Производное камптотецина, способ его получения, фармацевтическая композиция и ее применение











Формула / Реферат
1. Производное камптотецина, которое представляет собой фосфит CPT формулы I

где R1, R2, R3 и R4 независимо представляют собой водород, гидрокси, нитро, циано, галоген, карбокси, амино, диметиламино, 4-метилпиперазино, 2-триметилсилилэтил, трет-бутилдиметилсилил, С1-С6-алкокси, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С1-С6-алканоил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С1-С6-алкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, или С3-С6-циклоалкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино; или
R1 и R2 соединены посредством 1-3 других атомов с образованием гетероцикла, где гетероцикл представляет собой N-гетероцикл, S-гетероцикл, О-гетероцикл или гетероцикл, который содержит два гетероатома, которые выбраны из группы, включающей N, О и S, a R3 и R4 независимо представляют собой водород, гидрокси, нитро, циано, галоген, карбокси, амино, диметиламино, 4-метилпиперазино, 2-триметилсилилэтил, трет-бутилдиметилсилил, С1-С6-алкокси, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С1-С6-алканоил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С1-С6-алкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, или С3-С6-циклоалкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино; или
R1 и R2 независимо представляют собой водород, гидрокси, нитро, циано, галоген, карбокси, амино, диметиламино, 4-метилпиперазино, 2-триметилсилилэтил, трет-бутилдиметилсилил, С1-С6-алкокси, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С1-С6-алканоил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С1-С6-алкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, или С3-С6-циклоалкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, a R3 и R4 представляют собой атомы кислорода и соединены посредством -O-(СН2)n-O- с образованием цикла, где n равно 1 или 2,
или его фармацевтически приемлемые соли.
2. Производное камптотецина по п.1, которое представляет собой соль формулы II

где Xn+ представляет собой K+, Na+, Li+, Mg2+, Ca2+, Zn2+, Fe3+ или аммоний.
3. Производное камптотецина по п.1 или 2, где амино- или гидроксильная группа защищена защитной группой.
4. Производное камптотецина по п.2, где аммоний получен из одного из следующих оснований: NH3, монометиламина, диметиламина, триметиламина, моноэтиламина, диэтиламина, триэтиламина, метилэтиламина, диметилэтиламина, диизопропиламина, пирролидина, дигидроизоиндола, морфолина, N,N-диаллиламина, 4-метилпиперидина, этаноламина, 5-бромдигидроизоиндола, тиоморфолина, цис-2,6-диметилморфолина и этилендиамина.
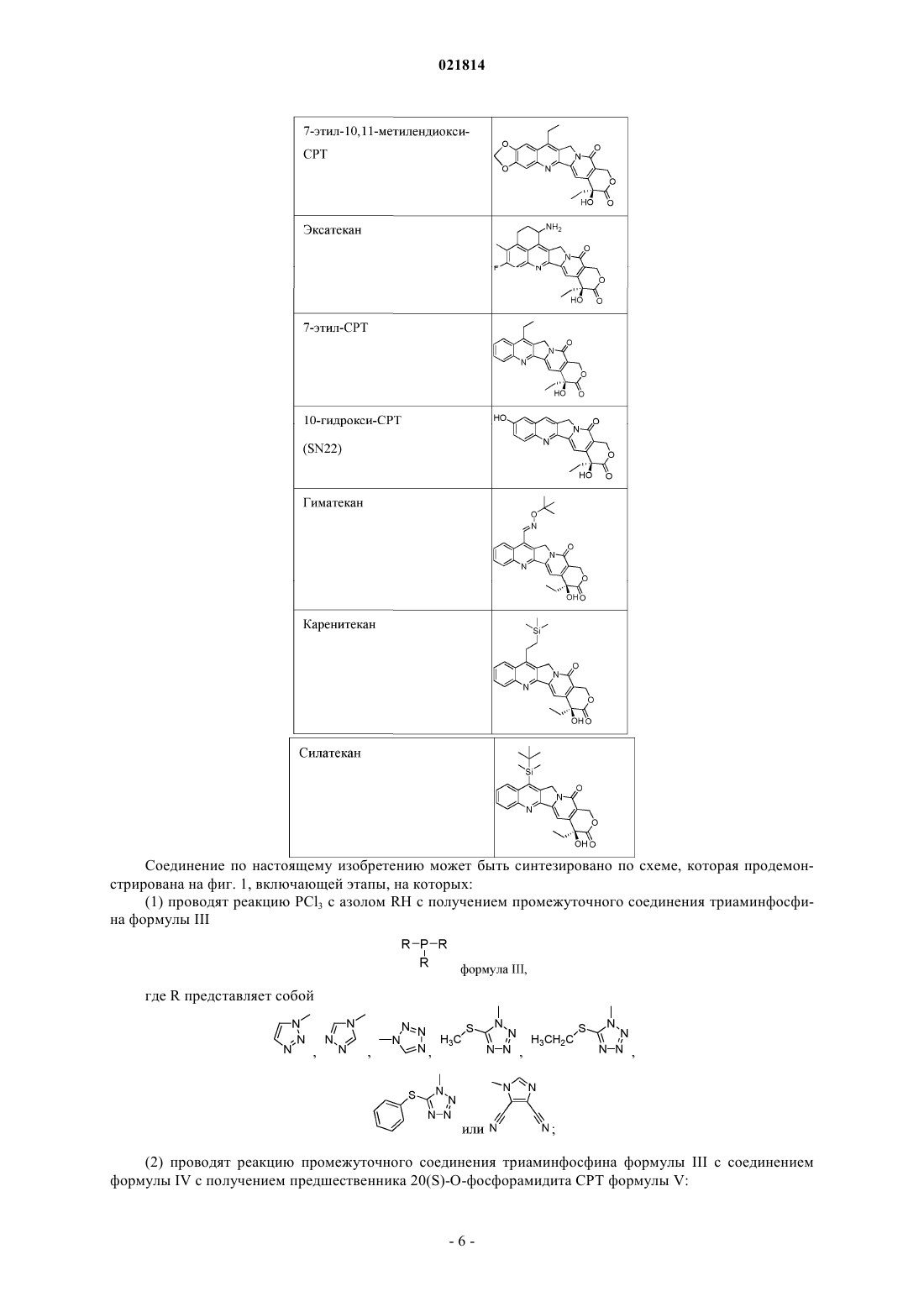
5. Производное камптотецина по любому из пп.1-4, которое представляет собой фосфит или его фармацевтически приемлемые соли, полученные из группы, включающей CPT, 7-этил-10-гидрокси-CPT, топотекан, 9-амино-CPT, иринотекан, 9-нитро-CPT, луртотекан, 7-этил-10,11-метилендиокси-CPT, эксатекан, 7-этил-CPT, 10-гидрокси-CPT, гиматекан, каренитекан и силатекан.
6. Способ получения производного камптотецина по п.1, включающий следующие этапы, на которых:
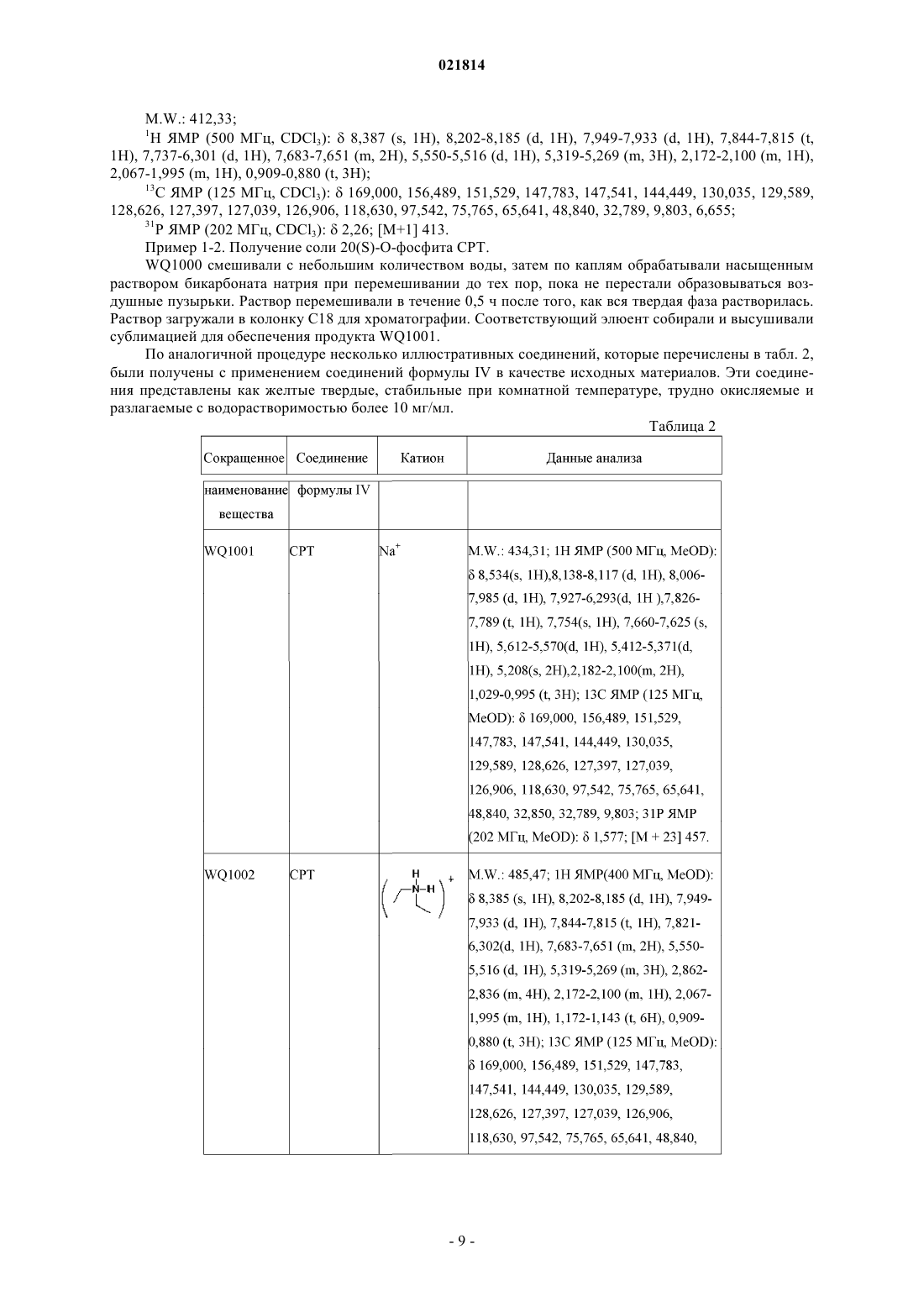
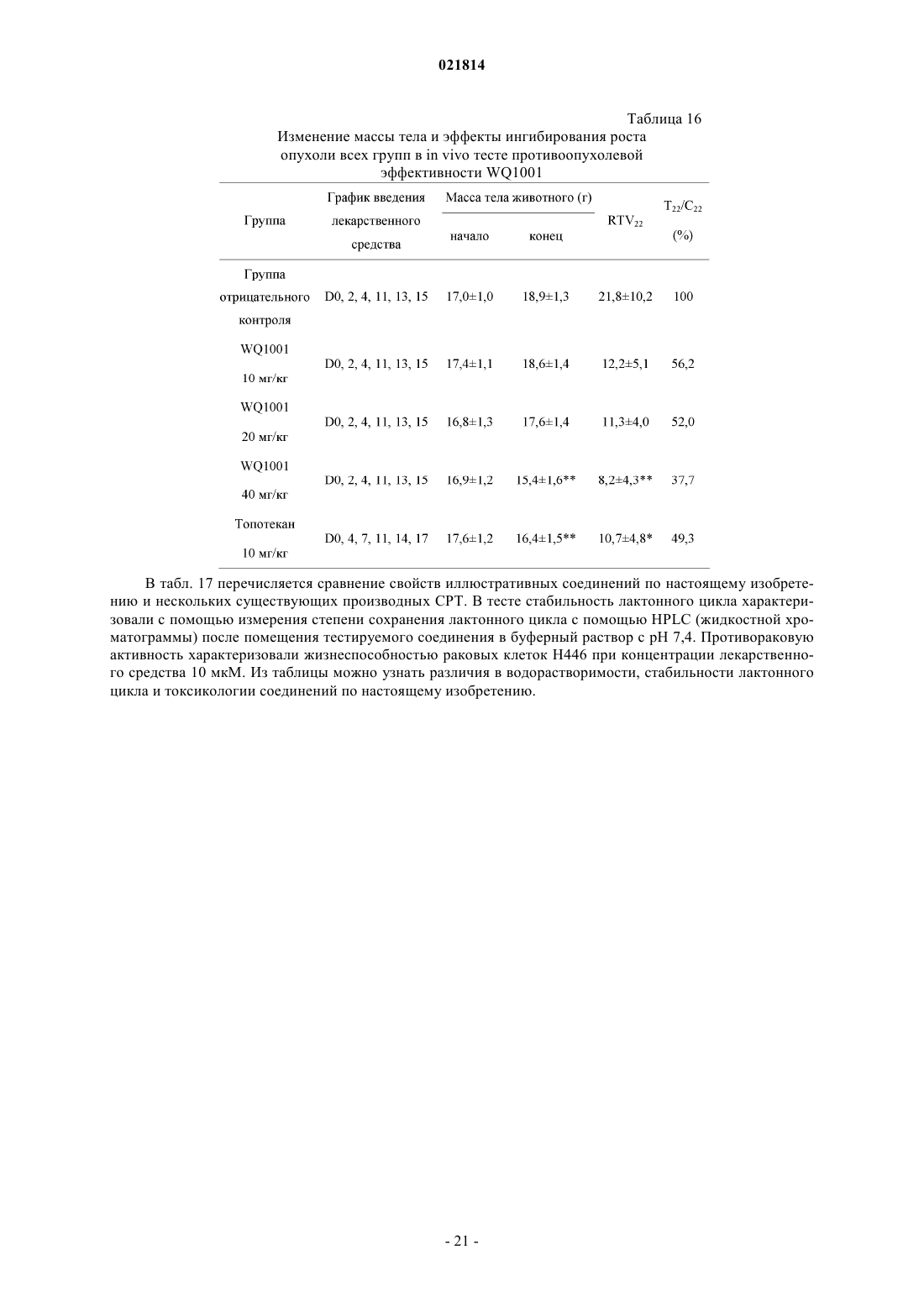
(1) проводят реакцию PCl3 с азольным соединением RH с получением промежуточного соединения триаминфосфина формулы III

где R представляет собой


(2) проводят реакцию промежуточного соединения триаминфосфина формулы III с соединением формулы IV с получением предшественника 20(S)-О-фосфорамидита CPT формулы V

где R1, R2, R3 и R4 являются такими, как определено в п.1, и, если R1, R2, R3 или R4 представляют собой или содержат гидроксильную группу или аминогруппу, гидроксильную группу или аминогруппу перед реакцией с соединением формулы III защищают защитной группой;
(3) проводят гидролиз предшественника формулы V с получением 20(S)-O-фосфита CPT формулы I

причем, если R1, R2, R3 или R4 представляют собой или содержат защищенную амино- или гидроксильную группу, защитную группу удаляют;
(4) необязательно проводят образование соли соединения формулы I с получением соответствующих солей.
