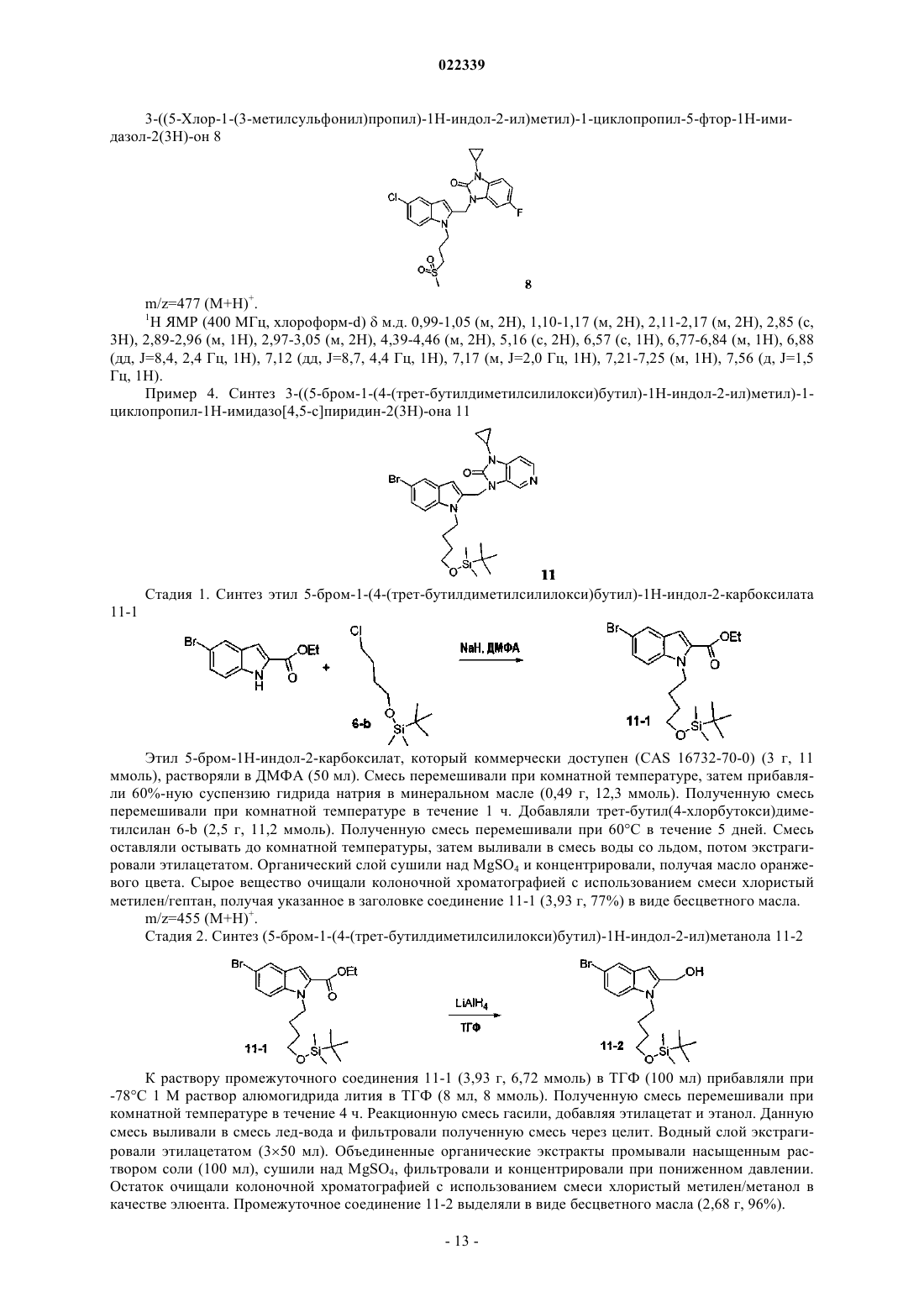
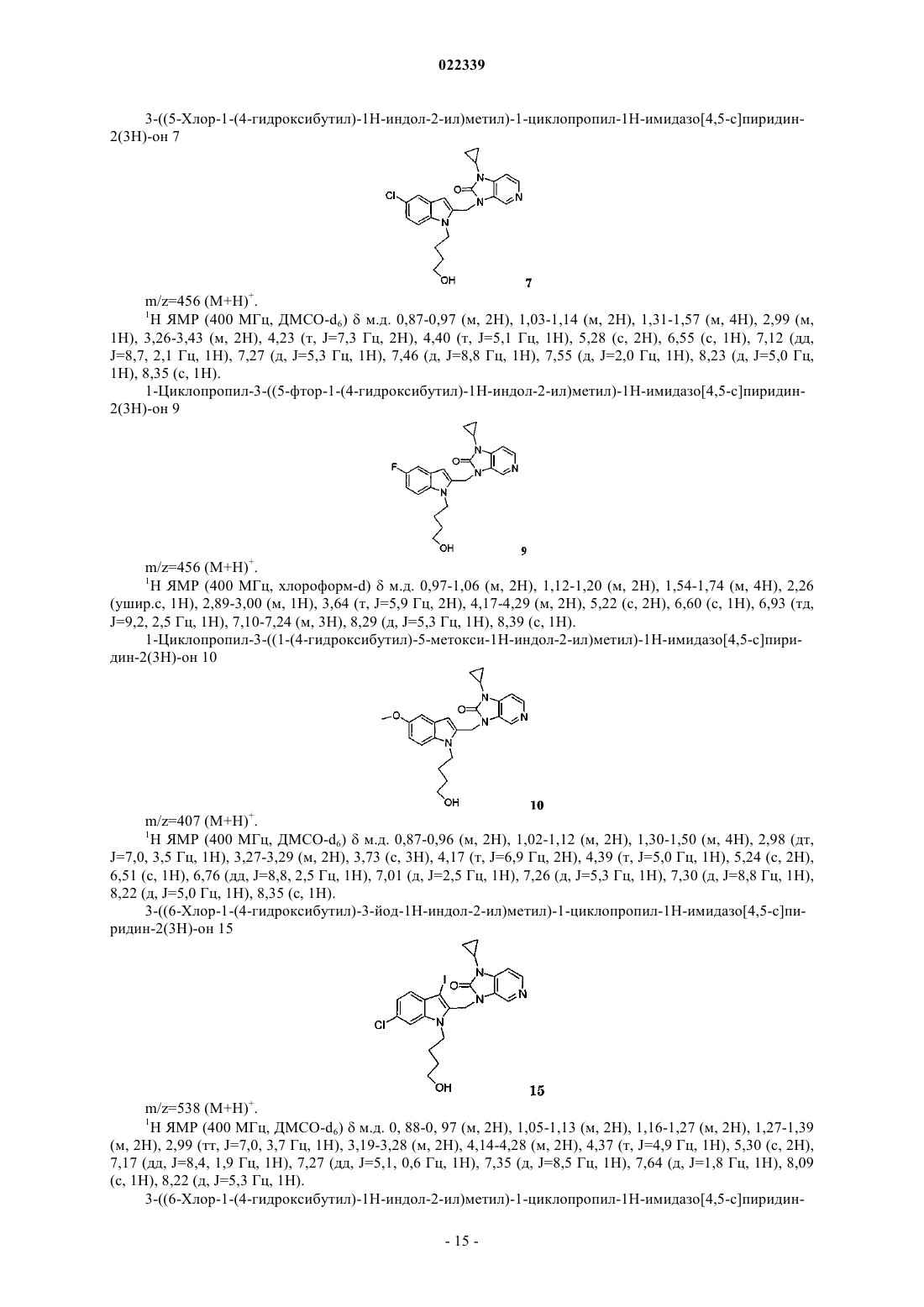
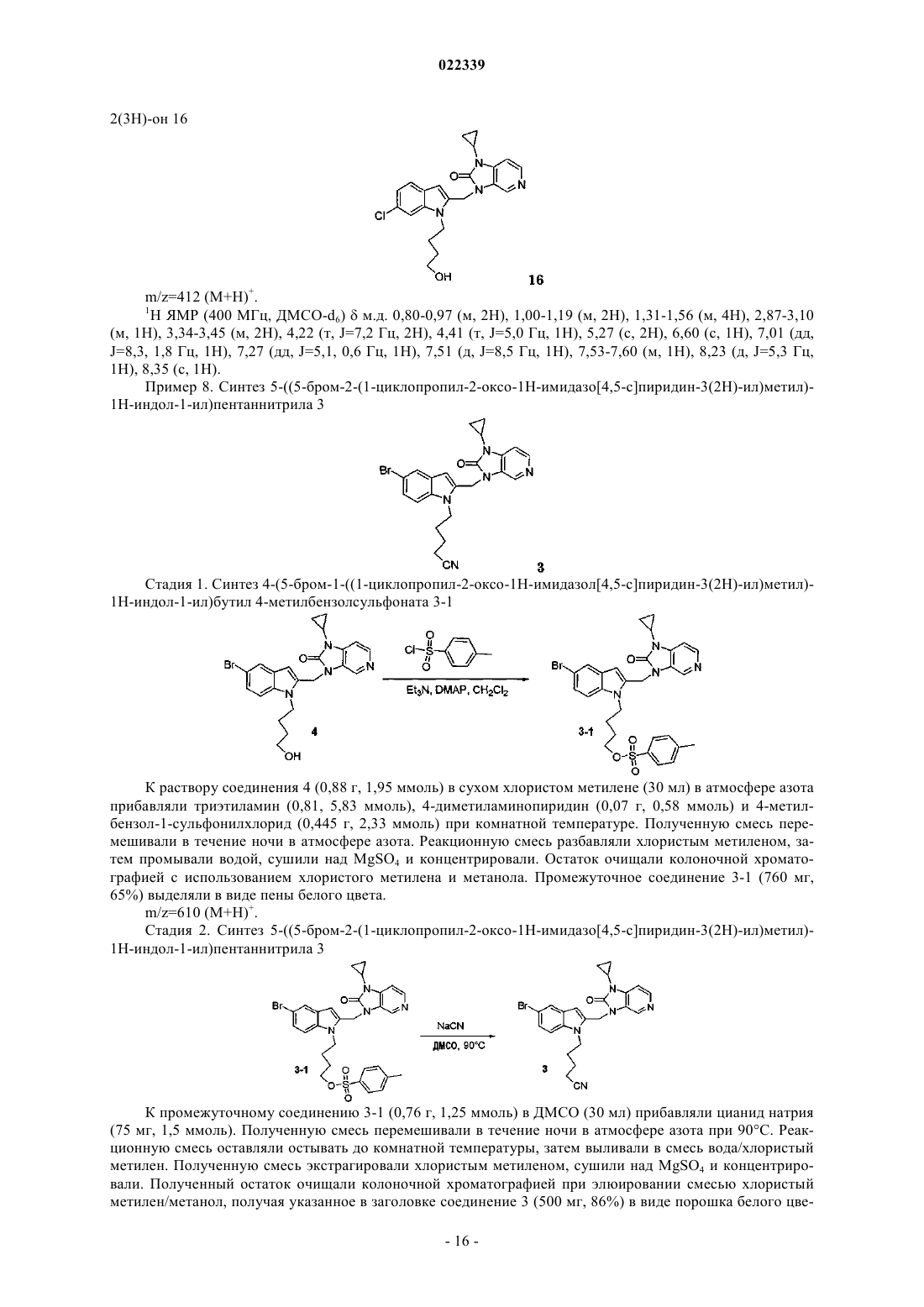
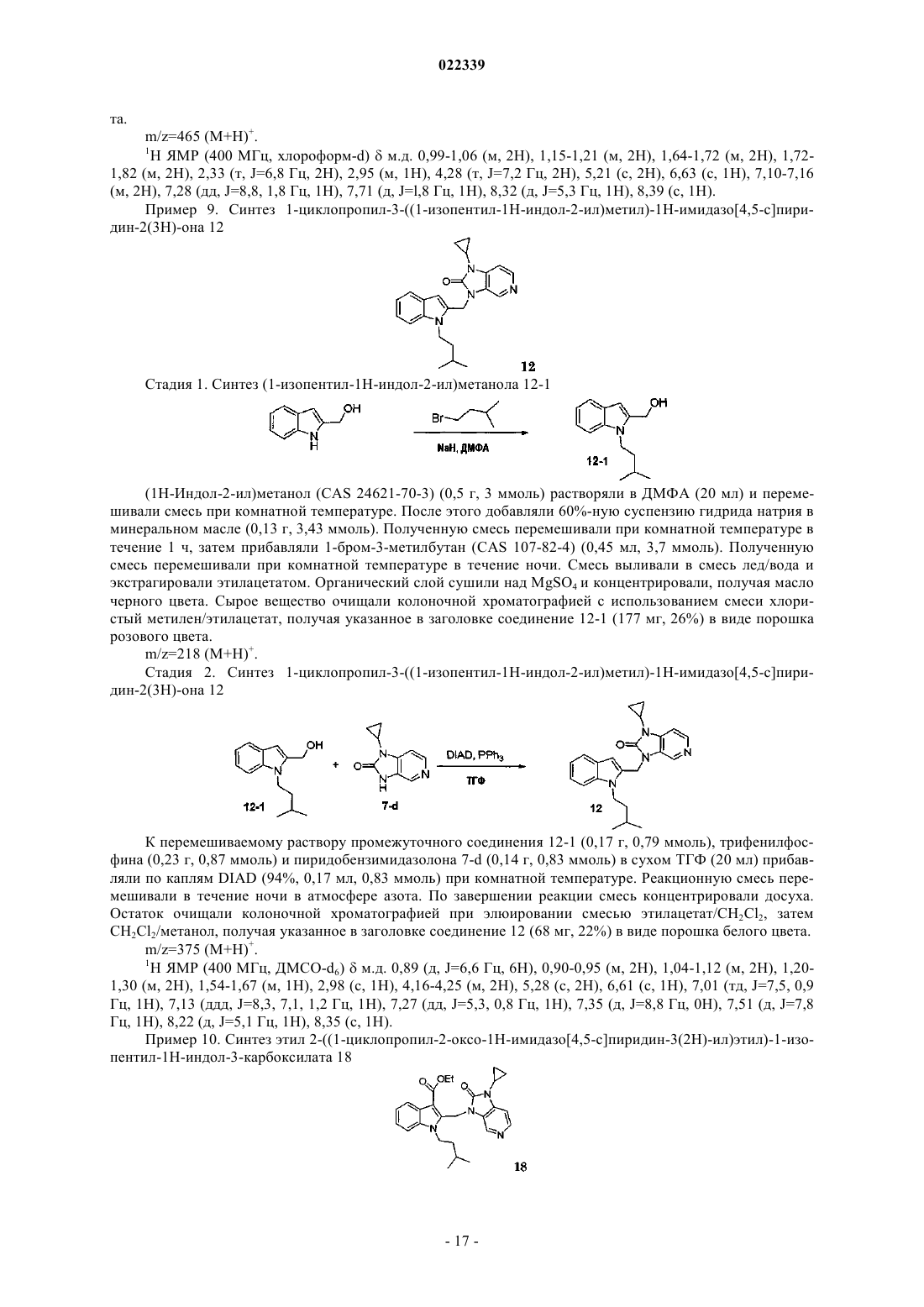
Индолы как противовирусные агенты в отношении респираторного синтициального вируса
Номер патента: 22339
Опубликовано: 30.12.2015
Авторы: Коиманс Людвиг Поль, Ху Лили, Рабуассон Пьер Жан-Мари Бернар, Тахри Абделлах, Вендевилль Сандрин Мари Элен, Йонкерс Тим Хьюго Мария, Демэн Самюэль Доминик


















Формула / Реферат
1. Соединение формулы I или его аддитивная соль

где каждый X независимо представляет собой С или N;
R1 выбирают из группы, включающей Н, галоген, С1-С6алкил, С3-С7циклоалкил, С1-С6алкокси, CF3 или OCF3;
R2 выбирают из группы, включающей Н и CO(R7);
R3 представляет собой -(CR8R9)n-R10;
R4 выбирают из группы, включающей С3-С7циклоалкил, SO2-R8, CH2CF3 или 4-6-членный насыщенный цикл, содержащий атом кислорода;
R5 присутствует в случае, когда X является С, и его выбирают из группы, включающей Н, CO(R7) и галоген;
R5 отсутствует в случае, когда X является N;
R7 выбирают из группы, включающей ОН, О(С1-С6алкил), NH2, NHSO2(С1-С6алкил)2, NR8R9;
n представляет собой целое число от 2 до 6;
каждый из R8 и R9 независимо выбирают из Н, С1-С10алкила, С3-С7циклоалкила;
R10 выбирают из группы, включающей С1-С6алкил, ОН, CN, F, CH2F, CF3, C(=NOH)NH2, CONR8R9, COOR8, SO2R8 или 6-членный насыщенный цикл, содержащий атомы кислорода и азота.
2. Соединение по п.1, где R4 выбирают из группы, включающей С3-С7циклоалкил, SO2-R8 или 4-6-членный насыщенный цикл, содержащий атом кислорода.
3. Соединение по п.1 или 2, где R1 выбирают из группы, включающей Н, галоген.
4. Соединение по любому из пп.1-3, где R1 в пара-положении к N-R3 выбирают из группы, включающей Н, галоген, а все остальные R1 представляют собой Н.
5. Соединение по любому из пп.1-4, где R1 выбирают из группы, включающей бром и хлор.
6. Соединение по любому из пп.1-5, где R2 представляет собой CO(R7).
7. Соединение по любому из пп.1-6, где R2 представляет собой Н.
