Производные 4,7-дигидропиразоло[1,5-a]пиразин-6-иламина, используемые в качестве ингибиторов бета-секретазы (васе)
Номер патента: 21723
Опубликовано: 31.08.2015
Авторы: Тресадерн Гэри Джон, Трабанко-Суарес Андрес Авелино, Дельгадо-Хименес Франсиска





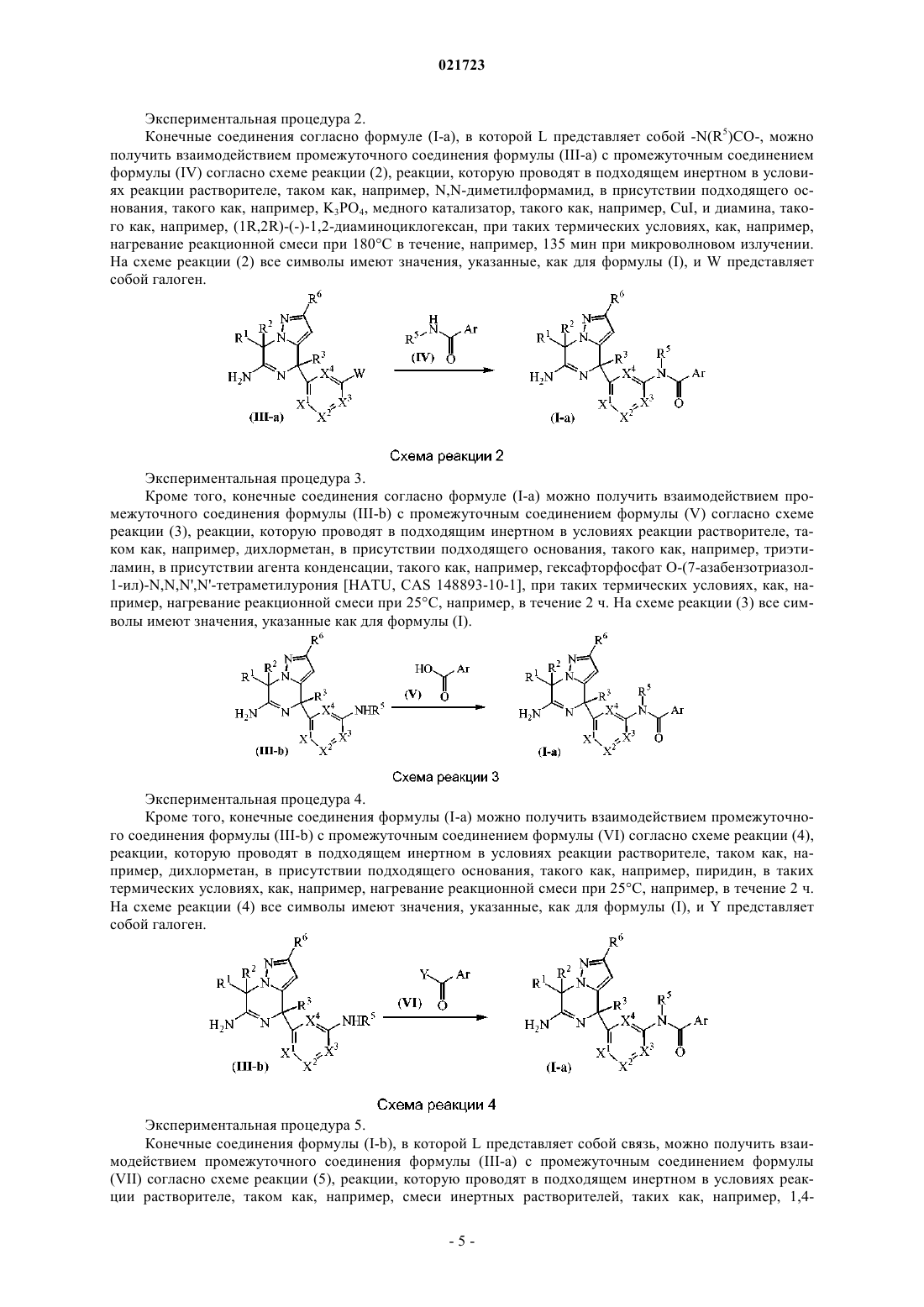



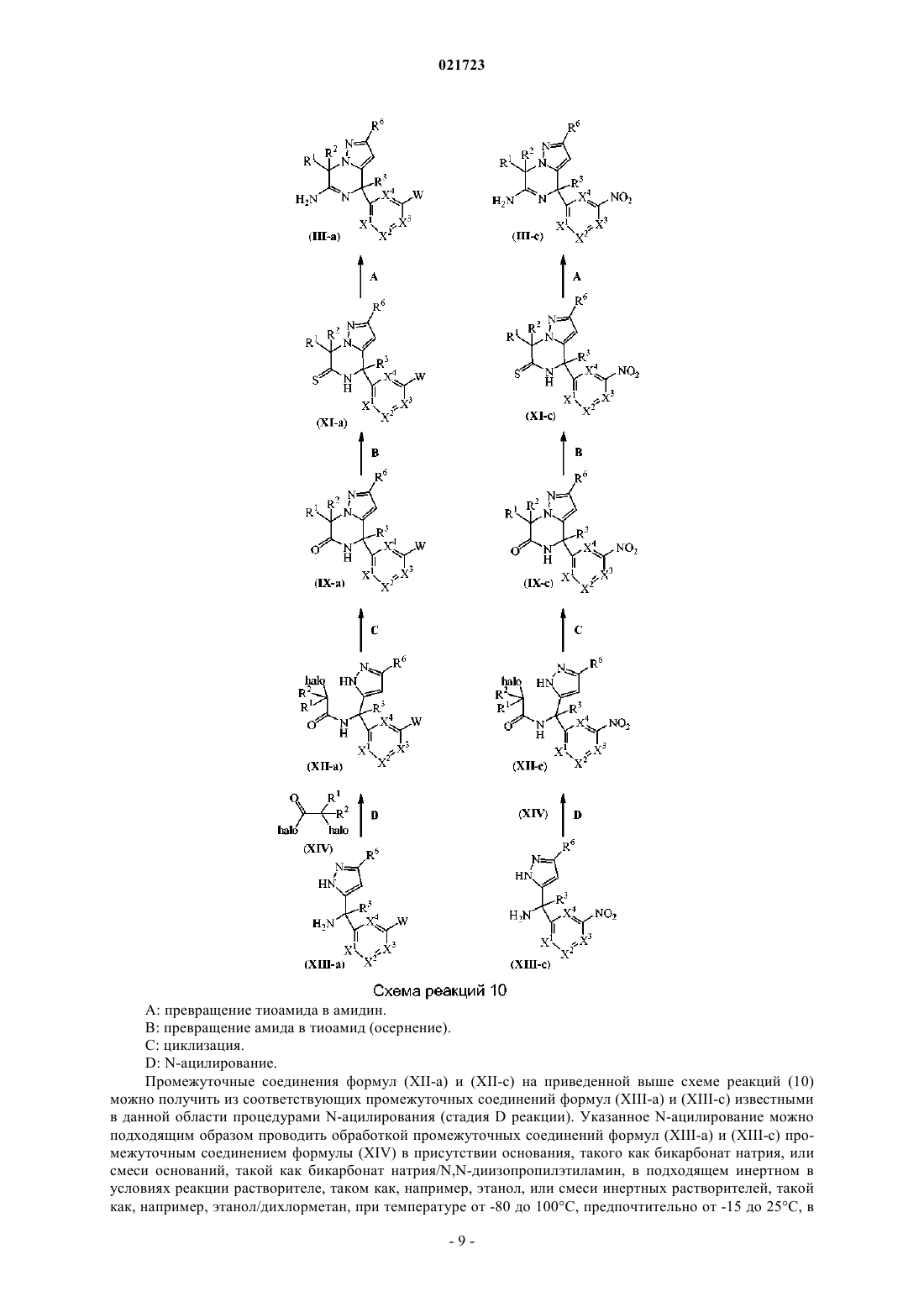



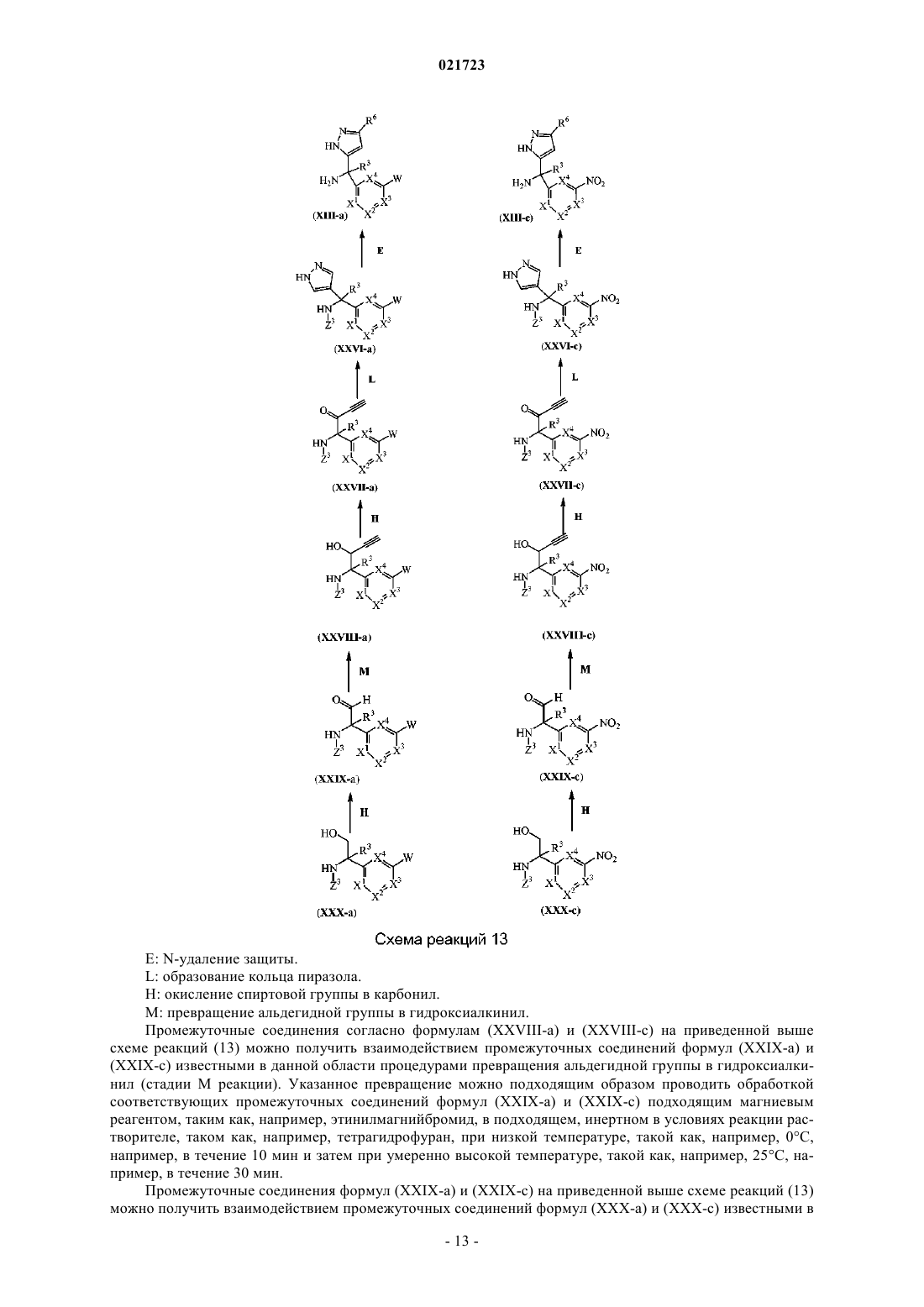
















Формула / Реферат
1. Соединение формулы (I)

или его таутомерная или стереоизомерная форма, у которого
R1 и R2 представляют собой водород;
R3 выбран из группы, состоящей из C1-3алкила и С3-6циклоалкила;
X1 и X3 представляют собой СН или F;
X2 и X4 представляют собой СН;
L представляет собой связь или -N(R5)CO-, где R5 представляет собой водород или C1-3алкил;
R6 представляет собой водород или трифторметил;
Ar представляет собой гомоарил или гетероарил;
гомоарил представляет собой фенил или фенил, замещенный одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена, циано, C1-3алкила, C1-3алкилокси, моно- и полигалогенС1-3алкила и моно- и полигалогенС1-3алкилокси;
гетероарил выбран из группы, состоящей из пиридила, пиримидила, пиразила, необязательно замещенных одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена, циано, C1-3алкила, C1-3алкилокси, моно- и полигалогенС1-3алкила и моно- и полигалогенС1-3алкилокси,
или его аддитивная соль или сольват.
2. Соединение по п.1, у которого
L представляет собой связь или -N(R5)CO-, где R5 представляет собой водород;
Ar представляет собой гомоарил или гетероарил;
гомоарил представляет собой фенил или фенил, замещенный одним или двумя заместителями, выбранными из группы, состоящей из галогена, циано, C1-3алкила и C1-3алкилокси;
гетероарил выбран из группы, состоящей из пиридила, пиримидила и пиразила, каждый из которых необязательно замещен одним или двумя заместителями, выбранными из группы, состоящей из галогена, циано, C1-3алкила и C1-3алкилокси;
или его аддитивная соль или сольват.
3. Соединение по п.1, у которого
X1, X2, X3, X4 представляют собой СН;
L представляет собой связь или -N(R5)CO-, где R5 представляет собой водород;
Ar представляет собой гомоарил или гетероарил;
гомоарил представляет собой фенил, замещенный хлором;
гетероарил выбран из группы, состоящей из пиридила и пиримидила, каждый из которых необязательно замещен одним или двумя заместителями, выбранными из группы, состоящей из хлора, фтора, циано, метила и метокси;
или его аддитивная соль или сольват.
4. Соединение по п.1, у которого атом углерода, замещенный R3, имеет R-конфигурацию.
5. Фармацевтическая композиция для лечения болезни Альцгеймера, умеренного когнитивного расстройства, старения, деменции, деменции с тельцами Леви, синдрома Дауна, деменции, связанной с инсультом, деменции, связанной с болезнью Паркинсона, и деменции, связанной с β-амилоидом, содержащая терапевтически эффективное количество соединения по любому из пп.1-4 и фармацевтически приемлемый носитель.
6. Способ получения фармацевтической композиции по п.5, отличающийся тем, что фармацевтически приемлемый носитель смешивают с терапевтически эффективным количеством соединения по любому из пп.1-4.
