, может вызывать трофическое воздействие на кишечный эпителий (Regulatory Peptides 90, 27-32 (2000. Введение GLP-2 приводит к увеличению массы тонкой кишки у грызунов и смягчает повреждение кишечника в моделях колита и энтерита у грызунов. Иммунодепрессия: ингибирование DP-IV можно применять для модуляции иммунной реакции, основываясь на исследованиях причастности фермента DP-IV к активации Т-клеток и к процессингу хемокинов, и на эффективности ингибиторов DP-IV в моделях заболевания in vivo. Установлено, что DP-IV идентичен CD26, маркеру клеточной поверхности активированных иммунных клеток. Экспрессия CD26 регулируется дифференцировкой и активированным статусом иммунных клеток. Общепринято, чтоCD26 функционирует как ко-стимулирующая молекула в моделях активации Т-клеток in vitro. Ряд хемокинов содержит пролин в предпоследнем положении, возможно для защиты их от деградации под действием неспецифических аминопептидаз. Установлено, что многие из них подвергаются процессингу invitro под действием DP-IV. В некоторых случаях (RANTES, бета-LD78, MDC, эотаксин, альфа-SDF-1) расщепление приводит к изменению активности при хемотаксисе и передаче сигнала. В некоторых случаях (RANTES), по-видимому, изменяется также селективность рецептора. В системах культивирования клеток in vitro идентифицированы многочисленные укороченные с N-конца формы ряда хемокинов,-6 006845 включая предсказанные продукты гидролиза DP-IV. На моделях трансплантации и артрита у животных установлено, что ингибиторы DP-IV являются эффективными иммунодепрессантами. Было показано, что необратимый ингибитор DP-IV продипин(Рrо-Pro-дифенилфосфонат) удваивает продолжительность функционирования сердечного аллотрансплантанта крыс с 7 до 14 дней (Transplantation 63, 1495-1500 (1997. Ингибиторы DP-IV исследовали на модели артрита, индуцированного коллагеном и алкилдиамином у крыс, и исследования показали статистически значительное уменьшение опухоли задней конечности в такой модели (Int. J. Immunopharmacology 19, 15-24 (1997), Immunopharmacology 40, 21-26 (1998. Уровень DP-IV повышается при ряде аутоиммунных заболеваний, включая ревматоидный артрит, рассеянный склероз, болезнь Грейвса и тиреоидит Хасимото (Immunology Today 20, 367, 375 (1999. ВИЧ-инфекция: ингибирование DP-IV можно применять для лечения или предотвращения ВИЧинфекции или СПИДа, потому что ряд хемокинов, которые ингибируют проникновение ВИЧ-клеток,являются потенциальными субстратами для DP-IV (Immunology Today 20, 367-375 (1999. В случае альфа-SDF-l расщепление уменьшает противовирусную активность (PNAS 95, 6331-6 (1998. Таким образом, при ингибировании DP-IV можно ожидать стабилизации альфа-SDF-l для уменьшения инвазивной способности ВИЧ. Гемопоэз: ингибирование DP-IV можно применять для лечения или предотвращения гемопоэза, потому что DP-IV может быть вовлечен в гемопоэз. Ингибитор DP-IV, Val-Boro-Pro, стимулировал гемопоэз в модели нейтропении мышей, индуцированной циклофосфамидом (WO 99/56753). Нейронные расстройства: ингибирование DP-IV можно применять для лечения или предотвращения различных нейронных или психиатрических расстройств, потому что ряд пептидов, вовлеченных в различные нейронные процессы, расщепляется DP-IV in vitro. Следовательно, ингибитор DP-IV может обладать терапевтическим преимуществом при лечении нейронных расстройств. Установлено, что эндоморфин-2, бета-казоморфин и вещество Р, все являются in vitro субстратами для DP-IV. Во всех случаях расщепление in vitro высокоэффективно, отношение kcat/Km 106 М-1 с-1 или больше. В модели анальгезии в тесте подпрыгивания под действием электрического тока крыс ингибиторDP-IV показал значительный эффект, который не зависел от присутствия экзогенного эндоморфина-2(Brain Research 815, 278-286 (1999. Инвазия (прорастание) опухоли и метастазов: ингибирование DP-IV можно применять для лечения или предотвращения инвазии опухоли и метастазов, потому что во время трансформации нормальных клеток в злокачественный фенотип наблюдается увеличение или уменьшение экспрессии некоторых эктопептидаз, включая DP-IV (J. Exp. Med. 190, 301-305 (1999. Повышение и понижение уровня таких протеинов, по-видимому, является специфичным в отношении различных типов тканей и клеток. Например, наблюдается повышенная экспрессия CD26/DP-IV при Т-клеточной лимфоме, Т-клеточном остром лимфобластном лейкозе, при карциномах щитовидной железы клеточного происхождения, базальноклеточных карциномах и карциномах молочной железы. Таким образом, ингибиторы DP-IV можно применять в лечении таких карцином. Доброкачественная гипертрофия предстательной железы (ВРН) : ингибирование DP-IV можно применять для лечения доброкачественной гипертрофии предстательной железы, так как у пациентов с ВРН отмечается повышенная активность DP-IV в ткани простаты (Eur. J. Clin. Chem. Clin. Biochem 30, 333-338(1992. Подвижность сперматозоидов/контрацепция мужчины: ингибирование DP-IV можно применять для изменения подвижности сперматозоидов и для контрацепции мужчины, так как в семенной жидкости важные для подвижности сперматозоида простатосомы, органеллы, производимые простатой, обладают сверхвысокими уровнями активности DP-IV (Eur. J. Clin. Chem. Clin. Biochem 30, 333-338 (1992. Гингивит: ингибирование DP-IV можно применять для лечения гингивита, так как в некоторых исследованиях, устанавливающих связь с тяжестью периодонтального заболевания, обнаружена активностьDP-IV в десневой жидкости, находящейся в десневой борозде (Arch. Oral Biol. 37, 167-173 (1992. Остеопороз: ингибирование DP-IV можно применять для лечения или профилактики остеопороза,так как в остеобластах присутствуют GIP-рецепторы. Соединения настоящего изобретения можно применять для лечения или предупреждения одного или более следующих состояний или заболеваний: (1) гипергликемия, (2) низкая переносимость глюкозы, (3) резистентность к инсулину, (4) ожирение, (5) расстройства липидного обмена, (6) дислипидемия,(7) гиперлипидемия, (8) гипертриглицеридемия, (9) гиперхолестеринемия, (10) низкие уровни HDL, (11) высокие уровни LDL, (12) атеросклероз и его последствия, (13) васкулярный рестеноз, (14) синдром раздраженной толстой кишки, (15) воспаление кишечника, включая болезнь Крона и неспецифический язвенный колит, (16) другие воспалительные состояния, (17) панкреатит, (18) ожирение брюшной полости,(19) нейродегенеративное заболевание, (20) ретинопатия, (21) нефропатия, (22) невропатия, (23) синдромX, (24) овариальный гиперандрогинизм (синдром поликистоза яичников), (25) диабет типа 2, (26) дефицит гормона роста, (27) нейтропения, (28) нейронные расстройства, (29) метастазирующая опухоль, (30) доброкачественная гипертрофия предстательной железы, (32) гингивит, (33) гипертония, (34) остеопороз и другие состояния, которые можно лечить или предупреждать путем ингибирования DP-IV.-7 006845 Обсуждаемые соединения также применяют для предупреждения или лечения вышеупомянутых заболеваний, расстройств и состояний в сочетании с другими агентами. Соединения настоящего изобретения можно применять в сочетании с одним или более других лекарственных средств для лечения, предупреждения, подавления или уменьшения интенсивности симптомов заболеваний или состояний, для которых применяются соединения формулы I или другие лекарственные средства, когда комбинация лекарственных средств более надежна или более эффективна, чем любое из лекарственных средств по отдельности. Другое такое лекарственное средство (средства) можно вводить в соответствии со способом применения и в количестве, в котором оно обычно применяется,одновременно или последовательно с соединением формулы I. Когда соединение формулы I применяют одновременно с одним или более других лекарственных средств, предпочтительна фармацевтическая композиция в виде дозированной лекарственной формы, содержащей такие другие лекарственные средства и соединение формулы I. Однако комбинированная терапия может также включать терапии, при которых соединение формулы I и одно или более других лекарственных средств вводятся по разным перекрывающимся схемам приема. Также рассматривается случай, когда соединения настоящего изобретения, применяемые в сочетании с одним или более других активных ингредиентов, и другие активные ингредиенты можно применять в более низких дозах, чем когда каждый из них применяется по отдельности. Соответственно, фармацевтические композиции настоящего изобретения включают композиции,которые кроме соединения формулы I содержат один или более других активных ингредиентов. Примеры других активных ингредиентов, которые можно вводить в сочетании с соединением формулы I, причем их можно вводить либо отдельно, либо в составе той же самой фармацевтической композиции, включают, но этим не ограничиваются:(a) другие ингибиторы дипептидилпептидазы IV (DP-IV);(b) сенсибилизаторы инсулина, включая (i) агонисты PPAR, такие как глитазоны (например, троглитазон, пиоглитазон, энглитазон, МСС-555, розиглитазон и т.