Производные 7-азаиндола в качестве селективных ингибиторов 11-бета-гидроксистероида дегидрогеназы 1 типа
Номер патента: 19707
Опубликовано: 30.05.2014
Авторы: Шультц Мелани, Роше Дидье, Аллаку-Бозек Софи, Карниато Денис







Формула / Реферат
1. Соединение формулы I

в которой R1, R2 представляют собой независимо друг от друга Н, А, С3-8циклоалкил;
R3, R4 представляют собой Н;
R5, R6 представляют собой Н;
X представляет собой -(С)m- или -S(O)2-;
Y представляет собой С3-8циклоалкил, фенилокси, фенил, необязательно моно-, ди- или тризамещенный Hal, А, С1-4алкилокси;
А представляет собой С1-6алкил;
n имеет значение 1 или 2;
m имеет значение 0 или 1,
и его физиологически приемлемые соли.
2. Соединение, выбранное из группы, состоящей из следующего:
а) (2-фторофенил)-[3-(1Н-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]метанон;
b) (4-метокси-2-метилфенил)-[3-(1H-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]метанон;
c) (циклогексил)-[3-(1H-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]метанон;
d) (пиридин-3-ил)-[3-(1H-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]метанон;
e) [3-(1Н-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]-о-толилметанон;
f) (2-метил-2-фенил-1)-[3-(1Н-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]пропан-1-он;
g) (4-диметиламинофенил)-[3-(1H-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]метанон;
h) (1-фенилциклопропил)-[3-(1Н-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]метанон;
i) 2-(4-хлорфенил)-2-метил-1-[3-(1Н-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]пропан-1-он;
j) 2-метил-2-фенокси-1-[3-(1H-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]пропан-1-он;
k) 1-(4-хлорофенил)циклобутил-[3-(1H-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]метанон;
l) 2-(4-хлорофенокси)-2-метил-1-[4-(1Н-пирроло[2,3-b]пиридин-3-ил)пиперидин-1-ил]пропан-1-он;
m) 2-метил-1-[4-(1Н-пирроло[2,3-b]пиридин-3-ил)пиперидин-1-ил]-2-[4-(5-трифторметилпиридин-2-ил)пиперазин-1-ил]пропан-1-он;
n) 4-(4-фторофенокси)-3,3-диметил-1-[4-(1Н-пирроло[2,3-b]пиридин-3-ил)пиперидин-1-ил]бутан-1-он;
о) 2-(4-хлорофенил)-2-метил-1-[4-(1Н-пирроло[2,3-b]пиридин-3-ил)пиперидин-1-ил]пропан-1-он;
р) [1-(4-хлорофенил)циклопропил]-[4-(1Н-пирроло[2,3-b]пиридин-3-ил)пиперидин-1-ил]метанон;
q) 4-(4-фторофенокси)-3,3-диметил-1-[3-(1H-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]бутан-1-он;
r) [1-(4-фторофенокси)циклопропил]-[3-(1Н-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]метанон;
s) [1-(4-хлорофенил)циклопропил]-[3-(1H-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]метанон;
t) 2-(4-хлоробензолсульфонил)-1-[3-(1Н-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]этанон и
их физиологически приемлемые соли.
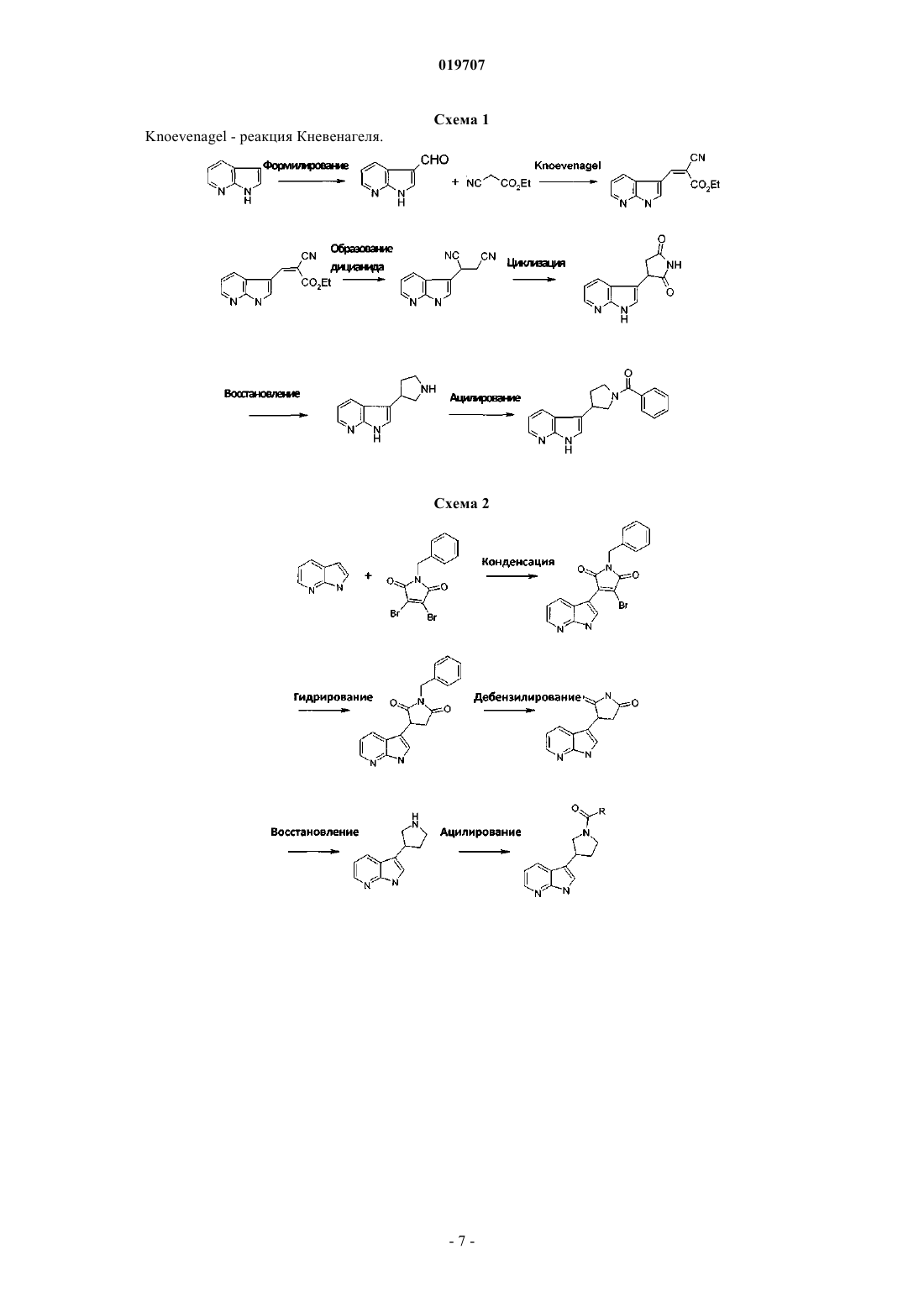
3. Способ получения соединения по п.1 или 2, который отличается тем, что азаиндол формулы II

в которой R5 и R6 являются такими, как определено выше,
а) формилируют для получения альдегида формулы III

в которой R5 и R6 являются такими, как определено выше,
указанный альдегид формулы III реагирует с этилцианоацетатом, что сопровождается реакцией Майкла с добавлением цианида, кислотной циклизацией и гидридным восстановлением для получения пирролидиноазаиндола формулы IV

в которой R5 и R6 являются такими, как определено выше, или
b) конденсируют с броммалеимидом VI

для получения пирролидиндиона формулы VII

в которой R5 и R6 являются такими, как определено выше,
гидрогенизация указанного пирролидиндиона формулы VII сопровождается последовательным снятием защитных бензиловых групп и гидридным восстановлением, приводит к получению пирролидиноазаиндола формулы IV

в которой R5 и R6 являются такими, как определено выше, или
с) реагирует в основной среде с кетоном VIII

где R3, R4 и n являются такими, как определено выше,
с получением смеси олефинов формул IX и X

в которых R3, R4, R5, R6 и n являются такими, как определено выше,
гидрогенизация указанных олефинов формул IX и X, в которых R3, R4, R5, R6 и n являются такими, как определено выше, сопровождается снятием защитных групп Boc и приводит к получению пирролидиноазаиндола формулы XI

в которой R3, R4, R5, R6 и n являются такими, как определено выше,
с последующим ацилированием полученных пирролидиноазаиндолов формул IV или XI, в которых R3, R4, R5, R6 и n являются такими, как определено выше, активированной карбоксильной кислотой формулы V

