Синтетический способ получения соединения эктеинасцидина
Номер патента: 6369
Опубликовано: 29.12.2005
Авторы: Фернандес Каролина, Гальего Пилар, Мансанарес Игнасио, Франсесч Андрес, Мант Саймон, Сарсуэло Мария, Перес Марта, Чичарро Хосе Луис, Мартин Мария Хесус, Куэвас Кармен
























Формула / Реферат
1. Стадия способа получения эктеинасцидинового соединения, где указанная стадия включает удаление обеих защитных групп в одну стадию в соответствии со следующей схемой:

где ProtNH представляет аминозащитную группу, а ProtOH представляет гидроксизащитную группу.
2. Способ по п.1, где ProtNH представляет трет-бутоксикарбонил и ProtOH представляет метоксиметокси.
3. Способ по п.1, где ProtNH представляет трет-бутоксикарбонил и ProtOH представляет метоксиэтоксиметокси.
4. Способ по п.1, включающий дополнительную стадию окисления a -аминолактона формулы (35) в соответствующий a -кетолактон формулы (36) путем трансаминирования

5. Способ по п.4, который включает дополнительную стадию стереоспецифического образования спиротетрагидроизохинолинового соединения Et770 из a-кетолактона формулы (36) по реакции Пикте-Шпенглера

6. Способ по п.5, который включает дополнительную стадию замещения нитрильной группы в положении С-21 соединения Et770 гидроксигруппой с получением соединения Et743

7. Стадия способа получения эктеинасцидинового соединения окислением a -аминолактона формулы (35) в соответствующий a-кетолактон формулы (36) путем трансаминирования

8. Способ по п.7, который включает дополнительную стадию стереоспецифического образования(получения) спиротетрагидроизохинолинового соединения Et770 из a-кетолактона формулы (36) по реакции Пикте-Шпенглера

9. Способ по п.8, который включает дополнительную стадию замещения нитрильной группы в положении С-21 соединения Et770 гидроксигруппой с получением соединения Et743

10. Стадия способа получения эктеинасцидинового соединения стереоспецифическим образованием спиротетрагидроизохинолинового соединения Et770 из a-кетолактона формулы (36) по реакции Пикте-Шпенглера

11. Способ по п.10, который включает дополнительную стадию замещения нитрильной группы в положении С-21 соединения Et770 гидроксигруппой с получением соединения Et743

12. Промежуточное соединение для синтеза эктеинасцидинового соединения формулы (35)

13. Промежуточное соединение для синтеза эктеинасцидинового соединения формулы (36)

