Соединения 4-замещенного-3-фенилсульфанилметилбицикло[3.1.0]гексана в качестве антагонистов mglur 2/3
Номер патента: 21724
Опубликовано: 31.08.2015
Авторы: Смит Стефон Корнелл, Ли Ренуа, Ветман Татьяна Натали, Митч Чарльз Говард







Формула / Реферат
1. Соединение формулы

где каждый из R1 и R2 независимо представляет собой водород, C1-C3 алкоксикарбонилоксиметил, C1-C5 алкилкарбонилоксиметил или C3-6 циклоалкилкарбонилоксиметил;
R3 независимо в каждом случае представляет собой метил, фтор или хлор;
R4 представляет собой гидроксил, амино, метилкарбониламино или 1,2,4-триазолилтио и
n представляет собой 1 или 2;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, отличающееся тем, что каждый из R1 и R2 представляет собой водород, или его фармацевтически приемлемая соль.
3. Соединение по п.1, отличающееся тем, что оба R1 и R2 отличны от водорода, или его фармацевтически приемлемая соль.
4. Соединение по п.1, отличающееся тем, что R1 и R2 одинаковы и отличны от водорода, или его фармацевтически приемлемая соль.
5. Соединение по п.4, отличающееся тем, что каждый из R1 и R2 представляет собой изопропилоксикарбонилоксиметил.
6. Соединение по любому из пп.1-5, отличающееся тем, что n представляет собой 2 и группы R3 находятся в 3 и 4 положениях фенила.
7. Соединение по любому из пп.1-6, отличающееся тем, что R3 независимо в каждом случае представляет собой хлор или фтор.
8. Соединение по п.1, представляющее собой (1S,2R,3S,4S,5R,6R)-2-амино-3-{[(3,4-дифторфенил)сульфанил]метил}-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновую кислоту, или его фармацевтически приемлемая соль.
9. Соединение по п.1, представляющее собой бис({[(1-метилэтокси)карбонил]окси}метил)-(1S,2R,3S,4S,5R,6R)-2-амино-3-{[(3,4-дифторфенил)сульфанил]метил}-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат, или его фармацевтически приемлемая соль.
10. Соединение по п.1, представляющее собой (1S,2R,3S,4S,5R,6R)-2-амино-3-{[(3-хлор-4-фторфенил)сульфанил]метил}-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновую кислоту, или его фармацевтически приемлемая соль.
11. Соединение по п.1, представляющее собой бис({[(1-метилэтокси)карбонил]окси}метил)-(1S,2R,3S,4S,5R,6R)-2-амино-3-{[(3-хлор-4-фторфенил)сульфанил]метил}-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат, или его фармацевтически приемлемая соль.
12. Применение соединения по любому из пп.1-11 или его фармацевтически приемлемой соли для лечения депрессивных расстройств.
13. Применение соединения по любому из пп.1-11 или его фармацевтически приемлемой соли для лечения расстройств чрезмерной сонливости.
14. Применение по п.12 или 13 у человека.
Текст
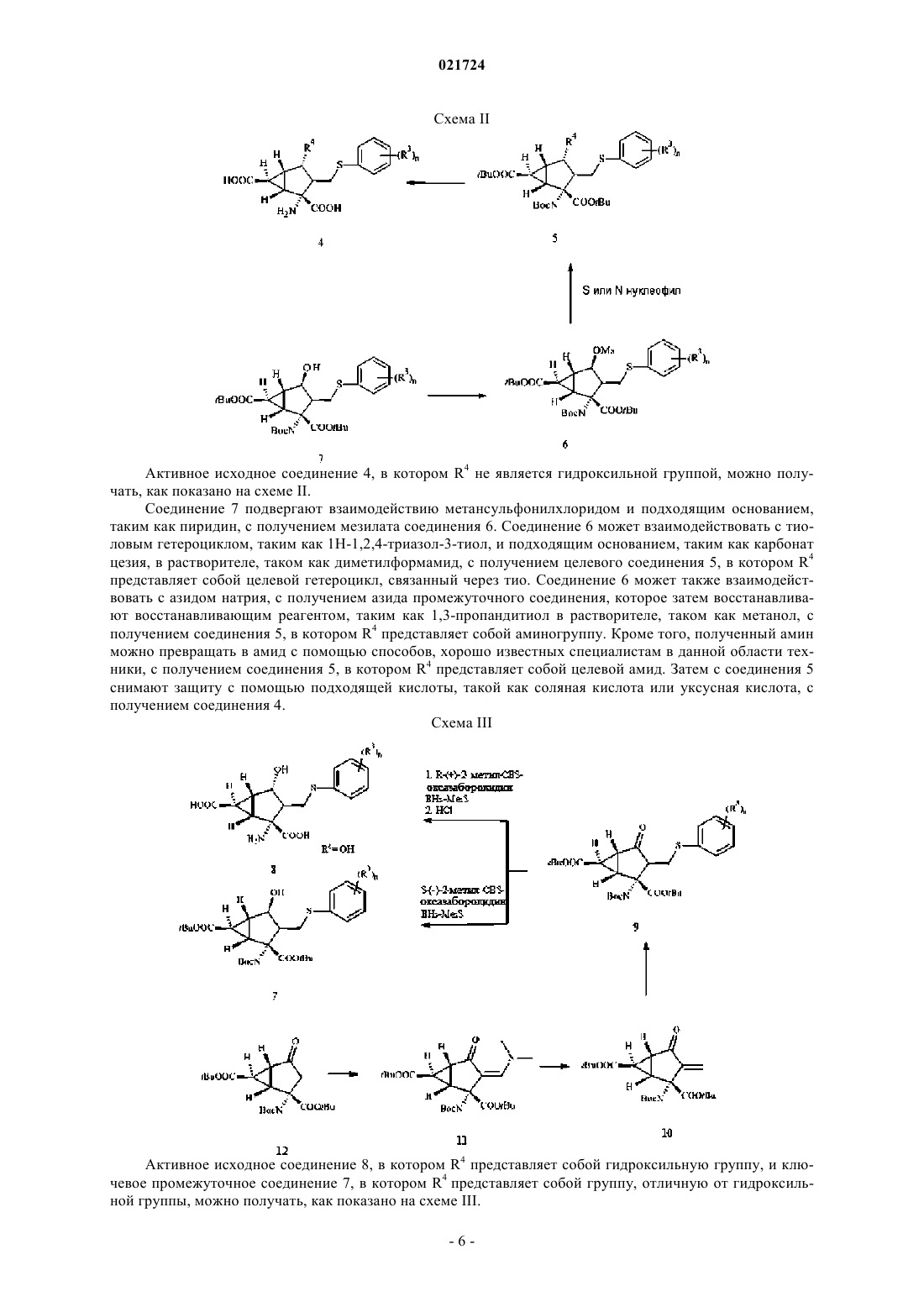
Предложен антагонист рецептора mGlu 2/3 формулы Смит Стефон Корнелл, Ли Ренуа,Митч Чарльз Говард, Ветман Татьяна Натали (US) Лыу Т.Н. (RU)(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Глутамат представляет собой основной возбуждающий нейромедиатор в головном мозге и вовлечен в большое количество физиологических процессов, опосредованных по меньшей мере 11 различными рецепторами, каждый со своей фармакологией. Подтипы 2 и 3 метаботропного глутаматного рецептора(также известные как mGlu2 и mGlu3) часто объединяют как mGlu рецепторы II группы на основании гомологии их последовательностей, сходного связывания вторичного мессенджера и сходных фармакологических характеристик. Антагонисты mGlu2/3 рецепторов обладали значительным фармакологическим действием на животных моделях в отношении депрессивных расстройств и расстройств чрезмерной сонливости. В связи с этим полагают, что антагонисты mGlu2/3 могут подходить для лечения депрессивных расстройств, таких как большое депрессивное расстройство (БДР), монополярная депрессия, дистимия и/или циклотимия, и/или подходить для лечения расстройств чрезмерной сонливости, таких как чрезмерная дневная сонливость (EDS), гиперсомния, связанная с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна (включая, но не ограничиваясь ими, расстройство сна, вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатическая гиперсомния и/или чрезмерная сонливость, связанная с невосстанавливающим сном (NRS). В патенте США 5916920 описаны некоторые соединения 3-монозамещенногобицикло[3.1.0]гексана в качестве модуляторов метаботропного рецептора глутамата, подходящие для лечения различных расстройств, в том числе в качестве антидепрессивных агентов. В патенте США 7157594 описаны различные соединения 3-монозамещенного-бицикло[3.1.0]гексана в качестве антагонистов mGlu рецептора II группы для применения для лечения различных расстройств, включающих депрессивные симптомы. В заявке на патент США 2007/0021394 А 1 описаны различные соединения 3 монозамещенного-бицикло[3.1.0]гексана в качестве антагонистов mGlu рецептора II группы и их пролекарства для применения для лечения различных расстройств, включая депрессию. В настоящем изобретении предложено семейство соединений 4-замещенного-3 фенилсульфанилметилбицикло[3.1.0]гексана, обладающих высокой антагонистической активностью в отношении mGlu2 и mGlu3 рецепторов. Соединения согласно настоящему изобретению также являются селективными по отношению к указанным mGlu2 и mGlu3 рецепторам, в частности, по сравнению с другими mGlu рецепторами. Также посредством изучения некоторых соединений на животных моделях было показано, что соединения согласно настоящему изобретению могут подходить для лечения депрессивных расстройств (которые могут включать большое депрессивное расстройство (БДР), монополярную депрессию, дистимию и/или циклотимию) и расстройств чрезмерной сонливости (которые могут включать чрезмерную дневную сонливость (EDS), гиперсомнию, связанную с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна (включая, но не ограничиваясь ими,расстройство сна, вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатическую гиперсомнию и/или чрезмерную сонливость, связанную с невосстанавливающим сном (NRS). Антидепрессивный и стимулирующие эффекты указанного механизма также позволяют предположить воздействие на симптомы депрессивных расстройств, таких как усталость, которые иначе трудно поддаются лечению существующими антидепрессантами. В настоящем изобретении предложены соединения формулы I где каждый из R1 и R2 независимо представляет собой водород, C1-C3 алкоксикарбонилоксиметил, C1-C5 алкилкарбонилоксиметил или C3-6 циклоалкилкарбонилоксиметил;R3 независимо в каждом случае представляет собой метил, фтор или хлор;n представляет собой 1 или 2; или их фармацевтически приемлемые соли. Особенностью настоящего изобретения является то, что соединения формулы I, где оба, R1 и R2,представляют собой водород (дикислотные соединения), являются терапевтически активными соединениями in vivo, тогда как соединения, в которых R1 или R2 или оба отличны от водорода, представляют собой пролекарства их терапевтически активных дикислотных аналогов. Соединения, в которых R1 илиR2 или оба отличны от водорода, гидролизуются in vivo с получением терапевтически активного дикислотного аналога. При пероральном введении указанные пролекарственные соединения, в частности дисложноэфирные пролекарства, обеспечивают повышенную биодоступность указанного дикислотного-1 021724 метаболита по сравнению с пероральным введением дикислотных соединений (оба R1 и R2 представляют собой водород), но указанные дикислотные соединения обеспечивают лучшую активность при внутривенном, внутримышечном или подкожном введении. В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации по меньшей мере с одним фармацевтически приемлемым носителем, разбавителем или наполнителем. Кроме того, в указанном аспекте изобретения предложена фармацевтическая композиция, подходящая для лечения депрессивных расстройств, например большого депрессивного расстройства, монополярной депрессии, дистимии и/или циклотимии, содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или более фармацевтически приемлемыми наполнителями, носителями или разбавителями. Согласно другому варианту реализации указанного аспекта изобретения предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации по меньшей мере с одним фармацевтически приемлемым носителем, наполнителем или разбавителями и возможно другие терапевтические ингредиенты. Согласно еще одному варианту реализации указанного аспекта изобретения указанная фармацевтическая композиция дополнительно содержит второй терапевтический агент, представляющий собой лекарственное средство, подходящее для лечения депрессивных расстройств, например ингибитор обратного захвата серотонина, например флуоксетин и/или циталопрам. Согласно еще одному варианту реализации указанного аспекта изобретения предложена фармацевтическая композиция, подходящая для лечения расстройств чрезмерной сонливости, например чрезмерной дневной сонливости (EDS), гиперсомнии, связанной с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна (включая, но не ограничиваясь ими, расстройство сна,вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатической гиперсомнии и/или чрезмерной сонливости, связанной с невосстанавливающим сном (NRS), содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или более