Аминоэфирные производные алкалоидов и их медицинские композиции
Номер патента: 20974
Опубликовано: 31.03.2015
Авторы: Риккабони Мауро, Калиджури Антонио, Амари Габриеле







Формула / Реферат
1. Соединение общей формулы (I)

где R1 выбран из группы, состоящей из Н, (С1-С10)-алкила, арила, (С3-С8)-циклоалкила, арилалкила и гетероарила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов, ОН, SH, CON(R5)2, NHCOR5, COR5, CO2R5, (C1-C10)-алкилсульфанила, (С1-C10)-алкилсульфинила, (С1-C10)-алкилсульфонила, (С1-C10)-алкила и (С1-C10)-алкоксила;
R2 выбран из группы, состоящей из (С1-C10)-алкила, арила, (С3-C8)-циклоалкила, арилалкила и гетероарила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов, ОН, SH, CON(R5)2, NHCOR5, COR5, CO2R5, (C1-C10)-алкилсульфанила, (С1-C10)-алкилсульфинила, (С1-C10)-алкилсульфонила, (С1-C10)-алкила и (С1-C10)-алкоксила,
R3 выбран из группы, состоящей из арила, гетероарила, арилалкила и гетероарилалкила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов, ОН, SH, NO2, CN, CON(R5)2, CO2R5, CF3, (С1-C10)-алкила и (С1-C10)-алкоксила;
R6 представляет собой группу формулы (i), или (ii), или (iii), или (iv)


где m = 1, 2 или 3;
n = 1, 2 или 3;
А- представляет собой физиологически приемлемый анион;
R4 представляет собой группу формулы (Y)

где р означает 0 или целое число от 1 до 4;
q означает 0;
Р отсутствует или выбран из группы, состоящей из О, S, SO, SO2, СО, СН=СН, SO2N(R5) и C(O)N(R5);
W выбран из группы, состоящей из Н, (С1-С6)-алкила, (С3-С8)-циклоалкила, арила и гетероарила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов, ОН, оксо (=O), SH, NO2, CN, CON(R5)2, NH2, NHCOR5, CO2R5, (C1-C10)-алкоксикарбонила, (С1-C10)-алкилсульфанила, (С1-C10)-алкилсульфинила, (С1-C10)-алкилсульфонила, (С1-C10)-алкила и (С1-C10)-алкоксила;
R5 выбран из группы, состоящей из Н, (С1-C10)-алкила, (С3-С7)-циклоалкила, (С3-С7)-циклоалкил-(С1-С6)-алкила, гетероарила и арила;
и его фармацевтически приемлемые соли,
где арил представляет собой фенил;
арилалкил относится к (С1-С4)алкилу, замещенному фенилом;
гетероарил относится к моно- или бициклическим кольцевым системам, которые имеют от 5 до 15 кольцевых атомов, где по меньшей мере одно кольцо является ароматическим и где по меньшей мере один кольцевой атом представляет собой гетероатом (N, NH, S или О);
гетероарилалкил относится к (С1-С4)алкилу, замещенному гетероарилом.
2. Соединение по п.1, где R1 представляет собой Н, R2 представляет собой арилалкил, или (С1-C10)-алкил, или арил, или (С3-С8)-циклоалкил, или гетероарил, R3 представляет собой арил или гетероарил, возможно замещенные одним или более атомами галогенов, и R6 представляет собой группу формулы (i)

согласно общей формуле (XIII)

3. Соединение по п.2, где R1 представляет собой Н, R2 и R3 представляют собой фенил.
4. Соединение по п.3, где R1 представляет собой Н, R2 и R3 представляют собой фенил и R4 представляет собой группу формулы (Y)

где р равно 1, Р представляет собой СО, q равно 0 и W представляет собой арил.
5. Соединение по п.3, где R4 представляет собой группу формулы (Y)

где р равно 1, Р представляет собой СО, q равно 0 и W представляет собой тиенил.
6. Соединение по п.3, где R4 представляет собой группу формулы (Y)
где р равно 2, Р представляет собой О, q равно 0 и W представляет собой фенил.
7. Соединение по п.3, где R4 представляет собой группу формулы (Y)

где р равно 3, Р представляет собой О, q равно 0 и W представляет собой фенил.
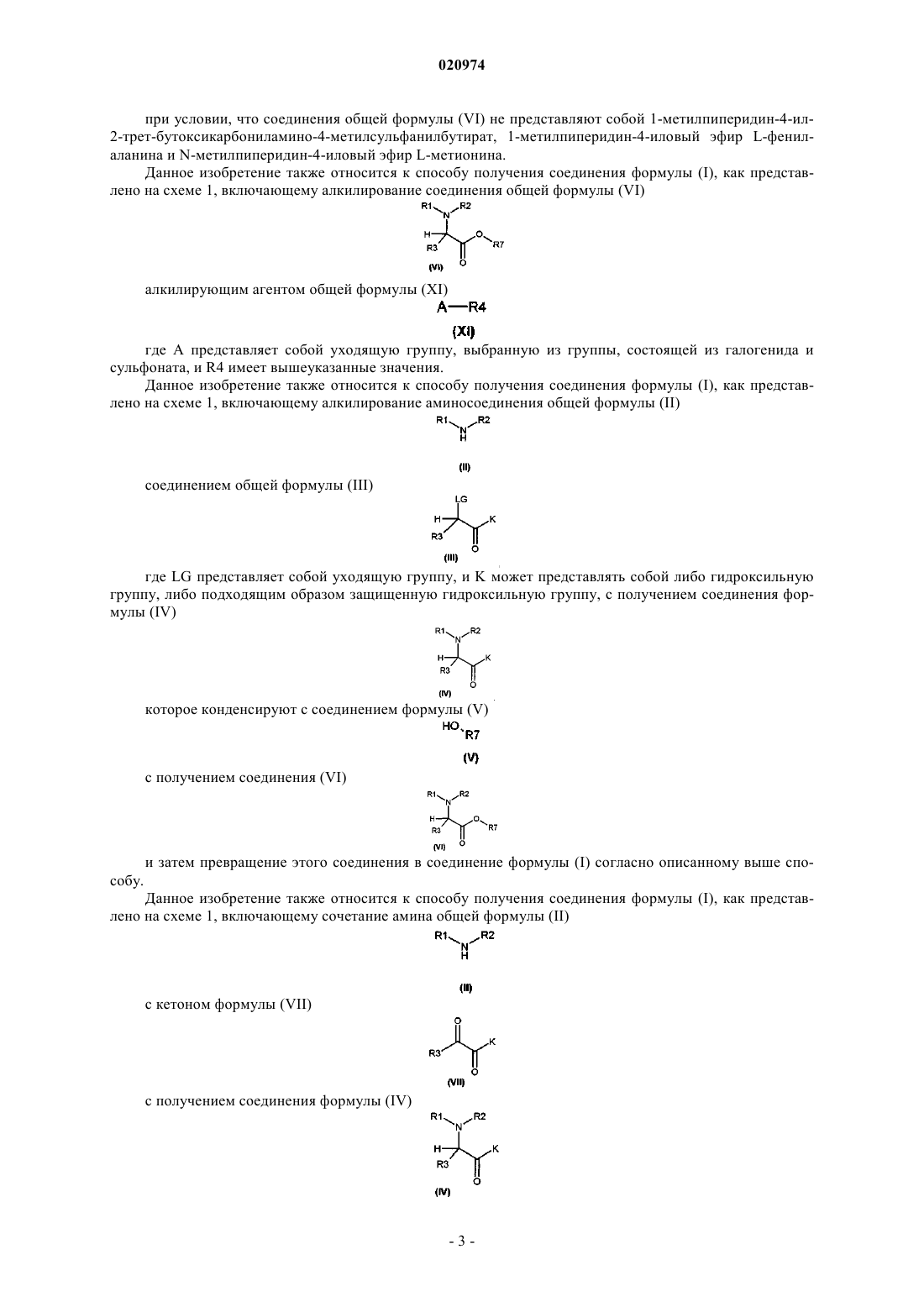
8. Соединение общей формулы (VI)

где R1, R2 и R3 такие, как определено в п.1;
R7 представляет собой группу формулы (vi), или (vii), или (viii), или (ix)

где m и n такие, как определено в п.1;
и его фармацевтически приемлемые соли,
при условии, что соединения общей формулы (VI) не представляют собой 1-метилпиперидин-4-ил-2-трет-бутоксикарбониламино-4-метилсульфанилбутират, 1-метилпиперидин-4-иловый эфир L-фенилаланина и N-метилпиперидин-4-иловый эфир L-метионина.
9. Соединение по п.8, где R1 представляет собой Н, R2 представляет собой (С1-C10)-алкил, или арил, или (С3-C8)-циклоалкил, или арилалкил, или гетероарил, R3 представляет собой арил, или арилалкил, или гетероарил, возможно замещенные одним или более атомами галогенов, и R7 представляет собой группу формулы (vi)

согласно общей формуле (XII)

10. Соединение по п.8, где R1 представляет собой Н, R2 представляет собой арилалкил, или (С1-C10)-алкил, или арил, или (С3-С8)-циклоалкил, или гетероарил, R3 представляет собой арил, или арилалкил, или гетероарил, возможно замещенные одним или более атомами галогенов, и R7 представляет собой группу формулы (vii)

где m=2 и n=1, согласно общей формуле (XIV)

11. Соединение по п.8, где R1 представляет собой Н, R2 представляет собой арилалкил, или (С1-C10)-алкил, или арил, или (С3-С8)-циклоалкил, или гетероарил, R3 представляет собой арил, или арилалкил, или гетероарил, возможно замещенные одним или более атомами галогенов, и R7 представляет собой группу формулы (vii), где m = 1 и n = 1, согласно общей формуле (XV)

12. Соединение по п.8, где R1 представляет собой Н, R2 представляет собой арилалкил, или (С1-C10)-алкил, или арил, или (С3-С8)-циклоалкил, или гетероарил, R3 представляет собой арил, или арилалкил, или гетероарил, возможно замещенные одним или более атомами галогенов, и R7 представляет собой группу формулы (vii), где m=2 и n=1, согласно общей формуле (XVI)

13. Соединение по п.8, где R1 представляет собой Н, R2 представляет собой арилалкил, или (С1-C10)-алкил, или арил, или (С3-C8)-циклоалкил, или гетероарил, R3 представляет собой арил, или арилалкил, или гетероарил, возможно замещенные одним или более атомами галогенов, и R7 представляет собой группу формулы (viii)

согласно общей формуле (XVII)

14. Соединение по п.8, где R1 представляет собой H, R2 представляет собой арилалкил, или (С1-С10)-алкил, или арил, или (С3-С8)-циклоалкил, или гетероарил, R3 представляет собой арил, или арилалкил, или гетероарил, возможно замещенные одним или более атомами галогенов, и R7 представляет собой группу формулы (ix)

согласно общей формуле (XVIII)

15. Соединение, выбранное из













16. Способ получения соединения формулы (I) по п.1, включающий алкилирование соединения общей формулы (VI)

алкилирующим агентом общей формулы (XI)

где А представляет собой подходящую уходящую группу, выбранную из группы, состоящей из галогенида и сульфоната, где R1, R2, R3, R4 и R7 такие, как определено в любом из пп.1 и 8.
17. Способ получения соединения формулы (I) по п.1, включающий алкилирование аминосоединения общей формулы (II)

соединением общей формулы (III)

где LG представляет собой подходящую уходящую группу, и K представляет собой либо гидроксильную группу, либо подходящим образом защищенную гидроксильную группу, с получением соединения формулы (IV)

которое конденсируют с соединением формулы (V)

с получением соединения формулы (VI)

и затем превращение соединения формулы (VI) в соединение формулы (I) согласно способу по п.16, и где R1, R2, R3 и R7 такие, как определено в любом из пп.1 и 8.
18. Способ получения соединения формулы (I) по п.1, включающий сочетание амина общей формулы (II)

с кетоном формулы (VII)

с получением соединения формулы (IV)

которое конденсируют с соединением формулы (V)

с получением соединения (VI)

и затем превращение соединения формулы (VI) в соединение формулы (I) согласно способу по п.16, где R1, R2, R3 и R7 такие, как определено в любом из пп.1 и 8, и K такой, как определено в п.17.
19. Способ получения соединения формулы (I) по п.1, включающий взаимодействие соединения формулы (IX)

с алкилирующим агентом общей формулы (VIII)

где z представляет собой карбонильную группу или подходящую уходящую группу, с получением соединения формулы (IV)

где R2 представляет собой водород, которое конденсируют с соединением формулы (V)

с получением соединения формулы (VI)