в которой R1, R2, X и Y являются такими, как определено выше,
для получения соединения формулы I, в которой R1, R2, R3, R4, R5, R6, X и Y являются такими, как определено выше, и при необходимости остатки X, Y, R1, R2, R3, R4, R5, R6, как заявлено в п.1, превращают в другие остатки X, Y, R1, R2, R3, R4, R5, R6, путем введения алкильной группы, или соединение формулы I выделяют и/или обрабатывают кислотой или основанием для получения соли.
4. Применение соединения по п.1 или 2 в качестве 11β-HSD1 ингибитора.
5. Применение соединения по п.1 или 2 для приготовления лекарственного средства, селективно ингибирующего энзим 11-бета-гидроксистероида дегидроксигеназы 1 типа.
6. Применение соединения по п.1 или 2 для приготовления лекарственного средства для лечения и/или предотвращения болезней, которые вызваны, опосредованы и/или репродуцированы высокими уровнями кортизола.
7. Применение соединения по п.1 или 2 для приготовления лекарственного средства для лечения и/или предотвращения одной или больше болезней или патологических состояний, выбранных из группы, которая состоит из следующего: метаболический синдром, сахарный диабет, особенно инсулиннезависимый сахарный диабет, преддиабет, резистентность к инсулину, низкая толерантность к глюкозе, гипергликемия, ожирение и связанные с весом нарушения, расстройства липидного обмена, такие как дислипидемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, низкие уровни HDL или высокие уровни LDL, глаукома, остеопороз, опосредованные глюкокортикоидом эффекты на нейронную функцию, такие как нарушение когнитивных функций, беспокойство или депрессия, нейродегенеративная болезнь, иммунные расстройства, такие как туберкулез, проказа или псориаз, артериальная гипертензия, атеросклероз и его осложнения, васкулярный рестеноз, сердечно-сосудистые болезни, панкреатит, ретинопатия, невропатия и нефропатия.
8. Фармацевтический состав, селективно ингибирующий энзим 11-бета-гидроксистероида дегидроксигеназы 1 типа, который отличается тем, что он содержит терапевтически эффективное количество одного или более соединений по п.1 или 2 и одно или более дополнительных соединений, выбранных из группы, состоящей из физиологически приемлемых наполнителей, вспомогательных веществ, разжижителей, носителей.
9. Способ получения фармацевтических составов, селективно ингибирующих энзим 11-бета-гидроксистероида дегидроксигеназы 1 типа, который отличается тем, что одно или больше соединений по п.1 или 2 и одно или более соединений, выбранных из группы, состоящей из твердых, жидких или полужидких наполнителей, вспомогательных средств, разжижителей, носителей, являются преобразованными в подходящую форму дозировки.
Текст
ПРОИЗВОДНЫЕ 7-АЗАИНДОЛА В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ 11 БЕТА-ГИДРОКСИСТЕРОИДА ДЕГИДРОГЕНАЗЫ 1 ТИПА Настоящее изобретение относится к производным 7-азаиндола формулы I в качестве селективных ингибиторов энзима 11-бета-гидроксистероида дегидрогеназы типа 1(11-HSD-1) и использованию таких соединений для лечения и предотвращения метаболического синдрома, сахарного диабета, резистентности к инсулину, ожирения, расстройства липидного обмена, глаукомы, остеопороза, когнитивных расстройств, беспокойства, депрессии, иммунных расстройств, артериальной гипертензии и других болезней и патологических состояний.(71)(73) Заявитель и патентовладелец: МЕРК ПАТЕНТ ГМБХ (DE) Область техники изобретения Настоящее изобретение относится к производным 7-азаиндола в качестве селективных ингибиторов энзима 11-бета-гидроксистероида дегидрогеназы 1 типа (11-HSD-1) и к применению таких соединений для лечения и предотвращения метаболического синдрома, сахарного диабета, резистентности к инсулину, ожирения, расстройства липидного обмена, глаукомы, остеопороза, когнитивных расстройств, беспокойства, депрессии, иммунологических нарушений, артериальной гипертензии и других заболеваний и состояний. Уровень техники изобретения Гидроксистероиды дегидрогеназы (HSDs) регулируют удерживание и активацию рецепторов стероидных гормонов путем превращения стероидных гормонов в их пассивные метаболиты. Из недавнего обзора см. Nobel et al., Eur. J. Biochem. 2001, 268: 4113-4125. Там существуют многочисленные классы HSDs. 11-Бета-гидроксистероиды дегидрогеназы (11HSDs) катализируют взаимопревращение активных глюкокортикоидов (таких как кортизол и кортикостерон) и их инертных форм (таких как кортизон и 11-дегидрокортикостерон). Изоформ 11-бетагидроксистероид дегидрогеназы 1 типа (11-HSD1) широко экспрессирован в печени, жировой ткани,головном мозге, легком и другой глюкокортикоидной ткани, в то время как экспрессия изоформа 2 (11HSD2) ограничена тканями, которые экспрессируют минералокортикоидный рецептор, такими как почка, кишка и плацента. В таком случае ингибирование 11-HSD2 связано с серьезными побочными эффектами, такими как артериальная гипертензия. Избыток кортизола связан с многочисленными нарушениями активности, включая диабет, ожирение, дислипидемию, резистентность к инсулину и артериальную гипертензию. Введение 11-HSD1 ингибиторов уменьшает уровень кортизола и других 11-гидроксистероидов в целевых тканях, таким образом, уменьшая эффекты избыточного количества кортизола и других 11-гидроксистероидов. Таким образом, 11-HSD1 представляет собой потенциальную цель для терапии, связанную с многочисленными нарушениями активности, которые могут быть улучшены путем ослабления глюкокортикоидного действия. Таким образом, ингибирование 11-HSD1 может быть использовано для предотвращения, лечения или регуляции болезней, опосредованных чрезвычайно высокими уровнями кортизола и других 11 гидроксистероидов, таких как диабет, ожирение, артериальная гипертензия или дислипидемия. Ингибирование 11-HSD1 активности в головном мозге, к примеру, до пониженных уровней кортизола может также быть полезным для лечения или ослабления беспокойства, депрессии, когнитивного расстройства или возрастной когнитивной дисфункции (Seckl et al., Endocrinology, 2001, 142: 1371-1376). Кортизол представляет собой важный и хорошо распознаваемый противовоспалительный гормон,который также действует как антагонист на действие инсулина в печени так, чтобы чувствительность к инсулину была уменьшена, приводя к повышенному глюконеогенезу и повышенным уровням глюкозы в печени. Пациенты, которые уже имеют нарушенную толерантность к глюкозе, имеют большую вероятность развивающегося диабета 2 типа в присутствии чрезвычайно высоких уровней кортизола (Long etal., J. Exp. Med. 1936, 63: 465-490; Houssay, Endocrinology 1942, 30: 884-892). Кроме того, было хорошо доказано, что 11-HSD1 играет важную роль в регулировании локального глюкокортикоидного эффекта и выработке глюкозы в печени (Jamieson et al., J. Endocrinol. 2000, 165: 685-692). В Walker, et al., J. Clin.Endocrinol. Metab. 1995, 80: 3155-3159, сообщалось, что введение неспецифичного 11-HSD1 ингибитора карбеноксолона привело к улучшенной печеночной чувствительности к инсулину у людей. Кроме того, предполагаемый механизм действия 11-HSD1 в лечении диабета был основан на различных экспериментах, которые проводились на мышах и крысах. Эти исследования показали, что уровни mRNA и активности двух ключевых энзимов в печеночной выработке глюкозы, фосфоенолпируват карбоксикиназа (PEPCK) и глюкозо-6-фосфатаза (G6Pase), были уменьшены после введения 11-HSD1 ингибиторов. Кроме того, уровни глюкозы крови и печеночная выработка глюкозы, как было показано,были уменьшены у 11-HSD1 нокаутных мышей. Дополнительные данные, собранные с применением этой модели мышиного нокаута, также подтверждают, что ингибирование 11-HSD1 не будет вызывать гипогликемию, так как основные уровни PEPCK и G6Pase являются отрегулированными независимо от глюкокортикоидов (Kotelevtsev et al., Proc. Natl. Acad. Sci. USA 1997, 94: 14924-14929). Таким образом, введение терапевтически эффективного количества 11-HSD1 ингибитора является эффективным при лечении, контроле и улучшении симптомов диабета, особенно неинсулинзависимого диабета (NIDDM, сахарный диабет 2 типа), и введение терапевтически эффективного количества 11HSD1 ингибитора на регулярной основе приостанавливает или предотвращает приступ диабета, особенно у людей. Эффект повышенных уровней кортизола также наблюдается у пациентов, которые имеют синдром Кушинга, который представляет собой нарушение обмена веществ, характеризующееся высокими уровнями кортизола в кровотоке. У пациентов с синдромом Кушинга часто развивается NIDDM. Чрезмерные уровни кортизола были связаны с ожирением, возможно из-за увеличенного печеночного глюконеогенеза. Брюшное ожирение тесно связано с нарушением толерантности к глюкозе, диабе-1 019707 том, гиперинсулинемией, гипертриглицеридемией и другими факторами метаболического синдрома, такими как высокое кровяное давление, повышение VLDL и понижение HDL (Montague et al., Diabetes,2000, 49: 883-888). У субъектов, страдающих ожирением, 11-HSD-1 активность в жировой ткани заметно увеличена и положительно коррелирована с массой тела. Было также сообщено, что ингибирование 11-HSD1 в преадипоцитах (стромальные клетки) привело к снижению нормы дифференцировки в адипоцитах. Прогнозировано, что это в результате приведет к уменьшенному увеличению в объеме (возможному сокращению) сальниковых жировых отложений, что может привести к уменьшенному центральному ожирению (Bujalska et al., Lancet 1997, 349: 1210-1213). Таким образом, введение эффективного количества 11-HSD1 ингибитора полезно при лечении или борьбе с ожирением. Долгосрочное лечение 11-HSD1 ингибитором также полезно при отсрочке или предотвращении симптомов ожирения, особенно если пациент использует 11-HSD1 ингибитор в комбинации с контролируемой диетой и упражнением. Уменьшая резистентность к инсулину и поддерживая глюкозную сыворотку при нормальных концентрациях, соединения настоящего изобретения также имеют пользу в лечении и предотвращении заболеваний, которые сопровождаются диабетом 2 типа и резистентностью к инсулину, включая метаболический синдром, ожирение, реактивную гипогликемию и диабетическую дислипидемию. Ингибирование 11-HSD1 в зрелых адипоцитах, как ожидалось, снижает секрецию ингибитора 1 активатора плазминогеля (PAI-1), который представляет собой независимый сердечно-сосудистый фактор риска, как сообщается в Halleux et al., J. Clin. Endocrinol. Metab. 1999, 84: 4097-4105. Кроме того, корреляция, как было показано, существовала между глюкокортикоидной активностью и определенными сердечно-сосудистыми факторами риска. Это предполагает, что сокращение глюкокортикоидных эффектов было бы выгодно в лечении или предотвращении определенных сердечно-сосудистых болезней(Walker et al., Hypertension 1998, 31: 891-895; и Fraser et al., Hypertension 1999, 33: 1364-1368). Так как артериальная гипертензия и дислипидемия способствуют развитию атеросклероза и ингибирование 11-HSD1 активности и сокращение количества кортизола выгодно в лечении или контролировании артериальной гипертензии, введение терапевтически эффективного количества 11-HSD1 ингибитора настоящего изобретения может также быть особенно выгодным в лечении, контролировании или замедлении симптома или предотвращении атеросклероза. 11-HSD1 был также привлечен в процессе контроля аппетита и поэтому, как полагают, играет дополнительную роль в связанных с весом заболеваниях. Известно, что удаление надпочечной железы уменьшает эффект голодания для увеличения как приема пищи, так и экспрессии гипоталамического нейропептида Y. Это означает, что глюкокортикоиды играют определенную роль в активировании приема пищи и что ингибирование 11-HSD1 в головном мозге может увеличить насыщение, что приведет в результате к уменьшенному рациону питания (Woods et al., Science, 1998, 280: 1378-1383). Другим возможным терапевтическим эффектом, связанным с модуляцией 11-HSD1, является тот,который связан с различными панкреатическими продуктами питания. Сообщается, что ингибирование 11-HSD1 в мышиных панкреатических -клетках, увеличение глюкозы стимулировало секрецию инсулина (Davani et al., J. Biol. Chem. 2000, 275: 34841-34844). Из предыдущего открытия следует, что глюкокортикоиды, как ранее было найдено, были ответственны за уменьшенную панкреатическую секрецию инсулина in vivo (Billaudel et al., Horm. Metab. Res. 1979, 11: 555-560). Таким образом, было предложено,что ингибирование 11-HSD1 привело бы к другим благоприятным воздействиям в лечении диабета кроме предполагаемых эффектов на печень и уменьшении жира. Чрезмерные уровни кортизола в головном мозге могут также привести к нейронной потере или дисфункции посредством потенцирования нейротоксинов. Введение эффективного количества 11-HSD1 ингибитора приводит к уменьшению, улучшению, контролю или предотвращению когнитивного нарушения, связанного со старением и нейронной дисфункцией. Когнитивное нарушение было связано со старением и излишними уровнями кортизола в головном мозге (см. J.R. Seckl и В.R. Walker, Endocrinology, 2001, 142: 1371-1376 и ссылки, приведенные там). 11-HSD1 также регулирует глюкокортикоидную активность в головном мозге и, таким образом, способствует нейротоксичности (Rajan et al., Neuroscience 1996, 16: 65- 70; Seckl et al., Necroendocrinol. 2000, 18: 49-99). Нагрузка и/или глюкокортикоиды, как известно, влияют на когнитивную функцию (de Quervain et al., Nature 1998, 394: 787-790), и неопубликованные результаты указывают на значительное улучшение памяти у крыс, которых лечили неспецифическим 11-HSD1 ингибитором. Эти сообщения, в дополнение к известным эффектам глюкокортикоидов в головном мозге, предполагают, что ингибирование 11-HSD1 в головном мозге может иметь положительный терапевтический эффект против беспокойства, депрессии и связанных с этим патологических состояний (Tronche et al., Nature Genetics 1999, 23: 99-103). 11-HSD1 реактивирует 11 дегидрокортикостерон к кортикостерону в гиппокампальных клетках и может усиливать действие нейротоксичности киназы, приводя к возрастным ухудшениям обучения. Таким образом, селективные ингибиторы 11-HSD1, как полагают, защищают против гиппокампального снижения функции с возрастом ингибирование 11-HSD1 в человеческом головном мозге защитит от вредных опосредованных глюкокортикоидом эффектов на нейронную функцию, таких как когнитивные нарушения, депрессия и повышенный аппетит. Кроме того, 11-HSD1, как полагают, играет роль в иммуномодуляции, основанную на общем восприятии, что глюкокортикоиды подавляют иммунную систему. Существует, как известно, динамическое взаимодействие между иммунной системой и НРА (гипоталамической гипофизарной надпочечниковой) системой (Rook, Baillier's Clin. Endocrinol. Metab. 2000, 13: 576-581), и глюкокортикоиды помогают балансировать между клеточно-опосредованными реакциями и гуморальными реакциями. Увеличенная глюкокортикоидная активность, которая может быть вызвана нагрузкой, связана с гуморальной реакцией и как таковое, ингибирование 11-HSD1 может привести к перемене реакции на основанную на клетке реакцию. На определенных стадиях заболевания, таких как туберкулез, проказа и псориаз, и даже при условиях чрезмерного напряжения, высокая глюкокортикоидная активность меняет иммунную реакцию на гуморальную реакцию, в то время когда фактически основанная на клетке реакция может быть более выгодной для пациента. Ингибирование 11-HSD1 активности и сопутствующее сокращение глюкокортикоидных уровней с другой стороны меняет иммунную реакцию на основанную на клетке реакцию (D. Mason, Immunology Today, 1991, 12: 57-60, и G.A.Vt. Rook, Baillier's Clin. Endocrinol. Metab., 1999, 13: 576-581). Из этого следует, что альтернативной пользой 11-HSD1 ингибирования было бы поддержание временной иммунной реакции в сочетании с иммунизацией, чтобы гарантировать,что будет получена основанная на клетке реакция. Последние описания предполагают, что уровни глюкокортикоидных целевых рецепторов и HSDs связаны с восприимчивостью к глаукоме (J. Stokes et al., Invest. Ophthalmol. 2000, 41: 1629-1638). Кроме того, недавно было сообщено о связи между ингибированием 11-HSD1 и понижением внутриглазного давления (Walker et al., poster P3-698 at the Endocrine society meeting June 12-15, 1999, San Diego). Было показано, что введение неспецифического 11-HSD1 ингибитора карбеноксолона привело к сокращению внутриглазного давления на 20% у нормальных пациентов. В глазу 11-HSD1 экспрессирован исключительно в базальных клетках роговичного эпителия, непигментированном эпителии роговой оболочки(участок выработки воды), ресничном мускуле, запирательной мышце и расширяющей мышце мускулов радужной оболочки. В отличие от этого, отдаленный изоэнзим 11-гидроксистероид дегидрогеназы 2 типа("11-HSD2") является чрезвычайно экспрессированным в непигментированном ресничном эпителии и роговичном эндотелии. Никакие HSDs не были найдены в трабекулярной сети, которая представляет собой участок оттока. Таким образом, было выдвинуто предположение, что 11-HSD1 играет роль в выработке воды, и ингибирование 11-HSD1 активности полезно в сокращении внутриглазного давления при лечении глаукомы. Глюкокортикоиды также играют существенную роль в скелетном развитии и функционировании,но вредны для такого развития и функционирования, когда присутствуют в избытке. Вызванная глюкокортикоидом потеря костной массы частично получена из ослабления полиферации остеобласта и синтеза коллагена, как сообщается в С.Н. Kim et al., J. Endocrinol. 1999, 162: 371-379. Было сообщено, что вредные действия глюкокортикоидов на формирование костных узлов могут быть уменьшены введением карбеноксолона, который представляет собой неспецифический 11-HSD1 ингибитор (С.G. Bellows et al.,Bone 1998, 23: 119-125). Дополнительные сообщения предполагают, что 11-HSD1 возможно является ответственным за обеспечение увеличенных уровней активного глюкокортикоида в остеокласте, и, таким образом, в увеличении резорбции кости (М.S. Cooper et al., Bone 2000, 27: 375-381). Эти данные предполагают, что ингибирование 11-HSD1 может иметь благоприятные действия против остеопороза посредством одного или более механизмов, которые могут действовать параллельно. Ингибиторы 11-HSD1 известны, к примеру, из WO 0410629, WO 03065983, WO 04089896, WO 04089380, WO 04065351, WO 04033427 или WO 04041264. В недавнем обозрении см. М. Wamil и J.R.in medicinal chemistry; 2006, 41, 127-140). Однако производные 7-азаиндола являются не раскрытыми в качестве активных 11-HSD1 ингибиторов. 3-Замещенные гетероциклоалкил-7-азаиндолы раскрыты, к примеру, в WO 2004106346 иWO 2004106298 в качестве связующих веществ допамина D2, D3 и рецепторов D4 и действующих в качестве 5-HT1A агонистов или частичных агонистов. 7-Азаиндолоны раскрыты в WO 2006099268, WO 2006044504, WO 2005013894 в качестве антагонистов для рецепторов CGRP. 5-Ацил-7-азаиндолы раскрыты в WO 2005085244 для ингибирования JNK3. 