Текст
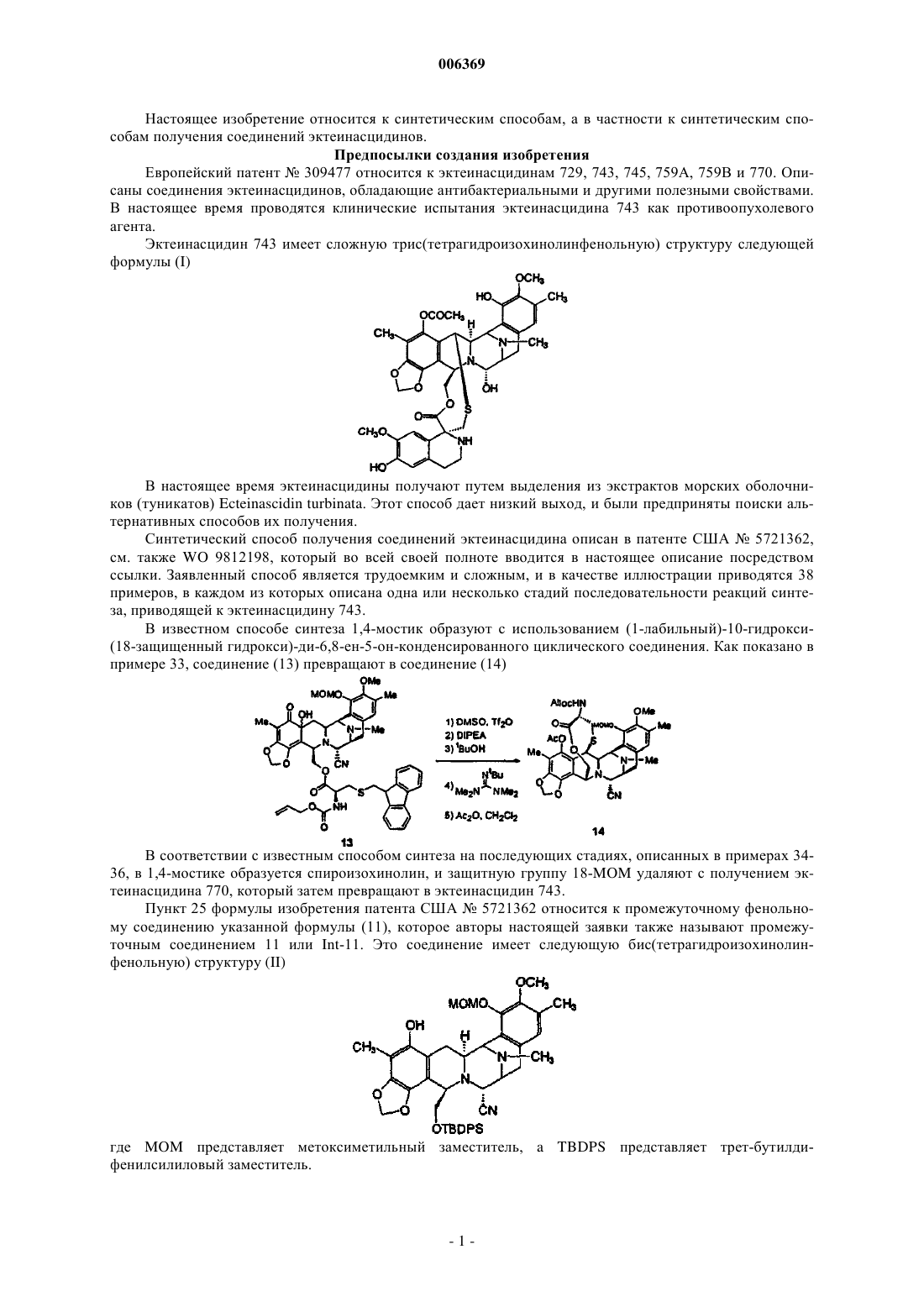
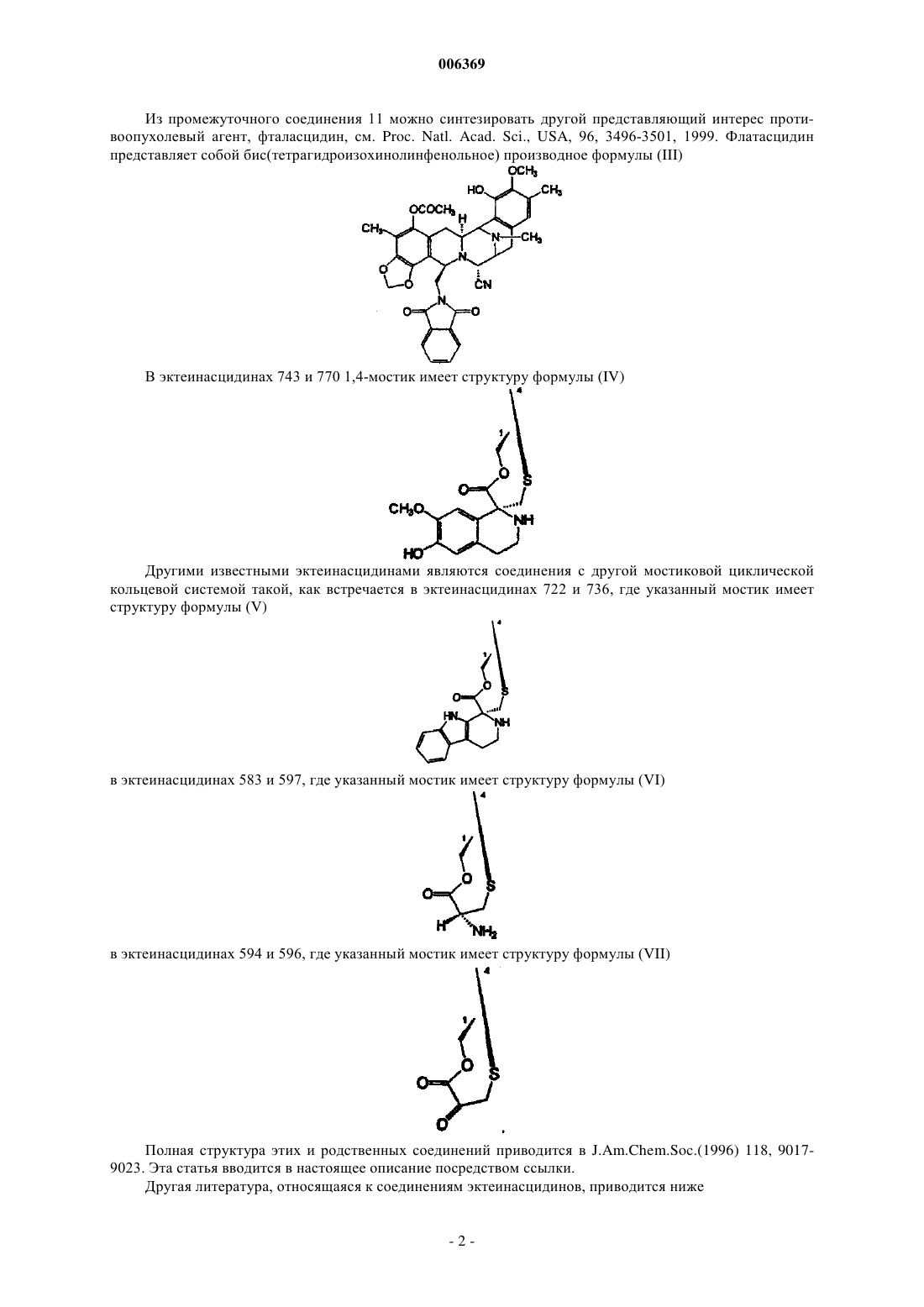
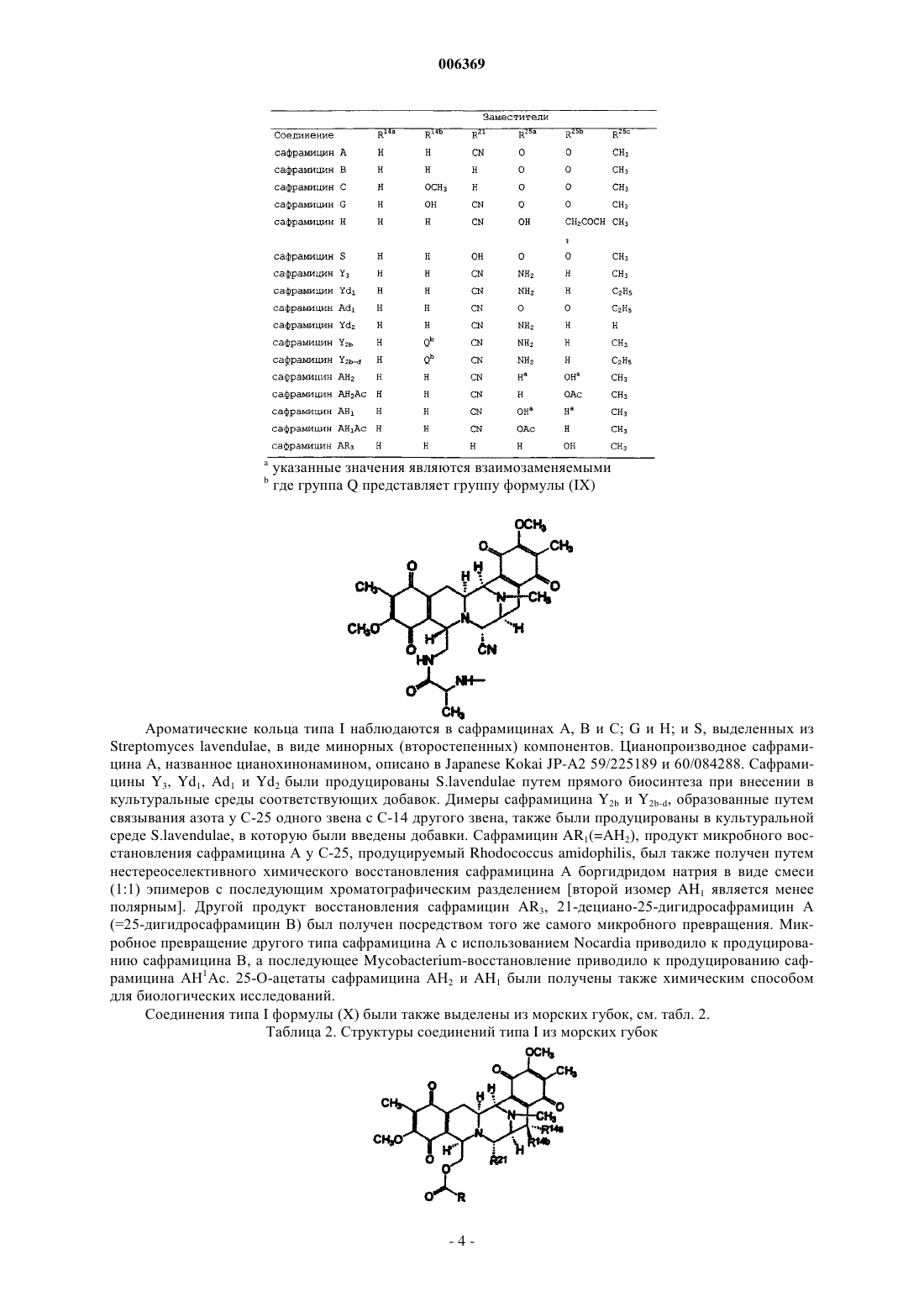
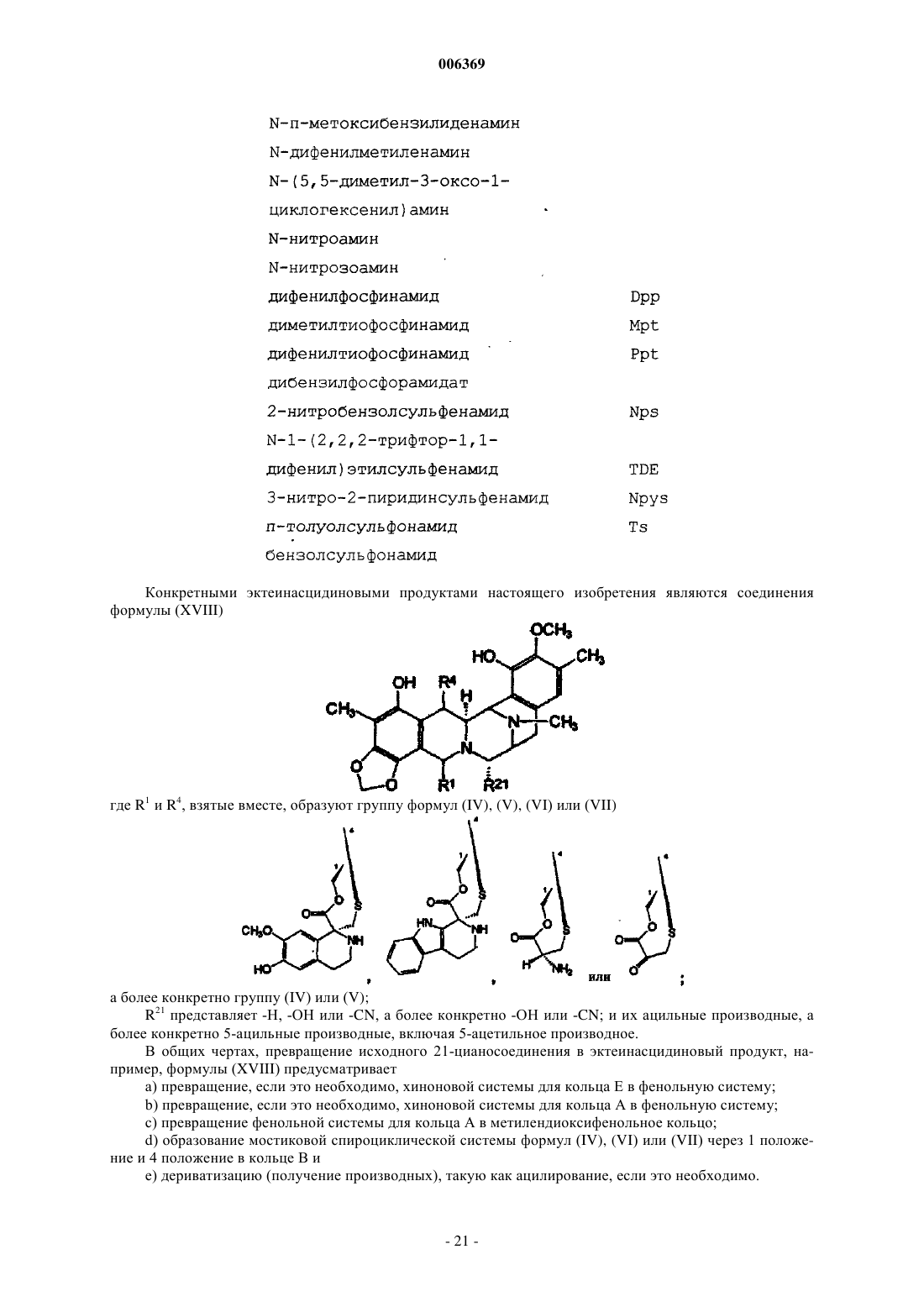
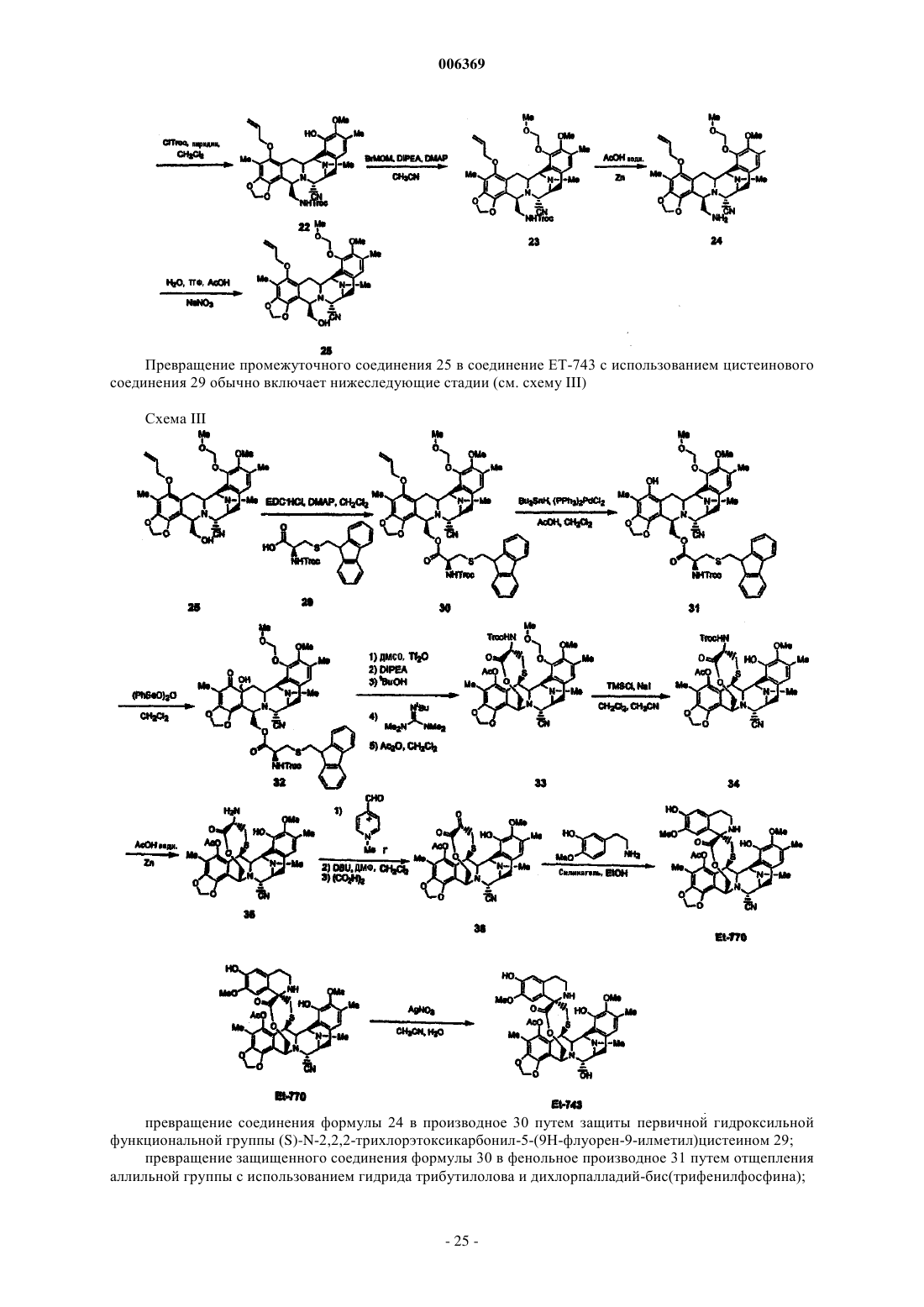
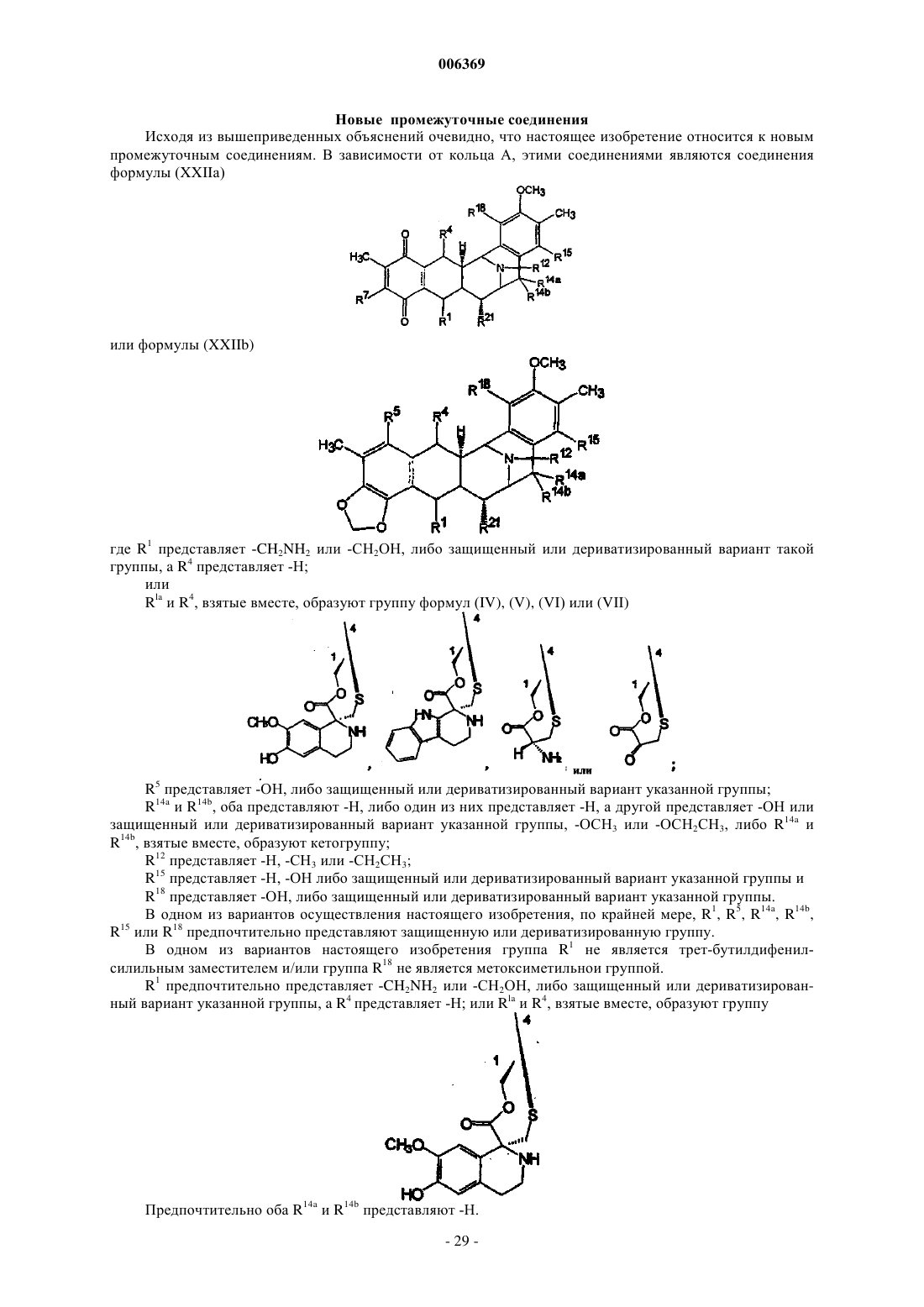
006369 Настоящее изобретение относится к синтетическим способам, а в частности к синтетическим способам получения соединений эктеинасцидинов. Предпосылки создания изобретения Европейский патент 309477 относится к эктеинасцидинам 729, 743, 745, 759 А, 759 В и 770. Описаны соединения эктеинасцидинов, обладающие антибактериальными и другими полезными свойствами. В настоящее время проводятся клинические испытания эктеинасцидина 743 как противоопухолевого агента. Эктеинасцидин 743 имеет сложную трис(тетрагидроизохинолинфенольную) структуру следующей формулы (I) В настоящее время эктеинасцидины получают путем выделения из экстрактов морских оболочников (туникатов) Ecteinascidin turbinata. Этот способ дает низкий выход, и были предприняты поиски альтернативных способов их получения. Синтетический способ получения соединений эктеинасцидина описан в патенте США 5721362,см. также WO 9812198, который во всей своей полноте вводится в настоящее описание посредством ссылки. Заявленный способ является трудоемким и сложным, и в качестве иллюстрации приводятся 38 примеров, в каждом из которых описана одна или несколько стадий последовательности реакций синтеза, приводящей к эктеинасцидину 743. В известном способе синтеза 1,4-мостик образуют с использованием (1-лабильный)-10-гидрокси(18-защищенный гидрокси)-ди-6,8-ен-5-он-конденсированного циклического соединения. Как показано в примере 33, соединение (13) превращают в соединение (14) В соответствии с известным способом синтеза на последующих стадиях, описанных в примерах 3436, в 1,4-мостике образуется спироизохинолин, и защитную группу 18-MOM удаляют с получением эктеинасцидина 770, который затем превращают в эктеинасцидин 743. Пункт 25 формулы изобретения патента США 5721362 относится к промежуточному фенольному соединению указанной формулы (11), которое авторы настоящей заявки также называют промежуточным соединением 11 или Int-11. Это соединение имеет следующую бис(тетрагидроизохинолинфенольную) структуру (II)-1 006369 Из промежуточного соединения 11 можно синтезировать другой представляющий интерес противоопухолевый агент, фталасцидин, см. Proc. Natl. Acad. Sci., USA, 96, 3496-3501, 1999. Флатасцидин представляет собой бис(тетрагидроизохинолинфенольное) производное формулы (III) В эктеинасцидинах 743 и 770 1,4-мостик имеет структуру формулы (IV) Другими известными эктеинасцидинами являются соединения с другой мостиковой циклической кольцевой системой такой, как встречается в эктеинасцидинах 722 и 736, где указанный мостик имеет структуру формулы (V) в эктеинасцидинах 583 и 597, где указанный мостик имеет структуру формулы (VI) в эктеинасцидинах 594 и 596, где указанный мостик имеет структуру формулы (VII) Полная структура этих и родственных соединений приводится в J.Am.Chem.Soc.(1996) 118, 90179023. Эта статья вводится в настоящее описание посредством ссылки. Другая литература, относящаяся к соединениям эктеинасцидинов, приводится нижеand Pommier et al., Biochemistry, 1996, 35: 13303-13309. Известны и другие соединения, у которых отсутствует циклическая кольцевая система с мостиками. Такими соединениями являются противоопухолевые-противомикробные бис(тетрагидроизохинолинхинон)содержащие антибиотики сафрацины и сафрамицины и природные морские продукты рениерамицины и ксестомицины, выделенные из культивированных микробов или губок. Все они имеют общий димерный тетрагидроизохинольный углеродный каркас. Эти соединения могут быть разделены на четыре типа, типы I-IV, в соответствии с характером окисления ароматических колец. Димерные изохинолинхиноны типа I представляют собой систему формулы (VIII), наиболее часто встречающуюся в соединениях этого класса, см. нижеследующую таблицу 1. Таблица 1. Структура сафрамициновых антибиотиков типа I указанные значения являются взаимозаменяемыми где группа Q представляет группу формулы (IX) Ароматические кольца типа I наблюдаются в сафрамицинах А, В и С; G и Н; и S, выделенных изStreptomyces lavendulae, в виде минорных (второстепенных) компонентов. Цианопроизводное сафрамицина А, названное цианохинонамином, описано в Japanese Kokai JP-A2 59/225189 и 60/084288. Сафрамицины Y3, Yd1, Ad1 и Yd2 были продуцированы S.lavendulae путем прямого биосинтеза при внесении в культуральные среды соответствующих добавок. Димеры сафрамицина Y2b и Y2b-d, образованные путем связывания азота у С-25 одного звена с С-14 другого звена, также были продуцированы в культуральной среде S.lavendulae, в которую были введены добавки. Сафрамицин AR1(=AH2), продукт микробного восстановления сафрамицина А у С-25, продуцируемый Rhodococcus amidophilis, был также получен путем нестереоселективного химического восстановления сафрамицина А боргидридом натрия в виде смеси(1:1) эпимеров с последующим хроматографическим разделением [второй изомер AH1 является менее полярным]. Другой продукт восстановления сафрамицин AR3, 21-дециано-25-дигидросафрамицин А(=25-дигидросафрамицин В) был получен посредством того же самого микробного превращения. Микробное превращение другого типа сафрамицина А с использованием Nocardia приводило к продуцированию сафрамицина В, а последующее Mycobacterium-восстановление приводило к продуцированию сафрамицина АН 1 Ас. 25-О-ацетаты сафрамицина АН 2 и AH1 были получены также химическим способом для биологических исследований. Соединения типа I формулы (X) были также выделены из морских губок, см. табл. 2. Таблица 2. Структуры соединений типа I из морских губок Рениерамицины A-D, наряду с биогенетически родственными мономерными изохинолинами, рениероном и родственными соединениями, были выделены из антимикробного экстракта губок видаReniera, собранных в Мексике. Структура рениерамицина А была первоначально отнесена к обращенной стереохимии при С-3, С-11 и С-13. Однако тщательная оценка данных 1 Н ЯМР для новых родственных соединений рениерамицинов Е и F, выделенных из тех же самых губок, собранных в Палау, выявила, что место конденсации колец рениерамицинов идентично месту конденсации колец сафрамицинов. Этот результат позволил сделать вывод, что первоначально определенная стереохимия рениерамицинов A-D должна быть такой же, как и у сафрамицинов. Ксестомицин был обнаружен в губках вида Xestospongia, собранных в водах, омывающих ШриЛанка. Соединения типа II формулы (XI) с восстановленным гидрохиноновым кольцом представляют собой сафрамицины D и F, выделенные из 5. lavendulae, и сафрамицины Мх-1 и Мх-2, выделенные из Структура типа III была обнаружена в антибиотиках сафрацинах А и В, выделенных из Pseudomonas fluorescens. Эти антибиотики формулы (XII) состоят из тетрагидроизохинолин-хиноновой субъединицы и тетрагидроизохинолинфенольной субъединицы. где R21 представляет -Н в сафрацине А и -ОН в сафрацине В. Сафрамицин R единственное соединение, классифицированное как соединение структуры типа IV,было также выделено из S.lavendulae. Это соединение формулы (XIII), состоящее из гидрохинонового кольца с боковой гликолевой сложноэфирной цепью у одного из атомов кислорода фенола, может служить пролекарством сафрамицина А из-за его умеренной токсичности. Все эти известные соединения имеют конденсированную систему из пяти колец (А)-(Е), как показано на представленной ниже структуре формулы (XIV) В эктеинасцидинах и в некоторых других соединениях кольца А и Е являются фенольными, тогда как в других соединениях, а в частности в сафрамицинах, указанные кольца А и Е являются хинольными. В известных соединениях кольца В и D представляют собой тетрагидро, а кольцо С представляет собой пергидро. Цели изобретения Необходимость в разработке альтернативных способов синтеза соединений эктеинасцидина и родственных соединений остается актуальной. Такие синтетические пути получения дадут возможность разработать более экономичные способы получения известных противоопухолевых агентов, а также позволят получить новые активные соединения. Краткое описание изобретения Настоящее изобретение относится к синтетическим способам получения промежуточных соединений, производных и родственных структур эктеинасцидина или других тетрагидроизохинолинфенольных соединений. В одном из своих аспектов настоящее изобретение относится к способу получения эктеинасцидинового продукта со спироамино-1,4-мостиком. Этот способ предусматривает образование 1,4 мостика с использованием (1-лабильный)-10-гидрокси-(18-защищенный гидрокси)-ди-6,8-ен-5-он-конденсированного циклического соединения, где конденсированное кольцо имеет формулу (XIV). В настоящем изобретении защиту в положении С-18 удаляют перед введением спироамина. Подходящими исходными веществами для новых синтетических способов являются соединения,родственные природным бис(тетрагидроизохинолин)алкалоидам. Такие исходные вещества могут быть получены из сафрамициновых или сафрациновых антибиотиков различных классов, доступных из различных культуральных бульонов, как подробно описано в WO 0069862, либо другими синтетическими или биохимическими способами. В этой связи WO 0069862 во всей своей полноте вводится в настоящее описание посредством ссылки. В настоящей заявке РСТ испрашивается приоритет с даты подачи заявкиPCT/GB00/01852, которая была опубликована как WO 0069862. Авторы настоящей заявки включают этот текст посредством ссылки на ту часть описания изобретения, которая не присутствует в описании настоящей заявки. Предпочтительные варианты осуществления настоящего изобретения В одном из своих аспектов настоящее изобретение относится к использованию промежуточного соединения 21 в ряде новых синтетических способов для получения эктеинасцидина 743 и родственных соединений Промежуточное соединение 21 имеет 5-аллилоксигруппу, где аллильная группа служит для защиты 5-гидроксигруппы. Следует отметить, что могут быть также легко использованы и другие защитные группы и что настоящее изобретение, в общих чертах, относится к использованию других таких 5 защищенных гидроксисоединений. Получение эктеинасцедина 743 и родственных соединений В общих чертах, превращение промежуточного соединения 21 или родственного соединения в эктеинасцидиновый продукт включает нижеследующие ключевые стадии превращений:(c) этерификация путем защиты первичной функциональной 1-гидроксигруппы, защищенной боковой цепью цистеина;(e) образование кольца с мостиком посредством реакции циклизации;(g) введение хинолина посредством транс-аминирования и реакции Пикте-Шпенглера. Большое число функциональных групп промежуточных соединений требует использования защитных групп для Е-кольцевого фенола и для цистеиновой боковой цепи в целях предупреждения нежелательных побочных реакций. Число самих альтернативных промежуточных соединений, которое может быть образовано, зависит от конкретного выбора защитных групп. Для комбинирования указанных превращений можно использовать другие возможные последовательности реакций в зависимости, главным образом, от защитных групп, выбранных для фенольного кольца и для амина цистеиновой боковой цепи. Общее число синтетических превращений также зависит от выбора защитных групп. В качестве иллюстрации ниже описано использование различных комбинаций защитных групп для шести типичных путей получения ЕТ-743 из промежуточного соединения 21, также называемого здесь Как очевидно каждому специалисту, описанные здесь реакционные схемы могут быть модифицированы и/или комбинированы различными способами, и альтернативные последовательности стадий, а также полученные при этом соединения являются частью настоящего изобретения. Кроме того, другие стратегии с использованием защитных групп, которые подробно здесь не описываются, также являются частью настоящего изобретения. Подробное описание шести типичных синтетических путей Полные реакционные схемы для каждого пути синтеза представлены в нижеследующих схемах 1-6.-7 006369 Схема 1 - ET-743: Полусинтетический альтернативный путь 1 Схема 2 - ET-743: Полусинтетический альтернативный путь 2-8 006369 Схема 3 - ET-743: Полусинтетический альтернативный путь 3 Схема 4 - ET-743: Полусинтетический альтернативный путь 4-9 006369 Схема 5 - ET-743: Полусинтетический альтернативный путь 5 Схема 6 - ET-743: Полусинтетический альтернативный путь 6 В пути синтеза 1 защита Е-кольцевого фенола достигается в три стадии, включающих защиту/снятие защиты амина SF21 с использованием Тrос. В путях синтеза 1 и 2 защита цистеиновой боковой цепи группой Воc позволяет снимать защиту у фенольных и цистеиновых групп в одну стадию, а не в две отдельные стадии. В остальных путях синтеза требуется дополнительная стадия снятия защиты. Путь синтеза 2 позволяет избежать образования промежуточного соединения 25 благодаря использованию методики прямой этерификации и последующей защиты фенола группой MEM. В путях синтеза 2 и 3 защиту Е-кольцевого фенола осуществляют лишь после стадий диазотирования и этерификации, что тем самым позволяет обеспечить защиту фенола в одну стадию, а не в три стадии, как в пути реакций 1. В путях синтеза 1, 2 и 3 прямая этерификация первичного спирта с использованием цистеинового производного позволяет избежать непродуктивных стадий защиты/снятия защиты у первичного спирта с использованием силильной группы (пути 4 и 5), что тем самым укорачивает эту последовательность реакций на две стадии. Путь 6 рассматривается здесь только как последние стадии получения из соединения 161, которое может быть легко получено из промежуточного соединения 21. В путях 4 и 5, первичный спирт, получаемый на начальной стадии диазотирования, защищают кремнием, что позволяет обеспечить селективную защиту Е-кольцевого фенола и избежать образования промежуточного соединения 25. После модификации А-кольца (снятия защиты/окисления) кремниевую группу удаляют и первичный спирт этерифицируют цистеиновым производым.