фармацевтически приемлемыми наполнителями, носителями или разбавителями. В настоящем изобретении также предложен способ лечения депрессивных расстройств, например большого депрессивного расстройства, монополярной депрессии, дистимии и/или циклотимии, у млекопитающего, включающий введение млекопитающему, которое нуждается в подобном лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Согласно другому варианту реализации указанного аспекта изобретения указанный способ дополнительно включает введение второго терапевтического агента в совместной, отдельной или последовательной комбинации,который представляет собой лекарственное средство, подходящее для лечения депрессивных расстройств, например ингибитор обратного захвата серотонина, например флуоксетин и/или циталопрам. Согласно другим вариантам реализации изобретения предложены способы лечения расстройств чрезмерной сонливости, включающие введение млекопитающему, которое нуждается в подобном лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Согласно другим вариантам реализации указанного аспекта изобретения указанная чрезмерная сонливость возникает вследствие любого одного или более следующих расстройств: чрезмерная дневная сонливость(EDS), гиперсомния, связанная с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна (включая, но не ограничиваясь ими, расстройство сна, вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатическая гиперсомния и/или чрезмерная сонливость, связанная с невосстанавливающим сном (NRS). Согласно одному конкретному варианту реализации указанных способов лечения указанное млекопитающее представляет собой человека. В настоящем изобретении также предложено соединение формулы I или его фармацевтически приемлемая соль для применения в терапии. В данном аспекте изобретения предложено соединение формулы I или его фармацевтически приемлемая соль для применения для лечения депрессивных расстройств. Согласно дополнительным вариантам реализации указанное депрессивное расстройство представляет собой любое из большого депрессивного расстройства (БДР), монополярной депрессии, дистимии и/или циклотимии. Согласно другим вариантам реализации аспекта настоящего изобретения предложено соединение формулы I или его фармацевтически приемлемая соль для применения в совместной, отдельной или последовательной комбинации с ингибитором обратного захвата серотонина, например флуоксетином и/или циталопрамом, для лечения депрессивных расстройств. Кроме того, указанный аспект изобретения включает соединение формулы I или его фармацевтически приемлемую соль для применения для лечения расстройств чрезмерной сонливости. Согласно конкретным вариантам реализации аспекта изобретения чрезмерная сонливость возникает вследствие любо-2 021724 го одного или более следующих расстройств: чрезмерная дневная сонливость (EDS), гиперсомния, связанная с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна(включая, но не ограничиваясь ими, расстройство сна, вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатическая гиперсомния и/или чрезмерная сонливость, связанная с невосстанавливающим сном (NRS). Согласно одному конкретному варианту реализации указанного аспекта изобретения указанные применения осуществляют у млекопитающих, в частности человека. Согласно другому аспекту настоящего изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения депрессивных расстройств, например большого депрессивного расстройства, монополярной депрессии, дистимии и/или циклотимии. Согласно другому варианту реализации указанного аспекта изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли и второго терапевтического агента, подходящего для лечения депрессивных расстройств, например, ингибитора обратного захвата серотонина, например флуоксетина и/или циталопрама, для получения лекарственного средства для лечения депрессивных расстройств. Согласно другому варианту реализации изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения расстройств чрезмерной сонливости. Согласно конкретным вариантам реализации указанного аспекта изобретения указанное лекарственное средство подходит для лечения любого одного или более следующих расстройств: чрезмерная дневная сонливость (EDS), гиперсомния, связанная с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна(включая, но не ограничиваясь ими, расстройство сна, вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатическая гиперсомния и/или чрезмерная сонливость, связанная с невосстанавливающим сном (NRS). Соединения согласно настоящему изобретению содержат основные и кислотные фрагменты и соответственно взаимодействуют с множеством органических и неорганических кислот и оснований с получением фармацевтически приемлемых солей. Фармацевтически приемлемые соли каждого из соединений согласно настоящему изобретению включены в объем настоящей заявки. В настоящей заявке термин"фармацевтически приемлемая соль" относится к любой соли соединения согласно изобретению, которая, по существу, нетоксична для живых организмов. Такие соли включают перечисленные в Journal ofPharmaceutical Science, 66, 2-19 (1977), известные специалистам в данной области. Предпочтительные классы соединений согласно настоящему изобретению представляют собой соединения, в которых: 1) оба, R1 и R2, представляют собой водород; 2) R1 или R2 или оба отличны от водорода; 3) оба, R1 и R2, отличны от водорода; 4) R1 и R2 одинаковы и отличны от водорода; 5) каждый из R1 и R2 представляет собой изопропоксикарбонилоксиметил; 6) n представляет собой 2; 7) R3 независимо в каждом случае представляет собой фтор или хлор; 8) n представляет собой 2 и группы R3 находятся в 3 и 4 положениях фенила. 9) n представляет собой 2 и каждая из групп R3 независимо представляет собой фтор или хлор и находится в 3 и 4 положениях фенила. 10) n представляет собой 2, обе группы R3 представляют собой фтор и указанные группы фтора находятся в 3 и 4 положениях фенила; 11) n представляет собой 2 и указанные группы R3 совместно с фенильным фрагментом, к которому они присоединены, образуют 3-хлор-4-фторфенил; 12) R4 представляет собой гидроксил. Следует понимать, что следующие предпочтительные соединения представляют собой комбинацию указанных выше предпочтительных выборок для заданных заместителей с предпочтительными выборками для других заместителей. Примеры таких комбинаций включают, но не ограничиваются ими, следующие предпочтительные классы соединений: 13) предпочтительные соединения согласно любому из абзацев 1-5 (предпочтительные выборки дляR1 и R2), в которых n представляет собой 2, обе группы R3 представляют собой фтор и указанные группы фтора находятся в 3 и 4 положениях фенила (абзац 10); 14) предпочтительные соединения согласно любому из абзацев 1-5 (предпочтительные выборки дляR1 и R2), в которых n представляет собой 2 и группы R3 совместно с фенильным фрагментом, к которому они присоединены, образуют 3-хлор-4-фторфенил (абзац 11); 15) предпочтительные соединения согласно любому из абзацев 1-5 (предпочтительные выборки дляR1 и R2), в которых R4 представляет собой гидроксил (абзац 12); 16) предпочтительные соединения согласно любому из абзацев 13-14, в которых R4 представляет собой гидроксил (абзац 12). Конкретные предпочтительные соединения представляют собой соединения, описанные в примерах, включая их свободные основания и фармацевтически приемлемые соли. Некоторыми предпочтительными соединениями являются(1S,2R,3S,4S,5R,6R)-2-амино-3-[(3,4-дифторфенил)сульфанил]метил-4-гидроксибицикло[3.1.0] гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль;[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль; бис([(1-метилэтокси)карбонил]оксиметил) (1S,2R,3S,4S,5R,6R)-2-амино-3-[(3,4-дифторфенил) сульфанил]метил-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемая соль и бис([(1-метилэтокси)карбонил]оксиметил)(1S,2R,3S,4S,5R,6R)-2-амино-3-[(3-хлор-4-фторфенил)сульфанил]метил-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемая соль (т.е. соединения примеров 1, 2, 12, 22 и 32 и их различные фармацевтически приемлемые соли). В настоящей заявке сокращения определяются следующим образом:"БСА" означает бычий сывороточный альбумин."DMEM" означает минимальную среду Игла, модифицированную по способу Дульбекко."HBSS" означает сбалансированный солевой раствор Хенкса."HPLC" означает жидкостную хроматографию высокого давления."IC50" означает концентрацию, при которой достигается 50% от максимального ингибирования."в.в." означает внутривенный или внутривенно."LC/MS" означает жидкостную хроматографию с последующей масс-спектроскопией."mFST" означает тест принудительного плавания на мышах; животная модель для антидепрессивной активности."