Способ получения соединений, применимых в качестве ингибиторов натрийзависимого переносчика глюкозы
Номер патента: 20209
Опубликовано: 30.09.2014
Авторы: Мехрман Стивен, Хонгу Мицуя, Чисхолм Морин, Уэллс Кеннет М., Абдель-Магид Ахмед Ф., Скотт Лоррейн, Чжан-Пласкет Фань, Номура Сумихиро, Кога Юити




























Формула / Реферат
1. Способ получения соединения формулы (I)

где кольцо А представляет собой бензольное кольцо, необязательно замещенное заместителями, выбранными из группы, состоящей из атома галогена, низшей алкильной группы, галогензамещенной низшей алкильной группы, низшей алкоксильной группы и фенильной группы;
кольцо В представляет собой гетероциклическое кольцо, выбранной из группы, содержащей тиофен, фуран, бензофуран, бензотиофен и бензотиазол, где гетероциклическое кольцо необязательно замещено заместителем, выбранным из группы, содержщей атом галогена, цианогруппу, низшую алкильную группу, галогензамещенную низшую алкильную группу, фенилзамещенную низшую алкильную группу, низшую алкоксильную группу, галогензамещенную низшую алкоксильную группу, фенильную группу, галогензамещенную фенильную группу, низшую алкилфенильную группу, низшую алкоксифенильную группу, тиенильную группу, галогензамещенную тиенильную группу, пиридильную группу, галопиридильную группу и тиазолильную группу;
X - атом углерода;
Y - группа -(СН2)n-; где n равно 1 или 2;
при условии, что в кольце А X участвует в ненасыщенной связи;
или его фармацевтически приемлемой соли,
включающий

взаимодействие соединения формулы (V) с соединением формулы (VI-S) в присутствии алкиллития, где соединение алкиллития выбрано из группы, состоящей из триметилсилилметиллития, 2,4,6-триметилфениллития и триэтилсилилметиллития, в органическом растворителе при температуре от приблизительно 0 до приблизительно -78°С с получением соответствующего соединения формулы (VII);
и где алкиллитий добавляют в смесь соединения формулы (V) и соединения формулы (VI-S)

взаимодействие соединения формулы (VII) с BF3OEt2 в присутствии триалкилсилана в органическом растворителе с образованием соответствующего соединения формулы (VIII)

взаимодействие соединения формулы (VIII) с уксусным ангидридом или ацетилхлоридом в присутствии органического основания, в чистом виде или в органическом растворителе, с образованием соответствующего соединения формулы (IX);

и снятие защитных групп в соединении формулы (IX) с образованием соответствующего соединения формулы (I).
2. Способ по п.1, где соединение формулы (VI-S) присутствует в количестве от приблизительно 1,0 до приблизительно 1,25 мол.экв. в расчете на соединение формулы (V).
3. Способ по п.1, где алкиллитий представляет собой (триметилсилил)метиллитий и где алкиллитий присутствует в количестве от приблизительно 2,0 до приблизительно 2,5 мол.экв. в расчете на соединение формулы (V).
4. Способ по п.1, где BF3OEt2 присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (VII) и где триалкилсилан представляет собой Et3SiH, который присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (V).
5. Способ по п.4, где молярное соотношение BF3OEt2 : Et3SiH составляет приблизительно 1:1.
6. Способ по п.1, где соединение формулы (VIII) вводят в реакцию с уксусным ангидридом, который присутствует в количестве от приблизительно 4,5 до приблизительно 5,0 мол.экв. в расчете на соединение формулы (VIII).
7. Способ по п.1, где органическим основанием является N-метилморфолин (NMM).
8. Способ по п.1, где соединение формулы (VIII) вводят в реакцию с уксусным ангидридом в присутствии каталитического количества 4-(N,N-диметиламино)пиридина (DMAP).
9. Способ по п.1, где из соединения формулы (IX) удаляют защитные группы посредством взаимодействия с основанием.
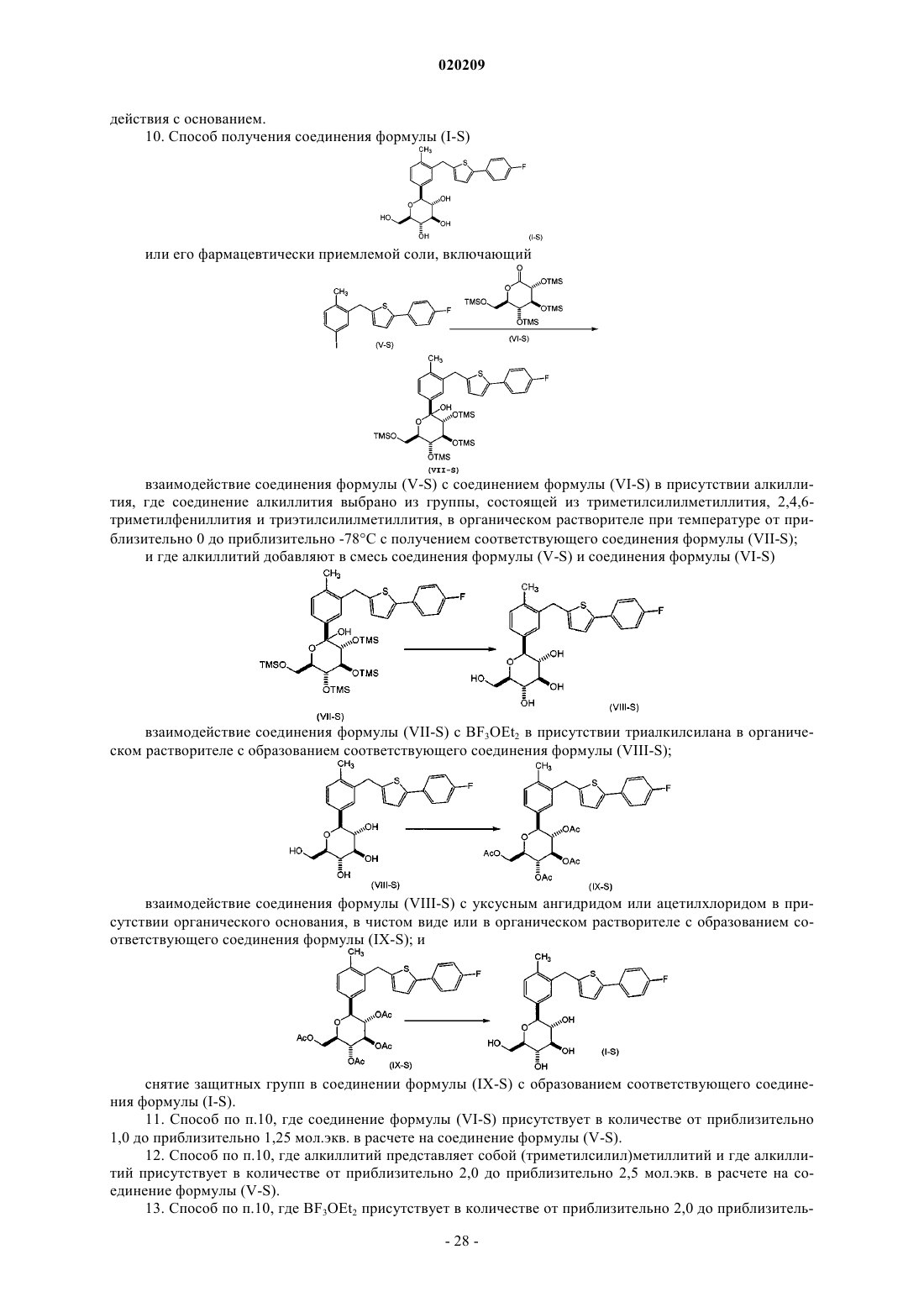
10. Способ получения соединения формулы (I-S)

или его фармацевтически приемлемой соли, включающий

взаимодействие соединения формулы (V-S) с соединением формулы (VI-S) в присутствии алкиллития, где соединение алкиллития выбрано из группы, состоящей из триметилсилилметиллития, 2,4,6-триметилфениллития и триэтилсилилметиллития, в органическом растворителе при температуре от приблизительно 0 до приблизительно -78°С с получением соответствующего соединения формулы (VII-S);
и где алкиллитий добавляют в смесь соединения формулы (V-S) и соединения формулы (VI-S)

взаимодействие соединения формулы (VII-S) с BF3OEt2 в присутствии триалкилсилана в органическом растворителе с образованием соответствующего соединения формулы (VIII-S)

взаимодействие соединения формулы (VIII-S) с уксусным ангидридом или ацетилхлоридом в присутствии органического основания, в чистом виде или в органическом растворителе с образованием соответствующего соединения формулы (IX-S);

и снятие защитных групп в соединении формулы (IX-S) с образованием соответствующего соединения формулы (I-S).
11. Способ по п.10, где соединение формулы (VI-S) присутствует в количестве от приблизительно 1,0 до приблизительно 1,25 мол.экв. в расчете на соединение формулы (V-S).
12. Способ по п.10, где алкиллитий представляет собой (триметилсилил)метиллитий и где алкиллитий присутствует в количестве от приблизительно 2,0 до приблизительно 2,5 мол.экв. в расчете на соединение формулы (V-S).
13. Способ по п.10, где BF3OEt2 присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (VII-S) и где триалкилсилан представляет собой Et3SiH, который присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (VII-S).
14. Способ по п.13, где молярное соотношение BF3OEt2 : Et3SiH составляет 1:1.
15. Способ по п.10, где соединение формулы (VIII-S) вводят в реакцию с уксусным ангидридом, который присутствует в количестве от приблизительно 4,5 до приблизительно 5,0 мол.экв. в расчете на соединение формулы (VIII-S).
16. Способ по п.10, где органическим основанием является NMM.
17. Способ по п.10, где соединение формулы (VIII-S) вводят в реакцию с уксусным ангидридом в присутствии каталитического количества DMAP.
18. Способ по п.10, где соединение формулы (IX-S) дополнительно суспендируют в метаноле и фильтруют.
19. Способ по п.10, где из соединения формулы (IX-S) удаляют защитные группы посредством взаимодействия с основанием.
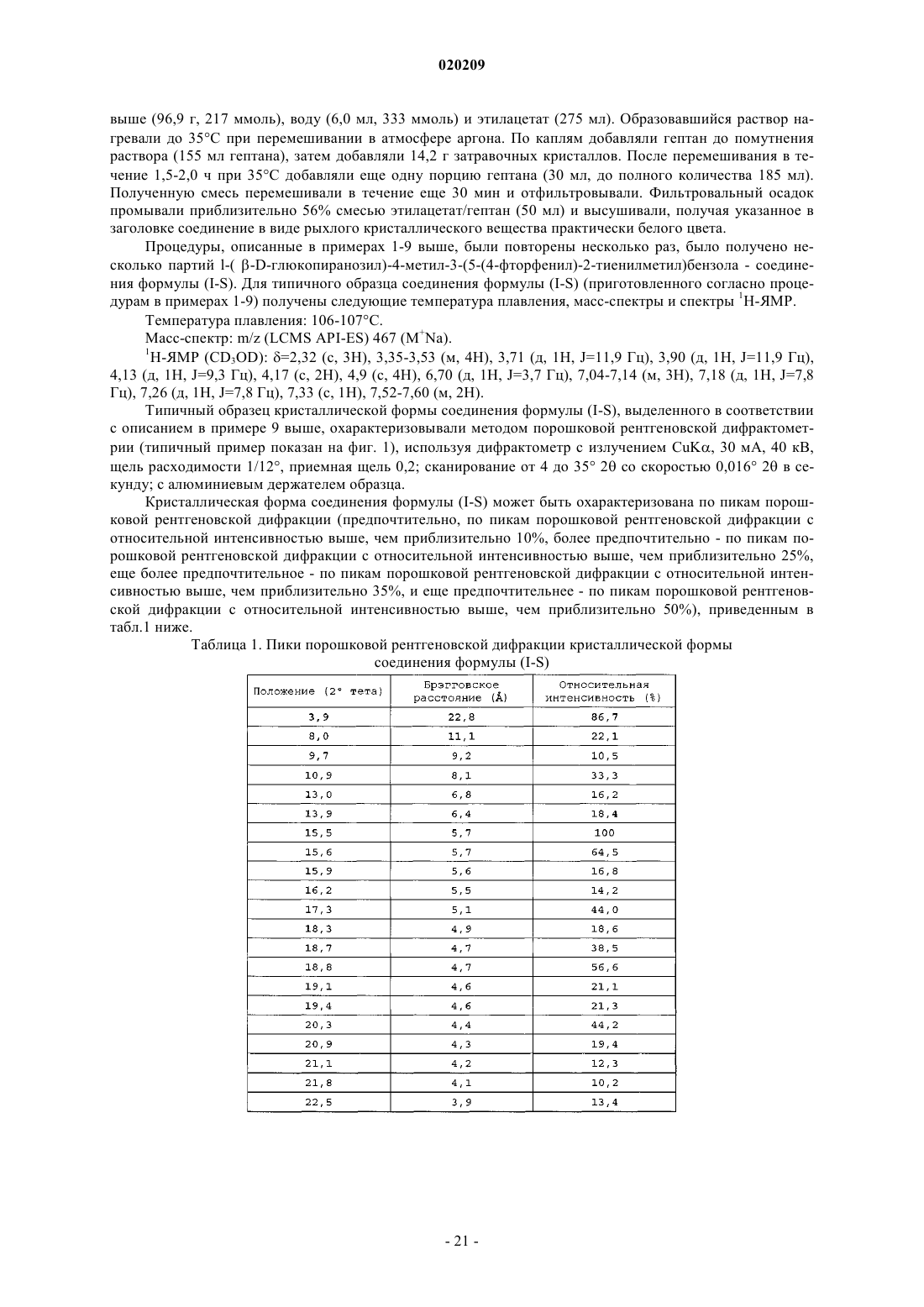
20. Способ перекристаллизации соединения формулы (I-S)

включающий
(a) растворение соединения формулы (I-S) в органическом растворителе с получением смеси А;
(b) нагревание смеси А до температуры от приблизительно 25 до приблизительно 45°С с образованием смеси В;
(c) добавление приблизительно от 1,0 до 2,0 мол.экв. воды в расчете на соединение формулы (I-S) в смесь В с получением смеси С;
(d) добавление гептана в смесь С с образованием суспензии соединения формулы (I-S); и
(e) выделение соединения формулы (I-S).
21. Способ по п.20, где органический растворитель представляет собой этилацетат.
22. Способ по п.20, где смесь А нагревают до температуры от приблизительно 30 до приблизительно 35°С.
23. Способ по п.20, где приблизительно 1,5 мол.экв. воды в расчете на соединение формулы (I-S) добавляют в смесь В.
24. Способ по п.21, где гептан добавляют в смесь С в количестве, дающем конечное объемное соотношение этилацетат:гептан, равное приблизительно 1,2:1,0.
25. Способ получения соединения формулы (I-K)

или его фармацевтически приемлемой соли;
включающий

взаимодействие соединения формулы (V-K) с соединением формулы (VI-S) в присутствии алкиллития, где соединение алкиллития выбрано из группы, состоящей из триметилсилилметиллития, 2,4,6-триметилфениллития и триэтилсилилметиллития, в органическом растворителе при температуре от приблизительно 0 до приблизительно -78°С с получением соответствующего соединения формулы (VII-K);
и где алкиллитий добавляют в смесь соединения формулы (V-K) и соединения формулы (VI-S);

снятие защитных групп в соединении формулы (VII-K) с образованием соответствующего соединения формулы (Х-K);

взаимодействие соединения формулы (Х-K) с BF3OEt2 в присутствии триалкилсилана в органическом растворителе с образованием соответствующего соединения формулы (VIII-K);

взаимодействие соединения формулы (VIII-K) с уксусным ангидридом или ацетилхлоридом в присутствии органического основания в чистом виде или в органическом растворителе с образованием соответствующего соединения формулы (IX-K); и