- 10006369 Эти изменения непосредственно связаны с проблемами, которые в более широком масштабе возникали при проведении последовательности реакций, описанной в WO 0069862. В результате этих изменений весь путь реакций 2 является короче на три стадии, а поэтому он потенциально является более подходящим и/или менее дорогостоящим для рутинного промышленного производства. Общее описание способа Таким образом, в соответствии с путями синтеза 1-6 настоящее изобретение относится к способу получения эктеинасцидинового продукта со спироамино-1,4-мостиком, причем указанный способ включает получение 1,4-мостика с использованием (1-лабильный)-10-гидрокси-(18-защищенный гидрокси)ди-6,8-ен-5-он-конденсированного циклического соединения, где защиту в положении С-18 удаляют перед введением спироамина. В одном из вариантов указанного способа, эктеинасцидиновый продукт имеет 21-гидроксигруппу, и этот способ предусматривает превращение 21-цианогруппы в 21-гидроксигруппу. Обычно спироамином является спирохинолин, а в частности спирохинолин эктеинасцидина 743. В предпочтительном способе 18-защищенную группу (1-лабильный)-10-гидрокси-(18-защищенный гидрокси)-ди-6,8-ен-5-он-конденсированного циклического соединения защищают группой MOM, метоксиметилом или MEM, метоксиэтоксиметильной группой. Подходящей 1-лабильной группой является N-защищенная цистеинилоксиметиленовая группа формулы -CH2-O-CO-CNProt1-CH2-S-H. В этой формуле Prot1 обычно представляет Воc, трет-бутоксикарбонил; Тrос, 2,2,2-трихлорэтилоксикарбонил; Cbz, бензилоксикарбонил или Alloc, аллилоксикарбонил. В некоторых вариантах осуществления указанного способа Prot1 удаляют на той же самой стадии,что и С-18-защиту. 1-лабильная группа может быть генерирована из 1-заместителя формулы -СH2-О-СО-CNHProt1 СН 2-3-Prot2. В этой формуле Prot2 обычно означает Fm, 9-флуоренилметил. 1-Заместитель формулы -CH2-O-CO-CNHProt1-CH2-S-Prot2 может быть образован посредством этерификации заместителя СН 2-О-Н. Такая этерификация может быть осуществлена до или после образования 10-гидрокси-ди-6,8-ен-5 он-структуры. В одном из вариантов заявленного способа исходным соединением является 1-аминометилен-(5 защищенный гидрокси)-7,8-диоксиметилен-18-гидрокси-21-цианоконденсированное циклическое соединение. 1-аминометиленовая группа может быть временно защищена для возможности защиты 18 гидроксигруппы, а затем эту временную защиту снимают. Альтернативно, С-18 гидроксигруппа может быть защищена после образования 1-сложноэфирной функциональной группы. В другом варианте 1-аминометиленовую группу превращают в 1-гидроксиметиленовую группу и указанную 1-гидроксиметиленовую группу временно защищают для возможности защиты 18 гидроксигруппы, а затем эту временную защиту снимают. Подходящая конденсированная кольцевая структура имеет формулу особенно, где R15 представляет Н. Один, или несколько, или все остальные заместители могут быть такими, как в эктеинасцидине 743. Полусинтез Настоящее изобретение позволяет использовать известное соединение, сафрацин В, также называемый хинонамином, в полусинтетическом способе. В более общих чертах, настоящее изобретение относится к полусинтетическому способу получения промежуточных соединений, производных и родственных структур эктеинасцидина или других тетрагидроизохинолинфенольных соединений из природных бис(тетрагидроизохинолин)алкалоидов. Подходящими исходными веществами для полусинтетического способа являются антибиотики класса сафрамицинов и сафрацинов, доступные из различных культуральных бульонов, и соединения класса рейнерамицинов и ксестомицинов, доступные из морских губок.- 11006369 Общая формула (XV) для исходных соединений представляет собой где R1 представляет амидометиленовую группу, такую как -CH2-NH-CO-CR25aR25bR25c, где R25a и R25b образуют кетогруппу, либо один из них представляет -ОН, -NH2 или -ОСОСН 3, а другой представляет СН 2 СОСН 3, -Н, -ОН или -ОСОСН 3, при условии, что если R25a представляет -ОН или -NH2, то R25b не является -ОН, а R25c представляет -Н, -СН 3 или -СН 2 СН 3, либо R1 представляет ацилоксиметиленовую группу, такую как -СН 2-O-CO-R, где R представляет -С(СН 3) =СН-СН 3 или -СН 3;R5 и R8 независимо выбраны из -Н, -ОН или -ОСОСН 2 ОН либо оба R5 и R8 представляют кето, а кольцо А представляет п-бензохиноновое кольцо;R14a и R14b оба представляют -Н, либо один из них представляет -Н, а другой представляет -ОН,-ОСН 3 или -ОСН 2 СН 3, либо R14a и R14b, взятые вместе, образуют кетогруппу;R15 и R18 независимо выбраны из -Н или -ОН, либо R5 и R8 оба представляют кето, а кольцо А представляет п-бензохиноновое кольцо иR21 представляет -ОН или -CN. Более общая формулы для соединений этого класса представлена ниже где каждая из замещающих групп, определенных R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, независимо выбрана из группы, состоящей из Н, ОН, ОСН 3, CN, =O, СН 3;X представляет другие амидные или сложноэфирные функциональные группы, содержащиеся в вышеупомянутых природных продуктах; и где каждое пунктирное кольцо представляет одну, две или три необязательных двойных связи. Таким образом, авторами настоящего изобретения были разработаны полусинтетические способы получения промежуточных соединений, включая промежуточные соединения 11 или 21, и способы получения эктеинасцидиновых соединений, а также фталасцидиновых и других соединений. Каждый из полусинтетических способов настоящего изобретения включает проведение ряда стадий превращений с получением нужного продукта. Каждая стадия сама по себе представляет собой процесс, осуществляемый в соответствии с настоящим изобретением. Настоящее изобретение не ограничивается указанными примерами путей синтеза, и, при необходимости, могут быть использованы альтернативные пути синтеза, предусматривающие, например, изменение порядка проведения стадий превращений, если это необходимо, или замену используемых защитных групп. В частности, настоящее изобретение включает получение исходного 21-циановещества общей формулы (XVI)- 12006369 Возможными исходными веществами могут быть также другие соединения формулы (XVI) с различными заместителями в 21-положении. В общих чертах, кандидатом является любое производное, которое может быть получено нуклеофильным замещением 21-гидроксигруппы в соединениях формулы(XV), где R21 представляет гидроксигруппу. Примерами подходящих 21-заместителей являются, но не ограничиваются ими: меркаптогруппа; алкилтиогруппа (алкильная группа имеет от 1 до 6 атомов углерода); арилтиогруппа (арильная группа имеет от 6 до 10 атомов углерода и является незамещенной или замещенной 1-5 заместителями, выбранными, например, из алкильной группы, имеющей 1-6 атомов углерода; алкоксигрупп, имеющих 1-6 атомов углерода; атомов галогена; меркаптогрупп и нитрогрупп); аминогруппа; моно- или диалкиламиногруппы (каждая из алкильных групп имеет от 1 до 6 атомов углерода); моно- или диариламиногруппы (каждая из арильных групп является такой, как она была определена выше для арилтиогрупп); -карбонилалкильная группа формулы -C(Ra) (Rb)-C(=O)RC, где Ra и Rb выбраны из атомов водорода, алкильных групп, имеющих от 1 до 20 атомов углерода, арильных групп(определенных выше в отношении арилтиогрупп) и аралкильных групп (в которых алкильная группа,имеющая от 1 до 4 атомов углерода, замещена арильной группой, определенной выше в отношении арилтиогрупп), при условии, что один из Ra и Rb представляет атом водорода; Rc выбран из атомов водорода, алкильной группы, имеющей от 1 до 20 атомов углерода, арильных групп (определенных выше в отношении арилтиогрупп) и аралкильных групп (в которых алкильная группа, имеющая от 1 до 4 атомов углерода, замещена арильной группой, определенной выше в отношении арилтиогрупп); алкоксигрупп,имеющих от 1 до 6 атомов углерода, аминогрупп или моно- или диалкиламиногрупп, определенных выше. Таким образом, в своем более общем аспекте настоящее изобретение относится к способам, где первую стадию проводят для образования 21-производного с использованием нуклеофильного реагента. Авторы настоящей заявки назвали такие соединения 21-Nuc-соединениями. Для некоторых конечных продуктов, а в частности эктеинасцидина 770 и фталасцидина, необходимо присутствие 21-цианогруппы, тогда как в других конечных продуктах она действует как защитная группа, которая может быть легко превращена в другой заместитель, такой как 21-гидроксигруппа эктеинасцидина 743 или 21-гидроксифталасцидина. Выбор 21-цианосоединения в качестве исходного вещества позволяет эффективно стабилизировать данную молекулу в процессе последующих стадий синтеза до тех пор, пока она, но необязательно, не удалится. Эти и другие преимущества можно достигнуть с использованием других 21-Nuc-соединений. В одном из важных аспектов своего осуществления настоящее изобретение относится к использованию 21-цианосоединения общей формулы (XVI) для получения бис- или трис(тетрагидроизохинолинфенольных) соединений. Продуктами, которые могут быть получены таким образом, являются промежуточные соединения, такие как промежуточное соединение 11 или 21, и эктеинасцидины, а также новые и известные соединения с родственной структурой. Предпочтительными исходными веществами являются соединения формулы (XV) или (XVI), гдеR14a и R14b оба представляют водород. Предпочтительными исходными веществами также являются соединения формулы (XV) или (XVI), где R15 представляет водород. Кроме того, предпочтительными исходными веществами являются соединения формулы (XV) или (XVI), где Е представляет фенольное кольцо. Предпочтительными исходными веществами также являются соединения формулы (XV) или(XVI), где по крайней мере один, а лучше два или три из R5, R8, R15 и R18 не являются водородом. Примерами подходящих исходных веществ настоящего изобретения являются сафрамицин А, сафрамицин В, сафрамицин С, сафрамицин G, сафрамицин Н, сафрамицин S, сафрамицин Y3, сафрамицинYd1, сафрамицин Ad1, сафрамицин Yd2, сафрамицин АН 2, сафрамицин АН 2 Ас, сафрамицин АН 1, сафрамицин AH1Ac, сафрамицин AR3, рениерамицин А, рениерамицин В, рениерамицин С, рениерамицин D,рениерамицин Е, рениерамицин F, ксестомицин, сафрамицин D, сафрамицин F, сафрамицин Мх-1, сафрамицин Мх-2, сафрацин А, сафрацин В и сафрамицин R. Предпочтительные исходные вещества имеют цианогруппу в положении 21, для группы R21. В особенно предпочтительном аспекте своего осуществления настоящее изобретение относится к полусинтетическому способу, где стадии превращения применяют по отношению к сафрацину В САФРАЦИН В Сафрацин В представляет собой кольцевую систему, близкородственную эктеинасцидинам. Это соединение имеет ту же самую пентациклическую структуру и тот же тип замещения в правом ароматическом кольце, кольце Е. Кроме того, сафрацин В имеет очень большое сходство с некоторыми синтетическими промежуточными соединениями в общем синтезе ЕТ-743, а в частности с промежуточными соединениями 11 или 21. Такое промежуточное соединение может быть превращено в Et-743 с использованием хорошо известного способа. Поэтому синтетическое превращение сафрацина В в промежуточные соединения 11 или 21 позволяет предложить полусинтетический способ получения ЕТ-743. Таким образом, из указанного соединения сафрацина В авторы настоящего изобретения получали промежуточные соединения 11 или 21, а из этих промежуточных соединений 11 или 21 получали конкретные соединения, в частности эктеинасцидины. Кроме того, авторами настоящего изобретения был получен фталасцидин из сафрацина В. Настоящее изобретение также относится к использованию сафрацина В для получения промежуточных соединений 11 или 21, соединений эктеинасцидина и других промежуточных соединений настоящего изобретения. Настоящее изобретение также относится к описанным здесь соединениям, полученным из других предлагаемых исходных веществ, и к их использованию для получения указанных соединений. Более предпочтительные исходные вещества настоящего изобретения имеют 21-цианогруппу. Наиболее предпочтительным соединением настоящего изобретения является соединение формулы 2. Это соединение получают непосредственно из сафрацина В и рассматривают как ключевое промежуточное соединение в данном полусинтетическом способе. В соответствующем аспекте настоящего изобретения авторами получен цианосафрацин В путем ферментации штамма Pseudomonas fluorescens, продуцирующего сафрацин В, и обработки культивированного бульона с использованием ионов цианида. Предпочтительным штаммом Pseudomonas fluorescens является штамм А 2-2, FERM ВР-14, который использовали в процедуре, описанной в ЕР 055299. Подходящим источником ионов цианида является цианид калия. В типичной процедуре обработки, указанный бульон фильтруют и добавляют избыток ионов цианида. После