Тетрагидроциклопента[b]индольные модуляторы рецепторов андрогенов
Номер патента: 19713
Опубликовано: 30.05.2014
Авторы: Ким Еуибонг Джемс, Джадхав Прабхакар Кондаджи, Кришнан Венкатеш







Формула / Реферат
1. Соединение формулы (I)

где R представляет собой заместитель, выбранный из группы, состоящей из

или фармацевтически приемлемая соль указанного соединения.
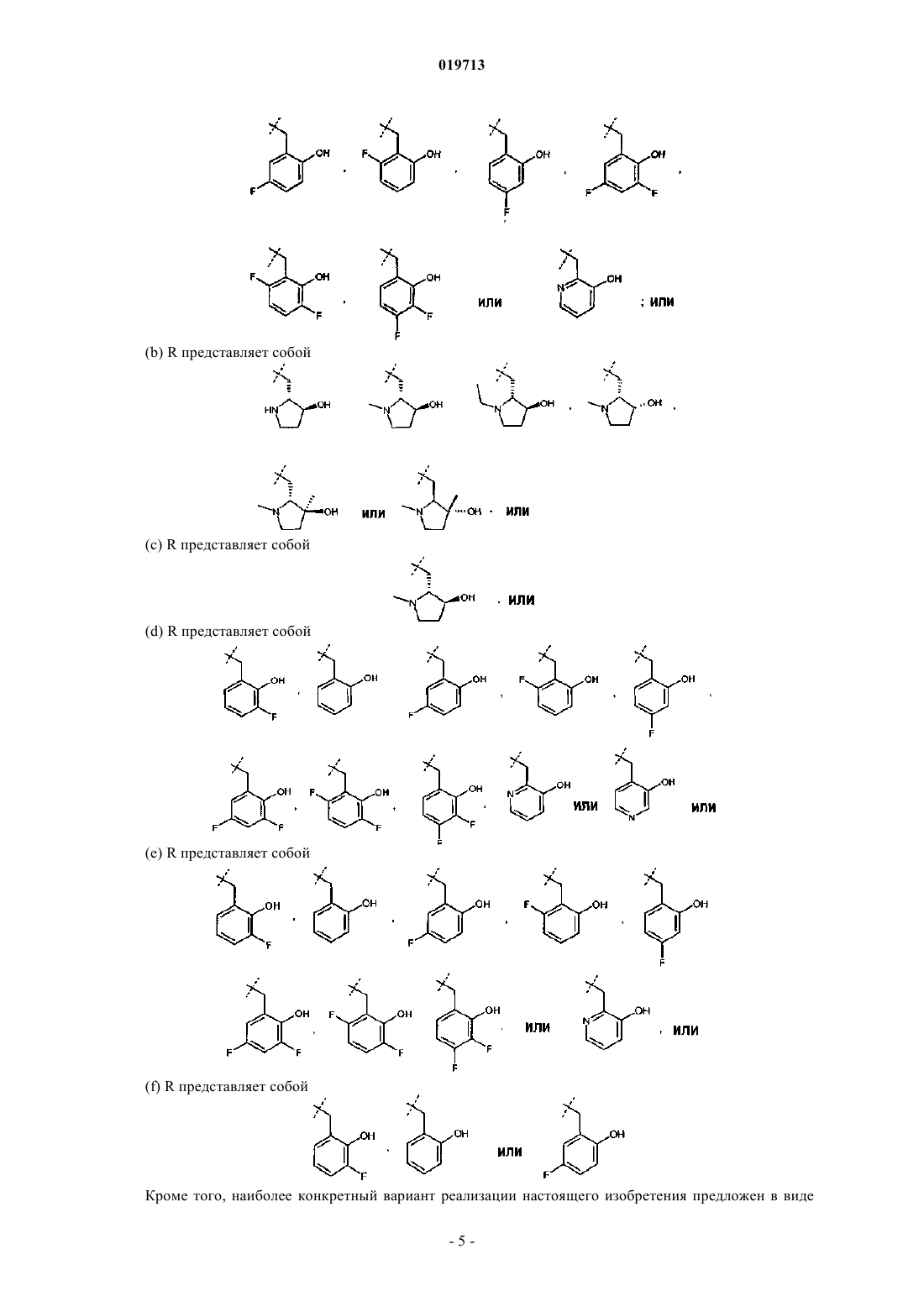
2. Соединение или соль по п.1, где R представляет собой

3. Соединение или соль по п.2, где R представляет собой

4. Соединение или соль по п.1, где R представляет собой

5. Соединение или соль по п.4, где R представляет собой

6. Соединение по п.1, выбранное из группы, состоящей из
изопропилового эфира [(S)-7-циано-4-((2R,3S)-3-гидрокси-1-метилпирролидин-2-илметил)-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты,
изопропилового эфира [(S)-7-циано-4-(3-фтор-2-гидроксибензил)-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты,
изопропилового эфира [(S)-7-циано-4-(2-гидроксибензил)-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты и
изопропилового эфира [(S)-7-циано-4-(5-фтор-2-гидроксибензил)-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты,
или фармацевтически приемлемой соли указанных соединений.
7. Применение соединения или соли по любому из пп.1-6 для лечения или профилактики снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы или эректильной дисфункции.
8. Применение соединения по п.7 для лечения или профилактики эректильной дисфункции.
9. Применение соединения или соли по любому из пп.1-6 для получения лекарственного средства для лечения снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы или эректильной дисфункции.
10. Фармацевтическая композиция, содержащая соединение или соль по любому из пп.1-6 в сочетании с одним или более фармацевтически приемлемыми носителями, разбавителями или наполнителями.
Текст
В настоящем изобретении предложено соединение формулы Джадхав Прабхакар Кондаджи,Кришнан Венкатеш, Ким Еуибонг Джемс (US) Медведев В.Н. (RU) или фармацевтически приемлемая соль указанного соединения; фармацевтические композиции,содержащие соединение формулы (I) в сочетании с подходящим носителем, разбавителем или наполнителем; и способы лечения или профилактики физиологических нарушений, в частности снижения костной массы, остеопороза, остеопении, снижения мышечной массы или мышечной силы или эректильной дисфункции, включающие введение соединения формулы (I) или фармацевтически приемлемой соли указанного соединения.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Эндогенные стероидные андрогены (например, тестостерон и 5-дигидротестостерон (ДГТ оказывают значительное влияние на многие физиологические функции. Известно применение терапии андрогенами в клинической практике при лечении гипогонадизма у мужчин. Представляется значимым, что,как было также показано, заместительная терапия андрогенами у мужчин с гипогонадизмом приводит к снижению резорбции костей и увеличению костной массы. Другие показания, при которых в клинической практике применяли андрогены, включают остеопороз и заболевания, связанные с мышечной атрофией. Кроме того, известны факты применения в последнее время заместительной терапии андрогенами у пожилых мужчин и для регуляции фертильности у мужчин. У женщин известно применение терапии андрогенами в клинической практике для лечения нарушений половой функции или сниженного либидо. Однако терапия андрогенами имеет свои ограничения. Например, нежелательные побочные эффекты терапии стероидными андрогенами включают стимуляцию роста предстательной железы и семенных пузырьков. Кроме того, была установлена взаимосвязь между стимуляцией опухолей предстательной железы, повышением уровня простат-специфического антигена (ПСА) (индикатора повышенного риска развития рака предстательной железы) и применением андрогенов. Также было установлено, что препараты стероидных андрогенов отрицательно характеризуются быстрым распадом в печени, что приводит к низкой биодоступности при пероральном введении и короткому периоду активности после парентерального введения, колебаниям уровня в плазме и гепатотоксичности или перекрестным взаимодействиям с рецепторами других стероидных гормонов (например, рецептором глюкокортикоидов (РГ), рецептором минералокортикоидов (РМ) и рецептором прогестерона (РП. Кроме того, у женщин применение стероидных андрогенов может приводить к оволосению и вирилизации. Таким образом, в данной области по-прежнему существует потребность в альтернативах терапии стероидными андрогенами. Желательно, чтобы такие альтернативы обладали благоприятными фармакологическими свойствами стероидных андрогенов, но при сниженной вероятности или частоте проявления типичных ограничений, связанных с терапией стероидными андрогенами. Следовательно, задачей настоящего изобретения является обеспечение нестероидных лигандов РА, обладающих активностью агонистов андрогенов. Более конкретно, задача состоит в обеспечении нестероидных агонистов андрогенов, которые связывались бы с РА с более высоким сродством по сравнению с рецепторами других стероидных гормонов. Еще более конкретно, задача состоит в обеспечении тканеспецифичных модуляторов рецепторов андрогенов (ТМРА), которые в мышцах или костях проявляли бы активность агонистов андрогенов, а в андрогенных тканях, таких как предстательная железа или семенные пузырьки, активность только частичных агонистов, частичных антагонистов или антагонистов. Следующие примеры отражают текущее состояние дел в данной области техники: в Cadilla et al.,Curr. Top. Med. Chem (2006); 6(3):245-270 предложен обзор модуляторов рецепторов андрогенов; в Segalet al., Expert Opin. Investig. Drugs (2006); 15(4):377-387 предложен обзор модуляторов рецепторов андрогенов; в международной заявке на патент PCT/US07/83745, находящейся на рассмотрении одновременно с заявкой на данный патент, предложены тетрагидроциклопента[b]индолы в качестве модуляторов рецепторов андрогенов; а в опубликованной международной заявке на патент WO 2007/83745 предложены тетрагидрокарбазолы в качестве модуляторов рецепторов андрогенов. Настоящее изобретение направлено на обеспечение некоторых тетрагидроциклопента[b]индолов,соответствующих формуле (I), приведенной ниже, которые обладают конкретными профилями активности в исследованиях in vitro и in vivo, позволяющими предположить, что они подходят для лечения нарушений, реагирующих на терапию стероидными андрогенами. Соответственно, в настоящем изобретении предложено соединение формулы (I) или их фармацевтически приемлемой соли. Согласно другому варианту реализации настоящего изобретения предложен способ лечения или профилактики гипогонадизма, снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы, нарушения половой функции у мужчин или у женщин, эректильной дисфункции или снижения либидо, включающий введение пациенту, который в этом нуждается, эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Согласно конкретному аспекту настоящего изобретения предложен способ лечения или профилактики снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы или эректильной дисфункции. Согласно более конкретному аспекту настоящего изобретения предложен способ лечения или профилактики снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы, вызванных иммобилизацией, бездействием или травмой, или эректильной дисфункции. Также в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения для лечения или профилактики гипогонадизма, снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы, нарушения половой функции у мужчин или у женщин, эректильной дисфункции или снижения либидо. Более конкретно, в изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения для лечения или профилактики снижения костной массы или плотности, остеопороза, остеопении,снижения мышечной массы или силы или эректильной дисфункции. Еще более конкретно, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения для лечения или профилактики снижения костной массы или плотности, остеопороза, остеопении,снижения мышечной массы или силы, вызванных иммобилизацией, бездействием или травмой, или эректильной дисфункции. Кроме того, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии. Согласно другому варианту реализации настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики гипогонадизма, снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы, нарушения половой функции у мужчин или у женщин,эректильной дисфункции или снижения либидо. Более конкретно, в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики снижения костной массы или плотности, остеопороза,остеопении, снижения мышечной массы или силы или эректильной дисфункции. Еще более конкретно, в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы, вызванных иммо-2 019713 билизацией, бездействием или травмой, или эректильной дисфункции. Кроме того, в настоящем изобретении предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в сочетании с одним или более фармацевтически приемлемыми носителями, разбавителями или наполнителями. Более конкретно, в настоящем изобретении предложена фармацевтическая композиция для лечения снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы или эректильной дисфункции, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в сочетании с одним или более фармацевтически приемлемыми носителями, разбавителями или наполнителями. Еще более конкретно, в настоящем изобретении предложена фармацевтическая композиция для лечения снижения костной массы или плотности, остеопороза, остеопении, снижения мышечной массы или силы,вызванных иммобилизацией, бездействием или травмой, или эректильной дисфункции. Настоящее изобретение также включает новые промежуточные соединения и способы, подходящие для синтеза соединения формулы (I). Также настоящее изобретение относится к фармацевтически приемлемым солям соединения формулы (I). Примеры фармацевтически приемлемых солей и способов их получения хорошо известны специалистам в данной области техники. Кроме того, настоящее изобретение также относится к сольватам соединения формулы (I) или фармацевтически приемлемых солей соединения формулы (I). Применяемый в настоящем описании термин "формула (I)" как таковой или любое конкретное соединение формулы (I) включает любой сольват данного соединения. Соединение формулы (I) может содержать один или более хиральных центров и, следовательно,может существовать в виде конкретных стереоизомерных конфигураций. Термины "R" и "S" в настоящем описании употребляют в значениях, принятых в органической химии для обозначения конкретных конфигураций хирального центра. Термины или "RS" относятся к конфигурации хирального центра, содержащей рацемат. Частичный перечень приоритетов и обсуждение стереохимии приведены в"Nomenclature of Organic Compounds: Principles и Practice" (J.H. Fletcher, et al., eds., 1974). Рентгенографический анализ и корреляция со временем удержания при хиральной ВЭЖХ. Конкретные стереоизомеры и энантиомеры соединений формулы (I) могут быть получены специалистом в данной области техники с помощью хорошо известных методик и способов, таких как предложенные в работе Eliel and Wilen, "Stereochemistry of Organic Compounds", John WileySons, Inc., 1994,Chapter 7; Separation of Stereoisomers, Resolution, Racemization; и Collet and Wilen, "Enantiomers, Racemates, and Resolutions", John WileySons, Inc., 1981. Например, конкретные стереоизомеры и энантиомеры можно получить при помощи стереоспецифического синтеза с применением энатиомерно и геометрически чистых или энантиомерно и геометрически обогащенных исходных веществ. Кроме того,конкретные стереоизомеры и энантиомеры можно разделить и выделить при помощи таких методов, как хроматография на хиральных неподвижных фазах, ферментативное разделение, фракционная перекристаллизация солей присоединения, а также методик, приведенных на схемах и в примерах в настоящем описании. Обозначение относится к связи, которая выступает вперед из плоскости страницы. Обозначение относится к связи, которая выступает назад из плоскости страницы. Обозначение относится к связи, которая существует в виде смеси связей, которые выдаются и вперед и назад из плоскости страницы. В настоящем описании термин "пациент" относится к человеку или млекопитающему, отличному от человека, такому как собака, кошка, корова, обезьяна, лошадь или овца. Более