и затем превращение соединения формулы (VI) в соединение формулы (I) согласно способу по п.16, где R1, R2, R3 и R7 такие, как определено в любом из пп.1 и 8, и K такой, как определено в п.17.
20. Способ по п.19, где уходящая группа представляет собой галогенид.
21. Способ получения соединения формулы (I) по п.1, включающий сочетание соединения общей формулы (IIIa)

с соединением общей формулы (V)

с получением соединения формулы (X)

которое подвергают взаимодействию с амином формулы (II)

с получением соединения формулы (VI)

и затем превращение соединения формулы (VI) в соединение формулы (I) согласно способу по п.16, где R1, R2, R3 и R7 такие, как определено в пп.1 и 8, K такой, как определено в п.17, и где W' представляет собой LG, как определено в п.17, или гидрокси.
22. Фармацевтическая композиция, обладающая активностью антагонистов М3-мускаринового рецептора, содержащая соединение по любому из пп.1-15 в смеси с одним или более фармацевтически приемлемыми носителями и/или эксципиентами.
23. Фармацевтическая композиция по п.22, пригодная для введения путем ингаляции.
24. Применение соединения по любому из пп.1-15 в качестве лекарственного средства, обладающего активностью антагонистов М3-мускаринового рецептора.
25. Применение любого из соединений по любому из пп.1-15 для изготовления лекарственного средства для предупреждения и/или лечения бронхо-обструктивных или воспалительных заболеваний.
26. Применение по п.25, где заболевание выбрано из группы, состоящей из астмы, или хронического бронхита, или хронического обструктивного заболевания легких (COPD).
27. Применение соединения общей формулы (XIX)

где R1 и R2 независимо выбраны из группы, состоящей из Н, (C1-C10)-алкила, арила, (С3-С8)-циклоалкила, арилалкила и гетероарила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов, ОН, SH, CON(R5)2, NHCOR5, COR5, CO2R5, (С1-C10)-алкилсульфанила, (С1-C10)-алкилсульфинила, (C1-C10)-алкилсульфонила, (С1-C10)-алкила и (С1-C10)-алкоксила;
R3 выбран из группы, состоящей из арила, гетероарила, арилалкила и гетероарилалкила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов, ОН, SH, NO2, CN, CON(R5)2, CO2R5, CF3, (С1-C10)-алкила и (С1-C10)-алкоксила;
R8 представляет собой группу формулы (i), или (ii), или (iii), или (iv), или (vi), или (vii), или (viii), или (ix)

m = 1, 2 или 3;
n = 1, 2 или 3;
А- представляет собой физиологически приемлемый анион;
R4 представляет собой группу формулы (Y)