2-Арил и 2-гетероарил-7-азаиндолы заявлены в качестве ингибиторов Itk. Азаиндол-3-пиперидины раскрыты в WO 2003082867 в качестве антагонистов гистаминового H1 рецептора. Однако ни одна из вышеупомянутых публикаций не охватывает ни производные 7-азаиндола настоящего изобретения, ни применение раскрытых соединений в качестве 11-HSD1 ингибиторов.WO 2007050381 раскрывает определенные 3-(N-ацилпиперидин-4-ил)-7-азаиндолы в качестве модуляторов рецептора ORL-1. Соединения являются полезными для лечения, предотвращения или улучшения рецептора ORL-1 опосредованного нарушением. Таким образом, так как по-прежнему существует постоянная потребность в эффективной терапии,преимущественная задача настоящего изобретения состоит в том, чтобы обеспечить новые фармацевтически активные соединения для лечения болезней, таких как сахарный диабет, ожирение, глаукома, остеопороз, когнитивные расстройства, иммунные расстройства, депрессия, артериальная гипертензия и др. Цитата любой ссылки в этом описании не является допущением того, что ссылка представляет собой уровень техники этого описания. Краткое описание изобретения К удивлению было найдено, что соединения настоящего изобретения являются очень активными 11-HSD1 ингибиторами. Поэтому воплощением настоящего изобретения являются соединения формулы I в которой R1, R2 представляют собой независимо друг от друга Н, А, С 3-8 циклоалкил;m имеет значение 0 или 1,и их физиологически приемлемые соли. Другое особенно преимущественное воплощение настоящего изобретения представляет собой соединения согласно формуле I, выбранные из группы, состоящей из следующего:t) 2-(4-хлорбензолсульфонил)-1-[3-(1 Н-пиррол[2,3-b]пиридин-3-ил)пирролидин-1-ил]этанон и их физиологически приемлемые соли. Спецификация, которая используется здесь для того, чтобы определить соединения, особенно соединения согласно изобретению, в основном основана на правилах IUPAC-организации по химическим соединениям и в особенности органическим соединениям. Термин "гидроксил" означает ОН группу. Термины "алкил" или "А", так же как другие группы, которые имеют приставку "алк", такие как алкенил, алкокси и алканоил, означают углеродистые цепи, которые могут быть линейными или разветвленными, и их комбинации, если углеродистая цепь не определена иначе. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, сек- и трет-бутил, пентил, гексил, гептил, октил, нонил и т.п. В тех случаях, где указанное количество атомов углерода позволит, к примеру от C3-C10, термин"алкил" также включает в себя циклоалкильные группы и комбинации линейных или разветвленных алкильных цепей, объединенных с циклоалкильными структурами. В тех случаях, когда число атомов углерода не определено, C1-C6 является заданным. Особенно предпочтительный C1-C4-алкил. Радикал C1C4-алкил представляет собой, к примеру, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил."Циклоалкил" представляет собой подмножество алкила и понимается в значении насыщенного моноциклического углеводорода, относительно термина "С 3-9 циклоалкил" имеющий 3-9 атомов углерода. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил,циклооктил и т.п. Циклоалкильная группа в основном является моноцикличной, если не заявлено чтолибо другое. Циклоалкильные группы являются насыщенными, если как-либо иначе не определено. Радикал C4-C8-циклоалкил представляет собой, к примеру, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил. Термины "арил" или "Ar" означают моно- или многоциклическую систему ароматического ядра,содержащую атомы углеродистых циклов. Преимущественные арилы являются моноцикличными или бицикличными 6-10-членных систем ароматических ядер. Примеры "арильных" групп включают, но не ограничены следующими, фенил, 2-нафтил, 1-нафтил, бифенил, инданил, так же как их замещенные производные. Самым преимущественным арилом является фенил. Термин "алкилокси" означает алкоксильные группы прямой или разветвленной конфигурации. "C1C4-алкилокси" означает алкоксильные группы прямой или разветвленной конфигурации, которые имеют обозначенное число атомов углерода. Использующийся здесь термин "арилокси" предпочтительно относится к группе АО-, в которой А представляет собой арил, как определено выше. C1-C4-алкилокси представляет собой, к примеру, метокси, этокси, пропокси, изопропокси и т.п. Термин "арилокси" означает алкоксильные группы, моно- или полициклическую систему ароматического ядра, содержащую циклические атомы углерода. Использующийся здесь термин "арилокси" предпочтительно относится к группе ArO-, в которой Ar представляет собой арил, как определено выше. Примеры "арилокси" групп включают, но не ограничены следующими, фенилокси, 2-нафтилокси, 1 нафтилокси, бифенилокси, инданилокси, так же как их замещенные производные. Самой предпочтительной арилокси является фенилокси."Гетероарил" или "Het" означают ароматический или частично ароматическое гетероциклическое соединение, которое содержит по крайней мере одно ядро гетероатома, выбранного из О, S и N. Гетероарилы, таким образом, включают гетероарилы, конденсированные в другие виды ядер, такие как арилы,циклоалкилы и гетероциклические соединения, которые не являются ароматическими. Примеры гетероарильных групп включают пирролил, изоксазолил, изотиазолил, пиразолил, пиридил, оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фуранил, триазинил, тиенил, пиримидил, бензисоксазонил, бензоксазонил, бензотиазолил, бензотиадиазолил, дигидробензофуранил,индолинил, пиридазинил, индазолил, изоксазолил, изоиндолил, дигидробензотиенил, индолизинил,циннолинил, фталазинил, квиназолинил, нафтиридинил, карбазолил, бенздиоксинил, бензодиоксолил,квиноксалинил, пуринил, фуразанил, тиофенил, изобензилфуранил, бензимидазолил, бензофуранил,бензотиенил, квинолил, индолил, изоквинолил, дибензофуранил и т.п. Для гетероциклических и гетероарильных групп ядра и системы ядер, содержащие от 3-15 атомов, включены, формируя 1-3 ядра. Использующийся здесь термин "карбонил" или "карбонильная часть" предпочтительно относится к группе С=О. Использующийся здесь термин "алкилкарбонил" предпочтительно относится к группе АС(О)-, в которой А представляет собой алкил, как определено здесь. Использующийся здесь термин "алкоксикарбонил" или "алкилоксикарбонил" предпочтительно относится к группе АОС(О)-, в которой А представляет собой алкил, как определено здесь. Гетероциклоалкил представляет собой моно-, би- или трициклический углеводород, содержащий от 3 до 18 кольцевых атомов, предпочтительно от 3 до 7 ядровых атомов, и содержит один или больше,предпочтительно 1-3 гетероатомов, выбранных из О, N или S. Гетероциклоалкил представляет собой, к примеру, пирролидинил, пиперидинил, пиперазинил, морфолинил, индолинилметил, имидазолинилметил и 2-азабицикло[2.2.2]октанил. Использующийся здесь термин "алкоксиалкил" предпочтительно относится к группе АОА-, в которой А представляет собой алкил, как определено здесь. Алкоксиалкил относится к углеводородной цепи,прерванной атомом кислорода. Использующийся здесь термин "гетероарилокси" предпочтительно относится к группе HetO-, в которой Het представляет собой гетероарил, как определено здесь. Термин "Hal" относится к фтору (F), хлору (Cl), брому (Br) или йоду (I). Бром и фтор являются в основном предпочтительными. Фтор является более всего предпочтительным в тех случаях, когда галогены являются замещенными на алкильную (галогеналкильную) или алкоксильную группу (к примеру,CF3 и CF3O). Использующийся здесь термин "галогеналкил" предпочтительно относится к алкильной группе, как определено выше, содержащей по крайней мере один атом углерода, замещенный по крайней мере од-5 019707 ним галогеном, галоген будет таким, как определено здесь. Примеры разветвленных или образующих прямую цепь "C1-C6-галогеналкильных" групп, полезные в настоящем изобретении, включают, но не ограничены следующими, метил, этил, пропил, изопропил, изобутил и n-бутил, замещенные независимо одним или более галогенами, к примеру фтором, хлором, бромом и йодом. Термин "состав", а именно фармацевтический состав, предназначен, чтобы охватить продукт,включающий активный компонент(ы) и инертный компонент(ы), которые составляют носитель, так же как любой продукт, который получается в результате, прямо или косвенно, комбинирования, комплексообразования или агрегации любых двух или больше компонентов, или диссоциации одного или большего количества компонентов, или других типов реакций или взаимодействий одного или большего количества компонентов. Соответственно, фармацевтические составы настоящего изобретения охватывают любой состав, полученный путем смешивания состава настоящего изобретения и фармацевтически приемлемого носителя. Термины "вводят" и "введение" состава необходимо понимать в значении предоставления состава изобретения или пролекарства состава изобретения для индивидуалистических потребностей. Использующийся здесь термин "эффективное количество" означает количество лекарства или фармацевтического агента, которое выявит биологическую или лекарственную реакцию ткани, системы,животного или человека, которая будет искомой, к примеру, исследователем или практикующим врачом. Кроме того, термин "терапевтически эффективное количество" означает любое количество, которое, по сравнению с соответствующим субъектом, который не получил такое количество, приведет в результате к улучшенному лечению, излечению, предотвращению или улучшению состояния болезни, расстройства или побочного эффекта либо уменьшения в норме продвижения болезни или расстройства. Термин также включает в свои рамки количества, эффективные для активизации нормальной физиологической функции. Соединения структурной формулы I могут содержать один или более асимметричных центров и могут, таким образом, проявляться в качестве рацематов и рацемических смесей, одинарных энантиомеров, диастереоизомерных смесей и отдельных диастереомеров. В настоящем изобретении подразумевается охватить все такие изомерные формы соединений структурной формулы I. Некоторые из соединений, которые описаны здесь, содержат олефиновые двойные связи, и если не определено иначе, подразумевается, что включают и Е, и Z геометрические изомеры. Некоторые из соединений, описанных здесь, могут существовать как таутомеры, такие как таутомеры кетоэнола. Отдельные таутомеры, так же как их смеси, охвачены в пределах соединений структурной формулы I. Соединения структурной формулы I могут быть разделены на отдельные диастереоизомеры путем, к примеру, фракционной кристаллизации из подходящего растворителя, к примеру метанола; или этилацетата или их смеси, или посредством хиральной хроматографии с применением оптически активной неподвижной фазы. Абсолютная стереохимия может быть определена путем рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных продуктов, которые являются полученными, в случае необходимости, с реактивом, содержащим асимметричный центр известной абсолютной конфигурации. Альтернативно, любой стереоизомер соединения общей структурной формулы I может быть получен путем стереоспецифического синтеза с применением оптически чистых исходных материалов или реактивов известной абсолютной конфигурации. В различном аспекте изобретения фармацевтический состав адресован I, включая соединения в соответствии со структурной формулой I, или его фармацевтически приемлемую соль или сольват, в комбинации с фармацевтически приемлемым носителем. Термин "сольват" означает гидрат, алкоголят или другой сольват кристаллизации. Соединения могут быть приготовлены общими способами согласно схемам 1-3, показанным ниже. Во всех способах приготовления весь исходный материал является известным или может быть легко приготовлен из известных исходных материалов. Таким образом, дополнительное воплощение настоящего изобретения представляет собой способ приготовления соединений настоящего изобретения, характеризующийся тем, что: а) азаиндол формулы II, в которой R5 и R6 являются такими, как определено выше, формилируют для получения альдегида формулы III, в которой R5 и R6 являются такими, как определено выше, указанный альдегид формулы III реагирует с этилцианоацетатом, что сопровождается реакцией Майкла цианидом, кислотной циклизацией и гидридным восстановлением для получения пирролидиноазаиндол формулы IV, в которой R5 и R6 являются такими, как определено выше, ацилирование указанного пирролидиноазаиндола формулы IV, в которой R5 и R6 являются такими, как определено выше, с активированной карбоксильной кислотой формулы V, в которой R1, R2, X и Y являются такими, как определено выше,выполнено для того, чтобы получить соединение формулы I, в которой R1, R2, R5, R6, X и Y являются такими, как определено вышеb) азаиндол формулы II, в которой R5 и R6 являются такими, как определено выше, конденсирован с бром-малеимидом VI для получения пирролидиндиона формулы VII, в которой R5 и R6 являются такими,как определено выше, гидрогенизация указанного пирролидиндиона формулы VII, что сопровождается последовательно снятием защитных бензиловых групп и гидридным восстановлением, производит пирролидиноазаиндол формулы IV, в которой R5 и R6 являются такими, как определено выше, ацилирование указанного пирролидиноазаиндола формулы IV, в которой R5 и R6 являются такими, как определено выше, с активированной карбоксильной кислотой формулы V, в которой R1, R2, X и Y являются такими, как определено выше, выполнено для того, чтобы получить соединение формулы I, в которой R1, R2, R5, R6,X и Y являются такими, как определено выше с) азаиндол формулы II, в которой R5 и R6 являются такими, как определено выше, реагирует под основной средой с кетоном VIII, где R3, R4 и n являются такими, как определено выше, чтобы получить смесь олефинов формулы IX и X, в которых R3, R4, R5, R6 и n являются такими, как определено выше,гидрогенизация указанных олефинов формулы IX и X, в которых R3, R4, R5, R6 и n являются такими, как определено выше, сопровождаемая снятием защитных групп Boc, что выдает пирролидиноазаиндол формулы XI, в которой R3, R4, R5, R6 и n являются такими, как определено выше, ацилированием указанного пирролидиноазаиндола формулы XI, в которой R3, R4, R5, R6 и n являются такими, как определено выше, реагирует с активированной карбоксильной кислотой формулы V, в которой R1, R2, X и Y являются такими, как определено выше, для того, чтобы получить соединение формулы I, в которой R1, R2, R3,R4, R5, R6, X и Y являются такими, как определено вышеd) остаточные X, Y, R1, R2, R3, R4, R5, R6, R7 и/или R8, как заявлено в п.1, преобразованы в другие остаточные X, Y, R1, R2, R3, R4, R5, R6, R7 и/или R8, к примеру, путем введения алкильной группы, илиe) соединение формулы I является изолированным и/или обработано кислотой или основой для того, чтобы получить его соль. Все неочищенные продукты были подвергнуты стандартной хроматографии, используя смеси растворителей, содержащие метанол, этанол, изопропиловый спирт, n-гексан, циклогексан или бензиновый эфир соответственно. Для дополнительного детального описания производственных процессов см. также примеры и следующее общее описание преимущественных условий. Физиологически приемлемая соль соединения согласно формуле I может также быть получена путем изолирования и/или обрабатывания соединения формулы I полученной путем описанной реакции с кислотой или основой. Соединения формулы I и также исходные материалы для их приготовления приготовлены способа-9 019707 ми, как описано в примерах, или способами, которые известны сами по себе, как описано в литературе (к примеру, в стандартных работах, таких как Houben-Weyl, Methoden der Organischen Chemie [Methods ofOrganic Chemistry], Georg Thieme Verlag, Stuttgart; Organic Reactions, John WileySons, Inc., New York),чтобы быть точным при реакционных условиях, которые являются известными и подходящими для упомянутых реакций. Здесь также может быть сделано применение вариантов, которые известны сами по себе, но не упомянуты здесь в особых деталях. Исходные материалы для требуемого процесса могут, если желательно, также быть сформированными in situ, не изолируя их от реакционной смеси, но вместо этого немедленно преобразовывая их далее в соединения формулы I. С другой стороны, возможно выполнить реакцию поэтапно. Предпочтительно реакция соединений выполнена в присутствии подходящего растворителя, который предпочтительно является инертным при соответствующих реакционных условиях. Примеры подходящих растворителей представляют собой углеводороды, такие как гексан, петролейный эфир, бензол,толуол или ксилол; хлорированные углеводороды, такие как трихлорэтилен, 1,2-дихлорэтан, тетрахлорметан, хлороформ или дихлорметан; спирты, такие как метанол, этанол, изопропиловый спирт, nпропанол, n-бутанол или трет-бутанол; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран (THF) или диоксан; гликолевые эфиры, такие как монометил этиленгликоля или моноэтиловый эфир или диметиловый эфир этиленгликоля (простой диметиловый эфир диэтиленгликоля); кетоны, такие как ацетон или бутанон; амиды, такие как ацетамид, диметилацетамид, диметилформамид(DMF) или N-метилпирролидинон (NMP); нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид (DMSO); нитросоединения, такие как нитрометан или нитробензол; сложные эфиры,такие как этилацетат, или смеси упомянутых растворителей или смеси с водой. Полярные растворители являются в основном предпочтительными. Примеры для подходящих полярных растворителей представляют собой хлорированные углеводороды, спирты, гликолевые эфиры, нитрилы, амиды и сульфоксиды или их смеси. Более предпочтительными являются амиды, особенно диметилформамид (DMF). Как указано выше, температура реакции составляет между приблизительно -100 и 300 С, в зависимости от этапа реакции и используемых условий. Время реакции находится в основном в диапазоне между несколькими минутами и несколькими днями, в зависимости от реактивности соответствующих соединений и соответствующих реакционных условий. Подходящее время реакции является без труда определимым методами, известными в данной области, к примеру мониторинг реакции. На основе реакционных температур, которые даны выше, подходящее время реакции в основном лежит в диапазоне между 10 мин и 48 ч. Основа формулы I может быть преобразована в ассоциированную соль присоединения кислоты, используя кислоту, к примеру, путем реакции эквивалентного количества основы и кислоты в предпочтительном инертном растворителе, таком как этанол, за которым следует выпаривание. Подходящими кислотами для этой реакции являются, в частности, те, которые дают физиологически приемлемые соли. Таким образом, возможно использовать неорганические кислоты, к примеру, такие как серная, сернистая, дитионовая, азотная, галогенводородные кислоты, такие как соляная или бромисто-водородная кислота, фосфорные кислоты, такие как, к примеру, ортофосфорная кислота, сульфаминовая кислота, кроме того, органические кислоты, в частности алифатические, ацикличные, аралифатические, ароматические или гетероциклические моноосновные или многоосновные карбоксильные, сульфоновые или серные кислоты, к примеру муравьиная кислота, уксусная, пропионовая, гексановая, октановая, декановая,гексадекановая, октадекановая, триметилуксусная, диэтилуксусная, малоновая, янтарная, пимелиновая,фумаровая, малеиновая, молочная, винная, яблочная, лимонная, глюконовая, аскорбиновая, никотиновая,изоникотиновая, метан- или этансульфоновая, этандисульфоновая, 2-гидроксиэтансульфоновая, бензолсульфоновая, триметоксибензойная, адамантанкарбоновая, р-толуолсульфоновая, гликолевая, эмбоновая,хлорфеноксиуксусная, аспарагиновая, глутаминовая кислоты, пролин, глиоксиловая кислота, пальмитиновая кислота, парахлорфеноксиизомасляная кислота, циклогексанкарбоновая кислота, 1-фосфат глюкозы, нафталинмоно- и -дисульфоновая кислоты или лаурилсерная кислота. Соли с физиологически неприемлемыми кислотами, к примеру пикраты, могут использоваться для того, чтобы изолировать и/или очистить соединения формулы I. С другой стороны, соединения формулы I могут быть преобразованы в соответствующие соли металлов, в частности соли щелочных металлов или соли щелочно-земельных металлов, или в соответствующие соли аммония, используя основания (к примеру, гидроксид натрия, гидроксид калия, карбонат натрия или карбонат калия). Подходящими солями являются, кроме того, замещенные соли аммония, к примеру диметил-, диэтил- и соли диизопропиламмония, соли моноэтанол-, диэтанол- и диизопропаноламмония, соли циклогексил- и дициклогексиламмония, соли дибензилэтилендиаммония, кроме того, к примеру, соли с аргинином или лизином. Если желательно, свободные основания формулы I могут быть выделены из их солей обработкой сильными основаниями, такими как гидроксид натрия, гидроксид калия, карбонат натрия или карбонат калия, до тех пор, пока никакие дополнительные кислотные группы не будут присутствовать в молекуле. В случаях, где соединения формулы I имеют свободные кислотные группы, солеобразование аналогично может быть достигнуто обработкой основаниями. Подходящими основаниями являются гидроксиды щелочных металлов, гидроксиды щелочно-земельных металлов или органические основания в форме первичных, вторичных или третичных аминов. Каждый этап реакции, описанный здесь, может не обязательно сопровождаться одной или более методикой обработки и/или методикой выделения. Такие подходящие процедуры известны в уровне технике, к примеру, из стандартных работ, таких как Houben-Weyl, Methoden der organischen Chemie [Methods of Organic Chemistry], Georg-Thieme-Verlag, Stuttgart). Примеры для таких процедур включают, но не ограничены испарением растворителя, дистилляцией, кристаллизацией, кристаллизацией с распадом на части, процедуры экстракции, процедуры мытья, процедуры дигерирования, процедуры фильтрации,хроматографии, хроматографии HPLC и процедуры высушивания, в особенности процедуры высушивания в вакууме и/или повышения температуры. Соединения, описанные здесь, представляют собой селективные ингибиторы 11-HSD1 энзима. Таким образом, настоящее изобретение относится к применению соединений настоящего изобретения в качестве ингибирования активности редуктазы 11-гидроксистероид дегидрогеназы 1, который является ответственным за превращение кортизона в кортизол. 