Модуляторы толлподобных рецепторов
Номер патента: 21377
Опубликовано: 30.06.2015
Авторы: Хуэй Хонь Чун, Халькомб Рэндэл Л., Ян Хун, Макфадден Райан, Десай Маной К., Хрватин Пол, Роусл Пол А.



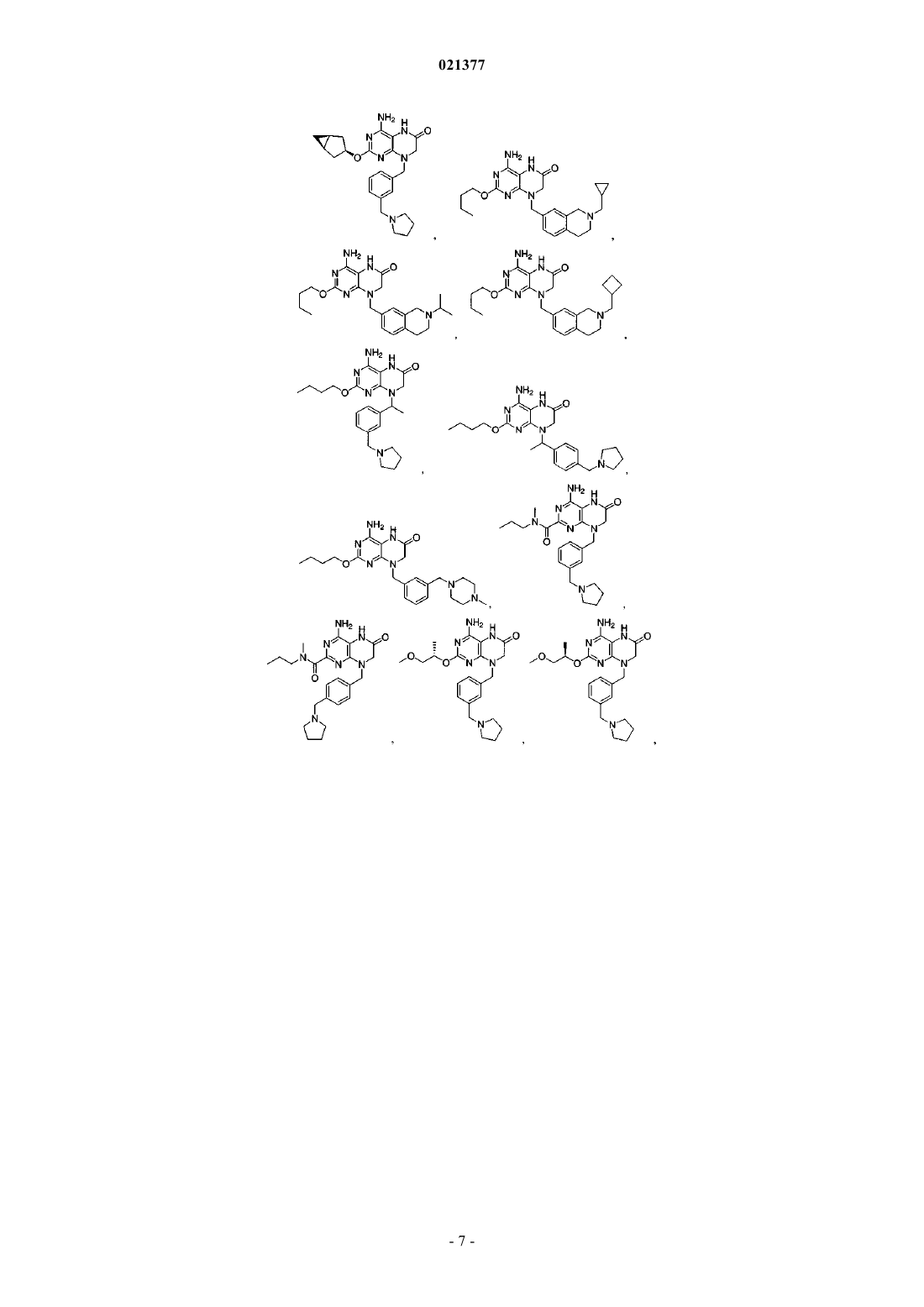
Формула / Реферат
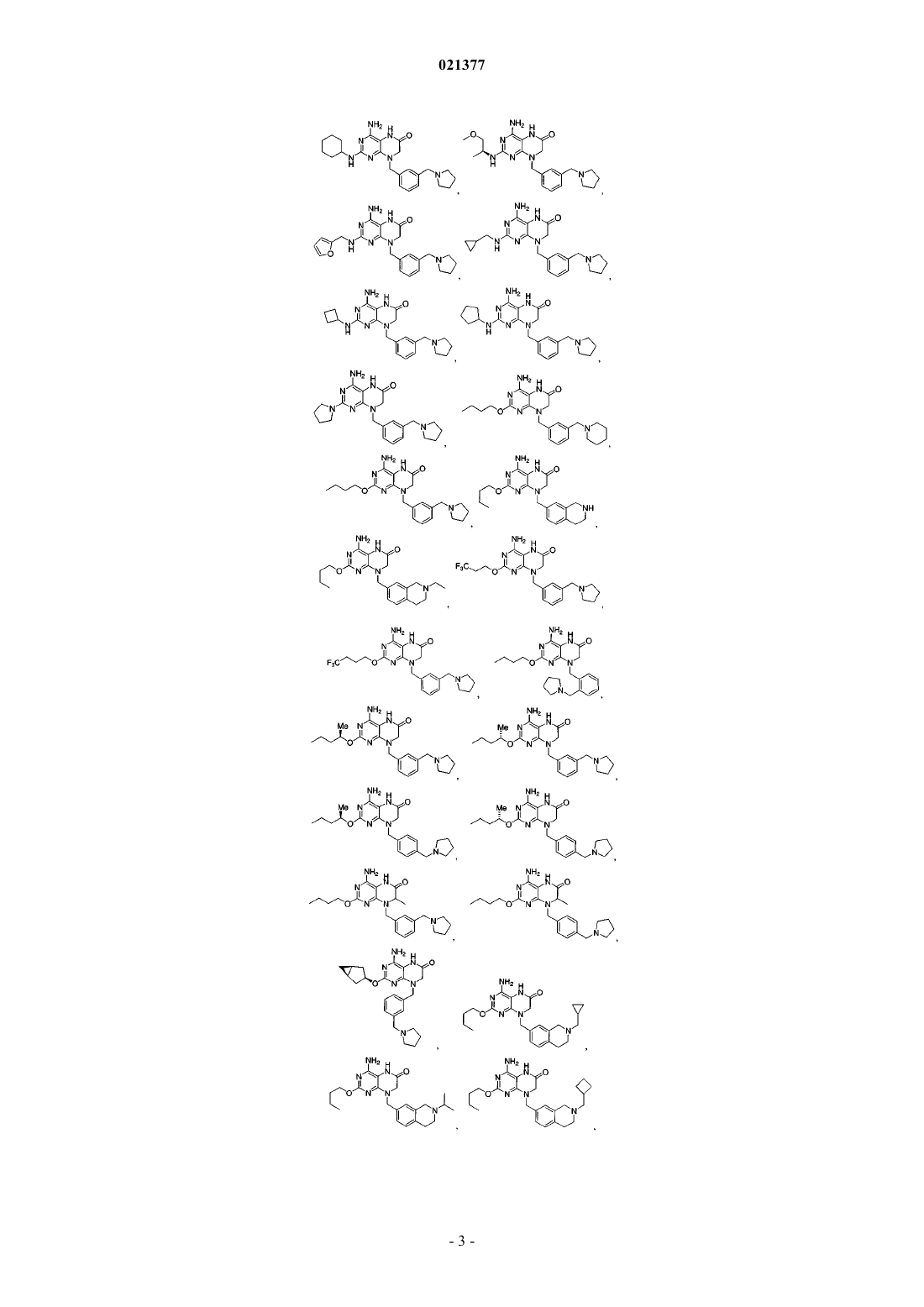
1. Соединение, выбранное из группы, состоящей из





или фармацевтически приемлемая соль указанного соединения.
2. Соединение по п.1, отличающееся тем, что указанное соединение представляет собой

или фармацевтически приемлемую соль указанного соединения.
3. Соединение по п.1, отличающееся тем, что указанное соединение представляет собой

или фармацевтически приемлемую соль указанного соединения.
4. Фармацевтическая композиция для применения в качестве модуляторов толлподобных рецепторов, содержащая терапевтически эффективное количество соединения по любому из пп.1-3 и фармацевтически приемлемый носитель или наполнитель.
Текст
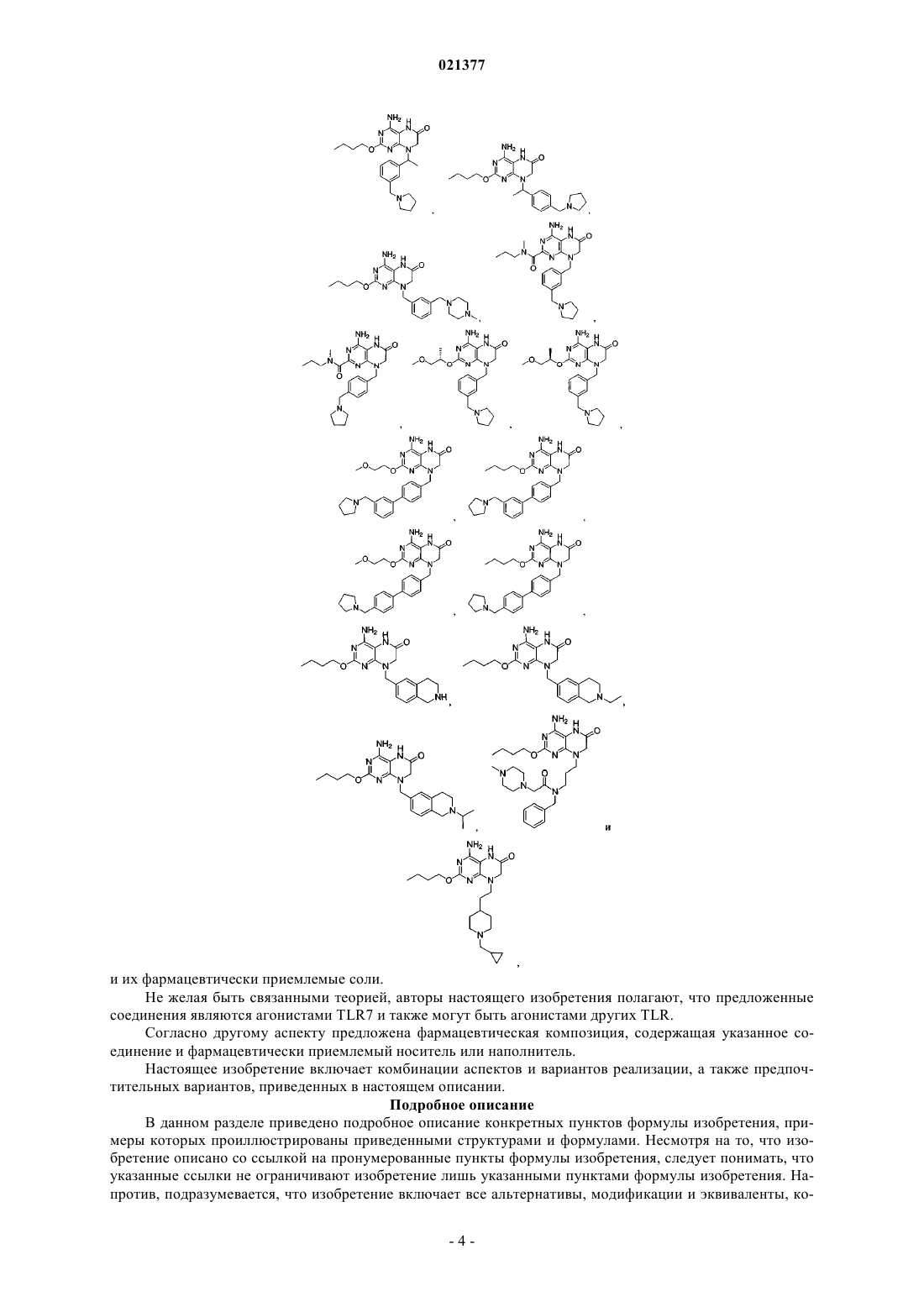
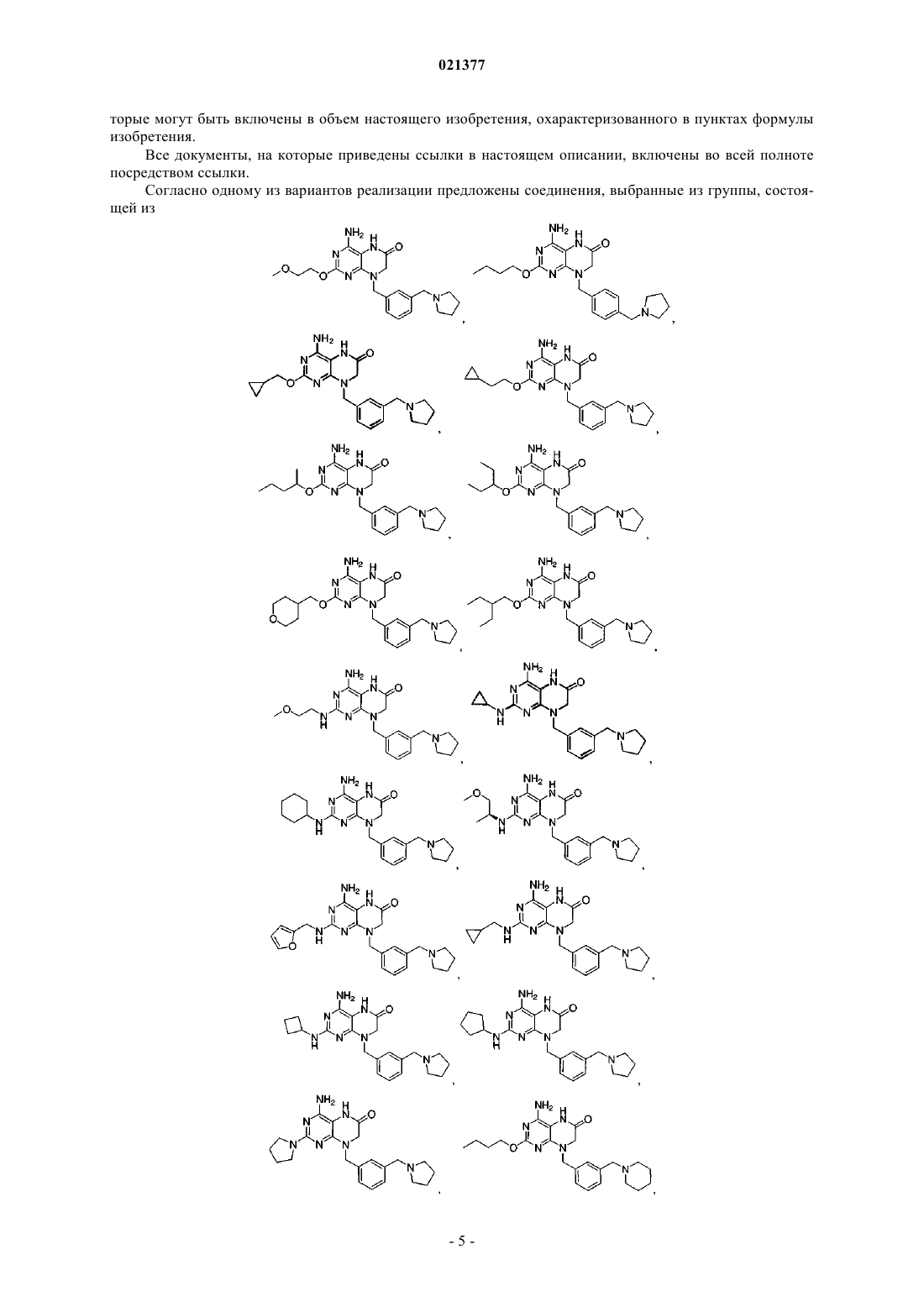
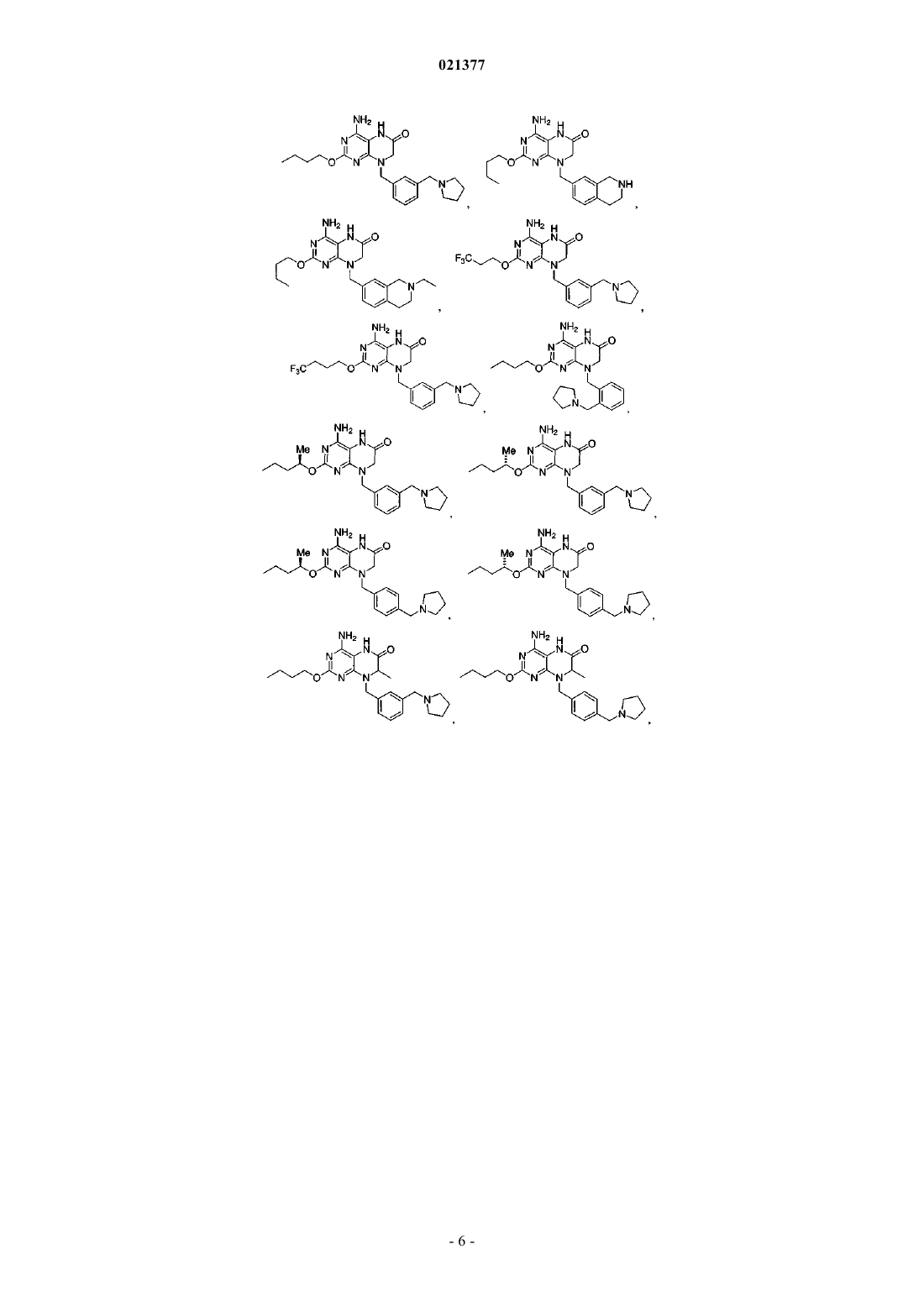
Предложены конкретные производные птеридинона, представляющие собой модуляторы TLR,фармацевтически приемлемые соли указанных соединений и фармацевтические композиции,содержащие указанные соединения. Область техники Настоящая заявка в целом относится к производным птеридинона и фармацевтическим композициям, которые селективно модулируют толлподобные рецепторы (например, TLR7). Уровень техники Врожденная иммунная система обеспечивает первую линию защиты организма от вторжения патогенных микроорганизмов. В случае врожденного иммунного ответа вторгающийся патоген распознается кодируемым в зародышевой линии рецептором, активация которого запускает сигнальный каскад, что приводит к индукции экспрессии цитокина. Рецепторы врожденной иммунной системы обладают широкой специфичностью, распознавая молекулярные структуры, которые являются высококонсервативными для различных патогенов. Одно из семейств указанных рецепторов известно как толлподобные рецепторы (TLR) благодаря их гомологичности рецепторам, впервые идентифицированным и получившим название у дрозофилы; указанные рецепторы присутствуют в клетках, таких как макрофаги, дендритные клетки и клетки эпителия. У млекопитающих существует по меньшей мере десять различных TLR. Для некоторых из указанных рецепторов были идентифицированы лиганды и соответствующие сигнальные каскады. Например,TLR2 активируется липопротеинами бактерий (например, Е. coli), TLR3 активируется двухцепочечной РНК, TLR4 активируется липополисахаридами (т.е. ЛПС или эндотоксинами) грамотрицательных бактерий (например, Salmonella и Е. coli O157:H7), TLR5 активируется флагеллином подвижных бактерий(например, Listeria), TLR7 распознает и реагирует на имиквимод, TLR9 активируется неметилированными последовательностями CpG ДНК патогена. Стимуляция каждого из указанных рецепторов приводит к активации транскрипционного фактора NK-В и других сигнальных молекул, которые участвуют в регуляции экспрессии генов цитокинов, включая гены, кодирующие фактор некроза опухолей альфа (TNF-),интерлейкин-1 (IL-1) и некоторые хемокины. Агонисты TLR7 представляют собой иммуностимуляторы и вызывают выработку эндогенного интерферона- in vivo. Существует ряд заболеваний, нарушений и состояний, связанных с TLR, для которых, как полагают, терапия с применением агониста TLR является перспективной, включая, но не ограничиваясь ими,меланому, немелкоклеточную карциному легких, почечноклеточную карциному, базальноклеточную карциному, светлоклеточную карциному, миелому, аллергические риниты, астму, хроническое обструктивное заболевание легких (ХОЗЛ), неспецифический язвенный колит, фиброз печени и вирусные инфекции, такие как вирус гепатита В (ВГВ), вирусы Flaviviridae, вирус гепатита С (ВГС), вирус папилломы человека (ВПЧ), респираторно-синцитиальный вирус (РСВ), тяжлый острый респираторный синдром (ТОРС), ВИЧ или грипп. Лечение вирусных инфекций, вызываемых Flaviviridae, при помощи агонистов TLR является особенно многообещающим. Вирусы семейства Flaviviridae включают по меньшей мере три различных рода, включая пестивирусы, флавивирусы и гепацивирусы (Кэлишер с соавт. (Calisher et al.), J. Gen. Virol.,1993, 70, 37-43). Пестивирусы вызывают многие отражающиеся на экономике заболевания у животных,такие как вирусная диарея крупного рогатого скота (BVDV), классическая чума свиней (CSFV, чума свиней) и пограничная болезнь овец (BDV), при этом их роль в заболеваниях человека охарактеризована в меньшей степени (В. Мэннинг с соавт. (Moenning, V., et al.), Adv. Vir. Res. 1992, 48, 53-98). Флавивирусы ответственны за серьезные заболевания у людей, такие как лихорадка денге и желтая лихорадка, а гепацивирусы вызывают вирусные инфекции гепатита С у человека. Другие значимые вирусные инфекции,вызываемые вирусами семейства Flaviviridae, включают лихорадку Западного Нила (ЛЗН), вирус японского энцефалита (ВЯЭ), вирус клещевого энцефалита, вирус Кунджин, энцефалит долины Муррея, энцефалит Сент-Луи, вирус омской геморрагической лихорадки и вирус Зика. В целом, инфекции, вызываемые семейством вирусов Flaviviridae, приводят к значительной смертности, заболеваемости и экономическим потерям по всему миру. Следовательно, существует необходимость в развитии эффективных способов лечения инфекций, вызываемых вирусами Flaviviridae. Вирус гепатита С (ВГС) является главной причиной хронических заболеваний печени по всему миру (Н. Бойер с соавт. (Boyer, N., et al.), J. Hepatol. 32:98-112, 2000), поэтому значительное внимание в проводящихся в настоящее время исследованиях, направленных на борьбу с вирусами, уделяется разработке улучшенных способов лечения хронических инфекций ВГС у человека (Ди Беселье A.M. и Бэйкон Б.Р. (Di Besceglie, A.M. and Bacon, B.R.), Scientific American, Oct.: 80-85 (1999); Гордон С.П. с соавт.Micro. 2007, 5(6), 453-463). Ряд способов лечения ВГС представлен в обзоре Баймока с соавт. (Bymock etal.) в Antiviral ChemistryChemiotherapy, 11:2; 79-95 (2000). В настоящее время для лечения хронических инфекций ВГС у человека применяют главным образом два противовирусных соединения: рибавирин, аналог нуклеозида, и интерферон- (ИФН). Рибавирин в отдельности не является эффективным для снижения уровней вирусной РНК, обладает значительной токсичностью и, как известно, вызывает анемию. Сообщалось, что комбинация ИФН и рибавирина является эффективной для лечения хронического гепатита С (Скотт Л.Дж. с соавт. (Scott, L.J., et al.), Drugs 2002, 62, 507-556), но длительный положительный эффект наблюдали менее чем у половины пациентов, получавших указанное лечение. ВГС распознается при помощи врожденных механизмов распознавания вирусов, которые индуцируют быстрый ИФН-ответ (Дастин с соавт. (Dustin et al.), Annu. Rev. Immunol. 2007, 25, 71-99). Вероятно,источники ИФН представляют собой, по меньшей мере, инфицированные гепатоциты и, в частности,плазмацитоидные дендритные клетки (ПДК), которые обеспечивают высокую экспрессию рецепторовTLR7 и секретируют большие количества ИФН. Хорсманс с соавт. (Horsmans, et al.) (Hepatology, 2005,42, 724-731) показали, что лечение в течение 7 дней агонистом TLR 7 изаторибином при введении один раз в день снижает концентрацию вируса в плазме у пациентов, инфицированных ВГС. Ли с соавт. (Lee,et al.) (Proc, Natl. Acad. Sci. USA, 2006, 103, 1828-1833) показали, что стимуляция TLR7 может индуцировать иммунитет к ВГС как за счет ИФН-механизма, так и за счет не связанных с ИФН механизмов. Указанные исследователи также обнаружили, что TLR7 экспрессируется как в нормальных, так и в инфицированных ВГС гепатоцитах. Представленные объединенные результаты подтверждают вывод о том, что стимуляция рецепторов TLR7, например, при помощи введения агониста TLR7, представляет собой перспективный механизм эффективного лечения естественных инфекций ВГС. С учетом существующей потребности в более эффективных способах лечения инфекций, вызываемых ВГС, необходимо разработать безопасные и терапевтически эффективные агонисты TLR7. Краткое описание изобретения Предложены соединения и их фармацевтически приемлемые соли. Не желая быть связанными теорией, авторы настоящего изобретения полагают, что предложенные соединения являются агонистами TLR7 и также могут быть агонистами других TLR. Согласно другому аспекту предложена фармацевтическая композиция, содержащая указанное соединение и фармацевтически приемлемый носитель или наполнитель. Настоящее изобретение включает комбинации аспектов и вариантов реализации, а также предпочтительных вариантов, приведенных в настоящем описании. Подробное описание В данном разделе приведено подробное описание конкретных пунктов формулы изобретения, примеры которых проиллюстрированы приведенными структурами и формулами. Несмотря на то, что изобретение описано со ссылкой на пронумерованные пункты формулы изобретения, следует понимать, что указанные ссылки не ограничивают изобретение лишь указанными пунктами формулы изобретения. Напротив, подразумевается, что изобретение включает все альтернативы, модификации и эквиваленты, ко-4 021377 торые могут быть включены в объем настоящего изобретения, охарактеризованного в пунктах формулы изобретения. Все документы, на которые приведены ссылки в настоящем описании, включены во всей полноте посредством ссылки. Согласно одному из вариантов реализации предложены соединения, выбранные из группы, состоящей из или их фармацевтически приемлемые соли. Определения Если не указано иное, следующие термины и фразы, используемые в настоящем описании, имеют представленные далее значения. Тот факт, что конкретный термин или фраза точно не определены, не следует соотносить с неопределенностью или недостаточной ясностью, а следует расценивать как то, что термины используют в их привычном