снятии защитных групп в соединении формулы (IX-K) с образованием соответствующего соединения формулы (I-K).
26. Способ по п.25, где соединение формулы (VI-S) присутствует в количестве от приблизительно 1,0 до приблизительно 1,25 мол.экв. в расчете на соединение формулы (V-K).
27. Способ по п.25, где алкиллитий представляет собой (триметилсилил)метиллитий и где алкиллитий присутствует в количестве от приблизительно 2,0 до приблизительно 2,5 мол.экв. в расчете на соединение формулы (V-K).
28. Способ по п.25, где BF3OEt2 присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (VII-K) и где триалкилсилан представляет собой Et3SiH, который присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (V-K).
29. Способ по п.28, где молярное соотношение BF3OEt2 :Et3SiH составляет примерно 1:1.
30. Способ по п.25, где соединение формулы (VIII-K) вводят в реакцию с уксусным ангидридом, который присутствует в количестве от приблизительно 4,5 до приблизительно 5,0 мол.экв. в расчете на соединение формулы (VIII-K).
31. Способ по п.25, где органическим основанием является NMM.
32. Способ по п.25, где соединение формулы (VIII-K) вводят в реакцию с уксусным ангидридом в присутствии каталитического количества DMAP.
33. Способ по п.25, где в соединении формулы (IX-K) удаляют защитные группы посредством взаимодействия с основанием.
Текст
СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ, ПРИМЕНИМЫХ В КАЧЕСТВЕ ИНГИБИТОРОВ НАТРИЙЗАВИСИМОГО ПЕРЕНОСЧИКА ГЛЮКОЗЫ Изобретение направлено на новый процесс получения соединений, оказывающих ингибирующее действие на натрийзависимые переносчики глюкозы (SGLT), присутствующие в кишечнике или почках.(BE); МИЦУБИСИ ТАНАБЕ ФАРМА КОРПОРЕЙШН (JP) Перекрестные ссылки на смежные изобретения Данная заявка истребует приоритет, заявленный в предварительной заявке на патент США 60/971067, поданной 10 сентября 2007 года, и в предварительной заявке на патент США 61/018822,поданной 3 января 2008 года, каковые полностью включаются в настоящий документ путем отсылки. Область изобретения Настоящее изобретение относится к новому процессу получения соединений, проявляющих ингибиторную активность в отношении натрий-зависимого переносчика глюкозы (НЗПГ), присутствующего в кишечнике и в почках. Предпосылки изобретения Диетотерапия и лечебная физкультура играют важную роль в лечении сахарного диабета. Если данные виды терапии не позволяют в достаточной мере контролировать состояние пациента, для лечения диабета дополнительно применяется инсулин или вводимый перорально противодиабетический препарат. В настоящее время в качестве противодиабетических препаратов применяются бигуанидные соединения, соединения сульфанил-мочевины, вещества, улучшающие резистентность к инсулину, и ингибиторы -глюкозидазы. Однако данные противодиабетические препараты имеют ряд побочных эффектов. Например, бигуанидные соединения вызывают лактоцидоз, соединения сульфанилмочевины вызывают значительную гипогликемию, препараты, повышающие резистентность к инсулину, приводят к отекам и сердечной недостаточности, а ингибиторы -глюкозидазы вызывают вздутие живота и диарею. Учитывая данные обстоятельства, желательно разработать новые лекарства для лечения сахарного диабета, не дающие подобных побочных эффектов. Недавно были получены данные о том, что с началом и прогрессивным развитием сахарного диабета связана гипергликемия (т.н. теория глюкозной токсичности). А именно, хроническая гипергликемия ведет к снижению секреции инсулина и далее к снижению чувствительности к инсулину; в результате увеличивается концентрация глюкозы в крови, что приводит к дальнейшему развитию сахарного диабета[Diabetologia, vol. 28, p. 119 (1985); Diabetes Care, vol. 13, p. 610 (1990), и т.д.]. Лечение гипергликемии позволяет разрушить данный цикл автоусиления, открывая возможность профилактики и лечения сахарного диабета. Одним из возможных методов лечения гипергликемии является экскретирование избыточной глюкозы непосредственно в мочу с нормализацией концентрации глюкозы в крови. Например, при ингибировании натрий-зависимого переносчика глюкозы, присутствующего в проксимальных извитых почечных канальцах, блокируется обратное поглощение глюкозы в почках, что способствует экскретированию глюкозы в мочу и снижению уровня глюкозы в крови. Надежно показано, что в моделях диабета на животных постоянное подкожное введение флоридзина, обладающего ингибиторной активностью в отношении НЗПГ, приводит к нормализации гипергликемии, и уровень глюкозы в крови животного может поддерживаться нормальным в течение длительного времени, что приводит к улучшению секреции инсулина и резистентности к инсулину [Journal of Clinical Investigation, vol. 79, p. 1510 (1987); там же., vol. 80, p. 1037 (1987); там же., vol. 87, p. 561 (1991), и т.д.]. Кроме того, при длительном лечении диабета ингибиторами НЗПГ в моделях на животных инсулиновый секреторный ответ и чувствительность к инсулину у животных улучшаются без отрицательных побочных эффектов на почки или нарушений ионного баланса крови, в результате удается избежать появления и развития нефропатии и диабетической нефропатии [Journal of Medicinal Chemistry, vol. 42, p. 5311 (1999); British Journal of Pharmacology, vol. 132, p. 578 (2001), Ueta, Ishihara, Matsumoto, Oku, Nawano, Fujita, Saito, Arakawa, Life Sci., в печати (2005), и т.д.]. На основании описанного выше можно ожидать, что ингибиторы НЗПГ могут улучшать секрецию инсулина и резистентность к инсулину, понижая уровень глюкозы в крови у пациентов с диабетом, и в дальнейшем предотвращая появление и развитие сахарного диабета и его осложнений. Краткое описание изобретения Изобретение относится к способу получения соединения формулы (I) где кольцо А представляет собой бензольное кольцо, необязательно замещенное заместителями,выбранными из группы, состоящей из атома галогена, низшей алкильной группы, галогензамещенной низшей алкильной группы, низшей алкоксильной группы и фенильной группы; кольцо В представляет собой гетероциклическое кольцо, выбранной из группы, содержащей тиофен, фуран, бензофуран, бензотиофен и бензотиазол, где гетероциклическое кольцо необязательно замещено заместителем, выбранным из группы, содержщей атом галогена, цианогруппу, низшую алкильную группу, галогензамещенную низшую алкильную группу, фенилзамещенную низшую алкильную группу,низшую алкоксильную группу, галогензамещенную низшую алкоксильную группу, фенильную группу,-1 020209Y - группа -(СН 2)n-; где n равно 1 или 2; при условии, что в кольце А X участвует в ненасыщенной связи; или его фармацевтически приемлемой соли, включающему взаимодействие соединения формулы (V) с соединением формулы (VI-S) в присутствии алкиллития, где соединение алкиллития выбрано из группы, состоящей из триметилсилилметиллития, 2,4,6 триметилфениллития и триэтилсилилметиллития, в органическом растворителе при температуре от приблизительно 0 С до приблизительно -78 С с получением соответствующего соединения формулы (VII); и где алкиллитий добавляют в смесь соединения формулы (V) и соединения формулы (VI-S); взаимодействие соединения формулы (VII) с BF3OEt2 в присутствии триалкилсилана в органическом растворителе с образованием соответствующего соединения формулы (VIII); взаимодействие соединения формулы (VIII) с уксусным ангидридом или ацетилхлоридом в присутствии органического основания, в чистом виде или в органическом растворителе, с образованием соответствующего соединения формулы (IX); и снятие защитных групп в соединении формулы (IX) с образованием соответствующего соединения формулы (I). В предпочтительном варианте предложенного способа соединение формулы (VI-S) присутствует в количестве от приблизительно 1,0 до приблизительно 1,25 мол.экв. в расчете на соединение формулы(V). В предпочтительном варианте предложенного способа алкиллитий представляет собой (триметилсилил)метиллитий, и где алкиллитий присутствует в количестве от приблизительно 2,0 до приблизительно 2,5 мол.экв. в расчете на соединение формулы (V). В предпочтительном варианте предложенного способа BF3OEt2 присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (VII), и где триалкилсилан представляет собой Et3SiH, который присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (V). В предпочтительном варианте предложенного способа молярное соотношение BF3OEt2:Et3SiH составляет приблизительно 1:1. В предпочтительном варианте предложенного способа соединение формулы (VIII) вводят в реакцию с уксусным ангидридом, который присутствует в количестве от приблизительно 4,5 до приблизительно 5,0 мол.экв. в расчете на соединение формулы (VIII). В предпочтительном варианте предложенного способа органическим основанием является Nметилморфолин(NMM). В предпочтительном варианте предложенного способа соединение формулы (VIII) вводят в реакцию с уксусным ангидридом в присутствии каталитического количества 4-(N,N-диметиламино)пиридина(DMAP). В предпочтительном варианте предложенного способа из соединения формулы (IX) удаляют за-2 020209 щитные группы посредством взаимодействия с основанием. Другим объектом настоящего изобретения является способ получения соединения формулы (I-S) или его фармацевтически приемлемой соли, включающий взаимодействие соединения формулы (V-S) с соединением формулы (VI-S) в присутствии алкиллития, где соединение алкиллития выбрано из группы, состоящей из триметилсилилметиллития, 2,4,6 триметилфениллития и триэтилсилилметиллития, в органическом растворителе при температуре от приблизительно 0 С до приблизительно -78 С с получением соответствующего соединения формулы (VIIS); и где алкиллитий добавляют в смесь соединения формулы (V-S) и соединения формулы (VI-S); взаимодействие соединения формулы (VII-S) с BF3OEt2 в присутствии триалкилсилана в органическом растворителе с образованием соответствующего соединения формулы (VIII-S); взаимодействие соединения формулы (VIII-S) с уксусным ангидридом или ацетилхлоридом в присутствии органического основания, в чистом виде или в органическом растворителе, с образованием соответствующего соединения формулы (IX-S); и снятие защитных групп в соединении формулы (IX-S) с образованием соответствующего соединения формулы (I-S). В предпочтительном варианте предложенного способа соединение формулы (VI-S) присутствует в количестве от приблизительно 1,0 до приблизительно 1,25 мол.экв. в расчете на соединение формулы (VS). В предпочтительном варианте предложенного способа алкиллитий представляет собой (триметилсилил)метиллитий, и где алкиллитий присутствует в количестве от приблизительно 2,0 до приблизительно 2,5 мол.экв. в расчете на соединение формулы (V-S). В предпочтительном варианте предложенного способа BF3OEt2 присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (VII-S), и где триалкилсилан представляет собой Et3SiH, который присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (VII-S). В предпочтительном варианте предложенного способа молярное соотношение BF3OEt2 : Et3SiH составляет 1:1. В предпочтительном варианте предложенного способа соединение формулы (VIII-S) вводят в реак-3 020209 цию с уксусным ангидридом, который присутствует в количестве от приблизительно 4,5 до приблизительно 5,0 мол.экв. в расчете на соединение формулы (VIII-S). В предпочтительном варианте предложенного способа органическим основанием является NMM. В предпочтительном варианте предложенного способа соединение формулы (VIII-S) вводят в реакцию с уксусным ангидридом в присутствии каталитического количества DMAP. В предпочтительном варианте предложенного способа соединение формулы (IX-S) дополнительно суспендируют в метаноле и фильтруют. В предпочтительном варианте предложенного способа из соединения формулы (IX-S) удаляют защитные группы посредством взаимодействия с основанием. Ещ одним другим объектом настоящего изобретения является способ перекристаллизации соединения формулы (I-S)(a) растворение соединения формулы (I-S) в органическом растворителе с получением смеси А;(b) нагревание смеси А до температуры от приблизительно 25 до приблизительно 45 С с образованием смеси В;(c) добавление приблизительно от 1,0 до 2,0 мол.экв. воды в расчете на соединение формулы (I-S) в смесь В с получением смеси С;(d) добавление гептана в смесь С с образованием суспензии соединения формулы (I-S); и(e) выделение соединения формулы (I-S). В предпочтительном варианте предложенного способа органический растворитель представляет собой этилацетат. В предпочтительном варианте предложенного способа смесь А нагревают до температуры от приблизительно 30 до приблизительно 35 С. В предпочтительном варианте предложенного способа где приблизительно 1,5 мол.экв. воды в расчете на соединение формулы (I-S) добавляют в смесь В. В предпочтительном варианте предложенного способа гептан добавляют в смесь С в количестве,дающем конечное объемное соотношение этилацетат:гептан, равное приблизительно 1,2:1,0. Настоящее изобретение также относится к способу получения соединения формулы (I-K) или его фармацевтически приемлемой соли, включающему взаимодействие соединения формулы (V-K) с соединением формулы (VI-S) в присутствии алкиллития, где соединение алкиллития выбрано из группы, состоящей из триметилсилилметиллития, 2,4,6 триметилфениллития и триэтилсилилметиллития, в органическом растворителе при температуре от приблизительно 0 до приблизительно -78 С с получением соответствующего соединения формулы (VII-K); и где алкиллитий добавляют в смесь соединения формулы (V-K) и соединения формулы (VI-S); снятие защитных групп в соединении формулы (VII-K) с образованием соответствующего соедине-4 020209 взаимодействие соединения формулы (Х-K) с BF3OEt2 в присутствии триалкилсилана в органическом растворителе с образованием соответствующего соединения формулы (VIII-K); взаимодействие соединения формулы (VIII-K) с уксусным ангидридом или ацетилхлоридом в присутствии органического основания, в чистом виде или в органическом растворителе, с образованием соответствующего соединения формулы (IX-K); и снятии защитных групп в соединении формулы (IX-K) с образованием соответствующего соединения формулы (I-K). В предпочтительном варианте предложенного способа соединение формулы (VI-S) присутствует в количестве от приблизительно 1,0 до приблизительно 1,25 мол.экв. в расчете на соединение формулы (VK). В предпочтительном варианте предложенного способа алкиллитий представляет собой (триметилсилил)метиллитий, и где алкиллитий присутствует в количестве от приблизительно 2,0 до приблизительно 2,5 мол.экв. в расчете на соединение формулы (V-K). В предпочтительном варианте предложенного способа BF3OEt2 присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (VII-K), и где триалкилсилан представляет собой Et3SiH, который присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (V-K). В предпочтительном варианте предложенного способа молярное соотношение BF3OEt2 : Et3SiH составляет примерно 1:1. В предпочтительном варианте предложенного способа соединение формулы (VTII-K) вводят в реакцию с уксусным ангидридом, который присутствует в количестве от приблизительно 4,5 до приблизительно 5,0 мол.экв. в расчете на соединение формулы (VIII-K). В предпочтительном варианте предложенного способа органическим основанием является NMM. В предпочтительном варианте предложенного способа соединение формулы (VIII-K) вводят в реакцию с уксусным ангидридом в присутствии каталитического количества DMAP. В предпочтительном варианте предложенного способа в соединении формулы (IX-K) удаляют защитные группы посредством взаимодействия с основанием. Краткое описание рисунков На фиг. 1 представлена типичная рентгеновская дифрактограмма кристаллической формы соединения с формулой (I-S). На фиг. 2 представлена типичная порошковая рентгеновская дифрактограмма для кристаллической формы соединения с формулой (I-K), полученная на рентгеновском дифрактометре RINT-ULTIMA3, Ригаку, Токио, Япония. На фиг. 3 представлена типичная порошковая рентгеновская дифрактограмма для кристаллической формы соединения с формулой (I-K), полученная на рентгеновском дифрактометре X'Pert Pro MPD, Philips. На фиг. 4 представлен типичный ИК спектр кристаллической формы соединения с формулой (I-K) в минеральном масле. На фиг. 5 представлен типичный ИК спектр кристаллической формы соединения с формулой (I-K) в таблетке KBr. Подробное описание изобретения Настоящее изобретение относится к процессу получения соединения с формулой (I) где X, Y, кольцо А и кольцо В определены в настоящем документе. Соединения с формулой (I) проявляют ингибиторную активность в отношении натрийзависимого переносчика глюкозы, присутствующего в кишечнике и почках млекопитающих, и могут применяться при лечении сахарного диабета или осложнений диабета, таких как диабетическая ретинопатия, диабетическая нейропатия, диабетическая нефропатия, ожирение и медленное заживление ран. Специалисты в данной области также определят,что любое из соединений или кристаллических форм, описанных в настоящем документе, при необходимости могут использоваться в сочетании с одним или несколькими противодиабетическими препаратами, противогипергликемическими препаратами и/или препаратами для лечения других заболеваний, и их введение может осуществляться в той же самой лекарственной форме, в отдельной лекарственной форме для перорального введения, или путем инъекции. В публикации РСТ WO 2005/012326 описывается класс соединений, являющихся ингибиторами натрийзависимого переносчика глюкозы (НЗПГ), в том числе соединение с формулой (I-K), также известное как 1-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-3-пиридил)-2-тиенилметил]бензол, и соединение с формулой (I-S), также известное как 1-(-D-глюкопиранозил)-4-метил-3-[5-(4-фторфенил)-2 тиенилметил]бензол. В публикации РСТ WO 2005/012326 также описывается использование названных соединений, в том числе соединения с формулой (I-K) и соединения с формулой (I-S), для лечения диабетов, ожирения, осложнений диабета и т.д. Настоящее изобретение также относится к способам получения соединения с формулой (I-S) или фармацевтически приемлемой соли данного соединения, и соединения с формулой (I-K) или фармацевтически приемлемой соли данного соединения. Термин "атом галогена" или "галоген" означает хлор, бром и фтор, причем предпочтительно хлор и фтор. Термин "алкильная группа" означает линейную или разветвленную одновалентную насыщенную углеводородную цепь, содержащую от 1 до 12 атомов углерода. Предпочтительной является алкильная группа с линейной или разветвленной цепью, содержащая от 1 до 6 атомов углерода, более предпочтительной является алкильная группа с линейной или разветвленной цепью, содержащая от 1 до 4 атомов углерода. Примером может служить метильная группа, этильная группа, пропильная группа, изопропильная группа, бутильная группа, трет-бутильная группа, изобутильная группа, пентильная группа, гексильная группа, изогексильная группа, гептильная группа, 4,4-диметилпентильная группа, октильная группа, 2,2,4-триметилпентильная группа, нонильная группа, децильная группа и различные изомеры этих групп с разветвленной цепью. Также в алкильную группу при необходимости может дополнительно и независимо вводиться от 1 до 4 перечисленных ниже заместителей. Термин "гетероциклил" означает одновалентную группу вышеупомянутого ненасыщенного моноциклического гетероциклического кольца или ненасыщенного конденсированного гетеробициклического кольца и одновалентную группу насыщенного варианта вышеупомянутого ненасыщенного моноциклического гетероциклического или ненасыщенного конденсированного гетеробициклического кольца. В гетероциклил при необходимости могут дополнительно и независимо вводиться от 1 до 4 заместителей,как указано ниже. Термин "низший", используемый в определениях формул в настоящем техническом описании, означает линейную или разветвленную углеродную цепь, содержащую от 1 до 6 атомов углерода, если иное не определено специально. Более предпочтительно, данный термин означает линейную или разветвленную углеродную цепь, содержащую от 1 до 4 атомов углерода. К фармацевтически приемлемым солям соединения с формулой I относятся, например, соль с щелочным металлом, таким как литий, натрий, калий и т.п.; соль с щелочно-земельным металлом, таким как кальций, магний и т.п.: соль с цинком или алюминием; соль с органическим основанием, таким как аммиак, холин, диэтаноламин, лизин, этилендиамин, трет-бутиламин, трет-октиламин, трис(гидроксиметил)аминометан, N-метилглюкозамин, триэтаноламин и дегидроабиэтиламин; соль с неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п.; или соль с органической кислотой, такой как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, и т.п.; или соль с аминокислотой, такой как аспарагиновая кислота, глутаминовая кислота и т.