11-HSD1 ингибиторы структурной формулы I в основном имеют константу ингибирования IC50 меньше чем приблизительно 500 нМ и предпочтительно меньше чем приблизительно 100 нМ. В основном IC50 отношение 11-HSD2 к 11-HSD1 соединения составляет по крайней мере приблизительно два или больше и предпочтительно приблизительно десять или больше. Даже более предпочтительными являются соединения с IC50 отношением 11-HSD2 к 11-HSD1 приблизительно 20 или больше. К примеру, соединения настоящего изобретения идеально демонстрируют константу ингибирования IC50 против 11-HSD2 больше чем приблизительно 1000 нМ и предпочтительно больше чем 5000 нМ. Настоящее изобретение включает применение 11-HSD1 ингибитора для лечения, контроля, улучшения состояния, предотвращения, отсрочки начала или уменьшения риска развития болезней и патологических состояний, которые описаны здесь, посредством излишка или нерегулируемого количества кортизола и/или других кортикостероидов у пациента, относящегося к млекопитающим, в особенности людей, путем введения эффективного количества соединения структурной формулы I или его фармацевтически приемлемой соли или сольвата. Ингибирование 11-HSD1 энзима ограничивает превращение кортизона, который, как правило, является инертным, в кортизол, который может вызвать или поспособствовать признакам этих болезней и патологических состояний, если присутствует в чрезмерном количестве. Таким образом, предпочтительное воплощение настоящего изобретения представляет собой применение соединения настоящего изобретения в качестве 11-HSD1 ингибитора. Дополнительное предпочтительное воплощение настоящего изобретения представляет собой применение соединения настоящего изобретения для приготовления лекарственного средства. Дополнительное предпочтительное воплощение настоящего изобретения представляет собой применение соединения настоящего изобретения для приготовления лекарственного средства для лечения и/или предотвращения болезней, которые вызваны, опосредованы и/или репродуцированы высокими уровнями кортизола. Дополнительное предпочтительное воплощение настоящего изобретения представляет собой применение соединения настоящего изобретения для приготовления лекарственного средства для лечения и/или предотвращения одной или больше болезней или патологических состояний, выбранных из группы, которая состоит из таких, как метаболический синдром, сахарный диабет, особенно инсулиннезависимый сахарный диабет, преддиабет, резистентность к инсулину, низкая толерантность к глюкозе, гипергликемия, ожирение и связанные с весом нарушения, расстройства липидного обмена, такие как дислипидемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, низкие уровни HDL или высокие уровни LDL, глаукома, остеопороз, опосредованные глюкокортикоидом эффекты на нейронную функцию, такие как нарушение когнитивных функций, беспокойство или депрессия, нейродегенеративная болезнь, иммунные расстройства, такие как туберкулез, проказа или псориаз, артериальная гипертензия, атеросклероз и его осложнения, васкулярный рестеноз, сердечно-сосудистые болезни, панкреатит,ретинопатия, невропатия и нефропатия. Соединения согласно настоящему изобретению могут применяться для лечения патологического состояния, выбранного из группы, которая состоит из следующего: гипергликемия, низкая толерантность к глюкозе, резистентность к инсулину, ожирение, расстройства липидного обмена, дислипидемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, низкие уровни HDL, высокие уровни LDL,атеросклероз и его осложнения, васкулярный рестеноз, панкреатит, брюшное ожирение, нейродегенеративная болезнь, ретинопатия, нефропатия, невропатия, метаболический синдром, артериальная гипертензия и другие патологические состояния и расстройства, где резистентность к инсулину представляет собой компонент, который у пациента, относящегося к млекопитающим, который нуждается в таком лечении, является обнаруженным, включает введение пациенту соединения в соответствии со структурной формулой I в количестве, которое является эффективным для лечения указанного патологического со- 11019707 стояния. Соединения согласно настоящему изобретению могут применяться для отсрочки начала патологического состояния, выбранного из группы, состоящей из следующего: гипергликемия, низкая толерантность к глюкозе, резистентность к инсулину, ожирение, расстройства липидного обмена, дислипидемия,гиперлипидемия, гипертриглицеридемия, гиперхолестеринемии, низкие уровни HDL, высокие уровниLDL, атеросклероз и его осложнения, васкулярный рестеноз, панкреатит, брюшное ожирение, нейродегенеративная болезнь, ретинопатия, нефропатия, невропатия, метаболический синдром, артериальная гипертензия и другие патологические состояния и расстройства, где резистентность к инсулину представляет собой компонент, который у пациента, относящегося к млекопитающим, который нуждается в таком лечении, является обнаруженным, включает введение пациенту соединения в соответствии со структурной формулой I в количестве, которое является эффективным для отсрочки начала патологического состояния. Дополнительное предпочтительное воплощение настоящего изобретения представляет собой фармацевтический состав, который отличается тем, что он содержит терапевтически эффективное количество одного или более соединений согласно изобретению. Соединения согласно настоящему изобретению могут входить в фармацевтический состав, который отличается тем, что он дополнительно содержит одно или более дополнительных соединений, выбранных из группы, состоящей из физиологически приемлемых наполнителей, вспомогательных веществ,вспомогательных лекарственных веществ, разжижителей, носителей и фармацевтически активных агентов за исключением соединений согласно изобретению. Соединения согласно настоящему изобретению могут быть использованы в виде набора (комплекта), который состоит из отдельных пакетов:a) терапевтически эффективное количество одного или более соединений согласно изобретению иb) терапевтически эффективное количество одного или более дополнительных фармацевтически активных агентов за исключением соединений согласно изобретению. Соединения структурной формулы I могут быть использованы в комбинации с одним или более другими лекарствами в лечении, предотвращении, ослаблении или улучшении состояния болезней или патологических состояний, для которых соединения структурной формулы I или других лекарств имеют пользу. Как правило, комбинация лекарств является более безопасной или более эффективной, чем один препарат в одиночку, или комбинация является более безопасной или более эффективной, чем это ожидалось бы на основе совокупных свойств отдельных лекарств. Такое другое лекарство(а) может быть применено, путем способа применения и в количестве, обычно используемом одновременно или последовательно с соединением структурной формулы I. Когда соединение структурной формулы I используется одновременно с одним или более другими лекарствами, продукт комбинации, содержащий такое другое лекарство(а) и соединение структурной формулы I, является предпочтительным. Тем не менее,комбинационная терапия также включает методы лечения, в которых соединение структурной формулы I и одно или более других лекарств вводят в различных порядках суперпозиций. Предусматривается, что при использовании в комбинации с другими активными компонентами соединение настоящего изобретения, или другой активный компонент, или оба могут использоваться эффективно в более низких дозах,чем тогда, когда каждый используется в одиночку. Соответственно, фармацевтические составы настоящего изобретения включают те, которые содержат один или более других активных компонентов, вдобавок к соединению структурной формулы I. Примеры других активных компонентов, которые можно вводить в комбинации с соединением структурной формулы I, или же вводятся отдельно или в том же самом фармацевтическом составе, включают, но не ограничены: ингибиторы дипептидилпептидазы IV (DP-IV); агенты, улучшающие чувствительность к инсулину, включая агонисты PPAR, такие как глитазоны (к примеру, троглитазон, пиоглитазон, энглитазон, МСС-555, росиглитазон и им подобные) и другие лиганды PPAR, включая двойные агонисты PPAR/, такие как KRP-297, и агонисты PPAR, такие как гемфиброзил, клофибрат, фенофибрат и безафибрат, и бигуаниды, такие как метформин и фенформин; инсулин или имитаторы инсулина; сульфомочевины и другие средства, усиливающие секрецию инсулина,такие как толбутамид,глипизид,меглитинид и связанные материалы; ингибиторы-глюкозидаза, такие как акарбоза; антагонисты рецептора глюкагона, такие как раскрытые вGLP-1, такие как раскрытые в WO 00/42026 и WO 00/59887; GIP, имитаторы GIP, такие как раскрытые вWO 00/58360, и агонисты рецептора GIP; РАСАР, имитаторы РАСАР, и агонисты рецептора 3 РАСАР,такие как раскрытые в WO 01/23420; агенты понижения холестерина, такие как ингибиторы редуктазыHMG-CoA (ловастатин, симвастатин, правастатин, церивастанин, флувастатин, аторвастатин, итавастатин, розувастатин и другие соответствующие), секвестранты жлчных кислот (холестирамин, колестипол и диалкиламиноалкил производные перекрестно сшитого декстрана), никотиновый спирт, никотиновая кислота или ее соль, ингибиторы абсорбции холестерина, такие как эзетимиб и бета-ситостерин, ацил СоА: ингибиторы холестерин ацилтрансфераза, такие как, к примеру, авазимиб, и антиоксиданты, такие как пробукол; агонисты PPAR, такие как раскрытые в WO 97/28149; соединения антиожирения, такие как фенфлурамин, декстенфлурамин, фентермин, сибутрамин, орлистат, антагонисты нейропептида Y1 или Y5, обратные агонисты и антагонисты рецептора СВ 1, адренергические агонисты рецепторов, агонисты рецептора меланокортина, в частности агонисты рецептора 4 меланокортина, антагонисты грелина, антагонисты рецептора меланиноконцентрирующего гормона (МСН); ингибиторы переносчика подвздошной желчной кислоты; агенты, предназначенные для использования в возбужденных патологических состояниях кроме глюкокортикоидов, такие как аспирин, нестероидные противовоспалительные лекарства, азулфидин, и ингибиторы селективного циклооксигеназа-2; ингибиторы протеинтирозинфосфатаза 1 В (РТР-1 В); противогипертонические средства, включая те, которые действуют на ангиотензин или системы ренина, такие как ангиотензин, преобразовывающий ингибиторы энзима, антагонисты рецептора ангиотензина II или ингибиторы ренина, такие как каптоприл, цилазаприл, эналаприл, фозиноприл, лизиноприл, квинаприл, рамиприл, зофеноприл, кандесартан, цилексетил, эпросартан, ирбесартан,лосартан, тазосартан, телмисатран и валзартан; и ингибиторы транспозиционных протеинов холестериловых эфиров (СЕТР). Вышеупомянутые комбинации включают соединение структурной формулы I, или их фармацевтически приемлемую соль или сольват, с одним или более другими активными соединениями. Неограничивающие примеры включают комбинации соединений структурной формулы I с двумя или больше активными соединениями, выбранными из таких, как бигуаниды, сульфонилмочевина, ингибиторы HMG-CoA редуктаза, агонисты PPAR, ингибиторы РТР-1 В, ингибиторы DP-IV и соединения антиожирения. Соединения согласно настоящему изобретению могут применяться в способе снижения риска развития патологических состояний, выбранных из группы, состоящей из следующего: гипергликемия, низкая толерантность к глюкозе, резистентность к инсулину, ожирение, расстройства липидного обмена,дислипидемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, низкие уровни HDL,высокие уровни LDL, атеросклероз и его осложнения, васкулярный рестеноз, панкреатит, брюшное ожирение, нейродегенеративная болезнь, ретинопатия, нефропатия, невропатия, метаболический синдром,артериальная гипертензия и другие патологические состояния и расстройства, где резистентность к инсулину представляет собой компонент у пациента, относящегося к млекопитающим, который нуждается в таком лечении, который включает введение пациенту соединения в соответствии со структурной формулой I в количестве, которое является эффективным для снижения риска развития указанного патологического состояния. Соединения согласно настоящему изобретению могут применяться для лечения патологического состояния, которое выбрано из группы, состоящей из следующего: гипергликемия, низкая толерантность к глюкозе, резистентность к инсулину, ожирение, расстройства липидного обмена, дислипидемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, низкие уровни HDL, высокие уровни LDL,атеросклероз и его осложнения, васкулярный рестеноз, панкреатит, брюшное ожирение, нейродегенеративная болезнь, ретинопатия, нефропатия, невропатия, метаболический синдром, артериальная гипертензия и другие патологические состояния и расстройства, где резистентность к инсулину представляет собой компонент у пациента, относящегося к млекопитающим, который нуждается в таком лечении, который включает введение пациенту эффективного количества соединения, которое определено в структурной формуле I, и соединения, выбранного из группы, состоящей из следующего: ингибиторы дипептидилпептидаза IV (DP-IV); агенты, улучшающие чувствительность к инсулину, выбранные из группы,состоящей из агонистов PPAR, агонистов PPAR, двойных агонистов PPAR/, и бигуаниды; инсулин или имитаторы инсулина; сульфомочевины и другие средства, усиливающие секрецию инсулина, ингибиторы -глюкозидаза; антагонисты рецептора глюкагона; GLP-1, аналоги GLP-1, агонисты рецептораGLP-1; GIP, имитаторы GIP, агонисты рецептора GIP; PACAP, имитаторы РАСАР, агонисты рецептора 3 РАСАР; агенты понижения холестерина, выбранные из группы, состоящей из следующего: ингибиторы редуктазы HMG-CoA, секвестранты, никотиновый спирт, никотиновая кислота или ее соль, ингибиторы абсорбции холестерина, ацил СоА: ингибиторы холестерин ацилтрансфераза, и антиоксиданты; агонистыPPAR; соединения антиожирения, ингибиторы переносчика подвздошной желчной кислоты; противовоспалительные средства кроме глюкокортикоидов; ингибиторы протеинтирозинфосфатаза 1 В (РТР-1 В); и противогипертонические средства, включая те, которые действуют на ангиотензин или системы ренина, такие как ангиотензин, преобразовывающий ингибиторы энзима, антагонисты рецептора ангиотензина II или ингибиторы ренина, такие как каптоприл, цилазаприл, эналаприл, фозиноприл, лизиноприл,квинаприл, рамиприл, зофеноприл, кандесартан, цилексетил, эпросартан, ирбесартан, лосартан, тазосартан, телмисатран и валзартан; указанные соединения вводятся пациенту в количестве, которое является эффективным для того, чтобы лечить указанное патологическое состояние. Ингибиторы дипептидилпептидаза IV, которые могут быть объединены с соединениями структурной формулы I, включают те, которые раскрыты в WO 03/004498, WO 03/004496; ЕР 1258476; WO 02/083128; WO 02/062764;WO 03/000181. Специфические соединения ингибитора DP-IV включают тиазолидид изолейцина; NVPDPP728; Р 32/98 и LAF237. Соединения против ожирения, которые могут быть объединены с соединениями структурной фор- 13019707 мулы I, включают в себя фенфлурамин, дексфенфлурамин, фентермин, сибутрамин, орлистат, антагонисты нейропептида Y1 или Y5, антагонисты или инверсивные агонисты каннабиноидного СВ 1 рецептора,агонисты рецепторов меланокортина, более конкретно агонисты рецепторов меланокортина-4, антагонисты грелина и антагонисты рецептора меланин-концентрирующего гормона (МСН). Для обзора соединений против ожирения, которые могут быть объединены с соединениями структурной формулы I, см. S.obesity", Expert Opin. Ther. Patents. 11: 1677-1692 (2001) and D. Spanswick and K. Lee, "Emerging antiobesity drugs," Expert Opin. Emerging Drugs, 8: 217-237 (2003). Антагонисты нейропептида Y5, которые могут быть объединены с соединениями структурной формулы I, включают в себя такие, которые описаны в US Patent No. 6335345 и WO 01/14376; и некоторые соединения, идентифицированные как GW 59884A; GW 569180 А; LY 366377 и CGP 71683A. Антагонисты каннабиноидного СВ 1 рецептора, которые могут быть объединены с соединениями структурной формулы I, включают в себя такие, которые описаны в РСТ Publication WO 03/007887; USUS Patent No. 5532237 и US Patent No. 5292736. Агонисты рецептора меланокортина, которые могут быть объединены с соединениями структурной формулы I, включают в себя такие, которые описаны в WO 03/009847; WO 02/068388; WO 99/64002;in the development of melanocortin-4 receptor agonists", Expert Opin. Ther. Patents, 12: 1631-1638 (2002). Соединения согласно настоящему изобретению могут применяться для лечения патологического состояния, выбранного из группы, которая состоит из следующего: гиперхолестеринемия, атеросклероз,низкие уровни HDL, высокие уровни LDL, гиперлипидемия, гипертриглицеридемия и дислипидемия, у пациента, относящегося к млекопитающим, который нуждается в таком лечении, который включает введение пациенту терапевтически эффективного количества соединения, которое определено в структурной формуле I, и ингибитора редуктазы HMG-CoA. Более подробно, соединения согласно настоящему изобретению могут применяться для лечения патологического состояния, выбранного из группы, состоящей из следующего: гиперхолестеринемия, атеросклероз, низкие уровни HDL, высокие уровни LDL, гиперлипидемия, гипертриглицеридемия и дислипидемия, у пациента, относящегося к млекопитающим, который нуждается в таком лечении, в котором ингибитор редуктазы HMG-CoA представляет собой статин. Еще более подробно, соединения согласно настоящему изобретению могут применяться для лечения патологического состояния, выбранного из группы, состоящей из следующего: гиперхолестеринемия, атеросклероз, низкие уровни HDL, высокие уровни LDL, гиперлипидемия, гипертриглицеридемия и дислипидемия, у пациента, относящегося к млекопитающим, который нуждается в таком лечении, в котором ингибитор редуктазы HMG-CoA представляет собой статин, выбранный из группы, состоящей из следующего: ловастатин, симвастатин, правастатин, церивастатин, флувастатин, аторвастатин, итивастатин и розувастатин. Соединения согласно настоящему изобретению могут применяться для снижения риска развития патологического состояния, выбранного из группы, состоящей из следующего: гиперхолестеринемия,атеросклероз, низкие уровни HDL, высокие уровни LDL, гиперлипидемия, гипертриглицеридемия и дислипидемия, и осложнения таких патологических состояний, который включает введение пациенту, относящемуся к млекопитающим, который нуждается в таком лечении, терапевтически эффективного количества соединения, которое определено в структурной формуле I, и ингибитора редуктазы HMG-CoA. Соединения согласно настоящему изобретению могут применяться для того, чтобы отсрочить начало