значении. При использовании в настоящем описании названий товарных знаков заявители независимо включают название товарного знака для продукта и активный(е) фармацевтический(е) ингредиент(ы) продукта, зарегистрированного под указанным товарным знаком. Термин "лечение" и его грамматические эквиваленты при использовании в контексте лечения заболевания означают замедление или остановку прогрессирования заболевания, или облегчение по меньшей мере одного симптома заболевания, более предпочтительно снижение более чем одного симптома заболевания. Например, лечение вирусной инфекции гепатита С может включать снижение концентрации ВГС в крови у инфицированного ВГС человека и/или снижение степени тяжести желтухи у инфицированного ВГС человека. Используемое в настоящем описании "соединение согласно настоящему изобретению" означает заявленное соединение, включая альтернативные формы указанного соединения, такие как сольватированные формы, гидратированные формы, этерифицированные формы или их физиологически функциональные производные. Соединения согласно настоящему изобретению также включают таутомерные формы,например таутомерные "енолы", представленные в настоящем описании. Аналогично, в отношении выделяемых промежуточных соединений фраза "соединение формулы (номер)" означает соединение, соответствующее указанной формуле, и его альтернативные формы. Специалистам в данной области техники станет очевидно, что соединения согласно настоящему изобретению могут находиться в сольватированной или гидратированной форме. Также специалистам в данной области техники станет очевидно, что соединения могут подвергаться этерификации. В рамки настоящего изобретения также включены таутомерные формы, а именно таутомерные "енолы", представленные в настоящем описании. Кроме того, настоящее изобретение включает формы пролекарств соединений, представленные в настоящем описании."Сложный эфир" означает любой сложный эфир соединения, в котором любая функциональная группа -СООН молекулы замещена на функциональную группу -C(O)OR или в котором любая функциональная группа -ОН замещена на функциональную группу -OC(O)R, в которой фрагмент R сложного эфира представляет собой углеродсодержащую группу, которая образует стабильный сложноэфирный фрагмент, включающий, но не ограничиваясь перечисленными, алкил, алкенил, алкинил, циклоалкил,циклоалкилалкил, арил, арилалкил, гетероциклил, гетероциклилалкил и их замещенные производные. Сложные эфиры также могут включать сложные эфиры, как определено выше, "таутомерных енолов",например, представленных ниже Термин "сложный эфир указанного соединения" включает, но не ограничивается ими, фармацевтически приемлемые эфиры указанного соединения. Термин "пролекарство", используемый в настоящем описании, относится к любому соединению,которое при введении в биологическую систему высвобождает лекарственное вещество, т.е. активный ингредиент, как результат самопроизвольной(ых) химической(их) реакции(й), катализируемой(ых) ферментами химической(их) реакции(й), фотолиза и/или метаболической(их) химической(их) реакции(й). Пролекарство, таким образом, представляет собой ковалентно модифицированный аналог или латентную форму терапевтически активного соединения. Специалистам в данной области техники станет очевидно, что соединения согласно настоящему изобретению могут содержать один или более хиральный центр. Указанные формы находятся в рамках настоящего изобретения. Также специалистам в данной области техники очевидно, что соединение способно к этерификации. В рамки настоящего изобретения включены сложные эфиры и другие физиологически функциональные производные. В рамки настоящего изобретения также включены таутомерные формы, а именно таутомерные енолы, представленные в настоящем описании. В дополнение, в рамки настоящего изобретения включены формы пролекарств соединений, представленные в настоящем описании. Предложенные соединения и их фармацевтически приемлемые соли могут существовать в виде различных полиморфов или псевдополиморфов. Используемый в настоящем описании термин "кристаллический полиморфизм" означает способность кристаллического соединения существовать в виде различных кристаллических структур. Полиморфизм, как правило, может происходить в результате изменений температуры, давления или обоих указанных факторов. Также полиморфизм может происходить в результате изменений процесса кристаллизации. Полиморфы можно различать при помощи различных физических характеристик, известных в данной области техники, таких как спектры рентгеновской дифракции, растворимость и температура плавления. Кристаллический полиморфизм может происходить в результате различий упаковки в кристаллах (упаковочный полиморфизм) или различий в упаковке различных конформеров одной и той же молекулы (конформационный полиморфизм). Используемый в настоящем описании термин "кристаллический псевдополиморфизм" означает способность гидрата или сольвата соединения существовать в виде различных кристаллических структур. Псевдополиморфы согласно настоящему изобретению могут существовать за счет различий упаковки в кристаллах (упаковочный псевдополиморфизм) или вследствие различий упаковки различных конформеров одной и той же молекулы (конформационный псевдополиморфизм). Настоящее изобретение охватывает все полиморфы и псевдополиморфы предложенных соединений и их фармацевтически приемлемых солей. Соединение согласно настоящему изобретению и его фармацевтически приемлемые соли также могут существовать в виде аморфного твердого вещества. Используемое в настоящем описании аморфное твердое вещество представляет собой твердое вещество, в котором отсутствует дальний порядок расположения атомов в твердом веществе. Указанное определение применяют, в случае если размер кристалла составляет два нанометра или менее. Для получения аморфных форм согласно настоящему изобретению можно применять добавки, включая растворители. Настоящее изобретение охватывает все аморфные формы соединений согласно настоящему изобретению и их фармацевтически приемлемых солей. Конкретные соединения, представленные в настоящем описании, содержат один или более хиральных центров или могут другим образом быть способными к существованию в виде множества стерео-9 021377 изомеров. В объем настоящего изобретения включены смеси стереоизомеров, а также очищенные энантиомеры или смеси, обогащенные одним из энантиомеров/диастереомеров. Также в объем настоящего изобретения включены индивидуальные изомеры соединений, представленных формулами, согласно настоящему изобретению, а также любые полностью или частично равновесные смеси указанных изомеров. В настоящее изобретение также включены индивидуальные изомеры соединений, соответствующих представленным выше формулам, в виде смесей с изомерами указанных соединений, в которых один или более хиральных центров являются обращенными. Термин "хиральный" относится к молекулам, которые обладают свойством несовместимости со своим зеркальным отображением, при этом термин "ахиральный" относится к молекулам, которые являются совместимыми со своим зеркальным отражением. Термин "стереоизомеры" относится к соединениям, которые имеют одинаковый химический состав,но отличаются взаимным расположением атомов или групп в пространстве."Диастереомер" относится к стереоизомеру, содержащему два или более центра хиральности, молекулы которого не являются зеркальными отражениями друг друга. Диастереомеры обладают различными физическими свойствами, например температурой плавления, температурой кипения, спектральными свойствами и реакционной способностью. Смеси диастереомеров можно разделять при помощи аналитических способов с высоким разрешением, например электрофореза и хроматографии."Энантиомеры" относятся к двум стереоизомерам соединения, которые не являются совместимыми зеркальными отображениями друг друга. Стереохимические определения и условные обозначения, используемые в настоящем описании, в целом соответствуют работам С.П. Паркера (S.P. Parker), Ed., McGraw-Hill Dictionary of Chemical Terms(1984) McGraw-Hill Book Company, New York; и E. Эльеля и С. Уилена (Eliel, E. and Wilen, S.) Stereochemistry of Organic Compounds (1994) John WileySons, Inc., New York. Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоскополяризованного света. При описании оптически активного соединения используют префиксы D и L или R и S для определения абсолютной конфигурации молекулы относительно ее хирального(ых) центра(ов). Префиксы d и l или (+) и (-) применяют для обозначения знака вращения соединением плоскополяризованного света, причем значения (-) или l обозначают, что соединение является левовращающим. Соединение, обозначенное префиксами (+) или d, является правовращающим. Для данной химической структуры указанные стереоизомеры являются одинаковыми за исключением того, что они являются зеркальными отражениями друг друга. Конкретный стереоизомер также может быть назван энантиомером, а смесь указанных изомеров часто называют энантиомерной смесью. Смесь, содержащую энантиомеры в соотношении 50:50, которая может существовать в случае, если в химической реакции или процессе отсутствовала стереоселективность или стереоспецифичность, называют рацемической смесью или рацематом. Терминами "рацемическая смесь" и "рацемат" называют эквимолярную смесь двух энантиомерных видов, лишенную оптической активности. В настоящее изобретение включены соль или сольват соединений, представленных в настоящем описании, включая их комбинации, такие как сольват соли. Соединения согласно настоящему изобретению могут существовать в сольватированной, например гидратированной, а также в несольватированной форме, настоящее изобретение охватывает все указанные формы. Как правило, но не исключительно, соли согласно настоящему изобретению представляют собой фармацевтически приемлемые соли. Соли, определенные в рамках термина "фармацевтически приемлемые соли", относятся к нетоксичным солям соединений согласно настоящему изобретению. Примеры подходящих фармацевтически приемлемых солей включают неорганические соли, полученные в результате добавления неорганической кислоты, такие как хлорид, бромид, сульфат, фосфат и нитрат; соли, полученные в результате добавления органической кислоты, такие как ацетат, галактарат,пропионат, сукцинат, лактат, гликолят, малат, тартрат, цитрат, малеат, фумарат, метансульфонат, птолуолсульфонат и аскорбат; соли с кислыми аминокислотами, такие как аспартат и глутамат; соли щелочных металлов, такие как натриевая соли и кальциевая соль; соли щелочно-земельных металлов, такие как магниевая соль и кальциевая соль; аммонийная соль; соли органических оснований, такие как соль триметиламина, соль триэтиламина, соль пиридина, соль пиколина, соль дициклогексиламина и сольN,N'-дибензилэтилендиамина; и соли с основными аминокислотами, такие как соль лизина и соль аргинина. Соли в некоторых случаях могут представлять собой гидраты или сольваты в этаноле. Защитные группы. В контексте настоящего изобретения защитные группы включают группы, образующие пролекарства, и химические защитные группы. Защитные группы являются доступными, общеизвестными и общепринятыми, их можно применять для предотвращения побочных реакций с защищенными группами в течение способов синтеза, т.