7. Применение соединения по любому из пп.1-4 для лечения, предупреждения или профилактики болезни Альцгеймера (AD), умеренного когнитивного расстройства, старения, деменции, деменции с тельцами Леви, синдрома Дауна, деменции, связанной с инсультом, деменции, связанной с болезнью Паркинсона, или деменции, связанной с β-амилоидом.
8. Способ лечения нарушения, выбранного из группы, состоящей из болезни Альцгеймера, умеренного когнитивного расстройства, старения, деменции, деменции с тельцами Леви, синдрома Дауна, деменции, связанной с инсультом, деменции, связанной с болезнью Паркинсона, и деменции, связанной с β-амилоидом, содержащий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп.1-4.
Текст
Настоящее изобретение относится к новым производным 4,7-дигидропиразоло[1,5-а]пиразин-6-иламина формулы I, где значения радикалов раскрыты в формуле изобретения используемым в качестве ингибиторов -секретазы, известной также в качестве фермента,расщепляющего -сайт амилоида, ВАСЕ, BACE1, Asp2 или мемапсина 2. Изобретение относится также к фармацевтическим композициям, содержащим такие соединения, к способам получения таких соединений и композиций и к применению таких соединений и композиций для предотвращения и лечения нарушений, в которых принимает участие -секретаза, таких как болезнь Альцгеймера (AD), умеренное когнитивное расстройство, старение, деменция, деменция с тельцами Леви, синдром Дауна, деменция, связанная с инсультом, деменция, связанная с болезнью Паркинсона, или деменция, связанная с -амилоидом.(71)(73) Заявитель и патентовладелец: ЯНССЕН ФАРМАЦЕВТИКА НВ (BE) Область техники, к которой относится изобретение Настоящее изобретение относится к новым производным 4,7-дигидропиразоло[1,5-а]пиразин-6-иламина, используемым в качестве ингибиторов -секретазы, известной также в качестве фермента, расщепляющего -сайт амилоида, ВАСЕ, ВАСЕ 1, Asp2 или мемапсина 2. Изобретение относится также к фармацевтическим композициям, содержащим такие соединения, к способам получения таких соединений и композиций и к применению таких соединений и композиций для предотвращения и лечения нарушений, в которых принимает участие -секретаза, таких как болезнь Альцгеймера (AD), умеренное когнитивное расстройство, старение, деменция, деменция с тельцами Леви, синдром Дауна, деменция, связанная с инсультом, деменция, связанная с болезнью Паркинсона, или деменция, связанная с -амилоидом. Уровень техники Болезнь Альцгеймера (AD) является нейродегенеративным заболеванием, связанным со старением. Пациенты с болезнью Альцгеймера страдают нарушением познавательной способности и потерей памяти, а также поведенческими проблемами, такими как тревога (страх). Более 90% пациентов, пораженныхAD, имеют спорадическую форму нарушения, в то время как менее 10% случаев таких нарушений являются семейными или наследственными. В Соединенных Штатах примерно 1 из 10 человек в возрасте 65 лет имеет AD, тогда как в возрасте 85 лет 1 человек из каждых двух лиц поражен AD. Средняя продолжительность жизни от первоначального диагноза составляет 7-10 лет, и пациентам с AD требуется тщательный уход для помощи в жизнеобеспечении пациента либо лицом, осуществляющим уход за больным, что очень дорого, либо членами семьи. В связи с ростом числа пожилых людей среди жителей AD является увеличивающейся медицинской проблемой. Доступные в настоящее время терапии для AD только лечат симптомы заболевания и включают в себя применение ингибиторов ацетилхолинэстеразы для улучшения когнитивных свойств, а также транквилизаторов и антипсихотических средств для ослабления поведенческих проблем, связанных с этим недугом. Отличительными патологическими признаками мозга пациентов с AD являются нейрофибриллярные клубки, которые генерируются гиперфосфорилированием -белка, и амилоидные бляшки, которые образуются агрегированием пептида -амилоида 1-42 (А 1-42). А 1-42 образует олигомеры и затем фибриллы и в конце концов амилоидные бляшки. Считается, что олигомеры и фибриллы являются особенно нейротоксичными и могут вызвать большинство неврологических повреждений, связанных с AD. Агенты, которые препятствуют образованию А 1-42, имеют потенциал быть модифицирующими заболевание агентами для лечения AD. А 1-42 образуется из амилоидного белка-предшественника (АРР),состоящего из 770 аминокислот. N-конец А 1-42 расщепляется -секретазой (ВАСЕ), а затем -секретаза расщепляет С-конец. Помимо А 1-42 -секретаза выделяет также А 1-40, которая является основным продуктом расщепления, а также А 1-38 и А 1-43. Эти формы А могут также агрегироваться с образованием олигомеров и фибрилл. Таким образом, можно предположить, что ингибиторы ВАСЕ предотвращают образование А 1-42, а также А 1-40, А 1-38 и А 1-43 и могут быть потенциальными терапевтическими агентами при лечении AD. Сущность изобретения Настоящее изобретение относится к соединению формулы (I) или его таутомерной или стереоизомерной форме, в которой R1 и R2 независимо выбраны из группы, состоящей из водорода, фтора, циано, C1-3 алкила, моно- и полигалогенС 1-3 алкила и С 3-6 циклоалкила, илиX1, X2, X3, X4 независимо представляют собой C(R4) или N, при условии, что не более чем два из них представляют собой N; каждый R4 выбран из группы, состоящей из водорода, галогена, C1-3 алкила,моно- и полигалогенС 1-3 алкила, циано, C1-3 алкилокси, моно- и полигалогенС 1-3 алкилокси;L представляет собой связь или -N(R5)CO-, где R5 представляет собой водород или C1-3 алкил;R6 представляет собой водород или трифторметил;Ar представляет собой гомоарил или гетероарил; гомоарил представляет собой фенил или фенил, замещенный одним, двумя или тремя заместителя-1 021723 ми, выбранными из группы, состоящей из галогена, циано, C1-3 алкила, C1-3 алкилокси, моно- и полигалогенС 1-3 алкила и моно- и полигалогенС 1-3 алкилокси; гетероарил выбран из группы, состоящей из пиридила, пиримидила, пиразила, пиридазила, фуранила, тиенила, пирролила, пиразолила, имидазолила, триазолила, тиазолила, тиадиазолила, оксазолила и оксадиазолила, каждый из которых необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена, циано, C1-3 алкила, C1-3 алкилокси, моно- и полигалогенС 1-3 алкила и моно- и полигалогенС 1-3 алкилокси,или его аддитивной соли или сольвату. Иллюстрацией изобретения является фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и любое из соединений, описанных выше. Иллюстрацией изобретения является фармацевтическая композиция, изготовленная смешиванием любого из соединений, описанных выше, и фармацевтически приемлемого носителя. Иллюстрацией изобретения является способ изготовления фармацевтической композиции, содержащий смешивание любого из соединений, описанных выше, и фармацевтически приемлемого носителя. Репрезентацией изобретения являются способы лечения нарушения, опосредуемого ферментом секретазой, включающие в себя введение субъекту, нуждающемуся в этом, терапевтически эффективного количества любого из описанных выше соединений или фармацевтических композиций. Следующей репрезентацией изобретения являются способы ингибирования фермента -секретазы,содержащие введение субъекту, нуждающемуся в этом, терапевтически эффективного количества любого из описанных выше соединений или фармацевтических композиций. Примером изобретения является способ лечения нарушения, выбранного из группы, состоящей из болезни Альцгеймера, умеренного когнитивного расстройства, старения, деменции, деменции с тельцами Леви, синдрома Дауна, деменции, связанной с инсультом, деменции, связанной с болезнью Паркинсона,и деменции, связанной с -амилоидом, предпочтительно болезни Альцгеймера, включающий в себя введение субъекту, нуждающемуся в этом, терапевтически эффективного количества любого из описанных выше соединений или фармацевтических композиций. Другим примером изобретения является любое из описанных выше соединений для применения при лечении (а) болезни Альцгеймера, (b) умеренного когнитивного расстройства, (с) старения, (d) деменции, (е) деменции с тельцами Леви, (f) синдрома Дауна, (g) деменции, связанной с инсультом, (h) деменции, связанной с болезнью Паркинсона, и (i) деменции, связанной с -амилоидом, у субъекта, нуждающегося в этом. Подробное описание изобретения Настоящее изобретение относится к соединениям формулы (I), в которой символы имеют значения,указанные выше, и их фармацевтически приемлемым солям и сольватам. Соединения формулы (I) являются ингибиторами фермента -секретазы (известной также как фермент расщепления -сайта, ВАСЕ,ВАСЕ 1, Asp2 или мемапсин 2) и являются применимыми при лечении болезни Альцгеймера, умеренного когнитивного нарушения, старения, деменции, деменции, связанной с инсультом, деменции с тельцами Леви, синдрома Дауна, деменции, связанной с болезнью Паркинсона, и деменции, связанной с амилоидом, предпочтительно болезни Альцгеймера, умеренного когнитивного нарушения или деменции,более предпочтительно болезни Альцгеймера. В варианте осуществления настоящего изобретения предложено соединение, у которогоR1 и R2 независимо выбраны из водорода и C1-3 алкила;X1, X2, X3, X4 независимо представляют собой C(R4), где каждый R4 выбран из водорода и галогена;L представляет собой связь или -N(R5)CO-, где R5 представляет собой водород;Ar представляет собой гомоарил или гетероарил; гомоарил представляет собой фенил или фенил, замещенный одним или двумя заместителями, выбранными из группы, состоящей из галогена, циано, C1-3 алкила и C1-3 алкилокси; гетероарил выбран из группы, состоящей из пиридила, пиримидила и пиразила, каждый из которых необязательно замещен одним или двумя заместителями, выбранными из группы, состоящей из галогена,циано, C1-3 алкила и C1-3 алкилокси; или его аддитивная соль или сольват. В другом варианте осуществления настоящего изобретения, предложено соединение, у которогоR1 и R2 независимо выбраны из водорода;X1, X2, X3, X4 независимо представляют собой СН;L представляет собой связь или -N(R5)CO-, где R5 представляет собой водород;Ar представляет собой гомоарил или гетероарил; гомоарил представляет собой фенил, замещенный хлором; гетероарил выбран из группы, состоящей из пиридила и пиримидила, каждый из которых необязательно замещен одним или двумя заместителями, выбранными из группы, состоящей из хлора, фтора,циано, метила и метокси; или его аддитивная соль или сольват.-2 021723 В другом варианте осуществления атом углерода, замещенный R3, имеет R-конфигурацию. В другом варианте осуществления настоящего изобретения предложено соединение, у которого R1 2 и R представляют собой водород;X1 представляет собой СН или CF и X2, X3, X4 представляют собой СН;Ar представляет собой пиридинил, замещенный одним или двумя атомами галогена, или пиразинил,замещенный метокси,или его соль или сольват. Определения"C1-3 алкил" будет означать неразветвленную или разветвленную насыщенную алкильную группу,имеющую 1, 2 или 3 атома углерода, например метил, этил, 1-пропил и 2-пропил;"C1-3 алкилокси" будет означать радикал простого эфира, у которого C1-3 алкил имеет значения, указанные выше;"моно- и полигалогенС 1-3 алкил" будет означать C1-3 алкил, который имеет значения, указанные выше, и замещен 1, 2, 3 или в случае, когда возможно, большим числом атомов галогена, которые имеют значения, указанные выше,"моно- и полигалогенС 1-3 алкилокси" будет означать радикал простого эфира, у которого моно- и полигалогенС 1-3 алкил имеет значения, указанные выше;"С 3-6 циклоалкандиил" будет означать двухвалентный радикал, такой как циклопропандиил, циклобутандиил, циклопентандиил и циклогександиил. Термин "субъект", применяемый в контексте, относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, который является или являлся объектом лечения, наблюдения или эксперимента. Термин "терапевтически эффективное количество", применяемый в контексте, означает, что количество активного соединения или фармацевтического агента, которое вызывает биологический или лекарственный ответ в системе ткани, организме животного или человека, которого добивается исследователь, ветеринар, врач или другой клиницист, и которое включает облегчение симптомов подвергаемой лечению болезни или нарушения. Предполагается, что в данном контексте термин "композиция" включает в себя продукт, содержащий указанные ингредиенты в указанных количествах, а также любой продукт, который получают непосредственно или опосредованно из комбинаций указанных ингредиентов в указанных количествах. Следует иметь в виду, что некоторые из соединений формулы (I) и их аддитивные соли, гидраты и сольваты могут содержать один или несколько центров хиральности и могут существовать в виде стереоизомерных форм. Термин "стереоизомерные формы", используемый в контексте ранее или позже, означает все возможные стереоизомерные формы, которые могут иметь соединения согласно формуле (I) и их аддитивные соли. Если не оговорено или не указано иное, химическое определение соединений означает смесь всех возможных стереохимически изомерных форм, причем указанные смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры, а также каждую из индивидуальных изомерных форм согласно формуле (I) и их солей, сольватов, по существу, индивидуальных, т.