8. Соединение по любому из пп.1-7, где R3 включает цепочку -(CR8R9)n, где R8 и R9 представляют собой Н, а n составляет 2-4.
9. Соединение по любому из пп.1-8, где R10 выбирают из группы, включающей F, CF3, ОН, SO2R8 и CN, при этом R8 предпочтительно представляет собой метил.
10. Соединение по любому из пп.1-9, где R4 представляет собой С3-С7циклоалкил, предпочтительно циклопропил или CH2CF3.
11. Соединение по любому из пп.1-10, где R4 представляет собой оксетан-3-ил.
12. Соединение по любому из пп.1-11, где X в пара-положении к N-R4 представляет собой С, a R5 в данном X является F.
13. Соединение по любому из пп.1-12, где X является N, а другие X представляют собой С, при этом N предпочтительно находится в пара-положении к N-R4.
14. Соединение по любому из пп.1-13, где самое большее один R5 выбирают из группы, включающей галоген, а другие R5 представляют собой Н.
15. Соединение по любому из пп.1-14, где все R5 представляют собой Н.
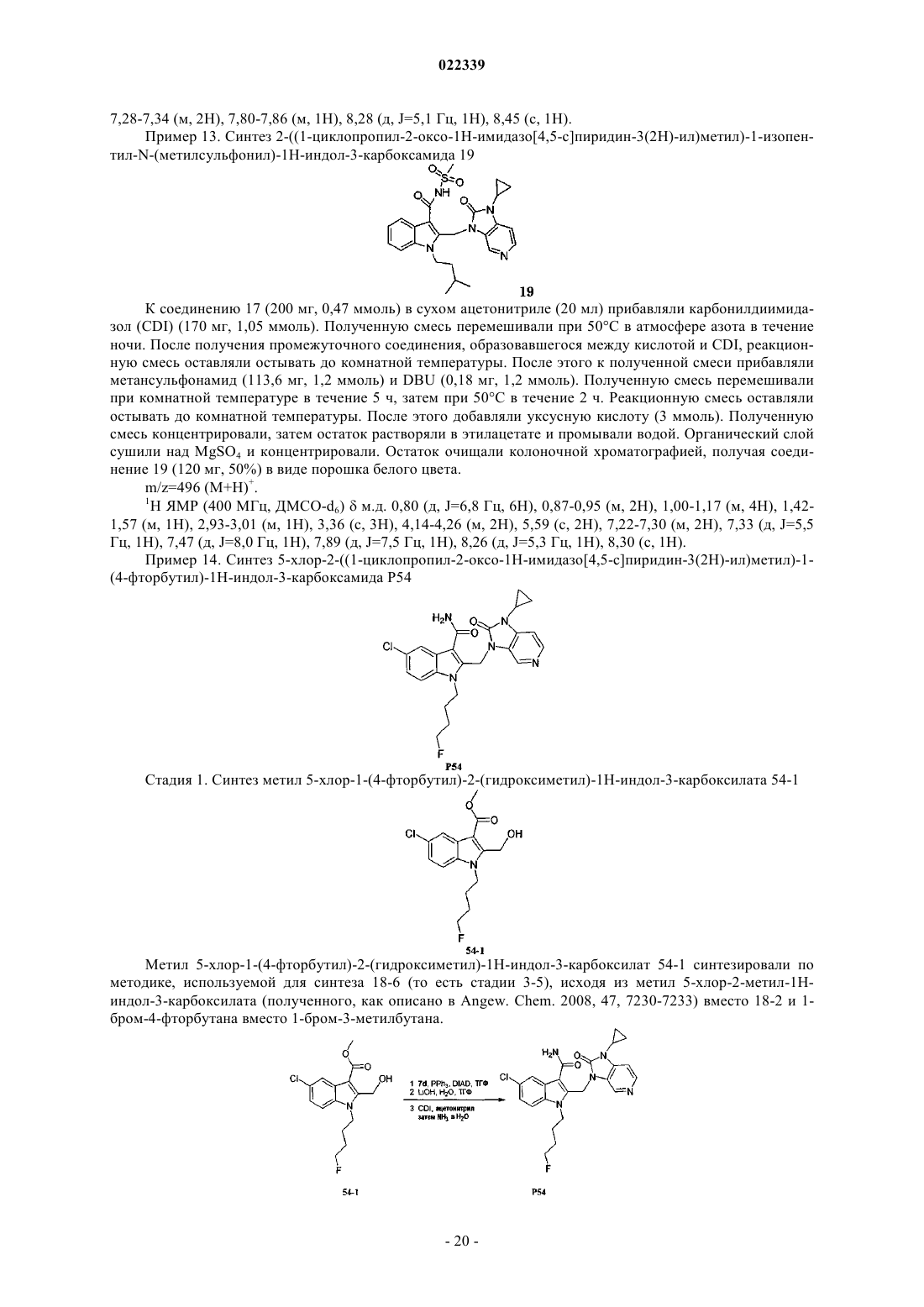
16. Соединение по п.1, где соединение представляет собой

или его аддитивная соль.
17. Соединение по п.16, где соединение представляет собой

18. Применение соединения по любому из пп.1-17 в качестве лекарственного средства для ингибирования репликации RSV.
19. Фармацевтическая композиция, содержащая в качестве активного ингредиента терапевтически эффективное количество соединения по любому из пп.1-17 и фармацевтически приемлемый носитель.
20. Способ получения фармацевтической композиции по п.19, включающий тщательное смешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения по любому из пп.1-17.
21. Способ получения соединения по любому из пп.1-17, включающий сочетание соединения, выбранного из группы, включающей II-a, II-b и II-с, с соединением III в соответствии с приведенной далее схемой 1, приводящее к получению производных формулы I, где все заместители R и X имеют значение согласно п.1 или 2.

22. Применение соединения по любому из пп.1-17 для получения лекарственного средства для ингибирования репликации RSV.