е. путей или способов получения соединений согласно настоящему изобретению. В большинстве случаев решение о выборе групп, которые необходимо защитить, времени, в которое нужно производить защиту, и природе химической защитной группы "ЗГ" зависит от химических условий реакции, от которых нужно производить защиту (например, кислотных, основных, окислительных, восстановительных или других условий) и требуемого направления синтеза. ЗГ не должны быть и, как правило, не являются одинаковыми в случае, если соединение замещено множеством ЗГ. В целом, ЗГ применяют для защиты функциональных групп, таких как карбоксил, гидроксил, тио- или аминогруппы, и для предотвращения, таким образом, побочных реакций или для увеличения другим способом эффективности синтеза. Порядок снятия защиты с получением свободных, незащищенных, групп зависит от требуемого направления синтеза и условий проведения реакции и может осуществляться в любом порядке, определенном специалистом в данной области техники. Могут быть защищены различные функциональные группы соединений согласно настоящему изобретению. Например, защитные группы для -ОН-групп (гидроксильных, карбоновых кислот, фосфоновых кислот или других функций) включают "группы, образующие простые или сложные эфиры". Группы, образующие простые или сложные эфиры, способны выступать в качестве химических защитных групп в схемах синтеза, представленных в настоящем описании. Тем не менее, некоторые гидроксил- и тиозащитные группы не являются группами, образующими простые или сложные эфиры, что понимают специалисты в данной области техники, и относятся к амидам, описанным ниже. Очень широкий ряд гидроксилзащитных групп, амидобразующих групп и соответствующие химические реакции их разложения описаны в Protective Groups in Organic Synthesis, Теодора В. Грин и Питер Г.М. Вутс (Theodora W. Greene and Peter G. M. Wuts) (John WileySons, Inc., New York, 1999, ISBN 0471-16019-9) ("Greene"). См. также Филип Дж. Коченски (Kocienski, Philip J.); Protecting Groups (GeorgThieme Verlag Stuttgart, New York, 1994), содержание которой включено в настоящее описание во всей полноте посредством ссылки. В частности, раздел 1, Protecting Groups: An Overview, стр. 1-20, раздела 2,Hydroxyl Protecting Groups, стр. 21-94, раздел 3, Diol Protecting Groups, стр. 95-117, раздел 4, CarboxylProtecting Groups, стр. 118-154, раздел 5, Carbonyl Protecting Groups, стр. 155-184. Для описания защитных групп для карбоновых кислот, фосфоновой кислоты, фосфоната, серной кислоты и других защитных групп для кислот, см. Greene, что представлено далее. Указанные группы включают в качестве примера и не для ограничения сложные эфиры, амиды, гидразиды и т.д. Защитные группы, образующие простые и сложные эфиры. Группы, образующие сложные эфиры, включают: (1) группы, образующие сложные эфиры с фосфонатами, такие как сложные эфиры с фосфонамидатами, сложные эфиры с фосфонотиоатами, сложные эфиры с фосфонатами и фосфон-бис-амидаты; (2) группы, образующие сложные эфиры с карбоксилом, и(3) группы, образующие сложные эфиры с серой, такие как сульфонат, сульфат и сульфинат. Метаболиты соединений согласно настоящему изобретению. Также в объем настоящего изобретения попадают продукты метаболизма in vivo соединений, представленных в настоящем описании. Указанные продукты могут быть получены, например, в результате окисления, восстановления, гидролиза, амидирования, этерификации и т.д., вводимого соединения, главным образом, вследствие ферментативных процессов. Соответственно в изобретение включены соединения, получаемые в результате процесса, включающего взаимодействие соединения согласно настоящему изобретению с млекопитающим в течение времени, достаточного для получения метаболического продукта указанного соединения. Указанные продукты, как правило, определяют при помощи получения соединения согласно настоящему изобретению с радиоактивной меткой (например, С или Н), парентерального введения полученного соединения в детектируемой дозе (например, равной более чем примерно 0,5 мг/кг) животному, такому как крыса, мышь, морская свинка, обезьяна, или человеку, выдерживания в течение времени, достаточного для прохождения метаболизма (как правило, от примерно 30 с до примерно 30 ч) и выделения продуктов превращения из мочи, крови или других биологических образцов. Указанные продукты легко выделить, так как они являются мечеными (другие продукты выделяют с применением антител, способных к связыванию эпитопов, выживающих в метаболите). Метаболитные структуры определяют подходящим способом, например при помощи МС или ЯМР анализа. В целом,анализ метаболитов проводят способом, аналогичным общепринятым исследованиям метаболизма, хорошо известным специалистам в данной области техники. Продукты конверсии, если они дополнительно не присутствуют in vivo в другом виде, подходят для диагностических исследований терапевтического дозирования соединений согласно настоящему изобретению, даже если они не сохраняют противоинфекционной активности сами по себе. Фармацевтические составы. Соединения согласно настоящему изобретению вводят в состав совместно с подходящими носителями и наполнителями, которые выбирают в соответствии с общепринятой практикой. Таблетки содержат наполнители, глиданты, вещества-наполнители, связующие вещества и т.д. Водные составы получают в стерильном виде, и в случае, если указанные составы предназначены для введения отличным от перорального способом, составы являются изотоническими. Все составы могут содержать наполнители,представленные, например, в Handbook of Pharmaceutical Excipients (1986), содержание которой включено в настоящее описание по всей полноте посредством ссылки. Наполнители включают аскорбиновую кислоту и другие антиоксиданты, хелатообразующие агенты, такие как ЭДТА, углеводы, такие как декстрин, гидроксиалкилцеллюлоза, гидроксиалкилметилцеллюлоза, стеариновую кислоту и т.д. рН составов находится в диапазоне от примерно 3 до примерно 11, но, как правило, составляет примерно от 7 до 10. Несмотря на то, что активные ингредиенты могут быть введены индивидуально, может быть предпочтительным их введение в виде фармацевтических составов. Составы согласно настоящему изобретению как для ветеринарного применения, так и для введения человеку, содержат по меньшей мере один активный ингредиент совместно с одним или более приемлемыми носителями и возможно другими терапевтическими ингредиентами. Носитель(и) должен быть "приемлемым" с точки зрения совместимости с другими ингредиентами состава и физиологически безопасным для потребителя указанного состава. Составы включают такие составы, которые подходят для представленных далее способов введения. Составы могут подходящим образом присутствовать в виде стандартной дозированной формы и могут быть получены любыми способами, известными в области фармации. Способы и составы в целом можно найти в Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, Pa.), содержание которой включено в настоящее описание по всей полноте посредством ссылки. Указанные способы включают стадию ассоциации активного ингредиента с носителем, который содержит один или более вспомогательных ингредиентов. В целом, составы получают при помощи однородной и тщательной ассоциации активного ингредиента с жидкими носителями или мелкодисперсными твердыми носителями или обоими типами носителей и затем, в случае необходимости, придания формы продукту. Составы согласно настоящему изобретению, подходящие для перорального введения, могут существовать в виде отдельных единиц, таких как капсулы, сашеты или таблетки, каждая из которых содержит предварительно определенное количество активного ингредиента; в виде порошка или гранул; в виде раствора или суспензии в водной или неводной жидкости или в виде жидкой эмульсии типа масло-вводе или жидкой эмульсии типа вода-в-масле. Также активный ингредиент можно вводить в виде болюса, электуария или пасты. Таблетку получают при помощи прессования или литья в форму, возможно с одним или более вспомогательными ингредиентами. Прессованные таблетки могут быть получены при помощи сжатия в подходящем аппарате активного ингредиента в свободнотекучей форме, такой как порошок или гранулы,возможно смешанного со связывающим веществом, смазкой, инертным разбавителем, консервантом,поверхностно активным или диспергирующим агентом. Прессованные таблетки получают при помощи прессования в подходящем аппарате смеси порошкообразного активного ингредиента, увлажненной инертным жидким разбавителем. Таблетки могут быть покрыты оболочкой или представлять собой таблетку с насечкой, а также могут быть приготовлены таким образом, чтобы обеспечивать замедленное или контролируемое высвобождение активного ингредиента. Для введения в глаз или другие внешние ткани, например, в рот или на кожу, составы, предпочтительно, применяют в виде топических мазей или кремов, содержащих активный(е) ингредиент(ы) в количестве, равном, например, от 0,075 до 20% мас./мас. (включая содержание активного(ых) ингредиента(ов) в диапазоне от 0,1 до 20% с границами, кратными 0,1% мас./мас., например 0,6% мас./мас., 0,7% мас./мас. и т.