Ингибиторы ns5a вгс










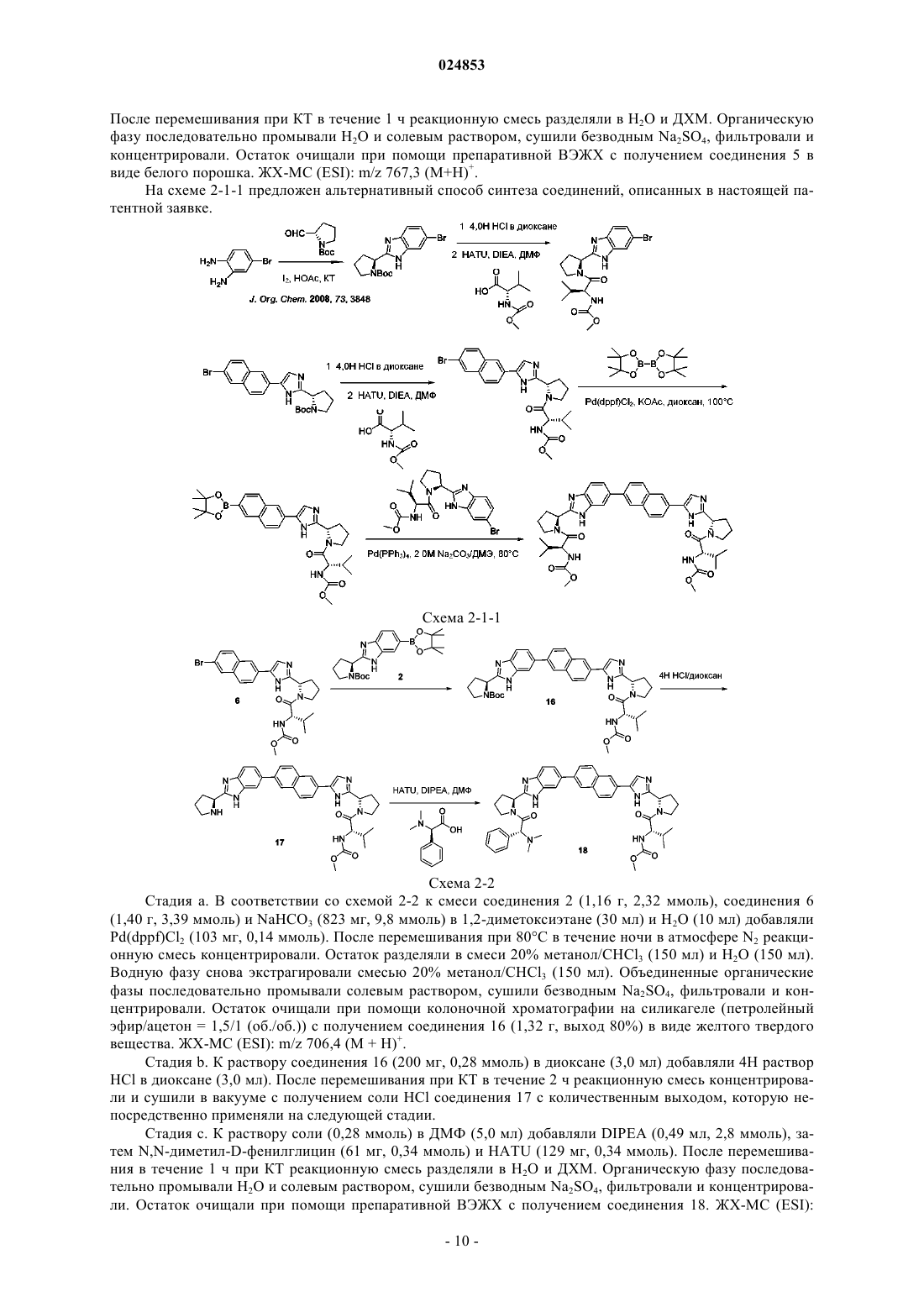



















Формула / Реферат
1. Соединение формулы IIIe

где каждый из X и X' независимо выбран из связи, -CH2-, -CH2-CH2- и -CH2O-;
Xb независимо представляет собой С или N, при условии, что не более двух Xb одновременно представляют собой N;
Xc независимо представляет собой С или N, при условии, что не более двух Xc одновременно представляют собой N;
Z и Z' независимо выбраны из остатка, состоящего из 1-3 аминокислот, -[U-(CR42)t-NR5-C(R42)t]u-U-(CR42)t-NR7-(CR42)t-R8, -U-(CR42)t-R8 и -[U-(CR42)t-NR5-(CR42)t]u-U-(CR42)t-O-(CR42)t-R8, где
U представляет собой -С(О)-,
каждый R4 независимо выбран из водорода, C1-C8 алкила, возможно замещенного ОН, C1-C8 гетероалкила, содержащего атом кислорода, С3-С12 циклоалкила, гетероцикла и С6-С10 арила,
каждый R5 и R7 независимо выбран из водорода и C1-C8 алкила,
R8 выбран из водорода, C1-C8 алкила, гетероцикла и С6-С10 арила,
или R7 и R8 совместно образуют 4-7-членное кольцо, возможно замещенное ОН,
каждый t независимо равен 0 или 1 и
u равен 0 или 1,
где гетероцикл означает ароматический или неароматический циклический радикал, содержащий по меньшей мере один гетероатом в качестве элемента кольца.
2. Соединение по п.1, отличающееся тем, что Z и Z' каждый представляет собой остаток, состоящий из 1-3 аминокислот.
3. Соединение по п.2, отличающееся тем, что указанные аминокислоты находятся в D-конфигурации.
4. Соединение по п.1, отличающееся тем, что Z и Z', каждый независимо, выбран из -[U-(CR42)t-NR5-(CR42)t]u-U-(CR42)t-NR7-(CR42)t-R8, -U-(CR42)t-R8 и -[U-(CR42)t-NR5-(CR42)t]u-U-(CR42)t-O-(CR42)t-R8.
5. Соединение по п.4, отличающееся тем, что один или оба Z и Z' представляют собой -[U-(CR42)t-NR5-(CR42)t]u-U-(CR42)t-NR7-(CR42)t-R8.
6. Соединение по п.5, отличающееся тем, что один или оба Z и Z' представляют собой -U-(CR42)t-NR5-(CR42)t-U-(CR42)t-NR7-(CR42)t-R8.
7. Соединение по п.5, отличающееся тем, что один или оба Z и Z' представляют собой -U-(CR42)t-NR7-(CR42)t-R8.
8. Соединение по п.4, отличающееся тем, что один или оба Z и Z' представляют собой -U-(CR42)t-R8.
9. Соединение по п.4, отличающееся тем, что один или оба Z и Z' представляют собой -[U-(CR42)t-NR5-(CR42)t]u-U-(CR42)t-O-(CR42)t-R8.
10. Соединение по п.9, отличающееся тем, что один или оба Z и Z' представляют собой -U-(CR42)t-NR5-(CR42)t-U-(CR42)t-O-(CR42)t-R8.
11. Соединение по п.9, отличающееся тем, что один или оба Z и Z' представляют собой -U-(CR42)t-O-(CR42)t-R8.
12. Соединение, выбранное из


13. Фармацевтическая композиция для лечения гепатита С, содержащая любое из соединений по пп.1-12.
14. Применение соединения по любому из пп.1-12 для получения лекарственного средства для лечения гепатита С.
15. Способ лечения гепатита С, включающий введение субъекту терапевтически эффективного количества любого из соединений по пп.1-12.
Текст
Предложены соединения формулы IIIe фармацевтические композиции и виды комбинированной терапии для подавления гепатита С. Указание родственных заявок Настоящая заявка испрашивает приоритет на основании предварительных заявок на патент США: 61/119723, поданной 3 декабря 2008 г.; 61/173590 и 61/214881, поданных 28 апреля 2009 г.; и 61/182958 и 61/182952, поданных 1 июня 2009 г. Область техники Настоящее изобретение относится к соединениям, подходящим для ингибирования репликации вируса гепатита С (ВГС), в частности функций неструктурного белка 5 А (NS5A) ВГС. Уровень техники ВГС представляет собой вирус с одноцепочечной РНК, который является членом семейства Flaviviridae. Данный вирус демонстрирует широкую генетическую гетерогенность, так как в настоящее время существует семь идентифицированных генотипов и более 50 идентифицированных подтипов. В инфицированных ВГС клетках вирусная РНК транслируется в полипротеин, который расщепляется на десять индивидуальных белков. На аминоконце находятся структурные белки: белок ядра (С) и гликопротеины оболочки, Е 1 и Е 2. За Е 1 и Е 2 следует р 7, интегральный белок мембраны. Кроме того, существует шесть неструктурных белков, NS2, NS3, NS4A, NS4B, NS5A и NS5B, которые играют функциональную роль в жизненном цикле ВГС (см., например, Lindenbach, B.D. and C.M. Rice, Nature. 436:933-938, 2005). Инфицирование ВГС представляет собой серьезную проблему в области здравоохранения. Согласно оценкам, 170 миллионов человек во всем мире хронически инфицированы ВГС. Инфекция ВГС может приводить к хроническому гепатиту, циррозу, печеночной недостаточности и гепатоцеллюлярной карциноме. Таким образом, хроническая инфекция ВГС во всем мире представляет собой основную причину преждевременной смертности, связанной заболеваниями печени. Существующий стандарт схемы лечения инфекции ВГС включает применение интерферона-альфа отдельно или в комбинации с рибавирином (ribavirin). Указанное лечение является тяжелым и иногда обладает изнурительными и тяжелыми побочными эффектами, и многие пациенты не проявляют долговременного ответа на лечение. Существует неотложная потребность в новых и эффективных способах лечения инфекции ВГС. Краткое описание изобретения Основные характеристики белка NS5A ВГС делают его идеальной мишенью для ингибиторов. В настоящем описании предложен класс соединений, действие которых нацелено на белок NS5A, и способы их применения для лечения инфекции ВГС у людей. В соответствии с первым вариантом реализации предложены соединения формулы IIIe где каждый из X и X' независимо выбран из связи, -CH2-, -CH2-CH2- и -CH2O-;Xb независимо представляет собой С или N, при условии, что не более двух Xb одновременно представляют собой N;Xc независимо представляет собой С или N, при условии, что не более двух Xc одновременно представляю собой N;Z и Z' независимо выбраны из остатка, состоящего из 1-3 аминокислот, -[U-(CR42)t-NR5-C(R42)t]u-U4U представляет собой -С(О)-,каждый R4 независимо выбран из водорода, C1-C8 алкила, возможно замещенного ОН, C1-C8 гетероалкила, содержащего атом кислорода, С 3-С 12 циклоалкила, гетероцикла, С 6-С 10 арила,каждый R5 и R7 независимо выбран из водорода и C1-C8 алкила,R8 выбран из водорода, C1-C8 алкила, гетероцикла, С 6-С 10 арила,или R7 и R8 совместно образуют 4-7-членное кольцо, возможно замещенное ОН,каждый t независимо равен 0 или 1 иu равен 0 или 1,где гетероцикл означает ароматический или неароматический циклический радикал, содержащий по меньшей мере один гетероатом в качестве элемента кольца. В другом варианте реализации Z и Z' каждый представляет собой остаток, состоящий из 1-3 аминокислоты. В другом варианте реализации указанные аминокислоты находятся в D-конфигурации. В другом варианте реализации Z и Z' каждый независимо выбран из -[U-(CR42)t-NR5-(CR42)t]u-U4-1 024853 В другом варианте реализации один или оба Z и Z' представляют собой -[U-(CR42)t-NR5-(CR42)t]u-U В шестом варианте реализации один или оба Z и Z' представляют собой -U-(CR42)t-NR5-(CR42)t-U В другом варианте реализации один или оба Z и Z' представляют собой -U-(CR42)t-NR7-(CR42)t-R8. В другом варианте реализации один или оба Z и Z' представляют собой -U-(CR42)t-R8. В другом варианте реализации один или оба Z и Z' представляют собой -[U-(CR42)t-NR5-(CR42)t]u-U4(CR 2)t-O-(CR42)t-R8. В другом варианте реализации один или оба Z и Z' представляют собой -U-(CR42)t-NR5-(CR42)t-U4(CR 2)t-O-(CR42)t-R8. В другом варианте реализации один или оба Z и Z' представляют собой -U-(CR42)t-O-(CR42)t-R8. В другом варианте реализации предложено соединение, выбранное из В другом варианте реализации предложена фармацевтическая композиция для лечения гепатита С,содержащая любое из соединений согласно настоящему изобретению. В другом первом варианте реализации предложено применение любого из соединений согласно настоящему изобретению для получения лекарственного средства для лечения гепатита С. В другом втором варианте реализации предложен способ лечения гепатита С, включающий введение субъекту терапевтически эффективного количества любого из соединений согласно настоящему изобретению. Подробное описание Если не указано иное, следующие термины, используемые в данной заявке, включая описание и формулу изобретения, имеют определения, приведенные ниже. Следует отметить, что в описании и прилагаемой формуле изобретения формы единственного числа включают множества объектов, если из контекста явным образом не следует иное. Определение стандартных химических терминов можно найти в справочных изданиях, включая Carey and Sundberg (2007) "Advanced Organic Chemistry 5th Ed". Vols. Aand B, Springer Science+Business Media LLC, New York. При реализации настоящего изобретения будут использованы, если не указано иное, традиционные методы синтетической органической химии, массспектроскопии, препаративные и аналитические методы хроматографии, химии белков, биохимии, технологии рекомбинантных ДНК и фармакологии. В настоящем описании термин алкил означает содержащие заместители или не содержащие заместители линейные и разветвленные алкильные радикалы, содержащие от одного до пятнадцати атомов углерода. В настоящем описании термин низший алкил означает как линейные, так и разветвленные-2 024853 алкильные радикалы, содержащие от одного до шести атомов углерода, и включает метил, этил, пропил,изопропил, бутил, изобутил, трет-бутил и т.д. Алкильная группа может содержать один или более заместителей, выбранных из галогена, -CN, -NO2, -C(O)2R, -C(O)R, -O-R, -N(RN)2, -N(RN)C(O)R,-N(RN)S(O)2R, -SR, -C(O)N(RN)2, -OC(O)R, -OC(O)N(RN)2, -SOR, -SO2R, -SO3R, -S(O)2N(RN)2, фосфата,фосфоната, циклоалкила, циклоалкенила, арила и гетероарила. В настоящем описании термин аминокислота означает группу, имеющую структуру либо в D-, либо L-конфигурации, и включает, но не ограничивается перечисленными: двадцать стандартных аминокислот: изолейцин, лейцин, лизин, метионин, фенилаланин, треонин, триптофан,валин, аланин, аспарагин, аспартат, цистеин, глутамат, глутамин, глицин, пролин, серин, тирозин, аргинин и гистидин. Настоящее изобретение также включает, без ограничения, аминокислоты Dконфигурации, бета-аминокислоты, аминокислоты с боковыми цепями, а также все неприродные аминокислоты, известные специалисту в данной области техники. В настоящем описании термины арил,ароматическая группа или ароматическое кольцо означают содержащие заместители или не содержащие заместители однокольцевые и многочисленные ароматические группы (например, фенил, пиридил и пиразол, и т.д.), и полициклические кольцевые системы (нафтил и хинолинил и т.д.). Полициклические кольца могут содержать два или более колец, в которых два атома являются общими для двух соседних колец (кольца конденсированы), при этом по меньшей мере одно из указанных колец является ароматическим, например, другие кольца могут представлять собой циклоалкилы, циклоалкенилы,арил, гетероциклы и/или гетероарилы. Арильная группа может содержать один или более заместителей,выбранных из галогена, алкила, -CN, -NO2, -CO2R, -C(O)R, -O-R, -N(RN)2, -N(RN)C(O)R, -N(RN)S(O)2R,-SR, -C(O)N(RN)2, -OC(O)R, -OC(O)N(RN)2, -SOR, -SO2R, -SO3R, -S(O)2N(RN)2, -SiR3, -P(O)R, фосфата,фосфоната, циклоалкила, циклоалкенила, арила и гетероарила. В настоящем описании термин циклоалкил означает содержащие заместители или не содержащие заместители циклические алкильные радикалы, содержащие от трех до двенадцати атомов углерода, и включает циклопропил, циклопентил, циклогексил и т.д. Термин циклоалкил также включает полициклические системы, содержащие два кольца, в которых два или более атомов являются общими для двух соседних колец (кольца конденсированы). Циклоалкильная группа может содержать один или более заместителей, выбранных из галогена, -CN, -NO2, -CO2R, -C(O)R, -O-R, -N(RN)2, -N(RN)C(O)R,-N(RN)S(O)2R, -SR, -C(O)N(RN)2, -OC(O)R, -OC(O)N(RN)2, -SOR, -SO2R, -S(O)2N(RN)2, фосфата, фосфоната, алкила, циклоалкенила, арила и гетероарила. В настоящем описании термин гетероалкил означает алкил, содержащий один или более гетероатомов. Термин гетероатом, в частности в пределах кольцевой системы, относится к N, О и S. В настоящем описании термин гетероциклическая группа, гетероцикл или гетероциклическое кольцо означает содержащие заместители или не содержащие заместители ароматические и неароматические циклические радикалы, содержащие по меньшей мере один гетероатом в качестве элемента кольца. Предпочтительные гетероциклические группы представляют собой группы, содержащие пять или шесть атомов кольца, которые включают по меньшей мере один гетероатом, и включают циклические амины, такие как морфолино, пиперидино, пирролидино и т.