конкретно, термин "пациент" относится к человеку. В настоящем описании термин "лечение" (или "лечить") включает препятствование, сдерживание, замедление, остановку или обращение прогрессирования или тяжести существующего симптома или нарушения. В настоящем описании термин "профилактика" (или "предотвращать" или "предотвращение") относится к препятствованию, сдерживанию или подавлению возникновения или появления симптома или нарушения. Как известно специалисту в данной области техники, физиологические нарушения могут иметь место в виде "хронического" состояния или в виде "острого" эпизода. Соответственно, лечение нарушений предполагает лечение и острых событий, и хронических состояний. При остром событии соединение вводят при появлении симптомов и прекращают введение,когда симптомы проходят, а хроническое состояние лечат на протяжении всего течения заболевания. Соединения, предложенные в настоящем изобретении, могут быть частью фармацевтической композиции. Как таковая, фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в сочетании с фармацевтически приемлемым носителем, разбавителем или наполнителем, является важным вариантом реализации изобретения. Примеры фармацевтических композиций и способов их получения хорошо известны в данной области техники. Соединение формулы(I) или композицию, содержащую соединение формулы (I), можно вводить любым путем, обеспечивающим биодоступность соединения, включая пероральный и парентеральный пути. В настоящем описании термин "эффективное количество" относится к количеству или дозе соединения формулы (I), которое при введении пациенту одной или нескольких доз обеспечивает желаемый эффект у пациента, подвергающегося диагностике или лечению. Эффективное количество может легко определить лечащий врач-диагност, а также специалист в данной области техники, принимая во внимание ряд факторов, таких как вид млекопитающего, его размер, возраст и общее состояние здоровья; рассматриваемое конкретное заболевание; степень тяжести заболевания; реакцию конкретного пациента; конкретное вводимое соединение; режим введения; характеристики биодоступности вводимого препарата; выбранный режим введения и применение каких-либо сопутствующих лекарственных средств. Соединения и композиции, предложенные в настоящем изобретении, можно вводить или применять в составе лекарственного средства либо в отдельности, либо в сочетании с традиционными терапевтическими агентами, применяемыми при конкретных нарушениях. Если соединения и композиции, предложенные в настоящем изобретении, применяют как часть комбинации, соединение или композицию, содержащую формулу (I), можно вводить либо в отдельности, либо как часть состава, содержащего терапевтический агент, с которым их комбинируют. В частности, традиционные терапевтические агенты для лечения эректильной дисфункции можно выгодно сочетать с соединениями формулы (I) или с композициями, содержащими соединения формулы(I). Традиционные агенты для лечения эректильной дисфункции включают ингибиторы фосфодиэстеразы 5 типа (ФДЭ 5) тадалафил (Циалис), силденафила цитрат (Виагра) и варденафила гидрохлорид(Левитра). Так, в настоящем изобретении также предложен способ лечения или профилактики эректильной дисфункции, включающий введение пациенту, который в этом нуждается, эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли в сочетании с агентом, выбранным из группы, состоящей из тадалафила, силденафил цитрата и варденафил гидрохлорида. Более конкретно, в настоящем изобретении предложен способ лечения или профилактики эректильной дисфункции, включающий введение пациенту, который в этом нуждается, эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли в сочетании с агентом, выбранным из группы, состоящей из Циалиса, Виагры и Левитры. Также в настоящем изобретении предложен состав для комбинированной терапии, содержащий:(b) один или более сопутствующих агентов, традиционно применяемых для лечения эректильной дисфункции, выбранных из группы, состоящей из тадалафила, силденафил цитрата и варденафил гидрохлорида; и, необязательно,(с) фармацевтически приемлемый носитель, разбавитель или наполнитель. Более конкретно, в настоящем изобретении предложен состав для комбинированной терапии, содержащий:(b) один или более сопутствующих агентов, традиционно применяемых для лечения эректильной дисфункции, выбранных из группы, состоящей из Циалиса, Виагры и Левитры; и, необязательно,(с) фармацевтически приемлемый носитель, разбавитель или наполнитель. Конкретные аспекты изобретения В следующем перечне указано несколько групп конкретных заместителей и конкретных переменных для соединений формулы (I). Следует понимать, что соединения формулы (I), содержащие такие конкретные заместители или переменные, а также способы и применения таких соединений представляют собой конкретные аспекты настоящего изобретения. Так, конкретным аспектом настоящего изобретения является соединение формулы (I), где: Кроме того, наиболее конкретный вариант реализации настоящего изобретения предложен в виде соединений формулы (I), примеры которых приведены в настоящем описании, а более конкретно изопропилового эфира [(S)-7-циано-4-2R,3S)-3-гидрокси-1-метилпирролидин-2-илметил)-1,2,3,4 тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты; изопропилового эфира[(S)-7-циано-4-(5-фтор-2-гидроксибензил)-1,2,3,4 тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты. Соединения формулы (I) можно получить при помощи различных способов, известных в данной области техники, а также представленных на схемах и описанных в примерах получения и примерах ниже. Однако предполагается, что следующее обсуждение никоим образом не ограничивает настоящее изобретение. Например, конкретные стадии синтеза для каждого из описываемых путей можно комбинировать различным образом или применять в сочетании со стадиями из других схем для получения дополнительных соединений формулы (I). Заместители, если не указано иное, являются такими, как определено ранее. Реагенты и исходные вещества легко доступны для специалиста в данной области техники или могут быть получены по способам, выбранным среди стандартных методик органической химии и химии гетероциклических соединений, методик, аналогичных синтезу известных близких по структуре соединений, и способов, описанных в примерах ниже, включая любые новые способы. В настоящем описании следующие термины имеют указанные значения: "МеОН" обозначает метанол; "EtOH" обозначает этанол; "i-PrOH" обозначает изопропанол; "EtOAc" обозначает этилацетат; "ДМФ" обозначает диметилфомамид; "ДМСО" обозначает метилсульфоксид; "NMP" обозначает 1-метил-2-пирролидинон; "ТФУ" обозначает трифторуксусную кислоту; "ТГФ" обозначает тетрагидрофуран; "ДЭАД" обозначает диэтилазодикарбоксилат; "ДИАД" обозначает диизопропилдиазокарбоксилат; "NBS" обозначает N-бромсукцинимид; "t-boc" или "boc" обозначает трет-бутоксикарбонил; "ТБДМС" обозначает трет-бутилдиметилсилил; а "Зг" обозначает защитную группу. Схема 1 Получение промежуточного соединения формулы (5) можно осуществлять в соответствии с реакциями, показанными на схеме 1. На схеме 1, стадия А, циклопентенон подвергают взаимодействию с фталимидом по реакции присоединения Михаэля с образованием (R,S)-2-(3-оксоциклопентил)изоиндол-1,3-диона (1). Реакцию проводят в метаноле/2 н. Na2CO3 в соотношении 10/1 по объему, предпочтительно при комнатной температуре, применяя условия, аналогичные условиям, описанным О. Nowitzki, et al. в Tetrahedron 1996, 52,11799-11810. Продукт выделяют путем добавления воды и (1) получают в виде белого твердого вещества. На стадии В (R,S)-2-(3-оксоциклопентил)изоиндол-1,3-дион (1) вступает в реакцию с 4 цианфенилгидразином в типичном синтезе индолов по Фишеру с образованием тетрагидроциклопента[b]индола формулы (2). Специалисты в данной области техники понимают, что существует множество кислых сред для проведения синтеза индолов по Фишеру, включая протонные кислоты и кислоты Льюиса. При предпочтительных условиях применяют смесь ледяной уксусной кислоты с 4 н. HCl в диоксане,-6 019713 при температуре от примерно 50 С до температуры кипения растворителя, примерно в течение от 4 до 24 ч. Продукт выделяют путем добавления воды, после чего отфильтровывают полученное твердое вещество. Твердое вещество подвергают обработке ультразвуком в метаноле с образованием вещества с достаточной степенью чистоты. В качестве альтернативы, реакцию проводят, применяя кислоту Льюиса, такую как хлорид цинка, в количестве от 2 до 4 экв. В соответствии с другими предпочтительными условиями для стадии В применяют этанол при температуре кипения в течение примерно от 4 до 24 ч. Продукт выделяют и его можно очистить путем фильтрования реакционной смеси с последующим хроматографированием на силикагеле фильтрата. На схеме 1, стадия С, фталимидную группу тетрагидроциклопента[b]индола формулы (2) отщепляют при помощи гидразина или гидрата гидразина с образованием аминотетрагидроциклопента[b]индола формулы (3) с применением условий, описанных у М. Alajarin, et al. (Eur. J. Org. Chem. 2002, 4222-4227). Реакция протекает в смеси растворителей тетрагидрофуран/этанол в соотношении примерно 5,5/1 по объему при температуре от 0 до 50 С, предпочтительно примерно при комнатной температуре, в течение от 4 до 72 ч. Образующийся фталгидразид удаляют путем фильтрования и продукт выделяют путем концентрирования фильтрата. Его можно очистить при помощи хроматографии с применением методов,известных в данной области техники. На схеме 1, стадия D, рацемический аминотетрагидроциклопента[b]индол формулы (3) разделяют с получением хирального (S)-аминотетрагидроциклопента[b]индола формулы (4). Амин растворяют в подходящем растворителе, таком как этанол, и перекристаллизовывают в виде соли D-пироглутаминовой кислоты. После выделения соли свободное основание формулы (4) получают путем растворения в воде и корректировки значения рН до 9 с применением концентрированного водного аммиака. Полученное твердое вещество отфильтровывают с получением (S)-2-амино-1,2,3,4-тетрагидроциклопента[b]индол-7 карбонила (4) с конкретным значением вращения: [a]D25-68,3 (MeOH). На стадии Е амин формулы (4) ацетилируют изопропилхлорформиатом с образованием карбамата формулы (5) с применением условий, хорошо известных специалистам в данной области техники. Амин объединяют с избытком основания в виде органического амина, такого как триэтиламин или диизопропилэтиламин, в инертном растворителе, таком как тетрагидрофуран, толуол, дихлорэтан или дихлорметан, N-метилпирролидинон или N,N-диметилформамид или их смесь. В соответствии с предпочтительными условиями применяют диизопропилэтиламин в дихлорметане в присутствии изопропилхлорформиата при температуре примерно от 0 до 40 С в течение от 1 до 72 ч. Продукт выделяют путем добавления воды и диэтилового эфира, после чего перемешивают и выделяют образующееся твердое вещество. Если продукт достаточно растворим в соответствующем органическом растворителе, его можно выделить при помощи методов экстракции. В качестве альтернативы, как показано на стадии F, рацемический амин формулы (3) защищают,получая защищенный трет-бутоксикарбонил-(boc) амин формулы (6), с применением условий, хорошо известных в данной области техники. На стадии G рацемическую смесь затем разделяют при помощи хиральной хроматографии и получают (S)-энантиомер формулы (7). Вос-группу затем удаляют с применением обычных кислых сред с помощью HCl или ТФУ и получают хиральный амин формулы (4). На схеме 2 показан альтернативный синтез промежуточного соединения формулы (5). На схеме 2,стадия A, (R,S)-2-(3-оксоциклопентил)изоиндол-1,3-дион (1) вступает в реакцию с гидрохлоридом 4 бромфенилгидразина в ходе обычного синтеза индолов по Фишеру. В соответствии с предпочтительными условиями применяют ледяную уксусную кислоту при примерно от 50 до 80 С в течение от 2 до 24 ч. На стадии В фталимидную группу тетрагидроциклопента[b]индола формулы (8) отщепляют при помощи гидразина или гидразин гидрата с образованием аминотетрагидроциклопента[b]индола формулы(9). В соответствии с предпочтительными условиями применяют тетрагидрофуран/этанол в смеси с соотношением примерно 20/1 по объему при температуре примерно от 40 до 70 С в течение от 1 до 12 ч. Образующийся фталгидразид удаляют путем фильтрования и продукт выделяют путем концентрирования фильтрата. На схеме 2, стадия С, амин формулы (9) защищают группой t-boc и получают защищенный амин формулы (10). В соответствии с предпочтительными условиями применяют ди-трет-бутилдикарбонат в инертном растворителе, таком как ТГФ или диоксан, в присутствии неорганического основания, такого как NaHCO3. На стадии D рацемическое вещество формулы (10), содержащее t-boc, разделяют при помощи хиральной ВЭЖХ с получением (S)-энантиомера формулы (11). На стадии Е (S)-бромкарбамат формулы (11) превращают в нитрил формулы (5). Реакцию проводят в инертном растворителе, таком как N,N'-диметилацетамид, в присутствии смеси ацетата цинка и формиата цинка, цианида цинка и цинковой пыли. Применяют палладиевый катализатор, такой как 1,1'-бис(дифенилфосфино)ферроцен)палладия(II) хлорид. Реакционную смесь нагревают при примерно от 80 до 120 С в течение от 2 до 24 ч. На стадии F группу boc удаляют с применением обычных кислых сред, таких как HCl/диоксан или ТФУ и получают хиральный амин формулы (4) (см. схему 1), который затем ацилируют, как описано на схеме 1, стадия Е, с применением изопропилхлорформиата с получением карбамата формулы (5). В качестве альтернативы, на схеме 2, стадия G, фталимидную группу тетрагидроциклопента[b]индола формулы (8) отщепляют при помощи гидразина или гидразина гидрата и получают аминотетрагидроциклопента[b]индол формулы (9), как описано ранее для стадии В. Далее образующийся свободный амин превращают в соль п-толуолсульфокислоты в этаноле. На стадии Н рацемическую тозилатную соль аминотетрагидроциклопента[b]индола формулы (13) высвобождают, а полученный свободный амин разделяют при помощи дибензил-D-винной кислоты, получая винно-кислую соль (5)-аминотетрагидроциклопента[b]индола формулы (14). Получение соли предпочтительно проводят в смеси растворителей из этанола и воды при кипячении с обратным холодильником в течение примерно от 1 до 6 ч, после чего смесь охлаждают и получают целевой энантиомер. На стадии I соль формулы (14) нейтрализуют с получением свободного основания и затем ацилируют, получая карбамат формулы (15). Соль нейтрализуют неорганическим основанием, таким как раствор гидроксида натрия. Свободное основание получают путем экстракции и последующего ацилирования при помощи изопропилхлорформиата в присутствии органического амина, такого как диизопропилэтиламин, в инертном растворителе или смеси растворителей, содержащей тетрагидрофуран и метил-третбутиловый эфир. На схеме 2, стадия J, бромкарбамат формулы (15) превращают в нитрил формулы (5), как описано ранее для стадии Е. Схема 3 Получение соединений, предложенных в изобретении, соответствующих формуле (20 а, b или с),можно проводить в соответствии с реакциями, показанными на схеме 3. На схеме 3, стадия А, циантетрагидроциклопента[b]индол формулы (5) алкилируют алкилмезилатом формулы (16) и получают алкилированный тетрагидроциклопента[b]индол формулы (17). Реакция протекает в инертном растворителе, таком как ДМФ, в присутствии неорганического основания, такого как карбонат цезия, с добавлением йодида калия. Реакция протекает при температуре примерно от 50 до 100 С в течение примерно от 8 до 72 ч. На схеме 3, стадия В, хиральную гидроксильную группу пирролидина формулы (17) переводят из(3S) в (3R). Хиральный переход проводят при помощи реакции Мицунобу и получают хлорацетоксипирролидин формулы (18). Как известно специалисту в данной области техники, существуют различные условия проведения реакции Мицунобу, применяемые в данной области техники. Например, спирт формулы (17), растворяют в подходящем безводном растворителе, таком как ТГФ, CH2Cl2 или толуол, и обрабатывают триалкил- или триарилфосфином, таким как Ме 3 Р, Bu3P или Ph3P, и диалкилазодикарбоксилатом, таким как ДЭАД или ДИАД. На стадии С 3R-хлорацетоксипирролидин формулы (18) подвергают гидролизу до 3Rгидроксипирролидина формулы (19 а). Гидролиз сопровождают применением неорганического основания, такого как гидроксид лития, в растворителе, таком как метанол, при от 0 до 50 С в течение от 4 до 24 ч. На стадии D защитную boc-группу удаляют с применением кислых сред, таких как ТФУ или предпочтительно 4 н. HCl в диоксане, и получают 3S-гидроксипирролидин формулы (19b). На схеме 3, стадия Е, гидроксипирролидин формулы (19 а или b) алкилируют с применением вос-9 019713 становительного аминирования и получают N-алкилпирролидин формулы (20 а, b или с). Реакция протекает в инертном растворителе, таком как ТГФ, дихлорметан или, предпочтительно, хлороформ, с применением восстановителя, такого как триацетоксиборгидрид натрия, при температуре примерно от 0 до 50 С в течение от 8 до 24 ч. Применяемым альдегидом может быть формальдегид или ацетальдегид с получением соединения формулы (20 а, b или с). трет-Бутиловый эфир (2R,3S)-3-гидрокси-2-метансульфонилоксиметилпирролидин-1-карбоновой кислоты (16) можно легко получить при помощи способов, аналогичных приведенным в настоящем описании, или при помощи методик, широко применяемых в данной области техники. Например, 3 гидрокси-L-пролин можно защитить при помощи boc с образованием (2S,3S)-1-(трет-бутоксикарбонил)3-гидроксипирролидин-2-карбоновой кислоты. Карбоновую кислоту восстанавливают до спирта и далее превращают в мезилат формулы (16). Схема 4 Получение соединений, предложенных в изобретении, соответствующих формуле (24 а и b) можно проводить в соответствии с реакциями, показанными на схеме 4. На схеме 4, стадия А, циантетрагидроциклопента[b]индол формулы (5) алкилируют алкилмезилатом формулы (21 а и b) и получают алкилированный тетрагидроциклопента[b]индол формулы (22 а и b). Алкилмезилат формулы (21 а и b) применяют в виде смеси двух транс-энантиомеров. Алкилирование проводят аналогично условиям, описанным на схеме 3, стадия А, выше. На стадии В защитную boc-группу удаляют при помощи кислой среды, такой как ТФУ или, предпочтительно, 4 н. HCl в диоксане. На схеме 4, стадия С, гидроксипролин формулы (23 а и b) алкилируют с применением условий восстановительного аминирования, которые приведены ранее для схемы 3, стадия Е, и получают смесь транс-энантиомеров формулы (24 а и b). Энантиомеры можно разделить при помощи хиральной хроматографии. трет-Бутиловый эфир транс-3-гидрокси-2-метансульфонилоксиметил-3-метилпирролидин-1 карбоновой кислоты (21 а и b) можно легко получить при помощи способов, аналогичных приведенным в настоящем описании или при помощи методик, широко применяемых в данной области техники. Например, транс-2-бензилоксиметил-3-метилпирролидин-3-ол можно получить путем однореакторного превращения соответствующего 2,3-азиридин-1-ола (J. Schomaker и S. Bhattacharjee, J. Am. Chem. Soc., 2007,129, 1996-2003). Азот в пирролидиноле затем защищают группой t-boc, а бензильную группу удаляют путем гидрогенизации. Образующийся спирт мезилируют и получают транс-энантиомеры формулы (21 а и b). Получение соединений, предложенных в изобретении, соответствующих формуле (27), можно проводить в соответствии с реакциями, показанными на схеме 5. На схеме 5, стадия А, циантетрагидроциклопента[b]индол формулы (5) алкилируют бензил- или пиридилметилгалогенидом формулы (25) и получают алкилированный тетрагидроциклопента[b]индол формулы (26). Фенильное или пиридильное кольцо замещают гидроксильной функциональной группой,которую защищают защитными группами (Зг), известными в данной области техники, например, такими как метиловый эфир или силиловый эфир. Алкилирование проводят аналогично условиям, описанным для схемы 3, стадия А, выше. На стадии В защитную группу удаляют и получают фенол или гидроксипиридин формулы (27). Например, метиловый эфир превращают в свободный гидроксил при помощи реакции с трибромидом бора в инертном растворителе, таком как дихлорметан, при температуре примерно от -20 до 25 С. В качестве альтернативы, силильную защитную группу можно удалить с помощью фторид-аниона с применением таких реагентов, как фторид цезия или, предпочтительно, фторид тетрабутиламмония. Бензилгалогениды формулы (25) можно легко получить при помощи способов, аналогичных приведенным в настоящем описании, или при помощи методик, широко применяемых в данной области техники. Например, фтор- или дифтор-2-гидроксибензальдегиды можно алкилировать йодметаном с получением 2-метоксибензальдегида. Бензальдегид можно восстановить до соответствующего бензилового спирта, а затем превратить в (хлорметил)-2-метоксибензен формулы (25), в которой Х=Cl, а Зг=Ме. В качестве альтернативы, фтор- или дифтор-2-метилфенолы можно силилировать при помощи третбутилдиметилхлорсилана, после чего провести реакцию с N-бромсукцимидом и получить бензилбромид формулы (25), в которой X=Br и Зг=ТБДМС. 2-(Хлорметил)-3-метоксипиридин можно получить из 3 метокси-2-пиколина путем хлорирования с применением POCl3. Определение биологической активности. По данным исследований in vitro и in vivo, приводимые в качестве примеров соединения формулы(I) обладают профилем активности, позволяющим предположить, что они подходят для лечения нарушений, восприимчивых к терапии стероидными андрогенами. В частности, соединения согласно примерам 1-10 и 12, соответствующие формуле (I), являются мощными лигандами РА, оказывающими агонистическое действие на рецептор андрогенов. Кроме того, соединения согласно примерам 1-10 и 12, соответствующие формуле (I), селективно связываются с РА по сравнению с каждым из РМ, РГ и РП. В настоящем описании термин "Kd" относится к равновесной константе диссоциации для комплекса лиганд-рецептор; термин "Ki" относится к равновесной константе диссоциации для комплекса лекарственный препарат-рецептор и является показателем концентрации препарата, при которой обеспечивается связывание с половиной сайтов связывания при равновесии; термин "Kb" относится к равновесной константе диссоциации для комплекса антагонист-рецептор; термин "IC50 " относится к концентрации агента, которая обеспечивает 50% от максимального ингибирующего ответа, возможного для этого агента, или, в качестве альтернативы, к концентрации агента, обеспечивающей 50% замещение лиганда, связывающегося с рецептором; термин "ЕС 50" относится к концентрации агента, которая обеспечивает 50% от максимального ответа, возможного для этого агента; а "ED50" относится к дозе вводимого терапевтического агента, обеспечивающей 50% от максимального ответа для данного агента. Исследование связывания ядерных рецепторов стероидных гормонов. Для исследования конкурентного связывания рецептор-лиганд с целью определения значения Ki применяют лизаты клеток эмбриональной почки человека HEK293, в избытке экспрессирующих РМ человека (рецептор минералокортикоидов), РГ (рецептор глюкокортикоидов), РА (рецептор андрогенов) или РП (рецептор прогестерона) человека. Типовые процедуры представлены ниже. Если кратко, исследования конкурентного связывания рецепторов стероидов проводят в буфере,содержащем 20 мМ буфера HEPES (рН 7,6), 0,2 мМ ЭДТА, 75 мМ NaCl, 1,5 мМ MgCl2, 20% глицерина,- 11019713(буфер для анализа). Обычно исследования связывания стероидных рецепторов включают применение радиоактивно меченных лигандов, таких как 0,25 нМ [3 Н]-альдостерона для связывания РМ, альдостерона для связывания РМ, 0,3 нМ [3 Н]-дексаметазона для связывания РГ, 0,36 нМ [3H]-метилтриенолона для связывания РА и 0,29 нМ [3 Н]-метилтриенолона для связывания РП, а также либо 20 мкг/мл лизата 293-РМ, 20 мкг лизата 293-РГ, 22 мкг лизата 293-РА, либо 40 мкг лизата 293-РП на лунку. Исследования обычно проводят в 96-луночном формате. Конкурирующие тестируемые соединения добавляют в разных концентрациях в диапазоне примерно от 0,01 нМ до 10 мкМ. Неспецифическое связывание определяют в присутствии 500 нМ альдостерона для связывания РМ, 500 нМ дексаметазона для связывания РГ или 500 нМ метилтриенолона для связывания РА и РГ. Реакционные смеси для оценки связывания (140 мкл) инкубируют в течение ночи при 4 С, затем в каждую реакционную смесь добавляют 70 мкл холодного угольно-декстранового буфера (содержащего на 50 мл буфера для анализа 0,75 г угля и 0,25 г декстрана). Планшеты подвергают перемешиванию в течение 8 мин на орбитальном шейкере при 4 С. Затем планшеты подвергают центрифугированию при 3000 об/мин при 4 С в течение 10 мин. Затем аликвоты по 120 мкл реакционной смеси для связывания переносят в другой 96-луночный планшет и в каждую лунку добавляют 175 мкл сцинтилляционной жидкости Wallac Optiphase Hisafe 3. Планшеты герметизируют и интенсивно встряхивают на орбитальном шейкере. После инкубации в течение 2 ч с планшетов считывают показания на счетчике Wallac Microbeta. Полученные данные применяют для вычислении теоретической IC50 и процента ингибирования при 10 мкМ. Kd для [3 Н]-альдостерона для связывания РМ, [3 Н]-дексаметазона для связывания РГ, [3 Н]метилтриенолона для связывания РА или [3 Н]-метилтриенолона для связывания РП определяют по связыванию насыщения. Значения IC50 для соединений переводят в Ki, применяя уравнение ЧенгаПрусоффа. В соответствии с протоколом, по существу соответствующим описанному выше, соединения согласно примерам 1-10 и 12 проявляют Ki в исследовании связывания РА 500 нМ. В частности, соединения согласно примерам 1, 2, 6 и 9 проявляли Ki в исследовании связывания РА примерно 51, 11, 13 и 0,8 нМ соответственно, тем самым демонстрируя, что соединения согласно настоящему изобретению являются мощными лигандами РА человека. Функциональные исследования модулирования ядерных рецепторов стероидных гормонов. Андрогены проявляют свое физиологическое действие посредством взаимодействия с рецепторами андрогенов. После связывания андрогена с РА в цитоплазме комплекс лиганд-рецептор переходит в ядро клетки, где он связывается с элементами гормонального ответа в ДНК, инициируя экспрессию геновмишеней. Действие андрогенов можно охарактеризовать как анаболическое или андрогенное по своей природе. Анаболическое (т.