Производные пиридино-пиридинонов, способ их получения и применение в терапии
Номер патента: 19362
Опубликовано: 31.03.2014
Авторы: Сави Пьер, Мартен Валери, Волль-Шаллье Сесиль, Маккорт Гари, Белльверг Патрис, Лассалль Жильбер








Формула / Реферат
1. Соединение формулы (I)
в которой R1 означает (C1-C4)алкильную группу;
R2 означает -(СН2)n'-В, где n'=0, 1, 2, 3, 4 и В является (С3-С5)циклоалкильной группой или (С1-С4)алкильной группой, необязательно замещенной одним или несколькими атомами фтора, (С1-С4)алкоксигруппой;
U означает карбонильную группу или группу -СН2-;
Y, Z, V и W обозначают, независимо один от другого, группу -СН- или атом углерода, замещенный группой R7, или гетероатом, такой как атом азота или атом серы, или не содержит его, причем цикл должен быть ароматическим и содержать от 5 до 6 звеньев;
R3, R4 означают, независимо один от другого, атом водорода или линейную (С1-С4)алкильную группу или R3 и R4 вместе с атомом углерода, с которым они связаны, образуют (С3-С5)циклоалкильную группу;
m является целым числом, равным 1, 2, 3, 4;
R5 означает атом водорода или (С1-С4)алкильную группу;
R6 означает -(CH2)n-L, где n=0, 1, 2, 3 и L является группой, выбранной независимо из следующих групп:
(С1-С5)алкильной группы, необязательно замещенной (С1-С4)алкоксигруппой;
(С3-С5)циклоалкильной группы;
арильной группы, содержащей 6 атомов углерода, необязательно замещенной одним или несколькими атомами галогена;
гетероарильной группы, содержащей 5-6 звеньев, в которой по меньшей мере один гетероатом, выбранный из атома азота или серы, необязательно замещен (С1-С4)алкильной группой;
гетероциклической насыщенной группы, в которой указанный гетероцикл содержит 4-7 звеньев, по меньшей мере один гетероатом которого выбран из атома азота, атома кислорода, и необязательно замещенной в любой позиции, в том числе на атоме азота, одним или несколькими заместителями, выбранными из атома фтора, фтор(С1-С4)алкильной группы, (С1-С4)алкильной группы, линейной или разветвленной, (С3-С5)циклоалкильной группы или (С1-С4)алкилсульфонамидной группы, причем абсолютная конфигурация углерода, замещенного на указанном гетероцикле, может быть R или S конфигурацией или рацемической;
R7 означает атом водорода, или (С1-С4)алкильную группу, или атом галогена,
в форме основания или аддитивной соли с кислотой, в форме сольвата, а также его энантиомеры и диастереоизомеры, включая его смеси.
2. Соединение формулы (I) по п.1, отличающееся тем, что
R1 означает (С1-С4)алкильную группу, и/или
R2 означает -(СН2)n'-В, где n'=0, 1 и В является (С3-С5)циклоалкильной группой или (С1-С4)алкильной группой, необязательно замещенной одним или несколькими атомами фтора, (С1-С4)алкоксигруппой, и/или
U означает карбонильную группу или группу -СН2-, и/или
Y, Z, V и W обозначают, независимо один от другого, группу -СН- или атом углерода, необязательно замещенный группой R7, или гетероатом, такой как атом азота или атом серы, причем цикл должен быть ароматическим и содержать от 5 до 6 звеньев, и/или
R3, R4 означают, независимо один от другого, атом водорода или линейную (С1-С4)алкильную группу или R3 и R4 вместе с атомом углерода, с которым они связаны, образуют (С3-С5)циклоалкильную группу, и/или
m является целым числом, равным 1, 2, 3, 4, и/или
R5 означает атом водорода или (С1-С4)алкильную группу, и/или
R6 означает -(CH2)n-L, где n=0, 1, 2, 3 и L является группой, выбранной независимо из следующих групп:
(С1-С5)алкильной группы, необязательно замещенной (С1-С4)алкоксигруппой;
(С3-С5)циклоалкильной группы;
арильной группы, содержащей 6 атомов углерода, необязательно замещенной одним или несколькими атомами галогена;
гетероарильной группы, содержащей 5-6 звеньев, в которой по меньшей мере один гетероатом, выбранный из атома азота или серы, необязательно замещен (С1-С4)алкильной группой;
гетероциклической насыщенной группы, в которой указанный гетероцикл содержит 5-7 звеньев, по меньшей мере один гетероатом которого выбран из атома азота, атома кислорода, и необязательно замещенной в любой позиции, в том числе на атоме азота, одним или несколькими заместителями, выбранными из атома фтора, фтор(С1-С4)алкильной группы, (С1-С4)алкильной группы, линейной или разветвленной, (С3-С5)циклоалкильной группы или (С1-С4)алкилсульфонамидной группы, причем абсолютная конфигурация углерода, замещенного на указанном гетероцикле, может быть R или S конфигурацией или рацемической, и/или
R7 означает атом водорода, или (С1-С4)алкильную группу, или атом галогена,
в форме основания или аддитивной соли с кислотой, в форме сольвата, а также его энантиомеры и диастереоизомеры, включая его смеси.
3. Соединение формулы (I) по п.1, отличающееся тем, что U означает карбонильную группу.
4. Соединение формулы (I) по п.1, отличающееся тем, что цикл, содержащий Y, Z, V и W, выбран из фенильной, пиридиновой, тиазольной, тиофеновой групп.
5. Соединение формулы (I) по п.1, отличающееся тем, что R3, и/или R4, и/или R5 означают атом водорода.
6. Соединение формулы (I) по п.1, отличающееся тем, что звено -[C(R3)(R4)]m-U-N(R5)(R6) находится в пара- или мета-положении к циклу, с которым оно связано.
7. Соединение формулы (I) по п.1, отличающееся тем, что оно выбрано из одного из следующих соединений:
2-амино-1-этил-N-метил-4-оксо-7-{4-[2-оксо-2-(фениламино)этил]фенил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-[4-(2-{[(1-этилпирролидин-2-ил)метил](метил)амино}-2-оксоэтил)фенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-{4-[2-оксо-2-(пиридин-3-иламино)этил]фенил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-{4-[2-оксо-2-(пиридин-2-иламино)этил]фенил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-(4-{2-[(2-хлорфенил)амино]-2-оксоэтил}фенил)-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-(4-{2-[(3,5-дифторфенил)амино]-2-оксоэтил}фенил)-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-(4-{2-оксо-2-[(пиридин-4-илметил)амино]этил}фенил)-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-(4-{2-оксо-2-[(пиридин-2-илметил)амино]этил}фенил)-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-(4-{2-[(2-метоксиэтил)амино]-2-оксоэтил}фенил)-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-{4-[2-(циклопропиламино)-2-оксоэтил]фенил}-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-7-(4-{2-[(1-метилэтил)амино]-2-оксоэтил}фенил)-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-{4-[2-(циклопентиламино)-2-оксоэтил]фенил}-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-{4-[2-оксо-2-(пиразин-2-иламино)этил]фенил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-{4-[2-({[(2R)-1-этилпирролидин-2-ил]метил}амино)-2-оксоэтил]фенил}-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-{4-[2-({[(2S)-1-этилпирролидин-2-ил]метил}амино)-2-оксоэтил]фенил}-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-l-этил-N-метил-4-оксо-7-{4-[2-оксо-2-(пиримидин-4-иламино)этил]фенил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-{4-[2-оксо-2-(пиридин-4-иламино)этил]фенил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-7-(4-{2-[(2-морфолин-4-илэтил)амино]-2-оксоэтил}фенил)-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-{5-[2-оксо-2-(пиридин-2-иламино)этил]пиридин-2-ил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-7-{4-[1-метил-2-оксо-2-(пиридин-2-иламино)этил]фенил}-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-{6-[2-оксо-2-(пиридин-2-иламино)этил]пиридин-3-ил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-[4-(2-{[(4-этилморфолин-3-ил)метил]амино}-2-оксоэтил)фенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-[6-(2-{[(4-этилморфолин-3-ил)метил]амино}-2-оксоэтил)пиридин-3-ил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-{4-[2-({[(2S)-1-этил-4,4-дифторпирролидин-2-ил]метил}амино)-2-оксоэтил]фенил}-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-[4-(2-{[(4-этилморфолин-3-ил)метил]амино}-1-метил-2-оксоэтил)фенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-{4-[2-({[(2S,4R)-1-этил-4-фторпирролидин-2-ил]метил}амино)-2-оксоэтил]фенил}-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-{4-[2-({[(2R)-1-(2-фторэтил)пирролидин-2-ил]метил}амино)-2-оксоэтил]фенил}-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-7-{4-[2-({[(2R)-1-(метилсульфонил)пирролидин-2-ил]метил}амино)-2-оксоэтил]фенил}-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-{5-[2-({[(2R)-1-(2-фторэтил)пирролидин-2-ил]метил}амино)-2-оксоэтил]пиридин-2-ил}-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-{4-[2-оксо-2-({[(2R)-1-(2,2,2-трифторэтил)пирролидин-2-ил]метил}амино)этил]фенил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-{4-[2-({[(2R)-1-(2,2-дифторэтил)пирролидин-2-ил]метил}амино)-2-оксоэтил]фенил}-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-7-{4-[2-({[4-(1-метилэтил)морфолин-3-ил]метил}амино)-2-оксоэтил]фенил}-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-[4-(2-{[(4-циклопропилморфолин-3-ил)метил]амино}-2-оксоэтил)фенил]-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-{5-[2-({[(2R)-1-(2,2-дифторэтил)пирролидин-2-ил]метил}амино)-2-оксоэтил]пиридин-2-ил}-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-{4-[2-оксо-2-(1,3-тиазол-2-иламино)этил]фенил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-7-[4-(2-{[(1-метил-1Н-имидазол-5-ил)метил]амино}-2-оксоэтил)фенил]-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-(4-{2-оксо-2-[(2-пиридин-3-илэтил)амино]этил}фенил)-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-7-(4-{2-[(3-морфолин-4-илпропил)амино]-2-оксоэтил}фенил)-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-(4-{2-оксо-2-[(2-пиридин-2-илэтил)амино]этил}фенил)-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-(4-{2-оксо-2-[(2-фенилэтил)амино]этил}фенил)-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-(4-{2-оксо-2-[(3-фенилпропил)амино]этил}фенил)-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-7-[4-(2-{[2-(1-метил-1Н-пиррол-2-ил)этил]амино}-2-оксоэтил)фенил]-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-[4-(2-{[(1-этилпирролидин-2-ил)метил]амино}-2-оксоэтил)-1,3-тиазол-2-ил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-(3-метоксипропил)-N-метил-4-оксо-7-{4-[2-оксо-2-(пиридин-2-иламино)этил]фенил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-[4-(2-{[1-(2,2-дифторэтил)пирролидин-3-ил]амино}-2-оксоэтил)фенил]-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-[4-(3-{[(1-этилпирролидин-2-ил)метил]амино}-3-оксопропил)фенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-[4-(4-{[(1-этилпирролидин-2-ил)метил]амино}-4-оксобутил)фенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-[4-(2-{[(1-этилпирролидин-2-ил)метил]амино}-2-оксоэтил)фенил]-4-оксо-N-пропил-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-[4-(2-{[(1-этилпирролидин-2-ил)метил]амино}-2-оксоэтил)фенил]-N-метил-4-оксо-1-(2,2,2-трифторэтил)-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-циклопентил-7-[4-(2-{[(1-этилпирролидин-2-ил)метил]амино}-2-оксоэтил)фенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-[4-(5-{[(1-этилпирролидин-2-ил)метил]амино}-5-оксопентил)фенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
1-этил-7-[5-(2-{[(1-этилпирролидин-2-ил)метил]амино}-2-оксоэтил)тиофен-2-ил]-N,2-диметил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-{4-[2-({[(2R)-1-этилпирролидин-2-ил]метил}амино)-2-оксоэтил]фенил}-N-метил-1-(2-метилпропил)-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-{4-[1-({[(2R)-1-этилпирролидин-2-ил]метил}карбамоил)циклопропил]фенил}-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-{2-[2-({[(2R)-1-этилпирролидин-2-ил]метил}амино)-2-оксоэтил]-1,3-тиазол-4-ил}-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-{4-[2-({[(2R)-1-этилпирролидин-2-ил]метил}амино)-2-оксоэтил]-2-фторфенил}-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-[3-хлор-4-(2-{[(1-этилпирролидин-2-ил)метил]амино}-2-оксоэтил)фенил]-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-[3-фтор-4-(2-{[(1-этилпирролидин-2-ил)метил]амино}-2-оксоэтил)фенил]-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-7-[3-метил-4-(2-{[(1-этилпирролидин-2-ил)метил]амино}-2-оксоэтил)фенил]-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-(циклопропилметил)-7-[4-(2-{[(1-этилпирролидин-2-ил)метил]амино}-2-оксоэтил)фенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-[4-(2-{[(1-этилпирролидин-2-ил)метил]амино}-2-оксоэтил)-2-метилфенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-7-[3-(2-{[(1-этилпирролидин-2-ил)метил]амино}-2-оксоэтил)фенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-{3-[2-оксо-2-(пиридин-2-иламино)этил]фенил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид;
2-амино-1-этил-N-метил-4-оксо-7-{4-[2-(пиридин-2-иламино)этил]фенил}-1,4-дигидро-1,8-нафтиридин-3-карбоксамид,
в форме основания или аддитивной соли с кислотой, в форме сольвата, а также его энантиомеры и диастереоизомеры, включая его смеси.