п.) и другие PPAR-лиганды, включая двойные агонисты /PPAR, такие как KRP-297, и агонисты PPAR, такие как производные фенофибровой кислоты (гемфиброзил, клофибрат, фенофибрат и безафибрат),(ii) бигуаниды, такие как метформин и фенформин, и (iii) ингибиторы (протеинтирозин)фосфатазы-1 В (РТР-1 В);(d) сульфонилмочевины и другие стимуляторы секреции инсулина, такие как толбутамид и глипизид, меглитинид и родственные материалы;(f) антагонисты глюкагоновых рецепторов, такие как описанные в заявках WO 98/04528, WO 99/01423, WO 00/39088 и WO 00/69810;(g) GLP-1, миметики GLP-1 и агонисты GLP-1-рецепторов, такие как описанные в заявках WO 00/42026 и WO 00/59887;(h) GIP и миметики GIP, такие как описанные в заявке WO 00/58360, и агонисты GIP-рецепторов;(i) РАСАР, миметики РАСАР, и агонисты рецептора-3 РАСАР, такие как описанные в заявке WO 01/23420;(j) агенты, понижающие холестерин, такие как (i) ингибиторы HMG-CoA-редуктазы (ловастатин,симвастатин, правастатин, флувастатин, аторвастатин, ривастатин, итавастатин, розувастатин и другие статины), (ii) агенты, усиливающие экскрецию (холестирамин, холестипол и диалкиламиноалкильные производные поперечно-сшитого декстрана), (iii) никотиниловый спирт, никотиновая кислота или ее соли, (iv) агонисты PPAR, такие как производные фенофибровой кислоты (гемфиброзил, клофибрат, фенофибрат и безафибрат), (v) двойные агонисты /PPAR, такие как KRP-297, (vi) ингибиторы абсорбции холестерина,такие как бета-ситостерол и эзетимиб,(vii) ингибиторы СоАацил:холестеролацилтрансферазы, такие как авасимиб, и (viii) антиоксиданты, такие как пробукол;(k) агонисты PPAR, такие как описанные в заявке WO 97/28149;(m) ингибитор транспортера желчной кислоты в подвздошную кишку; и(n) противовоспалительные агенты, такие как аспирин, нестероидные противовоспалительные лекарственные средства, глюкокортикоиды, азулфидин и селективные ингибиторы циклооксигеназы-2. Упомянутые выше комбинации включают комбинации соединения настоящего изобретения не только с одним из других активных соединений, но также и с двумя или более другими активными соединениями. Не ограничивающие примеры включают комбинацию соединений формулы I с двумя или более активными соединениями, выбранными из бигуанидов, сульфонилмочевин, ингибиторов HMGCoA-редуктазы, агонистов PPAR, ингибиторов РТР-1 В, других ингибиторов DP-IV и соединений, предназначенных против ожирения. Аналогично соединения настоящего изобретения можно применять в сочетании с другими лекарст-8 006845 венными средствами, применяемыми для лечения/профилактики/подавления или уменьшения интенсивности симптомов заболеваний или состояний, для которых применяются соединения настоящего изобретения. Другие такие лекарственные средства можно вводить в соответствии со способом применения и в количестве, в котором они обычно применяются, одновременно или последовательно с соединением настоящего изобретения. Когда соединение настоящего изобретения применяют одновременно с одним или более других лекарственных средств, предпочтительна фармацевтическая композиция, содержащая кроме соединения настоящего изобретения другие такие лекарственные средства. Соответственно, фармацевтические композиции настоящего изобретения включают композиции, которые кроме соединения настоящего изобретения содержат один или более других активных ингредиентов. Весовое соотношение соединения настоящего изобретения и второго активного ингредиента может варьироваться и будет зависеть от эффективной дозы каждого ингредиента. В общем случае, применяют эффективную дозу каждого из них. Таким образом, когда соединение настоящего изобретения объединено, например, с другим агентом, весовое соотношение соединения настоящего изобретения и другого агента в общем случае составляет приблизительно от 1000:1 до 1:1000, предпочтительно составляет приблизительно от 200:1 до 1:200. Комбинации соединения настоящего изобретения с другими активными ингредиентами в общем случае также находятся в пределах вышеупомянутого диапазона, однако в каждом случае следует применять эффективную дозу каждого активного ингредиента. В таких комбинациях соединение настоящего изобретения и другие активные агенты можно вводить отдельно или вместе. Кроме того, введение одного из элементов можно осуществить до, в то же самое время или последовательно после введения другого агента(ов). Соединения настоящего изобретения можно вводить перорально, парентерально (например, внутримышечно, внутрибрюшинно, внутривенно, ICV, внутриполостной инъекцией или вливанием, подкожной инъекцией или в виде имплантата), спреем для ингаляции, назальным, влагалищным, ректальным,подъязычным или локальным способом введения, можно приготовить из одного вещества или из веществ, взятых вместе, подходящую дозированную лекарственную форму, содержащую общепринятые нетоксичные фармацевтически приемлемые носители, вспомогательные вещества и связующие, соответствующие каждому способу введения. Кроме лечения теплокровных животных, таких как мыши, крысы,лошади, скот, овцы, собаки, кошки, обезьяны и т.д., соединения изобретения эффективны для лечения человека. Фармацевтические композиции для введения соединений настоящего изобретения можно представлять в общепринятой стандартной лекарственной форме и получить любым из известных в фармацевтике способов. Все способы включают стадию объединения активного ингредиента с носителем, который состоит из одного или более вспомогательных ингредиентов. В общем случае фармацевтические композиции получают путем унифицированного и однородного объединения активного ингредиента с жидким или тонкоизмельченным твердым носителем, или с тем и другим, и затем, в случае необходимости, придания продукту формы требуемого состава. Активное целевое соединение включают в фармацевтическую композицию в количестве, достаточном, чтобы оказать требуемое действие на процесс заболевания или состояние, связанное с заболеванием. Применяемый здесь термин композиция означает продукт,содержащий определенные ингредиенты в определенных количествах, также как любой продукт, который получается прямо или косвенно от комбинации определенных ингредиентов, взятых в определенных количествах. Фармацевтические композиции, содержащие активный ингредиент, можно получать в форме, подходящей для перорального применения, например таблетки, пастилки, лепешки, водные или масляные суспензии, дисперсии порошков или гранул, эмульсии, твердые или мягкие капсулы, сиропы или эликсиры. Композиции, предназначенные для перорального применения, можно приготовить согласно любому способу, известному в данной области для изготовления фармацевтических композиций, и такие композиции могут содержать один или более агентов, выбранных из группы, состоящей из подсластителей,корригентов, красителей и консервантов, чтобы обеспечить очень хорошие фармацевтические и аппетитные на вкус препараты. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, подходящими для изготовления таблеток. Такими наполнителями могут быть, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза,фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие агенты, например кукурузный крахмал или альгиновая кислота; связующие агенты, например крахмал, желатин или акация, и смазывающие агенты, например стеарат магния, стеариновая кислота или тальк. Таблетки могут быть без покрытия или их можно покрыть известными способами, чтобы замедлить дезинтеграцию и абсорбцию в желудочно-кишечном тракте и таким образом поддерживать их действие более длительный период. Например, для увеличения периода дезинтеграции и абсорбции можно применять такие материалы, как глицерилмоностеарат или глицерилдистеарат. В соответствии с методиками, описанными в патентах США 4256108; 4166452 и 4265874, можно также получить осмотические терапевтические таблетки с покрытием для контролируемого высвобождения. Препаративные формы для перорального применения можно также представить в виде твердых желатиновых капсул, где активный ингредиент смешан с инертным твердым разбавителем, например, кар-9 006845 бонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный ингредиент смешан с водой или масляной средой, например арахисовым маслом, вазелиновым маслом или оливковым маслом. Водные суспензии содержат активные материалы в смеси с наполнителями, подходящими для изготовления водных суспензий. Такие наполнители представляют собой суспендирующие агенты, например натрийкарбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия,поливинилпирролидон, трагакантовую камедь и аравийскую камедь; диспергирующие или смачивающие агенты могут быть представлены встречающимся в природе фосфатидом, например лецитином, или продуктами конденсации алкиленоксида с жирными кислотами, например, полиоксиэтиленстеаратом, или продуктами конденсации этиленоксида с алифатическими спиртами с длинной цепью, например гептадекаэтиленоксицетанолом, или продуктами конденсации этиленоксида с частичными сложными эфирами, получаемыми из жирных кислот и гексита, такими как полиоксиэтиленсорбитмоноолеат, или продуктами конденсации этиленоксида с частичными сложными эфирами, получаемыми из жирных кислот и ангидридов гексита, например полиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один консервант или более, например этил- или н-пропил-п-гидроксибензоат, один краситель или более, один корригент или более и один подсластитель или более, такой как сахароза или сахарин. Масляные суспензии можно получить путем суспендирования активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как вазелиновое масло. Масляные суспензии могут содержать загуститель,например пчелиный воск, твердый парафин или цетиловый спирт. Можно добавлять подсластители, такие как упомянутые выше, и корригенты, чтобы обеспечить приемлемый препарат для перорального приема. Такие композиции можно сохранять при добавлении антиоксиданта, такого как аскорбиновая кислота. В дисперсных порошках и гранулах, подходящих для получения водной суспензии при добавлении к ним воды, активный ингредиент смешивают с диспергатором или смачивающим агентом, суспендирующим агентом и одним или более консервантом. Подходящие диспергирующие или смачивающие агенты и суспендирующие агенты уже перечислены выше. Могут также присутствовать дополнительные наполнители, например подсластители, корригенты и красители. Фармацевтические композиции изобретения можно также получать в виде эмульсий масло в воде. Масляной фазой может быть растительное масло, например оливковое или арахисовое масло, или минеральное масло, например вазелиновое масло, или их смеси. Подходящими эмульгаторами могут стать встречающиеся в природе камеди, например аравийская камедь или трагакантовая камедь, встречающиеся в природе фосфатиды, например фосфатиды сои, лецитин и сложные эфиры или частичные сложные эфиры, получаемые из жирных кислот и ангидридов гексита, например сорбитанмоноолеат, и продукты конденсации упомянутых частичных сложных эфиров с этиленоксидом, например полиоксиэтиленсорбитанмоноолеат. Эмульсии могут также содержать подсластители и корригенты. Сиропы и эликсиры можно изготавливать с подсластителями, например глицерином, пропиленгликолем, сорбитом или сахарозой. Такие препаративные формы могут также содержать средство, уменьшающее раздражение, консервант, корригенты и красители. Фармацевтические композиции можно получать в форме стерильной водной или маслянистой суспензии для инъекции. Такую суспензию можно приготовить согласно известным способам, применяя подходящие диспергаторы или смачивающие агенты и суспендирующие агенты, упомянутые выше. Стерильный препарат для инъекций может также представлять стерильный раствор для инъекции или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например раствор в 1,3-бутандиоле. Среди приемлемых связующих и растворителей, которые можно применять, находится вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспензионной среды традиционно применяют стерильные нелетучие масла. Для такой цели можно применять любое жидкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для получения препаратов для инъекций применяют жирные кислоты, такие как олеиновая кислота. Соединения настоящего изобретения можно также вводить в виде суппозиториев для ректального введения лекарственного средства. Такие композиции можно получить путем смешения лекарственного средства с подходящим нераздражающим наполнителем, который является твердым при обычных температурах, но жидким при ректальной температуре и, следовательно, расплавится в прямой кишке для высвобождения лекарственного средства. Такими материалами являются масло какао и полиэтиленгликоли. Для местного применения используются кремы, мази, желе, растворы или суспензии и т.д., содержащие соединения настоящего изобретения (с целью такого применения местное применение должно включать жидкости для полоскания рта и горла). Фармацевтическая композиция и способ согласно настоящему изобретению могут также включать другие терапевтически активные соединения, как здесь отмечалось, которые обычно применяются при лечении упомянутых выше патологических состояний. Для лечения или предотвращения состояний, при которых требуется ингибирование активности- 10006845 фермента дипептидилпептидазы-IV, подходящий уровень дозировки обычно составляет приблизительно от 0,01 до 500 мг/кг массы пациента в день, при этом препарат можно вводить в виде разовых или многократных доз. Предпочтительный уровень дозировки составляет приблизительно от 0,1 до 250 мг/кг в день; более предпочтительный - приблизительно от 0,5 до приблизительно 100 мг/кг в день. Подходящий уровень дозировки может составлять приблизительно от 0,01 до 250 мг/кг в день, приблизительно от 0,05 до 100 мг/кг в день, или приблизительно от 0,1 до 50 мг/кг в день. В пределах такого диапазона дозировка может составлять от 0,05 до 0,5, от 0,5 до 5 или от 5 до 50 мг/кг в день. Для перорального приема композиции получают предпочтительно в форме таблеток, содержащих от 1,0 до 1000 мг активного ингредиента, в частности, 1,0, 5,0, 10,0, 15,0, 20,0, 25,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0, 400,0, 500,0,600,0, 750,0, 800,0, 900,0, и 1000,0 мг активного ингредиента для симптоматической коррекции дозировки для пациента, подвергаемого лечению. Соединения можно вводить в режиме от 1 до 4 раз в день,предпочтительно один раз или дважды в день. При лечении или профилактике сахарного диабета и/или гипергликемии или гипертриглицеридемии или других заболеваний, для которых предназначены соединения настоящего изобретения, удовлетворительные результаты обычно получают, когда соединения настоящего изобретения вводят ежедневно при дозе приблизительно от 0,1 до 100 мг/кг массы животного, препарат предпочтительно дают в виде разовой суточной дозы или в виде дозы, разделенной на два-шесть приемов в день, или в форме пролонгированного высвобождения лекарственного средства. Для самых больших млекопитающих общая суточная доза составляет приблизительно от 1,0 до 1000 мг, предпочтительно приблизительно от 1 до 50 мг. Для взрослого человека массой 70 кг общая суточная доза обычно составляет приблизительно от 7 до 350 мг. Для получения оптимальной терапевтической реакции указанный режим приема можно откорректировать. Однако понятно, что для любого из конкретных пациентов определенный уровень доз и частоту приема можно варьировать, они будут зависеть от ряда факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и продолжительность действия такого соединения, возраст, массу тела, общее состояние здоровья, пол, диету, режим и время введения, скорость выделения, комбинацию лекарственных средств, серьезность конкретного состояния и субъекта, подвергающегося терапии. На следующих схемах и примерах проиллюстрированы некоторые способы получения соединений настоящего изобретения. Исходные материалы получали согласно методикам, известным в данной области или как показано в описании. Соединения настоящего изобретения можно получить из промежуточных соединений аминокислот, таких как соединения формулы II, и замещенных гетероциклических промежуточных соединений, таких как соединения формулы III, при стандартных условиях реакции связывания пептидов, с последующим снятием защиты. Получение таких промежуточных соединений описано на следующих схемах. где Аr, X и R1 определены выше, а Р представляет подходящую защитную группу для азота, такую как трет-бутоксикарбонил, бензилоксикарбонил или 9-флуоренилметоксикарбонил. Схема 1 Соединения формулы II имеются в продаже, описаны в литературе или их можно получить с помощью ряда подходящих способов, известных в данной области. Один из таких способов проиллюстрирован на схеме 1. Кислоту 1, которая может иметься в продаже или легко получается из соответствующей аминокислоты с применением защитной группы, например ди-трет-бутилдикарбоната (Р=Вос), карбобензилоксихлорида (P=Cbz) или N-(9-флуоренилметоксикарбонилокси)сукцинимида (P=Fmoc) обрабатывают изобутилхлорформиатом и основанием, таким как триэтиламин или диизопропилэтиламин, и затем диазометаном. Для получения бета-аминокислоты II полученный диазокетон затем обрабатывают бензоатом серебра в растворителе, таком как метанол или водный раствор диоксана, его также можно подвергнуть обработке ультразвуком, следуя методике, описанной Sewald и др., Sinthesis, 837 (1997). Как понятно специалисту, для получения энантиомерно чистых бета-аминокислот II можно применять энантиомерно чистые альфа-аминокислоты 1. Описание альтернативных способов получения таких соедине- 11006845 ний можно найти в следующих обзорах: Е. Juaristi, Enantioselective Synthesis of -Amino Acids, Ed.,Wiley-VCH, New York: 1997, Juaristi и др., Aldrichimica Acta, 27, 3 (1994), Cole и др., Tetrahedron, 32, 9517 Соединения III имеются в продаже, описаны в литературе или их можно получить с помощью ряда подходящих способов, известных в данной области. Один из подходящих способов проиллюстрирован на схеме 2. Для получения соединения III ненасыщенное производное 2 восстанавливают, например, обработкой газообразным водородом в присутствии катализатора, такого как палладий на углероде или оксид платины в растворителе, таком как метанол или этанол. Схема 3 Промежуточный продукт 2 на схеме 2 сам по себе имеется в продаже, описан