ЯМР" означает ядерный магнитный резонанс."tBu" означает фрагмент третичного-бутила. Общая химия Соединения согласно настоящему изобретению можно получать согласно следующим схемам синтеза посредством способов, хорошо известных и применяемых в данной области техники. Подходящие условия реакций для стадий указанных схем хорошо известны в данной области техники и подходящие заменители для растворителей и сореагентов известны в рамках данной области техники. Кроме того,специалистам в данной области техники будет понятно, что синтетические промежуточные вещества можно выделять и/или очищать с помощью различных хорошо известных способов при необходимости или желании и настолько часто, чтобы было возможно применять различные промежуточные вещества непосредственно на последующих стадиях синтеза с небольшой очисткой или без очистки. Кроме того,специалисту в данной области техники будет понятно, что в некоторых случаях порядок включения фрагментов не важен. Конкретный порядок стадий, требуемый для получения соединений согласно настоящему изобретению, зависит от конкретного синтезируемого соединения, исходного соединения и относительной реакционной способности замещенных фрагментов, как хорошо известно квалифицированному химику. Все заместители, если не указано иное, такие как определено выше, и все реагенты хорошо известны и применяемы в данной области техники. Соединение 1, представляющее собой пролекарство, можно получать как показано на схеме I, где Соединение 4 подвергают взаимодействию с реагентом, защищающим аминогруппу, таким как дитрет-бутилдикарбонат, в условиях, хорошо известных специалистам в данной области техники, с получением соединения 3. Если группы R1 и R2 в соединении 2 одинаковы, соединение 3 подвергают взаимодействию с достаточным количеством подходящего хлорметилалкилкарбоната и подходящими реагентами, такими как йодид натрия и карбонат цезия, в подходящем растворителе, таком как диметилформамид, с получением целевого ди-сложноэфирного соединения 2 с одинаковыми R1 и R2. Если R1 и R2 в соединении 2 различны, путем регулирования количества первого хлорметилалкилкарбоната до примерно 1 экв. карбоновую кислоту на 5-членном кольце можно сначала превращать в R2 моносложный эфир. Указанное соединение R2 моносложного эфира можно дополнительно подвергать взаимодействию с 1 экв. другого хлорметилалкилкарбоната. Группу свободной карбоновой кислоты на 3-членном кольце можно затем превращать в R1 сложный эфир с получением целевого дисложного эфира с различными R1 и R2. Для получения R2 моносложного эфира на 5-членном кольце соединения 2 соединение 3 подвергают взаимодействию с примерно 1 экв. подходящего хлорметилалкилкарбоната и подходящими реагентами, такими как йодид натрия и карбонат цезия, в подходящем растворителе, таком как диметилформамид, с получением целевого R2 моносложного эфира соединения 2, в котором R1 представляет собой водород. Для получения R1 моносложного эфира на 3-членном кольце группу карбоновой кислоты на 5 членном кольце следует сначала защитить, поскольку она более реакционноспособна в основных условиях. Более конкретно, группа карбоновой кислоты на 5-членном кольце в соединении 3 может взаимодействовать с -хлор-4-метокситолуолом, йодидом натрия и бикарбонатом натрия в подходящем растворителе, таком как диметилформамид, с получением 4-метоксилбензил моносложного эфира. Затем указанную группу карбоновой кислоты на 3-членном кольце защищенного соединения 4-метоксилбензил моносложного эфира подвергают взаимодействию с подходящим хлорметилалкилкарбонатом с получением целевого R1 сложного эфира на указанном 3-членном кольце. Дисложный эфир обрабатывают подходящей кислотой, такой как трифторуксусная кислота, со снятием защиты с 4-метоксилбензиловой и Nтрет-бутоксикарбониловой групп, с получением целевого R1 моносложного эфира соединения 1, в котором R2 представляет собой водород. Затем с соединения 2, включая R2 моносложный эфир и дисложный эфир с одинаковыми или различными R1 и R2, снимают защиту с помощью подходящей кислоты, такой как соляная кислота в диоксане, с получением целевого соединения 1 или фармацевтически приемлемой соли. Активное исходное соединение 4, в котором R4 не является гидроксильной группой, можно получать, как показано на схеме II. Соединение 7 подвергают взаимодействию метансульфонилхлоридом и подходящим основанием,таким как пиридин, с получением мезилата соединения 6. Соединение 6 может взаимодействовать с тиоловым гетероциклом, таким как 1 Н-1,2,4-триазол-3-тиол, и подходящим основанием, таким как карбонат цезия, в растворителе, таком как диметилформамид, с получением целевого соединения 5, в котором R4 представляет собой целевой гетероцикл, связанный через тио. Соединение 6 может также взаимодействовать с азидом натрия, с получением азида промежуточного соединения, которое затем восстанавливают восстанавливающим реагентом, таким как 1,3-пропандитиол в растворителе, таком как метанол, с получением соединения 5, в котором R4 представляет собой аминогруппу. Кроме того, полученный амин можно превращать в амид с помощью способов, хорошо известных специалистам в данной области техники, с получением соединения 5, в котором R4 представляет собой целевой амид. Затем с соединения 5 снимают защиту с помощью подходящей кислоты, такой как соляная кислота или уксусная кислота, с получением соединения 4. Схема III Активное исходное соединение 8, в котором R4 представляет собой гидроксильную группу, и ключевое промежуточное соединение 7, в котором R4 представляет собой группу, отличную от гидроксильной группы, можно получать, как показано на схеме III. Соединение 12 (см. подробности синтеза в WO 03/104217 A2) подвергают взаимодействию с третбутоксибис(диметиламино)метаном в толуоле, с получением соединения 11. Соединение 11 в подходящем растворителе, таком как тетрагидрофуран, обрабатывают подходящим основанием, таким как триэтиламин, и подходящим восстанавливающим реагентом, таким как гидрид диизобутилалюминия, при пониженной температуре, с получением соединения 10. Затем соединение 10 подвергают взаимодействию с триэтиламином и соответствующим замещенным тиофенолом, таким как 3,4-дифтортиофенол, в подходящем растворителе, таком как толуол, с получением соединения 9. Кетоновую группу соединения 9 можно селективно восстановить до целевого (S) гидроксильного или (R) гидроксильного соединения путем применения (R)-метилоксазаборолидина или (S)-метилоксазаборолидина соответственно. С указанного (S) гидроксильного промежуточного соединения снимают защиту с помощью подходящей кислоты, такой как соляная кислота, в растворителе, таком как диоксан, с получением целевого активного исходного соединения 8, в котором R4 представляет собой (S)-гидроксильную группу. Указанное (R) гидроксильное промежуточное соединение 7 можно превратить в желаемый продукт посредством способа, показанного на схеме II. Пример получения 1. ди-трет-Бутил-(1S,2R,5R,6R)-2-(трет-бутоксикарбониламино)-3-(диметиламинометилен)-4-оксобицикло[3.1.0]гексан-2,6-дикарбоксилат(90,90 мл) добавляли трет-бутоксибис(диметиламино)метан (12,83 г, 73,63 ммоль). Затем указанную смесь нагревали до 80 С в течение 1 ч и затем оставляли охлаждаться до комнатной температуры. Объем растворителя уменьшали до примерно 35 мл. Смесь перемешивали во время добавления диэтилового эфира и гексана, чтобы вызвать образование осадка. Твердые вещества собирали посредством фильтрации, промывали гексанами и сушили на воздухе с получением титульного соединения (16,7 г, 35,79 ммоль, 97,2% выход). MS (m/z): 467.2 (М+Н). Пример получения 2. ди-трет-Бутил-(1S,2R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-метилиден 4-оксобицикло[3.1.0]гексан-2,6-дикарбоксилат К раствору ди-трет-бутил-(1S,2R,5R,6R)-2-(трет-бутоксикарбониламино)-3-(диметиламинометилен)-4-оксобицикло[3.1.0]гексан-2,6-дикарбоксилата (15,7 г, 33,8 ммоль) в тетрагидрофуране (340 мл) добавляли триэтиламин (6,6 мл, 47,32 ммоль). Смесь охлаждали до -78 С. Добавляли гидрид диизобутилалюминия (1N в толуоле, 50 мл, 50 ммоль) в течение 1 ч. Смесь перемешивали в течение двух дополнительных часов. Затем добавляли 30 мл насыщенного хлорида аммония. Смесь оставляли нагреваться до комнатной температуры. Смесь переносили в делительную воронку и промывали солевым раствором. Органический слой сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии (от 0 до 50% этилацетат/гексаны) с получением титульного соединения (12 г, 33,34 ммоль, 83,8% выход). MS (m/z): 422 (МН). Пример получения 3. ди-трет-Бутил-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4 дифторфенил)сульфанил]метил-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат ди-трет-Бутил-(1S,2R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-метилиден-4-оксобицикло[3.1.0] гексан-2,6-дикарбоксилат (1,03 г, 2,43 ммоль) в диэтиловом эфире (100 мл) барботировали газообразным азотом в течение 10 мин. Добавляли 3,4-дифтортиофенол (0,36 г, 2,43 ммоль) и триэтиламин (0,01 мл,0,05 мкмоль). Смесь нагревали до 40 С и перемешивали в течение 15 мин. Затем смесь оставляли охлаждаться до комнатной температуры, переносили в делительную воронку, разбавляли гексаном (40 мл),промывали 2N водным KOH (130 мл), сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением ди-трет-бутил-(1S,2R,3S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дифторфенил)сульфанил]метил-4-оксобицикло[3.1.0]гексан-2,6-дикарбоксилата (1,35 г, 2,37 ммоль ): MS (m/z): 567.8 (М-Н). Указанное вещество брали в 120 мл диэтилового эфира и медленно добавляли в течение 2 ч к 200 мл эфирного раствора при -10 С, содержащего R-(+)-2-метилCBS-оксазаборолидин (981,72 мг, 3,54 ммоль) и комплекс боран-метилсульфид (2 