где р означает 0 или целое число от 1 до 4;
q означает 0;
Р отсутствует или выбран из группы, состоящей из О, S, SO, SO2, СО, СН=СН, SO2N(R5) и C(O)N(R5);
W выбран из группы, состоящей из Н, (С1-С6)-алкила, (С3-С8)-циклоалкила, арила и гетероарила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов, ОН, оксо (=O), SH, NO2, CN, CON(R5)2, NH2, NHCOR5, CO2R5, (C1-C10)-алкоксикарбонила, (С1-C10)-алкилсульфанила, (С1-C10)-алкилсульфинила, (С1-C10)-алкилсульфонила, (С1-C10)-алкила и (С1-C10)-алкоксила;
R5 выбран из группы, состоящей из Н, (С1-C10)алкила, (С3-С7)циклоалкила, (С3-С7)циклоалкил-(С1-С6)алкила, гетероарила и арила;
где арил представляет собой фенил;
арилалкил относится к (С1-С4)алкилу, замещенному фенилом;
гетероарил относится к моно- или бициклическим кольцевым системам, которые имеют от 5 до 15 кольцевых атомов, где по меньшей мере одно кольцо является ароматическим и где по меньшей мере один кольцевой атом представляет собой гетероатом (N, NH, S или О);
гетероарилалкил относится к (С1-С4)алкилу, замещенному гетероарилом;
для изготовления лекарственного средства для предупреждения и/или лечения бронхо-обструктивных или воспалительных заболеваний.
28. Применение по п.27, где бронхо-обструктивное или воспалительное заболевание выбрано из астмы, или хронического бронхита, или хронического обструктивного заболевания легких (COPD).
29. Способ предупреждения и/или лечения бронхо-обструктивных или воспалительных заболеваний, включающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по любому из пп.1-15.
30. Способ по п.29, где бронхо-обструктивное или воспалительное заболевание выбрано из астмы, или хронического бронхита, или хронического обструктивного заболевания легких (COPD).
31. Способ предупреждения и/или лечения бронхо-обструктивных или воспалительных заболеваний, включающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (XIX) по п.27.
32. Способ по п.31, где бронхо-обструктивное или воспалительное заболевание выбрано из астмы, или хронического бронхита, или хронического обструктивного заболевания легких (COPD).
33. Устройство, которое представляет собой однодозовый или многодозовый сухой порошковый ингалятор, дозирующий ингалятор или небулайзер тонкого тумана, содержащее фармацевтическую композицию по п.22.
34. Устройство по п.33, которое представляет собой однодозовый или многодозовый сухой порошковый ингалятор.
35. Устройство по п.33, которое представляет собой дозирующий ингалятор или небулайзер тонкого тумана.
Текст
АМИНОЭФИРНЫЕ ПРОИЗВОДНЫЕ АЛКАЛОИДОВ И ИХ МЕДИЦИНСКИЕ КОМПОЗИЦИИ Настоящее изобретение относится к аминоэфирным производным алкалоидов формулы (XIX),обладающим действием антагонистов мускариновых рецепторов, к способам получения таких производных, к содержащим их композициям и к их терапевтическому применению. Область изобретения Настоящее изобретение относится к аминоэфирным производным алкалоидов, обладающих действием антагонистов мускариновых рецепторов, к способам их получения, к содержащим их композициям и к их терапевтическому применению. Предшествующий уровень техники Четвертичные аммониевые соли, обладающие действием антагонистом мускариновых (М) рецепторов, в настоящее время применяют в терапии для индуцирования бронходилатации для лечения респираторных заболеваний. Примерами широко известных антагонистов М-рецепторов являются ипратропия бромид и тиотропия бромид. Несколько классов химических веществ, действующих как лекарственные средства, являющиеся избирательными антагонистами М 3-рецептора, были разработаны для лечения воспалительных заболеваний или заболеваний дыхательных путей, таких как астма и хроническое обструктивное заболевание легких (COPD). Производные хинуклидинкарбамата и их применение в качестве антагонистов М 3 описаны в WO 02051841, WO 03053966 и WO 2008012290. Указанные антагонисты М- и М 3-рецепторов в настоящее время вводят ингаляцией с целью доставки лекарственного средства прямо в место действия и, следовательно, ограничения системного воздействия. Однако даже если системное воздействие может быть снижено путем введения ингаляцией, соединения предшествующего уровня техники все же могут проявлять нежелательные побочные эффекты вследствие системного всасывания. Следовательно, чрезвычайно желательно создать антагонисты М 3-рецептора, способные действовать местно и при этом иметь высокую эффективность и длительную продолжительность действия. Указанные лекарственные средства сразу после всасывания разлагаются до неактивных соединений, которые не оказывают никаких системных побочных эффектов, типичных для антагонистов мускариновых рецепторов. Согласно настоящему изобретению предложены аминоэфирные производные алкалоидов с такими терапевтически желательными характеристиками. Соединения общей формулы (I) действуют как мягкие лекарственные средства, поскольку они способны продуцировать персистентный бронходилатирующий эффект в легком, но постоянно и быстро превращаются в неактивные метаболиты после прохождения в плазму человека. Такое поведение дает большие преимущества с точки зрения безопасности. В US 2824106 раскрыты четвертичные алкильные производные тропинового ряда с повышенной спазмолитической активностью и, в частности, N-фениламиноацетильные производные. В J. Med. Chem. 1994, 37, 1712-1719, описаны пресинаптические холинергические модуляторы в качестве сильнодействующих агентов, усиливающих познавательную способность, и анальгетиков и, в частности, N-фениламиноацетильные производные. В US 2856407 раскрыты сложные эфиры аминокислот с гидроксипиперидинами, в частности Nметил-3-пиперидил-2'-N'-диметиламиноацетат и N-метил-4-пиперидил-2'-N'-диметиламиноацетат. В WO 99/20612 описаны соединения, которые ингибируют фарнезилирование продуктов мутантного гена ras и, в частности, производные 4-(метилсульфанил)бутаноата. В WO 2008/053158 описаны ингибиторы активности МАР-киназы р 38, полезные в лечении воспалительных и аутоиммунных заболеваний, в частности, производные метилпиперидин-4-ил-L-лейцината. В Khimiko-Farmatsevticheskii Zhurnal (1977), 11(7), 30-5, описан тропиновый эфир фенилглиоксиловой кислоты и, в частности, производные сложных эфиров фенилаланина. В ChemicalPharmaceutical Bulletin (1971), 19(12), 2603-8, описан одностадийный синтез атропина и других родственных алкалоидов из dl-фенилаланин-3-тропанилового эфира, в частности, производных сложных эфиров фенилаланина. Краткое изложение сущности изобретения Данное изобретение относится к аминоэфирным производным алкалоидов общей формулы (I) где R1 выбран из группы, состоящей из Н, (С 1-С 10)-алкила, арила, (С 3-С 8)-циклоалкила, арилалкила и гетероарила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов, ОН, SH, CON(R5)2, NHCOR5, COR5, CO2R5, (C1-C10)-алкилсульфанила, (С 1C10)-алкилсульфинила, (С 1-C10)-алкилсульфонила, (С 1-C10)-алкила и (С 1-C10)-алкоксила;R2 выбран из группы, состоящей из (С 1-C10)-алкила, арила, (С 3-С 8)-циклоалкила, арилалкила и гетероарила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей изR3 выбран из группы, состоящей из арила, гетероарила, арилалкила и гетероарилалкила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов,ОН, SH, NO2, CN, CON(R5)2, CO2R5, CF3, (С 1-C10)-алкила и (С 1-C10)-алкоксила;R6 представляет собой группу формулы (i), или (ii), или (iii), или (iv)n =1, 2 или 3; А- представляет собой физиологически приемлемый анион;R4 представляет собой группу формулы (Y) где р означает 0 или целое число от 1 до 4;q означает 0; Р отсутствует или выбран из группы, состоящей из О, S, SO, SO2, СО, СН=СН, SO2N(R5) иW выбран из группы, состоящей из Н, (С 1-С 6)-алкила, (С 3-С 8)-циклоалкила, арила и гетероарила,возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов, ОН, оксо (=O), SH, NO2, CN, CON(R5)2, NH2) NHCOR5, CO2R5, (C1-C10)-алкоксикарбонила,(С 1-C10)-алкилсульфанила, (С 1-C10)-алкилсульфинила, (С 1-C10)-алкилсульфонила, (С 1-С 10)-алкила и (С 1C10)-алкоксила;R5 выбран из группы, состоящей из Н, (С 1-C10)алкила, (С 3-С 7)циклоалкила, (С 3-С 7)циклоалкил-(С 1 С 6)алкила, гетероарила и арила; и к их фармацевтически приемлемым солям,где арил представляет собой фенил; арилалкил относится к (С 1-С 4)алкилу, замещенному фенилом; гетероарил относится к моно- или бициклическим кольцевым системам, которые имеют от 5 до 15 кольцевых атомов, где по меньшей мере одно кольцо является ароматическим и где по меньшей мере один кольцевой атом представляет собой гетероатом (N, NH, S или О); гетероарилалкил относится к (С 1-С 4)алкилу, замещенному гетероарилом. Данное изобретение также относится к соединениям общей формулы (VI)R7 представляет собой группу формулы (vi), или (vii), или (viii), или (ix) где m и n такие, как указано выше; и к их фармацевтически приемлемым солям,-2 020974 при условии, что соединения общей формулы (VI) не представляют собой 1-метилпиперидин-4-ил 2-трет-бутоксикарбониламино-4-метилсульфанилбутират, 1-метилпиперидин-4-иловый эфир L-фенилаланина и N-метилпиперидин-4-иловый эфир L-метионина. Данное изобретение также относится к способу получения соединения формулы (I), как представлено на схеме 1, включающему алкилирование соединения общей формулы (VI) алкилирующим агентом общей формулы (XI) где А представляет собой уходящую группу, выбранную из группы, состоящей из галогенида и сульфоната, и R4 имеет вышеуказанные значения. Данное изобретение также относится к способу получения соединения формулы (I), как представлено на схеме 1, включающему алкилирование аминосоединения общей формулы (II) соединением общей формулы (III) где LG представляет собой уходящую группу, и K может представлять собой либо гидроксильную группу, либо подходящим образом защищенную гидроксильную группу, с получением соединения формулы (IV) и затем превращение этого соединения в соединение формулы (I) согласно описанному выше способу. Данное изобретение также относится к способу получения соединения формулы (I), как представлено на схеме 1, включающему сочетание амина общей формулы (II) с получением соединения формулы (IV) с получением соединения формулы (VI) и затем превращение этого соединения в соединение формулы (I) согласно описанному выше способу. Данное изобретение также относится к способу получения соединения формулы (I), как представлено на схеме 1, включающему взаимодействие соединения (IX) с алкилирующим агентом общей формулы (VIII), где z представляет собой карбонильную группу или подходящую уходящую группу, предпочтительно галогенид, с получением соединения формулы (IV) с получением соединения формулы (VI) и затем превращение этого соединения в соединение формулы (I) согласно описанному выше способу. Данное изобретение также относится к способу получения соединения формулы (I), как представлено на схеме 1, включающему сочетание соединения общей формулы (III) с соединением общей формулы (V) с получением соединения формулы (X) которое подвергают взаимодействию с амином формулы (II) с получением соединения формулы (VI) и затем превращение этого соединения в соединение формулы (I) согласно описанному выше способу, где W' представляет собой LG или гидрокси. Согласно данному изобретению предложены также фармацевтические композиции соединений общей формулы (I) или общей формулы (VI), обладающие активностью антагонистов М 3-мускаринового рецептора, в комбинации или в смеси с одним, или более фармацевтически приемлемыми носителями и/или эксципиентами. Согласно данному изобретению предложено также применение соединений общей формулы (I) и(VI) в качестве лекарственного средства, обладающего активностью антагонистов М 3-мускаринового рецептора. В другом аспекте данного изобретения предложено применение соединения формулы (I) или общей формулы (VI) для изготовления лекарственного средства для предупреждения и/или лечения бронхообструктивных или воспалительных заболеваний, предпочтительно астмы, или хронического бронхита,или хронического обструктивного заболевания легких (COPD). В еще одном аспекте данного изобретения предложено применение соединения общей формулы где R1 и R2 независимо выбраны из группы, состоящей из Н, (C1-C10)-алкила, арила, (С 3-С 8)циклоалкила, арилалкила и гетероарила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов, ОН, SH, CON(R5)2, NHCOR5, COR5, CO2R5, (С 1C10)-алкилсульфанила, (С 1-C10)-алкилсульфинила, (C1-C10)-алкилсульфонила, (С 1-C10)-алкила и (С 1-C10)алкоксила;R3 выбран из группы, состоящей из арила, гетероарила, арилалкила и гетероарилалкила, возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов,ОН, SH, NO2, CN, CON(R5)2, CO2R5, CF3, (С 1-C10)-алкила и (С 1-C10)-алкоксила;R8 представляет собой группу формулы (i), или (ii), или (iii), или (iv), или (vi), или (vii), или (viii),или (ix)n =1, 2 или 3; А- представляет собой физиологически приемлемый анион;R4 представляет собой группу формулы (Y) где р означает 0 или целое число от 1 до 4;q означает 0; Р отсутствует или выбран из группы, состоящей из О, S, SO, SO2, СО, CH=CH, SO2N(R5) иW выбран из группы, состоящей из Н, (С 1-С 6)-алкила, (С 3-С 8)-циклоалкила, арила и гетероарила,возможно замещенных одним или более заместителями, выбранными из группы, состоящей из атомов галогенов, ОН, оксо (=O), SH, NO2, CN, CON(R5)2, NH2, NHCOR5, CO2R5, (C1-C10)-алкоксикарбонила,(С 1-C10)-алкилсульфанила, (С 1-C10)-алкилсульфинила, (С 1-C10)-алкилсульфонила, (С 1-C10)-алкила и (С 1C10)-алкоксила;R5 выбран из группы, состоящей из Н, (С 1-C10)-алкила, (С 3-С 7)-циклоалкила, (С 3-С 7)-циклоалкил(С 1-С 6)-алкила, гетероарила и арила,где арил, арилалкил, гетероарил и гетероарилалкил являются такими, как определено выше; для изготовления лекарственного средства для предупреждения и/или лечения бронхообструктивных или воспалительных заболеваний, предпочтительно астмы, или хронического бронхита,или хронического обструктивного заболевания легких (COPD). Согласно данному изобретению предложен также способ предупреждения и/или лечения бронхообструктивных или воспалительных заболеваний, предпочтительно астмы, или хронического бронхита,или хронического обструктивного заболевания легких (COPD), включающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения общей формулы (I),(VI) или (XIX). Согласно данному изобретению предложены также фармацевтические композиции для введения путем ингаляции. Данное изобретение также относится к устройству, которое представляет собой однодозовый или многодозовый сухой порошковый ингалятор, дозирующий ингалятор или небулайзер тонкого тумана,содержащий соединения общей формулы (I) или (VI). Определения Термин "атомы галогенов" охватывает фтор, хлор, бром и йод. Выражение "(С 1-C10)алкил" относится к алкильным группам с прямой или разветвленной цепью,где количество атомов углерода составляет от 1 до 10. Примерами указанных групп являются метил,этил, н-пропил, изопропил, трет-бутил, пентил, гексил, гептил, октанил, ноненил и деценил. Возможно, как указано выше, в алкильных группах один или более атомов водорода могут быть заменены атомами галогенов. Производные выражения "(С 1-C10)-алкоксикарбонил", "(С 1-C10)алкилсульфанил", "(C1-C10)-алкилсульфинил", "(С 1-C10)-алкилсульфонил" и "(С 1-C10)-алкоксил" следует толковать аналогичным образом. Производные выражения "(С 2-C10)алкенил" и "(С 2-C10)алкинил" следует толковать аналогичным образом как относящиеся к группам, содержащим по меньшей мере одну двойную или тройную связь. Выражение "арил" относится к моно-, би- или трициклическим кольцевым системам, которые имеют от 5 до 20 кольцевых атомов, предпочтительно от 5 до 15, и где по меньшей мере одно кольцо является ароматическим. В указанных кольцах один или более атомов водорода могут быть заменены одним или более атомами галогенов. Выражение "гетероарил" относится к моно-, би- или трициклическим кольцевым системам, которые имеют от 5 до 20 кольцевых атомов, предпочтительно от 5 до 15, где по меньшей мере одно кольцо является ароматическим и где по меньшей мере один кольцевой атом представляет собой гетероатом или гетероароматическую группу (например N, NH, S или О). В указанных кольцах один или более атомов водорода могут быть заменены одним или более атомами галогенов. Выражение "арилалкил" относится к "(С 1-С 4)алкилу", возможно замещенному арилом, как он определен выше. Примеры подходящих арилалкильных групп включают бензил и дифенилметил. Выражение "гетероарилалкил" относится к "(С 1-С 4)алкилу", возможно замещенному гетероарильной группой, как она определена выше. Примеры подходящих гетероарилалкильных групп включают тиофенилметил. Примеры подходящих моноциклических систем включают радикалы тиофена, циклопентадиена,бензола, пиррола, пиразола, имидазола, изоксазола, оксазола, изотиазола, тиазола, пиридина, имидазолидина, пиперидина и фурана. Примеры подходящих бициклических систем включают радикалы нафталина, бифенила, пурина,-6 020974 птеридина, бензотриазола, хинолина, изохинолина, индола, изоиндола и бензотиофена. Примеры подходящих трициклических систем включают радикалы флуорена. Подробное описание изобретения Данное изобретение относится к аминоэфирным производным алкалоидов формулы (I) и (VI), обладающим действием антагонистов мускариновых рецепторов, и к их солям, причем указанные производные предпочтительно действуют на М 3-рецепторах. Предпочтительно физиологически приемлемые анионы А- включают анионы, выбранные из хлорида, бромида, йодида, трифторацетата, формиата, сульфата, фосфата, метансульфоната, нитрата, малеата,ацетата, цитрата, фумарата, тартрата, оксалата, сукцината, бензоата и п-толуолсульфоната, предпочтительно хлорида, бромида и йодида. В предпочтительных соединениях общей формулы (I) R1 представляет собой водород, R2 представляет собой арилалкил, или (С 1-C10)-алкил, или арил, или (С 3-С 8)-циклоалкил, или гетероарил, и R3 представляет собой арил или гетероарил, возможно замещенные одним или более атомами галогенов, и гдеR6 такой, как определено выше. В предпочтительных соединениях общей формулы (VI) R1 представляет собой Н, R2 представляет собой арилалкил, или (С 1-C10)-алкил, или арил, или (С 3-С 8)-циклоалкил, или гетероарил, и R3 представляет собой арил или гетероарил, возможно замещенные одним или более атомами галогенов, и где R7 представляет собой группу формулы (vi) согласно общей формуле (XII) В предпочтительной группе соединений общей формулы (I) R1 представляет собой