Ингибиторы вируса гепатита с
Номер патента: 24173
Опубликовано: 31.08.2016
Авторы: Минвелл Николас А., Редучинтала Кишоре В., Скола Пол Майкл, Нагалакшми Пуличарла, Раджамани Рамкумар, Саркунам Кандхасами























Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемая соль,
где q имеет значение 1;
---- представляет собой двойную связь;
Rp представляет собой

Rp присоединен к основному молекулярному фрагменту через любой замещаемый атом углерода в группе;
n имеет значения 0, 1 или 2;
Х0 выбран из СН и N;
X1 выбран из СН и N;
X2 и X3 независимо выбраны из СН и N;
при условии, что по меньшей мере один из X1, X2 и X3 является отличным от N;
каждый Ra независимо выбран из алкокси и гало;
Rx представляет собой метил;
Ry и Rz, каждый, представляют собой водород;
R2 выбран из водорода, алкила и галоалкила;
R3 выбран из алкоксикарбонила и галоалкоксикарбонила;
термин "алкил" относится к группе, являющейся производной от насыщенного углеводорода с нормальной или разветвленной цепью, содержащей от 1 до 10 атомов углерода;
термин "галоалкил" относится к алкильной группе, замещенной 1, 2, 3 или 4 атомами галогена.
2. Соединение формулы (II)

или его фармацевтически приемлемая соль,
где n имеет значения 0, 1 или 2;
каждый R1 независимо выбран из алкокси и гало;
R2 выбран из водорода, алкила и галоалкила;
R3 выбран из алкоксикарбонила и галоалкоксикарбонила;
термин "алкил" относится к группе, являющейся производной от насыщенного углеводорода с нормальной или разветвленной цепью, содержащей от 1 до 10 атомов углерода;
термин "галоалкил" относится к алкильной группе, замещенной 1, 2, 3 или 4 атомами галогена.
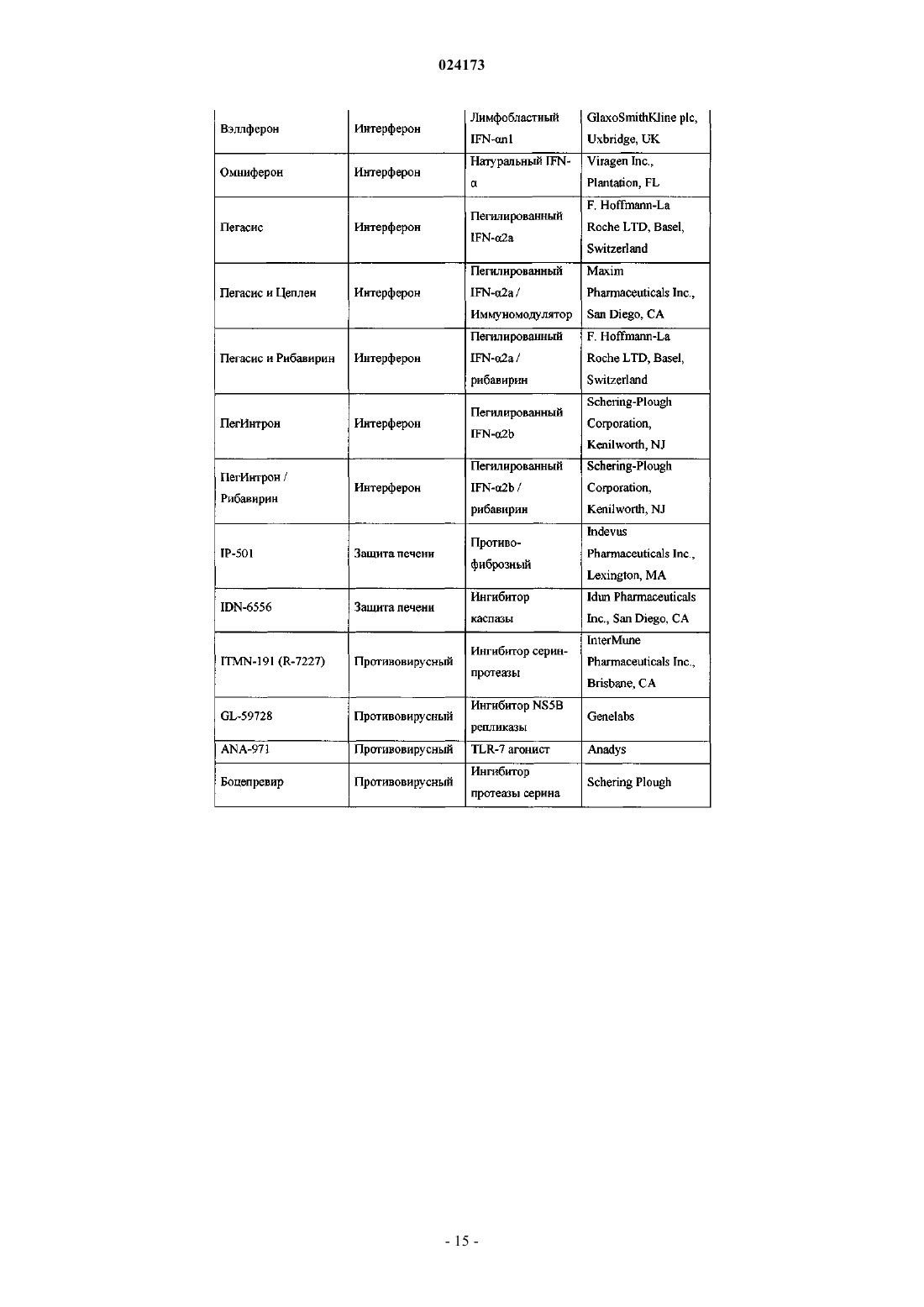
3. Соединение, выбранное из:





или его фармацевтически приемлемая соль.
Текст
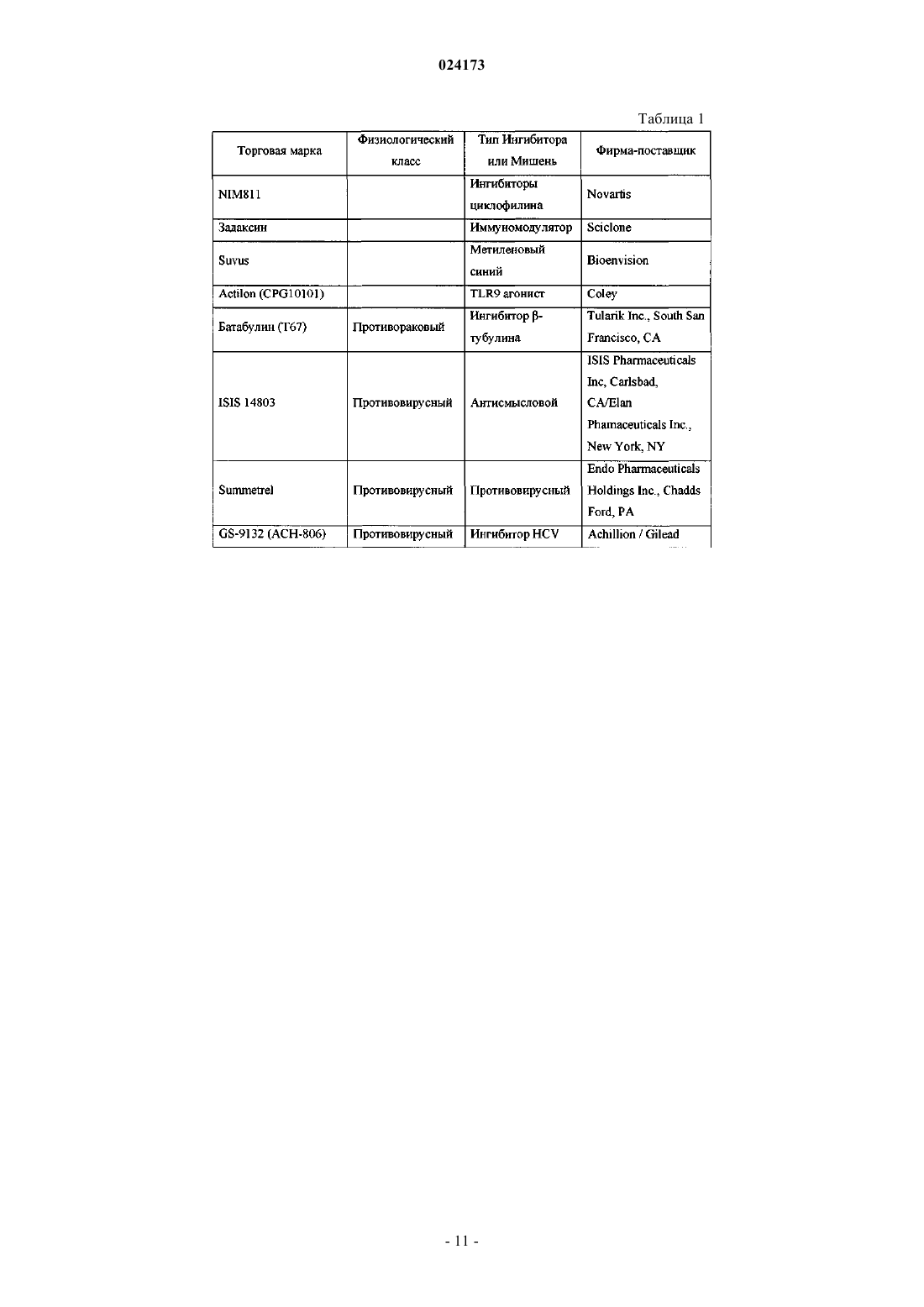
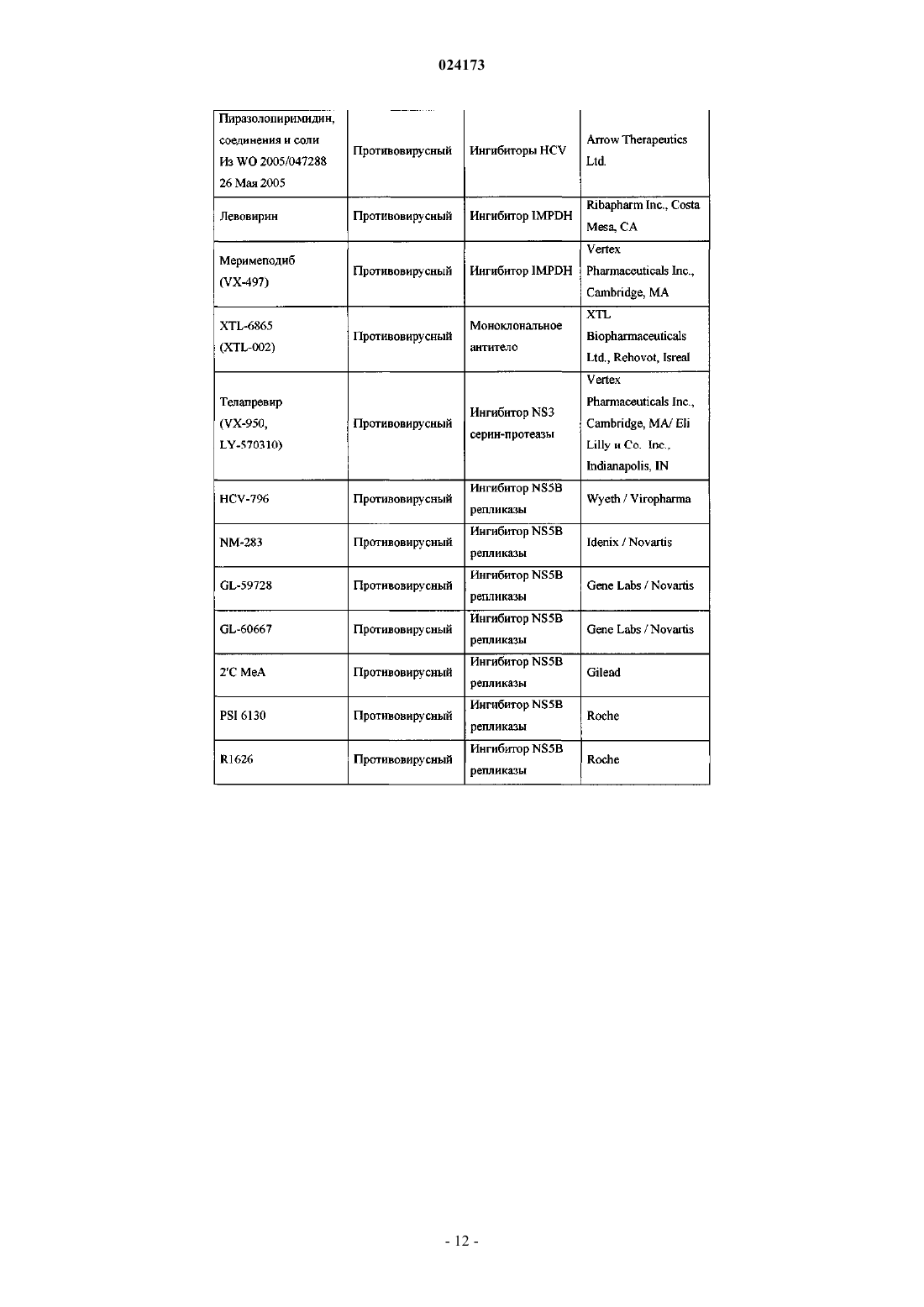
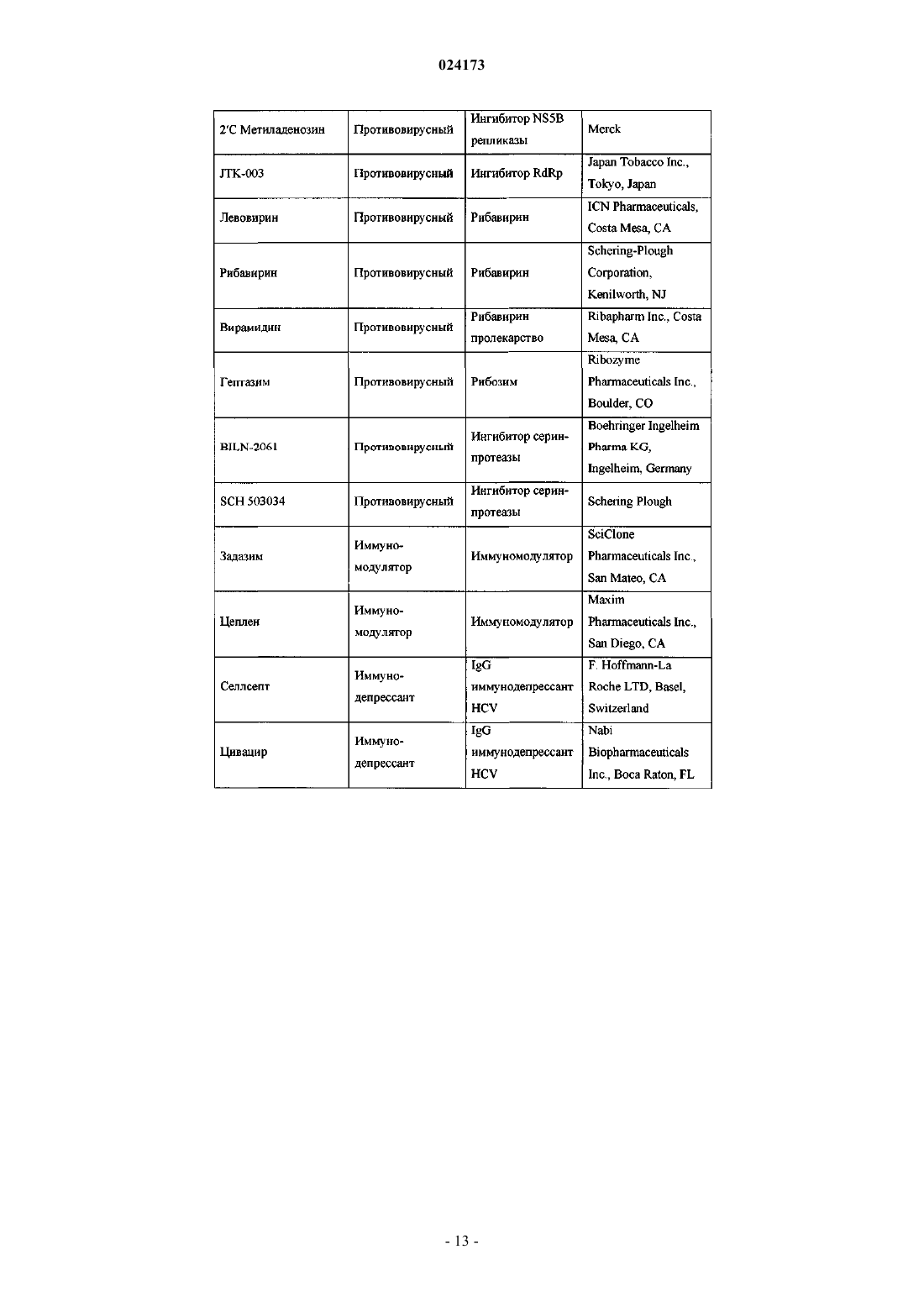
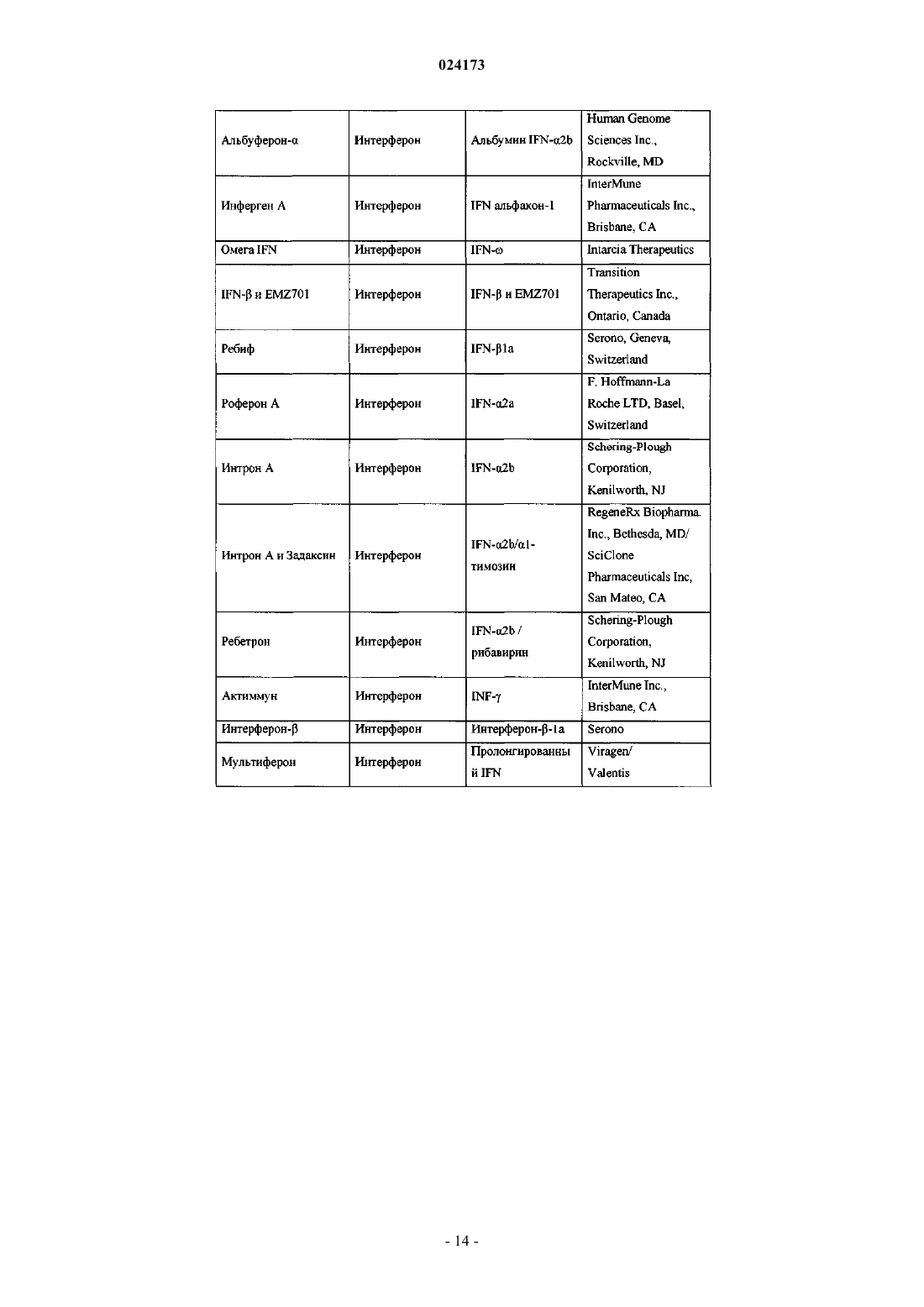
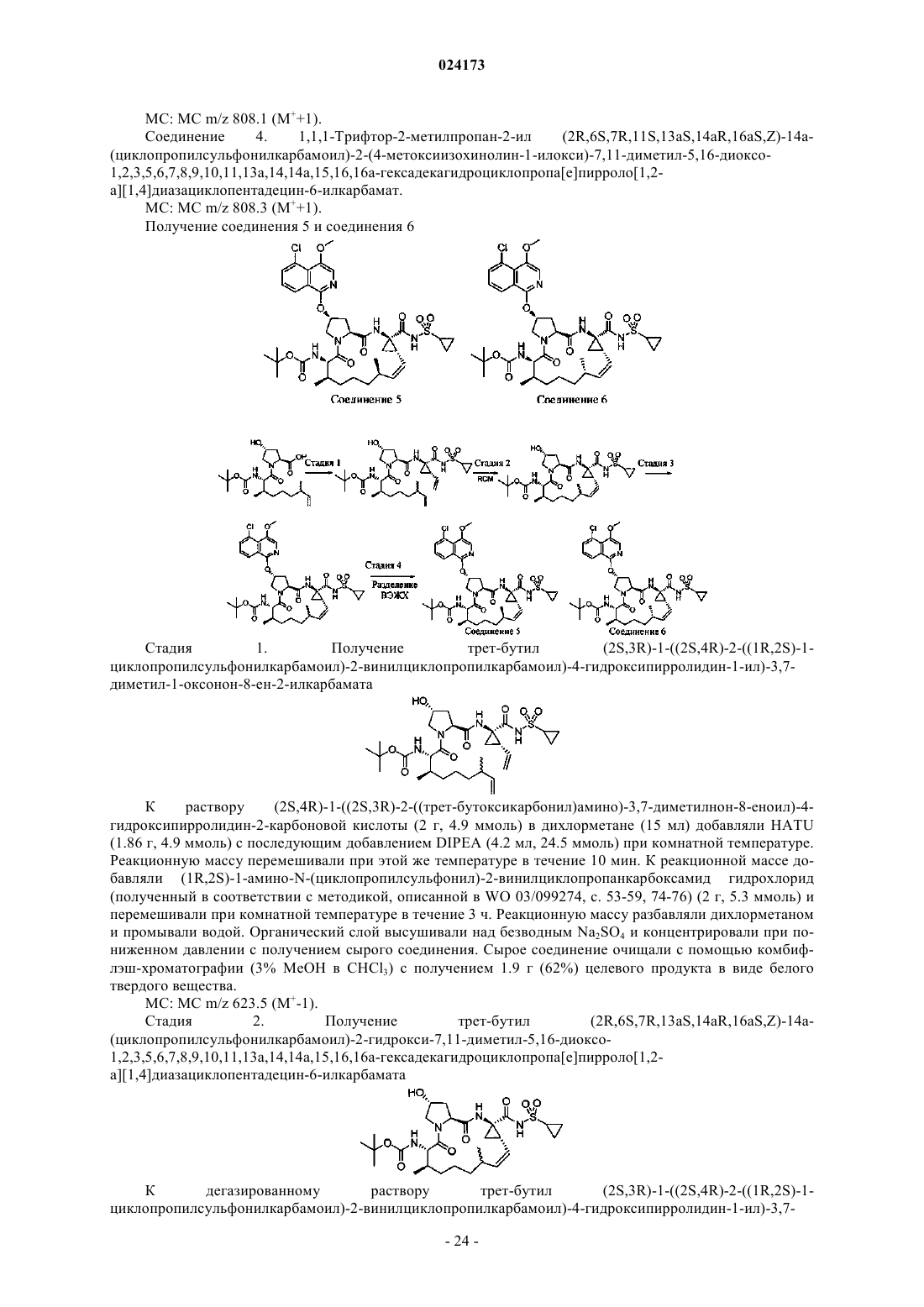
ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С Раджамани Рамкумар (US), Редучинтала Кишоре В., Саркунам Кандхасами,Нагалакшми Пуличарла (IN), Минвелл Николас А., Скола Пол Майкл (US) Представитель: Описаны ингибиторы вируса гепатита С, имеющие общую формулу (I) Также описаны композиции, содержащие соединения и способы применения этих соединений для ингибирования HCV. Перекрестная ссылка на родственные заявки Заявка на данное изобретение испрашивает приоритет согласно предварительной заявке на патент США, серийный номер 61/497141, поданной 15 июня 2011 г. Настоящее изобретение в целом относится к противовирусным соединениям и, более конкретно, к соединениям, которые могут ингибировать функцию протеазы NS3 (также называемой в данном документе как "сериновая протеаза"), кодируемой вирусом гепатита С (HCV), к композициям, содержащим указанные соединения, и к способам ингибирования функции протеазы NS3.HCV является самым распространенным патогеном человека, которым инфицировано приблизительно 170 млн человек во всем мире, что примерно в пять раз превышает количество инфицированных вирусом иммунодефицита человека типа 1. У существенной части HCV-инфицированных индивидуумов в дальнейшем развивается тяжелое прогрессирующее заболевание печени, включая цирроз печени и гепатоклеточную карциному. В настоящее время в наиболее эффективной терапии HCV используется комбинация альфаинтерферона и рибавирина, что приводит к устойчивой эффективности у 40% пациентов. Последние клинические результаты показывают, что пегилированный альфа-интерферон превосходит немодифицированный альфа-интерферон в качестве монотерапии. Однако, даже при экспериментальных терапевтических режимах с использованием комбинаций пегилированного альфа-интерферона и рибавирина, значительная часть пациентов не имеет устойчивого снижения вирусной нагрузки. Таким образом, существует очевидная и давно ощущаемая необходимость разработки эффективных терапевтических средств для лечения HCV-инфекции. HCV относится к вирусам, содержащим одноцепочечную (+)РНК. На основании сравнения расшифрованной аминокислотной последовательности и значительного сходства в нетранслируемой 5'-области РНК HCV относят к отдельному роду семейства флавивирусов (Flavivindae). Вирусные частицы всех членов семейства флавивирусов представляют собой покрытые оболочкой вирионы, геном которых состоит из одноцепочечной (+)РНК, которая кодирует все известные специфичные вирусные белки и транслируется как одна непрерывная открытая рамка считывания. Обнаруживается значительная гетерогенность нуклеотидной и кодируемой аминокислотной последовательности в различных областях генома HCV. Было охарактеризовано шесть основных генотипов и описано более 50 подтипов. Основные генотипы HCV различаются в их распределении по всему миру, и клиническое значение генетической гетерогенности HCV остается неясным несмотря на многочисленные исследования возможного влияния генотипов на патогенез и терапию. Геном HCV, представляющий собой одноцепочечную РНК, содержит приблизительно 9500 нуклеотидов и имеет единственную открытую рамку считывания (ORF), кодирующую один большой полипротеин, состоящий приблизительно из 3000 аминокислот. В инфицированных клетках данный полипротеин расщепляется на нескольких сайтах клеточными и вирусными протеазами, что приводит к образованию структурных и неструктурных (NS) белков. В случае HCV образование зрелых неструктурных белков (NS2, NS3, NS4A, NS4B, NS5A иNS5B) зависит от двух вирусных протеаз. Первая протеаза расщепляет по соединению NS2-NS3, вторая является сериновой протеазой, содержащейся в N-концевом домене NS3, и опосредует все последующие расщепления в направлении NS3, как в цис- на сайте расщепления NS3-NS4A, так и в транс- на оставшихся сайтах NS4A-NS4B, NS4B-NS5A, NS5A-NS5B. Белок NS4A, по-видимому, является многофункциональным белком, играет роль кофактора протеазы NS3 и участвует в мембранной локализации NS3 и других компонентов репликации вируса. Образование комплекса белка NS3 с NS4A является существенным для эффективного процессинга полипротеина, повышая протеолитическое расщепление на всех сайтах. Белок NS3 также проявляет нуклеозидтрифосфатазную и РНК геликазную активности. Белок NS5B является РНК-зависимой РНК-полимеразой, которая вовлекается в репликацию HCV. В настоящем изобретении предложены пептидные соединения, которые могут ингибировать функционирование протеазы NS3, например, в комбинации с протеазой NS4A. Кроме того, в настоящем изобретении описывается введение комбинированной терапии пациенту, согласно которой соединение в соответствии с настоящим изобретением, являющееся эффективным для ингибирования протеазы NS3HCV, может быть введено совместно с дополнительными соединениями, обладающими противовирусной активностью в отношении HCV. Согласно первому аспекту настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль,-1 024173 где q имеет значения 1 или 2; представляет собой одинарную или двойную связь; где Rp присоединен к основному молекулярному фрагменту через любой замещаемый атом углерода в группе;n имеет значения 0, 1, 2, 3, 4, 5 или 6; Х 0 выбран из СН и N;X1 выбран из СН и N;X2 и X3 независимо выбраны из СН, C(Ra) и N,при условии, что по меньшей мере один из X1, X2 и X3 является отличным от N; каждый Ra независимо выбран из алкенилокси, алкокси, алкоксиалкокси, алкила, бензодиоксанила,карбоксамидо, карбокси, карбоксиалкокси, циано, циклоалкилалкокси, циклоалкилокси, дейтероалкокси,диалкиламино, гало, галоалкила, галоалкокси, галоалкоксикарбонила, гидрокси, морфолинила, фенила,пиперазинила, пиразолила, пиридинила и пирролидинила, где указанные морфолинил, фенил, пиперазинил, пиридинил и пирролидинил необязательно замещены одной или двумя группами, независимо выбранными из алкокси, алкила, алкилсульфонила, гало, галоалкокси, галоалкила и морфолинила; и где две соседние группы Ra совместно с атомами углерода, к которым они присоединены, могут необязательно образовывать кольцо, выбранное из диоксанила, диоксоланила, морфолинила, пиранила и фенила, где данное кольцо необязательно замещено одной или двумя группами, независимо выбранными из алкила и гало;Rx выбран из метила и этила;Ry и Rz независимо выбраны из водорода и гидрокси при условии что, когда представляет собой двойную связь, Ry и Rz, каждый, представляют собой водород;R3 выбран из водорода, алкоксиалкоксикарбонила, алкоксикарбонила, алкиламинокарбонила, алкилкарбонила, циклоалкилалкоксикарбонила, циклоалкилкарбонила, циклоалкилоксикарбонила, дейтероалкоксикарбонила, дейтерогалоалкоксикарбонила, диалкиламинокарбонила, диалкиламинокарбонилкарбонила, галоалкоксикарбонила, галоалкиламинокарбонила, галоалкилкарбонила, гетероциклилкарбонила, гетероциклилоксикарбонила, фенилкарбонила и фенилоксикарбонила, где циклоалкильная часть указанных циклоалкилалкоксикарбонила, циклоалкилкарбонила и циклоалкилоксикарбонила, гетероциклильная часть указанных гетероциклилкарбонила и гетероциклилоксикарбонила и фенильная часть указанных фенилкарбонила и фенилоксикарбонила необязательно замещены одной, двумя или тремя группами, независимо выбранными из алкила, алкиламино, алкилкарбонила, циклоалкила, диалкиламино, гало, галоалкокси и галоалкила. Согласно первому варианту осуществления первого аспекта настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, гдеq имеет значение 1; представляет собой двойную связь;Ry и Rz, каждый, представляют собой водород. Согласно второму варианту осуществления первого аспекта настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, где Rp представляет собой: Согласно третьему варианту осуществления настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, гдеn имеет значения 0, 1, 2 или 3; каждый Ra независимо выбран из алкокси и гало;R3 выбран из алкоксикарбонила и галоалкоксикарбонила. Согласно второму аспекту настоящего