Производные 1-фенилсульфонил-1, 3-дигидро-2н-индол-2-она, их получение и их терапевтическое применение
Номер патента: 6019
Опубликовано: 25.08.2005
Авторы: Гарсия Жорж, Ру Ришар, Ди Мальта Ален, Тоннерр Бернар, Шентье Бруно, Серрадейль-Ле Галь Клодин, Ваньон Жан







Формула / Реферат
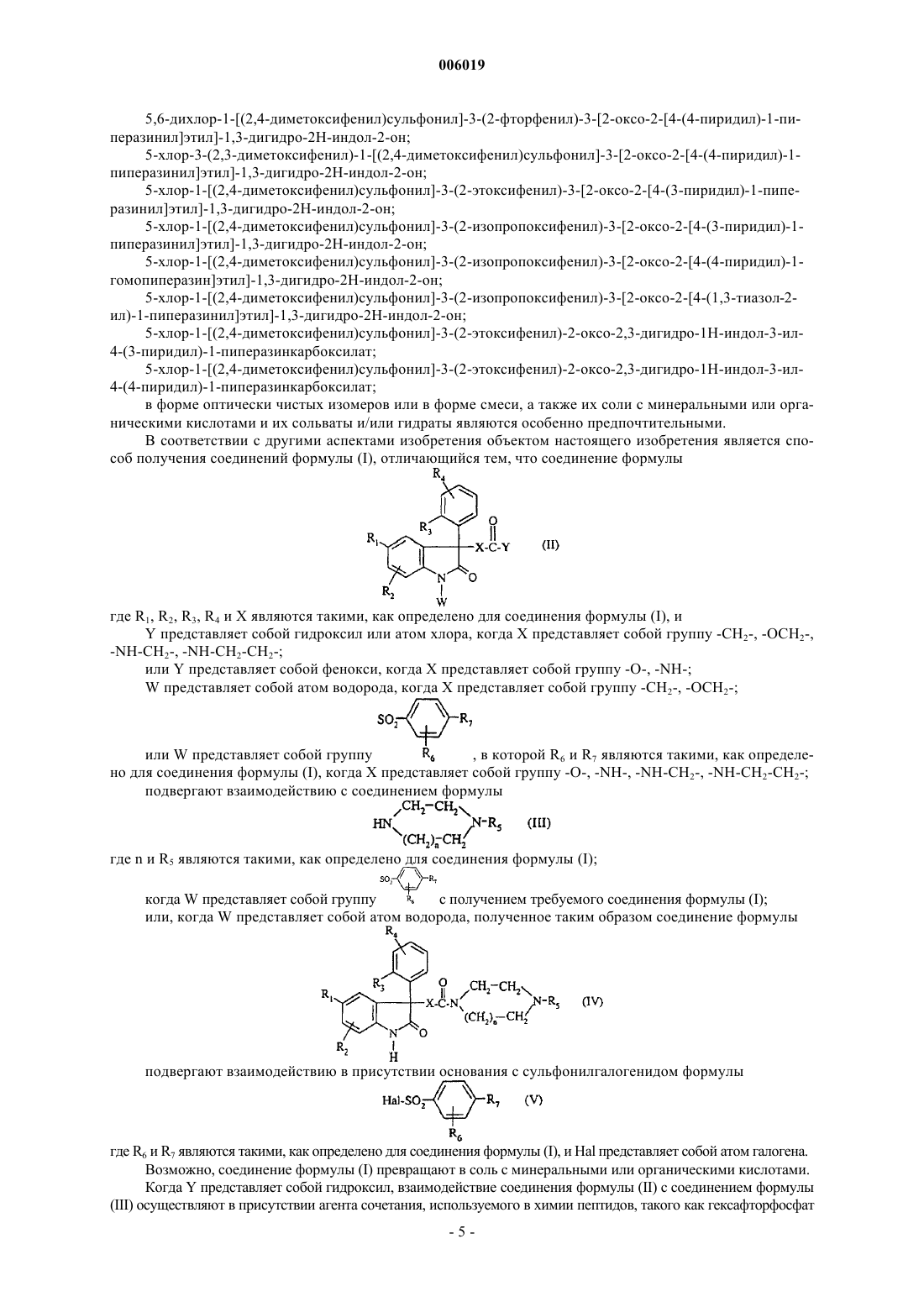
1. Соединение формулы

где n равно 1 или 2;
X представляет собой группу -CH2-, -O-, -NH-, -O-CH2-, -NH-CH2-, -NH-CH2-CH2-;
R1 представляет собой атом галогена, (C1-C4)алкил, (C1-C4)алкокси;
R2 представляет собой атом водорода, атом галогена, (C1-C4)алкил, (C1-C4)алкокси, радикал трифторметил;
R3 представляет собой атом галогена, (C1-C3)алкил, (C1-C3)алкокси, радикал трифторметил, радикал трифторметокси;
R4 представляет собой атом водорода, атом галогена, (C1-C3)алкил, (C1-C3)алкокси;
R5 представляет собой радикал, выбранный из

R6 представляет собой (C1-C4)алкокси;
R7 представляет собой (C1-C4)алкокси;
а также его соли с минеральными или органическими кислотами, их сольваты и/или их гидраты.
2. Соединение по п.1, где
n и X являются такими, как определено для соединения формулы (I) в п.1;
R1 представляет собой атом хлора или радикал метил;
R2 представляет собой атом водорода или находится в положении -6 индол-2-она и представляет собой атом хлора, радикал метил, радикал метокси или радикал трифторметил;
R3 представляет собой атом хлора, атом фтора, радикал метокси, радикал этокси, радикал изопропокси, радикал трифторметил или радикал трифторметокси;
R4 представляет собой атом водорода или радикал метокси;
R5 представляет собой 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 2-пиразинил, 3-пиридазинил или 1,3-тиазол-2-ил;
R6 находится в положении -2 фенила и представляет собой радикал метокси;
R7 представляет собой радикал метокси;
а также его соли с минеральными или органическими кислотами, их сольваты и/или гидраты.
3. Соединение, выбранное из
5-хлор-3-(2-этоксифенил)-1-[(2,4-диметоксифенил)сульфонил]-3-[2-оксо-2-[4-(4-пиридил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-она,
5-хлор-3-(2-изопропоксифенил)-1-[(2,4-диметоксифенил)сульфонил]-3-[2-оксо-2-[4-(4-пиридил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-она,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил-4-(2-пиридил)-1-пиперазинкарбоксилата,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил-4-(4-пиридил)-1-пиперазинкарбоксилата,
N-[5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил]-4-(4-пиридил)гомопиперазин-1-карбоксамида,
N-[5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил]-4-(3-пиридил)пиперазин-1-карбоксамида,
N-[5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил]-4-(4-пиридил)пиперазин-1-карбоксамида,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-3-[[3-оксо-3-[4-(2-пиридил)-1-пиперазинил]пропил]амино]-1,3-дигидро-2H-индол-2-она,
5,6-дихлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-3-[2-оксо-2-[4-(4-пиридил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-она,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-6-метил-3-[2-оксо-2-[4-(4-пиридил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-она,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-6-метил-2-оксо-2,3-дигидро-1H-индол-3-ил-4-(4-пиридил)-1-пиперазинкарбоксилата,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-6-метокси-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил-4-(4-пиридил)-1-пиперазинкарбоксилата,
N-[5-хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-2-оксо-2,3-дигидро-1H-индол-3-ил]-4-(2-пиридил)пиперазин-1-карбоксамида,
N-[5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-6-метил-2-оксо-2,3-дигидро-1H-индол-3-ил]-4-(4-пиридил)пиперазин-1-карбоксамида,
N-[6-хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-5-метил-2-оксо-2,3-дигидро-1H-индол-3-ил]-4-(4-пиридил)пиперазин-1-карбоксамида,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-фторфенил)-3-[2-оксо-2-[4-(4-пиридил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-она,
5,6-дихлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-фторфенил)-3-[2-оксо-2-[4-(4-пиридил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-она,
5-хлор-3-(2,3-диметоксифенил)-1-[(2,4-диметоксифенил)сульфонил]-3-(2-оксо-2-[4-(4-пиридил)-1-пиперазинил]этил)-1,3-дигидро-2H-индол-2-она,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-этоксифенил)-3-[2-оксо-2-[4-(3-пиридил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-она,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-3-[2-оксо-2-[4-(3-пиридил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-она,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-3-[2-оксо-2-[4-(4-пиридил)-1-гомопиперазин]этил]-1,3-дигидро-2H-индол-2-она,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-3-[2-оксо-2-[4-(1,3-тиазол-2-ил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-она,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-этоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил-4-(3-пиридил)-1-пиперазинкарбоксилата,
5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-этоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил-4-(4-пиридил)-1-пиперазинкарбоксилата
в форме оптически чистых изомеров или в форме смеси, и также их солей с минеральными или органическими кислотами и их сольватов и/или гидратов.
4. Способ получения соединений формулы (I) по п.1, отличающийся тем, что
соединение формулы

где R1, R2, R3, R4 являются такими, как определено для соединения формулы (I) в п.1, и
Y представляет собой гидроксил или атом хлора,
X представляет собой группу -CH2-, -OCH2-,
W представляет собой атом водорода,
подвергают взаимодействию с соединением формулы

где n и R5 являются такими, как определено для соединения формулы (I) в п.1;
и полученное таким образом соединение формулы

подвергают взаимодействию в присутствии основания с сульфонилгалогенидом формулы

где R6 и R7 являются такими, как определено для соединения формулы (I) в п.1, и Hal представляет собой атом галогена.
5. Способ получения соединений формулы (I) по п.1, отличающийся тем, что
соединение формулы

где R1, R2, R3, R4 являются такими, как определено для соединения формулы (I) в п.1, и
Y представляет собой гидроксил или атом хлора, когда X представляет собой группу -NH-CH2-, -NH-CH2-CH2-,
или Y представляет собой фенокси, когда X представляет собой группу -O-, -NH-;
W представляет собой группу  , в которой R6 и R7 являются такими, как определено для соединения формулы (I) в п.1,
, в которой R6 и R7 являются такими, как определено для соединения формулы (I) в п.1,
подвергают взаимодействию с соединением формулы

где n и R5 являются такими, как определено для соединения формулы (I) в п.1,
с получением требуемого соединения формулы (I).
6. Соединение формулы

где n равно 1 или 2;
X представляет собой группу -CH2-, -O-CH2-;
R1 представляет собой атом галогена, (C1-C4)алкил, (C1-C4)алкокси;
R2 представляет собой атом водорода, атом галогена, (C1-C4)алкил, (C1-C4)алкокси, радикал трифторметил;
R3 представляет собой атом галогена, (C1-C3)алкил, (C1-C3)алкокси, радикал трифторметил, радикал трифторметокси;
R4 представляет собой атом водорода, атом галогена, (C1-C3)алкил, (C1-C3)алкокси;
R5 представляет собой радикал, выбранный из