7. Фармацевтическая композиция для лечения рака, содержащая производное камптотецина по п.1 или 2, где рак выбран из группы, включающей рак легких, рак молочной железы, рак толстой кишки, рак предстательной железы, меланому, рак поджелудочной железы, рак желудка, рак печени, рак головного мозга, рак почек, рак матки, цервикальный рак, рак яичников, рак мочевых путей, гастроинтестинальный рак и лейкемию.
8. Фармацевтическая композиция по п.7, отличающаяся тем, что амино или гидроксил защищены защитной группой.
9. Фармацевтическая композиция по п.8, отличающаяся тем, что ион аммония получен из любого из следующих оснований: NH3, монометиламина, диметиламина, триметиламина, моноэтиламина, диэтиламина, триэтиламина, метилэтиламина, диметилэтиламина, диизопропиламина, пирролидина, дигидроизоиндола, морфолина, N,N-диаллиламина, 4-метилпиперидина, этаноламина, 5-бромдигидроизоиндола, тиоморфолина, цис-2,6-диметилморфолина и этилендиамина.
10. Применение производного камптотецина по п.1 или 2 при получении фармацевтического препарата для лечения рака, где указанный рак выбран из группы, включающей рак легких, рак молочной железы, рак толстой кишки, рак прямой кишки, рак предстательной железы, меланому, рак поджелудочной железы, рак желудка, рак печени, рак головного мозга, рак почек, рак матки, цервикальный рак, рак яичников, рак мочевых путей, гастроинтестинальный рак и лейкемию.
11. Применение по п.10, где указанный рак выбран из группы, включающей рак молочной железы, рак толстой кишки, рак прямой кишки и рак легких.
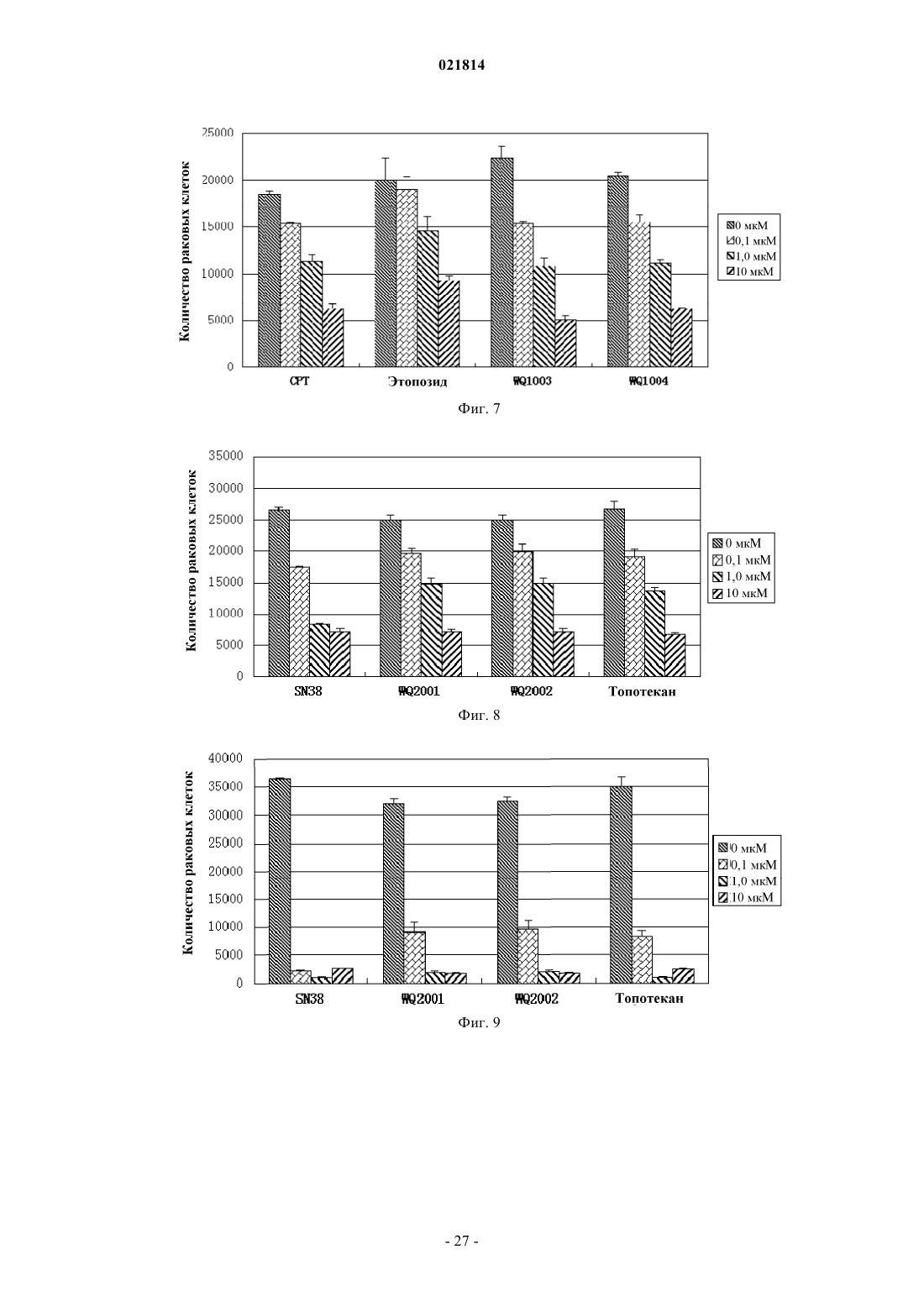
12. Применение по п.11, где указанный рак представляет собой мелкоклеточный рак легких.
