Триазолпиридиновые ингибиторы 11-бета-гидроксистероид-дегидрогеназы типа i
Номер патента: 19706
Опубликовано: 30.05.2014
Авторы: Ли Джеймс Дж., Канн Реджинальд О., Вэй Ченку, Робл Джеффри А., Цянь Синьхуа, Кеннеди Лоуренс Дж., Ван Хайся, Галелла Майкл, Колла Лаксма Р., Дешпанде Раджендра П., Ли Цзе Джек, Ли Цзюнь





























Формула / Реферат
1. Соединение формулы I, его энантиомеры, диастереомеры или фармацевтически приемлемые соли

где Q обозначает -алкил-ОН;
G обозначает циклопропил или циклобутил, каждый из которых может быть необязательно замещен одним или несколькими заместителями, выбранными из галогена, -ОН, -OR6, -SR6, -OCOR6, -CN, -NR5COR6,
-NR5SO2R6, -COR6, -CO2R6, -CO2H, -OCONR8R8a, -CONR8R8a, -NR5CO2R6, -SO2R6, алкила, алкокси, арила, амино, гетероциклила или гетероарила, где алкил, алкокси, арил, гетероарил или гетероциклил могут быть необязательно замещены R7, R7a, R7b и R7c, или
G обозначает изопропил, который может быть необязательно замещен одним или несколькими заместителями, выбранными из галогена, -ОН, -OR6, -SR6, -OCOR6, -CN, -NR5COR6, -NR5SO2R6, -COR6,
-CO2R6, -СО2Н, -OCONR8R8a, -CONR8R8a, -NR5CO2R6, -SO2R6, алкила, алкокси, арила, амино, гетероциклила или гетероарила, где алкил, алкокси, арил, гетероарил или гетероциклил могут быть необязательно замещены R7, R7a, R7b и R7c;
R5 в каждом случае независимо обозначает водород, алкил, циклоалкил, арил, галоидалкил, -COR8a, -CO2R8a, -SO2NR8R8a или -SO2R8a;
R6 в каждом случае независимо обозначает алкил, циклоалкил, арил или гетероарил, каждый из которых необязательно может быть замещен R7, R7a, R7b и R7c;
R7, R7a, R7b и R7c в каждом случае независимо обозначают галоген, алкил, галогеналкил, цианалкил, алкокси, галогеналкокси, арил, арилокси, ариларил, арилалкил, арилалкилокси, алкенил, циклоалкил, циклоалкилалкил, циклоалкилалкилокси, амино, -ОН, -CO2R8, -CONR8R8a, гидроксиалкил, ацил, гетероарил, гетероарилокси, гетероарилалкил, гетероарилалкокси, арилоксиалкил, алкилтио, арилалкилтио, арилоксиарил, алкиламидо, алканоиламино, арилкарбониламино, -SO2R9b, -NO2, -CN или тиол, где арил или гетероарил необязательно могут быть замещены R10, R10a, R10b или R10c;
R8 и R8a в каждом случае независимо обозначают водород, алкил, циклоалкил, арил или гетероарил, каждый из которых необязательно может быть замещен R10, R10a, R10b и R10c, или
альтернативно, R8 и R8a вместе с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо, содержащее 1, 2, 3 или 4 гетероатома, независимо выбранных из группы, состоящей из N, NH, О и S, которые могут быть необязательно замещены R10, R10a, R10b или R10c;
R9b в каждом случае независимо обозначает алкил, циклоалкил, арил или гетероарил, каждый из которых необязательно может быть замещен R10, R10a, R10b и R10c;
R10, R10a, R10b и R10c в каждом случае независимо обозначают галоген, алкил, галогеналкил, цианалкил, алкокси, арил, арилокси, галогеналкокси, ариларил, арилалкил, арилалкилокси, алкенил, циклоалкил, циклоалкилалкил, циклоалкилалкилокси, амино, -ОН, гидроксиалкил, ацил, гетероарил, гетероарилокси, гетероарилалкил, гетероарилалкокси, арилоксиалкил, алкилтио, арилалкилтио, арилоксиарил, алкиламидо, алканоиламино, арилкарбониламино, -NO2, -CN или тиол;
алкил сам по себе или как часть другой группы означает разветвленные и линейные насыщенные алифатические углеводородные группы, содержащие от 1 до 20 атомов углерода;
алкенил сам по себе или как часть другой группы относится к линейным или разветвленным радикалам, содержащим от 2 до 20 атомов углерода в цепи, которая включает от одной до шести двойных связей;
циклоалкил сам по себе или как часть другой группы включает насыщенные циклические углеводородные группы, содержащие от 1 до 10 колец, содержащих в целом от 3 до 20 атомов углерода, образующих кольцо, и которые могут быть конденсированы с 1 или 2 ароматическими кольцами, как указано для арила;
арил сам по себе или как часть другой группы относится к моноциклическим и бициклическим ароматическим группам, содержащим 6-10 атомов углерода в кольце, и необязательно могут включать 1-3 дополнительных кольца, конденсированных с карбоциклическим кольцом или гетероциклическим кольцом;
гетероциклил означает стабильное 3-14-членное моноциклическое, бициклическое или трициклическое гетероциклическое кольцо, которое является насыщенным, частично ненасыщенным или ненасыщенным и которое состоит из атомов углерода и 1, 2, 3 или 4 гетероатомов, независимо выбранных из группы, состоящей из N, NH, О или S;
гетероарил означает стабильное 5-7-членное моноциклическое или бициклическое или 7-10-членное бициклическое гетероциклическое ароматическое кольцо, которое состоит из атомов углерода и 1-4 гетероатомов, независимо выбранных из группы, состоящей из N, О или S, и является ароматическим по природе;
алкокси и арилокси сами по себе или как часть другой группы включают любую из групп алкила, арила, определенных выше, присоединенных к атому кислорода, и при условии, что Q не является -СН2ОН, когда G представляет собой незамещенный циклогептил.
2. Соединение по п.1, в котором
R6 в каждом случае независимо обозначает алкил или циклоалкил;
R7, R7a, R7b и R7c в каждом случае независимо обозначают галоген, алкил, галогеналкил, алкокси, арил, арилокси, арилалкил, циклоалкил, амино, -ОН, гидроксиалкил, гетероарил, гетероарилокси, алкилтио, -NO2 или -CN.
3. Соединение по п.1, выбранное из группы, состоящей из:










4. Соединение по п.1, выбранное из группы, состоящей из:

5. Соединение, его энантиомеры, диастереомеры или фармацевтически приемлемые соли, которое представляет собой

6. Соединение по любому из пп.1-5, которое представляет собой гидрохлорид или бисульфат.
7. Соединение по любому из пп.1-6, которое представляет собой кристаллическую форму гидрохлорида или бисульфата.
8. Фармацевтическая композиция, содержащая соединение по пп.1-7.
9. Фармацевтическая композиция по п.8, которая дополнительно содержит фармацевтически приемлемый носитель и, необязательно, по меньшей мере один дополнительный терапевтический агент.
10. Применение соединения по любому из пп.1-7 для изготовления лекарственного средства для лечения, профилактики или замедления развития диабета, гипергликемии, ожирения, дислипидемии, гипертонии, ухудшения познавательных способностей, ревматоидного артрита, остеоартрита, глаукомы, болезни Кушинга, атеросклероза, острого коронарного синдрома, инфаркта миокарда, стенокардии, заболевания периферических сосудов, перемежающейся хромоты, аномальной функции сердца, ишемии миокарда, инсульта и метаболического синдрома у млекопитающего.
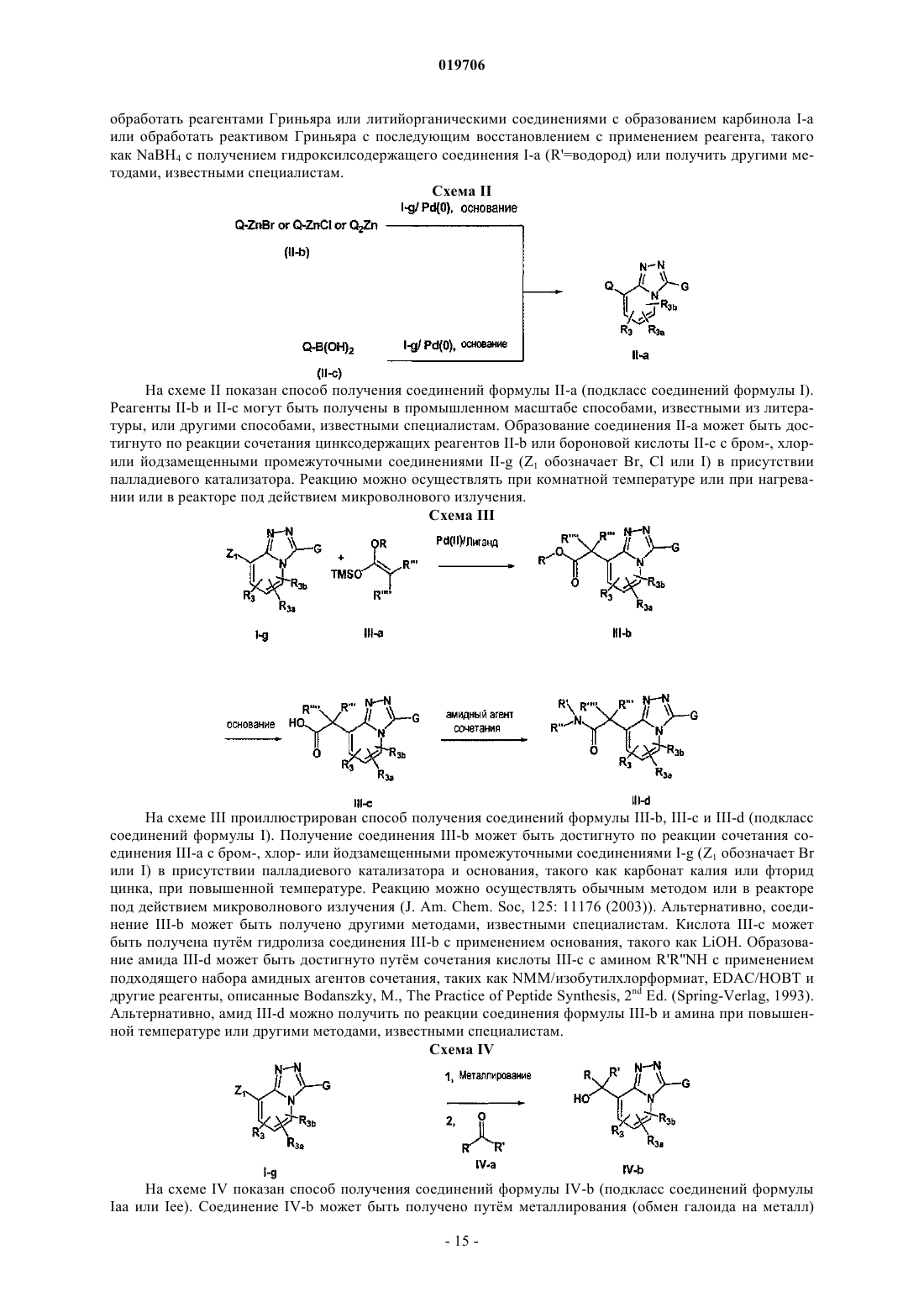
Текст
Предложены новые соединения, которые представляют собой ингибиторы 11-бетагидроксистероид-дегидрогеназы типа I. Ингибиторы 11-бета-гидроксистероид-дегидрогеназы типаI пригодны для лечения профилактики или замедления развития болезней, требующих применения терапии с использованием 11-бета-гидроксистероид-дегидрогеназы. Эти новые соединения имеют формулу I или являются стереомерами или фармацевтически приемлемыми солями этих соединений, где G и Q определены в описании. Предпосылки создания изобретения Стероидный гормон кортизол представляет собой ключевой регулятор многих физиологических процессов. Однако избыток кортизола, как, например, в случае болезни Кушинга, провоцирует серьзные метаболические аномалии, включая диабет типа 2, сердечно-сосудистую болезнь, ожирение и остеопороз. Тем не менее, у многих пациентов, страдающих этими заболеваниями, не наблюдается значительного увеличения уровня кортизола в плазме крови. Наряду с кортизолом в плазме крови отдельные ткани могут регулировать свой глюкокортикоидный фон при in situ превращении неактивного кортизола в активный гормон кортизол. В действительности, обычно высокая концентрация кортизола в плазме крови обеспечивает быструю подачу предшественника для конверсии кортизола при помощи внутриклеточного фермента 11-бета-гидроксистероид-дегидрогеназы типа I (11 бета-HSDI). 11 бета-HSDI является членом суперсемейства ферментов короткоцепочечных дегидрогеназ. Катализируя конверсию кортизона в кортизол, 11 бета-HSDI контролирует внутриклеточный глюкокортикоидный фон в соответствии со степенью е экспрессии и активности. Таким образом, 11 бета-HSDI может определять общий метаболический статус органа. 11 бета-HSDI экспрессируется с высокой степенью в печени и в низкой степени во многих метаболически активных тканях, включая жировую ткань, ЦНС,поджелудочную железу и гипофиз. Предполагается, что в печени высокий уровень активности 11 бетаHSDI будет стимулировать глюконеогенез и общую выработку глюкозы. В противоположность этому уменьшение активности 11 бета-HSDI будет подавлять глюконеогенез, что приводит к низким уровням глюкозы в плазме. Были проведены различные исследования, которые подтвердили эту гипотезу. Например, трансгенные мыши, экспрессирующие двукратный от нормального уровень 11 бета-HSDI только в жировой ткани,характеризуются ожирением в области брюшины, гипогликемией и резистентностью к инсулину (Masuzaki H. et al., "A Transgenic Model of Visceral Obesity and the Metabolic Syndrome", Science, 294: 2166-2170(2001. В противоположность этому, когда 11 бета-HSDI удаляется при гомологичной рекомбинации,мыши проявляют стойкость к ожирению, вызванному кормом, и к сопутствующему разрегулированию метаболизма глюкозы (Morton N.M. et al., "Novel Adipose Tissue-Mediated Resistance to Diet-induced Visceral Obesity in 11-beta-Hydroxysteroid-Dehydrogenase type I-Deficient Mice", Diabetes, 53: 931-938 (2004. Кроме того, лечение ожирения и диабета в случае генетических моделей мышей (мышей ob/ob, db/db иKKAy) при помощи специфичного ингибитора 11 бета-HSDI приводит к уменьшению выработки глюкозы печенью и к общему повышению чувствительности к инсулину (Alberts P. et al., "Selective Inhibition of 11-beta-Hydroxysteroid Dehydrogenase type I Improves Hepatic Insuling Sensitivity in Hyperglicemic MiceStrains", Endocrinology, 144: 4755-4762 (2003. Кроме того, ингибиторы 11 бета-HSDI оказались эффективными при лечении метаболического синдрома и атеросклероза у мышей, получавших корм с высоким содержанием жиров (Hermanowski-Vosatka et al., J. Exp. Med., 202 (4): 517-527 (2002. Основываясь на результатах части этих исследований, полагают, что локальный контроль уровней кортизола является важным в случае метаболических заболеваний у этих мышей. Кроме того, результаты этих исследований позволяют предположить, что ингибирование 11 бета-HSDI будет представлять собой эффективную стратегию лечения метаболических болезней, таких как диабет типа 2, ожирение и метаболический синдром. Эта идея подкрепляется также результатами ряда предварительных клинических исследований. Например, в нескольких работах было показано, что жировая ткань, отобранная у субъектов, страдающих ожирением, характеризуется повышенной степенью активности 11 бета-HSDI. Далее, исследования, проведнные с карбеноксолоном, природным продуктом, полученным из лакричника обыкновенного, который ингибирует как 11 бета-HSDI, так и 11 бета-HSD2 (превращает кортизол в кортизон в почках), привели к обнадживающим результатам. Семидневное, двойное слепое, перекрстное, использующее плацебо в качестве контроля исследование субъектов с умеренным избытком веса, больных диабетом типа 2, проведнное с применением карбеноксолона показало, что пациенты, принимавшие этот ингибитор (но не пациенты в группе, принимавшей плацебо), характеризовались снижением продуцирования глюкозы печенью (Andrews R.C. et al., J. Clin. Endocrinol. Metabol. 88: 285-291 (2003. Это наблюдение согласуется с ингибированием 11 бета-HSDI в печени. Результаты этих доклинических и ранних клинических исследований подкрепляют концепцию, что лечение активным и селективным ингибитором 11 бета-HSDI будет представлять собой эффективную терапию для пациентов, больных диабетом типа 2, ожирением и метаболическим синдромом. Сущность изобретения В соответствии с данным изобретением предусмотрены бициклические и родственные соединения общей формулы I где G и Q имеют значения, указанные ниже. Соединения согласно данному изобретению ингибируют активность фермента 11-бетагидроксистероид-дегидрогеназы типа I. Соответственно соединения согласно данному изобретению могут быть применены для лечения многих заболеваний и расстройств, ассоциируемых с 11-бета-1 019706 гидроксистероид-дегидрогеназой типа I, таких как диабет и связанные с ним состояния, микрососудистые осложнения, вызванные диабетом, макрососудистые осложнения, вызванные диабетом, сердечнососудистые болезни, метаболический синдром и его компоненты, воспалительные заболевания и другие болезни. Примеры болезней или расстройств, ассоциируемых с 11-бета-гидроксистероид-дегидрогеназой типа I, которые могут быть предотвращены, ингибированы или подвергнуты лечению согласно данному изобретению, включают, без ограничения, диабет, гипергликемию, ухудшенную переносимость глюкозы, резистентность к инсулину, гиперинсулинемию, ретинопатию, нейропатию, нефропатию, замедленное заживление ран, атеросклероз, его последствия (острый коронарный синдром, инфаркт миокарда,хроническая ангина, заболевание периферических сосудов, перемежающаяся хромота), аномальную функцию сердца, ишемию миокарда, инсульт, метаболический синдром, гипертонию, ожирение, дислипидемию, гиперлипидемию, гипертриглицеридонемию, гиперхолестеринемию, низкое содержание липидов высокой плотности, высокое содержание липидов низкой плотности, несердечную ишемию, инфекцию, рак, сосудистый рестеноз, панкреатит, нейродегенеративное расстройство, нарушение содержания липидов, ухудшение познавательных способностей, а также деменцию, заболевание костей, гиподистрофию, связанную с ВИЧ-протеазой, глаукому, воспалительные болезни, такие как ревматоидный артрит,болезнь Кушинга, болезнь Альцгеймера и остеоартрит. Данное изобретение предусматривает соединения общей формулы I, фармацевтические композиции на основе таких соединений и способы применения этих соединений. В частности, настоящее изобретение предусматривает