а также его соли с минеральными или органическими кислотами в форме оптически чистых изомеров или в форме смеси.
7. Лекарственное средство, отличающееся тем, что оно состоит из соединения по любому из пп.1-3 или из его фармацевтически приемлемой соли с минеральными или органическими кислотами и из их фармацевтически приемлемого сольвата и/или гидрата.
8. Фармацевтическая композиция, отличающаяся тем, что она содержит в качестве активного начала соединение по любому из пп.1-3 или его фармацевтически приемлемую соль с минеральными или органическими кислотами и их фармацевтически приемлемый сольват и/или гидрат, а также один или более чем один фармацевтически приемлемый эксципиент.
9. Применение соединения по любому из пп.1-3 или его фармацевтически приемлемых солей с минеральными или органическими кислотами и их фармацевтически приемлемых сольватов и/или гидратов для приготовления лекарственных средств для лечения сердечно-сосудистых состояний, стресса, тревоги, депрессии, обсессивно-компульсивного расстройства, панических атак, расстройств почечной системы, расстройств желудочной системы, мелкоклеточного рака легких, ожирения, диабета II типа, инсулинорезистентности, гипертриглицеридемии, атеросклероза, синдрома Кушинга, дисменореи и преждевременных родов.
Текст
006019 Настоящее изобретение относится к производным 1,3-дигидро-2H-индол-2-она, их получению и их терапевтическому применению. Соединения по настоящему изобретению проявляют сродство к аргинин-вазопрессиновым (AVP)V1b рецепторам и/или к окситоциновым (ОТ) рецепторам, и, кроме того, некоторые из них проявляют сродство к AVP V1a рецепторам.AVP представляет собой гормон, известный своим антидиуретическим действием и действием в отношении регулирования артериального давления. Он стимулирует некоторые типы рецепторов: V1(V1a, V1b), V2. Эти рецепторы расположены, в частности, в печени, кровеносных сосудах (коронарных сосудах, сосудах почек и мозга), тромбоцитах, почках, матке, надпочечниках, поджелудочной железе,центральной нервной системе и гипофизе. AVP, таким образом, оказывает сердечно-сосудистое действие, антидиуретическое и тромбоцит-агрегирующее действие и действия на печень, поджелудочную железу, центральную и периферическую нервную систему и матку. ОТ представляет собой нейрогипофизарный гормон циклической нонапетидной структуры, аналогичной структуре AVP. ОТ рецепторы обнаружены, главным образом, в гладкой мускулатуре матки и на миоэпителиальных клетках молочных желез. Таким образом, ОТ играет важную роль при родах, поскольку он участвует в сокращении мышцы матки и лактации. Кроме того, ОТ рецепторы также расположены в других периферических тканях и в центральной нервной системе; ОТ может, таким образом,оказывать действие на сердечно-сосудистую, почечную, эндокринную или поведенческую систему. Расположение различных рецепторов описано в S. Jard et al., Vasopressin and oxytocin receptors: anoverview, в Progress in Endocrinology, H. Imura and K. Shizurne ed., Experta Medica, Amsterdam, 1988, 11831188, и в следующих статьях: J. Lab. Clin. Med., 1989, 114 (6), 617-632 и Pharmacol. Rev., 1991, 43 (1), 73-108. Более детально, AVP V1a рецепторы расположены во многих периферических органах и в головном мозге. Их клонировали, в частности, в крысах и человеке, и они регулируют большинство известных действийAVP: агрегацию тромбоцитов, сокращения матки, сокращение кровеносных сосудов, секрецию альдостерона,кортизола, CRF (кортикотропинвысвобождающего фактора) и АСТН (адренокортикотропного гормона), гликогенолиз в печени, пролиферацию клеток и основные центральные действия AVP (гипотермию, память и т.д.).V1b рецепторы сначала были идентифицированы в аденогипофизе различных родов животных (крысы, свиньи, крупного рогатого скота, овцы и т.д.), включая человека (S. Jard et al., Mol. Pharmacol., 1986,30, 171-177; Y. Arsenijevic et al., J. Endocrinol., 1994, 141, 383-391; J. Schwartz et al., Endocrinology, 1991,129 (2), 1107-1109; Y. De Keyser et al., FEBS Letters, 1994, 356, 215-220), где они стимулируют высвобождение адренокортикотропного гормона при помощи AVP и усиливают влияния CRF на высвобождение АСТН (G. Е. Gillies et al., Nature, 1982, 299, 355). В гипоталамусе V1b рецепторы также индуцируют прямое высвобождение CRF (Neuroendocrinology, 1994, 60, 503-508) и являются в этих различных отношениях вовлеченными в стрессовые ситуации. Такие V1b рецепторы клонировали в крысах, человеке и мышах (Y. De Keyser, FEBS Letters, 1994, 356,215-220; Т. Sugimoto et al., J. Biol. Chem., 1994, 269 (43), 27088-27092; M. Saito et al., Biochem. Biophys.al., Journal of Molecular endocrinology, 1999, 22, 251-260), и различные исследования (in situ гибридизация,PCR (полимеразная цепная реакция) и т.д.) выявили повсеместное присутствие этих рецепторов в различных центральных тканях (в частности, в головном мозге, гипоталамусе и аденогипофизе), периферических тканях (почке, поджелудочной железе, надпочечниках, сердце, легких, кишечнике, желудке, печени, брыжейке, мочевом пузыре, тимусе, селезенке, матке, сетчатке, щитовидной железе и т.д.) и в некоторых опухолях (опухолях гипофиза, легких и т.д.), что позволяет предположить широкую биологическую и/или патологическую роль этих рецепторов и их возможное участие в различных заболеваниях. В качестве примера, исследования на крысах показали, что AVP регулирует поджелудочную железу при помощи V1b рецепторов, стимулируя секрецию инсулина и глюкагона (В. Lee et al., Am. J. Physiol. 269(Endocrinol. Metab. 32): E1095-E1100, 1995) или продуцирование катехоламинов в мозговом веществе надпочечников, которое является местом локального синтеза AVP (E. Grazzini et al., Endocrinology, 1996, 137 (а),3906-3914). Таким образом, полагают, что в ткани мозгового вещества надпочечников AVP при помощи этих рецепторов играет важную роль в некоторых типах феохромоцитом надпочечников, секретирующих AVP и, таким образом, индуцирующих длительное продуцирование катехоламинов, которые являются причиной гипертонических состояний, резистентных к антагонистам рецепторов ангиотензина II и к ингибиторам конвертирующих ферментов. Кора надпочечников также богата V1a рецепторами, вовлеченными в продуцирование глюкокортикоидов и минералокортикоидов (альдостерона и кортизола). При помощи этих рецепторов AVP (циркулирующий или синтезируемый местно) может индуцировать продуцирование альдостерона с эффективностью, сравнимой с эффективностью ангиотензина II (G. Guillon et al., Endocrinology,1995, 136 (3), 1285-1295). Кортизол является мощным регулятором продуцирования АСТН, гормона стресса. Недавние исследования также показали, что надпочечники способны высвобождать CRF и/или АСТН непосредственно через активацию V1b и/или V1a рецепторов, которые несут на себе медуллярные клетки (G.V1b рецепторы также считаются маркерами опухолей. АСТН-секретирующие опухоли, такие как некоторые опухоли гипофиза, некоторые бронхиальные карциномы (SCLC (виды мелкоклеточного рака лег-1 006019 ких, карциномы поджелудочной железы, надпочечников и щитовидной железы, включая в некоторых случаях синдром Кушинга (J. Bertherat et al., Eur. J. Endocrinol., 1996, 135, 173; G.A. Wittert et al., Lancet,1990, 335, 991-994; G. Dickstein et al., J. Clin. Endocrinol., Metab., 1996, 81 (8), 2934-2941), экспрессируют чрезмерное количество V1b рецепторов. Что касается V1a рецепторов, они являются маркерами, более характерными для видов мелкоклеточного рака легких (SCLC) (P.J. Woll et al., Biochem. Biophys. Res. Commun.,1989, 164 (1), 66-73). Таким образом, соединения по настоящему изобретению являются очевидными диагностическими средствами и представляют новый терапевтический подход к пролиферации и выявлению таких опухолей даже на ранних стадиях (мечение радиоактивными изотопами; SPECT (однофотонная эмиссионная компьютерная томография); PET сканирование (сканер позитронной эмиссионной томографии. Наличие большого количества мессенджеров V1b рецепторов в желудке и кишечнике предполагает участие AVP при помощи этого рецептора в высвобождении желудочно-кишечных гормонов, таких как холецистокинин, гастрин или секретин (Т. Sugimoto et al., Molecular cloning and functional expression of V1band S. Yoshida ed., Elvesier Science, 1995, 409-413). Производные 1,3-дигидро-2 Н-индол-2-она раскрыты в некоторых заявках на патент как лиганды аргинин-вазопрессиновых рецепторов и/или окситоциновых рецепторов: можно упомянуть заявки на патент WO 93/15051, ЕР 636608, ЕР 636609, WO 95/18105, WO 97/15556 и WO 98/25901. В настоящее время были обнаружены новые производные 1,3-дигидро-2 Н-индол-2-она, которые проявляют сродство и селективность по отношению к аргинин-вазопрессиновым V1b рецепторам и/или окситоциновым рецепторам, и, кроме того, некоторые из них проявляют сродство к V1a рецепторам. Эти производные не проявляют сродства к AVP V2 рецепторам. Таким образом, в соответствии с одним из аспектов данного изобретения одним из объектов изобретения являются соединения формулыX представляет собой группу -CH2-, -О-, -NH-, -O-CH2-, -NH-CH2-, -NH-CH2-СН 2-;R1 представляет собой атом галогена, (С 1-С 4)алкил, (С 1-С 4)алкокси;R2 представляет собой атом водорода, атом галогена, (С 1-С 4)алкил, (С 1-С 4)алкокси, радикал трифторметил;R3 представляет собой атом галогена, (С 1-С 3)алкил, (С 1-С 3)алкокси, радикал трифторметил, радикал трифторметокси;R4 представляет собой атом водорода, атом галогена, (С 1-С 3)алкил, (С 1-С 3)алкокси;R7 представляет собой (С 1-С 4)алкокси; а также их соли с минеральными или органическими кислотами, их сольваты и/или их гидраты. Соединения формулы (I) могут содержать один или более чем один асимметрический атом углерода. Они могут, таким образом, существовать в форме энантиомеров или диастереоизомеров. Эти энантиомеры и диастереоизомеры, а также их смеси, включая рацемические смеси, образуют часть данного изобретения. Соединения формулы (I) могут существовать в форме оснований или солей присоединения кислот. Такие соли присоединения образуют часть данного изобретения. Эти соли преимущественно получают с фармацевтически приемлемыми кислотами, но соли других кислот, которые пригодны для очистки или выделения соединений формулы (I), также образуют часть данного изобретения.-2 006019 Соединения формулы (I) также могут существовать в форме гидратов или сольватов, то есть в форме ассоциаций или комбинаций с одной или более чем одной молекулой воды или с растворителем. Такие гидраты или сольваты также образуют часть данного изобретения. Термин "галоген" означает атом хлора, брома, фтора или йода. Термин "алкил" означает линейный или разветвленный алкильный радикал из 1-3 атомов углерода или 1-4 атомов углерода, такой как радикал метил, этил, пропил, изопропил, бутил, изобутил, втop-бутил или трет-бутил. Термин "алкокси" означает линейный или разветвленный радикал алкокси из 1-3 атомов углерода или 1-4 атомов углерода, такой как радикал метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втop-бутокси или тpeт-бутокси. Преимущественно данное изобретение относится к соединениям формулы (I), где R5 представляет собой радикал, выбранный из В соответствии с настоящим изобретением предпочтительными соединениями формулы (I) являются те соединения, в которых R1 представляет собой атом хлора или радикал метил. В соответствии с настоящим изобретением предпочтительными соединениями формулы (I) являются те соединения, в которых R2 представляет собой атом водорода, атом хлора, радикал метил, радикал метокси или радикал трифторметил. В соответствии с настоящим изобретением предпочтительными соединениями формулы (I) являются те соединения, в которых R3 представляет собой атом хлора, атом фтора, радикал метокси, радикал этокси, радикал изопропокси, радикал трифторметокси или радикал трифторметил. В соответствии с настоящим изобретением предпочтительными соединениями формулы (I) являются те соединения, в которых R4 представляет собой атом водорода или радикал метокси. В соответствии с настоящим изобретением предпочтительными соединениями формулы (I) являются те соединения, в которых R5 представляет собой 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил,2-пиразинил, 3-пиридазинил или 1,3-тиазол-2-ил. В соответствии с настоящим изобретением предпочтительными соединениями формулы (I) являются те соединения, в которых R6 находится в положении -2 фенила и представляет собой радикал метокси. В соответствии с настоящим изобретением предпочтительными соединениями формулы (I) являются те соединения, в которых R7 представляет собой радикал метокси. В частности, предпочтительными соединениями формулы (I) являются те соединения, в которыхn и Х являются такими, как определено для соединения формулы (I);R1 представляет собой атом хлора или радикал метил;R2 представляет собой атом водорода или находится в положении -4 или -6 индол-2-она и представляет собой атом хлора, радикал метил, радикал метокси или радикал трифторметил;R3 представляет собой атом хлора, атом фтора, радикал метокси, радикал этокси, радикал изопропокси, радикал трифторметил или радикал трифторметокси;R4 представляет собой атом водорода или радикал метокси;R6 находится в положении -2 фенила и представляет собой радикал метокси;R7 представляет собой радикал метокси; а также их соли с минеральными или органическими кислотами, их сольваты и/или гидраты. Более детально, предпочтительными соединениями формулы (I) являются те соединения, в которыхn равно 1 или 2; Х представляет собой группу -CH2-, -О-, -NH-;R1 представляет собой атом хлора;R2 представляет собой атом водорода;R3 представляет собой радикал метокси, радикал этокси или радикал изопропокси;R4 представляет собой атом водорода;R6 находится в положении -2 фенила и представляет собой радикал метокси;R7 представляет собой радикал метокси; а также их соли с минеральными или органическими кислотами и их сольваты и/или гидраты.-3 006019 Более детально также предпочтительными соединениями формулы (I) являются те соединения, в которыхX представляет собой группу -СН 2-, -О-, -NH-;R1 представляет собой атом хлора или радикал метил;R2 представляет собой атом водорода или находится в положении -4 или -6 индол-2-она и представляет собой атом хлора, радикал метил, радикал метокси;R3 представляет собой атом хлора, атом фтора или радикал метокси;R4 представляет собой атом водорода;R6 находится в положении -2 фенила и представляет собой радикал метокси;R7 представляет собой радикал метокси; а также их соли с минеральными или органическими кислотами и их сольваты и/или гидраты. Наконец, более детально предпочтительными соединениями формулы (I) являются те соединения, в которыхn равно 1; Х представляет собой группу -О-, -NH-, -NH-CH2-CH2-;R1 представляет собой атом хлора;R2 представляет собой атом водорода;R3 представляет собой атом хлора или радикал метокси;R4 представляет собой атом водорода;R6 находится в положении -2 фенила и представляет собой радикал метокси;R7 представляет собой радикал метокси; а также их соли с минеральными или органическими кислотами и их сольваты и/или гидраты. Следующие соединения: 5-хлор-3-(2-этоксифенил)-1-[(2,4-диметоксифенил)сульфонил]-3-[2-оксо-2-[4-(4-пиридил)-1-пиперазинил]этил]-1,3-дигидро-2 Н-индол-2-он; 5-хлор-3-(2-изопропоксифенил)-1-[(2,4-диметоксифенил)сульфонил]-3-[2-оксо-2-[4-(4-пиридил)-1 пиперазинил]этил]-1,3-дигидро-2H-индол-2-он; 5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3-ил 4-(2-пиридил)-1-пиперазинкарбоксилат; 5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1H-индол 3-ил-4-(4-пиридил)-1-пиперазинкарбоксилат;-4 006019 5,6-дихлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-фторфенил)-3-[2-оксо-2-[4-(4-пиридил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-он; 5-хлор-3-(2,3-диметоксифенил)-1-[(2,4-диметоксифенил)сульфонил]-3-[2-оксо-2-[4-(4-пиридил)-1 пиперазинил]этил]-1,3-дигидро-2 Н-индол-2-он; 5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-этоксифенил)-3-[2-оксо-2-[4-(3-пиридил)-1-пиперазинил]этил]-1,3-дигидро-2 Н-индол-2-он; 5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-3-[2-оксо-2-[4-(3-пиридил)-1 пиперазинил]этил]-1,3-дигидро-2H-индол-2-он; 5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-3-[2-оксо-2-[4-(4-пиридил)-1 гомопиперазин]этил]-1,3-дигидро-2H-индол-2-он; 5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-3-[2-оксо-2-[4-(1,3-тиазол-2 ил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-он; 5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-этоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3-ил 4-(3-пиридил)-1-пиперазинкарбоксилат; 5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-этоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3-ил 4-(4-пиридил)-1-пиперазинкарбоксилат; в форме оптически чистых изомеров или в форме смеси, а также их соли с минеральными или органическими кислотами и их сольваты и/или гидраты являются особенно предпочтительными. В соответствии с другими аспектами изобретения объектом настоящего изобретения является способ получения соединений формулы (I), отличающийся тем, что соединение формулы где R1, R2, R3, R4 и X являются такими, как определено для соединения формулы (I), иY представляет собой гидроксил или атом хлора, когда Х представляет собой группу -СН 2-, -ОСН 2-,-NH-CH2-, -NH-CH2-CH2-; или Y представляет собой фенокси, когда Х представляет собой группу -О-, -NH-;W представляет собой атом водорода, когда Х представляет собой группу -СН 2-, -ОСН 2-;, в которой R6 и R7 являются такими, как определено для соединения формулы (I), когда Х представляет собой группу -О-, -NH-, -NH-CH2-, -NH-CH2-CH2-; подвергают взаимодействию с соединением формулы где n и R5 являются такими, как определено для соединения формулы (I); с получением требуемого соединения формулы (I); когда W представляет собой группу или, когда W представляет собой атом водорода, полученное таким образом соединение формулы где R6 и R7 являются такими, как определено для соединения формулы (I), и Hal представляет собой атом галогена. Возможно, соединение формулы (I) превращают в соль с минеральными или органическими кислотами. Когда Y представляет собой гидроксил, взаимодействие соединения формулы (II) с соединением формулы-5 006019 бензотриазол-1-илокситрис(диметиламино)фосфония или гексафторфосфат бензотриазол-1-илокситрипирролидинофосфония, и в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, в растворителе, таком как дихлорметан или N,N-диметилформамид, при температуре между 0 С и комнатной температурой. Когда Y представляет собой атом хлора, взаимодействие соединения (II) с соединением (III) осуществляют при отсутствии или в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, в растворителе, таком как дихлорметан или хлороформ, и при температуре между -60 С и комнатной температурой. При отсутствии основания используют избыток соединения (III). Когда Y представляет собой фенокси, взаимодействие соединения (II) с соединением (III) осуществляют в растворителе, таком как дихлорметан, хлороформ или тетрагидрофуран, или смеси таких растворителей и при температуре между комнатной температурой и температурой дефлегмации растворителя. Таким образом, это приводит к получению непосредственно либо соединения формулы (I), либо соединения формулы (IV). Взаимодействие соединения (IV) с сульфонилгалогенидом (V) осуществляют в присутствии сильного основания, например гидрида металла, такого как гидрид натрия, или алкоголята щелочного металла, такого как трет-бутилат калия, в растворителе, таком как N,N-диметилформамид или тетрагидрофуран, и при температуре между -70 С и комнатной температурой. Взаимодействие предпочтительно осуществляют, используя соединение формулы (V), в котором Hal представляет собой атом хлора. Соединения формулы (I), полученные таким образом, затем можно выделить из реакционной среды и очистить стандартными способами, например кристаллизацией или хроматографией. Соединения формулы (I), полученные таким образом, выделяют в форме свободного основания или соли в соответствии со стандартными методиками. Соединения формулы (II) получают в соответствии с различными методиками. Соединения формулы (II), в которых -Х- представляет собой группу -CH2-, Y представляет собой гидроксил и W представляет собой водород, получают в соответствии со схемой 1, приведенной ниже, где R1, R2,R3 и R4 являются такими, как определено для соединения формулы (I), и Alk представляет собой (С 1-С 2)алкил. Схема 1 На стадии а 1 схемы 1 соединение формулы (VIII) получают путем дегидроксилирования соответствующего соединения формулы (VI) под действием триэтилсилана в соответствии с Bioorganic and MedicinalChemistry Letters, 1997, 7 (10), 1255-1260. Соединение формулы (VIII) также можно получить путем реакции циклизации (стадия б 1) соединения формулы (VII) в среде сильной кислоты, например серной кислоты или полифосфорной кислоты, в соответствии со способом, описанным в WO 95/18105 или в J. Org. Chem., 1968, 33, 1640-1643.