д.), предпочтительно, от 0,2 до 15% мас./мас. и наиболее предпочтительно от 0,5 до 10% мас./мас. При вхождении в состав мазей активные ингредиенты можно применять вместе с парафиновой или смешиваемой с водой основой мази. Как альтернатива, активные ингредиенты могут входить в состав с основой крема типа масло-в-воде. При необходимости, водная фаза основы крема может содержать, например, по меньшей мере 30% мас./мас. полиспирта, т.е. спирта, содержащего две или более гидроксильные группы, такого как пропиленгликоль, бутан-1,3-диол, маннит, сорбит, глицерин и полиэтиленгликоль (включая ПЭГ 400) и их смеси. Составы для топического введения могут, при необходимости, включать соединение, которое усиливает абсорбцию или проницаемость активного ингредиента через кожу или другие пораженные зоны. Примеры указанных усилителей проницаемости через кожу включают диметилсульфоксид и аналоги на его основе. Масляная фаза эмульсий согласно настоящему изобретению может быть приготовлена из известных ингредиентов известными способами. Несмотря на то, что указанная фаза может содержать исключительно эмульсификатор (также известный как эмульгатор), предпочтительно фаза содержит смесь по меньшей мере одного эмульсификатора с жиром или маслом или и с жиром, и с маслом. Предпочтительно гидрофильный эмульсификатор содержится вместе с липофильным эмульсификатором, который действует как стабилизатор. Также предпочтительно, чтобы в состав фазы входили и масло, и жир. Взятые вместе, эмульсификатор(ы) с или без стабилизатора(ов) образуют так называемый эмульгирующий воск,а воск вместе с маслом и жиром образуют так называемую эмульгирующую основу мази, которая образует диспергированную в масле фазу составов кремов. Эмульгаторы и стабилизаторы эмульсии, подходящие для применения в составах согласно настоящему изобретению, включают Tween 60, Span 80, цетостеариловый спирт, бензиловый спирт, миристиловый спирт, глицерилмоностеарат и лаурилсульфат натрия. Выбор подходящих масел или жиров для состава основан на достижении требуемых косметических свойств. Крем предпочтительно должен быть нежирным, не оставляющим пятен и смываемым продуктом с подходящей консистенцией для предотвращения вытекания из туб или других контейнеров. Можно применять сложные эфиры одно- или двуосновных кислот и алкилов с линейной или разветвленной цепью, такие как диизоадипат, изоцетилстеарат, дисложный эфир жирных кислот кокосового масла и пропиленгликоля,изопропилмиристат,децилолеат,изопропилпальмитат,бутилстеарат,2 этилгексилпальмитат или смесь сложных эфиров с разветвленной цепью, известную как Crodamol CAP,причем последние три эфира являются предпочтительными. Указанные сложные эфиры можно применять индивидуально или в комбинации в зависимости от требуемых свойств. В качестве альтернативы применяют липиды с высокой температурой плавления, такие как белый мягкий парафин и/или жидкий парафин или другие минеральные масла. Фармацевтические составы согласно настоящему изобретению содержат одно или более соединений согласно настоящему изобретению вместе с одним или более фармацевтически приемлемыми носителями или наполнителями. Фармацевтические составы, содержащие активный ингредиент, могут находиться в любой форме, подходящей для требуемого способа введения. При применении для перорального введения можно получать, например, таблетки, формованные пастилки, пастилки, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, твердые или мягкие капсулы, сиропы или эликсиры. Композиции, предназначенные для перорального введения, можно получать в соответствии с любыми способами, известными в области производства фармацевтических композиций, причем указанные композиции могут содержать один или более агентов, включая подсластители, ароматизаторы, красители и консерванты, для получения приятного на вкус препарата. Приемлемыми являются таблетки, содержащие активный ингредиент в смеси с нетоксичным фармацевтически приемлемым наполнителем, который подходит для производства таблеток. Указанные наполнители могут представлять собой, например, инертные разбавители, такие как карбонаты кальция или натрия, лактоза, моногидрат лактозы, кроскармеллоза натрия, повидон, фосфаты кальция или натрия; гранулирующие агенты и вещества для улучшения распадаемости, такие как кукурузный крахмал или альгиновая кислота; связывающие агенты, такие как целлюлоза, микрокристаллическая целлюлоза, крахмал, желатин или камедь; и смазки, такие как стеарат магния, стеариновая кислота или тальк. Таблетки могут быть без оболочки или покрыты оболочкой при помощи известных способов, включая микроинкапсулирование, для задержки распадания и абсорбции в желудочно-кишечном тракте и обеспечения, таким образом, замедленного действия в течение длительного периода. Например, можно применять вещество, обеспечивающее задержку времени действия, такое как глицерилмоностеарат или глицерилдистеарат, индивидуально или вместе с воском. Составы для перорального введения также могут существовать в виде твердых желатиновых капсул, в которых активный ингредиент смешивают с инертным твердым разбавителем, например фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешивают с водой или масляной средой, такой как ореховое масло, жидкий парафин или оливковое масло. Водные суспензии согласно настоящему изобретению содержат активные вещества в смеси с наполнителями, подходящими для получения водных суспензий. Указанные наполнители включают суспендирующий агент, такой как карбоксиметилцеллюлоза натрия, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь, и диспергирующие или увлажняющие агенты, такие как фосфатиды природного происхождения (например, лецитин), продукт конденсации алкиленоксида и жирной кислоты (например, полиоксиэтиленстеарат), продукт конденсации этиленоксида и длинноцепочечного алифатического спирта (например, гептадекаэтиленоксицетанол), продукт конденсации этиленоксида и неполного сложного эфира, полученного из жирной кислоты и ангидрида гексита (например, полиоксиэтиленсорбитана моноолеат). Водная суспензия также может содержать один или более консервант, такой как этил- или н-пропил пгидроксибензоат, один или более красителей, один или более ароматизаторов и один или более подсластителей, таких как сахароза или сахарин. Масляные суспензии могут быть получены в результате суспендирования активного ингредиента в растительном масле, таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло, или в минеральном масле, таком как жидкий парафин. Суспензии для перорального введения могут содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Для получения приятных на вкус препаратов для перорального введения можно добавлять подсластители, представленные в настоящем описании, и ароматизаторы. Указанные композиции могут быть защищены при помощи добавления антиоксиданта, такого как аскорбиновая кислота. Диспергируемые порошки и гранулы согласно настоящему изобретению, подходящие для получения водных суспензий при помощи добавления воды, содержат активный ингредиент в смеси с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или более консервантом. Подходящие диспергирующие или увлажняющие агенты и суспендирующие агенты представлены веществами, описанными выше. Также могут присутствовать дополнительные наполнители, такие как подсластители, ароматизаторы и красители. Фармацевтические композиции согласно настоящему изобретению также могут находиться в виде эмульсий типа масло-в-воде. Масляная фаза может представлять собой растительное масло, такое как оливковое масло или арахисовое масло, минеральное масло, такое как жидкий парафин, или их смесь. Подходящие эмульгаторы включают камеди природного происхождения, такие как аравийская камедь и трагакантовая камедь, фосфатиды природного происхождения, такие как соевый лецитин, сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и ангидридов гексита, такие как сорбитана моноолеат, и продукты конденсации указанных неполных сложных эфиров и этиленоксидов, такие как полиоксиэтиленсорбитана моноолеат. Эмульсия также может содержать подсластители и ароматизаторы. В состав сиропов и эликсиров могут входить подсластители, такие как глицерин, сорбит или сахароза. Указанные составы также могут содержать успокоительное средство, консервант, ароматизатор или краситель. Фармацевтические композиции согласно настоящему изобретению могут находиться в виде стерильного инъецируемого препарата, такого как стерильная инъецируемая водная или масляная суспензия. Указанная суспензия может быть получена согласно известным в данной области техники способам с применением подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов,которые указаны в настоящем описании. Стерильный инъецируемый препарат также может представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например раствор в 1,3-бутандиоле, или указанный препарат может быть получен в виде лиофилизированного порошка. В число приемлемых носителей и растворителей, которые можно применять, входит