в литературе или его можно получить с помощью ряда подходящих способов, известных в данной области. Один из таких способов, когда X представляет CR2, проиллюстрирован на схеме 3. Для получения промежуточного соединения 2 а аминопиразин 3 обрабатывают 2-галогенкетоном, таким как 2-бромкетон 4 в растворителе,таком как метанол или этанол. С другой стороны, для получения промежуточного соединения 2 а, где R2 представляет Н, вместо промежуточного соединения 4 можно применять 2-бромдиметилацеталь 5 и каталитическое количество кислоты, такой как соляная кислота. Схема 4 Подходящий способ получения промежуточного соединения 2b, где X представляет N, проиллюстрирован на схеме 4. Для получения гидразинопиразина 7 хлорпиразин 6 обрабатывают гидразином. Соединение 2b можно получить конденсацией соединения 7 либо со сложным ортоэфиром, таким как сложный триэтиловый эфир ортокислоты 8, либо с карбоновой кислотой 9 в полифосфорной кислоте при повышенных температурах. Схема 5 Альтернативный способ получения соединения IIIb, где X представляет N, проиллюстрирован на схеме 5. Соединение 12 получают согласно способу, описанному выше, применяя вместо хлорпиразина 6- 12006845 дихлорпиразин 10. Затем для получения соединения IIIb в виде его монохлористо-водородной соли соединение 12 подвергают каталитическому гидрированию, применяя катализатор, такой как оксид платины. Схема 6 Для получения промежуточного соединения 13 промежуточные соединения II и III связывают при стандартных условиях реакции связывания пептидов, например, применяя 1-этил-3-(3 диметиламинопропил)карбодиимид (EDC), 1-гидроксибензотриазол (НОВТ), и основание, обычно диизопропилэтиламин, в растворителе, таком как N,N-диметилформамид (DMF) или дихлорметан, в течение от 3 до 48 ч при температуре окружающей среды, как показано на схеме 6. Затем удаляют защитную группу, например, при взаимодействии с трифторуксусной кислотой или с раствором хлористого водорода в метаноле в случае Воc, чтобы получить требуемый амин I. В случае необходимости продукт очищают от нежелательных побочных продуктов рекристаллизацией, растиранием, препаративной тонкослойной хроматографией, флэш-хроматографией на силикагеле, как описано в публикации W.С.Still, J.Org. Chem. 43, 2923 (1978), или ВЭЖХ. Соединения, которые очищены путем ВЭЖХ, можно выделить в виде соответствующих солей. Очищение промежуточных продуктов проводят таким же образом. В некоторых случаях промежуточный продукт 13, полученный в результате реакции связывания,описанной на схеме 6, можно далее модифицировать перед удалением защитной группы, например, путем обработки заместителей на X или R1. Такая обработка может включать, однако этим не ограничивается, реакции восстановления, окисления, алкилирования, ацилирования и гидролиза, которые обычно известны специалисту в данной области. В некоторых случаях порядок выполнения упомянутых выше на схемах реакций можно варьировать, чтобы облегчить протекание реакции или избежать нежелательных продуктов реакции. Следующие примеры приведены для того, чтобы можно было более полно понять изобретение. Данные примеры приведены только для иллюстрации и не должны истолковываться как ограничивающие изобретение каким-либо образом. Промежуточный продукт 1(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(2,5-дифторфенил)бутановая кислота Стадия A.(R,S)-N-(1,1-диметилэтоксикарбонил)-2,5-дифторфенилаланин К раствору 0,5 г (2,49 ммоль) 2,5-дифтор-DL-фенилаланина в 5 мл трет-бутанола добавляют последовательно 1,5 мл 2 н водного раствора гидроксида натрия и 543 мг ди-трет-бутилдикарбоната. Реакционную смесь перемешивают при температуре окружающей среды в течение 16 ч и разбавляют этилацетатом. Органическую фазу промывают последовательно 1 н раствором соляной кислоты и соляным раствором, сушат над сульфатом магния и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, дихлорметан:метанол:уксусная кислота в отношении 97:2:1), получая при этом 671 мг указанного в заголовке соединения. MS 302 (M+l) Стадия В. (R,S)-3-[(1,1-диметилэтоксикарбонил)амино]-1-диазо-4-(2,5-дифторфенил)бутан-2-он.- 13006845 К раствору 2,23 г (7,4 ммоль) (R,S)-N-(1,1-диметилэтоксикарбонил)-2,5-дифторфенилаланина в 100 мл простого диэтилового эфира при 0 С добавляют последовательно 1,37 мл (8,1 ммоль) триэтиламина и 0,931 мл (7,5 ммоль) изобутилхлорформиата, реакционную смесь перемешивают при указанной температуре в течение 15 мин. Добавляют охлажденный эфирный раствор диазометана до получения устойчивой желтой окраски и продолжают дальнейшее перемешивание в течение 16 ч. Избыток диазометана гасят,добавляя по каплям уксусную кислоту, разбавляют реакционную смесь этилацетатом и промывают последовательно 5%-ным раствором соляной кислоты, насыщенным водным раствором бикарбоната натрия и соляным раствором, сушат над сульфатом магния и концентрируют в вакууме. После очищения флэш-хроматографией (силикагель, гексан:этилацетат в отношении 4:1) получают 1,5 г диазокетона. ЯМР 1 Н (500 МГц, CDCl3)7,03-6,95 (м, 1 Н), 6,95-6,88 (м, 2 Н), 5,43 (уш.с, 1 Н), 5,18 (уш.с, 1 Н), 4,45(уш.с, 1 Н), 3,19-3,12 (м, 1 Н), 2,97-2,8 С (м, 1 Н), 1,38 (с, 9 Н). Стадия С. (3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(2, 5-дифторфенил)бутановая кислота К раствору 2,14 г (6,58 ммоль) (R,S)-3-[(1,1-диметилэтоксикарбонил)амино]-1-диазо-4-(2,5 дифторфенил)бутан-2-она, растворенного в 100 мл метанола, при -30 С добавляют последовательно 3,3 мл (19 ммоль) диизопропилэтиламина и 302 мг (1,32 ммоль) бензоата серебра. Реакционную смесь перемешивают в течение 90 мин перед разбавлением этилацетатом и промыванием последовательно 2 н раствором соляной кислоты, насыщенным водным раствором бикарбоната натрия и соляным раствором. Органическую фазу сушат над сульфатом магния, концентрируют в вакууме и разделяют энантиомеры препаративной хиральной ВЭЖХ (на колонке, Chiralpak AD, 5%-ный раствор этанола в гексане), получая при этом 550 мг требуемого (R)-энантиомера, который элюируется первым. Полученное вещество растворяют в 50 мл смеси тетрагидрофуран:метанол:1 н водный раствор гидроксида лития (3:1:1) и перемешивают при 50 С в течение 4 ч. Реакционную смесь охлаждают, подкисляют 5% разбавленной соляной кислотой и экстрагируют этилацетатом. Объединенные органические фазы промывают соляным раствором, сушат над сульфатом магния и концентрируют в вакууме, получая при этом 360 мг указанного в заголовке соединения в виде белого, вспененного твердого вещества. ЯМР 1 Н (500 МГц, CDCl3)7,21 (м, 1 Н), 6,98 (м, 2 Н), 6,10 (уш.с, 1 Н), 5,05 (м, 1 Н), 4,21 (м, 1 Н), 2,98 (м, 2 Н),2,60 (м, 2 Н), 1,38 (с, 9 Н). Промежуточный продукт 2(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(2-фтор-4-(трифторметил)фенил)бутановая кислота Стадия A. (2R,5S)-2,5-дигидро-3,6-диметокси-2-(2'-фтор-4'-(трифторметил)бензил)-5-изопропилпиразин К раствору 3,32 г (18 ммоль) коммерчески доступного (2S)-2,5-дигидро-3,6-диметокси-2 изопропилпиразина в 100 мл тетрагидрофурана при -70 С добавляют 12 мл (19 ммоль) 1,6 М раствора бутиллития в гексане. После перемешивания при указанной температуре в течение 20 мин добавляют 5 г(19,5 ммоль) 2-фтор-4-трифторметилбензилбромида в 20 мл тетрагидрофурана и продолжают перемешивание в течение 3 ч перед тем, как нагреть реакционную смесь до температуры окружающей среды. Реакционную смесь гасят водой, концентрируют в вакууме и экстрагируют этилацетатом. Объединенную органическую фазу промывают соляным раствором, сушат и концентрируют в вакууме. После очищения флэш-хроматографией (на силикагеле, 0-5% этилацетата в гексане) получают 5,5 г указанного в заголовке соединения. ЯМР 1 Н (500 МГц, CDCl3)7,33-7,25 (м, 3 Н), 4,35-4,31 (м, 1 Н), 3,75 (с, 3 Н), 3,65 (с, 3 Н),3,60 (т, 1 Н, J=3,4 Гц), 3,33 (дд, 1 Н, J=4,6, 13,5 Гц), 3,03 (дд, 1 Н, J=7, 13,5 Гц), 2,25-2,15 (м, 1 Н), 1,0 (д, 3 Н,J=7 Гц), 0,66 (д, 3 Н, J=7 Гц). Стадия В. Сложный метиловый эфир(2R,5S)-2,5-дигидро-3,6-диметокси-2-(2'-фтор-4'(трифторметил)бензил)-5-изопропилпиразина в 50 мл смеси ацетонитрил:дихлорметан (10:1) добавляют 80 мл 1 н водного раствора трифторуксусной кислоты. Реакционную смесь перемешивают в течение 6 ч и удаляют органические растворители в вакууме. Добавляют карбонат натрия до тех пор, пока раствор не станет основным (рН 8), затем реакционную смесь разбавляют 100 мл тетрагидрофурана и добавляют 10 г (46 ммоль) ди-трет-бутилдикарбоната. Полученную суспензию перемешивают в течение 16 ч, концентрируют в вакууме и экстрагируют этилацетатом. Объединенную органическую фазу промывают соляным раствором, сушат и концентрируют в вакууме. После очищения флэш-хроматографией (силикагель, 20%-ный раствор этилацетата в гексане) получают 5,1 г указанного в заголовке соединения. ЯМР Н (500 МГц, CDCl3)7,38-7,28 (м, 3 Н), 5,10 (уш.