-6 006019 На стадии в 1 соединение формулы (VIII) подвергают взаимодействию с соединением формулы HalCH2COOAlk, где Hal представляет собой атом галогена, предпочтительно бром или хлор, и Alk представляет собой (C1-С 2)алкил, в присутствии основания, такого как карбонат щелочного металла (например,карбонат калия) и иодид щелочного металла (например, иодид калия), или в присутствии сильного основания, такого как гидрид щелочного металла (например, гидрид натрия) или алкоголят щелочного металла (например, этилат натрия), с получением соединения формулы (IX). Взаимодействие осуществляют в растворителе, таком как ацетон или N,N-диметилформамид, и при температуре между 0 С и температурой дефлегмации растворителя. На стадии г 1 требуемое соединение формулы (II) получают путем гидролиза в щелочной среде соединения формулы (IX), используя гидроксид щелочного металла, такой как гидроксид натрия, гидроксид калия или гидроксид лития, в растворителе, таком как вода, метанол, этанол, тетрагидрофуран или диоксан или смесь этих растворителей, при температуре между 0 С и температурой дефлегмации растворителя. Соединения формулы (VI) известны и их получают известными способами, такими как способы,описанные в WO 95/18105. Например, соединение формулы (VI) получают путем взаимодействия производного 1 Н-индол-2,3 диона формулы где R1 и R2 являются такими, как определено для соединения формулы (I), с магнийорганическим производным формулы где R3 и R4 являются такими, как определено для соединения формулы (I), и Hal представляет собой атом галогена, предпочтительно бром или йод, в растворителе, таком как тетрагидрофуран или диэтиловый эфир, и при температуре между 0 С и температурой дефлегмации растворителя. Соединение формулы (VI), в которой R3 является таким, как определено для соединения формулы(I), и R4, отличный от водорода, находится в положении -3 или -6 фенила, также можно получить путем взаимодействия соединения формулы где R3 является таким, как определено для соединения формулы (I), и R4 находится в положении -2 или -5 фенила, с литиевым производным, таким как н-бутиллитий, и литиированное промежуточное соединение, полученное таким образом, затем подвергают взаимодействию с соединением формулы (X). Взаимодействие осуществляют в растворителе, таком как диэтиловый эфир, тетрагидрофуран, гексан или смесь этих растворителей, при температуре между -70 С и комнатной температурой. Производные 1H-индол-2,3-диона (X) имеются в продаже или их получают способами, описанными в Tetrahedron Letters, 1998, 39, 7679-7682; Tetrahedron Letters, 1994, 35, 7303-7306; J. Org. Chem., 1977, 42York, 1975,18, 2-58. Магнийорганические производные (XI) получают стандартными способами, хорошо известными специалистам в данной области техники. В частности, соединения формулы (VI), где R3=(С 1-С 2)алкокси и R4=Н, или R3=R4=(С 1-С 2)алкокси сR4 в положении -3 или -6 фенила, R2 не является атомом галогена, и R1 является таким, как определено для соединения формулы (I), можно получить способом, описанным в схеме 2. На стадии а 2 схемы 2 соединение формулы (XII) сначала подвергают взаимодействию с литиевым производным, таким как н-бутиллитий, при отсутствии или в присутствии основания, такого какN,N,N',N'-тетраметилэтилендиамин, и литиированное промежуточное соединение, полученное таким образом, затем подвергают взаимодействию с диэтилоксалатом с получением соединения формулы(XIII). Взаимодействие осуществляют в растворителе, таком как диэтиловый эфир, тетрагидрофуран,гексан или смесь этих растворителей, и при температуре между -70 С и комнатной температурой. На стадии б 2 соединение формулы (XIV) сначала подвергают взаимодействию с двумя эквивалентами литиевого производного, такого как тpeт-бутиллитий, и литиированное промежуточное соединение,полученное таким образом, затем подвергают взаимодействию с соединением формулы (XIII) с получением требуемого соединения формулы (VI). Взаимодействие осуществляют в растворителе, таком как диэтиловый эфир, тетрагидрофуран, пентан или смесь этих растворителей, и при температуре между-70 С и комнатной температурой. Соединения формулы (XII) имеются в продаже или их синтезируют традиционным способом. Соединения формулы (XIV) получают путем взаимодействия соответствующих производных анилина с ди-трет-бутилдикарбонатом традиционными способами. Соединения формулы (VII) известны и их получают известными способами, такими как способы,раскрытые в WO 95/18105 или в J. Оrg. Сhеm., 1968, 33, 1640-1643. Соединения формулы (II), где Х=-СН 2-, Y представляет собой атом хлора и W=Н, получают из соответствующих соединений формулы (II), где Y=ОН, путем взаимодействия с тионилхлоридом в растворителе, таком как толуол, и при температуре между 0 С и температурой дефлегмации растворителя. Соединения формулы (II), где -Х- представляет собой группу -О-, Y представляет собой фенокси и, получают в соответствии со схемой 3, приведенной ниже, где R1, R2, R3, R4, R6 и R7 являются такими, как определено для соединения формулы (I). На стадии а 3 схемы 3 гидроксил соединения формулы (VI) селективно защищают, используя, например, гексаметилдисилазан, способом, описанным в Synthetic Communications, 1993, 23 (12), 1633-1641. Соединение формулы (XV), полученное таким образом, подвергают взаимодействию на стадии б 3 с сульфонилгалогенидом формулы (V) в присутствии сильного основания в условиях, описанных выше. На стадии в 3 после удаления защиты триметилсилильной группы соединения формулы (XVI), полученного таким образом, получают соединение формулы (XVIII). Реакцию осуществляют под действием сильной кислоты, такой как соляная кислота или трифторуксусная кислота, в растворителе, таком как-9 006019 дихлорметан, ацетон, тетрагидрофуран или вода, или в смеси этих растворителей и при температуре между комнатной температурой и температурой дефлегмации растворителя. Соединение формулы (XVIII) также можно получить путем взаимодействия на стадии г 3 соединения формулы (X) с сульфонилгалогенидом формулы (V) в условиях, описанных выше, с последующим взаимодействием соединения формулы (XVII), полученного таким образом, с магнийорганическим производным формулы (XI) в условиях, описанных выше. На стадии е 3 соединение формулы (XVIII), полученное таким образом, подвергают взаимодействию с фенилхлорформиатом в присутствии основания, такого как пиридин, в растворителе, таком как дихлорметан,или без растворителя, при температуре между 0 и 100 С с получением требуемого соединения формулы (II). Соединение формулы (VI) получают способами, описанными выше. Соединение формулы (VI) также можно получить путем окисления воздухом соединения формулы (VIII) в присутствии основания,такого как гидрид натрия, и в присутствии диметилдисульфида. Соединение формулы (VI) также можно получить путем гидролиза галогенида формулы где R1, R2, R3 и R4 являются такими, как определено для соединения формулы (I), и Hal представляет собой атом галогена, предпочтительно бром или хлор. Взаимодействие осуществляют в растворителе, таком как тетрагидрофуран, и при температуре между комнатной температурой и температурой дефлегмации растворителя. Соединения формулы (XIX) известны, и их получают в соответствии с известными способами, такими как способы, описанные в WO 95/18105. Соединения формулы (II), в которых -Х- представляет собой группу -NH-, Y представляет собой фенокси, и W представляет собой группу, получают в соответствии со способами, описанными в WO 95/18105. Соединения формулы (II), где -Х- представляет собой группу -О-СН 2-, Y представляет собой гидроксил и W представляет собой водород, получают в соответствии со схемой 4, приведенной ниже, где R1,R2, R3 и R4 являются такими, как определено для соединения формулы (I). Схема 4- 10006019 На стадии а 4 схемы 4 соединение формулы (XIX) подвергают взаимодействию с метилгликолятом в присутствии сильного основания, такого как гидрид натрия, в растворителе, таком как тетрагидрофуран или дихлорметан, и при температуре между 0 С и температурой дефлегмации растворителя. Соединение формулы (XX), полученное таким образом, гидролизуют на стадии б 4 в щелочной среде в соответствии со способами, описанными выше на стадии г 1 схемы 1, с получением требуемого соединения формулы (II). Соединения формулы (II), в которых Х=-O-CH2-, Y представляет собой атом хлора и W=Н, получают из соответствующих соединений формулы (II), где Y=ОН путем взаимодействия с тионилхлоридом в соответствии со способом, упомянутым выше. Соединения формулы (II), где -Х- представляет собой группу -NH-CH2-, Y представляет собой гид, получают в соответствии со схемой 5, приведенной роксил и W представляет собой группу ниже, где R1, R2, R3, R4, R6 и R7 являются такими, как определено для соединения формулы (I). Схема 5 На стадии а 5 схемы 5 соединение формулы (XIX) подвергают взаимодействию с тpeт-бутиловым эфиром глицина в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, в растворителе, таком как дихлорметан, хлороформ или тетрагидрофуран, или в смеси этих растворителей и при температуре между 0 С и комнатной температурой. Соединение формулы (XXI), полученное таким образом, подвергают взаимодействию на стадии б 5 с сульфонилгалогенидом формулы (V) в присутствии сильного основания в условиях, описанных выше. На стадии в 5 соединение формулы (XXII), полученное таким образом, гидролизуют в кислой среде, используя сильную кислоту, такую как соляная кислота или трифторуксусная кислота, в растворителе, таком как дихлорметан, тетрагидрофуран, ацетон или вода, в смеси этих растворителей или без растворителя и при температуре между 0 С и комнатной температурой. Таким образом, получают требуемое соединение формулы (II). Соединения формулы (II), в которых Х=-NH-CH2-, Y представляет собой атом хлора и, получают из соответствующих соединений формулы (II), где Y=ОН, в соответствии соW= способами, описанными выше. Соединения формулы (I), где -Х- представляет собой группу -NH-CH2-СН 2-, Y представляет собой, получают в соответствии со схемой 6, приведенной гидроксил и W представляет собой группу ниже, где R1, R2, R3, R4, R6 и R7 являются такими, как описано для соединения формулы (I). На стадии а 6 схемы 6 соединение формулы (XIX) подвергают взаимодействию с тpeт-бутиловым эфиром -аланина в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, в растворителе, таком как дихлорметан, хлороформ или тетрагидрофуран, или в смеси этих растворителей и при температуре между 0 С и комнатной температурой. Соединение формулы (XXIII), полученное таким образом, подвергают взаимодействию на стадии б 6 с сульфонилгалогенидом формулы (V) в присутствии сильного основания, в условиях, описанных выше. На стадии в 6 соединение формулы (XXIV), полученное таким образом, гидролизуют в кислой среде, в условиях, описанных выше на стадии в 5 схемы 5. Таким образом, получают требуемое соединение формулы (II). Соединения формулы (II), где -Х-=-NH-CH2-CH2-, Y представляет собой атом хлора и W=,получают из соответствующих соединений формулы (II), где Y=-ОН, в соответствии со способами, описанными выше. Соединения формулы (III) имеются в продаже или их получают известными способами, описанными в J. Org. Chem., 1953, 18, 1484-1488, J. Med. Chem., 1978, 21 (6), 536-542, Chem. Pharm. Bull., 1991, 39(9), 2288-2300, Tetrahedron Letters, 1998, 39, 617-620 или в WO 97/28129. Например, соединение формулы (III) получают путем взаимодействия соединения формулы где n является таким, как определено для соединения формулы (I), и Z представляет собой водород или(XXVI) где R5 является таким, как определено для соединения формулы (I), и Hal представляет собой атом галогена, предпочтительно хлор, бром или йод. Взаимодействие осуществляют в присутствии или при отсутствии основания, в инертном растворителе, таком как этанол, 2-пропанол, н-бутанол или ацетонитрил, и при температуре между 0 С и температурой дефлегмации растворителя. Когда используют основание, его выбирают из органических оснований, таких как диизопропилэтиламин, или из карбонатов щелочных металлов, таких как карбонат натрия или карбонат калия. При отсутствии основания взаимодействие осуществляют, используя избыток соединения формулы (XXV). Это взаимодействие также можно осуществить без растворителя, нагревая смесь соединений (XXV) и (XXVI) до температур порядка 140 и 180 С. Где это уместно, в случае, когда Z представляет собой N-защитную группу, ее удаляют стандартными способами с получением требуемых соединений формулы (III).- 12006019 Соединения формулы (XXV) или формулы (XXVI) известны или их получают известными способами. Соединения формулы (V) известны или их получают известными способами, такими как способы,описанные в ЕР-0469984 и WO 95/18105. Например, соединения формулы (V) можно получить путем галогенирования соответствующих бензолсульфоновых кислот или их солей, например их натриевых или калиевых солей. Взаимодействие осуществляют в присутствии галогенирующего агента, такого как оксихлорид фосфора, тионилхлорид, трихлорид фосфора, трибромид фосфора или пентахлорид фосфора,без растворителя или в инертном растворителе, таком как галогенированный углеводород или N,Nдиметилформамид, и при температуре между -10 и 200 С. 2,4-Диметоксибензолсульфонилхлорид получают в соответствии с J. Am. Chem. Soc., 1952, 74,2008. 3,4-Диметоксибензолсульфонилхлорид имеется в продаже, или его получают в соответствии сJ. Med. Chem., 1977, 20 (10), 1235-1239. Для получения соединений формулы (I) в форме оптически чистых изомеров можно использовать стандартные методики разделения, например фракционную перекристаллизацию соли, образованной рацемическим основанием и оптически активной кислотой, принципы которой хорошо известны, или при помощи стандартных методик хроматографии на колонке с хиральной фазой. Оптически чистые соединения формулы (I) также можно получить из оптически чистого промежуточного соединения, которое используют для получения соединений формулы (I). Таким образом, когда желательно получить оптически чистое соединение формулы (I), в котором-Х-=-СН 2-, осуществляют оптическое разделение соединения формулы (II), в котором -Х-=-СН 2-, Y=ОН и W=Н, в соответствии со способом, описанным в J. Am. Chem. Soc., 1989, 111, 7650-7651, используя (R)пантолактон в качестве хирального реагента или используя хиральный амин, такой как (+)-цинхонин. Оптически чистое соединение формулы (I), в котором -Х-=-О-, можно получить способом, описанным на схеме 7, приведенной ниже, где R1, R2, R3, R4, n и R5 являются такими, как определено для соединения формулы (I), и R представляет собой фенил или изобутил. Схема 7 На стадии а 7 схемы 7 соединение формулы (VI) подвергают взаимодействию с фенилхлорформиатом в присутствии основания, такого как пиридин, в растворителе, таком как дихлорметан, или без растворителя и при температуре между 0 и 100 С.- 13006019 Соединение формулы (XXVII), полученное таким образом, подвергают взаимодействию на стадии б 7 с соединением формулы (III) в растворителе, таком как дихлорметан, хлороформ или тетрагидрофуран, или в смеси этих растворителей, при температуре между комнатной температурой и температурой дефлегмации растворителя с получением соединения формулы (XXVIII). Путем взаимодействия соединения (XXVIII) с L-лейцинолом (R=изобутил) или D-лейцинолом либо с (R)-(-)фенилглицинолом (R=фенил) или (S)-(+)фенилглицинолом получают на стадии в 7 смесь диастереоизомеров соединения формулы (XXIX), которую можно разделить, например, путем хроматографии или кристаллизации. Путем взаимодействия на стадии г 7 одного из диастереоизомеров соединения формулы (XXIX) с сильным основанием, таким как метилат натрия, в растворителе, таком как метанол или тетрагидрофуран, или в смеси этих растворителей, при температуре между 0 С и температурой дефлегмации растворителя получают оптически чистое соединение формулы (XXX). При взаимодействии соединения (XXX) с сульфонилгалогенидом формулы (V) в соответствии со способами, описанными выше, получают оптически чистое соединение формулы (I), в котором Х=-О-. Когда желательно получить оптически чистое соединение формулы (I), в котором -Х-=-NH-, соединение формулы (II), в котором -Х-=-NH-, Y=фенокси и W=, его получают в оптически чистой форме способом, описанным на схеме 8 ниже, где R1, R2, R3, R4, R6 и R7 являются такими, как определено для соединения формулы (I), и Hal представляет собой атом галогена, предпочтительно хлор или бром. Схема 8 На стадии а 8 схемы 8 соединение формулы (XIX) подвергают взаимодействию с (S)-(+)фенилглицинолом или (R)-(+)фенилглицинолом с получением смеси диастереоизомеров соединения формулы (XXXI), которую можно разделить при помощи хроматографии или кристаллизации. На стадии б 8 оптически чистое соединение формулы (XXXII) получают путем окисления тетраацетатом свинца и гидролиза в кислой среде одного из диастереоизомеров соединения (XXXI). На стадии в 8 в результате взаимодействия соединения (XXXII), полученного таким образом, с сульфонилгалогенидом формулы (V) с последующим взаимодействием соединения (XXXIII), полученного таким образом, с фенилхлорформиатом (стадия г 8) в соответствии со способами, описанными вWO 95/18105, получают требуемое оптически чистое соединение (II). В процессе любой из стадий получения соединений формулы (I) или промежуточных соединений формул (II), (III) или (IV) может быть необходимым и/или желательным защитить реакционноспособные или чувствительные функциональные группы, такие как аминная, гидроксильная или карбоксильная группы, присутствующие на любой из рассматриваемых молекул. Такую защиту можно осуществить,используя традиционные защитные группы, такие как группы, описанные в Protective Groups in OrganicThieme Verlag. Удаление защитных групп можно осуществить на подходящей последующей стадии, ис- 14006019 пользуя способы, известные специалистам в данной области техники, которые не влияют на остальную часть рассматриваемой молекулы.N-защитные группы, которые можно использовать, являются традиционными N-защитными группами, которые хорошо известны специалистам в данной области техники, такими как, например, третбутоксикарбонильная, флуоренилметоксикарбонильная, бензильная, бензгидрилиденовая или бензилоксикарбонильная группа. Соединения формулы (IV) являются новыми и образуют часть данного изобретения. Таким образом, в соответствии еще c одним аспектом объектом данного изобретения являются соединения формулы где n равно 1 или 2; Х представляет собой группу -CH2-, -O-CH2-;R1 представляет собой атом галогена, (С 1-С 4)алкил, (С 1-С 4)алкокси;R2 представляет собой атом водорода, атом галогена, (С 1-С 4)алкил, (С 1-С 4)алкокси, радикал трифторметил;R3 представляет собой атом галогена, (С 1-С 3)алкил, (С 1-С 3)алкокси, радикал трифторметил, радикал трифторметокси;R4 представляет собой атом водорода, атом галогена, (С 1-С 3)алкил, (С 1-С 3)алкокси; а также их соли с минеральными или органическими кислотами в форме оптически чистых изомеров или в форме смеси. Соединения формулы (I), указанной выше, также включают в себя соединения, в которых один или более чем один атом водорода или атом углерода замещен его радиоактивным изотопом, например тритием или углеродом-14. Такие меченые соединения применимы в научных исследованиях, исследованиях метаболизма и фармакокинетики и в биохимических тестах в качестве лигандов рецепторов. Следующие подготовительные примеры и примеры иллюстрируют данное изобретение, но не ограничивают его. В подготовительных примерах и примерах используют следующие сокращения: эфир: диэтиловый эфир изоэфир: диизопропиловый эфирDCC: 1,3-дициклогексилкарбодиимид НОВТ: гидрат 1-гидроксибензотриазола хлороводородный эфир: насыщенный раствор хлороводорода в диэтиловом эфире т.пл.: точка плавления- 15006019 Спектры протонного магнитного резонанса (1 Н ЯМР) записывают при 200 МГц в d6-DMSO, используя пик d6-DMSO в качестве стандарта. Химические сдвигивыражены в миллионных долях (м.д.). Наблюдаемые сигналы описывают таким образом: s: синглет; bs: широкий синглет; d: дублет; dd: двойной дублет; t: триплет; dt: двойной триплет; q: квартет; up: неразрешенный пик; mt: мультиплет. ЯМР спектры подтверждают структуры соединений. Подготовительные примеры Получение соединений формулы (II) Подготовительный пример 1.1. 2-[5-Хлор-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил] уксусная кислота.(II): R1=Cl; R2=Н; R3=ОСН 3; R4=Н; Х=-СН 2-; Y=ОН; W=Н. А) 5-Хлор-3-гидрокси-3-(2-метоксифенил)-1,3-дигидро-2 Н-индол-2-он. Это соединение получают способом, описанным в WO 95/18105. Раствор бромида 2-метоксифенилмагния получают из 16 г магния в 35 мл эфира и раствора 124 г 1-бром-2-метоксибензола в 175 мл эфира. Этот раствор добавляют по каплям в атмосфере аргона к смеси 30 г 5-хлор-1H-индол-2,3-диона в 250 млTHF, предварительно охлажденного на ледяной бане, и затем смесь оставляют перемешиваться и температуре позволяют достичь комнатной температуры. После перемешивания в течение 1 ч при комнатной температуре реакционную смесь медленно вливают в насыщенный раствор NH4Cl и THF выпаривают в вакууме. Образовавшийся осадок отфильтровывают путем отсасывания и промывают изоэфиром. Получают 42 г требуемого продукта и его используют без дополнительной очистки на следующей стадии. Б) 5-хлор-3-(2-метоксифенил)-1,3-дигидро-2 Н-индол-2-он. Смесь 5,8 г соединения, полученного на предыдущей стадии, 25 мл TFA и 10 мл триэтилсилана кипятят с обратным холодильником в течение 24 ч. Реакционную смесь концентрируют под вакуумом при 100 С, осадок переносят в 30 мл изоэфира и оставляют стоять при 20 С в течение 15 ч. Образовавшийся осадок отфильтровывают путем отсасывания и промывают изоэфиром и затем гептаном. Получают 4,3 г требуемого продукта. В) Этил-2-[5-хлор-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил]ацетат. Смесь 2,85 г соединения, полученного на предыдущей стадии, 2,05 г этилбромацетата, 2,05 г KI и 3,1 г K2 СО 3 в 15 мл ацетона кипятят с обратным холодильником в течение 20 ч. Минеральные соли отфильтровывают и фильтрат концентрируют в вакууме. Остаток экстрагируют ЕtOАс, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают в вакууме. Остаток растирают в изоэфире и образовавшийся осадок отфильтровывают путем отсасывания. Осадок подвергают хроматографии на силикагеле, элюируя DCM и затем ЕtOАс. 1,55 г требуемого продукта получают после перекристаллизации из изоэфира, точка плавления=184-187 С. Г) 2-[5-Хлор-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил]уксусная кислота. Раствор 0,5 г гранул NaOH в 10 мл воды добавляют при комнатной температуре к смеси 1,5 г соединения, полученного на предыдущей стадии, в 30 мл МеОН и смесь перемешивают в течение 20 ч при 20 С. Реакционную смесь концентрируют под вакуумом, осадок переносят в 20 мл воды, водную фазу промывают ЕtOАс и подкисляют до рН 1 путем добавления концентрированной HCl и образовавшийся осадок отфильтровывают путем отсасывания. Получают 1,15 г требуемого продукта, точка плавления=135-139 С. Подготовительный пример 1.2. 