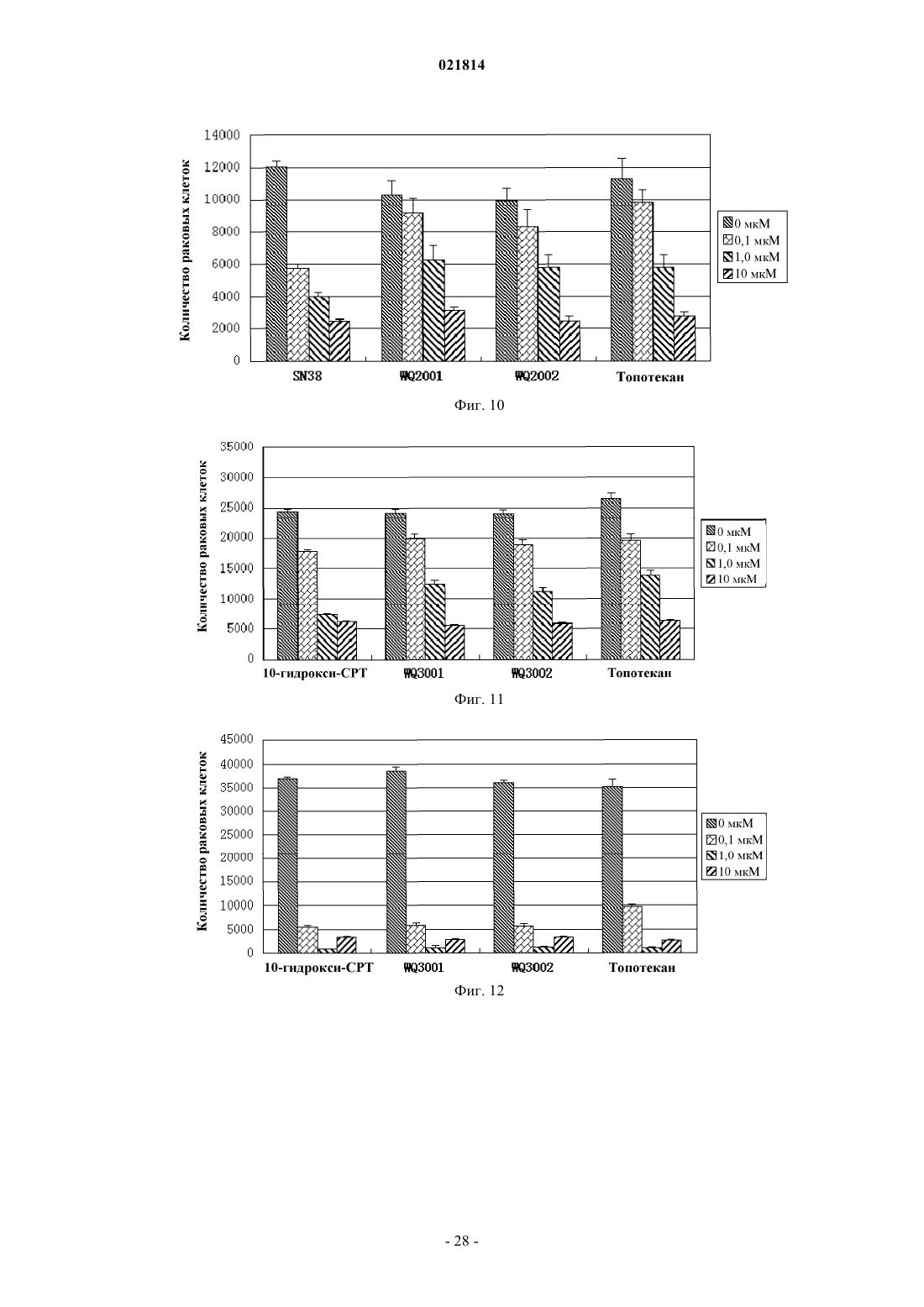
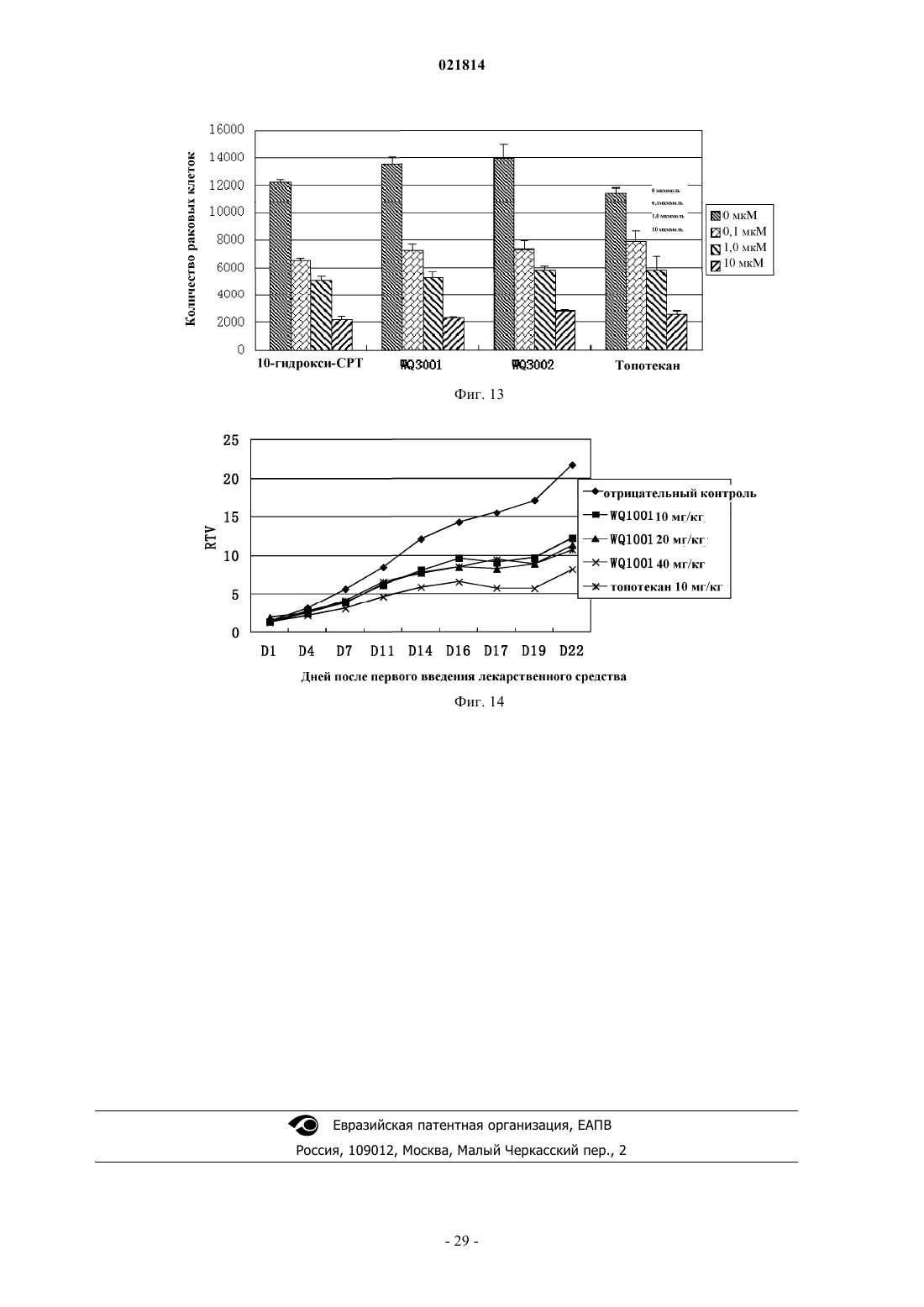
Текст
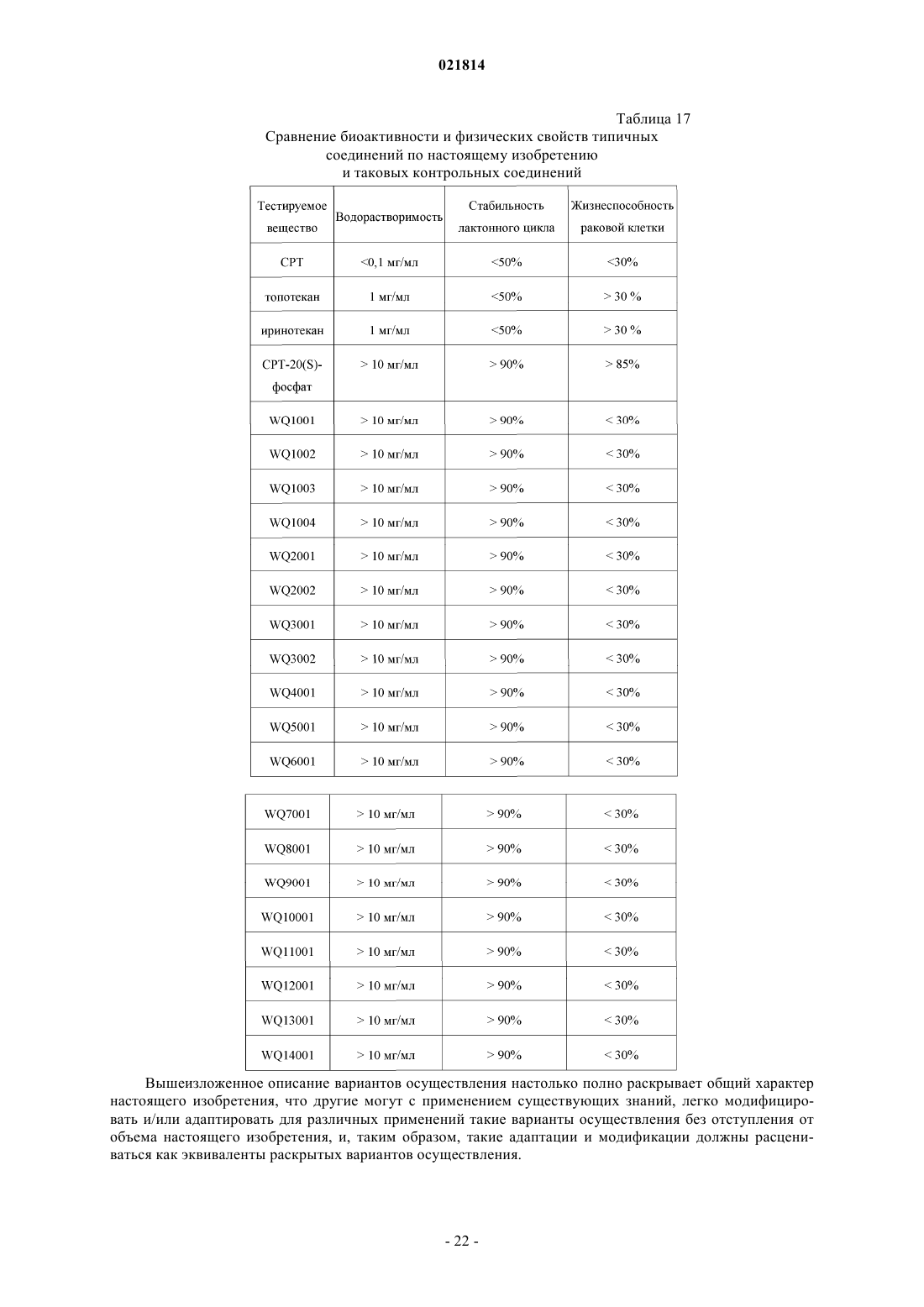
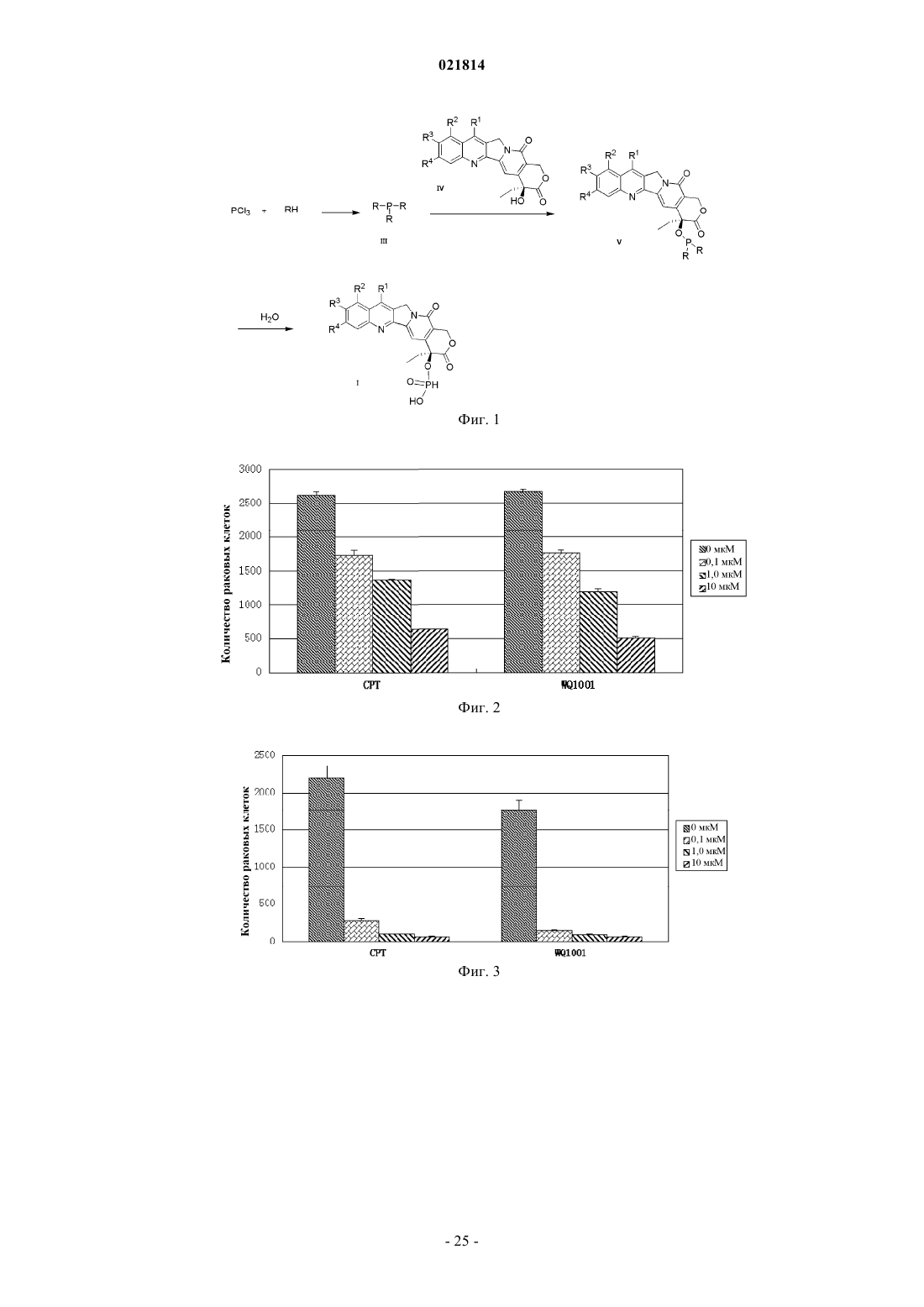
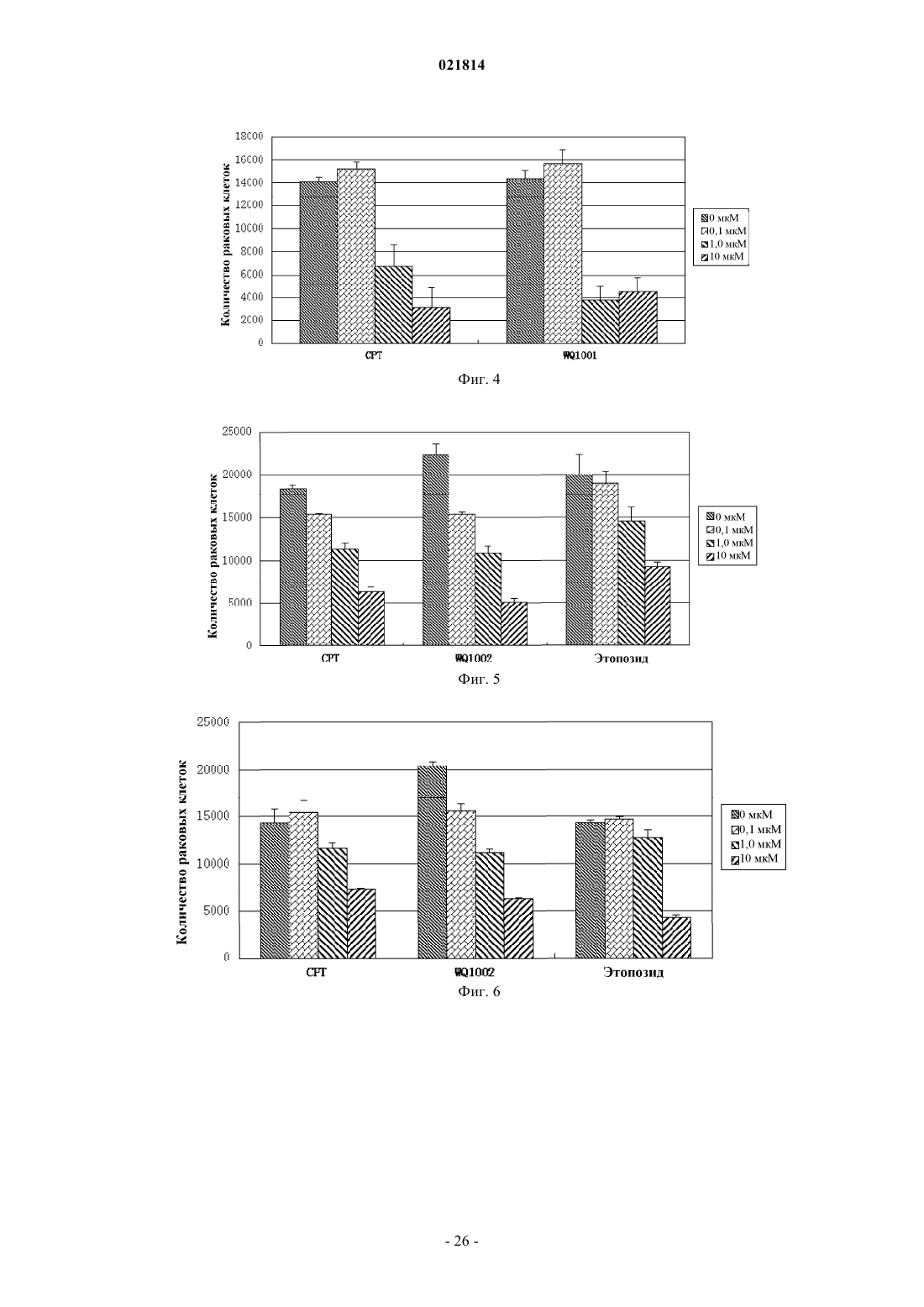
Изобретение относится к производному камптотецина, которое имеет такую структуру, как представлено формулой (II), в которой Xn+ выбран из H+, K+, Na+, Li+, Mg2+, Ca2+, Zn2+, Fe3+ и иона аммония, где R1, R2, R3 и R4 независимо представляют собой водород, гидроксильную группу, нитрогруппу, цианогруппу, галоген, карбоксильную группу, необязательно замещенную аминогруппу, кремнийсодержащую группу, моноциклическую арилоксигруппу, необязательно замещенную C1-C6-алкоксигруппу, необязательно замещенную C1-C6-алкилкарбонильную группу,необязательно замещенную C1-C6-алкильную группу или необязательно замещенную C3-C6 циклоалкильную группу; в качестве альтернативы R1 и R2 соединены посредством 1-3 других атомов с образованием гетероциклического кольца; в другом варианте осуществления R3 иR4 представляют собой атомы кислорода и соединены посредством -O-(CH2)n-O-, образуя кольцо, в котором n=1 или 2. Соединение обладает высокой водорастворимостью, химической стабильностью и высокой активностью при лечении рака. Область изобретения Настоящее изобретение относится к области фармацевтики, в частности к области противораковых лекарственных средств, более конкретно - к низкомолекулярным лекарственным средствам, способам их получения и фармацевтическим применениям. Предпосылки изобретения Природный камптотецин ("CPT") имеет пентациклическую структуру системы с конденсированными ядрами, состоящей из хинолиновых циклов (циклы А и В), пирролидинового цикла (цикл С), альфа-пиридонового цикла (цикл D) и 6-членного лактонного цикла (цикл Е). CPT имеет только один асимметрический центр в 20-й позиции и проявляет правостороннее вращение в соответствии сS-конфигурацией третичной гидроксильной группы. CPT представляет собой цитотоксический алкалоид,который был впервые выделен и охарактеризован Wall и его коллегами (J. Am. Chem. Soc. 88, 3888, 1966) из листьев и коры Camptotheca accuminata (NYSSACEAE), растения, которое присуще Китаю. Первичной клеточной мишенью для CPT является топоизомераза I (topo I), фермент, который участвует в релаксации сверхспиральной хромосомной ДНК во время репликации ДНК с помощью транзиторного одноцепочечного расщепления двуспиральной ДНК, расплетания и повторного лигирования. CPT связывается на поверхности ковалентного бинарного комплекса топоизомераза I-ДНК с образованием стабильного тройного комплекса, который предотвращает повторное лигирование ДНК после расплетания и, следовательно, приводит к репликационно-опосредованным двухцепочечным разрывам и повреждению ДНК. Так как ингибирование CPT может привести к гибели клеток во время S-фазы клеточного цикла, то CPT попадает в центр внимания интенсивных исследований по разработке противоракового лекарственного средства. (Nature Review/Cancer, October 2006, vol. 6, p. 789-802, Bioorg. Med. Chem., 2004, 12, p. 15851604). Природный CPT нерастворим в воде или в других водных растворителях, которые пригодны для парентерального введения. При рН 7 или выше структура лактонного Е-цикла CPT может быть гидролизована с образованием производного карбоксилата с разрывом кольца, которое является водорастворимым, но лишено необходимой биологической активности и проявляет высокую клиническую токсичность. При физиологических условиях реакция гидролиза лактонного Е-цикла может быть усилена из-за преимущественного связывания (в 150 раз выше CPT) производного карбоксилата с альбумином сыворотки крови (J. Med. Chem. 1993, 36, 2580; Anal. Biochem. 1993, 212, 285; Biochemistry, 1994, 33, 10325;Biochemistry, 1994, 33, 10325; Pharm. Sci. 1995, 84, 518). Нерастворимость в воде CPT и клиническая токсичность его производного карбоксилата являются двумя лимитирующими факторами, которые препятствуют применению CPT в качестве противоопухолевого химиотерапевтического средства в клинических применениях (Nature Review/Cancer, October 2006. Vol. 6, p. 789-802). Таким образом, было бы желательно обеспечить производные CPT с лучшей стабильностью лактона in vivo и водорастворимостью,чем у природного CPT (Bioorg. Med. Chem., 2004, 12, p. 1585-1604; Chem. Rev., 2009, 109(1), p. 213-235). В литературе попытки разработать биоактивные аналоги CPT с лучшей водорастворимостью были сосредоточены на введении гидрофильных групп в цикл(ы) А, В или/и С CPT (Bioorg. Med. Chem., 2004,12, p. 1585-1604; Chem. Rev., 2009, 109(1), p. 213-235). По сравнению с природным CPT присоединение химических модифицированных групп к системе сочлененных циклов будет в некотором смысле неблагоприятно сказываться на связывании CPT с поверхностью комплекса ковалентная бинарная топоизомераза I-ДНК с образованием стабильного третичного комплекса. В результате биоактивность этих аналогов CPT (например, топотекана, который используют в качестве стандартного противоракового лекарственного средства для ингибирования роста раковой клетки) обычно меньше, чем таковая у CPT (NatureReview/Cancer, October 2006. Vol.6, p. 789-802; Bioorg. Med. Chem., 2004, 12, p. 1585-1604). С другой стороны, химическая модификация циклов А, В, С CPT не может снизить степень гидролиза лактона Е-цикла. Считается, что гидролиз лактона Е-цикла облегчается образованием водородных связей между 20(S)-гидроксильной группой и соседней карбонильной группой (Bioorg. Med. Chem., 2004, 12, p. 15851604; Chem. Rev., 2009, 109(1), p. 213-235). Ранее в литературе было показано, что в целях повышения стабильности лактонного цикла CPT в одном подходе нарушается водородная связь между 20(S)-гидроксилом и соседним карбонилом, например с помощью реакции 20(S)-гидроксила с алкилом или ацилом с образованием простого эфира или сложного эфира, таким образом препятствуя ускорению гидролиза Е-цикла лактона. Однако 20(S)-гидроксильная группа является важной для фармакологической активности CPT. Доказано, что аналоги CPT без 20(S)-гидроксильной группы обычно лишены противоопухолевой активности (Organic Lett., 2004, 6(3), p. 321-324; Bioorg. Med. Chem., 2004, 12, p. 15851604; Chem. Rev., 2009, 109(1), p. 213-235). Из приведенного выше принцип присоединения водорастворимой группы пролекарства (например,ионизированной функциональной группы) в положении 20(S)-гидроксила станет практическим подходом для увеличения водорастворимости получаемой молекулы пролекарства (возможности введения лекарственного средства) при одновременном повышении стабильности лактона Е-цикла CPT пролекарства в крови во время циркуляции (клиническая безопасность лекарственного средства). Таким образом, в данном подходе к пролекарству можно преобразовывать водонерастворимую молекулу CPT в водорастворимое пролекарство CPT; поскольку такое водорастворимое пролекарство CPT может быстро распро-1 021814 страняться по всему телу человека после введения в кровоток, пролекарство CPT может находиться в очень низкой концентрации во время метаболизма, таким образом препятствуя осаждению CPT в кровяных сосудах. Кроме того, с помощью введения контрольной группы пролекарства в положение 20(S)-гидроксила взаимодействие водородной связи между 20(S)-гидроксилом и соседним карбонилом,которое будет способствовать гидролизу лактона Е-цикла, можно было бы предотвратить. Таким образом, стабильность лактона Е-цикла CPT пролекарства в кровотоке во время циркуляции может быть повышена, а также можно снизить проблемы клинической безопасности лекарственного средства, например гематотоксичность, которая относится к производному карбоксилата, полученному гидролизом CPT. Очевидно, что подход к пролекарству защиты положения 20(S)-гидроксила водорастворимой группой пролекарства является способом медицинской химии, с помощью которого можно поддерживать стабильность лактона, водорастворимость и биоактивность для того, чтобы способствовать разработке противоракового лекарственного средства на основе CPT. О попытках получить пролекарства на основе CPT или соединения на основе CPT с помощью химических модификаций в положении 20(S)-гидроксила сообщалось в литературе. Среди них большинство усилий были потрачены на введение различных защитных функциональных групп (включая липофильные и заряженные функциональные группы) с помощью этерификации 20(S)-гидроксильной группы(Chem. Rev., 2009, 109(1), p. 213-235). Превращение сложного эфира пролекарства в природный CPT опосредовано группой ферментов, известных как эстеразы, которые широко представлены в крови животных (включая людей). Недостатком пролекарства на основе сложного эфира является относительно плохая стабильность сложноэфирной связи в человеческом организме при физиологических условиях,которая легко разрушается эстеразами. Клиническая польза пролекарства на основе сложного эфира CPT не была очевидной (Chem. Rev., 2009, 109(1), p. 213-235). В результате другой попытки были получены 20(S)-O-фосфонат эфиры CPT (Organic Lett, 2004, 6(3), p. 321-324). Раскрытые 20(S)-O-фосфонаты могут улучшить водорастворимость и стабильность лактона CPT in vivo, но по результатам испытаний в экспериментах производные 20(S)-O-фосфонатов CPT лишены противоопухолевых активностей (Organic Lett.,2004, 6(3), p. 321-324). 