п. Предпочтительное соединение, которое может быть получено способом согласно настоящему изобетению, может быть выбрано из следующей группы: 1-(-D-глюкопиранозил)-4-хлор-3-(6-этилбензо[b]тиофен-2-ил-метил)бензол; 1-(-D-глюкопиранозил)-4-хлор-3-[5-(5-тиазолил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-хлор-3-(5-фенил-2-тиенил-метил)бензол; 1-(-D-глюкопиранозил)-4-метил-3-[5-(4-фторфенил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-хлор-3-[5-(2-пиримидинил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-метил-3-[5-(2-пиримидинил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-хлор-3-[5 цианофенил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-хлор-3-[5-(4-цианофенил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-метил-3-[5-(6-фтор-2-пиридил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-2-пиридил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-метил-3-[5 дифторметилфенил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-метил-3-[5-(3-цианофенил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-метил-3-[5-(4-цианофенил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-3-пиридил)-2-тиенилметил]бензол; 1-(-D-глюкопиранозил)-4-фтор-3-(5 цианофенил)-2-тиенилметил)бензол; фармацевтически приемлемых солей данных соединений; и пролекарств данных соединений. К особенно предпочтительным соединениям, составляющим предмет данного изобретения, относятся 1-(-D-глюкопиранозил)-4-метил-3-[5 цианофенил)-2-тиенилметил]бензол или его фармацевтически приемлемая соль или пролекарство; 1-(-D-глюкопиранозил)-4-метил-3-[5-(4-цианофенил)-2-тиенилметил]бензол или его фармацевтически приемлемая соль или пролекарство; 1-(-D-глюкопиранозил)-4-метил-3-[5-(4-фторфенил)-2-тиенилметил]бензол или его фармацевтически приемлемая соль или пролекарство; 1-(-D-глюкопиранозил)-4-хлор-3-[5 цианофенил)-2-тиенилметил]бензол или его фармацевтически приемлемая соль или пролекарство; 1-(-D-глюкопиранозил)-4-метил-3-[5-(6-фтор-2-пиридил)-2-тиенилметил]бензол или его фармацевтически приемлемая соль или пролекарство; 1-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-2-пиридил)-2-тиенилметил]бензол или его фармацевтически приемлемая соль или пролекарство; 1-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-3-пиридил)-2-тиенилметил]бензол или его фармацевтически приемлемая соль или пролекарство; 1-(-D-глюкопиранозил)-4-фтор-3-[5-(3-цианофенил)-2-тиенилметил]бензол или его фармацевтически приемлемая соль или пролекарство. В техническом описании и, в частности, на схемах и в примерах используются следующие сокращения:BF3OEt2 - диэтиловый эфират трехфтористого бора;TEA - триэтиламин; ТГФ - тетрагидрофуран. В целом для коммерческого применения желательно, чтобы продукт был достаточно удобен в обращении. Также для коммерческого применения предпочтительно, чтобы продукт производился в существенно чистой и кристаллической форме, чтобы обеспечить создание рецептур, удовлетворяющих точным фармацевтическим требованиям и спецификациям. Далее, для производства в коммерческих масштабах предпочтительно, чтобы продукт имел форму, легко допускающую фильтрацию и сушку. Наконец, предпочтительно, чтобы продукт был стабилен в течение длительного времени и не требовал специальных условий хранения. При использовании в настоящем документе, если иное не оговорено особо, термин "изолированная форма" означает, что соединение присутствует в форме, отделенной от твердых примесей других соединений, растворителей или биологической среды. В конкретной реализации соединение с формулой (I),соединение с формулой (I-S), соединение с формулой (I-K), кристаллическая форма соединения с формулой (I-S), и/или кристаллическая форма соединения с формулой (I-K) присутствует и/или получается в изолированной форме. При использовании в настоящем документе, если иное не оговорено особо, термин "существенно чистый" означает, что молярная процентная доля примесей в изолированном соединении составляет менее чем примерно 5 мол.%, предпочтительно менее чем примерно 2 мол.%, более предпочтительно менее чем примерно 0,5 мол.%, наиболее предпочтительно менее чем примерно 0,1 мол.%. В конкретной реализации соединение с формулой (I), соединение с формулой (I-S), соединение с формулой (I-K), кристаллическая форма соединения с формулой (I-S), и/или кристаллическая форма соединения с формулой (IK) присутствует и/или получается в существенно чистой форме. При использовании в настоящем документе, если иное не оговорено особо, термин "существенно свободный от соответствующих солевых форм" применительно к описанию соединения с формулой (I) означает, что молярная процентная доля соответствующей солевой формы (форм) в изолированном основании с формулой (I) составляет менее чем примерно 5 мол.%, предпочтительно менее чем примерно 2 мол.%, более предпочтительно менее чем примерно 0,5 мол.%, наиболее предпочтительно менее чем примерно 0,1 мол.%. В конкретной реализации соединение с формулой (I) соединение с формулой (I-S),соединение с формулой (I-K), кристаллическая форма соединения с формулой (I-S) и/или кристаллическая форма соединения с формулой (I-K) присутствует и/или получается существенно свободной от соответствующих солевых форм. В конкретной реализации настоящее изобретение относится к процессу получения соединения с формулой (I), где соединение с формулой (I) является существенно чистым. В другой конкретной реализации настоящее изобретение относится к процессу получения соединения с формулой (I-S), где соединение с формулой (I-S) является существенно чистым. В другой конкретной реализации настоящее изобретение относится к процессу получения соединений с формулой (I-K), где соединение с формулой (IK) является существенно чистым. В конкретной реализации настоящее изобретение относится к процессу получения соединений с формулой (I), где соединение с формулой (I) является существенно свободным от соответствующих солевых форм. В другой конкретной реализации настоящее изобретение относится к процессу получения соединений с формулой (I-S), где соединение с формулой (I-S) является существенно свободным от соответствующих солевых форм. В другой конкретной реализации настоящее изобретение относится к процессу получения соединений с формулой (I-K), где соединение с формулой (I-K) является существенно свободным от соответствующих солевых форм. Термин "субъект" при использовании в настоящем документе означает животное, предпочтительно млекопитающее, наиболее предпочтительно человека, которое является объектом лечения, наблюдения или эксперимента. Предпочтительно субъект испытывал и/или демонстрировал минимум один симптом заболевания или нарушения, лечение и/или профилактика которого осуществляется. Термин "терапевтически эффективное количество" при использовании в настоящем документе означает количество активного соединения или фармацевтического препарата, вызывающее биологический или медицинский ответ на тканевой системе, на животном или на человеке, ожидаемый исследователем,ветеринаром, врачом или иным клиническим специалистом, каковой ответ включает ослабление симптомов заболевания или нарушения, лечение которого осуществляется. Термин "состав" при использовании в настоящем документе предназначен для обозначения продукта, включающего указанные компоненты в указанных количествах, а также любого продукта, получающегося прямо или косвенно из сочетания указанных компонентов в указанных количествах. Соединение с формулой (I), полученное способом настоящего изобретения, проявляет очень высокую ингибиторную активность в отношении натрий-зависимого переносчика глюкозы, и очень сильный эффект в отношении снижения концентрации глюкозы в крови. Следовательно, соединение, составляющее предмет настоящего изобретения, может применяться для лечения или задержки развития или возникновения сахарного диабета, диабетической ретинопатии, диабетической нейропатии, диабетической нефропатии, медленного заживления ран, резистентности к инсулину, гипергликемии, гиперинсулинемии, повышенного уровня жирных кислот в крови, повышенного уровня глицерина в крови, гиперлипидемии, ожирения, гипертриглицеридемии, синдрома X, осложнений диабета, атеросклероза или повышенного давления. В частности, соединение, составляющее предмет изобретения, может применяться для лечения и профилактики сахарного диабета (сахарный диабет 1 и 2 типов и т.п.), осложнений диабета(например, диабетической ретинопатии, диабетической нейропатии, диабетической нефропатии) или ожирения, а также может применяться для лечения гипергликемии, возникающей после приема пищи. Соединение с формулой (I), полученное способом настоящего изобретения, или фармацевтически приемлемая соль этого соединения, может вводиться перорально или парэнтерально, и может применяться в форме соответствующей фармацевтического препарата. Соответствующие фармацевтические препараты для перорального введения включают такие препараты, как, например, твердые препараты,такие как таблетки, гранулы, капсулы, порошки и т.п., или препараты в виде растворов, суспензий,эмульсий и т.п. К соответствующим фармацевтическим препаратам для парэнтерального введения относятся, например, суппозитории, препараты для инъекций и препараты для внутривенного покапельного введения с применением дистиллированной воды для инъекций, физиологического солевого раствора или водного раствора глюкозы; а также препараты для ингаляции. Дозировка настоящего соединения с формулой (I) или его фармацевтически приемлемой соли может изменяться в зависимости от метода введения, возраста, массы тела, состояния пациента, от типа и тяжести заболевания, лечение которого производится, и обычно лежит в диапазоне от примерно 0,01 до 300 мг/кг/день или в любом меньшем диапазоне, предпочтительно в диапазоне от примерно 0,1 до 50 мг/кг/день или в любом меньшем диапазоне, предпочтительно в диапазоне от примерно 0,1 до 30 мг/кг/день или в любом меньшем диапазоне. Соединение с формулой I может при необходимости применяться в сочетании с одним или несколькими другими антидиабетическими препаратами, одним или несколькими препаратами для лечения осложнений диабета и/или одним или несколькими препаратами для лечения других заболеваний. Настоящее соединение и вышеназванные прочие препараты могут вводиться в одной и той же лекарственной форме или в отдельной лекарственной форме для перорального введения или путем инъекции. К другим противодиабетическим препаратам относятся, например, антидиабетические и антигипергликемические препараты, в том числе инсулин, стимуляторы секреции инсулина или средства, повышающие чувствительность к инсулину, другие противодиабетические препараты, механизм действия которых не связан с ингибированием НЗПГ, предпочтительно могут использоваться 1, 2, 3 или 4 вышеупомянутых других противодиабетических средства. Конкретными примерами таких препаратов являются бигуанидные соединения, соединения сульфанил-мочевины, ингибиторы -глюкозидазы, агонистыPPAR (например, тиазолидиндионовые соединения), двойные агонисты PPAR /, ингибиторы дипептидил-пептидазы IV (DPP4), митиглинидные и/или натеглинидные соединения, а также инсулин, глюкагонподобный пептид-1 (GLP-1), ингибиторы РТР 1 В, ингибиторы гликоген-фосфорилазы, модуляторы RXR и/или ингибиторы глюкозо-6-фосфатазы. К препаратам для лечения других заболеваний относятся, например, препарат против ожирения, антигипертензивный препарат, антитромбоцитарный препарат, антисклеротический препарат и/или гиполипидемический препарат. Ингибиторы НЗПГ с формулой I могут при необходимости использоваться в сочетании с препаратами для лечения осложнений диабета. К таким препаратам относятся, например, ингибиторы протеинкиназы С (РКС) и/или ингибиторы ангиотензин-превращающего фермента (АСЕ). Дозировка этих препаратов может изменяться в зависимости от возраста, массы тела и состояния пациентов, от метода введения, лекарственной формы и т.п. Указанные фармацевтические препараты могут перорально вводиться разным видам млекопитающих, включая людей, обезьян, собак и т.д., например, в лекарственной форме таблетки, капсулы, гранулы или порошка, парэнтерально, в форме препарата для инъекции, интраназально, или в форме трансдермального пластыря. Специалисты в данной области определят, что при отсутствии специальных указаний реакционные стадии получения требуемого продукта проводят в соответствующих условиях, следуя известным методикам. Примеры соответствующих растворителей, оснований, температур и других компонентов и параметров проведения реакций даны в подробных описаниях, приводимых далее в настоящем документе. Специалисты в данной области определят, что перечень упомянутых примеров не предполагает и не должен рассматриваться как какое-либо ограничение изобретения, излагаемого в приводимых далее пунктах с формулой изобретения. Специалисты в данной области также определят, что в техническом описании и в пунктах с формулой изобретения, представленных в настоящем документе, там, где реагент или класс/тип реагента (например, основание, растворитель и т.д.) упоминается для более чем одной стадии процесса, индивидуальные реагенты выбираются для каждой стадии независимо и могут быть одинаковыми или отличными друг от друга. Например, там, где органическое или неорганическое основание упоминается в качестве реагента на двух стадиях процесса, органическое или неорганическое основание, выбираемое для первой стадии, может быть тем же или отличаться от органического или неорганического основания, используемого на второй стадии. Для большей краткости описания некоторые количественные выражения, приводимые в настоящем документе, не содержат дополнительного термина "приблизительно". Подразумевается, что независимо от того, используется ли явным образом термин "приблизительно", каждое приводимое в настоящем документе количество означает реально приведенное значение, а также разумное для данной области приближение к приведенному значению, в том числе неточности приведенного значения, связанные с условиями проведения эксперимента и/или измерения. Для большей краткости описания некоторые количественные выражения в настоящем документе приводятся в виде диапазона, указанного от приблизительного количества X до приблизительного количества Y. Подразумевается, что упоминаемый диапазон не ограничивается указанной верхней и нижней границей, но включает весь диапазон от приблизительного количества X до приблизительного количества Y, и все меньшие входящие в него диапазоны. Если иное не оговорено особо, используемый в настоящем документе термин "защитная группа азота" означает группу, которая может присоединяться к атому азота с целью защиты упомянутого атома азота от участия в реакции и которая легко может быть снята после проведения реакции. Соответствующие защитные группы азота включают, но не ограничиваются ими, карбаматы - группы с формулой-C(O)O-R, где R представляет собой, например, метил, этил, трет-бутил, бензил, фенилэтил, СН 2=СНСН 2- и другие подобные группы; амиды - группы с формулой -C(O)-R', где R' представляет собой, например, метил, фенил, трифторметил и другие подобные группы; производные N-сульфонила группы с формулой -SO2-R", где R" представляет собой, например, толил, фенил, трифторметил, 2,2,5,7,8 пентаметилхроман-D-ил-, 2,3,6-триметил-4-метоксибензол и другие подобные группы. Другие соответствующие защитные группы для азота можно найти в таких книгах, как, например T.W. GreeneP.G.M.Wuts, Protective Groups in Organic Synthesis, John WileySons, 1991. Специалисты в данной области определят, что, если та или иная стадия реакции, составляющей предмет настоящего изобретения, может быть проведена в разных растворителях или системах растворителей, данная стадия также может быть проведена в смеси соответствующих растворителей или систем растворителей. Там, где в ходе процессов получения соединений в соответствии с данным изобретением образуется смесь стереоизомеров, данные стереоизомеры могут быть разделены с помощью традиционных методов,таких как препаративная хроматография. Соединения могут быть либо получены в рацемической форме,либо отдельные энантиомеры могут быть получены в ходе энантиоспецифичного синтеза или путем разделения. Соединения могут, например, быть разделены на составляющие их энантиомерные компоненты с использованием стандартных методик, например, образованием диастереоизомерных пар путем образования соли с оптически активной кислотой, такой как (-)-ди-п-толуоил-D-винной кислотой и/или (+)ди-п-толуоил-L-винной кислотой с последующей фракционной кристаллизацией и восстановлением свободного основания. Соединения также могут быть разделены путем образования диастереоизомерных эфиров или амидов с последующим хроматографическим разделением и удалением хирального вспомогательного соединения. Соединения могут также быть разделены с помощью хиральной ВЭЖХ-колонки. В ходе любого процесса получения соединений, составляющих предмет настоящего изобретения,может быть необходимо и/или желательно защитить чувствительные или реакционно-активные группы на участвующих в процессе молекулах. Этого можно достигнуть, используя традиционные защитные группы, например, описанные в книгах Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, PlenumPress, 1973; и T.W. GreeneP.G.M. Wuts, Protective Groups in Organic Synthesis, John WileySons, 1991. Защитные группы могут быть сняты на последующей удобной для этого стадии с применением известных в данной области методик. Специалисты в данной области определят, что в любом из процессов, описанных в настоящем документе, реакционноспособные заместители в соединениях с формулой (I), такие как гидроксигруппы,оксогруппы, карбоксильные группы и т.п. предпочтительно требуют защиты и затем снятия защиты в соответствии с известными методиками на соответствующих стадиях синтеза. Настоящее изобретение относится к процессу получения соединений с формулой (I), описанного в схеме 1 ниже. Схема 1 Соответственно, содержащее требуемые заместители соединение с формулой (V), которое либо является известным соединением, либо может быть получено известными методами, вводится в реакцию с соединением с формулой (VI-S), которое либо является известным соединением, либо может быть получено известными методами; где количество соединения с формулой (VI-S) предпочтительно находится в диапазоне от приблизительно 1,0 до приблизительно 2,0 мол.экв. или в любом входящем в него диапазо- 10020209 не, более предпочтительно находится в диапазоне от приблизительно 1,0 до приблизительно 1,25 мол.экв. или в любом входящем в него диапазоне, наиболее предпочтительно составляет приблизительно 1,2 мол.экв.; в присутствии алкиллития, например, триметилсилилметиллития, мезитиллития (т.е. 2,4,6 триметилфениллития), триэтилсилилметиллития, предпочтительно триметилсилилметиллития и подобных соединений, где количество алкиллития предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 3,0 мол.экв. или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 2,5 мол.экв. или в любом входящем в него диапазоне, наиболее предпочтительно составляет приблизительно 2,0 мол.экв.; в органическом растворителе, таком как ТГФ, гексан, пентан, МТВЕ, диоксан и подобных, предпочтительно ТГФ; при температуре в диапазоне от приблизительно 0 С до приблизительно -78 С, или в любом входящем в него диапазоне, предпочтительно при приблизительно -40 С; с получением соответствующего соединения с формулой (VII). Предпочтительно алкиллитий добавляется в смесь соединения с формулой (V) и соединения с формулой (VI-S). Специалисты в данной области определят, что соединение с формулой (V) может также быть введено в реакцию (как описано выше) с соединением с формулой (VI-S), в котором триметилсилильные(TMS) заместители заменены одним или несколькими соответственно выбранными альтернативными силильными группами, такими как триэтилсилил, фенилдиметилсилил и подобными. Соединение с формулой (VII) вводится в реакцию с BF3OEt2 в присутствии надлежащим образом выбранного триалкилсилана, такого как Et3SiH и подобных; где количество BF3OEt2 предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 10,0 мол.экв., или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 6,0 мол.экв., наиболее предпочтительно составляет приблизительно 3,0 мол.экв.; и где количество триалкилсилана предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 10,0 мол.экв. или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 6,0 мол.экв. или в любом входящем в него диапазоне, наиболее предпочтительно составляет приблизительно 3,0 мол.экв.; предпочтительно, чтобы соотношение BF3OEt2 и триалкилсилана составляло 1:1; в органическом растворителе, таком как ДХМ, DCE, ацетонитрил и им подобных, или в смеси упомянутых органических растворителей, предпочтительно в ДХМ; предпочтительно при температуре в диапазоне от приблизиблизительно 0 С до приблизительно -40 С, или в любом входящем в него диапазоне, более предпочтительно при приблизительно -30 С, с получением соответствующего соединения с формулой (VIII). Специалисты в данной области определят, что с соединения с формулой (VII) также могут быть сняты защитные группы с использованием известных методик (например, путем взаимодействия с выбранной надлежащим образом кислотой, например, HCl и ей подобных), с получением соответствующего соединения с формулой (X) которое затем вводится в реакцию с BF3OEt2 в присутствии надлежащим образом выбранного триалкилсилана, такого как Et3SiH, и ему подобных; где количество BF3OEt2 предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 10,0 мол.экв. или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 6,0 мол.экв., наиболее предпочтительно составляет приблизительно 3,0 мол.экв.; и где количество триалкилсилана предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 10,0 мол.экв.,или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 6,0 мол.экв., или в любом входящем в него диапазоне, наиболее предпочтительно, составляет приблизительно 3,0 мол.экв.; предпочтительно, чтобы соотношение BF3OEt2 и триалкилсилана составляло 1:1; в органическом растворителе, таком как ДХМ, DCE, ацетонитрил и им подобных, или в смеси упомянутых органических растворителей, предпочтительно в ДХМ; предпочтительно при температуре в диапазоне от приблизительно 0 С до приблизительно -40 С, или в любом входящем в него диапазоне,более предпочтительно при приблизительно -30 С; с получением соответствующего соединения с формулой (VIII). Соединение с формулой (VIII) вводится в реакцию с уксусным ангидридом или ацетилхлоридом,предпочтительно уксусным ангидридом, известным соединением; где количество уксусного ангидрида предпочтительно находится в диапазоне от приблизительно 4,0 до приблизительно 6,0 мол.экв., или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 4,5 до приблизительно 5,0 мол.экв., или в любом входящем в него диапазоне, наиболее предпочтительно составляет приблизительно 5,0 мол.