8. Способ получения соединения формулы (I) по любому из пп.1-7, отличающийся тем, что соединение формулы (XI)
вводят в реакцию с соединением формулы HNR5R6 в присутствии агента сочетания и основания, причем R1, R2, R3, R4, R6, V, W, Y, Z, m имеют значения, указанные в п.1.
9. Способ получения соединения формулы (I) по любому из пп.1-7, отличающийся тем, что соединение формулы (VII)
в которой X означает атом галогена,
вводят в реакцию с соединением формулы (IXa)
в которой G означает (С1-С4)алкоксигруппу или группу -NR5R6;
R1, R2, R3, R4, R5, R6, V, W, Y, Z, m имеют значения, указанные в п.1.
10. Способ получения соединения формулы (I) по любому из пп.1-7, отличающийся тем, что соединение формулы (VII)
вводят в реакцию с соединением формулы (IXb)
в которой G означает (С1-С4)алкоксигруппу или группу -NR5R6;
X означает атом галогена и
R1, R2, R3, R4, R5, R6, V, W, Y, Z, m имеют значения, указанные в п.1.
11. Лекарственное средство для лечения заболеваний, связанных с активностью протеинкиназ, отличающееся тем, что оно содержит соединение формулы (I) по любому из пп.1-7, или аддитивную соль этого соединения с фармацевтически приемлемой кислотой, или сольват соединения формулы (I).
12. Фармацевтическая композиция для лечения заболеваний, связанных с активностью протеинкиназ, отличающаяся тем, что она содержит соединение формулы (I) по любому из пп.1-7 или фармацевтически приемлемую соль, сольват этого соединения, а также по меньшей мере один фармацевтически приемлемый эксципиент.
13. Применение соединения формулы (I) по любому из пп.1-7 для получения лекарственного средства, предназначенного для лечения заболеваний, связанных с активностью протеинкиназ.
14. Применение соединения формулы (I) по любому из пп.1-7 для получения лекарственного средства, предназначенного для лечения пролиферативных заболеваний, таких как злокачественные опухоли в жидких средах, хронические или острые лейкозы, лимфоцитарные лимфомы, болезнь Ходжкина и миелопролиферативные синдромы и миелодиспластические синдромы.
15. Применение соединения формулы (I) по любому из пп.1-7 для получения лекарственного средства, предназначенного для лечения пролиферативных заболеваний, таких как злокачественные солидные опухоли, например злокачественные опухоли легких (NSCLC), костей, поджелудочной железы, кожи, синдром Капоши, внутриглазные меланомы, злокачественные опухоли молочной железы, матки, шейки матки, яичников, эндометрия, влагалища, вульвы, мочеиспускательного канала, полового члена, предстательной железы, карцинома фаллопиевых труб, такие злокачественные опухоли, как GIST и анального канала, прямой кишки, тонкого кишечника, толстой кишки, желудка, пищевода, эндокринных желез, щитовидной железы, паращитовидной железы или надпочечников, саркомы мягких тканей, саркомы Эвинга, остеосаркомы, фибросаркомы кожи, злокачественные опухоли мочевого пузыря или почек, неоплазмы центральной нервной системы, опухоли позвоночного столба или десмоиды, глиомы ствола головного мозга и глиобластомы, аденомы слизистой оболочки носа и их метастазы.
16. Применение соединения формулы (I) по любому из пп.1-7 для получения лекарственного средства, предназначенного для лечения незлокачественных пролиферативных заболеваний, таких как рестеноз, атеросклероз, тромбоз, сердечная недостаточность, гипертрофия сердца, артериальная легочная гипертензия, фиброз, диабетическая нефропатия, гломерулонефрит, хронический пиелонефрит, гемангиомы, аутоиммунные заболевания, такие как псориаз, склеродерматиты, иммуносупрессии.
17. Соединение формулы (I) по любому из пп.1-7 для получения лекарственного средства, предназначенного для лечения заболеваний, связанных с активностью протеинкиназ.
18. Соединение формулы (I) по любому из пп.1-7 для получения лекарственного средства, предназначенного для лечения пролиферативных заболеваний, таких как злокачественные опухоли в жидких средах, хронические или острые лейкозы, лимфоцитарные лимфомы, болезнь Ходжкина и миелопролиферативные синдромы и миелодиспластические синдромы.
19. Соединение формулы (I) по любому из пп.1-7 для получения лекарственного средства, предназначенного для лечения пролиферативных заболеваний, таких как злокачественные солидные опухоли, например злокачественные опухоли легких (NSCLC), костей, поджелудочной железы, кожи, синдром Капоши, внутриглазные меланомы, злокачественные опухоли молочной железы, матки, шейки матки, яичников, эндометрия, влагалища, вульвы, мочеиспускательного канала, полового члена, предстательной железы, карцинома фаллопиевых труб, такие злокачественные опухоли, как GIST и анального канала, прямой кишки, тонкого кишечника, толстой кишки, желудка, пищевода, эндокринных желез, щитовидной железы, паращитовидной железы или надпочечников, саркомы мягких тканей, саркомы Эвинга, остеосаркомы, фибросаркомы кожи, злокачественные опухоли мочевого пузыря или почек, неоплазмы центральной нервной системы, опухоли позвоночного столба или десмоиды, глиомы ствола головного мозга и глиобластомы, аденомы слизистой оболочки носа и их метастазы.
20. Соединение формулы (I) по любому из пп.1-7 для получения лекарственного средства, предназначенного для лечения незлокачественных пролиферативных заболеваний, таких как рестеноз, атеросклероз, тромбоз, сердечная недостаточность, гипертрофия сердца, артериальная легочная гипертензия, фиброз, диабетическая нефропатия, гломерулонефрит, хронический пиелонефрит, гемангиомы, аутоиммунные заболевания, такие как псориаз, склеродерматиты, иммуносупрессии.
21. Комбинация по меньшей мере одного соединения формулы (I) по любому из пп.1-7 по меньшей мере с одним химиотерапевтическим агентом, предназначенная для лечения заболеваний, связанных с активностью протеинкиназ.
Текст
ПРОИЗВОДНЫЕ ПИРИДИНО-ПИРИДИНОНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ТЕРАПИИ Изобретение относится к производным пиридино-пиридинонов общей формулы (I), в которой R1,R2, R3, R4, R5, R6, R7, V, W, Y, Z, m имеют значения, указанные в описании. Изобретение относится к способу получения соединений и к применению их в терапии. Настоящее изобретение относится к производным пиридино-пиридинонов, замещенных в положении 7 арилом или гетероарилом, который сам соответствующим образом замещен звеном типа-[C(R3)(R4)]m-U-N(R5)(R6), к способу их получения и к применению их в терапии в качестве ингибиторов киназной активности рецепторов к лигандам PDGFs (Platelet derived growth factors) и, необязательно,рецепторов к лиганду FLT3 (fms-like tyrosine kinase receptor). Рецепторы FLT3 и PDGF-R являются членами III класса семейства тирозинкиназных рецепторов(RTK), в которое входят также рецептор фактора стволовых клеток (c-kit) и рецептор фактора M-CSF (cfms). Они характеризуются внеклеточным доменом, состоящим из 5 иммуноглобулинподобных доменов,содержащих область связывания с лигандом, трансмембранный домен и внутриклеточную часть, состоящую из околомембранного домена, киназного домена, разделенного на две части инсерционным доменом (split domain)(UllrichSchlessinger, 1990). Связывание лигандов с рецепторами RTK индуцирует димеризацию рецепторов, активацию их тирозинкиназной части, которая приводит к трансфосфорилированию тирозиновых остатков (WeissSchlessinger, 1998). Таким образом, эти фосфорилированные остатки являются местом связывания с внутриклеточными белками, участвующими в передаче сигнала, которые in fine провоцируют различные клеточные ответы: сохранение, деление, пролиферацию, дифференцировку или же миграцию клеток(Claesson-Weish, 1994). Ген, кодирующий FLT3, находится на хромосоме 13q12 (Rosnet et al., 1992) и кодирует белок FLT3(антиген CD135), специфически экспрессируемый гематопоэтическими клетками, более точно, незрелыми клетками, такими как гематопоэтические клеточные линии и прогениторные мультипотентные клетки миелоидного и лимфоидного ряда, и его экспрессия исчезает по мере дифференцировки гематопоэтических клеток. Его лиганд, лиганд FLT3, индуцирует димеризацию рецептора и последующее аутофосфорилирование внутриклеточной части рецептора, которое приводит к активации сигнального каскада. Эффекты активации рецептора собственным лигандом означают выживаемость и увеличение числа прогениторных мультипотентных клеток. Были обнаружены две изоформы PDGFs-рецепторов: цепь PDGF-R-альфа и цепь PDGL-R-бета, которые, в ответ на связывание собственных лигандов, гомо- или гетеродимеризуются и индуцируют внутриклеточную сигнализацию. Рецепторы к PDGF экспрессируются, главным образом, клетками мезенхимного происхождения и обнаружены, в частности, на фибробластах, на гладкомышечных клетках, перицитах и в глиальных клетках (Ross et coll. 1986, Heldin, 1992). Фактор роста тромбоцитов "Platelet Derived Growth Factor", PDGF, белок с молекулярной массой около 30000 Да, секретируется, главным образом, тромбоцитами, и, дополнительно, эндотелием, гладкомышечными клетками сосудов и моноцитами. Фактор роста тромбоцитов образуется из двух полипептидных цепей, соединенных между собой сульфидными мостиками, образуя либо гомодимеры, либо гетеродимеры. Четыре гена (7 р 22, 22q13, 4q31 и 11q22) описаны как гены, способные кодировать 4 разные полипептидные цепи (А, В, С и D), которые после димеризации образуют пять биологически активных лигандов PDGF-AA, ВВ, СС, DD и АВ (в качестве обзора см. Yu et al. 2003). Была выявлена их специфичность, в частности лиганд PDGF-AA специфичен для изоформы альфа-рецептора, лиганд PDGF-D специфичен для формы ВВ и лиганд PDGF-C специфичен для формы альфа и альфа/бета. ЛигандыPDGFs являются мощными митогенами, а также задействованы в явлениях миграции, выживания, апоптоза и трансформации клеток. Ингибиторы активности альфа-, бета-рецепторов PDGF и FLT3 используются в различных терапевтических областях. Среди физиопатологических состояний, в которых могут быть задействованы эти рецепторы, можно назвать рак в жидких средах или лейкозы, злокачественные солидные опухоли с метастазами или без, где мишенями служат опухолевые клетки и/или клетки околоопухолевой зоны (сосудистые, фибробластовые), а также фиброзы и сосудистые заболевания. А. Рак в жидких средах. Лейкозы бывают различных типов и развиваются либо по миелоидному типу, либо по лимфиодному типу. Экспрессия FLT3 в лейкозных клетках, образующихся при остром миелоидном лейкозе (ОМЛ), составляет порядка 100% в зависимости от конкретного больного, таким образом, FLT3 способствует стимуляции выживания и пролиферации лейкозных клеток (Carow et al., 1996; Drexler et al., 1996, Stacchini etal., 1996). Кроме того, FLT3 является местом локализации активирующих мутаций, обнаруженных у 22-30% взрослых больных ОМЛ и у 11% детей. Чаще всего, речь идет о внутреннем тандемном удвоении (ITD),наблюдаемом на трансмембранном участке рецептора (более конкретно, между 14 и 15 экзонами). Эти мутации сохраняют рамку считывания, при этом размер мутаций может варьировать между 18 и 111 парами оснований. Реже, т.