Н, R2 представляет собой арилалкил, или (C1-C10)-алкил, или арил, или (С 3-С 8)-циклоалкил, или гетероарил, и R3 представляет собой арил, или арилалкил, или гетероарил, возможно замещенные одним или более атомами галогенов, и где R6 представляет собой группу формулы (i) согласно общей формуле (XIII) В предпочтительной группе соединений общей формулы (XII) R2 и R3 представляют собой фенил. В предпочтительной группе соединений общей формулы (XIII) R2 и R3 представляют собой фенил. В предпочтительной группе соединений общей формулы (XIII) R4 представляет собой группу формулы (Y) где р равно 1, Р представляет собой СО, q равно 0 и W представляет собой арил или гетероарил. В предпочтительной группе соединений общей формулы (XIII) R4 представляет собой группу формулы (Y), где р равно 1, Р представляет собой CO, q равно 0 и W представляет собой фенил. В предпочтительной группе соединений общей формулы (XIII) R4 представляет собой группу формулы (Y), где р равно 1, Р представляет собой СО, q равно 0 и W представляет собой тиенил или тиазолил. В предпочтительной группе соединений общей формулы (XIII) R4 представляет собой группу формулы(Y), где р равно 2, Р представляет собой О, q равно 0 и W представляет собой фенил. В предпочтительной группе соединений общей формулы (XIII) R4 представляет собой группу формулы(Y), где р равно 3, Р представляет собой О, q равно 0 и W представляет собой фенил. В предпочтительной группе соединений общей формулы (VI) R1 представляет собой Н, R2 представляет собой арилалкил, или (C1-C10)-алкил, или арил, или (С 3-С 8)-циклоалкил, или гетероарил, и R3 представляет собой арил, или арилалкил, или гетероарил, возможно замещенные одним или более атомами галогенов, и гдеR7 представляет собой группу формулы (vii) где m=2, и n=1, согласно общей формуле (XIV) В предпочтительной группе соединений общей формулы (XIV) R2 и R3 представляют собой фенил,возможно замещенный одним или более атомами галогенов. В предпочтительной группе соединений общей формулы (VI) R1 представляет собой Н, R2 представляет собой арилалкил, или (C1-C10)-алкил, или арил, или (С 3-С 8)-циклоалкил, или гетероарил, и R3 представляет собой арил, или арилалкил, или гетероарил, возможно замещенные одним или более атомами галогенов, и где R7 представляет собой группу формулы (vii), где m=1, и n=1, согласно общей формуле (XV) В предпочтительной группе соединений общей формулы (XV) R2 и R3 представляет собой фенил,возможно замещенный одним или более атомами галогенов. В предпочтительной группе соединений общей формулы (VI) R1 представляет собой Н, R2 представляет собой арилалкил, или (C1-C10)-алкил, или арил, или (С 3-С 8)-циклоалкил, или гетероарил, и R3 представляет собой арил, или арилалкил, или гетероарил, возможно замещенные одним или более атомами галогенов, и где R7 представляет собой группу формулы (vii), где m=1, и n=2, согласно общей формуле (XVI) В предпочтительной группе соединений общей формулы (XVI) R2 и R3 представляют собой фенил,возможно замещенный одним или более атомами галогенов. В предпочтительной группе соединений общей формулы (VI) R1 представляет собой Н, R2 представляет собой арилалкил, или (C1-C10)-алкил, или арил, или (С 3-С 8)-циклоалкил, или гетероарил, и R3 представляет собой арил, или арилалкил, или гетероарил, возможно замещенные одним или более атомами галогенов, и где R7 представляет собой группу формулы (viii) согласно общей формуле (XVII) В предпочтительной группе соединений общей формулы (XVII) R2 и R3 представляют собой фенил, возможно замещенный одним или более атомами галогенов. В предпочтительной группе соединений общей формулы (VI) R1 представляет собой Н, R2 представляет собой арилалкил, или (С 1-C10)-алкил, или арил, или (С 3-С 8)-циклоалкил, или гетероарил, и R3 представляет собой арил, или арилалкил, или гетероарил, предпочтительно замещенные одним или более атомами галогенов, и где R7 представляет собой группу формулы (ix) согласно общей формуле (XVIII) В предпочтительной группе соединений общей формулы (XVIII) R2 и R3 представляют собой фенил. Предпочтительную группу соединений составляют соответствующие четвертичные аммониевые соли соединений общей формулы (XII), (XIV), (XV), (XVII) и (XVIII). Очевидно, что соединения общей формулы (I) и (VI) могут содержать асимметрические центры. Поэтому данное изобретение также охватывает оптические стереоизомеры и их смеси. Если соединения по изобретению имеют по меньшей мере один асимметрический центр, то они соответственно могут существовать в виде энантиомеров. Если соединения по изобретению имеют два или более асимметрических центров, то они могут дополнительно существовать в виде диастереоизомеров. Следует иметь в виду, что все такие изомеры и их смеси в любом соотношении входят в объем настоящего изобретения. Активные соединения общей формулы (VI) и, в частности, соединения общей формулы (XII), (XIII),(XV) и (XVI), имеют по меньшей мере два хиральных центра, которые соответственно представляют собой атом углерода алкалоида, несущий аминоэфирную группу, и атом углерода, несущий Н, R3 и NHR2. Соединения общей формулы (XII), (XIII), (XV) и (XVI) могут быть получены в S-R, R-R, R-S, S-S конфигурации или в виде смеси диастереоизомеров (R-R и S-R конфигурация или R-S и S-S конфигурация). Согласно предпочтительному воплощению соединения общей формулы (XII), (XIII), (XV) и (XVI) имеют (R,R) конфигурацию. Активные соединения формулы (I) и (VI) имеют по меньшей мере один хиральный центр, который представляет собой атом углерода алкалоида, несущий аминоэфирную группу. Согласно еще одному воплощению соединение формулы (I) находится в форме (S)-энантиомера, когдаR6 представляет собой группу формулы (i). Согласно предпочтительному воплощению соединение формулы (I) находится в форме (R)энантиомера, когда R6 представляет собой группу формулы (i). Согласно конкретным воплощениям настоящего изобретения предложены соединения, указанные ниже. Соединения общей формулы (I) и (VI) могут быть получены способами, известными или очевидными специалисту в данной области техники. Некоторые способы, которые могут быть использованы, описаны ниже и представлены на схеме 1. Методика получения соединений формулы (I) Соединения общей формулы (IV) могут быть получены тремя разными путями: А, В и С. Путь А. Алкилирование аминосоединений общей формулы (II), где R1 и R2 такие, как указано выше, соединениями общей формулы (III), где LG представляет собой уходящую группу (например галогенид, такой как бромид), и K может представлять собой либо гидроксильную группу, либо подходящим образом защищенную гидроксильную группу (например, K = ОАлкил, такой как ОМе). Реакция может быть промотирована основанием, выбранным из группы, состоящей из триэтиламина, пиридина и 4 диметиламинопиридина, либо без растворителя, либо в подходящем растворителе (например, в ацетонитриле). Эту реакцию обычно осуществляют при температуре в пределах от 0 до 130 С в течение периода времени от 1 до 74 ч. Реакцию можно проводить в условиях обычного нагревания (используя масляную баню) или в условиях микроволнового нагревания. Реакцию можно проводить в открытом сосуде или в герметично закрытой пробирке. Реагенты общей формулы (III) имеются в продаже или без труда могут быть получены по стандартным методикам, широко освещенным в литературе. Например, соединения общей формулы (III), где LG представляет собой галоген, например бром, могут быть получены галогенированием возможно замещенного фенилового эфира уксусной кислоты (например, по методике, описанной в Epstein, J.W. inJ.Med. Chem., 1981, 24/5, 481). Альтернативно, соединения общей формулы (III) могут быть получены из соответствующим образом замещенного производного миндальной кислоты по методикам, известным специалистам в данной области техники (обзор подходящих реакций дан в Larock, L.C., ComprehensiveOrganic Transformation, Second edition (1999), John WileySon Inc, pg 689-700). Путь В. Сочетание амина общей формулы (II) и кетона общей формулы (VII) может быть осуществлено путем проведения реакции восстановительного аминирования по одной из разных методик, описанных в литературе (например, Suwa Т., Synthesis, 2000, 6, 789, или Fache, F. Tetrahedron, 1996, 52/29,9777, или Quiang, K., Adv. Synth. Catal. 2007, 349, 1657). Альтернативно, соединения общей формулы (VII) могут быть без труда превращены в соединения общей формулы (III) (путь F) в условиях, известных специалистам в данной области техники. В типичном способе кетоны общей формулы (VII) обрабатывают восстановителем, таким как боргидрид натрия и т.п., в подходящем растворителе (например, в этаноле и метаноле) с беспрепятственным получением соответствующего спиртового промежуточного соединения. Последующее превращение спиртовой группировки в уходящую группу (LG) дает соединения общей формулы (III). Эта активация может быть осуществлена согласно одной из стандартных методик, широко освещенных в литературе (обзор подходящих реакций дан в Carey, F.A. and Sundeberg, R.J. Advanced Organic Chemistry, Third Edition (1990),Plenum Press, New York and London, pg 121). Например, спиртовое промежуточное соединение может быть обработано метансульфонилхлоридом (LG = Ms) в присутствии основания, такого как триэтиламин,пиридин, 4-диметиламинопиридин и т.п., либо без растворителя, либо в апротонном растворителе (например, в дихлорметане). Эту реакцию обычно проводят при температуре в пределах от 0 до 130 С в течение периода времени от 1 до 74 ч. Путь С. Соединение формулы (IX) может быть подвергнуто взаимодействию с алкилирующим агентом общей формулы (VIII), где z представляет собой подходящую уходящую группу, такую как карбонильная группа, или галогенид (т.е. бром, йод, хлор), или сульфонатная эфирная группа (т.е. тозилат,трифлат, мезилат), по методикам, легко доступным специалистам в данной области техники (см., например, Huang, Tetrahedron, 1997, 53/37, 12391). В случае, когда z = О, тогда соединения формулы (IX) подвергают взаимодействию с альдегидами или кетонами общей формулы (VIII) с получением соответствующих иминов, которые восстанавливают до соединения формулы (IV) путем обработки подходящим восстановителем по одной из методик, описанных в литературе (обзор подходящих реакций приведен в Carey, F.A. and Sundeberg, R.J. AdvancedOrganic Chemistry, Third Edition (1990), Plenum Press, New York and London, chapter 5, 219, или Ando, A.,Tetrahedron, 1989, 45/16, 4969). В случае, когда R1 представляет собой арил или гетероарил и z представляет собой галоген (обычно йод или бром), тогда взаимодействие соединений общей формулы (VIII) и (IX) может быть промотировано подходящим катализатором. В типичном способе катализатор представляет собой медный катализатор (например йодид меди), и реакцию осуществляют в присутствии подходящего основания, выбранного из группы, состоящей из карбоната калия и карбоната цезия или аминов, таких как триэтиламин, в растворителях, выбранных из группы, состоящей из диметилсульфоксида (DMSO) и DMF (диметилформамид), при температуре в пределах от температуры окружающей среды до 110 С в течение периода времени от 1 до 48 ч. Эта реакцию можно проводить в условиях обычного нагревания (с использованием масляной бани) или под действием микроволнового излучения. Эту реакцию можно проводить либо в открытом сосуде, либо в герметично закрытой пробирке (Ma, D., Tetrahedron Asymmetry 1996, 7/11, 3075,или Kurokawa, M., Heterocycles, 2007, 71/4, 847). В случае, когда R1 представляет собой арил или гетероарил и z представляет собой галоген (обычно фтор или хлор), тогда соединение общей формулы (VIII) и соединение общей формулы (IX) могут быть подвергнуты взаимодействию в типичных условиях ароматического нуклеофильного замещения с получением соединения формулы (IV). Соединения общей формулы (VI) затем могут быть получены следующими двумя разными путями. Путь D. Соединения формулы (VI) могут быть получены в результате реакции сочетания спиртов формулы (V) с соединениями формулы (IV). Способ получения соединений формулы (VI) из спиртов формулы (V) и соединений формулы (IV) выбирают, исходя из реакционной способности спирта формулы (V), коммерческой доступности реагентов, таких как соединения формулы (IV), и совместимости с группами, присутствующими в обоих исходных веществах. Реакция сочетания соединения формулы (IV) и соединения формулы (V) может быть проведена несколькими способами, при которых K может представлять собой либо гидроксильную группу, либо галогенид, такой как хлор (обзор подходящих реакций приведен в Carey, F.A. and Sundeberg, R.J. AdvancedOrganic Chemistry, Third Edition (1990), Plenum Press, New York and London, pg 145, и Montalbetti, C., Tetrahedron, 2005, 61, 10827). В частности, в случае, когда K представляет собой защищенную гидроксильную группу, тогда защитную группу необходимо удалять до осуществления реакции сочетания с соединением формулы (V). Например, если K = ОМе, то гидролиз эфирной группировки может быть осуществлен путем обработки соединения формулы (IV) (K = ОМе) с подходящим водным основанием, выбранным из группы, состоящей из гидроксида натрия, гидроксида лития и гидроксида калия, в подходящих растворителях (например, в тетрагидрофуране, диоксане и т.д.). Эта реакция протекает при комнатной температуре (КТ) в течение периода времени от 1 до 36 ч. Альтернатива один. В типичном способе соединения формулы (VI) могут быть получены в результате реакции конденсации соединения формулы (V) с кислотой формулы (IV) (K = ОН) в стандартных условиях амидирования и пептидного сочетания. Например, в результате обработки кислоты формулы(IV) (K = ОН) одним или более эквивалентами коммерчески доступного агента конденсации, такого как карбодиимид (например, гидрохлорид 1-(3-диметиламино)пропил)-3-этилкарбодиимида (EDC) и т.п.),например, в присутствии N-гидроксибензотриазола (HOBt), с последующим взаимодействием активированного промежуточного соединения спиртом формулы (V) образуются соединения формулы (VI). Органическое основание, такое как триэтиламин, также может присутствовать в реакционной смеси. Активированное промежуточное соединение может быть либо выделено, либо предварительно образовано,либо образовано in situ. Подходящие растворители для реакции сочетания включают, но не ограничиваются ими, галогеноуглеродные растворители (например, дихлорметан), тетрагидрофуран, диоксан и ацетонитрил. Эта реакция протекает при температуре в пределах от 0 до 170 С в течение периода времени в пределах от примерно 1 до 72 ч. Эту реакцию можно проводить в условиях обычного нагревания (с использованием масляной бани) или под действием микроволнового излучения. Эту реакцию можно проводить либо в открытом сосуде, либо в герметично закрытой пробирке. Альтернатива два. В случае, когда K представляет собой галоген, такой как хлор, тогда спирт формулы (V) подвергают взаимодействию с подходящим ацилгалогенидом формулы (IV), используя методы,которые известны специалистам в данной области техники. Эта реакция может быть промотирована основанием, таким как триэтиламин, пиридин и 4-диметиламинопиридин, в подходящем растворителе (например, в дихлорметане). Эту реакцию осуществляют при температуре в пределах от 0 до 130 С в течение периода времени от 1 до 74 ч. Эту реакцию можно проводить в условиях обычного нагревания (с использованием масляной бани) или под действием микроволнового излучения. Эту реакцию можно проводить либо в открытом сосуде, либо в герметично закрытой пробирке. В некоторых воплощениях настоящего изобретения нужный ацилгалогенид формулы (IV) без труда может быть получен из соответствующей кислоты формулы (IV) (K = ОН). Эта активация может быть осуществлена по одной из стандартных методик, описанных в литературе. Например, обработка кислоты формулы (IV) (K = ОН) одним или более эквивалентами оксалилхлорида в присутствии каталитического количества диметилформамида (DMF) в галогеноуглеродном растворителе, таком как дихлорметан, при температуре в пределах от 0 до 35 С приводит к получению требуемого ацилхлорида (IV) (K = Cl). Альтернатива три. Альтернативно, ацилирование спирта формулы (V) с получением соединения общей формулы (VI) может быть осуществлено по методикам превращения in situ кислоты формулы (IV)(K = ОН) в соответствующие ацилгалогениды. Например, спирты формулы (V) подвергают взаимодействию с кислотами формулы (IV) (K = ОН) в присутствии трифенилфосфина и галогеноуглеродного растворителя, такого как тетрахлорид углерода или дихлорметан, при примерно комнатной температуре в течение максимального периода времени 16 ч (Lee, J.B. J. Am. Chem. Soc., 1966, 88, 3440). Альтернатива четыре. В другом способе получения соединений по настоящему изобретению кислота формулы (IV) (K = ОН) может быть активирована другими коммерчески доступными активирующими агентами, такими как гексафторфосфат бромтрипирролидинофосфония (PyBrOP) или карбонилимидазол,в подходящем апротонном растворителе (например в дихлорметане, тетрагидрофуране) при примерно комнатной температуре. В результате последующего взаимодействия активированного промежуточного соединения со спиртом формулы (V) образуется целевое соединение формулы (VI). Для осуществления этой реакции может потребоваться использование органического основания, такого как диизопропилэтиламин, и эта реакция обычно протекает при примерно комнатной температуре. Алтернатива пять. В еще одном способе получения соединений по настоящему изобретению соединения формулы (VI) могут быть эффективно получены в результате реакции конденсации кислоты формулы (IV) (K = ОН) со спиртом формулы (V) в типичных условиях Мицунобу (Kumara Swamy, K.C.,Chem. Rev. 2009, 109, 2551-2651). Например, кислоту формулы (IV) и спирт формулы (V) подвергают взаимодействию в присутствии фосфина (например, трифенилфосфина) и азадикарбоксилатного эфира(например, диэтилазодикарбоксилата или диизопропилазодикарбоксилата) в апротонном растворителе,таком как тетрагидрофуран. Эта реакция обычно протекает при температуре в пределах от 0 до 100 С в течение времени в пределах от примерно 30 мин до 72 ч. Альтернативно, соединения формулы (VI) могут быть получены путем Е.W может представлять собой LG или гидрокси. Соединения общей формулы (III) могут быть подвергнуты реакции сочетания с соединением общей формулы (V) с получением соединения формулы (X) по одной из методик, известных специалистам в данной области техники. Например, условия, используемые для осуществления этого взаимодействия, могут быть выбраны из условий, которые указаны на схеме 1 для реакции сочетания соединения формулы (IV) и соединения формулы (V). В случае, когда W представляет собой галогенид, тогда полученное промежуточное соединение формулы (X) затем может быть использовано в качестве алкилирующего агента для аминов общей формулы (II) с получением конечного целевого промежуточного соединения формулы (VI). Эта реакция может быть осуществлена в типичных условиях, всесторонне описанных в литературе, таких как условия,описанные для получения соединения формулы (IV), путем взаимодействия соединения формулы (II) с соединением формулы (III) (схема 1). В случае, когда W в соединении формулы (III) представляет собой гидрокси, тогда эта группа должна быть превращена в подходящую уходящую группу, выбранную из группы, состоящей из галогенида (т.