экв.; в присутствии органического основания, такого как N-метилморфолин (NMM), TEA, пиридин и им подобных, предпочтительно NMM; где количество органического основания предпочтительно находится в диапазоне от приблизительно 3,0 до приблизительно 6,0 мол.экв., или в любом входящем в него диапазоне, более предпочтительно составляет приблизительно 5,0 мол.экв.; дополнительно возможно в присутствии катализатора, такого как DMAP и ему подобных; предпочтительно в присутствии каталитических количеств DMAP; в чистой форме или в органическом растворителе, таком как ТГФ, ацетонитрил и им подобных,предпочтительно в ТГФ; при температуре в диапазоне от приблизительно -10 С до приблизительно комнатной температуры, или в любом входящем в него диапазоне, предпочтительно в диапазоне от приблизительно 0 С до приблизительно комнатной температуры; с получением соответствующего соединения с формулой (IX). Соединение с формулой (IX), предпочтительно, суспендируется или растворяется в растворителе,более предпочтительно, суспендируется; затем фильтруется, предпочтительно фильтруется при повышенной температуре, для удаления примесей и/или побочных продуктов. С соединения с формулой (IX) снимаются защитные группы с использованием известных методик. Например, соединение с формулой (IX) вводится в реакцию с надлежащим образом выбранным основанием, таким как LiOH, NaOH и им подобными, предпочтительно LiOH; где количество основания предпочтительно находится в диапазоне от приблизительно 0,1 до приблизительно 1,0 мол.экв., или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 0,25 до приблизительно 0,5 мол.экв., или в любом входящем в него диапазоне, наиболее предпочтительно составляет приблизительно 0,5 мол.экв. (например, каталитическое количество); в смеси воды ТГФ и метанола, где соотношение вода:ТГФ:метанол предпочтительно составляет примерно 1:2:3; предпочтительно при комнатной температуре с получением соответствующего соединения с формулой (I). Соединение с формулой (I) предпочтительно выделяется и/или перекристаллизовывается в соответствии с известными методиками. В конкретной реализации настоящее изобретение относится к процессу получения соединения с формулой (I-S), как показано на схеме 2 ниже. Схема 2 Соответственно, содержащее требуемые заместители соединение с формулой (V-S), которое либо является известным соединением, либо может быть получено известными методами, вводится в реакцию с соединением с формулой (VI-S), которое либо является известным соединением, либо может быть получено известными методами; где количество соединения с формулой (VI-S) предпочтительно находится в диапазоне от приблизительно 1,0 до приблизительно 2,0 мол.экв., или в любом входящем в него диапазоне, более предпочтительно, находится в диапазоне от приблизительно 1,0 до приблизительно 1,25 мол.экв., или в любом входящем в него диапазоне, наиболее предпочтительно составляет приблизительно 1,2 мол.экв.; в присутствии алкиллития, например, триметилсилилметиллития, мезитиллития (т.е. 2,4,6 триметилфениллития), триэтилсилилметиллития, предпочтительно триметилсилилметиллития и подобных соединений, где количество алкиллития предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 3,0 мол.экв., или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 2,5 мол.экв. или в любом входящем в него диапазоне, наиболее предпочтительно составляет приблизительно 2,0 мол.экв.; в органическом растворителе, таком как ТГФ, гексан, пентан, МТВЕ, диоксан и им подобных,предпочтительно ТГФ; при температуре в диапазоне от приблизительно 0 С до приблизительно -78 С,или в любом входящем в него диапазоне, предпочтительно при приблизительно -40 С; с получением соответствующего соединения с формулой (VII-S). Предпочтительно алкиллитий добавляется в смесь соединения с формулой (V-S) и соединения с формулой (VI-S). Специалисты в данной области определят, что соединение с формулой (V-S) может также быть введено в реакцию (как описано выше) с соединением с формулой (VI-S), в котором триметилсилильные(TMS) заместители заменены одним или несколькими соответственно выбранными альтернативными силильными группами, такими как триэтилсилил, фенилдиметилсилил и подобными. Соединение с формулой (VII-S) вводится в реакцию с BF3OEt2 в присутствии надлежащим образом выбранного триалкилсилана, такого как Et3SiH и подобных; где количество BF3OEt2 предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 10,0 мол.экв. или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 6,0 мол.экв., наиболее предпочтительно составляет приблизительно 3,0 мол.экв.; и где количество триалкилсилана предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 10,0 мол.экв. или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 2,0 до приблизительно 6,0 мол.экв. или в любом входящем в него диапазоне, наиболее предпочтительно составляет приблизительно 3,0 мол.экв.; предпочтительно, чтобы соотношение BF3OEt2 и триалкилсилана составляло 1:1; в органическом растворителе, таком как ДХМ, DCE, ацетонитрил, толуол и им подобных, или в смеси упомянутых органических растворителей, предпочтительно в ДХМ; предпочтительно при температуре в диапазоне от приблизительно 0 С до приблизительно -40 С, или в любом входящем в него диапазоне, более предпочтительно при приблизительно -30 С; с получением соответствующего соединения с формулой (VIII-S). Соединение с формулой (VIII-S) вводится в реакцию с уксусным ангидридом или ацетилхлоридом,предпочтительно уксусным ангидридом, известным соединением; где количество уксусного ангидрида предпочтительно находится в диапазоне от приблизительно 4,0 до приблизительно 6,0 мол.экв., или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 4,5 до приблизительно 5,0 мол.экв. или в любом входящем в него диапазоне, наиболее предпочтительно составляет приблизительно 5,0 мол.экв.; в присутствии органического основания, такого как N-метилморфолин (NMM), TEA, пиридин и им подобных, предпочтительно NMM; где количество органического основания предпочтительно находится в диапазоне от приблизительно 3,0 до приблизительно 6,0 мол.экв., или в любом входящем в него диапазоне, более предпочтительно составляет приблизительно 5,0 мол.экв.; дополнительно возможно в присутствии катализатора, такого как ОМАР и ему подобных; предпочтительно в присутствии каталитических количеств DMAP; в чистой форме или в органическом растворителе, таком как ТГФ, ацетонитрил и им подобных,предпочтительно в ТГФ; предпочтительно при температуре в диапазоне от приблизительно -10 С до приблизительно комнатной температуры, или в любом входящем в него диапазоне, предпочтительно в диапазоне от приблизительно 0 С до приблизительно комнатной температуры; с получением соответствующего соединения с формулой (IX-S). Соединение по формуле (IX-S) предпочтительно суспендируется или растворяется в растворителе,более предпочтительно - суспендируется; затем фильтруется, предпочтительно при повышенной температуре, для удаления примесей и/или побочных продуктов. Соединение с формулой (IX-S) предпочтительно суспендируется или растворяется в растворителе, более предпочтительно, суспендируется; затем фильтруется, предпочтительно фильтруется при повышенной температуре, для удаления примесей и/или побочных продуктов. С соединения с формулой (IX-S) снимаются защитные группы с использованием известных методик. Например, соединение с формулой (IX-S) вводится в реакцию с надлежащим образом выбранным основанием, таким как LiOH, NaOH и им подобными, предпочтительно, LiOH; где количество основания предпочтительно находится в диапазоне от приблизительно 0,1 до приблизительно 1,0 мол.экв. или в любом входящем в него диапазоне, более предпочтительно находится в диапазоне от приблизительно 0,25 до приблизительно 0,5 мол.экв. или в любом входящем в него диапазоне, наиболее предпочтительно составляет приблизительно 0,5 мол.экв. (например, каталитическое количество); в смеси воды, ТГФ и метанола, где соотношение вода:ТГФ:метанол предпочтительно составляет приблизительно 1:2:3; предпочтительно при комнатной температуре; с получением соответствующего соединения с формулой (I-S). Соединение по формуле (I-S) предпочтительно перекристаллизовывается. В конкретной реализации, соединение с формулой (I-S) перекристаллизовывается следующим образом: стадия А: соединение с формулой (I-S) растворяют в органическом растворителе, таком как этилацетат, метанол, этанол илиподобном, предпочтительно в этилацетате; и дополнительно фильтруют; стадия Б: полученную на стадии А смесь нагревают до температуры в диапазоне от приблизительно 25 С до приблизительно 45 С, предпочтительно в диапазоне от приблизительно 30 до приблизительно 35 С; и дополнительно фильтруют; стадия В: в приготовленную на стадии Б смесь добавляют воду, предпочтительно в количестве от приблизительно 1,0 до приблизительно 2,0 мол.экв., более предпочтительно приблизительно 1,5 мол.экв.; стадия Г: в смесь, приготовленную на стадии В, медленно добавляют гептан (чтобы началось осаждение, т.е. гептан играет роль осадителя), предпочтительно в таком количестве, чтобы конечное объемное отношение этилацетат:гептан находилось в диапазоне от приблизительно 1:1 до приблизительно 1,5:1, более предпочтительно - около 1,2:1; для получения осадка соединения с формулой (I-S); предпочтительно, с отделением указанного осадка фильтрацией с последующим высушиванием, следуя известным методикам. Предпочтительно в процессе перекристаллизации соединения с формулой (I-S), после добавления гептана в образовавшуюся смесь вносят зародыш желаемой полиморфной формы соединения с формулой (I-S). Специалисты в данной области определят, что существует несколько методов характеризации кристаллических форм, и настоящее изобретение не ограничивается методами или приборами, выбранными для характеризации соединений, составляющих предмет настоящего изобретения. Например, в порошковых рентгенограммах интенсивности дифракционных пиков в экспериментальных рентгенограммах могут варьироваться, как известно специалистам в данной области, в основном в связи с предпочтительной ориентацией (не полностью случайной ориентацией кристаллов) в приготовленном образце. Таким образом, предмет настоящего изобретения следует рассматривать в свете разнообразия возможных методов характеризации, известных специалистам в данной области. Настоящее изобретение обеспечивает получение кристаллической формы соединения с формулой В конкретной реализации настоящее изобретение обеспечивает кристаллическую форму соединения с формулой (I-S), полученную в соответствии с описанным в данном документе процессом перекристаллизации. В другой конкретной реализации настоящее изобретение обеспечивает получение кристаллической формы соединения по формуле (I-S), получаемого в соответствии со следующим процессом перекристаллизации. Стадия А: соединение с формулой (I-S) растворяют в этилацетате с получением смеси А; затем смесь А дополнительно фильтруют; стадия Б: смесь А нагревают до температуры в диапазоне от приблизительно 30 С до приблизительно 35 С с образованием смеси Б; затем смесь В дополнительно фильтруют В; стадия В: добавляют приблизительно 1,5 мол.экв. воды в смесь Б и получают смесь В; стадия Г: медленно добавляют гептан в смесь В с получением кристаллической формы соединения с формулой (I-S); стадия Д: кристаллическую форму соединения с формулой (I-S) выделяют фильтрацией и последующей сушкой. Настоящее изобретение далее обеспечивает кристаллическую форму соединения с формулой (I-K). Порошковую рентгеновскую дифрактограмму для типичного образца кристаллической формы соединения с формулой (I-K) сняли на порошковом рентгеновском дифрактометре RINT-ULTIMA3, Ригаку, Токио, Япония, используя линию излучения CuK при следующих параметрах съемки: (а) скорость сканирования: 1,00/мин; (б) мишень: CuK; (в) напряжение: 40 кВ/ (г) ток: 40 мА; (д) диапазон сканирования: от 3 до 40,0; (е) угловое разрешение: 0,0200; как показано на фиг. 2. Порошковую рентгеновскую дифрактограмму для типичного образца кристаллической формы со- 14020209 единения с формулой (I-K) также сняли на порошковом рентгеновском дифрактометре Philips X'Pert ProMPD, используя линию излучения CuK при следующих параметрах съемки: (а) скорость сканирования: 0,207/мин; (б) мишень: CuK; (в) напряжение: 45 кВ; (г) ток: 40 мА; (д) детектор: X'celerator; (e) диапазон сканирования: от 3 до 35; (ж) размер углового шага: 0,0165; и (з) время на один шаг: 10,16 с; как показано на фиг. 3. Инфракрасный спектр для типичных образцов кристаллической формы соединения с формулой (I-K) сняли в минеральном масле, как показано на фиг. 4, а также в таблетке K-Br, как показано на фиг. 5. В инфракрасных спектрах кристаллической формы соединения с формулой (I-K), приведенных на фиг. 4 и фиг. 5 ниже, по оси ординат отложено пропускание в процентах, а по оси абсцисс - волновое число в см-1. Фурье-ИК спектр кристаллической формы соединения с формулой (I-K) в минеральном масле сняли с разрешением 4 см-1. ИК спектр, показанный на фиг. 4, представляет собой сумму 4 сканов. В ИК спектре присутствуют основные характерные полосы поглощения на 1492, 1463, 1377, 1268, 1065 и 1023 см-1, что согласуется с функциональными группами, имеющимися в соединении (I-K). Фурье-ИК спектр кристаллической формы соединения с формулой (I-K) в таблетке KBr сняли с разрешением 4 см-1. ИК спектр, показанный на фиг. 5, представляет собой сумму 64 сканов. В ИК спектре присутствуют основные характерные полосы поглощения на 3431, 3321, 1493, 1269, 1065 и 1024 см-1. Провели термогравиметрический анализ на типичном образце кристаллической формы соединения с формулой (I-K). Термогравиметрический анализ выполняли следующим образом: 7,35 мг кристаллической формы соединения с формулой (I-K) взвесили и поместили в алюминиевый держатель кюветы ТГА-анализатораTG-8120 (RIGAKU, Japan). Затем сняли термогравиметрическую (ТГ) кривую для кристаллической формы соединения с формулой (I-K) при скорости нагрева 5 С в минуту в типичном диапазоне измерений от комнатной температуры до 200 С. В ходе термогравиметрического анализа кристаллическая форма соединения по формуле (I-K) в виде гидрата или сольвата не наблюдалась. Способ получения кристаллической формы соединения формулы (I-K)включает получение раствора соединения с формулой (I-K) и осаждение кристаллической формы из раствора. Кристаллическая форма соединения с формулой (I-K) может быть получена из раствора соединения с формулой (I-K) в надлежащем образом выбранном растворителе. Некоторые примеси могут действовать как ингибиторы кристаллизации, и такие примеси должны быть удалены одним из общепринятых методов, таким как хроматография на колонке с силикагелем, которые хорошо известны специалистам в данной области. Однако кристаллическая форма соединения с формулой (I-K) может быть получена из соединения с формулой (I-K), содержащего некоторое количество примесей. Кристаллическая форма соединения с формулой (I-K) может быть получена из раствора соединения с формулой (I-K) в надлежащем образом выбранном растворителе. Возможные растворители включают,но не ограничиваются, кетоны (например, ацетон, 2-бутанон), сложные эфиры (например, этилацетат,метилацетат), спирты (например, метанол, этанол, изопропанол) и смеси указанных растворителей. К наиболее предпочтительным растворителям относятся сложные эфиры, такие как этилацетат. В некоторых случаях в раствор соединения с формулой (I-K) может добавляться осадитель. Возможные осадители включают алканы (например, гексан, гептан), ароматические углеводороды (например, бензол, толуол),простые эфиры (например, диэтиловый эфир, диметиловый эфир, диизопропиловый эфир) и смеси указанных растворителей. Предпочтительный процесс осаждения кристаллической формы соединения с формулой (I-K) состоит из растворения в нагретом соответствующем растворителе (например, в сложных эфирах) неочищенного или аморфного соединения с формулой (I-K) (полученного, например, в соответствии с процедурами, описанными в публикации РСТ WO 2005/012326), и добавления в образующийся раствор осадителя по мере необходимости с последующим охлаждением получившегося раствора и фильтрацией. Точные условия, при которых образуется кристаллическая форма соединения (I-K), определяются эмпирически. Специалисты в данной области определят, что кристаллическую форму соединения с формулой (IK) выделить легче, чем соответствующую аморфную форму соединения с формулой (I-K), кроме того,после охлаждения ее можно отфильтровать из среды кристаллизации, затем промыть и высушить. Соедиенения формулы (I-S) (I-K), полученные способом по настоящему изобретению, вместе с фармацевтически приемлемым носителем можно включать в фармацевтические составы. Кристаллическая форма соединения с формулой (I-S) и кристаллическая форма соединения с формулой (I-K) далее могут использоваться в качестве ингибиторов натрийзависимых переносчиков глюкозы (SGLT2) и имеют очень высокую способность к снижению уровня глюкозы в крови. В конкретной реализации, кристаллическая форма соединения с формулой (I-S) и кристаллическая форма соединения с формулой (I-K), составляющие предмет настоящего изобретения, могут использоваться для лечения,профилактики и задержки развития или возникновения сахарного диабета (сахарного диабета 1 и 2 типа и т.д.), диабетических осложнений (таких как диабетическая ретинопатия, диабетическая нейропатия,диабетическая нефропатия), гипергликемии после приема пищи, замедленного заживления ран, рези- 15020209 стентности к инсулину, гипергликемии, повышенного содержания инсулина в крови, повышенного содержания жирных кислот в крови, повышенного содержания глицерина в крови, гиперлипидемии, ожирения, гипертриглицеридемии, синдрома X, атеросклероза или гипертонии. Фармацевтические составы, содержащие в качестве активного компонента одно или несколько соединений, описанных в данном документе, могут быть получены путем тщательного смешивания соединения или соединений с фармацевтическим носителем в соответствии с обычными методами приготовления фармацевтических составов. Носитель может быть выбран из широкого разнообразия в зависимости от нужного способа введения (например, перорального, парэнтерального). Так, для жидких препаратов для перорального применения, таких как суспензии, эликсиры и растворы, к возможным носителям и добавкам относятся вода, гликоли, масла, спирты, ароматизаторы, консерванты, стабилизаторы, красители и им подобные вещества; для твердых препаратов для перорального применения, таких как порошки,капсулы и таблетки, к возможным носителям относятся крахмалы, сахара, разбавители, гранулирующие агенты, смазки, связующие вещества, разрыхлители и им подобные вещества. Твердые препараты для перорального применения также могут иметь оболочку из таких веществ, как сахара, или иметь энтеросолюбильную оболочку для регулирования основного места всасывания препарата. В случае парэнтерального применения носитель обычно состоит из стерильной воды и других компонентов, добавляемых для повышения растворимости или сохраняемости препарата. Суспензии и растворы для инъекций также могут приготавливаться с использованием водных носителей с необходимыми добавками. Для приготовления фармацевтических составов одно или несколько соединений, составляющих предмет настоящего изобретения и являющихся активными компонентами, тщательно смешиваются с фармацевтическим носителем с помощью стандартных методов приготовления фармацевтических составов, при этом носитель может принимать различные формы в зависимости от предполагаемого способа введения препарата, например, перорального или парэнтерального (например, внутримышечного). Для приготовления состава в виде лекарственной формы для перорального применения можно использовать любой из обычных фармацевтических носителей. Так, для жидких препаратов для перорального применения, таких как, например, суспензии, эликсиры и растворы, к возможным носителям и добавкам относятся вода, гликоли, масла, спирты, ароматизаторы, консерванты, красители и им подобные вещества; для твердых препаратов для перорального применения, таких как порошки, капсулы, таблетки в форме капсулы, желатиновые капсулы и таблетки, к возможным носителям относятся крахмалы, сахара, разбавители, гранулирующие агенты, смазки, связующие вещества, разрыхлители и им подобные вещества. Ввиду простоты применения таблетки и капсулы представляют собой наиболее предпочтительную лекарственную форму, в которой, очевидно, используются твердые фармацевтические носители. При необходимости, на таблетки можно нанести сахарную или энтеросолюбильную оболочку с помощью стандартных методов. В случае парэнтеральных лекарственных форм носитель обычно состоит из стерильной воды, в которую могут добавляться другие компоненты, например, в целях повышения растворимости или сохраняемости. Возможно приготовление и составов для инъекций, в которых используются соответствующие жидкие носители, суспендирующие агенты и подобные им вещества. Описанные фармацевтические составы содержат в каждой стандартной лекарственной форме, например, таблетке, капсуле,порошке, составе для инъекций, чайной ложке и т.д., количество активного компонента, необходимое для получения эффективной дозы препарата, как описано выше. Описанные фармацевтические составы могут содержать в каждой стандартной лекарственной форме, например, в таблетке, капсуле, порошке,составе для инъекций, суппозитории, чайной ложке и т.