фармацевтическую композицию, содержащую терапевтическое количество соединения общей формулы I, одного или в комбинации с фармацевтически приемлемым носителем. Далее, в соответствии с данным изобретением предусмотрен способ профилактики, ингибирования и лечения развития или возникновения болезней или расстройств, ассоциируемых с активностью фермента 11-бета-гидроксистероид-дегидрогеназы типа I, таких как указанные выше и далее в данном описании, согласно которому млекопитающему, а именно человеку, пациенту, нуждающемуся в лечении,вводят терапевтическое количество соединения формулы I. Соединения по изобретению могут быть применены сами по себе или в комбинации с одним или более другими агентами. Кроме того, в соответствии с данным изобретением предусмотрен способ профилактики, ингибирования и лечения развития или возникновения болезней или расстройств, ассоциируемых с активностью фермента 11-бета-гидроксистероид-дегидрогеназы типа I, таких как указанные выше и далее в данном описании, согласно которому млекопитающему, а именно человеку, пациенту, нуждающемуся в лечении,вводят терапевтическое количество комбинации соединения формулы I и другого соединения формулы I и/или по меньшей мере одного терапевтического агента другого типа. Кроме того, настоящее изобретение предусматривает кристаллические формы соединений формулыI, фармацевтические композиции на основе таких кристаллических форм и способы применения этих форм. Краткое описание чертежей На фиг. 1 приведены наблюдавшиеся (экспериментальные, снятые при комнатной температуре) и имитированные (рассчитанные при комнатной температуре) порошковые рентгеновские дифрактограммы (Cu K =1,5418 ангстрем) кристаллической формы N-1 соли соединения формулы I. На фиг. 2 показана термограмма кристаллической формы N-1 соли соединения формулы I, полученная методом дифференциальной сканирующей калориметрии (DSC). На фиг. 3 приведена кривая для кристаллической формы N-1 соли соединения формулы I, полученная методом термогравиметрического анализа (TGA). Фиг. 4 показывает результаты анализа изотермы сорбции влаги кристаллической формой N-1 соли соединения формулы I. На фиг. 5 приведены наблюдавшиеся (экспериментальные, снятые при комнатной температуре) и имитированные (рассчитанные при комнатной температуре) порошковые рентгеновские дифрактограммы (Cu K =1,5418 ангстрем) кристаллической формы N-2 соли соединения формулы I. На фиг. 6 показана термограмма кристаллической формы N-2 соли соединения формулы I, полученная методом дифференциальной сканирующей калориметрии (DSC). На фиг. 7 приведена кривая для кристаллической формы N-2 соли соединения формулы I, полученная методом термогравиметрического анализа (TGA). На фиг. 8 приведены наблюдавшиеся (экспериментальные, снятые при комнатной температуре) и имитированные (рассчитанные при комнатной температуре) порошковые рентгеновские дифрактограммы (Cu K =1,5418 ангстрем) другой кристаллической формы N-1 соли соединения формулы I. Описание изобретения В соответствии с данным изобретением предусмотрены соединения формулы I их энантиомеры, диастереомеры или их соли, гдеG обозначает циклопропил или циклобутил, каждый из которых может быть необязательно замещен одним или несколькими заместителями, выбранными из галогена, -ОН, -OR6, -SR6, -OCOR6, -CN,-NR5COR6, -NR5SO2R6, -COR6, -CO2R6, -CO2H, -OCONR8R8a, -CONR8R8a, -NR5CO2R6, -SO2R6, алкила, алкокси, арила, амино, гетероциклила или гетероарила, где алкил, алкокси, арил, гетероарил или гетероциклил могут быть необязательно замещены R7, R7a, R7b и R7c; илиG обозначает изопропил, который может быть необязательно замещен одним или несколькими заместителями, выбранными из галогена, -ОН, -OR6, -SR6, -OCOR6, -CN, -NR5COR6, -NR5SO2R6, -COR6,-CO2R6, -CO2H, -OCONR8R8a, -CONR8R8a, -NR5CO2R6, -SO2R6, алкила, алкокси, арила, амино, гетероциклила или гетероарила, где алкил, алкокси, арил, гетероарил или гетероциклил могут быть необязательно замещены R7, R7a, R7b и R7c;R5 в каждом случае независимо обозначает водород, алкил, циклоалкил, арил, галоидалкил, -COR8a,-CO2R8a, -SO2NR8R8a или -SO2R8a;R6 в каждом случае независимо обозначает алкил, циклоалкил, арил или гетероарил, каждый из которых может быть замещн R7, R7a, R7b или R7c;R7, R7a, R7b и R7c в каждом случае независимо обозначают галоген, алкил, галогеналкил, цианалкил,алкокси, галогеналкокси, арил, арилокси, ариларил, арилалкил, арилалкилокси, алкенил, циклоалкил,циклоалкилалкил, циклоалкилалкилокси, амино, -ОН, -CO2R8, -CONR8R8a, гидроксиалкил, ацил, гетероарил, гетероарилокси, гетероарилалкил, гетероарилалкокси, арилоксиалкил, алкилтио, арилалкилтио, арилоксиарил, алкиламидо, алканоиламино, арилкарбониламино, -SO2R9b, -NO2, -CN или тиол, при этом арил или гетероарил могут быть замещены R10, R10a, R10b или R10c;R8 и R8a в каждом случае независимо обозначают водород, алкил, циклоалкил, арил или гетероарил,каждый из которых необязательно может быть замещен R10, R10a, R10b или R10c; или альтернативно, R8 и R8a вместе с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо, содержащее 1, 2, 3 или 4 гетероатома, независимо выбранные из группы, состоящей изN, NH, О или S, которые могут быть необязательно замещены R10, R10a, R10b или R10c;R9b в каждом случае независимо обозначает алкил, циклоалкил, арил или гетероарил, каждый из которых необязательно может быть замещен R10, R10a, R10b или R10c;R10, R10a, R10b или R10c в каждом случае независимо обозначают галоген, алкил, галогеналкил, цианалкил, алкокси, арил, арилокси, ариларил, арилалкил, арилалкилокси, алкенил, циклоалкил, циклоалкилалкил, циклоалкилалкилокси, амино, -ОН, гидроксиалкил, ацил, гетероарил, гетероарилокси, гетероарилалкил, гетероарилалкокси, арилоксиалкил, алкилтио, арилалкилтио, арилоксиарил, алкиламидо, алканоиламино, арилкарбониламино, -NO2, -CN или тиол. Согласно другому варианту изобретения соединения выбираются из числа соединений, описанных в примерах, предпочтительно в примерах 1, 11, 24 и 91, более предпочтительно в примере 1. Согласно другому варианту изобретения соединение по данному изобретению представляет собой гидрохлорид или бисульфат соединения, описанного в примере 1. Согласно ещ одному варианту соединение по изобретению представляет собой гидрохлорид соединения, описанного в примере 1. Согласно ещ одному варианту соединение по изобретению представляет собой кристаллическую форму гидрохлорида соединения, описанного в примере 1, предпочтительно форму N-1 или N-2, более предпочтительно форму N-1. Согласно одному из вариантов кристаллическая форма является практически чистой. Согласно одному из вариантов кристаллическая форма гидрохлорида соединения, описанного в примере 1, характеризуется параметрами элементарной ячейки, практически равными: размеры ячейки: а=13,5209 (3) Согласно одному из вариантов кристаллическая форма гидрохлорида соединения, описанного в примере 1, характеризуется следующими параметрами 2 порошковой рентгеновской дифракционной рештки (Cu K - 1,5418 ангстрем) 6,70,1, 11,10,1, 13,40,1, 13,70,1, 16,30,1, 19,10,1, 19,60,1,22,30,1 и 24,60,1, измеренными при комнатной температуре. Согласно одному из вариантов кристаллическая форма гидрохлорида соединения, описанного в примере 1, характеризуется параметрами 2 порошковой рентгеновской дифракционной рештки практически в соответствии с величинами, показанными на фиг. 1. Согласно одному из вариантов кристаллическая форма гидрохлорида соединения, описанного в примере 1, характеризуется термограммой дифференциальной сканирующей калориметрии, практически в соответствии с фиг. 2, при этом эндотермический переход наблюдается при температуре выше 150 С. Согласно одному из вариантов кристаллическая форма гидрохлорида соединения, описанного в примере 1, характеризуется кривой термического термогравиметрического анализа в соответствии с фиг. 3, при этом незначительная потеря веса наблюдается до примерно 100 С. Согласно одному из вариантов кристаллическая форма гидрохлорида соединения, описанного в примере 1, характеризуется параметрами элементарной ячейки, практически равными: размеры ячейки: а=12,908 (4)=90 Пространственная группа: Pca21 Молекулы/асимметричная единица (Z'): 1 Плотность, рассчит. г/см 3: 1,335. Согласно одному из вариантов кристаллическая форма гидрохлорида соединения, описанного в примере 1, характеризуется следующими параметрами 2 порошковой рентгеновской дифракционной рештки (Cu K - 1,5418 ангстрем) 6,90,1, 13,70,1, 15,40,1, 17,40,1, 16,30,1, 21,20,1 22,40,1 и 23,30,1, измеренными при комнатной температуре. Согласно одному из вариантов кристаллическая форма гидрохлорида соединения, описанного в примере 1, характеризуется параметрами 2 порошковой рентгеновской дифракционной рештки практически в соответствии с величинами, показанными на фиг. 5. Согласно одному из вариантов кристаллическая форма гидрохлорида соединения, описанного в примере 1, характеризуется термограммой дифференциальной сканирующей калориметрии, практически в соответствии с фиг. 6, при этом эндотермический переход наблюдается при температуре выше 150 С. Согласно одному из вариантов кристаллическая форма гидрохлорида соединения, описанного в примере 1, характеризуется кривой термического термогравиметрического анализа в соответствии с фиг. 7, при этом незначительная потеря веса наблюдается до примерно 100 С. Согласно другому варианту соединение по данному изобретению представляет собой бисульфат по примеру 1. Согласно ещ одному варианту соединение по изобретению представляет собой кристаллическую форму бисульфата соединения, предпочтительно форму N-1, более предпочтительно практически чистую форму. Согласно другому варианту кристаллическая форма бисульфата соединения, описанного в примере 1, характеризуется параметрами элементарной ячейки, практически равными: размеры ячейки: а=10,01 (1)=90 Пространственная группа: P21/c Молекулы/асимметричная единица (Z'): 1 Плотность, рассчит. г/см 3: 1,444. Согласно одному из вариантов кристаллическая форма бисульфата соединения, описанного в примере 1, характеризуется параметрами 2 порошковой рентгеновской дифракционной рештки практически в соответствии с величинами, показанными на фиг. 8. Согласно другому варианту настоящее изобретение относится к фармацевтическим композициям,содержащим терапевтически эффективное количество соединения, описанного в данном изобретении,предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевти-4 019706 чески приемлемым носителем и/или одним или более другими агентами. Согласно другому варианту настоящее изобретение относится к способам ингибирования активности фермента 11-бета-гидроксистероид-дегидрогеназы типа 1, включающим введение млекопитающему,например человеку, нуждающемуся в этом, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1H, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими терапевтическими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения болезней или расстройств, ассоциируемых с активностью фермента 11-бета-гидроксистероид-дегидрогеназы типа I, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими терапевтическими агентами. Примеры болезней или расстройств, ассоциируемых с 11-бета-гидроксистероид-дегидрогеназой типа I, которые могут быть предотвращены, ингибированы или подвергнуты лечению согласно данному изобретению, включают, без ограничения, диабет, гипергликемию, ухудшенную переносимость глюкозы, резистентность к инсулину, гиперинсулинемию, ретинопатию, нейропатию, нефропатию, замедленное заживление ран, атеросклероз, его последствия (острый коронарный синдром, инфаркт миокарда,хроническая ангина, заболевание периферических сосудов, перемежающаяся хромота), аномальную функцию сердца, ишемию миокарда, инсульт, метаболический синдром, гипертонию, ожирение, дислипидемию, гиперлипидемию, гипертриглицеридонемию, гиперхолестеринемию, низкое содержание липидов высокой плотности, высокое содержание липидов низкой плотности, несердечную ишемию, инфекцию, рак, сосудистый рестеноз, панкреатит, нейродегенеративное расстройство, нарушение содержания липидов, ухудшение познавательных способностей, а также деменцию, заболевание костей, гиподистрофию, связанную с ВИЧ-протеазой, глаукому, воспалительные болезни, такие как ревматоидный артрит,болезнь Кушинга, болезнь Альцгеймера и остеоартрит. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения диабета, гипергликемии, ожирения, дислипидемии,гипертонии, ухудшения познавательных способностей, ревматоидного артрита, остеоартрита, глаукомы,болезни Кушинга и метаболического синдрома, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более терапевтическими другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения диабета, включающему введение млекопитающему,например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1,1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими терапевтическими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения гипергликемии, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1H, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими терапевтическими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения ожирения, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения дислипидемии, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения гипертонии, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения ухудшения познавательных способностей, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения ревматоидного артрита, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении,терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения остеоартрита, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения метаболического синдрома, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении,терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения глаукомы, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относитсяк способу профилактики, ингибирования или лечения развития или возникновения болезни Кушинга, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к фармацевтическим композициям, содержащим терапевтически эффективное количество кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно другому варианту настоящее изобретение относится к способам ингибирования активности фермента 11-бета-гидроксистероид-дегидрогеназы типа I, включающим введение млекопитающему,например человеку, нуждающемуся в этом, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими терапевтическими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения болезней или расстройств, ассоциируемых с активностью фермента 11-бета-гидроксистероид-дегидрогеназы типа I, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими терапевтическими агентами. Примеры болезней или расстройств, ассоциируемых с 11-бета-гидроксистероид-дегидрогеназой типа I, которые могут быть предотвращены, ингибированы или подвергнуты лечению согласно данному изобретению, включают, без ограничения, диабет, гипергликемию, ухудшенную переносимость глюкозы, резистентность к инсулину, гиперинсулинемию, ретинопатию, нейропатию, нефропатию, замедленное заживление ран, атеросклероз, его последствия (острый коронарный синдром, инфаркт миокарда,хроническая ангина, заболевание периферических сосудов, перемежающаяся хромота), аномальную функцию сердца, ишемию миокарда, инсульт, метаболический синдром, гипертонию, ожирение, дислипидемию, гиперлипидемию, гипертриглицеридонемию, гиперхолестеринемию, низкое содержание липидов высокой плотности, высокое содержание липидов низкой плотности, несердечную ишемию, инфекцию, рак, сосудистый рестеноз, панкреатит, нейродегенеративное расстройство, нарушение содержания липидов, ухудшение познавательных способностей, а также деменцию, заболевание костей, гиподистрофию, связанную с ВИЧ-протеазой, глаукому, воспалительные болезни, такие как ревматоидный артрит,болезнь Кушинга, болезнь Альцгеймера и остеоартрит. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения диабета, гипергликемии, ожирения, дислипидемии,гипертонии, ухудшения познавательных способностей, ревматоидного артрита, остеоартрита, глаукомы,болезни Кушинга и метаболического синдрома, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более терапевтическими другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения диабета, включающему введение млекопитающему,например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими терапевтическими агентами. Согласно ещ одномуварианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения гипергликемии, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении,предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими терапевтическими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения ожирения, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения дислипидемии, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении,предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения гипертонии, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения ухудшения познавательных способностей, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения ревматоидного артрита, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении,терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения остеоартрита, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении,предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевти-7 019706 чески приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения метаболического синдрома, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении,терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения глаукомы, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к способу профилактики, ингибирования или лечения развития или возникновения болезни Кушинга, включающему введение млекопитающему, например человеку, нуждающемуся в профилактике, ингибировании или лечении, терапевтически эффективного количества кристаллической формы соединения, описанного в данном изобретении, предпочтительно в примерах 1, 1G, 1 Н, 1I, 24 и 91, одного или, возможно, в комбинации с фармацевтически приемлемым носителем и/или одним или более другими агентами. Согласно ещ одному варианту настоящее изобретение относится к соединению по данному изобретению, предпочтительно описанному в примерах 1, 1G, 1 Н, 1I, 24 и 91, для применения в терапии для лечения метаболической болезни. Согласно ещ одному варианту настоящее изобретение относится к совместному препарату соединения по данному изобретению, предпочтительно описанного в примерах 1, 1G, 1 Н, 1I, 24 и 91, и дополнительного терапевтического(их) агента(ов) для одновременного, раздельного или последовательного применения в терапии. Согласно ещ одному варианту настоящее изобретение относится к совместному препарату соединения по данному изобретению, предпочтительно описанного в примерах 1, 1G, 1 Н, 1I, 24 и 91, и дополнительного терапевтического агента(ов) для одновременного, раздельного или последовательного применения при лечении метаболической болезни. Согласно ещ одному варианту настоящее изобретение относится к кристаллическим формам соединений по изобретению, предпочтительно описанным в примерах 1, 1G, 1 Н, 1I, 24 и 91, для применения в терапии при лечении метаболической болезни. Согласно ещ одному варианту настоящее изобретение относится к совместному препарату на основе кристаллических форм соединений по данному изобретению, предпочтительно описанных в примерах 1, 1G, 1 Н, 1I, 24 и 91, и дополнительного терапевтического агента(ов) для одновременного, раздельного или последовательного применения в терапии. Согласно ещ одному варианту настоящее изобретение относится к совместному препарату на основе кристаллических форм соединений по данному изобретению, предпочтительно описанных в примерах 1, 1G, 1 Н, 1I, 24 и 91, и дополнительного терапевтического агента(ов) для одновременного, раздельного или последовательного применения при лечении метаболической болезни. Согласно ещ одному варианту настоящего изобретения предусмотрен способ получения соединения формулы VII-f который включает реакцию гидразида формулы VII-d с карбоновой кислотой илиPh3PCl2/диизопропилэтиламином в присутствии растворителя при повышенной температуре с получением 1,2,4-триазолпиридина формулы VII-е и последующее контактирование 1,2,4-триазолпиридина формулы VII-е с соответствующей кислотой с получением соединения формулы VII-f Согласно одному из вариантов предусмотрены способы, согласно которым гидразид формулы VII-d реагирует с карбоновой кислотой в присутствии растворителя. Согласно одному из вариантов предусмотрены способы, согласно которым гидразид формулы VII-d реагирует с карбоновой кислотой в присутствии растворителя с обратным холодильником. Согласно одному из вариантов предусмотрены способы, согласно которым карбоновая кислота выбрана из уксусной кислоты, п-тозиловой кислоты, бензойной кислоты, 2-, 3- или 4-хлорбензойной кислоты, 4-фторбензойной кислоты, 2-хлорбензолуксусной кислоты, 2- или 4-метилбензойной кислоты, 4 метоксибензойной кислоты, 2,6-диметоксибензойной кислоты, 2-метилпропионовой кислоты и 2,2 диметилпропановой кислоты. Согласно одному из вариантов предусмотрены способы, согласно которым растворитель выбран из толуола, 1-пропанола и их смесей, предпочтительно толуола. Согласно другому варианту предусмотрены способы, согласно которым гидразид формулы VII-d получают путм реакции гидрохлорида гидразинилпиридинила формулы VII-b и оксалилхлорида в среде растворителя в присутствии основания Согласно одному из вариантов предусмотрены способы, согласно которым реакцию гидрохлорида гидразинилпиридинила формулы VII-b с кислотой формулы VII-с и оксалилхлоридом проводят при комнатной температуре. Согласно одному из вариантов предусмотрены способы, согласно которым растворитель, применяемый при реакции гидрохлорида гидразинилпиридинила формулы VII-b с кислотой формулы VII-c и оксалилхлоридом, выбран из толуола, N,N-диметилформамида, диэтиламина, тетрагидрофурана, воды,дихлорметана, 2-метил-THF, метил-трет-бутилового эфира и их смесей, предпочтительно растворителем является N,N-диметилформамид, тетрагидрофуран и их смеси. Согласно одному из вариантов предусмотрены способы, согласно которым применяемое при реакции гидрохлорида гидразинилпиридинила формулы VII-b с кислотой формулы VII-c и оксалилхлоридом основание выбрано из гидроокиси натрия, карбоната калия, триэтиламина и дикалийфосфата, предпочтительно из гидроокиси натрия и карбоната калия, более предпочтительно оно является карбонатом калия. Согласно другому варианту предусмотрены способы, согласно которым гидрохлорид гидразинилпиридинила формулы VII-b получают путм реакции галоидпиридина формулы VII-а с гидразином при повышенной температуре с последующим получением хлористо-водородной соли при реакции с соляной кислотой с образованием гидрохлорида гидразинилпиридинила формулы VII-b Согласно одному из вариантов предусмотрены способы, согласно которым галоидпиридин формулы VII-а реагирует с гидразином в среде спирта, предпочтительно изопропанола, 1-пропанола, 1,4 диоксана, диметоксиэтана и их смесей или простого эфира. Согласно одному из вариантов предусмотрены способы, согласно которым галоидпиридин форму-9 019706 лы VII-a реагирует с гидразином при температуре 90-100 С, предпочтительно при температуре 92-94 С. Настоящее изобретение может быть осуществлено в виде других форм без выхода за рамки и объм его существенных признаков. Это изобретение охватывает также все комбинации альтернативных аспектов изобретения, описанного в данном изобретении. Очевидно, что любой и все варианты данного изобретения можно взять в сочетании с любым другим вариантом с целью описания дополнительных вариантов данного изобретения. Кроме того, любые элементы варианта могут объединяться с любым другими или всеми другими элементами любого из вариантов с целью описания дополнительных вариантов. Определения Соединения, описанные в данном изобретении, могут содержать асимметричные центры. Соединения по изобретению, содержащие асимметрично замещнный атом, могут быть выделены в оптически активной и рацемической формах. Из уровня техники хорошо известно, как получить оптически активные формы, например путм разделения рацемических форм или путм синтеза из оптически активных исходных веществ. В соединениях, описанных в данном изобретении, могут также содержаться многие геометрические изомеры олефинов, С=N двойные связи и т.п., и все такие стабильные изомеры охвачены настоящим изобретением. Описаны цис- и транс-геометрические изомеры соединения по изобретению и они могут быть выделены в виде смеси изомеров или отдельных изомерных форм.Подразумеваются все хиральные, диастереомерные, рацемические формы и все геометрические изомеры, если только специально не указаны конкретные стереохимия или изомерная форма. Один энантиомер соединения формулы I может обладать более высокой активностью, чем другой. Таким образом, считается, что все стереохимические формы являются частью данного изобретения. Если это требуется, разделение рацемата может быть достигнуто методом HPLC (ВЭЖХ) с применением хиральной колонки или путм разрешения с применением разрешающего агента, такого как хлорангидрид камфеновой кислоты, как описано Young S.D. et al., Antimicrobial Agents and Chemotherapy, 2602-2605(1995). Если соединения формулы I и их соли могут существовать в таутомерной форме, все такие таутомерные формы заявлены в данном изобретении как часть изобретения. Термин "замещнный" в данном изобретении означает, что любой атом водорода или несколько атомов водорода в указанном атоме или кольце могут быть замещены заместителем из указанной группы при условии, что нормальная валентность указанного атома или атома в кольце не превышена и что замещение приводит к получению стабильного соединения. Когда заместителем является кетогруппа (т.е.=О), тогда замещнными являются два водорода у данного атома. Когда какой-либо заместитель (например, R4) встречается более одного раза в составе какой-либо части общей формулы, его определение в каждом случае является независимым от его определения в каждом другом случае. Так, например, если показано, что какая-либо группа замещена (R4)m и m равен 03, это означает, что указанная группа может быть замещена заместителями R4 в количестве до трх и R4 в каждом случае независимо выбран из указанных значений R4. Кроме того, комбинация заместителей и/или переменных допустима только в том случае, когда такая комбинация приводит к образованию стабильного соединения. Когда показано, что связь заместителя пересекает связь, соединяющую два атома в кольце, тогда такой заместитель может быть присоединн к любому атому в кольце. Когда заместитель приведн без указания атома, при помощи которого такой заместитель присоединн к остальной части соединения данной формулы, тогда такой заместитель может быть присоединн через любой атом в таком заместителе. Комбинация заместителей и/или переменных допустима только в том случае, когда такая комбинация приводит к образованию стабильного соединения. Применяемый термин "алкил" охватывает и разветвлнные, и линейные насыщенные алифатические углеводородные группы, содержащие от 1 до 20 атомов углерода, предпочтительно от 1 до 10 атомов углерода, более предпочтительно от 1 до 8 атомов углерода, в нормальной цепи, такие как метил,этил, пропил, изопропил, бутил, трет-бутил, изобутил, пентил, гексил, изогексил, гептил, 4,4 диметилпентил, октил, 2,2,4-триметилпентил, нонил, децил, ундецил, додецил, различные их разветвлнные изомеры и т.п. Если иное не указано, термин "алкенил", используемый сам по себе или как часть другой группы,относится к линейным или разветвлнным радикалам, содержащим от 2 до 20 атомов углерода, предпочтительно от 2 до 12 атомов углерода, более предпочтительно от 1 до 8 атомов углерода, в нормальной цепи, которая включает от одной до шести двойных связей в нормальной цепи, таким как винил, 2 пропенил, 3-бутенил, 2-бутенил, 4-пентенил, 3-пентенил, 2-гексенил, 3-гексенил, 2-гептенил, 3-гептенил,4-гептенил, 3-октенил, 3-ноненил, 4-деценил, 3-ундеценил, 4-додеценил, 4,8,12-тетрадекатриенил и т.