Текст
ИНДОЛЫ КАК ПРОТИВОВИРУСНЫЕ АГЕНТЫ В ОТНОШЕНИИ РЕСПИРАТОРНОГО СИНТИЦИАЛЬНОГО ВИРУСА Описываются индолы, обладающие ингибирующей активностью в отношении репликации RSV и имеющие формулу I, их аддитивные соли, где R1, R2, R3, R4, R5 и X раскрыты в формуле изобретения, композиции, содержащие данные соединения в качестве активного ингредиента, и способы получения данных соединений и композиций. Коиманс Людвиг Поль, Демэн Самюэль Доминик, Ху Лили, Йонкерс Тим Хьюго Мария, Рабуассон Пьер Жан-Мари Бернар, Тахри Абделлах,Вендевилль Сандрин Мари Элен (BE) Медведев В.Н. (RU)(71)(73) Заявитель и патентовладелец: ЯНССЕН САЙЕНСИЗ АЙРЛЭНД ЮСи (IE) Область техники, к которой относится изобретение Данное изобретение относится к индолам, обладающим противовирусной активностью, в частности обладающим ингибирующей активностью в отношении респираторного синцитиального вируса (RSV). Кроме того, данное изобретение относится к получению таких индолов, композиций, содержащих данные индолы, и к соединениям для применения при лечении инфекции респираторного синцитиального вируса. Предшествующий уровень техникиRSV человека или респираторный синцитиальный вирус представляет собой крупный РНКсодержащий вирус, член семейства парамиксовирусов, подсемейства пневмовирусов, наряду с коровьим вирусом RSV. RSV человека ответственен за ряд заболеваний дыхательных путей у людей всех возрастов во всем мире. Он является основной причиной заболеваний нижних дыхательных путей в младенческом и детском возрасте. Почти половина всех младенцев сталкиваются с RSV на первом году жизни и почти все на протяжении первых двух лет жизни. Данная инфекция у маленьких детей может привести к поражению легких, сохраняющемуся в течение многих лет, и может способствовать развитию хронического легочного заболевания в последующей жизни (хроническая одышка, астма). Более старшие дети и взрослые в случае RSV-инфекции часто страдают обыкновенной простудой. В пожилом возрасте снова повышается восприимчивость, и RSV влечет за собой ряд вспышек пневмонии у пожилых людей, приводя к значительной смертности. Заражение вирусом из данной подгруппы не защищает от последующего инфицирования RSV, выделенного из той же подгруппы, в следующем зимнем сезоне. Таким образом, повторное заражение RSV является обычным несмотря на существование всего лишь двух подтипов А и В. В настоящее время для применения против RSV-инфекции принято только три лекарственных средства. Первое представляет собой рибавирин, аналог нуклеозида, предоставляющее аэрозольное лечение в случае серьезной RSV-инфекции у госпитализированных детей. Аэрозольный способ введения,токсичность (опасность тератогенности), стоимость и крайне непостоянная эффективность ограничивают его применение. Два других лекарственных средства, RespiGam (RSV-IG) и Synagis (паливизумаб),иммуностимуляторы на основе поликлональных и моноклональных антител, предназначены для профилактического применения. Оба они являются очень дорогостоящими и требуют парентерального введения. Все остальные попытки разработать безопасную и эффективную вакцину против RSV до сих пор оканчивались неудачей. Инактивированные вакцины не способны защитить от заболевания, а фактически, в некоторых случаях, усиливают заболевание при последующей инфекции. Попытки применения живых аттенуированных вакцин имели ограниченный успех. Очевидно, что существует необходимость в эффективном и простом для введения лекарственном средстве против RSV-инфекции. Было особенно предпочтительно предоставить лекарственные средства против репликации RSV, которые можно было бы вводить перорально. Ссылочный документ, озаглавленный "противовирусные агенты на основе имидазопиридина и имидазопиримидина", представляет собой WO 01/95910, который, фактически, относится к бензимидазольным противовирусным агентам. В нем представлены соединения, обладающие противовирусной активностью, однако со значениями EC50 в широком интервале от 0,001 до 50 мкМ (что обычно не представляет требуемую биологическую активность). Другой ссылочный документ, относящийся к RSVпротивовирусным агентам на основе 2-метилбензимидазола в том же интервале активности, представляет собой WO 03/053344. Следующий ссылочный документ из родственного предшествующего уровня техники, относящийся к соединениям в том же интервале активности, представляет собой WO 02/26228,в котором рассматриваются бензимидазолоновые противовирусные агенты (ссылочный документ по взаимосвязи структура-активность в отношении к ингибированию RSV, относящийся к соединениям 5 замещенного бензимидазола, представляет собой Wang Х.А. et al., Bioorganic and Medicinal Chemistry 17(2007), 4592-4598). Желательно предоставить новые лекарственные средства, обладающие противовирусной активностью. В частности, было бы желательно найти структуры соединений, которые дадут возможность получить противовирусную биологическую активность с порядком величины в более высоких областях предшествующего уровня техники (то есть у нижней границы вышеупомянутого интервала до 50 мкМ), а предпочтительно на уровне примерно наибольшей активности, более предпочтительно даже более высокой активности, чем у соединений, раскрытых в данной области. Следующая цель заключается в обнаружении соединений, обладающих пероральной противовирусной активностью. Краткое описание изобретения Для того чтобы лучше исследовать одну или более из вышеупомянутых целей, в данном изобретении в одном из аспектов предоставлены противовирусные соединения на основе индола, представленные формулой I, их пролекарство, N-оксид, аддитивная соль, четвертичный амин, комплекс с металлами или стереохимически изомерная форма где каждый X независимо представляет собой С или N;R5 присутствует в случае, когда X является С, и его выбирают из группы, включающей Н, C1 С 6 алкил, С 3-С 7 циклоалкил, С 1-С 6 алкокси, CO(R7), CF3 и галоген;n представляет собой целое число от 2 до 6; каждый из R8 и R9 независимо выбирают из Н, С 1-С 10 алкила, С 3-С 7 циклоалкила, или R8 и R9, взятые вместе, образуют 4-6-членный алифатический цикл, который необязательно содержит один или более гетероатомов, выбранных из группы N, S, О;R10 выбирают из группы, включающей Н, С 1-С 6 алкил, ОН, CN, F, CH2F, CF3, C(=NOH)NH2,CONR8R9, COOR8, CONR8SO2R9, CON(R8)SO2N(R8R9), NR8R9, NR8COOR9, OCOR8, NR8SO2R9, SO2NR8R9,SO2R8 или 4-6-членный насыщенный цикл, содержащий атом кислорода. В предпочтительном варианте осуществления R7 выбирают из группы, включающей ОН, О(C1 С 6 алкил), NH2, NHSO2N(С 1-С 6 алкил)2, NHSO2NHCH3, NHSO2(С 1-С 6 алкил), NHSO2(С 3-С 7 циклоалкил) иN(C1-С 6 алкил)2; каждый из R8 и R9 независимо выбирают из Н, С 1-С 10 алкила, С 3-С 7 циклоалкила,или R8 и R9, взятые вместе, образуют 4-6-членный алифатический цикл, который необязательно содержит гетероатом, выбранный из группы N, S, О;R10 выбирают из группы, включающей Н, С 1-С 6 алкил, ОН, CN, F, CH2F, CF3, CONR8R9, COOR8,CONR8SO2R9, CON(R8)SO2N(R8R9), NR8R9, NR8COOR9, OCOR8, NR8SO2R9, SO2NR8R9, SO2R8 или 4-6 членный насыщенный цикл, содержащий атом кислорода. Предпочтительно R4 выбирают из группы, включающей Н, С 1-С 10 алкил, С 3-С 7 циклоалкил, C2 С 10 алкенил, SO2-R8 или 4-6-членный насыщенный цикл, содержащий атом кислорода. В другом аспекте изобретение относится к вышеупомянутым соединениям для применения при лечении RSV-инфекции у теплокровных животных, предпочтительно людей. В еще одном аспекте изобретение предоставляет способ лечения вирусных RSV-инфекций у нуждающегося в этом субъекта, включающий введение указанному субъекту эффективного количества соединения, определенного выше. В еще одном аспекте данное изобретение состоит в применении определенного выше соединения для получения лекарственного средства для лечения RSV-инфекций. В следующем аспекте данное изобретение относится к фармацевтической композиции, содержащей определенное выше соединение и фармацевтически приемлемый эксципиент. В еще одном аспекте в изобретении предоставлены способы получения соединений, определенных выше. Подробное описание изобретения В молекулах формулы I в отличие от предшествующего уровня техники с одной из сторон (с левой стороны изображенной формулы) имеется замещенная индольная группа. Настоящее изобретение, в широком смысле, основано на здравом признании того факта, что данные соединения замещенных индолов,как правило, обладают интересной ингибирующей активностью по отношению к RSV. Кроме того, данные соединения позволяют добиться более высоких значений анти-RSV активности (то есть низшего уровня значений ЕС 50) в пределах, доступных в упомянутых выше ссылках. В частности, исходя из данных соединений, можно обнаружить молекулярные структуры, которые даже превосходят эталонные соединения в отношении биологической активности. Далее настоящее изобретение будет описано в отношении конкретных вариантов осуществления и со ссылкой на некоторые примеры, но изобретение ограничено не ими, а лишь формулой изобретения. В случае, когда термин "включает" используется в настоящем описании и формуле изобретения, он не исключает других элементов или стадий. При использовании существительного в единственном числе включает и множественное число данного существительного, если не установлено что-нибудь еще. Используемый в тексте термин "пролекарство" означает фармацевтически приемлемые производные, например сложные эфиры и амиды, такие, для которых конечный продукт биопревращения данного производного является активным лекарственным средством, определенным в случае соединений формулы (I). Ссылка by Goodman (The Pharmacological Basis of Therapeutics, 8th ed., MeGraw-Hill, Int. Ed. 1992,"biotransformation of Drugs", pp.13-15), в которой пролекарства описаны в общих чертах, настоящим включена в данное описание. Пролекарства характеризуются хорошей растворимостью в воде и биодоступностью и легко метаболизируются в активные ингибиторы in vivo. Как использовано в настоящем описании, С 1-С 6 алкил в качестве группы или части группы обозначает линейные или разветвленные насыщенные углеводородные радикалы, содержащие от 1 до 6 атомов углерода, такие как метил, этил, пропил, 1-метилэтил, бутил, пентил, гексил, 2-метилбутил и так далее. С 1-С 10 алкил в качестве группы или части группы обозначает линейные или разветвленные насыщенные углеводородные радикалы, содержащие от 1 до 10 атомов углерода, такие как группы, определенные для С 1-С 6 алкила, и гептил, октил, нонил, 2-метилгексил, 2-метилгептил, децил, 2-метилнонил и так далее. Подразумевается, что термин "С 2-С 10 алкенил", используемый в настоящем описании в качестве группы или части группы, включает линейные или разветвленные ненасыщенные углеводородные радикалы, содержащие по меньшей мере одну двойную связь, а предпочтительно содержащие одну двойную связь и от 2 до 10 атомов углерода, такие как этенил, пропенил, бутен-1-ил, бутен-2-ил, пентен-1-ил, пентен-2-ил, гексен-1-ил, гексен-2-ил, гексен-3-ил, 2-метилбутен-1-ил, гептен-1-ил, гептен-2-ил, гептен-3-ил,гептен-4-ил, 2-метилгексен-1-ил, октен-1-ил, октен-2-ил, октен-3-ил, октен-4-ил, 2-метилгептен-1-ил,нонен-1-ил, нонен-2-ил, нонен-3-ил, нонен-4-ил, нонен-5-ил, 2-метилоктен-1-ил, децен-1-ил, децен-2-ил,децен-3-ил, децен-4-ил, децен-5-ил, 2-метилнонен-1-ил и так далее. Всякий раз, когда С 2-С 10 алкенильная группа связана с гетероатомом, она предпочтительно связана через насыщенный атом углерода. С 1-С 6 алкокси