изобретения предложено соединение формулы (II) или его фармацевтически приемлемая соль,где n имеет значения 0, 1, 2, 3, 4, 5 или 6; каждый R1 независимо выбран из алкокси, алкила, карбоксамидо, карбокси, циано, циклоалкилокси,диалкиламино, гало, галоалкила, галоалкокси и фенила, где данный фенил необязательно замещен одной или двумя группами, независимо выбранными из алкокси, алкила, гало, галоалкокси и галоалкила;R3 выбран из алкоксикарбонила, алкилкарбонила, галоалкоксикарбонила, галоалкилкарбонила и фенилкарбонила, где данный фенил необязательно замещен одной или двумя группами, независимо выбранными из алкила и гало. Согласно третьему аспекту настоящего изобретения предложена композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Согласно первому варианту осуществления третьего аспекта настоящего изобретения предложена композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, по меньшей мере одно дополнительное соединение, обладающее противовирусной активностью в отношении HCV, и фармацевтический носитель. Согласно второму варианту осуществления по меньшей мере одно из указанных дополнительных соединений представляет собой интерферон или рибавирин. Согласно третьему варианту осуществления указанный интерферон выбран из интерферона альфа 2 В, пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа 2 А и лимфобластоидного интерферона тау. Согласно четвертому варианту осуществления третьего аспекта настоящего изобретения предложена композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, по меньшей мере одно дополнительное соединение, обладающее противовирусной активностью в отношении HCV, и фармацевтический носитель, в которой по меньшей мере одно из дополнительных соединений выбрано из интерлейкина 2, интерлейкина 6, интерлейкина 12, имиквимода (Imiquimod), рибавирина, ингибитора инозин-5'-монофосфат-дегидрогеназы, амантадина и римантадина. Согласно пятому варианту осуществления третьего аспекта настоящего изобретения предложена композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, по меньшей мере одно дополнительное соединение, обладающее противовирусной активностью в отношении HCV, и фармацевтический носитель, в которой по меньшей мере одно из указанных дополнительных соединений является эффективным ингибитором функции мишени, выбранной из металлопротеазы HCV, сериновой протеазы HCV,полимеразы HCV, хеликазы HCV, белка NS4B HCV, проникновения HCV в клетку, сборки вирионовHCV, выхода вирионов HCV из клетки, белка NS5A HCV и IMPDH (инозинмонофосфат-дегидрогеназы),для лечения HCV-инфекции. Согласно четвертому аспекту настоящего изобретения предложен способ лечения HCV-инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Согласно первому варианту осуществления четвертого аспекта изобретения данный способ дополнительно включает введение по меньшей мере одного дополнительного соединения, обладающего противовирусной активностью в отношении HCV, до введения, после или одновременно с соединением формулы (I) или его фармацевтически приемлемой солью. Согласно второму варианту осуществления четвертого аспекта изобретения по меньшей мере одно из указанных дополнительных соединений представляет собой интерферон или рибавирин. Согласно третьему варианту осуществления указанный интерферон выбран из интерферона альфа 2 В, пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа 2 А и лимфобластоидного интерферона тау. Согласно четвертому варианту осуществления четвертого аспекта настоящего изобретения предложен способ лечения HCV-инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного дополнительного соединения, обладающего противовирусной активностью в отношении HCV, до введения, после указанного введения или одновременно с соединением формулы (I) или его фармацевтически приемлемой солью, где по меньшей мере одно из указанных дополнительных соединений выбрано из интерлейкина 2, интерлейкина 6, интерлейкина 12, имиквимода, рибавирина,ингибитора инозин-5'-монофосфат-дегидрогеназы, амантадина и римантадина. Согласно пятому варианту осуществления четвертого аспекта настоящего изобретения предложен способ лечения HCVинфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного дополнительного соединения, обладающего противовирусной активностью в отношении HCV, до, после указанного введения или одновременно с соединением формулы (I) или его фармацевтически приемлемой солью,где по меньшей мере одно из указанных дополнительных соединений является эффективным ингибитором функции мишени, выбранной из металлопротеазы HCV, сериновой протеазы HCV, полимеразыHCV, хеликазы HCV, белка NS4B HCV, проникновения HCV в клетку, сборки вирионов HCV, выхода вирионов HCV из клетки, белка NS 5A HCV и IMPDH для лечения HCV-инфекции. Другие варианты осуществления настоящего изобретения могут включать подходящие комбинации вариантов осуществления, раскрытых в данном описании. В то же время другие аспекты и варианты осуществления изобретения будут очевидны из приведенного ниже описания. Описание настоящего изобретения следует истолковывать в соответствии с законами и принципами образования химических связей. В некоторых случаях может быть необходимым удалить атом водорода для того, чтобы поместить заместитель в любом данном положении. Необходимо понимать, что соединения согласно настоящему изобретению представляют собой соединения, которые являются достаточно стабильными для их использования в качестве фармацевтического вещества. Подразумевается, что определение любого заместителя или переменной в конкретном положении в молекуле не зависит от его определений в любых других положениях в данной молекуле. Например, когда n равно 2, каждая из двух R1 групп может быть одинаковой или различаться. Все патенты, заявки на патенты и литературные публикации, цитированные в описании изобретения, включены в данное описание посредством ссылки во всей своей полноте. В случае несоответствия настоящее описание, включая определения, имеет преимущественную силу. Термин "алкенил" в контексте данного описания относится к группе из 2-6 атомов углерода с нормальной неразветвленной или разветвленной цепью, содержащей по меньшей мере одну углеродуглеродную двойную связь. Термин "алкенилокси" в контексте данного описания относится к алкенильной группе, присоединенной к основному молекулярному фрагменту через атом кислорода. Термин "алкокси" в контексте данного описания относится к алкильной группе, присоединенной к основному молекулярному фрагменту через атом кислорода. Термин "алкоксиалкокси" в контексте данного описания относится к алкоксиалкильной группе,присоединенной к основному молекулярному фрагменту через атом кислорода. Термин "алкоксиалкоксикарбонил" в контексте данного описания относится к алкоксиалкоксигруппе, присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "алкоксиалкил" в контексте данного описания относится к алкильной группе, замещенной одной, двумя или тремя алкоксигруппами. Термин "алкоксикарбонил" в контексте данного описания относится к алкоксигруппе, присоединенной к основному молекулярному фрагменту посредством карбонильной группы. Термин "алкил" в контексте данного описания относится к группе, являющейся производной от насыщенного углеводорода с нормальной или разветвленной цепью, содержащей от 1 до 10 атомов углерода. Термин "алкилкарбонил" в контексте данного описания относится к алкильной группе, присоединенной к основному молекулярному фрагменту посредством карбонильной группы. Термин "алкиламино" в контексте данного описания относится к -NHRq, где Rq представляет собой алкил. Термин "алкиламинокарбонил" в контексте данного описания относится к алкиламиногруппе, присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "алкилсульфонил" в контексте данного описания относится к алкильной группе, присоединенной к основному молекулярному фрагменту посредством сульфонильной группы. Термин "карбонил" в контексте данного описания относится к -C(O-. Термин "карбоксамидо" в контексте данного описания относится к -C(O)NRxRy, где Rx и Ry независимо выбраны из водорода и алкила. Термин "карбокси" в контексте данного описания относится к -CO2H. Термин "карбоксиалкокси" в контексте данного описания относится к карбоксиалкильной группе,присоединенной к основному молекулярному фрагменту через атом кислорода. Термин "карбоксиалкил" в контексте данного описания относится к алкильной группе, замещенной одной, двумя или тремя карбоксильными группами. Термин "циано" в контексте данного описания относится к -CN. Термин "циклоалкил" в контексте данного описания относится к насыщенной моноциклической или бициклической углеводородной кольцевой системе, имеющей от 3 до 7 атомов углерода и не имеющей гетероатомов. Типичные примеры циклоалкильных групп включают, но не ограничиваясь ими, циклопропил, циклобутил и циклопентил. Термин "циклоалкилалкокси" в контексте данного описания относится к (циклоалкил)алкильной группе, присоединенной к основному молекулярному фрагменту через атом кислорода. Термин "циклоалкилалкоксикарбонил" в контексте данного описания относится к циклоалкилалкоксигруппе, присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "(циклоалкил)алкил" в контексте данного описания относится к алкильной группе, замещенной одной, двумя или тремя циклоалкильными группами. Термин "циклоалкилкарбонил" в контексте данного описания относится к циклоалкильной группе,присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "циклоалкилокси" в контексте данного описания относится к циклоалкильной группе, присоединенной к основному молекулярному фрагменту посредством атома кислорода Термин"циклоалкилоксикарбонил" в контексте данного описания относится к циклоалкилоксигруппе, присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "дейтероалкокси" в контексте данного описания относится к алкоксигруппе, где по меньшей мере