е. построение тканей) действие андрогенов включает увеличение мышечной массы и силы, и костной массы, а андрогенное (т.е. маскулинизирующее) действие андрогенов включает развитие вторичных мужских половых признаков, таких как ткани внутренних половых органов (т.е. предстательной железы и семенных пузырьков), наружных половых органов (полового члена и мошонки), либидо и участков роста волос. Чтобы продемонстрировать способность соединений, предложенных в настоящем изобретении, модулировать активность рецепторов стероидных гормонов (т.е. быть агонистами, частичными агонистами,частичными антагонистами или антагонистами), проводят биологические исследования, позволяющие определить функциональное модулирование экспрессии гена-мишени в клетках, временно трансфецированных белком ядерного рецептора и конструкцией (элемент гормонального ответа)-(ген-репортер). Специалист в данной области техники может легко получить из коммерческих источников или приготовить растворители, реагенты и лиганды, применяемые при функциональном анализе. Ниже приведены типовые процедуры для функционального исследования ядерных рецепторов гормонов. А. Панельный скрининг ядерных рецепторов гормонов. Клетки эмбриональной почки человека HEK293 трансфецируют плазмидами рецепторов стероидных гормонов и генов-репортеров при помощи подходящего реагента для трансфекции, такого какFugene. Если кратко, плазмиду-репортер, содержащую две копии пробазина ARE и промотор ТК (тимидинкиназы) выше к-ДНК-репортера люциферазы, трансфецируют в клетки HEK293 при помощи плазмиды, конститутивно экспрессирующей рецептор андрогенов человека (РА) с применением вирусного CMV (цитомегаловирус) промотора. Плазмиду-репортер, содержащую две копии GRE и промотор ТК выше к-ДНК-репортера люциферазы, трансфецируют при помощи плазмиды, конститутивно экспрессирующей рецептор глюкокортикоидов человека (РГ), рецептор минералокортикоидов человека(РМ) или рецептор прогестерона человека (РП) с применением вирусного промотора CMV. Клетки трансфецируют в колбах Т 150 см 2 в среде DMEM (модифицированная по способу Дульбеко среда игла) с 5% десорбированной с угля эмбриональной бычьей сывороткой (FBS). После инкубации в течение ночи трансфецированные клетки подвергают воздействию трипсина, высевают в планшеты с 96 лунками в среде DMEM, содержащей десорбированную с угля FBS, инкубируют в течение 4 ч и затем подвергают воздействию разных концентраций тестируемых соединений в диапазоне от 0,01 нМ до 10 мкМ. В режи- 12019713 ме определения антагонизма низкие концентрации агониста для каждого соответствующего рецептора добавляют в среду (0,08 нМ альдостерона для МР, 0,25 нМ дексаметазона для РГ, 0,66 нМ метилтриенолона для РА и 0,08 нМ промегесона для РП). После инкубации с тестируемыми соединениями в течение 24 ч клетки лизируют и активность люциферазы определяют при помощи стандартных методов. Данные аппроксимируют логистической кривой с четырьмя подгоночными параметрами, чтобы определить значения ЕС 50. Процент эффективности (соединения с насыщенными максимальными реакциями) или процент максимальной стимуляции (соединения с максимальными реакциями, которые не насыщались) определяют относительно максимальной стимуляции, полученной при следующих эталонных антагонистах: 30 нМ альдостерона для анализа РМ, 100 нМ метилтриенолона для анализа РА, 30 нМ промегестона для анализа РП и 100 нМ дексаметазона для анализа РГ. Значения IC50 определяют аналогично, применяя режим определения антагонизма. В режиме определения антагонизма процент ингибирования определяют путем сравнения активности тестируемого соединения в присутствии низкой концентрации агониста (0,08 нМ альдостерона для РМ, 0,25 нМ дексаметазона для РГ, 0,66 нМ метилтриенолона для РА и 0,08 нМ промегестона для РП) относительно реакции, производимой такой же низкой концентрацией агониста в отсутствие тестируемого соединения. В. Анализ по гену-репортеру С 2 С 12 AR/ARE. В качестве индикатора активности агонистов мышечной ткани проводят анализ по гену-репортеру С 2 С 12 AR/ARE. Если кратко, миобласты мыши С 2 С 12 совместно трансфецируют с применением реагента Fugene. Плазмиду-репортер, содержащую GRE/ARE (элемент ответа на глюкокортикоиды/элемент ответа на андрогены) и промотор ТК выше к-ДНК-репортера люциферазы, трансфецируют при помощи плазмиды, конститутивно экспрессирующей рецептор андрогенов человека (АР), с применением вирусного промотора CMV. Клетки трансфецируют во флаконах Т 150 см 2 в среде DMEM с 4% десорбированной с угля эмбриональной бычьей сывороткой (FBS). После инкубации в течение 5 ч трансфецированные клетки подвергают воздействию трипсина, высевают в планшеты с 96 лунками в среде DMEM, содержащей 4% десорбированную с угля FBS, инкубируют в течение 2 ч и затем подвергают воздействию разных концентраций тестируемых соединений в диапазоне от 0,01 нМ до 10 мкМ. После инкубации с тестируемыми соединениями в течение 48 ч клетки лизируют и активность люциферазы определяют при помощи стандартных методов. Данные аппроксимируют логистической кривой с четырьмя подгоночными параметрами, чтобы определить значения ЕС 50. % эффективности определяли относительно максимальной стимуляции, полученной при применении 10 нМ метилтриенолона. Специалист в данной области техники может легко разработать функциональный анализ модуляции ядерных рецепторов стероидных гормонов, аналогичный приведенным выше. В соответствии с протоколом, по существу описанным выше, соединения согласно примерам 1-10 и 12 проявляют ЕС 50 в анализе по гену-репортеру С 2 С 12 AR/ARE2000 нМ. Характерно, что соединения согласно примерам 1, 2, 6 и 9 проявляли ЕС 50 в анализе по гену-репортеру С 2 С 12 AR/ARE примерно 20,1,0, 0,3 и 0,3 соответственно, что демонстрирует, что соединения согласно настоящему изобретению являются агонистами РА человека. Модель для оценки эффективности и селективности. Самцов крыс Спраг-Доули (в возрасте 24 недели) кастрируют (подвергают гонадэктомии или"ГДЭ") в соответствии с одобренными процедурами и выжидают в течение восьми недель. Также готовят подобранных по возрасту фиктивно прооперированных мышей. Животных содержат в помещении с контролируемой температурой (24 С) с поддерживаемым 12-часовым циклом свет/темнота и неограниченным доступом к воде и пище. Животных рандомизируют на основании массы тела перед назначением тестируемой позиции. Соединения, предложенные в настоящем изобретении, вводят ежедневно перорально через зонд кастрированным тридцатидвухнедельным крысам (масса тела примерно 450-500 г) с применением традиционного носителя, такого как 1% натриевая соль карбоксиметилцеллюлозы (КМЦ)+0,25% Tween 80+0,05% AntiFoam в стерильной Н 2 О. Фиктивно прооперированных крыс, получавших один носитель, применяют в качестве положительного контроля лечения, а кастрированных крыс, получавших только носитель, применяют как отрицательный контроль лечения. Тестируемым животным ежедневно в течение двух или восьми недель вводят, например, 0,3, 1, 2, 3 или 6 мг/кг/день соединения, предложенного в настоящем изобретении. После периода лечения животных умерщвляют, и в каждой группе можно определить сырой вес мышцы, поднимающей задний проход(LA), и бульбокавернозной мышцы в каждой тестовой группе, и сравнить с сырым весом мышцы, поднимающей задний проход, и бульбокавернозной мышцы у контрольной группы кастрированных получавших только носитель животных (после взвешивания бульбокавернозную мышцу можно мгновенно заморозить в жидком азоте для дальнейшего применения при измерении мРНК синтазы окиси азота, как описано выше). В качестве показателя селективной активности в отношении тканей аналогично можно определить сырой вес предстательной железы у тестируемых животных и сравнить с сырым весом предстательной железы у контрольной группы кастрированных получавших только носитель животных. В соответствии с протоколом, по существу описанным выше, с применением восьминедельного пе- 13019713 риода лечения, исследования соединения, соответствующего примеру 2, дали следующие результаты: фиктивно прооперированные животные (только носитель) демонстрировали средний сырой вес LA примерно 0,255 г, средний сырой вес предстательной железы примерно 824,5 мг и средний сырой вес бульбокавернозной мышцы примерно 0,93 г; кастрированные, получавшие только носитель животные демонстрировали средний сырой вес LA примерно 0,094 г, средний сырой вес предстательной железы примерно 103,8 мг и средний сырой вес бульбокавернозной мышцы примерно 0,326 г; группа, получавшая исследуемое соединение в дозе 0,3 мг/кг, демонстрировала средний сырой вес LA примерно 0,137 г, средний сырой вес предстательной железы примерно 72,4 мг и средний сырой вес бульбокавернозной мышцы примерно 0,476 г; группа, получавшая исследуемое соединение в дозе 1,0 мг/кг, демонстрировала средний сырой вес LA примерно 0,182 г, средний сырой вес предстательной железы примерно 102,8 мг и средний сырой вес бульбокавернозной мышцы примерно 0,582 г; группа, получавшая исследуемое соединение в дозе 3,0 мг/кг, демонстрировала средний сырой вес LA примерно 0,205 г, средний сырой вес предстательной железы примерно 147,4 мг и средний сырой вес бульбокавернозной мышцы примерно 0,698 г; а группа, получавшая исследуемое соединение в дозе 6,0 мг/кг, демонстрировала средний сырой вес LA примерно 0,264 г, средний сырой вес предстательной железы примерно 271,8 мг и средний сырой вес бульбокавернозной мышцы примерно 0,955 г. Таким образом, лечение соединением, соответствующим примеру 2, вызывало дозозависимое увеличение веса мышцы, поднимающей задний проход, и бульбокавернозной мышцы по сравнению с контрольной группой кастрированных животных. Анализ эректильной активности in vitro. Для эректильной активности важное значение имеет путь синтазы окиси азота/циклического гуанозинмонофосфата (NOS/цГМФ). Экспрессия NOS приводит к выработке окиси азота (NO), который, в свою очередь, стимулирует выработку цГМФ посредством активации гуанилилциклазы. цГМФ стимулирует активность протеинкиназы G (PKG), которая опосредует расслабление гладких мышц пещеристых тел и облегчает эрекцию. Данные подтверждают роль андрогенов в регуляции экспрессии и активности изоформ NOS в пещеристом теле в экспериментальных моделях на животных. Traish et al., EuropeanUrology, 52; 54-70 (2007). Так, модуляторы рецепторов андрогенов, которые способны увеличивать экспрессию изоформ NOS, как предполагают, играют определенную роль в регулировании активности эрекции. Для определения способности соединений, предложенных в настоящем изобретении, к стимуляции экспрессии изоформ NOS, можно применять следующие способы in vitro. РНК выделяют из тканей луковицы и пещеристого тела, которые получают при вскрытии кастрированных крыс Спраг-Доули, которых готовили и которым вводили соединения, по существу, как описано выше для модели оценки эффективности и селективности. кДНК синтезируют из 2 мкг РНК с применением мощного набора кДНК в соответствии с инструкциями производителя. Затем проводят