в качестве группы или части группы обозначает О-С 1-С 6 алкильный радикал, гдеC1-6 алкил независимо имеет определенное выше значение. С 3-С 7 циклоалкил является общим термином для циклопропила, циклобутила, циклопентила, циклогексила или циклогептила. Используемый в настоящем описании термин -(CR8R9)n определяет n повторений подгруппы CR8R9,где каждая из данных подгрупп определена независимо. Термин "галоген" является общим для фтора, хлора, брома и йода. Следует отметить, что положения радикала в любой молекулярной группе, используемой в данных определениях, может быть любым в данной группе до тех пор, пока она является химически стабильной. Радикалы, используемые в определениях переменных, включают все возможные изомеры, если не установлено иначе. Например, пентил включает 1-пентил, 2-пентил и 3-пентил. Если какая-либо переменная встречается в каком-либо компоненте более одного раза, каждое определение является независимым. Всякий раз при использовании в дальнейшем подразумевается, что термин "соединения формулы(I) " или "настоящие соединения" включают соединения общей формулы (I), их пролекарства, N-оксиды,аддитивные соли, четвертичные амины, комплексы с металлами и стереохимически изомерные формы. Понятно, что некоторые соединения формулы (I) могут содержать один или более центров хиральности и существуют в виде стереохимически изомерных форм. Используемый выше термин "стереохимически изомерные формы" определяет все возможные соединения, состоящие из одинаковых атомов, связанных одинаковой последовательностью связей, но обладающие различными трехмерными структурами, не являющимися взаимопревращающимися, которые могут иметь соединения формулы (I). Если не упомянуто или не указано иначе, химическое обозначение соединения включает смесь всех возможных стереохимически изомерных форм, которые может иметь указанное соединение. Указанная смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Подразумевается, что все стереохимически изомерные формы соединения настоящего изобретения как в чистом виде, так и в смеси друг с другом, входят в объем настоящего изобретения. Чистые стереоизомерные формы упомянутых в настоящем описании соединений и промежуточных соединений определяют как изомеры, по существу, не содержащие других энантиомерных или диастереомерных форм одной и той же основной молекулярной структуры указанных соединений или промежуточных соединений. В частности, термин "стереохимически чистый" относится к соединениям или промежуточным соединениям со стереоизомерным избытком, составляющим по меньшей мере от 80% (то есть минимум 90% одного изомера и максимум 10% других возможных изомеров) до стереоизомерного избытка 100% (то есть 100% одного изомера и отсутствие другого), более конкретно, к соединениям или промежуточным соединениям со стереоизомерным избытком, составляющим по меньшей мере от 90 до 100%, еще более конкретно, со стереоизомерным избытком, составляющим по меньшей мере от 97 до 100%. Термины "энантиомерно чистый" и "диастереомерно чистый" следует понимать одинаково, но тогда в отношении энантиомерного избытка и соответственно диастереомерного избытка рассматриваемой смеси. Чистые стереоизомерные формы соединений и промежуточных соединений данного изобретения можно получить с использованием известных в данной области методов. Например, энантиомеры можно разделить друг от друга селективной кристаллизацией их диастереомерных солей с оптически активными кислотами или основаниями. Их примерами являются винная кислота, дибензоилвинная кислота, дитолуоилвинная кислота и камфорсульфоновая кислота. Альтернативным образом, энантиомеры можно разделить методом хроматографии с использованием хиральных носителей. Указанные чистые стереохимически изомерные формы можно также получить из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ, при условии, что реакция протекает стереоспецифически. Предпочтительно, если необходим определенный стереоизомер, указанное соединение можно синтезировать при помощи стереоспецифического способа получения. В данных способах преимущественно используются энантиомерно чистые исходные вещества. Индивидуальные диастереомерные рацематы формулы (I) можно получить стандартными способами. Подходящие способы физического разделения, которые предпочтительно можно использовать, представляют собой, например, селективную кристаллизацию и хроматографию, например колоночную хроматографию. Для некоторых соединений формулы (I), их пролекарств, N-оксидов, солей, сольватов, четвертичных аминов или комплексов с металлами и промежуточных соединений, применяемых при их получении, абсолютную стереохимическую конфигурацию экспериментально не определяли. Специалист в данной области может определить абсолютную конфигурацию подобных соединений при помощи известных в данной области способов, например, таких как рентгеноструктурный анализ. Подразумевается также, что настоящее изобретение включает все изотопы атомов, имеющихся в настоящем изобретении. Изотопы включают такие атомы, которые имеют одинаковый атомный номер,но разные массовые числа. При помощи общего примера и без ограничения, изотопы водорода включают тритий и дейтерий. Изотопы углерода включают С-13 и С-14. Для терапевтических целей соли соединений формулы (I) представляют собой соли, в которых противоион является фармацевтически приемлемым. Однако соли кислот и оснований, не являющихся фармацевтически приемлемыми, могут также найти применение, например, при получении или очистке фармацевтически приемлемого соединения. Все соли, являются ли они фармацевтически приемлемыми или нет, входят в объем настоящего изобретения. Подразумевается, что упомянутые выше фармацевтически приемлемые кислотно- и основноаддитивные соли включают терапевтически активные нетоксичные формы кислотно- и основноаддитивных солей, которые способны образовывать соединения формулы (I). Фармацевтически приемлемые кислотно-аддитивные соли можно легко получить, обрабатывая форму основания подобной подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогеноводородные кислоты, например хлористо-водородная или бромисто-водородная кислота, серная,азотная, фосфорная и тому подобные кислоты, или органические кислоты, например, такие как уксусная,пропионовая, гидроксиуксусная, молочная, пировиноградная, щавелевая (то есть этандиовая), малоновая,янтарная (то есть бутандиовая), малеиновая, фумаровая, яблочная (то есть гидроксибутандиовая), винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и тому подобные кислоты. Наоборот, указанные солевые формы можно перевести в форму свободного основания обработкой подходящим основанием. Соединения формулы (I), содержащие кислый протон, можно также перевести в их нетоксичные формы аддитивных солей с металлом или амином обработкой соответствующими органическими и неорганическими основаниями. Подходящие формы основных солей включают, например, соли аммония,соли щелочных и щелочно-земельных металлов, например соли лития, натрия, калия, магния, кальция и так далее, соли органических оснований, например соли бензатина, N-метил-D-глюкамина, гидрабамина,и соли аминокислот, например, таких как аргинин, лизин и так далее. Используемый выше термин "аддитивная соль" включает также сольваты, которые способны образовывать соединения формулы (I), а также их соли. Подобные сольваты представляют собой, например,гидраты, алкоголяты и так далее. Используемый выше термин "четвертичная соль" определяет четвертичные аммониевые соли, которые соединения формулы (I) способны образовывать по реакции основного атома азота соединения формулы (I) с соответствующим кватернизующим агентом, например, таким как необязательно замещенный алкилгалогенид, арилгалогенид или арилалкилгалогенид, например йодистый метил или йодистый бензил. Можно использовать также другие реагенты с хорошей уходящей группой, такие как алкилтрифторметансульфонаты, алкилметансульфонаты и алкил-п-толуолсульфонаты. Четвертичный амин имеет положительно заряженный атом азота. Фармацевтически приемлемые противоионы включают хлор, бром, йод, трифторацетат и ацетат. Выбранный противоион можно вводить при помощи ионообменных смол. Подразумевается, что N-оксидные формы настоящих соединений включают соединения формулы(I), в которых один или несколько атомов азота окислены до так называемого N-оксида. Понятно, что соединения формулы (I) могут иметь связь с металлом, хелатирующие и комплексообразующие свойства и, следовательно, могут существовать в виде комплексов с металлами или хелатов с металлами. Подразумевается, что подобные металлированные производные соединений формулы (I) включены в объем настоящего изобретения. Некоторые из соединений формулы (I) могут также существовать в виде таутомерных форм. Подразумевается, что подобные формы, хотя они явно не указаны в приведенных выше формулах, включены в объем настоящего изобретения. Понятно, что соединения данного изобретения, в том, что касается упомянутых выше левой и правой частей формулы I, предоставляют большое многообразие модификаций. Не уменьшая общего объема изобретения, некоторые варианты осуществления более подробно обсуждаются далее. В одном из предпочтительных вариантов изобретения R1 выбирают из группы, включающей Н, галоген, С 1-С 6 алкокси, CF3 и OCF3. В следующем предпочтительном варианте изобретения R1 в параположении к N-R3 выбирают из группы, включающей Н, галоген, а все остальные R1 являются Н. В другом предпочтительном варианте изобретения галоген представляет собой бром или хлор. В следующем предпочтительном варианте изобретения R3 включает цепь -(CR8R9)n, в которой R8 иR9 предпочтительно являются Н, а n равно 2-4. Предпочтительно R10 выбирают из группы, включающей ОН, F, CF2H, CF3, SO2R8 и CN. R8 предпочтительно представляет собой метил. В предпочтительном варианте осуществления R4 представляет собой С 3-С 7 циклоалкил, более предпочтительно циклопропил. В предпочтительном варианте осуществления, а более предпочтительно в связи с другими предпочтительными вариантами осуществления один X является N, а другие X являются С. В наиболее предпочтительном варианте осуществления один из X, представляющий собой N, является X в пара-положении кN-R4. Предпочтительно не более одного R5 выбирают из группы, включающей С 1-С 6 алкил, С 1-С 6 алкокси,галоген. Наиболее предпочтительно все R5 являются H. Предпочтительные соединения представляют собой соединения, перечисленные в приведенной далее табл. 1. Наиболее предпочтительными являются соединения 1, 2 и 3. Соединения формулы I можно получить описанными ниже способами с использованием методов синтеза, которые известны в области органической химии, или при помощи модификаций и получения производных, которые знакомы специалистам в данной области. Используемые здесь исходные вещества являются коммерчески доступными, или могут быть получены стандартными способами, которые известны в данной области, такими как способы, описанные в обычных справочниках. Предпочтительные способы включают, но не ограничиваются, описанные ниже способы. В ходе любой из следующих синтетических последовательностей может потребоваться и/или быть желательно защитить чувствительные или реакционноспособные группы, такие как группы, описанные уGreen T.W. и. Wuts P.G.M. (Protective Groups in Organic Chemistry, John WileySons, 1999), которые включены, таким образом, в виде ссылки. Соединения формулы I или их