один из атомов водорода замещен атомом дейтерия. Термин "дейтероалкоксикарбонил" в контексте данного описания относится к дейтероалкоксигруппе, присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "дейтерогалоалкокси" в контексте данного описания относится к галоалкоксигруппе, где по меньшей мере один из атомов водорода замещен атомом дейтерия. Термин "дейтерогалоалкоксикарбонил" в контексте данного описания относится к дейтерогалоалкоксигруппе, присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "диалкиламино" в контексте данного описания относится к -NRpRq, где Rp и Rq представляют собой алкильные группы. Алкильные группы могут быть одинаковыми или различными. Термин "диалкиламинокарбонил" в контексте данного описания относится к диалкиламиногруппе,присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "диалкиламинокарбонилкарбонил" в контексте данного описания относится к диалкиламинокарбонильной группе, присоединенной к основному молекулярному фрагменту через карбонильную группу. Термины "гало" и "галоген" в контексте данного описания относятся к F, Cl, Br и I. Термин "галоалкокси" в контексте данного описания относится к галоалкильной группе, присоединенной к основному молекулярному фрагменту посредством атома кислорода. Термин "галоалкоксикарбонил" в контексте данного описания относится к галоалкокси группе,присоединенной к основному молекулярному фрагменту посредством карбонильной группы. Термин "галоалкил" в контексте данного описания относится к алкильной группе, замещенной одним, двумя, тремя или четырьмя атомами галогена. Термин "галоалкилкарбонил" в контексте данного описания относится к галоалкильной группе,присоединенной к основному молекулярному фрагменту посредством карбонильной группы. Термин "галоалкиламино" в контексте данного описания относится к -NHRq, где Rq представляет собой галоалкильную группу. Термин "галоалкиламинокарбонил" в контексте данного описания относится к галоалкиламиногруппе, присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "галоалкилкарбонил" в контексте данного описания относится к галоалкильной группе,присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "гетероциклил" в контексте данного описания относится к 5-, 6- или 7-членному кольцу,содержащему один, два или три гетероатома, независимо выбранных из азота, кислорода и серы. 5-членное кольцо имеет от нуля до двух двойных связей, и 6- и 7-членное кольца имеют от нуля до трех двойных связей. Термин "гетероциклил" также включает бициклические группы, в которых гетероциклильное кольцо конденсировано с фенильной группой, моноциклической циклоалкенильной группой, моноциклической циклоалкильной группой или другой моноциклической гетероциклильной группой; и трициклические группы, в которых бициклическая система конденсирована с фенильной группой, моноциклической циклоалкенильной группой, моноциклической циклоалкильной группой или другой моноциклической гетероциклильной группой. Данные гетероциклильные группы по настоящему изобретению могут быть присоединены к основному молекулярному фрагменту через атом углерода или атом азота в группе. Примеры гетероциклильных групп включают, но не ограничиваются ими, бензотиенил, фурил, имидазолил, индолинил, индолил, изотиазолил, изоксазолил, морфолинил, оксазолил, пиперазинил, пиперидинил, пиразолил, пиридинил, пирролидинил, пирролопиридинил, пирролил, тиазолил, тиенил и тиоморфолинил. Термин "гетероциклилкарбонил" в контексте данного описания относится к гетероциклильной группе, присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "гетероциклилокси" в контексте данного описания относится к гетероциклильной группе,присоединенной к основному молекулярному фрагменту через атом кислорода. Термин "гетероциклилоксикарбонил" в контексте данного описания относится к гетероциклилоксигруппе, присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "гидрокси" в контексте данного описания относится к -OH. Термин "гидроксиалкил" в контексте данного описания относится к алкильной группе, замещенной одной, двумя или тремя гидроксигруппами. Термин "фенилкарбонил" в контексте данного описания относится к фенильной группе, присоединенной к основному молекулярному фрагменту посредством карбонильной группы. Термин "фенилокси" в контексте данного описания относится к фенильной группе, присоединенной к основному молекулярному фрагменту через атом кислорода. Термин "фенилоксикарбонил" в контексте данного описания относится к фенилоксигруппе, присоединенной к основному молекулярному фрагменту через карбонильную группу. Термин "сульфонил" в контексте данного описания относится к -SO2. Соединения согласно настоящему изобретению могут существовать в виде фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль" в контексте данного описания означает соли или цвиттер-ионные формы соединений согласно настоящему изобретению, которые являются растворимыми или диспергируемыми в воде или масле, являются, по результатам медицинской оценки,пригодными для использования в контакте с тканями пациентов, не обладают чрезмерной токсичностью,не вызывают чрезмерного раздражения, аллергической реакции или других проблем или осложнений,соизмеримых с разумным соотношением пользы и риска, и являются эффективными, если используются по назначению. Данные соли могут быть получены на заключительной стадии выделения и очистки соединений или отдельно в результате взаимодействия подходящей функциональной группы основания с подходящей кислотой. Типичные соли присоединения кислот включают ацетат, адипат, альгинат, цитрат,аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат; диглюконат, глицерофосфат, хемисульфат, гептаноат, гексаноат, формат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, мезитиленсульфонат, метансульфонат, нафтиленсульфонат, никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, трихлорацетат, трифторацетат, фосфат, глютамат, бикарбонат, пара-толуолсульфонат и ундеканоат. Примеры кислот, которые могут быть использованы для получения фармацевтически приемлемых солей присоединения, включают неорганические кислоты, такие как соляная, бромисто-водородная, серная и фосфорная, и органические кислоты, такие как щавелевая,малеиновая, янтарная и лимонная. Соли присоединения оснований могут быть получены на заключительных стадиях выделения и очистки соединений в результате взаимодействия кислотной группы с подходящим основанием, таким как гидроксид, карбонат или бикарбонат катиона металла, или с аммиаком или органическим первичным, вторичным или третичным амином. Катионы фармацевтически приемлемых солей включают катионы лития, натрия, калия, кальция, магния и алюминия, а также нетоксичные катионы четвертичных аминов, такие как аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, диэтиламин, этиламин, трибутиламин, пиридин, N,N-диметиланилин,N-метилпиперидин,N-метилморфолин,дициклогексиламин,прокаин,дибензиламин,N,N-дибензилфенэтиламин и N,N'-дибензилэтилендиамин. Другие типичные органические амины, которые могут быть использованы для получения солей присоединения оснований, включают этилендиамин,этаноламин, диэтаноламин, пиперидин и пиперазин. В контексте данного описания термин "противовирусная активность в отношении HCV" означает,что такое соединение является эффективным для лечения вируса HCV. Термин "соединения по настоящему изобретению" и эквивалентные выражения предназначены,чтобы охватить соединения формулы (I) и фармацевтически приемлемые энантиомеры, диастереомеры и их соли. Подобным образом, ссылки на промежуточные соединения предназначены, чтобы охватить их соли, где контекст позволяет это сделать. Термин "пациент" включает и человека и других млекопитающих. Термин "фармацевтическая композиция" обозначает композицию, содержащую соединение по настоящему изобретению в комбинации по меньшей мере с одним дополнительным фармацевтическим носителем, т.е. адъювантом, вспомогательным веществом или наполнителем лекарственной формы, такими как разбавители, консерванты, наполнители, вещества, регулирующие расход, разрыхлители, смачивающие вещества, эмульгаторы, суспендирующие вещества, подсластители, отдушки, ароматизаторы,антибактериальные вещества, противогрибковые вещества, смазывающие вещества и диспергирующие вещества в зависимости от характера способа введения форм дозирования. Могут, например, применяться ингредиенты, перечисленные в Remington's Pharmaceutical Sciences,18th ed., Mack Publishing Company, Easton, PA (1999). Фраза "фармацевтически приемлемый" в контексте данного описания относится к таким соединениям, веществам, композициям и/или лекарственным формам, которые, по результатам медицинской оценки, подходят для использования в контакте с тканями пациентов, т.е. не проявляют чрезмерной токсичности, не вызывают раздражения, аллергической реакции или других расстройств или осложнений,соизмеримых с приемлемым соотношением польза/риск. Термин "терапевтически эффективное количество" означает общее количество каждого активного компонента, которое является достаточным, чтобы оказать существенную пользу пациенту, например,устойчивое снижение вирусной нагрузки. Применительно к единичному активному ингредиенту, вводимому в чистом виде, термин означает, что этот ингредиент единственный. При применении к комбинации термин относится к объединенным количествам активных ингредиентов, которые приводят к терапевтическому эффекту, независимо от того, вводятся ли они в комбинации, последовательно или одновременно. Термины "лечить" и "лечение" относятся к (i) предотвращению заболевания, расстройства или патологического состояния у пациента, который может быть предрасположен к заболеванию, расстройству и/или патологическому состоянию, но еще не установлено, что таковое имеется; (ii) ингибированию заболевания, расстройства или патологического состояния, т.