перемешивания в течение соответствующего периода времени, такого как 1 ч, рН делают щелочным, то есть рН 9,5, а после экстракции органических веществ получают неочищенный экстракт, который может быть затем очищен с получением цианосафрацина В. Чтобы исключить всякие сомнения, следует отметить, что стереохимические структуры, указанные в описании настоящей патентной заявки, основаны на авторском представлении о точной стереохимии природных продуктов. Если принять во внимание тот факт, что если обнаружена ошибка в отнесении стереохимии, то необходимо внести соответствующую поправку в формулы, представленные в описании- 14006369 настоящей заявки. Кроме того, если учесть, что указанные способы синтеза могут быть модифицированы, то настоящее изобретение распространяется и на стереоизомеры. Продукты настоящего изобретения обычно представляют собой соединения формулы (XVIIb) где R1 и R4, взятые вместе, образуют группу формул (IV), (V), (VI) или (VII)R14a и R14b оба представляют -Н, либо один из них представляет -Н, а другой представляет -ОН,-ОСН 3 или -ОСН 2 СН 3, либо R14a и R14b, взятые вместе, образуют кетогруппу;R21 представляет -Н, -ОН или -CN; и производные, включая их ацильные производные, а особенно, где R5 представляет ацетилокси или другую ацилоксигруппу, имеющую вплоть до 4 атомов углерода. В формуле (XVIIb) группа R1, обычно вместе с R4, образует группу (IV) или (V). Группа R18 обычно является защищенной. R21 обычно представляет циано.R14a и R14b предпочтительно представляют водород. R15 предпочтительно представляет водород. Подходящими О-ацильными производными являются алифатические О-ацильные производные, а в частности ацильные производные с 1-4 атомами углерода, а обычно О-ацетильная группа, например, в 5 положении. Подходящими защитными группами для фенолов и гидроксигрупп являются простые и сложные эфиры, такие как алкил, алкоксиалкил, арилоксиалкил, алкоксиалкоксиалкил, алкилсилилалкоксиалкил,алкилтиоалкил, арилтиоалкил, азидоалкил, цианоалкил, хлоралкил, гетероциклил, арилацил, галогенарилацил, циклоалкилалкил, алкенил, циклоалкил, алкиларилалкил, алкоксиарилалкил, нитроарилалкил,галогенарилалкил, алкиламинокарбониларилалкил, алкилсульфиниларилалкил, алкилсилил и другие простые эфиры, и арилацил, арилалкилкарбонат, алифатический карбонат, алкилсульфиниларилалкилкарбонат, алкилкарбонат, арилгалогеналкилкарбонат, арилалкенилкарбонат, арилкарбамат, алкилфосфинил, алкилфосфинотиоил, арилфосфинотиоил, арилалкилсульфонат и другие сложные эфиры. Такие группы могут быть, но необязательно, замещены группами, упомянутыми ранее в определении R1. Подходящими защитными группами для аминов являются карбаматы, амиды или другие защитные группы, такие как алкил, арилалкил, сульфо- или галогенарилалкил, галогеналкил, алкилсилилалкил,арилалкил, циклоалкилалкил, алкиларилалкил, гетероциклилалкил, нитроарилалкил, ациламиноалкил,нитроарилдитиоарилалкил, дициклоалкилкарбоксамидоалкил, циклоалкил, алкенил, арилалкенил, нитроарилалкенил, гетероциклилалкенил, гетероциклил, гидроксигетероциклил, алкилдитио, алкокси- или галоген- или алкилсульфиниларилалкил, гетероциклилацил и другие карбаматы, и алканоил, галогеналканоил, арилалканоил, алкеноил, гетероциклилацил, ароил, арилароил, галогенароил, нитроароил и другие амиды, а также алкил, алкенил, алкилсилилалкоксиалкил, алкоксиалкил, цианоалкил, гетероциклил, алкоксиарилалкил, циклоалкил, нитроарил, арилалкил, алкокси- или гидроксиарилалкил и многие другие группы. Такие группы могут быть, но необязательно, замещены группами, упомянутыми ранее в определении R1.- 15006369 Примеры таких защитных групп представлены в нижеследующих таблицах. защита для группы -ОН Конкретными эктеинасцидиновыми продуктами настоящего изобретения являются соединения формулы (XVIII) где R1 и R4, взятые вместе, образуют группу формул (IV), (V), (VI) или (VII) а более конкретно группу (IV) или (V);a) превращение, если это необходимо, хиноновой системы для кольца Е в фенольную систему;b) превращение, если это необходимо, хиноновой системы для кольца А в фенольную систему;c) превращение фенольной системы для кольца А в метилендиоксифенольное кольцо;d) образование мостиковой спироциклической системы формул (IV), (VI) или (VII) через 1 положение и 4 положение в кольце В и- 21006369 Стадия (а), превращение, если это необходимо, хиноновой системы для кольца Е в фенольную систему, может быть осуществлена стандартными методами восстановления. Подходящей системой реагентов является водород вместе с катализатором "палладий на углероде", хотя могут быть использованы и другие системы восстановления. Стадия (b), превращение, если это необходимо, хиноновой системы для кольца А в фенольную систему, аналогична стадии (а) и не нуждается в более подробном описании. Стадия (с), превращение фенольной системы для кольца А в метилендиоксифенольное кольцо, может быть осуществлена несколькими способами, возможно, вместе со стадией (b). Так, например, хиноновое кольцо А может быть деметилировано по метоксизаместителю в 7-положении, восстановлено до дигидрохинона и акцептировано подходящим электрофильным реагентом, таким как СН 2 Вr2, ВrСН 2 Сl или аналогичным двухвалентным реагентом с получением непосредственно метилендиоксициклической системы, или двухвалентным реагентом, таким как тиокарбонилдиимидазол, с получением замещенной метилендиоксициклической системы, которая может быть превращена в нужное кольцо. Стадию (d) обычно осуществляют посредством соответствующего замещения в 1 положении мостикообразующим реагентом, который может способствовать формированию нужного мостика с образованием эксэндохинонметида в 4 положении и способствует взаимодействию метида с 1 заместителем,что приводит к образованию мостиковой структуры. Предпочтительные мостикообразующие реагенты имеют формулу (XIX) где Fu обозначает защищенную функциональную группу, такую как группа -NHProt4a, Prot3 представляет защитную группу, а пунктирная линия обозначает необязательную двойную связь. Обычно, метид образуется при первом введении гидроксигруппы в 10-положении в месте конденсации колец А и В, в результате чего образуется неполная структура формулы (XX) или более предпочтительно неполная структура формулы (XXI) где группу R" выбирают для нужной группы формул (IV), (V), (VI) или (VII). Для первых двух таких групп группа R" обычно имеет форму -CHFu-CH2-SProt3. Затем эти защитные группы могут быть удалены и модифицированы, если это необходимо, с получением нужного соединения. Типичная процедура стадии (d) описана в патенте США 5721362, который вводится в настоящее описание посредством ссылки. Более конкретно, см. текст в столбце 8, стадия (1), и пример 33 этого патента США, и аналогичные отрывки текста. Дериватизация (получение производных) на стадии (е) может включать ацилирование, например,группой Ra-CO-, где группа Ra может представлять различные группы, такие как алкил, алкокси, алкилен,арилалкил, арилалкилен, аминокислота-ацил или гетероциклил, каждый из которых необязательно замещен галогеном, циано, нитро, карбоксиалкилом, алкокси, арилом, арилокси, гетероциклилом, гетероциклилокси, алкилом, амино или замещенным амино. Другими ацилирующими агентами являются изотиоцианаты, такие как арилизотиоцианаты, а в частности фенилизоцианат. Алкильная, алкокси или алкиленовая группы Ra обычно имеют от 1 до 6 или 12 атомов углерода и могут быть прямыми, разветвленными или циклическими. Арильными группами обычно являются фенил, бифенил или нафтил. Гетероциклические группы могут быть ароматическими, либо частично или полностью ненасыщенными и обычно- 22006369 имеют от 4 до 8 атомов в кольце, а более предпочтительно 5 или 6 атомов в кольце с одним или несколькими гетероатомами, выбранными из азота, серы и кислорода. Не претендуя на исчерпывающее определение, можно указать, что типичными группами Ra являются алкил, галогеналкил, алкоксиалкил, галогеналкоксиалкил, арилалкилен, галогеналкиларилалкилен,ацил, галогенацил, арилалкил, алкенил и аминокислота. Так, например, Ra-CO может представлять ацетил, трифторацетил, 2,2,2-трихлорэтоксикарбонил, изовалерилкарбонил, транс-3-(трифторметил)циннамоилкарбонил, гептафторбутирилкарбонил, деканоилкарбонил, транс-циннамоилкарбонил, бутирилкарбонил, 3-хлорпропионилкарбонил, циннамоилкарбонил, 4-метилциннамоилкарбонил, гидроциннамоилкарбонил или транс-гексеноилкарбонил, или аланил, аргинил, аспартил, аспарагил, цистил, глутамил,глутаминил, глицил, гистидил, гидроксипропил, изолейцил, лейцил, лизил, метионил, фенилаланил, пролил, серил, треонил, тиронил, триптофил, тирозил, валил, а также другие менее известные аминокислотные ацильные группы, а также фталимидо и другие циклические амиды. Другие примеры можно найти среди вышеперечисленных защитных групп. Соединения, где -CO-Ra происходит от аминокислоты и включает аминогруппу, могут сами образовывать ацильные производные. Подходящими N-ацильными соединениями являются дипептиды, которые, в свою очередь, могут образовывать N-ацильные производные. В качестве примера можно отметить, что цианосафрацин В формулы 2 целесообразно превращать в ЕТ-743 более коротким и более прямым путем, чем в ранее описанных методах. Ретросинтетический анализ для получения ЕТ-743 с использованием соединения 29 проиллюстрирован на схеме I. Схема I В соответствии с вышеуказанной схемой I можно получить ЕТ-743 за 21 последовательную стадию. В этом способе цианосафрацин В превращают в промежуточное соединение 25 в соответствии с последовательностью реакций, которая, по существу, включает (1) удаление метоксигруппы, присутствующей в кольце А, (2) восстановление кольца А и образование метилендиоксигруппы в одном сосуде, (3) гидролиз амидной функциональной группы, присутствующей у углерода 1, (4) превращение полученной аминовой группы в гидроксильную группу. Кроме того, этот способ позволяет избежать защиты и снятия защиты у первичной спиртовой функциональной группы в положении 1 кольца В соединения 25, при использовании непосредственно цистеинового остатка 29 для образования промежуточного соединения 27. Цистеиновое производное 29 защищают по аминогруппе -трихлорэтоксикарбонильной защитной группой для того, чтобы сделать ее совместимой с присутствующей аллильной группой и группойMOM. Промежуточное соединение 27 подвергают прямому окислению и циклизации. Эти подходы, вместе с другой стратегией снятия защиты в последних стадиях синтеза, делают этот путь реакций новым и более приемлемым для промышленного применения, чем способ, описанный в патенте США 5721362. Превращение 2-цианосоединения в промежуточное соединение 25 обычно включает нижеследующие стадии (см. схему II): образование защищенного соединения формулы 14 посредством взаимодействия соединения 2 с трет-бутоксикарбонильным ангидридом; превращение соединения 14 в дизащищенное соединение формулы 15 посредством его взаимодействия с бромметилметиловым эфиром и диизопропилэтиламином в ацетонитриле; селективное удаление метоксигруппы из хиноновой системы в соединении 15 посредством взаимодействия с метанольным раствором гидроксида натрия с получением соединения формулы 16;- 23006369 превращение соединения 16 в метилендиоксисоединение формулы 18 в соответствии с нижеследующей предпочтительной последовательностью реакций: (1) восстановления хиноновой группы соединения 16 в присутствии 10% Pd/C в атмосфере водорода; (2) превращения гидрохинонового промежуточного соединения в метилендиоксисоединение формулы 17 посредством взаимодействия с бромхлорметаном и карбонатом цезия в атмосфере водорода; (3) превращения соединения 17 в соединение формулы 18 посредством защиты свободной гидроксильной группы в качестве группы OCH2R. Эту реакцию взаимодействия осуществляют с использованием BrCH2R и карбоната цезия, где R может представлять арил, СН=СН 2, OR' и т.