вода, раствор Рингера и изотонический раствор хлорида натрия. В дополнение в качестве растворителя или суспендирующей среды можно подходящим образом применять жирные масла. Для указанной цели можно применять любые мягкие жирные масла, включая синтетические моно- или диглицериды. В дополнение для получения инъецируемых лекарственных средств также можно применять жирные кислоты, такие как олеиновая кислота. Количество активного ингредиента, которое можно объединить с веществом-носителем для получения одной дозированной формы, зависит от принимающего лекарственное средство пациента и конкретного способа введения. Например, состав с замедленным высвобождением, предназначенный для перорального введения человеку, может содержать примерно от 1 до 1000 мг активного вещества, входящего в состав с подходящим и приемлемым количеством вещества-носителя, которое может изменяться от примерно 5 до примерно 95% от общей массы композиции (мас.:мас.). Фармацевтическая композиция может быть получена для обеспечения легко определяемых количеств вводимого вещества. Например, водный раствор, предназначенный для внутривенной инфузии, может содержать примерно от 3 до 500 мкг активного ингредиента на 1 мл раствора для того, чтобы могла происходить инфузия подходящего объема со скоростью, равной примерно 30 мл/ч. Составы, подходящие для введения в глаз, включают глазные капли, в которых активный ингредиент растворен или суспендирован в подходящем носителе, конкретно в водном растворителе активного ингредиента. Активный ингредиент предпочтительно содержится в указанных составах в концентрации,равной от 0,5 до 20%, преимущественно от 0,5 до 10%, конкретно равной примерно 1,5% (мас./мас.). Составы, подходящие для местного применения в ротовой полости, включают пастилки, содержащие активный ингредиент в ароматизированной основе, как правило, сахарозе или камеди или трагаканте; пастилы, содержащие активный ингредиент в инертной основе, как правило, желатине и глицерине,или сахарозе и камеди; и жидкости для полоскания рта, содержащие активный ингредиент в подходящем жидком носителе. Составы для ректального введения могут быть представлены в виде суппозиториев с подходящим основанием, включающим, например, масло какао или салицилат. Составы, подходящие для внутрилегочного или назального введения, содержат частицы с размером, находящимся в диапазоне, например, от 0,1 до 500 мкм (включая частицы с размером, находящимся в диапазоне от 0,1 и 500 мкм с границами, кратными, например, 0,5, 1, 30, 35 мкм и т.д.), которые вводят при помощи быстрой ингаляции через носовой канал или при помощи ингаляции через рот для достижения альвеолярных мешочков. Подходящие составы включают водные или масляные растворы активного ингредиента. Составы, подходящие для введения в виде аэрозоля или сухого порошка, могут быть получены подходящими способами и могут доставляться с другими терапевтическими агентами, такими как соединения, применяемые в настоящее время для лечения или предотвращения инфекций, представленных в настоящем описании. Составы, подходящие для вагинального введения могут существовать в виде пессариев, тампонов,кремов, гелей, паст, пен или спреев, содержащих в дополнение к активному ингредиенту известные в данной области техники носители, подходящие для указанного способа введения. Составы, подходящие для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворы,которые приводят к получению состава, изотонического с кровью пациента, которому вводят указанный состав; и водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загустители. Составы находятся в одноразовых контейнерах или контейнерах для многоразового введения, например в герметичных ампулах или пробирках, и могут храниться в сублимированном (лиофилизированном) состоянии, требующем исключительно добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед применением. Растворы и суспензии для немедленной инъ- 14021377 екции получают из стерильных порошков, гранул и таблеток, описанных ранее видов. Предпочтительные стандартные дозируемые составы представляют собой составы, которые содержат суточную дозу или единичную суточную поддозу, в соответствии с представленным выше описанием, или подходящую часть указанной дозы активного ингредиента. Следует понимать, что в дополнение к ингредиентам, конкретно указанным выше, составы согласно настоящему изобретению могут содержать другие агенты, применяемые в данной области техники, в зависимости от типа конкретного состава, например составы, подходящие для перорального введения,могут содержать ароматизаторы. Соединения согласно настоящему изобретению также могут входить в состав, обеспечивающий контролируемое высвобождение активного ингредиента, которое позволяет менее часто проводить введение дозы или улучшает фармакокинетические или токсикологические свойства активного ингредиента. Соответственно, в изобретении также предложены композиции, содержащие одно или более соединений согласно настоящему изобретению, представляющие собой составы, предназначенные для замедленного или контролируемого высвобождения. Эффективная дозировка активного ингредиента зависит, по меньшей мере, от природы состояния,требующего лечения, токсичности, от того, применяют ли соединение для профилактики (меньшие дозы) или против активного заболевания или состояния, способа доставки и фармацевтического состава, и определяется практикующими врачами с применением соответствующих исследований с повышением дозы. Полагают, что эффективная доза составляет от примерно 0,0001 до примерно 10 мг/кг массы тела в день, как правило, от примерно 0,001 до примерно 1 мг/кг массы тела в день, более типично от примерно 0,01 до примерно 1 мг/кг массы тела в день, еще более типично от примерно 0,05 до примерно 0,5 мг/кг массы тела в день. Например, возможная суточная доза для взрослого человека с массой тела, равной примерно 70 кг, находится в диапазоне от примерно 0,05 до примерно 100 мг, или от примерно 0,1 до примерно 25 мг, или от примерно 0,4 до примерно 4 мг, причем указанную дозу можно применять в виде единственной дозы или множественных доз. Согласно другому варианту реализации настоящее изобретение описывает фармацевтические композиции, содержащие соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или наполнитель. Способы введения. Одно или более соединений согласно настоящему изобретению (обозначенные в настоящем описании как активные ингредиенты) вводят любым способом, подходящим для состояния, требующего лечения. Подходящие способы введения включают пероральное, ректальное, назальное, местное (включая трансбуккальное и подъязычное), вагинальное и парентеральное (включая подкожное, внутримышечное,внутривенное, внутрикожное, интратекальное и эпидуральное) введение и т.д. Очевидно, что предпочтительный способ введения может изменяться в зависимости, например, от состояния пациента. Преимущество соединений согласно настоящему изобретению заключается в том, что они являются перорально биодоступными и могут дозироваться перорально. Способы лечения. Используемый в настоящем описании "агонист" представляет собой вещество, которое стимулирует связываемое вещество-партнер, как правило, рецептор. Специалисты в данной области техники понимают, что стимуляцию определяют в контексте конкретного исследования, также стимуляция может быть очевидна из приведенной в настоящем описании литературе, в результате сравнения с фактором или веществом, которое принято называть "агонистом" или "антагонистом" конкретного связываемого партнера, по существу, в одинаковых условиях. Стимуляцию можно определить как увеличение конкретного эффекта или функции, которое индуцируется взаимодействием агониста или частичного агониста и связываемого партнера, которое также может включать аллостерические эффекты. Используемый в настоящем описании "антагонист" представляет собой вещество, которое ингибирует связываемое вещество-партнер, как правило, рецептор. Специалисты в данной области техники понимают, что ингибирование определяют в контексте конкретного исследования, также ингибирование может быть определено в обсуждаемой в настоящем описании литературе, в результате сравнения с фактором или веществом, которое принято называть "агонистом" или "антагонистом" конкретного связываемого партнера, по существу, в одинаковых условиях. Ингибирование может быть определено как ослабление конкретного эффекта или функции, которое индуцируется взаимодействием антагониста и связываемого партнера, которое также может включать аллостерические эффекты. Используемые в настоящем описании "частичный агонист" или "частичный антагонист" представляют собой вещества, которые обеспечивают уровень стимуляции или ингибирования, соответственно,связываемого партнера, который не является целиком или полностью агонистическим или антагонистическим соответственно. Следует понимать, что стимуляцию, а также ингибирование определяют, по существу, для каждого вещества или категории веществ, определяемых как агонисты, антагонисты или частичные агонисты. Используемый в настоящем описании термин "внутренняя активность", или "эффективность" относится к некоторому определению биологической эффективности комплекса со связываемым партнером. С учетом фармакологии рецептора контекст, в котором следует определять внутреннюю активность или эффективность, зависят от свойств комплекса со связываемым партнером (например, рецептор/ли ганд) и рассмотрения активности, соответствующей конкретному биологическому результату. Например, в некоторых случаях, внутренняя активность может изменяться в зависимости от конкретного вторичного мессенджера, присутствующего в системе. Являются ли указанные специфические в зависимости от контекста оценки значимыми, и причины, по которым указанные оценки могут