д., и циклические эфиры, такие как тетрагидрофуран, тетрагидропиран и т.д. Термин ароматические гетероциклические группы, также называемые гетероарильными группами, означает однокольцевые гетероароматические группы, которые могут содержать от одного до трех гетероатомов, например, пиррол, фуран, тиофен, имидазол, оксазол, тиазол, триазол, пиразол, оксодиазол, тиадиазол, пиридин, пиразин, пиридазин, пиримидин и т.д. Термин гетероарил также включает полициклические гетероароматические системы, содержащие два или более колец, в которых два или более атомов являются общими для двух соседних колец (кольца конденсированы), при этом по меньшей мере одно из колец представляет собой гетероарил, например, другие кольца могут представлять собой циклоалкилы, циклоалкенилы, арил, гетероциклы и/или гетероарилы. Примеры полициклических гетероароматических систем включают хинолин, изохинолин, циннолин, тетрагидроизохинолин, хиноксалин, хиназолин, бензимидазол, бензофуран, бензотиофен, бензоксазол, бензотиазол,индазол, пурин, бензотриазол, пирролопиридин, пиразолопиридин и т.д. Гетероциклическая группа может содержать один или более заместителей, выбранных из группы, состоящей из галогена, алкила, -CN,-NO2, -CO2R, -C(O)R, -O-R, -N(RN)2, -N(RN)C(O)R, -N(RN)S(O)2R, -SR, -C(O)N(RN)2, -OC(O)R,-OC(O)N(RN)2, -SOR, -SO2R, -SO3R, -S(O)2N(RN)2, -SiR3, -P(O)R, фосфат, фосфонат, циклоалкил, циклоалкенил, арил и гетероарил. Термин фармацевтически приемлемый или фармакологически приемлемый означает вещество, которое не является биологически или иным образом нежелательным, т.е. указанное вещество может быть введено индивидууму, не вызывая при этом каких-либо нежелательных биологических эффектов или не взаимодействуя неблагоприятным образом с любым из компонентов композиции,-3 024853 в которой оно содержится. Соединения согласно настоящему изобретению можно применять для ингибирования или снижения активности ВГС, в частности, белка NS5A ВГС. В данных случаях ингибирование и снижение активности белка NS5A относится к более низкому уровню измеренной активности по сравнению с контрольным экспериментом, в котором на клетки или субъекты не действуют исследуемым соединением. В соответствии с конкретными аспектами ингибирование или снижение измеренной активности представляет собой по меньшей мере 10% снижение или ингибирование. Для специалиста в данной области техники очевидно, что снижение или ингибирование измеренной активности, составляющее по меньшей мере 20,50, 75, 90 или 100%, или любое число в данном диапазоне, может быть предпочтительным для конкретных видов применения. В соответствии с первым вариантом реализации предложены соединения формулы IIIe где каждый из X и X' независимо выбран из связи, -CH2-, -CH2-CH2- и -CH2O-;Xb независимо представляет собой С или N, при условии, что не более двух Xb одновременно представляю собой N;Xc независимо представляет собой С или N, при условии, что не более двух Xc одновременно представляю собой N;Z и Z' независимо выбраны из остатка, состоящего из 1-3 аминокислот, -[U-(CR42)t-NR5-C(R42)t]u-U4U представляет собой -С(О)-,каждый R4 независимо выбран из водорода, C1-C8 алкила, возможно замещенного ОН, C1-C8 гетероалкила, содержащего атом кислорода, С 3-С 12 циклоалкила, гетероцикла, С 6-С 10 арила,каждый R5 и R7 независимо выбран из водорода и C1-C8 алкила,R8 выбран из водорода, C1-C8 алкила, гетероцикла, С 6-С 10 арила,или R7 и R8 совместно образуют 4-7-членное кольцо, возможно замещенное ОН,каждый t независимо равен 0 или 1 иu равен 0 или 1,где гетероцикл означает ароматический или неароматический циклический радикал, содержащий по меньшей мере один гетероатом в качестве элемента кольца. В другом варианте реализации Z и Z' каждый представляет собой остаток, состоящий из 1-3 аминокислоты. В другом варианте реализации указанные аминокислоты находятся в D-конфигурации. В другом варианте реализации Z и Z' каждый независимо выбран из -[U-(CR42)t-NR5-(CR42)t]u-U4(CR 2)t-NR7-(CR42)t-R8, -U-(CR42)t-R8 и -[U-(CR42)t-NR5-(CR42)t]u-U-(CR42)t-O-(CR42)t-R8. В другом варианте реализации один или оба Z и Z' представляют собой -[U-(CR42)t-NR5-(CR42)t]u-U4(CR 2)t-NR7-(CR42)t-R8. В другом варианте реализации один или оба Z и Z' представляют собой -U-(CR42)t-NR5-(CR42)t-U4(CR 2)t-NR7-(CR42)t-R8. В другом варианте реализации один или оба Z и Z' представляют собой -U-(CR42)t-NR7-(CR42)t-R8. В другом варианте реализации один или оба Z и Z' представляют собой -U-(CR42)t-R8. В другом варианте реализации один или оба Z и Z' представляют собой -[U-(CR42)t-NR5-(CR42)t]u-U4(CR 2)t-O-(CR42)t-R8. В другом варианте реализации один или оба Z и Z' представляют собой -U-(CR42)t-NR5-(CR42)t-U4(CR 2)t-O-(CR42)t-R8. В другом варианте реализации один или оба Z и Z' представляют собой -U-(CR42)t-O-(CR42)t-R8. В другом варианте реализации предложено соединение, выбранное из В другом варианте реализации предложена фармацевтическая композиция для лечения гепатита С,содержащая любое из соединений согласно настоящему изобретению. В другом первом варианте реализации предложено применение любого из соединений согласно настоящему изобретению для получения лекарственного средства для лечения гепатита С. В другом втором варианте реализации предложен способ лечения гепатита С, включающий введение субъекту терапевтически эффективного количества любого из соединений согласно настоящему изобретению. Общий синтез Следующие схемы иллюстрируют некоторые способы синтеза, которые используют для получения соединений и их аналогов, включенных в настоящее изобретение. Специалисту в данной области техники понятно, что также могут быть использованы альтернативные способы для получения аналогично и подобным образом функционализированных промежуточных веществ и целевых молекул. Также возможны альтернативные реагенты для данного превращения. В настоящей заявке используют следующие сокращения:Boc трет-бутоксикарбонил ДХЭ Дихлорэтан ДХМ ДихлорметанLDA Диизопропиламид лития ЖХМС Жидкостная хроматография с масс-спектроскопиейMeI Метилиодид МеОН Метанол мин Минута (минуты) ммоль Миллимоль (миллимоли)PG Защитная группа РТТ Фенилтриметил трибромид Ру Пиридинrt Комнатная температура ТЭА ТриэтиламинTf Трифторметансульфонат ТФУ Трифторуксусная кислота ТФУА Трифторуксусный ангидрид ТГФ Тетрагидрофуран ТСХ Тонкослойная хроматография Реагенты и растворители, применяемые ниже, могут быть получены из коммерческих источников,таких как Aldrich Chemical Co. (Milwaukee, Wisconsin, USA). Спектры 1 Н-ЯМР регистрировали на спектрометре ЯМР Bruker 400 или 500 МГц. Значимые пики сводили в таблицы в следующем порядке: мультиплетность (s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; br s, широкий синглет), константа (константы) взаимодействия в герцах (Гц) и число протонов. Анализ методом масс-спектрометрии с ионизацией электрораспылением (ESI) проводили на масс-спектрометре с электрораспылением HewlettPackard 1100 MSD с использованием НР 1 100 ВЭЖХ для доставки образца. Результаты масс-спектрометрии описаны как отношение массы к заряду с последующей относительной распространенностью каждого иона (в скобках) или отдельным значением m/z для иона М+Н (или, как указано, М-Н), включая наиболее распространенные атомные изотопы. Изотопные образцы соответствуют ожидаемой формуле во всех случаях. Как правило, аналит растворяли в метаноле при 0,1 мг/мл и 5 микролитров вводили с помощью доставляющего растворителя в масс-спектрометр, который сканировал 100-1500 дальтон. Все соединения можно анализировать в положительном режиме ESI с применением градиента ацетонитрил/H2O (10-90%) ацетонитрила в H2O с 0,1% муравьиной кислотой в качестве растворителя для доставки. Соединения, приведенные ниже, также можно анализировать в отрицательном режиме ESI с использованием 2 мМ NH4OAc в смеси ацетонитрил/H2O в качестве растворителя для доставки. Энантиомерную чистоту определяли с применением системы Hewlett-Packard Series 1050, оснащенной хиральной колонкой ВЭЖХ (ChiralPak AD, 4,6 мм 150 мм), и изократического элюирования с использованием смеси изопропанол-гексан 5:95 в качестве подвижной фазы. Названия соединений были получены с применением программы ChemDraw Cambridge Soft Inc. Схема 1-1 Пример 1 - синтез соединений формулы IIc На схеме 1-1 описано получение целевых молекул и их аналогов с симметричными и несимметричными функциональными концевыми группами. Стадия а. К раствору 2-бромнафталина а (62,0 г, 300 ммоль) в ДХМ (1 л) добавляли AlCl3 (44,0 г,330 ммоль) и 2-хлорацетилхлорид (34,0 г, 330 ммоль) при 0 С. Реакционную смесь перемешивали при 0 С в течение 1 ч, затем добавляли H2O (500 мл) и экстрагировали. Органический слой промывали H2O,сушили безводным Na2SO4, выпаривали при пониженном давлении с получением 80 г неочищенного продукта, который очищали при помощи перекристаллизации из 10% смеси EtOAc-гексан (об./об.) с получением b (28 г, выход 36%) в виде белого твердого вещества: 1 Н ЯМР (500 МГц, CDCl3)8,44 (s, 1H),8,07 (s, 1H), 8,04 (d, J = 11,0 Гц, 1H), 7,84 (d, J = 8,5 Гц, 2 Н), 7,66 (d, J= 8,5 Гц, 1H), 4,81 (s, 2 Н) ppm; ЖХМС (ESI) m/z 282,9 (М + Н)+. Стадия b. К раствору b (28,0 г, 100 ммоль) в ДХМ (500 мл) добавляли N-Boc-L-Pro-OH (24,7 г, 115 ммоль) и Et3N (70,0 мл, 500 ммоль) и смесь перемешивали при комнатной температуре (КТ) в течение 2 ч. Смесь концентрировали при пониженном давлении с получением неочищенного с, которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI) m/z 462,1 (М + Н)+. Стадия с. К раствору с (46,0 г, 100 ммоль) в толуоле (500 мл) добавляли NH4OAc (77 г, 1,0 моль) и смесь перемешивали при 110 С в течение ночи и концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc 1:1 (об./об. с получением d (30 г, выход 68%) в виде желтого твердого вещества: ЖХ-МС (ESI) m/z 442,1 (М + Н)+. Стадия d. К раствору d (10,0 г, 23,0 ммоль) в безводном ДМЭ (200 мл) и эквимолярном количестве бороната е добавляли PPh3 (1,2 г, 4,6 ммоль), Pd(PPh3)4 (1,6 г, 2,3 ммоль) и 2,0 М раствор Na2CO3. Смесь кипятили с обратным холодильником в атмосфере аргона в течение ночи. Органический растворитель удаляли при пониженном давлении, и остаток обрабатывали H2O, экстрагировали EtOAc (2200 мл). Объединенные органические фазы сушили, фильтровали и концентрировали в вакууме с получением остатка, который очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc 3:1 (об./об. с получением f (10 г, выход 96%) в виде желтого твердого вещества. ЖХ-МС(ESI): m/z 709,3 (М+Н)+. Стадия е. К перемешиваемому раствору f (150 мг, 0,29 ммоль) в диоксане (3 мл) по каплям добавляли 4,0 Н раствор HCl в диоксане (3 мл). Смесь перемешивали при КТ в течение 4 ч, затем концентрировали с получением желтоватого твердого вещества (134 мг), которое непосредственно применяли на следующей стадии. Остаток (134 мг, 0,290 ммоль) суспендировали в ТГФ (5 мл), добавляли DIPEA (0,32 мл), а затем N-метоксикарбонил-L-Val-OH (151 мг, 0,860 ммоль). После перемешивания в течение 15 мин добавляли HATU (328 мг, 0,860 ммоль) и смесь дополнительно перемешивали при КТ в течение 2 ч,затем концентрировали. Остаток очищали при помощи препаративной ВЭЖХ с получением g (40 мг,выход 19%). Схема 1-2 Стадия а. В соответствии со схемой 1-2 к раствору соединения 3 (2,0 г, 4,5 ммоль) в диоксане (25 мл) добавляли 4,0 Н раствор HCl в диоксане (25 мл). После перемешивания при КТ в течение 4 ч реакционную смесь концентрировали, и остаток сушили в вакууме с получением желтоватого твердого вещества (2,1 г), которое непосредственно применяли на следующей стадии без дополнительной очистки. Стадия b. К остатку, полученному на стадии а (4,5 ммоль) добавляли ДМФ (25 мл), затем DIPEA(3,7 мл, 22,5 ммоль) и N-метилкарбамат-L-валина (945 мг, 5,4 ммоль). После перемешивания при КТ в течение 15 мин реакционную смесь медленно добавляли в H2O (400 мл). Отфильтровывали белое осажденное твердое вещество и сушили с получением соединения 6 (2,2 г, выход 98%). ЖХ-МС (ESI): m/z 499,1 (М+Н)+. Стадия с. К смеси соединения 6 (800 мг, 1,6 ммоль), соединения 7 (718 мг, 1,6 ммоль) и NaHCO3(480 мг, 5,7 ммоль) в 1,2-диметоксиэтане (15 мл) и H2O (5 мл) добавляли Pd(dppf)Cl2 (59 мг, 0,08 ммоль). После перемешивания в течение ночи при 80 С в атмосфере N2 реакционную смесь концентрировали. Осадок разделяли в смеси 20% метанол/CHCl3 (100 мл) и