п.; удаление трет-бутоксикарбонильных и метоксиметильных защитных групп соединения 18 посредством его взаимодействия с раствором НСl в диоксане с получением соединения формулы 19; указанную реакцию взаимодействия также осуществляют посредством смешивания соединения 18 с раствором трифторуксусной кислоты в дихлорметане; образование тиомочевинного соединения формулы 20 посредством взаимодействия соединения 19 с фенилизотиоцианатом; превращение соединения формулы 20 в аминовое соединение формулы 21 посредством реакции с раствором хлористого водорода в диоксане; превращение соединения формулы 21 в N-Troc-производное 22 посредством реакции с трихлорэтилхлорформиатом и пиридином; образование защищенного гидроксисоединения формулы 23 посредством взаимодействия соединения 22 с бромметилметиловым эфиром и диизопропилэтиламином; превращение соединения формулы 23 в N-H-производное 24 посредством реакции с уксусной кислотой и цинком; превращение соединения формулы 24 в гидрокси-соединение формулы 25 посредством реакции с нитритом натрия в уксусной кислоте; альтернативно может быть использован тетроксид азота в смеси уксусной кислоты и ацетонитрила с последующей обработкой гидроксидом натрия; кроме того, может быть использован нитрит натрия в смеси уксусного ангидрида-уксусной кислоты с последующей обработкой гидроксидом натрия. Схема II Превращение промежуточного соединения 25 в соединение ЕТ-743 с использованием цистеинового соединения 29 обычно включает нижеследующие стадии (см. схему III) Схема III превращение соединения формулы 24 в производное 30 путем защиты первичной гидроксильной функциональной группы (S)-N-2,2,2-трихлорэтоксикарбонил-5-(9 Н-флуорен-9-илметил)цистеином 29; превращение защищенного соединения формулы 30 в фенольное производное 31 путем отщепления аллильной группы с использованием гидрида трибутилолова и дихлорпалладий-бис(трифенилфосфина);- 25006369 превращение фенольного соединения формулы 31 в соединение формулы 32 путем окисления ангидридом бензолселениновой кислоты при низкой температуре; превращение гидроксисоединения формулы 32 в лактон 33 в соответствии со следующей последовательностью реакций: (1) взаимодействие соединения формулы 32 с 2 экв. ангидрида трифторметансульфоновой кислоты и 5 экв. ДМСО, (2) последующее взаимодействие с 8 экв. диизопропилэтиламина,(3) последующее взаимодействие с 4 экв. трет-бутилового спирта, (4) последующее взаимодействие с 7 экв. 2-трет-бутил-1,1,3,3-тетраметилгуанидина, (5) последующее взаимодействие с 10 экв. уксусного ангидрида; превращение лактонового соединения 33 в гидроксильное соединение 34 путем удаления защитной группы MOM с использованием TMSI; отщепление N-трихлорэтоксикарбонильной группы соединения формулы 34 в соединение 35 посредством реакции с Zn/AcOH; превращение аминового соединения 35 в соответствующее -кетолактоновое соединение 36 посредством реакции взаимодействия с карбоксальдегидхлоридом N-метилпиридиния, а затем с DBU; образование ЕТ-770 посредством реакции взаимодействия соединения формулы 36 с 3-гидрокси-4 метоксифенилэтиламином; превращение ЕТ-770 в ЕТ-743 посредством реакции взаимодействия с нитратом серебра в смесиAcN/H2O. Получение промежуточного соединения 11 и родственных промежуточных соединений Ретросинтетический анализ проиллюстрирован на нижеследующей реакционной схеме. В настоящем изобретении ключевой класс промежуточных соединений включает промежуточное соединение 11 и имеет общую формулу (XXI) где Prot1 и Prot2 представляют гидроксизащитные группы, предпочтительно различные. Для самого промежуточного соединения 11 группа Prot1 представляет метоксиметильную группу, a Prot2 представляет трет-бутилдифенилсилильную группу. Превращение 21-цианосоединения в промежуточное соединение 11 или родственное промежуточное соединение формулы (XXI) обычно включает следующие стадии:a) превращение, если это необходимо, хиноновой системы для кольца Е в фенольную систему;d) превращение, если это необходимо, хиноновой системы для кольца А в фенольную систему; е) превращение указанной фенольной системы кольца А в метилендиоксифенольное кольцо. На стадии (b) образование группы -OProt1 в 18-положении кольца Е является типичной реакцией защиты для фенольной группы, и в этом случае каких-либо конкретных комментариев не требуется. Подходящие условия выбирают в зависимости от природы защитной группы. Другие стадии аналогичны другим реакциям. На стадии (с) образование группы -CH2-OProt2 в 1 положении кольца В обычно осуществляют посредством образования группы -CH2NH2 в 1 положении с последующим превращением функциональной аминогруппы в функциональную гидроксигруппу и защитой. Таким образом, если исходное вещество имеет группу R1, которая представляет собой -CH2-NH-CO-CR25aR25bR25c, то следует удалить N-ацильную группу. Если исходное вещество имеет группу R1, которая представляет собой -CH2-O-CO-R, то для получения эктеинасцидинового продукта, в котором R1 имеет то же самое значение, каких-либо изменений- 26006369 не требуется. Для получения других продуктов следует удалить О-ацильную группу. Для осуществления таких реакций деацилирования существуют различные способы. В одном варианте деацилирование и превращение в функциональную гидроксигруппу осуществляют в одну стадию. Затем гидроксигруппа может быть ацилирована или каким-либо другим способом превращена в соответствующую группу R1. В патенте США 5721362 описаны синтетические способы получения ЕТ-743 посредством продолжительного многостадийного синтеза. Одним из промежуточных соединений этого синтеза является промежуточное соединение 11. Получение промежуточного соединения 11 может быть достигнуто с использованием цианосафрацина В в качестве исходного вещества, что дает более короткий способ получения такого промежуточного соединения, а поэтому позволяет усовершенствовать способ получения ЕТ-743. Цианосафрацин В может быть превращен в промежуточное соединение 25 описанными выше способами. Из промежуточного соединения 25 можно получить промежуточное соединение 11 с использованием нижеследующих стадий, см. схему VII: образование защищенного гидроксисоединения формулы 26 посредством взаимодействия соединения 25 с трет-бутилдифенилсилилхлоридом в присутствии основания; конечное отщепление аллильной группы под действием гидрида трибутилолова и дихлорпалладийбис(трифенилфосфина) в соединении 26 с образованием промежуточного соединения 11. Схема VII Один из вариантов синтетического способа настоящего изобретения для превращения сафрацина В в промежуточное соединение 11 предусматривает модификацию и расширение схемы VIII и включает последовательные стадии: стереоспецифического превращения соединения сафрацина В в соединение формулы 2 путем селективной замены ОН на CN посредством взаимодействия с KCN в кислой среде; образования соединения тиомочевины формулы 3 в результате взаимодействия соединения формулы 2 с фенилизотиоцианатом; превращения соединения тиомочевины формулы 3 в ацетамид формулы 5 путем гидролиза в кислой среде с последующим добавлением уксусного ангидрида; причем промежуточное аминовое соединение формулы 4 может быть выделено путем гашения гидролиза в кислой среде бикарбонатом натрия, но это промежуточное соединение является очень нестабильным и быстро превращается в пятичленный циклический имин, называемый соединением 6; образования защищенного соединения формулы 7 посредством взаимодействия с бромметилметиловым эфиром и диизопропилэтиламином в дихлорметане; селективного деметилирования метоксигруппы хиноновой системы соединения формулы 7 посредством реакции с метанольным раствором гидроксида натрия с получением соединения формулы 8; превращения соединения формулы 8 в метилендиоксисоединение формулы 9 в соответствии со следующей предпочтительной последовательностью реакций: (1) восстановления хиноновой группы соединения 8 с использованием 10% Pd/C в атмосфере водорода; (2) превращения гидрохинонового промежуточного соединения в метилендиоксисоединение формулы 9 посредством взаимодействия с бромхлорметаном и карбонатом цезия в атмосфере водорода; (3) превращения соединения формулы 9 в со- 27006369 единение формулы 10 путем защиты свободной гидроксильной группы как группы OCH2R посредством взаимодействия с BrCH2R и карбонатом цезия, где R может представлять арил, СН=СН 2, OR' и т.п.; превращения ацетамидной группы соединения формулы 10 в соответствующую гидроксильную группу формулы 25 посредством взаимодействия с тетроксидом азота в смеси уксусной кислоты и ацетата уксусной кислоты с последующей обработкой гидроксидом натрия; альтернативно может быть использован нитрит натрия в смеси уксусного ангидрида и уксусной кислоты с последующей обработкой гидроксидом натрия; альтернативно ацетамидная группа соединения формулы 10 может быть превращена в первичную аминовую группу посредством взаимодействия с гидразином или с Boc2O, DMAP, а затем с гидразином; причем такой первичный амин может быть превращен в соответствующую гидроксильную группу (соединение формулы 25) путем окислительного превращения первичного амина в соответствующий альдегид с использованием бензолсульфоната 4-формил-1-метилпиридиния или другого иона пиридиния, а затем с использованием DBU или обработки другим основанием с последующим гидролизом и восстановлением альдегида до соответствующей гидроксильной группы с использованием алюмогидрида лития или другого восстановителя; образования защищенного соединения формулы 26 в результате взаимодействия с третбутилдифенилсилилхлоридом и диметиламинопиридином в дихлорметане (схема VII); превращения силилированного соединения формулы 26 в промежуточное соединение 11 путем удаления защитной группы OCH2R и реакции в условиях восстановления или в кислых условиях. Обычно эти процедуры осуществляют с использованием палладиевой черни в атмосфере водорода или водной TFA или гидрида трибутилолова и дихлорбис(трифенилфосфинпалладия). Схема VIII Еще в одном предпочтительном варианте настоящего изобретения цианосоединение формулы 2 может быть превращено в промежуточное соединение 11 с использованием расширения схемы II, включающей дополнительные стадии: образование защищенного гидроксисоединения формулы 26 посредством взаимодействия соединения 25 с трет-бутилдифенилсилилхлоридом в присутствии основания; конечное отщепление аллильной группы под действием гидрида трибутилолова и дихлорпалладийбис(трифенилфосфина) в соединении 26 с образованием промежуточного соединения 11. Таким образом цианосафрацин В может быть превращен в ряд промежуточных соединений и производных с потенциальной противоопухолевой терапевтической активностью этими и другими способами. Эти промежуточные соединения могут быть получены из уже описанных соединений или с использованием альтернативных способов.- 28006369 Новые промежуточные соединения Исходя из вышеприведенных объяснений очевидно, что настоящее изобретение относится к новым промежуточным соединениям. В зависимости от кольца А, этими соединениями являются соединения формулы (XXIIа) где R1 представляет -CH2NH2 или -СН 2 ОН, либо защищенный или дериватизированный вариант такой группы, a R4 представляет -Н; илиRla и R4, взятые вместе, образуют группу формул (IV), (V), (VI) или (VII)R5 представляет -ОН, либо защищенный или дериватизированный вариант указанной группы;R14a и R14b, оба представляют -Н, либо один из них представляет -Н, а другой представляет -ОН или защищенный или дериватизированный вариант указанной группы, -ОСН 3 или -ОСН 2 СН 3, либо R14a иR15 представляет -Н, -ОН либо защищенный или дериватизированный вариант указанной группы иR18 представляет -ОН, либо защищенный или дериватизированный вариант указанной группы. В одном из вариантов осуществления настоящего изобретения, по крайней мере, R1, R5, R14a, R14b,15R или R18 предпочтительно представляют защищенную или дериватизированную группу. В одном из вариантов настоящего изобретения группа R1 не является трет-бутилдифенилсилильным заместителем и/или группа R18 не является метоксиметильнои группой.R1 предпочтительно представляет -CH2NH2 или -СН 2 ОН, либо защищенный или дериватизированный вариант указанной группы, a R4 представляет -Н; или Rla и R4, взятые вместе, образуют группу- 29006369 Один из предпочтительных классов промежуточных соединений включает соединение, которое было идентифицировано как соединение 25, имеющее формулу Предпочтительный класс соединений имеет общую формулу, где группа MOM заменена любой другой защитной группой. Другие предпочтительные промежуточные соединения включают соединения, которые авторы изобретения идентифицировали как соединения 45 и 43 (схема IX) Другие N-ацильные производные могут быть легко получены из соединения 45 и являются важной частью настоящего изобретения. Подходящими ацильными группами являются группы, упомянутые ранее. Также могут быть использованы соответствующие 21-гидроксисоединения и соединения из числа активных соединений, обнаруженных авторами настоящего изобретения. Новые активные соединения Кроме того, авторами настоящего изобретения было обнаружено, что некоторые соединения настоящего изобретения, которые первоначально были получены как промежуточные соединения, обладают исключительной активностью при лечении раковых заболеваний, таких как лейкозы, рак легких, рак толстой кишки, рак почек и меланома. Таким образом, настоящее изобретение относится к способу лечения млекопитающего, а в частности человека, с раковым заболеванием, который включает введение индивидууму, нуждающемуся в этом, терапевтически эффективного количества соединения настоящего изобретения или его фармацевтической композиции. Настоящее изобретение также относится к фармацевтическим композициям, которые содержат в качестве активного ингредиента соединение (или соединения) настоящего изобретения, а также к способам их получения. Примерами фармацевтических композиций являются любые твердые (таблетки, пилюли, капсулы,гранулы и т.п.) или жидкие (растворы, суспензии или эмульсии) композиции, имеющие подходящий состав или предназначенные для перорального, местного или парентерального введения, и эти композиции могут содержать чистое соединение или соединение в комбинации с любым носителем или с другими
МПК / Метки
МПК: A61K 35/00, C07D 515/22, C07D 471/18, C07D 491/22
Метки: соединения, эктеинасцидина, синтетический, получения, способ
Код ссылки
<a href="https://eas.patents.su/30-6369-sinteticheskijj-sposob-polucheniya-soedineniya-ekteinascidina.html" rel="bookmark" title="База патентов Евразийского Союза">Синтетический способ получения соединения эктеинасцидина</a>
Конъюгаты соединения, содержащего сульфгидрильную группу, и производного жирной кислоты, способ получения конюгатов, промежуточные соединения для их получения, способы повышения абсорбции и пролонгированного сохранения в крови и тканях млекопитающего соединения, содержащего сульфгидрильную группу