количественную ПЦР в режиме реального времени в соответствии с методологией флуоресцентного TaqMan (Applied Biosystems). Зонды Assays-on-Demand (Applied Biosystems) применяют для транскрипта синтазы окиси азота эпителия крысы (eNOS), в то время как были разработаны зонды для конкретной изоформы синтазы окиси азота полового члена у крыс (pnNOS), с применением программного обеспечения для разработки зондов (Applied Biosystems). Зонды разрабатывают для перекрывания области из 102 по гена нейрональной синтазы окиси азота у крыс (pnNOS), которые специфичны к pnNOS (положения 2865-2967). Последовательности праймеров MGB включают 5'CCGGAACCCTTGCGTTT 3' (SEQ ID NO:1) (прямая) и 5'CAGACTGTGGGCTTCAGAGTCA 3' (SEQ ID NO:2) (обратная),а последовательность зонда - 5'CCCGTAAAGGGCCT 3' (SEQ ID NO:3) (FAM NFQ). Набор зондов Assays-on-Demand для транскрипта PPIB (пептидил-пропил-цис-транс-изомер В) применяют в качестве внутреннего контроля. ПЦР проводят на системе детекции последовательностейABI Prism 7700 при следующих условиях термоциклирования: 2 мин при 50 С, 10 мин при 95 С, 40 циклов при 95 С в течение 30 с и 60 С в течение 1 мин. Все реакции проводят трижды. При применении процедур, по существу описанных в настоящем патенте, соединение, соответствующее примеру 2, демонстрирует дозозависимое увеличение количества мРНК синтазы окиси азота полового члена (pnNOS) в ткани бульбокавернозной мышцы, полученной от кастрированных крыс Спраг-Доули, получавших лечение в течение восьминедельного периода. Характерно, что группа, получавшая исследуемое соединение в дозе 0,3 мг/кг, демонстрировала экспрессию pnNOS примерно 97% от контроля; группа, получавшая исследуемое соединение в дозе 1,0 мг/кг, демонстрировала экспрессиюpnNOS примерно 93% от контроля; группа, получавшая исследуемое соединение в дозе 3,0 мг/кг, демонстрировала экспрессию pnNOS примерно 153% от контроля; а группа, получавшая исследуемое соединение в дозе 6,0 мг/кг, демонстрировала экспрессию pnNOS примерно 248% от контроля. Кроме того, в отдельной группе соединение, соответствующее примеру 2, демонстрирует увеличение количества мРНК синтазы окиси азота эпителия (eNOS) в ткани пещеристого тела, полученной от кастрированных крыс Спраг-Доули, получавших лечение в течение двухнедельного периода при дозе 1 мг/кг/день. Характерно, что экспрессия eNOS в группе, получавшей исследуемое соединение, составляла примерно 159% от контроля. Уменьшение мышечной массы или силы может происходить в результате иммобилизации или бездействия, например, после перелома кости, или замены тазобедренного или коленного сустава. Чтобы определить способность соединений, предложенных в настоящем изобретении, лечить или предотвращать потерю мышечной массы или силы, вызванную иммобилизацией, бездействием или травмой, можно применять следующие модели на животных. Модель потери мышечной массы при иммобилизации. Заднюю конечность 12-недельных самцов мыши ICR (Imprinting Control Region) иммобилизуют в положении сгибания подошвы при помощи гипсовой повязки на конечность. После иммобилизации в течение 7 дней мышей подвергают лечению путем ежедневного введения соединения, предложенного в настоящем изобретении, в течение разных периодов времени. Контрольным животным, с гипсом и без гипса, аналогичным образом вводят носитель в течение разных периодов времени. В конце протокола лечения мышей умерщвляют, определяют сырой вес икроножной мышцы, находившейся в гипсе, и индивидуальные лечебные группы сравнивают с контролем с применением носителя. В целом см. Am. J.Endocrinol. Metab. 289:969-980 (2005). Модель повреждения мышцы и травмы. Самцов мыши ICR кастрируют в возрасте 8 недель и выдерживают в течение дополнительных 8 недель. Мышей содержат в отдельных клетках и поддерживают 12-часовой цикл свет/темнота при 22 С с неограниченным доступом к пище и воде. Мышей анестезируют изофлюораном (1-5%) и производят инъекцию 100 мкл 10 мкМ кардиотоксина (naja naja atra; Sigma Aldrich) в икроножную мышцу с обеих сторон, чтобы вызвать повреждение мышцы. Животные восстанавливаются от анестезии, и их возвращают к нормальной активности в течение 5 мин. На 5-й день после инъекции животных лечат разными дозами соединения, предложенного в настоящем изобретении. Через 14 дней после инъекции подвергнутых лечению мышей подвергают эвтаназии, взвешивают, и ткань икроножной мышцы отбирают как из не подвергавшейся инъекциям, так и из подвергавшейся инъекции кардиотоксина задней лапы. Массу мышцы сравнивали с неподвергавшимся инъекции и обработке контролю, чтобы определить процент восстановления после травмы. Чтобы продемонстрировать, что соединения, предложенные в настоящем изобретении, способны лечить нарушения, связанные с потерей костной массы, такие как остеопороз и остеопения, можно применять другие модели на животных, хорошо известные специалистам в данной области техники. Примеры таких моделей приведены у Y.L. Ma et al., Japanese Journal of Bone и Mineral Metabolism 23 (Suppl.): 62-68 (2005); Y.L. Ma et al., Endocrinology 144:2008-2015 (2003); и K. Hanada et al., Biol. Pharm. Bull. 26(11):1563-1569 (2003). Конкретные упоминания сделаны относительно модели остеопении у самок крыс при дефиците эстрогенов, вызванном овариэктомией, и модели остеопении у самцов крыс при дефиците андрогенов, вызванном орхидэектомией. Модель остеопении при дефиците эстрогенов, вызванном овариэктомией. Неоплодотворенных самок крыс Спраг-Доули в возрасте 6 месяцев с массой примерно 220 г содержат с обеспечением неограниченного доступа к пище и воде. Животных подвергают двусторонней овариэкстомии (за исключением фиктивно оперированного контроля) (Дсо) и затем рандомизируют в лечебные группы, по 7-8 животных на группу. В каждом анализе обычно имеется по меньшей мере по 2 контроля, включая контроль фиктивно прооперированных (фиктивных) животных и контроль с овариэктомией (Дсо), которым вводят носитель. Крысам Дсо давали потерять костную массу в течение 1 месяца,чтобы достигнуть остеопении перед лечением тестируемым соединением. Тестируемые соединения вводят перорально через зонд животным Дсо в течение 8 недель. В качестве положительного контроля подгруппе животных Дсо можно вводить рекомбинантный ПТГ (паратгормон) (1-38) (примерно 10 мг/кг/д,подкожно). После завершения протокола тестирования для анализа волюметрической плотности костного вещества (BMD, мг/см 3) поясничного позвонка L-5 и бедренной кости применяют количественную компьютерную томографию. Биомеханический анализ изгиба в трех точках средней трети бедренной кости и нагрузки до отказа на проксимальную часть бедренной кости проводят при помощи аппарата для механических испытаний материалов и анализируют при помощи программного обеспечения TestWorks 4. Модель остеопении при дефиците андрогенов, вызванном орхиэктомией. Самцов крыс Спраг-Доули в возрасте 6 месяцев с массой примерно 485 г содержат с обеспечением неограниченного доступа к пище и воде. Животных подвергают двусторонней орхиэктомии (Доэ) (за исключением фиктивно оперированного контроля) и затем рандомизируют в лечебные группы, по 7-8 животных на группу. В каждом анализе обычно имеется по меньшей мере по 2 подгруппы контроля,включая контроль фиктивно прооперированных (фиктивных) животных и контроль с орхиэктомии (Доэ),которым вводят носитель. Крысам Доэ дают потерять костную массу в течение 2 месяцев, чтобы достигнуть остеопении перед началом лечения тестируемым соединением. Тестируемые соединения вводят перорально через зонд животным Доэ в течение 8 недель. В качестве положительного контроля подгруппе животных Доэ можно вводить рекомбинантный ПТГ (паратгормон) (1-38) (примерно 10 мг/кг/д, под- 15019713 кожно). После завершения протокола тестирования можно проводить анализ BMD позвонков и бедренной кости, а также биомеханический анализ бедренной кости, как описано выше для модели с самками крыс после овариэктомии (в целом см. Ma et al., JBMR 17:2256-2264 (2002) и Turner et al., Bone [Review] 14:595-608 (1993. Как очевидно для специалиста в данной области техники, протоколы с моделями на животных,описанные выше, можно легко адаптировать для применения в сочетании с соединениями и способами,предложенными в настоящем изобретении. Следующие примеры получения и примеры дополнительно иллюстрируют изобретение и представляют собой типичные способы синтеза соединений формулы (I), включая любые новые соединения, которые в общем виде описаны выше. Специалист в данной области техники может легко достать или синтезировать реагенты и исходные вещества. Следует понимать, что примеры получения и примеры приведены для иллюстрирования и не являются ограничивающими, и что специалистом в данной области техники могут быть сделаны различные модификации.R- или S-конфигурации соединений, предложенных в настоящем изобретении, можно определить при помощи стандартных методов, таких как рентгенографический анализ и корреляция со временем удержания при хиральной ВЭЖХ. Названия соединений, предложенных в настоящем изобретении, в общем случае даны с помощью Autonom 2000 для ISIS Draw. Пример получения 1. При интенсивном перемешивании при помощи механической мешалки к суспензии циклопентенона (100 г, 1,22 моль) и фталимида (180 г, 1,22 моль) в МеОН (886 мл) добавляют 2 М водного Na2CO3 (79 мл, 0,158 моль). Спустя приблизительно 2 ч должен образоваться плотный белый осадок. Смесь перемешивают при комнатной температуре в течение 24 ч. Твердое вещество белого цвета собирают путем вакуум-фильтрации и промывают метанолом (1 л). Твердое вещество суспендируют в воде (1 л) и смешивают в течение 3 ч. Твердое вещество собирают, высушивают в вакуумной печи при 40 С в течение ночи и получают 198 г (71%) указанное соединение в виде твердого вещества белого цвета. 1ES/MS (масс-спектрометрия с электрораспылением) m/z 230 (М+1, слабый). Примечание: продукт можно легко подвергнуть обратной реакции Михаэля путем обработке водным основанием. Пример получения 2. В 5-л колбе смешивают (R,S)-2-(3-оксоциклопентил)изоиндол-1,3-дион (295,3 г, 1,29 моль), 4 бромфенилгидразин-HCl (287,9 г, 1,29 моль) и ледяную уксусную кислоту (3 л) при механическом перемешивании. Реакционную смесь нагревают с обратным холодильником в течение 5 ч и затем охлаждают до комнатной температуры. Реакционную смесь выливают в воду (4 л) при быстром перемешивании. Твердое вещество собирают путем вакуум-фильтрации, промывают водой (4 л) и высушивают на воздухе в течение 30 мин. Твердое вещество суспендируют с применением МеОН (700 мл), собирают путем вакуум-фильтрации и промывают МеОН (100 мл). Твердое вещество серого цвета высушивают на воздухе в течение 2 ч, затем высушивают в течение ночи в вакуумной печи при 50 С и получают 414,67 г (84%) указанного соединения в виде темного твердого вещества. К раствору (R,S)-2-(7-бром-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)изоиндол-1,3-диона (150 г,393 ммоль) в ТГФ (1000 мл) и EtOH (150 мл) добавляют гидразин моногидрат (35,0 г, 34,0 мл, 699 ммоль). Реакционную смесь перемешивают путем механического перемешивания при комнатной температуре в течение 18 ч, а затем в течение 2 ч при 55 С, вследствие чего реакционная смесь становится очень вязкой, и добавляют ТГФ (425 мл) и EtOH (75 мл). Нагревание при 55 С продолжают еще в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры, фильтруют через диатомовую землю, промывают ТГФ и концентрируют до сухого состояния. Осадок смешивают с толуолом и EtOH и снова концентрируют до сухого состояния. Продукт подвергают воздействию высокого вакуума в течение 3 ч, получая 94 г (95%) указанного соединения в виде твердого вещества.