д., приблизительно от 0,01 до 1000 мг или любое количество в пределах этого диапазона, и могут вводиться в дозах приблизительно от 0,01 до 300 мг/кг/день или в любом количестве в пределах указанного диапазона, предпочтительно приблизительно от 0,1 до 50 мг/кг/день или в любом количестве в пределах указанного диапазона. В то же время дозировка может меняться в зависимости от потребностей пациента, степени тяжести заболевания и используемого соединения. Может применяться как ежедневное введение, так и пост-периодическое применение препарата. Составы предпочтительно должны быть в виде стандартных лекарственных форм для разового применения, таких как таблетки, пилюли, капсулы, порошки, гранулы, стерильные растворы или суспензии для парэнтерального применения, дозированные аэрозоли или жидкие спреи, капли, ампулы, автоинъекторы или суппозитории; для перорального, парэнтерального, интраназального, сублингвального или ректального применения, или ведения путем ингаляции или инсуффляции. Состав может также быть представлен в форме, предназначенной для применения раз в неделю или раз в месяц; например, нерастворимая соль активного соединения, такая как соль каприновой кислоты, может использоваться для получения препарата замедленного действия для внутримышечного введения. Для получения препарата в твердых составах, таких как таблетки, главный активный компонент смешивается с фармацевтическим носителем, т.е. с обычными компонентами для таблетирования, такими как крахмал, лактоза, сахароза,сорбит, тальк, стеариновая кислота, стеарат магния, дикальцийфосфат или смолы, а также с другими фармацевтическими разбавителями, такими как вода, для получения готового твердого состава, содержащего однородную смесь составляющего соединения. Термин "однородные" применительно к таким готовым составам означает, что активный компонент равномерно распределен по всему объему состава таким образом, что состав можно разделить на лекарственные формы равной эффективности, такие как таблетки, пилюли или капсулы. Затем готовый твердый состав разделяют на стандартные лекарственные формы для однократного применения описанного выше типа, содержащие от 0,01 до приблизительно 1000 мг активного компонента, составляющего предмет настоящего изобретения. Таблетки или пилюли с новым составом могут дополнительно иметь оболочку или быть обработаны иным образом для обеспечения желательного для лекарственной формы продолжительного действия. Например, таблетка или пилюля может содержать внутренний и внешний компоненты с дозой вещества, при этом внешний компонент полностью окружает внутренний. Два компонента могут быть разделены энтеросолюбильным слоем, не распадающимся в желудке и сохраняющим внутренний компонент до попадания в двенадцатиперстную кишку или задерживающим его высвобождение. В качестве подобных энтеросолюбильных слоев и оболочек могут использоваться разные вещества, в том числе ряд полимерных кислот с такими материалами, как шеллак, цетиловый спирт и ацетат целлюлозы. К жидким формам для перорального применения или инъекций, в которые могут входить новые составы, составляющие предмет настоящего изобретения, относятся водные растворы, сиропы с подходящими ароматизаторами, водные или масляные суспензии и ароматизированные эмульсии на основе съедобных масел, таких как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и подобные им фармацевтические жидкости. Возможные диспергирующие или суспендирующие агенты для водных суспензий включают в себя синтетические или натуральные смолы, такие как трагакантовая камедь, аравийская камедь, альгинат, декстран, натрийкарбоксиметилцеллюлоза, метилцеллюлоза, поливинилпирролидон и желатин. Методы лечения, описанные в настоящем изобретении, могут также применяться с использованием фармацевтического состава, в который входит любое из определенных в данном документе соединений и фармацевтически приемлемый носитель. Фармацевтический состав может содержать от 0,01 мг до 1000 мг соединения или любое количество в указанном диапазоне; предпочтительно приблизительно от 10 до 500 мг соединения, и может быть изготовлен в виде любой лекарственной формы, приемлемой для выбранного способа применения. Носители включают необходимые и инертные фармацевтические наполнители, включая, но не ограничиваясь, связующие вещества, суспендирующие агенты, любриканты, ароматизаторы, подсластители, консерванты, красители и материал оболочки. Составы, предназначенные для перорального применения,включают твердые формы, такие как пилюли, таблетки, каплеты, капсулы (каждый тип включает составы для немедленного, задержанного или продолжительного высвобождения активного вещества), гранулы и порошки, а также жидкие формы, такие как растворы, сиропы, эликсиры, эмульсии и суспензии. Составы для парэнтерального применения включают стерильные растворы, эмульсии и суспензии. Преимуществом соединений, составляющих предмет настоящего изобретения, является то, что их можно применять в виде разовой ежедневной дозы, или разделять суточную дозу на две, три или четыре меньших отдельных доз. Кроме того, соединения, составляющие предмет настоящего изобретения, могут вводиться интраназально с помощью соответствующих интраназальных носителей или через трансдермальные кожные пластыри, известные специалистам в данной области. В случае применения трансдермальной формы введение дозы будет осуществляться, конечно, непрерывным образом, а не отдельными меньшими дозами на протяжении всего срока применения пластыря. Например, в случае перорального применения в форме таблеток или капсул активный компонент лекарства может сочетаться с пероральным нетоксичным фармацевтически приемлемым носителем, таким как этанол, глицерин, вода и подобные им вещества. Кроме того, по желанию или при необходимости в состав смеси могут быть включены соответствующие связующие вещества, смазки, разрыхлители и красители. Возможные связующие вещества включают, без ограничений, крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, сахаристые вещества из кукурузы, природные и синтетические смолы, такие как аравийская камедь, трагакантовая камедь или олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и подобные им вещества. Разрыхлители включают, без ограничений, крахмал, метилцеллюлозу, агар, бентонит, ксантановуюя смолу и подобные им вещества. Жидкие формы с соответствующими ароматизаторами таких в суспендирующих или диспергирующих веществах, как синтетические или природные смолы, например, трагакантовая камедь, аравийская камедь, метилцеллюлоза и подобные им вещества. Для парэнтерального применения желательно использовать стерильные суспензии и растворы. Для внутривенного применения используются изотонические композиции, которые обычно содержат соответствующие консерванты. Для приготовления фармацевтического состава, составляющего предмет настоящего изобретения,соединение, полученное в соответствии с каким-либо из описанных в данном документе процессов и являющееся активным компонентом, тщательно смешивается с фармацевтическим носителем с помощью стандартных методов приготовления фармацевтических составов, при этом носитель может принимать различные формы в зависимости предполагаемого способа введения препарата, например перорального или парэнтерального. Соответствующие фаврмацевтически приемлемые носители хорошо известны специалистам в данной области. Описание некоторых из фармацевтически приемлемых носителей можно найти в справочнике фармацевтических наполнителей The Handbook of Pharmaceutical Excipients, опубликованном Американской фармацевтической ассоциацией и Фармацевтическим обществом Великобритании. Методы приготовления фармацевтических составов описаны в многочисленных книгах, таких какPharmaceutical Dosage Forms: Disperse Systems, Volumes 1-2, edited by Lieberman et al; опубликованных издательством Marcel Dekker, Inc. Соединения, описанные в настоящем документе, могут вводиться в виде любого из описанных выше составов и в соответствии с графиком применения, обычным для лечения описанных в данном документе заболеваний. Суточная доза может варьировать в широких пределах от 0,01 до 1000 мг для взрослого человека в день или в любых пределах внутри указанного диапазона. Для перорального применения составы предпочтительно изготавливаются в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0,25,0, 30,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0 и 500 мг активного компонента для коррекции дозы в соответствии с симптомами конкретного пациента. Эффективное количество лекарства, как правило,вводится при уровне дозировки от приблизительно 0,01 мг/кг до приблизительно 300 мг/кг в день, или в любых пределах внутри указанного диапазона, предпочтительно в количестве от приблизительно 0,01 мг/кг до приблизительно 100 мг/кг или в любых пределах внутри указанного диапазона. Более предпочтительно, уровень дозировки находится в диапазоне от приблизительно 0,01 до приблизительно 50,0 мг/кг в день или в любых пределах внутри указанного диапазона, еще более предпочтительно в диапазоне от приблизительно 0,01 до приблизительно 30,0 мг/кг в день или в любых пределах внутри указанного диапазона. Соединения можно принимать по схеме лечения от 1 до 4 раз в день. Оптимальные дозы применения могут быть без труда определены специалистами в данной области и будут варьироваться в зависимости от применяемого соединения, способа применения, эффективности конкретной композиции и развития болезни. Кроме того, на дозировку могут влиять факторы, зависящие от конкретного пациента, такие как возраст, вес, диета и время применения. Специалисты в данной области определят, что испытания, проводимые как in vivo, так и in vitro на должным образом выбранных и общепризнанных клеточных и/или животных моделях, позволяют прогнозировать возможности испытываемого соединения по лечению или профилактике того или иного заболевания. Специалисты в данной области также определят, что клинические испытания на людях, в том числе впервые проведенные на человеке, тесты дозировки и эффективности, на здоровых пациентах и/или лицах, страдающих тем или иным заболеванием, могут быть выполнены с использованием методов, известных специалистам в клинической и медицинской области. Приведенные ниже примеры призваны помочь в понимании сути изобретения и не предназначены и не должны рассматриваться как какое-либо ограничение изобретения, содержание которого изложено далее в формуле изобретения. В приведенных ниже примерах некоторые продукты синтеза указаны как выделенные в виде остатка. Специалисты в данной области определят, что термин "остаток" не подразумевает ограничения возможного физического состояния, в котором был выделен продукт, и может распространяться, например,на твердое вещество, масло, пену, смолу, сироп и подобные им формы. Пример 1. (5-Бром-2-метилфенил)-[5- (4-фторфенил)тиофен-2-ил]метанон Стадия А. В трехгорлую круглодонную колбу объемом 250 мл помещали 5-бром-2-метилбензойную кислоту (22,5 г, 0,10 моль), CH2Cl2 (100 мл) и ДМФА (0,25 мл) при температуре окружающей среды(20 С). Добавляли оксалилхлорид (12 мл, 0,13 моль) таким образом, чтобы внутренняя температура оставалась ниже 25 С. Наблюдалось интенсивное выделение газа. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды в атмосфере аргона, затем летучие вещества удаляли при пониженном давлении. Образовавшийся остаток (хлорангидридное соединение) растворяли в ДХМ (50 мл) и оставляли в атмосфере азота. Стадия Б. В отдельную трехгорлую круглодонную колбу объемом 500 мл помещали AlCl3 (15,0 г,0,11 моль) и 100 мл CH2Cl2. Суспензию охлаждали до -10 С на ледяной бане и добавляли 2-(4 фторфенил)тиофен (18,2 г, 0,10 моль), затем добавляли смесь, подготовленную на стадии А выше. Через 30 мин ледяную баню убирали и получившуюся смесь перемешивали при комнатной температуре в течение 2-3 ч. Образовавшуюся смесь охлаждали до -12 С и гасили медленным добавлением воды (20 мл),затем 2 н. HCl (20 мл) и гептана (100 мл). Выпадал осадок. Получившуюся смесь перемешивали в течение 1-2 ч и отфильтровывали, получая указанное в заголовке соединение в виде твердого вещества желтого В четырехгорлую круглодонную колбу объемом 3,0 л помещали соединение, полученное в примере 1 выше (119 г, 0,317 моль), триэтилсилан (148 мл, 0,926 моль), дихлорметан (700 мл) и ацетонитрил (700 мл). Получившуюся смесь охлаждали до -8 С на ледяной бане при перемешивании, а затем по каплям добавляли диэтилэфират трехфтористого бора (115 мл, 0,915 моль) таким образом, что температура не поднималась выше 0 С. Получившуюся смесь нагревали до комнатной температуры и перемешивали в течение ночи. Образовавшуюся смесь концентрировали при пониженном давлении, разбавляли изопропанолом (1,0 л), отфильтровывали и промывали водой, получая твердое вещество. После перекристаллизации твердого вещества из изопропанола получали указанное в заголовке соединение в виде твердого вещества желтого цвета. Пример 3. 2-(4-Фторфенил)-5-(5-йод-2-метилбензил)тиофен В четырехгорлую колбу емкостью 1,0 л помещали соединение, полученное в примере 2 выше (100 г, 276,80 ммоль), иодид натрия (82 г, 553,59 ммоль) и иодид меди(I) (2,6 г, 13,84 ммоль). Полученную смесь откачивали и продували аргоном, затем обрабатывали толуолом (261 мл), диглимом (56 мл) и N,N'диметилэтан-1,2-диамином (2,7 мл, 27,68 ммоль), получившуюся смесь оставляли на ночь при температуре 110 С. После расходования всего исходного материала получившуюся смесь охлаждали до комнатной температуры, отфильтровывали через Celite, промывали EtOAc и экстрагировали NH4OH. Органическую фазу высушивали (Na2SO4), отфильтровывали и концентрировали, получая твердое вещество. Твердые вещества отфильтровывали и перекристаллизовывали из гептана с получением искомого соединения в виде практически твердого вещества белого цвета (т.пл. 107 С). (См. также Klaper, A., Buchwald,S. L., "Copper-Catalyzed Halogen Exchange in Aryl Halides: An Aromatic Finkelstein Reaction", J. Am. Chem. В трехгорлую круглодонную колбу объемом 5,0 л помещали глюконолактон (155,2 г, 0,871 моль) и 4-метилморфолин (766 мл, 6,96 моль) в ТГФ (1,55 л). В охлажденную (-10 С) смесь добавляли хлортриметилсилан (660 мл, 5,21 моль) с такой скоростью, чтобы температура не поднималась выше 5 С. Через 1 ч реакционную смесь нагревали примерно до 35-40 С на 5 ч, затем в течение ночи перемешивали при температуре окружающей среды в атмосфере аргона. Образовавшуюся смесь охлаждали до -10 С и медленно добавляли воду (500-600 мл) до прекращения интенсивного выделения тепла. Получившуюся смесь разбавляли 4,0 л воды и 2,5 л гептана. Слои разделяли, и органическую фазу промывали водным раствором одноосновного фосфата натрия (1,5 л), водой (1,0 л) и солевым раствором (1,0 л). Органический слой высушивали над сульфатом магния и концентрировали в вакууме, получая указанное в заголовке соединение в виде жидкости светло-желтого цвета. Пример 5. В трехгорлую круглодонную колбу объемом 2,0 л помещали соединение, полученное в примере 3 выше (100 г, 232,68 ммоль), соединение, полученное в примере 4 выше (141 г, 302,49 ммоль), и тетрагидрофуран (750 мл). После охлаждения полученной смеси примерно до -40 С, в нее через капельную воронку добавляли 1,0 М раствор (триметилсилил)метиллития в гексане (489 мл, 489 ммоль), поддерживая температуру смеси около -40 С. Затем смесь гасили стандартным NaHCO3 (200 мл) и давали ей нагреться до комнатной температуры. Полученные фазы разделяли, высушивали (Na2SO4), отфильтровывали и концентрировали, получая указанное в заголовке соединение в виде густого маслянистого вещества. Пример 6. 1-(-D-Глюкопиранозил)-4-метил-3-(5-(4-фторфенил)-2-тиенилметил)бензол В трехгорлую круглодонную колбу объемом 2,0 л помещали соединение, полученное в примере 5 выше (232 г, 310 ммоль), и дихлорэтан (700 мл). Образовавшийся желтый раствор охлаждали до -30 С на ледяной бане при перемешивании. В раствор добавляли триэтилсилан (132 мл, 826 ммоль), затем медленно добавляли (в течение 1,75 ч) диэтилэфират трехфтористого бора (95,0 мл, 756 ммоль) таким образом, чтобы температура смеси не поднималась выше -20 С. Примерно через 30 мин после добавления эфирата ледяную баню убирали, и образовавшуюся желтую смесь перемешивали при температуре окружающей среды в атмосфере аргона в течение 1,0-1,5 ч. После завершения реакции образовавшуюся смесь выливали в холодную воду (800 мл), добавляли этилацетат (300 мл) и слои разделяли. Органический слой промывали насыщенным раствором бикарбоната, высушивали над сульфатом натрия и концентрировали, получая указанное в заголовке соединение в виде пены зеленого цвета. Пример 7. В трехгорлую круглодонную колбу объемом 2,0 л помещали соединение, полученное в примере 6 выше (119 г, 0,25 моль), 4-метилморфолин (145 мл, 1,30 моль), DMAP (3,25 г, 0,026 моль) и 1,0 л ТГФ. Получившуюся светло-зеленую смесь охлаждали до -10 С на ледяной бане при перемешивании, затем по каплям добавляли уксусный ангидрид (125 мл, 1,30 моль) таким образом, чтобы температура смеси не поднималась выше 0 С. Через 15 мин после введения ангидрида ледяную баню убирали. Получившуюся смесь перемешивали при температуре окружающей среды в течение 1,0 ч, затем концентрировали при пониженном давлении при 30-35 С, чтобы удалить большую часть растворителя. Получившуюся смесь разбавляли 10% фосфорной кислотой (приблизительно 300 мл), что приводило к образованию осадка кремового цвета. Полученную смесь растворяли в смеси этилацетата (600-800 мл), ТГФ (200-300 мл) и толуола (200-300 мл). После получения готового раствора слои разделяли, органический слой промывали насыщенным раствором бикарбоната и солевым раствором, затем высушивали и концентрировали, получая густой остаток. В остаток добавляли метанол, после чего из раствора выпадало практически белое твердое вещество. Кашицу перемешивали в течение 30 мин, затем отфильтровывали и получали указанное в заголовке соединение в виде твердого вещества практически белого цвета. Пример 8. 1-(-D-Глюкопиранозил)-4-метил-3-(5-(4-фторфенил)-2-тиенилметил)бензол В колбу помещали соединение, полученное в примере 7 выше (185 г, 302 ммоль), в смеси ТГФ (820 мл) и МеОН (1,23 л). В перемешиваемую суспензию добавляли раствор моногидрата гидроксида лития(6,33 г, 147 ммоль) в воде (410 мл). После перемешивания в течение ночи при температуре окружающей среды летучие вещества удаляли, а образовавшийся осадок разбавляли этилацетатом (500-600 мл). Слои разделяли, водный слой экстрагировали этилацетатом (3100 мл). Объединенный органический слой промывали солевым раствором (250 мл), высушивали над сульфатом натрия и концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде хрупкой пены. Пример 9. Кристаллизация 1-(-D-глюкопиранозил)-4-метил-3-(5-(4-фторфенил)-2-тиенилметил) бензола В трехгорлую круглодонную колбу объемом 1,0 л помещали соединение, полученное в примере 8 выше (96,9 г, 217 ммоль), воду (6,0 мл, 333 ммоль) и этилацетат (275 мл). Образовавшийся раствор нагревали до 35 С при перемешивании в атмосфере аргона. По каплям добавляли гептан до помутнения раствора (155 мл гептана), затем добавляли 14,2 г затравочных кристаллов. После перемешивания в течение 1,5-2,0 ч при 35 С добавляли еще одну порцию гептана (30 мл, до полного количества 185 мл). Полученную смесь перемешивали в течение еще 30 мин и отфильтровывали. Фильтровальный осадок промывали приблизительно 56% смесью этилацетат/гептан (50 мл) и высушивали, получая указанное в заголовке соединение в виде рыхлого кристаллического вещества практически белого цвета. Процедуры, описанные в примерах 1-9 выше, были повторены несколько раз, было получено несколько партий l-( -D-глюкопиранозил)-4-метил-3-(5-(4-фторфенил)-2-тиенилметил)бензола - соединения формулы (I-S). Для типичного образца соединения формулы (I-S) (приготовленного согласно процедурам в примерах 1-9) получены следующие температура плавления, масс-спектры и спектры 1 Н-ЯМР. Температура плавления: 106-107 С. Масс-спектр: m/z (LCMS API-ES) 467 (M+Na). 1H-ЯМР (CD3OD): =2,32 (с, 3 Н), 3,35-3,53 (м, 4 Н), 3,71 (д, 1 Н, J=11,9 Гц), 3,90 (д, 1 Н, J=11,9 Гц),4,13 (д, 1 Н, J=9,3 Гц), 4,17 (с, 2 Н), 4,9 (с, 4 Н), 6,70 (д, 1 Н, J=3,7 Гц), 7,04-7,14 (м, 3 Н), 7,18 (д, 1 Н, J=7,8 Гц), 7,26 (д, 1 Н, J=7,8 Гц), 7,33 (с, 1 Н), 7,52-7,60 (м, 2 Н). Типичный образец кристаллической формы соединения формулы (I-S), выделенного в соответствии с описанием в примере 9 выше, охарактеризовывали методом порошковой рентгеновской дифрактометрии (типичный пример показан на фиг. 1), используя дифрактометр с излучением CuK, 30 мА, 40 кВ,щель расходимости 1/12, приемная щель 0,2; сканирование от 4 до 35 2 со скоростью 0,016 2 в секунду; с алюминиевым держателем образца. Кристаллическая форма соединения формулы (I-S) может быть охарактеризована по пикам порошковой рентгеновской дифракции (предпочтительно, по пикам порошковой рентгеновской дифракции с относительной интенсивностью выше, чем приблизительно 10%, более предпочтительно - по пикам порошковой рентгеновской дифракции с относительной интенсивностью выше, чем приблизительно 25%,еще более предпочтительное - по пикам порошковой рентгеновской дифракции с относительной интенсивностью выше, чем приблизительно 35%, и еще предпочтительнее - по пикам порошковой рентгеновской дифракции с относительной интенсивностью выше, чем приблизительно 50%), приведенным в табл.1 ниже. Таблица 1. Пики порошковой рентгеновской дифракции кристаллической формы соединения формулы (I-S) Стадия А. В четырехгорлую круглодонную колбу объемом 5,0 л помещали 2-хлор-5-йодбензойную кислоту (470,8 г, 1,66 моль), CH2Cl2 (1,6 л) и ДМФА (5,0 мл, 0,03 моль) при температуре окружающей среды (20 С). Добавляли оксалилхлорид (170 мл, 1,94 моль) таким образом, чтобы температура смеси в колбе оставалась ниже 25 С. Наблюдался небольшой разогрев и интенсивное выделение газа. Полученную смесь перемешивали в течение ночи при температуре окружающей среды в атмосфере аргона, затем летучие вещества удаляли при пониженном давлении. Образовавшийся остаток (хлорангидридное соединение) разбавляли дихлорметаном (500 мл) и оставляли в атмосфере азота. Стадия Б. В отдельную трехгорлую круглодонную колбу объемом 5,0 л помещали AlCl3 (487,0 г,3,65 моль) и 1,5 л CH2Cl2. В охлажденную (-12 С) смесь добавляли 2-фтор-5-(2-тиенил)пиридин (299,0 г,1,66 моль) и затем смесь, полученную на стадии А выше. Через 20 мин ледяную баню убирали, реакционную смесь перемешивали при температуре окружающей среды в течение 2-3 ч. После завершения реакции образовавшуюся смесь охлаждали до -12 С и гасили медленным добавлением воды (400-500 мл), затем 2 н. HCl (100 мл) и гептана (100 мл). Во время гашения водой реакционной смеси не давали нагреваться выше 32 С. Полученную смесь перемешивали при температуре окружающей среды в течение ночи, в результате получая осадок. Полученную смесь отфильтровывали, промывали водой и высушивали, получая твердое вещество. Твердое вещество перекристаллизовывали из этилацетата, получая указанное в заголовке соединение в виде твердого вещества золотистого цвета. Пример 11. 