фармацевтически приемлемые соли можно получить в соответствии со схемами реакций, которые обсуждаются в настоящем описании далее. Если не указано иначе, заместители в схеме определены в соответствии с указанным выше. Выделение и очистку продуктов осуществляют стандартными методами, которые хорошо известны химику стандартной квалификации. На схеме 1 показан способ получения соединений формулы I, в которых радикалы от R1 до R5 и X определены, как указано выше. Обращаясь к схеме 1, соединение формулы I можно синтезировать сочетанием 2-гидроксиметилениндола II-а с N3-замещенным 2-оксоимидазопиридином или с N3-замещенным 2-оксоимидазобензолом III при помощи способа, известного в данной области, такого как реакция Мицунобу, в которой используется азадиизопропилдикарбоксилат и трифенилфосфин в подходящем растворителе, таком как ДМФА или ТГФ. Альтернативным образом, соединение формулы I можно получить замещением Y, который представляет собой галогенид, предпочтительно хлор II-b, или сульфонат, такой как мезилат II-с, в присутствии основания, такого как гидрид натрия, карбонат калия или карбонат цезия, в подходящем растворителе, таком как ДМФА или ТГФ. Получение соединения II-а Используемые в данном изобретении исходные вещества IV являются коммерчески доступными или могут быть синтезированы, без ограничения, способами, которые известны в данной области, такими как синтез Рейсерта или синтез Фишера, реакцией подобных индолов с R3-LG, где LG представляет собой уходящую группу, такую как галогенид, предпочтительно бром или сульфонат, в присутствии основания, такого как гидрид натрия, карбонат калия или карбонат цезия, в подходящем растворителе, таком как ДМФА или ТГФ, с образованием соединения V (схема 2). Превращение алкилового сложного эфира соединения V в спирт II-а проводили с использованием гидрида металла, такого как алюмогидрид лития или боргидрид натрия, в подходящем растворителе, таком как ТГФ, метанол или этанол. Схема 2 Обработка спирта II-а тионилхлоридом приводит к 2-хлорметилиндолу II-b. Альтернативным образом, спирт II-а можно превратить в промежуточное соединение II-с реакцией с метансульфонилхлоридом в присутствии органического основания, такого как триэтиламин или диизопропилэтиламин, в подходящем растворителе, таком как хлористый метилен (схема 3). Схема 3 Соединения III можно синтезировать с использованием методики, приведенной на схеме 4. Замещение амином Z, который представляет собой галогенид, предпочтительно фтор или алкоксигруппу,предпочтительно метокси, в нитропиридине или нитроариле VI, в подходящем растворителе, таком как ТГФ или ДМФА, в присутствии органического основания, такого как триэтиламин или диизопропилэтиламин, приводит к получению соединения VII. Восстановление нитрогруппы до амина VIII можно осуществить каталитическим способом с использованием водорода в присутствии катализатора, такого как палладий или платина, в подходящем растворителе, таком как метанол, или стехиометрическим способом, при использовании железа в присутствии хлорида аммония или хлорида олова в присутствии концентрированной хлористо-водородной кислоты. В результате циклизации полученного диамина VIII при помощи CDI, фосгена или трифосгена, в растворителе, таком как ацетонитрил или ТГФ, образуются N3 замещенные бензимидазолоны III. Альтернативным образом, соединение типа III можно получить, исходя из коммерчески доступных дианилинов IX, которые можно зациклизовать путем замыкания цикла при помощи CDI, фосгена или трифосгена, приводя к промежуточным соединениям типа X. Алкилирование или сульфонилирование атома азота мочевины X можно осуществить по реакции Мицунобу с использованием коммерчески доступных спиртов или замещением атома хлора в соединениях типа XI, получая соединения формулы III. Схема 4 Соединения формулы (I) можно перевести в соответствующие N-оксидные формы по известным в данной области методикам превращения трехвалентного атома азота в его N-оксидную форму. Как правило, указанную реакцию N-окисления можно осуществить, вводя исходное вещество формулы (I) во взаимодействие с соответствующей органической или неорганической перекисью. Подходящие неорганические перекиси включают, например, перекись водорода, перекиси щелочных или щелочноземельных металлов, например перекись натрия, перекись калия; подходящие органические перекиси могут включать пероксокислоты, например, такие как бензолкарбопероксокислота или галогензамещенная бензолкарбопероксокислота, например 3-хлорбензолкарбопероксокислота, пероксоалкановые кислоты, например пероксоуксусная кислота, алкилгидропероксиды, например трет-бутилгидропероксид. Подходящие растворители представляют собой, например, воду, низшие спирты, например этанол и так далее, углеводороды, например толуол, кетоны, например 2-бутанон, галогенсодержащие углеводороды,например хлористый метилен, и смеси подобных растворителей. Чистые стереохимически изомерные формы соединений формулы (I) можно получить, используя известные в данной области методы. Диастереомеры можно разделить физическими методами, такими как селективная кристаллизация, и хроматографическими методами, например методом противоточного распределения, жидкостной хроматографией и так далее. Соединения формулы (I), полученные описанными выше способами, обычно представляют собой рацемические смеси энантиомеров, которые можно отделить друг от друга при помощи известных в данной области методов разделения. Рацемические соединения формулы (I), являющиеся достаточно основными или кислыми, можно перевести в соответствующие им формы диастереомерных солей реакцией с подходящей хиральной кислотой, соответственно хиральным основанием. Указанные формы диастереомерных солей затем разделяют, например, селективной или дробной кристаллизацией, и высвобождают из них энантиомеры действия щелочи или кислоты. Альтернативный способ разделения энантиомерных форм соединений формулы (I) включает жидкостную хроматографию, в частности жидкостную хроматографию с использованием хирального носителя. Указанные чистые стереохимически изомерные формы можно также получить из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ, при условии, что реакция протекает стереоспецифично. Предпочтительно, если требуется конкретный стереоизомер, указанное соединение будет синтезировано стереоспецифическими способами получения. В данных способах предпочтительно используют энантиомерно чистые исходные вещества. В следующем аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I), определенного в настоящем описании, или соединения любой из подгрупп соединения формулы (I), определенного в настоящем описании, и фармацевтически приемлемый носитель. Терапевтически эффективное количество в данном контексте представляет собой количество, которого достаточно для оказания профилактического действия против, для стабилизации или для уменьшения вирусной инфекции, в частности вирусной RSVинфекции у инфицированных субъектов или субъектов с риском инфицирования. В еще одном аспекте данное изобретение относится к способу получения определенной в настоящем описании фармацевтической композиции, который включает тщательное перемешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы (I), определенного в настоящем описании, или соединения любой из подгрупп соединения формулы (I), определенного в настоящем описании. Таким образом, на основе соединений настоящего изобретения или любого варианта их осуществления можно получить различные фармацевтические формы для целей введения. В качестве подходящих композиций можно перечислить все композиции, которые обычно используют для системно вводимых лекарственных средств. Для получения фармацевтических композиций данного изобретения эффективное количество конкретного соединения, необязательно в форме аддитивной соли или комплекса с металлом в качестве активного ингредиента, объединяют при тщательном перемешивании с фармацевтически приемлемым носителем, при этом носитель может принимать разнообразные формы в зависимости от формы препарата, желательной для введения. Данные фармацевтические композиции желательны в виде единичной дозированной формы, подходящей, в частности, для введения пероральным, ректальным, чрескожным способом или путем парентеральной инъекции. Например, при получении композиций в пероральной дозированной форме можно применять любые обычные фармацевтические среды, например, такие как вода, гликоли, масла, спирты и так далее, в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы, или твердые носители, такие как крахмалы, сахара, каолин, лубриканты, связывающие вещества, дезинтегранты и так далее, в случае порошков, пилюль, капсул и таблеток. Благодаря простоте введения, таблетки и капсулы представляют собой наиболее предпочтительные пероральные единичные дозированные формы, в случае которых, очевидно, используются твердые фармацевтические носители. В случае парентеральных композиций носитель обычно будет включать стерильную воду, по меньшей мере, в значительной части, хотя может включать другие ингредиенты, например, для повышения растворимости. Например, можно получить растворы для инъекций, в которых носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Можно также получить суспензии для инъекций, в случае которых можно применять подходящие жидкие носители, суспендирующие агенты и так далее. Включены также препараты в твердой форме, которые предназначены для перевода, незадолго до применения, в препараты, имеющие жидкую форму. В композициях, подходящих для чрескожного введения, носитель необязательно включает агент, повышающий проникновение, и/или подходящий увлажняющий агент, необязательно в соединении с подходящими добавками любой природы в минорных количествах, при этом данные добавки не оказывают значительного вредного воздействия на кожу. Соединения настоящего изобретения можно также вводить путем оральной ингаляции или инсуффляции при помощи способов и препаратов, применяемых в данной области для введения таким методом. Так, в общем, соединения настоящего изобретения можно вводить в легкие в форме раствора, суспензии или сухого порошка, при этом предпочтительным является раствор. Для введения настоящих соединений подходит любая система, разработанная для доставки растворов, суспензий или сухих порошков путем оральной ингаляции или инсуффляции. Таким образом, в настоящем изобретении предоставлена также фармацевтическая композиция,приспособленная для введения путем ингаляции или инсуффляции через рот, содержащая соединение формулы (I) и фармацевтически приемлемый носитель. Предпочтительно соединения настоящего изобретения вводят путем ингаляции раствора в небулизированных или аэрозолизированных дозах. Для простоты введения и равномерности дозирования особенно предпочтительно составить упомянутые выше фармацевтические композиции в виде единичной дозированной формы. Единичная дозированная форма, как использовано в настоящем описании, относится к физически дискретным единицам,подходящим в качестве единичных доз, при этом каждая единица содержит определенное заранее количество активного ингредиента, рассчитанное для оказания требуемого терапевтического эффекта, в сочетании с необходимым фармацевтическим носителем. Примерами подобных единичных дозированных форм являются таблетки (включая таблетки с насечкой или с покрытием), капсулы, пилюли, суппозитории, пакетики с порошком, облатки, растворы или суспензии