е. бром, йод, хлор) и сульфонатной эфирной группы (т.е. тозилат, трифлат, мезилат), по методикам, доступным специалистам в данной области техники (общий обзор приведен в Carey, F.A. and Sundeberg, R.J. Advanced Organic Chemistry, Third Edition (1990), Plenum Press, New York and London, chapter 3,121), перед осуществлением реакции сочетания с аминами формулы (II). В случае, когда W представляет собой подходящим образом защищенную гидроксильную группу, тогда эта группа должна быть лишена защиты и активирована, как указано выше, перед осуществлением реакции сочетания с аминами формулы (II). Соединение общей формулы (VI), где R3 и R7 такие, как определено в данном описании выше, может быть получено либо в виде единственного диастереоизомера, либо в виде смеси диастереоизомеров. Например, в случае, когда R7 представляет собой группу формулы (vi), тогда спирт может иметь либо R,либо S конфигурацию. Если используют R-энантиомер, то соединение формулы (VI) может быть получено в S-R конфигурации, в R-R конфигурации или в виде смеси диастереоизомеров (R-R и S-R конфигурация). Смесь диастереоизомеров может быть превращена в соединения формулы (I), указанные на схеме 1,или без труда может быть разделена с получением двух индивидуальных диастереоизомеров, которые, в свою очередь, могут быть превращены в соединения формулы (I), указанные на схеме 1. Это разделение может быть осуществлено по методикам, известным специалистам в данной области техники. Эти методики включают, но не ограничиваются ими, хроматографическую очистку, очистку препаративной ВЭЖХ (высокоэффективная жидкостная хроматография) и кристаллизацию. Например, два диастереоизомера могут быть разделены флэш-хроматографией на силикагеле с элюированием подходящими растворителями или смесью растворителей, таких как DCM (дихлорметан) и метанол и т.п. В другом способе по настоящему изобретению разделение диастереоизомеров может быть осуществлено с использованием колонки, заполненной хиральной стационарной фазой, например Chiralpack AY, или Chiralcel OD,или Chiralcel OZ, и с элюированием, например ацетонитрилом и/или смесями ацетонитрила и спирта. Альтернативно, разделение диастереоизомеров без труда может быть осуществлено в результате кристаллизации из подходящего растворителя (например, диэтилового эфира), в форме свободного основания или после образования подходящей соли (например, с (+)-винной кислотой). Алкилирование соединений общей формулы (VI) алкилирующими агентами общей формулы (XI) где А представляет собой подходящую уходящую группу, выбранную из группы, состоящей из галогенида (т.е. брома, йода, хлора) и сульфонатной эфирной группы (т.е. тозилат, трифлат, мезилат),обеспечивает получение соединений общей формулы (I). Этот тип реакции в нескольких разных условиях подробно описан в литературе. Например, эта реакция может быть осуществлена в отсутствие растворителя или в подходящем растворителе, выбранном из группы, состоящей из ацетонитрила, DMF, DMSO и тетрагидрофурана. Эта реакция обычно протекает при температуре в пределах от 0 до 170 С в течение времени в пределах от нескольких мин до 72 ч. Эту реакцию можно проводить в условиях обычного нагревания (с использованием масляной бани) или под действием микроволнового излучения. Эту реакцию можно проводить либо в открытом сосуде, либо в герметично закрытой пробирке. Соединения общей формулы (I), указанные на схеме 1, можно рассматривать либо как конечные продукты, либо они могут быть дополнительно подвергнуты реакции с получением других соединений общей формулы (I). Так, группировка группы R1, R2, R3 или R6 в общей формуле (I) может быть подвергнута реакциям окисления, восстановления или расщепления (например, для удаления необходимой защитной группы) с получением других конечных соединений общей формулы (I). Согласно настоящему изобретению предложены также фармацевтические композиции соединений общей формулы (I) и общей формулы (VI) в смеси с одним или более фармацевтически приемлемыми носителями, например носителями, описанными в (см. Remington's Pharmaceutical Sciences Handbook,XVII Ed., Mack Pub., N.Y., U.S.A). В данном изобретении также раскрыты фармацевтические препараты для введения путем ингаляции, такие как ингалируемые порошки, содержащие пропеллент дозирующие аэрозоли или не содержащие пропеллент ингалируемые композиции. Введение соединений по настоящему изобретению может быть осуществлено в соответствии с тем,что требуется пациентам, например перорально, назально, парентерально (подкожно, внутривенно, внутримышечно, интрастернально и инфузией), ингаляцией, ректально, вагинально, наружно, местно, трансдермально и введением в глаза. Для введения соединений по изобретению могут быть использованы различные твердые пероральные лекарственные формы, в том числе такие твердые формы, как таблетки, желатиновые капсулы, капсулы, каплетки, гранулы, пастилки и порошки. Соединения по изобретению можно вводить сами по себе или совместно с различными фармацевтически приемлемыми носителями, разбавителями (такими как сахароза, маннит, лактоза, крахмалы) и эксципиентами, известными в данной области, включая, без ограничения, суспендирующие агенты, солюбилизаторы, буферные агенты, связывающие вещества, разрыхлители, консерванты, красители, корригенты, смазывающие вещества и т.п. Также предпочтительными для введения соединений по настоящему изобретению являются капсулы, таблетки и гели с высвобождением в течение определенного времени. Для введения соединений по изобретению также могут быть использованы различные жидкие пероральные лекарственные формы, в том числе водные и неводные растворы, эмульсии, суспензии, сиропы и эликсиры. Такие лекарственные формы могут также содержать подходящие инертные разбавители,известные в данной области, такие как вода, и подходящие эксципиенты, известные в данной области,такие как консерванты, смачивающие агенты, подсластители, корригенты, а также агенты для эмульгирования и/или суспендирования соединений по изобретению. Соединения по настоящему изобретению можно вводить инъекцией, например внутривенно, в форме изотонического стерильного раствора. Другие препараты также возможны. Суппозитории для ректального введения соединений по настоящему изобретению могут быть при- 19020974 готовлены путем смешивания соединения с подходящим эксципиентом, таким как масло какао, салицилаты и полиэтиленгликоли. Композиции для вагинального введения могут быть в форме крема, геля, пасты, пены или спрея,содержащих, в дополнение к активному ингредиенту, подходящие носители, известные в данной области. Для наружного введения фармацевтическая композиция может быть в форме крема, мази, линимента, лосьона, эмульсии, суспензии, геля, раствора, пасты, порошка, спрея и капель, подходящих для введения на кожу, в глаз, ухо или нос. Наружное введение может также включать трансдермальное введение с помощью таких средств, как трансдермальные пластыри. Для лечения заболеваний дыхательных путей соединения по изобретению предпочтительно вводят ингаляцией. Ингалируемые препараты включают ингалируемые порошки, дозирующие аэрозоли, содержащие пропеллент, или ингалируемые композиции, не содержащие пропеллент. Для введения в виде сухого порошка могут быть использованы однодозовые или многодозовые ингаляторы, известные из предшествующего уровня техники. В этом случае порошок может быть упакованным в желатиновых, пластиковых или иных капсулах, картриджах, или в блистерных упаковках, или в резервуаре. Разбавитель или носитель, как правило нетоксичный и химически инертный по отношению к соединениям по изобретению, например лактоза или любая другая добавка, подходящая для улучшения вдыхаемой фракции, могут быть добавлены к порошкообразным соединениям по изобретению. Ингаляционные аэрозоли, содержащие газ-пропеллент, такой как гидрофторалканы, могут содержать соединения по изобретению либо в растворе, либо в дисперсной форме. Композиции, доставляемые с помощью пропеллента, могут также содержать другие ингредиенты, такие как сорастворители, стабилизаторы и возможно другие эксципиенты. Не содержащие пропеллент ингалируемые композиции, содержащие соединения по изобретению,могут быть в форме растворов или суспензий в водной, спиртовой или водноспиртовой среде, и их можно доставлять с помощью струйных или ультразвуковых небулайзеров или небулайзеров тонкого тумана. Соединения по изобретению можно вводить в виде только одного активного агента или в комбинации с другими фармацевтическими активными ингредиентами, включая ингредиенты, в настоящее время используемые в лечении респираторных расстройств, например бета 2-агонисты, кортикостероиды и антихолинергические или антимускариновые агенты. Дозировки соединений по изобретению зависят от многочисленных факторов, включая конкретное заболевание, которое лечат, тяжесть симптомов, путь введения, частоту введения доз, конкретное используемое соединение, эффективность, токсикологический профиль и фармакокинетический профиль соединения. Предпочтительно соединения формулы (I) можно вводить, например, в дозировке, составляющей от 0,001 до 1000 мг/сутки, предпочтительно от 0,1 до 500 мг/сутки. При введении соединений формулы (I) ингаляционным путем их предпочтительно вводят в дозировке от 0,001 до 500 мг/сутки, предпочтительно от 0,1 до 200 мг/сутки. Соединения формулы (I) можно вводить для предупреждения и/или лечения любого заболевания,при котором активны антагонисты М 3. Указанное заболевание включает заболевания, в которые вовлечено воспаление, такие как астма и COPD, острый ринит; заболевания, затрагивающие желудочнокишечный тракт, такие как пептическая язва; заболевания, затрагивающие сердечно-сосудистую систему, такие как острый инфаркт миокарда; заболевания, затрагивающие мочеполовой тракт, такие как почечные колики; отравление антихолинэстеразными средствами и грибами; применение для анестезии; применение в офтальмологии. Они также включают неврологические и психиатрические расстройства, такие как болезнь Паркинсона и морская болезнь. Предпочтительно соединения формулы (I) можно вводить для предупреждения и/или лечения респираторных заболеваний, таких как состояния астмы и COPD от легкой до острой тяжести. Другие респираторные заболевания включают бронхит, бронхиолит, бронхоэктаз, острый назофарингит, острый и хронический синусит, гайморит, фарингит, тонзилит, ларингит, трахеит, эпиглоттит,круп, хроническое заболевание миндалин и аденоидов, гипертрофию миндалин и аденоидов, перитозиллярный абсцесс, ринит, абсцесс или язву носа, пневмонию, вирусную и бактериальную пневмонию,бронхопневмонию, грипп, экзогенный аллергический альвеолит, пневмокониоз шахтеров, асбестоз,пневмокониоз, пневмонопатию, респираторные состояния вследствие воздействия химических газов,паров и других внешних агентов, эмфизему, плеврит, пневмоторакс, абсцесс легкого и средостения, легочную гиперемию и гипостаз, поствоспалительный легочный фиброз, иную альвеолярную и париетоальвеолярную пневмонопатию, идиопатический фиброзный альвеолит, синдром Хаменна-Рича, ателектаз, ARDS (респираторный дистресс-синдром взрослых), острую дыхательную недостаточность, медиастинит. В данном изобретении также ракрыт набор, включающий в себя фармацевтические композиции со- 20020974 единений общей формулы (I) или (VI), одних или в комбинации, или в смеси с одним или более фармацевтически приемлемыми носителями и/или эксципиентами, и устройство, которое может представлять собой однодозовый или многодозовый сухой порошковый ингалятор, дозирующий ингалятор и небулайзер тонкого тумана, содержащий соединения общей формулы (I) или (VI). Настоящее изобретение далее дополнительно описано приведенными ниже примерами.-Бромфенилуксусную кислоту (5,01 г, 23,2 ммоль) растворяют в анилине (25 мл, 274 ммоль), и эту смесь подвергают взаимодействию в закрытом сосуде под действием микроволнового излучения (MB) при 120 С в течение 5 мин (ЖХ-МС (жидкостная хроматография/масс-спектрометрия) мониторинг: полная конверсия). Дихлорметан (DCM) (100 мл) добавляют в реакционную смесь, и полученное твердое вещество фильтруют. В раствор добавляют 2 М Ма 2 СО 3 (50 мл), и водный слой промывают DCM (3100 мл). Водный слой подкисляют 12 н. HCl (36 мл), и указанное в заголовке соединение выделяют в виде рацемической смеси фильтрованием (5,1 г, выход 97%, белое твердое вещество). Получение (R)-хинуклидин-3-ил-2-фенил-2-(фениламино)ацетата (диастереоизомеры 1 и 2 соединения С 2) В раствор 2-фенил-2-(фениламино)уксусной кислоты (I1) (1,90 г, 8,41 ммоль) в диоксане добавляют 4 М раствор HCl в диоксане (5 мл), и эту реакционную смесь перемешивают при КТ в течение 1 ч. Растворитель выпаривают при пониженном давлении с получением белого твердого вещества, которое растворяют в сухом THF (тетрагидрофуран) (40 мл). К полученному раствору добавляют DCC (дициклогексилкарбодиимид) (2,12 г, 10,0 ммоль), HOBt (1,33 г, 10,0 ммоль) и 3(R)-хинуклидинол (3,10 г, 25,1 ммоль), и эту смесь перемешивают в течение 96 ч при КТ в потоке азота (ЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, к полученному неочищенному остатку добавляют 1 н. HCl (50 мл), и водный слой промывают EtOAc (2100 мл); насыщенный раствор NaHCO3 добавляют к водному слою (рН 7-8), и целевое соединение экстрагируют DCM. Полученное неочищенное вещество очищают флэш-хроматографией (DCM/MeOH = 95/5, 0,1% NH3 (водн., выделяя диастереоизомер 1 соединения С 2 в виде белого твердого вещества (0,8 г; выход 28%, единственный диастереоизомер) и затем диастереоизомер 2 соединения С 2 в виде белого твердого вещества (0,4 г, выход 14%, единственный диастереоизомер). Диастереоизомер 1 соединения С 2 (80 мг) дополнительно очищают препаративной ЖХ-МС с получением 55,3 мг бледно-желтого масла (TFA-соль (TFA-трифторуксусная кислота. Диастереоизомер 2 соединения С 2 (65 мг) дополнительно очищают препаративной ЖХ-МС с получением 28,3 мг бледно-желтого масла (TFA-соль). Диастереоизомер 1 соединения С 2 1 Н ЯМР (300 МГц, DMSO-d6) м.д. (миллионные доли): 9.38 (br. s., 1H), 7.50-7.72 (m, 2 Н), 7.26-7.50 (m,3 Н), 6.98-7.25 (m, 2 Н), 6.68-6.83 (m, 2 Н), 6.52-6.68 (m, 1 Н), 5.34 (s, 1 Н), 4.92-5.18 (m, 1 Н), 3.57-3.67 (m, 1 Н),3.05-3.33 (m, 3 Н), 2.57-2.81 (m, 2 Н), 2.08-2.30 (m, 1 Н), 1.63-1.94 (m, 4 Н); ЖХ-МС (ЭРИ положит. (электрораспылительная ионизация с регистрацией положительных ионов: 337,3 (МН+);[]D = 2,4 (с = 0,5, МеОН). Соединения, указанные в табл.1, получены в результате очистки методом ЖХ-МС, как описано выше для С 2, с использованием в качестве исходного вещества коммерчески доступных производных 2 бромфенилуксусной кислоты и анилинов. Таблица 1 Пример 2 Альтернативное получение (R)-хинуклидин-3-ил-2-фенил-2-(фениламино)ацетата (диастереоизомеры 1 и 2 соединения С 2) К раствору 2-фенил-2-(фениламино)уксусной кислоты (I1) (3,40 г, 14,9 ммоль) в THF (600 мл) добавляют DCC (4,02 г, 19,4 ммоль), HOBt (3,06 г, 19,44 ммоль) и 3(R)-хинуклидинол (3,80 г, 29,9 ммоль). Полученную смесь перемешивают в течение 16 ч при КТ (ЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, остаток переносят в EtOAc, и нерастворимое вещество отфильтровывают. Прозрачный раствор промывают 1 М K2CO3 и затем рассолом, сушат над Na2SO4, фильтруют и упаривают досуха. Полученное неочищенное вещество очищают флэш-хроматографией (DCM/MeOH = 95/5, 0,1%NH3 (водн. с выделением сначала диастереоизомера 1 соединения С 2 в виде белого твердого вещества(1,13 г, выход 22,5%, единственный диастереоизомер) и затем диастереоизомера 2 соединения С 2 в виде белого твердого вещества (0,69 г, выход 13,7%, единственный диастереоизомер). Диастереоизомер 1 соединения С 2 1 Н ЯМР (300 МГц, DMSO-d6) м.д.: 7.48-7.59 (m, 2 Н), 7.26-7.46 (m, 3 Н), 7.02-7.14 (m, 2 Н), 6.67-6.79 Получение (S)-метил-2-(метилсульфонилокси)-2-фенилацетата (I5) В раствор (S)-метил-2-гидрокси-2-фенилацетата (20,0 г, 120 ммоль) в DCM (240 мл), поддерживаемый при 0 С в потоке N2, добавляют мезилхлорид (11,2 мл, 144 ммоль) и TEA (триэтиламин) (20,1 мл,144 ммоль), и эту смесь перемешивают при КТ в течение 1 ч (УЭЖХ-МС (ультраэффективная жидкостная хроматография/масс-спектрометрия) мониторинг: полная конверсия). Смесь охлаждают до 0 С, добавляют 0,1 н. HCl (240 мл), и целевое соединение экстрагируют DCM (2200 мл). Объединенные DCMфазы сушат над Na2SO4, фильтруют и упаривают досуха. Полученное неочищенное вещество очищают фильтрованием через слой диоксида кремния, элюируя DCM, с получением 22,4 г указанного в заголовке соединения в виде белого твердого вещества (выход 76%). Получение (R)-метил-2-фенил-2-(фениламино)ацетата (I6) Анилин (16,8 мл, 184 ммоль) добавляют к раствору (S)-метил-2-(метилсульфонилокси)-2-фенилацетата (I5) (22,4 г, 92,0 ммоль) в CH3CN (50 мл). Эту смесь нагревают под действием микроволнового излучения при 120 С в течение 5 мин (УЭЖХ-МС мониторинг: полная конверсия). Полученную неочищенную реакционную смесь распределяют между EtOAc (200 мл) и 1 н. HCl (200 мл), и водную фазу экстрагируют EtOAc (3200 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и упаривают под вакуумом с получением 20,3 г бледно-желтого твердого вещества (выход 92%), которое используют на следующей стадии без дополнительной очистки. Получение (R)-2-фенил-2-(фениламино)уксусной кислоты (I7) 12 н. HCl (85 мл) добавляют к раствору (R)-метил-2-фенил-2-(фениламино)ацетата (I6) (20,3 г, 84,0 ммоль) в диоксане (85 мл), и эту смесь нагревают при 70 С в течение 18 ч (УЭЖХ-МС мониторинг: полная конверсия). Диоксан выпаривают, смесь охлаждают до 0 С, и полученное твердое вещество собирают фильтрованием с получением 19,1 г промежуточного соединения I7 в виде белого твердого вещества(140 мл) в инертной атмосфере. Добавляют 3S-хинуклидинол (4,0 г, 31,6 ммоль), трифенилфосфин (8,90 г, 43,0 ммоль) и диэтилазодикарбоксилат (6,8 мл, 43,0 ммоль), и полученную смесь кипятят с обратным холодильником в течение 30 мин (УЭЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, и остаток распределяют между водой (100 мл) и EtOAc (100 мл). Водную фазу дополнительно экстрагируют EtOAc (3100 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и упаривают под вакуумом. Полученное неочищенное вещество очищают флэш-хроматографией (DCM/MeOH = 95/5), выделяя 8,7 г диастереоизомера 1 соединения С 2 в виде бледно-желтого твердого вещества (выход 90%, единственный диастереоизомер). Диастереоизомер 1 соединения С 2 1 Н ЯМР (300 МГц, DMSO-d6) м.