H2O (100 мл). Органическую фазу отделяли и водную фазу снова экстрагировали смесью 20% метанол/CHCl3 (100 мл). Объединенные органические фазы последовательно промывали солевым раствором, сушили безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 15:1 (об./об. с получением соединения 8 (1,0 г, выход 85%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 732,4 (М+Н)+. Стадия d. К раствору соединения 8 (200 мг, 0,27 ммоль) в диоксане (3,0 мл) добавляли 4 Н растворHCl в диоксане (3,0 мл). После перемешивания при КТ в течение 2 ч реакционную смесь концентрировали, остаток сушили в вакууме с получением соли HCl с количественным выходом, которую применяли непосредственно на следующей стадии без дополнительной очистки. Стадия е. К раствору соли (0,27 ммоль) в ДМФ (5,0 мл) добавляли DIPEA (0,47 мл, 2,7 ммоль), затем N,N-диметил-О-фенилглицин (59 мг, 0,33 ммоль) и HATU (125 мг, 0,33 ммоль). После перемешивания при КТ в течение 1 ч реакционную смесь разделяли в H2O и ДХМ. Органическую фазу последовательно промывали H2O и солевым раствором, сушили безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ с получением соединения 9. ЖХ-МС (ESI): m/z 793,4 (М+Н)+.KOAc (1,85 г, 18,8 ммоль) в 1,4-диоксане (100 мл) добавляли Pd(dppf)Cl2 (440 мг, 0,6 ммоль). После перемешивания при 80 С в течение 3 ч в атмосфере N2 реакционную смесь концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 2/1 (об./об. с получением соединения 11 (2,8 г, выход 80%) в виде белого твердого вещества. ЖХ-МС (ESI): m/z 490,3NaHCO3 (420 мг, 4,99 ммоль) в 1,2-диметоксиэтане (30 мл) и H2O (10 мл) добавляли Pd(dppf)Cl2 (139 мг,0,19 ммоль). После перемешивания при 80 С в течение ночи в атмосфере N2 реакционную смесь концентрировали. Остаток разделяли в смеси 20% метанол/CHCl3 (100 мл) и H2O (100 мл). Водную фазу снова экстрагировали смесью 20% метанол/CHCl3 (100 мл). Объединенные органические фазы последовательно промывали солевым раствором, сушили Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 2/1 (об./об. с получением соединения 13 (635 мг, выход 68%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 732,4HCl в диоксане (3,0 мл). После перемешивания при КТ в течение 2 ч реакционную смесь концентрировали, и остаток сушили в вакууме с получением соли HCl соединения 14 с количественным выходом, которую непосредственно применяли на следующей стадии без дополнительной очистки. Стадия d. К раствору соли (0,27 ммоль) в ДМФ (5,0 мл) добавляли DIPEA (0,47 мл, 2,7 ммоль), затем N,N-диметил-О-фенилглицин (59 мг, 0,33 ммоль) и HATU (125 мг, 0,33 ммоль). После перемешивания при КТ в течение 1 ч реакционную смесь разделяли в H2O и ДХМ. Органическую фазу последовательно промывали H2O и солевым раствором, сушили безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ с получением соединения 15. ЖХ-МС (ESI): Схема 2-1 Пример 2 - синтез соединений формулы IIIe Стадия а. В соответствии со схемой 2-1 к смеси соединения 1 (5,05 г, 13,8 ммоль), бис(пинаколато)дибора (7,1 г, 27,9 ммоль) и KOAc (3,2 г, 32,5 ммоль) в 1,4-диоксане (100 мл) добавлялиPd(dppf)Cl2 (400 мг, 0,5 ммоль). После перемешивания при 80 С в течение 3 ч в атмосфере N2 реакционную смесь концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле(петролейный эфир/EtOAc = 2/1 (об./об. с получением соединения 2 (3,0 г, выход 53%) в виде коричневого твердого вещества. ЖХ-МС (ESI): m/z 414,2 (М+Н)+. Стадия b. К смеси соединения 2 (522 мг, 1,26 ммоль), соединения 3 (500 мг, 1,13 ммоль) и NaHCO3(333 мг, 3,96 ммоль) в 1,2-диметоксиэтане (30 мл) и H2O (10 мл) добавляли Pd(dppf)Cl2 (74 мг, 0,1 ммоль). После перемешивания при 80 С в течение ночи в атмосфере N2 реакционную смесь концентрировали. Остаток разделяли в смеси 20% метанол/CHCl3 (100 мл) и H2O (100 мл). Отделяли органическую фазу, а водную фазу снова экстрагировали смесью 20% метанол/CHCl3 (100 мл). Объединенные органические фазы последовательно промывали солевым раствором, сушили безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (ДХМ/МеОН= 50:1 (об./об. с получением соединения 4 (450 мг, выход 55%) в виде желтого твердого вещества. ЖХМС (ESI): m/z 649,3 (М+Н)+. Стадия с. К перемешиваемому раствору соединения 4 (160 мг, 0,25 ммоль) в диоксане (2,0 мл) добавляли 4 Н раствор HCl в диоксане (2,0 мл). После перемешивания при КТ в течение 3 ч реакционную смесь концентрировали, и остаток сушили в вакууме с получением соли HCl с количественным выходом,которую непосредственно применяли на следующей стадии без дополнительной очистки. Стадия d. К раствору полученной ранее соли (0,25 ммоль) в ДМФ (4,0 мл) добавляли DIPEA (0,44 мл, 2,5 ммоль), затем N-метилкарбамат L-треонина (110 мг, 0,62 ммоль) и HATU (240 мг, 0,63 ммоль).-9 024853 После перемешивания при КТ в течение 1 ч реакционную смесь разделяли в H2O и ДХМ. Органическую фазу последовательно промывали H2O и солевым раствором, сушили безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ с получением соединения 5 в виде белого порошка. ЖХ-МС (ESI): m/z 767,3 (М+Н)+. На схеме 2-1-1 предложен альтернативный способ синтеза соединений, описанных в настоящей патентной заявке. Схема 2-2 Стадия а. В соответствии со схемой 2-2 к смеси соединения 2 (1,16 г, 2,32 ммоль), соединения 6Pd(dppf)Cl2 (103 мг, 0,14 ммоль). После перемешивания при 80 С в течение ночи в атмосфере N2 реакционную смесь концентрировали. Остаток разделяли в смеси 20% метанол/CHCl3 (150 мл) и H2O (150 мл). Водную фазу снова экстрагировали смесью 20% метанол/CHCl3 (150 мл). Объединенные органические фазы последовательно промывали солевым раствором, сушили безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 1,5/1 (об./об. с получением соединения 16 (1,32 г, выход 80%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 706,4 (М + Н)+. Стадия b. К раствору соединения 16 (200 мг, 0,28 ммоль) в диоксане (3,0 мл) добавляли 4 Н растворHCl в диоксане (3,0 мл). После перемешивания при КТ в течение 2 ч реакционную смесь концентрировали и сушили в вакууме с получением соли HCl соединения 17 с количественным выходом, которую непосредственно применяли на следующей стадии. Стадия с. К раствору соли (0,28 ммоль) в ДМФ (5,0 мл) добавляли DIPEA (0,49 мл, 2,8 ммоль), затем N,N-диметил-D-фенилглицин (61 мг, 0,34 ммоль) и HATU (129 мг, 0,34 ммоль). После перемешивания в течение 1 ч при КТ реакционную смесь разделяли в H2O и ДХМ. Органическую фазу последовательно промывали H2O и солевым раствором, сушили безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ с получением соединения 18. ЖХ-МС (ESI): Схема 2-3 Стадия а. В соответствии со схемой 2-3 к раствору соединения 1 (4,0 г, 10,9 ммоль) в диоксане (40 мл) добавляли 4 Н раствор HCl в диоксане (40 мл). После перемешивания при КТ в течение ночи реакционную смесь концентрировали. Остаток промывали ДХМ, фильтровали и сушили в вакууме с получением гидрохлоридной соли с количественным выходом, которую применяли на следующей стадии без дополнительной очистки. Стадия b. К раствору соли (10,9 ммоль) в ДМФ (30 мл) добавляли DIPEA (5,8 мл, 33,0 ммоль), затемN-метоксикарбонил-D-валин (2,1 г, 12,1 ммоль) и HATU (4,6 г, 12,1 ммоль). После перемешивания при КТ в течение 1 ч реакционную смесь разделяли в H2O и ДХМ. Органическую фазу последовательно промывали Н 2 О и солевым раствором, сушили безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (ДХМ/петролейный эфир = 4/4 (об./об. с получением соединения 19 (3,0 г, выход 65%). ЖХ-МС (ESI): m/z 423,1 (М+Н)+. Стадия с. К смеси соединения 11 (800 мг, 1,9 ммоль), соединения 19 (700 мг, 1,7 ммоль) и NaHCO3(561 мг, 6,6 ммоль) в 1,2-диметоксиэтане (60 мл) и Н 2 О (20 мл) добавляли Pd(dppf)Cl2 (183 мг, 0,25 ммоль). После перемешивания при 80 С в течение ночи в атмосфере N2 реакционную смесь концентрировали. Затем остаток разделяли в смеси 20% метанол/CHCl3 (100 мл) и H2O (100 мл). Водную фазу снова экстрагировали смесью 20% метанол/CHCl3 (100 мл). Объединенные органические фазы последовательно промывали солевым раствором, сушили Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 2/1 (об./об. с получением соединения 20 (600 мг, выход 52%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 706,4 (М+Н)+. Стадия d. К раствору соединения 20 (200 мг, 0,28 ммоль) в диоксане (3,0 мл) добавляли 4 Н растворHCl в диоксане (3,0 мл). После перемешивания при КТ в течение 2 ч реакционную смесь концентрировали и сушили остаток в вакууме с получением соли HCl соединения 21 с количественным выходом, которую непосредственно применяли на следующей стадии без дополнительной очистки. Стадия е. К раствору соединения 21 (0,28 ммоль) в ДМФ (5,0 мл) добавляли DIPEA (0,49 мл, 2,8 ммоль), затем N,N-диметил-О-фенилглицин (64 мг, 0,36 ммоль) и HATU (129 мг, 0,34 ммоль). После перемешивания при КТ в течение 1 ч реакционную смесь разделяли в H2O и ДХМ. Органическую фазу последовательно промывали H2O и солевым раствором, сушили безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ с получением соединения 22. ЖХ-МС(111 мг, 0,140 ммоль) при КТ в атмосфере N2. После перемешивания при 80 С в течение ночи в атмосфере N2 реакционную смесь концентрировали, остаток разбавляли EtOAc (100 мл) и H2O (25 мл). Органическую фазу промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель, остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 1/2(об./об. с получением соединения 142 (360 мг, выход 75%) в виде желтого твердого вещества. ЖХ-МС(ESI): m/z 707,4 (М+Н)+. Стадия b. К раствору соединения 142 (115 мг, 0,16 ммоль) в диоксане (2,0 мл) при КТ добавляли 4 Н раствор HCl в диоксане (2,0 мл). После перемешивания при КТ в течение ночи реакционную смесь концентрировали, остаток сушили в вакууме с получением соли HCl, которую применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI) m/z 607,3 (М + Н)+. Стадия с. Затем соль HCl растворяли в ДМФ (2 мл), к полученной смеси добавляли DIEA (0,28 мл,1,6 ммоль), N-Мос-L-(тетрагидро-2 Н-пирин-4-ил)глицин (41 мг, 0,19 ммоль) и HATU (73 мг, 0,19 ммоль). После перемешивания при КТ в течение 15 мин реакционную смесь концентрировали, остаток очищали при помощи препаративной ВЭЖХ с получением соединения 143. ЖХ-МС: (ESI) m/z 806,4 (М+Н)+. Пример 3 - синтез дополнительных соединений формулы IIc Схема 3-1 Стадия а. В соответствии со схемой 3-1 к раствору соединения 1 (49,7 г, 0,25 моль) в ДМСО по каплям при КТ добавляли 40% водный раствор HBr (0,50 моль). После перемешивания при 90 С в течение 3 ч реакционную смесь выливали в Н 2 О и полученную смесь выдерживали при 5060 С. Желтое твердое вещество собирали при помощи фильтрования и двукратной перекристаллизации из смеси ацетон/Н 2 О(1/19 (об./об. с получением соединения 2 (50 г, выход 87%). ЖХ-МС (ESI): m/z 212,9 (М+Н)+. Стадия b. Смесь соединения 2 (19,0 г, 80,0 ммоль) и 4-бромбензол-1,2-диамина (15,0 г, 80,0 ммоль) в НОАс (180 мл) кипятили с обратным холодильником в течение ночи Затем реакционную смесь выливали в ледяную H2O. Собирали твердое вещество при помощи фильтрования и очищали при помощи колоночной хроматографии на силикагеле с получением соединений 3 и 3' (2,8 г, выход 10%) в виде пары региоизомеров. ЖХ-МС (ESI): m/z 362,9 (М+Н)+. Стадия с. Смесь соединения 3 (4,8 г, 5,4 ммоль), бис(пинаколато)дибора (9,6 г, 38 ммоль), ацетата калия (3,8 г, 38 ммоль) и Pd(dppf)Cl2CH2Cl2 (524 мг, 0,54 ммоль) в диоксане (100 мл) перемешивали при 80 С в течение 17 ч в атмосфере Ar. Затем реакционную смесь фильтровали Отфильтрованный осадок несколько раз промывали EtOAc (50 мл 3). Фильтрат промывали H2O и сушили безводным Na2SO4. Удаляли растворитель и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон =101 (об./об. с получением соединений 4 и 4' (2,2 г, выход 89%) в виде пары региоизомеров. ЖХ-МС (ESI): m/z 459,3 (М+Н)+. (Соответствующую бороновую кислоту также выделяли и применяли в качестве активного промежуточного соединения на следующей стадии.) Стадия d. Смесь соединений 4 и 4' (1,0 г, 2,2, ммоль), (S)-трет-бутил-2-(5-йод-1-Н-имидазол-2- 12024853 ил)пирролидин-1-карбоксилата (2,0 г, 5,4 ммоль), бикарбоната натрия (1,5 г, 18 ммоль) и Pd(dppf)Cl2CH2Cl2 (427 мг, 0,44 ммоль) в ДМЭ/Н 2 О (3/1 (об./об. (80 мл) перемешивали при 80 С в течение 17 ч в атмосфере Ar. Затем реакционную смесь концентрировали, и остаток разбавляли EtOAc (100 мл) Органический слой промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель, остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон =101m/z 677,3 (М+Н)+. Стадия