е. прекращению его развития; и/или (iii) облегчению заболевания, расстройства или патологического состояния, т.е., вызывать регрессию заболевания, расстройства и/или патологического состояния. В случае использования в наименовании соединений по настоящему изобретению обозначения Р 1',Р 1, Р 2, Р 2, Р 3 и Р 4 в контексте данного описания отображают относительные положения аминокислотных остатков связывания ингибитора протеазы по отношению к связыванию природного пептидного субстрата расщепления. Расщепление происходит в природном субстрате между Р 1 и Р 1', где неосновные положения обозначают аминокислоты, начиная с С-конца пептидного природного сайта расщепления, в направлении N-конца, тогда как основные положения исходят из N-конца обозначения сайта расщепления и продолжаются по направлению к С-концу. Например, Р 1' относится к первому положению от правого конца С-конца сайта расщепления (т.е. первой позиции N-конца), тогда как с Р 1 начинается нумерация с левой стороны С-концевого сайта расщепления, Р 2: второе положение от С-конца и т.д.). (См.Berger A.Schechter I., Transactions of the Royal Society London series (1970), B257, 249-264]. В соединениях по настоящему изобретению существуют центры асимметрии. Например, соединения могут включать циклопропильный элемент Р 1 формулы где каждый С 1 и С 2 представляет собой асимметричный атом углерода в позициях 1 и 2 циклопропильного кольца. Следует понимать, что настоящее описание охватывает все стереохимические формы или их смеси,которые обладают способностью ингибировать протеазу HCV. Некоторые соединения по настоящему изобретению также могут существовать в различных устойчивых конформационных формах, которые могут быть разделены. Торсионная асимметрия в связи с ограниченным вращением вокруг асимметричной одинарной связи, например, из-за стерических препятствий или напряжения кольца, может позволить разделение различных конформеров. Настоящее изобретение включает каждый конформационный изомер этих соединений и их смеси. Некоторые соединения по настоящему изобретению могут существовать в цвиттер-ионной форме, и настоящее изобретение включает каждую цвиттер-ионную форму этих соединений и их смесей. Когда является возможным, что для использования в терапии могут быть введены терапевтически эффективные количества соединения формулы (I), а также его фармацевтически приемлемых солей в виде исходного химического продукта, то можно представить активный ингредиент в виде фармацевтической композиции. Соответственно, изобретение дополнительно относится к фармацевтическим композициям, которые включают терапевтически эффективные количества соединения формулы (I) или его фармацевтически приемлемых солей, и один или более фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ. Соединения формулы (I) и их фармацевтически приемлемые соли являются такими, как описано выше. Носитель(и), разбавитель(и) или вспомогательное(ые) вещество(а) должны быть приемлемыми, в смысле быть совместимыми с другими ингредиентами композиции и не вредными для реципиента. В соответствии с другим аспектом настоящее изобретение относится также к способу получения фармацевтической композиции, включающей смешивание соединения формулы (I) или его фармацевтически приемлемой соли с одним или более фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами. Фармацевтические композиции могут быть представлены в форме стандартной дозы, содержащей определенное количество активного ингредиента на единицу дозы. Уровни дозирования от около 0,01 до около 150 мг/кг массы тела в сутки, предпочтительно от около 0,05 до около 100 мг/кг массы тела в сутки соединений по настоящему изобретению являются типичными при монотерапии предупреждения и лечения HCV опосредованного заболевания. Как правило, фармацевтические композиции по изобретению вводятся от 1 до 5 раз в сутки или в виде альтернативы как непрерывная инфузия. Такое введение может применяться в качестве терапии хронического или острого состояния. Количество активного ингредиента, которое может быть соединено с материалами носителя для получения единичной лекарственной формы, будет меняться в зависимости от состояния пациента, тяжести состояния, времени введения, способа введения, скорости выведения принимаемого соединения, продолжительности лечения и возраста, пола, веса и состояния пациента. Предпочтительные единичные дозировки композиций представляют собой такие, которые содержат суточную дозу или часть дозы так, как изложено здесь выше,или подходящую часть ее как активного ингредиента. Как правило, лечение может начинаться с маленьких доз, существенно меньших, чем оптимальная доза соединения. Затем доза увеличивается небольшими порциями до тех пор, пока при данных обстоятельствах не будет достигнут оптимальный эффект. В общем, соединение, наиболее желательно, вводится при уровне концентрации, который, как правило,окажет эффективные противовирусные воздействия, не вызывая никаких вредных или разрушительных побочных эффектов. Когда композиции по настоящему изобретению содержат комбинацию соединения по настоящему изобретению и одного или более дополнительных терапевтических и/или профилактических средств,тогда как соединение, так и дополнительное средство могут присутствовать в дозе, которая меньше или равна дозе, обычно вводимой при режиме монотерапии. Композиции по данному изобретению могут быть объединены с одним или более дополнительными терапевтическими или профилактическими веществами, например, в виде монолитной и/или би/многослойной таблетки или могут вводиться отдельно от терапевтического или профилактического средства (средств). Фармацевтические композиции могут быть адаптированы для введения любым подходящим способом, например, перорально (в том числе буккально или сублингвально), ректально, назально, местно (в том числе буккально, сублингвально или трансдермально), вагинально или парентерально (в том числе подкожно, внутрикожно, внутримышечно,внутрисуставно, надчревно, интратекально, внутрь пораженных тканей, внутривенно или внутрикожно посредством инъекции или инфузии). Такие композиции могут быть получены любым способом, известным в области фармацевтики, например, путем введения в композицию активного ингредиента с носителем (носителями) или вспомогательным веществом (веществами). Фармацевтические композиции для перорального введения могут быть представлены в виде дискретных единиц, таких как капсулы или таблетки; порошки или гранулы; в виде растворов или суспензий в водных или неводных жидкостях; съедобных пен или взбитых масс; или жидких эмульсий масло-вводе или эмульсий вода-в-масле. Например, для перорального введения в форме таблеток или капсул активный лекарственный компонент может быть соединен с пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и т.п. Порошки получают путем измельчения соединения до соответствующего тонкого размера частиц и смешивания с подобным образом измельченным фармацевтическим носителем, таким как съедобный углевод, как, например, крахмал или маннит. Также могут присутствовать ароматизатор, консервант, диспергирующее и окрашивающее вещества. Капсулы изготавливаются путем приготовления порошковой смеси, как описано выше, и наполнения ею сформированных желатиновых оболочек. К порошковой смеси до заправки капсул могут быть добавлены вещества, способствующие скольжению, и смазывающие вещества, такие как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль. Также может быть добавлено дезинтегрирующее или растворяющее вещество, такое как агар-агар, карбонат кальция или карбонат натрия, для улучшения доступности лекарственного средства, когда капсула проглатывается. Кроме того, при желании или необходимости, в смесь также могут быть введены подходящие связующие, смазывающие, дезинтегрирующие агенты и окрашивающие вещества. Подходящие связующие вещества включают крахмал, желатин, натуральные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические смолы, такие как камедь, трагакант или натрия альгинат, карбоксиметилцеллюлозу, полиэтиленгликоль и т.п. Смазывающие вещества, используемые в этих лекарственных формах, включают олеат натрия, хлорид натрия и т.п. Дезинтеграторы включают,без ограничения, крахмал, метилцеллюлозу, агар, бетонит, ксантановую смолу и т.п. Таблетки изготавливаются, например, путем получения порошковой смеси, гранулирования или брикетирования с добавлением скользящего вещества и разрыхлителя и прессования в таблетки. Порошковая смесь готовится путем смешивания соединения, подходящим образом измельченного, с разбавителем или основой, как описано выше, и, необязательно, со связующим веществом, таким как карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, замедлителем растворения, таким как парафин, ускорителем резорбции, таким как четвертичная соль, и/или поглощающим веществом, таким как бетонит, каолин или дикальцийфосфат. Порошковая смесь может быть гранулирована путем смачивания со связующим, таким как сироп, крахмальный клейстер, клейкое вещество на основе камеди, или с растворами целлюлозных или полимерных материалов и пропускания через сито. В качестве альтернативы гранулированию порошковая смесь может быть пропущена через таблеточную машину и образовывать не полностью сформированные заготовки, измельчаемые в гранулы. Гранулы могут быть смазаны, чтобы предотвратить прилипание к формирующему таблетки штампу, путем добавления стеариновой кислоты, стеаратной соли, талька или минерального масла Смазанная смесь затем прессуется в таблетки. Соединения по настоящему изобретению могут быть также объединены с сыпучим инертным носителем и спрессованы в таблетки непосредственно, минуя стадии гранулирования или получения заготовок. Могут применяться прозрачное или непрозрачное защитное покрытие, состоящее из изолирующего слоя шеллака, покрытие из сахара или полимерного материала и полирующее покрытие воском. К этим покрытиям могут быть добавлены красящие вещества, чтобы сделать различия между лекарственными формами. Пероральные жидкости, такие как растворы, сиропы и эликсиры, могут быть изготовлены в форме единицы дозирования, так что данная величина содержит определенное количество соединения. Сиропы могут быть получены путем растворения соединения в соответствующим образом ароматизированном водном растворе, при этом эликсиры готовятся с использованием нетоксичных носителей. Также могут быть добавлены солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и полиоксиэтиленовые простые эфиры сорбита, консерванты, ароматизирующие добавки, такие как масло мяты перечной, или натуральные подсластители, или сахарин, или другие искусственные подсластители,и т.