д.: 7.49-7.58 (m, 2 Н), 7.25-7.47 (m, 3 Н), 7.00-7.12 (m, 2 Н), 6.66-6.78 Схема 4 Получение этил-2-(3-фторфенил)ацетата (I8) В раствор 3-фторфенилуксусной кислоты (10,0 г, 64,9 ммоль) в этаноле (300 мл) добавляют каталитическое количество H2SO4 (98%, 1 мл), и эту смесь подвергают взаимодействию в течение 12 ч при 100 С (ЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, и остаток распределяют между EtOAc и водой. Органический слой отделяют и сушат над Na2SO4 с получением 10,1 г соединения(I8) в виде белого твердого вещества (выход 85%). Получение этил-2-бром-2-(3-фторфенил)ацетата (I9) В раствор этил-2-(3-фторфенил)ацетата (I8) (10,1 г, 55,0 ммоль) в CCl4 (250 мл) добавляют NBS (Nбромсукцинимид) (9,70 г, 55,0 ммоль), и эту смесь кипятят с обратным холодильником в течение 2 ч при 80 С (ЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, и полученный неочищенный остаток растворяют в DCM и очищают фильтрованием через слой диоксида кремния с получением 7,80 г соединения (I9) в виде бесцветного масла (выход 54%). Получение этил-2-(3-фторфенил)-2-(3-фторфениламино)ацетата (I10) Раствор этил-2-бром-2-(3-фторфенил)ацетата (19) (1,80 г, 6,84 ммоль) в 3-фторанилине (6,50 мл,68,0 ммоль) нагревают при 100 С в течение 5 мин под действием микроволнового излучения (ЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, и полученный неочищенный остаток очищают флэш-хроматографией (гексан/EtOAc = 9/1) с получением 2,00 г соединения 110 в виде желтого масла (выход 99%). Получение 2-(3-фторфенил)-2-(3-фторфениламино)уксусной кислоты (I11) В раствор этил-2-(3-фторфенил)-2-(3-фторфениламино)ацетата (2,00 г, 6,81 ммоль) в THF/H2O (1:1 смесь, 10 мл) добавляют LiOH (816 мг, 34,0 ммоль), и эту смесь подвергают взаимодействию в течение 2 ч при КТ (ЖХ-МС мониторинг: полная конверсия). Добавляют 1 н. HCl (15 мл), и целевое соединение экстрагируют EtOAc. Полученное неочищенное вещество очищают флэш-хроматографией (гексан/EtOAc = 1/1) с получением 1,79 г соединения I11 в виде бледно-желтого твердого вещества (выход 99%). Получение (R)-хинуклидин-3-ил-2-(3-фторфенил)-2-(3-фторфениламино)ацетата (диастереоизомеры 1 и 2 соединения С 12) В раствор 2-(3-фторфенил)-2-(3-фторфениламино)уксусной кислоты (I11) (1,79 г, 6,80 ммоль) в диоксане (70 мл) добавляют 4 М раствор HCl в диоксане (5 мл). Эту реакционную смесь перемешивают при КТ в течение 1 ч, затем растворитель выпаривают при пониженном давлении с получением белого твердого вещества. Это вещество растворяют в сухом THF (70 мл) и добавляют DCC (1,71 г, 8,20 ммоль),HOBt (1,10 г, 8,20 ммоль) и 3(R)-хинуклидинол (2,60 г, 20,4 ммоль). Реакционную смесь перемешивают при КТ в течение 96 ч в потоке азота (ЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, и остаток распределяют между EtOAc и водой. Органический слой отделяют, сушат над Na2SO4,фильтруют и упаривают досуха. Неочищенный остаток очищают флэш-хроматографией (DCM/MeOH = 97/3), выделяя сначала 150 мг диастереоизомера 1 соединения С 51 в виде бледно-желтого твердого вещества (выход 6%, единственный диастереоизомер) и затем 830 мг смеси диастереоизомеров 1 и 2 соединения С 12 в виде бледно-желтого масла (выход 32%, смесь диастереоизомеров). Диастереоизомер 1 соединения С 12 (80 мг) дополнительно очищают препаративной ЖХ-МС с получением 53,0 мг бледно-желтого масла (TFA соль). Диастереоизомер 1 соединения С 12 1 Н ЯМР (300 МГц, DMSO-d6) м.д.: 9.48 (br. s., 1 Н), 7.31-7.58 (m, 3 Н), 7.15-7.26 (m, 1 Н), 7.10 (td, 1 Н),6.77 (d, 1 Н), 6.47-6.65 (m, 2 Н), 6.27-6.47 (m, 1 Н), 5.48 (d, 1 Н), 4.95-5.20 (m, 1 Н), 3.63 (ddd, 1 Н), 3.07-3.31 Схема 5 В раствор 2-фенил-2-(фениламино)уксусной кислоты (I1) (234 мг, 0,91 ммоль) в диоксане добавляют 4 М раствор HCl в диоксане (5 мл), и эту реакционную смесь перемешивают при КТ в течение 1 ч. Растворитель выпаривают при пониженном давлении с получением белого твердого вещества, которое растворяют в сухом THF (10 мл). Добавляют DCC (222 мг, 1,12 ммоль), HOBt (144 мг, 1,14 ммоль) и Nметил-4-пиперидинол (307 мг, 2,71 ммоль), и эту смесь перемешивают в течение 96 ч при КТ в потоке азота (ЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, и остаток распределяют между DCM и водой. Органический слой отделяют и сушат над Na2SO4. Неочищенное соединение очищают препаративной ЖХ-МС, и собранные фракции распределяют между 2 М K2CO3 и EtOAc. Органическую фазу сушат над Na2SO4, фильтруют и упаривают досуха с получением 20,6 мг указанного в заголовке соединения в виде белого твердого вещества (выход 7%, рацемическая смесь). 1 Н ЯМР (300 МГц, DMSO-d6) м.д.: 7.45-7.60 (m, 2 Н), 7.20-7.45 (m, 3 Н), 6.95-7.17 (m, 2 Н), 6.69 (d,2 Н), 6.47-6.63 (m, 1 Н), 6.23 (d, 1H), 5.20 (d, 1H), 4.71 (tt, 1 Н), 2.34-2.48 (m, 1 Н), 2.11-2.25 (m, 2 Н), 2.09 (s,3 Н), 1.94-2.08 (m, 1 Н), 1.71-1.90 (m, 1 Н), 1.51-1.71 (m, 2 Н), 1.29-1.51 (m, 1 Н); ЖХ-МС (ЭРИ положит.): 325,2 (МН+). Пример 6 Получение (R)-1-метилпирролидин-4-ил-2-фенил-2-(фениламино)ацетата (С 14)(10 мл) перемешивают при комнатной температуре в течение ночи в потоке азота (ЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, и остаток переносят в водный раствор HCl (рН примерно 2) и промывают DCM. Водную фазу подщелачивают NaHCO3 и экстрагируют DCM (три раза). Органические слои объединяют, сушат над Na2SO4, фильтруют и упаривают досуха. Полученный неочищенный остаток сначала очищают флэш-хроматографией (от DCM до DCM/MeOH = 95/5) и затем препаративной ЖХ-МС. Очищенное вещество распределяют между насыщенным раствором NaHCO3 и DCM, органическую фазу сушат над Na2SO4, фильтруют и упаривают под вакуумом с получением 90,8 мг указанного в заголовке соединения в виде коричневого масла (выход 33%, смесь диастереоизомеров). 1 Н ЯМР (300 МГц, ХЛОРОФОРМ-d) м.д. Диастереоизомер 1 соединения С 14: 7.46-7.57 (m, 2 Н), 7.29-7.45 (m, 3 Н), 7.08-7.21 (m, 2 Н), 6.67-6.81(m, 1 Н), 6.50-6.67 (m, 2 Н), 5.20-5.37 (m, 1 Н), 5.12 (d, 1 Н), 4.84-5.05 (m, 1 Н), 2.46-3.04 (m, 4 Н), 2.33 (s, 3 Н),2.26-2.40 (m, 1 Н), 1.86-2.05 (m, 1 Н); ЖХ-МС (ЭРИ положит.): 311,3 (МН+). Соединения, указанные в табл. 2, получены, как описано выше для диастереоизомеров 1 и 2 соединения С 14, подвергая кислоту I1 реакции сочетания с коммерчески доступными (S)-1-метилпирролидин 3-олом и (R)-1-метилпиперидин-3-олом соответственно. Таблица 2(10 мл) нагревают под действием микроволнового излучения при 100 С в течение 1 ч (ЖХ-МС мониторинг: полная конверсия). Реакционную смесь концентрируют при пониженном давлении, и остаток распределяют между EtOAc и водой. Органическую фазу сушат над Na2SO4, фильтруют и упаривают досуха. Полученный неочищенный остаток очищают препаративной ВЭЖХ. Собранное вещество распределяют между 1 н. NaHCO3 и DCM, органическую фазу отделяют, сушат над Na2SO4, фильтруют и упаривают под вакуумом с получением 20,1 мг указанного в заголовке соединения в виде бесцветного масла(выход 7%, смесь диастереоизомеров). 1 Н ЯМР (300 МГц, ХЛОРОФОРМ-d) м.д.: 7.45-7.57 (m, 2 Н), 7.29-7.45 (m, 3 Н), 7.04-7.21 (m, 2 Н),6.65-6.77 (m, 1 Н), 6.51-6.65 (m, 2 Н), 5.02-5.13 (m, 2 Н), 5.00 (d, 1 Н), 3.07-3.30 (m, 1 Н), 2.81-3.07 (m, 1 Н),2.28 (s, 3 Н), 2.19-2.27 (m, 1 Н), 1.84-2.18 (m, 3 Н), 1.70-1.82 (m, 2 Н), 1.45 (d, 1 Н), 1.17 (ddd, 1 Н); ЖХ-МС (ЭРИ положит.): 351,3 (МН+). Соединение С 18, указанное в табл. 3, получено, как описано выше для соединения С 17. Таблица 3THF (70 мл) добавляют DCC (1,03 г, 4,91 ммоль), HOBt (660 мг, 4,90 ммоль) и 3(R)-хинуклидинол (1,18 г,9,48 ммоль), и полученную смесь перемешивают в течение 12 ч при КТ в потоке азота (ЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, и остаток распределяют между EtOAc (100 мл) и 2 МK2CO3 (50 мл). Органический слой отделяют, промывают рассолом и сушат над Na2SO4. Полученное неочищенное вещество очищают флэш-хроматографией (DCM/MeOH = 95/5, 0,1% NH3 (водн. с получением 1,12 г соединения I19 (выход 80%). Получение дитрифторацетата (S)-(R)-хинуклидин-3-ил)-2-амино-3-фенилпропаноата (диастереоизомер 1 соединения С 20) В раствор соединения I19 (220 мг, 0,58 ммоль) в DCM (5 мл) добавляют трифторуксусную кислоту(0,50 мл, 4,41 ммоль), и эту смесь перемешивают при КТ в течение ночи. Растворитель выпаривают, и полученный неочищенный остаток очищают препаративной ЖХ-МС с получением 59,4 мг диастереоизомера 1 соединения С 20 в виде коричневого смолистого твердого вещества (выход 56%, соль дитрифторацетат, единственный диастереоизомер). 1 Н ЯМР (300 МГц, DMSO-d6) м.д.: 9.81 (br. s., 1 Н), 8.52 (br. s., 3 Н), 7.24-7.41 (m, 5H), 5.02-5.09 (m,1 Н), 4.20-4.46 (m, 1 Н), 3.60-3.73 (m, 1 Н), 3.00-3.33 (m, 7 Н), 2.01 (br. s., 1 Н), 1.79-1.95 (m, 1 Н), 1.76 (br. s.,1H), 1.60 (br. s., 2H);- 26020974 ЖХ-МС (ЭРИ положит.): 275,3 (МН+). Диастереоизомер 2 соединения С 20, указанный в табл. 4, получен, как описано ниже для диастереоизомера 1 соединения С 20, с использованием в качестве исходного вещества коммерчески доступной Схема 9 Получение 3-фенил-2-(фениламино)пропионовой кислоты (I21) В раствор (S)-2-амино-3-фенилпропионовой кислоты (1,52 г, 9,12 ммоль) в смеси DMF/Н 2 О (10/1, 16,5 мл) в потоке азота добавляют йодбензол (1,00 мл, 9,12 ммоль), дихлорбис(три-о-толилфосфин)палладий(II)(286 мг, 1,51 ммоль) и триэтиламин (TEA) (2,50 мл, 18,0 ммоль), и эту смесь перемешивают в течение 24 ч при 100 С в потоке азота (ЖХ-МС мониторинг: полная конверсия). Реакционную смесь распределяют междуEtOAc и водой. Добавляют 1 н. HCl до рН 1, и органический слой отделяют, промывают рассолом и сушат над Na2SO4. Полученное неочищенное вещество очищают флэш-хроматографией (DCM/MeOH = 99/1) с получением 1,21 г соединения I21 (выход 56%). Получение (R)-1-аза-бицикло[2.2.2]окт-3-илового эфира (S)-3-фенил-2-фениламинопропионовой кислоты (диастереоизомер 1 соединения С 22) В раствор 3-фенил-2-(фениламино)пропионовой кислоты (I21) (1,21 г, 5,01 ммоль) в диоксане добавляют 4 М раствор HCl в диоксане (5 мл), и эту реакционную смесь перемешивают при КТ в течение 1 ч. Растворитель выпаривают при пониженном давлении с получением белого твердого вещества, которое растворяют в сухом THF (70 мл). К полученному раствору добавляют DCC (1,22 г, 6,01 ммоль), HOBt(0,8 г, 6,0 ммоль) и 3(R)-хинуклидинол (1,31 г, 10,1 ммоль), и эту смесь перемешивают в течение 24 ч при КТ в потоке азота (ЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают. К полученному неочищенному остатку добавляют 1 н. HCl (20 мл), и водный слой промывают EtOAc (250 мл). NaHCO3 добавляют к водному слою (рН 7-8), и продукт экстрагируют DCM и сушат над Na2SO4 с получением 530 мг (35%-й выход) диастереоизомера 1 соединения С 22, который дополнительно очищают препаративной ЖХ-МС с получением указанного в заголовке продукта в виде коричневого масла (смесь диастереоизомеров, которая содержит 20% диастереоизомера 2 соединения С 22, TFA-соль). 1 Н ЯМР (300 МГц, DMSO-d6) м.д.: 9.58 (br. s., 1H), 7.19-7.38 (m, 5H), 6.91-7.15 (m, 2 Н), 6.55-6.68 (m,3 Н), 4.94 (ddd, 1H), 4.34 (t, 1H), 3.62 (ddd, 1 Н), 2.92-3.25 (m, 7 Н), 1.63-1.95 (m, 3 Н), 1.36-1.63 (m, 2 Н); ЖХ-МС (ЭРИ положит.): 351,3 (МН+). Диастереоизомер 2 соединения С 22, указанный в табл.5, получен, как описано ниже для диастереоизомера 1 соединения С 22, с использованием в качестве исходного вещества коммерчески доступной Схема 10 Получение 2-(бензиламино)-2-фенилуксусной кислоты (I23) Раствор метил-2-(бензиламино)-2-фенилацетата (1,00 г, 3,90 ммоль) в THF (90 мл) и 1 н. NaOH (10 мл) перемешивают при комнатной температуре в течение ночи (ЖХ-МС мониторинг: полная конверсия). Растворитель удаляют при пониженном давлении, и неочищенный остаток распределяют между EtOAc и водой. Водную фазу подкисляют концентрированной HCl (рН примерно 3) и затем экстрагируют EtOAc(три раза). Органическую фазу сушат над Na2SO4, фильтруют и упаривают досуха с получением указанного в заголовке соединения в виде белого твердого вещества (940 мг, количественный выход), которое используют на следующей стадии без дополнительной очистки. Получение (R)-хинуклидин-3-ил-2-(бензиламино)-2-фенилацетата (диастереоизомеры 1 и 2 соединения С 24) Смесь 2-(бензиламино)-2-фенилуксусной кислоты (I23) (0,94 г, 3,90 ммоль), DCC (0,97 г, 4,70 ммоль), HOBt (0,63 г, 4,07 ммоль) и 3(R)-хинуклидинола (1,51 г, 11,7 ммоль) в сухом THF (30 мл) перемешивают при комнатной температуре в течение ночи в потоке азота (ЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, и остаток переносят в EtOAc и промывают дважды водой. Органическую фазу сушат над Na2SO4, фильтруют и упаривают досуха. Полученное неочищенное вещество очищают флэш-хроматографией (DCM/MeOH = от 98/2, 0,2% NH3(водн.) до 95/5, 0,5% NH3(водн. с получением смеси диастереоизомеров 1 и 2 соединения С 24 (231 мг, выход 52%, смесь диастереоизомеров). Смесь диастереоизомеров 1 и 2 соединения С 24 (55 мг) дополнительно очищают препаративной ВЭЖХ. 1 Н ЯМР (300 МГц, DMSO-d6 +Na2CO3) м.д.: 7.20-7.45 (m, 11 Н), 4.55-4.79 (m, 1H), 4.35 (s, 1H), 3.67 Схема 11 Получение дигидрохлорида (R)-хинуклидин-3-ил-2-амино-2-фенилацетата (С 25) Смесь 2-(трет-бутоксикарбониламино)-2-фенилуксусной кислоты (0,90 г, 4,18 ммоль), HOBt (0,68 г,12,5 ммоль), DCC (1,29 г, 6,27 ммоль) и 3(R)-хинуклидинола (1,59 г, 12,5 ммоль) в сухом THF (50 мл) перемешивают при комнатной температуре в течение 16 ч (ЖХ-МС мониторинг: полная конверсия). Растворитель удаляют при пониженном давлении, остаток переносят в EtOAc и промывают дважды 2 МK2CO3. Органическую фазу сушат над Na2SO4, фильтруют и упаривают досуха. Соединение растирают с гексаном с получением (R)-хинуклидин-3-ил-2-(трет-бутоксикарбониламино)-2-фенилацетата в виде белого твердого вещества (1,46 г; выход 97%, смесь диастереоизомеров). Это соединение растворяют вDCM (50 мл) и добавляют 4 н. HCl в диоксане (5 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 ч (ЖХ-МС мониторинг: полная конверсия) и затем растворитель отделяют. Смолистое твердое вещество растирают с гексаном с получением соединения С 25 в виде белого твердого вещества (1,26 г, выход 94%, дигидрохлорид, смесь диастереоизомеров). Получение дитрифторацетата (R)-хинуклидин-3-ил-2-(циклопентиламино)-2-фенилацетата (С 26)(10 мл) и обрабатывают триацетоксиборгидридом натрия (508 мг, 2,40 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 30 мин, затем добавляют циклопентанон (106 мкл, 1,20 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение еще 16 ч (ЖХ-МС мониторинг: полная конверсия). Реакционную смесь разбавляют DCM и промывают дважды 1 н. NaOH,органический слой сушат над Na2SO4, фильтруют и упаривают досуха. Неочищенное соединение очищают препаративной ВЭЖХ с получением соединения С 26 в виде белого твердого вещества (53,1 мг,- 28020974(m, 1 Н), 1.78-2.14 (m, 4 Н), 1.61-1.77 (m, 5 Н), 1.31-1.57 (m, 3 Н); ЖХ-МС (ЭРИ положит.): 329,2 (МН+). Соединение С 27, указанное в табл. 6, получено, как описано выше для С 26, с использованием циклогексанона вместо циклопентанона. Таблица 6 Схема 12 Получение (4-хлорфениламино)фенилуксусной кислоты (I28) В раствор -бромфенилуксусной кислоты (1,00 г, 4,65 ммоль) в ацетонитриле (20 мл) добавляют 4 хлорфениламин (1,18 г, 9,30 ммоль), и эту смесь подвергают взаимодействию в закрытом сосуде под действием микроволнового излучения при 100 С в течение 1 ч (УЭЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, и остаток распределяют между EtOAc и 1 н. HCl. Органическую фазу сушат над Na2SO4, фильтруют и упаривают досуха с получением промежуточного соединения I76 в виде желтого твердого вещества (0,57 г; выход 47%). Получение (R)-1-аза-бицикло[2.2.2]окт-3-илового эфира (4-хлорфениламино)фенилуксусной кислоты (С 29) В раствор (4-хлорфениламино)фенилуксусной кислоты (I28) (574 мг, 2,20 ммоль) в сухом THF (20 мл) добавляют DCC (543 мг, 2,64 ммоль), HOBt (359 мг, 2,64 ммоль) и 3(R)-хинуклидинол (558 мг, 4,40 ммоль). Полученную смесь перемешивают при КТ в течение ночи (УЭЖХ-МС мониторинг: полная конверсия). Растворитель выпаривают, и остаток распределяют между EtOAc и 2 М K2CO3. Органическую фазу сушат над Na2SO4, фильтруют и упаривают досуха. Полученный неочищенный остаток очищают флэш-хроматографией (DCM/MeOH = от 99/1 до 85/15) с получением указанного в заголовке соединения в виде белого твердого вещества (306 мг, выход 37%, смесь диастереоизомеров). 1 Н ЯМР (300 МГц, DMSO-d6) м.д.: 7.46-7.59 (m, 2 Н), 7.22-7.45 (m, 3 Н), 6.99-7.18 (m, 2 Н), 6.65-6.80(m, 2 Н), 6.52 (d, 1 Н), 5.28 (d, 1 Н), 4.60-4.82 (m, 1 Н), 3.04 (ddd, 1 Н), 2.54-2.70 (m, 4 Н), 2.03-2.36 (m, 1 Н),1.67-1.97 (m, 1 Н), 1.36-1.68 (m, 2 Н), 1.05-1.35 (m, 2 Н); ЖХ-МС (ЭРИ положит.): 371,1 (МН+). Соединения, указанные в табл. 7, получены, как описано выше для С 29, с использованием в качестве исходного вещества подходящих коммерчески доступных производных 2-бромфенилуксусной кислоты и анилинов.
МПК / Метки
МПК: C07D 207/12, C07D 453/02, C07D 451/10, A61P 11/08, A61K 31/4465, A61K 31/401, C07D 211/42, A61K 31/439, C07D 211/46
Метки: алкалоидов, производные, аминоэфирные, композиции, медицинские
Код ссылки
<a href="https://eas.patents.su/30-20974-aminoefirnye-proizvodnye-alkaloidov-i-ih-medicinskie-kompozicii.html" rel="bookmark" title="База патентов Евразийского Союза">Аминоэфирные производные алкалоидов и их медицинские композиции</a>
Производные хинуклидина карбоната и их медицинские композиции