е. приблизительно у 7% больных ОМЛ, обнаружена точечная мутация по остатку D835, расположенному в киназном домене. В большинстве случаев, формы FLT3 ITD создают более высокий риск развития рецидива болезни и являются прогностическими маркерами низкой выживаемости больных. Эти два типа мутаций приводили к конститутивной активации киназного домена, не зависящей от стимуляции со стороны лиганда, и показали себя в качестве фактора, трансформирующего ге-1 019362 матопоэтические клетки in vitro и in vivo (Mizuki et al., 2000; Tse et al., 2000). Авторы Kelly et coll. (2002) с помощью тонкого эксперимента на реконструкционной модели костного мозга у мышей показали, что форма FLT3 ITD вызывает миелопролиферативный синдром. Интерес к использованию ингибиторов тирозинкиназной активности был подтвержден одновременно в тестах in vitro и in vivo несколькими исследовательскими группами, а также исследованиямиFLT3 ITD недавнего времени на реконструкционной модели костного мозга; этот ингибитор показал себя в качестве продукта, способного индуцировать регрессию опухоли и увеличивать продолжительность жизни животных (Ofarrel, 2003). Более того, последние данные показали интерес использования комбинации таких ингибиторов с цитотоксическими препаратами, каким является, например, даунорубицин (Levis et al., 2004). Интересным представляется и тот факт, что бластические клетки типа ОМЛ могут также суперэкспрессировать другие рецепторы с киназной активностью, такие как c-kit или же PDGF-R. Миелопролиферативный/диспластический синдромы. Довольно часто цитогенетические аномалии, вызванные хромосомными транслокациями, связывают с миелопролиферативными синдромами. Эти хромосомные перестройки приводят к генерированию разрегулированных слитых белков с тирозинкиназной активностью, которые принимают участие в пролиферации миелоидных бластных клеток. Слитые белки с киназной активностью рецептора PDGF-R бета. Слитые белки с киназной активностью рецептора PDGF-R бета состоят из внутриклеточной части рецептора PDGF-R бета и, с другой стороны, из N-концевого домена другого белка (обычно фактора транскрипции). В частности, они имеют отношение к хроническому миеломоноцитарному лейкозуTel/PDGF-R бета. Наиболее показателен последний белок. Он образуется при транслокацииt(5;12)(q31;p12) и кодирует слитый белок, состоящий из N-концевой области фактора транскрипции Tel и С-концевой области рецептора PDGF-R бета. Олигомеризационный домен, находящийся в зоне Tel, приводит к образованию димерной формы слитого белка и к конститутивной активации киназного домена. Этот белок показал себя in vitro как соединение, способное трансформировать в несколько приемов гематопоэтические клетки, в частности, более подробно об этом описано в статье M. Carrol et coll. PNAS,1996, 93, 14845-14850. In vivo этот слитый белок приводит к синдрому гиперпролиферации миелоидных клеток (Ritchie et coll. 1999). Кроме того, у животных и, как показали клинические испытания, у человека ингибиторы тирозинкиназной активности ингибируют пролиферацию бластных клеток и позволяют затормозить процесс лейкогенеза. Слитые белки с киназной активностью рецептора PDGF-R альфа. К двум слитым белкам, в состав которых входит рецептор PDGF-R альфа, относят белки: bcr-PDGFR альфа, присутствующий в клетках атипичной хронической миелоидной лейкемии (ХМЛ), и FIP1L1PDGF-R альфа, обнаруженный в субпопуляции лейкемических клеток, причем источником происхождения LEC "эозинофильной лейкемии" является синдром гиперэозинофилии (Griffin et coll. 2003). Этот слитый белок обладает конститутивной активностью киназного домена в рецепторе PDGF-R альфа и является ответственным за беспорядочную пролиферацию упомянутых клеток. Ингибиторы киназной активности рецептора PDGF-R альфа показали себя эффективными в отношении пролиферации FIP1L1-PDGF-R альфа положительных клеток, поэтому в последнее время соединение, являющееся ингибитором такого рода, было рекомендовано для лечения HES/CEL (гиперэозинофильного синдрома/хронической эозинофильной лейкемии). Таким образом, ингибирование киназной активности рецепторов PDGF-R альфа и бета и активности FLT3wt и FLT3ITD, как это показали соединения согласно изобретению, представляет терапевтический интерес для лечения ОМЛ. Помимо ОМЛ и миелопролиферативных синдромов другие виды лейкозов также могут представлять интерес в качестве мишеней для таких ингибиторов, в том числе лейкозы B-LAL и T-LAL (острые лимфоидные Т-клеточные или В-клеточные лейкозы), при которых также экспрессируется FLT3. Кроме того, в связи с тем, что FLT3 в норме экспрессирован на клетках гематопоэтических клеточных линий, и тем, что экспрессия этого рецептора обнаружена на клетках лейкемических клеточных линий, ингибиторы киназной активности FLT3 могут оказаться интересными для лечения лейкозов любого типа (в том числе ХМЛ, хронический миелоидный лейкоз), при которых клетки лейкемических клеточных линий задействованы в устойчивости рецидива заболевания. В. Солидные злокачественные опухоли. Ингибиторы тирозинкиназной активности альфа- и бета-рецепторов PDGF-R могут быть интересны для лечения солидных злокачественных опухолей, выбирая в качестве мишени либо непосредственно опухолевую клетку, которая благодаря аутокринному или паракринному механизму становится чувствительной к ингибитору тирозинкиназной активности PDGF-R, либо клетки, окружающие опухоль, в этом случае дестабилизируется сосудистая сеть опухоли, что благоприятствует соединению ингибитора с другими терапевтическими агентами. Примеры солидных злокачественных опухолей, где в качестве мишени выбрана опухолевая клетка. Рак мягких тканей: саркома Эвинга. Саркома Эвинга представляет собой вид костного рака, возникающий главным образом у детей и молодых людей (средний возраст 13 лет). Этот вид рака составляет 10% от числа примитивных костных опухолей и имеет высокий риск метастазирования. Это редко встречающаяся опухоль, которая затрагивает 2-3 человека на миллион жителей в год. Опухолевые клетки характеризуются транслокацией хромосомы t(11;12), кодирующей слитый белок EWS/FLI1. Ответственными клетками являются клетки мезенхимы, которые экспрессируют рецептор PDGF-R бета, индуцирующий подвижность и рост клеток саркомы Эвинга при стимулирующем участии PDGFBB (Uren et al. 2003). Кроме того, Zwerner and May (2001) доказали существование экспрессии PDGF-C клетками саркомы Эвинга. Эти же клетки экспрессируют также рецептор RTK c-kit и было показано, что ингибитор киназной активности рецептора PDGF-R и c-kit способен ингибировать опухолевый рост клеток саркомы Эванса в мышиной модели ксенотрансплантата (Merchant et coll., 2002). Опухоль соединительной ткани (GIST, фибросаркома кожи).GIST (стромальные гастроинтестинальные опухоли). Группа под руководством Fletcher (2004) заинтересовалась тем, что у 15% больных GIST ген KIT не подвергается мутации и не проявляет суперэкспрессию (KIT-wt). Исследователи наблюдали сильную гиперэкспрессию рецептора PDGF-R альфа. Эта ситуация наблюдалась приблизительно в одной трети случаев заболеваний GIST KIT-wt. Что касается мутаций рецепторов PDGF-RA, то исследователи наблюдали эти мутации (составившие 35%) в случаях, когда KIT экспрессировался в норме. Мутации рецептора PDGF-RA увеличивали тирозинкиназную и конститутивную активность и затрагивали аспартамовую кислоту в положении 842 аминокислотной последовательности. Также как и в случае саркомы Эвинга, два ингибитора киназной активности рецепторов c-kit и PDGF-R показали свою эффективностьin vitro и in vivo в отношении пролиферации клеток мутированного PDGF-R альфа (Le Tourneau et coll. 2007; Cortess et coll., 2005). Фибросаркомы кожи (Дарье-Феррана или выбухающие или DFSP). Фибросаркома кожи Дарье-Феррана (или DFSP) представляет собой клеточную фузиформную опухоль кожи с промежуточной степенью злокачественности, характеризующуюся медленным развитием и высоким риском рецидива в случае неполного удаления. Генетическая аномалия, отмеченная в 95% случаев заболевания, открыта в 1990 году, когда была обнаружена, в частности, транслокация хромосом 17 и 22 t(17-22) (q22;q13), которая привела к слиянию генов COL1A1 и PDGF В и большому количеству белка PDGFB, гиперэкспрессирующего свой тирозинкиназный рецептор PDGFR. Ингибирование киназной активности рецептора PDGF-R является многообещающей терапией, поскольку in vitro это приводит к снижению роста опухоли в моделях опухолевого трансплантата у иммунодефицитных мышей (SjoblomT. et coll., 2001). Кроме того, клинические исследования показали эффективность (полная или частичная ремиссия) такой молекулы при заболеваниях DFSP (в качестве обзора см. Мс Arthur, 2007). Глиомы и глиобластомы. Глиобластома является наиболее распространенной и наиболее агрессивной опухолью головного мозга при средней продолжительности жизни больного около 1 года. PDGF и его рецепторы (альфа и бета) чаще всего экспрессируются в глиомах. Существует предположение, что аутокринная/паракринная петля может способствовать патогенезу этих опухолей. Рецептор PDGF-R альфа экспрессируется предпочтительно в клетках опухоли, тогда как рецептор PDGF-R бета экспрессируется предпочтительно в сосудистых эндотелиальных клетках опухоли. Блокировка киназной активности рецептора PDGF-R показала свою эффективность: 1) in vitro путем уменьшения числа колоний в мягком агаре и ингибирования пролиферации клеточных линий, 2) путем снижения опухолевого роста в моделях трансплантата у голых мышей, 3) в возможности сочетать указанную блокировку с облучением на модели внутричерепной трансплантации клеточных линий глиобластом (Oerbel et al., 2006; Geng et coll., 2005, Strawn et coll.,1994, Chin et al., 1997). Таким образом, соединения согласно изобретению представляют интерес для лечения саркомы Эванса, GIST, фибросарком кожи, а также демоидных опухолей, гемангиом и других фибросарком, при которых наблюдается экспрессия PDGF-R. С. PDGF-R рецептор в качестве терапевтической мишени в околоопухолевой зоне. Ангиогенез. Клетки околоопухолевой зоны составляют неотъемлемую часть развития рака независимо от того,первичная опухоль или вторичная (метастазы). Среди клеток этой зоны, которые экспрессируют рецептор PDGF и в отношении которых выявлена роль этого рецептора, встречаются клетки стенок сосудов,т.