быть значимыми в контексте настоящего изобретения, очевидны специалисту в данной области техники. Используемый в настоящем описании термин "модуляция рецептора" включает агонизм, частичный агонизм, антагонизм, частичный антагонизм или обратный агонизм рецептора. Согласно одному из вариантов реализации можно применять способ индукции иммунного ответа на множество эпитопов вирусной инфекции у человека. Индукцию иммунного ответа на вирусную инфекцию можно определить при помощи любых способов определения наличия иммунного ответа, известных специалистам в данной области техники. Подходящие способы определения иммунного ответа согласно настоящему изобретению включают, помимо прочего, определение снижения концентрации вируса или антигена в сыворотке пациента, определение ИФНсекретирующих пептидспецифических Т-клеток и определение повышенного содержание одного или более фермента печени, такого как аланинтрансфераза (АЛТ) и аспартаттрансфераза (ACT). Согласно одному из вариантов реализации определение ИФН-секретирующих пептидспецифических Т-клеток завершают с применением исследования ELISPOT. Другой вариант реализации включает снижение концентрации вируса ВГВ, включая снижение, определенное при помощи исследования ПЦР. Лечение согласно настоящему изобретению, как правило, приводит к стимуляции иммунного ответа на ВГВ или ВГС у человека, инфицированного ВГВ или ВГС, соответственно, и соответствующее снижение концентрации вируса ВГВ или ВГС у инфицированного человека. Примеры иммунных ответов включают продуцирование антител (например, антител IgG) и/или продуцирование цитокинов, таких как интерфероны, которые модулируют активность иммунной системы. Ответ иммунной системы может представлять собой заново индуцированный ответ или может представлять собой усиление существующего иммунного ответа. В частности, ответ иммунной системы может представлять собой сероконверсию одного или более антигенов ВГВ или ВГС. Содержание вируса в крови можно определять при помощи измерения содержания ДНК ВГВ или ДНК ВГС в крови. Например, содержание ДНК ВГВ в сыворотке крови можно определять с применением исследования ПЦР Roche COBAS Amplicor Monitor (версия 2.0; нижний предел количественной оценки, 300 копий/мл [57 IU/мл]) и исследования b ДНК Quantiplex (нижний предел количественной оценки,0,7 MEq/мл; Bayer Diagnostics, ранее Chiron Diagnostics, Emeryville, CA). Содержание антител против специфических антигенов ВГВ или ВГС (например, поверхностного антигена гепатита В (HBsAG можно измерить с применением описанных в данной области техники способов, таких как ферментативный иммуноанализ и исследование ферментативной иммуноабсорбции. Например, содержание антител против специфических антигенов ВГВ или ВГС можно измерить с применением системы ферментативного иммуноанализа с применением микрочастиц Abbott AxSYM (Abbott Laboratories, North Chicago, IL). Соединение согласно настоящему изобретению можно вводить при помощи любого подходящего способа и любых подходящих средств, в том числе путем перорального или парентерального (например,внутривенного) введения. Терапевтически эффективное количество соединения согласно настоящему изобретению составляет от примерно 0,00001 до примерно 10 мг/кг массы тела в день, например от примерно 0,0001 до примерно 10 мг/кг массы тела в день, или, например, от примерно 0,001 до примерно 1 мг/кг массы тела в день, или, например, от примерно 0,01 до примерно 1 мг/кг массы тела в день, или,например, от примерно 0,05 до примерно 0,5 мг/кг массы тела в день, или, например, от примерно 0,3 мкг до примерно 30 мг в день, или, например, от примерно 30 до примерно 300 мкг в день. Частоту дозирования соединения согласно настоящему изобретению определяют в зависимости от требований для конкретного пациента, частота дозирования может составлять, например, один раз в день или два или более раз в день. Введение соединения согласно настоящему изобретению продолжают в течение периода, необходимого для лечения инфекции ВГВ или ВГС. Например, соединение согласно настоящему изобретению можно вводить человеку, инфицированному ВГВ или ВГС в течение периода,равного от 20 до 180 дней, или, например, в течение периода, равного от 20 до 90 дней, или, например, в течение периода, равного от 30 до 60 дней. Введение может быть прерываемым, с периодом в несколько или более дней, в течение которых пациент получает дневную дозу соединения согласно настоящему изобретению, и последующего периода, в течение которого пациент не получает дневную дозу соединения согласно настоящему изобретению. Например, пациент может получать дозу соединения согласно настоящему изобретению через день или три раза в неделю. В качестве примера пациенту могут вводить дозу соединения согласно настоящему изобретению каждый день в течение периода, равного от 1 до 14 дней, с последующим периодом,равным от 7 до 21 дней, в течение которого пациенту не вводят дозу соединения согласно настоящему изобретению, с последующим периодом (например, от 1 до 14 дней), в течение которого пациенту снова вводят дневную дозу соединения согласно настоящему изобретению. Перемежающиеся периоды введения согласно настоящему изобретению, чередуемые с периодами отсутствия введения соединения согласно настоящему изобретению, можно повторять в соответствии с клиническими требованиями для лечения пациента. Индукция сероконверсии ВГС или ВГВ у пациентов, хронически инфицированных каждым из указанных вирусов, является неожиданным свойством соединений согласно настоящему изобретению. Согласно лечебной практике пациента, страдающего от ВГВ, или пациента, страдающего от ВГС, лечат соединением согласно настоящему изобретению, применяемым индивидуально или в комбинации с одним или более другими терапевтическими агентами, до возникновения индукции или усиления иммунного ответа на ВГВ или ВГЦ и снижения концентрации вируса ВГВ или ВГС в крови. Затем, несмотря на то, что вирусы ВГВ или ВГС могут присутствовать в организме пациента в латентной форме, можно остановить лечение соединением согласно настоящему изобретению, при этом собственная иммунная система пациента способна подавлять дальнейшую репликацию вируса. У пациентов, которых лечили согласно настоящему изобретению и которые проходили лечение противовирусным агентом, который подавляет репликацию вируса ВГВ или вируса ВГС, в организме в течение лечения противовирусным(и) агентом(ами) могут содержаться небольшие, или недетектируемые, частицы вируса. Для указанных пациентов очевидно прохождение сероконверсии в случае, если введение противовирусного(ых) агента(ов) остановлено, но при этом не наблюдается увеличение концентрации ВГВ или ВГС в крови. При реализации настоящего изобретения индуцируется иммунный ответ на один или более из антигенов ВГВ или ВГС. Например, может быть индуцирован иммунный ответ на поверхностный антиген ВГВ (HBsAG) или на небольшой участок поверхностного антигена ВГВ (малый S антиген), или на средний участок поверхностного антигена ВГС (средний S антиген), или на их комбинацию. В качестве примера, может быть индуцирован иммунный ответ на поверхностный антиген ВГВ (HBsAG), а также на другие антигены ВГВ, например ядерную полимеразу или х-белок. Индукцию иммунного ответа на ВГС или ВГВ можно определить при помощи любых способов определения наличия иммунного ответа, известных специалистам в данной области техники. Подходящие способы детектирования иммунного ответа согласно настоящему изобретению включают, среди прочих,определение снижения концентрации вируса в сыворотке пациента, например, при помощи измерения содержания ДНК ВГВ или ДНК ВГС в крови пациента с применением исследования ПЦР, и/или при помощи измерения содержания антител анти-ВГВ или антител анти-ВГС, в крови пациента с применением способа, например ELISA. Дополнительно соединения согласно настоящему изобретению пригодны для лечения раковых заболеваний или опухолей (включая дисплазии, такие как дисплазия шейки матки). Такие заболевания включают гематобластозы, карциномы ротовой полости (например, губ, языка или глотки), пищеварительных органов (например, пищевода, желудка, тонкой кишки, толстой кишки, задней кишки или прямой кишки), печени и желчных протоков, поджелудочной железы, дыхательной системы, например гортани или легких (мелкоклеточные и немелкоклеточные), костей, соединительных тканей, кожи (например, меланому), груди, репродуктивных органов (матки, шейки матки, яичек, яичников или предстательной железы), мочевыводящих путей (например, мочевого пузыря или почек), мозга и эндокринных желез, например, щитовидной железы. В целом, соединения согласно настоящему изобретению применяют для лечения любых новообразований, включая не только гематобластозы, но и солидные опухоли любых видов. Гематобластозы, в целом, определены как пролиферативные нарушения кровяных клеток и/или их предшественников, при которых указанные клетки пролиферируют неконтролируемым способом. Анатомически, гематобластозы делят на две главные группы: лимфомы - злокачественные скопления лимфоцитов, главным образом, но не исключительно, в лимфатических узлах, и лейкемии - неоплазмы, получаемые, как правило, из лимфоцитов или миелоцитов и поражающие, главным образом, костный мозг и периферическую кровь. Лимфомы подразделяют на болезнь Ходжкина и неходжкинскую лимфому(НХЛ), причем последняя включает несколько различных видов, которые могут различаться по клиническим свойствам (например, агрессивная лимфома, индолентная лимфома), гистологически (например,фолликулярная лимфома, лимфома из клеток зоны мантии) или на основании природы злокачественных клеток (например, В лимфоцитов, Т лимфоцитов). Лейкемии и связанные с ними злокачественные опухоли включают острую миелогенную лейкемию (ОМЛ), хроническую миелогенную лейкемию (ХМЛ),острую