п. Если иное не указано, термин "циклоалкил", используемый сам по себе или как часть другой группы, включает насыщенные циклические углеводородные группы, содержащие от 1 до 10 колец, предпочтительно 1-3 кольца, включая моноциклический алкил, бициклический алкил (или бициклоалкил) или трициклический алкил, содержащие в целом от 3 до 20 атомов углерода, образующих кольцо, предпочтительно от 3 до 15 атомов, более предпочтительно от 3 до 10 атомов, образующих кольцо, которые могут быть конденсированы с 1 или 2 ароматическими кольцами, как описано для арила, включающие цик- 10019706 лопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклодецил, циклододецил, циклогексенил, Когда алкильные группы, определнные выше, содержат простые связи для присоединения к другим группам у двух различных атомов углерода, они называются "алкиленовыми" группами. Когда алкенильные группы, определнные выше, содержат простые связи для присоединения к другим группам у двух различных атомов углерода, они называются "алкениленовыми" группами. Термин "гало" или "галоген" относится к фтору, хлору, брому и йоду; термин "галогеналкил" включает разветвлнные и линейные насыщенные алифатические углеводородные группы, например, CF3,содержащие указанное число атомов углерода, замещнные 1 или более атомами галоида (например,-CvFw, где v равен 1-3 и w равен 1-(2v+1. Если иное не указано, термин "арил", используемый сам по себе или как часть другой группы, относится к моноциклическим и бициклическим ароматическим группам, содержащим 6-10 атомов углерода в кольце (таким как фенил или нафтил, включая 1-нафтил и 2-нафтил), которые могут включать 1-3 дополнительных кольца, конденсированные с карбоциклическим кольцом или гетероциклическим кольцом (таким как арил, циклоалкил, гетероарил или циклогетероалкил), например Если иное не указано, термины "низший алкокси", "алкокси", "арилокси" или "аралкокси", используемые сам по себе или как часть другой группы, включают любой из указанных выше алкилов, аралкилов или арилов, присоединнных к атому кислорода. Если иное не указано, термин "амино", используемый сам по себе или как часть другой группы, относится к аминогруппам, которые могут быть замещены одним или двумя заместителями, которые могут быть одинаковыми или разными, такими как алкил, арил, арилалкил, гетероарил, гетероарилалкил, циклогетероалкил, циклогетероалкилалкил, циклоалкил, циклоалкилалкил, галоидалкил, гидроксиалкил,алкоксиалкил или тиоалкил. Эти заместители, в свою очередь, могут быть далее замещены карбоксильной группой и/или любой из групп R1 или заместителей для R1. Как указано выше, заместители у аминогрупп вместе с атомом азота, к которому они присоединены, могут образовывать 1-пирролидинил, 1 пиперидинил, 1-азепинил, 4-морфолинил, 4-тиаморфолинил, 1-пиперазинил, 4-алкил-1-пиперазинил, 4 арилалкил-1-пиперазинил, 4-диарилалкил-1-пиперазинил, 1-пирролидинил, 1-пиперидинил или 1 азепинил, которые могут быть замещены алкилом, алкокси, алкилтио, галоидом, трифторметилом или гидрокси. Если иное не указано, термины "низший алкилтио", "алкилтио", "арилтио" или "аралкилтио", используемые сами по себе или как часть другой группы, включают любой из указанных выше алкилов,аралкилов или арилов, присоединнных к атому серы. Если иное не указано, термины "низший алкиламино", "алкиламино", "ариламино" или "аралкиламино", используемые сам по себе или как часть другой группы, включают любой из указанных выше алкилов, аралкилов или арилов, присоединнных к атому азота. Если иное не указано, термины "гетероциклил", "гетероциклическая система", "гетероциклическое кольцо" означают стабильное 3-14-членное моноциклическое, бициклическое или трициклическое гетероциклическое кольцо, которое является насыщенным, частично ненасыщенным или ненасыщенным(ароматическим), которое содержит атомы углерода и 1, 2, 3 или 4 гетероатома, независимо выбранные из группы, состоящей из N, NH, О или S, включая любую бициклическую группу, в которой любое из указанных гетероциклических колец конденсировано с образованием бензольного кольца. Атомы азота и серы могут быть окисленными. Гетероциклическое кольцо может быть присоединено к боковой группе через любой гетероатом или атом углерода с образованием стабильной структуры. Гетероциклические кольца, описанные в данном изобретении, могут быть замещены у атома углерода или атома азота, если получающееся соединение является стабильным. Если это специально указано, азот в гетероцикле может быть кватернизованным. Предпочтительно, чтобы общее число атомов серы и кислорода в гетероцикле превышало 1 и эти гетероатомы не являлись соседними. Применяемый в данном изобретении термин "ароматическая гетероциклическая система" или "гетероарил" означает стабильное 5-7-членное моноциклическое или бициклическое или 7-10-членное бициклическое гетероциклическое ароматическое кольцо, которое состоит из атомов углерода и 1-4 гетероатомов, независимо выбранных из группы, состоящей из N, О или S, и является ароматическим по природе. Примеры гетероциклов включают, без ограничения, 1 Н-имидазолил, 1-пирролидонил, 2 Н,6 Н-1,5,2 дитиазинил, 2 Н-пирролил, 1 Н-индолил, 4-пиперидонил, 4 аН-карбазол, 4 Н-хинолизинил, 6 Н-1,2,5 тиадиазинил, акридинил, азоцинил, бензимидазолил, бензофуранил, бензотиофуранил, бензотиофенил,бензоксазолил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, бензимидазалонил, карбазолил, 4aH-карбазолил, Р-карболинил, хроманил, хроменил, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро-(2,3-6)фуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, индазолил, индоленил, индолинил, индолизинил, индолил, изобензофуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил (бензимидазолил), изотиазолил, изоксазолил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3 оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, 1,2,4-оксадиазолил, оксазолил,оксазолидинилперимидинил, фенантридинил, фенантролинил, фенарсазинил, феназинил, фенотиазинил,феноксатиинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, птеридинил, пиперидонил, 4 пиперидонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазолил, пиридоимидазолил, пиридотиазолил, пиридинил, пиридил, пиримидинил,пирролидинил, пирролинил, пирролил, хиназолинил, хинолинил, 4 Н-хинилизинил, хиноксалинил, хинуклидинил, карболинил, тетрагидрофуранил, тетраизохинолинил, тетрагидрохинолинил, 6H-1,2,5 тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил,тиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,3 триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил, тетразолил и ксантенил. Согласно другому аспекту изобретения гетероциклы включают, без ограничения, пиридинил, тиофенил, фуранил, индазолил, бензотиазолил, бензимидазолил, бензотиафенил, бензофуранил, бензоксазолил, бензизоксазолил,хинолинил, изохинолиниол, имидазолил, индолил, изоиндолил, пиперидинил, пиперидонил, 4 пиперидонил, пиперонил, пирразолил, 1,2,4-триазолил, 1,2,3-триазолил, тетразолил, тиазолил, оксазолил,пиразинил или пиримидинил. Данное изобретение охватывает также конденсированные кольца и спиросоединения, содержащие, например, указанные выше гетероциклы. Примерами гетероарилов являются 1 Н-индазолил, 2 Н,6 Н-1,5,2-дитиазинил, индолил, 4 Нкарбазолил, 4 Н-хинолизинил, 6 Н-1,2,5-тиадиазинил, акридинил, азоцинил, бензимидазолил, бензофуранил, бензотиофуранил, бензотиофенил, бензоксазолил, бентиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, бензимидазалонил, карбазолил, 4aH-карбазолил, -карболинил, хроманил, хроменил, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро- (2,3b)тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, индазолил,индоленил, индолинил, индолизинил, индолил, изобензофуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил (бензимидазолил), изотиазолил, изоксазолил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4 оксадиазолил, оксазолидинил, оксазолил, оксазолидинилперимидинил, фенантридинил, фенантролинил,фенарсазинил, феназинил, фенотиазинил, феноксатиинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, птеридинил, пиперидонил, 4-пиперидонил, птеридинил, пуринил, пиранил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиразотриазинил, пиридазинил, пиридооксазолил, пиридоимидазолил, пиридотиазолил, пиридинил, пиридил, пиримидинил, пирролидинил, пирролинил, пирролил, хиназолинил, хинолинил, 4H-хинолизинил, хиноксалинил, хинуклидинил, карболинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, 6 Н-1,2,5-тиадиазинил, 1,2,3-тиадиазинил,1,2,4-тиадиазинил, 1,2,5-тиадиазинил, 1,3,4-тиадиазинил, тиантренил, тиазолил, тиенил, тиенотиазолил,тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5 триазолил, 1,3,4-триазолил, тетразолил и ксантенил. Согласно другому аспекту данного изобретения примерами гетероарилов являются индолил, бензимидазолил, бензофуранил, бензотиофуранил, бензоксазолил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, бензимидазалонил, циннолинил, фуранил, имидазолил, индазолил, пиразолотриазинил, пиридазинил, пиридил, пиридинил, пиримидинил, пирролил, хиназолинил, хинолинил, тиазолил, тиенил и тетразолил. Применяемый в данном изобретении термин "гетероциклоалкил" или "гетероциклил", используемый сам по себе или как часть другой группы, относится к гетероциклическим группам, определение которых приведено выше, присоединнным при помощи атома углерода или гетероатома к алкильной цепи. Применяемый в данном изобретении термин "гетероарилалкил" или "гетероарилалкенил", используемый сам по себе или как часть другой группы, относится к гетероарильным группам, определение которых приведено выше, присоединнным при помощи атома углерода или гетероатома к алкильной цепи, алкилену или алкенилену, определение которых приведено выше. Применяемый в данном изобретении термин "циано" относится к группе -CN. Применяемый в данном изобретении термин "нитро" относится к группе -NO2. Применяемый в данном изобретении термин "гидрокси" относится к группе -ОН. Выражение "фармацевтически приемлемое(ый),(ая)"относится к таким соединениям, материалам,композициям и/или лекарственным формам, которые, с точки здравого медицинского суждения, приемлемы для контакта с тканями в организме людей и животных без проявления токсичности, раздражения,аллергической реакции или других проблем или осложнений при разумном отношении польза/риск. Применяемое в данном изобретении выражение "фармацевтически приемлемые соли" относится к производным описанных соединений, когда родительское соединение модифицируется путм получения его кислых или основных солей. Примеры фармацевтически приемлемых солей включают, без ограничения, соли основных остатков, таких как амины, с минеральными и органическими кислотами; щелочные или органические соли кислот и основных остатков, например, соли карбоновых кислот; и т.п. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные аммониевые соли родительского соединения, например, соли нетоксичных неорганических и органических кислот. Например, такие соли включают соли, полученные из неорганических кислот, таких как соляная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и т.п.; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная,лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфокислота, метансульфокислота, этандисульфокислота, щавелевая, изэтионовая, это может быть кислая серно-кислая соль и т.п. Фармацевтически приемлемые соли по изобретению могут быть получены из родительского соединения, которое содержит основный или кислый фрагмент, путм проведения обычных химических реакций. Обычно такие соли получают путм взаимодействия свободной кислоты или основания этого соединения со стехиометрическим количеством соответствующих основания или кислоты в воде или в среде органического растворителя или в их смеси; обычно предпочтительными являются неводная среда,например, простой эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечень приемлемых солей приведн в Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, PA, p. 1418(1985), содержание этого источника включено в данное изобретение в качестве ссылки. Любое соединение, которое может быть превращено in vivo с получением биоактивного агента (т.е. соединения формулы I), представляет собой пролекарство, охватываемое данным изобретением. Термин "пролекарства" включает сложные эфиры и карбонаты, полученные при реакции одной или более гидроксильных групп соединений формулы I с алкил-, алкоксил- или арилзамещнными ацилирующими агентами с применением методик, известных специалистам в данной области, с получением ацетатов, пивалатов, метилкарбонатов, бензоатов и т.п. Известны различные формы пролекарств и они описаны в: а) The Practice of Medicinal Chemistry, Camille G. Wermuth et al., Ch. 31 (Academic Press, 1996); б) Design of Prodrugs, edited by H. Bundgaard (Elsevier, 1985); в) A Textbook of Drug Design and Development, P. Krogsgaard-Larson and H. Bundgaard, eds. Ch. 5, p.p. 113-191 (Harwood Academic Publishers, 1991); и г) Hydrolysis in Drug and Prodrug Metabolism, Bernard Testa and Joachim M. Mayer (Wiley-VCH,2003). Указанные источники включены в данное изобретение в качестве ссылок. Кроме того, соединения формулы I после их образования предпочтительно выделяют и очищают с получением композиции, содержащей соединение формулы I в количестве, по весу равном или превышающем 99% ("практически чистое" соединение I), которое затем применяют или на основе которого получают состав, как описано в данном изобретении. Все стереомеры соединений согласно данному изобретению входят в объм данного изобретения или в виде смесей, или в чистой или практически чистой форме. Соединения по изобретению могут содержать асимметричные центры у любого атома углерода, включая любой такой атом в заместителях R и/или проявляют полиморфизм. Соответственно, соединения формулы I могут существовать в энантиомерной или диастереомерной формах или в виде их смесей. Способы получения в качестве исходных веществ могут использовать рацематы, энантиомеры или диастереомеры. Когда получают диастереомерные или энантиомерные продукты, они могут быть разделены обычными методами, например, методом хроматографии или методом фракционной кристаллизации. Кроме того, соединения формулы I могут существовать в таутомерной форме. Такие таутомерные формы соединений формулы I также охвачены данным изобретением. Термины "стабильное соединение" и "стабильная структура" означают соединение, которое является достаточно объмным, чтобы выдержать выделение из реакционной смеси при приемлемой степени чистоты, обработку и хранение и получение на его основе приемлемого эффективного терапевтического агента. Данное изобретение также охватывает такие стабильные соединения. Термин "терапевтически эффективное количество" включает такое количество одного соединения по изобретению или такое количество соединения по изобретению в комбинации с другими активными ингредиентами, которые являются эффективными для ингибирования активности фермента 11-бетагидроксистероид-дегидрогеназы типа 1 или эффективными для лечения или профилактики метаболической и других болезней. Применяемый в данном изобретении термин "лечение" охватывает лечение болезни или состояния у млекопитающего, в особенности у человека, и включает: а) профилактику болезни или состояния у млекопитающего, в особенности когда такое млекопитающее предрасположено к такой болезни или к такому состоянию, но ещ не был поставлен диагноз о наличии болезни/состояния; б) ингибирование болезни/состояния, т.