е. перициты, и гладкомышечные клетки, а также активированные фибробласты. Ангиогенез является процессом генерирования новых капилляров из предсуществующих кровеносных сосудов или же путем мобилизации и дифференцировки клеток костного мозга. Так, была отмечена в процессах неоваскуляризации опухолей одновременная неконтролируемая пролиферация эндотелиальных клеток и мобилизация ангиобластов из костного мозга. Было показано in vitro и in vivo, что многие факторы роста стимулируют эндотелиальную пролиферацию, такие как, например, факторы VEGF иFGF. Помимо указанных механизмов было также установлено, что клетки стенок, такие как перициты и гладкомышечные клетки, принимают участие в стабилизации новообразованной сети сосудов. Неправильная экспрессия рецептора PDGF-R бета является причиной дефицита перицитов у мышей и приводит к смерти животных в конце беременности, вызванной микрокровоизлияниями и отеками (Hellstrom etal., 1999, Hellstrom et al., 2001). Согласно тонкому эксперименту с трансплантацией клеток экспрессияPDGF-R бета перицитами необходима для привлечения перицитов к сосудам опухоли путем удерживания PDGF-B эндотелиальными клетками, а также PDGF-B, секретируемого опухолевыми клетками(Abramsson et al., 2003). На трансгенной модели RiplTag2 рака поджелудочной железы, описанной Songet coll., также была показана экспрессия PDGF-R бета на прогениторных периваскулярных клетках головного мозга, происходящих из костного мозга, при этом прогениторные клетки дифференцировались в зрелые перициты вокруг опухоли. Интерес блокировки активности рецептора PDGF-R на опухолевых перицитах был продемонстрирован на примере использования ингибитора тирозинкиназной активности рецептора PDGF-R в животных моделях (трансгенная модель опухоли поджелудочной железы и модель имплантированной глиомы) и оказалось, что воздействие на рост опухоли был значительным при сочетании с ингибитором киназной активности рецептора VEGF-R (Bergers et coll., 2003). Литературные данные (Cao et al., 2002, Fons et coll.,2004) подтверждают также участие PDGF-R альфа и PDGF-C в ангиогенезе и в дифференцировке прогениторных эндотелиальных клеток в перициты и гладкомышечные клетки. Активированные фибробласты. Рецептор PDGF-R находится в избытке в опухолевой строме и обнаруживается на активированных фибробластах (миофибробласты). Было показано на примере двух опытов, что ассоциация ингибиторов или антагонистов рецептора PDGF-R с цитотоксическими агентами приводит к снижению микроплотности сосудов раковых опухолей яичников (Apte et coll., 2004) и раковых опухолей поджелудочной кислоты (Hwang et coll., 2003). Рецептор PDGF-R бета регулирует давление в интерстициальных тканях опухоли (Heuchel et coll., 1999), а совместное использование ингибиторов PDGF-R и химиотерапевтических агентов улучшает их доставку в опухолевые клетки и одновременно уменьшает внутриопухолевое давление (Griffon-Etienne, 1999). И, наконец, на мышиной модели продемонстрировано, что введение ингибитора киназной активности PDGF-R улучшает усвоение химиотерапевтических агентов опухолью и,таким образом, увеличивает их эффективность (Griffon-Etienne, 1999; Pietras et coll., 2002; Pietras et coll.,2003). Эти эффекты несомненно являются эффектом TAF (tumour associated fibroblastes, опухольассоциированных фибробластов), называемых также CAF (carcinoma associated fibroblastes, карциномаассоциированными фибробластами), активированных фибробластов, находящихся вокруг опухоли, которые экспрессируют PDGF-R, как это подтверждено недавними работами Hwang et coll., (2008), Kain et al.(2008), Pietras et al. (2008) на моделях vivo рака поджелудочной железы и цервикального канцерогенеза. Стимуляция, вызванная PDGF-лигандом, продуцируемым опухолевыми клетками, стимулирует фибробласты, которые производят внеклеточный матрикс и, таким путем, увеличивают интерстициальное давление. Кроме того, уменьшая это давление, можно облегчить доставку лекарств к опухоли молочной железы и, таким образом, увеличить их эффективность. Это означает, что активированные фибробласты,находящиеся в опухолевой строме, представляют новую терапевтическую мишень в онкологии (в качестве обзора см. BouzinFeron, 2007). Метастазы. Многие работы указывают на то, что пара рецептор PDGFR и его PDGF-лиганд участвует в развитии метастазирования, и утверждают, что они оказывают воздействие на ангиогенез и на метастазирование через кровяное русло, а также оказывают прямое воздействие на лимфангиогенез и, следовательно,на метастазирование через лимфатическое русло. В частности, в одном научном журнале документально подтверждается прямое участие PDGF-BB в лимфангиогенезе и в лимфатическом метастазировании (Caoet coll., 2005). Но в большинстве работ говорится об экспрессии PDGF-R в ткани, окружающей метастазы, которая способствует возникновению и развитию вторичных опухолей. Чаще всего в качестве примера ссылаются на развитие метастаз в кости. Пример рака предстательной железы. Кость часто является местом возникновения метастаз. 85-100% больных, которые умирают от рака предстательной железы, имеют метастазы в костях. Химиотерапия улучшает как показатель выживаемости без прогрессии заболевания, так и общий показатель выживаемости, однако из-за очень высокой гетерогенности метастаз в кости у одного и того же пациента химиотерапия оказывается неэффективной как лечебное средство. Было показано на модели иммунодефицитной мыши, что PDGF-BB играет важную роль в развитии остеобластических метастаз костной ткани in vivo (Yu et coll., 2003). Что касаетсяPDGF-DD, то он ускоряет рост опухолевых клеток предстательной железы и увеличивает их взаимодействие с клетками стромы. Экспрессия рецептора PDGF альфа и бета обнаружена соответственно в 6275% случаев рака предстательной железы. Кроме того, иммуногистохимический анализ показал, что опухоль предстательной железы и ее метастазы экспрессировали PDGF-R (Hwang et coll., 2003). АвторыKim et coll., (2003) показали, что PDGF-R экспрессируется в костных метастазах и эндотелиальных клетках кровеносных сосудов, зависимых от метастаз. Ингибитор тирозинкиназного PDGF-R, соединенный с цититоксическим агентом, значительно снижает количество костных метастаз рака предстательной железы в мышиной модели (Uehara et coll., 2003). Кроме того, эта же самая ассоциация приводит к апоптозу опухолевых клеток, эпителиальных клеток кровеносных сосудов и к ингибированию роста опухолевых клеток в кости. Блокировка этих рецепторов и их путей сигнализации в кости представляет собой новое терапевтическое решение (Hwang et coll., 2003; Uehara et coll., 2003). Клинические испытания на людях показали преимущество, которое дает ассоциация ингибитора PDGF-R и цитотоксического агента пациентам, болеющим гормонорезистентным раком предстательной железы с костными метастазами. Уменьшение уровня маркера (предстательная железа-специфический антиген) PSA50% наблюдалось у 38% пациентов. Средняя продолжительность ответа PSA составляла 8 месяцев, а продолжительность периода выживания без прогрессии заболевания составляла 11 месяцев. Исходя из этих различных публикаций ясно, что соединения согласно изобретению представляют интерес для лечения солидных раковых опухолей в связи с их эффектом, производимым на клетки окружающей опухоль ткани, и с эффектом, достигаемым при использовании ассоциации с другими терапевтическими агентами, таким как цитотоксические агенты, или ингибиторами ангиогенеза.D. Фиброзы. Причиной фиброзов часто является одно первичное событие, такое как раковая опухоль, радиотерапия, гепатиты, алкоголизм. Участие в них PDGF ясно доказано на примере фиброза легких (включая асбестоз), фиброза почек (гранулонефрит), фиброза костного мозга (часто ассоциированного с мегакариоцитарным лейкозом), индуцированного радиотерапией, а также на примере фиброза печени и поджелудочной железы (связанного с алкоголизмом или с гепатитами) (в качестве обзора см. J.C. Bonner,2004). В частности, была четко показана суперэкспрессия PDGF, а также представлены связанные с ней результаты испытания, полученные на моделях in vivo с ингибиторами ТК-активности PDGF-R. Среди этих испытаний одно из них, осуществленное исследователями Einter et coll. (2002), доказало, что PDGFCC является мощным индуктором почечного фиброза. Исследователи провели тест на эффективность нейтрализующих антител в моделях с односторонним перевязыванием уретры, при котором особенно быстро развивается фиброз. Они отметили ярко выраженный антифиброзный эффект с одновременным снижением скопления миофибробластов, снижением скопления внеклеточного матрикса и снижением слоев коллагена IV типа. Другое исследование, проведенное на модели фиброза легких, индуцированного блеомицином у мышей, показало эффективность ингибитора ТК-активности PDGF-R в отношении предупреждения фиброза благодаря ингибированию пролиферации мезенхимальных клеток (Aono etcoll., 2005). На модели фиброза, индуцированного асбестом, ингибитор PDGF-R TK снизил прогрессию фиброза в легочной паренхиме и уменьшил коллагеновый слой (Vuorinen K., Gao F., Oury T.D., KinnulaLung Res. 2007 Sep; 33(7): 357-73). Многими исследовательскими группами было подтверждено участиеPDGF-R в фиброзе печени. Было четко доказано, что PDGF ВВ и DD обладают профиброгенным действием на печеночные звездчатые клетки (Rovida et coll., 2008; Borkham-Kamphorst et coll., 2007). В тесте invivo ингибитор PDGF-R TK оказался способным снизить фиброгенез на ранней стадии на модели с перевязанным желчным протоком у крысы (Neef et coll., 2006). Таким образом, на основании опубликованных в литературе данных, соединения согласно изобретению представляют терапевтический интерес для лечения различных типов фиброза. Е. Сосудистые заболевания: атеросклероз и рестеноз, артериосклероз. Пролиферация и миграция гладкомышечных клеток сосудов способствуют гипертрофии интимы артерий и таким образом играют доминирующую роль в формировании атеросклероза и рестеноза после ангиопластики и эндоартерэктомии. Было ясно показано in vitro и in vivo на моделях животных, чтоPDGF задействован в указанных явлениях. В частности, in vivo было показано увеличение экспрессииPDGF-R последовательно снижает размер поражений торакальной и абдоминальной артерии у диабетических мышей АроЕ-КО (животные обработаны стрептозотоцином). Другое исследование показало, что ингибирование сигнальных путей, индуцированное PDGF (TK или антисмысловой PDGF-A), приводит к снижению образования неоинтимы в моделях "ballon injury" и "coronary artery restenosis" (Deguchi J.,1999, Ferns et coll. 1991, Sirois et al., 1997, Lindner et coll. 1995). Таким образом, ингибиторы тирозинкиназной активности PDGF-R, такие как соединения согласно изобретению, представляют собой терапию выбора, либо в индивидуальном виде, либо в сочетании с антагонистами других факторов роста, задействованных в названных патологиях, таких как FGF, при лечении патологий, связанных с пролиферацией гладкомышечных клеток сосудов, таких как атеросклероз, пост-ангиопластический рестеноз, или последствия установки эндоваскулярных протезов (стентов) или в процессе аортокоронарного шунтирования. Соединения согласно изобретению, обладающие ингибирующей активностью в отношении ТК активности PDGF-R, представляют интерес для лечения указанных сосудистых заболеваний.F. Другие заболевания. Соединения согласно изобретению могут быть показаны для лечения других патологий, в частности артериальной легочной гипертензии (НТАР) в идиопатической форме. НТАР характеризуется стойким повышенным давлением в легочной артерии, приводящим к недостаточности правого желудочка и, часто, к смерти пациента. Она ассоциируется с ростом пролиферации и миграции гладкомышечных клеток сосудов легких. Авторы Schermuly et coll., (2005) показали, что ингибирование тирозинкиназной активности рецепторов PDGF значительно улучшает течение болезни. Для этого авторы использовали среди прочего модель экспериментальной легочной артериальной гипертензии у крысы, созданной введением монокроталина в течение 28 дней. Все крысы, обработанные лекарственным средством, выжили, а 50% крыс из контрольной необработанной группы умерли. Настоящее изобретение относится к соединениям формулы (I)R2 означает -(СН 2)n'-В, где n'=0, 1, 2, 3, 4 и В является (С 3-С 5)циклоалкильной группой или (С 1 С 4)алкильной группой, необязательно замещенной одним или несколькими атомами фтора, (С 1 С 4)алкоксигруппой;U означает карбонильную группу или группу -СН 2-;Y, Z, V и W обозначают, независимо один от другого, группу -СН- или атом углерода, замещенный группой R7, или гетероатом, такой как атом азота или атом серы, или не содержит его, причем цикл должен быть ароматическим и содержать от 5 до 6 звеньев;R3, R4 означают, независимо один от другого, атом водорода или линейную (С 1-С 4)алкильную группу или R3 и R4 вместе с атомом углерода, с которым они связаны, образуют (С 3-С 5)циклоалкильную группу;R5 означает атом водорода или (С 1-С 4)алкильную группу;L является группой, выбранной независимо из следующих групп:(С 3-С 5)циклоалкильной группы; арильной