д, 1 Н), 4,65-3,98 (м, 1 Н), 3,76 (с, 3 Н), 3,32-3,25 (м, 1 Н),3,13-3,05 (м, 1 Н), 1,40 (с, 9 Н). Стадия С. (R)-N-1,1-диметилэтоксикарбонил)-2-фтор-4-трифторметил)фенилаланин Раствор 5,1 г (14 ммоль) сложного метилового эфира (R,S)-N-1,1-диметилэтоксикарбонил)-2 фтор-4-трифторметил)фенилаланина в 350 мл смеси тетрагидрофуран:метанол:1 н гидроксид лития(3:1:1) перемешивают при 50 С в течение 4 ч. Реакционную смесь' охлаждают, подкисляют 5% разбавленной соляной кислотой и экстрагируют этилацетатом. Объединенные органические фазы промывают соляным раствором, сушат над сульфатом магния и концентрируют в вакууме, получая при этом 4,8 г указанного в заголовке соединения. ЯМР 1 Н (500 МГц, CD3OD)7,45-7,38 (м, 3 Н), 4,44-4,40 (м, 1 Н),3,38-3,33 (м, 1 Н), 2,98 (дд, 1 Н, J=9,6, 13,5 Гц), 1,44 (с, 9 Н). Стадия(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-[2-фтор-4-(трифторметил)фенил]бутановая кислота К раствору 3,4 г (9,7 ммоль) продукта со стадии С в 60 мл тетрагидрофурана при 0 С добавляют последовательно 2,3 мл (13 ммоль) диизопропилэтиламина и 1,7 мл (13 ммоль) изобутилхлорформиата и перемешивают реакционную смесь при указанной температуре в течение 30 мин. Затем добавляют охлажденный эфирный раствор диазометана до получения устойчивой желтой окраски и продолжают дальнейшее перемешивание в течение 16 ч. Избыток диазометана гасят, добавляя по каплям уксусную кислоту, разбавляют реакционную смесь этилацетатом и промывают последовательно 5%-ным раствором соляной кислоты, насыщенным водным раствором бикарбоната натрия и соляным раствором, сушат над сульфатом магния и концентрируют в вакууме. После очищения флэш-хроматографией (на силикагеле,гексан:этилацетат в отношении 9:1) получают 0,5 г диазокетона. К раствору 0,5 г (1,33 ммоль) диазокетона, растворенного в 100 мл метанола при 0 С, добавляют последовательно 0,7 мл (4 ммоль) диизопропилэтиламина и 32 мг (0,13 ммоль) бензоата серебра. Реакционную смесь перемешивают в течении 2 ч прежде, чем разбавить этилацетатом и промыть последовательно 2 н раствором соляной кислоты, насыщенным водным раствором бикарбоната натрия и соляным раствором. Органическую фазу сушат над сульфатом магния, концентрируют в вакууме и растворяют в 50 мл смеси тетрагидрофуран:метанол:1 н водный раствор гидроксида лития (3:1:1) и перемешивают при 50 С в течение 3 ч. Реакционную смесь охлаждают, подкисляют 5% разбавленной соляной кислотой и экстрагируют этилацетатом. Объединенные органические фазы промывают соляным раствором, сушат над сульфатом магния и концентрируют в вакууме, получая при этом 410 мг указанного в заголовке соединения в виде белого вспененного твердого вещества. ЯМР 1 Н (500 МГц, CD3OD)7,47-7,33 (м, 3 Н), 4,88 (уш.с, 1 Н), 4,26-3,98 (м, 1 Н), 3,06-3,01(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутановая кислота Стадия A. (2S,5R)-2,5-дигидро-3,6-диметокси-2-изопропил-5-(2', 4', 5'-трифторбензил) пиразин Указанное в заголовке соединение (3,81 г) получают из 3,42 г (18,5 ммоль) (2S)-2, 5-дигидро-3,6 диметокси-2-изопропилпиразина согласно методике, описанной для получения промежуточного соединения 2, стадия А. ЯМР 1 Н (500 МГц, CDCl3)7,01 (м, 1 Н), 6,85 (м, 1 Н), 4,22 (м, 1 Н), 3,78 (м, 3 Н), 3,64(м, 3 Н), 3,61 (м, 1 Н), 3,20 (м, 1 Н), 2,98 (м, 1 Н), 2,20 (м, 1 Н), 0,99 (д, 3 Н, J=8 Гц), 0,62 (д, 3 Н, J=8 Гц). Стадия В. Сложный метиловый эфир (R)-N-(l,l-диметилэтоксикарбонил)-2,4,5-трифторфенилаланина К раствору 3,81 г (11,6 ммоль) (2S, 5R)-2,5-дигидро-3,6-диметокси-2-изопропил-5-(2',4',5'трифторбензил) пиразина в 20 мл ацетонитрила добавляют 20 мл 2 н соляной кислоты. Реакционную смесь перемешивают в течении 72 ч и концентрируют в вакууме. Остаток растворяют в 30 мл дихлорметана и добавляют 10 мл (72 ммоль) триэтиламина и 9,68 г (44,8 ммоль) ди-трет-бутилдикарбоната. Реакционную смесь перемешивают в течение 16 ч, разбавляют этилацетатом и последовательно промывают 1 н соляной кислотой и соляным раствором. Органическую фазу сушат над сульфатом натрия, концентрируют в вакууме и очищают флэш-хроматографией (на силикагеле, гексан:этилацетат в отношении 9:1), получая при этом 2,41 г указанного в заголовке соединения. ЯМР 1 Н (500 МГц, CDCl3)6,99 (м,1 Н), 6,94 (м, 1 Н), 5,08 (м, 1 Н), 4,58 (м, 1 Н), 3,78 (м, 3 Н), 3,19 (м, 1 Н) , 3,01 (м, 1 Н), 1,41 (с, 9 Н). Стадия С. (R)-N-(1,1-диметилэтоксикарбонил)-2,4,5-трифторфенилаланин Указанное в заголовке соединение (2,01 г) получают из 2,41 г (7,5 ммоль) сложного метилового эфира (R)-N-(l,l-диметилэтоксикарбонил)-2,4,5-трифторфенилаланина согласно методике, описанной для получения промежуточного соединения 2, стадия С. MS (M+1)-BOC 220,9.- 15006845 Стадия D. (3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-[2,4,5-трифторфенил]бутановая кислота К раствору 0,37 г (1,16 ммоль) (R)-N-(l,l-диметилэтоксикарбонил)-2,4,5-трифторфенилаланина в 10 мл простого диэтилового эфира при -20 С добавляют последовательно 0,193 мл (1,3 ммоль) триэтиламина и 0,18 мл (1,3 ммоль) изобутилхлорформиата и перемешивают реакционную смесь при указанной температуре в течение 15 мин. Затем добавляют охлажденный эфирный раствор диазометана до получения устойчивой желтой окраски и продолжают дальнейшее перемешивание в течение 1 ч. Избыток диазометана гасят, добавляя по каплям уксусную кислоту, разбавляют реакционную смесь этилацетатом и промывают последовательно насыщенным водным раствором бикарбоната натрия и соляным раствором,сушат над сульфатом магния и концентрируют в вакууме. После очищения флэш-хроматографией (силикагель, гексан:этилацетат в отношении 3:1) получают 0,36 г диазокетона. К раствору 0,35 г (1,15 ммоль) диазокетона, растворенного в 12 мл смеси 1,4-диоксан:вода (5:1), добавляют 26 мг (0,113 ммоль) бензоата серебра. Полученный раствор в течение 2 ч обрабатывают ультразвуком, после чего растворяют в этилацетате и последовательно промывают 1 н раствором соляной кислоты и соляным раствором, сушат над сульфатом магния и концентрируют в вакууме. После очищения флэш-хроматографией (на силикагеле, дихлорметан:метанол:уксусная кислота в отношении 97:2:1) получают 401 мг указанного в заголовке соединения. ЯМР 1 Н (500 МГц, CDCl3)7,06 (м, 1 Н), 6,95 (м, 1 Н), 5,06 (уш.с, 1 Н), 4,18 (м, 1 Н), 2,98(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-[4-бром-2,5-дифторфенил]бутановая кислота Стадия А. 4-Бром-2,5-дифторбензилбромид К раствору 2 г (8,44 ммоль) 4-бром-2,5-дифторбензойной кислоты (полученной согласно методике,описанной в публикации Ishikawa и др., Коgуо Kagaku Zasshi, стр. 972-979, 1970) в 20 мл тетрагидрофурана добавляют 40 мл 1 М раствора комплекса борана с тетрагидрофураном. Раствор кипятят с обратным холодильником в течение 64 ч, охлаждают до комнатной температуры и добавляют 100 мл метанола. Реакционную смесь нагревают в течение следующих 2 ч, охлаждают и концентрируют в вакууме. После очищения флэш-хроматографией (на силикагеле, гексан:этилацетат в отношении 9:1) получают 1,6 г 4 бром-2,5-дифторбензилового спирта. К раствору 1,3 г (5,6 ммоль) 4-бром-2,5-дифторбензилового спирта в 20 мл дихлорметана при 0 С добавляют 2,27 г (6,7 ммоль) тетрабромида углерода и 1,8 г (6,7 ммоль) трифенилфосфина. Реакционную смесь перемешивают в течение 2 ч при указанной температуре, растворитель удаляют в вакууме и остаток смешивают с 100 мл простого диэтилового эфира. Раствор фильтруют, концентрируют в вакууме и очищают флэш-хроматографией (на силикагеле, гексан:этилацетат в отношении 9:1), получая при этом 1,5 г указанного в заголовке соединения. Стадия В. (2S,5R)-2,5-дигидро-3,6-диметокси-2-изопропил-5-(4'-бром-2', 5'-дифторбензил) пиразин Указанное в заголовке соединение (1,61 г) получают из 0,865 г (4,7 ммоль) (2S)-2,5-дигидро-3,6 диметокси-2-изопропилпиразина и 1,5 г (5,2 ммоль) 4-бром-2,5-дифторбензилбромида согласно методике, описанной для промежуточного соединения 2, стадия А. ЯМР 1 Н (400 МГц, CDCl3)7,21 (м, 1 Н), 6,97 (м, 1 Н), 4,25 (м, 1 Н), 3,78 (с, 3 Н), 3,70-3,64 (м, 4 Н),3,25-3,18 (м, 1 Н), 2,96-2,90 (м, 1 Н), 2,25-2,16 (м, 1 Н), 1,01 (д, 3 Н, J=8 Гц), 0,65 (д, 3 Н, J=8 Гц). Стадия С. Сложный метиловый эфир(R)-N-(1,1-диметилэтоксикарбонил)-4-бром-2,5 дифторфенилаланина К раствору 1,61 г (4,14 ммоль) (2S,5R)-2,5-дигидро-3,6-диметокси-2-изопропил-5-(4'-бром-2',5'дифторбензил) пиразина в 10 мл ацетонитрила добавляют 10 мл 2 н соляной кислоты. Реакционную смесь перемешивают в течение 16 ч и концентрируют в вакууме. Остаток растворяют в 30 мл дихлорметана и добавляют 5,6 мл (40 ммоль) триэтиламина и 2,2 г (10 ммоль) ди-трет-бутилдикарбоната. Реакционную смесь перемешивают в течение 16 ч, разбавляют этилацетатом и промывают последовательно насыщенным водным раствором бикарбоната натрия и соляным раствором. Органическую фазу сушат над сульфатом магния, концентрируют в вакууме и очищают флэш-хроматографией (на силикагеле, гексангэтилацетат в отношении 9:1), получая при этом 1,22 г указанного в заголовке соединения. ЯМР 1 Н (400 МГц,CDCl3)7,27-7,15 (м, 1 Н), 6,98-6,93 (м, 1 Н), 5,08 (уш.