е. остановку их развития; и/или в) облегчение болезни/состояния, т.е. появление регрессии болезненного состояния. Синтез Соединения по изобретению могут быть получены по следующим реакционным схемам согласно приведнному описанию, а также согласно релевантной литературе, которая может быть использованы специалистом в данной области. Примеры реагентов и методик для проведения этих реакций описаны ниже и в рабочих примерах. Схема I На схеме I проиллюстрирован способ получения соединений формулы I-а (подкласс соединений формулы I). Промежуточные фтор-, хлор- или бромпиридин могут быть получены в промышленном масштабе способами, известными из литературы, или другими способами, известными специалистам. Реакцию соединения формулы I-b с гидразином проводили при повышенной температуре с получением промежуточного соединения I-c. Ацилирование промежуточного соединения I-c при помощи кислоты I-d с применением подходящих амидных агентов сочетания, таких как NMM/изобутилхлорформиат,EDAC/HOBT и другие реагенты, описанное Bodanszky, M., The Practice of Peptide Synthesis, 2nd Ed.(Spring-Verlag, 1993) привело к получению промежуточного гидразида I-f. Или же гидразид I-c может быть получен по реакции I-c и хлорангидрида кислоты I-e в присутствии подходящего основания, такого как DIEA или TEA. Образование 1,2,4-триазолпиридина I-g может быть достигнуто путм взаимодействия I-f с Et3P/CCl4 в присутствии основания, такого как ТЕА/основания Hunig, или с Ph3PCl2 в присутствии основания, такого как TEA. Образование 1,2,4-триазолпиридина I-g может быть также достигнуто с применением соединения 1,2,4-триазолпиридина I-g в качестве исходного соединения в присутствии уксусной кислоты при повышенной температуре или путм обычного нагревания, или с использованием микроволнового излучения. Альтернативно, получение 1,2,4-триазолпиридина I-g может быть осуществлено путм реакции I-f с POCl3 при повышенной температуре или другими методами, известными специалистам. Получение метилкарбоксилата 1,2,4-триазолпиридина I-h можно осуществить по реакции карбоксилирования, катализируемой палладием, в присутствии СО и метанола. Сложный эфир I-h можно обработать реагентами Гриньяра или литийорганическими соединениями с образованием карбинола I-а или обработать реактивом Гриньяра с последующим восстановлением с применением реагента, такого как NaBH4 с получением гидроксилсодержащего соединения I-a (R'=водород) или получить другими методами, известными специалистам. Схема II На схеме II показан способ получения соединений формулы II-а (подкласс соединений формулы I). Реагенты II-b и II-с могут быть получены в промышленном масштабе способами, известными из литературы, или другими способами, известными специалистам. Образование соединения II-а может быть достигнуто по реакции сочетания цинксодержащих реагентов II-b или бороновой кислоты II-c с бром-, хлорили йодзамещенными промежуточными соединениями II-g (Z1 обозначает Br, Cl или I) в присутствии палладиевого катализатора. Реакцию можно осуществлять при комнатной температуре или при нагревании или в реакторе под действием микроволнового излучения. Схема III На схеме III проиллюстрирован способ получения соединений формулы III-b, III-с и III-d (подкласс соединений формулы I). Получение соединения III-b может быть достигнуто по реакции сочетания соединения III-a с бром-, хлор- или йодзамещенными промежуточными соединениями I-g (Z1 обозначает Br или I) в присутствии палладиевого катализатора и основания, такого как карбонат калия или фторид цинка, при повышенной температуре. Реакцию можно осуществлять обычным методом или в реакторе под действием микроволнового излучения (J. Am. Chem. Soc, 125: 11176 (2003. Альтернативно, соединение III-b может быть получено другими методами, известными специалистам. Кислота III-с может быть получена путм гидролиза соединения III-b с применением основания, такого как LiOH. Образование амида III-d может быть достигнуто путм сочетания кислоты III-с с амином R'RNH с применением подходящего набора амидных агентов сочетания, таких как NMM/изобутилхлорформиат, EDAC/HOBT и другие реагенты, описанные Bodanszky, М., The Practice of Peptide Synthesis, 2nd Ed. (Spring-Verlag, 1993). Альтернативно, амид III-d можно получить по реакции соединения формулы III-b и амина при повышенной температуре или другими методами, известными специалистам. Схема IV На схеме IV показан способ получения соединений формулы IV-b (подкласс соединений формулыIaa или Iee). Соединение IV-b может быть получено путм металлирования (обмен галоида на металл) соединения Ig с применением алкилметаллических реагентов, таких как BuLi или изопропилмагнийхлорид, с последующим добавлением альдегида или кетона IV-a с образованием соединения IV-b. Или же соединение IV-b может быть получено другими методами, известными специалистам. Схема V На схеме V показан способ получения соединений формулы V-d (подкласс соединений формулы Iaa или Iee). Фтор-, хлор- или бромпиридины могут быть получены в промышленном масштабе способами,известными из литературы, или другими способами, известными специалистам. Реакцию соединения формулы V-a с гидразином в подходящем растворителе, таком как 1-пропанол или изопропиловый спирт, можно проводить при повышенной температуре с получением промежуточного соединения V-b. Соединение V-b может быть выделено непосредственно или в виде соответствующей соли (например,гидрохлорида). Ацилирование промежуточного соединения V-b кислотой V-d с применением подходящего набора амидных агентов сочетания, таких как NMM/изобутилхлорформиат, EDAC/HOBT и другие реагенты, описанные Bodanszky, M., The Practice of Peptide Synthesis, 2nd Ed. (Spring-Verlag, 1993) обеспечивает получение промежуточного гидразида V-c. Или же гидразид V-c может быть получен по реакции соединения формулы V-b и хлорангидрида кислоты I-c (который может быть получен по реакции кислоты I-d с оксалилхлоридом) в присутствии подходящего растворителя, такого как толуол, DMF или THF, в присутствии соответствующего основания, такого как NaOH, K2CO3, DIEA или TEA. 1,2,4 триазолпиридин IIa может быть получен по реакции V-c с Et3P/CCl4 в присутствии основания, такого какTEA или DIEA. Получение 1,2,4-триазолпиридина IIa можно также осуществить из соединения V-c в присутствии карбоновой кислоты, такой как уксусная кислота, п-тозиловая кислота, бензойная кислота,2-, 3- или 4-хлорбензойная кислота, 4-фторбензойная кислота, 2-хлорбензолуксусная кислота, 2- или 4 метилбензойная кислота, 4-метоксибензойная кислота, 2,6-диметилбензойная кислота, 2,6 диметоксибензойная кислота, 2-метилпропионовая кислота и 2,2-диметилпропионовая кислота, в среде растворителя, такого как толуол, при повышенной температуре, например, с обратным холодильником,или по обычной методике, или в реакторе под действием микроволнового излучения с получением 1,2,4 триазолпиридина V-d. Или же 1,2,4-триазолпиридин V-d можно получить по реакции соединения V-c сPOCl3 при повышенной температуре или другими методами, известными специалистам. Схема VI На схеме VI показан способ получения соединений формулы VI-е (подкласс соединений формулыIdd). Промежуточный бромпиридин VI-а можно получить в промышленном масштабе способами, известными из литературы, или другими способами, известными специалистам. Обработка соединения формулы VI-а н-BuLi или другими металлирующими агентами с последующим добавлением соединенияVI-b обеспечивает получение промежуточного кетона VI-c. Получение 1,2,3-триазолпиридина VI-d можно осуществить по реакции соединения VI-c и бензолсульфонгидразида в присутствии основания, такого как морфолин (Tetrahedron, 53: 8257-8268 (1997, или другими методами, известными специалистам, с последующей циклизацией при помощи такого реагента, как йодбензолдиацетат. Образование соединения VI-е может быть достигнуто с применением таких же превращений, которые описаны на схемах I-IV,или другими методами, известными специалистам. Примеры Следующие рабочие примеры служат для лучшей иллюстрации, но не ограничения, некоторых предпочтительных вариантов данного изобретения. Общие положения Термин HPLC относится к высокоэффективной жидкостной хроматографии Shimadzu, которая проводится одним из следующих методов. Метод А: колонка YMC или Phenomex С 18, 5 мкм, 4,650 мм, градиент 4 мин 0-100% растворителя В [90% МеОН: 10% Н 2 О: 0,2% Н 3 РО 4] и 100-0% растворителя А [10% МеОН: 90% Н 2 О: 0,2% Н 3 РО 4] со скоростью истечения 4 мл/мин и выдержкой в течение 1 мин, детектор ультрафиолетового света (УФ) работал при длине волны, равной 220 нм. Метод В: колонка Phenomenex S5 ODS 4,630 мм, градиент элюирования 0-100% В/А в течение 2 мин (растворитель А=10% МеОН/Н 2 О, содержит 0,1% TFA, растворитель В=90% МеОН/Н 2 О, содержит 0,1% TFA), со скоростью истечения 4 мл/мин и выдержкой в течение 1 мин, детектор ультрафиолетового света (УФ) работал при длине волны 220 нм. Метод С: колонка YMS S7 ODS 3,050 мм, градиент элюирования 0-100% В/А в течение 2 минTFA), со скоростью истечения 4 мл/мин и выдержкой в течение 1 мин, детектор ультрафиолетового света(УФ) работал при длине волны 220 нм. Термин "преп. хроматография" относится к автоматизированной системе Shimadzu, где применяется смесь растворителя А (10% МеОН/H2O, содержит 0,1% TFA, растворитель В=90% МеОН/Н 2 О, содержит 0,1% TFA). Колонки были загружены YMC или Phenomenex ODS С 18, 5 мкм или эквивалентной смолой. Характеристика кристаллических форм Монокристалл. Для получения данных рентгенограмм применяли дифрактометр Bruker SMART, излучение CuK,графитовый монохроматор (=1,54056 ангстрем), данные получали при комнатной температуре. Полный набор данных получали, используя -сканирование в интервале 2 при расстоянии от детектора до кристалла 4,98 см. Для эмпирической коррекции абсорбции применяли SADABS, соединнный с дифрактометром (Bruker AXS. 1998. SMART и SAINTPLUS. Area Detector Control and Integration Softwear, BrukerAXS, Madison, Wisconsin, USA). Конечные параметры элементарной ячейки определяли, используя полный набор данных. Все структуры были решены прямыми методами и уточнены методом наименьших квадратов с матрицей полного ранга с применением пакета программ SHELXTL (Sheldrick, G.M. 1997, SHELXTL.Structure Determination Programs. Version 5.10, Bruker AXS, Madison, Wisconsin, USA). При уточнении применяли функцию w(Fo-Fc)2. R определяли как Fo-Fc/Fo, a Rw=[w(Fo-Fc2/wFo2)]1/2, гдеw обозначает соответствующую весовую функцию на основе ошибок, наблюдавшихся при определении величин интенсивности. Карты, полученные с помощью преобразования Фурье, изучали на всех стадиях уточнения. Параметры атомов, отличных от водорода, уточняли методом анизотропного термического замещения. Атомы водорода с водородными связями располагались на конечных картах Фурье, а положения других атомов водорода были рассчитаны на основе идеальной геометрии с применением стандартных длин связей и углов. Им были присвоены изотропические температурные факторы и они были включены при расчте структурных факторов с фиксированными параметрами. Альтернативно данные для монокристалла получали с применением прибора серийного дифрактометра Bruker-Nonius1 CAD4. Параметры элементарной ячейки определяли методом наименьших квадратов для экспериментальных величин отражений под большим углом 25. Величины интенсивности измеряли методом сканирования при переменном значении -2 с использованием излучения CuK(=1,5418 ангстрем) при постоянной температуре и были скорректированы только для факторов поляризации Лоренца. Фоновые сигналы регистрировали при экстремальных значениях сканирования в течение половины времени сканирования. Или же данные для монокристалла получали, используя системуBruker-Nonius Kappa с использованием излучения CuK (=1,5418 ангстрем). Индексирование и обработку измеренных величин интенсивности осуществляли при помощи программы HKL20002 в наборе программ Collect program3. Или же данные для монокристалла получали, используя систему Braker-AXSAPEX2 CCD с использованием излучения CuK (=1,5418 ангстрем). Индексирование и обработку измеренных величин интенсивности осуществляли при помощи программы АРЕХ 2 в наборе программ. Там, где это указано, кристаллы охлаждали холодным потоком системы Oxford cryo system5 во время сбора данных. 