п. В случае необходимости, единица дозирования композиций для перорального введения может находиться в форме микрокапсул. Композиция может быть изготовлена таким образом, чтобы продлить или замедлить высвобождение, например, посредством покрытия или закладывания дисперсного материала в полимеры, воск и т.п. Соединения формулы (I) и их фармацевтически приемлемые соли могут также вводиться в форме липосомных систем доставки, таких как маленькие моноламеллярные везикулы, большие моноламеллярные везикулы и мультиламеллярные везикулы. Липосомы могут быть сформированы из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть доставлены с использованием моноклональных антител как отдельных носителей, к которым присоединяются молекулы соединения. Соединения также могут быть присоединены к растворимым полимерам, как наводимым на цель носителям лекарственных средств. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный группами палитоила. Кроме того, соединения могут быть связаны с биологически разлагаемыми полимерами, полезными в достижении контролируемого высвобождения лекарственного средства, например с полимерами молочной кислоты, полиэпсилон-капролактоном,полигидроксимасляной кислотой, поли-(орто)-сложными эфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и сшитыми или амфипатичными блок-сополимерами гидрогелей. Фармацевтические композиции, адаптированные для трансдермального введения, могут быть представлены в виде дискретных пластырей, которые предназначены оставаться в тесном контакте с эпидермисом реципиента в течение продолжительного периода времени. Например, активный ингредиент может быть доставлен из пластыря посредством ионтофореза, как, в общем, описано в Pharmaceutical Фармацевтические композиции, адаптированные для местного введения, могут быть составлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Для лечения глаз или других внешних тканей, например рта и кожи, композиции предпочтительно применять в качестве наружной мази или крема. Когда композиция приготовлена в виде мази, активный ингредиент может быть использован либо с парафиновой, либо со смешивающейся с водой мазевой основой. В качестве альтернативы активный ингредиент может быть введен в крем с основой для крема масло-в-воде или вода в масле. Фармацевтические композиции, адаптированные для местного введения в глаз, включают глазные капли, в которых активный ингредиент растворяют или суспендируют в подходящем носителе, главным образом, в водном растворителе. Фармацевтические композиции, адаптированные для местного введения в рот, включают таблетки,пастилки и жидкости для полоскания рта. Фармацевтические композиции, адаптированные для ректального введения, могут быть представлены в виде свечей или в виде клизм. Фармацевтические композиции, адаптированные для назального введения, где носитель представляет собой твердое вещество, включают порошок для курсового приема, который вводят посредством вдыхания через нос, т.е. посредством быстрого вдоха через носовой проход из контейнера с порошком,который удерживается вплотную к носу. Подходящие композиции, в которых носитель представляет собой жидкость для введения в виде назального спрея или назальных капель, включают водные или масляные растворы активного ингредиента. Фармацевтические композиции, адаптированные для введения путем ингаляции, включают частицы в виде мелкой пыли или тумана, которые могут быть получены с помощью различных типов дозирующих, нагнетающих аэрозоли ингаляторов, небулайзеров или инсуфляторов. Фармацевтические композиции, адаптированные для вагинального применения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или распыляемых составов. Фармацевтические композиции, адаптированные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворы, которые переводят композицию в состояние, изотоническое с кровью предполагаемого реципиента; водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например, в запаянных ампулах и флаконах, и могут храниться в сублимированном (лиофилизированном) состоянии, требуя лишь добавления стерильного жидкого носителя, например воды, для инъекций, непосредственно перед применением. Изготовленные для немедленного введения растворы для инъекций и суспензии могут быть получены из стерильных порошков, гранул и таблеток. Следует иметь в виду, что в дополнение к ингредиентам, в частности, упомянутым выше, композиции могут включать другие вещества, общепринятые в данной области, с учетом типа рассматриваемой композиции, например композиции, подходящие для перорального введения, могут включать ароматизаторы. В табл. 1 приведены некоторые иллюстративные примеры соединений, которые могут быть введены с соединениями по настоящему изобретению. Соединения по изобретению могут вводиться с другими соединениями, обладающими противовирусной активностью в отношении HCV, в комбинированной терапии либо совместно, либо отдельно, либо путем объединения соединений в композицию. Соединения по настоящему изобретению могут также применяться в качестве лабораторных реагентов. Соединения могут играть важную роль в обеспечении исследовательских инструментов при разработке анализов вирусной репликации, проверки правильности животных тест-систем и структурных биологических исследований для дальнейшего расширения знаний о механизмах заболевания вирусным гепатитом С. Кроме того, соединения по настоящему изобретению полезны в установлении или определении сайтов связывания других противовирусных соединений, например, путем конкурентного ингибирования. Соединения по настоящему изобретению могут также применяться для обработки или предупреждения вирусного загрязнения материалов и, следовательно, снижать риск вирусного заражения лабораторных или медицинских работников и пациентов, которые вступают в контакт с такими материалами,например, с кровью, тканями, хирургическими инструментами и одеждой, лабораторными инструментами и одеждой, и аппаратами и материалами для сбора или переливания крови. Это изобретение предназначено охватить соединения, имеющие формулу (I), при получении синтетическим способом или в процессах обмена веществ, включая те, которые происходят в организме человека или животного (in vivo), или в процессах, происходящих in vitro. Настоящее изобретение будет описано в связи с некоторыми вариантами осуществления изобретения, которые не предназначены для ограничения его объема. Напротив, настоящее изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем изобрете- 16024173 ния. Таким образом, следующие примеры, которые включают конкретные варианты осуществления, будут иллюстрировать одно применение на практике настоящего изобретения при том понимании, что примеры приводятся для целей иллюстрации конкретных вариантов осуществления и представлены для того, чтобы предложить то, что считается наиболее полезным и легко понимаемым описанием приемов и концептуальных аспектов. Сокращения, применяемые в настоящем описании, включая, в частности, иллюстративные примеры, которые следуют далее, хорошо известны специалистам в данной области. Некоторые из используемых сокращений представляют собой следующие:rt, или RT, или Rt - комнатная температура или время удерживания (в соответствии с контекстом);TFA - трифторуксусная кислота. Исходные материалы, используемые для синтеза соединений по настоящему изобретению, известны специалистам в данной области и могут быть легко приготовлены или являются коммерчески доступными. Следующие нижеизложенные методы приведены с иллюстративными целями и не предназначены для ограничения объема формулы изобретения. Следует иметь в виду, что может быть необходимо для получения такого соединения, в котором функциональная группа защищена с использованием общепринятой защитной группы, затем удалить эту защитную группу для получения соединения по настоящему изобретению. Особенности, касающиеся применения защитных групп по настоящему изобретению, известны специалистам в данной области. ляли уксусную кислоту (5 г, 83 ммоль) при комнатной температуре. Реакционную смесь нагревали до 125C в течение 4 ч. Побочный продукт этанол перегоняли путем нормальной дистилляции с получением сырого соединения этил 3-метилпент-4-еноат (70 г, 71.0%) в виде бесцветного масла Его переносили на следующую стадию без дополнительной очистки. 1 Н ЯМР (DMCO-d6): ppm 5.82-5.73 (m, 1H), 5.03-4.92 (m, 2H), 4.08-4.02 (m, 2H), 2.50 (m, 1H), 2.312.26 (m, 2H), 1.17 (t, J=8 Гц, 3 Н), 1.00 (d, J=8 Гц, 3 Н). Стадия 2. Получение 3-метилпент-4-еноевой кислоты К раствору этил 3-метилпент-4-еноата (70 г, 492 ммоль) в метаноле (500 мл) добавляли KOH (41.4 г,738 ммоль) при комнатной температуре. Реакционную массу нагревали с обратным холодильником в течение 18 ч. Реакционную массу дистиллировали при пониженном давлении. К остатку добавляли эфир(750 мл), промывали насыщенным раствором бикарбоната натрия (3350 мл). Объединенный водный слой подкисляли концентрированной HCl (250 мл), экстрагировали DCM (500 мл 3). Объединенный органический слой высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением сырого соединения 3-метилпент-4-еноевая кислота (52 г, 93%) в виде коричневого масла. 