20(S)-О-Фосфонат эфиры не могут быть превращены в CPT при физиологических условиях (Organic Lett., 2004, 6(3), p. 321-324). Таким образом, по-прежнему была бы желательной разработка производных CPT, которые обладают приемлемой водорастворимостью и стабильностью лактона Е-цикла, а также хорошей противораковой активностью. Краткое описание изобретения Одной целью настоящего изобретения является обеспечение нового производного камптотецина с идеальной противоопухолевой активностью, водорастворимостью и стабильностью лактона Е-цикла. Другой целью настоящего изобретения является обеспечение способа получения вышеупомянутого производного CPT. Одной дополнительной целью настоящего изобретения является обеспечение применений вышеупомянутого производного CPT при получении фармацевтического препарата для лечения рака. В одном аспекте настоящего изобретения обеспечивают CPT-фосфит формулы I где R1, R2, R3 и R4 независимо представляют собой водород, гидрокси, нитро, циано, галоген, карбокси, необязательно замещенный амино, кремнийсодержащую группу (например, силил, силоксил, например, содержащую C1-C6, но настоящее изобретение не ограничивается этим), моноциклический арилокси, С 1-С 6-алкокси, необязательно замещенный гидрокси, нитро, циано, галогеном или амино,С 1-С 6-алканоил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С 1-С 6-алкил,необязательно замещенный гидрокси, нитро, циано, галогеном или амино, или С 3-С 6-циклоалкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино; илиR1 и R2 соединены посредством 1-3 других атомов с образованием гетероцикла, где гетероцикл представляет собой N-гетероцикл, S-гетероцикл, О-гетероцикл или гетероцикл, содержащий два гетероатома, которые выбраны из группы, состоящей из N, О и S, a R3 и R4 определены выше; илиR1 и R2 определены выше, a R3, R4 представляют собой атомы кислорода и соединены посредством Во втором аспекте настоящего изобретения обеспечивают соль фосфита CPT формулы IIXn+ выбран из K+, Na+, Li+, Mg2+, Ca2+, Zn2+, Fe3+ и аммония. Настоящее изобретение также относится к получению вышеописанных соединений и фармацевтических композиций, содержащих вышеописанные соединения, и их применению при получении фармацевтических препаратов. Кроме того, обладая хорошей биоактивностью, соединения производных CPT по настоящему изобретению обладают идеальной водорастворимостью и высоким уровнем стабильности лактонного цикла при физиологических условиях. Производные CPT по настоящему изобретению также демонстрируют относительно низкую токсичность. Краткое описание графических материалов На фиг. 1 продемонстрирована схема синтеза производных камптотецина по настоящему изобретению; на фиг. 2 продемонстрировано как соединение WQ1001 инициирует дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких); на фиг. 3 продемонстрировано как соединение WQ1001 инициирует дозозависимую клеточную гибель клеток MDAMB231 (рака молочной железы); на фиг. 4 продемонстрировано как соединение WQ1001 инициирует дозозависимую клеточную гибель клеток НСТ 116 (рака толстой кишки); на фиг. 5 продемонстрировано как соединение WQ1002 инициирует дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких); на фиг. 6 продемонстрировано как соединение WQ1002 инициирует дозозависимую клеточную гибель клеток НСТ 116 (рака толстой кишки); на фиг. 7 продемонстрировано как соединения WQ1003 и WQ1004 инициируют дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких); на фиг. 8 продемонстрировано как соединения WQ2001 и WQ2002 инициируют дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких); на фиг. 9 продемонстрировано как соединения WQ2001 и WQ2002 инициируют дозозависимую клеточную гибель клеток НСТ 116 (рака толстой кишки); на фиг. 10 продемонстрировано как соединения WQ2001 и WQ2002 инициируют дозозависимую клеточную гибель клеток MDAMB231 (рака молочной железы); на фиг. 11 продемонстрировано как соединения WQ3001 и WQ3002 инициируют дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких); на фиг. 12 продемонстрировано как соединения WQ3001 и WQ3002 инициируют дозозависимую клеточную гибель клеток НСТ 116 (рака толстой кишки); на фиг. 13 продемонстрировано как соединения WQ3001 и WQ3002 инициируют дозозависимую клеточную гибель клеток MDAMB231 (рака молочной железы); на фиг. 14 продемонстрировано относительное изменение объемов опухолей в противоопухолевых экспериментах in vivo WQ1001. Подробное описание изобретения Если не указано другое, выражения, которые используют в контексте настоящего изобретения, определены в следующем тексте. Другие выражения, которые не определены в следующем тексте, как правило, имеют значения известные в области, к которой относится настоящее изобретение. Выражение "пролекарство на основе CPT" относится к производному камптотецина с 20(S)-гидроксильной группой, защищенной биоразрушаемой защитной группой. При физиологических условиях биоразрушаемая защитная группа 20(S)-гидроксильной группы является медленно расщепляемой специфическими ферментами с образованием фармацевтически активного камптотецина. В данном контексте млекопитающее включает, без ограничения, примата, в частности человека; грызун включает мышь, крысу и хомяка; домашнее животное включает кролика, лошадь, корову, собаку и кошку и т.д. В некоторых вариантах осуществления млекопитающее относится к человеку. Один аспект настоящего изобретения относится к фосфористо-кислому камптотецину формулы I где R1, R2, R3 и R4 независимо представляют собой водород, гидрокси, нитро, циано, галоген, карбокси, необязательно замещенный амино, кремнийсодержащую группу (например, силил, силоксил, например, содержащий С 1-С 6, но настоящее изобретение не ограничивается этим), моноциклический арилокси, С 1-С 6-алкокси, необязательно замещенный гидрокси, нитро, циано, галогеном или амино,С 1-С 6-алканоил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С 1-С 6-алкил,необязательно замещенный гидрокси, нитро, циано, галогеном или амино, или С 3-С 6-циклоалкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино; илиR1 и R2 соединены посредством 1-3 других атомов с образованием гетероцикла, где гетероцикл представляет собой N-гетероцикл, S-гетероцикл, О-гетероцикл или гетероцикл, который содержит два гетероатома, выбранных из группы, включающей N, О и S, а R3, R4 независимо представляют собой водород, гидрокси, нитро, циано, галоген, карбокси, необязательно замещенный амино, кремнийсодержащую группу (например, силил, силоксил, например, содержащий С 1-С 6, но настоящее изобретение не ограничивается этим), моноциклический арилокси, С 1-С 6-алкокси, необязательно замещенный гидрокси,нитро, циано, галогеном или амино, С 1-С 6-алканоил, необязательно замещенный гидрокси, нитро, циано,галогеном или амино, С 1-С 6-алкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, или С 3-С 6-циклоалкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино; илиR1, R2 независимо представляют собой водород, гидрокси, нитро, циано, галоген, карбокси, необязательно замещенный амино, кремнийсодержащую группу (например, силил, силоксил, например, содержащий С 1-С 6, но настоящее изобретение не ограничивается этим), моноциклический арилокси,С 1-С 6-алкокси, необязательно замещенный гидрокси, нитро, циано, галогеном или амино,С 1-С 6-алканоил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С 1-С 6-алкил,необязательно замещенный гидрокси, нитро, циано, галогеном или амино, или С 3-С 6-циклоалкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, a R3 и R4 представляют собой атомы кислорода и соединены посредством -O-(СН 2)n-O- с образованием цикла, где n равно 1 или 2. В приведенных выше вариантах осуществления, когда замещающая группа содержит амино или гидроксильную группу, причем амино или гидроксильная группа может быть защищена защитной группой, как обычно применяется в данной области техники. Предпочтительно защитная группа амина выбрана из бензоила, изобутирила, трет-бутилоксикарбонила, тритила, формила и т.д. Предпочтительно защитная группа гидроксила выбрана из метила, метоксиметила, бензилоксиметила, бензила, триметилсилила, т-бутилдиметилсилила, ацетила, трифторацетила, триметилацетила, бензоила, алкилацила и т.д. Другие подходящие защитные группы, известные специалистам в данной области техники, раскрыты в(1999). Дополнительно, предпочтительно защитная группа представляет собой группу, которая может быть ферментативно расщеплена в физиологических условиях, такую как ацил. Предпочтительно для обеспечения проявления биоактивности аналогами CPT по настоящему изобретению R1, R2, R3 и R4 выбраны из групп меньшего стерического препятствия для CPT, обычно таких,которые имеют меньшую молекулярную массу, например менее 100. Как показано в экспериментах, соединения обладают хорошей активностью лекарственного средства и водорастворимостью. Другой аспект настоящего изобретения относится к фосфористо-кислой соли камптотецина формулы IIXn+ представляет собой K+, Na+, Li+, Mg2+, Ca2+, Zn2+, Fe3+ или аммоний, где аммоний может быть получен из одного из следующих оснований: NH3, монометиламина, диметиламина, триметиламина, моноэтиламина, диэтиламина, триэтиламина, метилэтиламина, диметилэтиламина, диизопропиламина,пирролидина, дигидроизоиндола, морфолина, N,N-диаллиламина, 4-метилпиперидина, этаноламина,5-бромдигидроизоиндола, тиоморфолина, цис-2,6-диметилморфолина и этилендиамина. В дополнение к хорошей фармацевтической активности соль формулы II обладает желаемой стабильностью и хорошей водорастворимостью при физиологических условиях. Предпочтительно соединения формулы I и формулы II получены из соединения формулы IV, как перечислено в табл. 1, путем присоединения фосфористо-кислого фрагмента к положению С-20 Соединение по настоящему изобретению может быть синтезировано по схеме, которая продемонстрирована на фиг. 1, включающей этапы, на которых:(1) проводят реакцию PCl3 с азолом RH с получением промежуточного соединения триаминфосфина формулы III(2) проводят реакцию промежуточного соединения триаминфосфина формулы III с соединением формулы IV с получением предшественника 20(S)-О-фосфорамидита CPT формулы V: где R1, R2, R3 и R4 являются такими, как ранее определено для формулы I, и, если R1, R2, R3 или R4 представляют собой или содержат гидроксильную группу или аминогруппу, гидроксильную группу или аминогруппу перед реакцией с соединением формулы III защищают защитной группой;(3) проводят гидролиз предшественника 20(S)-О-фосфорамидита формулы V с получением 20(S)-О-фосфита CPT формулы I причем если R1, R2, R3 или R4 представляют собой или содержат защищенную амино или гидроксильную группу, защитную группу удаляют;(4) проводят образование соли соединения формулы I с применением основания, которое обеспечивает соответствующую соль. Основания, которые можно применять на данном этапе