1Oxford Cryosystems Cryostream cooler Cosier, J. et al., J. Appl. Cryst., 19, 105 (1986). Структуры решали прямыми методами и уточняли на основе полученных отражений, применяя или пакет программ SDP6 с незначительными модификациями или кристаллографические пакеты MAXUS или SHELXTL. Производные параметров атомов (координаты и температурные факторы) уточняли методом полноматричных наименьших квадратов. Минимизированная функция при уточнении представляла собойw(Fo-Fc)2. R определяли как Fo-Fc/Fo, в то время как Rw=[w(Fo-Fc2/wFo2)]1/2, где w обозначает соответствующую весовую функцию с учетом ошибок в наблюдаемых интенсивностях. Карты различия изучали на всех стадиях уточнения. Атомы водорода располагали в идеальных положениях с изотропическими температурными факторами, но параметры атомов водорода не меняли.PXD. Данные дифрактограммы получали с применением системы Bruker C2 GADDS (General Area Detector Diffraction System). Использовали излучение CuK (40 кВ, 50 мА). Расстояние от образца до детектора составляло 15 см. Образцы порошка помещали в герметичные стеклянные капилляры диаметром 1 мм или менее, капилляр вращался во время сбора данных. Данные регистрировали для 3235 при времени экспонирования образца по меньшей мере 2000 с. Полученные двухразмерные дифракционные дуги интегрировали для создания традиционной одноразмерной дифрактограммы PXRD с размером шага 0,02 2 в интервале 3-35 2. Альтернативно, данные дифрактограммы получали с применением гониометра Bruker GADDS с приводимой в движение вручную платформой из керамического материала. Образцы порошка помещали в тонкостенные стеклянные капилляры диаметром 17 мм или менее, капилляр вращался во время сбора данных. Расстояние от образца до детектора составляло 1 см. Использовали излучение CuK (=1,5418 ангстрем). Данные регистрировали для 3235 при времени экспонирования образца по меньшей мере 00 с. 6DSC. Дифференциальную сканирующую калориметрию проводили в приборе ТА Instruments модельQ2000, Q1000 или 2920. Образец (2-6 г) взвешивали в алюминиевом поддоне и записывали вес с точностью до сотой доли миллиграмма и проводили DSC. Прибор продували азотом со скоростью 50 мл/мин. Данные регистрировали в интервале температур между комнатной температурой и 300 С со скоростью нагрева 10 С/мин. Был построен график с эндотермическими пиками, направленными вниз.TGA. Термический гравиметрический анализ (TGA) проводили на приборе ТА Instruments модель Q500 или 2920. Образец (около 10-30 г) помещали в предварительно взвешенный платиновый тигель. Измеряли точно вес образца и записывали его с точностью до тысячной доли миллиграмма на приборе. Печь продували газообразным азотом со скоростью 100 мл/мин. Данные регистрировали в интервале температур между комнатной температурой и 300 С со скоростью нагрева 10 С/мин. Сорбция влаги. Изотермы сорбции влаги строили на приборе VTI SGA-100 Symmetric Vapor Analyser, используя примерно 10 мг образца. Образец высушивали при температуре 60 С до тех пор, пока потеря веса за 10 мин не составила 0,0005%/мин. Образец испытывали при температуре 25 С и при относительной влажности или 4, 5, 15, 25, 35, 45, 50, 65, 75, 85 и 95%. Уравновешивание при каждом значении относительной влажности достигали, когда была достигнута скорость 0,0003% вес./мин в течение 35 мин или максимум 600 мин. 7 В примерах и в описании далее используются следующие сокращения:Cbz - карбобензилокси или карбобензокси или бензилоксикарбонил; СО - окись углерода;FMOC - флуоренилметоксикарбонил; НОАс или АсОН - уксусная кислота; НОАТ - 1-гидрокси-7-азобензотриазол; НОВТ - 1-гидроксибензотриазол;IPA - изопропиловый спирт или изопропанол; К раствору 2-хлор-3-бромпиридина (14,5 г, 75,1 ммоль) в 100 мл диоксана добавляли безводный гидразин (35,4 мл, 1130 ммоль) при RT. Реакционную смесь нагревали с обратным холодильником в течение 15 ч и затем охлаждали до RT. После удаления большей части растворителя при пониженном давлении, полученный остаток разбавляли этилацетатом, промывали водой, сушили над Na2SO4 и концентрировали с получением остатка. Перекристаллизация остатка из этилацетата и гексанов обеспечила получение соединения 1 А (12,9 г, 91%) в виде твердого вещества. Раствор 1-(4-хлорфенил)циклопропанкарбоновой кислоты (8,71 г, 44,3 ммоль) и NMM в THF (200 мл) охлаждали до 0 С. При этой температуре по каплям добавляли изобутилхлорформиат в течение 10 мин и полученную белую суспензию перемешивали в течение 1 ч. Затем добавляли раствор соединения 1 А (8,33 г, 44,3 ммоль) в THF в течение 10 мин. После окончания добавления реакционную смесь перемешивали в течение 15 мин, затем реакционную смесь нагревали до RT и перемешивали в течение 2 ч. Реакционную смесь распределяли между этилацетатом и водой. Органическую фазу отделяли, сушили над Na2SO4 и затем концентрировали под вакуумом с получением соединения 1 В (10,2 г, 27,8 ммоль, выход 63%) в виде почти белого цвета. Перемешиваемый раствор соединения 1 В (10 г, 27,3 ммоль), четыреххлористого углерода (31,6 мл,327 ммоль) и основания Хунига (28,6 мл, 167 ммоль) в 275 мл THF охлаждали до 0 С. По каплям добавляли триэтилфосфин (12,1 мл, 82 ммоль в течение 10 мин. После окончания добавления реакционную смесь медленно нагревали до RT и перемешивали в течение 16 ч. Затем реакционную смесь обрабатыва- 20019706 ли водой (100 мл) и этилацетатом (2200 мл). Органические фазы промывали рассолом, сушили надNa2SO4 и концентрировали под вакуумом с получением желтого твердого вещества. Это вещество желтого цвета растирали со 100 мл смеси этилацетат/гексан (2:1, об./об.) и полученную смесь отфильтровывали. Полученное твердое вещество промывали этилацетатом (50 мл) и путем фильтрации выделяли соединение 1 С (7,2 г) в виде вещества бледно-желтого цвета. В реакционную колбу помещали соединение 1 С (2,5 г, 7,17 ммоль), 1,3-бис(дифенилфосфино)пропан (0,592 г, 1,43 ммоль), триэтиламин (3,00 мл, 21,5 ммоль) и МеОН (50 мл). Через смесь пропускали в течение 2 мин СО, затем колбу герметизировали и пропускали снова СО (газ) под давлением 25 ф/дюйм 2. Реакционную смесь нагревали до 80 С и перемешивали ее в течение 24 ч. Затем под вакуумом удаляли МеОН и добавляли 20 мл рассола. Полученную смесь экстрагировали EtOAc(330 мл). Соединенные органические слои промывали рассолом, сушили над Na2SO4, отфильтровывали и концентрировали с получением технического продукта. Этот продукт очищали методом хроматографии на колонке, элюировали смесью EtOAc/гексан (0 до 100%) с получением соединения 1D (2,02 г) в виде пены светло-желтого цвета.LC/MS (m/z)=328 (М+Н)+. Пример 1. При RT в атмосфере азота в трубчатый приемник, облучаемый микроволновым излучением, загружали раствор соединения 1D (131 мг, 0,40 ммоль) в 3 мл THF с последующим добавлением метилмагнийхлорида (400 мкл, 3 М в THF, 1,2 ммоль). После окончания добавления реакционную смесь перемешивали при RT в течение 1 ч и затем нагревали до 65 С и перемешивали в течение 6 ч. Затем охлаждали смесь до RT. Добавляли рассол (3 мл) и экстрагировали EtOAc (35 мл). Соединенные органические слои концентрировали и очищали методом колоночной хроматографии (колонка с 40 г двуокиси кремния, от 0% до 100% смеси EtOAc/гексан), получая соединение по примеру 1 (45 мг) в виде белой пены. В круглодонную колбу объемом 2,0 л последовательно загружали 2-(2-фторпиридин-3-ил)пропан-2 ол (60,0 г, 386,7 ммоль, 1 экв.), 1-пропанол (120 мл) и Na2CO3 (43,0 г, 405,7 ммоль, 1,05 экв.). Моногидрат гидразина (96,8 г, 1930 ммоль, 5,2 экв.) добавляли в колбу и полученную смесь нагревали до 92-94 С. После достижения указанной температуры осуществляли мониторинг реакции при помощи HPLC до израсходования исходного 2-(2-фторпиридин-3-ил)пропан-2-ола (примерно через 24-26 ч). Полученную двухфазную суспензию охлаждали до 20 С и затем добавляли воду и толуол (900 мл). По окончании добавления полученную смесь перемешивали при 20 С в течение примерно 10-15 мин. Затем давали органической и водным фазам осесть в течение 10 мин и отделяли водную фазу. Органический слой промывали 20%-ным водным рассолом (450 мл) и концентрировали под вакуумом при температуре примерно 55-60 С с получением раствора в толуоле (240 мл). Этот толуольный раствор охлаждали до 25 С и добавляли 1-пропанол (60 мл). К полученному раствору добавляли 5-6 N HCl в среде IPA (0,6 экв.) в течение 30-40 мин. После окончания добавления добавляли затравку полученного ранее 2-(2 гидразинпиридин-3-ил)пропан-2-ола моногидрата гидрохлорида (1,0 вес.%) и дополнительную порцию раствора 5-6 N HCl в IPA (0,7 экв.). Полученную суспензию перемешивали при 20 С в течение по мень- 21019706 шей мере 12 ч и фильтровали. Остаток на фильтре промывали смесью 1-пропанола и толуола (9:1,об./об., 200 мл 3) и затем сушили под вакуумом при комнатной температуре в течение 4-5 ч. Затем остаток от фильтрования высушивали в атмосфере азота при температуре 40 С в течение 2-3 ч и затем при комнатной температуре в течение примерно 16 ч, получали сырой продукт, соединение 1E (52,1 г, выход 60,7%, степень чистоты по данным HPLC составляла более 99%). Соединение 1E также перекристаллизовывали путем смешения сырого 2-(2-гидразинилпиридин-3 ил)пропан-2-ола моногидрата гидрохлорида (25 г, 113 ммоль), IPA (150 мл) и 1-пропанола (100 мл) в круглодонной колбе объемом 500 мл. Полученную суспензию нагревали в атмосфере азота до 55-60 С и перемешивали в течение примерно 5-10 мин. Затем добавляли толуол (100 мл). В полученную смесь добавляли затравочные кристаллы 2-(2-гидразинпиридин-3-ил)пропан-2-ола моногидрата гидрохлорида(1,0 вес.%) и дополнительное количество толуола (100 мл, общее количество толуола составило 200 мл)(общее время добавления толуола составило 30 мин). После окончания добавления смесь перемешивали в течение примерно 16 ч и затем полученную суспензию отфильтровывали. Осадок на фильтре промывали толуолом и изопропиловым спиртом (2:1, об./об., 25 мл 2) и высушивали в вакуумной печи при комнатной температуре в атмосфере азота в течение примерно 16 ч для получения перекристаллизованного соединения 1E (13,4 г, выход 53%, точка плавления 88,1-111,2 С (разлHRMS соединения 1E (M+I): рассчитано 168.1137, найдено 168.1137. Аналитический расчет для соединения 1E: С, 43.35; Н, 7.27; N, 18.95; С 1, 15.99. Найдено: С, 43.53; Н, 7.29; N, 19.06, Cl, 15.99. Соединение 1F. 1-(4-Хлорфенил)-N'-(3-(2-гидроксипропан-2-ил)пиридин-2 ил)циклопропанкарбогидразид К суспензии 1-(4-хлорфенил)циклопропанкарбоновой кислоты (34,7 г, 177 ммоль, 1,1 экв.) в толуоле (142 мл) добавляли DMF (124 мкл, 1,6 ммоль, 0,01 экв.). После окончания добавления суспензию перемешивали при температуре около 25 С в течение примерно 5 мин и затем при комнатной температуре в течение 20 мин добавляли чистый оксалилхлорид (14,8 мл, 167 ммоль, 1,0 экв.). Полученную суспензию перемешивали в течение примерно 4 ч (по мере протекания реакции твердые вещества постепенно растворялись). В отдельной колбе охлаждали до примерно 5 С смесь соединения 1E (35,5 г, 160 ммоль,1,0 экв.) и карбоната калия (51 г, 369 ммоль, 2,3 экв.) в THF (355 мл). Сразу же после достижения указанной температуры добавляли воду (248 мл) и перемешивали двухфазную смесь в течение 15 мин. Затем к двухфазному раствору добавляли раствор хлорангидрида кислоты в течение 20 мин. После окончания добавления реакционную смесь перемешивали в течение 30 мин. Реакционную смесь анализировали методом HPLC, по данным HPLC реакция была завершена. Водный слой удаляли и органический слой промывали водой (180 мл 2) и затем концентрировали путем вакуумной перегонки при температуре около 60 С с получением раствора продукта (178 мл). К этому раствору продукта добавляли толуол (70 мл) и полученный раствор нагревали до примерно 70 С. После достижения указанной температуры добавляли н-гептан (425 мл) в течение 30 мин с получением суспензии. Суспензию охлаждали до температуры около 20 С и перемешивали в течение 3 ч. Затем суспензию отфильтровывали. Остаток на фильтре промывали н-гептаном (142 мл) и высушивали под вакуумом при RT в течение 48 ч, получая технический продукт 1F (51 г, выход 92%). Этот технический продукт 1F суспендировали в ацетонитриле (770 мл) и воде (35 мл) и полученную смесь нагревали до температуры около 75 С, перемешивали в течение 5 мин для полного растворения продукта. Полученный раствор затем охлаждали до температуры около 60 С. После достижения этой температуры в течение 1 ч добавляли воду (280 мл). Полученную суспензию охлаждали до примерно 20 С в течение 2 ч. Затем при этой температуре суспензию перемешивали в течение 2 ч. Полученный твердый продукт отфильтровывали, промывали водой (142 мл) и затем сушили под вакуумом при температуре около 70 С, получая перекристаллизованное соединение 1F (46 г, выход 83%). По данным HPLC степень чистоты равнялась 99,64%. Соединение 1F также перекристаллизовывали путем суспендирования сырого соединения 1F (17 г) в метаноле (170 мл) и последующего нагревания полученной суспензии с обратным холодильником с целью полного растворения. Затем добавляли воду (8,5 мл) и полученную суспензию охлаждали до температуры около 20 С. При этой температуре суспензию перемешивали в течение примерно 16 ч и затем отфильтровывали. Остаток на фильтре промывали водой (68 мл) и затем высушивали под вакуумом при температуре около 70 С, получали перекристаллизованное соединение 1F (14,1 г, выход 83%, точка плавления 191 С). ИК (KBr) 3355, 3226, 2978, 1633, 1591, 1574, 1541, 1494, 1456, 1384, 1316, 1269, 1240, 1139, 1098,978, 766, 534 см-1. 1 Н NMR (400 MHz, DMSO-d6):1.07 (dd, J=6.9, 4.1 Hz, 2H), 1.44 (dd, J=6.8, 4.0 Hz, 2H), 1.51 (s, 6H),5.72 (s, 1H), 6.69 (dd, J=7.4, 4.9 Hz, 1H), 7.39 (dd, J=7.6, 1.5 Hz, 1H), 7.43 (dt, J=5.6, 2.3 Hz, 2H), 7.50 (dt,J=5.5, 2.3 Hz, 2H), 7.95 (dd, J=4.9, 1.6 Hz, 1H), 8.97 (d, J=3.2 Hz, 1H), 9.06 (dd, J=3.3 Hz, 1H). 