(800 мл) и насыщенного водного NaHCO3 (200 мл) обрабатывают ди-трет-бутилдикарбонатом (80,3 г, 357 ммоль) порциями и перемешивают при комнатной температуре в течение 1 ч. Реакционную смесь разбавляют EtOAc (300 мл) и солевым раствором (100 мл). Разделяют слои, органический слой высушивают над MgSO4, фильтруют и концентрируют до образования темного маслянистого твердого вещества. Твердое вещество смешивают с CH2Cl2 (400 мл), охлаждают на ледяной бане и фильтруют. Твердое вещество промывают CH2Cl2 и гексанами с получением 28,1 г (34%) указанного соединения в виде твердого вещества. Еще 78,8 г (60%) указанного соединения получают путем концентрирования фильтрата и очищения при помощи хроматографии (1 л силикагеля, загружают как концентрированный раствор трет-Бутиловый эфир R,S)-7-бром-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты (635 г, 1810 ммоль) сначала растирают с холодным Et2O, а затем очищают при помощи хиральной ВЭЖХ (колонка: Chiralcel OJ 832 см; элюент: 100% МеОН), получая 310 г указанного соединения (элюируемый вторично изомер) в виде твердого вещества желтовато-коричневого цвета. Хиральная ВЭЖХ В ДМФ (250 мл) вместе перемешивают следующие реагенты при 100 С в течение 18 ч: третбутиловый эфир S)-7-бром-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты (50,0 г,142 ммоль), цианид цинка (11,9 г, 99,7 ммоль), ацетат цинка (5,22 г, 28,5 ммоль), (1,1'-бис(дифенилфосфино)ферроцен)палладия (II) хлорид (Pd(dppf)2Cl2) (1,74 г, 2,14 ммоль) и цинк (3,72 г, 56,9 ммоль). Реакционную смесь концентрируют до сухого состояния и разделяют между водой и этилацетатом. Органические вещества промывают водой и солевым раствором, затем концентрируют, получая твердое вещество. Твердое вещество хроматографируют в двух равных частях силикагеля (1600 мл) следующим образом: загружают в виде раствора в CH2Cl2 и элюируют при помощи 2% EtOAc в CH2Cl2(3 л), затем при помощи 5% EtOAc в CH2Cl2. Продукт выделяют из двух колонок с образованием 29 г(69%) указанного соединения в виде твердого вещества белого цвета. 1 трет-Бутиловый эфир 5)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты (10 г, 33,63 ммоль) растворяют в 1,4-диоксане (102 мл) и обрабатывают 4 М HCl/диоксаном (102 мл) при комнатной температуре. Через 18 ч твердое вещество отфильтровывают, промывают Et2O (50 мл), а затем высушивают под вакуумом. Твердое вещество суспендируют в дихлорметане (168 мл) и обрабатывают диизопропилэтиламином (9,13 г, 12,3 мл, 70,1 ммоль) и изопропилхлорформиатом (1,0 М в толуоле, 34,0 мл, 34,0 ммоль) при комнатной температуре. Через 4 ч реакционную смесь обрабатывают водой (50 мл), концентрируют и получают водную суспензию продукта. Далее реакционную смесь разбавляют водой (500 мл) и обрабатывают ультразвуком в течение 15 мин на ультразвуковой бане. Твердое вещество желто-коричневого цвета отфильтровывают и высушивают под вакуумом при 40 С. Твердое вещество суспендируют в Et2O (100 мл), обрабатывают ультразвуком в течение 10 мин на ультразвуковой бане, фильтруют, промывают Et2O (50 мл), а затем высушивают под вакуумом и получают 8,20 г(выход 86%) указанного соединения в виде твердого вещество желто-коричневого цвета. Суспензию транс-3-гидрокси-L-пролина (5 г, 37,56 ммоль) в метаноле (100 мл) обрабатывают диизопропилэтиламином (6,55 мл, 37,56 ммоль) и далее ди-трет-бутилдикарбонатом (8,87 г, 39,44 ммоль) при комнатной температуре. Образующуюся суспензию перемешивают в течение 2 ч при комнатной температуре, пока она становится однородным раствором. Растворитель удаляют под вакуумом, а осадок растворяют в этилацетате (120 мл). Раствор органических веществ промывают 1 н. водным хлористым водородом. Водный слой отбрасывают, а органический слой промывают солевым раствором, высушивают над сульфатом натрия, концентрируют, высушивают под вакуумом и получают 8,0 г (90%) указанного соединения. 1 Раствор 1-трет-бутилового эфира 3-гидроксипирролидин-1,2-дикарбоновой кислоты (13,29 г, 57,47 ммоль) в тетрагидрофуране (130 мл) по каплям обрабатывают комплексом боран-метилсульфид (16,06 мл, 172,41 ммоль) и перемешивают в течение ночи при комнатной температуре. Смесь охлаждают до 5 С на водно-ледяной бане и обрабатывают капельно 3 н. водным раствором хлористого водорода (5 мл), пока не прекратит выделяться газ. Образующуюся суспензию далее перемешивают в течение 30 мин и разбавляют 5 н. водным раствором гидроксида натрия, пока не растворится твердое вещество белого цвета. Его экстрагируют при помощи этилацетата (3100 мл). Органический слой промывают водой (2100 мл) и солевым раствором, высушивают над сульфатом натрия, фильтруют и концентрируют до сухого состояния. Образующееся масло высушивают под вакуумом и получают 10,03 г (80%) указанного соединения. Суспензию (2R,3S)-трет-бутил-3-гидрокси-2-(гидроксиметил)пирролидин-1-карбоксилата (5,04 г,23,20 ммоль) и дибутилоксоолова (7,07 г, 27,84 ммоль) в толуоле (50 мл) нагревают на масляной бане с обратным холодильником при 130 С в течение 2 ч. Смесь охлаждают до 0 С в ледяной ванне в течение 30 мин и обрабатывают метансульфонилхлоридом (2,15 мл, 27,84 ммоль) один раз. Реакционную смесь перемешивают в течение 30 мин при 0 С, а затем смеси дают постепенно нагреваться до комнатной температуры в течение ночи. Раствор концентрируют и образовавшийся осадок очищают при помощи жидкостной хроматографии среднего давления, элюируя при помощи этилацетата:гексана (8:2). Фракции,содержащие чистый продукт, объединяют, концентрируют и получают 6,14 г (96%) указанного соединения в виде густого масла.(3,45 г, 10,59 ммоль) и йодида калия (88 мг, 529 мкмоль) в диметилформамиде (50 мл) нагревают на масляной бане при 60 С в течение двух дней. После охлаждения до комнатной температуры реакционную смесь разбавляют этилацетатом (120 мл) и промывают водой (3100 мл) и солевым раствором. Органическую часть высушивают над сульфатом натрия, фильтруют и концентрируют до сухого состояния. Образующийся осадок очищают при помощи жидкостной хроматографии среднего давления, элюируют при помощи этилацетата:хлороформа (2:8). Фракции, содержащие чистый продукт, объединяют, концентрируют и получают 1,59 г (62%) указанного соединения.(20 мл) капельно обрабатывают диэтилазодикарбоксилатом (374 мкл, 2,36 ммоль) при комнатной температуре. Реакционную смесь перемешивают в течение ночи. Реакционную смесь разбавляют этилацетатом(50 мл) и затем промывают водой и солевым раствором. Органическую часть высушивают над сульфатом натрия и концентрируют. Образующийся осадок очищают при помощи жидкостной хроматографии среднего давления, элюируют при помощи этилацетата:гексана (1:1). Фракции, содержащие чистый продукт, объединяют, концентрируют и получают 0,59 г (54%) указанного соединения.LC-ES/MS m/z 581,0 [M+Na]+, TR=4,46 мин. Пример получения 13. Гидрохлорид изопропилового эфира [(S)-7-циано-4-2R,3R)-3-гидроксипирролидин-2-илметил)1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты(2R,3R)-3-(2-хлорацетокси)-2-S)-7-циано-2 изопропоксикарбониламино-2,3-дигидро-1 Н-циклопента[b]индол-4-илметил)пирролидин-1-карбоновой кислоты (500 мг, 894,4 мкмоль) в МеОН (2 мл) обрабатывают 2 н. раствором LiOH (2 мл) и суспензию перемешивают в течение 4 ч при комнатной температуре. Суспензию разбавляют этилацетатом (60 мл) и промывают водой и солевым раствором. Органическую часть высушивают над сульфатом натрия, фильтруют и концентрируют. Осадок (0,39 г) растворяют в 10 мл МеОН (10 мл) и обрабатывают 4 н. HCl в растворе 1,4-диоксана (10 мл). Раствор перемешивают в течение 2 ч, концентрируют под вакуумом и получают 0,37 г (92%) указанного соединения.(100 мл) обрабатывают стружками магния (5,26 г, 214,4 ммоль) и обрабатывают ультразвуком в течение 45 мин на водяной бане. Образующуюся мутную суспензию перемешивают при 40 С на масляной бане в течение 20 ч. К реакционной смеси добавляют силикагель (30 мл) и разбавляют метанолом (50 мл), пока суспензия не станет смешиваемой. Суспензию перемешивают в течение 30 мин и концентрируют. Остаток высушивают под вакуумом в течение ночи. Остаток, содержащий диоксид кремния, загружают на картридж DASI65 и элюируют при помощи хлороформа:метанола:водного аммиака (90:9:1). Исходный транс-1-бензосульфонил-2-бензилоксиметил-3-метилпирролидин-3-ол (4,0 г) получают также в форме целевого незащищенного вещества (5,47 г). Растворяют незащищенное вещество в тетрагидрофуране (20 мл) и обрабатывают ди-трет-бутилдикарбонатом (5,7 г, 26,1 ммоль). Раствор перемешивают в течение 1 ч и концентрируют. Осадок разбавляют этилацетатом (100 мл) и промывают 0,5 н. водным раствором гидроксида натрия (50 мл) и солевым раствором. Органическую часть высушивают над сульфатом натрия,фильтруют и концентрируют. Образующийся осадок очищают при помощи жидкостной хроматографии среднего давления и элюируют при помощи этилацетата:гексана (1:1). Фракции, содержащие чистый продукт, объединяют и концентрируют с получением 8,15 г (71%) указанного соединения. Раствор трет-бутилового эфира транс-3-гидрокси-2-гидроксиметил-3-метилпирролидин-1 карбоновой кислоты (8,12 г, 25,26 ммоль) в метаноле (100 мл) обрабатывают 5% палладием на угле (50% влажности, 3 г, 28,19 ммоль) и гидрогенизируют при 350 кПа в течение ночи. Катализатор удаляют путем фильтрации, а фильтрат концентрируют. Образующееся масло высушивают под вакуумом и получа- 20019713 Смесь трет-бутилового эфира транс-3-гидрокси-2-метансульфонилоксиметил-3-метилпирролидин 1-карбоновой кислоты (2,69 г, 11,63 ммоль) и триэтиламина (1,78 мл, 12,79 ммоль) в дихлорметане (100 мл) охлаждают на бане ацетонитрил/сухой лед (-40 С) в течение 30 мин и обрабатывают метансульфонилхлоридом (945,21 мкл, 12,21 ммоль) один раз. Раствор перемешивают в течение 1 ч при -40 С и выливают в делительную воронку, содержащую дихлорметан (50 мл) и воду (100 мл). Раствор органических веществ промывают водой и солевым раствором, высушивают над сульфатом натрия, фильтруют и концентрируют. Осадок очищают при помощи жидкостной хроматографии среднего давления и элюируют при помощи этилацетата:гексана (8:2). Фракции, содержащие чистый продукт, объединяют и концентрируют с получением 3,30 г (91%) указанного соединения.S)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2 ил)карбаминовой кислоты (2,00 г, 7,06 ммоль), трет-бутилового эфира транс-3-гидрокси-2 метансульфонилоксиметил-3-метилпирролидин-1-карбоновой кислоты (3,28 г, 10,59 ммоль), цезия карбоната (5,11 г, 15,53 ммоль) и йодида калия (118 мг, 705,9 мкмоль) в диметилформамиде (40 мл) перемешивают в атмосфере аргона на масляной бане при 60 С в течение 48 ч. Образующуюся суспензию охлаждают до комнатной температуры и разбавляют этилацетатом (120 мл), затем промывают водой (3100 мл) и солевым раствором. Органическую часть высушивают над сульфатом натрия, фильтруют и концентрируют до сухого состояния. Образующийся осадок очищают при помощи жидкостной хроматографии среднего давления и элюируют при помощи этилацетата:гексана (1:1). Фракции, содержащие чистый продукт, объединяют и концентрируют с получением 2,5 г (71%) указанного соединения.N,N-диметилформамиде (30 мл) обрабатывают йодметаном (3,13 мл, 50,2 ммоль) и перемешивают при комнатной температуре в атмосфере азота в течение 2 ч. Реакционную смесь разбавляют диэтиловым эфиром (150 мл) и промывают 0,5 М соляной кислотой (2150 мл). Органическую часть высушивают над безводным сульфатом натрия, фильтруют, концентрируют и получают 3,42 г (88%) указанного соединения в виде прозрачного масла. 1 Н ЯМР (400 МГц, CDCl3)7,59 (д, 1 Н), 7,32 (дд, 1 Н), 7,04 (м, 1 Н), 4,04 (с, 3 Н). К раствору 3-фтор-2-метоксибензальдегида (3,41 г, 22,1 ммоль) в метаноле (20 мл) медленно порциями добавляют боргидрид натрия (1,00 г, 26,6 ммоль). Реакционную смесь перемешивают при комнатной температуре в атмосфере азота в течение ночи. Реакционную смесь разбавляют диэтиловым эфиром,промывают водой дважды, высушивают над безводным сульфатом натрия, фильтруют и концентрируют с получением 3,02 г (87%) указанного соединения в виде прозрачного бесцветного масла. 1 Н ЯМР (400 МГц, CDCl3)7,09-6,92 (м, 3 Н), 4,65 (д, 2 Н), 3,97 (с, 3 Н), 2,14 (т, 1 Н, OH). Пример получения 20. 1-(Хлорметил)-3-фтор-2-метоксибензол Раствор (3-фтор-2-метоксифенил)метанола (3,02 г, 19,3 ммоль) и триэтиламина (6,74 мл, 48,4 ммоль) в дихлорметане (20 мл) обрабатывают метансульфонилхлоридом (2,99 мл, 38,7 ммоль) медленно при комнатной температуре и перемешивают в атмосфере азота в течение 3 ч. Реакционную смесь разбавляют диэтиловым эфиром, промывают дважды 0,5 М соляной кислотой, высушивают над безводным сульфатом натрия, фильтруют и концентрируют с получением указанного соединения в виде масла желтого цвета (2,71 г, 80%). 