включают, без ограничения, NaOH, Na2CO3, NaHCO3, KOH, KHCO3, K2CO3, LiOH, LiHCO3, Li2CO3, NH4HCO3, Ca(OH)2,CaCO3, Ca(HCO3)2, Mg(HCO3)2, Zn(HCO3)2, Zn(OH)2 и Fe(OH)3, где желательна соль четвертичного аммония, а также возможно применение основания четвертичного аммония соответственно. Соединения формул I и II по настоящему изобретению являются эффективными при лечении рака млекопитающих, в частности рака человека (также называемого злокачественной опухолью), включая все формы рака на недифференцированной, среднедифференцированной и высокодифференцированной стадии. При введении соединения по настоящему изобретению пациентам, нуждающимся в таком лечении, вводят эффективное количество соединения или состава, содержащего одно или несколько соединений по настоящему изобретению. Как применяется в данном документе, выражение "эффективное количество" предназначено для обозначения количества, при котором соединение по настоящему изобретению будет приводить к желаемому эффекту. Например, для лечения рака/злокачественной опухоли"эффективное количество" относится к количеству, которое будет ингибировать или замедлять развитие рака, или уничтожать рак или клетки злокачественной опухоли, и/или служить причиной регрессии и/или временной ремиссии рака, такого как злокачественные опухоли, например уменьшать объем или размер таких опухолей или абсолютно удалять опухоли. Фармацевтически эффективным количеством или дозировкой предпочтительно является 0,1-100 мг соединения по настоящему изобретению на кг массы тела. Более предпочтительно фармацевтически эффективным количеством или дозировкой предпочтительно является 0,1-50 мг соединения по настоящему изобретению на 1 кг массы тела. При необходимости или целесообразности, как определяется врачом или ветеринаром, эффективное количество может находиться за пределами, упомянутыми выше. Когда соединение по настоящему изобретению вводят в виде его фармацевтически приемлемой соли, сольвата или гидрата, эффективное количество относится к количеству свободного соединения. Соединение или фармацевтическая композиция в соответствии с настоящим изобретением могут применяться при лечении ряда опухолей и/или форм рака, включая, без ограничения, солидные опухоли,такие как рак легких, молочной железы, толстой кишки, предстательной железы, меланомы, поджелудочной железы, желудка, печени, головного мозга, почек, матки, цервикальный рак, рак яичников, мочевых путей, гастроинтестинальный рак и т.д., а также переносимые с кровью опухоли, такие как лейкемия. Предпочтительные солидные опухоли включают, без ограничения, рак толстой кишки и прямой кишки, рак молочной железы и рак легких, в частности мелкоклеточный рак легких. Соединение в соответствии с настоящим изобретением может быть применено в комбинации с одним или несколькими другими противораковыми лекарственными средствами. Другие противораковые лекарственные средства в контексте включают 1) модулятор рецептора эстрогена, например тамоксифен,ралоксифен, идоксифен; 2) модулятор рецептора андрогена, например финастерид, нулатимид, флутамид, бикалутамид; 3) модулятор рецептора ретиноида, например бексаротен, кислота витамина А,13-цис-ретиноевая кислота, 9-цис-ретиноевая кислота; 4) цитотоксические вещества, включающие алки-7 021814 лирующие средства, фактор некроза опухоли, ингибитор тубулина, ингибиторы топоизомеразы, например ифосфамид, карбоплатин, ранимустин, фотемустин, оксалиплатин, митоксантрон, паклитаксел и топотекан; 5) антипролиферативные средства, например триметрексат, флударабин и капецитабин; 6) ингибиторы ацилтрансферазы; 7) ингибитор редуктазы HMG-СоА; 8) ингибитор ВИЧ-протеазы и 9) ингибитор обратной транскриптазы и т.д. Соединение по настоящему изобретению также применимо в качестве ингибитора фермента топоизомеразы I. Соединение по настоящему изобретению можно вводить в дозе, которая является эффективной при ингибировании фермента топоизомеразы I. Количество обычно составляет приблизительно 0,1100 мг/кг массы тела в неделю, предпочтительно приблизительно 1-50 мг/кг в неделю. Соединение по настоящему изобретению может также действовать как противовирусное средство(например, против ВИЧ) и противопаразитарное средство. Соединение по настоящему изобретению можно вводить само по себе или в виде его фармацевтической композиции. Помимо соединения и фармацевтически приемлемых носителей композиция по настоящему изобретению может включать другие активные материалы, которые не ухудшают желаемое действие и/или дополняют желаемое действие. Соединения/активные материалы в соответствии с настоящим изобретением можно вводить любым путем, например перорально, назально, парентерально, внутривенно, внутрикожно, подкожно или местно в жидкой или твердой форме. Для целей парентерального терапевтического введения активный ингредиент может быть включен в раствор или суспензию. Растворы или суспензии могут также включать следующие компоненты для инъекции: стерильный растворитель, такой как вода; суспензии липосомальных частиц, в результате чего частицы содержат стабильное активное лекарственное средство в ядре частицы в рН-регулируемой и защищенной среде; суспензии липосомальных частиц, активное лекарственное средство в которых прикреплено к внешней поверхности частицы или любому из бислоев частицы; физиологический раствор,нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие средства, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты, и средства для регуляции тоничности, такие как хлорид натрия или декстроза. Препарат для парентерального введения может содержаться в ампулах,одноразовых шприцах или флаконах, изготовленных из стекла или пластика, которые содержат множество доз. Композиции для перорального применения обычно включают инертный растворитель или съедобный носитель. Они могут содержаться в желатиновых капсулах или быть спрессованными в таблетку. С целью перорального терапевтического введения вышеупомянутые соединения можно получить в форме таблеток, пилюль, капсул, пастилок, настоев, суспензий, сиропов, облаток, жевательных резинок и т.п Таблетки, пилюли, капсулы и т.п. могут содержать следующие ингредиенты: связывающее вещество,такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатины; наполнитель, такой как крахмал или лактоза, средство для улучшения распадаемости, такое как альгиновая кислота, примогель, кукурузный крахмал и т.п.; смазывающее вещество, такое как стеарат магния или стероты; скользящее вещество, такое как коллоидный диоксид кремния; и подсластитель, такой как сахароза или сахарин; или может быть добавлена вкусовая добавка, такая как мята перечная, метилсалицилат или апельсиновый ароматизатор. Когда единица дозирования находится в форме капсулы, кроме материала указанного выше типа, она может содержать жидкий носитель, такой как жирное масло. Другие формы единицы дозирования могут содержать другие различные материалы, которые изменяют физическую форму дозированной единицы, например покрытия. Таким образом, таблетки или пилюли, например, могут быть покрыты сахаром, шеллаком, или другими энтеросолюбильными покрывающими средствами. Сироп может содержать, помимо активного соединения, сахарозу в качестве подсластителя, а также консервант, контрастное вещество или краситель и ароматизатор. Материалы, которые применяют при получении данных композиций, должны быть фармацевтически или ветеринарно чистыми и нетоксичными в применяемых количествах. Пример 1-1. Получение 20(S)-O-фосфита CPT (WQ1000). 0,69 г 1,2,4-1H-триазола (15 ммоль) растворяли в безводном пиридине (20 мл) и охлаждали до 0 С на ледяной бане с последующим добавлением 0,69 г трихлорида фосфора (5 ммоль). После удаления ледяной бани раствор 3,48 г CPT в 30 мл пиридина добавляли при перемешивании при комнатной температуре. Перемешивание реакционной смеси продолжали до тех пор, пока CPT не был полностью израсходован, затем при перемешивании добавляли 10 мл воды. После окончания реакции реакционный раствор выпаривали при пониженном давлении и очищали остаток на силикагеле. Соответствующий элюент собирали и выпаривали досуха при пониженном давлении. Твердый остаток повторно растворяли в метаноле или этаноле, затем по каплям обрабатывали ацетоном или простым эфиром для осаждения твердого вещества. Целевой продукт WQ1000 получали в виде светло-желтого порошка.WQ1000 смешивали с небольшим количеством воды, затем по каплям обрабатывали насыщенным раствором бикарбоната натрия при перемешивании до тех пор, пока не перестали образовываться воздушные пузырьки. Раствор перемешивали в течение 0,5 ч после того, как вся твердая фаза растворилась. Раствор загружали в колонку С 18 для хроматографии. Соответствующий элюент собирали и высушивали сублимацией для обеспечения продукта WQ1001. По аналогичной процедуре несколько иллюстративных соединений, которые перечислены в табл. 2,были получены с применением соединений формулы IV в качестве исходных материалов. Эти соединения представлены как желтые твердые, стабильные при комнатной температуре, трудно окисляемые и разлагаемые с водорастворимостью более 10 мг/мл. Таблица 2 1 Пример 2. Оценка противораковой активности WQ1001 in vitro. Для оценки способности соединения убивать раковые клетки в in vitro экспериментах на раковых клеточных линиях осуществляли анализ жизнеспособности клеток с применением набора CellTiter-Glo,предоставленного Promega Corporation. С помощью набора измеряют уровни АТФ при помощи ферментативного анализа люциферазы. Нормальные жизнеспособные клетки будут продуцировать определенный уровень АТФ при метаболизме. Ферментативная реакция между продуктом АТФ и люциферазой будет излучать определенный уровень люминесцентного сигнала, который захватывается люминометром и записывается как определенное люминесцентное считывание. Мертвые клетки с их ослабленными метаболическими функциями и отсутствием продуцирования АТФ не генерируют люминесцентные сигналы в тех же условиях измерения, таким образом, считывания люминесцентного сигнала будут равны нулю. При применении данного способа оценки