М в тетрагидрофуране,5,02 мл, 10,04 ммоль). Смесь перемешивали в течение дополнительного часа после последнего добавления ди-трет-бутил-(1S,2R,3S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дифторфенил)сульфанил] метил-4-оксобицикло[3.1.0]гексан-2,6-дикарбоксилата. Силикагель (30 г) добавляли в течение 30 мин и реакционную смесь постепенно нагревали до комнатной температуры. Суспензию фильтровали и промывали 300 мл диэтилового эфира. Растворитель концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии, элюируя (от 0 до 15% этилацетат/гексаны), с получением титульного соединения (0,844 г, 1,48 ммоль, 60,7% выход): MS (m/z): 569.8(М-Н). Следующие соединения получали, по существу, согласно способу примера получения 3. ди-трет-Бутил-(1S,2R,5R,6R)-2-(трет-бутоксикарбониламино)-3-метилен-4-оксобицикло[3.1.0]гексан-2,6-дикарбоксилат (8 г, 18,9 ммоль) в диэтиловом эфире (80 мл) барботировали газообразным азотом в течение 10 мин. Добавляли 4-фтор-3-метилтиофенол (2,7 г, 18,9 ммоль) и триэтиламин (0,26 мл, 1,89 ммоль). Смесь нагревали до 40 С и перемешивали в течение 15 мин. Затем смесь оставляли охлаждаться до комнатной температуры, переносили в капельную воронку и добавляли медленно в течение 2 ч при-10 С к 200 мл эфирного раствора, содержащего S-(-)-2-метил-CBS-оксазаборолидин (1 М в тетрагидрофуране) (5,67 мл, 5,67 ммоль) и комплекс боран-метилсульфид (2 М в тетрагидрофуране, 8,5 мл, 17 ммоль). Смесь перемешивали в течение дополнительного часа после последнего добавления. Силикагель(40 г) добавляли в течение 30 мин и реакционную смесь постепенно нагревали до комнатной температуры. Суспензию фильтровали и промывали 300 мл диэтилового эфира. Растворитель концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии, элюируя (от 0 до 25% этилацетат/гексаны), с получением титульного соединения (9,8 г, 17,3 ммоль, 91,5% выход). MS (m/z): 565.8 (М-Н). Пример получения 11. ди-трет-Бутил-(1S,2R,3S,4R,5R,6R)-2-(трет-бутоксикарбониламино)-3-[(4 фтор-3-метилфенил)сульфанилметил]-4-метилсульфонилоксибицикло[3.1.0]гексан-2,6-дикарбоксилат ди-трет-Бутил-(1S,2R,3S,4R,5R,6R)-2-(трет-бутоксикарбониламино)-3-[(4-фтор-3-метилфенил) сульфанилметил]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат (4,6 г, 8,10 ммоль) в пиридине (60 мл) охлаждали до 0 С. К указанной смеси добавляли метансульфохлорид (1,88 мл, 24,31 ммоль). Смесь нагревали до 40 С и перемешивали в течение 1 ч, охлаждали до комнатной температуры и оставляли перемешиваться в течение 18 ч. Смесь концентрировали при пониженном давлении с получением остатка. Остаток разделяли между этилацетатом и 1N водной HCl (250 мл). Органический слой отделяли,сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением титульного соединения (5,2 г, 8,05 ммоль, 99,4% выход): MS (m/z): 643.6 (М-Н). ди-трет-Бутил-(1S,2R,3S,4R,5R,6R)-2-(трет-бутоксикарбониламино)-3-[(4-фтор-3-метилфенил) сульфанилметил]-4-метилсульфонилоксибицикло[3.1.0]гексан-2,6-дикарбоксилат (5,3 г, 8,21 ммоль) растворяли в диметилформамиде (100 мл). К указанной смеси добавляли карбонат цезия (5,40 г, 16,41 ммоль), 1,2,4-триазол-3-тиол (3,42 г, 32,83 ммоль) и триацетоксиборгидрид натрия (906 мг, 4,10 ммоль). Смесь перемешивали при 40 С в течение 72 ч. Реакцию охлаждали и гасили водой и водным NH4Cl. Смесь переносили в делительную воронку и экстрагировали диэтиловым эфиром, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии (от 0 до 50% этилацетат/гексаны) с получением титульного соединения (0,88 г, 1,35 ммоль, 16,5% выход). MS (m/z): 651 (М+Н). Пример получения 13. ди-трет-Бутил-(1S,2R,3R,4S,5R,6S)-4-азидо-2-(трет-бутоксикарбониламино)3 -[(4-фтор-3-метилфенил)сульфанилметил]бицикло[3.1.0]гексан-2,6-дикарбоксилат ди-трет-Бутил-(1S,2R,3S,4R,5R,6R)-2-(трет-бутоксикарбониламино)-3-[(4-фтор-3-метилфенил) сульфанилметил]-4-метилсульфонилоксибицикло[3.1.0]гексан-2,6-дикарбоксилат (5,9 г, 9,14 ммоль) растворяли в диметилсульфоксиде (30 мл). К указанной смеси добавляли азид натрия (2,5 г, 38,37 ммоль). Смесь перемешивали при 100 С в течение 18 ч. Растворитель удаляли при пониженном давлении с получением остатка. Остаток суспендировали в диэтиловом эфире (100 мл) и фильтровали. Органический слой переносили в делительную воронку и промывали водой и солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии (от 0 до 15% этилацетат/гексаны) с получением титульного соединения (3,14 г, 5,30 ммоль, 58% выход). MS (m/z): 591 (М-Н). Пример получения 14. ди-трет-Бутил-(1S,2R,3R,4S,5R,6S)-4-амино-2-(трет-бутоксикарбониламино)3-[(4-фтор-3-метилфенил)сульфанилметил]бицикло[3.1.0]гексан-2,6-дикарбоксилат ди-трет-Бутил-(1S,2R,3R,4S,5R,6S)-4-азидо-2-(трет-бутоксикарбониламино)-3-[(4-фтор-3 метилфенил)сульфанилметил]бицикло[3.1.0]гексан-2,6-дикарбоксилат (1,88 г, 3,17 ммоль) растворяли в метаноле (15,86 мл). К указанной смеси добавляли триэтиламин (1,77 мл, 12,7 ммоль) и 1,3 пропандитиол (1,28 мл, 12,69 ммоль). Смесь перемешивали при комнатной температуре в течение 18 ч. Смесь выливали в воду и экстрагировали этилацетатом, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэшхроматографии (от 50 до 100% этилацетат/гексаны), с получением титульного соединения (1,2 г, 2,12 ммоль, 66,76% выход). MS (m/z): 567.2 (M+1). Пример получения 15. ди-трет-Бутил-(1S,2R,3R,4S,5R,6S)-4-ацетамидо-2-(трет-бутоксикар- 10021724 ди-трет-Бутил-(1S,2R,3R,4S,5R,6S)-4-амино-2-(трет-бутоксикарбониламино)-3-[(4-фтор-3 метилфенил)сульфанилметил]бицикло[3.1.0]гексан-2,6-дикарбоксилат (0,15 г, 264,67 мкмоль) растворяли в дихлорметане (10 мл). К указанной смеси добавляли триэтиламин (55,34 мкл, 397,01 мкмоль) и ацетилхлорид (28,25 мкл, 397,01 мкмоль). Смесь перемешивали при комнатной температуре в течение 10 мин. Растворитель удаляли при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии (от 10 до 100% этилацетат/гексаны) с получением титульного соединения (100 мг,164,27 мкмоль, 62,06% выход); 1 Н ЯМР (CD3Cl)1.44 (t, 27 Н), 1.95 (s, 3H), 2.16 (m, 1H), 2.22 (s, 3H), 2.60 (dd, 1H), 2.78 (bs, 1H),3.10 (dd, 1H), 4.59 (m, 1H), 5.50 (d, 1H), 6.92 (t, 1H), 7.06 (m, 1H), 7.11 (d, 1H). Пример получения 16. (1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дифторфенил)сульфанил] метил-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновая кислота ди-трет-Бутил-(1S,2R,3R,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дифторфенил)сульфанил]метил-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат (4,11 г, 7,19 ммоль) взвешивали в однолитровой круглодонной колбе с мешалкой. Добавляли хлороводород (4N в диоксане, 120 мл, 480,0 ммоль). Смесь нагревали до 70 С в течение 2 ч и затем оставляли охлаждаться до комнатной температуры. Растворитель удаляли при пониженном давлении с получением остатка. Остаток растворяли в дихлорметане (200 мл) и растворитель удаляли при пониженном давлении с получением остатка. Указанную процедуру повторяли еще два раза с получением гидрохлорида (1S,2R,3S,4S,5R,6R)-2-амино-3-[(3,4 дифторфенил)сульфанилметил]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты. Указанное вещество брали в тетрагидрофуране (100 мл) в виде суспензии. К указанной суспензии добавляли триэтиламин (40,08 мл, 287,57 ммоль). Суспензию перемешивали в течение 10 мин и затем добавляли метанол (50 мл). К реакционной смеси добавляли ди-трет-бутилдикарбонат (4,71 г, 21,57 ммоль) и смесь нагревали до 80 С в течение 2 ч. Смесь оставляли дойти до комнатной температуры и растворитель удаляли при пониженном давлении с получением остатка. Остаток растворяли в ацетонитриле (50 мл), переносили в делительную воронку и промывали гексанами. Слой ацетонитрила отделяли и удаляли при пониженном давлении с получением остатка. Остаток суспендировали в диэтиловом эфире, переносили в делительную воронку, промывали 1N водной HCl, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением титульного соединения (3 г, 6,53 ммоль, 90,82% выход). MS (m/z): 457.8 (М-Н). Следующие соединения получали, по существу, согласно способу примера получения 16. К перемешиваемой смеси (1S,2R,3S,4S,5R,6R)-2-(трет-бутоксикарбониламино)-3-[(4-фтор-3 метилфенил)сульфанилметил]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (2,4 г, 5,27 ммоль), бисульфата тетра(н-бутил)аммония (178,90 мг, 526,89 мкмоль) и бикарбоната натрия (3,54 г,42,15 ммоль) в дихлорметане (13,2 мл), и воды (13,2 мл) добавляли хлорметиловый эфир хлорсульфоновой кислоты (1,20 мл, 11,59 ммоль). Смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь выливали в воду и экстрагировали дихлорметаном. Объединенные органические вещества сушили над сульфатом магния, фильтровали и концентрировали с получением остатка. Остаток очищали с помощью флэш-хроматографии (20-35% этилацетат/гексан) с получением титульного соединения (1,37 г, 2,48 ммоль, 47% выход). MS (m/z): 574.0 (M+Na). Следующие соединения получали, по существу, согласно способу примера получения 21:(1S,2R,3S,4S,5R,6R)-2-(трет-бутоксикарбониламино)-3-[(3,4-дифторфенил)сульфанилметил]-4 гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (1 г, 2,18 ммоль) в диметилформамиде (13,06 мл) добавляли карбонат калия (668,43 мг, 4,79 ммоль), йодид натрия (75,03 мг, 500,58 мкмоль). Смесь перемешивали в