5-[5-(2-Хлор-5-йодбензил)тиофен-2-ил]-2-фторпиридин В четырехгорлую круглодонную колбу объемом 5,0 л помещали соединение, полученное в примере 10 выше (350 г, 0,787 моль), триэтилсилан (650 мл, 4,07 моль) и ацетонитрил (1,75 л). Полученную смесь нагревали до 30 С и по каплям добавляли диэтилэфират трехфтористого бора (500 мл, 3,98 моль) таким образом, чтобы температура смеси не поднималась выше 58 С. Перемешивание продолжали при температуре окружающей среды. После завершения полученную смесь добавляли в охлажденный (5 С) водный раствор бикарбоната натрия (400 г в 2,0 л воды). Водную смесь перемешивали при температуре окружающей среды в течение часа, а затем разбавляли этилацетатом (500 мл). Слои разделяли, водный слой экстрагировали этилацетатом (2400 мл). Объединенный органический слой промывали солевым раствором, высушивали и концентрировали, получая твердое вещество светло-коричневого цвета. Твердое вещество растворяли в горячем толуоле (приблизительно 1,5-1,75 л), обрабатывали силикагелем (250 г), разбавляли гептаном (1,0 л), перемешивали в течение 30-40 мин и отфильтровывали при повышенной температуре. Объем смеси уменьшался, к смеси добавляли новую порцию гептана. При охлаждении до комнатной температуры из раствора выпадало твердое вещество. Полученную смесь отфильтровывали,получая указанное в заголовке соединение в виде твердого вещества желтого цвета. Пример 12. В коническую колбу объемом 1 л помещали соединение, полученное в примере 11 выше (94,4 г,219,70 ммоль), соединение, полученное в примере 4 выше (102 г, 219,70 ммоль), и тетрагидрофуран (585 мл). Полученную смесь отфильтровывали через стеклянный фильтр с Celite и молекулярными ситами 4 АЕ (10 г) в трехгорлую круглодонную колбу объемом 2,0 л, на которую установили мешалку, выход для азота, термопару и капельную воронку с вакуумным адаптером. Затем полученную смесь охлаждали до-70 С на бане с сухим льдом/ацетоном. В капельную воронку помещали 1,0 М раствор (триметилсилил)метиллития в гексане (450 мл; 450 ммоль), температуру смеси поддерживали ниже приблизительно-60 С. По завершении добавления алкиллития полученной смеси давали нагреться до -30 С, гасили, выливая в перемешиваемую смесь NaHCO3 (400 мл, 50% насыщенный) в делительной воронке объемом 2 л,разбавляли гептаном (200 мл) и фазы разделяли. Органическую фазу промывали водой (20 мл) и солевым раствором (50 мл), затем отделяли и высушивали (Na2SO4), отфильтровывали и концентрировали, получая указанное в заголовке соединение в виде густого маслянистого вещества. Пример 13 В трехгорлую круглодонную колбу объемом 2 л, оборудованную холодной баней, капельной воронкой, датчиком температуры, выходом для азота и мешалкой, помещали вещество, полученное в примере 11 выше (100 г, 232,73 ммоль), и вещество, полученное в примере 4 выше (130,4 г, 325,8 ммоль),затем добавляли ТГФ (660 мл). Затем полученную смесь охлаждали до -70 С на бане с сухим льдом/ацетоном. В капельную воронку помещали триметилсилилметиллитий (210 мл; 413,70 ммоль),который медленно добавляли в реакционную смесь, чтобы ее температура оставалась ниже приблизительно -70 С. После добавления алкиллития полученную смесь перемешивали еще 20 минут. В полученную смесь через капельную воронку добавляли 2 М раствор HCl (250 мл; 500,00 ммоль). Полученной смеси давали нагреться до комнатной температуры, переносили ее в делительную воронку, и экстрагировали этилацетатом (2200 мл). Органическую фазу отделяли и высушивали (MgSO4), полученную смесь отфильтровывали и концентрировали, получая указанное в заголовке соединение в виде густого маслянистого вещества. Пример 14. В четырехгорлую круглодонную колбу объемом 3,0 л помещали соединение, полученное в примере 13 выше (112 г, 0,23 моль), и ацетонитрил (1,0 л). Образовавшуюся смесь охлаждали до -20 С на ледяной бане при перемешивании. В раствор добавляли триэтилсилан (185 мл, 1,16 моль), затем медленно добавляли диэтилэфират трехфтористого бора (150 мл, 1,20 моль) таким образом, чтобы температура смеси не поднималась выше -20 С. После добавления эфирата образовавшейся смеси темно-оранжевого цвета давали медленно нагреться до 0 С. Затем медленно добавляли водный раствор бикарбоната натрия (200 г в 500 мл дистиллированной воды) и слои разделялись. Органический слой концентрировали, чтобы удалить большую часть ацетонитрила, а затем разбавляли этилацетатом (350 мл). Водный слой насыщали хлоридом натрия и экстрагировали этилацетатом (350 мл). Объединенный органический слой промывали насыщенным раствором хлорида натрия (100 мл), высушивали над сульфатом натрия (135 г) и концентрировали, получая указанное в заголовке соединение в виде пены желтого цвета. Пример 15 В трехгорлую круглодонную колбу объемом 500 мл помещали соединение, полученное в примере 14 выше (23,56 г, 50,0 ммоль), 4-метилморфолин (27,5 мл, 250 ммоль) и DMAP (0,60 г, 4,86 ммоль) в ТГФ(160 мл). Получившуюся смесь желтого цвета охлаждали до -10 С на ледяной бане при перемешивании,затем по каплям добавляли уксусный ангидрид (23,6 мл, 250 ммоль) таким образом, чтобы температура смеси не поднималась выше 0 С. Через 15 мин после введения ангидрида ледяную баню убирали. Получившуюся смесь перемешивали при температуре окружающей среды в течение 1,5 ч, затем концентрировали при пониженном давлении при температуре приблизительно 30-35 С, чтобы удалить большую часть растворителя. Получившийся остаток растворяли в этилацетате (100-150 мл) и разбавляли 1 н. HCl(100-150 мл). Слои разделяли, водный слой экстрагировали этилацетатом (2x30 мл). Объединенный органический слой промывали водой, насыщенным раствором бикарбоната и солевым раствором, используя по 100 мл каждого раствора, и концентрировали, получая влажное твердое вещество. Твердое вещество перекристаллизовывали из горячего метанола (300-425 мл), получая указанное в заголовке соединение в виде твердого вещества светло-желтого цвета. Пример 16. 1-(-D-глюкопиранозил)-4-метил-3-(5-(6-фторпирид-3-ил)-2-тиенилметил)бензол В одногорлую круглодонную колбу объемом 250 мл помещали соединение, полученное в примере 15 выше (8,52 г, 13,4 ммоль) в смеси ТГФ (50 мл) и метанола (50 мл). В перемешиваемую суспензию добавляли 3 н. бикарбонат натрия (1,2 мл, 3,60 ммоль). Полученную смесь перемешивали в течение 1 ч при температуре окружающей среды. Летучие вещества удаляли, полученный остаток разбавляли этилацетатом (50 мл). Слои разделяли, водный слой экстрагировали этилацетатом (310 мл). Объединенный органический слой промывали солевым раствором, высушивали над сульфатом натрия, отфильтровывали и концентрировали до половины объема, получая твердый осадок. Указанное в заголовке соединение выделяли фильтрованием в виде твердого вещества кремового цвета. Для типичного образца соединения формулы (I-K) (приготовленного согласно процедурам в примерах выше) получены следующие температура плавления, масс-спектры и спектры 1 Н-ЯМР. Температура плавления: 130-132 С. Масс-спектр: m/z (LCMS API-ES) 466 (М+Н). 1 Н-ЯМР (DMSO-d6): =3,05-3,31 (м, 4 Н), 3,45 (дт, 1 Н, J=5,3 Гц, J=12,2 Гц), 3,70 (дд, 1 Н, J=5,3 Гц,J=ll,4 Гц), 4,02 (д, 1 Н, J=9,7 Гц), 4,28 (д, 2 Н, J=3,5 Гц), 4,46 (т, 1 Н, J=6,2 Гц), 4,89 (д, 1 Н, J=6,2 Гц), 4,99 (д,2 Н, J=5,3 Гц), 6,93 (д, 1 Н, J=3,5 Гц), 7,21 (дд, 1 Н, J=3,5 Гц, J=8,3 Гц), 7,28 (дд, 1 Н, J=2,0 Гц, J=8,3 Гц),7,39-7,48 (м, 3 Н), 8,17 (ддд, 1 Н, J=16,2 Гц, J=8,3 Гц, J=2,6 Гц), 8,46 (с, 1 Н). Соединение формулы (I-K), приготовленное, например, в соответствии с описанием в примере 16 выше, можно охарактеризовать по пикам порошковой рентгеновской дифракции (предпочтительно, по пикам порошковой рентгеновской дифракции с относительной интенсивностью выше, чем приблизительно 10%, более предпочтительно - по пикам порошковой рентгеновской дифракции с относительной интенсивностью выше, чем приблизительно 25%, еще более предпочтительно - по пикам порошковой рентгеновской дифракции с относительной интенсивностью выше, чем приблизительно 35%, еще более предпочтительно - по пикам порошковой рентгеновской дифракции с относительной интенсивностью выше, чем приблизительно 50%), приведенным в табл.2 ниже. Таблица 2. Пики порошковой рентгеновской дифракции кристаллической формы соединения формулы (I-K) Пример 17. Кристаллизация 1-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-3-пиридил)-2 тиенилметил]бензола. 1-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-3-пиридил)-2-тиенилметил]бензол (пена, 23,1 г; получен как описано в публикации РСТ WO 2005/012326) растворяли в этилацетате (345 мл) и добавляли в раствор затравочный кристалл l-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-3-пиридил)-2-тиентилметил] бензола. Смесь кипятили с обратным холодильником в течение 30 мин, затем перемешивали при 50 С в течение 14 ч. После охлаждения до комнатной температуры осадок собирали фильтрованием, промывали этилацетатом (100 мл) и высушивали, получая бесцветные кристаллы 1-(-D-глюкопиранозил)-4-хлор-3[5-(6-фтор-3-пиридил)-2-тиенилметил]бензола (20,34 г). Т.пл. 131-134 С. Элементный анализ рассчитано для C22H21CIFNO5S: С 56,71; Н 4,54; N 3,01; F 4,08; Cl 7,61; S 6,88; получено: С 56,59; Н 4,55; N 3,01; F 4,00; Cl 7,60; S 6,94. Пример 18.1-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-3-пиридил)-2-тиенилметил]бензол. К раствору 1-(2,3,4,6-тетра-О-ацетил-1D-глюкопиранозил)-4-хлор-3-(5-(6-фтор-3-пиридил)-2 тиенилметил)бензола (9,64 г; получен как описано в публикации РСТ WO 2005/012326) в смеси метанол:тетрагидрофуран (75:75 мл) добавляли раствор метоксида натрия в метаноле (28%, 0,09 мл), полученную смесь перемешивали при комнатной температуре в атмосфере аргона в течение 1,5 ч. Органический растворитель выпаривали при пониженном давлении и добавляли солевой раствор (200 мл). Смесь экстрагировали этилацетатом (500 мл) и высушивали органический слой над сульфатом магния. После обработки активированным углем нерастворимые вещества отфильтровывали и фильтрат выпаривали при пониженном давлении. Остаток растворяли в этилацетате (60 мл) и добавляли затравочные кристаллы 1-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-3-пиридил)-2-тиенилметил]бензола. Смесь перемешивали при 50 С в течение 2,5 ч, кипятили с обратным холодильником в течение 45 мин и перемешивали при комнатной температуре в течение ночи. Осажденные кристаллы измельчали и смесь снова перемешивали при 50 С в течение 30 мин, кипятили с обратным холодильником в течение 45 мин и перемешивали при комнатной температуре в течение ночи. Осажденные кристаллы собирали, дважды промывали этилацетатом (40 мл) и высушивали, получая бесцветные кристаллы 1-(-D-глюкопиранозил)-4-хлор-3-[5-(6 фтор-3-пиридил)-2-тиенилметил]бензола (5,59 г). Т.пл. 131-133 С. Пример 19. Справочный пример А Стадия (1). Получение 1-(2,3,4,6-тетра-О-ацетилD-глюкопиранозил)-4-хлор-3-[5-(6-фтор-3 пиридил)-2-тиенилметил]бензола. Суспензию 1-(2,3,4,6-тетра-O-ацетилD-глюкопиранозил)-4-хлор-3-(5-бром-2-тиенилметил)бензола (13,5 г; получен как описано в публикации РСТ WO 2005/012326), 2-фторпиридин-5-борной кислоты (Frontier Scientific, 4,63 г), фторида цезия (19,96 г) и тетракис(трифенилфосфин)палалдия(0) (2,53 г) в 1,2-диметоксиэтане (200 мл) кипятили с обратным холодильником в течение 1,5 ч. Реакционную смесь выливали в насыщенный водный раствор гидрокарбоната натрия и экстрагировали этилацетатом. Органический слой промывали солевым раствором, высушивали над сульфатом натрия и выпаривали растворитель при пониженном давлении. Остаток растворяли в этилацетате и смесь обрабатывали активированным углем, а затем отфильтровывали через сигикагель, обработанный аминосиланом (27 мл). Фильтрат выпаривали при пониженном давлении, остаток очищали хроматографией на колонке с силикагелемF, 3,0; N, 2,21; S, 5,06. Получено: С, 56,8; Н, 4,47; Cl, 5,6; F, 2,91; N, 2,29; S, 4,93. Стадия (2). Получение 1-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-3-пиридил)-2-тиенилметил] бензола. Соединение, полученное на стадии (1) выше (8,33 г), растворяли в смеси метанол (200 мл) - тетрагидрофуран (100 мл), добавляли метоксид натрия (28% раствор в метаноле, 5 капель), и перемешивали смесь при комнатной температуре в течение 4 ч. Растворитель выпаривали при пониженном давлении,остаток очищали хроматографией на колонке с силикагелем (хлороформ:метанол 100:0-88:12) и растирали в смеси изопропиловый эфир - 2-пропанол, получая l-(-D-глюкопиранозил)-4-хлор-3-[5-(6-фтор-3 пиридил)-2-тиенилметил]бензол (4,61 г) в виде бесцветного порошка.H-ЯМР (ДМСО-d6)3,07-3,27 (м, 4 Н), 3,38-3,49 (м, 1 Н), 3,67-3,80 (м, 1 Н), 4,02 (д, J=9,4 Гц, 1 Н),4,27 (вид. д, J=3,1 Гц, 2 Н), 4,33 (д, J=4,2 Гц, 1 Н), 4,85 (д, J=5,7 Гц, 1 Н), 4,95 (дд, J=5,0, 3,8 Гц, 2 Н), 6,92 (д,J=3,7 Гц, 1 Н), 7,18-7,22 (м, 1 Н), 7,26-7,29 (м, 1 Н), 7,40-7,44 (м, 3 Н), 8,13-8,19 (м, 1 Н), 8,44-8,45 (м, 1 Н). Пример 20. В качестве конкретной реализации состава для перорального применения, 100 мг соединения, полученного в примере 9, смешивают с достаточным количеством мелкоизмельченной лактозы, получая общее количество от 580 до 590 мг для заполнения твердой гелевой капсулы размера 0. Приведенное выше техническое описание содержит сведения о принципах, лежащих в основе настоящего изобретения, и иллюстрирующие его примеры, однако следует понимать, что практическое применение изобретения распространяется на все обычные вариации, адаптации и/или модификации, в соответствии с нижеприведенными пунктами и их эквивалентами. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы (I) где кольцо А представляет собой бензольное кольцо, необязательно замещенное заместителями,выбранными из группы, состоящей из атома галогена, низшей алкильной группы, галогензамещенной низшей алкильной группы, низшей алкоксильной группы и фенильной группы; кольцо В представляет собой гетероциклическое кольцо, выбранной из группы, содержащей тиофен, фуран, бензофуран, бензотиофен и бензотиазол, где гетероциклическое кольцо необязательно замещено заместителем, выбранным из группы, содержщей атом галогена, цианогруппу, низшую алкильную группу, галогензамещенную низшую алкильную группу, фенилзамещенную низшую алкильную группу,низшую алкоксильную группу, галогензамещенную низшую алкоксильную группу, фенильную группу,галогензамещенную фенильную группу, низшую алкилфенильную группу, низшую алкоксифенильнуюY - группа -(СН 2)n-; где n равно 1 или 2; при условии, что в кольце А X участвует в ненасыщенной связи; или его фармацевтически приемлемой соли,включающий взаимодействие соединения формулы (V) с соединением формулы (VI-S) в присутствии алкиллития, где соединение алкиллития выбрано из группы, состоящей из триметилсилилметиллития, 2,4,6 триметилфениллития и триэтилсилилметиллития, в органическом растворителе при температуре от приблизительно 0 до приблизительно -78 С с получением соответствующего соединения формулы (VII); и где алкиллитий добавляют в смесь соединения формулы (V) и соединения формулы (VI-S) взаимодействие соединения формулы (VII) с BF3OEt2 в присутствии триалкилсилана в органическом растворителе с образованием соответствующего соединения формулы (VIII); взаимодействие соединения формулы (VIII) с уксусным ангидридом или ацетилхлоридом в присутствии органического основания, в чистом виде или в органическом растворителе, с образованием соответствующего соединения формулы (IX); и снятие защитных групп в соединении формулы (IX) с образованием соответствующего соединения формулы (I). 2. Способ по п.1, где соединение формулы (VI-S) присутствует в количестве от приблизительно 1,0 до приблизительно 1,25 мол.экв. в расчете на соединение формулы (V). 3. Способ по п.1, где алкиллитий представляет собой (триметилсилил)метиллитий и где алкиллитий присутствует в количестве от приблизительно 2,0 до приблизительно 2,5 мол.экв. в расчете на соединение формулы (V). 4. Способ по п.1, где BF3OEt2 присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (VII) и где триалкилсилан представляет собой Et3SiH, который присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (V). 5. Способ по п.4, где молярное соотношение BF3OEt2 : Et3SiH составляет приблизительно 1:1. 6. Способ по п.1, где соединение формулы (VIII) вводят в реакцию с уксусным ангидридом, который присутствует в количестве от приблизительно 4,5 до приблизительно 5,0 мол.экв. в расчете на соединение формулы (VIII). 7. Способ по п.1, где органическим основанием является N-метилморфолин (NMM). 8. Способ по п.1, где соединение формулы (VIII) вводят в реакцию с уксусным ангидридом в присутствии каталитического количества 4-(N,N-диметиламино)пиридина (DMAP). 9. Способ по п.1, где из соединения формулы (IX) удаляют защитные группы посредством взаимо- 27020209 действия с основанием. 10. Способ получения соединения формулы (I-S) или его фармацевтически приемлемой соли, включающий взаимодействие соединения формулы (V-S) с соединением формулы (VI-S) в присутствии алкиллития, где соединение алкиллития выбрано из группы, состоящей из триметилсилилметиллития, 2,4,6 триметилфениллития и триэтилсилилметиллития, в органическом растворителе при температуре от приблизительно 0 до приблизительно -78 С с получением соответствующего соединения формулы (VII-S); и где алкиллитий добавляют в смесь соединения формулы (V-S) и соединения формулы (VI-S) взаимодействие соединения формулы (VII-S) с BF3OEt2 в присутствии триалкилсилана в органическом растворителе с образованием соответствующего соединения формулы (VIII-S); взаимодействие соединения формулы (VIII-S) с уксусным ангидридом или ацетилхлоридом в присутствии органического основания, в чистом виде или в органическом растворителе с образованием соответствующего соединения формулы (IX-S); и снятие защитных групп в соединении формулы (IX-S) с образованием соответствующего соединения формулы (I-S). 11. Способ по п.10, где соединение формулы (VI-S) присутствует в количестве от приблизительно 1,0 до приблизительно 1,25 мол.экв. в расчете на соединение формулы (V-S). 12. Способ по п.10, где алкиллитий представляет собой (триметилсилил)метиллитий и где алкиллитий присутствует в количестве от приблизительно 2,0 до приблизительно 2,5 мол.экв. в расчете на соединение формулы (V-S). 13. Способ по п.10, где BF3OEt2 присутствует в количестве от приблизительно 2,0 до приблизитель- 28020209 но 6,0 мол.экв. в расчете на соединение формулы (VII-S) и где триалкилсилан представляет собой Et3SiH,который присутствует в количестве от приблизительно 2,0 до приблизительно 6,0 мол.экв. в расчете на соединение формулы (VII-S). 14. Способ по п.13, где молярное соотношение BF3OEt2 : Et3SiH составляет 1:1. 15. Способ по п.10, где соединение формулы (VIII-S) вводят в реакцию с уксусным ангидридом, который присутствует в количестве от приблизительно 4,5 до приблизительно 5,0 мол.экв. в расчете на соединение формулы (VIII-S). 16. Способ по п.10, где органическим основанием является NMM. 17. Способ по п.10, где соединение формулы (VIII-S) вводят в реакциюс уксусным ангидридом в присутствии каталитического количества DMAP. 18. Способ по п.10, где соединение формулы (IX-S) дополнительно суспендируют в метаноле и фильтруют. 19. Способ по п.10, где из соединения формулы (IX-S) удаляют защитные группы посредством взаимодействия с основанием. 20. Способ перекристаллизации соединения формулы (I-S)(a) растворение соединения формулы (I-S) в органическом растворителе с получением смеси А;(b) нагревание смеси А до температуры от приблизительно 25 до приблизительно 45 С с образованием смеси В;(c) добавление приблизительно от 1,0 до 2,0 мол.экв. воды в расчете на соединение формулы (I-S) в смесь В с получением смеси С;(d) добавление гептана в смесь С с образованием суспензии соединения формулы (I-S); и(e) выделение соединения формулы (I-S). 21. Способ по п.20, где органический растворитель представляет собой этилацетат. 22. Способ по п.20, где смесь А нагревают до температуры от приблизительно 30 до приблизительно 35 С. 23. Способ по п.20, где приблизительно 1,5 мол.экв. воды в расчете на соединение формулы (I-S) добавляют в смесь В. 24. Способ по п.21, где гептан добавляют в смесь С в количестве, дающем конечное объемное соотношение этилацетат:гептан, равное приблизительно 1,2:1,0. 25. Способ получения соединения формулы (I-K) или его фармацевтически приемлемой соли; включающий взаимодействие соединения формулы (V-K) с соединением формулы (VI-S) в присутствии алкиллития, где соединение алкиллития выбрано из группы, состоящей из триметилсилилметиллития, 2,4,6 триметилфениллития и триэтилсилилметиллития, в органическом растворителе при температуре от при- 29
МПК / Метки
МПК: A61K 31/381, A61P 3/00, C07D 409/10
Метки: соединений, получения, способ, натрийзависимого, качестве, переносчика, ингибиторов, глюкозы, применимых
Код ссылки
<a href="https://eas.patents.su/30-20209-sposob-polucheniya-soedinenijj-primenimyh-v-kachestve-ingibitorov-natrijjzavisimogo-perenoschika-glyukozy.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения соединений, применимых в качестве ингибиторов натрийзависимого переносчика глюкозы</a>
Способ получения промежуточных соединений, применимых для получения противораковых соединений