для инъекции и так далее, и их разделенные кратные количества. Соединения формулы (I) проявляют противовирусные свойства. Вирусные инфекции, которые можно вылечить при помощи соединений и способов настоящего изобретения, включают вирусные инфекции, вызванные орто- и парамиксовирусами, в частности человеческим и коровьим респираторным синцитиальным вирусом (RSV). Кроме того, ряд соединений данного изобретения проявляют активность в отношении мутировавших штаммов RSV. Помимо этого, многие соединения данного изобретения проявляют благоприятный фармакокинетический профиль и обладают привлекательными свойствами в отношении биодоступности, включая приемлемый период полувыведения, AUC и пиковые значения и отсутствие неблагоприятных явлений, таких как недостаточно быстрое начало действия и удержание в тканях. Противовирусная активность в отношении RSV настоящих соединений in vitro была протестирована в эксперименте, описанном в экспериментальной части описания, и ее также можно продемонстрировать при помощи анализа на снижение выхода вируса. Противовирусная активность в отношении RSV настоящих соединений in vivo можно продемонстрировать на экспериментальной модели с использованием хлопковых крыс, как описано у Wyde et al. (Antiviral Research (1998), 38, 31-42). Благодаря своим противовирусным свойствам, в частности своим свойствам против RSV, соединения формулы (I) или любой вариант их осуществления, их пролекарства, N-оксиды, аддитивные соли,четвертичные амины, комплексы с металлами и стереохимически изомерные формы применимы для лечения индивидуумов, страдающих вирусной инфекцией, в частности RSV-инфекцией, и для профилактики данных инфекций. В целом, соединения настоящего изобретения могут применяться для лечения теплокровных животных, зараженных вирусами, в частности респираторным синцитиальным вирусом. Следовательно, соединения настоящего изобретения или любой вариант их осуществления можно применять в качестве лекарственных средств. Указанное применение в качестве лекарственного средства или способ лечения включает системное введение зараженным вирусом субъектам или субъектам, восприимчивым к вирусным инфекциям, эффективного количества для борьбы с состояниями, связанными с вирусной инфекцией, в частности RSV-инфекцией. Настоящее изобретение относится также к применению настоящих соединений или любого варианта их осуществления для получения лекарственного средства для лечения или предупреждения вирусных инфекций, в частности RSV-инфекции. Кроме того, настоящее изобретение относится к способу лечения теплокровного животного, зараженного вирусом или имеющего риск заражения вирусом, в частности RSV, при этом указанный способ включает введение эффективного в отношении вируса количества соединения формулы (I), определенного в настоящем описании, или соединения любой из подгрупп соединения формулы (I), определенных в настоящем описании. Обычно предполагается, что эффективное в отношении вируса суточное количество составляет от 0,01 до 500 мг/кг массы тела, более предпочтительно от 0,1 до 50 мг/кг массы тела. Может оказаться уместным вводить необходимую дозу в виде двух, трех, четырех или более субдоз с соответствующими интервалами в течение суток. Указанные субдозы могут быть составлены в виде единичных дозированных форм, например, содержащих от 1 до 1000 мг, а, в частности, от 5 до 200 мг активного ингредиента в единичной дозированной форме. Точная дозировка и частота введения зависят от конкретного применяемого соединения формулы(I), конкретного состояния, подлежащего лечению, тяжести данного состояния, подлежащего лечению,возраста, пола, степени нарушения и общего физического состояния конкретного пациента, а также других лекарственных средств, которые может принимать индивидуум, как хорошо известно специалисту в данной области. Кроме того, очевидно, что указанное эффективное суточное количество можно уменьшить или увеличить в зависимости от реакции получающего лечение субъекта и/или в зависимости от оценки врача, прописывающего соединения настоящего изобретения. Таким образом, интервалы эффективного суточного количества, упомянутые выше, представляют собой лишь методические указания. Кроме того, в качестве лекарственного средства можно применять комбинацию другого противовирусного агента и соединения формулы (I). Таким образом, настоящее изобретение относится к продукту,содержащему (а) соединение формулы (I) и (b) другое противовирусное соединение в качестве объединенного препарата для одновременного, отдельного или последовательного применения для противовирусного лечения. В одном препарате с фармацевтически приемлемыми носителями можно объединить различные лекарственные средства. Например, в целях лечения или предупреждения RSV-инфекций соединения настоящего изобретения можно соединить с интерфероном-бета или фактором некроза опухоли-альфа. Далее изобретение будет иллюстрировано со ссылкой на следующие неограничивающие примеры. Пример 1. Синтез промежуточных соединений Все промежуточные соединения, необходимые для синтеза целевых соединений формулы I, синтезируют, как описано на следующих схемах 5-9. Схема 5. Синтез 1-бром-3-(метилсульфонил)пропана 5-с. Стадия 1. Синтез 3-(метилсульфонил)пропан-1-ола 5-b. Спирт 5-а (200 г, 1900 ммоль) растворяли в CH2Cl2 (2000 мл). Смесь охлаждали до 0 С. Прибавляли порциями 85%-ный раствор м-СРВА в воде (970 г, 5700 ммоль), поддерживая температуру в интервале от 0 до 5 С. После прибавления смесь оставляли нагреваться до 25 С и перемешивали в течение 15 ч. Смесь фильтровали через слой целита. Фильтрат очищали флэш-хроматографией (элюент: петролейный эфир:этилацетат=3:1, а затем смесью этилацетат:метанол=10:1), получая промежуточное соединение 5-b(75 г, 29%). Стадия 2. Синтез 1-бром-3-(метилсульфонил)пропан 5-с. Промежуточное соединение 5-b (75 г, 543 ммоль) растворяли в CH2Cl2 (750 мл). Смесь охлаждали до 0 С. Прибавляли по каплям трехбромистый фосфор (53,6 мл, 570 ммоль), поддерживая температуру в интервале от 0 до 5 С. После прибавления смесь оставляли нагреваться до 25 С и перемешивали в течение 15 ч. Смесь выливали в ледяную воду. Отделенный органический слой промывали раствором соли Схема 6. Синтез трет-бутил(4-хлорбутокси)диметилсилана 6-b. Спирт 6-а (100 г, 920 ммоль) растворяли в CH2Cl2 (1000 мл) при комнатной температуре. Смесь охлаждали до 0 С, затем прибавляли имидазол (81,5 г, 1200 ммоль) и TBDMS-Cl (152 г, 1010 ммоль). Полученную смесь перемешивали в течение 4 ч при комнатной температуре, затем фильтровали. Фильтрат последовательно промывали 10%-ной HCl и раствором соли. Полученный раствор сушили над MgSO4,фильтровали, потом концентрировали, получая указанное в заголовке соединение 6-b (100 г, 50%) в виде бесцветного масла. 3250 ммоль) и диизопропилэтиламин (336 г, 2600 ммоль) в сухом этаноле (800 мл) кипятили в течение 3 ч. Смесь охлаждали до 0 С. Осадок выделяли фильтрованием. Остаток на фильтре промывали холодным этанолом (150 мл). Твердое вещество сушили, получая указанное в заголовке соединение 7-b (167 г, выход 72%) в виде порошка белого цвета. Стадия 2. Синтез N4-циклопропилпиридин-3,4-диамина 7-с. Промежуточное соединение 7-b (167 г, 932 ммоль) в этаноле (1400 мл) гидрировали (50 фунтов/кв. дюйм) при 20 С с использованием влажного 10%-ного Pd/C (34 г) в качестве катализатора в течение ночи. После поглощения Н 2 (3 экв.) катализатор отфильтровывали, а фильтрат упаривали. Остаток промывали метилтретбутиловым эфиром, получая указанное в заголовке соединение 7-с (133 г, 95%) в виде порошка желтого цвета. Стадия 3. Синтез 1-циклопропил-1 Н-имидазо[4,5-с]пиридин-2(3H)-она 7-d. Карбонилдиимидазол (151,8 г, 936 ммоль) прибавляли к раствору промежуточного соединения 7-с(133 г, 891,4 ммоль) в CH3CN (1800 мл) при 0 С. Реакционной смеси давали нагреться до 10 С и перемешивали в течение 1 ч. Осадок выделяли фильтрованием и промывали CH3CN (200 мл), получая указанное в заголовке соединение 7-d (101 г, 65%) в виде порошка белого цвета. Схема 8. Синтез 1-(оксетан-3-ил)-1 Н-имидазо[4,5-с]пиридин-2(3H)-она 8-d. Соединение 8-d получали таким же образом, как соединение 7-d, используя 3-аминооксетан в качестве исходного соединения. Схема 9. Синтез 1-циклопропил-5-фтор-1 Н-бензо[d]имидазол-2(3H)-она 9-d. Стадия 1. Синтез N-циклопропил-4-фтор-2-нитроанилина 9-b. 1,4-Дифтор-2-нитробензол 9-а (CAS 364-74-9) (15 г, 94,3 ммоль) растворяли в ДМФА (500 мл). Прибавляли циклопропиламин (7 мл, 100 ммоль), затем триэтиламин (30 мл, 217 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в воду и экстрагировали хлористым метиленом, сушили над MgSO4 и концентрировали. Твердое вещество оранжевого цвета очищали колоночной хроматографией с использованием хлористого метилена и метанола, получая промежуточное соединение 9-b (16 г, 86%) в виде твердого вещества оранжевого цвета.H ЯМР (400 МГц, хлороформ-d)м.д. 0,63-0,68 (м, 2H), 0,88-0,95 (м, 2H), 2,54-2,55 (м, 1H), 7,277,34 (м, 2 Н), 7,84-7,90 (м, 1 Н), 7,93-8,02 (м, 1 Н). Стадия 2. Синтез N4-циклопропил-4-фторбензол-1,2-диамин 9-с. Промежуточное соединение 9-b (16 г, 82 ммоль) в этаноле (200 мл) гидрировали при комнатной температуре с использованием влажного 10%-ного Pd/C в качестве катализатора в течение ночи. После поглощения Н 2 (3 экв.) катализатор отфильтровывали, а фильтрат упаривали. Остаток промывали этанолом, получая указанное в заголовке соединение 9-с (12,8 г, 94%) в виде твердого вещества белого цвета.m/z=167 (M+H)+. Стадия 3. Синтез 1-циклопропил-5-фтор-1H-бензо[d]имидазол-2(3H)-она 9-d. Карбонилдиимидазол (13,15 г, 81 ммоль) прибавляли к раствору промежуточного соединения 9-с(12,8 г, 77,3 ммоль) в CH3CN (150 мл) при 0 С. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 4 ч. Растворитель удаляли, затем остаток очищали колоночной хроматографией с использованием смеси CH2Cl2/метанол, получая твердое вещество светло-коричневого цвета, которое растирали с диэтиловым эфиром, получая соединение 9-d (7,4 г, 50%) в виде твердого вещества белого цвета. Этил 5-бром-1H-индол-2-карбоксилат (CAS 16732-70-0) (2,3 г, 8,6 ммоль) растворяли в ДМФА (50 мл). Смесь перемешивали при комнатной температуре, затем прибавляли 60%-ную суспензию гидрида натрия в минеральном масле (0,52 г, 12,8 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 ч, затем добавляли 1-бром-3-(метилсульфонил)пропан 5-с (2,6 г, 12,8 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в смесь ледвода и экстрагировали этилацетатом. Органический слой сушили над MgSO4 и концентрировали, получая сырое масло коричневого цвета. Сырое вещество очищали колоночной хроматографией с использованием смеси хлористый метилен/метанол, получая указанное в заголовке соединение 2-1 (3,2 г, 96%) в виде твердого вещества белого цвета. К раствору промежуточного соединения 2-1 (3,2 г, 8,24 ммоль) в ТГФ (100 мл) прибавляли при комнатной температуре алюмогидрид лития (2 М раствор в ТГФ, 5,2 мл, 10,4 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили, добавляя этилацетат и этанол. Полученную смесь выливали в смесь лед-вода, затем фильтровали через целит. Водный слой экстрагировали этилацетатом (350 мл). Объединенные органические экстракты промывали насыщенным раствором соли (100 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией с использованием смеси хлористый метилен/метанол в качестве элюента. Продукт 2-2 выделяли (2,5 г, 88%) в виде твердого вещества белого