лимфобластную лейкемию (ОЛЛ) и хроническую лимфоцитарную лейкемию (ХЛЛ). Другие гематобластозы включают дискразию клеток плазмы, включая множественную миелому, и миелодиспластические синдромы. Примеры синтеза. Для подробного описания экспериментов используют конкретные сокращения и аббревиатуры. Несмотря на то, что многие из них понятны специалисту в данной области техники, в табл. 1 представлен список указанных сокращений и аббревиатур. Общая схема получения производных птеридонона Соединение F. Соединение D растворяли в метаноле (2 мл), к полученному раствору добавляли раствор оксона(1,08 г) в Н 2 О (3 мл). Смесь перемешивали в течение 30 мин, после чего окисление, по существу, завершалось. Смесь добавляли в воду и экстрагировали в CH2Cl2. Сушили органическую фазу Na2SO4, фильтровали и концентрировали в вакууме с получением требуемого сульфонового промежуточного соединения, которое применяли на следующей стадии. Помещали сульфон и CS2CO3 (384 мг) в CH2Cl2 (4 мл), к указанной смеси по каплям добавляли 2-метоксиэтанол (880 мкл). После перемешивания в течение 1 ч оставалось некоторое количество исходного вещества, что было определено при помощи ЖХ/МС, дополнительно добавляли 200 мкл 2-метоксиэтанола и реакционную смесь дополнительно перемешивали в течение 30 мин. Реакционную смесь разбавляли CH2Cl2 и промывали водой. Органический слой сушилиNa2SO4, фильтровали и концентрировали в вакууме. Продукт очищали при помощи флэш-хроматографии на силикагеле, элюируя 20% МеОН в CH2Cl2, с получением соединения F с выходом 40%. 1 Н ЯМР (CD3OD):7,40-7,15 (m, 4 Н), 4,69 (br s, 2 Н), 4,33 (t, J = 4,8 Гц, 2 Н), 4,17 (q, J = 6,9 Гц, 2 Н),4,04 (s, 2 Н), 3,68 (s, 2 Н), 3,03 (t, J = 4,2 Гц, 2 Н), 3,68 (s, 3H), 2,60 (s, 4H), 1,81 (s, 4H), 1,24 (t, J = 7,2 Гц,ЗН); МС: 489,2 (М + Н+). Пример 2. Смесь соединения F (33 мг), железных опилок (56 мг) и уксусной кислоты (1 мл) перемешивали при КТ в течение 4 ч. За указанное время конверсия проходила не до конца, поэтому добавляли дополнительную порцию железных опилок (20 мг) и реакционную смесь дополнительно перемешивали в течение 6 ч. Добавляли третью порцию железных опилок (30 мг) и дополнительно перемешивали смесь в течение 12 ч. Смесь фильтровали через силикагель и удаляли растворитель в вакууме. Продукт очищали от остальных веществ при помощи препаративной ВЭЖХ на колонке С 18, элюируя с градиентом 2-98% ацетонитрила в Н 2 О, с получением соединения согласно примеру 2. 1 Н ЯМР (CD3OD):7,62 (s, 1H), 7,50 (s, 3 Н), 4,95 (s, 2H), 4,60-4,53 (m, 2H), 4,39 (s, 2H), 4,15 (s, 2H),3,95-3,67 (m, 2H), 3,60-3,42 (m, 2H), 3,68 (s, 3 Н), 3,25-3,12 (m, 2H), 2,23-1,95 (m, 4H); МС: 413,2 (М+Н+). Схема 3 Способ I: 3-(пирролидин-1'-ил)метилбензонитрил. К раствору 3-(бромметил)бензонитрила (30,0 г, 1,00 экв.) в абсолютном EtOH (600 мл) добавляли пирролидин (13,3 мл, 1,00 экв.), затем K2CO3 (безводный 63,5 г, 3,00 экв.). Реакционную смесь интенсивно перемешивали при 65 С до завершения израсходования бромида (реакцию контролировали на пластинах для ТСХ Merck 254 нм, покрытых оксидом кремния, с применением комбинации EtOAc/гексан в качестве элюента). Реакционную смесь (которая может быть оранжевого цвета) охлаждали до 23 С и фильтровали через крупнопористые стеклянные фритты, фильтрат концентрировали. Полученный остаток разделяли в Н 2 О и EtOAc (по 300 мл каждого растворителя), собирали органическую фазу. Водный слой экстрагировали (2200 мл EtOAc). Все полученные органические слои объединяли, сушили(Na2SO4), фильтровали и концентрировали в вакууме с получением титульного нитрила (21,1 г, выход 74%) в виде оранжевого остатка. 1 Н ЯМР (CDCl3, 300 МГц):(ppm) 7,65 (s, 1H), 7,59 (d, J = 7,7 Гц, 1 Н), 7,54 (d, J = 7,6 Гц, 1 Н), 7,41 Способ II: 3-(пирролидин-1'-ил)метилбензальдегид. Суспензию K2CO3 (2,09 г, 15,2 ммоль, 3,00 экв.) в абсолютном этаноле (20 мл) обрабатывали пирролидином (439 мкл, 5,05 ммоль, 1,00 экв.). Добавляли 3-(бромметил)бензальдегид (1,00 г, 5,05 ммоль,1,00 экв.) и нагревали реакционную смесь до 65 С в течение 1 ч. Реакционную смесь охлаждали и фильтровали. Осадок промывали дополнительным количеством этанола. Концентрировали фильтрат с получением мутной маслянистой жидкости и разделяли в ДХМ (50 мл) и 2% (мас./об.) водном раствореNaHCO3 (50 мл). Собирали органическую фазу, водный слой экстрагировали в ДХМ (250 мл). Объединяли все органические слои, сушили (Na2SO4), фильтровали и концентрировали с получением 3(пирролидин-1-илметил)бензальдегида (846 мг, выход 88%) в виде светло-желтой маслянистой жидкости, которую применяли без дополнительной очистки. 1 Н ЯМР: 300 МГц, (CDCl3) : 10,00 (s, 1H), 7,84 (s, 1 Н), 7,76 (d, J = 7,6 Гц, 1 Н), 7,62 (d, J = 7,6 Гц,1H), 7,47 (dd, J = 7,6 Гц, 7,6 Гц, 1 Н), 3,69 (s, 2 Н), 2,52 (m, 4 Н), 1,79 (m, 4 Н). ЖХМС-ИЭР+: рассчитанная для C12H16NO: 190,1 (М+Н+); экспериментальная: 190,1 (М+Н+). Схема 5 В круглодонную колбу вместимостью 1 л загружали LiAlH4 (7,55 г) и безводный Et2O (230 мл). После охлаждения до 0 С медленно в течение 5 мин добавляли 3-(пирролидин-1-илметил)бензонитрил(18,55 г) в ТГФ (30 мл). Цвет реакционной смеси менялся с оранжевого на зеленый. После завершения реакции (определенного при помощи ТСХ с применением пластин Merck 254 нм, покрытых оксидом кремния с элюентом ДХМ/МеОН/водн. NH4OH или при помощи ЖХМС), реакционную смесь медленно обрабатывали сначала Н 2 О (7,5 мл) в течение времени, достаточного для прекращения выделения газа,затем (через 5 мин после окончания выделения газа) 15% (мас./об.) водным раствором NaOH (7,5 мл)(снова до окончания выделения газа, с последующей паузой, равной 5 мин) и, наконец, дополнительным количеством Н 2 О (26,5 мл). Фильтровали реакционную смесь через стеклянные фритты для удаления всех содержащихся твердых веществ, осадок промывали Et2O (100 мл). Сушили фильтрат большим количеством MgSO4, фильтровали и концентрировали с получением титульного амина (17,0 г, выход 90%) в виде маслянистой жидкости. 1 Н ЯМР (CDCl3, 300 МГц):(ppm) 7,32-7,17 (m, 4 Н), 3,86 (s, 2 Н), 3,62 (s, 2 Н), 2,52 (m, 4 Н), 1,79 (m,4 Н), 1,61 (s, шир., 2 Н). ЖХМС-ИЭР+: рассчитанная для C12H19N2: 191,1 (М+Н+); экспериментальная: 191,0 (М+Н+). Схема 6Et3N (27,4 мл, 2,20 экв.). К указанному раствору при 23 С по каплям в течение 10 мин добавляли этилбромацетат (9,90 мл, 1,00 экв.). Через 24 ч реакционную смесь разбавляли Н 2 О (600 мл) и экстрагировали в EtOAc (3150 мл). Объединяли органические слои, сушили (MgSO4), фильтровали и концентрировали с получением титульного продукта в виде желтой маслянистой жидкости (21,2 г, 86%). 1 Н ЯМР (CDCl3, 300 МГц):(ppm) 7,32-7,18 (m, 4 Н), 4,19 (q, J = 7,0 Гц, 2 Н), 3,80 (s, 2H), 3,61 (s,2H), 2,51 (m, 4 Н), 1,79 (m, 4 Н), 1,28 (t, J = 7,0 Гц, 3 Н). ЖХМС-ИЭР+: рассчитанная для C16H25N2O: 277,2 Способ V: 4,6-дигидрокси-2-метилтио-5-нитропиримидин. Раствор 4,6-дигидрокси-2-метилтиопиримидина (42 г, 0,257 моль) в трифторуксусной кислоте (91 мл, 1,186 моль) перемешивали при 23 С и нагревали до растворения всех твердые веществ. Реакционную смесь перемешивали в течение 5 ч при 23 С. Затем при 0 С в течение 25 мин по частям добавляли дымящую азотную HNO3 (15 мл, 350 ммоль). Перемешивали реакционную смесь в течение 20 ч при 23 С,обрабатывали Н 2 О (при 23 С) до достижения 80% конверсии (согласно данным ЖХ-МС). Собирали твердый осадок при помощи фильтрования с получением 4,6-дигидрокси-2-метилтио-5-нитропиримидина в виде рыжеватого твердого вещества. Перегоняли твердое вещество в азеотропе с толуолом с получением 35 г светло-коричневого порошкового твердого вещества. 1 Н ЯМР 300 МГц, (CD3OD, 300 МГц):(ppm) 2,63 (s, 3 Н). ЖХМС-ИЭР-: рассчитанная для Способ VI: 4,6-дихлор-2-метилтио-5-нитропиримидин. В круглодонную колбу вместимостью 500 мл помещали POCl3 (89,5 мл, 0,960 моль, 5,00 экв.) иN,N-диметиланилин (73,0 мл, 0,576 моль, 3,00 экв.). Реакционную смесь охлаждали до 0 С и по частям,контролируя выделение тепла, добавляли 4,6-дигидрокси-2-метилтио-5-нитропиримидин (39,0 г, 0,192 моль, 1,00 экв.). После снижения выделения тепла реакционную смесь осторожно нагревали до 100 С в течение 2 ч. Затем переносили реакционную смесь в верхнюю камеру экстрактора непрерывного действия для фаз с более низкой плотностью и непрерывно экстрагировали горячим гексаном, который выли- 21021377 вали в нижнюю камеру. В течение экстракции поддерживали температуру нижней камеры, равной 140 С. После достижения минимума УФ-активности (254 нм) гексановой фазы верхней камеры систему охлаждали. Концентрировали гексановую фазу в вакууме с получением маслянистой жидкости. Очищали остаток при помощи хроматографии на силикагеле (1 г остатка/3 г оксида кремния) (элюент: ДХМ). В течение помещения в колонку (для достижения текучести к остатку добавляли 20 мл ДХМ) наблюдали небольшое выделение тепла. После хроматографии получали кристаллический 4,6-дихлор-2-метилтио-5 нитропиримидин (34,9 г, выход 76%). 1 Н ЯМР: 300 МГц, (CDCl3):(ppm) 2,62 (s, 3 Н). ЖХМС-ИЭР+: соединение не ионизировалось. Схема 9 Способ VII, стадия 1: 4-амино-6-хлор-2-метилтио-5-нитропиримидин. К раствору полученного ранее дихлорида (2,46 г, 10,2 ммоль) в ТГФ (34 мл) при -20 С добавлялиEt3N (3,14 мл, 22,5 ммоль), затем раствор NH3 (2,0 М раствор в МеОН, 5,4 мл, 11 ммоль). Перемешивали смесь при нагревании до 0 С в течение 1,5 ч (данные ЖХ/МС показывали израсходование исходных веществ. Наблюдали незначительное прохождение бис-добавления). Реакционную смесь применяли далее без обработки. Схема 10 Способ VII, стадия 2: этил-N-[4-амино-2-метилтио-5-нитропиримидин-6-ил],N-[3'-(пирролидин 1"-илметил)бензил]глицинат. К полученной ранее реакционной смеси при 0 С в течение 5 мин добавляли вторичный амин (2,82 г,10,2 ммоль) в ТГФ (10 мл). Перемешивали реакционную смесь до израсходования исходных веществ, определенного при помощи ЖХ/МС, в течение примерно 30 мин. Фильтровали реакционную смесь через стеклянные фритты; промывали осадок EtOAc. Концентрировали фильтрат и разделяли в EtOAc (30 мл) и 5% водном растворе Na2CO3 (30 мл). Собирали органическую фазу, водную фазу дважды экстрагировали в EtOAc (каждый раз в 30 мл). Сушили объединенные органические слои MgSCO4, фильтровали и концентрировали в вакууме. Затем при 70 С добавляли абсолютный EtOH (12 мл), затем оставляли раствор постепенно охлаждаться до 23 С. Отфильтровывали кристаллы через стеклянные фритты и промывали гексаном, затем сушили в вакууме. Продукт представлял собой желтовато-зеленое твердое вещество. 