Номер патента: 5561
Опубликовано: 28.04.2005
Авторы: Лехнер Ричард Шелтон, Сантафьянос Динос Пол, Норрис Тимоти
МПК: C07D 239/94
Метки: применимых, соединений, промежуточных, получения, противораковых, способ
Формула / Реферат:
1. Способ получения соединения формулы 3 где R1 и R2, каждый независимо, выбраны из C1-C10алкила и C1-C10алкокси, причем указанные алкил и алкокси необязательно имеют до 2 заместителей, независимо выбранных из гидрокси и C1-C6алкокси; где соединение формулы 3 получают обработкой соединения формулы 5 где R1 и R2 определены как указано выше, тионилхлоридом в безводном дихлорметане. 2. Способ по п.1, где как R1, так и R2 являются...
Производные 1,2-дизамещенного 4-бензиламинопирролидина в качестве ингибиторов белка-переносчика холестерилового эфира, используемые для лечения заболеваний, таких как гиперлипидемия или артериосклероз

Номер патента: 17939
Опубликовано: 30.04.2013
Авторы: Омори Осаму, Моги Мунето, Мияке Такахиро, Ясошима Кайо, Имасе Хидетомо, Каванами Тошио, Ямада Кен
МПК: A61K 31/40, A61P 3/06, C07D 207/14...
Метки: используемые, артериосклероз, заболеваний, качестве, холестерилового, белка-переносчика, производные, таких, 1,2-дизамещенного, 4-бензиламинопирролидина, гиперлипидемия, лечения, эфира, ингибиторов
Формула / Реферат:
1. Соединение формулы (I)в которой R1 обозначает (C1-C7)алкил-O-C(O)-, (C1-C7)алканоил, пиримидинил или тетразолил, где указанный пиримидинил необязательно содержит от 1 до 3 заместителей, выбранных из группы, включающей галоген, ди(C1-C7)алкиламиногруппу, (C1-C7)алкоксигруппу, гетероциклил, где заместитель гетероциклил дополнительно необязательно содержит от 1 до 3 заместителей, выбранных из группы, включающей (C1-C7)алкил, гидроксигруппу или...
Производные пиримидона в качестве промежуточных соединений для получения ингибиторов gsk3&beta или gsk3&beta и cdk5/p25