или уменьшить риск развития атеросклероза в пациенте-человеке, который нуждается в таком лечении, которые включает введение указанному пациенту эффективного количества соединения, которое определено в структурной формуле I, и ингибитора редуктазы HMG-CoA. Более подробно, соединения согласно настоящему изобретению могут применяться для того, чтобы отсрочить начало или уменьшить риск развивающегося атеросклероза в пациенте-человеке, который нуждается в таком лечении, в котором ингибитор редуктазы HMG-CoA представляет собой статин. Еще более подробно, соединения согласно настоящему изобретению могут применяться для того,чтобы отсрочить начало или уменьшить риск I развивающегося атеросклероза в пациенте-человеке, который нуждается в таком лечении, в котором ингибитор редуктазы HMG-CoA представляет собой статин, выбранный из группы, состоящей из следующего: ловастатин, симвастатин, правастатин, церивастатин, флувастатин, аторвастатин, итавастатин и розувастатин. Еще более подробно, соединения согласно настоящему изобретению могут применяться для того,чтобы отсрочить начало или уменьшить риск развивающегося атеросклероза в пациенте-человеке, который нуждается в таком лечении, в котором статин представляет собой симвастатин. Соединения согласно настоящему изобретению могут применяться для того, чтобы отсрочить начало или уменьшить риск развивающегося атеросклероза в пациенте-человеке, который нуждается в таком лечении, в которой ингибитор редуктазы HMG-CoA представляет собой статин и дополнительно вклю- 14019707 чает введение ингибитора абсорбции холестерина. Более подробно, соединения согласно настоящему изобретению могут применяться для того, чтобы отсрочить начало или уменьшить риск развивающегося атеросклероза в пациенте-человеке, который нуждается в таком лечении, в котором ингибитор редуктазы HMG-CoA представляет собой статин и ингибитор абсорбции холестерина представляет собой эзетимиб. Соединения согласно настоящему изобретению могут применяться в виде фармацевтического состава, который включает соединение согласно структурной формуле I, соединение выбрано из группы,состоящей из следующего: ингибиторы DP-IV; агенты, улучшающие чувствительность к инсулину, выбранные из группы, состоящей из агонистов PPAR, агонистов PPAR, двойных агонистов PPAR/, и бигуаниды; инсулин или имитаторы инсулина; сульфомочевины и другие средства, усиливающие секрецию инсулина, ингибиторы -глюкозидазы; антагонисты рецептора глюкагона; GLP-1, аналоги GLP-1 и агонисты рецептора GLP-1; GIP, имитаторы GIP и агонисты рецептора GIP; РАСАР, имитаторы РАСАР и агонисты рецептора 3 РАСАР; агенты понижения холестерина, выбранные из группы, состоящей из таких, как ингибиторы редуктазы HMG-CoA, секвестранты, никотиновый спирт, никотиновая кислота или ее соль, ингибиторы абсорбции холестерина, ацил СоА: ингибиторы холестерин ацилтрансфераза и антиоксиданты; агонисты PPAR; соединения антиожирения, ингибиторы переносчика подвздошной желчной кислоты; противовоспалительные средства кроме глюкокортикоидов; ингибиторы протеинтирозинфосфатаза 1 В (РТР-1 В); и противогипертонические средства, включая те, которые действуют на ангиотензин или системы ренина, такие как ангиотензин, преобразовывающий ингибиторы энзима, антагонисты рецептора ангиотензина II или ингибиторы ренина, такие как каптоприл, цилазаприл, эналаприл,фозиноприл, лизиноприл, квинаприл, рамиприл, зофеноприл, кандесартан, цилексетил, эпросартан, ирбесартан, лосартан, тазосартан, телмисатран и валзартан; ингибиторы транпозиционного протеина холестерилового эфира (СЕТР); и фармацевтически приемлемый носитель. Дополнительное воплощение настоящего изобретения представляет собой процесс для производства указанных фармацевтических составов, который отличается тем, что одно или больше соединений согласно изобретению и одно или более соединений, выбранных из группы, состоящей из твердых, жидких или полужидких наполнителей, вспомогательных средств, вспомогательных лекарственных веществ,разжижителей, носителей и фармацевтически активных агентов кроме соединений согласно изобретению, являются преобразованными в подходящую форму дозировки. Фармацевтические соединения настоящего изобретения могут быть введены каким-либо образом,которым они достигнут намеченного назначения. К примеру, введение может быть оральным, парентеральным, наружным, брюшным, внутривенным, внутримышечным, ингаляционным, назальным, внутрисуставным, интраспинальным, транстрахеальным, трансокулярным, подкожным, внутрибрюшинным,трансдермальным или ротовым путем. Альтернативно, или одновременно, введение может быть оральным путем. Дозировка введения будет зависеть от возраста, здоровья и веса реципиента, типа параллельного лечения, если таковые имеют место, частоты лечения и природы желаемого эффекта. Парентеральное введение является предпочтительным. Оральное введение является особенно предпочтительным. Подходящие формы дозировки включают, но не ограничены такими, как капсулы, таблетки, окатыши, драже, полутвердые частицы, порошки, гранулы, суппозитории, мази, крема, лосьоны, ингалянты,инъекционные растворы, припарки, гели, ленты, глазные капли, раствор, сиропы, аэрозоли, суспензия,эмульсия, которые могут быть произведены согласно способам, которые известны в данной области, к примеру, как описано ниже: таблетки: смешивание активного ингредиента(ов) и вспомогательных веществ, прессование указанной смеси в таблетки (прямое прессование), не обязательно гранулирование части смеси перед прессованием; капсулы: смешивание активного ингредиента(ов) и вспомогательных веществ для получения жидкого порошка, не обязательно гранулируя порошок, заполняя порошки/гранулят в открытые капсулы, закупоривая капсулы; полутвердые частицы (мази, гели, крема): растворение/диспергирование активного ингредиента(ов) в водном или жирном носителе; последующее смешивание водной/жирной фазы с дополнительной жирной/водной фазой, гомогенизация(только крема); суппозитории (ректальные и вагинальные): растворение/диспергирование активного ингредиента(ов) в материале носителя, превращенном в жидкость путем нагревания (ректальные: материал носителя обычно воск; влагалищные: носитель обычно нагретый раствор гелеобразного агента), отливание указанной смеси в суппозиторные формы, нормализация и извлечение суппозитория из форм; аэрозоли: диспергирование/растворение активного агента(ов) в пропелленте, бутилирование указанной смеси в аэрозольный ингалятор. В основном нехимические пути для производства фармацевтических составов и/или фармацевтических препаратов включают этапы обработки на подходящих механических средствах, известных в данной области, которые перемещают одно или более соединений согласно изобретению в форму дозировки, подходящую для введения пациенту, который нуждается в таком лечении. Обычно, перемещение одного или более соединений согласно изобретению в такую форму дозировки включает добавление одного или более соединений, выбранных из группы, состоящей из носителей, наполнителей, вспомогательных веществ и фармацевтически активных компонентов, кроме соединений согласно изобретению. Подходящие этапы обработки включают, но не ограничены такими, как комбинирование, размалывание,смешивание, гранулирование, растворение, диспергирование, гомогенизация, отливание и/или прессование соответствующих активных и неактивных компонентов. Механические средства для исполнения указанных этапов обработки известны в данной области, к примеру, из Ullmann's Encyclopedia of Industrial Chemistry, 5th Edition. В этом отношении активные компоненты представляют собой предпочтительно по крайней мере одно соединение согласно этому изобретению и одно или более дополнительных соединений кроме соединений согласно изобретению, которые демонстрируют ценные фармацевтические свойства, предпочтительно те фармацевтические активные агенты, кроме соединений согласно изобретению, которые раскрыты здесь. Особенно подходящими для орального использования являются таблетки, пилюли, таблетки с покрытием, капсулы, порошки, гранулы, сиропы, соки или капли, подходящими для ректального использования являются свечи, подходящими для парентерального использования являются растворы, предпочтительно основанные на масле или водные растворы, кроме того, суспензии, эмульсии или имплантирующие примеси, и подходящими для внешнего использования являются мази, кремы или порошки. Новые соединения могут также быть лиофизированными и продуктом реакции использования лиофизата, к примеру, для приготовления инъекционных препаратов. Обозначенные препараты могут быть стерилизоваными и/или содержать вспомогательные вещества, такие как смазки, консерванты, стабилизаторы и/или смачивающие агенты, эмульгаторы, соли для изменения осмотическое давление, буферные вещества, красители, ароматизаторы и/или множество дополнительных активных компонентов, к примеру,один или более витаминов. Подходящие наполнители представляют собой органические или неорганические вещества, которые являются подходящими для брюшного (к примеру, орального), парентерального или наружного введения и не реагируют с новыми соединениями, к примеру вода, растительные масла, бензиловые спирты,алкиленгликоли, полиэтиленгликоли, глицеринтриацетат, желатин, углеводы, такие как лактоза, сахароза, маннит, сорбитол или крахмал (крахмал кукурузы, крахмал пшеницы, крахмал риса, картофельный крахмал), целлюлозные препараты и/или фосфаты кальция, к примеру трикальцийфосфат или вторичный кислый фосфат кальция, стеарат магния, тальк, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза натрия, поливинилпирролидон и/или вазелин. Если желательно, агенты для улучшения распадаемости таблеток могут быть добавлены, такие как вышеупомянутые крахмалы и также карбоксиметилкрахмал, поперечно сшитый поливинилпирролидон,агар или альгиновая кислота либо ее соль, такая как альгинат натрия. Вспомогательные вещества включают, без ограничения, регулирующие поток вещества и смазки, к примеру кварц, тальк, стеариновую кислоту или ее соли, такие как стеарат магния или стеарат кальция, и/или полиэтиленгликоль. Ядра драже обеспечены подходящим покрытием, которое, если желательно, является стойким к желудочным сокам. С этой целью концентрированные растворы сахарида могут использоваться, которые могут не обязательно содержать гуммиарабик, тальк, поливинилпирролидон, полиэтиленгликоль и/или диоксид титана, летучие растворы и подходящие органические растворители или растворяющие смеси. Чтобы получить покрытия, стойкие к желудочным сокам или обеспечить форму дозировки, обеспечивающую преимущество длительного действия, таблетка, драже или пилюля могут содержать компонент внутренней дозировки и внешней дозировки, последний находится в форме оболочки по-прежнему. Эти два компонента могут быть отделены энтеросолюбильным слоем, который служит для сопротивления распаду в животе и позволяет внутреннему компоненту проходить неповрежденным в двенадцатиперстную кишку или быть отсроченным в выходе. Множество материалов может быть использовано для таких энтеросолюбильных слоев или покрытий, используются материалы, включающие многие полимерные кислоты и смеси полимерных кислот с такими материалами, как шеллак, ацетиловый спирт, растворы подходящих целлюлозных препаратов, такие как фталат ацетилцеллюлозы, ацетат целлюлозы или фталат гидроксипропиметилцеллюлозы. Красящие вещества или пигменты могут быть добавлены к таблеткам или покрытиям драже, к примеру, для идентификации или чтобы характеризовать комбинации доз активных соединений. Подходящие вещества-носители представляют собой органические или неорганические вещества,которые являются подходящими для брюшного (к примеру, орального) или парентерального введения или наружного нанесения и не реагируют с новыми соединениями, к примеру вода, растительные масла,бензиловые спирты, полиэтиленгликоли, желатин, углеводы, такие как лактоза или крахмал, стеарат магния, тальк и вазелин. В частности, таблетки, покрытые таблетки, капсулы, сиропы, суспензии, эмульсии или свечи используются для брюшного введения, растворы, предпочтительно масляные или водные рас- 16019707 творы, кроме того, суспензии, эмульсии или имплантирующие примеси используются для парентерального введения и мази, крема или порошки используются для наружного нанесения. Новые соединения могут также быть лиофилизированы, полученные лиофизаты могут использоваться, к примеру, для производства инъекционных препаратов. Обозначенные препараты могут быть стерилизованными и/или могут содержать наполнители, такие как смазки, консерванты, стабилизаторы и/или смачивающие агенты, эмульгаторы, соли для воздействия на осмотическое давление, буферные вещества, красители, приправы и/или ароматизаторы. Они могут,если желательно, также содержать одно или более дополнительных активных соединений, к примеру,один или более витаминов. Другие фармацевтические препараты, которые могут быть использованы орально, включают капсулы плотной посадки, сделанные из желатина, так же как мягкие, запечатанные капсулы, сделанные из желатина и пластификатора, такого как глицерин или сорбитол. Капсулы плотной насадки могут содержать активные соединения в форме гранул, которые могут быть смешаны с наполнителями, такими как лактоза, связующие вещества, такие как крахмалы, и/или смазки, такие как тальк или стеарат магния и,необязательно, стабилизаторы. В мягких капсулах активные соединения предпочтительно растворены или суспендированы в подходящих жидкостях, таких как жирные масла, или жидкий парафин. Кроме того, могут быть добавлены стабилизаторы. Жидкие формы, в которых новые соединения настоящего изобретения могут быть внедрены для введения орально, включают водные растворы, соответственно приправленные сиропы, водные или масляные суспензии, и приправленные эмульсии с пищевыми маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, так же как эликсиры и подобные фармацевтические среды. Подходящие диспергирующие или суспендирующие агенты для водных суспензий включают синтетические и натуральные резины, такие как трагакант, гуммиарабик, альгинат, декстран, карбоксиметилцеллюлоза натрия, метилцеллюлоза, поливинилпирролидон или желатин. Подходящие составы для парентерального введения включают водные растворы активных соединений в водорастворимой форме, к примеру водорастворимые соли и щелочные растворы. Кроме того,суспензии активных соединений в качестве соответствующих масляных инъекционных суспензий могут быть введены. Подходящие липофильные растворители или среды для лекарства включают жирные масла, к примеру кунжутное масло, или сложные эфиры синтетической жирной кислоты, к примеру этилолеат, или триглицериды, или полиэтиленгликоль-400 (соединения являются растворимыми в PEG400). Водные инъекционные суспензии могут содержать вещества, которые увеличивают вязкость суспензии, включая, к примеру, карбоксиметилцеллюлозу натрия, сорбитол и/или декстран, необязательно суспензия может также содержать стабилизаторы. Для введения в качестве ингаляционного распыления можно использовать распылители, в которых активный компонент или растворен, или суспендирован в газ-пропеллент или смеси газа-пропеллена (к примеру СО 2 или хлорфторуглероды). Активный компонент полезно используется здесь в тонкодисперсной форме, когда один или более дополнительных физиологически приемлемых растворителей могут присутствовать, к примеру этанол. Растворы для ингаляции могут быть введены при помощи обычных ингаляторов. Возможные фармацевтические препараты, которые могут использоваться ректально, включают, к примеру, суппозитории, которые состоят из комбинации одного или больше активных соединений с основанием суппозитория. Подходящими основаниями суппозитория являются, к примеру, натуральные или синтетические триглицериды или парафиновые углеводороды. Кроме того, также возможно использовать желатиновые ректальные капсулы, которые состоят из комбинации активных соединений с основанием. Возможные основные материалы включают, к примеру, жидкие триглицериды, полиэтиленгликоли или парафиновые углеводороды. Для использования в медицине соединения настоящего изобретения будут в форме фармацевтически приемлемых солей. Другие соли могут, однако, быть полезными в приготовлении соединений согласно изобретению или их фармацевтически приемлемых солей. Подходящие фармацевтически приемлемые соли соединений этого изобретения включают соли, полученные посредством добавления кислоты, которые могут, к примеру, быть сформированными путем смешивания раствора соединения согласно изобретению с раствором фармацевтически приемлемой кислоты, такой как соляная, серная, метансульфоновая, фумаровая, малеиновая, янтарная, уксусная, бензойная, щавелевая, лимонная, винная, угольная или фосфорная кислоты. Кроме того, когда соединения изобретения имеют в себе кислотную часть, их подходящие фармацевтически приемлемые соли могут включать соли щелочного металла, к примеру соли калия или натрия; соли щелочно-земельного металла, к примеру соли кальция или магния; и соли,сформированные с подходящими органическими основаниями, к примеру четвертичная аммониевая соль. Соединения согласно настоящему изобретению могут применяться в виде пролекарства соединений настоящего изобретения выше. В основном, такие пролекарства будут функциональными производными соединений настоящего изобретения, которые являются без труда конвертируемыми in vivo в необходи- 17019707 мое соединение настоящего изобретения. Общепринятые методы для селекции и препараты подходящих производных пролекарства описаны, к примеру, в Design of Prodrugs, ed. H. Bundgaard, Elsevier, 1985. Фармацевтические препараты могут быть использованы в качестве медикаментов для человеческой и ветеринарной медицины. Использующийся здесь термин "эффективное количество" означает количество препарата или фармацевтического агента, которое выявит биологическую или лечебную реакцию ткани, системы, животного или человека, которое будет найдено, к примеру, исследователем или практикующим врачом. Кроме того, термин "терапевтически эффективное количество" означает любое количество, которое, по сравнению с соответствующим субъектом, который не получил такое количество,приведет в результате к улучшению лечения, исцелению, предотвращению или улучшению состояния болезни, расстройства или побочного эффекта или уменьшению в норме развития болезни или расстройства. Термин также включает в свои рамки количества, которые являются эффективными для улучшения нормальной физиологической функции. Указанное терапевтически эффективное количество одного или больше соединений согласно изобретению являются известными для квалифицированного специалиста или может быть легко определено стандартными способами, известными в данной области. Вещества согласно изобретению в основном вводят по аналогии с коммерческими препаратами. Как правило, подходящие дозы, которые являются терапевтически эффективными, лежат в диапазоне между 0.0005 и 1000 мг, предпочтительно между 0.005 и 500 мг и особенно между 0.5 и 100 мг за одну дозу. Ежедневная доза составляет предпочтительно между приблизительно 0.001 и 10 мг/кг массы тела. Специалисты в данной области смогут без труда оценить, что уровни дозы могут изменяться в зависимости от конкретного соединения, тяжести симптомов и восприимчивости субъекта кпобочным эффектам. Некоторые из конкретных соединений являются более мощными по сравнению с другими. Преимущественные дозировки для данного соединения являются без труда определимыми специалистом в данной области посредством множества способов. Предпочтительный способ заключается в том, чтобы измерить физиологическую действенность данного соединения. Реципиент, или пациент, может быть из любых разновидностей, относящихся к млекопитающим, к примеру примат sp., в особенности люди; грызуны, включая мышей, крыс и хомяков; кролики; лошади,жвачные животные, собаки, животные из семейства кошачьих и т.