е. Смесь соединений 5 и 5' (200 мг, 0,3 ммоль) в 4,0 Н растворе HCl в диоксане (10 мл) перемешивали при КТ в течение ночи. Удаляли растворитель, остаток сушили в вакууме с получением солиHCl, которую применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 477,2(М+Н)+. Стадия f. Затем соль HCl растворяли в ДМФ (3 мл), к полученной смеси последовательно добавлялиEt3N (304 мг, 3,0 ммоль), N-Moc-L-Val-OH (116 мг, 0,66 ммоль) и HATU (251 мг, 0,66 ммоль). После перемешивания при КТ в течение 2 ч реакционную смесь выливали в Н 2 О (50 мл), полученную суспензию несколько раз экстрагировали ДХМ (20 мл 3). Экстракты объединяли, промывали солевым раствором,сушили безводным MgSO4. Растворитель удаляли и остаток очищали при помощи препаративной ВЭЖХ с получением соединений 6 и 6' в виде пары региоизомеров. ЖХ-МС (ESI): m/z 791,4 (М+Н)+. Схема 3-2 Стадия а. В соответствии со схемой 3-2 к раствору соединения 7 (909 мг, 1,86 ммоль), (S)-третбутил 2-(5-(6-бромпиридин-3-ил)пирролидин)-1-карбоксилата (800 мг, 2,04 ммоль) и NaHCO3 (625 мг,7,44 ммоль) в 1,2-диметоксиэтане (100 мл) и H2O (30 мл) добавляли Pd(dppf)Cl2 (152 мг, 0,186 ммоль) при КТ в атмосфере Ar. После перемешивания при 80 С в течение ночи в атмосфере Ar реакционную смесь концентрировали. Остаток разбавляли CH2Cl2 (200 мл). Органический слой промывали H2O и сушили безводным Na2SO4. Удаляли растворитель и остаток очищали при помощи колоночной хроматографии на силикагеле (ДХМ/МеОН = 50:1 (об./об. с получением соединения 8 (700 мг, выход 55%). ЖХ-МС(ESI): m/z 676,4 (М+Н)+. Стадия b. К перемешиваемому раствору соединения 8 (200 мг, 0,296 ммоль) в диоксане (3 мл) добавляли 4 Н раствор HCl в диоксане (3 мл). После перемешивания при КТ в течение 4 ч реакционную смесь концентрировали, и остаток сушили в вакууме с получением соли HCl, которую применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 476,2 (М+Н)+. Стадия с. Затем соль HCl растворяли в ДМФ (3 мл), в полученную смесь последовательно добавляли DIEA (388 мг, 3,0 ммоль), N-Moc-L-Val-OH (116 мг, 0,66 ммоль) и HATU (251 мг, 0,66 ммоль). После перемешивания при КТ в течение 2 ч реакционную смесь выливали в H2O (50 мл), полученную суспензию несколько раз экстрагировали в ДХМ (20 мл 3). Экстракты объединяли, промывали солевым раствором и сушили безводным MgSO4. Удаляли растворитель и остаток очищали при помощи препаративной ВЭЖХ с получением соединения 9. 1 Н ЯМР (500 МГц, CDCl3)9,09 (s, 1H), 8,67 (s, 1H), 8,31-8,34 Схема 3-3 Стадия а. В соответствии со схемой 3-3 к раствору соединения 10 (45,0 г, 247 ммоль) в МеОН (500 мл) при КТ добавляли NaOMe (1,4 г, 25 ммоль). После перемешивания при КТ в течение 48 ч в реакционную смесь добавляли NH4Cl (13,4 г, 250 ммоль) и полученную смесь дополнительно перемешивали в течение 24 ч. Удаляли растворитель и остаток сушили в вакууме с получением соединения 11, которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 199,0 (М+Н)+. Стадия b. К раствору соединения 11 (15 г, 75 ммоль) в CH3CN (500 мл) добавляли K2CO3 (11,4 г,83,0 ммоль), затем 2-фтор-5-нитробензальдегид (12,7 г, 75,0 ммоль). После кипячения с обратным холодильником в течение 12 ч реакционную смесь концентрировали, и остаток промывали МеОН с получением неочищенного соединения 12 (12 г), которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС: (ESI) m/z = 330,0 (М+Н)+. Стадия с. К раствору соединения 12 (5,0 г, 15 ммоль) в МеОН (500 мл) добавляли хлорид олова (II)(14,3 г, 75,0 ммоль) и концентрированную соляную кислоту (17 мл). После перемешивания при КТ в течение 3,5 ч в реакционную смесь осторожно добавляли насыщенный водный раствор NaHCO3 (470 мл). Полученную смесь экстрагировали в этилацетате (100 мл 3). Экстракты объединяли и промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и остаток сушили в вакууме с получением неочищенного соединения 13 (2,5 г). ЖХ-МС: (ESI) m/z = 300,0 (М+Н)+. Стадия d. К раствору соединения 13 (300 мг, 1,0 ммоль) в концентрированной HCl (0,25 мл) по каплям при 0 С добавляли раствор NaNO2 (76 мг, 1,1 ммоль) в H2O (1 мл). После перемешивания при 0 С в течение 30 мин реакционную смесь добавляли к раствору K2CO3 (207 мг, 1,5 ммоль) и Et2NH (0,11 г, 1,5 ммоль) в ледяной H2O (1 мл). Затем к смеси добавляли диэтиловый эфир (100 мл). Отделяли органический слой, промывали H2O (15 мл) и сушили безводным Na2SO4. Удаляли растворитель и остаток сушили в вакууме с получением неочищенного соединения 14 (350 мг), которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 384,1 (М+Н)+. Стадия е. К раствору соединения 14 (1,8 г, 4,7 ммоль) и LiBr (834 мг, 9,6 ммоль) в ацетонитриле (10 мл) при КТ добавляли TMSCl (782 мг, 7,2 ммоль). После перемешивания при 60 С в течение 15 мин реакционную смесь охлаждали до КТ и обрабатывали 5% водным раствором NaHCO3 (30 мл). Смесь концентрировали и остаток экстрагировали CH2Cl2 (50 мл 3). Объединяли экстракты, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и остаток очищали при помощи колоночной хроматографии на силикагеле (пентан/диэтиловый эфир = 1/19 (об./об. с получением соединения 15(1,0 г, выход 59%). ЖХ-МС: (ESI) m/z = 362,9 (М+Н)+. Стадия f. К раствору соединения 15 (300 мг, 0,82 ммоль) в диоксане (20 мл) последовательно добавляли бис(пинаколато)дибор (915 мг, 3,63 ммоль), ацетат калия (403 мг, 4,12 ммоль) и Pd(dppf)Cl2 (134 мг,0,160 ммоль) при КТ в атмосфере Ar. После перемешивания при 80 С в течение 17 ч в атмосфере Ar реакционную смесь разбавляли EtOAc (100 мл). Полученную смесь промывали Н 2 О и сушили безводнымNa2SO4. Удаляли растворитель и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 3/1 (об./об. с получением соединения 16 (227 мг, выход 60%). ЖХ-МС(ESI): m/z 459,3 (М+Н)+. (Соответствующую бороновую кислоту также отделяли и применяли в качестве- 14024853 активного промежуточного соединения на следующей стадии.) Стадия g. К раствору соединения 16 (300 мг, 0,65 ммоль) в ДМЭ/H2O (3/1 (об./об.), 30 мл) последовательно добавляли (S)-трет-бутил-2-(5-йод-1H-имидазол-2-ил)пирролидин-1-карбоксилат (595 мг, 1,64 ммоль), NaHCO3 (443 мг, 5,28 ммоль) и Pd(dppf)Cl2CH2Cl2 (126 мг, 0,13 ммоль) при КТ в атмосфере Ar. После перемешивания при 80 С в течение 17 ч в атмосфере Ar реакционную смесь разбавляли EtOAc(150 мл). Отделяли органический слой, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 2/1 (об./об. с получением соединения 17 (151 мг, выход 34%) в виде желтоватого твердого вещества ЖХ-МС (ESI): m/z 677,3 (М+Н)+. Стадия h. К раствору соединения 17 (100 мг, 0,15 ммоль) в диоксане (2 мл) при КТ добавляли 4 Н раствор HCl в диоксане (2 мл). После перемешивания при КТ в течение ночи удаляли растворитель, остаток сушили в вакууме с получением соли HCl, которую применяли на следующей стадии без дополнительной очистки ЖХ-МС (ESI): m/z 477,2 (М+Н)+. Стадия i. К раствору соли HCl в ДМФ (2 мл) добавляли DIPEA (0,24 мл, 1,5 ммоль), затем N-Moc-LVal-OH (65 мг, 0,37 ммоль) и HATU (141 мг, 0,37 ммоль). После перемешивания при КТ в течение 30 мин реакционный раствор выливали в H2O (50 мл). Суспензию фильтровали и твердое вещество очищали при помощи препаративной ВЭЖХ с получением соединения 18. 1 Н ЯМР (500 МГц, CD3OD)9,69 (s, 1H Схема 3-4 Стадия а. В соответствии со схемой 3-4 к раствору соединения 19 (5,00 г, 19,8 ммоль) в CH3CN (200 мл) при КТ последовательно добавляли EDCI (9,10 г, 47,6 ммоль), HOBt (1,34 г, 5,95 ммоль),MeNH(OMe)HCl (2,93 г, 30 ммоль) и Et3N (6,6 г, 65,3 ммоль) После перемешивания при КТ в течение 3 ч реакционную смесь концентрировали, и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 5/1 (об./об. с получением соединения 20 (5,1 г, выход 87%) в виде белого твердого вещества ЖХ-МС (ESI): m/z 295,0 (М + Н)+. Стадия b. К раствору соединения 20 (2,0 г, 6,8 ммоль) в ТГФ (200 мл) медленно добавляли 3 М раствор MeMgCl в ТГФ (4,5 мл) при 0 С в атмосфере N2. После перемешивания при 0 С в течение 1 ч, а затем при КТ в течение 1 ч реакционную смесь гасили при помощи добавления нескольких капель водного раствора NH4Cl. Реакционную смесь концентрировали и остаток разбавляли водным раствором NaHCO3(5 мл) и EtOAc (100 мл). Органическую фазу промывали солевым раствором и сушили безводнымNa2SO4. Удаляли растворитель и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/AcOEt = 10:1 (об./об. с получением соединения 21 (1,0 г, 59%) в виде белого твердого вещества. ЖХ-МС (ESI): m/z 250,0 (М+Н)+. Стадия с. К раствору соединения 21 (500 мг, 2,0 ммоль) в НОАс (20 мл) и 48% водном растворе HBr(0,5 мл) при КТ медленно добавляли Br2 (320 мг, 2,0 ммоль) в 48% водном растворе HBr (0,5 мл). После перемешивания при КТ в течение 2 ч реакционную смесь концентрировали и остаток разбавляли H2O(100 мл). Смесь экстрагировали в EtOAc (100 мл 3). Объединяли экстракты и промывали насыщенным раствором NaHCO3 (30 мл 3) и сушили безводным Na2SO4. Удаляли растворитель и остаток сушили в вакууме с получением неочищенного соединения 22 (440 мг) в виде белого твердого вещества, которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 327,9 (М+Н)+. Стадия d. К раствору соединения 22 (415 мг, 1,26 ммоль) в CH3CN (15 мл) при КТ последовательно- 15024853 добавляли N-Boc-L-Pro-OH (300 мг, 1,36 ммоль) и Et3N (382 мг, 3,78 ммоль). После перемешивания при КТ в течение 2 ч реакционную смесь концентрировали и остаток сушили в вакууме с получением соединения 23 (580 мг), которое применяли на следующей стадии без дополнительной очистки; ЖХ-МС (ESI):m/z 463,1 (М+Н)+. Стадия е. Смесь соединения 23 (580 мг, 1,25 ммоль) и NH4OAc (962 мг, 12,5 ммоль) в толуоле (25 мл) перемешивали при 110 С в течение ночи. Реакционную смесь концентрировали и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 9/1 (об./об. с получением соединения 24 (400 мг, 72%) в виде белого твердого вещества. ЖХ-МС (ESI): m/z 443,1 (М+Н)+. Стадия f. К смеси соединения 24 (380 мг, 0,86 ммоль), (S)-трет-бутил 2-(5-(4-(4,4,5,5-тетраметил 1,3,2-диоксаборолан-2-ил)-1 Н-имидазол-2-ил)пирролидин-1-карбоксилата (378 мг, 0,860 ммоль) и NaHCO3 (253 мг, 3,01 ммоль) в 1,2-диметоксиэтане (15 мл) добавляли Pd(dppf)Cl2 (35 мг, 0,04 ммоль) в атмосфере N2. После перемешивания при 80 С в течение ночи в атмосфере N2 реакционную смесь концентрировали. Остаток разбавляли Н 2 О (50 мл) и экстрагировали EtOAc (50 мл 3) Экстракты объединяли и промывали солевым раствором и сушили безводным Na2SO4 Удаляли растворитель и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 5/2 (об./об. с получением соединения 25 (550 мг, выход 95%) в виде желтого твердого вещества ЖХ-МС (ESI): m/z 676,4(М+Н)+. Стадия g. К раствору соединения 26 (150 мг, 0,22 ммоль) в диоксане (2 мл) при КТ добавляли 4 Н раствор HCl в диоксане (2 мл). После перемешивания при КТ в течение ночи удаляли растворитель и остаток сушили в вакууме с получением соли HCl, которую применяли на следующей стадии без дополнительной очистки ЖХ-МС (ESI): m/z 476,2 (М+Н)+. Стадия h. К смеси соли HCl в ДМФ (2 мл) добавляли DIPEA (0,37 мл, 2,3 ммоль), затем N-Moc-LVal-OH (101 мг, 0,58 ммоль) и HATU (218 мг, 0,58 ммоль). После перемешивания при КТ в течение 30 мин реакционную смесь концентрировали и очищали остаток при помощи препаративной ВЭЖХ с получением соединения 26. 