е. связанных менее чем с 10%, предпочтительно менее чем с 5%, в частности менее чем с 2% и наиболее предпочтительно менее чем с 1% других изомеров. Когда соединения согласно данному изобретению имеют по крайней мере один хиральный центр,они могут соответственно существовать в виде энантиомеров. Когда соединения обладают двумя или более хиральными центрами, они могут дополнительно существовать в виде диастереомеров. Должно быть понятно, что все такие изомеры и их смеси включены в объем настоящего изобретения. Когда соединение присутствует в виде энантиомера, энантиомер предпочтительно присутствует при энантиомерном избытке, который превышает или равен приблизительно 80%, более предпочтительно при энантиомерном избытке, который превышает или равен приблизительно 90%, еще более предпочтительно при энантиомерном избытке, который превышает или равен приблизительно 95%, еще более предпочтительно при энантиомерном избытке, который превышает или равен приблизительно 98%, наиболее предпочтительно при энантиомерном избытке, который превышает или равен приблизительно 99%. Аналогично этому, когда соединение присутствует в виде диастереомера, диастереомер присутствует при диастереомерном избытке, который превышает или равен приблизительно 80%, более предпочтительно при диастереомерном избытке, который превышает или равен приблизительно 90%, еще более предпочтительно при диастереомерном избытке, который превышает или равен приблизительно 95%, еще более предпочтительно при диастереомерном избытке, который превышает или равен приблизительно 98%, наиболее предпочтительно при диастереомерном избытке, который превышает или равен приблизительно 99%. Кроме того, некоторые из кристаллических форм соединений настоящего изобретения могут существовать в виде полиморфов, и предполагается, что как таковые они включены в настоящее изобретение. Кроме того, некоторые из соединений настоящего изобретения могут образовывать сольваты с водой(т.е. гидраты) или обычными органическими растворителями, и предполагается, что такие сольваты также включены в объем данного изобретения. Для применения в медицине соли соединений данного изобретения относятся к нетоксичным "фармацевтически приемлемым солям". Другие соли, однако, могут быть применимыми при получении соединений согласно данному изобретению или их фармацевтически приемлемых солей. Подходящие фармацевтически приемлемые соли соединений включают в себя аддитивные соли кислот, которые можно, например, получить смешиванием раствора соединения с раствором фармацевтически приемлемой кислоты, такой как хлористо-водородная кислота, серная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, бензойная кислота, лимонная кислота, винная кислота,угольная кислота или фосфорная кислота. Кроме того, когда соединения по изобретению содержат кислотный остаток, их подходящие фармацевтически приемлемые соли могут включать в себя соли щелочных металлов, например соли натрия или калия, соли щелочно-земельных металлов, например соли кальция или магния, и соли, образованные с подходящими органическими лигандами, например соли четвертичного аммония. Репрезентативные кислоты, которые можно применять при получении фармацевтически приемлемых солей, включают, но не ограничиваются перечисленным, уксусную кислоту, 2,2-дихлоруксусную кислоту, ацилированные аминокислоты, адипиновую кислоту, альгиновую кислоту, аскорбиновую кислоту, L-аспарагиновую кислоту, бензолсульфоновую кислоту, бензойную кислоту, 4 ацетамидобензойную кислоту, (+)-камфорную кислоту, камфорасульфоновую кислоту, каприновую кислоту, капроновую кислоту, каприловую кислоту, коричную кислоту, лимонную кислоту, цикламовую кислоту, этан-1,2-дисульфоновую кислоту, этансульфоновую кислоту, 2-гидроксиэтансульфоновую кислоту, муравьиную кислоту, фумаровую кислоту, галактаровую кислоту, гентизиновую кислоту, глюкогептоновую кислоту, D-глюконовую кислоту, D-глюкуроновую кислоту, L-глутаминовую кислоту, бетаоксоглутаровую кислоту, гликолевую кислоту, гиппуровую кислоту, бромистоводородную кислоту, хлористо-водородную кислоту, (+)-L-молочную кислоту, -DL-молочную кислоту, лактобионовую кислоту, малеиновую кислоту, (-)-L-яблочную кислоту, малоновую кислоту, -DL-миндальную кислоту, метансульфоновую кислоту, нафталин-2-сульфоновую кислоту, нафталин-1,5-дисульфоновую кислоту, 1 гидрокси-2-нафтойную кислоту, никотиновую кислоту, азотную кислоту, олеиновую кислоту, оротовую кислоту, щавелевую кислоту, пальмитиновую кислоту, памовую кислоту, фосфорную кислоту, Lпироглутаминовую кислоту, салициловую кислоту, 4-аминосалициловую кислоту, себациновую кислоту,стеариновую кислоту, янтарную кислоту, серную кислоту, дубильную кислоту, (+)-L-винную кислоту,тиоциановую кислоту, п-толуолсульфоновую кислоту, трифторметилсульфоновую кислоту и ундециленовую кислоту. Репрезентативные основания, которые можно применять при получении фармацевтически приемлемых солей, включают, но не ограничиваются перечисленным, аммиак, L-аргинин, бенетамин, бензатин, гидроксид кальция, холин, диметилэтаноламин, диэтаноламин, диэтиламин, 2(диэтиламино)этанол, этаноламин, этилендиамин, N-метилглюкамин, гидрабамин, 1 Н-имидазол, Lлизин, гидроксид магния, 4-(2-гидроксиэтил)морфолин, пиперазин, гидроксид калия, 1-(2 гидроксиэтил)пирролидин, вторичный амин, гидроксид натрия, триэтаноламин, трометамин и гидроксид цинка. Химические названия соединений настоящего изобретения создавали согласно правилам номенклатуры, принятым Chemical Abstpacts Service. Соединений согласно формуле (I) могут также существовать в их таутомерной форме. Предполагается, что такие формы, хотя они и не указаны непосредственно в приведенной выше формуле, включены в объем настоящего изобретения. А. Получение конечных соединений. Экспериментальная процедура 1. Конечные соединения формулы согласно формуле (I) можно получить взаимодействием промежуточного соединения формулы (II) с подходящим источником аммиака, таким как, например, хлорид аммония или водный раствор аммиака, согласно схеме реакции (1), реакции, которую проводят в подходящем инертном в условиях реакции растворителе, таком как, например, вода или метанол, при таких термических условиях, как, например, нагревание реакционной смеси при 60 С, например, в течение 6 ч. На схеме реакции (1) все символы имеют значения, указанные, как для формулы (I). Экспериментальная процедура 2. Конечные соединения согласно формуле (I-a), в которой L представляет собой -N(R5)CO-, можно получить взаимодействием промежуточного соединения формулы (III-а) с промежуточным соединением формулы (IV) согласно схеме реакции (2), реакции, которую проводят в подходящем инертном в условиях реакции растворителе, таком как, например, N,N-диметилформамид, в присутствии подходящего основания, такого как, например, K3PO4, медного катализатор, такого как, например, CuI, и диамина, такого как, например, (1R,2R)-(-)-1,2-диаминоциклогексан, при таких термических условиях, как, например,нагревание реакционной смеси при 180 С в течение, например, 135 мин при микроволновом излучении. На схеме реакции (2) все символы имеют значения, указанные, как для формулы (I), и W представляет собой галоген. Экспериментальная процедура 3. Кроме того, конечные соединения согласно формуле (I-a) можно получить взаимодействием промежуточного соединения формулы (III-b) с промежуточным соединением формулы (V) согласно схеме реакции (3), реакции, которую проводят в подходящим инертном в условиях реакции растворителе, таком как, например, дихлорметан, в присутствии подходящего основания, такого как, например, триэтиламин, в присутствии агента конденсации, такого как, например, гексафторфосфат О-(7-азабензотриазол 1-ил)-N,N,N',N'-тетраметилурония [HATU, CAS 148893-10-1], при таких термических условиях, как, например, нагревание реакционной смеси при 25 С, например, в течение 2 ч. На схеме реакции (3) все символы имеют значения, указанные как для формулы (I). Экспериментальная процедура 4. Кроме того, конечные соединения формулы (I-a) можно получить взаимодействием промежуточного соединения формулы (III-b) с промежуточным соединением формулы (VI) согласно схеме реакции (4),реакции, которую проводят в подходящем инертном в условиях реакции растворителе, таком как, например, дихлорметан, в присутствии подходящего основания, такого как, например, пиридин, в таких термических условиях, как, например, нагревание реакционной смеси при 25 С, например, в течение 2 ч. На схеме реакции (4) все символы имеют значения, указанные, как для формулы (I), и Y представляет собой галоген. Экспериментальная процедура 5. Конечные соединения формулы (I-b), в которой L представляет собой связь, можно получить взаимодействием промежуточного соединения формулы (III-а) с промежуточным соединением формулы(VII) согласно схеме реакции (5), реакции, которую проводят в подходящем инертном в условиях реакции растворителе, таком как, например, смеси инертных растворителей, таких как, например, 1,4-5 021723 диоксан/этанол, в присутствии подходящего основания, такого как, например, водный K3CO3, катализатора, комплексного соединения Pd, такого как, например, тетракис(трифенилфосфин)палладий(0) [CAS 14221-01-3], при таких термических условиях, как, например, нагревание реакционной смеси при 80 С,например, в течение 20 ч или, например, нагревание реакционной смеси при 150 С в течение от 10 до 30 мин в условиях микроволнового излучения. В схеме реакции (5) все символы имеют значения, указанные, как для формулы (I), и W представляет собой галоген. R7 и R8 могут быть водородом или алкилом или могут быть взяты вместе с образованием, например, двухвалентного радикала формулы -СН 2 СН 2-,-СН 2 СН 2 СН 2- или -C(СН 3)2C(СН 3)2-. Ряд промежуточных соединений и исходных веществ в предыдущих получениях являются известными соединениями, которые можно получить согласно известным в данной области методикам получения указанных или аналогичных соединений, и некоторые промежуточные соединения являются новыми соединениями. Ряд таких способов получения будет описан ниже более подробно. В. Получение промежуточных соединений. Экспериментальная процедура 6. Промежуточные продукты согласно формуле (II) можно получить взаимодействием промежуточного соединения формулы (VIII) с подходящим, являющимся донором серы реагентом для синтеза тиоамидов, таким как, например, пентасульфид фосфора или 2,4-бис-(4-метоксифенил)-1,3-дитиа-2,4 дифосфетан-2,4-дисульфид [реагент Лавессона, CAS 19172-47-5], согласно схеме реакции (6), реакции,которую проводят в инертном в условиях реакции растворителе, таком как, например, тетрагидрофуран или толуол, в присутствии подходящего основания, такого как, например, пиридин, при таких термических условиях, как, например, нагревание реакционной смеси при 90 С, например, в течение 18 ч. На схеме реакции (6) все символы имеют значения, указанные, как для формулы (I). Экспериментальная процедура 7. Промежуточные соединения формулы (VIII), в которой L представляет собой связь, можно получить взаимодействием промежуточного соединения формулы (IX-а) с промежуточным соединением формулы (VII) согласно схеме реакции (7), реакции, которую проводят в подходящей смеси инертных растворителей, такой как, например, 1,4-диоксан/вода, в присутствии подходящего основания, такого как, например, водный раствор Na2CO3, катализатора, комплексного соединения Pd, такого как, например, тетракис(трифенилфосфин)палладий(0) [CAS 14221-01-3], при таких термических условиях, как,например, нагревание реакционной смеси при 80 С, например, в течение 20 ч, или, например, нагревание реакционной смеси при 150 С, например, в течение 15 мин в условиях микроволнового излучения. На схеме реакции (7) все символы имеют значения, указанные, как для формулы (I), и W представляет собой галоген. R7 и R8 могут быть водородом или алкилом или могут быть взяты вместе с образованием, например, двухвалентного радикала формулы -СН 2 СН 2-, -СН 2 СН 2 СН 2- или -С (СН 3)2 С (СН 3)2-. Экспериментальная процедура 8. Промежуточные соединения согласно формуле (III-b) можно получить из соответствующих промежуточных соединений формулы (III-а) известными в данной области процедурами сочетания типа Бухвальд-Хартвига согласно схеме реакции (8). Указанное сочетание можно проводить обработкой промежуточных соединений формулы (III-а) промежуточным соединением формулы (X) в подходящем инертном в условиях реакции растворителе, таком как, например, этанол, или смеси инертных растворителей,такой как, например, 1,2-диметоксиэтан/вода/этанол, в присутствии подходящего основания, такого как,например, водный K3PO4 или Cs2CO3, катализатора, комплексного соединения Pd, такого как, например,[1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) [CAS 72287-26-4] или диацетат трансбис(дициклогексиламин)палладия [DAPCy, CAS 628339-96-8] в термических условиях, таких как, например, нагревание реакционной смеси при 80 С, например, в течение 20 ч, или, например, нагревание реакционной смеси при 130 С, например, в течение 10 мин в условиях микроволнового излучения. На схеме реакции (8) все символы имеют значения, указанные, как для формулы (I), и W представляет собой галоген. R5 представляет собой водород или C1-3 алкил. Экспериментальная процедура 9. Кроме того, промежуточные соединения согласно формуле (III-b), в которой R5 представляет собой водород, можно получить из соответствующих промежуточных соединений формулы (III-C)известными в данной области процедурами восстановления нитрогруппы в аминогруппу по схеме реакции (9). Указанное восстановление можно преимущественно проводить известными в данной области процедурами каталитического гидрирования. Например, указанное восстановление можно проводить перемешиванием реагентов в атмосфере водорода и в присутствии подходящего