д. Моделями животных являются представляющие интерес для экспериментальных исследований, обеспечивая модель для лечения человеческой болезни. Конкретная доза для отдельного пациента зависит, тем не менее, от множества факторов, к примеру от эффективности используемых конкретных соединений, от возраста, массы тела, общего состояния здоровья, пола, вида диеты, от времени и пути введения, от нормы экскреции, типа введения и формы дозировки, которая будет вводиться, фармацевтического сочетания и серьезности специфического расстройства, к которому имеет отношение терапия. Специальная терапевтически эффективная доза для отдельного пациента может без труда быть определенной путем обычного экспериментирования, к примеру, доктором или практикующим врачом, который консультирует или принимает участие в терапевтическом лечении. В случае многих расстройств восприимчивость специфической клетки для лечения с обусловленными соединениями может быть определена путем in vitro тестирования. Типично посев клетки объединен с обусловленным соединением при переменных концентрациях на время, которое является достаточным для того, чтобы позволить активным агентам показать соответствующую реакцию, обычно приблизительно между одним часом и одной неделей. Для in vitro тестирования могут быть использованы культивируемые клетки из образца биопсии. Даже без дополнительных деталей предполагается, что квалифицированный специалист в данной области может использовать приведенное выше описание в максимально широкой области. Преимущественные воплощения, таким образом, должны просто быть расценены как описательное раскрытие, которое абсолютно не ограничивает в любом случае. Выше и ниже, все температуры указаны в С. В следующих примерах "обычное выделение продукта реакции" означает, что, в случае необходимости, растворитель удален, вода добавлена в случае необходимости, рН отрегулирован, в случае необходимости, до между 2 и 10, в зависимости от разбавления конечного продукта, смесь экстрагирована этилацетатом или дихлорметанон, фазы отделены, органическая фаза вымыта сатурированным раствором NaHCO3, если желательно водой, и сатурирована раствором NaCl, высушена над сульфатом натрия, отфильтрована и выпарена, и продукт очищен хроматографией на силикагеле путем препаративной HPLC и/или кристализации. Очищенные соединения, если желательно, лиофилизированы. Масс-спектрометрия (MS): ESI (электрораспылительная ионизация) (М+Н)+. Список сокращений и акронимов: АсОН - уксусная кислота; anh - обезвоженный; atm (атм) - атмосфера(ы); ВОС - третбутоксикарбонил; CDI - 1,1'-карбонилдиимидазол; conc - концентрированый; d - день(дни); dec - разложение; DMAC - NN-диметилацетамид; DMPU - 1,3-диметил-3,4,5,6-тетрагидро-2(1H)пиримидон; DMF NN-диметилформамид; DMSO - диметилсульфоксид; DPPA - дифенилфосфорил азид; EDCI - 1-(3 диметиламинопропил)-3-этилкарбодиимид; EtOAc - этилацетат; EtOH - этанол (100%); Et2O - диэтило- 18019707 вый эфир; Et3N - триэтиламин; ч - час(ы); МеОН - метанол; pet. ether - петролейный эфир (интервал кипения 30-60 С); temp - температура; THF - тетрагидрофуран; TFA - трифторАсОН; Tf - трифторметансульфонил. Соединения общей формулы I настоящего изобретения могут быть приготовлены в соответствии с процедурами следующих схем 1 и 2 и примеров. Во всех препаративных методах весь исходный материал является известным или может легко быть приготовлен из известных исходных материалов. Схема 1POCl3 (45 мл) был добавлен по капле при 0 С под азотом, чтобы высушить DMF (28 мл), смесь была перемешана при 0 С в течение 15 мин. 7-Азаиндол (25 г, 0.21 моль) был медленно добавлен. Тогда реакционная смесь была перемешана до RT и затем при 80 С в течение 24 ч. После завершения реакции реакционная смесь была разлита в лед, нейтрализована охлажденным раствором NaOH 1 N, экстрагирована этилацетатом, затем CH2Cl2/MeOH (90/10), отлужена раствором NaOH 1 N и экстрагированы сноваCH2Cl2/MeOH (90/10). Органические фазы были высушены над Na2SO4 и концентрированы досуха, чтобы получить оранжевое твердое вещество (16.3 г).H NMR (DMSO-d6) 7.30 (dd, 1H), 8.36-8.43 (m, 2H), 8.49 (s, 1H), 9.94 (s, 1H), 12.82 (s, 1H). Этап 2. Реакция Кневенагеля. 2-Циано-3-(1 Н-пиррол[2,3-b]пиридин-3-ил)сложный метиловый эфир акриловой кислоты. 7-Азаиндол-3-карбоксальдегид (3.36 г, 22.99 ммоль) был взят в 60 мл метанола и охлажден до 0 С. Был добавлен пиперидин (2.5 мл, 25.28 ммоль), за которым следует этилцианоацетат (2.23 мл, 25.27 ммоль). Реакция была перемешана при RT в течение 5 ч, затем концентрирована, гашена водой и отфильтрована, чтобы получить желтый твердый продукт (4.07 г).H NMR (DMSO-d6) 3.85 (s, 3 Н), 7.33 (dd, 1H), 8.42 (d, 1H), 8.55 (d, 1H), 8.60 (s, 1H), 8.65 (s, 1H). Этап 3. Образование дицианида. 2-(1H-Пиррол[2,3-b]пиридин-3-ил)сукцинонитрил. Цианоэфир (4.42 г, 19.45 ммоль) был взят в метаноле (60 мл) и воде (12 мл). Цианид калия (2.53 г,38.85 ммоль) был добавлен. Реакционная смесь была дефлегмирована в течение 2.5 ч. Реакционная масса была концентрирована, затем гашена водой, экстрагирована этилацетатом, высушена над Na2SO4. Очищение путем колоночной хроматографии, используя CH2Cl2/MeOH (95/05) в качестве элюента дало коричневатый твердый продукт (2.67 г). Дицианид (0.64 г, 3.26 ммоль) был взят в (3 мл) кристаллизованной уксусной кислоте и серная кислота (0.64 мл) была добавлена по капле. Реакционная смесь была нагрета при 120 С в течение 1.5 ч. Смесь была концентрирована и гашена водой, далее следует экстракция этилацетатом. Органический слой был концентрирован насухо и остаток очищен путем колоночной хроматографии, используя гептан/этилацетат (10/90), затем этилацетат в качестве элюента, чтобы получить оранжевое твердое вещество (0.368 г).LAH (0.211 г, 5.5 ммоль) был взят в RB фляге и охлажден до 0-5 С, сухой THF (10 мл) был медленно добавлен, далее следует дион (0.25 г, 1.1 ммоль). Реакционная смесь была нагрета до рефлюкса в течение всей ночи, гашена водой, 15%-ным NaOH и водой. Полученный продукт был экстрагирован этилацетатом и концентрирован, чтобы получить масло (187 мг) как неочищенный продукт, который был использован в следующем этапе без дополнительной очистки. 1(2-Фторфенил)-[3-(1 Н-пиррол[2,3-b]пиридин-3-ил)пирролидин-1-ил]метанон. Предшествующий амин (0.2 г, 1.08 ммоль) был растворен в DMF (3 мл) и охлажден до 0 С. Затем НОВТ (0.0432 г, 0.318 ммоль), EDCI (0.303 г, 1.59 ммоль) были добавлены, за которыми следовали триэтиламин (0.44 мл) и 2-фторбензойная кислота (0.178 г, 1.27 ммоль). Смесь была перемешана при RT в течение всей ночи, гашена 20 мл 10% раствора бикарбоната натрия и экстрагирована этилацетатом. Этилацетат был концентрирован и беспримесный продукт получен путем колоночной хроматографии как от белого твердого вещества (0.050 г).H NMR (CDCl3) 2-2.5 (m, 2H), 3.4-4 (m, 4H), 4.2 (m, 1H), 7-7.5 (m, 6H), 7.9 (dd, 1H), 8.30 (s, 1H),11.25 (s, 1H). Следующие соединения были сделаны подобным способом, как описано в примере 1. Пример 2.H NMR (MeOD) 1.8-2.5 (m, 6H), 2.7-3.25 (m, 4H), 3.5-4 (m, 3 Н), 6.9-7.5 (m, 6H), 8.0 (dd, 1H), 8.15 (sl,1H). Последующие соединения были получены подобным способом, как описано в примере 1, используя 3-пиперидин-4-ил-1H-пиррол[2,3-b]пиридин вместо 3-пирролидин-3-ил-1 Н-пиррол[2,3-b]пиридина. Пример 12. 2-(4-Хлорфенокси)-2-метил-1-[4-(1 Н-пиррол[2,3-b]пиридин-3-ил)пиперидин-1-ил]пропан-1-он. Пример 21. Этап 1. Конденсация. 1-Бензил-3-бромо-4-(1 Н-пиррол[2,3-b]пиридин-3-ил)пиррол-2,5-дион. Соединение было приготовлено в соответствии с Bioorg. Med. Chem., 2004, 12, 3167. Раствор EtMgBr (86.96 мл, 86.96 ммоль, 1 M в THF) был добавлен по капле под азотом, раствор 7 азаиндола (10.273 г, 86.96 ммоль) в толуоле 240 мл - при RT. После 1.5 ч раствор 2,3-дибромо-Nбензилмалеимида (10 г, 28.98 ммоль) в толуоле (240 мл) был добавлен по капле. Затем реакционная смесь перемешивалась в течение 0.3 ч, CH2Cl2 (360 мл) был добавлен и смесь нагрета до 45 С в течение 72 ч. Гидролиз был выполнен насыщенным раствором NH4Cl. После экстракции этилацетатом и концентрации под вакуумом желаемое соединение было осаждено из CH2Cl2 и высушено (6.92 г как желтый порошок).H NMR (DMSO-d6) 4.72 (s, 2H), 7.15-7.45 (м. 7H), 8.24 (s, 1H), 8.33 (s, 1H), 12,75 (s, 1H). Этап 2. Гидрирование. 1-Бензил-3-(1H-пиррол[2,3-b]пиридин-3-ил)пирролидин-2.5-дион. Винилбромид (6.8 г, 17.79 ммоль) в МеОН (100 мл) был гидрогенизирован на 5% Pd/C (0.68 г) под грузилом (10 бар) в течение ночи. Затем реакционная смесь была отфильтрована и концентрирована насухо. Неочищенный остаток был очищен путем флэш-хроматографии, используя CH2Cl2, затемCH2Cl2/MeOH (97/03) в качестве элюента, чтобы получить масло, который был растерт в порошок с ацетоном, чтобы получить желаемый продукт как светло-желтый порошок 2.23 г.(d, 1H), 8.4 (d, 1H), 12.4 (s, 1H). Этап 3. Дебензилирование. 1H-Пиррол[2,3-b]пиридин-3-ил-пирролидин-2,5-дион. К вышеупомянутому соединению (0.5 г, 1.638 ммоль) в толуоле (30 мл) был добавлен AlCl3 (1.093 г,8.2 ммоль). Реакционная смесь была нагрета до рефлукса в течение ночи. Толуол был выпарен, вода была добавлена и остаток экстрагирован этилацетатом. После выпаривания неочищенный продукт был очищен путем флэш-хроматографии, используя CH2Cl2/MeOH (95/05) в качестве элюента и осажден из изопропилового эфира, чтобы получить коричневое тело 0.26 г. Пример 22. Этап 1. Конденсация. 5-(1 Н-Пиррол[2,3-b]пиридин-3-ил)-3,4-дигидро-2 Н-пиридин-1-карбоновой кислоты трет-бутиловый сложный эфир и 5-(1 Н-пиррол[2,3-b]пиридин-3-ил)-3,6-дигидро-2 Н-пиридин-1-карбоновой кислоты трет-бутиловый сложный эфир. 7-Азаиндол (0.5 г, 4.23 ммоль) и N-Boc-пиперидин-3-он (2.1 г, 10.5 ммоль) и 2 М KOH в МеОН (11 мл) были нагреты до 60 С в течение 17 ч. Затем реакционная смесь была концентрирована в вакууме,отреагирована в этилацетате, органическая фаза была вымыта водой и морской водой, высушена над сульфатом магния и концентрирована насухо. Остаток был растерт в порошок со смесью хлорида метилена и гептана. Осадок был отфильтрован и высушен, чтобы получить смесь 75/25 желаемых продуктов как светло-желтое твердое вещество (0.356 г). Исходный раствор был очищен путем флэшхроматографии, используя гептан/этилацетат от 60/40 до 40/60 в качестве элюента, чтобы получить смесь 65/35 желаемых продуктов как светло-желтый порошок (0.455 г). 1H NMR (DMSO-d6) основной продукт 1.51 (d, 9 Н), 1.92 (м., 2 Н), 2.44 (м., 2 Н), 3.57 (м., 2 Н), 7.14 (м.,1 Н), 7.36 (dd, 1 Н), 7.49 (d, 1 Н), 8.05 (м., 1 Н), 8.25 (d, 1H), 11.64 (sl, 1 Н), побочный продукт 1.46 (s, 9 Н),2.30 (м., 2 Н), 3.52 (м., 2 Н), 4.24 (sl, 2H), 6.31 (м., 1 Н), 7.11 (м., 1 Н), 7.56 (sl, 1 Н), 8.20 (м., 2 Н), 11.75 (sl,1H). Этап 2. Гидрирование. 3-(1 Н-Пиррол[2,3-b]пиридин-3-ил)пиперидин-1-карбоновой кислоты трет-бутиловый сложный эфир. Смесь вышеупомянутых изомеров (0.428 г, 1.43 ммоль) в смеси 1/1 MeOH/THF (10 мл) была гидрогенизирована на Pd(OH)2/C 10-20% (0.01 г) под грузилом (10 бар) в течение 4 ч. Затем реакционная смесь была отфильтрована и концентрирована насухо. Нечищеный остаток был очищен путем флэшхроматографии, используя гептан/этилацетат 60/40 в качестве элюента, чтобы получить желаемый продукт как масляное твердое вещество (0.17 г). 1H NMR (DMSO-d6) 1.43 (s, 9H), 1.75-1.85 (м., 3 Н), 2.04 (dl, 1H), 2.89 (м., 3 Н), 3.93 (dl, 1 Н), 4.1 (sl,1 Н), 7.06 (dd, 1 Н), 7.40 (s, 1 Н), 8.01 (м., 1 Н), 8.23 (м., 1 Н), 11.45 (sl, 1H). Этап 3. Снятие защитных групп. 3-Пиперидин-3-ил-1 Н-пиррол[2,3-b]пиридин. К вышеупомянутому соединению (93 мг, 0.31 ммоль) в диоксане (1 мл) было добавлено 0.232 мл 4 М раствора HCl в диоксине. Реакционная смесь была перемешана при RT в течение 2 ч и концентрирована насухо. Остаток был очищен путем флэш-хроматографии, используя CH2Cl2/MeOH/NH3 (90/10/3) в качестве элюента, чтобы получить желаемый продукт как масляное твердое вещество (10 мг). 1(11 бета-HSD2) энзимы типа 2 были экспрессированы в мышинной кишечной палочке и микросомные фракции печени крысы были приобретены у TEBU. Анализы 11 бета-HSD1 энзима были выполнены в 96 лунках пластин микротитратора в суммарном объме 100, содержащем 30 мМ буфер Hepes, pH 7,4 с 1 мМ EDTA, смесь субстрата кортизона/NADPH(200 нМ /200 мкМ), G-6-P (1 мМ) и ингибиторы в последовательных растворениях. Реакции были инициированы добавлением 10 мкл 11 бета-HSD1 (3 мкг) из кишечной палочки, либо в качестве микросомных фракций печени крыс и мышей (2,5 мкг). После смешивания пластины встряхивали в течение 150 мин при 37 С. Реакции были завершены с 10 мкл стоп-раствором 18 бета-глицирретиновой кислоты. Определения уровней кортизола в 11 бета-HSD1 препаратах были проверены HTRF (HTRF анализ кортизола из Cis bio international). Активность выражена в % контроля или концентрации для ингибирования 50% активности энзима(IC50). Этот анализ был также применен к 11 бета-HSD2 энзиму, кортизолу, NAD и карбеноксолон использовались как основание, кофактор и ингибитор соответственно. Пример 24. Инъекционные ампулы. Раствор 100 г активного соединения настоящего изобретения и 5 г двунатриевого гидрогенфосфата были доведены до рН 6.5 в 3 л дважды дистиллированной воды с применением 2 N соляной кислоты,стерильно отфильтрованы, распределены в инъекционные ампулы, лиофилизированы в стерильных условиях и стерильно запечатаны. Каждая инъекционная ампула содержит 5 мг активного соединения. Пример 25. Суппозитории. Смесь 20 г активного соединения настоящего изобретения конденсирована с 100 г лецитина сои и 1400 г масла какао, разлиты в формы и оставлены охлаждаться. Каждый суппозиторий содержит 20 мг активного соединения. Пример 26. Раствор. Раствор 1 г активного соединения настоящего изобретения, 9.38 г NaH2PO42 Н 2 О, 28.48 гNa2HPO412 Н 2 О и 0.1 г хлорида бензалкония приготовлен в 940 мл дистиллированной дважды воды. Он доведен до рН 6.8, добавлен до 1 л и стерилизуется облучением. Этот раствор может использоваться в форме глазных капель. Пример 27. Мазь. 500 мг активного соединения настоящего изобретения смешаны с 99.5 г вазелина при стерильных патологические состояниях. Пример 28. Таблетки. Смесь 1 кг активного соединения настоящего изобретения, 4 кг лактозы, 1.2 кг картофельного крахмала, 0.2 кг талька и 0.1 кг стеарата магния сжата, чтобы дать таблетки в общепринятой манере таким образом, чтобы каждая таблетка содержала 10 мг активного соединения. Пример 29. Покрытые таблетки. Аналогично к предыдущему примеру, таблетки нажаты и тогда покрыты в общепринятой манере,используя покрытие сахарозы, картофельного крахмала, талька, tragacanth и красителя. Пример 30. Капсулы. 2 кг активного соединения настоящего изобретения распределены в твердые капсулы желатина в общепринятой манере таким образом, чтобы каждая капсула содержала 20 мг активного соединения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы I в которой R1, R2 представляют собой независимо друг от друга Н, А, С 3-8 циклоалкил;m имеет значение 0 или 1,и его физиологически приемлемые соли. 2. Соединение, выбранное из группы, состоящей из следующего: а) (2-фторофенил)-[3-(1 Н-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]метанон;t) 2-(4-хлоробензолсульфонил)-1-[3-(1 Н-пирроло[2,3-b]пиридин-3-ил)пирролидин-1-ил]этанон и их физиологически приемлемые соли. 3. Способ получения соединения по п.1 или 2, который отличается тем, что азаиндол формулы II в которой R5 и R6 являются такими, как определено выше,а) формилируют для получения альдегида формулы III в которой R5 и R6 являются такими, как определено выше,указанный альдегид формулы III реагирует с этилцианоацетатом, что сопровождается реакцией Майкла с добавлением цианида, кислотной циклизацией и гидридным восстановлением для получения пирролидиноазаиндола формулы IV для получения пирролидиндиона формулы VII в которой R5 и R6 являются такими, как определено выше,гидрогенизация указанного пирролидиндиона формулы VII сопровождается последовательным снятием защитных бензиловых групп и гидридным восстановлением, приводит к получению пирролидиноазаиндола формулы IV где R3, R4 и n являются такими, как определено выше,с получением смеси олефинов формул IX и X в которых R3, R4, R5, R6 и n являются такими, как определено выше,- 26019707 гидрогенизация указанных олефинов формул IX и X, в которых R3, R4, R5, R6 и n являются такими,как определено выше, сопровождается снятием защитных групп Boc и приводит к получению пирролидиноазаиндола формулы XI в которой R3, R4, R5, R6 и n являются такими, как определено выше,с последующим ацилированием полученных пирролидиноазаиндолов формул IV или XI, в которыхR3, R4, R5, R6 и n являются такими, как определено выше, активированной карбоксильной кислотой формулы V в которой R1, R2, X и Y являются такими, как определено выше,для получения соединения формулы I, в которой R1, R2, R3, R4, R5, R6, X и Y являются такими, как определено выше, и при необходимости остатки X, Y, R1, R2, R3, R4, R5, R6, как заявлено в п.1, превращают в другие остатки X, Y, R1, R2, R3, R4, R5, R6, путем введения алкильной группы, или соединение формулы I выделяют и/или обрабатывают кислотой или основанием для получения соли. 4. Применение соединения по п.1 или 2 в качестве 11-HSD1 ингибитора. 5. Применение соединения по п.1 или 2 для приготовления лекарственного средства, селективно ингибирующего энзим 11-бета-гидроксистероида дегидроксигеназы 1 типа. 6. Применение соединения по п.1 или 2 для приготовления лекарственного средства для лечения и/или предотвращения болезней, которые вызваны, опосредованы и/или репродуцированы высокими уровнями кортизола. 7. Применение соединения по п.1 или 2 для приготовления лекарственного средства для лечения и/или предотвращения одной или больше болезней или патологических состояний, выбранных из группы, которая состоит из следующего: метаболический синдром, сахарный диабет, особенно инсулиннезависимый сахарный диабет, преддиабет, резистентность к инсулину, низкая толерантность к глюкозе, гипергликемия, ожирение и связанные с весом нарушения, расстройства липидного обмена, такие как дислипидемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, низкие уровни HDL или высокие уровни LDL, глаукома, остеопороз, опосредованные глюкокортикоидом эффекты на нейронную функцию, такие как нарушение когнитивных функций, беспокойство или депрессия, нейродегенеративная болезнь, иммунные расстройства, такие как туберкулез, проказа или псориаз, артериальная гипертензия, атеросклероз и его осложнения, васкулярный рестеноз, сердечно-сосудистые болезни, панкреатит,ретинопатия, невропатия и нефропатия. 8. Фармацевтический состав, селективно ингибирующий энзим 11-бета-гидроксистероида дегидроксигеназы 1 типа, который отличается тем, что он содержит терапевтически эффективное количество одного или более соединений по п.1 или 2 и одно или более дополнительных соединений, выбранных из группы, состоящей из физиологически приемлемых наполнителей, вспомогательных веществ, разжижителей, носителей. 9. Способ получения фармацевтических составов, селективно ингибирующих энзим 11-бетагидроксистероида дегидроксигеназы 1 типа, который отличается тем, что одно или больше соединений по п.1 или 2 и одно или более соединений, выбранных из группы, состоящей из твердых, жидких или полужидких наполнителей, вспомогательных средств, разжижителей, носителей, являются преобразованными в подходящую форму дозировки.
МПК / Метки
МПК: A61P 25/00, C07D 471/04, A61K 31/33
Метки: производные, селективных, 7-азаиндола, качестве, дегидрогеназы, типа, ингибиторов, 11-бета-гидроксистероида
Код ссылки
<a href="https://eas.patents.su/28-19707-proizvodnye-7-azaindola-v-kachestve-selektivnyh-ingibitorov-11-beta-gidroksisteroida-degidrogenazy-1-tipa.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 7-азаиндола в качестве селективных ингибиторов 11-бета-гидроксистероида дегидрогеназы 1 типа</a>
Спироциклы в качестве ингибиторов 11-бета гидроксилстероиддегидрогеназы типа 1