2-[5-Хлор-3-(2-этоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3-ил]уксусная кислота.A) 1-Бром-2-этоксибензол. Смесь 17,5 г 2-бромфенола, 66 мл диэтилсульфата и 170 мл 10%-ного раствора NaOH кипятят с обратным холодильником в течение 2 ч. После охлаждения реакционной смеси до комнатной температуры ее экстрагируют ЕtOАс и органическую фазу промывают 2 н раствором NaOH, сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 19,6 г требуемого продукта. Б) 5-Хлор-3-(2-этоксифенил)-3-гидрокси-1,3-дигидро-2 Н-индол-2-он. Раствор бромида 2-этоксифенилмагния получают из 2,2 г магния в 10 мл эфира и из раствора 16,5 г соединения, полученного на предыдущей стадии, в 40 мл эфира. Этот раствор добавляют по каплям и в атмосфере азота к смеси 5 г 5-хлор-1 Н-индол-2,3-диона в 20 мл THF, поддерживая температуру реакционной среды ниже 35 С. После перемешивания в течение 2 ч при комнатной температуре реакционную смесь вливают в 200 мл 2 н HCl, экстрагируют ЕtOАс, органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток переносят в горячий изоэфир и оставляют кристаллизоваться. Образовавшийся кристаллический продукт отфильтровывают путем отсасывания, промывают изоэфиром и сушат. Получают 5,7 г требуемого продукта, точка плавления=251 С.B) 5-Хлор-3-(2-этоксифенил)-1,3-дигидро-2 Н-индол-2-он. Это соединение получают способом, описанным на стадии Б подготовительного примера 1.1, начиная с 4 г соединения, полученного на предыдущей стадии, 15 мл TFA и 5,6 мл триэтилсилана. Получают 3,6 г требуемого продукта. Г) Этил-2-[5-хлор-3-(2-этоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил]ацетат.- 16006019 Это соединение получают способом, описанным на стадии В подготовительного примера 1.1, начиная с 3,5 г соединения, полученного на предыдущей стадии, 2,2 г этилбромацетата, 2,2 г KI и 3,5 г К 2 СО 3 в 20 мл ацетона. Получают 1,7 г требуемого продукта после осаждения из изоэфира, точка плавления=181 С. Д) 2-[5-Хлор-3-(2-этоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3-ил]уксусная кислота. Смесь 1,65 г соединения, полученного на предыдущей стадии, и 2 мл 30%-ного раствора NaOH в 50 мл воды и 20 мл THF перемешивают при комнатной температуре в течение 24 ч. Смесь концентрируют под вакуумом, осадок переносят в 100 мл воды, водную фазу промывают ЕtOАс и подкисляют до рН 1 путем добавления концентрированной HCl и экстрагируют ЕtOАс, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 1,2 г требуемого продукта после кристаллизации из изоэфира, точка плавления=212 С. Подготовительный пример 1.3. 2-[5-Хлор-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3 ил]уксусная кислота, правовращающий изомер.(II): R1=Cl; R2=Н; R3=ОСН(СН 3)2; R4=Н; Х=-CH2-; Y=ОН; W=Н. А) 1-Бром-2-изопропоксибензол. Раствор этилата натрия получают из 280 мл ЕtOН и 7,3 г натрия, добавляют 50 г 2-бромфенола и смесь перемешивают при комнатной температуре в течение 30 мин. Затем добавляют 43 г изопропилбромида и смесь кипятят с обратным холодильником в течение 15 ч. Реакционную смесь концентрируют под вакуумом, осадок переносят в 1 н раствор NaOH и экстрагируют эфиром, органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Полученное в результате масло перегоняют при пониженном давлении с получением 49,3 г требуемого продукта в виде бесцветной жидкости, точка кипения=113-115 С при 3366 Па. Б) 5-Хлор-3-гидрокси-3-(2-изопропоксифенил)-1,3-дигидро-2 Н-индол-2-он. Раствор бромида 2-изопропоксифенилмагния получают из 6,3 г магния в 20 мл эфира и 49,3 г соединения, полученного на предыдущей стадии. Этот раствор добавляют в течение 3 мин к смеси 13,3 г 5 хлор-1 Н-индол-2,3-диона в 30 мл THF, затем кипятят с обратным холодильником в течение 90 мин. После охлаждения до комнатной температуры реакционную смесь вливают в смесь лед/концентрированная HCl и экстрагируют ЕtOАс, органическую фазу промывают 1 н раствором NaOH и водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток переносят в горячий изоэфир и образовавшийся осадок отфильтровывают путем отсасывания после растирания. Получают 17,4 г требуемого продукта. В) 5-Хлор-3-(2-изопропоксифенил)-1,3-дигидро-2 Н-индол-2-он. Смесь 3,5 г соединения, полученного на предыдущей стадии, 15 мл TFA и 5,5 мл триэтилсилана кипятят с обратным холодильником в течение 8 ч. Смесь концентрируют под вакуумом и остаток подвергают хроматографии на силикагеле, элюируя изоэфиром и затем DCM. Получают 2,2 г требуемого продукта после кристаллизации из гептана, точка плавления=154 С. Г) Этил-2-[5-хлор-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3-ил]ацетат. Это соединение получают способом, описанным на стадии В подготовительного примера 1.1, начиная с 2,55 г соединения, полученного на предыдущей стадии, 1,67 г этилбромацетата, 1,66 г KI и 2,52 г К 2 СО 3 в 12 мл ацетона. Полученный продукт подвергают хроматографии на оксиде алюминия, элюируяDCM и затем ацетоном. Получают 1,6 г требуемого продукта после кристаллизации из изоэфира, точка плавления=193 С. Д) 2-[5-Хлор-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3-ил]уксусная кислота. Раствор 1 г гранул NaOH в 10 мл воды добавляют при 40 С к раствору 1,6 г соединения, полученного на предыдущей стадии, в 10 мл ЕtOН и смесь перемешивают в течение 20 ч при 20 С. Полученную в результате смесь концентрируют под вакуумом, осадок переносят в 30 мл воды и нагревают до 40 С и нерастворившееся вещество отфильтровывают. Фильтрат подкисляют до рН 1 путем добавления концентрированной HCl и образовавшийся осадок отфильтровывают путем отсасывания и сушат. Получают 1,15 г требуемого продукта, точка плавления=146-148 С. Е) (3R)-4,4-Диметил-2-оксотетрагидро-3-фуранил-2-[5-хлор-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил]ацетат, более полярный изомер. Смесь 2 г соединения, полученного на предыдущей стадии, и 10 мл тионилхлорида перемешивают при 20 С в течение 20 ч. Смесь концентрируют под вакуумом, остаток переносят в DCM и растворитель концентрируют под вакуумом при комнатной температуре с получением 2 г хлорангидрида. Хлорангидрид растворяют в 20 мл DCM, этот раствор добавляют к смеси 0,8 г (R)-пантолактона и 1 г N-метилпирролидина в 10 мл DCM, предварительно охлажденного до 10 С, и полученную в результате смесь перемешивают при комнатной температуре в течение 2 ч. Реакционную смесь концентрируют под вакуумом,осадок переносят в воду и экстрагируют ЕtOАс, органическую фазу промывают водой и сушат надNa2SO4 и растворитель выпаривают под вакуумом. Осадок подвергают хроматографии на силикагеле Н,элюируя DCM. Диастереоизомеры разделяют и более полярное соединение собирают. Получают 0,6 г требуемого продукта. Ж) 2-[5-Хлор-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3-ил]уксусная кислота, правовращающий изомер.- 17006019 Раствор 0,8 г моногидрата гидроксида лития в 10 мл воды добавляют к смеси 0,8 г соединения, полученного на предыдущей стадии, в 30 мл МеОН, и смесь перемешивают при 20 С в течение 20 ч. Реакционную смесь концентрируют под вакуумом, осадок экстрагируют водой и водную фазу промывают эфиром, подкисляют до рН 1 путем добавления концентрированной HCl и экстрагируют DCM, органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 0,5 г требуемого продукта. D20=+84(с=0,25; хлороформ). Подготовительный пример 1.4. 2-[3-(2-Xлорфенил)-5-метил-2-оксо-2,3-дигидро-1H-индол-3-ил]уксусная кислота.(II): R1=СН 3; R2=Н; R3=Cl; R4=Н; Х=-СН 2-; Y=ОН; W=Н. А) 3-(2-Хлорфенил)-5-метил-1,3-дигидро-2 Н-индол-3-он. Это соединение получают способом, описанным в WO 95/18105 на стадиях А и Б подготовительного примера 65. Б) Метил-2-[3-(2-хлорфенил)-5-метил-2-оксо-2,3-дигидро-1H-индол-3-ил]ацетат. Смесь 5 г соединения, полученного на предыдущей стадии, в 10 мл DMF охлаждают до температуры ниже 10 С, добавляют по частям 0,87 г 60%-ного гидрида натрия в масле и смесь перемешивают в течение 30 мин. Затем добавляют 3,15 мл метилбромацетата, смесь перемешивают при комнатной температуре в течение 4 ч, затем дополнительно добавляют 1 мл метилбромацетата и смесь перемешивают при комнатной температуре в течение 16 ч. Реакционную смесь вливают в воду и экстрагируют ЕtOАс, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя DCM и затем смесью DCM/ЕtOАс (95/5; об./об.). Получают 0,57 г требуемого продукта. В) 2-[3-(2-Хлорфенил)-5-метил-2-оксо-2,3-дигидро-1 Н-индол-3-ил]уксусная кислота. Раствор 0,16 г гранул NaOH в 40 мл воды добавляют к смеси 0,57 г соединения, полученного на предыдущей стадии, в 10 мл диоксана и полученную в результате смесь перемешивают при комнатной температуре в течение 24 ч. К реакционной смеси добавляют 50 мл воды, водную фазу промывают ЕtOАс, подкисляют до рН 1 путем добавления 1 н HCl и экстрагируют ЕtOАс, органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируют смесью DCM/ЕtOАс (95/5; об./об.). Получают 0,27 г требуемого продукта. Подготовительный пример 1.5. 2-[5,6-Дихлор-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3 ил]уксусная кислота.(II): R1=Cl; R2=6-Cl; R3=ОСН 3; R4=Н; Х=-CH2-; Y=ОН; W=Н. А) 5,6-Дихлор-1H-индол-2,3-дион. Это соединение получают способом, описанным в J. Am. Chem. Soc., 1946, 68, 2697-2703, или способом, описанным в J. Org. Chem., 1952, 17, 149-156. Б) 5,6-Дихлор-3-гидрокси-3-(2-метоксифенил)-1,3-дигидро-2 Н-индол-2-он. 7,5 г 1-бром-2-метоксибензола добавляют по каплям к суспензии 1,1 г магния в 20 мл эфира, содержащей несколько кристаллов йода, при этом температуру дефлегмации поддерживают с того момента,как ее достигли. В конце добавления смесь нагревают при температуре дефлегмации в течение 2 ч. Затем добавляют суспензию 4,3 г 5,6-дихлор-1 Н-индол-2,3-диона в 20 мл THF и полученную в результате смесь кипятят с обратным холодильником в течение 30 мин. После охлаждения до комнатной температуры реакционную смесь вливают в смесь вода/лед/концентрированная НСl и экстрагируют ЕtOАс, органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток растирают в горячем изоэфире и образовавшийся осадок отфильтровывают путем отсасывания и промывают эфиром. Получают 5,2 г требуемого продукта, точка плавления=245-246 С. С) 5,6-Дихлор-3-(2-метоксифенил)-1,3-дигидро-2H-индол-2-он. Смесь 5,1 г соединения, полученного на предыдущей стадии, 25 мл TFA и 10 мл триэтилсилана кипятят с обратным холодильником в течение 15 ч. Смесь концентрируют под вакуумом, остаток растирают в гептане и образовавшийся осадок отфильтровывают путем отсасывания. Получают 4,8 г требуемого продукта после кристаллизации из изоэфира, точка плавления=195-197 С. Г) Этил-2-[5,6-дихлор-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил]ацетат. Смесь 4,8 г соединения, полученного на предыдущей стадии, 2,7 г этилбромацетата, 2,7 г KI и 4,4 г К 2 СО 3 в 25 мл ацетона кипятят с обратным холодильником в течение 16 ч. Минеральные соли отфильтровывают и фильтрат концентрируют под вакуумом. Остаток подвергают хроматографии на силикагеле,элюируя DCM и затем ЕtOАс. Получают 2,6 г требуемого продукта после кристаллизации из изоэфира,точка плавления=214-219 С. Д) 2-[5,6-Дихлор-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3-ил]уксусная кислота. 3 мл 2 н раствора NaOH добавляют к смеси 2,5 г соединения, полученного на предыдущей стадии, в 50 мл МеОН и смесь перемешивают в течение 48 ч при 20 С. Полученную смесь концентрируют под вакуумом, остаток переносят в 30 мл воды, нерастворившееся вещество отфильтровывают, фильтрат подкисляют до рН 1 путем добавления концентрированной HCl и экстрагируют ЕtOАс, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 2 г требуемого продукта после кристаллизации из изоэфира, точка плавления=222-224 С.(II): R1=Cl; R2=6-СН 3; R3=ОСН 3; R4=Н; Х=-СН 2-; Y=ОН; W=Н. А) Этил-2-(2-метоксифенил)-2-оксоацетат. Раствор 27 г 1-бром-2-метоксибензола в 270 мл эфира охлаждают до -70 С в атмосфере аргона, добавляют по каплям 90 мл 1,6 М раствора н-бутиллития в пентане и смесь затем перемешивают в течение 45 мин. Быстро добавляют 78 мл диэтилоксалата и смесь перемешивают, позволяя температуре достичь комнатной. После перемешивания в течение 1 ч при комнатной температуре к реакционной смеси добавляют насыщенный раствор NH4Cl, фазы разделяют отстаиванием, водную фазу экстрагируют эфиром,объединенные органические фазы промывают водой и насыщенным раствором NaCl и сушат над Na2SO4 и растворители выпаривают под вакуумом. Избыток диэтилоксалата удаляют перегонкой под вакуумом(точка кипения=87 С при 2000 Па). Полученный в результате продукт подвергают хроматографии на силикагеле, элюируя смесью DCM/гексан (50/50; об./об.) и затем DCM. Полученный продукт очищают перегонкой под вакуумом. Получают 13 г требуемого продукта, точка кипения=110 С при 3 Па. Б) 5-Хлор-3-гидрокси-3-(2-метоксифенил)-6-метил-1,3-дигидро-2H-индол-2-он. а) трет-Бутил-4-хлор-3-метилфенилкарбамат. Смесь 10 г 4-хлор-3-метиланилина и 15,26 г ди-трет-бутилдикарбоната в 50 мл диоксана перемешивают при комнатной температуре в течение 24 ч. Реакционную смесь концентрируют под вакуумом и остаток подвергают хроматографии на силикагеле, элюируя градиентом смеси DCM/гексан от (50/50; об./об.) до (70/70; об./об.). Получают 5,6 г требуемого продукта и их используют без дополнительной очистки. б) Раствор 5 г трeт-бутил-4-хлор-3-метилфенилкарбамата в 45 мл эфира охлаждают до -70 С, в атмосфере аргона, добавляют по каплям 30 мл 1,5 М раствора тpeт-бутиллития в пентане, смесь перемешивают в течение 1 ч, позволяя температуре подняться до -10 С, и перемешивают в течение 1 ч 45 мин при-10 С. Реакционную смесь охлаждают до -70 С, добавляют по каплям раствор 5 г соединения, полученного на стадии А в 25 мл THF, смесь перемешивают в течение 1 ч и оставляют нагреваться до -30 С и затем перемешивают в течение ночи, позволяя температуре достичь комнатной температуры. К реакционной смеси добавляют насыщенный раствор NH4Cl, THF выпаривают, полученную в результате водную фазу экстрагируют 3 раза ЕtOАс, органическую фазу промывают водой, насыщенным раствором NaCl и сушат над Na2SO4, растворитель частично выпаривают и кристаллический продукт отфильтровывают путем отсасывания. Получают 2,6 г требуемого продукта, точка плавления=254-256 С. В) 5-Хлор-3-(2-метоксифенил)-6-метил-1,3-дигидро-2 Н-индол-2-он. Смесь 3 г соединения, полученного на предыдущей стадии, 15 мл TFA и 6 мл триэтилсилана кипятят с обратным холодильником в течение 15 ч. Смесь концентрируют под вакуумом и осадок растирают в пентане с получением 2,85 г требуемого продукта, точка плавления=193 С. Г) Этил-2-[5-хлор-3-(2-метоксифенил)-6-метил-2-оксо-2,3-дигидро-1 Н-индол-3-ил]ацетат. Смесь 2,8 г соединения, полученного на предыдущей стадии, 1,7 г этилбромацетата, 1,7 г KI и 2,7 г К 2 СО 3 в 15 мл ацетона кипятят с обратным холодильником в течение 16 ч. Минеральные соли отфильтровывают и фильтрат концентрируют под вакуумом. Получают 2,35 г требуемого продукта после кристаллизации из изоэфира, точка плавления=170 С. Д) 2-[5-Хлор-3-(2-метоксифенил)-6-метил-2-оксо-2,3-дигидро-1 Н-индол-3-ил]уксусная кислота. Смесь 2,3 г соединения, полученного на предыдущей стадии, и 3,5 г 12 н раствора NaOH в 100 мл МеОН и 5 мл воды перемешивают при 20 С в течение 20 ч. Смесь концентрируют под вакуумом, остаток переносят в 30 мл воды, подкисляют до рН 1 путем добавления концентрированной HCl и экстрагируют ЕtOАс, органические фазы промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 1,3 г требуемого продукта после кристаллизации из изоэфира, точка плавления=214-216 С. Подготовительный пример 1.7. 2-[5-Хлор-3-(2-изопропоксифенил)-6-метил-2-оксо-2,3-дигидро-1 Ниндол-3-ил]уксусная кислота.A) Этил-2-[2-[(трет-бутоксикарбонил)амино]-5-хлор-4-метилфенил]-2-оксоацетат. Раствор 5,9 г трет-бутил-4-хлор-3-метилфенилкарбамата в 60 мл эфира охлаждают до -50 С в атмосфере азота, добавляют по каплям 42 мл раствора тpeт-бутиллития в пентане и смесь перемешивают в течение 2 ч, позволяя температуре подняться до -20 С. Реакционную смесь охлаждают до -70 С, добавляют по каплям раствор 4,5 г диэтилоксалата в 30 мл THF и полученную смесь перемешивают в течение 1 ч, позволяя температуре достичь 20 С. Реакционную смесь вливают в насыщенный раствор NH4Cl, фазы разделяют отстаиванием, водную фазу экстрагируют EtOAc, объединенные органические фазы промывают 1 н HCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесью изоэфир/пентан (50/50; об./об.). Получают 8 г требуемого продукта в виде масла, которое кристаллизуется, точка плавления=111 С. Б) 5-Хлор-6-метил-1H-индол-2,3-дион. Смесь 8 г соединения, полученного на предыдущей стадии, и 60 мл 3 н раствора HCl в 80 мл THF кипятят с обратным холодильником в течение 2 ч. Реакционную смесь концентрируют под вакуумом,остаток переносят в ЕtOН и растворитель опять концентрируют под вакуумом. Остаток переносят в- 19006019 ЕtOН и образовавшийся осадок отфильтровывают путем отсасывания и промывают изоэфиром. Получают 1,9 г требуемого продукта.B) 5-Хлор-3-гидрокси-3-(2-изопропоксифенил)-6-метил-1,3-дигидро-2H-индол-2-он. 3,2 г 1-бром-2-изопропоксибензола добавляют по каплям к суспензии 0,36 г магния в 10 мл эфира,поддерживания дефлегмацию с момента ее начала. В конце добавления смесь нагревают при температуре дефлегмации в течение 2 ч. Затем добавляют одной порцией раствор 1,8 г соединения, полученного на предыдущей стадии, в 30 мл THF и смесь кипятят с обратным холодильником в течение 30 мин. После охлаждения до комнатной температуры реакционную смесь вливают в смесь лед/концентрированная НСl и экстрагируют ЕtOАс, органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток растирают в горячем изоэфире и образовавшийся осадок отфильтровывают путем отсасывания и промывают горячим ЕtOАс. Получают 1,8 г требуемого продукта и его используют без дополнительной очистки. Г) 5-Хлор-3-(2-изопропоксифенил)-6-метил-1,3-дигидро-2 Н-индол-2-он. Смесь 2,3 г соединения, полученного на предыдущей стадии, 15 мл TFA и 3,8 мл триэтилсилана кипятят с обратным холодильником в течение 10 ч. Полученную в результате смесь концентрируют под вакуумом (13 Па), остаток растирают в пентане и образовавшийся осадок отфильтровывают путем отсасывания. Осадок подвергают хроматографии на силикагеле, элюируя DCM. Получают 0,85 г требуемого продукта, точка плавления=145-146 С. Д) Этил-2-[5-хлор-3-(2-изопропоксифенил)-6-метил-2-оксо-2,3-дигидро-1H-индол-3-ил]ацетат. Смесь 0,8 г соединения, полученного на предыдущей стадии, 0,53 г этилбромацетата, 0,53 г KI и 0,8 г К 2 СО 3 в 6 мл ацетона кипятят с обратным холодильником в течение 15 ч. Минеральные соли отфильтровывают, фильтрат концентрируют под вакуумом, остаток экстрагируют DCM, органическую фазу промывают водой и нерастворившееся вещество отфильтровывают. Фильтрат подвергают хроматографии на силикагеле, элюируя DCM и затем смесью DCM/ЕtOАс (70/30; об./об.). Получают 0,55 г требуемого продукта, точка плавления=152-154 С. Е) 2-[5-Хлор-3-(2-изопропоксифенил)-6-метил-2-оксо-2,3-дигидро-1H-индол-3-ил]уксусная кислота. Смесь 0,53 г соединения, полученного на предыдущей стадии, и 0,25 г гранул NaOH в 20 мл МеОН и 5 мл воды перемешивают при температуре 25 С в течение 20 ч. Смесь концентрируют под вакуумом,остаток растворяют в 20 мл воды и подкисляют до рН 1 путем добавления концентрированной HCl и образовавшийся осадок отфильтровывают путем отсасывания. Получают 0,45 г требуемого продукта после кристаллизации из смеси изоэфир/пентан (50/50; об./об.), точка плавления=148-150 С. Подготовительный пример 1.8. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-2 оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбонат.. А) 5-Хлор-3-(2-метоксифенил)-3-[(триметилсилил)окси]-1,3-дигидро-2H-индол-2-он. Смесь 2,9 г соединения, полученного на стадии А подготовительного примера 1.1, и 0,16 г безводного хлорида цинка в 40 мл ацетонитрила нагревают до температуры дефлегмации, добавляют 1,7 гHMDS и смесь кипятят с обратным холодильником в течение 1 ч. Полученную смесь концентрируют под вакуумом, остаток экстрагируют ЕtOАс, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 2,4 г требуемого продукта после кристаллизации из изоэфира, точка плавления=185-187 С. Б) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-3-[(триметилсилил)окси]-1,3-дигидро-2 Н-индол-2-он. 0,17 г 60%-ного гидрида натрия в масле добавляют к смеси 2,4 г соединения, полученного на предыдущей стадии, в 30 мл THF и смесь перемешивают в течение 20 мин при комнатной температуре. Затем добавляют 1,7 г 2,4-диметоксибензолсульфонилхлорида и смесь перемешивают в течение 1 ч при комнатной температуре. Полученную в результате смесь концентрируют под вакуумом, остаток экстрагируют ЕtOАс, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 2,65 г требуемого продукта после кристаллизации из изоэфира, точка плавления=157-158 С. В) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-гидрокси-3-(2-метоксифенил)-1,3-дигидро-2 Н-индол-2-он. Смесь 2,4 г соединения, полученного на предыдущей стадии, и 10 мл TFA в 20 мл DCM перемешивают в течение 4 ч при температуре 20 С. Смесь концентрируют под вакуумом, остаток экстрагируют ЕtOАс, органическую фазу промывают 10%-ным раствором Na2CO3 и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 1,75 г требуемого продукта после кристаллизации из изоэфира,точка плавления=153-160 С. Г) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1H-индол-3 ил-фенилкарбонат.- 20006019 Смесь 1,6 г соединения, полученного на предыдущей стадии, и 1 г фенилхлорформиата в 20 мл пиридина перемешивают в течение 3 суток при температуре 20 С. Смесь концентрируют под вакуумом,остаток экстрагируют ЕtOАс, органическую фазу промывают 10%-ным раствором АсОН и сушат надNa2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле,элюируя изоэфиром. Получают 1,05 г требуемого продукта после кристаллизации из изоэфира, точка плавления=212-213 С. Подготовительный пример 1.9. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбонат.DMF и смесь перемешивают в течение 30 мин при 20 С. Затем добавляют 4,8 г 2,4-диметоксибензолсульфонилхлорида и смесь перемешивают при 20 С в течение 1 ч. Полученную смесь концентрируют под вакуумом (1,3 Па), остаток экстрагируют ЕtOАс, органическую фазу промывают водой и сушат надNa2SO4 и растворитель выпаривают под вакуумом. Остаток растирают в изоэфире и образовавшийся осадок отфильтровывают путем отсасывания. Получают 2,9 г требуемого продукта после кристаллизации из горячего ЕtOАс, точка плавления=194,5 С. Б) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-гидрокси-3-(2-изопропоксифенил)-1,3-дигидро-2Hиндол-2-он. Раствор бромида 2-изопропоксифенилмагния получают из 0,5 г магния в 20 мл THF и 4 г 1-бром-2 изопропоксибензола. Этот раствор добавляют по каплям и при комнатной температуре к смеси 4,8 г соединения, полученного на предыдущей стадии, в 50 мл THF, затем кипятят с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь вливают в смесь лед/6 нHCl и экстрагируют ЕtOАс, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесьюDCM/MeOH (99/1; об./об.). Получают 3,8 г требуемого продукта. В) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбонат. Смесь 1 г соединения, полученного на предыдущей стадии, и 1,4 г фенилхлорформиата в 10 мл пиридина перемешивают при комнатной температуре в течение 48 ч. Смесь концентрируют под вакуумом,остаток экстрагируют ЕtOАс, органическую фазу промывают водой, 1 н раствором HCl, водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 0,86 г требуемого продукта после кристаллизации из изоэфира, точка плавления=197 С. Подготовительный пример 1.10. Фенил-5-хлор-3-[(2-изопропоксифенил)-2-оксо-3-[(феноксикарбонил)окси]-1-индолинкарбоксилат.(XXVII): R1=Cl; R2=Н; R3=ОСН(СН 3)2; R4=Н. Смесь 6,34 г соединения, полученного на стадии Б подготовительного примера 1.3, в 60 мл пиридина нагревают до 70 С, добавляют по каплям 7 г фенилхлорформиата и смесь затем перемешивают при 70 С в течение 3 ч и при комнатной температуре в течение ночи. Реакционную смесь вливают в воду и экстрагируют DCM, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя DCM. Полученный продукт растирают в изоэфире и образовавшийся осадок отфильтровывают путем отсасывания. Получают 8 г требуемого продукта. Подготовительный пример 1.11. 5-Хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбонат.. А) 5-Хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-3-гидрокси-1,3-дигидро-2H-индол-2-он. Раствор бромида 2-хлорфенилмагния получают из 0,4 г магния, 0,01 г йода, 20 мл эфира и 2,9 г 1-бром-2-хлорбензола. Смесь 2,8 г соединения, полученного на стадии А подготовительного примера 1.9,в 30 мл THF добавляют к этому раствору при температуре дефлегмации за 1 мин и полученную смесь поддерживают при температуре дефлегмации в течение 20 мин. После охлаждения до комнатной температуры реакционную смесь вливают в смесь лед/концентрированная HCl и экстрагируют ЕtOАс, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя изоэфиром и затем DCM. Получают 1,55 г требуемого продукта в виде пены, которую используют без дополнительной очистки. Б) 5-Хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-2-оксо-2,3-дигидро-1H-индол-3-илфенилкарбонат. Смесь 0,65 г соединения, полученного на предыдущей стадии, и 0,3 г фенилхлорформиата в 5 мл пиридина перемешивают при 20 С в течение 20 ч. Смесь концентрируют под вакуумом, остаток экстра- 21006019 гируют ЕtOАс, органическую фазу промывают 1 н раствором HCl, 1 н раствором NaOH и водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 0,6 г требуемого продукта после кристаллизации из изоэфира, точка плавления=222-223 С. Подготовительный пример 1.12. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-фторфенил)-2 оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбонат.. А) D,L-2-Фторминдальная кислота. Это соединение получают способом, описанным в J. Org. Chem., 1968, 33, 2565-2566. Это соединение можно также получить способом, приведенным ниже. Смесь 17,4 г 2-фторбензальдегида и 9,6 г цианида калия в 30 мл эфира охлаждают до температуры ниже 10 С, добавляют за 30 мин 15 мл концентрированной HCl и полученную в результате смесь перемешивают при комнатной температуре в течение 2 ч. После разделения фаз отстаиванием органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Неочищенный продукт, полученный таким образом, переносят в 20 мл концентрированной HCl и кипятят с обратным холодильником в течение 5 ч. После охлаждения до комнатной температуры реакционную смесь экстрагируют эфиром, органическую фазу промывают водой и сушат надNa2SO4 и растворитель выпаривают под вакуумом. Получают 17,5 г требуемого продукта после кристаллизации из изоэфира. Б) N-Парахлорфенил- DL-2-фторманделамид. Смесь 17,5 г соединения, полученного на предыдущей стадии, и 13 г парахлоранилина в 100 мл 1,2 дихлорбензола кипятят с обратным холодильником в течение 3 ч, удаляя образующуюся воду с помощью аппарата Дина-Старка. После охлаждения до комнатной температуры смесь оставляют кристаллизоваться. Образовавшийся осадок отфильтровывают путем отсасывания и растворяют в ЕtOАс, органическую фазу промывают дважды 4 н раствором НСl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 16,2 г требуемого продукта после кристаллизации из изоэфира. В) 5-Хлор-3-(2-фторфенил)-1,3-дигидро-2 Н-индол-2-он. 16,1 г соединения, полученного на предыдущей стадии, добавляют при комнатной температуре к смеси 64 мл концентрированной (95%) H2SO4 и 16 мл дымящей серной кислоты (30%-ный олеум) и смесь затем перемешивают в течение 8 ч. Реакционную смесь вливают в смесь лед/вода и экстрагируют ЕtOАс, органическую фазу промывают дважды водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 12,2 г требуемого продукта после кристаллизации из изоэфира. Г) 3-Бром-5-хлор-3-(2-фторфенил)-1,3-дигидро-2 Н-индол-2-он. Раствор 0,78 мл брома в 20 мл хлороформа добавляют медленно при комнатной температуре к раствору 4 г соединения, полученного на предыдущей стадии, в 100 мл хлороформа. Реакционную смесь концентрируют под вакуумом с получением 4 г требуемого продукта после кристаллизации из изоэфира. Д) 5-Хлор-3-(2-фторфенил)-5-гидрокси-1,3-дигидро-2 Н-индол-2-он. Смесь 2 г соединения, полученного на предыдущей стадии, в 5 мл воды и 20 мл THF кипятят с обратным холодильником в течение 20 ч. Смесь концентрируют под вакуумом, остаток переносят в 10%ный раствор Nа 2 СО 3 и образовавшийся осадок отфильтровывают путем отсасывания. Получают 1,45 г требуемого продукта после кристаллизации из изоэфира, точка плавления=265 С (с разложением). Е) 5-Хлор-3-(2-фторфенил)-3-[(триметилсилил)окси]-1,3-дигидро-2 Н-индол-2-он. Смесь 1,4 г соединения, полученного на предыдущей стадии, и 0,12 г хлорида цинка в 32 мл ацетонитрила нагревают до температуры дефлегмации, добавляют 6 мл HMDS и смесь кипятят с обратным холодильником в течение 8 ч. Полученную в результате смесь концентрируют под вакуумом, остаток экстрагируют ЕtOАс, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 1,35 г требуемого продукта после кристаллизации из гептана, точка плавления=125-127 С. Ж) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-фторфенил)-3-[(триметилсилил)окси]-1,3-дигидро 2 Н-индол-2-он. Это соединение получают способом, описанным на стадии Б подготовительного примера 1.8, начиная с 1,3 г соединения, полученного на предыдущей стадии, 0,1 г 60%-ного гидрида натрия в масле, 15 млTHF и 1 г 2,4-диметоксибензолсульфонилхлорида. Получают 1,75 г требуемого продукта после кристаллизации из изоэфира, точка плавления=184-186 С. З) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-фторфенил)-3-гидрокси-1,3-дигидро-2 Н-индол-2-он. Смесь 1,7 г соединения, полученного на предыдущей стадии, 2 мл 12 н HCl и 2 мл воды в 30 мл ацетона перемешивают в течение 4 ч. Смесь концентрируют под вакуумом и остаток переносят в МеОН и снова концентрируют под вакуумом. Остаток растирают в изоэфире и образовавшийся осадок отфильтровывают путем отсасывания. Получают 1,3 г требуемого продукта, точка плавления=144-146 С. И) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-фторфенил)-2-оксо-2,3-дигидро-1H-индол-3-илфенилкарбонат.- 22006019 0,7 г фенилхлорформиата добавляют при комнатной температуре к смеси 1 г соединения, полученного на предыдущей стадии, и 0,5 мл пиридина в 20 мл DCM и смесь перемешивают в течение 48 ч. Реакционную смесь промывают дважды водой, органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесью DCM/MeOHA) 5-Хлор-3-гидрокси-3-(2-трифторметилфенил)-1,3-дигидро-2 Н-индол-2-он. Смесь 2,3 г магния и 0,01 г йода в 20 мл эфира нагревают до температуры дефлегмации, добавляют по каплям раствор 26 г 1-бром-2-трифторметилбензола в 20 мл эфира и полученную в результате смесь нагревают с обратным холодильником в течение 60 мин. Затем добавляют раствор 9 г 5-хлор-1H-индол 2,3-диона в 40 мл THF и кипячение с обратным холодильником продолжают в течение 30 мин. После охлаждения до комнатной температуры реакционную смесь вливают в смесь лед/концентрированная HCl и экстрагируют эфиром, органическую фазу промывают 1 н раствором NaOH и водой и сушат надNa2SO4 и растворитель выпаривают под вакуумом. Получают 5,3 г требуемого продукта после кристаллизации из гептана, точка плавления=205-208 С. Б) 5-Хлор-3-(2-трифторметилфенил)-1,3-дигидро-2H-индол-2-он. Смесь 3 г соединения, полученного на предыдущей стадии, и 0,1 г хлорида цинка в 20 мл ацетонитрила нагревают до температуры дефлегмации, добавляют 6 мл HMDS и полученную в результате смесь кипятят с обратным холодильником в течение 1 ч. Смесь концентрируют под вакуумом, остаток экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 3,9 г требуемого продукта в виде масла, которое кристаллизуется, точка плавления=175-176 С.B) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-трифторметилфенил)-3-[(триметилсилил)окси]1,3-дигидро-2H-индол-2-он. Это соединение получают способом, описанным на стадии Б подготовительного примера 1.8, начиная с 3,8 г соединения, полученного на предыдущей стадии, 0,45 г 60%-ного гидрида натрия в масле, 30 млTHF и 2,5 г 2,4-диметоксибензолсульфонилхлорида. Получают 5 г требуемого продукта после кристаллизации из гептана, точка плавления=144-145 С. Г) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-гидрокси-3-(2-трифторметилфенил)-1,3-дигидро-2Hиндол-2-он. Смесь 4,9 г соединения, полученного на предыдущей стадии, 3 мл 12 н HCl и 5 мл воды в 50 млTHF перемешивают при температуре 20 С в течение 15 ч. Полученную в результате смесь концентрируют под вакуумом при температуре ниже 40 С, остаток переносят в 30 мл 10%-ного раствора NaHCО 3 и экстрагируют ЕtOАс, экстракты сушат над Nа 2SO4 и растворитель выпаривают под вакуумом. Получают 3,8 г требуемого продукта после кристаллизации из смеси изоэфир/гептан (80/20; об./об.), точка плавления=143 С. Д) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-трифторметилфенил)-2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбонат. 2 г фенилхлорформиата добавляют по каплям при 20 С к смеси 1,7 г соединения, полученного на предыдущей стадии, в 20 мл пиридина и смесь перемешивают при 20 С в течение 7 ч. Полученную в результате смесь концентрируют под вакуумом, остаток переносят в 30 мл 10%-ного раствора АсОН и экстрагируют эфиром, органическую фазу промывают 10%-ным раствором NаНСО 3 и водой и сушат надNa2SO4 и растворитель выпаривают под вакуумом. Получают 1,8 г требуемого продукта после кристаллизации из гептана, точка плавления=191 С. Подготовительный пример 1.14. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-трифторметоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3-ил-фенилкарбонат.(II): R1=Cl; R2=Н; R3=OCF3; R4=Н; Х=-О-; Y= А) 5-Хлор-3-гидрокси-3-(2-трифторметоксифенил)-1,3-дигидро-2 Н-индол-2-он. Раствор 25 г 1-бром-2-трифторметоксибензола в 130 мл эфира добавляют по каплям к смеси 2,8 г магния в 20 мл эфира, при этом дефлегмацию поддерживают с момента ее начала. В конце добавления смесь нагревают с обратным холодильником в течение 1 ч. Затем добавляют смесь 7,5 г 5-хлор-1 Ниндол-2,3-диона в 100 мл THF при температуре ниже 40 С, затем кипятят с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь вливают в смесь лед/концентрированная HCl и экстрагируют ЕtOАс, органическую фазу промывают водой, 1 н растворомNaOH и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 6,5 г требуемого продукта после кристаллизации из смеси DCM/изоэфир (20/80; об./об.), точка плавления=214 С. Б) 5-Хлор-3-(2-трифторметоксифенил)-3-[(триметилсилил)окси]-1,3-дигидро-2H-индол-2-он.- 23006019 Смесь 2 г соединения, полученного на предыдущей стадии, 0,05 г хлорида цинка и 1,4 г HMDS в 100 мл ацетонитрила кипятят с обратным холодильником в течение 2 ч. Смесь концентрируют под вакуумом, остаток экстрагируют EtOAc, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 2,5 г требуемого продукта и его используют без дополнительной очистки. В) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-трифторметоксифенил)-3-[(триметилсилил)окси]1,3-дигидро-2 Н-индол-2-он. Это соединение получают способом, описанным на стадии Б подготовительного примера 1.8, начиная с 2,5 г соединения, полученного на предыдущей стадии, 0,3 г 60%-ного гидрида натрия в масле, 50 млTHF и 1,7 г 2,4-диметоксибензолсульфонилхлорида. Получают 2,7 г требуемого продукта после кристаллизации из изоэфира, точка плавления=181 С. Г) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-гидрокси-3-(2-трифторметоксифенил)-1,3-дигидро 2H-индол-2-он. Смесь 2,7 г соединения, полученного на предыдущей стадии, и 1 мл концентрированной HCl в 50 мл ацетона перемешивают в течение 1 ч при температуре ниже 40 С. Полученную в результате смесь концентрируют под вакуумом, остаток экстрагируют EtOAc, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 1,66 г требуемого продукта после кристаллизации из изоэфира, точка плавления=81 С. Д) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-трифторметоксифенил)-2-оксо-2,3-дигидро-1 Ниндол-3-ил-фенилкарбонат. 1,15 г фенилхлорформиата добавляют при комнатной температуре к смеси 1,6 г соединения, полученного на предыдущей стадии, и 0,8 мл пиридина в 20 мл DCM и смесь перемешивают при комнатной температуре в течение 1 ч. Реакционную смесь промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 1,34 г требуемого продукта после кристаллизации из изоэфира,точка плавления=203-204 С. Подготовительный пример 1.15. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-6 метил-2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбонат.A) 5-Хлор-3-(2-метоксифенил)-6-метил-3-[(триметилсилил)окси]-1,3-дигидро-1 Н-индол-2-он. Смесь 0,65 г соединения, полученного на стадии Б подготовительного примера 1.6, и 0,05 г хлорида цинка в 10 мл ацетонитрила нагревают до температуры дефлегмации, добавляют 2,1 мл HMDS и кипение с обратным холодильником поддерживают в течение 1 ч. Полученную в результате смесь концентрируют под вакуумом, остаток экстрагируют ЕtOАс, органическую фазу промывают водой и сушат надNa2SO4 и растворитель выпаривают под вакуумом. Получают 0,7 г требуемого продукта после кристаллизации из гептана, точка плавления=227 С. Б) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-6-метил-3-[(триметилсилил)окси]1,3-дигидро-2H-индол-2-он. Это соединение получают способом, описанным на стадии Б подготовительного примера 1.8, начиная с 0,85 г соединения, полученного на предыдущей стадии, 0,06 г 60%-ного гидрида натрия в масле, 20 млTHF и 0,6 г 2,4-диметоксибензолсульфонилхлорида. Получают 0,95 г требуемого продукта после кристаллизации из изоэфира, точка плавления=190-191 С.B) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-гидрокси-3-(2-метоксифенил)-6-метил-1,3-дигидро 2H-индол-2-он. 0,5 мл 12 н раствора HCl добавляют к раствору 0,85 г соединения, полученного на предыдущей стадии, в 20 мл ацетона и смесь перемешивают при 20 С в течение 4 ч. Полученную смесь концентрируют под вакуумом, остаток переносят в 50 мл DCM и растворитель снова концентрируют под вакуумом. Получают 0,75 г требуемого продукта после кристаллизации из изоэфира, точка плавления=207-208 С. Г) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-6-метил-2-оксо-2,3-дигидро-1 Ниндол-3-ил-фенилкарбонат. 1 г фенилхлорформиата добавляют к раствору 0,7 г соединения, полученного на предыдущей стадии, в 10 мл пиридина и смесь перемешивают при 20 С в течение 48 ч. Полученную в результате смесь концентрируют под вакуумом, остаток экстрагируют ЕtOАс, органическую фазу промывают 1 н раствором HCl, водой, 1 н раствором NaOH и водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 0,75 г требуемого продукта после кристаллизации из изоэфира, точка плавления=196 С (с разложением). Подготовительный пример 1.16. 5-Хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-6 метокси-2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбонат. 006019 Смесь 36 г 2-хлор-5-нитроанизола и никеля Ренея в 150 мл МеОН и 200 мл THF гидрируют в аппарате Парра в течение 4 ч при 35 С и под давлением 1,3 бар (1,3 х 105 Па). Катализатор отфильтровывают на Celite и фильтрат концентрируют под вакуумом. Получают 28 г требуемого продукта и его используют без дополнительной очистки. Б) N-(4-Хлор-3-метоксифенил)-D,L-2-хлорманделамид. Смесь 28 г соединения, полученного на предыдущей стадии, и 33,13 г D,L-2-хлорминдальной кислоты в 128 мл 1,2-дихлорбензола нагревают при 230 С в течение 4 ч, удаляя образующуюся воду с использованием аппарата Дина-Старка. Реакционную смесь частично концентрируют под вакуумом и оставляют кристаллизоваться. Образовавшийся кристаллический продукт отфильтровывают путем отсасывания и промывают изоэфиром. Получают 40 г требуемого продукта. В) 5-Хлор-3-(2-хлорфенил)-6-метокси-1,3-дигидро-2H-индол-2-он. 40 г соединения, полученного на предыдущей стадии, быстро добавляют к 550 г полифосфорной кислоты и смесь затем нагревают при 60 С в течение 8 ч и оставляют перемешиваться в течение ночи,позволяя температуре достичь комнатной температуры. К реакционной смеси добавляют ледяную воду и образовавшийся осадок отфильтровывают путем отсасывания и промывают водой. Осадок переносят в ЕtOАс и полученный белый продукт после растирания отфильтровывают путем отсасывания и промывают изоэфиром. Получают 17,2 г требуемого продукта, точка плавления=243-247 С. Г) 5-Хлор-3-(2-хлорфенил)-3-гидрокси-6-метокси-1,3-дигидро-2 Н-индол-2-он. 2,56 г 60%-ного гидрида натрия в масле добавляют при комнатной температуре в атмосфере аргона к раствору 17,2 г соединения, полученного на предыдущей стадии, в 220 мл THF. После прекращения выделения газа добавляют 6,85 г диметилдисульфида, в реакционную смесь барботируют воздух и смесь перемешивают при комнатной температуре в течение 72 ч. К реакционной смеси добавляют воду, THF выпаривают под вакуумом, оставшуюся водную фазу экстрагируют ЕtOАс, органическую фазу промывают водой, насыщенным раствором NaCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Полученный продукт растворяют в DCM, растворитель частично концентрируют, смесь оставляют кристаллизоваться и образовавшийся кристаллический продукт отфильтровывают путем отсасывания. Получают 6 г требуемого продукта, точка плавления=237-240 С. Д) 5-Хлор-3-(2-хлорфенил)-6-метокси-3-[(триметилсилил)окси]-1,3-дигидро-2 Н-индол-2-он. Смесь 1 г соединения, полученного на предыдущей стадии, и 0,07 г хлорида цинка в 10 мл ацетонитрила нагревают до температуры дефлегмации, добавляют 3 мл HMDS и кипячение с обратным холодильником поддерживают в течение 1 ч. Полученную смесь концентрируют под вакуумом, остаток экстрагируют ЕtOАс, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 1,25 г требуемого продукта после кристаллизации из гептана, точка плавления=211-212 С. Е) 5-Хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-6-метокси-3-[(триметилсилил)окси]1,3-дигидро-2H-индол-2-он. Это соединение получают способом, описанным на стадии Б подготовительного примера 1.8, начиная с 1,2 г соединения, полученного на предыдущей стадии, 0,08 г 60%-ного гидрида натрия в масле, 15 млTHF и 0,8 г 2,4-диметоксибензолсульфонилхлорида. Получают 1,6 г требуемого продукта после кристаллизации из изоэфира, точка плавления=217-219 С. Ж) 5-Хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-3-гидрокси-6-метокси-1,3-дигидро 2H-индол-2-он. Смесь 1,55 г соединения, полученного на предыдущей стадии, и 2 мл 12 н HCl в 30 мл ацетона перемешивают при 20 С в течение 4 ч. Полученную в результате смесь концентрируют под вакуумом и остаток переносят в ацетон и снова концентрируют под вакуумом. Остаток переносят в DCM и концентрируют под вакуумом. Остаток растирают в изоэфире и образовавшийся осадок отфильтровывают путем отсасывания. Получают 1,3 г требуемого продукта, точка плавления=233-235 С. З) 5-Хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-6-метокси-2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбонат. Смесь 1,3 г соединения, полученного на предыдущей стадии, в 10 мл пиридина охлаждают до 10 С,добавляют 0,8 г фенилхлорформиата и смесь перемешивают при 20 С в течение 20 мин. Добавляют еще 0,7 г фенилхлорформиата и смесь перемешивают при 20 С в течение 20 ч. Полученную смесь концентрируют под вакуумом, осадок экстрагируют ЕtOАс, органическую фазу промывают 1 н раствором HCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя DCM. Получают 0,7 г требуемого продукта после кристаллизации из смеси пентан/изоэфир, точка плавления=162-163 С. Подготовительный пример 1.17. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2,5-диметоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбонат.- 25006019 Раствор бромида 2,5-диметоксифенилмагния получают из 2,2 г магния, 18 г 1-бром-2,5-диметоксибензола и 50 мл эфира. Этот раствор добавляют по каплям к смеси 5 г 5-хлор-1H-индол-2,3-диона в 50 млTHF при температуре ниже 30 С, затем кипятят с обратным холодильником в течение 3 ч. После охлаждения до комнатной температуры реакционную смесь вливают в 1 н раствор HCl и экстрагируют ЕtOАс,органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 7,1 г требуемого продукта после кристаллизации из горячего изоэфира. Б) 5-Хлор-3-(2,5-диметоксифенил)-3-[(триметилсилил)окси]-1,3-дигидро-2H-индол-2-он. Смесь 4 г соединения, полученного на предыдущей стадии, и 0,085 г хлорида цинка в 45 мл ацетонитрила нагревают до температуры дефлегмации, добавляют 2,8 мл HMDS и температуру дефлегмации поддерживают в течение 1 ч. Полученную смесь концентрируют под вакуумом, остаток переносят в воду и экстрагируют эфиром, органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 5 г требуемого продукта после кристаллизации из изоэфира.B) 5-Хлор-3-(2,5-диметоксифенил)-1-[(2,4-диметоксифенил)сульфонил]-3-[(триметилсилил)окси]-1,3 дигидро-2 Н-индол-2-он. Это соединение получают способом, описанным на стадии Б подготовительного примера 1.8, начиная с 2 г соединения, полученного на предыдущей стадии, 0,135 г 60%-ного гидрида натрия в масле, 40 млTHF и 1,3 г 2,4-диметоксибензолсульфонилхлорида. Получают 2,2 г требуемого продукта после кристаллизации из изоэфира. Г) 5-Хлор-3-(2,5-диметоксифенил)-1-[(2,4-диметоксифенил)сульфонил]-3-гидрокси-1,3-дигидро-2H-индол-2-он. Смесь 1 г соединения, полученного на предыдущей стадии, и 0,5 мл 12 н HCl в 20 мл ацетона перемешивают при комнатной температуре в течение 4 ч. Смесь концентрируют под вакуумом и остаток переносят в DCM и снова концентрируют под вакуумом. Остаток растирают в изоэфире и образовавшийся осадок отфильтровывают путем отсасывания. Получают 0,84 г требуемого продукта. Д) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2,5-диметоксифенил)-2-оксо-2,3-дигидро-1 Н-индол 3-ил-фенилкарбонат. Смесь 0,8 г соединения, полученного на предыдущей стадии, и 0,4 мл фенилхлорформиата в 5 мл пиридина перемешивают при комнатной температуре в течение ночи. К реакционной смеси добавляют воду, полученную смесь экстрагируют DCM, органическую фазу промывают 2 н раствором HCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 0,84 г требуемого продукта после кристаллизации из изоэфира. Подготовительный пример 1.18. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-2 оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбамат.. А) 3-Амино-5-хлор-3-(2-метоксифенил)-1,3-дигидро-2 Н-индол-2-он. Это соединение получают способом, описанным в WO 95/18105, в подготовительном примере 7. Б) 3-Амино-5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-1,3-дигидро-1 Н-индол 2-он. Раствор 1,64 г соединения, полученного на предыдущей стадии, в 20 мл DMF охлаждают до 4 С,добавляют 0,25 г 60%-ного гидрида натрия в масле и смесь перемешивают при 4 С в течение 30 мин. Затем добавляют 1,34 г 2,4-диметоксибензолсульфонилхлорида и смесь перемешивают при комнатной температуре в течение 4 ч. К реакционной смеси добавляют 50 мл воды, полученную в результате смесь экстрагируют EtOAc, органическую фазу промывают насыщенным раствором NaCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесью DCM/EtOAc (97/3; об./об.). Получают 2 г требуемого продукта после кристаллизации из смесиDCM/изоэфир. В) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1 Н-индол 3-ил-фенилкарбамат. Раствор 2 г соединения, полученного на предыдущей стадии, и 10 мл пиридина в 10 мл DCM охлаждают до 4 С, добавляют 0,77 мл фенилхлорформиата и смесь перемешивают при комнатной температуре в течение 1 ч. Полученную в результате смесь концентрируют под вакуумом, остаток экстрагируютEtOAc, органическую фазу промывают насыщенным раствором NaCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесьюDCM/EtOAc (96/4; об./об.). Получают 2,6 г требуемого продукта после кристаллизации из смеси 006019 Смесь 9 г соединения, полученного на стадии А подготовительного примера 1.1, и 3,74 мл пиридина в 100 мл DCM охлаждают до 0 С, добавляют по каплям в течение 3 мин раствор 3,45 мл тионилхлорида в 3 мл DCM и смесь перемешивают в течение 30 мин. К реакционной смеси добавляют воду и DCM выпаривают под вакуумом. Образовавшийся осадок отфильтровывают путем отсасывания, промывают 4 раза водой и затем холодным изоэфиром и сушат. Получают 8,8 г требуемого продукта. Б) 5-Хлор-3-(1S)-2-гидрокси-1-фенилэтил]амино]-3-(2-метоксифенил)-1,3-дигидро-2H-индол-2-он,изомеры А и Б. Смесь 7 г соединения, полученного на предыдущей стадии, и 7,65 г (S)-(+)фенилглицинола в 100 мл хлороформа перемешивают при комнатной температуре в течение 2 ч. Затем добавляют 2,9 г DIPEA и смесь перемешивают при комнатной температуре в течение 48 ч. Образовавшийся осадок отфильтровывают путем отсасывания и собирают изомер А. Фильтрационные жидкости промывают 5%-ным раствором К 2 СО 3, органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесью DCM/EtOAc (70/30; об./об.). Разделяют два диастереоизомера: менее полярный изомер А: получают 4,55 г (осаждают и подвергают хроматографии),D20=+193 (с=0,16; хлороформ); более полярный изомер Б.B) 3-Амино-5-хлор-3-(2-метоксифенил)-1,3-дигидро-2 Н-индол-2-он, правовращающий изомер. Смесь 4,55 г соединения, полученного на предыдущей стадии (изомер А), и 5,5 г тетраацетата свинца в 75 мл DCM и 35 мл МеОН перемешивают при комнатной температуре в течение 1 ч 30 мин. Смесь концентрируют под вакуумом, остаток переносят в насыщенный раствор NаНСО 3 и экстрагируют ЕtOАс,органическую фазу промывают водой и насыщенным раствором NaCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Полученное масло переносят в 100 мл 3 н раствора HCl, добавляют 10 мл МеОН и смесь перемешивают при комнатной температуре в течение 2 ч. Органические растворители концентрируют под вакуумом, водную фазу промывают дважды эфиром, подщелачивают путем добавления гранул КОН и экстрагируют ЕtOАс, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 1,178 г требуемого продукта после кристаллизации из смеси DСМ/изоэфир/ТНF, точка плавления=202 С.D20=+83,3 (с=0,16; хлороформ). Г) 3-Амино-5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-1,3-дигидро-2H-индол 2-он, правовращающий изомер. Раствор 1,178 г соединения, полученного на предыдущей стадии, в 10 мл DMF охлаждают до 0 С,добавляют 0,188 г 60%-ного гидрида натрия в масле в атмосфере аргона и смесь перемешивают до прекращения выделения газа. Затем добавляют 1,02 г 2,4-диметоксибензолсульфонилхлорида и смесь перемешивают при комнатной температуре в течение 3 ч. Реакционную смесь вливают в 5%-ный раствор К 2 СО 3 и экстрагируют ЕtOАс, органическую фазу промывают водой, 5%-ным раствором K2 СО 3 и насыщенным раствором NaCl и сушат над Nа 2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесью DCM/ЕtOАс (95/5; об./об.). Получают 1,254 г требуемого продукта после кристаллизации из смеси DCM/эфир/изоэфир, точка плавления=172-173 С.D20=+113 (с=0,18; хлороформ). Д) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-2-оксо-2,3-дигидро-1 Н-индол 3-ил-фенилкарбамат, индивидуальный изомер. Раствор 1,1 г соединения, полученного на предыдущей стадии, в 10 мл DCM охлаждают в ледяной бане, добавляют 1,8 мл пиридина, затем по каплям добавляют 0,37 мл фенилхлорформиата и смесь оставляют в холодных условиях на ночь. Реакционную смесь разбавляют DCM, органическую фазу промывают 3 раза водой, сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесь DCM/ЕtOАс (98/2; об./об.). Получают 1,136 г требуемого продукта. Подготовительный пример 1.20. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1 Н-индол-3 З-ил-фенилкарбамат, индивидуальный изомер.. А) 3,5-Дихлор-3-(2-изопропоксифенил)-1,3-дигидро-2 Н-индол-2-он. Смесь 12 г соединения, полученного на стадии Б подготовительного примера 1.3, и 8,35 мл пиридина в 165 мл DCM охлаждают до 0 С, добавляют по каплям 7,62 мл тионилхлорида и смесь перемешивают в течение 15 мин. Полученную смесь концентрируют под вакуумом, остаток экстрагируют ЕtOАс,органическую фазу промывают водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесью DCM/EtOAc (90/10; об./об.). Получают 15,22 г требуемого продукта. Б) 5-Хлор-3-(1S)-2-гидрокси-1-фенилэтил]амино]-3-(2-изопропоксифенил)-1,3-дигидро-2 Н-индол 2-он, изомер А и изомер Б.- 27006019 Смесь 8,17 г соединения, полученного на предыдущей стадии, и 3,66 г (S)-(+)фенилглицинола в 265 мл хлороформа перемешивают при комнатной температуре в течение 1 ч. Затем добавляют 4,66 млDIPEA, смесь перемешивают при комнатной температуре в течение 3 ч и затем нагревают при 50 С в течение 18 ч. Полученную в результате смесь концентрируют под вакуумом, осадок экстрагируют ЕtOАс, органическую фазу промывают 5%-ным раствором K2 СО 3 и водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесьюDCM/EtOAc (90/10; об./об.), и отделяют менее полярный изомер, изомер А: получают 4,98 г после кристаллизации из изоэфира, точка плавления=212,7 С.D20=+212,4 (с=0,2; хлороформ). Хроматографию продолжают, элюируя смесью DCM/EtOAc (70/30; об./об.) и отделяют более полярный изомер, изомер Б: получают 3,49 г после кристаллизации из изоэфира, точка плавления=241,3 С.D20=-6,6 (с=0,2; хлороформ). В) 3-Амино-5-хлор-3-(2-изопропоксифенил)-1,3-дигидро-2 Н-индол-2-он, правовращающий изомер. Раствор 3,9 г соединения, полученного на предыдущей стадии (изомер А), и 75 мл DCM и 37 мл МеОН охлаждают до 0 С, добавляют 4,35 г тетраацетата свинца и смесь перемешивают в течение 2 ч. Добавляют три капли 5%-ного раствора NаНСО 3, смесь концентрируют под вакуумом, остаток экстрагируют ЕtOАс, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Полученный продукт переносят в 120 мл концентрированнойHCl и промывают эфиром, водную фазу подщелачивают путем добавления К 2 СО 3, экстрагируют ЕtOАс и оставляют кристаллизоваться. Образовавшиеся кристаллы отфильтровывают путем отсасывания и сушат. Фильтрационные жидкости концентрируют под вакуумом, остаток подвергают хроматографии на силикагеле, элюируя смесь DCM/EtOAc (65/35; об./об.), и полученный продукт кристаллизуют из изоэфира. Получают всего 2,03 г требуемого продукта, точка плавления=193 С.D20=+44 (с=0,18; хлороформ). Г) 3-Амино-5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-1,3-дигидро-2 Н-индол-2-он, правовращающий изомер. Это соединение получают способом, описанным на стадии Г подготовительного примера 1.19, начиная с 1,86 г соединения, полученного на предыдущей стадии, 0,282 г 60%-ного гидрида натрия в масле,20 мл DMF и 1,395 г 2,4-диметоксибензолсульфонилхлорида. Получают 2,68 г требуемого продукта после кристаллизации из гексана.D20=+79,5 (с=0,2; хлороформ). Д) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбамат, индивидуальный изомер. Смесь 2,68 г соединения, полученного на предыдущей стадии, и 4,5 мл пиридина в 25 мл DCM охлаждают до 0 С, добавляют в течение 30 с 1,06 г фенилхлорформиата и смесь оставляют в холодных условиях в течение 18 ч. Реакционную смесь подвергают хроматографии непосредственно на силикагеле в смеси DCM/гексан (80/20; об./об.) и элюируют смесью DCM/EtOAc (95/5; об./об.). Получают 2,88 г требуемого продукта и его используют без дополнительной очистки. Подготовительный пример 1.21. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбамат, индивидуальный изомер.. А) 3-Амино-5-хлор-3-(2-изопропоксифенил)-1,3-дигидро-2H-индол-2-он, левовращающий изомер. Раствор 3,4 г соединения, полученного на стадии Б подготовительного примера 1.20 (изомер Б) в 65 млDCM и 32,4 мл МеОН охлаждают до 0 С, добавляют 3,8 г тетраацетата свинца и смесь перемешивают в течение 40 мин. Добавляют три капли 5%-ного раствора NaHCO3, смесь концентрируют под вакуумом,остаток экстрагируют ЕtOАс, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Полученный продукт переносят в 120 мл 0,5 н раствора HCl и промывают эфиром, водную фазу подщелачивают путем добавления К 2 СО 3 и образовавшийся осадок отфильтровывают путем отсасывания. Получают 1,9 г требуемого продукта после кристаллизации из изоэфира, точка плавления=192 С.D20=-42 (с=0,2; хлороформ). Б) 3-Амино-5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-изопропоксифенил)-1,3-дигидро-2H-индол-2-он, левовращающий изомер. Это соединение получали способом, описанным на стадии Г подготовительного примера 1.19, начиная с 1,67 г соединения, полученного на предыдущей стадии, 0,253 г 60%-ного гидрида натрия в масле,20 мл DMF и 1,249 г 2,4-диметоксибензолсульфонилхлорида. Получают 2,453 г требуемого продукта после кристаллизации из гексана.- 28006019 Смесь 2,103 г соединения, полученного на предыдущей стадии, и 3,5 мл пиридина в 20 мл DCM охлаждают до 0 С, добавляют 0,665 мл фенилхлорформиата и смесь оставляют в холодных условиях на 36 ч. Полученную в результате смесь концентрируют под вакуумом, остаток переносят в 5%-ный раствор К 2 СО 3 и экстрагируют ЕtOАс, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над Nа 2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесью DCM/гексан (80/20; об./об.). Получают 1,88 г требуемого продукта и его используют без дополнительной очистки. Подготовительный пример 1.22. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2,5-диметоксифенил)2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбамат.(II): R1=Cl; R2=Н; R3=ОСН 3; R4=5-ОСН 3, Х=-NH-; Y= А) 3,5-Дихлор-3-(2,5-диметоксифенил)-1,3-дигидро-1 Н-индол-2-он. Смесь 3 г соединения, полученного на стадии А подготовительного примера 1.17, и 1,2 мл пиридина в 50 мл DCM охлаждают до температуры ниже 20 С, добавляют 0,8 мл тионилхлорида и смесь перемешивают в течение 1 ч. Реакционную смесь промывают водой, органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируяDCM. Получают 1,9 требуемого продукта и его используют без дополнительной очистки. Б) 3-Амино-5-хлор-3-(2,5-диметоксифенил)-1,3-дигидро-2H-индол-2-он. Смесь 1,25 г соединения, полученного на предыдущей стадии, в 7 мл THF охлаждают до 0 С, барботируют газообразный аммиак 4 раза по 10 мин в течение 6 ч и смесь перемешивают при комнатной температуре в течение 24 ч. Полученную в результате смесь концентрируют под вакуумом, остаток переносят в DCM и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя DCM и затем смесью DCM/EtOAC (60/40; об./об.). Получают 0,808 г требуемого продукта и его используют без дополнительной очистки. В) 3-Амино-5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2,5-диметоксифенил)-1,3-дигидро-2H-индол-2-он. Это соединение получают способом, описанным на стадии Г подготовительного примера 1.19, начиная с 0,749 г соединения, полученного на предыдущей стадии, 0,113 г 60%-ного гидрида натрия в масле, 7 мл DMF и 0,612 г 2,4-диметоксибензолсульфонилхлорида. Получают 0,9 г требуемого продукта после кристаллизации из смеси DCM/изоэфир, точка плавления=191 С. Г) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2,5-диметоксифенил)-2-оксо-2,3-дигидро-1 Н-индол 3-ил-фенилкарбамат. Смесь 0,52 г соединения, полученного на предыдущей стадии, и 1 мл пиридина в 5 мл DCM охлаждают до 0 С, добавляют 0,16 мл фенилхлорформиата и смесь перемешивают в течение 16 ч. К реакционной смеси добавляют воду, DCM выпаривают под вакуумом, остаток экстрагируют ЕtOАс, органическую фазу промывают 5%-ным раствором К 2 СО 3, водой и насыщенным раствором NaCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируяDCM. Получают 0,46 г требуемого продукта и его используют без дополнительной очистки. Подготовительный пример 1.23. 5-Хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-2-оксо 2,3-дигидро-1H-индол-3-ил-фенилкарбамат.. А) 3-Амино-5-хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-1,3-дигидро-2 Н-индол-2-он. Это соединение получают способом, описанным в WO 95/18105 в примере 3. Б) 5-Хлор-3-(2-хлорфенил)-1-[(2,4-диметоксифенил)сульфонил]-2-оксо-2,3-дигидро-1 Н-индол-3-илфенилкарбамат. Раствор 3 г соединения, полученного на предыдущей стадии, в 25 мл пиридина охлаждают до 0 С,по каплям добавляют раствор 1,25 г фенилхлорформиата в 2 мл DCM и смесь перемешивают при 0 С в течение 3 ч и затем при комнатной температуре в течение ночи. Смесь снова охлаждают до 0 С, добавляют 0,96 г фенилхлорформиата и смесь оставляют в холодных условиях на 18 ч. Полученную в результате смесь концентрируют под вакуумом, остаток переносят в 5%-ный раствор К 2 СО 3 и экстрагируют ЕtOАс, органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесью DCM/EtOAc (95/5; об./об.). Получают 2,88 г требуемого продукта. Подготовительный пример 1.24. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-6 метил-2-оксо-2,3-дигидро-1 Н-индол-3-ил-фенилкарбамат.A) 3,5-Дихлор-3-(2-метоксифенил)-6-метил-1,3-дигидро-2 Н-индол-2-он. Смесь 2,0 г соединения, полученного на стадии Б подготовительного примера 1.6, в 45 мл DCM охлаждают до 0 С, добавляют 0,77 мл пиридина, затем 1,17 г тионилхлорида и смесь перемешивают в- 29006019 течение 2 ч, затем позволяют температуре достичь комнатной температуры. К реакционной смеси добавляют воду и DCM и после разделения фаз отстаиванием органическую фазу промывают 4 раза водой и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают требуемый продукт и его используют без дополнительной очистки. Б) 3-Амино-5-хлор-3-(2-метоксифенил)-6-метил-1,3-дигидро-2 Н-индол-2-он. Газообразный аммиак барботируют в течение 30 мин при комнатной температуре в смесь 3,4 г соединения, полученного на предыдущей стадии, в 25 мл DCM и полученную в результате смесь перемешивают в течение 18 ч. Смесь концентрируют под вакуумом, остаток экстрагируют ЕtOАс, органическую фазу промывают насыщенным раствором NaCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя смесью DCM/MeOH (95/5; об./об.). Получают 2 г требуемого продукта.B) 3-Амино-5-хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-6-метил-1,3-дигидро-2Hиндол-2-он. Это соединение получают способом, описанным на стадии Б подготовительного примера 1.18, начиная с 2 г соединения, полученного на предыдущей стадии, 0,29 г 60%-ного гидрида натрия в масле, 15 млDMF и 1,54 г 2,4-диметоксибензолсульфонилхлорида. Получают 3,2 г требуемого продукта. Г) 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-6-метил-2-оксо-2,3-дигидро-1Hиндол-3-ил-фенилкарбамат. Раствор 3,2 г соединения, полученного на предыдущей стадии, и 10 мл пиридина в 30 мл DCM охлаждают до 4 С, по каплям добавляют 1,2 мл фенилхлорформиата и смесь перемешивают при комнатной температуре в течение 2 ч. Полученную в результате смесь концентрируют под вакуумом, остаток экстрагируют ЕtOАс, органическую фазу промывают 5%-ным раствором KHSO4, 5%-ным растворомNa2CO3, водой и насыщенным раствором NaCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. 2,3 г требуемого продукта получают после кристаллизации из смеси DCM/изоэфир. Подготовительный пример 1.25. 5-Хлор-1-[(2,4-диметоксифенил)сульфонил]-3-(2-метоксифенил)-6 трифторметил-2-оксо-2,3-дигидро-1H-индол-3-ил-фенилкарбамат, индивидуальный изомер.. А) 5-Хлор-3-гидрокси-3-(2-метоксифенил)-6-трифторметил-1,3-дигидро-2 Н-индол-2-он. а) трет-Бутил-4-хлор-3-трифторметилфенилкарбамат. Это соединение получают способом, описанным на стадии Б а) подготовительного примера 1.6, начиная с 4-хлор-3-трифторметиланилина и ди-трет-бутилдикарбоната в диоксане. Требуемый продукт получают в виде масла, которое затвердевает, точка плавления=90 С. б) Раствор 4 г трет-бутил-4-хлор-3-трифторметилфенилкарбамата в 30 мл эфира охлаждают до -70 С в атмосфере аргона, добавляют по каплям 22 мл 1,5 М раствора тpeт-бутиллития в пентане и смесь перемешивают в течение 1 ч, позволяя температуре достичь -10 С, и перемешивают в течение 2 ч 30 мин при-10 С. Реакционную смесь охлаждают до -70 С, добавляют по каплям раствор 3,05 г соединения, полученного на стадии А подготовительного примера 1.6, в 15 мл THF и смесь перемешивают в течение 1 ч,позволяя температуре достичь -30 С, и затем в течение 16 ч, позволяя температуре достичь комнатной температуры. К реакционной смеси добавляют насыщенный раствор NH4Cl эфир и THF выпаривают,полученную в результате водную фазу экстрагируют ЕtOАс, органическую фазу промывают водой и насыщенным раствором NaCl и сушат над Na2SO4 и растворитель выпаривают под вакуумом. Остаток подвергают хроматографии на силикагеле, элюируя DCM и затем смесью DCM/EtOAc (90/10; об./об.). Получают 1,48 г требуемого продукта после кристаллизации из смеси изоэфир/гексан, т.пл. 230-231 С. Б) 3,5-Дихлор-3-(2-метоксифенил)-6-трифторметил-1,3-дигидро-2H-индол-2-он. Суспензию 1,3 г соединения, полученного на стадии А, в 8 мл DCM охлаждают до 0 С, добавляют 0,43 мл пиридина, затем добавляют 0,4 мл тионилхлорида и смесь перемешивают в течение 15 мин. Реакционную смесь промывают 3 раза водой, органическую фазу сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 1,2 г требуемого продукта и его используют без дополнительной очистки. В) 5-Хлор-3-(1S)-2-гидрокси-1-фенилэтил]амино]-3-(2-метоксифенил)-6-трифторметил-1,3-дигидро-2H-индол-2-он, изомер А и изомер Б. Смесь 1,29 г соединения, полученного на предыдущей стадии, и 0,47 г (S)-(+)фенилглицинола в 35 мл хлороформа перемешивают при комнатной температуре в течение 4 ч. Затем добавляют 0,6 мл DIPEA, растворитель концентрируют наполовину и смесь перемешивают при комнатной температуре в течение 22 ч. Реакционную смесь концентрируют под вакуумом и остаток подвергают хроматографии на силикагеле, элюируя смесью DCM/EtOAc (85/15; об./об.). Один диастереоизомер отделяют: менее полярный изомер А: 0,712 г получают после кристаллизации из смеси DCM/изоэфир, точка плавления=205 С.
МПК / Метки
МПК: A61K 31/496, C07D 401/12, A61P 13/00
Метки: терапевтическое, 3-дигидро-2н-индол-2-она, получение, применение, 1-фенилсульфонил-1, производные
Код ссылки
<a href="https://eas.patents.su/30-6019-proizvodnye-1-fenilsulfonil-1-3-digidro-2n-indol-2-ona-ih-poluchenie-i-ih-terapevticheskoe-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 1-фенилсульфонил-1, 3-дигидро-2н-индол-2-она, их получение и их терапевтическое применение</a>
Производные бензимидазола, их получение и терапевтическое применение