противораковой активности соединения определенную концентрацию противоракового лекарственного средства добавляют в то же самое количество жизнеспособных раковых клеток и считывание люминесцентного сигнала получают в определенный момент времени посредством наборов CellTiter-Glo. Считывание более низкого люминесцентного сигнала означает более низкий уровень жизнеспособных раковых клеток после обработки противораковым лекарственным средством и, следовательно, более сильную способность лекарственного средства убивать раковые клетки. Подробнее процедура представляет собой следующее: определенное количество клеток мелкоклеточного рака легких (АТСС каталожныйН 446), клеток рака молочной железы (АТСС каталожныйMDAMB231) или клеток рака толстой кишки (АТСС каталожный . НСТ 116) высевают в 96 лунок с одинаковой средой для клеточной культуры, затем соответственно обрабатывают WQ1001 и другим противораковым лекарственным средством в течение 24, 48 и 72 ч. В определенные моменты времени раковые клетки перемешивали с реагентами CellTiter-Glo в течение 1 ч и записывали соответствующие люминесцентные сигналы. Исходя из того, что считанные люминесцентные сигналы пропорциональны количеству жизнеспособных раковых клеток, считанный люминесцентный сигнал, соответственно, можно перевести в количество жизнеспособных раковых клеток. Оценку клеточной жизнеспособности получали делением количества жизнеспособных раковых клеток после обработки определенной концентрацией противоракового лекарственного средства на количество жизнеспособных раковых клеток контрольной группы, которую не обрабатывали лекарственным средством. Противораковые активности соединения WQ1001 обобщены на фиг. 2-4 и в табл. 3-5. На фиг. 2 продемонстрировано как соединение WQ1001 инициирует дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких). По х-координате блок "CPT" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч с применением CPT (растворенным в DMSO) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно); по х-координате, блок "WQ1001" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч с применениемWQ1001 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Таблица 3 Жизнеспособность клеток Н 446 после обработки лекарственным средством в течение 48 ч Можно наблюдать, что водорастворимое WQ1001 инициирует дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких), и этот эффект лучше такового для CPT. На фиг. 3 продемонстрировано как соединение WQ1001 инициирует дозозависимую клеточную гибель клеток MDAMB231 (рака молочной железы). По х-координате блок "CPT" представляет обработку клеток MDAMB231 (рака молочной железы) в течение 48 ч CPT (растворенным в DMSO) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ1001" представляет обработку клеток MDAMB231 (рака молочной железы) в течение 48 ч WQ1001 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Таблица 4 Жизнеспособность клеток MDAMB231 после обработки лекарственным средством Можно наблюдать, что водорастворимое WQ1001 инициирует дозозависимую клеточную гибель клеток MDAMB231 (рака молочной железы), и этот эффект сопоставим с таковым для CPT. На фиг. 4 продемонстрировано как соединение WQ1001 инициирует дозозависимую клеточную гибель клеток (рака толстой кишки). По х-координате блок "CPT" представляет обработку клеток НСТ 116(0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ1001" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч WQ1001 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Таблица 5 Жизнеспособность клеток НСТ 116 после обработки лекарственным средством в течение 48 ч Можно наблюдать, что водорастворимое WQ1001 инициирует дозозависимую клеточную гибель клеток НСТ 116 (рака толстой кишки), и при концентрации 1,0 мкМ жизнеспособность клеток после обработки тестируемым образцом значительно ниже, чем таковая для соответствующего контроля. Пример 3. Оценка противораковой активности WQ1002 in vitro. С помощью способа примера 2 измеряли противораковую активность соединения WQ1002, а результаты измерений показаны на фиг. 5, 6 и в табл. 6, 7. На фиг. 5 продемонстрировано как соединение WQ1002 инициирует дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких). По х-координате блок "CPT" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч CPT (растворенным в DMSO) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ1002" представляет обработку клеток Н 446 (мелкоклеточный рак легких) в течение 48 ч WQ1002 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок"этопозид" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч этопозидом (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Таблица 6 Жизнеспособность клеток Н 446 после обработки лекарственным средством в течение 48 ч Можно наблюдать, что водорастворимое WQ1002 инициирует дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких), и этот эффект лучше такового для CPT и этопозида (который уже используется в клиническом применении при нацеливании на топоизомеразу II). На фиг. 6 продемонстрировано как соединение WQ1002 инициирует дозозависимую клеточную гибель клеток НСТ 116 (рака толстой кишки). По х-координате блок "CPT" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч CPT (растворенным в DMSO) в 4 различных концентрациях(0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ1002" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч WQ1002 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "этопозид" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч этопозидом (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Таблица 7 Жизнеспособность клеток НСТ 116 после обработки лекарственным средством в течение 48 ч Можно наблюдать, что водорастворимое WQ1002 инициирует дозозависимую клеточную гибель клеток НСТ 116 (рака толстой кишки), и этот эффект лучше такового для CPT и этопозида (который уже используется в клиническом применении при нацеливании на топоизомеразу II). Примеры 4 и 5. Оценка противораковой активности WQ1003 и WQ1004 in vitro. С помощью способа примера 2 измеряли противораковую активность соединений WQ1003 иWQ1004, а результаты измерений показаны на фиг. 7 и в табл. 8. На фиг. 7 продемонстрировано как соединения WQ1003 и WQ1004 инициируют дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких). По х-координате блок "CPT" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч CPT (растворенным в DMSO) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "этопозид" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч этопозидом (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ1003" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч WQ1003 (растворенным в физиологическом растворе) в 4 различных концентрациях(0, 0,1, 1,0, 10 мкМ соответственно). По х-координате, блок "WQ1004" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч WQ1004 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Таблица 8 Жизнеспособность клеток Н 446 после обработки лекарственным средством в течение 48 ч Можно наблюдать, что водорастворимые WQ1003 и WQ1004 инициируют дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких) и их эффекты лучше таковых для CPT и таковых для этопозида (который уже используется в клиническом применении при нацеливании на топоизомеразу II). Примеры 6 и 7. Оценка противораковой активности WQ2001 и WQ2002 in vitro С помощью способа примера 2 измеряли противораковую активность соединений WQ2001 иWQ2002, а результаты измерений показаны на фиг. 8-10 и в табл. 9-11. На фиг. 8 продемонстрировано как соединения WQ2001 и WQ2002 инициируют дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких). По х-координате блок "SN38" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч SN38 (растворенным в DMSO) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "топотекан" представляет обработку клеток Н 446 (мелкоклеточного рака легких) клетки в течение 48 ч топотеканом(растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ2001" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч WQ2001 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ2002" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч WQ2002 (растворенным в физиологическом рас- 16021814 творе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Таблица 9 Жизнеспособность клеток Н 446 после обработки лекарственным средством в течение 48 ч Можно наблюдать, что водорастворимые WQ2001 и WQ2002 инициируют дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких) и их эффекты сопоставимы с таковыми для SN38 и топотекана (который уже используется в клиническом применении при нацеливании на топоизомеразу I). На фиг. 9 продемонстрировано как соединения WQ2001 и WQ2002 инициируют дозозависимую клеточную гибель клеток НСТ 116 (рака толстой кишки). По х-координате блок "SN38" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч SN38 (растворенным в DMSO) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "топотекан" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч топотеканом (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ2001" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч WQ2001(растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ2002" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч WQ2002 (растворенным в физиологическом растворе) в 4 различных концентрациях(0, 0,1, 1,0, 10 мкМ соответственно). Таблица 10 Жизнеспособность клеток НСТ 116 после обработки лекарственным средством в течение 48 ч Можно наблюдать, что соединения WQ2001 и WQ2002 инициируют дозозависимую клеточную гибель клеток НСТ 116 (рака толстой кишки) и их эффекты сопоставимы с таковыми для SN38 и топотекана(который уже используется в клиническом применении при нацеливании на топоизомеразу I), при этом оба обладают хорошим эффектом. На фиг. 10 продемонстрировано как соединения WQ2001 и WQ2002 инициируют дозозависимую клеточную гибель клеток MDAMB231 (рака молочной железы). По х-координате блок "SN38" представляет обработку клеток MDAMB231 в течение 48 ч SN38 (растворенным в