Номер патента: 16017
Опубликовано: 30.01.2012
Авторы: Чжан Колин, Яо Вэньцин, Чжо Цзиньцун
МПК: C07D 471/10, A61K 31/444, A61P 3/04...
Метки: спироциклы, 11-бета, гидроксилстероиддегидрогеназы, типа, качестве, ингибиторов
Формула / Реферат:
1. Соединение, представляющее собой 5-{3-фтор-4-[(5S)-2-(цис-4-гидроксициклогексил)-1-оксо-2,7-диазаспиро[4,5]дец-7-ил]фенил}-N,N-диметилпиридин-2-карбоксамид или его фармацевтически приемлемую соль.2. Соединение, представляющее собой 5-{3-фтор-4-[(5S)-2-(цис-4-гидроксициклогексил)-1-оксо-2,7-диазаспиро[4,5]дец-7-ил]фенил}-N-метилпиридин-2-карбоксамид или его фармацевтически приемлемую соль.3. Соединение, представляющее собой...
Производные пирролидин-2-она и пиперидин-2-она, используемые в качестве ингибиторов 11-бета-гидроксистероид-дегидрогеназы

Номер патента: 11097
Опубликовано: 30.12.2008
Авторы: Никифоров Тео Теофанов, Ван Дер Векен Луи Йозеф Элизабет, Димитров Владимир Димчев, Яроскова Либуше, Линдерс Йоаннес Теодорус Мария, Бюйк Кристоф Франсис Роберт Нестор
МПК: A61K 31/45, A61K 31/4015, A61P 3/04...
Метки: пирролидин-2-она, пиперидин-2-она, производные, используемые, 11-бета-гидроксистероид-дегидрогеназы, качестве, ингибиторов
Формула / Реферат:
1. Соединение формулы его N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где n равно 1 или 2; L представляет собой C1-3алкильный линкер, необязательно замещенный одним или двумя заместителями, выбранными из C1-4алкила, C1-3алкилоксиС1-4алкила, гидроксиС1-4алкила, гидрокси, C1-3алкилокси или фенилС1-4алкила; M представляет собой прямую связь или C1-3алкильный линкер, необязательно замещенный...
Производные n-2-адамантанил-2-феноксиацетамида в качестве ингибиторов 11-бета- гидроксистероид-дегидрогеназы