Номер патента: 584
Опубликовано: 29.12.1999
Авторы: Шен Вей Чанг, Икрами Хуссейн М.
МПК: C07H 19/048, C07D 213/70, A61K 31/44...
Метки: соединения, млекопитающего, конюгатов, сохранения, способы, сульфгидрильную, промежуточные, конъюгаты, группу, тканях, повышения, содержащего, получения, пролонгированного, производного, способ, крови, жирной, абсорбции, кислоты
Формула / Реферат:
1. Соединение общей формулы VI где Р является фрагментом соединения, содержащего сульфгидрильную группу, выбранного из группы, включающей пептиды, белки или олигонуклеотиды; R1 представляет собой водород, низший алкил или арил; R2 представляет собой фрагмент, содержащий липидную группу; а R3 представляет собой гидроксил, фрагмент, содержащий липидную группу или аминокислотную последовательность, включающую 1 или 2 аминокислоты и...
Хиральные бифосфиновые соединения, комплексы на их основе и способ получения хирального бифосфинового соединения

Номер патента: 1377
Опубликовано: 26.02.2001
Авторы: Пай Филип, Воланте Ральф П., Россен Кай
МПК: C07F 9/50, C07C 25/22, C07B 53/00...
Метки: комплексы, получения, соединения, основе, бифосфинового, бифосфиновые, хирального, способ, хиральные
Формула / Реферат:
1. Хиральные бифосфины формулы где R представляет C1-4-алкил, С3-6-циклоалкил или арил, незамещенный или замещенный -F, -СН3, -СF3 или СН3О-; и Х1 и Х2 связывают два R2Р-замещенные бензола и независимо образуют 2-4-членную связывающую группу, состоящую из 2-4 атомов углерода и одного незамещенного или замещенного гетероатома, выбранных из О, S, SO, SO2 или 2. Соединение по п.1, в котором число атомов в связи X1 является таким же, как и...
Нафтильные соединения, промежуточные соединения для их получения, применение нафтильных соединений, способ снижения холестерина