группы, содержащей 6 атомов углерода, необязательно замещенной одним или несколькими атомами галогена; гетероарильной группы, содержащей 5-6 звеньев, в которой по меньшей мере один гетероатом, выбранный из атома азота или серы, необязательно замещен (С 1-С 4)алкильной группой; гетероциклической насыщенной группы, в которой указанный гетероцикл содержит 4-7 звеньев, по меньшей мере один гетероатом которого выбран из атома азота, атома кислорода, и необязательно замещенной в любой позиции, в том числе на атоме азота, одним или несколькими заместителями, выбранными из атома фтора, фтор(С 1-С 4)алкильной группы, (С 1-С 4)алкильной группы, линейной или разветвленной, (С 3-С 5)циклоалкильной группы или (С 1-С 4)алкилсульфонамидной группы, причем абсолютная конфигурация углерода, замещенного на указанном гетероцикле, может быть R или S конфигурацией,или рацемической;R7 означает атом водорода, или (С 1-С 4)алкильную группу, или атом галогена. Соединения формулы (I) могут содержать один или несколько асимметрических атомов углерода. Следовательно, они могут находиться в форме энантиомеров или диастереоизомеров. Эти энантиомеры,диастереоизомеры, а также их смеси, в том числе рацемические, составляют часть изобретения. Например, когда L означает гетероциклическую группу, абсолютная конфигурация атома углерода,замещенного на указанном гетероцикле, может быть R или S. Соединения формулы (I) могут находиться, в частности, в виде оснований или аддитивных солей с кислотами. Такие аддитивные соли составляют часть изобретения. Эти соли можно получать с помощью фармацевтически приемлемых кислот, а также с помощью других кислот, полезных, например, для очистки или выделения соединений формулы (I), они также составляют часть изобретения. Соединения формулы (I) могут также существовать в форме сольватов, а именно в форме ассоциаций или комбинаций с одним или несколькими молекулами растворителя. Такие сольваты также составляют часть изобретения. В рамках настоящего изобретения подразумевают под: алкильной группой: алифатическую насыщенную группу, содержащую 1-4 атома углерода, причем цепь является линейной или, когда алкильная цепь содержит не менее 3 атомов углерода, она может быть разветвленной или циклической (включая циклизацию только частичную). В качестве примеров можно назвать группы метильную, этильную, пропильную, изопропильную, бутильную, изобутильную,трет-бутильную, метилциклопропильную и т.д., а также циклоалкильные группы, описанные ниже; циклоалкильной группой: циклическую алкильную группу, содержащую 3-5 атомов углерода, все атомы углерода которой заняты в цикле. Можно назвать циклопропильную, циклобутильную, циклопентильную группы; алкоксигруппой: -О-алкильную группу, в которой алкильная группа имеет значение, описанное выше; атомом галогена: фтор, хлор, бром или йод; галогеналкильной группой: группу, содержащую алкильную группу, которая определена выше, в которой один или несколько атомов водорода замещены атомом галогена, который определен выше; гетероатомом: атом азота, кислорода или серы; арильной группой: ароматическую моноциклическую группу, содержащую 6 звеньев, например фенильную группу; гетероарильной группой: ароматическую моноциклическую группу, содержащую 5-6 звеньев, в которой 1-3 гетероатома являются такими, как они определены выше. В качестве примера можно назвать пиридиновую, пиразиновую, пиримидиновую, имидазольную, пиррольную, тиофеновую, тиазольную группы; гетероциклической группой: алкильную циклическую группу, содержащую 4-7 звеньев, в которой один или несколько гетероатомов определены выше. В качестве примеров можно назвать пирролидиновую, морфолиновую, пиперидиновую, пиперазиновую группы. Среди соединений формулы (I) согласно изобретению можно назвать группу соединений, которые определены, как указано ниже:R2 означает -(СН 2)n'-В, где n'=0, 1 и В является (С 3-С 5)циклоалкильной группой или (С 1 С 4)алкильной группой, необязательно замещенной одним или несколькими атомами фтора, (С 1 С 4)алкоксигруппой, и/илиU означает карбонильную группу или группу -СН 2-, и/илиY, Z, V и W обозначают, независимо один от другого, группу -СН- или атом углерода, необязательно замещенный группой R7, или гетероатом, такой как атом азота или атом серы, причем цикл должен быть ароматическим и содержать от 5 до 6 звеньев, и/илиR3, R4 означают, независимо один от другого, атом водорода или линейную (С 1-С 4)алкильную группу или R3 и R4 вместе с атомом углерода, с которым они связаны, образуют (С 3-С 5)циклоалкильную группу, и/илиR5 означает атом водорода или (С 1-С 4)алкильную группу, и/илиR6 означает -(CH2)n-L, в которой n=0, 1, 2, 3 и L является группой, выбранной независимо из следующих групп:(С 3-С 5)циклоалкильной группы; арильной группы, содержащей 6 атомов углерода, необязательно замещенной одним или несколькими атомами галогена; гетероарильной группы, содержащей 5-6 звеньев, в которой по меньшей мере один гетероатом, выбранный из атома азота или серы, необязательно замещен (С 1-С 4)алкильной группой; гетероциклической насыщенной группы, в которой указанный гетероцикл содержит 5-7 звеньев, по меньшей мере один гетероатом которого выбран из атома азота, атома кислорода, и необязательно замещенной в любой позиции, в том числе на атоме азота, одним или несколькими заместителями, выбранными из атома фтора, фтор(С 1-С 4)алкильной группы, (С 1-С 4)алкильной группы, линейной или разветвленной, (С 3-С 5)циклоалкильной группы или (С 1-С 4)алкилсульфонамидной группы, причем абсолютная конфигурация углерода, замещенного на указанном гетероцикле, может быть R или S конфигурацией,или рацемической, и/илиR7 означает атом водорода или (С 1-С 4)алкильную группу или атом галогена. Среди соединений формулы (I) согласно изобретению первую подгруппу соединений составляют соединения, в которых U означает карбонильную группу. Среди соединений формулы (I) согласно изобретению вторую подгруппу соединений составляют соединения, в которых цикл, включающий Y, Z, V и W, выбран из фенильной, пиридиновой, тиазольной,тиофеновой группы. Среди соединений формулы (I) согласно изобретению третью подгруппу соединений составляют соединения, в которых R3 означает атом водорода. Среди соединений формулы (I) согласно изобретению четвертую подгруппу соединений составля-7 019362 ют соединения, в которых R4 означает атом водорода. Среди соединений формулы (I) согласно изобретению пятую подгруппу соединений составляют соединения, в которых R5 означает атом водорода. Среди соединений формулы (I) согласно изобретению шестую подгруппу соединений составляют соединения, в которых m равен 1. Среди соединений формулы (I) согласно изобретению седьмую подгруппу соединений составляют соединения, в которых цепь -[С(R3R4)]m-U-N(R5)(R6) находится в пара-положении по отношению к циклу, с которым она связана. Среди соединений формулы (I) согласно изобретению восьмую подгруппу соединений составляют соединения, в которых цепь -[С(R3R4)]m-U-N(R5)(R6) находится в мета-положении по отношению к циклу, с которым она связана. Все группы и подгруппы могут быть независимо друг от друга использованы в виде комбинаций для получения соединений согласно изобретению. Среди комбинаций групп и подгрупп, соответствующих соединениям согласно изобретению, можно назвать первую комбинацию, соответствующую соединениям, в которыхR1, R2, Y, Z, V, W, R6, n имеют значения, которые определены выше. Можно назвать вторую комбинацию, соответствующую соединениям согласно изобретению, в которыхY, Z, V, W означают, независимо один от другого, гетероатом, атом углерода, замещенный группойR7 означает атом водорода или галогена или (С 1-С 4)алкильную группу;R4 означает атом водорода или (С 1-С 4)алкильную группу;R6 означает -(CH2)n-L, где L означает группу, выбранную из следующих групп: арильной группы, содержащей 6 атомов углерода, необязательно замещенной одним или несколькими атомами галогена; гетероарильной группы, содержащей 5-6 звеньев, по меньшей мере один гетероатом которой выбран из атома азота или серы; насыщенной гетероциклической группы, в которой указанный гетероцикл содержит 5-6 звеньев, по меньшей мере один гетероатом которого выбран из атома азота, атома кислорода, и необязательно замещенной в каком-либо положении, включая атом азота, одним или несколькими заместителями, выбранными из атома фтора, фтор (С 1-С 4)алкильной группы, (С 1-С 4)алкильной группы или (С 3 С 5)циклоалкильной группы;n имеет значения, которые определены выше. Можно назвать третью комбинацию, соответствующую соединениям согласно изобретению, в которыхY, Z, V, W означают, независимо один от другого, гетероатом, атом углерода, замещенный группойR7 означает атом водорода или галогена или (С 1-С 4)алкильную группу;R6 означает -(CH2)n-L, где L означает группу, выбранную из следующих групп: арильной группы, содержащей 6 атомов углерода, необязательно замещенной одним или несколькими атомами галогена; гетероарильной группы, содержащей 5-6 звеньев, по меньшей мере один гетероатом которой выбран из атома азота или серы; насыщенной гетероциклической группы, в которой указанный гетероцикл содержит 5-6 звеньев, по меньшей мере один гетероатом которого выбран из атома азота, атома кислорода, и необязательно замещенной в каком-либо положении, включая атом азота, одним или несколькими заместителями, выбранными из атома фтора, фтор(С 1-С 4)алкильной группы или (С 1-С 4)алкильной группы;n имеет значения, которые определены выше. Можно назвать четвертую комбинацию, соответствующую соединениям согласно изобретению, в которыхY, Z, V, W означают, независимо один от другого, гетероатом или группу -СН-;R6 означает -(CH2)nL, где L означает группу, выбранную из следующих групп: арильной группы, содержащей 6 атомов углерода, необязательно замещенной одним или несколькими атомами галогена; гетероарильной группы, содержащей 5-6 звеньев, по меньшей мере один гетероатом которой выбран из атома азота или серы; насыщенной гетероциклической группы, в которой указанный гетероцикл содержит 5-6 звеньев, по меньшей мере один гетероатом которого выбран из атома азота, атома кислорода, и необязательно замещенной в каком-либо положении, включая атом азота, одним или несколькими заместителями, выбранными из атома фтора, (С 1-С 4)алкильной группы или (С 3-С 5)циклоалкильной группы;m и n имеют значения, указанные выше. Можно назвать пятую комбинацию, соответствующую соединениям согласно изобретению, в которыхn имеет значения, указанные выше. Можно назвать шестую комбинацию, соответствующую соединениям согласно изобретению, в которыхR6 означает -(CH2)n-L, где L означает арильную группу, необязательно замещенную одним или несколькими атомами галогена;n имеет значения, указанные выше. Седьмая комбинация соответствует соединениям согласно изобретению, в которыхR2 означает -(СН 2)n'-В, где n'=0 или 1 и В означает (С 3-С 5)циклоалкильную группу или (С 1 С 4)алкильную группу, необязательно замещенную одним или несколькими атомами фтора;Y, Z, V, W означают, независимо один от другого, гетероатом, группу -СН-, атом углерода, необязательно замещенный группой R7, или без него;R3, R4 означают атом водорода или R3 и R4 образуют вместе с углеродом, с которым они связаны,(С 3-С 5)циклоалкильную группу;R6 означает -(CH2)n-L, где L означает насыщенную гетероциклическую группу, необязательно замещенную в каком-либо положении, включая атом азота, одним или несколькими заместителями, вы-9 