цвета. К перемешиваемому раствору промежуточного соединения 2-2 (0,5 г, 1,3 ммоль), трифенилфосфина(0,37 г, 1,4 ммоль) и пиридобензимидазолона 7-d (0,34 г, 2 ммоль) в сухом ТГФ (30 мл) прибавляли по каплям DIAD (94%, 0,71 мл, 1,36 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение ночи. По завершении реакции смесь концентрировали досуха и очищали остаток колоночной хроматографией при элюировании смесью этилацетат/CH2Cl2, затем CH2Cl2/метанол, получая указанное в заголовке соединение 2 (458 мг, 70%) в виде твердого вещества белого цвета. Этил 5-бром-1H-индол-2-карбоксилат, который коммерчески доступен (CAS 16732-70-0) (3 г, 11 ммоль), растворяли в ДМФА (50 мл). Смесь перемешивали при комнатной температуре, затем прибавляли 60%-ную суспензию гидрида натрия в минеральном масле (0,49 г, 12,3 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли трет-бутил(4-хлорбутокси)диметилсилан 6-b (2,5 г, 11,2 ммоль). Полученную смесь перемешивали при 60 С в течение 5 дней. Смесь оставляли остывать до комнатной температуры, затем выливали в смесь воды со льдом, потом экстрагировали этилацетатом. Органический слой сушили над MgSO4 и концентрировали, получая масло оранжевого цвета. Сырое вещество очищали колоночной хроматографией с использованием смеси хлористый метилен/гептан, получая указанное в заголовке соединение 11-1 (3,93 г, 77%) в виде бесцветного масла. К раствору промежуточного соединения 11-1 (3,93 г, 6,72 ммоль) в ТГФ (100 мл) прибавляли при-78 С 1 М раствор алюмогидрида лития в ТГФ (8 мл, 8 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь гасили, добавляя этилацетат и этанол. Данную смесь выливали в смесь лед-вода и фильтровали полученную смесь через целит. Водный слой экстрагировали этилацетатом (350 мл). Объединенные органические экстракты промывали насыщенным раствором соли (100 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией с использованием смеси хлористый метилен/метанол в качестве элюента. Промежуточное соединение 11-2 выделяли в виде бесцветного масла (2,68 г, 96%). К перемешиваемому раствору промежуточного соединения 11-2 (0,77 г, 1,86 ммоль), трифенилфосфина (0,54 г, 2,05 ммоль) и пиридобензимидазолона 7-d (0,34 г, 2 ммоль) в сухом ТГФ (30 мл) прибавляли по каплям DIAD (94%, 0,38 мл, 1,96 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение ночи. По завершении реакции смесь концентрировали досуха и очищали остаток колоночной хроматографией при элюировании смесью этилацетат/CH2Cl2, затем CH2Cl2/метанол, получая указанный в заголовке продукт 11 (1,06 г, 61%) в виде бесцветного масла. Соединение 13 получали так же, как и соединение 11, исходя из коммерчески доступного индола. Промежуточное соединение 11 (1,06 г, 1,14 ммоль) растворяли в метаноле (30 мл), а затем прибавляли фторид аммония (0,172 г, 4,6 ммоль). Полученную смесь перемешивали при 60 С в течение ночи. Реакционную смесь оставляли остывать до комнатной температуры, потом удаляли растворитель. Остаток очищали колоночной хроматографией с использованием смеси хлористый метилен/метанол, получая данный продукт в виде твердого вещества белого цвета (323 мг, 62%).H ЯМР (400 МГц, ДМСО-d6)м.д. 0,88-0,97 (м, 2 Н), 1,03-1,13 (м, 2 Н), 1,32-1,53 (м, 4 Н), 2,99 (дт,J=7,0, 3,4 Гц, 1 Н), 3,34-3,40 (м, 2 Н), 4,23 (т, J=7,4 Гц, 2 Н), 4,40 (т, J=5,0 Гц, 1 Н), 5,28 (с, 2 Н), 6,55 (с, 1 Н),7,23 (дд, J=8,7, 1,9 Гц, 1 Н), 7,27 (д, J=5,3 Гц, 1 Н), 7,42 (д, J=8,8 Гц, 1 Н), 7,70 (д, J=1,8 Гц, 1 Н), 8,23 (д,J=5,3 Гц, 1 Н), 8,34 (с, 1 Н). Пример 7. Соединения 7, 9, 10, 15 и 16 получали так же, как и соединение 4, исходя из соответствующих коммерчески доступных индолов. К раствору соединения 4 (0,88 г, 1,95 ммоль) в сухом хлористом метилене (30 мл) в атмосфере азота прибавляли триэтиламин (0,81, 5,83 ммоль), 4-диметиламинопиридин (0,07 г, 0,58 ммоль) и 4-метилбензол-1-сульфонилхлорид (0,445 г, 2,33 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение ночи в атмосфере азота. Реакционную смесь разбавляли хлористым метиленом, затем промывали водой, сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией с использованием хлористого метилена и метанола. Промежуточное соединение 3-1 (760 мг,65%) выделяли в виде пены белого цвета.(75 мг, 1,5 ммоль). Полученную смесь перемешивали в течение ночи в атмосфере азота при 90 С. Реакционную смесь оставляли остывать до комнатной температуры, затем выливали в смесь вода/хлористый метилен. Полученную смесь экстрагировали хлористым метиленом, сушили над MgSO4 и концентрировали. Полученный остаток очищали колоночной хроматографией при элюировании смесью хлористый метилен/метанол, получая указанное в заголовке соединение 3 (500 мг, 86%) в виде порошка белого цве- 16022339(1 Н-Индол-2-ил)метанол (CAS 24621-70-3) (0,5 г, 3 ммоль) растворяли в ДМФА (20 мл) и перемешивали смесь при комнатной температуре. После этого добавляли 60%-ную суспензию гидрида натрия в минеральном масле (0,13 г, 3,43 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 ч, затем прибавляли 1-бром-3-метилбутан (CAS 107-82-4) (0,45 мл, 3,7 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в смесь лед/вода и экстрагировали этилацетатом. Органический слой сушили над MgSO4 и концентрировали, получая масло черного цвета. Сырое вещество очищали колоночной хроматографией с использованием смеси хлористый метилен/этилацетат, получая указанное в заголовке соединение 12-1 (177 мг, 26%) в виде порошка розового цвета. К перемешиваемому раствору промежуточного соединения 12-1 (0,17 г, 0,79 ммоль), трифенилфосфина (0,23 г, 0,87 ммоль) и пиридобензимидазолона 7-d (0,14 г, 0,83 ммоль) в сухом ТГФ (20 мл) прибавляли по каплям DIAD (94%, 0,17 мл, 0,83 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение ночи в атмосфере азота. По завершении реакции смесь концентрировали досуха. Остаток очищали колоночной хроматографией при элюировании смесью этилацетат/CH2Cl2, затемCH2Cl2/метанол, получая указанное в заголовке соединение 12 (68 мг, 22%) в виде порошка белого цвета. К раствору фенилгидразина (125 г, 1150 ммоль) и этил 3-оксобутаноата (100 г, 770 ммоль) в третбутилметиловом эфире (1000 мл) прибавляли уксусную кислоту (2 мл). Полученную смесь перемешивали при 0 С в течение 1 ч. Растворитель выпаривали в вакууме. Остаток (220 г) использовали на следующей стадии как есть. Стадия 2. Синтез этил 2-метил-1H-индол-3-карбоксилата 18-2. Промежуточное соединение 18-1 (160 г) прибавляли порциями к конц. H2SO4 (800 мл) при -10 С при энергичном перемешивании. Раствор перемешивали в течение 1 ч при -10 С и в течение 2 ч при 15 С. Раствор выливали в смесь лед-вода и экстрагировали трет-бутилметиловым эфиром. После удаления растворителя твердое вещество промывали петролейным эфиром. Получали промежуточное соединение 18-2 (80 г, 70%). Стадия 3. Синтез этил 1-изопентил-2-метил-1H-индол-3-карбоксилата 18-3. К раствору промежуточного соединения 18-2 (38 г, 187 ммоль) в CH3CN (1000 мл) прибавляли 1 бром-3-метилбутан (94 мл, 747 ммоль) и Cs2CO3 (121 г, 374 ммоль). Полученную смесь нагревали при кипении в течение 2 ч. Осадок отфильтровывали, а фильтрат упаривали в вакууме. Остаток очищали высокоэффективной жидкостной хроматографией (С 18, элюент: CH3OH/H2O от 15/85 до 45/55 в присутствии 0,1% ТФУ в качестве буфера). Собирали чистые фракции и удаляли летучие вещества в вакууме, а рН водного раствора доводили до 8 добавлением NaHCO3. Остаток экстрагировали CH2Cl2 (2100 мл). Органический слой промывали насыщенным раствором соли (100 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме, получая требуемое промежуточное соединение 18-3 (20 г, 40%). Стадия 4. Синтез этил 2-формил-1-изопентил-1H-индол-3-карбоксилата 18-4. К раствору промежуточного соединения 18-3 (9,8 г, 35,8 ммоль) в уксусной кислоте (150 мл) прибавляли SeO2 (14 г, 71,6 ммоль). Полученную смесь нагревали при кипении в течение 12 ч, затем оставляли остывать до комнатной температуры. После этого добавляли воду (200 мл) и CH2Cl2 (200 мл). Органический слой промывали насыщенным раствором соли (150 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток использовали на следующей стадии без дополнительной очистки. Получали смесь продуктов (10 г, 70% 18-5 и 10% 18-4). Стадия 5. Синтез этил 2-(гидроксиметил)-1-изопентил-1H-индол-3-карбоксилата 18-6. Смесь промежуточных продуктов 18-4 и 18-5 (10 г) растворяли в метаноле (100 мл) и охлаждали до-15 С. Порциями прибавляли NaBH4 (0,4 г, 10,4 ммоль). Смесь перемешивали при -15 С в течение 10 мин и нагревали до 15 С в течение 0,5 ч. Добавляли насыщенный раствор NaHCO3. Растворитель удаля- 18022339 ли в вакууме. Добавляли CH2Cl2 (100 мл) и Н 2 О (100 мл). Органический слой промывали насыщенным раствором соли и сушили над Na2SO4. Полученный остаток растворяли в метаноле (150 мл). ПрибавлялиK2CO3 (9,8 г, 71,6 ммоль). Смесь перемешивали при 15 С в течение 2 ч. рН доводили до 4 добавлением 1 н. HCl. Смесь экстрагировали CH2Cl2 (200 мл). Органический слой промывали насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат=1:3), получая промежуточное соединение 18-6 (3,63 г, 35% от 18-3) в виде порошка белого цвета. Стадия 6. Синтез этил 2-1-циклопропил-2-оксо-1H-имидазо[4,5-с]пиридин-3(2H)-ил)метил)-1-изопентил-1H-индол-3-карбоксилата 18. Для синтеза соединения 18 использовали ту же методику, что и для получения соединения 12. Соединение 18 (0,5 г, 1 ммоль) растворяли в ТГФ (25 мл), прибавляли гидроксид лития (48 мг, 2 ммоль), растворенный в воде (5 мл). Полученную смесь перемешивали при 60 С в течение ночи. Реакционную смесь оставляли остывать до комнатной температуры, затем выливали в воду. рН полученной смеси доводили до рН 4, добавляя 1 М раствор хлористо-водородной кислоты. После этого смесь экстрагировали этилацетатом. Органический слой сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией с использованием хлористого метилена и метанола. Указанное в заголовке соединение 17 (400 мг, 94%) выделяли в виде порошка белого цвета. К соединению 17 (150 мг, 0,36 ммоль) в сухом ацетонитриле (20 мл) прибавляли карбонилдиимидазол (CDI) (145 мг, 0,9 ммоль). Полученную смесь перемешивали при 50 С в атмосфере азота в течение ночи. После получения промежуточного соединения, образовавшегося между кислотой и CDI, реакционную смесь оставляли остывать до комнатной температуры. После этого прибавляли раствор аммиака в воде (448 мг, 3,5 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Осадок отфильтровывали, затем последовательно промывали водой и ацетонитрилом. Полученное твердое вещество сушили в печи, получая соединение 14 (150 мг, 94%) в виде твердого вещества белого цвета. К соединению 17 (200 мг, 0,47 ммоль) в сухом ацетонитриле (20 мл) прибавляли карбонилдиимидазол (CDI) (170 мг, 1,05 ммоль). Полученную смесь перемешивали при 50 С в атмосфере азота в течение ночи. После получения промежуточного соединения, образовавшегося между кислотой и CDI, реакционную смесь оставляли остывать до комнатной температуры. После этого к полученной смеси прибавляли метансульфонамид (113,6 мг, 1,2 ммоль) и DBU (0,18 мг, 1,2 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 5 ч, затем при 50 С в течение 2 ч. Реакционную смесь оставляли остывать до комнатной температуры. После этого добавляли уксусную кислоту (3 ммоль). Полученную смесь концентрировали, затем остаток растворяли в этилацетате и промывали водой. Органический слой сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией, получая соединение 19 (120 мг, 50%) в виде порошка белого цвета. Метил 5-хлор-1-(4-фторбутил)-2-(гидроксиметил)-1 Н-индол-3-карбоксилат 54-1 синтезировали по методике, используемой для синтеза 18-6 (то есть стадии 3-5), исходя из метил 5-хлор-2-метил-1 Ниндол-3-карбоксилата (полученного, как описано в Angew. Chem. 2008, 47, 7230-7233) вместо 18-2 и 1 бром-4-фторбутана вместо 1-бром-3-метилбутана. Требуемый продукт Р 54 синтезировали, следуя стадиям, описанным для синтеза Р 14, исходя из 54-1 вместо 18-6.