Номер патента: 12263
Опубликовано: 28.08.2009
Авторы: Линдерс Йоаннес Теодорус Мария, Яроскова Либуше, Виллемсенс Густаф Хенри Мария, Бисхофф Франсуа Пол, Ван Дер Векен Луи Йозеф Элизабет
МПК: A61P 19/10, A61K 31/165, A61P 25/28...
Метки: гидроксистероид-дегидрогеназы, 11-бета, производные, ингибиторов, качестве, n-2-адамантанил-2-феноксиацетамида
Формула / Реферат:
1. Применение соединения формулы (I) его N-оксидных форм, фармацевтически приемлемых аддитивных солей и стереохимически изомерных форм, где n=1, 2, 3 или 4; Z представляет О, S, NR6, SO или SO2; R1 представляет водород, циано, гидрокси или C1-4алкил, необязательно замещенный галогеном; R2 представляет водород, C1-4алкил или C1-4алкилокси-; R3 представляет водород, С1-4алкил, C1-4алкилокси- или R3 в комбинации с R2 вместе образуют двухвалентный...
Производные адамантилпирролидин-2-она в качестве ингибиторов 11-бета-гидроксистероид-дегидрогеназы

Номер патента: 11021
Опубликовано: 30.12.2008
Авторы: Яроскова Либуше, Бюйк Кристоф Франсис Роберт Нестор, Линдерс Йоаннес Теодорус Мария, Ван Дер Векен Луи Йозеф Элизабет
МПК: A61K 31/402, C07D 207/26, C07D 211/76...
Метки: ингибиторов, производные, качестве, 11-бета-гидроксистероид-дегидрогеназы, адамантилпирролидин-2-она
Формула / Реферат:
1. Соединение, имеющее формулу его N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где n равно 1 или 2; М представляет собой прямую связь или C1-3алкильный линкер, необязательно замещенный одним или двумя заместителями, выбранными из С1-4алкила, C1-3алкилокси-С1-4алкила, гидрокси-С1-4алкила, гидрокси, C1-3алкилокси- или фенил-С1-4алкила; каждый из R1 и R2 независимо представляет собой водород,...
Производные лактама циклогексилимидазола в качестве ингибиторов 11-бета-гидроксистероид-дегидрогеназы 1

Номер патента: 14717
Опубликовано: 28.02.2011
Авторы: Уоллэйс Оуэн Брендан, Саеед Ашраф, Гюнтер Ребекка Линн, Снайдер Нэнси Джун, Сюй Яньпин, Мэбри Томас Эдвард
МПК: A61K 31/4015, A61P 3/00, C07D 401/14...
Метки: лактама, 11-бета-гидроксистероид-дегидрогеназы, ингибиторов, циклогексилимидазола, качестве, производные
Формула / Реферат:
1. Соединение, структура которого представлена формулойв которой R1 представляет собой -Н, -галоген, -О-СН3(необязательно, имеющий в качестве заместителей от одного до трех атомов галогена) или -СН3 (необязательно, имеющий в качестве заместителей от одного до трех атомов галогена);R2 представляет собой -Н, -галоген, -О-СН3(необязательно, имеющий в качестве заместителей от одного до трех атомов галогена) или -СН3 (необязательно, имеющий в...
Предыдущий патент: Триазолпиридиновые ингибиторы 11-бета-гидроксистероид-дегидрогеназы типа i
Следующий патент: Соединения-аналоги 6-окса-8α-стероидных эстрогенов, способ их получения и их применение
Случайный патент: Электрохимическое восстановление оксидов металлов