Номер патента: 1600
Опубликовано: 25.06.2001
Авторы: Брайант Генри У., Кроуелл Томас А., Палковиц Алан Д., Джонс Чарльз Д.
МПК: A61P 19/10, C07C 47/546, A61K 31/33...
Метки: получения, соединений, применение, способ, нафтильных, холестерина, нафтильные, соединения, снижения, промежуточные
Формула / Реферат:
1. Соединение формулы I где R1 является -Н, -ОН, -O(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr; где Аr является фенилом или замещенным фенилом, OCO(С1-С6-алкилом), -O(СО)O(С1-С6-алкилом) или -ОSО2(С4-С6-алкилом); R2 является -Н, -F, -Cl, -ОН, -О(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr, где Аr является фенилом или замещенным фенилом, -OCO(С1-С6-алкилом),...
Промежуточное соединение для получения паклитаксела и способ получения промежуточного соединения

Номер патента: 678
Опубликовано: 28.02.2000
Авторы: Свинделл Чарльз С., Чандер Мадхави С., Систи Николас Дж.
МПК: C07D 305/14
Метки: получения, соединения, промежуточного, способ, промежуточное, соединение, паклитаксела
Формула / Реферат:
1. Промежуточное соединение для получения паклитаксела, где промежуточное соединение имеет общую формулу где P1 представляет гидрирующуюся бензильную защитную группу. 2. Промежуточное соединение паклитаксела по п.1, где гидрирующуюся бензильную защитную группу выбирают из группы, включающей бензилоксиметил и бензил. 3. Способ получения промежуточного соединения для получения паклитаксела, где промежуточное соединение имеет общую формулу ...
Трициклические соединения, обладающие активностью в отношении интегринов, в частности, в отношении интегрина альфаvбета 3, способ их получения и промежуточные соединения, используемые в этом способе,их применение в качестве медикаментов и содержащие их фармацевтические композиции.