1 Н ЯМР (500 МГц, CD3OD)7,96 (d, 2H, J =11,5), 7,83-7,78 (m, 4H), 7,72 (d, 2H,J=8,0), 5,56 (m, 1H), 5,38-5,32 (m, 2H), 4,46-4,42 (m, 1H), 4,27-4,26 (m, 1H), 4,21-4,13 (m, 2H), 3,97-3,94 (m,1H), 3,66 (s, 6H), 2,89-2,86 (m, 1H), 2,64-2,62 (m, 2H), 2,34-2,25 (m, 3 Н), 2,01-1,96 (m, 2H), 0,94-0,87 (m,12H) ppm, ЖХ-МС (ESI): m/z 790,4 (M + H)+. Схема 3-5 Стадия а. В соответствии со схемой 3-5 к смеси трихлорацетальдегида (7,2 г, 48 ммоль) в воде (120 мл) добавляли Na2SO4 (104 г), затем 4-бромбензамин (35) в конц. водном растворе HCl (10 мл) и NH2OHHCl (8,8 г, 0,13 моль) в Н 2 О (100 мл). После кипячения с обратным холодильником в течение 1 ч реакционную смесь охлаждали до КТ. Твердое вещество собирали при помощи фильтрования и сушили в вакууме с получением соединения 36 (8,0 г, 91%) в виде желтого твердого вещества. ЖХ-МС (ESI) m/z: 243,0 (М+Н)+. Стадия b. В круглодонную колбу помещали 20 мл H2SO4 (98%) и нагревали раствор до 50 С. Затем добавляли соединение 36 (4,8 г, 20 ммоль) с такой скоростью, чтобы температура находилась в диапазоне от 60 до 70 С. После завершения добавления соединения 36 полученную смесь нагревали до 80 С и дополнительно перемешивали в течение 10 мин. Смесь охлаждали до КТ и выливали в лед (200 г). Твердое вещество собирали при помощи фильтрования, промывали несколько раз водой и сушили в вакууме с получением соединения 37 (3,6 г, выход 80%) в виде оранжевого твердого вещества ЖХ-МС (ESI) m/z 225,9 (М+Н)+. Стадия с. Смесь соединения 37 (1,35 г, 6,0 ммоль), 1-(4-бромфенил)этанона (1,14 г, 5,7 ммоль) и- 16024853 гидроксида калия (1,02 г, 18,3 ммоль) в этаноле (50 мл) кипятили с обратным холодильником в течение ночи. Реакционную смесь концентрировали и остаток разбавляли петролейным эфиром (100 мл) и водой(200 мл) Водную фазу отделяли, подкисляли при помощи добавления 1 Н раствора HCl, затем экстрагировали в этилацетате (50 мл 3). Объединяли экстракты, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и остаток сушили в вакууме с получением неочищенного соединения 38 (1,2 г) в виде красного твердого вещества, которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI) m/z 405,9 (М+Н)+. Стадия d. Колбу, содержащую соединение 5 (1,2 г, 2,95 ммоль), нагревали до 300 С в течение 30 мин в атмосфере Ar. Затем твердое вещество очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 19 1 (об./об. с получением соединения 39 (160 мг, выход 15%) в виде желтого твердого вещества. ЖХ-МС (ESI) m/z 361,9 (М+Н)+. Стадия е. Смесь соединения 39 (0,11 г, 0,30 ммоль), бис(пинаколато)дибора (0,34 г, 1,3 ммоль), ацетата калия (0,15 г, 1,5 ммоль) и Pd(dppf)Cl2 (50 мг, 0,06 ммоль) и диоксане (20 мл) перемешивали при 80 С в течение ночи в атмосфере Ar. Затем реакционную смесь разбавляли EtOAc (100 мл). Полученную смесь промывали H2O (50 мл) и сушили безводным Na2SO4. Удаляли растворитель и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 10/1 (об./об. с получением соединения 40 (0,12 г, выход 86%). ЖХ-МС (ESI) m/z 458,3 (М+Н)+. (Соответствующую бороновую кислоту также отделяли и применяли в качестве активного промежуточного соединения на следующей стадии.) Стадия f. К раствору соединения 40 (120 мг, 0,26 ммоль) в ДМЭ/H2O (3/1 (об./об.), 24 мл) последовательно добавляли (S)-трет-бутил-2-(5-йод-1 Н-имидазол-2-ил)пирролидин-1-карбоксилат (290 мг, 0,80 ммоль), NaHCO3 (220 мг, 2,6 ммоль) и Pd(dppf)Cl2CH2Cl2 (62 мг, 0,064 ммоль) при КТ в атмосфере Ar. После перемешивания при 80 С в течение ночи в атмосфере Ar реакционную смесь разбавляли EtOAc(100 мл). Отделяли органический слой, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 2/1 (об./об. с получением соединения 17 (151 мг, выход 86%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 676,4 (М+Н)+. Стадия g. К перемешиваемому раствору соединения 41 (120 мг, 0,18 ммоль) в диоксане (2 мл) добавляли смесь 4 Н HCl/диоксан (2 мл). После перемешивания при КТ в течение ночи реакционную смесь концентрировали, остаток сушили в вакууме с получением соли HCl, которую применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 476,2 (М+Н)+. Стадия h. К смеси соли HCl в ДМФ (2 мл) добавляли DIPEA (0,3 мл, 1,8 ммоль), затем N-Moc-LVal-OH (79 мг, 0,45 ммоль) и HATU (169 мг, 0,45 ммоль). После перемешивания при КТ в течение 30 мин реакционную смесь медленно выливали в H2O. Собирали твердое вещество при помощи фильтрования и очищали при помощи препаративной ВЭЖХ с получением соединения 42. 1 Н ЯМР (500 МГц,CD3OD)8,96 (d, 2H, J= 9,5 Гц), 8,63 (s, 1H), 8,53 (d, 2H, J= 10,0 Гц), 8,40-8,39 (m, 3H), 8,18 (s, 1H), 8,08- 17024853 Стадия а. В соответствии со схемой 3-6 к раствору метилата натрия (0,4 г, 7,7 ммоль) в метаноле (10 мл) при КТ добавляли смесь 4-метокси-2-нитробензальдегида (42) (1,4 г, 7,7 ммоль) и 4-метоксифенилацетонитрила (1,13 г, 7,7 ммоль). После перемешивания при КТ в течение 5 ч реакционную смесь фильтровали. Твердое вещество последовательно промывали водой и 95% этанолом, и сушили в вакууме с получением соединения 43 (1,82 г, выход 77%) в виде желтого порошка. 1 Н ЯМР (500 МГц, CDCl3)7,90 (d, J= 8,5 Гц, 1H), 7,85 (s, 1H), 7,70 (d, J= 2,0 Гц, 1H), 7,63 (d, J= 9,0 Гц, 2 Н), 7,28 (m, 1H), 6,98 (d, J= 9,0 Гц, 2 Н), 3,94 (s, 3 Н), 3,87 (s, 3 Н) ppm. ЖХ-МС (ESI): m/z 311,1 (М + Н)+. Стадия b. К раствору соединения 43 (15,5 г, 50 ммоль) в смеси ТГФ/метанол (5/1 (об./об.), 240 мл) при КТ добавляли NaBH4 (2,8 г, 75 ммоль). После перемешивания при КТ в течение 4 ч реакционную смесь выливали в ледяную воду и обрабатывали 1 Н водным раствором HCl. Полученную смесь экстрагировали в EtOAc (50 мл 2). Объединяли экстракты, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и сушили остаток в вакууме с получением неочищенного соединения 44 (9,8 г), которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 335,1 (M+Na)+. Стадия с. Смесь соединения 44 (9,0 г, 29 ммоль) и 10% Pd/C (4,5 г) в ТГФ (240 мл) и МеОН (60 мл) перемешивали при 45 С в течение 48 ч в атмосфере H2. Полученную смесь фильтровали черезCELITE545; отфильтрованный осадок промывали МеОН (50 мл 3). Фильтрат концентрировали и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 9:1) с получением соединения 45 (5,5 г, выход 71%) в виде белого твердого вещества. 1 Н ЯМР (500 МГц,CDCl3)7,16 (d, J= 8,5 Гц, 2 Н), 6,91-6,87 (m, 3 Н), 6,25 (d, J= 8,5 Гц, 1H), 6,12 (s, 1H), 3,80 (s, 3 Н), 3,75 (s,3 Н), 3,41 (d, J = 11,0 Гц, 1H), 3,27 (t, J = 11,0 Гц, 1H), 3,11-3,05 (m, 1H), 2,90 (d, J= 8,0 Гц, 2H) ppm; ЖХМС (ESI): m/z 270,1 (M + H)+. Стадия d. Смесь соединения 45 (2,7 г, 10 ммоль) и 10% Pd/C (1,4 г) перемешивали при 270280 С в течение 30 мин в атмосфере Ar. Смесь очищали при помощи колоночной хроматографии на силикагеле(петролейный эфир/EtOAc = 6/1 (об./об. с получением соединения 46 (1,8 г, 68%) в виде белого твердого вещества. ЖХ-МС (ESI): m/z 266,1 (М+Н)+. Стадия е. К раствору соединения 46 (0,80 г, 3,0 ммоль) в CH2Cl2 (30 мл) при -40 С добавляли смесь 4 Н BBr3/CH2Cl2 (4,5 мл, 18 ммоль). После перемешивания при КТ в течение ночи реакционную смесь разбавляли водой (30 мл). Полученную смесь обрабатывали 1H водным раствором NaOH для доведения до значения рН, равного 8, и экстрагировали в EtOAc (60 мл 2). Объединяли экстракты, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи хроматографии на силикагеле (петролейный эфир/EtOAc = 2/1 (об./об. с получением соединения 47 (0,7 г, 99%) в виде белого твердого вещества. ЖХ-МС (ESI): m/z 238,1 (М+Н)+. Стадия f. К раствору соединения 47 (0,82 г, 3,5 ммоль) и пиридина (1,3 г, 16 ммоль) в CH2Cl2 (45 мл) при 0 С добавляли Tf2O (3,6 г, 13 ммоль). После перемешивания при КТ в течение 30 мин реакционную смесь концентрировали, остаток очищали при помощи хроматографии на силикагеле (петролейный эфир/EtOAc = 10/1 (об./об. с получением соединения 48 (0,40 г, 23%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 502,1 (М+Н)+. Стадия g. Смесь соединения 48 (0,40 г, 0,80 ммоль), бис(пинаколато)дибора (1,0 г, 4,0 ммоль), ацетата калия (0,55 г, 5,6 ммоль) и Pd(dppf)Cl2 (200 мг, 0,24 ммоль) и диоксана (20 мл) перемешивали при 80 С в течение ночи в атмосфере Ar. Затем реакционную смесь разбавляли EtOAc (100 мл) Полученную смесь промывали Н 2 О (50 мл) и сушили безводным Na2SO4 Удаляли растворитель и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 10/1 (об./об. с получением соединения 49 (0,20 г, выход 54%). ЖХ-МС (ESI) m/z 458,3 (М+Н)+. (Соответствующую бороновую кислоту отделяли и применяли в качестве активного промежуточного соединения на следующей стадии.) Стадия h. К раствору соединения 49 (160 мг, 0,35 ммоль) в ДМЭ/H2O (3/1 (об./об.), 40 мл) последовательно при КТ в атмосфере Ar добавляли (S)-трет-бутил-2-(5-йод-1H-имидазол-2-ил)пирролидин-1 карбосилат (388 мг, 1,07 ммоль), NaHCO3 (289 мг, 3,44 ммоль) и Pd(dppf)Cl2CH2Cl2 (71 мг, 0,090 ммоль). После перемешивания при 80 С в течение ночи в атмосфере Ar реакционную смесь разбавляли EtOAc(100 мл). Отделяли органический слой, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 2/1 (об./об. с получением соединения 50 (151 мг, выход 64%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 676,4 (М+Н)+. Стадия i. К перемешиваемому раствору соединения 50 (140 мг, 0,21 ммоль) в диоксане (2 мл) при КТ добавляли 4 Н раствор HCl в диоксане (2 мл). После перемешивания при КТ в течение ночи реакционную смесь концентрировали, остаток сушили в вакууме с получением соли HCl, которую применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 476,2 (М+Н)+. Стадия j. К смеси соли HCl в ДМФ (2 мл) добавляли DIPEA (0,35 мл, 2,1 ммоль), затем N-Boc-LVal-OH (92 мг, 0,53 ммоль) и HATU (200 мг, 0,530 ммоль). После перемешивания при КТ в течение 30 мин реакционную смесь выливали в воду. Собирали твердое вещество при помощи фильтрования и очищали при помощи препаративной ВЭЖХ с получением соединения 51. 1H ЯМР (500 МГц, CD3OD) Схема 3-7 Стадия а. В соответствии со схемой 3-7 смесь соединения 52 (9,35 г, 50 ммоль), TMS-ацетиленаPPh3 (2,62 г, 10,0 ммоль) в безводном ТГФ (100 мл) кипятили с обратным холодильником в течение ночи в атмосфере Ar. Реакционную смесь концентрировали и остаток разбавляли водой (50 мл) и EtOAc (150 мл). Органический слой промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 10/1 (об./об. с получением соединения 53 (10,0 г, 98%) в виде желтой маслянистой жидкости. ЖХ-МС (ESI): m/z 205,1 (М+Н)+. Стадия b. Смесь соединения 53 (2,4 г, 11,7 ммоль) и K2CO3 (4,9 г, 35,3 ммоль) в ТГФ (20 мл) и МеОН (20 мл) перемешивали при КТ в течение 3 ч. Удаляли растворитель и остаток разбавляли EtOAc (150 мл), промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 10/1 (об./об. с получением соединения 54 (1,3 г, 84%) в виде желтой маслянистой жидкости. ЖХ-МС (ESI): m/z 133,1(М+Н)+. Стадия с. К раствору соединения 55 (25,0 г, 184 ммоль) в АсОН (125 мл) добавляли Br2 (11,0 мл, 220 ммоль). После перемешивания при КТ в течение 4 ч реакционную смесь фильтровали. Твердое вещество промывали Н 2 О и сушили в вакууме с получением соединения 56 (38 г, 96%) в виде белого твердого вещества. ЖХ-МС (ESI): m/z 215,0 (М+Н)+. Стадия d. Смесь соединения 54 (17,9 г, 83,3 ммоль), соединения 56 (11,0 г, 83,3 ммоль), CuI (1,59 г,0,25 ммоль), Et3N (23,00 мл, 166,6 ммоль). Pd(PPh3)2Cl2 (2,95 г, 4,20 ммоль) и PPh3 (4,40 г, 16,7 ммоль) в ДМФ (100 мл) перемешивали при 40 С в течение ночи в атмосфере N2. Реакционную смесь концентрировали и разбавляли остаток EtOAc (500 мл). Полученную смесь промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 10/1 (об./об. с получением соединения 57 (9,8 г, 45%). ЖХ-МС (ESI): m/z 267,1 (М+Н)+. Стадия е. К раствору соединения 57 (5,5 г, 21 ммоль) в ЕЮН (100 мл) последовательно добавляли гидрохлорид гидроксиламина (1,73 г, 25,0 ммоль) и NaOAc (2,05 г, 25,0 ммоль). После перемешивания при 60 С в течение 2 ч к реакционной смеси добавляли K2CO3 (4,3 г, 31 ммоль) и Н 2 О (15 мл). Полученную смесь кипятили с обратным холодильником в течение 12 ч, затем концентрировали. Остаток растворяли в EtOAc и полученную смесь промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и остаток сушили в вакууме с получением неочищенного соединения 58 (5,8 г). ЖХ-МС(ESI): m/z 282,1 (М+Н)+. Стадия f. Смесь соединения 58 (100 мг, 0,36 ммоль) и 5% Pd/C (75 мг) в EtOH (25 мл) перемешивали при КТ в течение ночи в атмосфере H2. Реакционную смесь фильтровали через CELITE545. Отфильтрованный осадок промывали МеОН (25 мл 3). Концентрировали фильтрат, очищали остаток при помощи колоночной хроматографии на силикагеле с получением соединения 59 (50 мг, 53%). ЖХ-МС(ESI): m/z 266,1 (М+Н)+. Стадия g. К раствору соединения 59 (2,0 г, 7,5 ммоль) в CH2Cl2 (75 мл) при -40 С в атмосфере N2 добавляли 4 Н раствор BBr2 в CH2Cl2 (12 мл, 45 ммоль). После перемешивания при КТ в течение ночи реакционную смесь гасили при помощи добавления воды (10 мл). Затем реакционную смесь обрабатывали насыщенным водным раствором NaHCO3 для достижения значения рН, равного 8. Промывали органический слой солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 2/1 (об./об. с получением соединения 60 (1,36 г, 76%) в виде белого твердого вещества. ЖХ-МС (ESI): m/z 238,1(М+Н)+. Стадия h. К раствору субстрата 7 (1,36 г, 5,7 ммоль) и пиридина (2,03 г, 25,7 ммоль) в CH2Cl2 (120 мл) при 0 С добавляли Tf2O (5,84 г, 20,7 ммоль) в CH2Cl2 (30 мл). После перемешивания при 0 С в течение 30 мин, реакционную смесь концентрировали и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 10/1 (об./об. с получением соединения 61 (2,4 г,84%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 502,0 (М+Н)+. Стадия i. Смесь соединения 61 (2,0 г, 4,0 ммоль), бис(пинаколато)дибора (5,1 г, 20 ммоль), ацетата калия (2,7 г, 28 ммоль) и Pd(dppf)Cl2 (0,98 г, 1,2 ммоль) и диоксана (80 мл) перемешивали при 80 С в течение ночи в атмосфере Ar. Затем реакционную смесь разбавляли EtOAc (100 мл). Полученную смесь промывали H2O (50 мл) и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 10/1 (об/об с получением соединения 62 (986 мг, выход 54%). ЖХ-МС (ESI) m/z: 458,3 (М+Н)+. (Соответствующую бороновую кислоту отделяли и применяли в качестве активного промежуточного соединения на следующей стадии.) Стадия j. К раствору соединения 62 (1,7 г, 3,7 ммоль) в ДМЭ/H2O (3/1 (об./об.), 40 мл) при КТ в атмосфере Ar последовательно добавляли (S)-трет-бутил-2-(5-йод-1H-имидазол-2-ил)пирролидин-1-карбоксилат (3,70 г, 10,0 ммоль), NaHCO3 (2,7 г, 32 ммоль) и Pd(dppf)Cl2 (0,65 мг, 0,80 ммоль). После перемешивания при 80 С в течение ночи в атмосфере Ar реакционную смесь разбавляли EtOAc (150 мл). Отделяли органический слой, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 2/1 (об./об. с получением соединения 63 (650 мг, 26%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 676,4 (М+Н)+. Стадия k. К перемешиваемому раствору соединения 63 (200 мг, 0,3 ммоль) в диоксане (3 мл) при КТ добавляли 4 Н раствор HCl в диоксане (3 мл). После перемешивания при КТ в течение ночи реакционную смесь концентрировали и сушили остаток в вакууме с получением соли HCl, которую применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 476,2 (М+Н)+. Стадия l. Затем к смеси соли HCl в ДМФ (3 мл) добавляли DIPEA (0,5 мл, 3,0 ммоль), затем N-MocL-Val-OH (130 мг, 0,740 ммоль) и HATU (281 мг, 0,740 ммоль). После перемешивания при КТ в течение 30 мин реакционную смесь выливали в H2O. Собирали твердое вещество при помощи фильтрования и очищали при помощи препаративной ВЭЖХ с получением соединения 64. 