катализатора, такого как, например, палладий на угле, платина на угле, никель Ренея, и подобных катализаторов. Подходящими растворителями являются, например, вода, спирты, например метанол, этанол и тому подобное, сложные эфиры, например этилацетат и тому подобное. Для того чтобы повысить скорость указанной реакции восстановления,можно подходящим образом повысить температуру и/или давление реакционной смеси. Нежелательное дальнейшее гидрирование некоторых функциональных групп в реагентах и продуктах реакций можно предотвратить добавлением каталитического яда, такого как, например, тиофен и тому подобное, к реакционной смеси. На схеме реакции (9) все символы имеют значения, указанные, как для формулы (I). Экспериментальная процедура 10. Промежуточные соединения формул (III-а) и (III-с) обычно получают по стадиям реакций, показанным ниже на схемах реакций (10), (11), (12) и (13). Производные амидинов формул (III-а) и (III-с) на схеме реакции (10) можно преимущественно получить из соответствующих производных тиоамидов формул (XI-а) и (XI-с)известными в данной области процедурами превращения тиоамида в амидин (стадия А реакции). Указанное превращение можно подходящим образом проводить обработкой промежуточных соединений формул (XI-а) и (XI-с) источником аммиака, таким как, например, хлорид аммония или водный раствор аммиака, в подходящем, инертном в условиях реакции растворителе, таком как, например, вода или метанол и тому подобное, в таких термических условиях, как, например, нагревание реакционной смеси при 60 С, например, в течение 6 ч. Производные тиоамидов формул (XI-а) и (XI-с) на схеме реакции (10) можно получить из производных амидов формулы (IX-а) и (IX-с) известными в данной области процедурами осернения (стадия В реакции). Указанное превращение можно подходящим образом проводить обработкой промежуточных соединений формул (IX-а) и (IX-c) агентом осернения, таким как, например, пентасульфид фосфора или 2,4-бис-(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфид [реагент Лавессона, CAS 19172-475], в инертном в условиях реакции растворителе, таком как, например, тетрагидрофуран или 1,4-диоксан и тому подобное, при таких термических условиях, как, например, нагревание реакционной смеси при 50 С, например, в течение 50 мин. Производные амидов формул (IX-а) и (IX-с) на схеме реакции (10) можно получить из соответствующих промежуточных соединений формул (XII-a) и (XII-c) известными в данной области процедурами циклизации (стадия с реакции). Указанную циклизацию можно подходящим образом проводить обработкой промежуточных соединений формул (XII-a) и (XII-c) подходящим основанием, таким как гидрид натрия, в подходящем инертном в условиях реакции растворителе, таком как, например, тетрагидрофуран и тому подобное, при температуре от -80 до 100 С, предпочтительно от -15 до 25 С в течение от 30 мин до 100 ч, предпочтительно от 1 до 24 ч.D: N-ацилирование. Промежуточные соединения формул (XII-а) и (XII-c) на приведенной выше схеме реакций (10) можно получить из соответствующих промежуточных соединений формул (XIII-а) и (XIII-с) известными в данной области процедурами N-ацилирования (стадия D реакции). Указанное N-ацилирование можно подходящим образом проводить обработкой промежуточных соединений формул (XIII-а) и (XIII-с) промежуточным соединением формулы (XIV) в присутствии основания, такого как бикарбонат натрия, или смеси оснований, такой как бикарбонат натрия/N,N-диизопропилэтиламин, в подходящем инертном в условиях реакции растворителе, таком как, например, этанол, или смеси инертных растворителей, такой как, например, этанол/дихлорметан, при температуре от -80 до 100 С, предпочтительно от -15 до 25 С, в течение от 30 мин до 100 ч, предпочтительно от 1 до 24 ч. Промежуточные соединения формул (XIII-а) и (XIII-с) на схеме реакций (11) можно получить из соответствующих промежуточных соединений формул (XV-a) и (XV-c), в которых Z1 представляет собой подходящую защитную группу системы пиразола, такую как, например, диметилсульфамоильная группа,и Z2 представляет собой подходящую защитную группу аминов, такую как, например, третбутансульфинильная группа, известными в данной области процедурами N-снятия защиты (стадия Е реакции). Указанное N-снятие защиты можно подходящим образом проводить обработкой соответствующих промежуточных соединений формул (XV-а) и (XV-с) подходящим кислотным агентом, таким как,например, хлористо-водородная кислота, в подходящем инертном растворителе, таком как, например,1,4-диоксан, при умеренно высокой температуре, такой как, например, 25 С, например, в течение 1 ч. Промежуточные соединения согласно формулам (XV-a) и (XV-c) на схеме реакции (11) можно получить взаимодействием промежуточных соединений формул (XVII-a) и (XVII-c) известными в данной области процедурами превращения имина в алкиламин (стадия F реакции). Указанное превращение можно подходящим образом проводить обработкой соответствующих промежуточных соединений формул (XVII-a) и (XVII-c) промежуточным соединением формулы (XVI), в которой Y представляет собой галоген, в подходящем, инертном в условиях реакции растворителе, таком как, например, тетрагидрофуран, при низкой температуре, такой как, например, 0 С, например, в течение 2 ч.G: превращение кетона в имин. Н: окисление спиртовой группы в карбонил.I: орто-литиирование-алкилирование. Промежуточные соединения согласно формулам (XVII-a) и (XVII-c) на приведенной выше схеме реакций (11) можно получить взаимодействием промежуточных соединений формул (XIX-а) и (XIX-с) по известным в данной области методикам превращения кетона в имин (стадия G реакции). Указанное превращение можно подходящим образом проводить обработкой соответствующих промежуточных соединений формул (XIX-а) и (XIX-с) промежуточным соединением формулы (XVIII), в которой Z2 представляет собой алкилсульфинильную группу, такую как, например, трет-бутансульфинильная группа, в присутствии подходящего катализатора, кислоты Льюиса, такой как изопропоксид титана(IV), в подходящем инертном в условиях реакции растворителе, таком как, например, толуол, при таких термических условиях, как, например, нагревание реакционной смеси при 110 С, например, в течение 24 ч. Промежуточные соединения формул (XIX-а) и (XIX-с) в приведенной выше схеме реакций (11) можно получить взаимодействием промежуточных соединений формул (ХХ-а) и (XX-с) известными в данной области процедурами окисления спиртовой группы в карбонил (стадии Н реакции). Указанное окисление можно подходящим образом проводить обработкой соответствующих промежуточных соединений формул (ХХ-а) и (ХХ-с) окисляющим агентом, таким как, например, периодинан Десс-Мартина[CAS: 87413-09-0], в подходящем, инертном в условиях реакции растворителе, таком как, например, дихлорметан, при низких температурах, таких как, например, 0 С, например, в течение 10 мин и затем при умеренно высокой температуре, такой как, например, 25 С, например, в течение 1 ч. Промежуточные соединения согласно формулам (ХХ-а) и (ХХ-с) на приведенной выше схеме реакции (11) можно получить взаимодействием промежуточных соединений формул (XXII-а) и (XXII-c) известными в данной области процедурами ортолитиирования-алкилирования (стадия I реакции). Указанное превращение можно подходящим образом проводить обработкой соответствующих промежуточных соединений формул (XXII-а) и (XXII-c) подходящим литийорганическим реагентом, таким как, например, н-бутиллитий, в подходящем инертном в условиях реакции растворителе, таком как, например, тетрагидрофуран, при низких температурах, таких как, например, -78 С, например, в течение 45 мин, с последующей обработкой промежуточными соединениями формул (XXI-а) и (XXI-с) при низкой температуре, такой, как, например, -78 С, например, в течение 45 мин. Промежуточные соединения формул (XXII-a) и (XXII-c), в которых Z1 представляет собой подходящую защитную группу системы пиразола, такую как, например, диметилсульфамоильную группу,обычно получают известными в данной области процедурами типа N-защиты, описанным в литературе.J: превращение амида Вейнреба в кетон.- 11021723 Кроме того, промежуточные соединения формул (XIX-а) и (XIX-c), в которых R6 представляет собой водород, на приведенной выше схеме реакций (12) можно получить взаимодействием промежуточных соединений формул (XXIV-a) и (XXIV-с) известными в данной области процедурами превращения амида Вейнреба в кетон (стадия J реакции). Указанное превращение можно подходящим образом проводить обработкой соответствующих промежуточных соединений формул (XXIV-a) и (XXIV-с) промежуточными соединениями формул (XXIII-a) и (XXIII-с), в которых Y представляет собой галоген, в подходящем инертном в реакционных условиях растворителе, таком как, например, тетрагидрофуран, при низкой температуре, такой как, например, -78 С, например, в течение 1 ч и затем при умеренно высокой температуре, такой как, например, 25 С, например, в течение 5 ч. Промежуточные соединения формул (XXIV-a) и (XXIV-c) на приведенной выше схеме реакций (12) можно получить взаимодействием промежуточных соединений формул (XXV-a) и (XXV-с) по известным процедурам получения амида Вейнреба (стадия K реакции). Указанное превращение можно подходящим образом проводить обработкой соответствующих промежуточных соединений формул (XXV-a) и(XXV-c) N,O-диметилгидроксиламином в присутствии подходящего основания, такого как, например,изопропилмагнийхлорид, в подходящем инертном в условиях реакции растворителе, таком как, например, дихлорметан, при низкой температуре, такой как, например, -78 С, например, в течение 1 ч и затем при умеренно высокой температуре, такой как, например, 25 С, например, в течение 24 ч. Промежуточные соединения формул (XXV-a) и (XXV-c), в которых Z1 представляет собой подходящую защитную группу системы пиразола, такую как, например, диметилсульфамоильная группа, являются коммерчески доступными. Кроме того, промежуточные соединения формул (XIII-а) и (XIII-с), в которых R6 представляет собой водород, на схеме реакции (13) можно получить из соответствующих промежуточных соединений формул (XXVI-a) и (XXVI-с), в которых Z3 представляет собой защитную группу для аминов, такую как,например, трет-бутоксикарбонильная группа (стадия Е реакции), известными в данной области процедурами N-снятия защиты, таким как процедуры, описанные на схеме реакции (11) (стадия Е реакции). Промежуточные соединения формул (XXVI-a) и (XXVI-с) на схеме реакций (13) можно получить взаимодействием промежуточных соединений формул (XXVII-a) и (XXVII-c) известными в данной области процедурами образования кольца пиразола (стадия L реакции). Указанное образование кольца пиразола можно подходящим образом проводить обработкой соответствующих промежуточных соединений формул (XXVII-a) и (XXVII-c) в подходящем инертном растворителе, таком как, например, этанол, в присутствии гидразина, при умеренно высокой температуре, такой как, например, 25 С, например, в течение 1 ч. Промежуточные соединения формул (XXVII-a) и (XXVII-c) на схеме реакций (13) можно получить взаимодействием промежуточных соединений формул (XXVIII-a) и (XXVIII-c) известными в данной области процедурами окисления спиртовой группы в карбонил, таким как процедуры, описанные на схеме реакций (11) (стадия Н реакции).L: образование кольца пиразола. Н: окисление спиртовой группы в карбонил. М: превращение альдегидной группы в гидроксиалкинил. Промежуточные соединения согласно формулам (XXVIII-a) и (XXVIII-c) на приведенной выше схеме реакций (13) можно получить взаимодействием промежуточных соединений формул (XXIX-а) и(XXIX-c) известными в данной области процедурами превращения альдегидной группы в гидроксиалкинил (стадии М реакции). Указанное превращение можно подходящим образом проводить обработкой соответствующих промежуточных соединений формул (XXIX-а) и (XXIX-c) подходящим магниевым реагентом, таким как, например, этинилмагнийбромид, в подходящем, инертном в условиях реакции растворителе, таком как, например, тетрагидрофуран, при низкой температуре, такой как, например, 0 С,например, в течение 10 мин и затем при умеренно высокой температуре, такой как, например, 25 С, например, в течение 30 мин. Промежуточные соединения формул (XXIX-а) и (XXIX-c) на приведенной выше схеме реакций (13) можно получить взаимодействием промежуточных соединений формул (ХХХ-а) и (XXX-с) известными в данной области процедурами окисления спиртовой группы в карбонил, такими как процедуры, описанные на схеме реакций (11) (стадия Н реакции). Промежуточные соединения формул (ХХХ-а) и (ХХХ-с), в которых Z3 представляет собой защитную группу для аминов, такую как, например, трет-бутоксикарбонильная группа, обычно получают известными в данной области процедурами типа Strecker, описанными в литературе. Фармацевтические композиции Настоящее изобретение также относится к композициям для предотвращения или лечения заболеваний, при которых ингибирование -секретазы является благоприятным, таких как болезнь Альцгеймера (AD), умеренное когнитивное расстройство, старение, деменция, деменция с тельцами Леви, синдром Дауна, деменция, связанная с инсультом, деменция, связанная с болезнью Паркинсона, и деменция, связанная с -амилоидом. Указанные композиции содержат терапевтически эффективное количество соединения формулы (I) и фармацевтически приемлемый носитель или разбавитель. Хотя активный ингредиент можно вводить отдельно, предпочтительным является его применение в составе фармацевтической