Номер патента: 5950
Опубликовано: 25.08.2005
Авторы: Бишон Даниэль, Болкениус Франк, Ван Дорсселер Вивиан, Барт Франсис
МПК: C07D 471/06, A61K 31/4375, A61P 9/00...
Метки: производные, получение, бензимидазола, применение, терапевтическое
Формула / Реферат:
1. Соединения, соответствующие формуле (I) в которой R1 представляет собой атом водорода, группу C1-C4алкил, атом галогена, нитрогруппу или группу C1-C4алкокси, R2 и R2' независимо друг от друга представляют собой атом водорода или группу C1-C4алкил, X представляет собой атом азота или атом углерода, n равно 1 или 2, m равно 1 или 2 и, когда X представляет собой атом азота, R3 представляет собой атом водорода или группу C1-C4алкил, давая...
Новые производные 1,3-дигидро-2н-индол-2-она и их применение в качестве лигандов рецепторов аргинин-вазопрессина v1b и v1a

Номер патента: 4628
Опубликовано: 24.06.2004
Авторы: Ру Ришар, Тоннерр Бернар, Серрадель-Ле Галь Клодин, Ваньон Жан
МПК: A61K 31/404, A61P 43/00, C07D 403/04...
Метки: аргинин-вазопрессина, новые, применение, рецепторов, 1,3-дигидро-2н-индол-2-она, производные, лигандов, качестве
Формула / Реферат:
1. Соединение формулы где R1 представляет собой атом галогена; (C1-C4)алкил; (C1-C4)алкокси; трифторметильный радикал; трифторметокси радикал; R2 представляет собой атом водорода; атом галогена; (C1-C4)алкил; (C1-C4)алкокси; трифторметильный радикал; или R2 находится в положении -6-индол-2-онового кольца и R1 и R2 вместе представляют собой двухвалентный триметиленовый радикал; R3 представляет собой атом галогена; гидроксил; (C1-C2)алкил;...
Производные бензофурана, их получение и применение