1 Н ЯМР (DMCO-d6):ppm 12.04 (s, br, 1H), 5.84-5.75 (m, 1H), 5.03-4.92 (m, 2H), 2.56-2.50 (m, 1H),2.28-2.22 (m, 2H), 1.00 (d, J=8 Гц, 3 Н). Стадия 3. Получение 3-метилпент-4-ен-1-ола К суспензии алюмогидрида лития (6.03 г, 0.159 моль) в 150 мл сухого Et2O при 0C добавляли по каплям раствор 3-метилпент-4-еноевой кислоты (17.4 г, 0.122 моль) в 100 мл сухого Et2O. После завершения добавления реакцию оставляли нагреваться до комнатной температуры и перемешивали в течение 2 ч. Реакционную массу выливали в а 1 л химический стакан. Осторожно добавляли воду (6.03 мл) и 15%NaOH (6.03 мл) при хорошем перемешивании на протяжении 30 мин. Выделившееся белое твердое вещество удаляли путем фильтрации и хорошо промывали Et2O. Объединенные фильтраты высушивали и концентрировали перегонкой при атмосферном давлении. Остаток перегоняли с получением целевого соединения 9.04 г (74%). 1 Н ЯМР (CDCl3):ppm 5.65 (m, 1 Н), 4.90 (m, 2H), 3.55 (m, 2H), 3.30 (m, 1H), 2.25 (m, 1H), 1.50 (q,J=7 Гц, 2 Н), 1.00 (d, J=7 Гц, 3 Н). Стадия 4. Получение 5-бром-3-метилпент-1-ена К перемешанному раствору 4.504 г (44.97 ммоль) 3-метилпент-4-ен-1-ола в 100 мл дихлорметана при 0C последовательно добавляли 3.8 мл (49.1 ммоль) метансульфонилхлорида и 6.91 мл (49.6 ммоль) триэтиламина и полученную в результате смесь перемешивали при 0C в течение 15 мин. Реакционную смесь затем выливали в 100 мл насыщенного водного NaHCO3, и полученную в результате смесь энергично перемешивали в течение 10 мин. Органическую фазу отделяли, высушивали (Na2SO4), и затем концентрировали при пониженном давлении с получением сырого мезилатного соединения. Сырое соединение растворяли в 130 мл сухого THF и к раствору добавляли 5.87 г (67.6 ммоль) безводного бромида лития. Полученную в результате смесь нагревали с обратным холодильником в течение 4 ч. Реакционную массу охлаждали до комнатной температуры и разбавляли 300 мл пентана. Органическую фазу промывали двумя 100-мл порциями насыщенного NaHCO3 и пятью 100-мл порциями воды, и затем высушивали (Na2SO4). Растворитель удаляли путем перегонки через 30-см колонку Vigreux при атмосферном давлении. Выпарной дистилляцией остатка (70C, 80 мм Hg) получали 6.28 г (86%) бромида. 1 Н ЯМР (CDCl3): ppm 1.01 (d, J=5 Гц, 3 Н), 1.80 (dt, J=J'=5 Гц, 2 Н), 2.35 (m, J=5 Гц, 1H), 3.36 (t,J=5 Гц, 2H), 5.02 (m, 2H), 5.64 (m, 1H). Стадия 5. Получение (2R)-6-метилокт-7-ен-2-ола Магниевую стружку (1.3 г, 55.2 ммоль) суспендировали в сухом THF (50 мл) и к смеси добавляли щепотку йода (20 мг) при комнатной температуре. К этой реакционной массе добавляли раствор 5-бром 3-метилпент-1-ена (6 г, 36.8 ммоль) в THF (200 мл). Реакционную массу нагревали с помощью термофена для инициирования реакции. Когда реакция, по оценке, завершилась, раствор канюлировали к раствору (S)-пропиленоксида (3.21 г, 55.2 ммоль) и бромида меди (0.528 г, 3.68 ммоль) в THF (50 мл) при -78C. Реакционную массу оставляли нагреваться до комнатной температуры и перемешивали на протяжении ночи. Реакционную массу быстро охлаждали насыщенным раствором хлорида аммония и экстрагировали диэтиловым эфиром (3200 мл). Объединенный органический слой высушивали над безводным Na2SO4 и концентрировали при комнатной температуре с получением сырого соединения. Сырое соединение очищали с помощью колоночной хроматографии (силикагель, 10% ТВМЕ в петролейном эфире) с получением (2R)-6-метилокт-7-ен-2-ола (12.4 г, 92%) в виде маслянистой жидкости. 1 Н ЯМР (400 МГц, CDCl3):ppm 5.82-5.69 (m, 1H), 4.98-4.91 (m, 2 Н), 4.27-4.26 (m, 1 Н), 3.65-3.39(0.51 г, 4.22 ммоль), и раствор перемешивали в течение 10 мин. К реакционной массе добавляли паратолуолсульфонилхлорид (18.5 г, 97 ммоль) при 0C. Реакционную массу оставляли нагреваться до комнатной температуры и перемешивали на протяжении ночи. Пиридин удаляли при пониженном давлении,и остаток разбавляли этилацетатом (200 мл). Органический раствор промывали водным 1.5 н. растворомHCl, насыщенным раствором бикарбоната, концентрированным соляным раствором, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого соединения(14.5 г, 32%). Сырое соединение переносили на следующую стадию без дополнительной очистки. Стадия 7. Получение (3R)-этил 2-(дифенилметиленамино)-3,7-диметилнон-8-еноатаLiHMDS (60.7 мл, 60.7 ммоль, 1 М раствор в THF) при 0C. Реакционную массу оставляли нагреваться до комнатной температуры, нагревали при 110C в течение 2 ч. Реакционную массу охлаждали до комнатной температуры, быстро охлаждали водой и экстрагировали этилацетатом (3200 мл). Объединенный органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого соединения (14.5 г). Сырое соединение переносили на следующую стадию без дополнительной очистки. МС: МС m/z 392.1 (М 1). Стадия 8. Получение (3R)-этил 2-амино-3,7-диметилнон-8-еноата К раствору (3R)-этил 2-(дифенилметиленамино)-3,7 диметилнон-8-еноата (14.5 г, 37 ммоль) в диэтиловом эфире (25 мл) добавляли водный 1.5 н. раствор HCl (125 мл) и реакционную массу перемешивали при комнатной температуре на протяжении ночи. Реакционную массу промывали диэтиловым эфиром(100 мл). Водный раствор ощелачивали, применяя насыщенный раствор бикарбоната натрия, и экстрагировали этилацетатом (3100 мл). Объединенный органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого соединения (2.1 г, 23%). Сырое соединение переносили на следующую стадию без дополнительной очистки. 1 Н ЯМР (400 МГц, CDCl3):ppm 5.85-5.60 (m, 1H), 4.98-4.85 (dd, J=1.6, 2.8 Гц, 2H), 4.10-4.06 (m,2H), 3.15-3.14 (m, 1H), 2.15-2.02 (m, 1H), 1.94-1.75 (m, 3H), 1.32-1.17 (m, 9H), 0.95-0.93 (m, 3H), 0.84-0.74DIPEA (1.7 г, 13.2 ммоль) с последующим добавлением (Вос)2 О (2.4 г, 11.4 ммоль) при комнатной температуре. Реакционную массу перемешивали при комнатной температуре на протяжении ночи. Реакционную массу разбавляли DCM и промывали водой. Органический слой высушивали над безводнымNa2SO4 и концентрировали при пониженном давлении с получением сырого соединения. Сырое соединение очищали с помощью колоночной хроматографии (силикагель, 20% этилацетата в петролейном эфире) с получением 2.7 г (84%) (3R)-этил 2-(трет-бутоксикарбониламино)-3,7-диметилнон-8-еноата в виде маслянистой жидкости. 1 Н ЯМР (400 МГц, CDCl3):ppm 5.85-5.60 (m, 1H), 4.98-4.85 (dd, J=1.6, 2.8 Гц, 2 Н), 4.10-4.06 (m,2 Н), 2.15-2.02 (m, 1H), 1.55-1.44 (m, 8 Н), 1.39-1.30 (s, 9 Н), 1.27-1.21 (m, 4 Н), 0.95-0.93 (m, 3 Н), 0.84-0.74THF/вода (40 мл, 1:1) добавляли метанол (20 мл) с последующим добавлением LiOH (0.987 г, 41.2 ммоль) при комнатной температуре. Реакционную массу перемешивали при комнатной температуре на протяжении ночи. Растворитель выпаривали при пониженном давлении и остаток разбавляли водой (100 мл). Водный раствор подкисляли водным 1.5 н. раствором HCl до рН 3 и экстрагировали этилацетатом(3100 мл). Объединенный органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого соединения. Сырое соединение очищали с помощью колоночной хроматографии (силикагель, 3% метанола в DCM) с получением 2.1 г (85%) целевого соединения в виде клейкой жидкости. 1 Н ЯМР (400 МГц, DMCO-d6):ppm 12.2-12.02 (bs, 1H), 6.92-6.85 (m, 1H), 5.72-5.66 (m, 1H), 4.984.85 (dd, J=1.6, 2.8 Гц, 2H), 4.03-3.95 (m, 1H), 2,58-2.56 (m, 1H), 2.15-2.02 (m, 1H), 1.45-1.36 (m, 10H), 1.271.21 (m, 5H), 0.95-0.93 (m, 3H), 0.84-0.74 (m, 3H). К кашице (3R)-2-трет-бутоксикарбонил)амино)-3,7-диметилнон-8-еноевой кислоты (6.5 г,21.7 ммоль), L-4-гидроксипролин метилового эфира гидрохлорида (3.9 г, 21.7 ммоль) и о-(7 азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (8.36 г, 21.7 ммоль) в DCM (50 мл) при 0C добавляли по каплям N,N-диизопропилэтиламин (11.5 мл, 66.6 ммоль). Полученную в результате светло-желтую смесь перемешивали при комнатной температуре на протяжении ночи, промывали 1 МHCl (35 мл) и концентрированным соляным раствором, высушивали над Na2SO4, фильтровали, концентрировали под вакуумом. Оставшееся масло (12 г) очищали с помощью обычно применяемой колоночной хроматографии (60-120 г силикагельная колонка), элюировали 4%-8% IPA-гексан с получением целевого продукта (2S,4R)-метил 1-2S,3R)-2-(трет-бутоксикарбониламино)-3,7-диметилнон-8-еноил)-4 гидроксипирролидин-2-карбоксилат (9 а, 4.3 г, выход 46%) в виде белой пены и нецелевого диастереомера К раствору (2S,4R)-метил 1-2S,3R)-2-(трет-бутоксикарбониламино)-3,7-диметилнон-8-еноил)-4 гидроксипирролидин-2-карбоксилата (4.3 г, 10 ммоль) в THF/вода (30 мл, 1:1) добавляли метанол (10 мл) с последующим добавлением LiOH (1.3 г, 0.030 моль) при комнатной температуре. Реакционную массу перемешивали при комнатной температуре на протяжении ночи. Растворитель выпаривали при пониженном давлении, и остаток разбавляли водой (100 мл). Водный раствор подкисляли водным 1.5 н. раствором HCl до рН 3 и экстрагировали этилацетатом (3100 мл). Объединенный органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением целевого продукта(2S,4R)-1-2S,3R)-2-(трет-бутоксикарбониламино)-3,7-диметилнон-8-еноил)-4 гидроксипирролидин-2-карбоновая кислота (3.2 г, выход 78 %) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3):ppm 5.70-5.61 (m, 1H), 5.14-5.12 (d, J=8.8 Гц, 1H), 4.96-4.88 (m, 2 Н),4.82-4.78 (t, J=8.4 Гц, 1 Н), 4.52 (bs, 1H), 4.27-4.23 (m, 1 Н), 3.57-3.53 (m, 3H), 2.48-2.42 (m, 1H), 2.33-2.28(2S,4R)-1-3R)-2-трет-бутоксикарбонил)амино)-3,7-диметилнон-8-еноил)-4 гидроксипирролидин-2-карбоновой кислоты (1.5 г, 3.6 ммоль) в DMCO добавляли 1-хлор-4 метоксиизохинолин (840 мг, 4.4 ммоль) с последующим добавлением t-BuOK (1 M раствор в THF, 18 мл) при комнатной температуре в атмосфере азота. Реакционную массу перемешивали при комнатной температуре в течение 4 ч. Реакционную массу быстро охлаждали водным раствором лимонной кислоты и экстрагировали этилацетатом. Объединенный органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого соединения. Сырое соединение очищали с помощью комбифлэш-хроматографии (Combiflash) с получением целевого продукта (800 мг,40%) в виде не совсем белого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3):ppm 8.11-8.09 (m, 2 Н), 7.70-7.66 (t, J=7.4 Гц, 1H), 7.56-7.51 (t, J=8.0 Гц,1H), 7.47 (s, 1H), 5.78 (bs, 1H), 5.69-5.65 (m, 1H), 5.14-5.11 (m, 1H), 4.96-4.88 (m, 3H), 4.43-4.41 (m, 1H),4.27-4.24 (m, 1H), 3.99 (s, 4H), 2.75-2.73 (m, 1H), 2.67-2.65 (m, 1H), 2.10 (bs, 1H), 1.88-1.85 (m, 2H), 1.31 (s,9H), 1.24 (s, 2H), 0.98-0.96 (m, 4H), 0.91-0.86 (m, 4H);(1R,2S)-1-амино-N-(циклопропилсульфонил)-2-винилциклопропанкарбоксамида гидрохлорида (полученного в соответствии с методикой, описанной в WO 03/099274, стр. No. 53-59, 74-76) (600 мг,1.5 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную массу разбавляли DCM и промывали водой. Органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого соединения. Сырое соединение очищали с помощью комбифлэш-хроматографии с получением целевого продукта(900 мг, 90%) в виде не совсем белого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3):ppm 8.13-8.07 (m, 2 Н), 7.72-7.67 (t, J=7.2 Гц, 1 Н), 7.57-7.53 (t, J=7.2 Гц,1 Н), 7.47 (s, 1 Н), 6.80 (bs, 1 Н), 5.84-5.79 (m, 2 Н), 5.73-5.64 (m, 1 Н), 5.39-5.38 (m, 1 Н), 5.28-5.23 (d, J=17.2 Гц, 1 Н), 5.15-5.12 (d, J=10.4 Гц, 1H), 4.97-4.88 (m, 2 Н), 4.49-4.45 (m, 2 Н), 4.19-4.17 (m, 2 Н), 4.00 (s, 3 Н),2.90-2.81 (m, 1H), 2.54-2.48 (m, 1 Н), 2.23-2.07 (m, 2 Н), 2.05-1.99 (m, 2 Н), 1.49-1.42 (m, 1 Н), 1.33 (s, 12H),1.22-1.15 (m, 2 Н), 1.07-1.02 (m, 2 Н), 0.98-0.96 (d, J=6.8 Гц, 4 Н), 0.86-0.81 (d, J=6.8 Гц, 4 Н); МС: МС m/z 782.2 (М 1). Стадия 5. Получение соединения 1 и соединения 2(100 мл) добавляли Катализатор Граббса II поколения (24 мг, 10% массовая доля) при комнатной температуре в атмосфере азота. Реакционную массу нагревали при 95C на протяжении ночи. Растворитель выпаривали при пониженном давлении, и остаток очищали с помощью комбифлэш-хроматографии с получением (500 мг, 58%) целевого продукта в виде смеси диастереомеров. Смесь диастереомеров отделяли, используя препаративную ВЭЖХ, с получением Соединения 1 (12 мг, 8%) и соединения 2 (8 мг,3%). Соединение 1. трет-Бутил(500 мг,0.66 ммоль) в диоксане, HCl, перемешивали при комнатной температуре в течение 30 мин. Растворитель выпаривали при пониженном давлении с получением сырого соединения (350 мг, 77%). Сырое соединение промывали диэтиловым эфиром и переносили на следующую стадию без дополнительной очистки. МС: МС m/z 652.2 (М+-1). Получение пиридин-2-ил-1,1,1-трифтор-2-метилпропан-2-ил карбоната К кашице NaH (1.03 г, 25.8 ммоль) в THF (70 мл) добавляли 1,1,1-трифтор-2-метилпропан-2-ол (3 г,23.42 ммоль). Реакцию перемешивали при 0C в течение 20 мин. Затем к смеси добавляли раствор дипиридин-2-ил карбоната (5.06 г, 23.42 ммоль) в THF (30 мл). Полученный в результате раствор перемешивали при комнатной температуре на протяжении ночи. Отфильтровывали полученные в результате твердые побочные продукты и промывали EtOAc (220 мл). Объединенные органические слои промывали водой, высушивали над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный в результате белый пиридин-2-ил (1,1,1-трифтор-2-метилпропан-2-ил) карбонат (1.24 г,21%) применяли непосредственно в качестве реагента. Получение соединения 3 и соединения 4. К раствору(350 мг, 0.53 ммоль) в DCM (4 мл) добавляли DIPEA (0.3 мл, 1.8 ммоль) с последующим добавлением пиридин-2-ил-2,2,2-трифтор-1,1-диметил этилового эфира углекислоты (180 мг, 0.72 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную массу разбавляли DCM и промывали водой. Органический слой высушивали над безводнымNa2SO4 и выпаривали при пониженном давлении с получением сырого соединения в виде смеси диастереомеров. Смесь диастереомеров отделяли, используя препаративную ВЭЖХ, с получением соединения 3 (50 мг, 12%) и соединения 4 (30 мг, 7%) в виде белого твердого вещества. Соединение 3. 1,1,1-Трифтор-2-метилпропан-2-ил(1.86 г, 4.9 ммоль) с последующим добавлением DIPEA (4.2 мл, 24.5 ммоль) при комнатной температуре. Реакционную массу перемешивали при этой же температуре в течение 10 мин. К реакционной массе добавляли (1R,2S)-1-амино-N-(циклопропилсульфонил)-2-винилциклопропанкарбоксамид гидрохлорид(полученный в соответствии с методикой, описанной в WO 03/099274, с. 53-59, 74-76) (2 г, 5.3 ммоль) и перемешивали при комнатной температуре в течение 3 ч. Реакционную массу разбавляли дихлорметаном и промывали водой. Органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого соединения. Сырое соединение очищали с помощью комбифлэш-хроматографии (3% МеОН в CHCl3) с получением 1.9 г (62%) целевого продукта в виде белого твердого вещества. МС: МС m/z 623.5 (М+-1). Стадия 2. Получение трет-бутил диметил-1-оксонон-8-ен-2-илкарбамата (0.9 г, 1.4 ммоль) в дихлорэтане (150 мл) добавляли катализатор Граббса II поколения (90 мг, 10% массовые доли) при комнатной температуре в атмосфере азота. Реакционную массу нагревали при 95C на протяжении ночи. Растворитель выпаривали при пониженном давлении, и остаток очищали с помощью ISCO с получением целевого соединения (600 мг, 69%). МС: МС m/z 595.2 (М+-1). Стадия 3. Получение трет-бутил (2R,6S,7R,13aS,14aR,16aS,Z)-2-(5-хлор-4-метоксиизохинолин-1 илокси)-14 а-(циклопропилсульфонилкарбамоил)-7,11-диметил-5,16-диоксо 1,2,3,5,6,7,8,9,10,11,13 а,14,14 а,15,16,16 а-гексадекагидроциклопропа[е]пирроло[1,2 а][1,4]диазациклопентадецин-6-илкарбамата(1M раствор в THF, 450 мг, 4.0 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную массу быстро охлаждали водным раствором лимонной кислоты и экстрагировали этилацетатом (3150 мл). Объединенный органический слой промывали водой, концентрированным соляным раствором, высушивали над безводнымNa2SO4 и концентрировали при пониженном давлении с получением сырого соединения в виде смеси диастереомеров. МС: МС m/z 789.3 (М 1). Стадия 4. Получение соединения 5 и соединения 6. Диастереомерную смесь (2R,6S,7R,13aS,14aR,16aS,Z)-2-(5-хлор-4-метоксиизохинолин-1-илокси)14 а-(циклопропилсульфонилкарбамоил)-7,11-диметил-5,16-диоксо 1,2,3,5,6,7,8,9,10,11,13 а,14,14 а,15,16,16 а-гексадекагидроциклопропа[е]пирроло[1,2 а][1,4]диазациклопентадецин-6-илкарбамата разделяли с помощью препаративной ВЭЖХ с получением целевого соединения 5 и соединения 6.(300 мг,3.3 ммоль) в диоксане.HCl перемешивали при комнатной температуре в течение 30 мин. Растворитель выпаривали при пониженном давлении с получением сырого соединения (250 мг, 96%). Сырое соединение промывали диэтиловым эфиром и переносили на следующую стадию без дополнительной очистки.MC: MC m/z 688.2 (М+-36). Соединение 7 и соединение 8 получали, используя промежуточные соединения, описанные здесь, и следуя общей методике, описанной для синтеза соединения 3. Диастереомерную смесь отделяли с помощью препаративной ВЭЖХ. Соединение 7. 1,1,1-Трифтор-2-метилпропан-2-ил (2R,6S,7R,11R,13aS,14aR,16aS,Z)-2-(5-хлор-4 метоксиизохинолин-1-илокси)-14 а-(циклопропилсульфонилкарбамоил)-7,11-диметил-5,16-диоксо 1,2,3,5,6,7,8,9,10,11,13 а,14,14 а,15,16,16 а-гексадекагидроциклопропа[е]пирроло[1,2 а][1,4]диазациклопентадецин-6-илкарбамат. 1 Н ЯМР (400 МГц, CD3OD):ppm 8.18-8.15 (d, J=7.6 Гц, 1 Н), 7.78-7.76 (d, J=6.4 Гц, 1H), 7.70 (s,1H), 7.49-7.45 (t, J=8.0 Гц, 1H), 5.81 (m, 1H), 5.37-5.35 (t, J=10.8 Гц, 1 Н), 5.04-4.94 (m, 1H), 4.78-4.75 (d,J=11.2 Гц, 1H), 4.70-4.66 (m, 1H), 4.01-3.97 (m, 1H), 3.96 (s, 3H), 3.80-3.77 (d, J=10.4 Гц, 1H), 2.98-2.94 (m,1H), 2.77-2.64 (m, 3H), 2.45-2.38 (m, 1H), 1.84-1.82 (m, 1H), 1.74-1.71 (m, 1H), 1.65-1.60 (m, 3H), 1.43-1.29 Соединения 9 и 10 получали, используя промежуточные соединения, описанные здесь, и следуя общей методике, описанной для синтеза соединения 7 и соединения 8. Соединение 9. 1,1,1-Трифтор-2-метилпропан-2-ил Стадия 1. Получение 1-хлор-4-метоксиизохинолина. К раствору 1-хлоризохинолин-4-ола (5.0 г, 27.8 ммоль) в ацетонитриле (50 мл) добавляли ТМСдиазометан (12.73 г, 111.2 ммоль) при 0C. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 2 ч. Растворитель выпаривали при пониженном давлении с получением сырого соединения. Сырое соединение очищали с помощью хроматографии на силикагеле с получением 1-хлор-4-метоксиизохинолина (2.5 г, 46.4%) в виде не совсем белого твердого вещества. 1 Н ЯМР (400 МГц, CD3OD):ppm 8.29-8.17 (m, 2 Н), 7.97 (s, 1 Н), 7.91-7.82 (m, 2H), 4.05 (s, 3H);(4.01 г, 25.82 ммоль) при комнатной температуре. Реакционный сосуд (пробирка под давлением) герметично закрывали и нагревали при 145C в течение 18 ч. Реакционную массу разбавляли водой и экстрагировали этилацетатом. Органический слой высушивали над безводным Na2SO4 и выпаривали при пониженном давлении с получением сырого соединения. Сырое соединение очищали с помощью хроматографии на силикагеле с получением целевого соединения (700 мг, 62%) в виде белого твердого вещества. 1 Стадия 1. Получение (Е)-3-(2-хлорфенил)акрилоил азида. К раствору (Е)-3-(2-хлорфенил) акриловой кислоты (13 г, 71.2 ммоль) в бензоле (100 мл) добавляли триэтиламин (14.4 г, 141 ммоль) с последующим добавлением DPPA (19.5 г, 71.2 ммоль) при комнатной температуре. Реакционную массу перемешивали при этой же температуре в течение 18 ч. Растворитель выпаривали при пониженном давлении, и остаток разбавляли водой и экстрагировали дихлорметаном. Органический слой высушивали над безводным сульфатом натрия, фильтровали и выпаривали с получением сырого соединения. Сырое соединение очищали с помощью обычно применяемой колоночной хроматографии (силикагель, 60-120 ме 1H) с использованием 10% этилацетата в петролейном эфире в качестве подвижной фазы с получением целевого соединения в виде бледно-желтого твердого вещества(Е)-3-(2-хлорфенил)акрилоил азид (2 г, 9.63 ммоль). Реакцию нагревали при 250C в течение 2 ч. Реакционную массу охлаждали до комнатной температуры и разбавляли петролейным эфиром. Осажденное твердое вещество фильтровали, промывали петролейным эфиром с получением сырого соединения(1.1 г, 63.6%) в виде желтого твердого вещества. Сырое соединение переносили на следующую стадию без дополнительной очистки. 