течение 10 мин при комнатной температуре. Добавляли хлорметилизопропилкарбонат(1 г, 6,53 ммоль). Смесь перемешивали при комнатной температуре в течение 18 ч. Добавляли уксусную кислоту (4 мл) и смесь перемешивали в течение 10 мин. Объем растворителя понижали до примерно 10 мл при пониженном давлении с получением вязкого остатка. Остаток разбавляли диэтиловым эфиром и перемешивали в течение 10 мин. Раствор пропускали через фильтр и растворитель удаляли при пониженном давлении с получением остатка. Остаток оставляли в высоком вакууме в течение 1 ч. Остаток очищали с помощью флэш-хроматографии, элюировали (от 0 до 35% тетрагидрофуран/гексаны), с получением титульного соединения (0,88 г, 1,27 ммоль, 58,5% выход). MS (m/z): 714.2 (M+Na). Следующие соединения получали, по существу, согласно способу примера получения 24. Изомасляную кислоту (0,21 г, 2,42 ммоль) растворяли в диметилформамиде (10 мл). К указанному раствору добавляли карбонат калия (0,54 г, 3,87 ммоль). Смесь перемешивали при 50 С в течение 3 ч и затем охлаждали до комнатной температуры. К смеси добавляли бис(хлорметил)-(1S,2R,3S,4S,5R,6R)-2(трет-бутоксикарбониламино)-3-[(4-фтор-3-метилфенил)сульфанилметил]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат (535 мг, 0,97 ммоль). Смесь перемешивали при комнатной температуре в течение 18 ч. Смесь разбавляли этилацетатом, переносили в делительную воронку, промывали солевым раствором, сушили над сульфатом магния и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии (10-40% этилацетат/гексаны) с получением титульного соединения (230 мг, 0,36 ммоль, 37%). MS (m/z): 678.2 (M+Na). Следующие соединения получали, по существу, согласно способу примера получения 35. ди-трет-Бутил-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дифторфенил)сульфанил]метил-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат (0,58 г, 7,19 ммоль) взвешивали в 100 мл круглодонной колбе с мешалкой. Добавляли хлороводород (4N в диоксане, 33 мл, 132,0 ммоль). Смесь нагревали до 70 С в течение 2 ч и затем оставляли охлаждаться до комнатной температуры. Растворитель удаляли при пониженном давлении с получением остатка. Остаток растворяли в дихлорметане(50 мл) и растворитель удаляли при пониженном давлении с получением остатка. Данную процедуру повторяли еще три раза с получением титульного соединения (567 мг, 1,43 ммоль, 97% выход). MS (m/z): 360.0 (M+1). Следующие соединения получали, по существу. согласно способу примера 1. ди-трет-Бутил-(1S,2R,3S,4S,5R,6R)-2-(трет-бутоксикарбониламино)-4-гидрокси-3-(п-толилсульфанилметил)бицикло[3.1.0]гексан-2,6-дикарбоксилат (300 мг, 545,73 мкмоль) помещали в виалу для микроволнового реактора. В виалу добавляли воду (2 мл, 110 ммоль) и уксусную кислоту (2 мл, 34,9 ммоль). Смесь нагревали в микроволновом реакторе до 140 С в течение 20 мин. Растворитель удаляли при пониженном давлении с получением титульного соединения (165 мг, 489,04 мкмоль, 89,6%). MS(m/z): 338.0 (М+Н). Следующие соединения получали, по существу, согласно способу примера 8. бис([(1-Метилэтокси)карбонил]оксиметил)-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дифторфенил)сульфанил]метил-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат (0,88 г,1,27 ммоль) растворяли в хлороводороде (4N в диоксане, 30 мл, 120,00 ммоль) и перемешивали при комнатной температуре в течение 1,5 ч. Растворитель удаляли при пониженном давлении с получением остатка. Остаток растворяли в дихлорметане и растворитель удаляли при пониженном давлении. Указанный процесс повторяли 8 раз. Остаток оставляли в высоком вакууме в течение ночи с получением титульного соединения (0,692 г, 1,10 ммоль, 86,61% выход). MS (m/z): 591.8 (М+Н). Следующие соединения получали, по существу, согласно способу примера 12. трет-Бутоксибис(диметиламино)метан (481,1 мл, 2,33 моль) добавляли к суспензии ди-трет-бутил(1S,2R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-4-оксобицикло[3.1.0]гексан-2,6-дикарбоксилата (600 г,1,46 моль) в сухом толуоле (3,6 л) при комнатной температуре в атмосфере азота. Смесь нагревали при 80 С в течение 3 ч и 45 мин, затем охлаждали до комнатной температуры и перемешивали в течение ночи. Реакционный объем уменьшали в вакууме, разбавляли метил-трет-бутиловым эфиром (1,8 л) и гексаном (1,8 л) и перемешивали в течение 3 ч при 15 С. Через 3 ч полученное твердое вещество собирали посредством фильтрации, промывали холодным гексаном (21,8 л) и сушили в вакууме с получением титульного соединения (620,4 г, выход 91%). HPLC-MS: 98%. К раствору ди-трет-бутил-(1S,2R,5R,6R)-2-(трет-бутоксикарбониламино)-3-(диметиламинометилен)-4-оксобицикло[3.1.0]гексан-2,6-дикарбоксилата (620,4 г, 1,33 моль) в сухом тетрагидрофуране (12 л) добавляли триэтиламин (277,3 мл, 1,99 моль) при комнатной температуре в атмосфере азота. Смесь охлаждали до -47 С и по каплям в течение 2 ч добавляли гидрид диизобутилалюминия (1 М в гексане,2,06 л, 2,06 моль). Полученную смесь перемешивали при -47 С. Через 1 ч 15 мин по каплям при -47 С добавляли уксусную кислоту (118 мл, 2,06 моль), нагревали до комнатной температуры и затем перемешивали в течение ночи. Добавляли 20% Н 3 РО 4 в воде до рН 2. Отделяли органическую фазу и экстрагировали водную фазу этилацетатом (21,7 л). Объединенные органические фазы последовательно промывали 10% водной HCl (1,5 л), водой (1,5 л) и солевым раствором (1,5 л), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением твердого вещества. Полученное твердое вещество растирали с водой (3,2 л), собирали посредством фильтрации и затем сушили с получением титульного соединения (558,2 г, выход 99%). HPLC-MS: 97.4%. Стадия 3. ди-трет-Бутил-(1S,2R,3S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дифторфенил) сульфанил]метил-4-оксобицикло[3.1.0] гексан-2,6-дикарбоксилат Суспензию ди-трет-бутил-(1S,2R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-метилиден-4-оксобицикло[3.1.0]гексан-2,6-дикарбоксилата (350,00 г, 826,43 ммоль) в толуоле (2,95 л) обрабатывали 3,4 дифтортиофенолом (172,49 г, 1,18 моль) и триэтиламином (205,61 мл, 149,28 г, 1,48 моль) при 25 С. Смесь перемешивали при 80 С. Через 12 ч реакцию охлаждали до комнатной температуры, последовательно промывали 2N водным NaOH (рН 10) и водной 1N HCl (рН 4), сушили над MgSO4 и концентрировали в вакууме с получением остатка. Растирали остаток с гексаном (1 л) и удаляли растворитель с получением титульного соединения (664 г, 100% выход). Стадия 4. ди-трет-Бутил-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дифторфенил)сульфанил]метил-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат(86,68 г, 101,98 мл, 1,14 моль) в безводном метил-трет-бутиловом эфире (4,56 л) охлаждали до -40 С. К указанному раствору добавляли ди-трет-бутил-(1S,2R,3S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3[(3,4-дифторфенил)сульфанил]метил-4-оксобицикло[3.1.0]гексан-2,6-дикарбоксилат (650,00 г, 1,14 моль) в метил-трет-бутиловом эфире (3,42 л) через капельную воронку в течение 2 ч, после чего реакционную смесь нагревали до 0 С. Через 1 ч добавляли метанол (461,80 мл, 11,41 моль) и внутреннюю температуру поддерживали ниже 15 С. Реакционную смесь промывали 2N водным NaOH (2 л), сушили надMgSO4 и концентрировали в вакууме с получением остатка. Остаток очищали с помощью хроматографии на силикагеле (от 8:1 до 1:1 гексан/этилацетат) с получением титульного соединения (580 г, 89% выход). К раствору ди-трет-бутил-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4 дифторфенил)сульфанил]метил-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилата (550,00 г, 962,07 ммоль) в 1,4-диоксане (192,41 мл) добавляли воду (1,10 л) и 12,18 М хлороводород в воде (789,88 мл,9,62 моль). Полученную суспензию перемешивали при 100 С. Затем через 12 ч реакционную смесь охлаждали до 25 С, перемешивали в течение 12 ч и затем подщелачивали NaOH (50% мас./мас.) до рН 2,65. Полученную смесь перемешивали при 10 С в течение 30 мин, после чего осадок собирали посредством фильтрации, промывали водой (1 л), метил-трет-бутиловым эфиром (1 л) и сушили в течение 2 ч при 25 С и затем при 60 С в печи до постоянной массы с получением титульного соединения (300 г, 87% выход). MS (m/z): 360 (M+1). Стадия 6. (1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дифторфенил)сульфанил] метил-4-гидроксибицикло[3.1.0] гексан-2,6-дикарбоновая кислота(500,9 мл) при 25 С добавляли триэтиламин (407,27 мл, 2,92 моль) и [2-(третбутоксикарбонилоксиимино)-2-фенилацетонитрил] (308,39 г, 1,25 моль). Смесь нагревали до 50 С. Через 12 ч реакционную смесь охлаждали до 25 С, разбавляли водой (2,5 л) и промывали метил-третбутиловым эфиром (61 л). Подщелачивали водную фазу раствором водной 1N HCl до рН 2 и экстрагировали этилацетатом (32 л). Объединенные экстракты этилацетата промывали солевым раствором, сушили над MgSO4 и концентрировали в вакууме с получением титульного соединения (250 г, 65% выход).