Номер патента: 3781
Опубликовано: 28.08.2003
Авторы: Руланд Томас, Педерсен Хенрик, Роттлендер Марио, Дансер Роберт, Андерсен Ким, Бегесе Клаус Петер
МПК: A61K 31/343, C07D 307/87
Метки: производные, применение, бензофурана, получение
Формула / Реферат:
1. Изобензофуран общей формулы I где R1 представляет собой водород, галоген, трифторметил, трифторметилсульфонилокси, C1-6-алкил, C2-6-алкенил, C2-6-алкинил, C3-8-циклоалкил, C1-6-алкокси, гидрокси, формил, ацил, амино, C1-6-алкиламино, C2-12-диалкиламино, ациламино, C1-6-алкоксикарбониламино, аминокарбониламино, C1-6-алкиламинокарбониламино, C2-12-диалкиламинокарбониламино, нитро, циано, COOH или COO-C1-6-алкил; R2 и R3, каждый независимо,...
Производные 4-, 5-, 6- и 7-замещенного индола и индолина, их получение и применение

Номер патента: 4248
Опубликовано: 26.02.2004
Авторы: Мольтсен Эйнер Кнуд, Крог-Енсен Христиан, Миккельсен Иван
МПК: A61K 31/495, A61P 25/24, C07D 405/14...
Метки: производные, индола, применение, получение, индолина, 7-замещенного
Формула / Реферат:
1. Производное 4-, 5-, 6- или 7-замещенного индола или индолина формулы где W представляет собой N, C, CH или COH и пунктирные линии обозначают необязательные связи, и A представляет собой группу формулы где пунктирная линия обозначает связь; n равно 0, 1, 2, 3, 4 или 5 и m равно 0, 1, 2, 3, 4 или 5; Z представляет собой CH2, O, S, CO, SO или SO2, при условии, что если n равно 0, то Z представляет собой CH2; R3-R9 и R11-R12 независимо...
Аминированные производные дигидро-1,3,5-триазина и их применение в терапии

Номер патента: 5378
Опубликовано: 24.02.2005
Авторы: Доар Лилиан, Муане Жерар, Кергоа Мишлин, Краво Даниель, Мезанжо Дидье
МПК: C07D 251/10, A61P 3/10, A61K 31/53...
Метки: применение, аминированные, дигидро-1,3,5-триазина, терапии, производные
Формула / Реферат:
1. Соединение общей формулы (I) где R1, R2, R3 и R4 выбираются независимо друг от друга из групп H, (C1-C20)алкил, замещенный или не замещенный галогеном, (C1-C5)алкилом, (C1-C5)алкоксилом, (C3-C8)циклоалкилом, (C2-C20)алкенил, замещенный или не замещенный галогеном, (C1-C5)алкилом, (C1-C5)алкоксилом, (C2-C20)алкинил, замещенный или не замещенный галогеном, (C1-C5)алкилом, алкоксилом, (C3-C8)циклоалкил, замещенный или не замещенный...