Номер патента: 9456
Опубликовано: 28.12.2007
Авторы: Маргери Северен, Локхид Алистер, Галле Тьерри, Саади Мурад, Неделек Ален, Йеш Филипп, Словински Франк, Ларденуа Патрик
МПК: A61K 31/519, A61P 25/28, C07D 235/00...
Метки: ингибиторов, промежуточных, соединений, gsk3&beta, пиримидона, получения, производные, качестве
Формула / Реферат:
1. Производное пиримидона, представленное формулой (III) где R1 представляет собой пиримидиновое кольцо, возможно замещенное С3-6циклоалкильной группой, С1-4алкильной группой, С1-4алкоксигруппой, бензильной группой или атомом галогена; R3 и R4 представляют собой, каждый независимо, атом водорода, С1-6алкильную группу, гидроксигруппу, С1-4алкоксигруппу или атом галогена; R5 представляет собой атом водорода, С1-6алкильную группу или атом...
Способы и соединения, предназначенные для получения ингибиторов котранспортера натрий-глюкозы 2 типа

Номер патента: 17411
Опубликовано: 28.12.2012
Авторы: Иимура Синия, Гудвин Николь Кэтлин, Чжан Хаймин, Харрисон Брайс Олден, Сонг Цюлин, Янь Цзе, Чжао Мэттью Манчжу, Мейбон Росс, У Вэньсюэ
МПК: C07H 5/04, C07H 15/04, C07H 7/04...
Метки: соединения, типа, способы, натрий-глюкозы, котранспортера, предназначенные, ингибиторов, получения
Формула / Реферат:
1. Способ получения соединения формулы Iили его соли, который включает взаимодействие соединения формулы IIс основанием с получением соединения формулы I,где Y представляет собой О, S, NR4 или C(R4)2;Z1 представляет собой О, S, SO или SO2;каждый Р1 независимо представляет собой гидроксизащитную группу, устойчивую в кислых условиях;каждый R1 независимо представляет собой водород, галоген, циано, OR1A SR1A или C1-10алкил;каждый R1A независимо...
Новое применение гетероциклических соединений, активных в качестве ингибиторов бета-лактамаз, и содержащие их фармацевтические композиции

Номер патента: 7220
Опубликовано: 25.08.2006
Авторы: Фроментэн Клод, Роулендс Дэвид Ален, Лампила Максим, Асзоди Жозеф
МПК: A61K 31/4995, A61K 31/55, A61K 31/439...
Метки: новое, применение, качестве, ингибиторов, соединений, фармацевтические, композиции, активных, бета-лактамаз, гетероциклических, содержащие
Формула / Реферат:
1. Применение соединений общей формулы (I) где R1 означает атом водорода, радикал COOR, CONR6, R7, R выбирают из группы, образованной алкильным радикалом, содержащим от 1 до 6 атомов углерода, не обязательно замещенным пиридильным или карбамоильным радикалом, -СН2-алкенильным радикалом, содержащим в сумме от 3 до 9 атомов углерода, арилом, содержащим от 6 до 10 атомов углерода, или аралкилом, содержащим от 7 до 11 атомов углерода, при этом ядро...
Предыдущий патент: Устройство доставки в кожу с изолирующим слоем in situ
Следующий патент: Влажное гранулирование с использованием вещества, связывающего воду
Случайный патент: Способ изготовления мембранно-электродных устройств и их комплектов