композиции. Соответственно этому настоящее изобретение относится далее к фармацевтической композиции, содержащей соединение согласно настоящему изобретению вместе с фармацевтически приемлемым носителем или разбавителем. Носитель или разбавитель должен быть"приемлемым" в смысле совместимости с другими ингредиентами композиции и должен быть невредным для его реципиентов. Фармацевтические композиции данного изобретения можно получить любыми способами, хорошо известными в области фармации. Терапевтически эффективное количество конкретного соединения в форме основания или в форме аддитивной соли, в качестве активного ингредиента объединяют с образованием однородной смеси с фармацевтически приемлемым носителем, который может принимать различные формы в зависимости от формы препарата, требуемого для введения. Эти фармацевтические композиции являются желательными в виде дозированной лекарственной формы, подходящей предпочтительно для системного введения, такого как пероральное, чрескожное или парентеральное введение,или местного введения, такого как введение посредством ингаляции, спрея для носа, глазных капель или посредством крема, геля, шампуня или тому подобного. Например, при получении композиций в пероральной лекарственной форме можно применять любую из обычных фармацевтических сред, таких как,например, вода, гликоли, масла, спирты и тому подобное, в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара,каолин, смазывающие вещества, связывающие вещества, дезинтегрирующие агенты и тому подобное в случае порошков, пилюль, капсул и таблеток. Из-за легкости их введения таблетки и капсулы представляют собой наиболее подходящую пероральную дозированную лекарственную форму, в этом случае,несомненно, применяют твердые фармацевтические носители. Для парентеральных композиций носитель обычно содержит стерильную воду, по меньшей мере, в значительном количестве, хотя могут быть включены другие ингредиенты, например, для повышения растворимости. Например, можно получить растворы для инъекций, в которых носитель содержит солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Можно также получить суспензии для инъекций, в этом случае можно применять подходящие жидкие носители, суспендирующие агенты и тому подобное. В композициях,подходящих для подкожного введения, носитель необязательно содержит повышающий проникновение агент и/или подходящий смачивающий агент, необязательно в сочетании с подходящими добавками любой природы в небольших пропорциях, такие добавки не оказывают никакие существенные вредные действия на кожу. Указанные добавки могут облегчить введение в кожу и/или могут быть полезными при получении желаемых композиций. Эти композиции можно вводить различными путями, например в виде чрескожного пластыря, в виде пятна или в виде мази. Особенно подходящим является изготовление вышеуказанных фармацевтических композиций в дозированной лекарственной форме для простоты введения и однородности дозировки. Дозированная лекарственная форма, применяемая в описании и формуле изобретения, относится к физически дискретным единицам, подходящим в качестве единичных доз, причем каждая единица содержит заданное количество активного ингредиента, рассчитанное для получения желаемого терапевтического действия, в сочетании с требуемым фармацевтическим носителем. Примерами таких дозированных лекарственных форм являются таблетки (включая таблетки с насечками или таблетки, покрытые оболочкой), капсулы,пилюли, пакетики с порошком, облатки, растворы или суспензии для инъекции, чайные ложки с препаратом, столовые ложки с препаратом и тому подобное, и их сегрегированные многократные дозы. Точная дозировка и частота введения зависят от конкретного применяемого соединения формулы(I), конкретного подвергаемого лечению состояния и тяжести состояния, подвергаемого лечению, возраста, массы, пола, степени нарушения и общего физического состояния конкретного пациента, а также другой лекарственной терапии, которую может принимать индивидуум, что хорошо известно специалистам в данной области. Кроме того, очевидно, что указанное эффективное суточное количество можно уменьшить или увеличить в зависимости от реакции подвергаемого лечению субъекта и/или в зависимости от оценки врача, прописывающего соединения настоящего изобретения. В зависимости от способа введения фармацевтическая композиция будет содержать от 0,05 до 99 мас.%, предпочтительно от 0,1 до 7 0 мас.%, более предпочтительно от 0,1 до 50 мас.% активного ингредиента и от 1 до 99,95 мас.%, предпочтительно от 30 до 99,9% мас.%, более предпочтительно от 50 до 99,9 мас.% фармацевтически приемлемого носителя, причем все проценты основаны на общей массе композиции. Данные соединения можно применять для системного введения, такого как пероральное, чрескожное или парентеральное введение, или местного введения, такого как введение посредством ингаляции,спрея для носа, глазных капель или посредством крема, геля, шампуня или тому подобного. Соединения предпочтительно вводят перорально. Точная дозировка и частота введения зависят от конкретного применяемого соединения согласно формуле (I), конкретного подвергаемого лечению состояния, тяжести состояния, подвергаемого лечению, возраста, массы, пола, степени нарушения и общего физического состояния конкретного пациента, а также другой лекарственной терапии, которую может принимать индивидуум, что хорошо известно специалистам в данной области. Кроме того, очевидно, что указанное эффективное суточное количество можно уменьшить или увеличить в зависимости от реакции подвергаемого лечению субъекта и/или в зависимости от оценки врача, прописывающего соединения настоящего изобретения. Количество соединения формулы (I), которое может быть объединено с веществом-носителем для изготовления лекарственной формы для разового применения, будет изменяться в зависимости от подвергаемого лечению заболевания, вида млекопитающего и конкретного способа введения. Тем не менее,в качестве общего руководства подходящие единичные дозы соединений настоящего изобретения могут,например, предпочтительно содержать от 0,1 до 1000 мг активного вещества. Предпочтительная единичная доза составляет от 1 до приблизительно 500 мг. Более предпочтительной единичной дозой является доза от 1 до приблизительно 300 мг. Еще более предпочтительной дозой является доза от 1 до 100 мг. Такие единичные дозы можно вводить более одного раза в день, например 2, 3, 4, 5 или 6 раз в день, но предпочтительно 1 или 2 раза в день, так, чтобы общая доза для взрослого пациента массой 70 кг была в диапазоне от 0,001 до приблизительно 15 мг на 1 кг массы субъекта на одно введение. Предпочтительная доза составляет от 0,01 до приблизительно 1,5 мг на 1 кг массы субъекта на одно введение, и такая терапия может продолжаться в течение ряда недель или месяцев, а в некоторых случаях лет. Однако должно быть понятно, что конкретный уровень дозы для любого конкретного пациента будет зависеть от целого ряда факторов, включающих в себя активность конкретного используемого соединения, возраст, массу тела, общее состояние здоровья, пол и питание подвергаемого лечению пациента; время и путь введения; скорость выведения; другие лекарственные средства, которые ранее были введены, и тяжести конкретного заболевания, подвергаемого лечению, что хорошо знают специалисты в данной области. В некоторых случаях может быть необходимо применять дозы вне этих диапазонов, что должно быть очевидно специалистам в данной области. Кроме того, обращается внимание на то, что клиницист или лечащий врач должен знать, как и когда начать, прервать, регулировать или прекратить терапию в зависимости от реакции отдельного пациента. Имеется в виду, что следующие примеры иллюстрируют, но не ограничивают объем настоящего изобретения. Экспериментальная часть В дальнейшем "т.пл" означает точку плавления, "вод." означает водный, "р.с." означает реакционную смесь, "к.т." означает комнатную температуру, "DIPEA" означает диизопропилэтиламин, "DIPE" означает диизопропиловый простой эфир, "Et2O" означает диэтиловый простой эфир, "ТГФ" означает тетрагидрофуран, "ДМФА" означает диметилформамид, "DCM" означает дихлорметан, "AcOEt" означает этилацетат, "АсОН" означает уксусную кислоту, "МеОН" означает метанол, "EtOH" означает этанол,"рац." означает рацемический, "нас." означает насыщенный, "SFC" означает сверхкритическую жидкостную хроматографию, "SFC-MC" означает сверхкритическую жидкостную хроматографию/массспектрометрию, "LCMS" означает жидкостную хроматографию/масс-спектрометрию, "ВЭЖХ" означает высокоэффективную жидкостную хроматографию, "DMTMM" означает хлорид 4-(4,6-диметокси-1,3,5 триазин-2-ил)-4-метилморфолиния, "HATU" означает гексафторфосфат О-(7-азабензотриазол-1-ил)N,N,N',N'-тетраметилурония. А. Получение промежуточных соединений. Пример А 1. Получение промежуточного соединения 1: рац-2-амино-2-(3-бромфенил)пропаннитрила Триметилсилилцианид (20 г, 200 ммоль) добавляли к перемешиваемому раствору 3 бромацетофенона (20 г, 100 ммоль) и NH4Cl (11 г, 200 ммоль) в растворе NH3/MeOH (400 мл). Смесь перемешивали при комнатной температуре в течение 4 дней. Затем растворитель упаривали в вакууме и остаток помещали в AcOEt (100 мл). Твердое вещество отфильтровывали и фильтрат упаривали в вакуу- 15021723 ме, получая при этом рац-2-амино-2-(3-бромфенил)пропионитрил (20 г, выход 86%), который применяли на следующей стадии без дополнительной очистки. Пример А 2. Получение промежуточного соединения 2: рац-метил-2-амино-2-(3-бромфенил)пропаноата(500 мл) и смесь кипятили с обратным холодильником в течение 4 дней. После охлаждения до комнатной температуры добавляли AcOEt (100 мл) и воду (100 мл) и смесь экстрагировали AcOEt (2100 мл). Объединенные водные слои подщелачивали водным раствором аммиака до рН 8 и экстрагировали AcOEt(5100 мл). Объединенные органические слои сушили (Na2SO4), фильтровали и растворители выпаривали в вакууме, получая при этом метиловый эфир рац-2-амино-2-(3-бромфенил)пропионовой кислоты(10,6 г, 46% выход) в виде масла. Пример A3. Получение промежуточного соединения 3: рац-2-амино-2-(3-бромфенил)пропан-1-ола Литийалюминийгидрид (1M в ТГФ; 22 мл, 22 ммоль) добавляли по каплям к перемешиваемому раствору метилового эфира рац-2-амино-2-(3-бромфенил)пропионовой кислоты (7,5 г, 29,1 ммоль) в ТГФ(200 мл) при -15 С. Смесь оставляли для медленного прогревания до 0 С в течение 1 ч. Затем по каплям добавляли еще ТГФ (150 мл) и нас. раствор Na2SO4 до тех пор, пока больше не образовывался водород. Затем добавляли безводный Na2SO4 и смесь перемешивали на протяжении ночи при комнатной температуре. Смесь фильтровали через диатомовую землю, промывали ТГФ и растворитель выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель, 7 М раствор аммиака в растворе метанола в DCM, от 0/100 до 3/97). Целевые фракции собирали и концентрировали в вакууме,получая при этом рац-2-амино-2-(3-бромфенил)пропан-1-ол (5,70 г, выход 85%) в виде масла. Пример А 4. Получение промежуточного соединения 4: (R)-2-амино-2-(3-бромфенил)пропан-1-ола Образец рац-2-амино-2-(3-бромфенил)пропан-1-ола (15,4 г) разделяли на соответствующие энантиомеры препаративной SFC (на Chiralpak Daicel AD250 мм). Подвижная фаза (СО 2, МеОН с 0,2%iPrNH2), получая при этом (R)-2-амино-2-(3-бромфенил)пропан-1-ол (7,21 г, выход 40%).D=-14,9 (589 нм, с 0,2946 мас./об.%, МеОН, 20 С). Пример А 5. Получение промежуточного соединения 5: рац-трет-бутил-N-[1-(3-бромфенил)-2 гидрокси-1-метилэтил]карбамат ди-трет-Бутилдикарбонат (4,84 г, 22,16 ммоль) добавляли порциями к перемешиваемому раствору рац-2-амино-2-(3-бромфенил)пропан-1-ола (1,7 г, 7,39 ммоль) в смеси нас. NaHCO3 (15 мл) и ТГФ (15 мл) при 0 С. Смесь перемешивали при 0 С в течение 10 мин и при комнатной температуре в течение 15 ч. Смесь охлаждали на бане с ледяной водой и подкисляли при перемешивании до рН 1-2 посредствомKHSO4. Органический слой отделяли и водный слой далее экстрагировали AcOEt. Объединенные органические слои отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (диоксид кремния; AcOEt в DCM, от 0/100 до 20/80). Целевые фракции собирали и концентрировали в вакууме, получая при этом трет-бутиловый эфир рац-[1-(3-бромфенил)-2-гидрокси-1-метилэтил]карбаминовой кислоты (2,36 г, выход 93%) в виде бесцветного масла. Пример А 6. Получение промежуточного соединения 6: рац-трет-бутил-N-[1-(3-бромфенил)-1 метил-2-оксоэтил]карбамата Периодинан Десса-Мартина (3,55 г, 8,36 ммоль) добавляли порциями на протяжении 5 мин к раствору трет-бутилового эфира рац-[1-(3-бромфенил)-2-гидрокси-1-метилэтил]карбаминовой кислоты (2,3 г, 6,97 ммоль) в сухом DCM (45 мл) при 0 С. Смесь перемешивали при 0 С в течение 10 мин и при комнатной температуре в течение 1 ч. Реакционную смесь гасили NaHCO3 (водн. нас. раствор) с последующим гашением NaHSO3 (водн. нас. раствор). Затем добавляли Et2O и смесь перемешивали при комнатной температуре в течение 30 мин. Органический слой отделяли и водный слой далее экстрагировали Et2O. Объединенные органические слои отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; DCM). Целевые фракции собирали и концентрировали в вакууме, получая при этом трет-бутиловый эфир рац-[1-(3 бромфенил)-1-метил-2-оксоэтил]карбаминовой кислоты (2 г, выход 80%) в виде бесцветного масла. Пример А 7. Получение промежуточного соединения 7: рац-трет-бутил-N-[1-(3-бромфенил)-2 гидрокси-1-метилбут-3-инил]карбамат 0,5 М раствор этинилмагнийбромида в ТГФ (23,89 мл, 