1 Н ЯМР (500 МГц, CD3OD) Схема 3-8 Стадия а. В соответствии со схемой 3-8 к раствору соединения 132 (3,70 г, 14,7 ммоль) в ДМФ (50 мл) при КТ добавляли гидрохлорид N,О-диметилгидроксиламина (1,46 г, 15,0 ммоль), HATU (6,15 г, 16,2 ммоль) и Et3N (2,22 г, 22,0 ммоль). После перемешивания при КТ в течение 24 ч реакционную смесь концентрировали и разбавляли остаток ДХМ (150 мл). Смесь последовательно промывали насыщенным водным раствором NH4Cl и солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 4/1(об./об. с получением соединения 133 (3,78 г, выход 87%) в виде желтого твердого вещества. ЖХ-МС(ESI): m/z 295,0 (М+Н)+. Стадия b. К раствору соединения 133 (3,53 г, 12,0 ммоль) в ТГФ (80 мл) при 0 С медленно добавляли 3 М раствор MeMgCl в ТГФ (6 мл). После перемешивания при 0 С в течение 1 ч, а затем при КТ в течение еще одного ч реакционную смесь гасили при помощи добавления насыщенного водного раствораNH4Cl. Реакционную смесь концентрировали и к остатку добавляли насыщенный водный раствор NaHCO3 (25 мл) и EtOAc (100 мл). Промывали органическую фазу солевым раствором и сушили безводнымNa2SO4. Удаляли растворитель и сушили остаток в вакууме с получением соединения 134 (3,0 г, 100%) в виде белого твердого вещества. ЖХ-МС (ESI): m/z 250,0 (М+Н)+. Стадия с. К раствору соединения 134 (2,80 г, 11,2 ммоль) в ДХМ (80 мл) добавляли i-Pr2NEt (5,79 г,44,8 ммоль). Смесь охлаждали до 0 С и по каплям добавляли TMSOTf (7,47 г, 33,6 ммоль). После перемешивания при 0 С в течение 30 мин, а затем при КТ в течение 1 ч реакционную смесь промывали последовательно насыщенным водным раствором NaHCO3 и солевым раствором и сушили безводнымNa2SO4. Удалял и растворитель и сушили остаток в вакууме с получением неочищенного соединения 135(3,6 г), которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 322,0(М+Н)+. Стадия d. К раствору соединения 135 (3,60 г, 11,2 ммоль) в ТГФ (60 мл) при 0 С по каплям добавляли раствор NBS (1,79 г, 10,1 ммоль) в ТГФ (20 мл). После перемешивания при 10 С в течение 1 ч реакционную смесь концентрировали и разбавляли остаток ДХМ (150 мл). Промывали смесь солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и сушили остаток в вакууме с получением неочищенного соединения 136 (3,6 г), которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 327,9 (М+Н)+. Стадия е. К раствору соединения 136 (3,6 г, 10,9 ммоль) в EtOAc (100 мл) при КТ добавляли (S)-NBoc-Pro-OH (2,47 г, 11,5 ммоль) и Et3N (3,31 г, 32,7 ммоль). После перемешивания при КТ в течение 5 ч реакционную смесь последовательно промывали насыщенным водным раствором NaHCO3 и сушили безводным Na2SO4. Удаляли растворитель и сушили остаток в вакууме с получением неочищенного соединения 137 (5,0 г), которое применяли на следующей стадии без дополнительной очистки ЖХ-МС(ESI): m/z 463,1 (М+Н)+. Стадия f Смесь неочищенного соединения 137 (5,0 г) и NH4OAc (8,39 г, 109 ммоль) в толуоле (100 мл) перемешивали при 115 С в течение ночи Удаляли растворитель и разбавляли остаток EtOAc (200 мл) Промывали смесь водой и сушили безводным Na2SO4 Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 3/1 (об./об. с получением- 21024853 соединения 138 (1,2 г, 25%) в виде белого твердого вещества. ЖХ-МС (ESI): m/z 443,1 (М+Н)+. Стадия g. К смеси соединения 138 (442 мг, 1,00 ммоль), соединения 139 (546 мг, 1,10 ммоль) и NaHCO3 (336 мг, 4,00 ммоль) в 1,2-диметоксиэтане (8 мл) и H2O (2 мл) в атмосфере N2 добавлялиPd(dppf)Cl2CH2Cl2 (163 мг, 0,20 ммоль). После перемешивания при 80 С в течение ночи реакционную смесь концентрировали и разбавляли остаток EtOAc (50 мл) и H2O (10 мл). Промывали органическую фазу солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 1/2 (об./об. с получением соединения 140 (500 мг, выход 68%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 733,4HCl в диоксане (2,0 мл). После перемешивания при КТ в течение 2 ч реакционную смесь концентрировали и растворяли остаток в воде (5 мл), и добавляли насыщенный водный раствор NaHCO3 для достижения значения рН, равного 8. Полученную смесь насыщали NaCl и экстрагировали в ДХМ (15 мл 5). Объединяли экстракты и сушили безводным Na2SO4. Удаляли растворитель и сушили остаток в вакууме с получением свободного основания, которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 633,3 (М+Н)+. Стадия i. Затем свободное основание растворяли в ДХМ (5 мл) и к смеси добавляли N-Moc-L-ValOH (40 мг, 0,23 ммоль) и DIC (29 мг, 0,23 ммоль). После перемешивания при КТ в течение 20 мин реакционную смесь концентрировали и остаток очищали при помощи препаративной ВЭЖХ с получением соединения 141. ЖХ-МС (ESI): m/z 790,4 (М+Н)+. Схема 4-1 Пример 4 - синтез соединения формулы IIe Стадия а. В соответствии со схемой 4-1 к раствору соединения 27 (5,0 г, 20 ммоль) в CH3CN (200 мл) при КТ добавляли EDCI (5,8 г, 30 ммоль), HOBt (675 мг, 30 ммоль), MeNH(OMe)HCl (2,93 г, 30 ммоль) и Et3N (6,1 г, 60 ммоль). После перемешивания при КТ в течение 3 ч реакционную смесь концентрировали и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 5/1 (об./об. с получением соединения 28 (5,4 г, выход 92%) в виде белого твердого вещества. ЖХ-МС (ESI): m/z 294,0 (М+Н)+. Стадия b. К раствору соединения 28 (2,9 г, 10 ммоль) в ТГФ (100 мл) при 0 С в атмосфере N2 медленно добавляли 3 М раствор MeMgCl в ТГФ (20 ммоль). После перемешивания при 0 С в течение 1 ч, а затем при КТ в течение 1 ч реакционную смесь гасили при помощи добавления нескольких капель водного раствора NH4Cl. Реакционную смесь концентрировали и разбавляли остаток EtOAc (100 мл). Промывали органическую фазу насыщенным водным раствором NaHCO3 и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 10 1 (об./об. с получением соединения 29 (2,3 г, выход 92%) ЖХ-МС (ESI): m/z 249,0(М+Н)+. Стадия с. К раствору соединения 29 (1,84 г, 7,4 ммоль) в ДХМ (100 мл) при 0 С по каплям добавляли Br2 (18,8 г, 14,7 ммоль). После перемешивания при 0 С в течение 30 мин реакционную смесь нагревали до КТ при перемешивании в течение 2 ч. Затем реакционную смесь последовательно промывали водой, насыщенным водным раствором NaHCO3 и сушили органическую фазу безводным Na2SO4. Удаляли растворитель и сушили остаток в вакууме с получением неочищенного соединения 30 (2,0 г) в виде желтого твердого вещества, которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС(ESI): m/z 326,9 (М+Н)+. Стадия d. К раствору соединения 30 (1,95 г, 5,9 ммоль) в ДХМ (50 мл) при КТ добавляли N-Boc-LPro-OH (1,6 г, 7,3 ммоль) и Et3N (1,7 мл, 12,2 ммоль). После перемешивания при КТ в течение 2 ч реакционную смесь последовательно промывали насыщенным раствором NH4Cl и солевым раствором, суши- 22024853 ли органическую фазу безводным Na2SO4. Удаляли растворитель и сушили остаток в вакууме с получением неочищенного соединения 31 (2,4 г), которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 462,1 (М+Н)+. Стадия е. Смесь соединения 31 (2,4 г, 5,2 ммоль) и NH4OAc (4,0 г, 52 ммоль) в толуоле (52 мл) перемешивали при 110 С в течение ночи. Затем реакционную смесь охлаждали до КТ и разбавляли EtOAc(100 мл). Последовательно промывали смесь насыщенным водным раствором Na2CO3 (50 мл 2) и солевым раствором; сушили органическую фазу Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 1/1 (об./об. с получением соединения 32 (1,4 г, 62%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 442,1 (М+Н)+. Стадия f. К смеси соединения 32 (1,0 г, 2,3 ммоль), (S)-трет-бутил 2-(5-(4-(4,4,5,5-тетраметил-1,3,2 диоксаборолан-2-ил)-1H-имидазол-2-ил)пирролидин-1-карбоксилата (1,0 г, 2,3 ммоль) и NaHCO3 (0,76 г,9,0 ммоль) в 1,2-диметоксиэтане (30 мл) и H2O (10 мл) в атмосфере N2 добавляли Pd(dppf)Cl2 (277 мг,0,34 ммоль). После перемешивания при 80 С в течение ночи в атмосфере N2 реакционную смесь концентрировали. Разбавляли остаток H2O (50 мл) и экстрагировали водную фазу в EtOAc (50 мл 3). Объединяли экстракты и промывали солевым раствором, и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 5/2(об./об. с получением соединения 33 (1,0 г, выход 78%) в виде желтого твердого вещества. ЖХ-МС(ESI): m/z 675,4 (М+Н)+. Стадия g. К перемешиваемому раствору соединения 33 (250 мг, 0,37 ммоль) в диоксане (3 мл) по каплям при КТ добавляли 4,0 Н раствор HCl в диоксане (3 мл). После перемешивания при КТ в течение 4 ч реакционную смесь концентрировали и сушили остаток в вакууме с получением соли HCl, которую применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 475,3 (М+Н)+. Стадия h. Затем соль HCl суспендировали в ТГФ (5 мл) и DIPEA (0,35 мл) и N-Moc-L-Val-OH (130 мг, 0,74 ммоль) при КТ. После перемешивания при КТ в течение 15 мин добавляли HATU (340 мг, 0,89 ммоль) и полученную реакционную смесь дополнительно перемешивали при КТ в течение 2 ч. Удаляли растворитель и очищали остаток при помощи препаративной ВЭЖХ с получением соединения 34. 