(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дифторфенил)сульфанил] метил-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (150,00 г, 326,46 ммоль) в диметилформамиде (3,38 л) последовательно обрабатывали карбонатом калия (180,48 г, 1,3 моль), хлорметилизопропилкарбонатом (149,43 г, 979,39 ммоль) и йодидом натрия (9,79 г, 65,29 ммоль), смесь перемешивали в атмосфере азота при 25 С. Через 12 ч к смеси добавляли воду (1,5 л), твердые вещества отфильтровали и фильтрат экстрагировали метил-трет-бутиловым эфиром (31,5 л). Объединенные органические вещества последовательно промывали водой, солевым раствором, сушили над MgSO4 и концентрировали в вакууме. Полученный остаток очищали с помощью хроматографии на силикагеле (от 2:1 до 1:1 гексан/этилацетат) с получением титульного соединения (225 г, 70% выход). MS (m/z): 592 (М+-Вос). Стадия 8. Гидрохлорид бис-([(1-метилэтокси)карбонил]оксиметил) (1S,2R,3S,4S,5R,6R)-2-амино- 24021724 бис([(1-Метилэтокси)карбонил]оксиметил)(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]3-[(3,4-дифторфенил)сульфанил]метил-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат (124,9 г,180,57 ммоль) обрабатывали 4N хлороводородом в 1,4-диоксане (1,12 л, 4,50 моль) при 25 С. Через 90 мин растворитель удаляли в вакууме и остаток суспендировали в метил-трет-бутиловом эфире (1 л) в течение 30 мин. Полученный осадок собирали посредством фильтрации, промывали метил-третбутиловым эфиром (500 мл) и сушили в печи при 45 С в течение 16 ч. Полученную соль растворяли в дихлорметане и воде, затем нейтрализовали триэтиламином. Органическую фазу отделяли, сушили надMgSO4 и концентрировали в вакууме с получением остатка. Остаток очищали с помощью хроматографии на силикагеле (от 3:1 до 1:1 гексан/этилацетат) с получением свободного основания, которое обрабатывали 4N HCl в 1,4-диоксане (950 мл) при 25 С. Через 15 мин растворитель выпаривали в вакууме и остаток суспендировали в метил-трет-бутиловом эфире (1 л) и гексане (250 мл). Полученное твердое вещество фильтровали, промывали метил-трет-бутиловым эфиром (500 мл) и сушили в вакууме при 45 С до постоянной массы с получением титульного соединения (98,5 г, 87% выход). MS (m/z): 592 (M+1). Литературные данные (Witkin, Jeffrey M., и Eiler, William J.A. (2006), Antagonism of MetabotropicReceptors: Potential Drug Targets for Psychiatric Disorders, The Open Medicinal Chemistry Journal, т. 4, с. 2036) и данные, полученные в неклинических исследованиях на животных, подтверждают роль антагонистов mGlu2/3 в лечении депрессивных расстройств и расстройств чрезмерной сонливости. В частности, с помощью грызунов, контролируемых ЭЭГ, было обнаружено, что антагонисты рецепторов mGlu2/3 эффективны на моделях депрессивных расстройств у грызунов и стимулируют бодрствование без проявления непропорциональной или клинически значимой гиперактивности или чрезмерной компенсационной гиперсомнии. Повышенная концентрация внимания свидетельствует о повышенной внимательности,улучшенной когнитивной функции и, вероятно, уменьшенной усталости. Поскольку описанные ранее расстройства представляют собой распространнные сопутствующие клинические состояния, антагонисты mGlu2/3 рецептора могут быть особенно эффективны у конкретных групп пациентов, таких как пациенты с большим депрессивным расстройством, монополярной депрессией, дистимией и/или циклотимией или любым расстройством чрезмерной сонливости. Расстройства чрезмерной сонливости могут включать, но не ограничиваются ими, чрезмерную дневную сонливость (EDS), гиперсомнию, связанную с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна (включая, но не ограничиваясь ими, расстройство сна, вызванное сменным режимом работы, расстройство сна,вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сонбодрствование), идиопатическую гиперсомнию и/или чрезмерную сонливость, связанную с невосстанавливающим сном (NRS). Для демонстрации характеристик настоящих соединений типичные соединения могут быть протестированы в следующих in vitro и in vivo анализах. Анализы цАМФ антагонистов mGlu2 и mGlu3 рецепторов. Антагонистическую активность анализировали на рекомбинантных клетках AV12, стабильно экспрессирующих человеческие mGlu2 или mGlu3 рецепторы и крысиный глутаматный транспортер ЕААТ 1(транспортер возбуждающих аминокислот 1 типа). Клеточную линию выдерживали путем культивирования в DMEM с высоким уровнем глюкозы и гидрохлорида пиридоксина, с добавлением 5% диализированной фетальной бычьей сыворотки (FBS), 1 мМ пирувата натрия, 1 мМ HEPES и 1 мМ L-глутамина; генетицин и гигромицин В применяли в качестве антибиотиков. Сливные культуры выращивали при 37 С в атмосфере, содержащей 6,5% СО 2, и пассажировали дважды в неделю. Клетки собирали с помощью 0,25% трипсина, суспендировали в замораживающей среде (FBS с 10% ДМСО) в концентрации 107 клеткок/мл, аликвоты хранили в жидком азоте. За 24 ч до анализа клетки высеивали с плотностью 8,00010,000 клеток на лунку в 96-луночные черные планшеты для культуры тканей с половинным объемом лунок (Costar 3875) в 50 мкл DMEM с высоким уровнем глюкозы и гидрохлорид пиридоксина с добавлением 5% диализированой FBS, 1 мМ пирувата натрия, 1 мМ HEPES, 100 мкг/мл ампициллина и 250 мкМ(mGlu2) или 125 мкМ (mGlu3) L-глутамина. Обращение ингибирования выработки цАМФ, стимулированной форсколином, с помощью исследуемых соединений измеряли с применением технологии гомогенных исследований флуоресценции с разрешением по времени (HTRF; Cisbio cat 62 АМ 4 РЕВ). Среду удаляли и клетки инкубировали со 100 мкл цАМФ стимулирующего буфера (STIM) в течение 30 мин при 37 С. (STIM буфер содержал 500 млHBSS (сбалансированный солевой раствор Хенкса), 1000 мл DPBS (фосфатно-солевой буфер Дульбекко),0,034% БСА, 1,67 мМ HEPES и 500 мкМ IBMX (изобутилметилксантин) (Sigma I5879).) Соединения исследовали с помощью 10-точечной кривой концентрация-эффект с применением 3 Х последовательных разведений, с последующим 40-кратным разбавлением в STIM буфере. DCG IV (Tocris 0975) служит в качестве контрольного агониста. Конечная реакционная смесь содержала 1 мкМ (для mGlu2) или 3 мкМ(для mGlu3) форсколина (Sigma F6886), DCG IV при его ЕС 90 и до 25 мкМ исследуемого соединения. Клетки инкубировали при 37 С в течение 20 мин. Для измерения уровня цАМФ конъюгат цАМФ-d2 и конъюгат анти-цАМФ-криптата в лизисном буфере инкубировали с обработанными клетками при комнатной температуре в течение 1 ч (mGlu2) или 1,5 ч (mGlu3). Сигнал HTRF детектировали с применением планшет-ридера EnVision (Perkin-Elmer) с вычислением отношения флуоресценции при 665-620 нМ. Необработанные данные преобразовывали в количество цАМФ (пикомоль/лунка) с применением стандартной кривой цАМФ, полученной для каждого эксперимента. Относительные значения IC50 вычисляли сверху вниз кривой концентрация-эффект с применением программного обеспечения для четырехпараметрического логистического построения кривой (ActivityBase v5.3.1.22).FLIPR и цАМФ анализы на селективность к mGlu рецептору. Относительную антагонистическую активность соединений согласно изобретению в отношении других человеческих mGlu рецепторов можно оценить с помощью либо цАМФ анализа, либо флуорометрического аназиза кальциевого ответа (см., например, Fell et al., JPET (в печати. Вкратце, в указанных исследованиях применяли индивидуальные AV12 клеточные линии, содержащие крысиный глутаматный транспортер ЕААТ 1 и стабильно экспрессирующие человеческие mGlu1, 2, 3, 4, 5 и 8 рецепторы. Рецепторы mGlu 1 и 5 являются Gq-сопряженными, поэтому они естественно передают сигнал на фосфолипазу С, приводя к выработке ответа в виде кальциевого потока, который можно применять для измерения активации рецептора с использованием флюориметрического планшетного ридера (FLIPR, Molecular Devices). Клеточные линии, экстпрессирующие mGlu2, 3, 4 и 8 рецепторы, разработаны для экспрессирования субъединицы G15, чтобы указанные Gi-сопряженные рецепторы вырабатывали ответ в виде потока кальция, аналогично клеточным линиям, экспрессирующим рецепторы mGlu1 и 5. mGlu6 рецептор тестировали в цАМФ формате с использованием способов, аналогичных способам, разработанным для mGlu2 и mGlu3, приведенным выше. Указанные клеточные линии выдерживали так же, как описано выше, за исключением того, что количества L-глутамина и подборки агентов (генетицин, гигромицин В, зеоцин и бластицидин) могут зависеть от клеточной линии. Сливные культуры пассажировали дважды в неделю. Уровни внутриклеточного кальция контролировали с помощью FLIPR до и после добавления тестируемых соединений и красителей Fluo-3 AM (Invitrogen) или Calcium 4 (Molecular Devices), в зависимости от клеточной линии. Клетки высеивали в планшет за 24 ч до анализа в различных концентрациях глутамина и при различной плотности клеток на лунку, в зависимости от клеточной линии. Среду удаляли и клетки инкубировали с 8 мкМ красителя (50 мкл на лунку) в течение 90 или 120 мин (в зависимости от клеточной линии) при 25 С. FLIPR анализ с двумя добавлениями, генерирующий 11-точечную кривую концентрация-эффект для агониста глутамата, проводили перед каждым экспериментом для подтверждения соответствующей восприимчивости клеток. Результаты анализировали с применениемGraphPad Prism v4.03 для расчета концентраций глутамата, необходимых для получения ЕС 90 (анализ антагониста) и ЕС 10 (анализ потенциирующего фактора) ответов. Соединение исследовали на каждом mGlu рецепторе в FLIPR анализе с двумя добавлениями с применением 