019362 бранными из атома фтора, фтор (С 1-С 4)алкильной группы, (С 1-С 4)алкильной группы, линейной, или разветвленной, или (С 3-С 5)циклоалкильной группы или (С 1-С 4)алкилсульфонамидной группы,R7, m и n имеют значения, указанные выше. Восьмая комбинация соответствует соединениям согласно изобретению, в которыхY, Z, V, W означают, независимо один от другого, гетероатом или атом углерода или группу -СН-;n имеет значения, указанные выше. Девятая комбинация соответствует соединениям согласно изобретению, в которыхn имеет значения, указанные выше. Среди соединений формулы (I) согласно изобретению можно, в частности, назвать следующие соединения: 2-амино-1-этил-N-метил-4-оксо-7-4-[2-оксо-2-(фениламино)этил]фенил-1,4-дигидро-1,8 нафтиридин-3-карбоксамид (соединение 1); 2-амино-1-этил-7-[4-(2-[(1-этилпирролидин-2-ил)метил](метил)амино-2-оксоэтил)фенил]-Nметил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 2); 2-амино-1-этил-N-метил-4-оксо-7-4-[2-оксо-2-(пиридин-3-иламино)этил]фенил-1,4-дигидро-1,8 нафтиридин-3-карбоксамид (соединение 3); 2-амино-1-этил-N-метил-4-оксо-7-4-[2-оксо-2-(пиридин-2-иламино)этил]фенил-1,4-дигидро-1,8 нафтиридин-3-карбоксамид (соединение 4); 2-амино-7-(4-2-[(2-хлорфенил)амино]-2-оксоэтилфенил)-1-этил-N-метил-4-оксо-1,4-дигидро-1,8 нафтиридин-3-карбоксамид (соединение 5); 2-амино-7-(4-2-[(3,5-дифторфенил)амино]-2-оксоэтилфенил)-1-этил-N-метил-4-оксо-1,4-дигидро 1,8-нафтиридин-3-карбоксамид (соединение 6); 2-амино-1-этил-N-метил-4-оксо-7-(4-2-оксо-2-[(пиридин-4-илметил)амино]этилфенил)-1,4 дигидро-1,8-нафтиридин-3-карбоксамид (соединение 7); 2-амино-1-этил-N-метил-4-оксо-7-(4-2-оксо-2-[(пиридин-2-илметил)амино]этилфенил)-1,4 дигидро-1,8-нафтиридин-3-карбоксамид (соединение 8); 2-амино-1-этил-7-(4-2-[(2-метоксиэтил)амино]-2-оксоэтилфенил)-N-метил-4-оксо-1,4-дигидро 1,8-нафтиридин-3-карбоксамид (соединение 9); 2-амино-7-4-[2-(циклопропиламино)-2-оксоэтил]фенил-1-этил-N-метил-4-оксо-1,4-дигидро-1,8 нафтиридин-3-карбоксамид (соединение 10); 2-амино-1-этил-N-метил-7-(4-2-[(1-метилэтил)амино]-2-оксоэтилфенил)-4-оксо-1,4-дигидро-1,8 нафтиридин-3-карбоксамид (соединение 11); 2-амино-7-4-[2-(циклопентиламино)-2-оксоэтил]фенил-1-этил-N-метил-4-оксо-1,4-дигидро-1,8 нафтиридин-3-карбоксамид (соединение 12); 2-амино-1-этил-N-метил-4-оксо-7-4-[2-оксо-2-(пиразин-2-иламино)этил]фенил-1,4-дигидро-1,8 нафтиридин-3-карбоксамид (соединение 13); 2-амино-1-этил-7-4-[2-([(2R)-1-этилпирролидин-2-ил]метиламино)-2-оксоэтил]фенил-N-метил 4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 14); 2-амино-1-этил-7-4-[2-([(2S)-1-этилпирролидин-2-ил]метиламино)-2-оксоэтил]фенил-N-метил 4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 15); 2-амино-1-этил-N-метил-4-оксо-7-4-[2-оксо-2-(пиримидин-4-иламино)этил]фенил-1,4-дигидро 1,8-нафтиридин-3-карбоксамид (соединение 16); 2-амино-1-этил-N-метил-4-оксо-7-4-[2-оксо-2-(пиридин-4-иламино)этил]фенил-1,4-дигидро-1,8 нафтиридин-3-карбоксамид (соединение 17); 2-амино-7-[4-(2-[(1-этилпирролидин-2-ил)метил]амино-2-оксоэтил)фенил]-N-метил-4-оксо-1(2,2,2-трифторэтил)-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 49); 2-амино-1-циклопентил-7-[4-(2-[(1-этилпирролидин-2-ил)метил]амино-2-оксоэтил)фенил]-Nметил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 50); 2-амино-1-этил-7-[4-(5-[(1-этилпирролидин-2-ил)метил]амино-5-оксопентил)фенил]-N-метил-4 оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 51); 1-этил-7-[5-(2-[(1-этилпирролидин-2-ил)метил]амино-2-оксоэтил)тиофен-2-ил]-N,2-диметил-4 оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 52); 2-амино-7-4-[2-([(2R)-1-этилпирролидин-2-ил]метиламино)-2-оксоэтил]фенил-N-метил-1-(2 метилпропил)-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 53); 2-амино-1-этил-7-4-[1-([(2R)-1-этилпирролидин-2-ил]метилкарбамоил)циклопропил]фенил-Nметил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 54); 2-амино-1-этил-7-2-[2-([(2R)-1-этилпирролидин-2-ил]метиламино)-2-оксоэтил]-1,3-тиазол-4-илN-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 55); 2-амино-1-этил-7-4-[2-([(2R)-1-этилпирролидин-2-ил]метиламино)-2-оксоэтил]-2-фторфенил-Nметил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 56); 2-амино-7-[3-хлор-4-(2-[(1-этилпирролидин-2-ил)метил]амино-2-оксоэтил)фенил]-1-этил-Nметил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 57); 2-амино-7-[3-фтор-4-(2-[(1-этилпирролидин-2-ил)метил]амино-2-оксоэтил)фенил]-1-этил-Nметил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 58); 2-амино-7-[3-метил-4-(2-[(1-этилпирролидин-2-ил)метил]амино-2-оксоэтил)фенил]-1-этил-Nметил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 59); 2-амино-1-(циклопропилметил)-7-[4-(2-[(1-этилпирролидин-2-ил)метил]амино-2 оксоэтил)фенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 60); 2-амино-1-этил-7-[4-(2-[(1-этилпирролидин-2-ил)метил]амино-2-оксоэтил)-2-метилфенил]-Nметил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 61); 2-амино-1-этил-7-[3-(2-[(1-этиллирролидин-2-ил)метил]амино-2-оксоэтил)фенил]-N-метил-4 оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 62); 2-амино 1-этил-N-метил-4-оксо-7-3-[2-оксо-2-(пиридин-2-иламино)этил]фенил-1,4-дигидро-1,8 нафтиридин-3-карбоксамид (соединение 63); 2-амино-1-этил-N-метил-4-оксо-7-4-[2-(пиридин-2-иламино)этил]фенил-1,4-дигидро-1,8 нафтиридин-3-карбоксамид (соединение 64). Следует отметить, что указанные выше соединения имеют наименования, соответствующие номенклатуре IUPAC, использованной с применением программного обеспечения ACDLABS 10.0ACD/наименование (Advanced Chemistry development). В дальнейшем описании под защитной группой Pg подразумевают группу, которая способна, с одной стороны, защищать реакционноспособную функциональную группу, такую как гидрокси или амино группа, в процессе синтеза, и, с другой стороны, регенерировать сохранившуюся функциональную группу в конце синтеза. Примеры защитных групп, а также способы защиты и снятия защиты раскрыты в публикации "Protective Groups in Organic Synthesis". Green et coll., 2nd Edition (JohnSons, Inc., NewYork). 1991. Под уходящей группой подразумевают в дальнейшем описании группу, которая легко отщепляется благодаря гетеролитическому разрыву связи с уходом электронной пары. Поэтому такая группа может быть легко заменена другой группой, например, во время реакции замещения. Такими уходящими группами являются, например, галогены или активированная гидроксигруппа, такая как метансульфонатная,бензолсульфонатная, пара-толуолсульфонатная, трифлатная, ацетатная и т.д. Примеры уходящих групп,а также способы их получения приведены в публикации "Advances in Organic Chemistry", J.March, 3rd Edition, Wiley Interscience, 1985, c. 310-316. В соответствии с изобретением соединения общей формулы (I) можно получить благодаря следующему способу. Следуя схеме 1, 2,6-дигалогенникотиновую кислоту формулы (II) монозамещают в положении 2 амином формулы R2-NH2 (где R2 имеет значения, указанные выше в отношении соединения формулы (I при комнатной температуре или при температуре в диапазоне 50-100 С при классическом нагревании или при нагревании в микроволновой печи, в протонном растворителе, таком как спирт, например этанол, н-бутанол, трет-бутанол или вода. Затем кислоту (III), выходящую со стадии (i), активируют до получения производного формулы (IV), либо в форме фторангидрида кислоты при взаимодействии с цианурилфторидом при комнатной температуре в присутствии основания, такого как триэтиламин или пиридин и в растворителе, таком как дихлорметан или ТГФ, как это описано G.OLAH et coll., в журналеSynthesis (1973), 487, либо в форме имидазолида при взаимодействии с карбодиимидазолом в растворителе, таком как ДМФ или ТГФ, или в соответствии с другими способами, известными специалисту, таким как способ, описанный MUKAIYAMA и TANAKA в журнале Chem. Lett. (1976), 303 или ISHIKAWA и SASAKI в Chem. Lett. (1976), 1407. Фторангидрид кислоты или имидазолид формулы (IV), полученные на выходе со стадии (ii), представляющие собой высокореакционные, но стабильные продукты, затем подвергают взаимодействию сN-замещенным цианоацетамидом формулы (V) согласно способам А или В. В соответствии со способом А используют два эквивалента основания, такого как гидрид натрия или трет-бутоксид калия, для осуществления стадии (iv) конденсации N-замещенного производного цианацетамида (V) с соединением формулы (IV) и, после выдерживания в течение ночи при комнатной температуре, получают -кетоцианацетамид формулы (VI), который затем циклизуют до получения пиридино-пиридинона формулы (VII) при нагревании с температурой в диапазоне 90-125 С в полярном растворителе, таком как н-бутанол, ДМСО или ДМФ. Способ В аналогичен способу А включая стадию конденсации (iv), но к реакционной смеси добавляется третий эквивалент используемого основания, и полученное соединение формулы (VI) циклизуютin situ при комнатной температуре и получают сразу пиридино-пиридиноновое соединение формулыN-Алкилцианоацетамиды формулы (V) получают согласно стадии (iii) при взаимодействии этилцианоацетамида с избытком амина формулы R1-NH2 (где R1 имеет значения, указанные в отношении соединений формулы (I) согласно изобретению) в растворителе, таком как ТГФ или этанол, при температуре от комнатной до температуры рефлюкса растворителя. Для получения соединений формулы (I) согласно изобретению используются два способа, где исходным соединением является галогенированное промежуточное соединение формулы (VII). В соответствии со способом 1, описанным на схеме 2, промежуточное соединение (VII) вводят на стадии (vi) в реакцию сочетания Сузуки с использованием бороновой кислоты или сложного боронового эфира биспинасола (IXa), где m, R3 и R4 и V, W, Y, Z определены выше для соединений формулы (I) согласно изобретению, при условии, что цикл содержит от 5 до 6 звеньев, a G является группой (С 1 С 4)алкокси, такой как OEt, или звено -NR5R6, в котором R5 и R6 имеют значения, указанные выше для соединений формулы (I). Эту реакцию (vi) осуществляют в присутствии комплекса на основе палладия (в состоянии окисления(0) или (II, такого как, например, комплекс Pd(PPh3)4, PdCl2(PPh3)2, Pd2dba3, Xphos или PdCl2(dppf), в полярном, протонном или не протонном растворителе, таком как ДМЭ, этанол, ДМФ,диоксан, или смесь этих растворителей, в присутствии основания, такого как карбонат цезия, водный гидрокарбонат натрия или K3PO4, при классическом нагревании с температурой в диапазоне 80-120 С или при микроволновом нагревании в диапазоне 130-170 С. В случае, когда G означает звено -NR5R6, в котором R5 и R6 имеют значения, указанные выше для соединений формулы (I), т.е. путь 1, соединение формулы (I) согласно изобретению получают непосредственно после окончания реакции сочетания на стадии (vi). В случае, когда G является (С 1-С 4)алкоксигруппой, такой как OEt, т.