1 Н ЯМР (500 МГц, CDCl3)8,04-8,06 (m, 1H), 7,96-7,99 (m, 2 Н), 7,91-7,92 (m, 2 Н), 7,79 (s, 1H), 7,70-7,71 (m,2 Н), 7,66-7,67 (m, 2 Н), 7,60-7,61 (m, 2 Н), 5,29-5,31 (m, 2 Н), 4,27 (s, 2H), 4,13 (s, 2 Н), 3,92 (s, 2 Н), 3,68 (s,6 Н), 2,63 (s, 2 Н), 2,17-2,32 (m, 6 Н), 2,12 (s, 2H), 0,93-0,97 (m, 12 Н) ppm; ЖХ-МС (ESI): m/z 789,4 (М+Н)+. Пример 5 - синтез соединений формулы IIIl Схема 5-1 Стадия а. В соответствии со схемой 5-1 к смеси соединения 65 (300 мг, 1,05 ммоль), (S)-трет-бутил 2-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-бензо[d]имидазол-2-ил)пирролидин-1-карбоксилата (1,14 г, 2,75 ммоль) и NaHCO3 (740 мг, 8,80 ммоль) в 1,2-диметоксиэтане (30 мл) и воде (10 мл) при КТ в атмосфере N2 добавляли Pd(dppf)Cl2 (179 мг, 0,220 ммоль). После перемешивания при 80 С в течение ночи реакционную смесь концентрировали. Разбавляли остаток ДХМ (100 мл) и водой (25 мл). Промывали органическую фазу солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 2/1 (об./об. с получением соединения 66 (650 мг, 86%). ЖХ-МС (ESI): m/z 699,4 (М+Н)+. Стадия b. К раствору соединения 66 (110 мг, 0,16 ммоль) в диоксане (2 мл) при КТ добавляли 4,0 Н раствор HCl в диоксане (2 мл). После перемешивания при КТ в течение 3 ч реакционную смесь концентрировали и сушили остаток в вакууме с получением соли HCl, которую непосредственно применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 499,3 (М+Н)+. Стадия с. Затем соль HCl растворяли в ДМФ (2 мл), затем при КТ добавляли DIPEA (207 мг, 16 ммоль), N-Moc-L-Val-OH (68 мг, 0,39 ммоль) и HATU (148 мг, 0,39 ммоль). После перемешивания при КТ в течение 15 мин реакционную смесь добавляли в воду. Собирали твердое вещество при помощи фильтрования и очищали при помощи препаративной ВЭЖХ с получением соединения 67. ЖХ-МС (ESI)m/z 813,4 (М+Н)+. Пример 6 - синтез соединений формулы IIId Схема 6-1 Стадия а. В соответствии со схемой 6-1 к смеси соединения 70 (8,00 г, 35,7 ммоль, приобретено в(10,50 г, 107,1 ммоль) в 1,4-диоксане (600 мл) при КТ в атмосфере N2 добавляли Pd(dppf)Cl2 (2,9 г, 3,6 ммоль). После перемешивания в течение 3 ч в атмосфере N2 реакционную смесь охлаждали до КТ и фильтровали через Celite545. Отфильтрованный осадок промывали EtOAc (100 мл 3). Фильтрат концентрировали и разбавляли остаток EtOAc (500 мл). Полученную смесь промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 1/1 (об./об. с получением соединения 71 (8,28 г, выход 86%) в виде светло-коричневого твердого вещества. ЖХ-МС (ESI) m/z 272,1 (М+Н)+. Стадия b. К смеси соединения 71 (5,90 г, 21,8 ммоль), (S)-трет-бутил 2-(5-йод-1H-имидазол-2 ил)пирролидин-1-карбоксилата (9,50 г, 26,2 ммоль), NaHCO3 (7,30 г, 87,2 ммоль) в 1,2-диметоксиэтане(500 мл) в воде (150 мл) в атмосфере N2 добавляли Pd(dppf)Cl2 (3,6 г, 4,4 ммоль). После перемешивания при 80 С в течение ночи реакционную смесь концентрировали и разбавляли остаток EtOAc (250 мл) и водой (50 мл). Промывали органический слой солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (ДХМ/МеОН = 5/1 (об./об. с получением соединения 72 (5,30 г, выход 64%) в виде желтого твердого вещества. ЖХ-МС(ESI) m/z 381,2 (М+Н)+. Стадия с. К раствору соединения 72 (2,0 г, 5,26 ммоль) в 40 мл пиридина по каплям при 0 С добавляли Tf2O (3,71 г, 13,1 ммоль). После перемешивания при 0 С в течение 1 ч и при КТ в течение 3 ч реакционную смесь концентрировали. Очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 4/1 (об./об. с получением соединения 73 (2,04 г, выход 60%) в виде желтого твердого вещества. ЖХ-МС (ESI) m/z 645,1 (М+Н)+. Стадия d. К смеси соединения 73 (500 мг, 0,78 ммоль), метил (S)-3-метил-1-оксо-1-S)-2-(6-(4,4,5,5 тетраметил-1,2,3-диоксаборолан-2-ил)-1H-бензо[d]имидазол-2-ил)бутан-2-илкарбамата (74) (419 мг, 0,89 ммоль) и NaHCO3 (299 г, 3,56 ммоль) в 1,2-диметоксиэтане (60 мл) и воде (20 мл) при КТ в атмосфере N2 добавляли Pd(dppf)Cl2 (147 мг, 0,18 ммоль). После перемешивания при 80 С в течение ночи в атмосфереN2 реакционную смесь концентрировали. Разбавляли остаток EtOAc (100 мл) и водой (25 мл). Промывали органический слой солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 1/1 (об./об. с получением соединения 75 (0,40 г, выход 64%) в виде желтого твердого вещества. ЖХ-МС (ESI) m/z 707,4 (М+Н)+. Стадия е. К раствору соединения 75 (114 мг, 0,161 ммоль) в диоксане (2 мл) при КТ добавляли 4 Н раствор HCl в диоксане (2 мл). После перемешивания при КТ в течение 3 ч реакционную смесь концентрировали и сушили остаток в вакууме с получением соли HCl, которую применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI) m/z 607,3 (М+Н)+. Стадия f. Затем соль HCl растворяли в ДМФ (2 мл), затем при КТ добавляли Et3N (0,11 мл, 0,81 ммоль), N-Moc-L-Val-OH (32 мг, 0,18 ммоль) и HATU (69 мг, 0,18 ммоль). После перемешивания при КТ в течение 1 ч реакционную смесь концентрировали и очищали остаток при помощи препаративной Схема 6-2 Стадия а. В соответствии со схемой 6-2 к раствору соединения 78 (50,0 г, 0,30 моль) в ТГФ (500 мл) и Н 2 О (500 мл) добавляли K2CO3 (83 г, 0,60 моль) и (Вос)2 О (73,0 г, 0,330 моль). После перемешивания при КТ в течение ночи реакционную смесь концентрировали и экстрагировали остаток EtOAc (250 мл 3). Объединяли экстракты, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и сушили остаток в вакууме с получением неочищенного соединения 78 (62 г), которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI) m/z 230,1 (М+Н)+. Стадия b. К раствору соединения 78 (60,0 г, 260 ммоль) в EtOH (1 л) при КТ медленно добавлялиNaBH4 (50,0 г, 1,30 моль). После перемешивания при КТ в течение ночи реакционную смесь гасили при помощи добавления ацетона (10 мл). Полученную смесь концентрировали и разбавляли остаток EtOAc(500 мл). Промывали смесь солевым раствором и сушили в вакууме. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 1/1 (об./об. с получением соединения 79 (42,0 г, выход 80%) в виде белого твердого вещества. ЖХ-МС (ESI) m/z 202,1 (М+Н)+. Стадия с. К раствору соединения 79 (30,0 г, 150 ммоль) и ДМСО (35,0 г, 450 ммоль) в ДХМ (1 л) при -78 С добавляли оксалилхлорид (28,0 г, 220 ммоль). После перемешивания при -78 С в течение 4 ч к реакционной смеси добавляли Et3N (60,0 г, 600 ммоль) и полученную смесь дополнительно перемешивали в течение 1 ч при -78 С. Затем реакционную смесь гасили при помощи добавления H2O. Отделяли органический слой, а водный слой экстрагировали ДХМ (200 мл 2). Объединяли экстракты, промывали солевым раствором и сушили Na2SO4. Удаляли растворитель и сушили в вакууме с получением неочищенного соединения 80 (22,0 г) в виде бесцветной маслянистой жидкости, которое немедленно применяли без дополнительной очистки. ЖХ-МС (ESI) m/z 200,1 (М+Н)+. Стадия d. Смесь соединения 80 (7,7 г, 38,5 ммоль), 6-бромпиридин-2,3-диамина (8,0 г, 42,8 ммоль)(публикация международной заявки РСТ WO 2008021851) и йода (1,08 г, 4,28 ммоль) в АсОН (30 мл) перемешивали при КТ в течение ночи. Нейтрализовали реакционную смесь при помощи добавления насыщенного водного раствора NaHCO3. Полученную смесь экстрагировали EtOAc (200 мл 3). Объединяли экстракты, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (ДХМ/МеОН = 80/1 (об./об. с получением соединения 81 (7,8 г, выход 55%). ЖХ-МС (ESI) m/z 367,1 (М+Н)+. Стадия е. Смесь соединения 82 (10,0 г, 20,1 ммоль), бис(пинаколато)дибора (7,65 г, 30,1 ммоль),ацетата калия (6,89 г, 70,3 ммоль) и Pd(dppf)Cl2CH2Cl2 (886 мг, 1,0 ммоль) в 1,4-диоксане (200 мл) перемешивали при 80 С в течение 3 ч в атмосфере N2. Фильтровали реакционную смесь через CELITE545 и отфильтрованный осадок промывали EtOAc (200 мл 3). Промывали фильтрат солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (ДХМ/МеОН = 50/1 (об./об. с получением соединения 83 (9,8 г, выход 89%) в виде белого твердого вещества. ЖХ-МС (ESI) m/z 547,3 (М+Н)+. Стадия f. Смесь соединения 81 (2,0 г, 5,4 ммоль), соединения 83 (2,9 г, 5,4 ммоль), NaHCO3 (1,60 г,18,9 ммоль) и Pd(dppf)Cl2CH2Cl2 (239 мг, 0,27 ммоль) в 1,2-диметоксиэтане (90 мл) и воде (30 мл) перемешивали при 80 С в течение ночи в атмосфере N2. Концентрировали реакционную смесь и к остатку добавляли ДХМ (200 мл) и воду (50 мл). Промывали органическую фазу солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии- 25024853 на силикагеле (ДХМ/МеОН = 80/1 (об./об. с получением соединения 84 (1,5 г, выход 40%) в виде желтого твердого вещества. ЖХ-МС (ESI) m/z 707,4 (М+Н)+. Стадия g. К раствору соединения 84 (200 мг, 0,28 ммоль) в 3 мл диоксана добавляли 4 Н раствор HCl в диоксане (3 мл) После перемешивания при КТ в течение 3 ч реакционную смесь концентрировали и сушили остаток в вакууме с получением соли HCl, которую применяли наследующей стадии без дополнительной очистки ЖХ-МС (ESI) m/z 607,3 (М+Н)+. Стадия h. Затем соль HCl растворяли в ДМФ (3 мл) и к полученной смеси добавляли Et3N (0,20 мл,1,4 ммоль), N-Moc-L-Val-OH (55 мг, 0,31 ммоль) и HATU (118 мг, 0,31 ммоль). После перемешивания при КТ в течение 1 ч реакционную смесь концентрировали и очищали остаток при помощи препаративной ВЭЖХ с получением соединения 85. ЖХ-МС (ESI): m/z 764,4 (М+Н)+. Схема 6-3 Стадия а. В соответствии со схемой 6-3 к раствору N-Boc-L-Pro-OH (29 г, 135 ммоль) и DIPEA (29 г,225 ммоль) в ТГФ (500 мл) при КТ добавляли HATU (51 г, 135 ммоль). После перемешивания при КТ в течение 10 мин добавляли 4-бромбензо-1,2-диамин (95) (25 г, 135 ммоль) и полученный раствор дополнительно перемешивали при КТ в течение нескольких ч. Затем реакционную смесь концентрировали и разбавляли остаток EtOAc (500 мл). Полученную смесь несколько раз промывали водой (100 мл 3) и сушили безводным Na2SO4. Удаляли растворитель и сушили остаток в вакууме с получением смеси неочищенных соединений 96 и 96', которые применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI): m/z 384,1 (М+Н)+. Стадия b. Смесь неочищенных соединений 96 и 96', полученных в представленной выше реакции, в АсОН (1000 мл) перемешивали при 40 С в течение 12 ч. Затем реакционную смесь осторожно нейтрализовали при помощи добавления насыщенного водного раствора бикарбоната натрия до достижения значения рН, равного 8. Полученную смесь несколько раз экстрагировали в EtOAc (250 мл 3). Объединяли экстракты, промывали водой и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи хроматографии на силикагеле (петролейный эфир/EtOAc = 4/1 (об./об. с получением соединения 97 (35 г, выход 71%, получено в две стадии из соединения 95) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 366,1 (М+Н)+. Стадия с. Смесь соединения 97 (10,0 г, 27,3 ммоль), триметилсилилацетилена (4,0 г, 41,0 ммоль),DIPEA (3,5 г, 27,3 ммоль), CuI (220 мг, 1,15 ммоль), PPh3 (1,2 г, 4,6 ммоль) и Pd(PPh3)2Cl2 (1,6 г, 2,3 ммоль) в безводном ТГФ (200 мл) кипятили с обратным холодильником в течение ночи в атмосфере N2. Концентрировали реакционную смесь и разбавляли остаток EtOAc (250 мл). Промывали смесь солевым- 26024853 раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 3/1 (об./об. с получением соединения 98(150 мл) перемешивали при КТ в течение 3 ч. Фильтровали реакционную смесь через CELITE545 и отфильтрованный осадок промывали EtOAc (100 мл 3). Концентрировали фильтрат и разбавляли остаток ДХМ (250 мл). Промывали смесь солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 2/1 (об./об. с получением соединения 99 (4,7 г, выход 75%). ЖХ-МС (ESI): m/z 312,2(М+Н)+. Стадия е. К раствору метагидроксибензальдегида (100) (30,0 г, 0,24 моль) в сухом CHCl3 (245 мл) медленно в течение 40-45 мин при КТ добавляли бром (12,36 мл, 0,24 моль). После завершения добавления реакционную смесь перемешивали при КТ в течение 3 ч. Затем для нейтрализации смеси осторожно добавляли насыщенный водный раствор NaHCO3. Промывали органический слой солевым раствором и сушили Na2SO4. Удаляли растворитель и сушили остаток в вакууме с получением неочищенного соединения 101 (37 г) в виде коричневого твердого вещества. ЖХ-МС (ESI): m/z 200,9 (М+Н)+. Стадия f. К раствору соединения 101 (10 г, 49,8 моль) в безводной смеси ТГФ/ДМФ (5/1 (об./об.),120 мл) при 0 С в атмосфере N2 добавляли NaH (2,0 г, 51 ммоль, 60% дисперсия в минеральном масле). После перемешивания при КТ в течение 30 мин к реакционной смеси в течение 20-25 мин добавляли бензилбромид (8,7 мл, 73 ммоль). Полученную смесь перемешивали при КТ в течение ночи и гасили реакционную смесь при помощи добавления насыщенного водного раствора NH4Cl (50 мл). Концентрировали реакционную смесь и разбавляли остаток EtOAc (150 мл) и водой (50 мл). Промывали органическую фазу солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 10/1 (об./об. с получением соединения 102 (11 г, выход 77%). ЖХ-МС (ESI): m/z 291,0 (М+Н)+. Стадия g. Смесь соединения 99 (2,80 г, 9,0 ммоль), соединения 102 (2,6 г, 9,0 ммоль), Pd(PPh3)2Cl2(6,3 г, 0,9 ммоль), CuI (2,55 г, 1,34 ммоль), Et3N (2,5 мл, 18 ммоль) и PPh3 (4,7 г, 1,8 ммоль) в ДМФ (100 мл) перемешивали при 60 С в течение 12 ч. Затем реакционную смесь концентрировали. Остаток разбавляли EtOAc (150 мл) и водой (50 мл). Промывали органическую фазу солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 10/1 (об./об. с получением соединения 103 (4,0 г, выход 86%). ЖХ-МС (ESI): m/z 522,2 (М+Н)+. Стадия h К раствору соединения 103 (4,1 г, 7,9 ммоль) в EtOH (100 мл) при КТ последовательно добавляли гидрохлорид гидроксиламина (650 мг, 9,4 ммоль) и NaOAc (770 мг, 9,4 ммоль) После перемешивания при 60 С в течение 2 ч к реакционной смеси добавляли K2CO3 (1,64 г, 11,85 ммоль) и воду (20 мл) Полученную смесь кипятили с обратным холодильником в течение 12 ч Затем реакционную смесь концентрировали и разбавляли остаток EtOAc (200 мл) и водой (20 мл) Промывали органическую фазу солевым раствором и сушили безводным Na2SO4 Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 5/1 (об./об.) до ДХМ/МеОН = 5/1(об./об. с получением соединения 104 (1,5 г, выход 36%). ЖХ-МС (ESI): m/z 537,2 (М+Н)+. Стадия i. Смесь соединения 104 и 10% Pd/C (1,5 г) в МеОН (50 мл) перемешивали при КТ в течение ночи в атмосфере H2. Затем реакционную смесь фильтровали через CELITE545 и отфильтрованный осадок промывали МеОН (50 мл 3). Концентрировали фильтрат и очищали остаток при помощи колоночной хроматографии на силикагеле с получением соединения 105 (670 мг, выход 