Номер патента: 19166
Опубликовано: 30.01.2014
Авторы: Амари Габриеле, Дельканале Маурицио
МПК: A61P 11/00, C07D 453/02, A61K 31/439...
Метки: производные, медицинские, карбоната, композиции, хинуклидина
Формула / Реферат:
1. Соединение общей формулы (I)где А может представлять собой арил, или гетероарил, или арилалкил, или гетероарилалкил либо группу формулы (а)где R3 и R4 являются одинаковыми или разными и могут быть независимо выбраны из группы, состоящей из Н, (С3-С8)циклоалкила, арила или гетероарила, где указанные арил или гетероарил могут быть, возможно, замещены атомом галогена или одним или несколькими заместителями, независимо выбранными из групп ОН, SH,...
Производные 2н-пиридазин-3-она, фармацевтические композиции, содержащие эти производные, и способ получения активного ингредиента

Номер патента: 6246
Опубликовано: 27.10.2005
Авторы: Рацне Байногель Юдит, Баркоци Йожеф, Котаи Надь Петер, Леваи Дьёрди, Сенаши Габор, Вельман Янош, Шмидт Эва, Шимиг Дьюла, Миклошне Ковач Анико, Паллаги Каталин, Гачальи Иштван, Эдьед Андраш
МПК: A61K 31/50, C07D 413/14
Метки: ингредиента, эти, содержащие, активного, получения, производные, способ, фармацевтические, композиции, 2н-пиридазин-3-она
Формула / Реферат:
1. Производное 2H-пиридазин-3-она формулы где R означает атом водорода или C1-4-алкильную группу, X и Y независимо представляют собой атом водорода, атом галогена или группу формулы при условии, что один из X и Y всегда представляет собой группу формулы II, а другой радикал представляет собой атом водорода или атом галогена, где в формуле II n имеет значение 1 или 2, и его фармацевтически приемлемые соли присоединения кислот. 2. Производное...
Производные 2,3-бензодиазепина и фармацевтические композиции, содержащие эти производные в качестве активного ингредиента