13MS соединения 1F (M+1): m/e, 346.15. Рассчитано для соединения 1F: С, 62.52; H, 5.827; N, 12.15; Cl, 10.25. Найдено: С, 62.58; Н, 5.78; N, 12.27, Cl, 10.26. Пример 1G. В трехгорлую круглодонную колбу объемом 50 мл помещали смесь соединения 1F (1,5 г, 4,34 ммоль, 1,0 экв.), толуола (15 мл) и уксусной кислоты (3 мл, 3,2 г, 52,4 ммоль, 12,1 экв.) и нагревали до температуры около 100-105 С и перемешивали в течение не менее 35,0 ч. Затем смесь анализировали методом HPLC, данные показали, что реакция была завершена на 97%. Реакционную смесь перегоняли при атмосферном давлении, получая остаток (примерно 10,0 мл). К этому остатку добавляли толуол (15 мл) и полученную смесь снова концентрировали путем перегонки при атмосферном давлении до объема около 16,5 мл. Затем реакционную смесь охлаждали до 20 С и перемешивали в течение примерно 16 ч. По окончании этого периода смесь разбавляли водой (12 мл) и затем добавляли конц. HCl (560 мкл, 6,5 ммоль, 1,5 экв.). Полученную смесь перемешивали в течение примерно 10 мин и затем отделяли органический слой и выгружали его. Водный слой обрабатывали смесью толуол/гептан (2:1, об./об., 6 мл). Затем снова отделали органический слой и выгружали его. К водному слою добавляли толуол (15 мл) и полученную водную двухфазную смесь охлаждали до примерно 5 С. При этой температуре для достижения рН примерно 14 добавляли 10 N NaOH (водная, 950 мкл, 9,5 ммоль, 2,2 экв.). По окончании добавления полученную смесь перемешивали в течение 15 мин и затем разделяли органический и водный слои. Органический слой промывали 10 вес.% NaCl (водный раствор, 7,5 мл) и разбавляли толуолом (7,5 мл). Полученную смесь концентрировали путем перегонки при атмосферном давлении до конечного объема, составляющего около 13,5 мл. Этот раствор охлаждали до примерно 20 С и затем медленно добавляли смесь (0,5 мл, 0,4 экв.) конц. HCl (560 мкл, водная, 6,5 ммоль, 1,5 экв.) в среде IPA (1,5 мл) с последующим добавлением затравочных кристаллов соединения по примеру 1G (7,9 мг, 0,5%). К полученной суспензии добавляли остальную часть (1,5 мл, 1,1 экв.) смеси конц. HCl/IPA в течение 10 мин. По окончании добавления полученную суспензию перемешивали при температуре около 20 С в течение примерно 16 ч. Затем суспензию отфильтровывали. Остаток на фильтре промывали толуолом (5 мл) и затем высушивали под вакуумом, получая сырой продукт по примеру 1G со степенью чистоты 99,29% по данным HPLC, 1,2 г, в виде твердого вещества белого цвета. Выход составил 72,8%. Альтернативно, соединение по примеру 1G получали следующим образом: в трехгорлую круглодонную колбу объемом 50 мл добавляли смесь соединения 1F (1,0 г, 2,95 ммоль, 1 экв.), толуола (5,1 мл) и уксусной кислоты (1,7 мл, 1,8 г, 29,5 ммоль, 10 экв.) и нагревали до примерно 100 С в течение 24 ч. Эту смесь разбавляли толуолом (15 мл). Полученную смесь концентрировали путем перегонки при температуре около 110 С с получением раствора объемом около 8,0 мл. Этот раствор охлаждали до примерно 20 С и затем добавляли раствор (0,5 мл, 0,5 экв.) конц. HCl (540 мкл, водная, 6,3 ммоль, 2,1 экв.) в изопропиловом спирте (1,5 мл). После окончания добавления добавляли затравочные кристаллы соединения из примера 1G (11,8 мг), получали светлую суспензию. К этой суспензии в течение 3 мин добавляли остальную часть (1,5 мл, 1,6 экв.) смеси конц. HCl/IPA. Полученную суспензию перемешивали при температуре около 20 С в течение примерно 16 ч. По окончании этого периода суспензию отфильтровывали. Остаток на фильтре промывали толуолом (4 мл) и затем высушивали под вакуумом с получением сырого продукта примера 1G, 1,0 г, степень чистоты по данным HPLC составляла 99,13%, продукт имел белый цвет. Получение кристаллической формы N-1 соединения из примера 1G. Сырой продукт из примера 1G (1,15 г) растворяли в EtOH (7,5 мл) при температуре около 65 С. После окончания растворения для образования слоя затравочных кристаллов добавляли н-гептан (7,5 мл) и затравочные кристаллы соединения из примера 1G, и полученную светлую суспензию перемешивали в течение примерно 10 мин, затем добавляли еще н-гептан (15 мл) в течение 15 мин. Эту суспензию перемешивали при температуре 65 С в течение 30 мин и затем охлаждали до примерно 20 С и перемешивали в течение не менее 16 ч. Затем суспензию отфильтровывали. Остаток на фильтре промывали 10% раствором этанола в н-гептанах (5 мл) и затем высушивали в вакуумной печи при температуре 45 С в течение примерно 16 ч с получением соединения из примера 1G (1,0 г, выход 63,9%) в виде твердого вещества белого цвета. Продукт анализировали методами, описанными выше, он оказался кристаллическим, формой N-1.C NMR (125.8 MHz, CD3OD-d4):16.1, 20.6, 30.5, 72.6, 119.9, 125.0, 129.6, 130.4, 133.6, 134.6,136.3, 138.3, 144.5, 150.0. Альтернативно кристаллическую форму N-1 соединения из примера 1G получали из сырого соединения по примеру 1G следующим образом. Сырой продукт из примера 1G (около 1,0 г) растворяли в EtOH (5 мл) при температуре 65 С. После окончания растворения добавляли н-гептан (5 мл) и полученную смесь перемешивали в течение примерно 10 мин для получения слоя затравочных кристаллов. Полученную светлую суспензию перемешивали в течение примерно 10 мин и затем снова добавляли н-гептан (7,5 мл) в течение 15 мин. После окончания добавления суспензию перемешивали в течение 30 мин, затем охлаждали до температуры около 20 С и перемешивали в течение примерно 16 ч. Затем суспензию отфильтровывали. Осадок на фильтре промывали 10% этанола в н-гептанах (5 мл) и затем высушивали в вакуумной печи при 45 С в течение примерно 16 ч, получали соединение по примеру 1G (0,8 г, выход 72,4%) в виде твердого вещества белого цвета. Степень чистоты по данным HPLC составляла 99,83%. Физические свойства этого соединения были похожи на физические свойства продукта, описанного выше. Получение кристаллической формы N-2 соединения по примеру 1G. Сырой продукт из примера 1G (318 мг) растворяли в толуоле (1,8 л) при температуре 90 С. Полученный раствор перемешивали при температуре 90 С в течение 30 мин и затем медленно охлаждали до 20 С в течение 3 ч. После достижения этой температуры охлаждающую баню удаляли и полученную суспензию перемешивали в течение примерно 16 ч. Затем полученный твердый продукт белого цвета собирали путем фильтрования и сушили при 30 С в вакуумной печи в течение примерно 16 ч, получали 220 мг продукта. Этот продукт анализировали методом PXRD, он оказался кристаллическим. Он представлял собой форму N-2. Альтернативно, кристаллическую форму N-2 соединения по примеру 1G получали из сырого продукта по примеру 1G следующим образом. Сырое соединение по примеру 1G (100 мг) растворяли в МТВЕ (0,6 л) при температуре 75 С. Полученный раствор перемешивали при 75 С в течение 20 мин и затем медленно охлаждали до 20 С в течение 2,5 ч. При этой температуре охлаждающую баню удаляли и полученную суспензию перемешивали в течение примерно 16 ч. Затем полученный твердый продукт белого цвета отфильтровывали и высушивали при температуре 30 С в вакуумной печи в течение примерно 16 ч, получали твердый продукт. Этот продукт имел физические свойства, похожие на физические свойства формы N-2, описанной выше. Получали кристаллические формы соединения по примеру 1G и их получение описано ниже в табл. 1. Указанные кристаллические формы содержат кристаллы форм N-1 и N-2. Эти формы анализировали одним или несколькими методами, описанными выше. Таблица 1 Пример 1G (а). Рентгенограмма монокристалла. Ниже приведены примерные параметры элементарной ячейки в ангстремах , измеренные при комнатной температуре методом, описанным выше, а также объем кристаллической ячейки (V), пространственная группа (sg), молекулы в асимметричной ячейке и плотность кристалла для формы N-1 соединения по примеру 1G. Размеры ячейки:=90 Пространственная группа: Р 21/с Молекулы/асимметричная единица (Z1): 1 Плотность, рассчитанная в г/см 3: 1.358 Параметры элементарной ячейки получены методом рентгеновского анализа единичного кристалла,описанным в Stout et al., X-Ray Structure Determination: A Practical Guide (MacMillian, 1968), этот источник ранее включен в данное изобретение в качестве ссылки. Анализ сорбции влаги показывает, что форма N-1 не является гигроскопичной в интервале величинRH от примерно 25 до примерно 75% при температуре 25 С. На фиг. 4 показана изотерма сорбции влаги кристаллической формы N-1 соединения по примеру 1G. Порошковая рентгеновская дифракция. Данные порошковой рентгеновской дифракции (PXRD) получали по методу, описанному выше. В табл. 2 а и на фиг. 1 показаны эти данные для кристаллической формы N-1 соединения по примеру 1G. Таблица 2 а Характеристические положения пиков дифракции (200.1) при RT, полученные при помощи дифрактометра (CuK) с вращающимся капилляром с 2, калиброванным с применением стандарта NIST Дифференциальная сканирующая калориметрия (DSC). Метод дифференциальной сканирующей калориметрии осуществляли для каждой кристаллической формы с использованием модели Q1000TA Instruments. Для каждого анализа камеру для образца промывали газообразным азотом ультравысокой степени чистоты сверху вниз. Прибор калибровали при помощи индия с высокой степенью чистоты. Скорость нагрева составляла 10 С/мин в интервале температур между 25 и 300 С. Поток тепла, который нормализовали по весу образца, измеряли в зависимости от измеренной температуры образца. Данные приведены в Вт/г ("W/g"). График имеет эндотермические пики, направленные вниз. На фиг. 2 приведена термограмма DSC для кристаллической формы N-1 соединения по примеру 1G, на ней наблюдается эндотермический переход выше примерно 150 С. Термогравиметрический анализ (TGA). Термогравиметрический анализ проводили, как описано выше. На фиг. 3 приведена кривая TGA для кристаллической формы N-1 соединения по примеру 1G, которое характеризуется незначительной потерей веса до температуры около 100 С. Пример 1G (b). Параметры монокристалла. Ниже приведены примерные размеры элементарной ячейки в ангстремах , измеренные при комнатной температуре методом, описанным выше, а также объем кристаллической ячейки (V), пространственная группа (sg), молекулы в асимметричной ячейке и плотность кристалла для формы N-2 соединения по примеру 1G. Размеры элементарной ячейки:=90 Пространственная группа: Pca21 Молекулы/асимметричная ячейка (Z'): 1 Плотность, рассчитанная в г/см 3: 1.335 Параметры элементарной ячейки определяли методом рентгеновского кристаллографического анализа, как описано в Stout et al., X-Ray Structure Determination: A Practical Guide (MacMillian, 1968), включенном в качестве ссылки в данное изобретение. Рентгеновская порошковая дифракция. Данные по определению рентгеновской порошковой дифракции (PXRD) были получены с применением метода PXRD, описанного выше. В табл. 2b и на фиг. 5 приведены эти данные для кристаллической формы N-2 соединения по примеру 1G. Таблица 2b Характеристические положения пиков дифракции (200.1) при RT, полученные при помощи дифрактометра (CuK) с вращающимся капилляром с 2, калиброванным с применением стандарта NIST Дифференциальная сканирующая калориметрия (DSC). Метод дифференциальной сканирующей калориметрии осуществляли для каждой кристаллической формы с использованием модели Q1000TA Instruments. Для каждого анализа камеру для образца промывали газообразным азотом ультравысокой степени чистоты сверху вниз. Прибор калибровали при помощи индия с высокой степенью чистоты. Скорость нагрева составляла 10 С/мин в интервале температур между 25 и 300 С. Поток тепла, который нормализовали по весу образца, измеряли в зависимости от измеренной температуры образца. Данные приведены в Вт/г ("W/g"). График имеет эндотермические пики, направленные вниз. На фиг. 6 приведена термограмма DSC для кристаллической формы N-2 соединения по примеру 1G, на ней наблюдается эндотермический переход выше примерно 150 С. Термогравиметрический анализ (TGA). Термогравиметрический анализ проводили, как описано выше. На фиг. 7 показана кривая TGA для кристаллической формы N-2 соединения по примеру 1G, которая характеризовалась незначительной потерей веса до температуры около 100 С. Пример 1H. 2-(3-(1-(4-Хлорфенил)циклопропил)-[1,2,4]триазоло[4,3-а]пиридин-8-ил)пропан-2-ола бисульфат Для ускорения растворения в сосуд, содержащий соединение по примеру 1, при комнатной температуре добавляли водный раствор серной кислоты (5:1, об./об.). В течение минуты добавления водного раствора серной кислоты без перемешивания осаждалось твердое вещество. Это твердое вещество отфильтровывали, получая соединение по примеру 1H в виде продукта белого цвета. Это вещество анализировали одним или несколькими методами, описанными выше, оно оказалось кристаллическим. Это была кристаллическая форма N-1. Параметры монокристалла. Ниже приведены примерные размеры элементарной ячейки в ангстремах, измеренные при комнатной температуре методом, описанным выше, а также объем кристаллической ячейки (V), пространственная группа (sg), молекулы в асимметричной ячейке и плотность кристалла для формы N-1 соединения по примеру 1 Н. Размеры элементарной ячейки:=90 Пространственная группа: Pca21/c Молекулы/асимметричная ячейка (Z'): 1 Плотность, рассчитанная в г/см 3: 1.444 Параметры элементарной ячейки определяли методом рентгеновского кристаллографического анализа, как описано в Stout et al., X-Ray Structure Determination: A Practical Guide (MacMillian, 1968). Порошковая рентгеновская дифракция. Данные были получены методом PXRD, описанным выше. На фиг. 8 показаны эти данные для кристаллической формы N-1 соединения по примеру 1 Н. трифтор К суспензии соединения 1 С (70 мг, 0,2 ммоль) в 2 мл THF при температуре -10 С медленно добавляли комплекс изопропилмагнийхлорида с хлоридом лития (0,602 мл, 0,602 ммоль). После окончания добавления смесь перемешивали при температуре -10 С (нагревали до 0 С на короткое время) в течение 1 ч и затем добавляли быстро циклобутанон (70,4 мг, 1,0 ммоль). Реакционную смесь перемешивали при-10 С в течение 30 мин и затем медленно нагревали до rt и перемешивали при этой температуре в течение 3 ч. Затем реакционную смесь обрабатывали водой и экстрагировали EtOAc (35 мл). Соединенные органические слои концентрировали и сырой продукт очищали методом prep-HPLC (H2O/CH3CN/TFA,20-100% В, колонка Luna 30100), получая соединение по примеру 2 (15 мг, 16%) в виде масла (сольLC/MS (m/z)=340 (М+Н)+. Степень чистоты по данным HPLC составляла 95%. Пример 3. 