(м, 2 Н), 2,92-3,01 (м, 1 Н), 4,26 (с, 3 Н), 4,39 (т, J=6,00 Гц, 1 Н), 5,63 (с, 2 Н), 7,23-7,28 (м, 2 Н), 7,57 (д, J=8,78 Гц, 1 Н), 7,86 (д, J=2,01 Гц, 1 Н), 8,21 (д, J=5,27 Гц, 1 Н), 8,39 (с, 1 Н). Пример 15. Характеристика соединений 1-19 и Р 20-Р 81 и проверка на ингибирующую активность в отношении RSV приведены в табл. 1-3. Пример 16. Производные Р 82-Р 105 получены согласно описанным выше способам или в комбинации с методами, которые известны в данной области (табл. 4). Общие подробности эксперимента Анализ методом ВЭЖХ-МС проводили с использованием любого из следующих способов. Способ 1 Измерение методом ВЭЖХ проводили с использованием модуля Agilent 1100, включающего насос,детектор на диодной матрице (DAD) (используемая длина волны 220 нм), нагреватель колонок и колонку, описанную далее. Поток из колонки разделяли на Agilent MSD серии G1946C и G1956A. На МС детектор устанавливали интерфейс API-ES (ионизация при атмосферном давлении типа электроспрей). Масс-спектры получали путем сканирования от 100 до 1000. Напряжение на капиллярной игле составляло 2500 В в режиме положительной ионизации и 3000 В в режиме отрицательной ионизации. Напряжение фрагментации составляло 50 В. Температуру осушающего газа поддерживали при 350 С при потоке 10 л/мин. ВЭЖХ с обращенной фазой осуществляли на YMC-Pack ODS-AQ колонке 502,0 мм 5 мм при скорости потока 0,8 мл/мин. Использовали две подвижные фазы (подвижная фаза А: вода с 0,1% ТФУ; подвижная фаза В: ацетонитрил с 0,05% ТФУ). Сначала выдерживали 100% А в течение 1 мин. Затем применяли градиент до 40% А и 60% В в течение 4 мин и выдерживали в течение 2,5 мин. Использовали обычные объемы инъекций, равные 2 мл. Температура печи составляла 50 С (полярность МС: положительная). Способ 2 Измерение методом ВЭЖХ проводили с использованием модуля Agilent 1100, включающего насос,детектор на диодной матрице (DAD) (используемая длина волны 220 нм), нагреватель колонок и колонку, описанную далее. Поток из колонки разделяли на Agilent MSD серии G1946C и G1956A. На МС детектор устанавливали интерфейс API-ES (ионизация при атмосферном давлении типа электроспрей). Масс-спектры получали путем сканирования от 100 до 1000. Напряжение на капиллярной игле составляло 2500 В в режиме положительной ионизации и 3000 В в режиме отрицательной ионизации. Напряжение фрагментации составляло 50 В. Температуру осушающего газа поддерживали при 350 С при потоке 10 л/мин. ВЭЖХ с обращенной фазой осуществляли на YMC-Pack ODS-AQ колонке 502,0 мм 5 мм при скорости потока 0,8 мл/мин. Использовали две подвижные фазы (подвижная фаза А: вода с 0,1% ТФУ; подвижная фаза В: ацетонитрил с 0,05% ТФУ). Сначала выдерживали 90% А и 10% В в течение 0,8 мин. Затем применяли градиент до 20% А и 80% В в течение 3,7 мин и выдерживали в течение 3 мин. Использовали обычные объемы инъекций, равные 2 мл. Температура печи составляла 50 С (полярность МС: положительная). Способ 3 Колонка: XTerra MS C18 2,5 мкм, 4,650 мм, подвижная фаза А: 10 мМ NH4OOCH + 0,1% НСООН в Н 2 О, подвижная фаза В: МеОН при работе при температуре колонки 50 С с использованием скорости потока 1,5 мл/мин. Условия градиента: t=0 мин: 65% А, 35% В; t=3,5 мин: 5% А, 95% В; t=5,5 мин: 5% А,95% В; t=5,6 мин: 65% А, 35% В; t=7 мин: 65% А, 35% В. Способ 4 Колонка: SunFire C18 3,5 мкм, 4,6100 мм, подвижная фаза А: 10 мМ NH4OOCH + 0,1% НСООН вH2O, подвижная фаза В: МеОН при работе при температуре колонки 50 С с использованием скорости потока 1,5 мл/мин. Условия градиента: t=0 мин: 65% А, 35% В; t=7 мин: 5% А, 95% В; t=9,6 мин: 5% А,95% В; t=9,8 мин: 65% А, 35% В; t=12 мин: 65% А, 35% В. Спектры ЯМР регистрировали на спектрометре Bruker Avance 400, работающем при частоте 400 МГц в случае 1 Н. Химические сдвиги приведены в м.д., а значения J в Гц. Мультиплетность указана с использованием следующих сокращений: д - в случае дублета, т - в случае триплета, м - в случае мультиплета и так далее. Тонкослойную хроматографию (ТСХ) осуществляли на алюминиевых пластинках 510 см, покрытых силикагелем 60 F254 (Merck KGaA). Противовирусная активность Черные 96-луночные микротитровочные планшеты (Corning, Амстердам, Нидерланды) заполняли в двух экземплярах с использованием заказной роботизированной системы с серийными 4-кратными разбавлениями соединения в конечном объеме 50 мкл питательной среды (среда RPMI без фенолового красного, 10% FBS, 0,04% гентамицина (50 мг/мл) и 0,5% ДМСО). Затем в каждую лунку добавляли 100 мкл суспензии клеток HeLa (5104 клеток/мл) в питательной среде, после чего добавляли 50 мкл вирусаrgRSV224 (MOI=0,02) в питательной среде при помощи дозатора-диспенсера Multidrop (ThermoScientific, Эрембодегем, Бельгия). Вирус rgRSV224 представляет собой искусственно созданный вирус,включающий дополнительный ген GFP (Hallak et al., 2000) и лицензированный от NIH (Bethesda, MD,США). В каждый тест включали холостые опыты со средой, вирус- и mock-инфицированием. Клетки инкубировали при 37 С в атмосфере 5% СО 2. Через три дня после подвергания действию вируса проводили количественную оценку репликации вируса, определяя экспрессию GFP в клетках при помощи лазерного микроскопа MSM (Tibotec, Берсе, Бельгия). EC50 определяли как 50% ингибирующую концентрацию в отношении экспрессии GFP. Параллельно с этим соединения инкубировали в течение трех дней в наборе белых 96-луночных микротитровочных планшетов (Corning) и определяли цитотоксичность соединений в клетках HeLa, определяя содержание АТФ данных клеток при помощи набора реактивов
МПК / Метки
МПК: A61K 31/437, C07D 403/06, C07D 471/04, A61P 31/12
Метки: агенты, противовирусные, отношении, синтициального, индолы, вируса, респираторного
Код ссылки
<a href="https://eas.patents.su/30-22339-indoly-kak-protivovirusnye-agenty-v-otnoshenii-respiratornogo-sinticialnogo-virusa.html" rel="bookmark" title="База патентов Евразийского Союза">Индолы как противовирусные агенты в отношении респираторного синтициального вируса</a>
Азабензимидазолы в качестве противовирусных средств в отношении респираторного синцитиального вируса

Номер патента: 21613
Опубликовано: 30.07.2015
Авторы: Рабуассон Пьер Жан-Мари Бернар, Тахри Абделлах, Ху Лили, Йонкерс Тим Хьюго Мария, Коиманс Людвиг Поль, Вендевилль Сандрин Мари Элен, Демэн Самюэль Доминик
МПК: C07D 471/04, A61P 31/12, A61K 31/437...
Метки: вируса, синцитиального, противовирусных, качестве, средств, отношении, азабензимидазолы, респираторного
Формула / Реферат:
1. Соединение формулы I или его аддитивная сольгде каждый X независимо представляет собой С или N, по меньшей мере один X = N;каждый Y независимо представляет собой С или N;R1 присутствует в тех случаях, когда X = С, и R1 выбран из группы Н, галогена, N(R5)2, CO(R6), C(=NH)NH2, CF3 и B(OH)2;R1 отсутствует в тех случаях, когда X = N;R2 представляет собой -(CR7R8)n-R9;R3 выбран из группы, состоящей из Н, C1-C10-алкила, C3-C7-циклоалкила, CH2CF3...
Макроциклические пептиды, обладающие активностью в отношении вируса гепатита с

Номер патента: 7738
Опубликовано: 29.12.2006
Авторы: Лина-Брюне Монсе, Гори Вида Ж.
МПК: A61K 38/06, A61P 31/14, C07K 5/08...
Метки: обладающие, гепатита, активностью, вируса, пептиды, макроциклические, отношении
Формула / Реферат:
1. Соединение формулы (I) в котором R1 обозначает гидрокси или NHSO2R1A, где R1A обозначает метил, этил, циклопропил, циклобутил, циклопентил, циклопропилметил, циклогексил, ССl3, СF3, фенил, 2-фторфенил или 4-метилфенил, R2 обозначает С5-С6циклоалкил и R3 обозначает циклопентил; или его фармацевтически приемлемая соль. 2. Соединение по п.1, в котором R1A обозначает циклопропил. 3. Соединение по п.1 или 2, в котором R1 обозначает гидрокси. 4....
Производные 5-гидрокси-4-аминометил-1-циклогексил(или циклогептил)-3-алкоксикарбонилиндола и их фармацевтически приемлемые соли, обладающие противовирусной активностью в отношении вируса гриппа а

Номер патента: 18346
Опубликовано: 30.07.2013
Авторы: Трофимов Фёдор Александрович, Верховский Юрий Григорьевич, Гончарова Анна Яковлевна, Цыб Анатолий Фёдорович, Цышкова Нина Гавриловна, Розиев Рахимджан Ахметджанович, Подгородниченко Владимир Константинович
МПК: A61K 31/404, C07D 209/42
Метки: фармацевтически, противовирусной, соли, приемлемые, активностью, обладающие, циклогептил)-3-алкоксикарбонилиндола, производные, отношении, 5-гидрокси-4-аминометил-1-циклогексил(или, гриппа, вируса
Формула / Реферат:
1. Средство, обладающее противовирусной активностью в отношении вируса гриппа А и представляющее собой производное 5-гидрокси-4-аминометил-1-циклогексил(или циклогептил)-3-алкоксикарбонилиндола общей формулы (I)где n=1 или 2;R3 представляет собой С1-С3-алкил;Alk представляет собой C1-С6-алкильную группу;R1 и R2, каждый независимо, представляет собой С1-С4-алкил илигруппа NR1R2 означает группы, соответствующие формуламили его фармацевтически...
Композиции для лечения респираторного дистресс-синдрома взрослых или респираторного дистресс-синдрома новорожденных, содержащие 3-(циклопропилметокси)-n-(3,5-дихлор-4-пиридинил)-4-(дифторметокси) бензамид и легочное поверхностно-активное вещество

Номер патента: 1758
Опубликовано: 27.08.2001
Авторы: Амшлер Херманн, Бойме Рольф, Килиан Ульрих, Крюгер Уве, Германн Паул-Георг
МПК: A61P 11/00, A61K 31/683
Метки: поверхностно-активное, вещество, новорожденных, дистресс-синдрома, взрослых, бензамид, содержащие, 3-(циклопропилметокси)-n-(3,5-дихлор-4-пиридинил)-4-(дифторметокси, респираторного, легочное, лечения, композиции
Формула / Реферат:
1. Композиция, предназначенная для лечения респираторного дистресс-синдрома новорожденных (РДСН) и респираторного дистресс-синдрома взрослых (РДСВ), включающая N-(3,5-дихлорпирид-4-ил)-3-циклопропилметокси-4-дифторметоксибензамид и/или его фармакологически переносимые соли и легочное поверхностно-активное вещество (ПАВ). 2. Композиция по п.1, содержащая в качестве легочного ПАВ смеси фосфолипидов. 3. Композиция по п.2, содержащая фосфолипиды,...
Антивирусная фармацевтическая композиция, содержащая глицирризиновую кислоту и, по меньшей мере, один белок, имеющий активность в отношении вируса простого герпеса типа 1

Номер патента: 3126
Опубликовано: 27.02.2003
Авторы: Пинца Марио, Помпей Раффаэло
МПК: A61P 31/22, A61K 38/47, A61K 38/40...
Метки: отношении, активность, белок, имеющий, содержащая, композиция, мере, один, вируса, меньшей, антивирусная, глицирризиновую, типа, кислоту, герпеса, простого, фармацевтическая
Формула / Реферат:
1. Антивирусная фармацевтическая композиция, отличающаяся тем, что она содержит глицирризиновую кислоту и, по меньшей мере, один белок, имеющий активность в отношении вируса простого герпеса типа 1, при условии, что когда указанный выше белок является лизоцимом и композиция находится в форме водного раствора, она не содержит соли щелочного или щелочно-земельного металла. 2. Композиция по п.1, отличающаяся тем, что указанный белок является...