56%). ЖХ-МС (ESI):m/z 431,2 (M+H)+. Стадия j. К раствору соединения 105 (650 мг, 1,5 ммоль) в безводном пиридине (711 мг, 9,0 ммоль) при 0 С добавляли Tf2O (1,07 г, 3,8 ммоль). После перемешивания при КТ в течение ночи реакционную смесь концентрировали и разбавляли остаток EtOAc (100 мл). Промывали смесь солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 5/1 (об./об. с получением соединения 106 (720 мг,выход 69%). ЖХ-МС (ESI): m/z 695,1 (М+Н)+. Стадия k. Смесь соединения 106 (410 мг, 0,6 ммоль), бис(пинаколато)дибора (227 мг, 0,9 ммоль),PdCl2(dppf)CH2Cl2 (100 мг, 0,12 ммоль) и KOAc (235 мг, 2,4 ммоль) в диоксане (15 мл) перемешивали при 80 С в течение 1 ч в атмосфере N2 Реакционную смесь применяли на следующей стадии без какой-либо обработки ЖХ-МС (ESI): m/z 673,2 (М+Н)+. Стадия l. К полученной выше реакционной смеси в атмосфере N2 добавляли (S)-трет-бутил-2-(5 йод-1H-имидазол-2-ил)пирролидин-1-карбоксилат (370 мг, 1,02 ммоль), затем NaHCO3 (201 мг, 2,4 ммоль), 1,2-диметоксиэтан (4 мл), воду (2 мл) и Pd(dppf)Cl2CH2Cl2 (100 мг, 0,12 ммоль). После перемешивания при 80 С в течение 2 ч в атмосфере N2 к реакционной смеси добавляли K2CO3 (691 мг, 5 ммоль) и МеОН (20 мл). После перемешивания при КТ в течение 30 мин реакционную смесь концентрировали. Разбавляли остаток EtOAc (150 мл) и водой (50 мл). Промывали органический слой солевым раствором и- 27024853 сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 5/1 (об./об. с получением соединения 108 (140 мг,выход 36%, получено в две стадии из соединения 107). ЖХ-МС (ESI) m/z 650,3 (М+Н)+. Стадия m. К раствору соединения 108 (135 мг, 0,2 ммоль) в диоксане (2 мл) при КТ добавляли 4 Н раствор HCl в диоксане (2 мл). После перемешивания при КТ в течение ночи реакционную смесь концентрировали и сушили остаток в вакууме с получением соли HCl, которую применяли на следующей стадии без дополнительной очистки. ЖХМС (ESI): m/z 450,2 (М+Н)+. Стадия n. Затем соль HCl растворяли в ДМФ (2 мл) и к полученной смеси добавляли DIPEA (0,33 мл, 2,0 ммоль), N-THPoc-L-Val-OH (108 мг, 0,50 ммоль) и HATU (190 мг, 0,50 ммоль). После перемешивания при КТ в течение 15 мин реакционную смесь добавляли в ледяную воду. Собирали твердое вещество при помощи фильтрования и очищали при помощи препаративной ВЭЖХ с получением соединения 109. ЖХ-МС (ESI): m/z 848,4 (М+Н)+. Схема 6-4 Стадия а. В соответствии со схемой 6-4 к раствору (S)-4-(трет-бутоксикарбонил)морфин-3 карбоновой кислоты (4,1 г, 22,0 ммоль) и DIPEA (4,3 г, 33,0 ммоль) в ТГФ (100 мл) при КТ добавляли соединение 95 (4,6 г, 20,0 ммоль). После перемешивания в течение 5 мин к реакционной смеси добавлялиHATU (7,6 г, 20,0 ммоль) и полученную смесь перемешивали при КТ в течение 2 ч. Концентрировали реакционную смесь и разбавляли остаток EtOAc (200 мл) и водой (50 мл). Промывали органическую фазу солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и сушили остаток в вакууме с получением неочищенной смеси соединений 110 и 110' (10 г), которую применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI) m/z 400,1 (М+Н)+. Стадия b. Смесь соединений 110 и 110' (10 г) в АсОН (50 мл) перемешивали при 40 С в течение 16 ч. Затем реакционную смесь добавляли в ледяную воду (200 мл) и нейтрализовали при помощи добавления насыщенного водного раствора Na2CO3 для достижения значения рН, равного 8. Полученную смесь экстрагировали в EtOAc (100 мл 3), объединяли экстракты, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 1/3 (об./об. с получением соединения 111 (4,5 г, выход 60%; получено в две стадии из соединения 95) в виде желтого твердого вещества. ЖХ-МС (ESI) m/z 382,1(М+Н)+. Стадия с. К раствору 1-(6-бромнафталин-2-ил)-2-хлорэтанона (112) (27,0 г, 95,2 ммоль) в ДХМ (200 мл) последовательно добавляли (S)-4-(трет-бутоксикарбонил)морфин-3-карбоновую кислоту (20,0 г, 86,6 ммоль) и Et3N (60,0 мл, 433 ммоль). После перемешивания при 45 С в течение ночи реакционную смесь последовательно промывали насыщенным водным раствором NaHCO3 (50 мл), насыщенным водным- 28024853 раствором NH4Cl (50 мл) и солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и сушили остаток в вакууме с получением неочищенного соединения 113 (41,4 г), которое применяли на следующей стадии без дополнительной очистки. ЖХ-МС (ESI) m/z 478,1 (М+Н)+. Стадия d. Смесь неочищенного соединения 113 (41,4 г) и NH4OAc (100 г, 1,30 моль) в толуоле (300 мл) перемешивали при 120 С в течение ночи. Концентрировали реакционную смесь и разбавляли остаток EtOAc (500 мл). Промывали смесь водой и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = градиент от 6/1 до 1/1 (об./об. с получением соединения 114 (24 г, 61%; получено в две стадии из соединения 112) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 458,1 (М+Н)+. Стадия е. К смеси соединения 114 (3 г, 6,55 ммоль), бис(пинаколато)дибора (1,83 г, 7,2 ммоль) иK2CO3 (1,67 г, 17,03 ммоль) в 1,4-диоксане (100 мл) в атмосфере N2 добавляли Pd(dppf)Cl2 ДХМ (0,8 г,0,98 ммоль). После перемешивания при 80 С в течение ночи в атмосфере N2 реакционную смесь фильтровали через CELITE545 и отфильтрованный осадок промывали EtOAc (100 мл 3). Промывали фильтрат солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (ДХМ/МеОН = 50/1 (об./об. с получением соединения 115 (2,0 г, выход 61%). ЖХ-МС (ESI): m/z 506,3 (М+Н)+. Стадия f. К смеси соединения 111 (500 мг, 1,3 ммоль), соединения 115 (900 мг, 1,78 ммоль) и NaHCO3 (328 мг, 3,9 ммоль) в ДМЭ (15 мл) и воде (5 мл) в атмосфере N2 добавляли Pd(dppf)Cl2 ДХМ (106 мг, 0,13 ммоль). После перемешивания при 80 С в течение ночи в атмосфере N2 реакционную смесь концентрировали и разбавляли остаток EtOAc (100 мл) и водой (25 мл). Промывали органическую фазу солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 4/1 (об./об. с получением соединения 116 (310 мг, выход 35%) в виде желтого твердого вещества. ЖХ-МС (ESI): m/z 703,3 (M+Na)+. Стадия g. К перемешиваемому раствору соединения 116 (150 мг, 0,31 ммоль) в диоксане (3,0 мл) при КТ добавляли 4 Н раствор HCl в диоксане (3,0 мл). После перемешивания при КТ в течение 3 ч реакционную смесь концентрировали и сушили остаток в вакууме с получением соли HCl, которую применяли на следующей стадии без дополнительной очистки. Стадия h. Затем соль HCl растворяли в ДМФ (3,0 мл) и к полученной смеси последовательно добавляли DIPEA (0,43 мл, 2,5 ммоль), N-Moc-L-Val-OH (136 мг, 0,78 ммоль) и HATU (353 мг, 0,93 ммоль). После перемешивания при КТ в течение 2 ч реакционную смесь концентрировали и очищали остаток при помощи препаративной ВЭЖХ с получением соединения 117. ЖХ-МС (ESI): m/z 795,4 (М+Н)+. Пример 7 - синтез соединений формулы IIIg Схема 7-1 Стадия а. В соответствии со схемой 7-1 к раствору 6-бромхинолин-2(1 Н)-она (70) (0,40 г, 1,8 ммоль) в безводном пиридине (12 мл) при 0 С по каплям добавляли Tf2O (0,81 г, 2,9 ммоль). После перемешивания при 0 С в течение 1 ч и при КТ в течение 3 ч реакционную смесь концентрировали. Растворяли остаток в ДХМ (100 мл); полученную смесь промывали водой (25 мл х 3) и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/ацетон = 2/1 (об./об. с получением соединения 86 (0,54 г, выход 84%) в виде желтого твердого вещества. ЖХ-МС (ESI) m/z 355,9 (М+Н)+. Стадия b. К смеси соединения 86 (0,54 г, 1,5 ммоль), (S)-трет-бутил 2-(6-(4,4,5,5-тетраметил-1,3,2 диоксаборолан-2-ил)-1H-бензо[d]имидазол-2-ил)пирролидин-1-карбоксилата (1,24 г, 3,0 ммоль) и NaHCO3 (1,01 г, 12,0 ммоль) в 1,2-диметоксиэтане (30 мл) и воде (10 мл) при КТ в атмосфере N2 добавлялиPd(dppf)Cl2CH2Cl2 (0,27 г, 0,3 ммоль). После перемешивания при 80 С в течение ночи реакционную смесь концентрировали. Остаток разбавляли EtOAc (100 мл) и водой (25 мл). Отделяли органическую фазу, промывали солевым раствором и сушили безводным Na2SO4. Удаляли растворитель и очищали остаток при помощи колоночной хроматографии на силикагеле (петролейный эфир/EtOAc = 1/1 (об./об. с получением соединения 87 (1,0 г, выход 95%) в виде желтого твердого вещества. ЖХ-МС (ESI) m/z 700,4 (М+Н)+. Стадия с. К раствору соединения 87 (100 мг, 0,14 ммоль) в диоксане (2 мл) при КТ добавляли 4 Н
МПК / Метки
МПК: A01N 57/00, A61K 31/675
Метки: ингибиторы, вгс
Код ссылки
<a href="https://eas.patents.su/30-24853-ingibitory-ns5a-vgs.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы ns5a вгс</a>
Имидазопиразиновые ингибиторы syk, фармацевтическая композиция, содержащая имидазопиразиновые ингибиторы syk, и способы их применения

Номер патента: 21293
Опубликовано: 29.05.2015
Авторы: Ли Сёнг Х., Сюй Цзяньцзюнь, Митчелл Скотт А., Бломгрен Питер А., Стэффорд Дуглас Г., Карри Кевин С., Кропф Джеффри И.
МПК: A61K 31/535
Метки: композиция, фармацевтическая, применения, способы, содержащая, ингибиторы, имидазопиразиновые
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль,где R1 означает (2-метил-2-гидроксипропокси)пиридин-6-ил, (2-метоксиэтокси)пиридинил, 2-(диметиламино)этокси-3-пиридинил, гидроксиэтокси-5-пиридинил, (3-метил-3-гидроксиазетидин)пиридин-3-ил, (3-метил-3-гидроксиазетидин)пиридин-2-ил, (3-гидроксиазетидин)пиридин-2-ил, (гидрокси(диметилэтил))-5-пиридинил, (4-метил-4-гидроксипиперидин)пиридин-2-ил,...
Ингибиторы акт и p70 s6-киназы

Номер патента: 18947
Опубликовано: 29.11.2013
Авторы: Шеферд Тимоти Алан, Джозеф Саджан, Дэлли Роберт Дин
МПК: A61P 35/00, A61K 31/52, C07D 473/00...
Метки: ингибиторы, s6-киназы, акт
Формула / Реферат:
1. Соединение формулыгде X представляет собой F, Cl, CF3, CN или Н;Y представляет собой F, Н или Cl;R1 и R2 независимо представляют собой Н, С1-С4алкил или СН2СН2ОН; или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют пирролидиновое кольцо, необязательно замещенное гидроксиметилом в положении 2 или гидроксилом в положении 3, или азетидиновое кольцо, замещенное гидроксилом в положении 3;R3 представляет собой Н или ОН;R6...
Ингибиторы диацилглицеролацилтрансферазы и фармацевтическая композиция, включающая их

Номер патента: 20128
Опубликовано: 29.08.2014
Авторы: Шаус Джон Менерт, Хембр Эрик Джеймс, Арнольд Маклин Брайан, Ши Цин, Люй Цзяньлян, Риззо Джон Роберт, Бьючемп Томас Джеймс, Кэнада Эмили Джейн
МПК: A61K 31/5517, A61P 3/00, C07D 487/04...
Метки: ингибиторы, включающая, композиция, фармацевтическая, диацилглицеролацилтрансферазы
Формула / Реферат:
1. Соединение формулыили его фармацевтически приемлемая соль;где R1 выбран из группы, состоящей из Н, F, Cl, -CF3, -CH3, -ОН, -O(C1-C4 алкила), -О-циклопропила, -О-СН2-фенила, -OC(H)F2 и -OCF3;R2 выбран из группы, состоящей из Н и -СН3;R3 выбран из группы, состоящей из Н и -СН3, при условии, что по меньшей мере один из группы, состоящей из R2 и R3, представляет собой Н; иn равен 1, 2 или 3.2. Соединение или его соль по п.1, где n равен 1.3....
Ингибиторы пролилгидроксилаз

Номер патента: 18220
Опубликовано: 28.06.2013
Авторы: Лю Жунган, Уиггалл Кеннет, Фитч Дюк М., Даффи Кевин Дж., Цзинь Цзянь, Шо Энтони Н.
МПК: A61K 31/47, C07D 487/08, A61K 31/495...
Метки: пролилгидроксилаз, ингибиторы
Формула / Реферат:
1. Соединение N-[(1,3-дициклогексил-6-гидрокси-2,4-диоксо-1,2,3,4-тетрагидро-5-пиримидинил)карбонил]глицин или его фармацевтически приемлемая соль.2. Соединение по п.1, которое представляет собой N-[(1,3-дициклогексил-6-гидрокси-2,4-диоксо-1,2,3,4-тетрагидро-5-пиримидинил)карбонил]глицин.3. Соединение по п.1, которое представляет собой фармацевтически приемлемую соль...
Ингибиторы seh и их применение

Номер патента: 18414
Опубликовано: 30.07.2013
Авторы: Марино Джозеф Пол, Дин Юнь, Тхалджи Реема К.
МПК: A01N 43/66, A61K 31/53
Метки: применение, ингибиторы
Формула / Реферат:
1. Соединение, отвечающее формуле IФормула Iгде А представляет фенил или пиридил;R1 представляет собой CF3, галоген, OCF3, CN, O-C1-C6-алкил, морфолино, СО2Н или N(CH3)2;R2 означает Н;m равно 1;n означает 1, 2 или 3;В означает циклогексил;R4 означает Н;Z означает О;Y означает C1-С3-алкил, фенил, тиофенил или пиридил; где указанные фенил, тиофенил или пиридил могут быть замещены -СО2Н, SO2Me, CF3, галогеном или CN;R5 означает Н или C1-С6-алкил...
Предыдущий патент: Способ и устройство для сжигания топлива при высокой температуре и высоком давлении и соответствующие система и средства
Следующий патент: Способ и система бурения морских скважин
Случайный патент: Соединение {5-[(1r)-1-(2,6-дихлор-3-фторфенил)этокси]-6-аминопиридазин-3-ил}-n-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)карбоксамид в качестве киназного ингибитора