1 Н ЯМР (CDCl3, 300 МГц):(ppm) 7,32-7,16 (m, 4 Н), 4,69 (s, 2 Н), 4,19 (q, J = 7 Гц, 2 Н), 4,07 (s, 2 Н),3,60 (s, 2 Н), 2,49 (m, 4 Н), 2,40 (s, 3 Н), 1,78 (m, 4 Н), 1,23 (t, 3 Н, J = 7 Гц). ЖХМС-ИЭР+: рассчитанная для Способ VIII: этил-N-[4-амино-2-метансульфонил-5-нитропиримидин-6-ил],N-[3'-(пирролидин 1"-илметил)бензил]глицинат. К раствору (суспензии) сульфида (3,68 г, 8,00 ммоль) в EtOH (40 мл) при 0 С последовательно добавляли дигидрат вольфрамата натрия (792 мг, 2,40 ммоль), уксусную кислоту (4,6 мл, 80 ммоль) и пероксид водорода (3,4 мл, 40 ммоль, 35% мас./мас. в Н 2 О). Через 3 ч дополнительно добавляли уксусную кислоту (4,6 мл) и пероксид водорода (3,4 мл). Поддерживали температуру реакционной смеси, равную 0 С, в течение 16 ч. Осторожно при 0 С добавляли насыщенный раствор Na2SO3 (50 мл), затем CH2Cl2(75 мл). Разделяли слои, водный слой экстрагировали в CH2Cl2 (450 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме, затем применяли без дополнитель- 22021377 Способ X: к раствору сульфона (1,0 г, 2,0 ммоль) в спирте (R-OH) (10 мл) добавляли ТФУ (470 мкл,6,1 ммоль). Реакционную смесь перемешивали при 100 С в течение 1 ч. Выливали реакционную смесь в насыщенный раствор NaHCO3 (20 мл) и CH2Cl2 (30 мл). Разделяли слои, водный слой экстрагировали вCH2Cl2 (30 мл). Объединенные органические слои сушили MgSO4, фильтровали и концентрировали в вакууме. Проводили очистку при помощи хроматографии на силикагеле (1 г субстрата/10 г SiO2) (2-15% МеОН/CH2Cl2). Схема 14 Способ XI: к раствору сульфона (1,0 г, 2,0 ммоль) в спирте (R-OH) (10 мл) добавляли ДМФ (1,0 мл) и ТФУ (470 мкл, 6,1 ммоль). Перемешивали реакционную смесь при 90-100 С в течение 1 ч. Выливали реакционную смесь в насыщенный раствор NaHCO3 (20 мл) и CH2Cl2 (30 мл). Разделяли слои, водный слой экстрагировали в CH2Cl2 (30 мл). Объединенные органические слои сушили MgSO4, фильтровали и концентрировали в вакууме. Проводили очистку при помощи хроматографии на силикагеле (1 г субстрата/10 г SiO2) (2-15% МеОН/CH2Cl2). Схема 15 Способ XII: к раствору нитросоединения (730 мг, 1,5 ммоль) в МеОН (10 мл) добавляли никель Ренея (-200 мкл, суспензия в Н 2 О). Продували реакционный сосуд Н 2, затем перемешивали в атмосфере Н 2 в течение 1,5 ч. Фильтровали смесь через Celite с CH2Cl2 и МеОН (1:1). Концентрировали фильтрат в вакууме и оставляли в лиофилизаторе на ночь. Получали указанный в заголовке продукт в виде свободного основания, представляющего собой белое твердое вещество. Схема 16 Способ XIII: суспензию сульфона (50 мг), ТГФ (1,0 мл) и амина (R1R2NH) (100 л) нагревали до 60 С в течение 3 ч. Охлаждали реакционную смесь до 23 С и непосредственно помещали в С 18-колонку с обращенной фазой (50 мг/4 г наполнителя) и очищали при помощи ЖХ (элюент: нейтральная смесь Способ XIV: раствор нитросоединения (50 мг) в МеОН (4,0 мл) обрабатывали никелем Ренея (200 мкл, суспензия в Н 2 О). Реакционный сосуд продували Н 2, затем перемешивали в атмосфере Н 2 в течение 1,5 ч. Фильтровали смесь через Celite с CH2Cl2 и МеОН (1:1). Фильтрат концентрировали и сушили в вакууме с получением продукта в виде свободного основания. При необходимости перед концентрированием к фильтрату добавляли 1,0 М водный раствор HCl (200 л), что приводило к получению соли HCl,которая обладает более выраженными резонансами 1 Н ЯМР. Схема 18-10 С добавляли Et3N (474 мкл, 3,40 ммоль, затем раствор NH3 (2,0 М раствор в МеОН, 750 мкл, 1,5 ммоль). Смесь перемешивали при нагревании до 0 С в течение 1,5 ч (данные ЖХ/МС показывали израсходование исходных веществ). Реакционную смесь применяли далее без обработки. Схема 19 Способ XV, стадия 2: метил-,-(1'",2'"-этилиден),N-[4-амино-2-метилтио-5-нитропиримидин-6 ил],N-[3'-(пирролидин-1"-илметил)бензил]глицинат. К полученной ранее смеси при 0 С добавляли неочищенный вторичный амин (-1,5 ммоль) в ТГФ(1,5 мл). Перемешивали реакционную смесь при КТ в течение 18 ч, затем при 60 С в течение 6 ч. Добавляли насыщенный раствор NH4Cl (10 мл). Разделяли слои, водный слой экстрагировали в EtOAc (210 мл). Объединенные органические слои сушили MgSO4, фильтровали и концентрировали в вакууме. Очистка при помощи хроматографии на силикагеле (1 г субстрата/15 г SiO2) (2-20% МеОН/ДХМ) приводила к получению продукта. ЖХМС-ESI+: рассчитанная для C22H29N6O4S: 473,6 (М+Н+); экспериментальная: 473,1 (М+Н). Схема 20(34 мл) при 0 С добавляли Et3N (3,14 мл, 22,5 ммоль), затем по каплям метилбромацетат (1,04 мл, 22,3 ммоль). Перемешивали реакционную смесь до израсходования исходных веществ, определенного при помощи ЖХ/МС, в течение примерно 2 ч. Смесь, содержащую продукт, применяли далее без дополнительной обработки. ЖХМС-ИЭР+: рассчитанная для C15H23N2O2: 263,4 (М+Н+); экспериментальная: 263,1
МПК / Метки
МПК: A61P 31/12, A61P 35/00, C07D 487/04, C07D 475/06, A61K 31/519, C07D 475/08, C07D 475/12
Метки: толлподобных, рецепторов, модуляторы
Код ссылки
<a href="https://eas.patents.su/30-21377-modulyatory-tollpodobnyh-receptorov.html" rel="bookmark" title="База патентов Евразийского Союза">Модуляторы толлподобных рецепторов</a>
Модуляторы функций рецепторов семейства рецепторов tnf/ngf и других белков

Номер патента: 4062
Опубликовано: 25.12.2003
Авторы: Коваленко Андрей, Уоллах Дэвид, Ли Йонган, Хорвиц Маршалл С.
МПК: C07K 14/47, C12N 15/12, A61K 38/17...
Метки: других, семейства, модуляторы, функций, рецепторов, белков
Формула / Реферат:
1. Последовательность ДНК, кодирующая RIP-ассоциированный белок (RAP-2), его изоформы, фрагменты или аналоги, включающая последовательность, показанную на фиг. 1, причем указанные белок RAP-2, его изоформы, фрагменты или аналоги способны связываться с белком RIP или способны модулировать или опосредовать внутриклеточную активность RIP. 2. Последовательность ДНК по п.1, включающая последовательность, показанную на фиг. 2. 3. Последовательность...
Модуляторы сск-1 рецепторов

Номер патента: 9479
Опубликовано: 28.02.2008
Авторы: Гомес Лоран, Брайтенбухер Дж.Гай, Макклур Келли Дж., Шэнкли Найджел П., Хуан Лимин, Хэк Майкл Д., Мортон Магда Ф., Барретт Терренс Д., Сехорн Кларк А.
МПК: C07D 231/12, C07D 401/06, C07D 401/04...
Метки: модуляторы, рецепторов, сск-1
Формула / Реферат:
1. Антагонисты CCK-1 рецепторов общей формулы где R1 представляет заместитель в 1 или 2 положении, который выбирают из группы, состоящей из водорода, a) фенила, необязательно моно-, ди- или тризамещенного Rp или дизамещенного у соседних атомов углерода -OC1-4алкиленО-, -(СН2)2-3NH-, -(CH2)1-2NH(CH2)-, -(СН2)2-3N(C1-4алкил)- или -(СН2)1-2N(C1-4алкил) (СН2)-; Rp выбирают из группы, состоящей из -ОН, -C1-6алкила, -OC1-6алкила, фенила, -Офенила,...
Модуляторы рецепторов глюкокортикоидов

Номер патента: 4886
Опубликовано: 26.08.2004
Авторы: Свик Эндрю Гордон, Дау Роберт Ли, Морган Брэдли Пол, Лиу Кевин Кун-Чин
МПК: A61K 31/05, C07C 35/42, C07D 213/40...
Метки: модуляторы, глюкокортикоидов, рецепторов
Формула / Реферат:
1. Соединение формулы II его изомер, пролекарство указанного соединения или изомера или фармацевтически приемлемая соль указанного соединения, изомера и пролекарства; где R1 представляет собой фенил, незамещенный или замещенный одним из следующих заместителей: -OH, -NR12R13, -NR12-C(O)-(C1-C4)алкил, -CN, -(C0-C2)алкил-het, -O-(C1-C3)алкил-C(O)-NR12R13, -NR12-(C0-C2)алкил-C(O)-NR12R13, -(C0-C2)алкил-NR12-SO2-R13, -NR12-SO2-het,...
Модуляторы рецепторов андрогенов

Номер патента: 9902
Опубликовано: 28.04.2008
Авторы: Лей Хуангсу, Ху Лейн-Ен, Ду Дэниел Юнлонг, Лефкер Брюс Аллен
МПК: A61K 31/277, A61P 17/14, A61P 5/28...
Метки: рецепторов, модуляторы, андрогенов
Формула / Реферат:
1. Соединение формулы гидрат указанного соединения или фармацевтически приемлемая соль указанного соединения, где X1 представляет собой галоген или галогеноалкил; X2 представляет собой -CR3R4R5, -CH=CH2 или -CуCH; R1 и R2, каждый независимо, представляет собой заместитель, выбранный из группы, состоящей из водорода, С1-6алкила, галогена, галогеноалкила, гидроксиалкила, тиола и тиоалкила; R3, R4 и R5, каждый независимо, представляет собой...
Модуляторы никотиновых ацетилхолиновых рецепторов

Номер патента: 16948
Опубликовано: 30.08.2012
Авторы: Варроне Маурицио, Ботманн Хендрик, Маккари Лаура, Гирон Кьяра, Микко Иоланда, Гаррисон Бойд, Прателли Кармела, Заналетти Риккардо, Хайдар Симон, Ненчини Арианна
МПК: A61P 25/00, C07D 231/40, A61K 31/4155...
Метки: модуляторы, рецепторов, ацетилхолиновых, никотиновых
Формула / Реферат:
1. Соединение формулы (I)где Т представляет собой (C3-С5)алкан-α,w-диил, возможно содержащий группу оксо и возможно замещенный одним или более чем одним галогеном; гидроксигруппами; группами (С1-С5)алкил, (С1-С5)алкокси, фтор(С1-С5)алкил, гидрокси(С1-С5)алкил, (С1-С5)алкилиден, фтор(С1-С5)алкилиден; группами (С3-С6)циклоалкан-1,1-диил, окса(С3-С6)циклоалкан-1,1-диил; группами (С3-С6)циклоалкан-1,2-диил, окса(С3-С6)циклоалкан-1,2-диил, где...
Предыдущий патент: Станция для подготовки медикаментов
Следующий патент: Способы измерения сигнала помехи в минисотовой системе
Случайный патент: Способ электропорации свекловичной стружки и устройство для его осуществления