Номер патента: 2271
Опубликовано: 28.02.2002
Авторы: Кнолле Йохен, Гадек Томас Р., Бернар Серж, Штильц Ханс-Ульрих, Бодари Сара С., Тетш Жан-Жорж, Питти Роберт М., Гурвест Жан-Франсуа, Венер Фолькмар, Макдауэлл Роберт С., Карниато Дени
МПК: A61P 9/10, C07C 281/12, A61K 31/19...
Метки: соединения, трициклические, способе,их, частности, альфаvбета, этом, отношении, качестве, получения, интегринов, применение, интегрина, способ, промежуточные, фармацевтические, активностью, содержащие, композиции, используемые, медикаментов, обладающие
Формула / Реферат:
1. Соединения общей формулы (I) в которой R1 обозначает группу -О-[А]-[В]-COR6, в которой R6 обозначает -ОН, C1-С6алкокси, -О-СН2-СН(ОН)-СН2OН, [A] обозначает группу C1-С6алкилен, возможно замещенный оксогруппой, [B] обозначает радикал -CH(Z)- или простую связь, Z обозначает группу -NHCO2Rc, или -NHSO2Rc, где Rc обозначает радикал фенил(C1-С4)алкил-, хинолинил или пиридинилимидазолил(C1-С4)алкил-, R2 и R3, одинаковые или разные, обозначают атом...
Предыдущий патент: Химически модифицированные коньюгаты прогенипоэтина
Следующий патент: Синергические гербицидные смеси
Случайный патент: Устройство для регулирования напряжения питания элементов компьютерной системы в режиме ожидания