с, 1 Н), 4,61-4,55 (м, 1 Н), 3,78 (с, 3 Н), 3,23-3,18 (м,1 Н), 3,05-2,95 (м, 1 Н), 1,41 (с, 9 Н). Стадия D. (R)-N-(1,1-диметилэтоксикарбонил)-4-бром-2,5-дифторфенилаланин Указанное в заголовке соединение (1,34 г) получают из 1,4 г (3,5 ммоль) сложного метилового эфира (R)-N-(l,l-диметилэтоксикарбонил)-4-бром-2,5-дифторфенилаланина согласно методике, описанной- 16006845 для получения промежуточного соединения 2, стадия С. MS (M+1) 380,3 и 382,3. Стадия Е. (3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-[4'-бром-2',5'-дифторфенил]бутановая кислота Указанное в заголовке соединение (0,36 г) получают из 0,6 г (1,57 ммоль) (R)-N-(1,1 диметилэтоксикарбонил)-4-бром-2,5-дифторфенилаланина согласно методике, описанной для получения промежуточного соединения 3, стадия D. MS (М+1) 394,1 и 396,1. Пример 1. Дигидрохлорид 7-[(3R)-3-амино-4-(3,4-дифторфенил)бутаноил]-2-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина Стадия А. 2-(Трифторметил)имидазо[1,2-а]пиразин К раствору 2-аминопиразина (5,25 г, 55,2 ммоль) в этаноле (120 мл) добавляют 1-бром-3,3,3 трифторацетон (5,73 мл, 55,2 ммоль). Реакционную смесь кипятят с обратным холодильником при перемешивании в течение 20 ч. После выпаривания растворителя остаток распределяют между этилацетатом и насыщенным водным раствором бикарбоната натрия. Водный слой экстрагируют этилацетатом (х 3). Объединенную органическую фазу промывают соляным раствором, сушат над сульфатом магния и концентрируют. Остаток очищают флэш-хроматографией (на силикагеле, этилацетат:гексан в отношении 1:1, затем 100% этилацетат), получая при этом 2,35 г указанного в заголовке соединения в виде твердого вещества. ЯМР 1 Н (500 МГц, CDCl3)8,02 (м, 2 Н), 8,13 (м, 1 Н), 9,22 (с, 1 Н) ESI-MS 188 (М+1). Стадия В. 2-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин К раствору 2-(трифторметил)имидазо[1,2-а]пиразина (2,0 г, 10,46 ммоль, со стадии А) в метаноле(100 мл) добавляют 10% палладия на углероде (400 мг). Смесь перемешивают в атмосфере водорода при температуре окружающей среды в течение 14 ч. Смесь фильтруют через целит (Celite) и промывают метанолом (хЗ). Фильтрат концентрируют и очищают флэш-хроматографией (на силикагеле, 10% метанола в этилацетате, затем 15% метанола в хлороформе с 1% водного раствора гидроксида аммония), получая при этом 1,33 г указанного в заголовке соединения в виде твердого вещества. ЯМР 1 Н (500 МГц, CDCl3)1,93 (уш.с, 1 Н), 3,26 (т, 2 Н, J=5,5 Гц), 3,99 (т, 2 Н, J=5,5 Гц), 4,10 (с, 1 Н), 7,16 (с, 1 Н). ESI-MS 192 (М+1). Стадия С. 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(3,4-дифторфенил)бутаноил]-2(трифторметил)-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин К раствору 2-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина (64,3 мг, 0,34 ммоль, со стадии В) и (3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-[3,4-дифторфенил]бутановой кислоты (105,9 мг, 0,34 ммоль) в дихлорметане (5 мл) добавляют НОВТ (54,5 мг, 0,42 ммоль) при 0 С. Реакционную смесь перемешивают при 0 С в течение 10 мин, затем добавляют EDC (96,6 мг, 0,50 ммоль). После удаления бани со льдом реакционную смесь перемешивают при температуре окружающей среды в течение 14 ч. Смесь концентрируют и очищают ВЭЖХ (Gilson; колонка YMC-Pack Pro C18, 100x20 мм I.D.; градиент растворителей от смеси: 10% ацетонитрила, 90% воды и 0,1% трифторуксусной кислоты до смеси: 90% ацетонитрила, 10% воды и 0,1% трифторуксусной кислоты), получая при этом 115 мг указанного в заголовке соединения в виде вспененного твердого вещества. ЯМР 1 Н (500 МГц, CDCl3)1,36 (с, 9 Н),2,62 (м, 2 Н), 2,86 (м, 2 Н), 3,34 (уш.с, 1 Н), 3,86 (м, 1 Н), 4,05 (м, 4 Н), 4,85 (м, 1 Н), 5,30-5,38 (м, 1 Н), 6,97 (м,3 Н), 7,28 (м, 1 Н). LC/MS 489 (М+1). Стадия D. Дигидрохлорид 7-[(3R)-3-амино-4-(3,4-дифторфенил)бутаноил]-2-(трифторметил)5,6,7,8-тетрагидроимидазо[1,2-а]пиразина К 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(3,4-дифторфенил)бутаноил]-2-(трифторметил)5,6,7,8-тетрагидроимидазо[1,2-а]пиразину (110,8 мг, 0,226 ммоль, со стадии С) добавляют 2 мл метанола,насыщенного хлористым водородом. Реакционную смесь перемешивают при температуре окружающей среды в течение 1 ч. После концентрирования получают 89,5 мг указанного в заголовке соединения в виде вспененного твердого вещества. ЯМР 1 Н (500 МГц, CD3OD)2,97-3,10 (м, 4 Н), 3,91-4,34 (м, 5 Н),4,90-5,04 (м, 2 Н), 7,16-7,33 (м, 2 Н), 8,01-8,08 (м, 1 Н). ESI-MS 389 (М+1). Пример 2.- 17006845 Дигидрохлорид 7-[(3R)-3-амино-4-(2,5-дифторфенил)бутаноил]-2-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина Стадия А. 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(2,5-дифторфенил)бутаноил]-5,6,7,8 тетрагидроимидазо[1,2-а]пиразин Указанное в заголовке соединение получают из 2-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,2 а]пиразина (277 мг, 1,45 ммоль, по примеру 1, стадия В), (3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4(2,5-дифторфенил)бутановой кислоты (промежуточный продукт 1, 416 мг, 1,32 ммоль), DIPEA (226 мг,1,58 моль), НОВТ (216 мг, 1,98 моль) и HATU (753 мг, 1,98 моль) в ДМФ (6 мл) по методике, аналогичной описанной в примере 1 для стадии С, исключая способ очищения. Соединение очищают препаративной ТСХ (на силикагеле, 20%-ный раствор гексана в этилацетате, затем 10%-ный раствор метанола в дихлорметане), получая при этом 360 мг указанного в заголовке соединения в виде вспененного твердого вещества. ЯМР 1 Н (500 МГц, CDCl3)1,35 (с, 9 Н), 2,62 (м, 2 Н), 2,88 (м, 2 Н), 3,88-4,16 (м, 5 Н), 4,73 (с,1 Н), 4,85 (м, 1 Н), 5,26-5,39 (м, 1 Н), 6,90 (уш.с, 1 Н), 7,06 (м, 2 Н), 7,24 (м, 1 Н). ESI-MS 489 (М+1). Стадия В. Дигидрохлорид 7-[(3R)-3-амино-4-(2,5-дифторфенил)бутаноил]-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина Указанное в заголовке соединение получают из 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4(2,5-дифторфенил)бутаноил]-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина (349,8 мг, 0,72 моль, со стадии А) в 1,5 мл метанола, насыщенного хлористым водородом, по методике, аналогичной описанной в примере 1 для стадии D. После выпаривания растворителя получают 299 мг указанного в заголовке соединения в виде вспененного твердого вещества. ЯМР 1 Н (500 МГц, CD3OD)3,10-3,17 (м, 2 Н), 2,89-2,99 (м, 2 Н), 3,94-4,22 (м, 4 Н), 4,33 (м, 1 Н),4,91-5,48 (м, 2 Н), 7,07-7,23 (м, 3 Н), 8,05 (м, 1 Н). ESI-MS 389 (М+1). Пример 3. Дигидрохлорид 7-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-2-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина Стадия А. 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутаноил]-5,6,7,8 тетрагидроимидазо[1,2-а]пиразин Указанное в заголовке соединение получают из 2-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,2 а]пиразина (31,7 мг, 0,166 ммоль, по примеру 1, стадия В), (3R)-3-[(1,1-диметилэтоксикарбонил)амино]4-(2,4,5-трифторфенил)бутановой кислоты (промежуточное соединение 3,57 мг, 0,166 ммоль), НОВТ(26,9 мг, 0,199 ммоль) и EDC (47,8 мг, 0,249 ммоль) в 4 мл дихлорметана по методике, аналогичной описанной в примере 1 для стадии С. После очищения препаративной ТСХ (на силикагеле, 100% этилацетат,затем 10%-ный раствор метанола в дихлорметане) получают 40 мг указанного в заголовке соединения в виде вспененного твердого вещества. ЯМР 1 Н (500 МГц, CDCl3)1,35 (с, 9 Н), 3,00 (м, 2 Н), 3,30 (м, 2 Н),3,93 (м, 1 Н), 4,04-4,24 (м, 2 Н), 4,23 (с, 1 Н), 4,35 (м, 1 Н), 4,97-5,48 (м, 2 Н), 7,22 (м, 1 Н), 7,44 (м, 1 Н), 8,04(м, 1 Н). ESI-MS 507 (М+1). Стадия В. Дигидрохлорид 7-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина Указанное в заголовке соединение получают из 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4(2,4,5-трифторфенил)бутаноил]-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина (38 мг, 0,075 ммоль, со стадии А) в 1,5 мл метанола, насыщенного хлористым водородом, по методике, аналогичной описанной в примере 1 для стадии D). После выпаривания растворителя получают 34 мг указанного в заголовке соединения в виде вспененного твердого вещества. ЯМР 1 Н (500 МГц, CD3OD)2,59-2,66 (м, 2 Н), 2,92 (м, 2 Н),3,89-4,16-4,22 (м, 5 Н), 4,70-4,84 (м, 2 Н), 5,42 (м, 1 Н), 6,86 (м, 1 Н), 7,06 (м, 1 Н), 7,24 (м, 1 Н). ESI-MS 407- 18006845 Стадия А. Имидазо[1,2-а]пиразин К раствору 2-аминопиразина (2,0 г, 21,03 ммоль) в этаноле (40 мл) добавляют 2-бром-1,1 диметоксиэтан (2,5 мл, 21,03 ммоль), затем 5 капель концентрированной соляной кислоты. После кипячения с обратным холодильником в течение 14 ч растворитель выпаривают. Остаток распределяют между этилацетатом и насыщенным водным раствором бикарбоната натрия. Водный слой экстрагируют этилацетатом (хЗ). Объединенную органическую фазу промывают соляным раствором, сушат над сульфатом магния и концентрируют. Остаток очищают флэш-хроматографией (100% этилацетат, 10%-ный раствор метанола в этилацетате, затем 10%-ный раствор метанола в дихлорметане), получая при этом 536 мг указанного в заголовке соединения в виде твердого вещества. ЯМР 1 Н (500 МГц, CDCl3)7,70 (уш.с,1 Н), 7,82 (уш.с, 1 Н), 7,89 (д, 1 Н, J=4,4 Гц), 8,10 (д, 1 Н, J=4,6 Гц), 9,12 (с, 1 Н). Стадия В. 5,6,7,8-Тетрагидроимидазо[1,2-а]пиразин Указанное в заголовке соединение получают из имидазо[1,2-а]пиразина (500 мг, 4,20 ммоль, со стадии А) и оксида платины (250 мг) в метаноле (50 мл) по методике, аналогичной описанной в примере 1 для стадии В. После концентрирования получают указанное в заголовке соединение (512 мг) в виде вязкого маслянистого вещества. ЯМР 1 Н (500 МГц, CD3OD)3,37 (т, 1 Н, J=5,5 Гц), 4,18 (т, 2 Н, J=5,6 Гц),4,88 (с, 1 Н), 7,27 (д, J=l,6 Гц, 1 Н), 7,33 (д, 1 Н). Стадия С. 