е. путь 2, после проведения реакции сочетания Сузуки на стадии (vi) получают соединение (X). Затем соединение (X) омыляют на стадии (vii) в присутствии нуклеофильного агента, такого как LiOH или NaOH в водном растворе, в растворителе, таком как полярный растворитель, например, ТГФ, ДМФ, МеОН или EtOH при температуре от комнатной до 80 С с получением соединения (XI), которое затем на стадии (viii) вводят в реакцию пептидного сочетания с выбранным амином HNR5R6, в котором R5 и R6 имеют значения, которые определены для соединения формулы (I), в присутствии агента сочетания, такого как TBTU, HBTU или CDI, и основания, например, диизопропилэтиламина, триэтиламина или NaHCO3, в апротонном растворителе,таком как дихлорметан, ТГФ или ДМФ, или другими способами, известными специалисту, такими как способы, описанные в публикации "Princiales of Peptide Synthesis", 2nd Ed 1993, M. Bodanszky, SpringerLaboratory, для получения соединения формулы (I). Для получения соединений формулы (I) согласно изобретению используют второй способ, где исходным соединением является промежуточное галогенированное соединение формулы (VII), этот способ 2 представлен на схеме 3. Промежуточное галогенированное соединение формулы (VII) превращают в производное бис-пинаколового эфира бороновой кислоты или в производное бороновой кислоты формулы (VIII) в соответствии со стадией (ix) путем взаимодействия с бис-(пинаколят)дибораном в присутствии [1,1'-бис-(дифенилфосфино)ферроцен]дихлорпалладия(II) и ацетата калия или карбоната калия в полярном растворителе, таком как ДМСО, ДМФ, ДМЭ или диоксан, при температуре в диапазоне 50100 С, согласно способу, описанному ISHIYAMA, Т. et coll. в журнале J. Org. Chem., 1995, 60, 7508-7510 и GIROUX, A. et coll. в журнале Tet. Lett., 1997, 38, 3841-3844. На следующей стадии (х) соединение(VIII) в форме бороновой кислоты или сложного эфира бороновой кислоты вводят в реакцию типа Сузуки с галогенированным ароматическим соединением формулы (IXb), в которой X, m, R3 и R4, U и V, W,Y, Z имеют значения, указанные выше для соединений формулы (I) согласно изобретению, при условии,что цикл содержит от 5 до 6 звеньев и G является (С 1-С 4)алкоксигруппой, такой как OEt или звено-NR5R6, в котором R5 и R6 имеют значения, указанные для соединений формулы (I). Для проведения этой реакции (х) подходят условия осуществления сочетания Сузуки, описанные для стадии (vi) схемы 2. В случае, когда G является звеном -NR5R6, в котором R5 и R6 имеют значения, указанные выше, aU является либо карбонилом, либо метиленовым звеном, соединение формулы (I) согласно изобретению получают непосредственно по окончании стадии сочетания (х) (путь 1). В случае, когда G является группой OEt, a U является карбонилом, т.е. следуя путем 2, в результате сочетания Сузуки (стадия х) получают соединение (X). Затем это соединение (X) вводят на стадию (xi) для получения соединения (XI) в кислотной форме, которое, в свою очередь, превращают на стадии (xii) в соединение формулы (I) согласно изобретению. Стадии (xi) и (xii) идентичны, соответственно, стадиям(vii) и (viii), описанным выше. Если способы получения исходных продуктов, реактивов, таких как амины HNR5R6, или соединений формулы IXa и IXb, используемых согласно схемам 1, 2 и 3, не описаны, то значит, что они являются коммерчески доступными или могут быть получены методами, описанными в литературе или известными специалисту. Если необходимо, некоторые реакционноспособные группы, находящиеся в группах R2, R5 или R6,могут быть защищены на время протекания этих реакций защитными группами, таким как группы, описанные в публикации "Protective Groups in Organic Synthesis", Green et coll., 2nd Edition (John Wiley Согласно другому аспекту изобретение относится также к соединениям формул (VIII), (X), (XI). Эти соединения полезны в качестве промежуточных соединений получения соединений формулы (I). Следующие примеры иллюстрируют получение некоторых соединений согласно изобретению. Эти примеры не ограничивают объем изобретения, а только иллюстрируют его. Номера соединений, полученных в примерах, соответствуют номерам, которые приведены в таблице, представленной ниже, которая иллюстрирует химическую структуру и физические свойства некоторых соединений согласно изобретению. В тексте использованы следующая аббревиатура и брутто-формулы: Используемые микроволновые приборы: Biotage, Initiator. Условия для проведения анализов. Условия проведения анализов с использованием жидкостной хроматографии LC/УФ-спектроскопииH2O+AcNH4 (5 нМ)+3% CH3CN/B: CH3CN. Градиент 0-20 мин от 0 до 100%В; УФ-детектирование: 210 нм. Условие GC Cl/CH4+: ионизация Cl/CH4+, 30 мин. Колонка: Agilent HP-5MS 30 м 250 мкм пленка толщиной 0,25 мкм. Температура 250 С, газ-вектор: гелий, постоянная скорость потока 1,4 мл/мин. Масс-спектрометрия (MS). Режим ионизации: электроспрей с положительной ионизацией ESI+, диапазон масс: 90-1500 е.а.м. Спрей-камера: температура газа: 350 С; подача сухого газа (N2): 10,0 л/мин; давление распыления: 30 psig; напряжение улавливателя ионов: 4000 В. Спектры 1 Н ЯМР получали с использованием ЯМР-спектрометров фирмы Bruker с частотой 250,300 или 400 МГц в ДМСО-d6, используя пик сигнала ДМСО-d5 в качестве стандарта. Химические смещениявыражены в миллионных долях (м.д.). Регистрируемые сигналы были выражены следующим образом: с=синглет, д=дуплет, т=триплет, м=массив или широкий синглет; Н=протон. Точки плавления ниже 260 С были измерены на стенде Кофлера, а точки плавления выше 260 С измерялись на приборе Бюхи В-545. Вращающая способность замерялась на полярометре типа полярометра Perkin-Elmer с силой тока 55 мкА. Пример 1: гидрохлорид 2-амино-1-этил-7-4-[2-([(2R)-1-(2-фторэтил)пирролидин-2 ил]метиламино)-2-оксоэтил]фенил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 27). 1.1: 6-хлор-2-(этиламино)пиридин-3-карбоновая кислота. Раствор 18,0 г (84,4 ммоль) 2,6-дихлорникотиновой кислоты в 180 мл (3,45 моль) 70% раствора этиламина в воде нагревают при 50 С в течение 10 ч. Избыток амина выпаривают при пониженном давлении, затем прибавляют 10% водный раствор уксусной кислоты до выпадения осадка. Твердый бежевый продукт центрифугируют, промывают холодной водой и сушат в сушильной камере. Получают 10,5 г целевого продукта. Выход=62%. Точка плавления: 158-160 С. МН+: 201,1 (время удерживания: 7,7 мин, условие 1). 1.2: 6-хлор-2-(этиламино)пиридин-3-карбонилфторид. К суспензии 10,5 г (52,3 ммоль) соединения, полученного на стадии 1.1, в дихлорметане (250 мл) последовательно прибавляют 4,2 мл (52,3 ммоль) пиридина и 8,4 мл (99,6 ммоль) цианурилхлорида. Смесь перемешивают в течение 3 ч при комнатной температуре, затем фильтруют. Твердый продукт споласкивают дихлорметаном (100 мл) и фильтрат промывают два раза ледяной водой (60 мл). Органический слой сушат над Na2SO4, затем концентрируют при пониженном давлении. Получают 10,44 г продукта в виде масла оранжевого цвета. Выход=99%. Продукт используют на следующей стадии без очистки. 1.3: 2-циано-N-метилацетамид. К 10,9 г (353,6 ммоль) раствора метиламина в ТГФ, охлажденного до 0 С, прибавляют по каплям 20 г (176,8 ммоль) этилцианацетата, затем реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворители выпаривают при пониженном давлении и полученный продукт очищают путем перекристаллизации из толуола. Получают 16,8 г продукта в виде твердого бежевого вещества. Выход=96%. Точка плавления: 99 С. Способ А (1.4 и 1.5, изложенные ниже). 1.4: 3-[6-хлор-2-(этиламино)пиридин-3-ил]-2-циано-3-гидрокси-N-метилпроп-2-енамид. К раствору 9,80 г (100 ммоль) соединения, полученного на стадии 1.3, в 100 мл безводного ДМФ,охлажденному до 0-5 С, прибавляют небольшими порциями 3,98 г (100 ммоль) 60% гидрида натрия в минеральном масле. После окончания выделения водорода смесь перемешивают в течение 10 мин при комнатной температуре, затем снова охлаждают до 0-5 С. Прибавляют раствор 10,1 г (49,8 ммоль) соединения, полученного на стадии 1.2, в 60 мл ДМФ и смесь перемешивают при комнатной температуре в течение ночи, затем прибавляют 2,85 мл (49,8 ммоль) уксусной кислоты. Выпаривают ДМФ при пониженном давлении, затем остаток обрабатывают водой и полученный продукт экстрагируют два раза сме- 17019362 сью дихлорметан:метанол в соотношении 95:5, затем один раз смесью этилацетат:ТГФ (2:1). Объединенные органические слои сушат над MgSO4, затем растворители выпаривают при пониженном давлении. Получают 19,0 г продукта, используемого в полученном виде на следующей стадии. 1.5: 2-амино-7-хлор-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид. Раствор 19,0 г (49,8 ммоль) неочищенного продукта, полученного на стадии 1.4, в 600 мл нбутанола нагревают в течение 48 ч при 110 С. Растворитель выпаривают при пониженном давлении и полученный твердый продукт растирают в метаноле. Затем твердый продукт отфильтровывают и сушат в сушильной камере. Получают 7,9 г целевого продукта в виде твердого вещества бледно-желтого цвета. Выход=57%. Точка плавления: 283-286 С. МН+: 281,2; (время удерживания=6,99 мин, условие 1). Способ В (1.6, изложенный ниже, вместо 1.4 и 1.5). 1.6: 2-амино-7-хлор-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид. К раствору 0,48 г (4,9 ммоль) соединения, полученного на стадии 1.3, в безводном ДМФ (7 мл), охлажденному до 0-5 С, прибавляют небольшими порциям 0,4 г (9,95 ммоль) 60% гидрида натрия в минеральном масле. Смесь перемешивают при этой температуре в течение 10 мин, затем прибавляют раствор 1,0 г (4,93 ммоль) соединения, полученного на стадии 1.2, в безводном ДМФ (5 мл). Реакционную смесь перемешивают в течение 1 ночи при комнатной температуре, затем снова прибавляют маленькими порциями 0,2 г (4,9 ммоль) 60% гидрида натрия. Перемешивание продолжают при этой же температуре в течение 30 мин, затем добавляют 0,56 мл (9,8 ммоль) уксусной кислоты с последующим добавлением 60 мл воды и отфильтровывают твердый продукт, затем споласкивают водой и сушат в сушильной камере. Получают 1,30 г целевого продукта. Выход=94%. Точка плавления: 283-284 С. МН+: 281,2 (время удерживания=6,99 мин, условие 1). 1.7: [7-амино-8-этил-6-(метилкарбамоил)-5-оксо-5,8-дигидро-1,8-нафтиридин-2-ил]бороновая кислота. Суспензию 8 г (0,03 моль) соединения, полученного на стадии 1.5 или 1.6 (в зависимости от используемого способа А или В), 8,0 г (0,03 моль) бис-(пинаколят)диборана и 8,5 г (0,08 моль) ацетата калия в диметилсульфоксиде (130 мл) дегазируют аргоном в течение 15 мин, добавляют 1,4 г (1,7 ммоль) комплекса [1,1'-бис-(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (1:1) и смесь нагревают при 80 С в течение 30 мин в атмосфере аргона, затем охлаждают, разбавляют 1,1 л воды и подкисляют до рН 4 добавлением уксусной кислоты (50 мл). Смесь фильтруют и осадок черного цвета промывают водой (40 мл), затем простым эфиром (60 мл). Черный остаток обрабатывают 575 мл раствораNaOH (1 н.) и смесь фильтруют на целите 545. Фильтрат подкисляют 60 мл уксусной кислоты и осадок отфильтровывают, промывают водой и простым эфиром, затем сушат в сушильном шкафу. Получают 6,85 г продукта в виде порошка белого цвета. Выход=83%. Точка плавления: 335 С, МН+: 291,2 (время удерживания=5,3 мин, условие 1). 1H ЯМР (250 МГц, ДМСО-d6),(м.д.): 11,69 (с,1 Н); 11,12 (кв, 1 Н, 4,67 Гц); 8,47 (с, 2 Н); 8,44 (д, 1 Н,7,7 Гц); 7,9 (с, 1 Н); 7,75 (д, 1 Н, 7,7 Гц); 4,72 (м, 2 Н); 2,8 (д, 3 Н, 4,67 Гц); 1,22 (т, 3 Н, 6,9 Гц). 