Номер патента: 5867
Опубликовано: 30.06.2005
Авторы: Гиглер Габор, Баркоци Йожеф, Леваи Дьердь, Грефф Зольтан, Линг Иштван, Сенаши Габор, Раткаи Зольтан, Вег Миклош, Сабо Геза, Шимиг Дьюла, Мартонне Марко Бернадетт, Харшинг Ласло Габор
МПК: A61K 31/551, A61P 25/00, C07D 243/02...
Метки: композиции, эти, фармацевтические, 2,3-бензодиазепина, производные, содержащие, ингредиента, активного, качестве
Формула / Реферат:
1. Производное 2,3-бензодиазепина формулы I где X - водород, хлор или метоксигруппа, Y - водород или галоген, Z - метил или хлор, R - C1-4 алкил или группа формулы -NR1R2, где R1 и R2, независимо, представляют собой водород, C1-4 алкил, C1-4 алкоксил или C3-6 циклоалкил, и его фармацевтически приемлемые соли с кислотами. 2. Производное 2,3-бензодиазепина по п.1, где X - хлор, Y - водород, хлор или бром, R - C1-4 алкил, Z - определен в п.1, и...
Способы получения алкалоидов хиназолина

Номер патента: 9047
Опубликовано: 26.10.2007
Авторы: Хоффман Ханс-Райнер, Матуш Рудольф, Моорманн Йохим
МПК: C07D 487/04
Метки: получения, алкалоидов, способы, хиназолина
Формула / Реферат:
1. Способ получения соединения, имеющего следующую формулу (I) путем преобразования соединения, имеющего формулу (II), при помощи 2-пирролидона, отличающийся тем, что используется избыточное количество 2-пирролидона относительно соединения (II). 2. Способ по п.1, отличающийся тем, что на последующей стадии продукт (I) реакции выделяют непосредственно из реакционной смеси методом кристаллизации. 3. Способ по п.1 или 2, отличающийся тем, что...
Композиция на основе алкалоидов эбурнаменинового типа, стимулирующая церебральную активность

Номер патента: 189
Опубликовано: 24.12.1998
Авторы: Кальво Ласаро Элена, Кальво Мондело Фернандо, Ласаро Флорес Консуэло, Манреса Ферреро Мария Тереса, Кальво Ласаро Паула
МПК: A61K 31/52
Метки: активность, основе, алкалоидов, эбурнаменинового, композиция, стимулирующая, типа, церебральную
Формула / Реферат:
1. Композиция для перорального использования, которая стимулирует активность мозга, отличающаяся тем, что она содержит смесь, состоящую из а) алкалоидов эбурнаменинового типа общей формулы I: где R обозначает Н или -ОН; или между С-14 и С-15 имеется двойная связь, R' обозначает -ОН или CO2R", a R" содержит от 1 до 6 атомов углерода, R'" обозначает Н, галоген (такой как бром или хлор) или нитрогруппу, имеющих природное происхождение и...
Предыдущий патент: Кристаллическая форма трипептидного кетоэпоксидного соединения и способ его получения
Следующий патент: Система и способ для изготовления самоохлаждающегося контейнера
Случайный патент: Препараты для перорального применения с непрерывным (постоянным) высвобождением при приёме фармацевтически активного ингредиента (api) вместе с пищей