1(S)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2 илкарбаминовой кислоты (1,50 г, 5,29 ммоль) и 1-(хлорметил)-3-фтор-2-метоксибензола (1,11 г, 6,35 ммоль) в N,N-диметилформамиде (10 мл) обрабатывают карбонатом цезия (2,59 г, 7,94 ммоль) при комнатной температуре и перемешивают в течение ночи в атмосфере азота. Реакционную смесь разбавляют водой и эфиром/гексанами. Твердые вещества белого цвета, которые выпадают в осадок, отфильтровывают, промывают водой, эфиром, гексаном и высушивают под вакуумом при 50 С в течение 48 ч и получают указанное соединение в виде твердого вещества белого цвета (2,18 г, 98%).S)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-илкарбаминовой кислоты (2,0 г, 7,06 ммоль) и карбоната цезия (3,22 г, 9,88 ммоль) в ДМФ (40 мл) обрабатывают хлоридом 2-метоксибензила (1,16 г, 7,41 ммоль). Реакционную смесь нагревают при 50 С в течение 18 ч. Реакционную смесь охлаждают до комнатной температуры и разбавляют водой (300 мл). Твердое вещество белого цвета собирают и промывают водой. Твердое вещество высушивают при 40 С в вакуумной печи. После высушивания получают 2,80 г (98%) продукта в виде твердого вещества белого цвета. трет-Бутилдиметилхлорсилан (14,34 г, 93,24 ммоль) соединяют с 4-фтор-2-метилфенолом (10,00 г,77,70 ммоль) и 1H-имидазолом (13,32 г, 194,24 ммоль) в диметилформамиде (100 мл) и перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют эфиром (200 мл) и промывают водой (2100 мл) и солевым раствором. Органическую часть высушивают над сульфатом натрия,фильтруют, концентрируют и получают 18,9 г (100%) указанного соединения. Смесь трет-бутил-(4-фтор-2-метилбензил)диметилсилана (6,20 г, 25,79 ммоль) и Nбромсукцинимида (4,75 г, 26,31 ммоль) в четыреххлористом углероде (50 мл) нагревают с обратным холодильником на масляной бане при 90 С в течение 10 мин и обрабатывают пероксидом бензоила (64 мг,258 мкмоль). Образующуюся суспензию нагревают с обратным холодильником в течение 2 ч и охлаждают до комнатной температуры. Твердое вещество удаляют путем фильтрации, а фильтрат концентрируют под вакуумом. Образующийся осадок загружают на картридж ReadySep (25 г) и элюируют при помощи гексана (300 мл) с получением 6,4 г (78%) указанного соединения в виде прозрачного масла. Смесь 3-бром-4-(хлорметил)пиридина (3,16 г, 11,5 ммоль), изопропилового эфира S)-7-циано 1,2,3,4-тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты (2,50 г, 8,82 ммоль) и карбоната цезия (4,31 г, 13,2 ммоль) в диметилформамиде (20 мл) перемешивают при комнатной температуре в атмосфере азота в течение ночи. Затем реакционную смесь разбавляют этилацетатом, дихлорметаном и водой. Органическую фазу отделяют, дважды промывают водой, высушивают над безводным сульфатом натрия, фильтруют, концентрируют под вакуумом и получают масло коричневого цвета (4,75 г). Масло дважды растирают с гексанами, затем дважды с эфиром. Затем его очищают при помощи флэшхроматографии, элюируют 30-60% этилацетатом/гексаном и получают твердое вещество грязно-белого цвета (3,23 г, 81%). К раствору (S)-изопропил-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-илкарбамата (680 мг,2,40 ммоль) и 4-хлор-2-(хлорметил)-3-метоксипиридина (553 мг, 2,88 ммоль) в диметилформамиде (5 мл) добавляют карбонат цезия (1,17 г, 3,60 ммоль) и смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют водой и отфильтровывают твердое вещество бежевого цвета. Его промывают водой, затем высушивают в вакуумной печи при 50 С в течение 48 ч и получают твердое вещество серого цвета (970 мг, 92%). Смесь (S)-изопропил-4-4-хлор-3-метоксипиридин-2-ил)метил)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-илкарбамата (12,53 г, 28,55 ммоль) и 10% палладиевого катализатора на угле (1,00 г) в метаноле (150 мл) гидрогенизируют при 50 фунт/кв.дюйм при комнатной температуре в течение ночи. Свежую партию 10% палладиевого катализатора на угле (1,2 г) суспендируют в воде (2 мл) и добавляют к реакционной смеси, которую гидрогенизируют (50 фунт/кв.дюйм) при комнатной температуре в течение ночи. Катализатор отфильтровывают, раствор концентрируют при пониженном давлении и получают твердое вещество бледно-желтого цвета. Твердое вещество дважды растирают с эфиром и высушивают под высоким вакуумом с получением вещества бледно-желтого цвета (10,80 г, 94%). Раствор трет-бутилового эфира (2R,3S)-2-S)-7-циано-2-изопропоксикарбониламино-2,3-дигидро 1 Н-циклопента[b]индол-4-илметил)-3-гидроксипирролидин-1-карбоновой кислоты (0,50 г, 1,04 ммоль) в метаноле (20 мл) обрабатывают 4 н. хлористым водородом в 1,4-диоксане (20 мл), перемешивают при комнатной температуре в течение 3 ч и концентрируют. Осадок суспендируют в этилацетате (200 мл) и обрабатывают 2 н. водным раствором гидроксида натрия (20 мл). Органический слой промывают солевым раствором, высушивают над сульфатом натрия и концентрируют. Образующийся осадок очищают при помощи жидкостной хроматографии среднего давления и элюируют при помощи метанола:хлороформа (5:95). Фракции, содержащие чистый продукт, объединяют, концентрируют и получают 0,38 г (96%) указанного соединения. Раствор трет-бутилового эфира (2R,3S)-2-S)-7-циано-2-изопропоксикарбониламино-2,3-дигидро 1 Н-циклопента[b]индол-4-илметил)-3-гидроксипирролидин-1-карбоновой кислоты (3,10 г, 6,42 ммоль) в этаноле (20 мл) обрабатывают 4 н. хлористым водородом в 1,4-диоксане (20 мл) и перемешивают при комнатной температуре в течение 3 ч. Образующийся раствор концентрируют под вакуумом, суспендируют в этилацетате (200 мл) и обрабатывают 2 н. водным раствором гидроксида натрия (70 мл). Органический слой промывают солевым раствором, высушивают над сульфатом натрия и концентрируют. Образующийся осадок растворяют в хлороформе (100 мл) и обрабатывают формальдегидом (1,45 мл, 19,27 ммоль) и триацетоксиборгидридом натрия (4,25 г, 19,27 ммоль). Образующуюся суспензию перемешивают в течение ночи при комнатной температуре. Суспензию обрабатывают насыщенным раствором бикарбоната натрия (40 мл) и перемешивают в течение 30 мин. Образующуюся суспензию разбавляют дихлорметаном (100 мл) и промывают водой (3100 мл) и солевым раствором. Органическую часть высушивают над сульфатом натрия, фильтруют и концентрируют до сухого состояния. Образующийся осадок очищают при помощи жидкостной хроматографии среднего давления и элюируют при помощи метанола:хлороформа (5:95). Фракции, содержащие чистый продукт, объединяют, концентрируют и получают Раствор изопропилового эфира [(S)-7-циано-4-2R,3S)-3-гидроксипирролидин-2-илметил)-1,2,3,4 тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты (300 мг, 784,4 мкмоль) в ацетонитриле (20 мл) обрабатывают ацетальдегидом (1,0 мл, 17,80 ммоль) и триацетоксиборгидридом натрия (520 мг, 2,35 ммоль) и перемешивают при комнатной температуре в течение ночи. Суспензию обрабатывают насыщенным раствором бикарбоната натрия (40 мл) и перемешивают в течение 30 мин. Образующуюся суспензию разбавляют дихлорметаном (100 мл) и промывают водой (3100 мл) и солевым раствором. Органическую часть высушивают над сульфатом натрия, фильтруют и концентрируют до сухого состояния. Образующийся осадок очищают при помощи жидкостной хроматографии среднего давления, элюируя при помощи метанола:хлороформа (5:95). Фракции, содержащие чистый продукт, объединяют, концентрируют и получают 0,1 г (31%) указанного соединения. Раствор гидрохлорида изопропилового эфира [(S)-7-циано-4-2R,3R)-3-гидроксипирролидин-2 илметил)-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты (380 мг, 907,1 мкмоль) в ацетонитриле (50 мл) обрабатывают метанолом (5 мл), пока не растворится большая часть твердого вещества, и затем обрабатывают формальдегидом (136,3 мкл, 1,81 ммоль) и триацетоксиборгидридом натрия (400,5 мг, 1,81 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Добавляют насыщенный водный раствор бикарбоната натрия (50 мл) и смешивают в течение 30 мин. Смесь разбавляют этилацетатом (120 мл) и образующуюся суспензию промывают водой (3100 мл) и солевым раствором. Органическую часть высушивают над сульфатом натрия, фильтруют и концентрируют до сухого состояния. Осадок очищают при помощи жидкостной хроматографии среднего давления,элюируя при помощи метанола:хлороформа (5:95). Фракции, содержащие чистый продукт, объединяют,концентрируют и получают 0,2 г (61%) указанного соединения.(15 мл) и перемешивают при комнатной температуре в течение 3 ч. Реакционная смесь становится суспензией из белого твердого вещества и красноватого раствора. Суспензию концентрируют, осадок суспендируют в ацетонитриле (30 мл) и обрабатывают 30% водным раствором формальдегида (1,13 мл,15,10 ммоль) и триацетоксиборгидридом натрия (3,33 г, 15,10 ммоль). Образующуюся суспензию перемешивают в течение ночи. Суспензию обрабатывают насыщенным раствором бикарбоната натрия и пе- 25019713 ремешивают в течение 30 мин. Разбавляют суспензию этилацетатом (120 мл) и промывают водой (3100 мл) и солевым раствором. Высушивают органическую часть над сульфатом натрия, фильтруют и концентрируют до сухого состояния. Очищают осадок при помощи жидкостной хроматографии среднего давления и элюируют при помощи этилацетата:гексана (1:1). Фракции, содержащие чистый продукт, объединяют, концентрируют и получают 1,75 г (84%) указанного соединения.[(S)-7-циано-4-(транс-3-гидрокси-1-метил-3 метилпирролидин-2-илметил)-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты (2,15 г,5,24 ммоль) разделяют на колонке chiralpak AD-H, 4,6150 мм и элюируют со скоростью 1 мл/мин при помощи ацетонитрила:метанола:диметилэтиламина (30:70:0,2), проводя детектирование на длине волны 225 нм. Собирают пик 1 с 2,0 до 2,5 мин и пик 2 с 4,0 до 7,0 мин. Фракции, содержащие чистый продукт, объединяют, концентрируют и получают 0,926 г (43%) изомера 1 и 0,822 г (38%) изомера 2. Пример 8. Изопропиловый эфир К раствору (S)-изопропил 7-циано-4-(3-фтор-2-метоксибензил)-1,2,3,4-тетрагидроциклопента[b]индол-2-илкарбамата (1,88 г, 4,46 ммоль) в дихлорметане (50 мл) добавляют 1 М трибромида бора в дихлорметане (13,4 мл, 13,4 ммоль) при 0 С и перемешивают в течение ночи. Реакцию гасят слегка влажным ацетонитрилом. Летучие вещества удаляют при пониженном давлении, а осадок высушивают под высоким вакуумом. Затем его очищают при помощи колоночной хроматографии с 5% этилацетатом в хлороформе и получают твердое вещество желтого цвета. Твердое вещество суспендируют в минимальном количестве дихлорметана и дают отстояться при комнатной температуре. Твердые вещества белого цвета собирают. Их отфильтровывают и быстро промывают дихлорметаном. Маточный раствор концентрируют и процедуру повторяют с осадком три раза. Объединяют твердые вещества белого цвета и получают указанное соединение (1,17 г, 64%).[(S)-7-циано-4-(2-метоксибензил)-1,2,3,4 тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты (1,50 г, 3,72 ммоль) в дихлорметане (37 мл) при 0 С добавляют BBr3 (1,0 М в дихлорметане, 11,2 мл, 11,2 ммоль). Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 18 ч. Реакцию гасят насыщенным водным раствором NH4Cl. Добавляют воду (70 мл) и неочищенный продукт экстрагируют при помощи дихлорметана(75 мл) и EtOAc (275 мл). Объединенные органические соединения высушивают над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают на силикагеле (40 г, 30% EtOAc/гексаны), а затем повторно очищают на силикагеле (40 г, 5% ацетонитрил/дихлорметан) и получают 1,00 г продукта с чистотой примерно 90%. Реакцию повторяют на 1,77 г (4,38 ммоль) и проводят тем же способом. Неочищенный продукт объединяют с продуктом с 90% чистотой из предыдущего цикла, и объединенный продукт очищают на силикагеле (240 г, 2:33:65 МеОН/дихлорметан/гексаны) и получают 991 мг (совокупный выход для двух реакций 37%) указанного соединения в виде твердого вещества белого цвета. 1S)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2 ил)карбаминовой кислоты (1,15 г, 4,06 ммоль) и карбоната цезия (1,60 г, 4,87 ммоль) в диметилформамиде (15 мл) перемешивают при -40 С (баня с сухим льдом) в течение 30 мин и обрабатывают (2 бромметил-4-фторфенокси)-трет-бутилдиметилсиланом (1,56 г, 4,87 ммоль). Реакционную смесь перемешивают при -40 С (баня с сухим льдом) в течение 5 ч и нагревают до комнатной температуры в течение 6 ч. Образующуюся суспензию разбавляют этилацетатом (120 мл) и промывают водой (3100 мл) и солевым раствором. Органическую часть высушивают над сульфатом натрия, фильтруют и концентрируют до сухого состояния. Образующийся осадок очищают при помощи жидкостной хроматографии среднего давления и элюируют при помощи этилацетата:гексана (1:1). Фракции, содержащие чистый продукт, объединяют, концентрируют и получают 0,6 г вещества, защищенного силильными группами. Его растворяют в тетрагидрофуране (10 мл) и добавляют раствор фторида тетрабутиламмония (1 М, 5 мл) при комнатной температуре. Смесь перемешивают в течение 1 ч и концентрируют. Осадок разбавляют этилацетатом (100 мл) и промывают водой и солевым раствором. Органическую часть высушивают над сульфатом натрия и концентрируют. Образующийся осадок очищают при помощи жидкостной хроматографии среднего давления и элюируют при помощи ацетонитрила:дихлорметана (8:92). Фракции,содержащие чистый продукт, объединяют и концентрируют. Получают твердое вещество белого цвета и перекристаллизовывают при помощи ацетонитрила с получением 0,35 г (21%) указанного соединения.