10-точечного профиля концентрация-эффект, начиная с конечной концентрации 25 мкМ для анализа агониста и 12,5 мкМ для анализов потенциирующего фактора и антагониста. В первом добавлении выявляли какую-либо агонистическую активность и во втором добавлении, состоящем из 100 мкл выбранных концентраций (в зависимости от клеточной линии) глутамата в буфере для анализа, получали ЕС 10 или ЕС 90 глутаматный ответ. Агонистические эффекты агониста количественно определяли как процентное отношение стимуляции, вызванной только соединением, к максимальному глутаматному ответу. Антагонистические эффекты количественно определяли путем расчета процентного отношения ингибирования ЕС 90 глутаматного ответа, вызванного соединением. Потенциирующие эффекты количественно определяли как процентное отношение увеличения при наличии ЕС 10 ответа в глутамате и ECmax ответа. Все данные рассчитывали в виде относительных значений IC50 или ЕС 50 с применением программного обеспечения для 4-параметрического логистического построения кривой (ActivityBasev5.3.1.22). Антагонистическую активность в mGlu6 клетках измеряли с использованием цАМФ согласно способу, аналогичному описанному выше для mGlu2 и mGlu3 активности, за исключением того, что контрольным агонистом был L-AP4 (Tocris). Для измерения mGlu6 агонистической активности рассчитыва- 26021724 ют степень, в которой соединение ингибирует выработку цАМФ, стимулированную форсколином. Относительные значения IC50 и EC50 вычисляли сверху вниз кривой концентрация-эффект с применением программного обеспечения для 4-параметрического логистического построения кривой (ActivityBasev5.3.1.22). Соединения, приведенные в примерах, в которых оба R1 и R2 представляют собой водород, тестировали, по существу, как описано выше, и обнаружили, что они обладают высокой антагонистической активностью в отношении mGlu2 и mGlu3 рецепторов. Также обнаружили, что соединения, приведенные в примерах, в которых оба R1 и R2 представляют собой водород, являются селективными антагонистамиmGlu2 и mGlu3 рецепторов в отличие от других подтипов mGlu рецепторов. Значения IC50 в отношенииmGlu2 и mGlu3 рецепторов для соединений, приведенных в примерах, в которых оба R1 и R2 представляют собой водород, были менее 70 и 140 нМ соответственно, в то время как значения IC50 в отношении других тестируемых mGlu рецепторов были значительно выше. Соединения согласно примерам 1 и 2 тестировали, по существу. как описано выше, и обнаружили, что они обладают профилями активности,приведенными в табл. 1. Таблица 1 Данные по селективности Кроме того, некоторые соединения по настоящему изобретению показали отсутствие существенной активности в отношении других физиологически значимых рецепторов, таких как, но не ограничиваясь ими, hERG канал, серотониновые рецепторы (особенно 5-HT2A и 5-HT2B), мускариновые рецепторы (особенно М 2) и iGluR рецепторы (особенно iGluR5). Соединение согласно примеру 1 тестировали с использованием известных способов анализа и обнаружили, что оно не обладает заметной активностью в отношении указанных рецепторов. Следовательно, полагают, что физиологически значимые дозы соединений согласно изобретению обеспечивают существенное ингибирование mGlu2 и mGlu3 рецепторов in vivo, при этом, по существу,не взаимодействуя с другими mGlu рецепторами или другими физиологически значимыми рецепторами,и, таким образом, обеспечивают требуемую фармакологию, предотвращая возникновение нежелательных побочных эффектов, связанных с нецелевой активностью. Тест вынужденного плавания у мышей (mFST).mFST является доказанным in vivo анализом на антидепрессивную активность (Li et al. J. Pharmacol.Exp. Ther. 319(l):254-9, 2006). Мыши, обработанные известными клинически эффективными антидепрессантами (селективными ингибиторами обратного захвата серотонина и/или трициклическими антидепрессантами), демонстрировали поведение, выраженное в уменьшении времени неподвижности после помещения в сосуд с водой, поведение, связанное с отчаяньем. mFST применяли для определения потенциальной антидепрессивной активности новых mGlu2/3 антагонистов, в частности, так как описано в ранее опубликованных способах (см., например, Li et al. J. Pharmacol. Exp. Ther. 319(l):254-9, 2006). Вкратце, применяли мышей NIH-Swiss мужского пола (Harlan Sprague-Dawley, Indianapolis, IN), весящих 25-30 г. Животных, размещенных в одной группе, изымали из вивария в исследовательскую зону в отдельные клетки и позволяли адаптироваться к новой среде в течение по меньшей мере 1 ч перед тестированием. Соединения, в которых оба R1 и R2 представляют собой водород, растворяли в воде с минимальным количеством NaOH, добавленным для растворения, и вводили и.п. Соединения, в которых R1 и/илиR2 отличны от водорода, получали в день использования в 2,0-2,5% N-метилпирролидиноне и затем суспендировали в 1% НЕС, 0,25% Tween 80 и 0,05% Dow antifoam и вводили перорально. Мышей помещали в цилиндр (диаметр: 10 см; высота: 25 см), наполненный 6 см воды (22-25 С), на 6 мин. Подсчитывали продолжительность неподвижности в течение последних 4 мин из 6 мин периода исследования. Мышь записывали как неподвижную, если отсутствовали плавательные движения или совершались только те движения, которые необходимы для поддержания головы над водой. Типичные соединения тестировали, по существу, как описано выше, и обнаружили, что они значительно уменьшают время неподвижности у мышей дикого типа. Соединения, приведенные в примерах, в которых оба, R1 и R2, представляют собой водород, анализировали, по существу, как описано выше, и обнаружили, что они обладают значениями ED60 менее 30 мг/кг и.п., с максимальным уменьшением времени неподвижности по меньшей мере на 25%. Соединения согласно примерам 1, 2, 12/32 и 22 анализировали, по существу, как описано выше, и обнаружили, что они обладают активностями, приведенными в табл. 2. Следовательно, полагают, что соединения согласно настоящему изобретению обладают антидепрессивной активностью in vivo. Таблица 2 Тест вынужденного плавания у мышей (mFST) Согласно другим исследованиям изучали мышей с делециями рецепторов (mGlu2 нокаутные мыши); указанных мышей разводили путем применения скрещивания "гетерозиготагетерозигота" и применяли в качестве однопомтников для -/- и +/+ мышиных контрольных групп (Taconic Farms). Обнаружили, что соединения согласно примерам 1 и 2 (10 мг/кг, и.п., за 30 мин) значительно уменьшают время неподвижности у mGlu2+/+ мышей, но не у mGlu2-/- мышей. Также обнаружили, что соединения согласно примеру 12/32 (30 мг/кг, п.о., за 120 мин) уменьшают время неподвижности у mGlu2+/+ мышей, но не у mGlu2-/- мышей. Данные результаты также демонстрируют, что mGlu2 рецепторы участвуют в антидепрессивном действии соединений согласно изобретению. Соединения согласно изобретению могут также быть протестированы с другими соединениями,подходящими для лечения депрессивных расстройств, например СИОЗС, на их способность усиливать антидепрессивное действие по сравнению с каждым соединением в отдельности. Соединение согласно примеру 12 (10 мг/кг п.о.) исследовали в тесте вынужденного плавания у мышей в отдельности и в комбинации с флуоксетином (10 мг/кг, и.п.) или циталопрамом (1 мг/кг, и.п.) и обнаружили значительное увеличение антидепрессивного действия относительно действия любого соединения по отдельности, как показано в табл. 3, приведенной ниже. Кроме того, исследование уровня активного дикислотного фрагмента соединения согласно примеру 12 (т.е. того же соединения, что и свободное основание согласно примеру 1) в мозгу и плазме, а также уровней флуоксетина и циталопрама в плазме, показало отсутствие увеличения степени воздействия, что подтверждает обнаружение того, что повышенная антидепрессивная активность обеспечивается не только благодаря увеличению воздействия соединений в центральной нервной системе. Таблица 3 3 начительно отличается от любого соединения согласно примеру 12 или флуоксетина в отдельности, р 0.05 Значительно отличается от любого соединения согласно примеру 12 или циталопрама в отдельности, р 0.05 Контроль бодрствования и поведения у крыс. Типичные соединения согласно настоящему изобретению тестировали на крысах на способность увеличивать количество времени в состоянии бодрствования без нежелательных побочных эффектов,таких как БДГ-фаза, двигательная недостаточность в состоянии бодрствования (непропорциональная гипер- или гиполокомоция) и/или рикошетная гиперсомния. Тестируемых животных постоянно контролировали с помощью электроэнцефалограмм (ЭЭГ), электромиограмм (ЭМГ) и измерения общего времени бодрствования, рикошетной гиперсомнии и интенсивности двигательной активности в течение бодрствования. Способы проведения подобных исследований хорошо известны в данной области техники (см., например, способы, описанные в Edgar DM, Seidel WF. Modafinil induces wakefulness without intensifying motor activity or subsequent rebound hypersomnolence in the rat. J. PharmacologyExperimentalAA, Booth V, Mashour GA, Рое GR. Open-source logic-based automated sleep scoring software using electrophysiological recordings in rats. J. Neurosci Methods. 