1 Н ЯМР (400 МГц, CD3OD):ppm 8.29-8.27 (d, J=8 Гц, 1 Н), 7.84-7.82 (d, J=8 Гц, 1H), 7.49 (t, J=8 Гц,1H), 7.32-7.30 (d, J=8 Гц, 1H), 6.98-6.96 (d, J=8 Гц, 1H);MC: MC m/z 180.7 (M1). Стадия 3. Получение 5-хлор-4-метоксиизохинолш-1-(2 Н)-она. К раствору 5-хлоризохинолин-1(2 Н)-она (3.8 г, 21.2 ммоль) в метаноле (70 мл) добавляли иодозобензолдиацетат (7.5 г, 23.4 ммоль) с последующим добавлением метансульфокислоты (2.45 г,25.5 ммоль) при комнатной температуре. Реакционную массу нагревали с обратным холодильником в течение 3 ч. Растворитель выпаривали, и остаток разбавляли холодной водой. Осажденное твердое вещество фильтровали, промывали водой с получением сырого соединения (3.9 г, 88%) в виде твердого вещества светло-красного цвета. 1 Н ЯМР (400 МГц, CDCl3):ppm 8.06-8.03 (d, J=12 Гц, 1 Н), 7.60-7.57 (d, J=12 Гц, 1H), 7.42 (d,J=8 Гц, 1H), 7.01-6.99 (br s, 1H), 3.51 (s, 3H);MC: MC m/z 209.1 (M1). Стадия 4. Получение 1,5-дихлор-4-метоксиизохинолина. Раствор 5-хлор-4-метоксиизохинолин-1-(2 Н)-она (6.4 г, 30.5 ммоль) в POCl3 (45 мл) нагревали с обратным холодильником в течение 18 ч. Растворитель выпаривали при пониженном давлении, и остаток разбавляли холодной водой. Осажденное твердое вещество фильтровали, промывали водой с получением сырого соединения (4 г, 57.4%) в виде твердого вещества светло-коричневого цвета. 1 Н ЯМР (400 МГц, CDCl3):ppm 8.28-8.25 (d, J=12 Гц, 1H), 7.87 (s, 1H), 7.79-7.76 (d, J=12 Гц, 1H),7.58-7.54 (m, 1H), 4.03 (s, 3H); Стадия 1. Получение 7-фтор-4,6-диметоксиизохинолин-1(2 Н)-она. К раствору 7-фтор-6-метоксиизохинолин-1(2 Н)-она (6.3 г, 32.6 ммоль) в метаноле (70 мл) добавляли йодозобензолдиацетат (10.5 г, 32.6 ммоль) с последующим добавлением метансульфокислоты (3.76 г,39.1 ммоль) при комнатной температуре. Реакционную массу нагревали с обратным холодильником в течение 3 ч. Растворитель выпаривали, и остаток разбавляли холодной водой. Осажденное твердое вещество фильтровали, промывали водой с получением сырого соединения (6.6 г, 91%) в виде твердого вещества светло-красного цвета. МС: МС m/z 224.0 (М 1). Стадия 2. Получение 1-хлор-7-фтор-4,6-диметоксиизохинолина. Раствор 7-фтор-4,6-диметоксиизохинолин-1(2 Н)-она (7.6 г, 34.1 ммоль) в POCl3 (50 мл) нагревали с обратным холодильником в течение 18 ч. Растворитель выпаривали при пониженном давлении, и остаток разбавляли холодной водой. Водный раствор ощелачивали твердым карбонатом натрия и экстрагировали этилацетатом. Органический слой высушивали над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении с получением сырого соединения. Сырое соединение очищали с помощью хроматографии на силикагеле (20% этилацетата в петролейном эфире) с получением целевого соединения (2 г, 24.3%) в виде бледно-желтого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3):ppm 7.88-7.85 (d, J=12 Гц, 1H), 7.75 (s, 1H), 7.54-7.52 (d, J=8 Гц, 1 Н),4.05 (s, 3 Н); МС: МС m/z 242.0 (М 1). Стадия 3. Получение 1,7-дифтор-4,6-диметоксиизохинолина. К раствору 1-хлор-7-фтор-4,6-диметоксиизохинолина (2 г, 8.28 ммоль) в DMCO (5 мл) добавляли фторид цезия (2.51 г, 16.55 ммоль) при комнатной температуре. Реакционный сосуд (пробирка под давлением) герметично закрывали и нагревали при 145C в течение 18 ч. Реакционную массу разбавляли водой и экстрагировали этилацетатом. Объединенный органический слой высушивали над безводнымNa2SO4 и выпаривали при пониженном давлении с получением сырого соединения. Сырое соединение очищали с помощью хроматографии на силикагеле с получением целевого соединения (475 мг, 25.5%) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3):ppm 7.69-7.67 (d, J=8 Гц, 1H), 7.53-7.51 (d, J=8 Гц, 1H), 7.45-7.44 (m,1H), 4.05 (s, 3H), 4.03 (s, 3H); 19 Стадия 1. Получение (Е)-3-(3-метоксифенил) акрилового азида. К раствору (Е)-3-(3-метоксифенил) акриловой кислоты (20 г, 112 ммоль) в бензоле (200 мл) добавляли триэтиламин (22.72 г, 224 ммоль) с последующим добавлением DPPA (30.9 г, 112 ммоль) при комнатной температуре. Реакционную массу перемешивали при этой же температуре в течение 18 ч. Растворитель выпаривали при пониженном давлении, и остаток разбавляли водой и экстрагировали дихлорметаном. Объединенный органический слой высушивали над безводным сульфатом натрия, фильтровали и выпаривали с получением сырого соединения. Сырое соединение очищали с помощью обычно применяемой колоночной хроматографии (силикагель, 60-120 меш) с использованием 10% этилацетата в петролейном эфире в качестве подвижной фазы с получением целевого соединения в виде белого твердого вещества (18 г, 79%). 1 Н ЯМР (400 МГц, CDCl3):ppm 7.73-7.69 (d, J=16 Гц, 1H), 7.33-7.29 (t, J=8 Гц, 1H), 7.26 (s, 1H),7.14-7.12 (d, J=8 Гц, 1H), 7.05 (s, 1H), 6.98-6.96 (d, J=4 Гц, 1H), 6.43-6.39 (d, J=16 Гц, 1H), 3.86 (s, 3H). Стадия 2. Получение 6-метоксиизохинолин-1(2 Н)-она. К раствору (Е)-3-(3-метоксифенил)акрилоил азида (7 г, 34.4 ммоль) в 1,2-дихлорбензоле (70 мл) добавляли ацетат ртути (0.11 г, 0.34 ммоль). Реакцию нагревали при 150C в течение 10 мин и температуру повышали до 180C в течение 1 ч. Реакционную массу охлаждали до комнатной температуры и разбавляли петролейным эфиром. Осажденное твердое вещество фильтровали, промывали петролейным эфиром с получением сырого соединения (3.5 г, 58%) в виде желтого твердого вещества. Сырое соединение переносили на следующую стадию без дополнительной очистки. 1 Н ЯМР (400 МГц, DMCO):ppm 8.09-8.06 (d, J=12 Гц, 1H), 7.14-7.11 (m, 2H), 7.05-7.03 (d, J=8 Гц,1 Н), 6.48-6.46 (d, J=4 Гц, 1H), 3.86 (s, 3H);
МПК / Метки
МПК: A61P 31/14, C07D 237/32, C07D 217/24, C07K 5/08
Метки: гепатита, ингибиторы, вируса
Код ссылки
<a href="https://eas.patents.su/30-24173-ingibitory-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы вируса гепатита с</a>
Ингибиторы вируса гепатита с

Номер патента: 21194
Опубликовано: 30.04.2015
Авторы: Тимонко Стивен, Кеннет Дж., Белема Маконен, Пател Бхарат П., Пак Шон К., Натали Джр.
МПК: C07D 209/54
Метки: ингибиторы, гепатита, вируса
Формула / Реферат:
1. Способ получения соединения формулы (III)или его фармацевтически приемлемой соли,где R4 и R5 независимо выбраны из атома водорода, (С1-С6)алкила, (C6-C10)арила, (С6-С10)арил(С1-С6)алкила, гетероциклила и гетероциклил(С1-С2)алкила, где гетероциклил представляет собой 5-6-членный гетероциклил, включающий 1-2 гетероатома, выбранных из N, S и О,включающий обработку соединения формулы (IV)ацетатом аммония в присутствии основания.2. Способ по п.1,...
Ингибиторы вируса гепатита с

Номер патента: 21260
Опубликовано: 29.05.2015
Авторы: Хаманн Лоренс Г., Чэнь Ци, Белема Маконен, Лопез Омар Д.
МПК: A61K 31/422, A61P 31/00, A61K 31/4184...
Метки: ингибиторы, гепатита, вируса
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, гдеs равно 0 или 1;L представляет собой -L1-L2-, гдеL1 и L2 независимо выбраны изпри условии, что по меньшей мере один из L1 и L2 отличается отY и Y' независимо представляют собой кислород (О) или NH;R1 представляет собой -C(O)Rx;R2 представляет собой -C(O)Ry;Rx и Ry независимо выбраны из алкила, независимо замещенного одним или более заместителями, независимо выбранными из арила,...
Ингибиторы вируса гепатита c

Номер патента: 22624
Опубликовано: 29.02.2016
Авторы: Сунь Ли-Цян, Бабу П.В.К.Суреш, Хиберт Шелдон, Минвелл Николас А., Рендучинтала Кишоре В., Мулл Эрик, Баушер Майкл С., Скола Пол Майкл, Нагалакшми Пуличарла, Саркунам Кандхасами, Гиллис Эрик П., Чжао Цянь, Раджамани Рамкумар
МПК: A61K 38/12, C07D 487/04, C07K 5/12...
Метки: гепатита, вируса, ингибиторы
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль,где р представляет собой 1 или 2; представляет собой одинарную или двойную связь;R1 выбран изгде R1 присоединен к основному молекулярному фрагменту через любой замещаемый атом углерода в группе;m имеет значения 1;n имеет значения 0, 1, 2, 3, 4, 5 или 6;Х0 выбран из СН и N;X1 выбран из СН и N;X2 и X3 независимо выбраны из СН, C(Ra) и N при условии, что по меньшей мере один из X1, X2...
Ингибиторы вируса гепатита с

Номер патента: 24171
Опубликовано: 31.08.2016
Авторы: Сринивасу Потхуканури, Минвелл Николас А., Лопез Омар Д., Бендер Джон А., Белема Маконен, Гупта Самаямунтула Венката Сатиа Арун Кумар, Чен Ки, Рампулла Ричард А.
МПК: C07D 403/14, A61K 31/433, A61K 31/4178...
Метки: гепатита, вируса, ингибиторы
Формула / Реферат:
1. Соединение формулы (II)или его фармацевтически приемлемая соль,где каждый D представляет собой NH;L представляет собой связь или фенил;каждый из Z1 и Z2 независимо выбран из СН и N;каждый из Z3 и Z4 независимо выбран из С и N, при условии, что не более двух из Z1, Z2, Z3 и Z4 представляют собой N;A представляет собой четырех-шестичленное кольцо, необязательно содержащее один, два или три гетероатома, независимо выбранных из атома азота и...
Ингибиторы вируса гепатита с

Номер патента: 18793
Опубликовано: 30.10.2013
Авторы: Кэдоу Джон Ф., Белема Маконен
МПК: A61K 31/4178, A61K 31/439, A61P 31/14...
Метки: вируса, ингибиторы, гепатита
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, где L выбран из связи,R1 и R2 представляют собойили R1 представляет собойи R2 выбран изгде обозначает точку присоединения к исходной молекуле;R3 и R4 независимо выбраны из водорода и гало;каждый R5 независимо выбран из водорода;каждый R6 независимо выбран из алкила;R6a представляет собой алкил, где алкил необязательно может образовывать конденсированное 3-членное кольцо со смежным...
Предыдущий патент: Способ изготовления основы с маркировкой и основа с маркировкой, изготовленная указанным способом
Следующий патент: Способ алкоксикарбонилирования замещенных алкенов
Случайный патент: Ингибиторы протеаз