(50 мл) нагревают при 100 С в течение 1 ч. Добавляют трис-(дибензилиденацетон)дипалладий (110 мг,120 мкмоль) и 2-ди-трет-бутилфосфино-3,4,5,6-тетраметил-2',4',6'-триизопропил-1,1'-бифенил (230 мг,479 мкмоль) и смесь нагревают в течение 3 ч. Реакционную смесь разбавляют водой и дихлорметаном. Затем слои разделяют и водный слой промывают дважды дихлорметаном. Водную часть подкисляют до рН 1-2, добавляя по капле 5 М HCl, и затем нейтрализуют до рН 7 карбонатом калия. Добавляют этилацетат и фильтруют двухслойную смесь. Органическую фазу разделяют, высушивают над безводным сульфатом натрия, фильтруют и концентрируют под вакуумом с получением осадка коричневого цвета. Осадок растирают с эфиром два раза и высушивают под вакуумом. Затем его очищают при помощи флэш-хроматографии, элюируют при помощи 1-6% метанола в хлороформе и получают неочищенный продукт (2,8 г). Его очищают при помощи препаративной сверхкритической жидкостной хроматографии (колонка: этилпиридин 20150 мм (Princeton Chromatography, Inc.); расход: 70 мл/мин 35 С и 100 фунт/кв.дюйм; элюент: 5-50% МеОН с 0,1% изопропиламином: углекислый газ), и получают указанное соединение в виде твердого вещества белого цвета (548 мг, 20%). Изопропиловый эфир (S)-4-3-метоксипиридин-2-ил)метил)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-илкарбаминовой кислоты (4,59 г, 11,4 ммоль) и пиридин гидрохлорид (21,0 г, 182 ммоль) смешивают с магниевой стружкой в герметичной колбе объемом 75 мл, которую погружают в нагретую масляную баню (170 С). Твердые вещества плавят приблизительно в течение 30 с и перемешивают в течение 60 мин. Смесь охлаждают до комнатной температуры, переносят в другую колбу и растворяют в тетрагидрофуране (100 мл) и воде (100 мл). Медленно по частям добавляют карбонат калия(40 г) и после перемешивания в течение 5 мин при комнатной температуре добавляют 1 М раствор изопропилхлорформиата в толуоле (34,0 мл, 34,0 ммоль). Часть растворителя удаляют при пониженном давлении, а осадок разбавляют дихлорметаном и этилацетатом. Коричневый нерастворимый осадок отфильтровывают и разделяют фазы. Органическую фазу промывают 10% карбонатом калия один раз, высушивают над безводным сульфатом натрия, фильтруют, концентрируют под вакуумом и получают пену коричневого цвета. Пену растворяют в метаноле (20 мл), добавляют 2 М гидроксид натрия (10 мл) и реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Растворитель удаляют при пониженном давлении. Осадок растворяют в 0,5 М HCl/EtOAc/CHCl3 и фильтруют двухслойную смесь. Затем его подкисляют до рН 7-8 5% карбонатом калия. Фазы разделяют и органическую фазу один раз промывают 5% карбонатом калия, высушивают над безводным сульфатом натрия, фильтруют, концентрируют под вакуумом и получают пену оранжевого цвета. Пену очищают при помощи флэшхроматографии на силикагеле и элюируют 10-40% этилацетатом/хлороформом, получая твердое вещество красного цвета (3,33 г). Твердое вещество растворяют в 0,5 М гидроксиде натрия (30 мл). Водный слой два раза промывают дихлорметаном, а органические слои отбрасывают. В водном слое корректируют рН до 4-5 при помощи 5 М соляной кислоты. Твердое вещество грязно-белого цвета собирают из раствора. рН корректируют до 8-9 карбонатом калия, путем фильтрации собирают твердое вещество желтого цвета и сушат на воздухе в течение 1 ч. Затем его суспендируют в минимальном количестве ацетонитрила, фильтруют и промывают минимальным количеством ацетонитрила. Маточный раствор концентрируют, процедуру повторяют два раза и получают белый порошок, который высушивают под высоким вакуумом в течение ночи и получают указанное соединение в виде твердого вещества белого цвета (1,45 г, 33%).LC-ES/MS m/z 391,2 [М+Н]+, TR=3,32 мин. Альтернативный синтез для примера 12. Пример получения 27. Гидрокси-2-(гидроксиметил)пиридина гидрохлорид (50 г, 339,6 ммоль) и метанол (250 мл) соединяют в атмосфере азота и охлаждают до 22 С. К интенсивно перемешиваемой смеси добавляют 4,5 М метоксида натрия в МеОН (170,8 мл, 747,2 ммоль). Смесь перемешивают при комнатной температуре в течение 2 ч и затем растворитель выпаривают. Образующееся вещество растворяют в ДМСО (250 мл),добавляют алкилбромид (41 г, 339,6 ммоль) и смесь перемешивают в течение 18 ч при комнатной температуре. Реакционную смесь добавляют в воду (800 мл) и экстрагируют при помощи метиленхлорида(380 мл). Органические части объединяют и высушивают над сульфатом натрия, фильтруют, а растворитель выпаривают и получают указанное соединение (34,4 г, 62%).(3-Аллилоксипиридин-2-ил)метанол (34 г, 205,8 ммоль) растворяют в дихлорметане (340 мл) в атмосфере азота и добавляют триэтиламин (34,4 мл, 247 ммоль) при 22 С. Смесь нагревают до комнатной температуры на водяной бане и добавляют метансульфонилхлорид (16,7 мл, 216 ммоль), поддерживая температуру ниже 30 С. Реакционной смеси дают перемешиваться в течение ночи при комнатной температуре. Добавляют воду (500 мл) и смесь перемешивают в течение 10 мин. Органическую часть разделяют и промывают раствором, насыщенным NaHCO3 (100 мл). Органическую часть высушивают над сульфатом натрия, фильтруют и растворитель выпаривают с получением указанного соединения. Образующийся осадок высушивают под вакуумом до постоянного веса и получают указанное соединение (29 г,79%). Изопропиловый эфир S)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-ил)карбаминовой кислоты (25 г, 88,2 ммоль) растворяют в диметилформамиде (150 мл) в атмосфере азота. Последовательно добавляют карбонат цезия (57,5 г, 176,5 ммоль) и 3-аллилокси-2-хлорметилпиридин (16,2 г, 88,2 ммоль). Реакционную смесь перемешивают при 40 С в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры и добавляют к воде (1250 мл). После перемешивания в течение 1 ч твердое вещество светло-кремового цвета отфильтровывают. Неочищенное вещество очищают путем перекристаллизации из i-PrOH и затем высушивают под вакуумом до постоянного веса, и получают указанное соединение (32 г; 84%). Изопропиловый эфир [(S)-4-(3-аллилоксипиридин-2-илметил)-7-циано-1,2,3,4-тетрагидроциклопента[b]индол-2-ил]карбаминовой кислоты (40 г, 87,3 ммоль) и 1,4-диоксан (400 мл) соединяют и смесь дегазируют путем продувания азотом в течение 1 ч. Затем добавляют триэтиламин (24,4 мл, 174,7 ммоль) и муравьиную кислоту (6,60 мл, 174,7) и смесь нагревают в атмосфере азота при 80 С. тетра-бис(Трифенилфосфин)палладий (1,02 г, 0,87 ммоль) добавляют при поддержании атмосферы азота. Реакционную смесь нагревают при 80 С в течение 3 ч и затем охлаждают до комнатной температуры. Реакционную смесь фильтруют через пластину из диатомовой земли. Фильтрат выпаривают и неочищенное вещество высушивают под вакуумом с получением твердого вещество коричневого цвета. Твердое вещество растворяют в NMP (100 мл) и затем добавляют к воде (1,2 л). Твердое вещество светло-кремового цвета собирают путем фильтрации и высушивают в течение ночи под вакуумом с получением 41 г неочищенного вещества. Вещество очищают путем перекристаллизации из ацетонитрила и затем высушивают под вакуумом до постоянного веса с получением указанного соединения (29,5 г, 86%).
МПК / Метки
МПК: A61P 21/00, A61K 31/403, C07D 403/06, C07D 401/06, A61P 5/26, A61P 19/10, C07D 209/94, A61K 31/4439
Метки: модуляторы, рецепторов, андрогенов, тетрагидроциклопента[b]индольные
Код ссылки
<a href="https://eas.patents.su/30-19713-tetragidrociklopentabindolnye-modulyatory-receptorov-androgenov.html" rel="bookmark" title="База патентов Евразийского Союза">Тетрагидроциклопента[b]индольные модуляторы рецепторов андрогенов</a>
Модуляторы рецепторов андрогенов

Номер патента: 9902
Опубликовано: 28.04.2008
Авторы: Лей Хуангсу, Ху Лейн-Ен, Ду Дэниел Юнлонг, Лефкер Брюс Аллен
МПК: A61K 31/277, A61P 17/14, A61P 5/28...
Метки: рецепторов, андрогенов, модуляторы
Формула / Реферат:
1. Соединение формулы гидрат указанного соединения или фармацевтически приемлемая соль указанного соединения, где X1 представляет собой галоген или галогеноалкил; X2 представляет собой -CR3R4R5, -CH=CH2 или -CуCH; R1 и R2, каждый независимо, представляет собой заместитель, выбранный из группы, состоящей из водорода, С1-6алкила, галогена, галогеноалкила, гидроксиалкила, тиола и тиоалкила; R3, R4 и R5, каждый независимо, представляет собой...
Соединения тетрагидроциклопента[b]индола в качестве модуляторов рецептора андрогенов

Номер патента: 15627
Опубликовано: 31.10.2011
Авторы: Мэттьюз Дональд Пол, Грин Джонатан Эдвард, Гавардинас Константинос, Джадхав Прабхакар Кондаджи
МПК: A61K 31/404, A61P 11/00, A61P 19/02...
Метки: тетрагидроциклопента[b]индола, соединения, рецептора, качестве, андрогенов, модуляторов
Формула / Реферат:
1. Соединение формулы (I)где углеродный центр С* может быть в R, S конфигурации;R1 представляет циано, -CH=NOCH3, -OCHF2 или -OCF3;R2 представляет -COR2a или -SO2R2b;R2a представляет (С1-С4)алкил, (C1-C4)алкокси, циклопропил или -NRaRb;R2b представляет (С1-С4)алкил, циклопропил или -NRaRb;Ra и Rb, каждый независимо, представляют собой в каждом случае Н или (C1-C4)алкил иR3 представляет гетероарильную группу, выбранную из группы, состоящей из...
Полизамещённые селективные модуляторы рецептора андрогенов и способы их применения

Номер патента: 13818
Опубликовано: 30.08.2010
Авторы: Чен Дзюн, Хванг Донг Дзин, Долтон Джеймс Т., Миллер Дуэйн Д., Веверка Карен А., Стейнер Митчелл С.
МПК: A61K 31/275, A61K 31/655, A61K 31/165...
Метки: применения, рецептора, способы, модуляторы, полизамещённые, селективные, андрогенов
Формула / Реферат:
1. Соединение - селективный модулятор рецептора андрогенов (SARM), представленное структурой формулы IVгде X представляет собой О;G представляет собой О или S;Т представляет собой ОН;R представляет собой (C1-C6)алкил, галоген(C1-C6)алкил, дигалоген(С1-C6)алкил, тригалоген(C1-C6)алкил, CH2F, CHF2, CF3, CF2CF3, фенил, галоген, (С2-С6)алкенил или ОН;R1 представляет собой CH3;R3 представляет собой F, Cl, Br, I, CN, NO2, COR, COOH, CONHR, CF3,...
Модуляторы функций рецепторов семейства рецепторов tnf/ngf и других белков

Номер патента: 4062
Опубликовано: 25.12.2003
Авторы: Хорвиц Маршалл С., Коваленко Андрей, Уоллах Дэвид, Ли Йонган
МПК: C12N 15/12, A61K 38/17, C07K 14/47...
Метки: рецепторов, других, белков, модуляторы, функций, семейства
Формула / Реферат:
1. Последовательность ДНК, кодирующая RIP-ассоциированный белок (RAP-2), его изоформы, фрагменты или аналоги, включающая последовательность, показанную на фиг. 1, причем указанные белок RAP-2, его изоформы, фрагменты или аналоги способны связываться с белком RIP или способны модулировать или опосредовать внутриклеточную активность RIP. 2. Последовательность ДНК по п.1, включающая последовательность, показанную на фиг. 2. 3. Последовательность...
Селективные модуляторы эстрогенных рецепторов, содержащие фенилсульфонильную группу

Номер патента: 8024
Опубликовано: 27.02.2007
Авторы: Джоунз Скотт Алан, Фрэнк Скотт Алан, Фонг Кин Чиу, Льюис Джордж Сал, Дэлли Роберт Дин, Додж Джеффри Алан, Уоллас Оуэн Брендан, Хаммел Конрад Уилсон, Шеперд Тимоти Алан
МПК: A61K 31/4453, C07D 295/08, A61P 5/32...
Метки: рецепторов, группу, селективные, модуляторы, эстрогенных, содержащие, фенилсульфонильную
Формула / Реферат:
1. Соединение формулы I где m, q и r независимо равны 0, 1 или 2; n равно 0 или 1; R обозначает Н или COR2; R0 в каждом случае независимо обозначает ОН, CF3, галоген, C1-С6алкил или C1-С6алкокси; R1 и R1' независимо обозначают C1-С6алкил, C1-С6алкокси, NR3R3a, CF3 или CH2CF3; или если n и q равны 0, то фрагмент -SO2R1 вместе с фенильным кольцом, к которому он присоединен, может образовывать фрагмент формулы (а) или (b): где t и v равны 0, 1...
Предыдущий патент: Комплектационное устройство для комплектации изделий в заказные контейнеры
Следующий патент: Устройство для ухода за полостью рта
Случайный патент: Способ получения технологической вкусовой добавки с пониженным содержанием акриламида, добавка, полученная этим способом, и ее применение