2009; 184(1): 10-8). Исследования проводили следующим образом. Подготовка животного. Взрослых крыс Wistar мужского пола (приблизительно 270-300 г на момент операции) хирургически подготавливали для постоянной регистрации ЭЭГ, ЭМГ, температуры тела и проводили следующие действия. Крыс хирургически снабжали черепным имплантатом, состоящим из четырех ввинчивающихся электродов из нержавеющей стали для регистрации ЭЭГ (два фронтальных[3,9 мм перед теменем и 2,0 мм медиолатерально], два затылочных [6,4 мм за теменем, 5,5 мм медиолатерально]) и двух проводов из нержавеющей стали, покрытых тефлоном, для регистрации ЭМГ (расположенных под затылочной трапециевидной мышцей). Перед операцией вся проводка была припаяна к миниатюрному разъему (Microtech, Boothwyn, PA). Устройство имплантата фиксировали к черепу с помощью комбинации ввинчивающихся электродов из нержавеющей стали для ЭЭГ, цианакрилата, нанесенного между разъемом имплантата и черепом, и стоматологического акрила. Температуру тела и двигательную активность контролировали посредством миниатюрного передатчика (Minimitter PDT4000 г,Philips Respironics, Bend, OR), хирургически помещенного в брюшную полость. На восстановление отводили по меньшей мере 3 недели. Условия при регистрации. Каждую крысу размещали отдельно внутри микроизолирующей клетки,модифицированной вставленной поликарбонатной крышкой-фильтром, позволяющей увеличить вертикальный габарит. Гибкий кабель, минимально затрудняющий движения, подсоединяли одним концом к коммутатору, размещенному вверху клетки, и другим концом к черепному имплантату животного. Каждая клетка размещена в отдельном вентилируемом отсеке камеры для регистрации сна-бодрствования из нержавеющей стали. Еда и вода были доступны ad libitum и комнатную температуру поддерживали примерно 231 С. В ходе исследования поддерживали 24-часовой цикл свет-темнота (LD 12:12) с помощью люминесцентного света. Относительная влажность составляла приблизительно 50%. Животных не тревожили в течение по меньшей мере 30 ч до и после каждой обработки. Дизайн клинического исследования и дозировки. Соединения, в которых оба R1 и R2 представляют собой водород, растворяли в воде с минимальным количеством NaOH, добавленного для растворения, и вводили и.п. в объеме 1,0 мл на 1 кг массы тела. Соединения, в которых R1 и/или R2 отличны от водорода, вводили п.о. в объеме 2 мл на 1 кг массы тела в одном из двух возможных носителях: i) 2,5% Nметил-2-пирролидинона в гидроксиэтилцеллюлозе или ii) 10% камедь с 0,05% Dow Corning Antifoam в воде. Носитель или одну из доз соединения вводили псевдо-случайно, чтобы ни одна из крыс не получила одинаковую обработку дважды и ни одна из крыс не получила более двух из 8 обработок в каком-либо исследовании. Каждую крысу забирали из ее клетки на примерно 1 мин для взвешивания и обработки. По меньшей мере 6-дневный "промывочный" период предшествовал и следовал после каждой обработки. Сбор данных. Распознавание сна и бодрствования может быть автоматизировано (например., Van(Х 10,000, полоса пропускания 1-30 Гц), ЭМГ усиливали и интегрировали (полоса пропускания 110-100 Гц, RMS интегрирование) и одновременно контролировали неспецифическую двигательную активность(LMA). Состояния возбуждения разделяли на 10-секундные периоды не-REM сна, REM сна, бодрствования или тета-доминирующего бодрствования. Двигательную активность (LMA) регистрировали в виде числа отсчтов в минуту и определяли с помощью коммерчески доступных телеметрических примников(ER4000, Minimitter, Bend, OR). Статистический анализ. Возраст и массу тела суммировали по среднему, минимуму и максимуму относительно экспериментальных групп. Все животные, обладающие по меньшей мере одним выходным данным, включали в обобщающие результаты (например, мы включали подходящие данные из экспериментальных групп животных, для которых были применимы телеметрические данные, но не применимы ЭЭГ данные). Период наблюдений после обработки разделяли на 2 пост-дозировочных интервала (первые 7 ч и первые 19 ч), при этом время дозирования определяли как начальное время = 0. Выходные данные объединяли по каждому периоду путем вычисления либо среднечасовых, либо суммарных значений
МПК / Метки
МПК: C07C 323/58, A61K 31/10
Метки: mglur, соединения, качестве, 4-замещенного-3-фенилсульфанилметилбицикло[3.1.0]гексана, антагонистов
Код ссылки
<a href="https://eas.patents.su/30-21724-soedineniya-4-zameshhennogo-3-fenilsulfanilmetilbiciklo310geksana-v-kachestve-antagonistov-mglur-2-3.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения 4-замещенного-3-фенилсульфанилметилбицикло[3.1.0]гексана в качестве антагонистов mglur 2/3</a>
Производные 3-азабицикло (3.1.0) гексана в качестве антагонистов опиоидного рецептора

Номер патента: 6708
Опубликовано: 24.02.2006
Авторы: Лирас Спирос, Макхарди Стэнтон Ферст, Хек Стивен Дональд
МПК: A61K 31/403, A61K 31/4178, C07D 209/52...
Метки: 3-азабицикло, гексана, качестве, опиоидного, 3.1.0, рецептора, антагонистов, производные
Формула / Реферат:
1. Соединение, описываемое формулой I, где X представляет собой H, галоген, -OH, -CN, -C1-C4алкил, замещенный атомами галогена в количестве от одного до трех, либо -O(C1-C4алкил), где C1-C4алкил в -O(C1-C4алкиле) необязательно замещен атомами галогена в количестве от одного до трех; Q представляет собой галоген, -OH, -O(C1-C4алкил), -NH2, -N(C1-C4алкил)(C1-C4алкил), -C(=O)NH2, -C(=O)NH(C1-C4алкил), C(=O)N(C1-C4алкил) (C1-C4алкил) или...
Бензамиды 4-(аминометил) пиперидина, замещённого гидроксикарбонилфенилом, в качестве антагонистов 5нт4-рецепторов

Номер патента: 9464
Опубликовано: 28.12.2007
Авторы: Гейсен Хенрикус Якобус Мария, Босман Жан-Поль Рене Мари Андре, Мевеллек Лоранс Анн
МПК: A61P 1/00, C07D 405/12
Метки: качестве, гидроксикарбонилфенилом, пиперидина, 5нт4-рецепторов, 4-(аминометил, антагонистов, бензамиды, замещённого
Формула / Реферат:
1. Бензамиды 4-(аминометил)пиперидина, замещенного гидроксикарбонилфенилом формулы (I) их стереоизомеры или фармацевтически приемлемые кислотно-аддитивные и основно-аддитивные соли, в которых -Rl-R2- является двухвалентным радикалом формулы R3 представляет водород, галоген, C1-6алкил или C1-6алкилоксигруппу; R4 представляет водород, галоген, C1-6алкил; R5 представляет водород или C1-6алкил, и радикал -OR5, расположенный в 3- или 4-положении...
Спиропиперидиновые соединения в качестве антагонистов рецептора orl-1

Номер патента: 20391
Опубликовано: 30.10.2014
Авторы: Толедо Эскрибано Мигель Анхель, Педрегал-Терсеро Консепсьон, Хименес-Агуадо Альма Мария, Мартинес-Грау Мария Анхелес, Лафуэнте Бланко Селия, Бенито Колладо Ана Белен, Диас Буэсо Нурия
МПК: C07D 495/20, A61P 25/00, A61K 31/438...
Метки: рецептора, качестве, спиропиперидиновые, orl-1, соединения, антагонистов
Формула / Реферат:
1. Соединение формулыгдеR1 представляет собой фтор или хлор;каждый из R2a и R2b представляет собой водород или фтор;R3 представляет собой водород, метил, гидроксиметил или (С1-С3)алкоксиметил;R4 выбран из группы, состоящей из фтора, хлора, циано, цианометила, (C1-С3)алкила, циклопропила, гидроксиметила, метокси, метоксиметила, аминокарбонилоксиметила, метиламинокарбонилоксиметила, диметиламинокарбонилоксиметила, метилкарбонила, аминокарбонила,...
Спиропиперидиновые соединения в качестве антагонистов рецептора orl-1

Номер патента: 20848
Опубликовано: 27.02.2015
Авторы: Лафуэнте Бланко Селия, Педрегал-Терсеро Консепсьон, Диас Буэсо Нурия, Хименес-Агуадо Альма Мария, Бенито Колладо Ана Белен, Мартинес-Грау Мария Анхелес, Толедо Эскрибано Мигель Анхель
МПК: A61P 25/00, C07D 495/20, A61K 31/444...
Метки: спиропиперидиновые, рецептора, соединения, качестве, антагонистов, orl-1
Формула / Реферат:
1. Соединение формулыгде R1 представляет собой фтор или хлор;каждый из R2a и R2b представляет собой водород или фтор;R3 представляет собой водород, метил, гидроксиметил или (C1-С3) алкоксиметил;R4 выбран из группы, состоящей из фтора, хлора, циано, цианометила, (С1-С3) алкила, циклопропила, гидроксиметила, метокси, циклопропилметокси, аминокарбонилметокси, (C1-С3) алкоксиметила, циклопропилоксиметила, циклопропилметоксиметила,...
Соединения пиперазинилпиразинов в качестве антагонистов серотонин 5-ht2 рецептора

Номер патента: 5975
Опубликовано: 25.08.2005
Авторы: Скоби Мартин, Нильссон Бьерн М.
МПК: A61K 31/497, C07D 241/18, A61P 25/00...
Метки: пиперазинилпиразинов, рецептора, серотонин, качестве, соединения, 5-ht2, антагонистов
Формула / Реферат:
1. Соединение общей формулы (I) в которой R1 является водородом, C1-C4-алкилом, C3-C4-алкенилом, C1-C4-ацилом, C1-C4-алкоксикарбонилом, 2-гидроксиэтилом, 2-цианоэтилом, тетрагидропиран-2-илом или группой, защищающей азот; R2 является водородом, C1-C4-алкилом, гидроксиметилом, C1-C4-алкоксиметилом или фторметилом; R3 и R4, каждый независимо, являются водородом, метилом, C1-C4-алкилом, арилом, гетероарилом, где арильные и гетероарильные остатки,...
Предыдущий патент: Производные 4,7-дигидропиразоло[1,5-a]пиразин-6-иламина, используемые в качестве ингибиторов бета-секретазы (васе)
Следующий патент: Новый способ бурения подземных полостей
Случайный патент: Способ изготовления слябов для слоистых металлических изделий и слябы для слоистых металлических изделий