3-(1-(4-Хлорфенил)циклопропил-8-(проп-1-ен-2-ил-[1,2,4]триазоло[4,3-а]пиридина соль TFA Через смесь соединения 1 С (0,070 г, 0,201 ммоль), боронового эфира 4,4,5,5-тетраметил-2-(проп-1 ен-2-ил)-1,3,2-диоксаборолана (0,038 мл, 0,201 ммоль) и K3PO4 (1,107 г, 0,502 ммоль) при перемешивании пропускали сильный ток аргона в течение 5 мин. Затем добавляли PdCl2 (dppf)-CH2Cl2 (0,016 г, 0,020 ммоль). После окончания добавления реакционный сосуд промывали аргоном, закрывали и затем нагревали до 90 С в течение 20 ч. По окончании этого периода реакционную смесь охлаждали до rt и отфильтровывали, получая сырой продукт. Этот сырой продукт очищали методом prep-HPLC(H2O/CH3CN/TFA, 20-100% В, колонка Luna 30100), получая соединение по примеру 3 (30 мг, 48%) в виде масла (соль TFA).LC/MS (m/z)=310 (М+Н)+. Степень чистоты по данным HPLC составляла 95%. Пример 3 В. 3-(1-(4-Хлорфенил)циклопропил-8-(проп-1-ен-2-ил-[1,2,4]триазоло[4,3-а]пиридин К суспензии соединения 1F (15,1 г, 43,6 ммоль, 1 экв.) в толуоле (135 мл) добавляли фосфорилхлорид (21 мл, 224 ммоль, 5,1 экв.) в течение 5 мин. После окончания добавления реакционную смесь нагревали до 95 С и выдерживали при этой температуре в течение 22 ч. Затем реакционную смесь концентрировали путем вакуумной перегонки до объема равного 45 мл. К оставшемуся раствору добавляли ацетонитрил (150 мл). Полученный раствор концентрировали путем перегонки при атмосферном давлении до минимального объема и затем добавляли ацетонитрил (60 мл) до конечного объема около 105 мл. Полученный органический раствор охлаждали до температуры около 5 С и для получения суспензии добавляли раствор карбоната калия (водный, 13,3 г, 95,3 ммоль, 2,2 экв.) в воде (260 мл). Суспензию нагревали до температуры около 20 С и при этой температуре перемешивали в течение 18 ч. Потом суспензию отфильтровывали. Остаток на фильтре промывали водой (75 мл) и высушивали под вакуумом в течение примерно 3 ч, получая сырой продукт (12,1 г). Сырой продукт добавляли в колбу, затем добавляли толуол (50 мл). Полученную смесь перемешивали в течение 20 мин для полного растворения. Затем к раствору в течение 15 мин добавляли смесь концентрированной соляной кислоты (4,3 мл, 50,1 ммоль, 1,2 экв.) и изопропилового спирта (15 мл). Полученную суспензию гидрохлоридной соли перемешивали при температуре около 20 С в течение 18 ч. Полученное вещество отфильтровывали, промывали толуолом (25 мл) и затем сушили под вакуумом в течение 3 ч, получая соединение по примеру 3 В в виде гидрохлоридной соли (11,2 г). Гидрохлоридную соль помещали в колбу и затем добавляли ацетонитрил (45 мл) и воду (45 мл). Полученную смесь перемешивали в течение 5 мин для полного растворения и затем охлаждали до 5 С. При этой температуре в течение 15 мин добавляли раствор карбоната калия (7 г, 50,1 ммоль, 1,2 экв.) и воду (90 мл). Полученную суспензию перемешивали в течение 30 мин при комнатной температуре. Полученное твердое веще- 27019706 ство выделяли путем фильтрования, промывали водой (60 мл) и затем сушили под вакуумом при комнатной температуре в течение 48 ч с получением соединения по примеру 3 В в виде свободного основания (9,5 г). Свободное основание (9,5 г) растворяли в ацетонитриле (45 мл) при температуре 40 С. После окончания растворения раствор охлаждали до 20 С и добавляли воду (90 мл, 6 об.). После окончания добавления полученную суспензию перемешивали в течение 3 ч. Затем полученное твердое вещество отфильтровывали, промывали водой (45 мл) и затем высушивали под вакуумом при 70 С в течение примерно 18 ч с получением перекристаллизованного продукта по примеру 3 В (выход: 8,5 г, 63%). Степень чистоты 99,61%, точка плавления 128,9 С. ИК (KBr) 3099, 3025, 1630, 1601, 1484, 1451, 1368, 1325, 1107, 1091, 1044, 1007, 924, 916, 749, 675,531 см-1. 1 Н NMR (400 MHz, CDC13):1.47-1.60 (m, 2 Н), 1.66-1.78 (m, 2 Н), 2.30 (s, 3H), 5.63 (br s, 1H), 6.75 (t,J=6.9 Hz, 1H), 6.78 (s, 1H), 7.18 (s, 1H), 7.16-7.27 (m, 3H), 6.99-7.10 (m, 2H), 7.74 (d, J=6.1, 1H). 13 К раствору хлорида цинка (274 мг, 2,0 ммоль) в THF (4 мл) добавляли 2-(1,3-диоксан-2 ил)этилмагнийбромид (8,8 мл, 4,4 ммоль, 0,5 М ЕРА) с получением бис-(2-(1,3-диоксан-2-ил)этил)цинка. В отдельном реакционном сосуде при rt в течение 3 ч перемешивали смесь соединения 1 С (348,6 мг, 1,0 ммоль) и карбоната калия (276 мг, 2,0 ммоль) в DMF (5 мл). Затем через эту смесь пропускали аргон в течение 5 мин и затем добавляли бис-(2-(1,3-диоксан-2-ил)этилцинк и Pd(dppf)Cl2[CH2Cl2] (81,7 мг, 0,10 ммоль). После окончания добавления реакционный сосуд промывали аргоном, закрывали и затем нагревали при 90 С в течение 16 ч. Затем реакционную смесь распределяли между диэтиловым эфиром и водой и энергично перемешивали в течение 15 мин. Органическую фазу отделяли, сушили над Na2SO4 и концентрировали под вакуумом, получая остаток. Остаток очищали методом флэш-хроматографии (SiO2,0-100% этилацетата/гексанов) с последующей очисткой методом prep-HPLC (колонка Phenomenex AxiaLuna (30100 мм); 0-100% В в течение 15 мин, затем 3 мин выдержки в В со скоростью истечения 40 мл/мин; растворитель А=10% MeCN, 90% Н 2 О; растворитель В=90% MeCN, 10% Н 2 О), получали соединение 4 А (289 мг, 75%) в виде бледно-желтой пены. К раствору соединения 4 А (96,0 мг, 0,25 ммоль) в ацетоне (2,5 мл) добавляли серную кислоту (111 мкл, 1,0 ммоль, 9 М). Полученную смесь нагревали с обратным холодильником и перемешивали в течение 2 ч. Затем реакционную смесь распределяли между этилацетатом и 50%-ным насыщенным водным раствором бикарбоната натрия и затем добавляли твердый хлористый натрий до насыщения водной фазы. Органическую фазу отделяли, высушивали над Na2SO4 и концентрировали под вакуумом с получением остатка. Этот остаток очищали методом флэш-хроматографии (SiO2, 0-100% смеси этилацетат/гексаны), получали соединение 4 В (95,6 мг, 73%) в виде пены неправильного белого цвета. Пример 4. При 0 С к раствору соединения 4 В (32,6 мг, 0,1 ммоль) в THF (1 мл) добавляли одной порцией боргидрид натрия (3,9 мг, 0,1 ммоль). После окончания добавления реакционную смесь перемешивали в течение 1 ч. Затем добавляли 50%-ный насыщенный водный раствор хлорида аммония (1 мл) и полученную смесь энергично перемешивали в течение 1 ч. Затем добавляли этилацетат (2 мл) и органический слой отделяли, сушили над Na2SO4 и концентрировали под вакуумом, получая остаток. Этот остаток очищали методом препаративной HPLC (колонка Phenomenex Axia Luna (30100 мм); 0-100% В в течение 15 мин, затем выдержка в В со скоростью истечения 40 мл/мин; растворитель А=10% MeCN, 90% Н 2 О с 0,1% TFA; растворитель В=90% MeCN, 10% Н 2 О с 0,1% TFA), отбирали нужные фракции, промывали насыщенным водным раствором NaHCO3 и концентрировали с получением соединения по примеру 4 (9,5 мг, выход 29%) в виде бледно-желтой пены.LC/MS (m/z)=384 (М+Н)+. Степень чистоты по данным HPLC95%. Пример 5. Метил-2-(3-(1-(4-хлорфенил)циклобутил)-[1,2,4]триазоло[4,3-а]пиридин-8-ил)-2-метилпропионат В сосуд, облучаемый микроволнами, добавляли 8-бром-3-(1-(4-хлорфенил)циклобутил)[1,2,4]триазоло[4,3-а]пиридин (50 мг, 0,14 ммоль) (получен, как в примере 1), ZnF2 (7,2 мг, 0,069 ммоль) и Pd(dba)2 (7,9 мг, 0,014 ммоль). Сосуд промывали аргоном и затем добавляли P(tBu)3 (28 мкл, 1,0 М,0,028 ммоль), (1-метокси-2-метилпроп-I-енилокси)триметилсилан (36 мг, 0,207 ммоль) и DMF (1 мл). Сосуд закрывали и нагревали в течение 16 ч при температуре 80 С. Затем реакционную смесь охлаждали до комнатной температуры и распределяли между этилацетатом/Et2O (1:1) и водой. Органическую фазу отделяли, сушили над Na2SO4 и концентрировали под вакуумом, получая остаток. Остаток очищали методом prep-HPLC (колонка Phenomenex Axia Luna (30100 мм); 50-70% В в течение 15 мин, затем выдержка в В, 40 мл/мин; растворитель А=10% МеОН, 90% Н 2 О, 0,1% TFA; растворитель В=90% МеОН,10% H2O, 0,1% TFA). Фракции, содержащие полученный продукт, нейтрализовали пропусканием (сила тяжести) через картридж с бикарбонатом (PolymerLabs, PL-НСО 3 МР-смола, 0,36 ммоль, один картридж в цилиндре объемом 18 мл) и затем концентрировали, получая соединение по примеру 5 (16 мг, 30%) в виде белой пены.LC/MS (m/z)=384 (М+Н)+. Степень чистоты по данным HPLC составляла 95%. Пример 6. 2-(3-(1-(4-Хлорфенил)циклопропил)-[1,2,4]триазоло[1,5-а]пиридин-7-ил)пропан-2-ол В сухую круглодонную колбу объемом 250 мл добавляли 15 мл THF и 2,5 М н-BuLi (8,4 мл, 21,1 ммоль). Колбу охлаждали до -78 С и по каплям добавляли в нее раствор 2,6-дибромпиридина (5 г, 21,1 ммоль) в 40 мл THF через воронку в атмосфере азота. После окончания добавления смесь перемешивали еще 15 мин при температуре -78 С. К полученному темно-зеленому раствору при -78 С добавляли 1-(4 хлорфенил)циклопропанкарбонитрил (4,5 г, 25,3 ммоль) в течение 1 мин. Затем давали реакционной смеси нагреться до комнатной температуры и добавляли в нее 6 N HCl (27,5 мл, 165 ммоль) и нагревали с обратным холодильником в течение 10 мин с последующим перемешиванием при комнатной температуре в течение 1,5 ч. Затем подщелачивали раствор путем добавления 1 N NaOH при температуре равной 0 С. Органический слой отделяли и экстрагировали водный слой EtOAc (350 мл). Объединенные органические слои высушивали (MgSO4) и концентрировали под вакуумом, получая 8,7 г сырого продукта в виде оранжевого масла, которое очищали методом флэш-хроматографии над 330 г силикагеля (элюировали смесью гексаны:EtOAc 95:5), получали соединение 6 А (3,3 г, выход 47%) в виде твердого вещества белого цвета. 1 Н NMR:7.62 (d, J=8 Hz, 1 Н), 7.47 (t, J=8 Hz, 1H), 7.37 (d, J=8 Hz, 1H), 7.24 (d, J=8 Hz, 2H), 7.13 (d,J=8 Hz, 2H), 1.79-1.76 (m, 2H), 1.30-1.28 (m, 2H).
МПК / Метки
МПК: A61P 3/10, A61K 31/437, A61P 19/02, C07D 471/04
Метки: ингибиторы, типа, 11-бета-гидроксистероид-дегидрогеназы, триазолпиридиновые
Код ссылки
<a href="https://eas.patents.su/30-19706-triazolpiridinovye-ingibitory-11-beta-gidroksisteroid-degidrogenazy-tipa-i.html" rel="bookmark" title="База патентов Евразийского Союза">Триазолпиридиновые ингибиторы 11-бета-гидроксистероид-дегидрогеназы типа i</a>
Ингибиторы 11-бета-гидроксистероид-дегидрогеназы типа 1

Номер патента: 16415
Опубликовано: 30.04.2012
Авторы: Уоллэйс Оуэн Брендан, Снайдер Нэнси Джун, Красутский Алексей Павлович, Сюй Яньпин, Йорк Джереми Шуленбург
МПК: C07D 207/27, A61K 31/4015, A61P 3/00...
Метки: ингибиторы, типа, 11-бета-гидроксистероид-дегидрогеназы
Формула / Реферат:
1. Соединение, представленное структурной формулойгде R0a представляет собой -галоген;R0b представляет собой -Н или -галоген;R1 представляет собой -Н, -галоген;R2 представляет собой -Н, -галоген;R3 представляет собой -Н;R4 представляет собой -ОН, -галоген, -(C1-С6)алкокси (необязательно замещенный от одного до трех атомами галогена),где пунктирная линия представляет собой место присоединения к позиции R4;R5 представляет собой -Н, -галоген,...
Ингибиторы 11-бета-гидроксистероид дегидрогеназы типа 1

Номер патента: 15499
Опубликовано: 31.08.2011
Авторы: Мэбри Томас Эдвард, Сюй Яньпин, Виннероски Леонард Лэрри, Снайдер Нэнси Джун, Уоллэйс Оуэн Брендан
МПК: C07D 209/44, A61P 3/10, A61K 31/4015...
Метки: дегидрогеназы, типа, 11-бета-гидроксистероид, ингибиторы
Формула / Реферат:
1. Соединение, представленное структурной формулойили его фармацевтически приемлемая соль,где Ra представляет собой -Н или -ОН;Rb представляет собой -Н илиRa и Rb в комбинации с циклогексильным кольцом, к которому они присоединены, образуютгде звездочка представляет собой атом углерода, который является общим с лактамным кольцом;R1 представляет собой -Н или -галоген;R2 представляет собой -Н или -галоген;R3 представляет собой -Н или -галоген;R4...
Ингибиторы 11-бета-гидроксистероид дегидрогеназы 1

Номер патента: 15106
Опубликовано: 30.06.2011
Авторы: Ли Жэньхуа, Уоллэйс Оуэн Брендан, Снайдер Нэнси Джун, Хансен Марвин Мартин, Мэбри Томас Эдвард, Буш Джули Кей, Сюй Яньпин
МПК: A61P 3/10, A61K 31/4025, A61K 31/4015...
Метки: дегидрогеназы, 11-бета-гидроксистероид, ингибиторы
Формула / Реферат:
1. Соединение структурной формулыгде R0 представляет собойгде пунктирная линия указывает место присоединения к положению R0;R1 представляет собой -галоген;R2 представляет собой -Н, -галоген;R3 представляет собой -Н;R4 представляет собой -ОН,где пунктирная линия обозначает место присоединения к положению R4;R5 представляет собой -Н, -галоген, -ОН, -(C1-C4) алкил, замещенный 1-3 атомами галогена, -SO2-(С1-С4) алкил,где пунктирная линия обозначает...
Ингибиторы 11-бета-гидроксистероид дегидрогеназы 1

Номер патента: 15516
Опубликовано: 31.08.2011
Авторы: Сюй Яньпин, Уоллэйс Оуэн Брендан, Мэбри Томас Эдвард, Виннероски Леонард Лэрри, Ли Жэньхуа
МПК: A61K 31/4025, A61P 3/10, A61K 31/4015...
Метки: ингибиторы, дегидрогеназы, 11-бета-гидроксистероид
Формула / Реферат:
1. Соединение, имеющее структурную формулугде R0 представляет собойпри этом пунктирная линия обозначает точку присоединения к положению R0;причем Ra представляет собой -H или -галоген; Rb представляет собой -H или галоген; Rc представляет собой -H;R1 представляет собой -галоген;R2 представляет собой -галоген;R3 представляет собой -H;R4 представляет собой -OH, -галоген, -(C1-C6)алкокси илигде пунктирная линия обозначаем точку присоединения к...
Ингибиторы 11-бета-гидроксистероид дегидрогеназы 1, фармацевтическая композиция на их основе и их применение

Номер патента: 16360
Опубликовано: 30.04.2012
Авторы: Сюй Яньпин, Уоллэйс Оуэн Брендан, Ли Жэньхуа
МПК: A61K 31/4545, C07D 401/14, A61P 3/10...
Метки: дегидрогеназы, основе, композиция, 11-бета-гидроксистероид, фармацевтическая, ингибиторы, применение
Формула / Реферат:
1. Соединение структурной формулыили его фармацевтически приемлемая соль,где R1 представляет собой -H;R2 представляет собой -галоген;R3 представляет собой -галоген;R4 представляет собой -H;R5 представляет собойгде пунктирной линией обозначена точка присоединения к положению заместителя, обозначенного R5;где n равно 1;R9 представляет собой -OH или -CH3 (необязательно замещенный 1-3 атомами галогена);R10 представляет собой -H иR22 представляет...
Предыдущий патент: Пищевая композиция, обладающая диетическими и профилактическими свойствами (варианты), и включающие ее функциональные пищевые продукты, в том числе напитки
Следующий патент: Производные 7-азаиндола в качестве селективных ингибиторов 11-бета-гидроксистероида дегидрогеназы 1 типа
Случайный патент: Способ получения частицеобразующих композиций, содержащих конденсированные пирролокарбазолы