DMSO) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Блок "топотекан" представляет обработку клетокMDAMB231 в течение 48 ч топотеканом (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ2001" представляет обработку клеток MDAMB23 в течение 48 ч WQ2001 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ2002" представляет обработку клеток MDAMB23 в течение 48 ч WQ2002 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Результаты измерений приведены в табл. 11. Таблица 11 Жизнеспособность клеток MDAMB231 после обработки лекарственным средством в течение 48 ч Можно наблюдать, что водорастворимые WQ2001 и WQ2002 инициируют дозозависимую клеточную гибель клеток MDAMB23 и их эффекты несколько ниже, чем таковые для SN38, но сопоставимы с таковыми для топотекана (который уже используется в клиническом применении при нацеливании на топоизомеразу I). Примеры 8 и 9. Оценка противораковой активности WQ3001 и WQ3002 in vitro. С помощью способа примера 2 измерили противораковую активность соединений WQ3001 иWQ3002, а результаты измерений показаны на фиг. 11-13 и в табл. 12-14. На фиг. 11 и в табл. 12 продемонстрировано как соединения WQ3001 и WQ3002 инициируют дозозависимую клеточную гибель клеток Н 446 (мелкоклеточного рака легких). По х-координате блок"10-гидрокси-CPT" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч"10-гидрокси-CPT" (растворенным в DMSO) в 4 различных концентрациях (О, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "топотекан" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч топотеканом (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ3001" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч WQ3001 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок"WQ3002" представляет обработку клеток Н 446 (мелкоклеточного рака легких) в течение 48 ч WQ3002(растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Таблица 12 Жизнеспособность клеток Н 446 после обработки лекарственным средством в течение 48 ч Можно наблюдать, что WQ3001 и WQ3002 инициируют дозозависимую клеточную гибель клеток Н 446 (мелкоклеточный рак легких) и их эффекты сопоставимы с таковыми для 10-гидрокси-CPT и топотекана (который уже используется в клиническом применении при нацеливании на топоизомеразу I). На фиг. 12 и в табл. 13 продемонстрировано как соединения WQ3001 и WQ3002 инициируют дозозависимую клеточную гибель клеток НСТ 116 (рака толстой кишки). По х-координате блок "10-гидроксиCPT" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч 10-гидрокси-CPT(растворенным в DMSO) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "топотекан" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч топотеканом (растворенным в физиологическом растворе) в 4 различных концентрациях(0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ3001" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч WQ3001 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ3002" представляет обработку клеток НСТ 116 (рака толстой кишки) в течение 48 ч WQ3002 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Таблица 13 Жизнеспособность клеток НСТ 116 после обработки лекарственным средством в течение 48 ч Можно наблюдать, что WQ3001 и WQ3002 инициируют дозозависимую клеточную гибель клеток НСТ 116 (рака толстой кишки) и их эффекты сопоставимы с таковыми для 10-гидрокси-CPT и топотекана(который уже используется в клиническом применении при нацеливании на топоизомеразу I). На фиг. 13 и в табл. 14 продемонстрировано как соединения WQ3001 и WQ3002 инициируют дозозависимую клеточную гибель клеток MDAMB231 (рака молочной железы). По х-координате блок"10-гидрокси-CPT" представляет обработку клеток MDAMB231 (рака молочной железы) в течение 48 ч 10-гидрокси-CPT (растворенным в DMSO) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "топотекан" представляет обработку клеток MDAMB231 (рака молочной железы) в течение 48 ч топотеканом (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ3001" представляет обработку клеток MDAMB23 в течение 48 ч WQ3001 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). По х-координате блок "WQ3002" представляет обработку клеток MDAMB23 в течение 48 ч WQ3002 (растворенным в физиологическом растворе) в 4 различных концентрациях (0, 0,1, 1,0, 10 мкМ соответственно). Таблица 14 Жизнеспособность клеток MDAMB231 после обработки лекарственным средством в течение 48 ч Можно наблюдать, что водорастворимые WQ3001 и WQ3002 инициируют дозозависимую клеточную гибель клеток MDAMB23, и их эффекты сопоставимы с таковыми для 10-гидрокси-CPT и топотекана (который уже используется в клиническом применении при нацеливании на топоизомеразу I). Пример 10: исследование на животных для оценки противораковой активности с применением клеток NCI-H446 мелкоклеточного рака легких человека в ксенографтной модели "голой" мыши. Клетки NCI-H446 мелкоклеточного рака легких человека пересаживали "голым" мышам. Когда объем опухоли вырос приблизительно до 100 мм, мышей рандомизированно разделяли на 5 групп по стратифицированным объемам опухоли: группа отрицательного контроля, три группы для WQ1001(группа низкой дозировки, группа средней дозировки и группа высокой дозировки), а также группа положительного контроля (топотекан). Лекарственные средства вводили путем внутривенных инъекций. Подробнее схема введения лекарственного средства показана в табл. 15. День первого введения лекарственного средства записан как DO. Массу тела измеряли перед каждым введением лекарственного средства и устанавливали количество лекарственного средства в соответствии с массой тела. После прекращения введения лекарственного средства массы тела измеряли дважды в неделю. В конце теста (D22) массы тела измеряли непосредственно перед умерщвлением животных. Таблица 15 Схема введения лекарственного средства при исследовании активности противоопухолевого лекарственного средства WQ1001 in vivo Лекарственное средство вводили сразу после разделения групп. Результаты изменения массы тела демонстрируют, что массы тела группы 10 мг/кг WQ1001 были нормальные; массы тела животного групп 20 и 40 мг/кг WQ1001 значительно снизились через одну неделю после введения лекарственного средства, но вернулись к нормальным в течение 5 дней после прекращения введения лекарственного средства; массы тела животного группы 40 мг/кг WQ1001 и группы 10 мг/кг топотекана снизились значительно в конце теста (D22), но ни одно животное не умерло. Дозировки лекарственного средства группы 40 мг/кг WQ1001 и группы 10 мг/кг топотекана достигли своей MTD (дозировки максимальной токсичности) соответственно. В конце теста увеличения опухолей всех групп, которые обрабатывали лекарственным средством,были ниже, чем таковые в группе отрицательного контроля. WQ1001 вводили путем внутривенных инъекций один раз через день 3 раза подряд, после чего 7 дней лекарственное средство не вводили, затем осуществляли еще один цикл, всего 6 введений. При всех введениях, 10 г, 20 и 40 мг/кг, ингибировался рост опухоли, а активность при 40 мг/кг была наилучшей. При введении 10 мг/кг топотекана дважды в неделю с интервалом в 2-3 дня в течение 3 недель (6 раз) значительно ингибировался рост опухоли с активностью, которая сопоставима с 10 или 20 мг/кг WQ1001. Из исследования токсичности на животных (изменение массы тела животного) и противоопухолевой активности можно сделать вывод, что при условии одинаковой противоопухолевой активностиWQ1001 менее токсично для животных, чем топотекан. Результаты измерений приведены в табл. 16 и на фиг. 14. В табл. 16 RTV означает относительный объем опухоли, который рассчитывается как Vt/V0, где V0 представляет собой объем опухоли, измеренный в день D0, a Vt представляет собой объем опухоли каждого измерения. Индикатор оценки противоопухолевой активности представляет собой относительную скорость роста опухоли T/C(%)=(TRTV/CRTV)100%, где TRTV представляет собой RTV группы обработки,a CRTV представляет собой RTV группы отрицательного контроля. Таблица 16 Изменение массы тела и эффекты ингибирования роста опухоли всех групп в in vivo тесте противоопухолевой эффективности WQ1001 В табл. 17 перечисляется сравнение свойств иллюстративных соединений по настоящему изобретению и нескольких существующих производных CPT. В тесте стабильность лактонного цикла характеризовали с помощью измерения степени сохранения лактонного цикла с помощью HPLC (жидкостной хроматограммы) после помещения тестируемого соединения в буферный раствор с рН 7,4. Противораковую активность характеризовали жизнеспособностью раковых клеток Н 446 при концентрации лекарственного средства 10 мкМ. Из таблицы можно узнать различия в водорастворимости, стабильности лактонного цикла и токсикологии соединений по настоящему изобретению. Таблица 17 Сравнение биоактивности и физических свойств типичных соединений по настоящему изобретению и таковых контрольных соединений Вышеизложенное описание вариантов осуществления настолько полно раскрывает общий характер настоящего изобретения, что другие могут с применением существующих знаний, легко модифицировать и/или адаптировать для различных применений такие варианты осуществления без отступления от объема настоящего изобретения, и, таким образом, такие адаптации и модификации должны расцениваться как эквиваленты раскрытых вариантов осуществления. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Производное камптотецина, которое представляет собой фосфит CPT формулы I где R1, R2, R3 и R4 независимо представляют собой водород, гидрокси, нитро, циано, галоген, карбокси, амино, диметиламино, 4-метилпиперазино, 2-триметилсилилэтил, трет-бутилдиметилсилил,С 1-С 6-алкокси, необязательно замещенный гидрокси, нитро, циано, галогеном или амино,С 1-С 6-алканоил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С 1-С 6-алкил,необязательно замещенный гидрокси, нитро, циано, галогеном или амино, или С 3-С 6-циклоалкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино; илиR1 и R2 соединены посредством 1-3 других атомов с образованием гетероцикла, где гетероцикл представляет собой N-гетероцикл, S-гетероцикл, О-гетероцикл или гетероцикл, который содержит два гетероатома, которые выбраны из группы, включающей N, О и S, a R3 и R4 независимо представляют собой водород, гидрокси, нитро, циано, галоген, карбокси, амино, диметиламино, 4-метилпиперазино,2-триметилсилилэтил, трет-бутилдиметилсилил, С 1-С 6-алкокси, необязательно замещенный гидрокси,нитро, циано, галогеном или амино, С 1-С 6-алканоил, необязательно замещенный гидрокси, нитро, циано,галогеном или амино, С 1-С 6-алкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, или С 3-С 6-циклоалкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино; илиR1 и R2 независимо представляют собой водород, гидрокси, нитро, циано, галоген, карбокси, амино,диметиламино, 4-метилпиперазино, 2-триметилсилилэтил, трет-бутилдиметилсилил, С 1-С 6-алкокси, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С 1-С 6-алканоил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, С 1-С 6-алкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, или С 3-С 6-циклоалкил, необязательно замещенный гидрокси, нитро, циано, галогеном или амино, a R3 и R4 представляют собой атомы кислорода и соединены посредством -O-(СН 2)n-O- с образованием цикла, где n равно 1 или 2,или его фармацевтически приемлемые соли. 2. Производное камптотецина по п.1, которое представляет собой соль формулы II где Xn+ представляет собой K+, Na+, Li+, Mg2+, Ca2+, Zn2+, Fe3+ или аммоний. 3. Производное камптотецина по п.1 или 2, где амино- или гидроксильная группа защищена защитной группой. 4. Производное камптотецина по п.2, где аммоний получен из одного из следующих оснований:NH3, монометиламина, диметиламина, триметиламина, моноэтиламина, диэтиламина, триэтиламина, метилэтиламина, диметилэтиламина, диизопропиламина, пирролидина, дигидроизоиндола, морфолина,N,N-диаллиламина, 4-метилпиперидина, этаноламина, 5-бромдигидроизоиндола, тиоморфолина,цис-2,6-диметилморфолина и этилендиамина. 5. Производное камптотецина по любому из пп.1-4, которое представляет собой фосфит или его фармацевтически приемлемые соли, полученные из группы, включающей CPT, 7-этил-10-гидрокси-CPT,топотекан, 9-амино-CPT, иринотекан, 9-нитро-CPT, луртотекан, 7-этил-10,11-метилендиокси-CPT, эксатекан, 7-этил-CPT, 10-гидрокси-CPT, гиматекан, каренитекан и силатекан. 6. Способ получения производного камптотецина по п.1, включающий следующие этапы, на которых:(1) проводят реакцию PCl3 с азольным соединением RH с получением промежуточного соединения триаминфосфина формулы III(2) проводят реакцию промежуточного соединения триаминфосфина формулы III с соединением формулы IV с получением предшественника 20(S)-О-фосфорамидита CPT формулы V где R1, R2, R3 и R4 являются такими, как определено в п.1, и, если R1, R2, R3 или R4 представляют собой или содержат гидроксильную группу или аминогруппу, гидроксильную группу или аминогруппу перед реакцией с соединением формулы III защищают защитной группой;(3) проводят гидролиз предшественника формулы V с получением 20(S)-O-фосфита CPT формулы I причем, если R1, R2, R3 или R4 представляют собой или содержат защищенную амино- или гидроксильную группу, защитную группу удаляют;(4) необязательно проводят образование соли соединения формулы I с получением соответствующих солей. 7. Фармацевтическая композиция для лечения рака, содержащая производное камптотецина по п.1 или 2, где рак выбран из группы, включающей рак легких, рак молочной железы, рак толстой кишки, рак предстательной железы, меланому, рак поджелудочной железы, рак желудка, рак печени, рак головного мозга, рак почек, рак матки, цервикальный рак, рак яичников, рак мочевых путей, гастроинтестинальный рак и лейкемию. 8. Фармацевтическая композиция по п.7, отличающаяся тем, что амино или гидроксил защищены защитной группой. 9. Фармацевтическая композиция по п.8, отличающаяся тем, что ион аммония получен из любого из следующих оснований: NH3, монометиламина, диметиламина, триметиламина, моноэтиламина, диэтиламина, триэтиламина, метилэтиламина, диметилэтиламина, диизопропиламина, пирролидина, дигидроизоиндола, морфолина, N,N-диаллиламина, 4-метилпиперидина, этаноламина, 5-бромдигидроизоиндола,тиоморфолина, цис-2,6-диметилморфолина и этилендиамина. 10. Применение производного камптотецина по п.1 или 2 при получении фармацевтического препарата для лечения рака, где указанный рак выбран из группы, включающей рак легких, рак молочной железы, рак толстой кишки, рак прямой кишки, рак предстательной железы, меланому, рак поджелудочной железы, рак желудка, рак печени, рак головного мозга, рак почек, рак матки, цервикальный рак, рак яичников, рак мочевых путей, гастроинтестинальный рак и лейкемию. 11. Применение по п.10, где указанный рак выбран из группы, включающей рак молочной железы,рак толстой кишки, рак прямой кишки и рак легких. 12. Применение по п.11, где указанный рак представляет собой мелкоклеточный рак легких.
МПК / Метки
МПК: C07D 491/22, A61P 35/00, A61K 31/675, C07F 9/6561
Метки: получения, композиция, применение, камптотецина, фармацевтическая, способ, производное
Код ссылки
<a href="https://eas.patents.su/30-21814-proizvodnoe-kamptotecina-sposob-ego-polucheniya-farmacevticheskaya-kompoziciya-i-ee-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Производное камптотецина, способ его получения, фармацевтическая композиция и ее применение</a>
Липидная композиция, содержащая производное камптотецина (варианты), способ её получения и применение

Номер патента: 6741
Опубликовано: 28.04.2006
Авторы: Чжанг Цзя-Ай, Ахмад Имран, Рахман Аквилар
МПК: A61K 9/127, A61K 9/48, A61K 9/133...
Метки: применение, способ, липидная, производное, содержащая, камптотецина, композиция, варианты, получения
Формула / Реферат:
1. Композиция, содержащая производное камптотецина и по меньшей мере один липид, отличающаяся тем, что в качестве производного камптотецина она содержит соединение, выбранное из SN-38 или химического вещества, находящегося в равновесии с SN-38, при этом приблизительно 70 мас.% или более данного соединения находится в комплексе с частью липида, а указанный липид включает в себя кардиолипин. 2. Композиция по п.1, отличающаяся тем, что комплекс...
Конъюгат, включающий белок и полимер или его производное (варианты), способ его получения, применение конъюгата и содержащая его фармацевтическая композиция

Номер патента: 14103
Опубликовано: 29.10.2010
Авторы: Цандер Норберт, Шиммел Мартин, Хаккет Франк, Айхнер Вольфрам, Краус Эльмар, Лангер Клаус, Орландо Микеле, Конрадт Харальд, Зоммермейер Клаус, Франк Рональд
МПК: A61K 47/48
Метки: полимер, производное, белок, варианты, применение, включающий, композиция, получения, содержащая, способ, конъюгат, конъюгата, фармацевтическая
Формула / Реферат:
1. Способ получения конъюгата, содержащего белок и полимер или его производное, в котором полимер представляет собой гидроксиалкилкрахмал (HAS), предпочтительно гидроксиэтилкрахмал, предпочтительно гидроксиэтилкрахмал, имеющий молекулярную массу от 2 до 200 кДа, предпочтительно от 4 до 130 кДа, более предпочтительно от 4 до 70 кДа, причем способ включает введение в реакцию по меньшей мере одной функциональной группы А полимера или его...
(аза)индольное производное, замещенное по положению 5, содержащая его фармацевтическая композиция, промежуточные соединения и способ их получения

Номер патента: 20373
Опубликовано: 30.10.2014
Авторы: Мауджери Катерина, Мангано Джорджина, Каццолла Никола, Драгоне Патриция, Гарофало Барбара, Колетта Изабелла, Гарроне Беатриче, Ализи Мария Алессандра, Фурлотти Гвидо
МПК: A61P 29/00, A61K 31/404, A61K 31/437...
Метки: композиция, замещенное, положению, фармацевтическая, аза)индольное, получения, соединения, содержащая, способ, промежуточные, производное
Формула / Реферат:
1. (Аза)индольное производное, замещенное по положению 5, формулы (I)в которой X представляет собой атом галогена или метил, трифторметильную, амино-, циано-, гидроксигруппу;Y и Z, которые могут быть одинаковыми или различными, представляют собой атом водорода или галогена либо (C1-C3)алкил, трифторметил, нитро, амино, гидрокси, (C1-C3)алкокси;G1 представляет собой атом азота или группу CH;G2 представляет собой группу CH;G3 представляет собой...
Метансульфонат производного камптотецина, способы его получения и фармацевтическая композиция на его основе

Номер патента: 36
Опубликовано: 26.02.1998
Авторы: Ногути Сигеру, Китаока Хироаки, Камихара Синдзи, Терасава Хирофуми, Канаи Казуаки
МПК: C07D 491/22, A61K 31/47
Метки: метансульфонат, получения, способы, основе, камптотецина, композиция, производного, фармацевтическая
Формула / Реферат:
1. Метансульфонат производного камптотецина формулы ( 1 ) или его гидрат. 2. Фармацевтическая композиция, обладающая противоопухолевой активностью, включающая производное камптотецина, отличающаяся тем, что в качестве указанного производного она включает метансульфонат производного камптотецина формулы (1) или его гидрат, или фармацевтически приемлемую соль. 3. Способ получения метансульфоната производного камптотецина формулы ( 1...
Производное адамантилбензамида, фармацевтическая композиция, включающая его, и его применение

Номер патента: 20496
Опубликовано: 28.11.2014
Авторы: Полисетти Дхарма Рао, Гупта Супарна, Эбдруп Серен
МПК: A61K 31/44, A01N 43/42
Метки: включающая, композиция, его, адамантилбензамида, производное, применение, фармацевтическая
Формула / Реферат:
1. Соединение, которое представляет собой 3-(5-хлорпиридин-2-илокси)-N-(5-гидроксиадамантан-2-ил)бензамид или его фармацевтически приемлемую соль, где по меньшей мере 90% присутствующего соединения имеет форму E-изомера.2. Соединение по п.1, где по меньшей мере 95% присутствующего соединения имеет форму E-изомера.3. Соединение по п.1, где по меньшей мере 98% присутствующего соединения имеет форму E-изомера.4. Применение соединения по любому из...
Предыдущий патент: Ударный плиометрический эспандер (варианты)
Следующий патент: Спасательное транспортное средство
Случайный патент: Способ получения пищевого продукта и пищевой продукт