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(3,4-дифторфенил)бутаноил]-5,6,7,8 тетрагидроимидазо[1,2-а]пиразин Указанное в заголовке соединение получают из 5,6,7,8-тетрагидроимидазо[1,2-а]пиразина (31,3 мг,0,254 ммоль, со стадии В), (3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(3,4-дифторфенил)бутановой кислоты (80 мг, ммоль), DIPEA (32,8 мг, 0,254 ммоль), НОВТ (41,2 мг, 0,305 ммоль) и EDC (73 мг, 0,381 ммоль) в 5 мл дихлорметана по методике, аналогичной описанной в примере 1 для стадии С. После очищения ВЭЖХ (Gilson; колонка YMC-Pack Pro C18, 100x20 мм I.D.; система растворителей с градиентом от смеси: 10% ацетонитрила, 90% воды и 0,1% трифторуксусной кислоты до смеси: 90% ацетонитрила,10% воды и 0,1% трифторуксусной кислоты) получают 75 мг указанного в заголовке вещества в виде вязкого маслянистого вещества. ЯМР 1 Н (500 МГц, CDCl3)1,38 (с, 9 Н), 2,05 (уш.с, 1 Н), 2,62 (м, 2 Н),2,89 (м, 2 Н), 3,81-4,04 (м, 5 Н), 4,64-4,88 (м, 2 Н), 5,38 (м, 1 Н), 6,88 (м, 2 Н), 7,05 (м, 3 Н). ESI-MS 421D. Дигидрохлорид 7-[(3R)-3-амино-4-(3,4-дифторфенил)бутаноил]-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина Указанное в заголовке соединение получают из 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4(3,4-дифторфенил)бутаноил]-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина (72 мг, 0,171 ммоль, со стадии С) в 1,5 мл метанола, насыщенного хлористым водородом, по методике, аналогичной описанной в примере 1 для стадии D. После концентрирования получают 66 мг указанного в заголовке соединения в виде вспененного твердого вещества. ЯМР 1 Н (500 МГц, CD3OD)2,96-3,13 (м, 4 Н), 3,93 (м, 1 Н), 4,13 (м, 2 Н),4,26-4,38 (м, 2 Н), 4,90-5,04 (м, 2 Н), 7,19-7,36 (м, 3 Н), 7,58 (м, 1 Н). ESI-MS 321 (М+1). Пример 5. Дигидрохлорид 7-[(3R)-3-амино-4-(3,4-дифторфенил)бутаноил]-3-этил-5,6,7,8-тетрагидро-1,2,4 триазоло[4,3-а]пиразина Стадия А. 8-Хлор-3-этил-1,2,4-триазоло[4,3-а]пиразин К 3-хлор-2-гидразинопиразину (3,0 г, 20,75 ммоль), полученному из 2,3-дихлорпиразина и гидразина по методике, аналогичной описанной в литературе (Huynh-Dinh и др., J. Оrg. Chem. 1979, 44, 1028),добавляют 8 мл триэтилортопропионата. После кипячения с обратным холодильником в течение 10 ч реакционную смесь охлаждают до температуры окружающей среды и отфильтровывают осадок. Твердое вещество очищают флэш-хроматографией (100% этилацетат, затем 10% метанола в этилацетате), получая при этом 2,73 г указанного в заголовке соединения в виде твердого вещества. ЯМР 1 Н (500 МГц,CDCl3)1,54 (т, 3 Н, J=7,6 Гц), 3,16 (кв, 2 Н, J=7,8 Гц), 7,70 (д, 1 Н, J=4,5 Гц), 7,83 (д, 1 Н, J=4,8 Гц). Стадия В. Гидрохлорид 3-этил-5,6,7,8-тетрагидро-1,2,4-триазоло[4,3-а]пиразина Указанное в заголовке соединение получают из 8-хлор-3-этил-1,2,4-триазоло[4,3-а]пиразина (2,70 г,14,8 ммоль, со стадии А) и оксида платины (0,4 г) в 200 мл метанола на встряхивающем смесителе в атмосфере водорода (50 фунт/кв. дюйм) в течение 14 ч. После фильтрования через целит и последующего концентрирования получают указанное в заголовке соединение в виде твердого вещества. ЯМР 1 Н (500 МГц, CD3OD)1,36 (т, 3 Н, J=6,0 Гц), 2,84 (кв, 2 Н, J=6,0 Гц), 3,70 (т, 2 Н, J=8,0 Гц), 4,28 (т, 2 Н, J=8,0 Гц),4,06 (с,2 Н). ESI-MS 153 (М+1).- 19006845 Стадия С. 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(3,4-дифторфенил)бутаноил]-3-этил 5,6,7,8-тетрагидро-1,2,4-триазоло[4,3-а]пиразин Указанное в заголовке соединение получают из гидрохлорида 3-этил-5,6,7,8-тетрагидро-1,2,4 триазоло[4,3-а]пиразина (400 мг, 2,12 ммоль, со стадии В), (3R)-3-[(1,1-диметилэтоксикарбонил)амино]4-(3,4-дифторфенил)бутановой кислоты (668 мг, 2,12 ммоль), DIPEA (1,1 мл, 4,24 ммоль), НОВТ (343,8 мг, 2,54 ммоль), EDC (609,6 мг, 3,18 ммоль) в 20 мл дихлорметана по методике, аналогичной описанной в примере 1 для стадии С. Неочищенный продукт очищают ВЭЖХ (Gilson; колонка YMC-Pack Pro C18,100x20 мм I.D.; градиент растворителей от смеси: 10% ацетонитрила, 90% воды и 0,1% трифторуксусной кислоты до смеси: 90% ацетонитрила, 10% воды и 0,1% трифторуксусной кислоты), получая при этом 366,3 мг указанного в заголовке соединения в виде вязкого маслянистого вещества. ЯМР 1 Н (500 МГц,CDCl3)1,31-1,34 (м, 12 Н), 2,67-2,92 (м, 6 Н), 4,03-4,12 (м, 4 Н), 5,03-5,31 (м, 3 Н), 6,93 (с, 1 Н), 7,05 (м, 2 Н)D. Дигидрохлорид 7-[(3R)-3-амино-4-(3,4-дифторфенил)бутаноил]-3-этил-5,6,7,8 тетрагидро-1,2,4-триазоло[4,3-а]пиразина Указанное в заголовке соединение получают из 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4(3,4-дифторфенил)бутаноил]-3-этил-5,6,7,8-тетрагидро-1,2,4-триазоло[4,3-а]пиразина (30 мг, 0,067 ммоль, со стадии С) в 1,5 мл метанола, насыщенного хлористым водородом, по методике, аналогичной описанной в примере 1 для стадии D. После выпаривания растворителя получают 28 мг указанного в заголовке соединения в виде вязкого маслянистого вещества. ЯМР 1 Н (500 МГц, CD3OD)1,45 (т, 3 Н),2,93-3,07 (м, 6 Н), 3,90-4,31 (м, 5 Н), 5,08 (м, 2 Н), 7,16 (с, 1 Н), 7,31 (м, 2 Н). ESI-MS 350 (М+Н). Пример 6. Гидрохлорид 7-[(3R)-3-амино-4-(2,5-дифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидро 1,2,4-триазоло[4,3-а]пиразина Стадия А. 3-(Трифторметил)-1,2,4-триазоло[4,3-а]пиразин Смесь 2-гидразинопиразина (820 мг, 7,45 ммоль), полученного из 2-хлорпиразина и гидразина по методике, аналогичной описанной в литературе (P.J. Nelson и К.Т. Potts, J. Org. Chem. 1962, 27, 324 3 за исключением того, что неочищенный продукт экстрагируют смесью 10% метанол/дихлорметан и фильтруют, а фильтрат концентрируют и очищают флэш-хроматографией на силикагеле, элюируя 100% этилацетатом, а затем 10%-ным раствором метанола в дихлорметане), TFA (2,55 г, 22,4 ммоль) и полифосфорной кислоты (10 мл) нагревают до 140 С при перемешивании в течение 18 ч. Раствор добавляют ко льду и нейтрализуют добавлением гидроксида аммония. Водный раствор экстрагируют этилацетатом(х 3), промывают соляным раствором и сушат над обезвоженным сульфатом магния. После концентрирования и последующей флэш-хроматографии (на силикагеле, гексан:этилацетат в отношении 1:1, затем 100% этилацетат) получают указанное в заголовке соединение в виде твердого вещества (861 мг). ЯМР 1 Н (500 МГц, CDCl3)8,17-8,20 (м, 2 Н), 9,54 (с, 1 Н). LC/MS (М+1) 189. Стадия В. 3-(Трифторметил)- 5,6,7,8-тетрагидро-1,2, 4-триазоло[4,3-а]пиразин 3-(Трифторметил)-1,2,4-триазоло[4,3-а]пиразин (540 мг, 2,87 ммоль, со стадии А) гидрируют в атмосфере водорода с 10% Pd/C (200 мг) в качестве катализатора в этаноле (10 мл) при температуре окружающей среды в течение 18 ч. После фильтрования через целит и последующего концентрирования получают маслянистое вещество темного цвета. К полученному маслянистому веществу добавляют дихлорметан, и нерастворимый черный осадок отфильтровывают. При концентрировании фильтрата получают указанное в заголовке соединение в виде маслянистого вещества (495 мг). ЯМР 1H (500 МГц, CDCl3)2,21 (уш., 1 Н), 3,29 (т, 2 Н, J=5,5 Гц), 4,09 (т, 2 Н, J=5,5 Гц), 4,24 (с, 2 Н). LC/MS (М+1) 193. Стадия С. 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(2,5-дифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидро-1,2,4-триазоло[4,3-а]пиразин Указанное в заголовке соединение получают из (3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(2,5 дифторфенил)бутановой кислоты (промежуточное соединение 1,50 мг, 0,16 ммоль) и 3-(трифторметил)5,6,7,8-тетрагидро-1,2,4-триазоло [4,3-а]пиразина (30 мг, 0,16 ммоль) по методике, аналогичной описанной в примере 1 для стадии С. Неочищенный продукт очищают препаративной ТСХ (на силикагеле,100% этилацетата, затем 10% метанол/дихлорэтан (х 2, получая при этом указанное в заголовке соединение (38,1 мг) в виде твердого вещества. ЯМР 1 Н (500 МГц, CDCl3)1,38 (с, 9 Н), 2,57-3,05 (м, 4 Н), 3,854,30 (м, 5 Н), 4,90 (с, 1 Н), 4,95-5,15 (м, 1 Н), 5,22-5,40 (уш., 1 Н), 6,86-7,24 (м, 3 Н). LC/MS (M+1-t-Boc) 390.- 20006845 Стадия D. Гидрохлорид 7-[(3R)-3-амино-4-(2,5-дифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8 тетрагидро-1,2,4-триазоло[4,3-а]пиразина Указанное в заголовке соединение получают из 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4(2,5-дифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидро-1,2,4-триазоло[4,3-а]пиразина(19,1 мг, 0,039 ммоль, со стадии С) по методике, аналогичной описанной в примере 1 для стадии D. При концентрировании получают указанное в заголовке соединение (16,1 мг) в виде твердого вещества. ЯМР 1 Н Гидрохлорид 7-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидро-1,2,4-триазоло[4,3-а]пиразина Стадия А. 