11,94 ммоль) по каплям добавляли к раствору трет-бутилового эфира рац-[1-(3-бромфенил)-1-метил-2-оксоэтил]карбаминовой кислоты (1,96 г, 5,97 ммоль) в ТГФ (60 мл) при 0 С в атмосфере азота. Смесь перемешивали при 0 С в течение 15 мин и при комнатной температуре в течение 30 мин. Смесь разбавляли NH4Cl (водн. нас. раствор) и экстрагировалиDCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме, получая при этом трет-бутиловый эфир рац-[1-(3-бромфенил)-2-гидрокси-1-метил-бут-3-инил]карбаминовой кислоты (2,11 г, выход 99%) в виде масла, которое применяли на следующей стадии без дополнительной очистки. Пример А 8. Получение промежуточного соединения 8: рац-трет-бутил-N-[1-(3-бромфенил)-1 метил-2-оксобут-3-инил]карбамата Периодинан Десса-Мартина (3,04 г, 7,16 ммоль) добавляли порциями на протяжении 5 мин к раствору трет-бутилового эфира рац-[1-(3-бромфенил)-2-гидрокси-1-метилбут-3-инил]карбаминовой кислоты (2,12 г, 5,97 ммоль) в сухом DCM (20 мл) при 0 С. Смесь перемешивали при 0 С в течение 10 мин и при комнатной температуре в течение 1 ч. Реакционную смесь гасили NaHCO3 (водн. нас. раствор) с последующим гашением NaHSO3 (водный нас. раствор). Затем добавляли Et2O и смесь перемешивали при комнатной температуре в течение 30 мин. Органический слой отделяли и водный слой дополнительно экстрагировали Et2O. Объединенные органические слои отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель, DCM). Целевые фракции собирали и концентрировали в вакууме, получая при этом трет-бутиловый эфир рац-[1-(3-бромфенил)-1-метил-2-оксобут-3-инил]карбаминовой кислоты (1,89 г, выход 90%) в виде масла. Пример А 9. Получение промежуточного соединения 9: рац-трет-бутил-N-[1-(3-бромфенил)-1-(1 Нпиразол-3-ил)этил]карбамата Гидразингидрат (2,48 мл, 51,10 ммоль) добавляли к раствору трет-бутилового эфира рац-[1-(3 бромфенил)-1-метил-2-оксо-бут-3-инил]карбаминовой кислоты (1,8 г, 5,11 ммоль) в EtOH (30 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляли в вакууме и остаток растворяли в DCM и промывали водой. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силика- 17021723 гель; AcOEt в DCM, от 0/100 до 50/50). Целевые фракции собирали и концентрировали в вакууме, получая при этом трет-бутиловый эфир рац-[1-(3-бромфенил)-1-(1 Н-пиразол-3-ил)этил]карбаминовой кислоты (1,62 г, выход 87%) в виде твердого вещества белого цвета. Пример А 10. Получение промежуточного соединения 10: рац-1-(3-бромфенил)-1-(1 Н-пиразол-3 ил)этанамина 4 М хлористо-водородную кислоту в диоксане (7,88 мл, 31,54 ммоль) добавляли к трет-бутиловому эфиру рац-[1-(3-бромфенил)-1-(1 Н-пиразол-3-ил)этил]карбаминовой кислоты (1,65 г, 4,51 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель выпаривали в вакууме. Остаток суспендировали в DCM и промывали NaHCO3 (водн. нас. раствор). Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме, получая при этом рац-1-(3-бромфенил)-1-(1 Н-пиразол-3-ил)этиламин (1,2 г, выход 100%) в виде твердого вещества белого цвета, которое применяли на следующей стадии без дополнительной очистки. Пример А 11. Получение промежуточного соединения 11: рац-N-[1-(3-бромфенил)-1-(1 Н-пиразол-3 ил)этил]-2-хлорацетамидаDIPEA (1,18 мл, 6,77 ммоль) добавляли к раствору рац-1-(3-бромфенил)-1-(1 Н-пиразол-3 ил)этиламина (1,2 г, 4,51 ммоль) в DCM (20 мл) и смесь охлаждали на ледяной бане. Затем добавляли хлорацетилхлорид (0,40 мл, 4,96 ммоль) и смесь перемешивали при 0 С в течение 3 ч. Смесь разбавлялиNH4Cl (водн. нас. раствор) и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэшхроматографией (силикагель, AcOEt в DCM, от 0/100 до 20/80). Целевые фракции собирали и концентрировали в вакууме. Остаток растворяли в этаноле (10 мл) и NaHCO3 (водн. нас. раствор) (1 мл) и смесь перемешивали при комнатной температуре в течение 30 мин. Смесь разбавляли водой и продукт экстрагировали DCM. Объединенные органические слои концентрировали в вакууме, получая при этом рац-N[1-(3-бромфенил)-1-(1 Н-пиразол-3-ил)этил]-2-хлорацетамид (1,22 г, выход 79%) в виде бесцветного масла, которое применяли на следующей стадии без дополнительной очистки. Пример А 12. Получение промежуточного соединения 12: рац-4-(3-бромфенил)-4-метил-4,5 дигидропиразоло[1,5-а]пиразин-6-она Раствор рац-N-[1-(3-бромфенил)-1-(1 Н-пиразол-3-ил)этил]-2-хлорацетамида (1,22 г, 3,56 ммоль) в ТГФ (40 мл) по каплям добавляли к суспензии гидрида натрия (0,28 г, 7,12 ммоль) в ТГФ (40 мл) при 0 С в атмосфере азота. Смесь перемешивали при 0 С в течение 1 ч. Смесь разбавляли водой и продукт экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель: AcOEt в DCM,от 50/50 до 100/0). Целевые фракции собирали и концентрировали в вакууме, получая при этом рац-4-(3 бромфенил)-4-метил-4,5-дигидропиразоло[1,5-а]пиразин-6-он (0,7 г, выход 64%) в виде твердого вещества белого цвета. Пример А 13. Получение промежуточного соединения 13: рац-4-(3-бромфенил)-4-метил-4,5 дигидропиразоло[1,5-а]пиразин-6-тиона Пентасульфид фосфора (1,02 г, 4,57 ммоль) добавляли к раствору рац-4-(3-бромфенил)-4-метил-4,5 дигидропиразоло [1,5-а]пиразин-6-она (0,7 г, 2,29 ммоль) в пиридине (10 мл) и смесь нагревали при 95 С в течение 18 ч. Затем растворитель выпаривали в вакууме и остаток очищали колоночной флэшхроматографией (силикагель; раствор AcOEt в DCM, от 0/100 до 10 0/0). Целевые фракции собирали и концентрировали в вакууме, получая при этом рац-4-(3-бромфенил)-4-метил-4,5-дигидропиразоло[1,5- 18021723 а]пиразин-6-тион (0,45 г, выход 61%) в виде твердого вещества белого цвета. Пример А 14. Получение промежуточного соединения 14: рац-4-(3-бромфенил)-4-метил-4,7 дигидропиразоло[1,5-а]пиразин-6-аминаNH4Cl (0,15 г, 2,79 ммоль) добавляли к перемешиваемому раствору рац-4-(3-бромфенил)-4-метил 4,5-дигидропиразоло[1,5-а]пиразин-6-тиона (0,45 г, 1,40 ммоль) в EtOH (50 мл) и смесь нагревали при 80 С в течение 28 ч. Растворитель удаляли в вакууме и остаток растворяли в DCM и промывали водой. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; 7 М раствор аммиака в растворе метанола в AcOEt, от 0/100 до 20/80). Целевые фракции собирали и концентрировали в вакууме, получая при этом рац-4-(3-бромфенил)-4-метил-4,7-дигидропиразоло[1,5-а]пиразин-6-иламин (0,42 г, выход 99%) в виде желтого твердого вещества. Пример А 15. Получение промежуточного соединения 15: рац-4-[3-(бензгидрилиденамино)фенил]-4 метил-4,7-дигидропиразоло[1,5-а]пиразин-6-амина Толуол (10 мл) добавляли к смеси рац-4-(3-бромфенил)-4-метил-4,7-дигидропиразоло[1,5 а]пиразин-6-иламина (0,39 г, 1,28 ммоль), трис(дибензилиденацетон)дипалладия(0) (0,12 г, 0,13 ммоль),рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (0,24 г, 0,38 ммоль) и трет-бутоксида натрия (0,22 г, 2,3 ммоль) в запаянной трубке в атмосфере азота при комнатной температуре. Смесь продували азотом в течение нескольких минут и затем добавляли имин бензофенона (0,43 мл, 2,56 ммоль) и смесь перемешивали при 100 С в течение 2 ч. После охлаждения смесь разбавляли водой и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (диоксид кремния; 7 М раствор аммиака в растворе метанола в DCM, от 0/100 до 3/97). Требуемые фракции собирали и концентрировали в вакууме, получая при этом рац-4-[3-(бензгидрилиденамино)фенил]-4-метил-4,7-дигидропиразоло[1,5-а]пиразин-6-иламин(0,37 г, выход 70%) в виде желтой пены. Пример А 16. Получение промежуточного соединения 16: рац-4-(3-аминофенил)-4-метил-4,7 дигидропиразоло[1,5-а]пиразин-6-амина 37% хлористо-водородную кислоту в Н 2 О (0,14 мл) добавляли к раствору рац-4-[3(бензгидрилиденамино)фенил]-4-метил-4,7-дигидропиразоло[1,5-а]пиразин-6-иламина (0,37 г, 0,9 ммоль) в изопропаноле (10 мл). Смесь перемешивали при комнатной температуре в течение 3 ч. Добавляли Et2O и смесь перемешивали в течение 15 мин. Осажденное твердое вещество отделяли фильтрованием, промывали Et2O и сушили в вакууме. Остаток суспендировали в DCM и промывали NaHCO3 (водн. нас. раствор). Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме, получая при этом рац-4-(3-аминофенил)-4-метил-4,7-дигидропиразоло[1,5-а]пиразин-6-амин (0,21 г,выход 97%) в виде твердого вещества белого цвета, которое применяли на следующей стадии без дополнительной очистки. Пример А 17. Получение промежуточного соединения 17: 1 Н-пиразол-3-карбоновой кислоты Раствор перманганата калия (16,17 г, 102,31 ммоль) в воде (150 мл) добавляли к раствору 3 метилпиразола (4,2 г, 51,15 ммоль) в воде (100 мл) и смесь кипятили с обратным холодильником на протяжении ночи. После охлаждения до комнатной температуры нерастворимое вещество удаляли фильтрованием. Фильтрат концентрировали до 30 мл и 2 н. HCl добавляли до осаждения твердого вещества. Твердое вещество отделяли фильтрованием, промывали холодной водой и сушили в вакууме, получая при этом 1 Н-пиразол-3-карбоновую кислоту (3,1 г, выход 54%) в виде твердого вещества белого цвета,которое применяли на следующей стадии без дополнительной очистки. Пример А 18. Получение промежуточного соединения 18: метил-1 Н-пиразол-3-карбоксилата Серную кислоту (5,8 мл) добавляли по каплям к перемешиваемому раствору 1 Н-пиразол-3 карбоновой кислоты (1 г, 8,92 ммоль) в МеОН (65 мл) при 0 С. После завершения добавления смеси предоставляли возможность для нагревания до комнатной температуры и перемешивали в течение 18 ч. Смесь концентрировали в вакууме и остаток растворяли в воде и подщелачивали NaHCO3 (водн. нас. раствор). Смесь экстрагировали AcOEt. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме, получая при этом метиловый эфир 1 Н-пиразол-3-карбоновой кислоты (0,7 г, выход 62%) в виде твердого вещества белого цвета, которое применяли на следующей стадии без дополнительной очистки. Пример А 19. Получение промежуточного соединения 19: метил-1-(диметилсульфамоил)-1 Нпиразол-3-карбоксилата Гидрид натрия (1,57 г, 41,03 ммоль) добавляли к раствору метилового эфира 1 Н-пиразол-3 карбоновой кислоты (3,45 г, 27,36 ммоль) в ТГФ (20 мл) при 0 С. Смесь перемешивали при 0 С в течение 30 мин. Затем добавляли диметилсульфамоилхлорид (4,41 мл, 41,03 ммоль) и смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 18 ч. Смесь разбавляли водой и продукт экстрагировали AcOEt. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; AcOEt вDCM, от 0/100 до 10/90). Целевые фракции собирали и концентрировали в вакууме, получая при этом метиловый эфир 1-диметилсульфамоил-1 Н-пиразол-3-карбоновой кислоты (4,8 г, выход 75%) в виде бесцветного масла. Пример А 20. Получение промежуточного соединения 20: 1-(диметилсульфамоил)-N-метокси-Nметил-1 Н-пиразол-3-карбоксилата Метиловый эфир 1-диметилсульфамоил-1 Н-пиразол-3-карбоновой кислоты (4 г, 17,15 ммоль) и гидрохлорид N,O-диметилгидроксиламина (2,18 г, 22,29 ммоль) суспендировали в DCM (20 мл). Смесь продували азотом и охлаждали до -78 С. Затем по каплям добавляли раствор изопропилмагнийхлорида(2M В ТГФ) (24,01 мл, 48,02 ммоль). Когда добавление завершали, смесь оставляли для нагревания до комнатной температуры и перемешивали на протяжении ночи. Смесь гасили NH4Cl (водн. нас. раствор) и продукт экстрагировали AcOEt. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель,AcOEt в DCM, от 0/100 до 100/0). Целевые фракции собирали и концентрировали в вакууме, получая при этом метоксиметиламид 1-диметилсульфамоил-1 Н-пиразол-3-карбоновой кислоты (3,2 г, выход 71%) в виде бледно-желтого цвета масла. Пример А 21. Получение промежуточного соединения 21: 3-(3-хлорфенил)карбонил-N,N-диметил 1 Н-пиразол-1-сульфонамида(20 мл) при -78 С в атмосфере азота. Смесь перемешивали при -78 С в течение 1 ч и затем дополнительно перемешивали при комнатной температуре в течение 5 ч. Смесь гасили NH4Cl (водн. нас. раствор) и продукт экстрагировали AcOEt. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель;AcOEt в DCM, от 0/100 до 10/90). Целевые фракции собирали и концентрировали в вакууме, получая при этом диметиламид 3-(3-хлорбензоил)-1H-пиразол-1-сульфоновой кислоты (1,68 г, выход 88%) в виде бледно-желтого твердого вещества. Пример А 22. Получение промежуточного соединения 22: 3-[(трет-бутилсульфинил)имино]-(3 хлорфенил)метил-N,N-диметил-1 Н-пиразол-1-сульфонамида(0,71 г, 5,89 ммоль) в толуоле (32 мл) в атмосфере азота. Смесь перемешивали при 110 С в течение 24 ч. Смесь охлаждали и выливали в солевой раствор при быстром перемешивании. Смесь фильтровали через диатомовую землю и осадок на фильтре промывали AcOEt. Фильтрат переносили в делительную воронку, где органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; AcOEt в DCM, от 0/100 до 10/90). Целевые фракции собирали и концентрировали в вакууме, получая при этом диметиламид 3-[(3 хлорфенил)-(2-метилпропан-2-сульфинилимино)метил]пиразол-1-сульфоновой кислоты (2,17 г, выход 97%) в виде желтого масла. Пример А 23. Получение промежуточного соединения 23: 3-[1-(трет-бутилсульфиниламино)-1-(3 хлорфенил)этил]-N,N-диметил-1 Н-пиразол-1-сульфонамида Метилмагнийбромид (15,08 мл, 21,11 ммоль) добавляли к раствору диметиламида 3-[(3 хлорфенил)-(2-метилпропан-2-сульфинилимино)метил]пиразол-1-сульфоновой кислоты (2,2 г, 5,28 ммоль) в ТГФ (25 мл) при 0 С в атмосфере азота. Смесь перемешивали при 0 С в течение 2 ч, гасили(MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; AcOEt в DCM, от 0/100 до 100/0). Целевые фракции собирали и концентрировали в вакууме, получая при этом диметиламид 3-[1-(3-хлорфенил)-1-(2-метилпропан-2 сульфиниламино)этил]пиразол-1-сульфоновой кислоты (2,28 г, выход 99%) в виде бесцветного масла,которое затвердевает при стоянии. Пример А 24. Получение промежуточного соединения 24: рац-1-(3-хлорфенил)-1-(1 Н-пиразол-3 ил)этанамина 4 М хлористо-водородную кислоту в диоксане (19,79 мл, 79,15 ммоль) добавляли к раствору диметиламида 3-[1-(3-хлорфенил)-1-(2-метилпропан-2-сульфиниламино)этил]пиразол-1-сульфоновой кислоты (2,29 г, 5,28 ммоль) в МеОН (5 мл) и смесь перемешивали при 80 С в запаянной трубке в течение 18 ч. Растворитель выпаривали в вакууме. Остаток выливали в NaHCO3 (водн. нас. раствор) и экстрагировалиDCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме, получая при этом рац-1-(3-хлорфенил)-1-(1 Н-пиразол-3-ил)этиламин (1 г, выход 86%) в виде бледножелтого твердого вещества, которое применяли на следующей стадии без дополнительной очистки. Пример А 25. Получение промежуточного соединения 25: рац-2-хлор-N-[1-(3-хлорфенил)-1-(1 Нпиразол-3-ил)этил]ацетамида Промежуточное соединение 25 синтезировали таким же способом, как способ, описанный в примере A11. Исходя из промежуточного соединения 24 (1 г, 4,51 ммоль), промежуточное соединение 25 получили (0,73 г, выход 54%) в виде твердого вещества белого цвета. Пример А 26. Получение промежуточного соединения 26: рац-4-(3-хлорфенил)-4-метил-4,5 дигидропиразоло[1,5-а]пиразин-6-она Промежуточное соединение 26 синтезировали таким же способом, как способ, описанный в примере А 12. Исходя из промежуточного соединения 25 (0,73 г, 2,43 ммоль), промежуточное соединение 26 получили (0,45 г, выход 71%) в виде твердого вещества белого цвета. Пример А 27. Получение промежуточного соединения 27: рац-4-метил-4-(3-пиримидин-5-илфенил)4,5-дигидропиразоло[1,5-а]пиразин-6-он Ацетат палладия(II) (0,017 г, 0,075 ммоль) добавляли к перемешиваемой суспензии рац-4-(3 хлорфенил)-4-метил-4,5-дигидропиразоло[1,5-а]пиразин-6-она (0,13 г, 0,50 ммоль), пиримидин-5 бороновой кислоты (0,19 г, 1,49 ммоль), 2-дициклогексилфосфино-2',6'-диметоксибифенила (0,061 г,0,149 ммоль) и фосфата калия (0,21 г, 0,99 ммоль) в толуоле (5 мл) и EtOH (0,5 мл) при комнатной температуре и в атмосфере азота. Смесь перемешивали при 150 С в течение 30 мин в условиях микроволнового излучения. Затем смесь фильтровали через диатомовую землю и промывали AcOEt. Фильтрат выпаривали в вакууме. Остаток очищали колоночной флэш-хроматографией (силикагель; AcOEt). Целевые фракции собирали и концентрировали в вакууме, получая при этом рац-4-метил-4-(3-пиримидин-5 илфенил)-4,5-дигидропиразоло[1,5-а]пиразин-6-он (0,09 г, выход 59%) в виде твердого вещества белого цвета. Пример А 28. Получение промежуточного соединения 28: рац-4-метил-4-(3-пиримидин-5-илфенил)4,5-дигидропиразоло[1,5-а]пиразин-6-тиона Реагент Лавессона (0,14 г, 0,35 ммоль) добавляли к перемешиваемому раствору рац-4-метил-4-(3 пиримидин-5-илфенил)-4,5-дигидропиразоло[1,5-а]пиразин-6-тиона (0,09 г, 0,30 ммоль) в пиридине (2 мл) при комнатной температуре. Смесь нагревали при 95 С в течение 18 ч. Растворитель выпаривали в вакууме и неочищенный продукт очищали колоночной флэш-хроматографией (силикагель; AcOEt вDCM, от 0/100 до 20/80). Целевые фракции собирали и концентрировали в вакууме, получая при этом рац-4-метил-4-(3-пиримидин-5-ил-фенил)-4,5-дигидропиразоло[1,5-а]пиразин-6-тион (0,02 г, выход 21%) в виде твердого вещества белого цвета. Пример А 29. Получение промежуточного соединения 29: рац-4-[3-(5-метоксипиридин-3-ил)фенил]4-метил-4,5-дигидропиразоло[1,5-а]пиразин-6-она пературе и в атмосфере азота. Смесь перемешивали при 150 С в течение 30 мин в условиях микроволнового излучения. Затем смесь фильтровали через диатомовую землю и промывали AcOEt. Фильтрат выпаривали в вакууме. Остаток очищали колоночной флэш-хроматографией (силикагель; AcOEt). Целевые фракции собирали и концентрировали в вакууме, получая при этом рац-4-[3-(5-метоксипиридин-3 ил)фенил]-4-метил-4,5-дигидропиразоло[1,5-а]пиразин-6-он (0,11 г, выход 51%) в виде твердого вещества белого цвета. Пример A30. Получение промежуточного соединения 30: рац-4-[3-(5-метоксипиридин-3-ил)фенил]4-метил-4,5-дигидропиразоло[1,5-а]пиразин-6-тиона Пиридин (3 мл) добавляли к смеси рац-4-[3-(5-метоксипиридин-3-ил)фенил]-4-метил-4,5 дигидропиразоло[1,5-а]пиразин-6-она (0,11 г, 0,31 ммоль) и пентасульфида фосфора (0,07 г, 0,31 ммоль),смесь нагревали при 80 С в течение 5 ч. Затем добавляли еще пентасульфид фосфора (0,07 г, 0,31 ммоль) и смесь нагревали при 100 С в течение 18 ч. Затем растворитель выпаривали в вакууме и остаток очищали колоночной флэш-хроматографией (силикагель; МеОН в DCM, от 0/100 до 3/97). Целевые фракции собирали и концентрировали в вакууме, получая при этом рац-4-[3-(5 метоксипиридин-3-ил)фенил]-4-метил-4,5-дигидропиразоло[1,5-а]пиразин-6-тион (0,1 г, выход 93%) в виде твердого вещества белого цвета. Пример А 31. Получение промежуточного соединения 31: рац-4-(5-бром-2,4-дифторфенил)-4-метил 4,5-дигидропиразоло[1,5-а]пиразин-6-тиона Пентасульфид фосфора (2,53 г, 11,40 ммоль) добавляли к раствору рац-4-(5-бром-2,4 дифторфенил)-4-метил-4,5-дигидропиразоло[1,5-а]пиразин-6-она (3 г, 8,77 ммоль), полученного по такой же методике, как описана ранее для получения промежуточного рац-4-(3-бромфенил)-4-метил-4,5 дигидропиразоло[1,5-а]пиразин-6-она, в пиридине (30 мл) и смесь нагревали при 95 С в течение 18 ч. Затем растворитель выпаривали в вакууме и остаток очищали колоночной флэш-хроматографией (силикагель; AcOEt в DCM, от 0/100 до 40/60). Целевые фракции собирали и концентрировали в вакууме, получая при этом рац-4-(5-бром-2,4-дифторфенил)-4-метил-4,5-дигидропиразоло[1,5-а]пиразин-6-тион (2,4 г, выход 76%) в виде твердого вещества белого цвета. Пример A32. Получение промежуточного соединения 32: рац-4-(5-бром-2,4-дифторфенил)-4-метил 4,5-дигидропиразоло[1,5-а]пиразин-6-аминаNH4Cl (0,72 г, 13,4 ммоль) добавляли к перемешиваемой суспензии рац-4-(5-бром-2,4 дифторфенил)-4-метил-4,5-дигидропиразоло[1,5-а]пиразин-6-тиона (2,4 г, 6,7 ммоль) в 2 М растворе аммиака в EtOH (67 мл) и смесь нагревали при 85 С в течение 18 ч. Растворитель удаляли в вакууме и остаток суспендировали в DCM и промывали водой. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали в вакууме. Сырой продукт очищали колоночной флэшхроматографией (силикагель; 7 М раствор аммиака в растворе метанола в AcOEt, от 0/100 до 20/80). Целевые фракции собирали и концентрировали в вакууме, получая при этом рац-4-(5-бром-2,4 дифторфенил)-4-метил-4,5-дигидропиразоло[1,5-а]пиразин-6-иламин (1,8 г, выход 78%) в виде желтого твердого вещества. Пример А 33. Получение промежуточного соединения 33: (R)-трет-бутил-N-[1-(3-бромфенил)-2 гидрокси-1-метилэтил]карбамата Промежуточное соединение 33 синтезировали по такой же методике, как описана в примере А 5. Исходя из (R)-2-амино-2-(3-бромфенил)пропан-1-ола (4,7 г, 20,43 ммоль), промежуточное соединение 33 получали (6,4 г, выход 95%) в виде бесцветного масла, которое отверждалось при стоянии. Пример A34. Получение промежуточного соединения 34: (R)-трет-бутил-N-[1-(3-бромфенил)-1 метил-2-оксоэтил]карбамата Промежуточное соединение 34 синтезировали по такой же методике, как описана в примере А 6. Исходя из промежуточного соединения 33 (6,4 г, 19,38 ммоль), промежуточное соединение 34 получали(5,7 г, выход 90%) в виде бесцветного масла, которое отверждалось при стоянии. Пример A35. Получение промежуточного соединения 35: диастереоизомерной смеси (1R,2R)- и Промежуточное соединение 35 синтезировали по такой же методике, как методика, описанная в примере А 7. Исходя из промежуточного соединения 34 (5,7 г, 17,38 ммоль), промежуточное соединение 35 получали (5,4 г, выход 88%) в виде диастереоизомерной смеси в форме масла, которое использовали в следующей стадии без дополнительной очистки. Пример A36. Получение промежуточного соединения 36: (R)-трет-бутил-N-[1-(3-бромфенил)-1 метил-2-оксобут-3-инил]карбамата Промежуточное соединение 36 синтезировали такой же методикой, как методика, описанная в примере А 8. Исходя из промежуточного соединения 35 (5,4 г, 15,24 ммоль), промежуточное соединение 36 получали (5,3 г, выход 99%) в виде масла бледно-желтого цвета. Пример A37. Получение промежуточного соединения 37: (R)-трет-бутил-N-[1-(3-бромфенил)-1(1 Н-пиразол-3-ил)этил]карбамата Промежуточное соединение 37 синтезировали такой же методикой, как методика, описанная в примере А 9. Исходя из промежуточного соединения 36 (5,3 г, 15,05 ммоль), промежуточное соединение 37 получали (5 г, выход 91%) в виде пены. Пример A38. Получение промежуточного соединения 38: (R)-1-(3-бромфенил)-1-(1 Н-пиразол-3 ил)этанамина Промежуточное соединение 38 синтезировали такой же методикой, как методика, описанная в примере А 10. Исходя из промежуточного соединения 37 (5 г, 13,65 ммоль), промежуточное соединение 38 получали (3,5 г, выход 96%) в виде твердого вещества белого цвета, которое использовали в следующей стадии без дополнительной очистки. Пример A39. Получение промежуточного соединения 39: (R)-N-[1-(3-бромфенил)-1-(1 Н-пиразол-3 ил)этил]-2-хлорацетамида Промежуточное соединение 39 синтезировали такой же методикой, как методика, описанная в примере A11. Исходя из промежуточного соединения 38 (3,5 г, 13,15 ммоль), промежуточное соединение 39 получали (3,5 г, выход 78%) в виде бесцветного масла. Пример А 40. Получение промежуточного соединения 40: (R)-4-(3-бромфенил)-4-метил-4,5 дигидропиразоло[1,5-а]пиразин-6-она Промежуточное соединение 40 синтезировали такой же методикой, как методика, описанная в примере А 12. Исходя из промежуточного соединения 39 (3,5 г, 10,22 ммоль), промежуточное соединение 40 получали (2,15 г, выход 69%) в виде твердого вещества белого цвета. Пример А 41. Получение промежуточного соединения 41: (R)-4-(3-бромфенил)-4-метил-4,5 дигидропиразоло[1,5-а]пиразин-6-тиона Промежуточное соединение 41 синтезировали такой же методикой, как методика, описанная в примере А 13. Исходя из промежуточного соединения 40 (2,1 г, 6,86 ммоль), промежуточное соединение 41 получали (1,8 г, выход 81%) в виде пены. Пример А 42. Получение промежуточного соединения 42: (R)-4-(3-бромфенил)-4-метил-4,7 дигидропиразоло[1,5-а]пиразин-6-амина 32% водный раствор аммиака (11,9 мл, 201,1 ммоль) добавляли к перемешиваемой смеси (R)-4-(3 бромфенил)-4-метил-4,5-дигидропиразоло[1,5-а]пиразин-6-тиона (1,8 г, 5,59 ммоль) в 7 н. растворе аммиака в метаноле (11,97 мл, 83,79 ммоль) в запаянной трубке. Смесь перемешивали при 60 С в течение 90 мин. После охлаждения до комнатной температуры смесь разбавляли водой и Na2CO3 (водн. нас. раствор) и экстрагировали DCM. Органический слой отделяли, сушили (Na2SO4, фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; 7 М раствор аммиака в растворе метанола в DCM, от 0/100 до 2/98, до 3/97, до 10/90). Требуемые фракции собирали и концентрировали в вакууме, получая при этом (R)-4-(3-бромфенил)-4-метил-4,7 дигидропиразоло[1,5-а]пиразин-6-иламин (1,4 г, выход 82%) в виде желтого твердого вещества. Пример А 43. Получение промежуточного соединения 43: N,N-диметил-3-(трифторметил)-1 Нпиразол-1-сульфонамида 1,4-Диазабицикло[2.2.2]октан (5,44 г, 48,5 ммоль) и диметилсульфамоилхлорид (4,76 мл, 44,46 ммоль) добавляли к раствору 3-(трифторметил)пиразола (5,5 г, 40,42 ммоль) в ацетонитриле (50 мл) при 0 С. Смеси предоставляли возможность для нагревания до комнатной температуры и перемешивали в течение 18 ч. Смесь концентрировали в вакууме и остаток разбавляли водой. Продукт экстрагировалиAcOEt. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; DCM). Целевые фракции собирали и концентрировали в вакууме, получая при этом промежуточное соединение 43 (9,4 г, выход 95%) в виде бесцветного масла. Пример 44. Получение промежуточного соединения 44: 5-[(3-бромфенил)(гидрокси)метил]-N,Nдиметил-3-(трифторметил)-1H-пиразол-1-сульфонамида Раствор бутиллития (2,5 М в гексанах) (15,2 мл, 37,9 ммоль) добавляли к раствору промежуточного соединения 43 (8,4 г, 34,54 ммоль) в ТГФ (125 мл) при -78 С в атмосфере азота. Смесь перемешивали при -78 С в течение 45 мин и затем по каплям добавляли 2-бромбензальдегид (6 мл, 51,8 ммоль). Реакционную смесь перемешивали при -78 С в течение 30 мин и давали ей возможность нагреться до комнатной температуры и перемешивали в течение 1 ч. Смесь гасили NH4Cl (водн. нас. раствор) и продукт экстрагировали AcOEt. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; DCM в гептанах, от 0/100 до 10/90). Целевые фракции собирали и концентрировали в вакууме, получая при этом промежуточное соединение 44 (13,2 г, выход 89%) в виде бесцветного масла. Пример А 45. Получение промежуточного соединения 45: 5-[(3-бромфенил)карбонил]-N,N-диметил 3-(трифторметил)-1 Н-пиразол-1-сульфонамида Диоксид марганца (15,4 г, 169,3 ммоль) добавляли к раствору промежуточного соединения 44 (14,5 г, 33,86 ммоль) в 1,4-диоксане (150 мл). Смесь перемешивали при 120 С в течение 3 ч. Реакционную смесь охлаждали до 40 С и фильтровали через диатомовую землю. Растворитель выпаривали в вакууме,получая при этом промежуточное соединение 45 (25,6 г, выход 97%) в виде твердого вещества белого цвета, которое использовали как таковое на следующей стадии. Пример А 46. Получение промежуточного соединения 46: 5-[(3-бромфенил)[(трет-бутилсульфинил) имино]метил]-N,N-диметил-3-(трифторметил)-1 Н-пиразол-1-сульфонамида Изопропоксид титана(IV) (11,35 мл, 46,9 ммоль) добавляли к смеси промежуточного соединения 45(10 г, 23,46 ммоль) и 2-метил-2-пропансульфинамида (3,128 г, 25,81 ммоль) в толуоле (140 мл) в атмосфере азота. Смесь перемешивали при 110 С в течение 8 ч. Смесь охлаждали и выливали в солевой раствор при быстром перемешивании. Смесь фильтровали через диатомовую землю и осадок на фильтре промывали AcOEt. Фильтрат переносили в делительную воронку и органический слой отделяли, сушили(MgSO4), фильтровали и растворитель выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; AcOEt в DCM, от 0/100 до 10/90). Целевые фракции собирали и концентрировали в вакууме, получая при этом промежуточное соединение 46 (4 г, выход 32%) в виде желтого масла. Пример А 47. Получение промежуточного соединения 47: рац-5-[1-(3-бромфенил)-1-[(третбутилсульфинил)амино]этил]-N,N-диметил-3-(трифторметил)-1 Н-пиразол-1-сульфонамида Метилмагнийбромид (3 М раствор в диэтиловом простом эфире, 6,3 мл, 18,89 ммоль) добавляли к раствору промежуточного соединения 46 (4 г, 7,56 ммоль) в ТГФ (56 мл) при -78 С в атмосфере азота. Смесь перемешивали при -78 С в течение 30 мин и затем давали ей возможность нагреться до комнатной температуры и перемешивали в течение 18 ч. Реакционную смесь гасили NH4Cl (водн. нас. раствор) и продукт экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель;AcOEt в DCM, от 0/100 до 100/0). Целевые фракции собирали и концентрировали в вакууме, получая при этом промежуточное соединение 47 (3,6 г, выход 87%) в виде бесцветного масла, которое отверждается при стоянии. Пример А 48. Получение промежуточного соединения 48: рац-5-[1-амино-1-(3-бромфенил)этил]N,N-диметил-3-(трифторметил)-1 Н-пиразол-1-сульфонамида 4 М хлористо-водородную кислоту в диоксане (24,7 мл, 99 ммоль) добавляли к раствору промежуточного соединения 47 (3,6 г, 6,6 ммоль) в МеОН (5 мл) и смесь перемешивали при 80 С в запаянной трубке в течение 18 ч. Растворитель выпаривали в вакууме. Остаток выливали в NaHCO3 (водн. нас. раствор) и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; раствор метанола в DCM, от 0/100 2/98). Целевые фракции собирали и концентрировали в вакууме, получая при этом промежуточное соединение 48 (1,5 г, выход 54%) в виде бледно-желтого твердого вещества. Пример А 49. Получение промежуточного соединения 49: рац-N-1-(3-бромфенил)-1-[3(трифторметил)-1 Н-пиразол-5-ил]этил-2-хлорацетамидаDIPEA (1,9 мл, 11,2 ммоль) добавляли к раствору промежуточного соединения 48 (1,5 г, 4,49 ммоль) в DCM (20 мл) и смесь охлаждали на ледяной бане. Затем добавляли хлорацетилхлорид (0,429 мл, 5,38 ммоль) и смесь перемешивали при 0 С в течение 3 ч. Смесь разбавляли NH4Cl (водн. нас. раствор) и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель,AcOEt в DCM, от 0/100 до 30/70). Целевые фракции собирали и концентрировали в вакууме, получая при этом промежуточное соединение 49 (1,1 г, выход 60%) в виде бледно-желтого твердого вещества. Пример А 50. Получение промежуточного соединения 50: рац-4-(3-бромфенил)-4-метил-2(трифторметил)-4,5-дигидропиразоло[1,5-а]пиразин-6-она Раствор промежуточного соединения 49 (1,1 г, 2.68 ммоль) в ТГФ (40 мл) добавляли по каплям к суспензии гидрида натрия (0,214 г, 5,36 ммоль) в ТГФ (40 мл) при 0 С в атмосфере азота. Смесь перемешивали 0 С в течение 1 ч. Смесь разбавляли водой и продукт экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; AcOEt в DCM, от 0/100 до 20/80). Целевые фракции со- 27021723 бирали и концентрировали в вакууме, получая при этом промежуточное соединение 50 (0,92 г, выход 92%) в виде твердого вещества белого цвета. Пример А 51. Получение промежуточного соединения 51: рац-4-(3-бромфенил)-4-метил-2(трифторметил)-4,5-дигидропиразоло[1,5-а]пиразин-6-тиона Пентасульфид фосфора (0,82 г, 3,69 ммоль) добавляли к раствору промежуточного соединения 50(0,92 г, 2,46 ммоль) в пиридине (10 мл) и смесь нагревали при 90 С в течение 18 ч. Реакционную смесь концентрировали в вакууме и остаток очищали колоночной флэш-хроматографией (силикагель; AcOEt вDCM, от 0/100 до 100/0). Целевые фракции собирали и концентрировали в вакууме, получая при этом промежуточное соединение 51 (0,27 г, выход 28%) в виде бледно-желтого твердого вещества. Пример А 52. Получение промежуточного соединения 52: рац-4-(3-бромфенил)-4-метил-2(трифторметил)-4,5-дигидропиразоло[1,5-а]пиразин-6-аминаNH4Cl (0,148 г, 2,77 ммоль) добавляли к перемешиваемой суспензии промежуточного соединения 51 (0,27 г, 0,69 ммоль) в 2 М растворе аммиака в EtOH (6 мл) и смесь нагревали при 80 С в течение 18 ч. Растворитель удаляли в вакууме и остаток суспендировали в DCM и промывали водой. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; 7 М раствор аммиака в растворе метанола вDCM, от 0/100 до 2/98). Целевые фракции собирали и концентрировали в вакууме, получая при этом промежуточный продукт 52 (0,195 г, выход 75%) в виде бежевого твердого вещества. Пример А 53. Получение промежуточного соединения 53: рац-5-[(5-бром-2-фторфенил)(гидрокси) метил]-N,N-диметил-3-(трифторметил)-1 Н-пиразол-1-сульфонамида Промежуточное соединение 53 синтезировали такой же методикой, как методика, описанная в примере А 44. Исходя из промежуточного соединения 43 (17 г, 69,9 ммоль) промежуточное соединение 53 получали (28 г, выход 76%) в виде бесцветного масла. Пример А 54. Получение промежуточного соединения 54: рац-5-[(5-бром-2-фторфенил)карбонил]N,N-диметил-3-(трифторметил)-1 Н-пиразол-1-сульфонамида Промежуточное соединение 54 синтезировали такой же методикой, как методика, описанная в примере А 45. Исходя из промежуточного соединения 53 (28 г, 53,3 ммоль) промежуточное соединение 54 получали (25 г, выход 97%) в виде твердого вещества белого цвета, которое применяли как таковое на следующей стадии. Пример А 55. Получение промежуточного соединения 55: рац-5-[(5-бром-2-фторфенил)[(трет- 28021723 Промежуточное соединение 55 синтезировали такой же методикой, как методика, описанная в примере А 46. Исходя из промежуточного соединения 54 (25,6 г, 57,6 ммоль) промежуточное соединение 55 получали (21 г, выход 67%) в виде бледно-желтого твердого вещества. Пример А 56. Получение промежуточного соединения 56: рац-5-[1-(5-бром-2-фторфенил)-1-[(третбутилсульфинил)амино]этил]-N,N-диметил-3-(трифторметил)-1 Н-пиразол-1-сульфонамида Промежуточное соединение 56 синтезировали такой же методикой, как методика, описанная в примере А 47. Исходя из промежуточного соединения 55 (21 г, 38,36 ммоль) промежуточное соединение 56 получали (19 г, выход 88%) в виде бесцветного масла, которое отверждается при стоянии. Пример А 57. Получение промежуточного соединения 57: рац-1-(5-бром-2-фторфенил)-1-[3(трифторметил)-1 Н-пиразол-5-ил]этанамина 1,25 М хлористо-водородную кислоту в метаноле (159 мл, 199 ммоль) добавляли к промежуточному соединению 56 (18,7 г, 33,2 ммоль) и смесь перемешивали при 60 С в запаянной трубке в течение 3 ч. Растворитель выпаривали в вакууме. Остаток выливали в NaHCO3 (вод. нас. раствор) и экстрагировалиDCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворители выпаривали в вакууме, получая при этом промежуточное соединение 57 (11,5 г, выход 98%) в виде бледно-желтого твердого вещества, которое применяли как таковое на следующей стадии без дополнительной очистки. Пример А 58. Получение промежуточного соединения 58: рац-N-1-(5-бром-2-фторфенил)-1-[3(трифторметил)-1 Н-пиразол-5-ил]этил-2-хлорацетамида Промежуточное соединение 58 синтезировали такой же методикой, как методика, описанная в примере А 49. Исходя из промежуточного соединения 57 (11,5 г, 32,66 ммоль) промежуточное соединение 58 получали (6,6 г, выход 47%) в виде бледно-желтого твердого вещества. Пример А 59. Получение промежуточного соединения 59: рац-4-(5-бром-2-фторфенил)-4-метил-2(трифторметил)-4,5-дигидропиразоло[1,5-а]пиразин-6-она Промежуточное соединение 59 синтезировали такой же методикой, как методика, описанная в примере А 50. Исходя из промежуточного соединения 58 (6,6 г, 15,4 ммоль) промежуточное соединение 59
МПК / Метки
МПК: A61K 31/4985, A61P 25/28, C07D 487/04
Метки: васе, производные, бета-секретазы, качестве, 4,7-дигидропиразоло[1,5-a]пиразин-6-иламина, ингибиторов, используемые
Код ссылки
<a href="https://eas.patents.su/30-21723-proizvodnye-47-digidropirazolo15-apirazin-6-ilamina-ispolzuemye-v-kachestve-ingibitorov-beta-sekretazy-vase.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 4,7-дигидропиразоло[1,5-a]пиразин-6-иламина, используемые в качестве ингибиторов бета-секретазы (васе)</a>
Производные 5,6-дигидро-2h-[1,4]оксазин-3-иламина в качестве ингибиторов бета-секретазы (bace)

Номер патента: 21240
Опубликовано: 29.05.2015
Авторы: Мартинес Ламенка Каролина, Тресадерн Гэри Джон, Гейсен Хенрикус Якобус Мария, Макдональд Грегор Джеймс, Ван Гол Михиль Люк Мария, Ромбаутс Фредерик Ян Рита, Трабанко-Суарес Андрес Авелино
МПК: A61K 31/535, A61P 25/16, A61K 31/5355...
Метки: бета-секретазы, bace, 5,6-дигидро-2h-[1,4]оксазин-3-иламина, ингибиторов, производные, качестве
Формула / Реферат:
1. Соединение формулы (I)или его таутомерная или стереоизомерная форма, гдеR1, R2 и R3 независимо выбраны из группы, состоящей из водорода и фтора;R4 представляет собой фтор или трифторметил;R5 выбран из группы, состоящей из C1-3-алкила и циклопропила;X1, X2, X3, X4 независимо представляют собой C(R6);каждый R6 выбран из группы, состоящей из водорода и галогена;L представляет собой связь или -N(R7)CO-, где R7 представляет собой водород или...
Производные пирролидин-2-она и пиперидин-2-она, используемые в качестве ингибиторов 11-бета-гидроксистероид-дегидрогеназы

Номер патента: 11097
Опубликовано: 30.12.2008
Авторы: Ван Дер Векен Луи Йозеф Элизабет, Димитров Владимир Димчев, Никифоров Тео Теофанов, Бюйк Кристоф Франсис Роберт Нестор, Яроскова Либуше, Линдерс Йоаннес Теодорус Мария
МПК: A61K 31/45, A61P 3/04, A61K 31/4015...
Метки: используемые, пиперидин-2-она, ингибиторов, пирролидин-2-она, 11-бета-гидроксистероид-дегидрогеназы, качестве, производные
Формула / Реферат:
1. Соединение формулы его N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где n равно 1 или 2; L представляет собой C1-3алкильный линкер, необязательно замещенный одним или двумя заместителями, выбранными из C1-4алкила, C1-3алкилоксиС1-4алкила, гидроксиС1-4алкила, гидрокси, C1-3алкилокси или фенилС1-4алкила; M представляет собой прямую связь или C1-3алкильный линкер, необязательно замещенный...
Производные 1,2-дизамещенного 4-бензиламинопирролидина в качестве ингибиторов белка-переносчика холестерилового эфира, используемые для лечения заболеваний, таких как гиперлипидемия или артериосклероз

Номер патента: 17939
Опубликовано: 30.04.2013
Авторы: Ясошима Кайо, Мияке Такахиро, Ямада Кен, Каванами Тошио, Омори Осаму, Моги Мунето, Имасе Хидетомо
МПК: C07D 207/14, A61P 3/06, A61K 31/40...
Метки: производные, белка-переносчика, эфира, качестве, таких, гиперлипидемия, ингибиторов, холестерилового, 4-бензиламинопирролидина, заболеваний, лечения, используемые, 1,2-дизамещенного, артериосклероз
Формула / Реферат:
1. Соединение формулы (I)в которой R1 обозначает (C1-C7)алкил-O-C(O)-, (C1-C7)алканоил, пиримидинил или тетразолил, где указанный пиримидинил необязательно содержит от 1 до 3 заместителей, выбранных из группы, включающей галоген, ди(C1-C7)алкиламиногруппу, (C1-C7)алкоксигруппу, гетероциклил, где заместитель гетероциклил дополнительно необязательно содержит от 1 до 3 заместителей, выбранных из группы, включающей (C1-C7)алкил, гидроксигруппу или...
Производные адамантилпирролидин-2-она в качестве ингибиторов 11-бета-гидроксистероид-дегидрогеназы

Номер патента: 11021
Опубликовано: 30.12.2008
Авторы: Линдерс Йоаннес Теодорус Мария, Яроскова Либуше, Ван Дер Векен Луи Йозеф Элизабет, Бюйк Кристоф Франсис Роберт Нестор
МПК: A61K 31/402, C07D 207/26, C07D 211/76...
Метки: адамантилпирролидин-2-она, ингибиторов, качестве, производные, 11-бета-гидроксистероид-дегидрогеназы
Формула / Реферат:
1. Соединение, имеющее формулу его N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где n равно 1 или 2; М представляет собой прямую связь или C1-3алкильный линкер, необязательно замещенный одним или двумя заместителями, выбранными из С1-4алкила, C1-3алкилокси-С1-4алкила, гидрокси-С1-4алкила, гидрокси, C1-3алкилокси- или фенил-С1-4алкила; каждый из R1 и R2 независимо представляет собой водород,...
Производные циклогексилпиразол-лактама в качестве ингибиторов 11-бета-гидроксистероиддегидрогеназы 1

Номер патента: 15675
Опубликовано: 31.10.2011
Авторы: Сюй Яньпин, Мэбри Томас Эдвард, Айкер Томас Дэниел, Стефенсон Грегори Алан, Снайдер Нэнси Джун, Саеед Ашраф, Тянь Хунци, Анцевено Петер Бьяджо, Уоллэйс Оуэн Брендан, Красутский Алексей Павлович, Виннероски Леонард Лэрри, Ли Жэньхуа
МПК: A61P 9/10, A61P 3/10, A61K 31/416...
Метки: качестве, производные, ингибиторов, циклогексилпиразол-лактама, 11-бета-гидроксистероиддегидрогеназы
Формула / Реферат:
1. Соединение, структура которого представлена формулойгде R1представляет собой -H, -галоген;R2 представляет собой -галоген;R3 представляет собой -H;R4 представляет собой -OH, -галоген, -(C1-C6)алкокси, -O-CH2-C(O)NH2, -C(O)N(R10)(R11),при этом пунктирная линия указывает на точку присоединения к положению R4в формуле I;где n равно 1;R5 представляет собой -H, -галоген, -OH, -CN, -(C1-C4)алкил (необязательно замещенный 1-3 атомами галогена),...
Предыдущий патент: Способ получения удобрений, содержащих диспергированную микронизированную серу
Следующий патент: Соединения 4-замещенного-3-фенилсульфанилметилбицикло[3.1.0]гексана в качестве антагонистов mglur 2/3
Случайный патент: Трубы нефтяного сортамента для развальцовки в скважине и дуплексная нержавеющая сталь, используемая для труб нефтяного сортамента для развальцовки