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутаноил]-3(трифторметил)-5,6,7,8-тетрагидро-1,2,4-триазоло[4,3-а]пиразин Указанное в заголовке соединение получают из (3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4(2,4,5-трифторфенил)бутановой кислоты (промежуточное соединение 3, 50,1 мг, 0,15 ммоль) и 3(трифторметил)-5,6,7,8-тетрагидро-1,2,4-триазоло[4,3-а]пиразина (39,2 мг, 0,20 ммоль) по методике, аналогичной описанной в примере 1 для стадии С. Неочищенный продукт очищают препаративной ТСХ (на силикагеле, 100% этилацетат), получая при этом указанное в заголовке соединение (29 мг) в виде твердого вещества. ЯМР 1 Н (500 МГц, CDCl3)1,37 (с, 9 Н), 2,61-3,00 (м, 4 Н), 3,92-4,30 (м, 5 Н), 4,93 (с, 1 Н),4,95-5,12 (м, 1 Н), 5,22-5,35 (уш., 1 Н), 6,83-6,95 (м, 1 Н), 7,02-7,12 (м, 1 Н). LC/MS (M+1-t-Bu) 452. Стадия В. Гидрохлорид 7-[(3R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)5,6,7,8-тетрагидро-1,2,4-триазоло[4,3-а]пиразина Указанное в заголовке соединение получают из 7-[(3R)-3-[(1,1-диметилэтоксикарбонил)амино]-4(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидро-1,2,4-триазоло[4,3-а]пиразина (22 мг, 0,039 ммоль, со стадии А) по методике, аналогичной описанной в примере 1 для стадии D. После концентрирования получают указанное в заголовке соединение (16,5 мг) в виде твердого вещества. ЯМР 1 Н (500 МГц, CD3OD)2,75-3,15 (м, 4 Н), 3,82-4,35 (м, 5 Н), 4,90-5,05 (м, 2 Н), 7,16-7,25 (м, 1 Н), 7,30-7,42 После того, как изобретение описано и проиллюстрированы некоторые из его конкретных воплощений, специалисту в данной области очевидно, что можно делать различные адаптации, изменения,модификации, замены, исключения или добавления процедур и условий (протоколов) без отступления от сущности и объема изобретения. Например, для любого из показаний, связанных с соединениями изо- 22006845 бретения, указанных выше, можно применять эффективные дозировки, отличающиеся от определенных,указанных в описании дозировок из-за вариаций в восприимчивости подвергаемого лечению млекопитающего. Наблюдаемые специфические фармакологические реакции могут изменяться в соответствии и в зависимости от конкретно выбранных активных соединений или от того, присутствуют ли фармацевтические носители, также как в зависимости от типа композиции и применяемого способа введения, и в соответствии с целями и практикой настоящего изобретения предусматриваются ожидаемые вариации или различия в результатах. Поэтому подразумевается, что изобретение определяется объемом формулы изобретения, которая следует дальше, и что такая формула изобретения интерпретируется настолько широко, насколько приемлемо. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, выбранное из группы, состоящей из и их фармацевтически приемлемых солей. 2. Соединение по п.1, выбранное из группы, состоящей из- 27006845 или его фармацевтически приемлемая соль. 3. Соединение по п.2, которое представляет или его фармацевтически приемлемая соль. 4. Соединение по п.2, которое представляет или его фармацевтически приемлемая соль. 5. Соединение по п.2, которое представляет или его фармацевтически приемлемая соль. 6. Соединение формулы Ib где Аr представляет фенил, незамещенный или замещенный 1-5 заместителями, которые независимо выбраны из группы, состоящей из(1) водорода,(2) C1-6-алкила с линейной или разветвленной цепью, незамещенного или замещенного фенилом или 1-5 атомами фтора,(3) фенила. 7. Соединение по п.6, где Аr выбран из группы, состоящей из где Аr представляет фенил, незамещенный или замещенный 1-5 заместителями, которые независимо выбраны из группы, состоящей из(1) водорода,(2) C1-6-алкила с линейной или разветвленной цепью, незамещенного или замещенного фенилом или 1-5 атомами фтора и(1) водорода,(2) C1-6-алкила с линейной или разветвленной цепью, незамещенного или замещенного 1-5 атомами фтора,(3) фенила, незамещенного или замещенного 1-3 заместителями, независимо выбранными из фтора,ОСН 3 и OCF3. 12. Соединение по п.11, где R2 выбран из группы, состоящей из(11) 3,4-дифторфенила. 13. Соединение по п.12, где R2 представляет CF3 или CF2CF3. 14. Фармацевтическая композиция, содержащая инертный носитель и соединение по п.6 или 11. 15. Применение соединения по п.6 или 11 для изготовления лекарственного средства для применения для лечения состояния, выбранного из группы, выбранной из гипергликемии, диабета типа 2, ожирения, и липидных расстройств у млекопитающих.
МПК / Метки
МПК: C07D 487/04, A61P 3/10, A61K 31/4985
Метки: пиразины, предотвращения, лечения, бета-аминотетрагидроимидазо, диабета, дипептидилпептидазы, ингибиторы, тетрагидротриазоло
Код ссылки
<a href="https://eas.patents.su/30-6845-beta-aminotetragidroimidazo-1-2-a-piraziny-i-tetragidrotriazolo-4-3-a-piraziny-kak-ingibitory-dipeptidilpeptidazy-dlya-lecheniya-ili-predotvrashheniya-diabeta.html" rel="bookmark" title="База патентов Евразийского Союза">Бета-аминотетрагидроимидазо – (1, 2 – а) – пиразины и тетрагидротриазоло – (4, 3 – а ) – пиразины как ингибиторы дипептидилпептидазы для лечения или предотвращения диабета</a>
Арилзамещенные пиридины, пиримидины, пиразины или триазины (варианты), фармацевтическая композиция, способ лечения нарушения у млекопитающего, связанного с блокадой натриевых каналов, способ лечения,профилактики или снижения интенсивности различных нарушений или состояний, способ снижения интенсивности или профилактики приступов судороги у животного

Номер патента: 5770
Опубликовано: 30.06.2005
Авторы: Хогенкэмп Дерк Дж., Нгуйен Фонг, Шао Бин
МПК: A61P 25/08, C07D 213/81, A61K 31/505...
Метки: арилзамещенные, связанного, приступов, животного, нарушения, пиридины, фармацевтическая, лечения, профилактики, пиримидины, судороги, способ, композиция, натриевых, различных, снижения, варианты, пиразины, интенсивности, млекопитающего, состояний, блокадой, нарушений, лечения,профилактики, триазины, каналов
Формула / Реферат:
1. Арилзамещенные пиридины, пиримидины, пиразины или триазины формулы I или их фармацевтически приемлемые соль, пролекарство или сольват, где Y означает или R7; при условии, что если Y означает радикал R7, то R1 означает аминокарбонил; A1, A2 и A3 независимо выбирают из группы, включающей в себя CR2 или N, при условии, что A1, A2 и A3 все одновременно не означают N; R1 выбирают из группы, включающей в себя C1-6алкил, необязательно замещенный...
Способ лечения диабета росиглитазоном и инсулином

Номер патента: 4800
Опубликовано: 26.08.2004
Автор: Смит Стефен Элистэр
МПК: A61P 3/10, A61K 31/4439
Метки: способ, росиглитазоном, инсулином, лечения, диабета
Формула / Реферат:
1. Комбинация, включающая от 2 до 12 мг 5-[4-[2-(N-метил-N-(2-пиридил)амино)этокси]бензил]тиазолидин-2,4-диона (соединение (I)) или его фармацевтически приемлемой формы и инсулин для лечения диабета типа 2 и состояний, связанных с диабетом типа 2. 2. Комбинация по п.1, включающая от 2 до 4 мг, от 4 до 8 мг или от 8 до 12 мг соединения (I) или его фармацевтически приемлемой формы. 3. Комбинация по п.1 или 2, включающая от 2 до 4 мг соединения (I)...
Способ лечения диабета тиазолидиндионом и метформином

Номер патента: 3144
Опубликовано: 27.02.2003
Автор: Смит Стефен Элистэр
МПК: A61P 3/10, A61K 31/44
Метки: способ, метформином, лечения, тиазолидиндионом, диабета
Формула / Реферат:
- формула изобретения, действующая на территории Договаривающихся государств, для которых ниже не указана особая редакция формулы1. Способ лечения сахарного диабета и состояний, связанных с сахарным диабетом, у млекопитающих, включающий введение от 2 до 12 мг 5-[4-[2-(N-метил-N-(2-пиридил)амино)этокси]бензил]тиазолидин-2,4-диона (cоединение I) или его фармацевтически приемлемой формы и бигуанидного антигипергликемического агента млекопитающему,...
Ингибиторы 11-бета-гидроксистероиддегидрогеназы типа 1

Номер патента: 5274
Опубликовано: 30.12.2004
Авторы: Курц Гвидо, Барф Тьерд, Валлгорда Йерк, Нильссон Марианне, Эмонд Рикард
МПК: A61P 3/00, C07D 277/52, A61K 31/426...
Метки: 11-бета-гидроксистероиддегидрогеназы, типа, ингибиторы
Формула / Реферат:
1. Соединение формулы (II) где T представляет фенил, нафтил, гетероарил, означающий моноциклическую, би- или трициклическую ароматическую кольцевую систему, в которой только одно кольцо является ароматическим, имеющую от 5 до 14 атомов в кольце, в которой одно или несколько атомов кольца являются иными, чем углерод, такими как азот, сера, кислород и селен; фенил-C2-алкенил или нафтил-C2-алкенил, необязательно независимо замещенные [R]n, где n...
Способ лечения диабета тиазолидиндионом и сульфонилмочевиной

Номер патента: 3303
Опубликовано: 24.04.2003
Авторы: Смит Стефен Элистэр, Бакингэм Робин Эдвин
МПК: A61P 3/10, A61K 31/64
Метки: способ, сульфонилмочевиной, тиазолидиндионом, лечения, диабета
Формула / Реферат:
1. Способ лечения сахарного диабета и состояний, связанных с сахарным диабетом, у млекопитающих, включающий введение эффективного нетоксичного и фармацевтически приемлемого количества от 2 до 12 мг в день 5-[4-[2-(N-метил-N-(2-пиридил)амино)этокси]бензил]тиазолидин-2,4-диона (соединение I) или его таутомерной формы, и/или его фармацевтически приемлемого производного и субмаксимального количества сульфонилмочевины (средства, усиливающего секрецию...
Предыдущий патент: Окрашенные твердые капсулы
Следующий патент: Болеутоляющая композиция для перорального применения с регулируемым высвобождением опиоида
Случайный патент: 3 - гетероциклилзамещённые производные бензойной кислоты