1.8: 1-тритил-D-пролинамид. При охлаждении (ледяная баня) и в инертной атмосфере прибавляют 10 мл триэтиламина (69,7 ммоль) к 5 г (33,2 ммоль) гидрохлорида пролинамида, суспензированного в хлороформе (50 мл), затем добавляют порцией 9,7 г (34,9 ммоль) тритилхлорида. Реакцию оставляют перемешиваться при комнатной температуре в течение 2 суток. Реакционную смесь растворяют в дихлорметане (200 мл) и последовательно промывают 2 раза раствором HCl (1 н.) (150 мл), водой (150 мл), насыщенным раствором NaHCO3 (150 мл) и насыщенным раствором NaCl (150 мл). Органический слой сушат (Na2SO4), фильтруют и упаривают при пониженном давлении. Остаток перекристаллизовывают из 20 мл горячего этанола. Получают 11 г соединения в виде кристаллов белого цвета. Выход=93%. Точка плавления: 162 С. 1.9: 1-[(2R)-1-тритилпирролидин-2-ил]метанамин. При охлаждении (ледяная баня) и в инертной атмосфере прибавляют по каплям 60 мл (61,6 ммоль) раствора литийалюминийгидрида (1 н.) в ТГФ к 11 г (30,8 ммоль) соединения, полученного на стадии 1.8,суспензированного в ТГФ (60 мл). После добавления реакционную смесь нагревают 3 ч при 70 С, затем охлаждают до 0 С. Затем по каплям последовательно прибавляют 2,8 мл воды (с образованием осадка),2,8 мл раствора NaOH (1 н.) и 8,3 мл воды. Полученную смесь перемешивают 30 мин, затем фильтруют,споласкивают твердый продукт ТГФ (10 мл) и фильтрат концентрируют при пониженном давлении. Получают 10 г соединения в виде порошка ярко-желтого цвета и используют в полученном виде на следующей стадии. Выход=95%. Точка плавления: 100 С. 1.10: 2-(4-иодофенил)-N-[(2R)-пирролидин-2-илметил]ацетамид. В инертной атмосфере при комнатной температуре прибавляют 4 мл (43,8 ммоль) оксалилхлорида к 3,8 г (14,6 ммоль) 4-иодофенилуксусной кислоты, суспензированной в дихлорметане (54 мл). Прибавляют две капли ДМФ и реакционную смесь перемешивают 1 ч при комнатной температуре, затем концентрируют при пониженном давлении. Остаток обрабатывают толуолом и снова концентрируют. Полученный таким путем хлорангидрид кислоты растворяют в дихлорметане (10 мл) и прикапывают при охлаж- 18019362 дении (ледяная баня) и в инертной атмосфере к 5 г (14,6 ммоль) соединения, полученного на стадии 1.9,суспензированного в дихлорметане (30 мл) в присутствии 2,45 г (29,2 ммоль) NaHCO3. Реакционную смесь перемешивают в течение ночи при комнатной температуре, затем фильтруют на целите и концентрируют при пониженном давлении. Получают 8,5 г продукта в виде пенистого продукта желтого цвета,который используют непосредственно на следующей стадии без очистки. МН+: 586. 8,5 г (14,6 ммоль) соединения, полученного на предыдущей стадии, обрабатывают при охлаждении(ледяная баня) смесью этанол/HCl 35% (41 мл/3,2 мл). Реакционную среду перемешивают 2 ч при комнатной температуре. Реакционную среду концентрируют при пониженном давлении, обрабатывают водой (30 мл) и экстрагируют простым эфиром (30 мл 2). Водный слой охлаждают, подщелачивают с помощью 35% раствора NaOH, вливаемого по каплям до достижения рН 10. Затем продукт экстрагируют дихлорметаном (100 мл 6). Органический слой сушат (Na2SO4), фильтруют и упаривают при пониженном давлении. Получают 4,6 г соединения в виде белого порошка. Выход=92% в конце трех стадий. Точка плавления: 120 С. МН+: 345 (время удерживания: 4,65 мин,условие 1). 1.11: N-[(2R)-1-(2-фторэтил)пирролидин-2-ил]метил-2-(4-иодофенил)ацетамид. В герметизируемый сосуд вводят 0,53 г NaHCO3 (6,4 ммоль) и 0,49 г 1-иод-2-фторэтана (2,8 ммоль) к 0,88 г (2,6 ммоль) соединения, полученного на стадии 1.10, растворенного в ДМФ (6 мл). Сосуд герметично закрывают и нагревают при 90 С в течение 5 ч. Реакционную смесь охлаждают, затем выпаривают ДМФ при пониженном давлении, полученный остаток обрабатывают водой (10 мл) и экстрагируют этилацетатом (20 мл 2). Органический слой сушат над сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Полученный остаток очищают флэш-хроматографией на силикагеле (элюент: дихлорметан/метанол, 1% NH4OH) с градиентом метанола от 0 до 10%. Получают 0,82 г соединения в виде порошка бежевого цвета. Выход=85%. Точка плавления: 96 С. МН+: 391 (время удерживания: 5,1 мин, условие 1). 1.12: гидрохлорид 2-амино-1-этил-7-4-[2-([(2R)-1-(2-фторэтил)пирролидин-2-ил]метиламино)-2 оксоэтил]фенил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 27). В герметизируемый сосуд вводят 2,1 мл насыщенного водного раствора NaHCO3 к 0,19 г (0,51 ммоль) соединения, полученного на стадии 1.11, и к 0,15 г (0,53 ммоль) соединения, полученного на стадии 1.7, растворенного в смеси 1,2-диметоксиэтана/этанола 2/1 (6 мл). После дегазирования аргоном в течение 15 мин прибавляют 0,03 г (0,03 ммоль) тетракис-(трифенилфосфин)палладия. Реакционную среду нагревают при 100 С в течение 3 ч. Реакционную среду охлаждают до комнатной температуры, затем прибавляют воду (10 мл), образовавшийся осадок отфильтровывают, споласкивают водой (5 мл 2), затем дихлорметаном (5 мл 2) и сушат в сушильной камере. Затем продукт очищают флэш-хроматографией на силикагеле (элюент: дихлорметан/метанол, 1% NH4OH) с градиентом метанола от 0 до 10%. Получают 0,05 г продукта в виде белого порошка. Полученные 0,05 г (0,1 ммоль) соединения суспензируют в 3 мл метанола и прибавляют 0,1 мл (0,1 ммоль) раствора HCl (1 н.) в простом эфире. Реакционную среду перемешивают при комнатной температуре 30 мин, затем прибавляют 5 мл простого эфира, образовавшийся осадок выделяют фильтрацией и сушат в сушильной камере. Получают 0,031 г продукта в виде белого порошка. Выход 19%. Точка плавления: 180 С, МН+: 509,3 (время удерживания 4,6 мин, условие 1). 1(22,2 ммоль) порошкообразного гидрокарбоната и 0,7 мл (7,0 моль) изопропилиодида, затем смесь нагревают при 150 С в микроволновой печи в течение 30 мин. Реакционную смесь охлаждают до комнатной температуры, разбавляют дихлорметаном (30 мл) и фильтруют на целите. Фильтрат концентрируют при пониженном давлении и получают 1,1 г соединения в виде порошка бежевого цвета, используемого на следующей стадии без очистки. Выход: 97%. МН+: 173,2 (время удерживания: 0,72 мин, условие 1). 2.2: гидрохлорид 1-[4-(1-метилэтил)морфолин-3-ил]метанамин. К раствору 1,1 г (6,15 ммоль) соединения, полученного на стадии 2.1, в безводном ТГФ, в инертной атмосфере прибавляют по каплям 15,4 мл раствора (1 М) литийалюминийгидрида в ТГФ, затем реакционную смесь нагревают при 70 С в течение 2 ч, затем охлаждают до комнатной температуры. Затем мед- 19019362 ленно и последовательно прибавляют 0,6 мл воды, 0,6 мл едкого натра (1 н.) и 1,8 мл воды, затем реакционную смесь перемешивают 30 мин при комнатной температуре, выпавший осадок удаляют фильтрацией, споласкивают ТГФ (20 мл), фильтрат концентрируют при пониженном давлении, затем добавляют 6,2 мл раствора HCl (1 М) в диэтиловом эфире, смесь перемешивают при комнатной температуре в течение 30 мин, затем концентрируют при пониженном давлении. Получают 1,0 г гидрохлорида соединения в виде порошка каштанового цвета, который используют без очистки на следующей стадии. Выход: 86%. МН+: 159,4 (время удерживания 0,53 мин, условие 1). 2.3: этил-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]ацетат. Смесь 9,5 г (32,8 ммоль) этил(4-иодофенил)ацетата и 9,16 г (36,1 ммоль) бис-(пинаколят)диборана,растворенного в безводном диметилсульфоксиде (65 мл), дегазируют аргоном 15 мин, затем добавляют 27,8 г (98,4 ммоль) ацетата калия и 1,34 г (1,64 моль) дихлор(фосфиноферроцен)палладия и реакционную смесь нагревают при 55 С в течение 1 ч 30 мин в атмосфере аргона. Реакционную смесь разбавляют 220 мл этилацетата, затем органический слой промывают три раза водой (200 мл), затем сушат над Na2SO4 и концентрируют при пониженном давлении. Получают 10,8 г соединения в виде неочищенного масла каштанового цвета, которое используют в полученном виде на следующей стадии. МН+: 291,2 (время удерживания: 9,4 мин, условие 1). 2.4: этил 4-[7-амино-8-этил-6-(метилкарбамоил)-5-оксо-5,8-дигидро-1,8-нафтиридин-2 ил]фенилацетат. К 6,16 г (21,9 ммоль) соединения, полученного на стадии 1.5 или 1.6 (в зависимости от используемого способа А или В) и 7,64 г (26,3 ммоль) соединения, полученного на стадии 2.3, растворенного в смеси 1,2-диметоксиэтана/этанола 2/1 (275 мл), прибавляют 88 мл водного насыщенного раствораNaHCO3. После дегазирования смеси аргоном в течение 15 мин прибавляют 1,27 г (1,1 ммоль) тетракис(трифенилфосфин)палладия. Реакционную среду нагревают при 100 С в течение 4 ч. Реакционную среду охлаждают до комнатной температуры, затем добавляют воду (300 мл), образовавшийся осадок отфильтровывают, споласкивают водой (20 мл 2), промывают этилацетатом (50 мл), затем сушат в вакууме над Р 2 О 5. Получают 5,3 г продукта в виде белого порошка. Выход: 59%. Точка плавления: 195 С. МН+: 409,1 (время удерживания: 5,89 мин, условие 1). 2.5: 4-[7-амино-8-этил-6-(метилкарбамоил)-5-оксо-5,8-дигидро-1,8-нафтиридин-2 ил]фенилуксусная кислота. К суспензии 0,87 г (2,1 ммоль) соединения, полученного на стадии 2.4, в смеси 1/1/1 ТГФ/метанол/вода (12 мл), прибавляют одной порцией 0,134 г (3,2 ммоль) гидроксида лития. Реакционную смесь нагревают при 70 С в течение 3 ч, затем охлаждают до комнатной температуры. Добавляют воду (10 мл) и подкисляют реакционную среду раствором HCl (1 н.) до рН 2, выпавший осадок отделяют фильтрацией и сушат в вакууме над Р 2 О 5. Получают 0,78 г продукта в виде белого порошка. Выход: 96%. Точка плавления: 260 С. МН+: 381,1 (время удерживания: 6,74 мин, условие 1). 2.6: гидрохлорид 2-амино-1-этил-N-метил-7-4-[2-([4-(1-метилэтил)морфолин-3-ил]метиламино)2-оксоэтил]фенил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 32). В инертной атмосфере 0,18 г N,N-карбодиимидазола прибавляют к суспензии 0,4 г (1,1 ммоль) соединения, полученного на стадии 2.5, в ДМФ (5 мл), реакционную среду перемешивают при комнатной температуре 1,5 ч, затем прибавляют 0,225 г (1,2 ммоль) соединения, полученного на стадии 2.2, растворенного в ДМФ (1 мл) и предварительно перемешанного в присутствии 0,13 г (1,3 ммоль) карбоната натрия, затем смесь нагревают при 80 С в течение 2 ч. Удаляют ДМФ в процессе выпаривания смеси при пониженном давлении, затем остаток очищают флэш-хроматографией на силикагеле (элюент: дихлорметан/метанол/1% NH4OH) с градиентом метанола от 0 до 5%. Получают 0,3 г продукта в виде белого порошка. Выход: 57%. МН+: 521,3. К суспензии 0,28 г (0,54 ммоль) полученного соединения в метаноле (2 мл) добавляют 0,05 мл концентрированного раствора HCl (35%). Смесь перемешивают в течение 1 ч, затем прибавляют диэтиловый эфир (10 мл), образовавшийся твердый продукт отделяют фильтрацией и сушат в вакууме. Получают 0,26 г гидрохлоридной соли продукта в виде порошка оранжеватого цвета. Выход: 87%. Точка плавления: 192 С. МН+: 521,3 (время удерживания: 9,1 мин, условие 2). 1H ЯМР (400 МГц, ДМСО-d6),(м.д.): 11,75 (с, 1 Н); 11,13 (кв, 1 Н); 10,86 (с, 1 Н); 8,52 (д, 2 Н, 8,1 Гц); 8,16 (д, 2 Н, 8,3 Гц); 8,01 (с, 1 Н); 7,95 (д, 1 Н, 8,1 Гц); 7,46 (д, 2 Н, 8,3 Гц); 4,61 (кв, 2 Н, 6,8 Гц); 3,96 (м,3 Н); 3,83-3,02 (м очень шир., 9 Н); 2,8 (с, 3 Н); 1,24 (м, 9 Н). Пример 3: гидрохлорид 2-амино-1-этил-N-метил-4-оксо-7-5-[2-оксо-2-(пиридин-2 иламино)этил]пиридин-2-ил-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 19). 3.1: 2-(6-хлорпиридин-3-ил)-N-пиридин-2-илацетамид. В инертной атмосфере 6,23 г (38,5 ммоль) N,N-карбодиимидазола прибавляют к суспензии 6 г (35 ммоль) 2-хлорпиридилуксусной кислоты в безводном ТГФ (90 мл) при комнатной температуре. Реакционную среду перемешивают при этой же температуре в течение 2 ч, затем прибавляют 5,43 г (57,7 ммоль) 2-аминопиридина и смесь нагревают в течение 2 ч с рефлюксом. К реакционной смеси прибав- 20019362 ляют 200 мл дихлорметана, охлаждают до комнатной температуры, полученный органический слой промывают насыщенным раствором хлорида аммония, затем водным раствором едкого натра (1 н.), сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток очищают флэшхроматографией на силикагеле (элюент: дихлорметан/этилацетат, градиент этилацетата от 0 до 79%). Получают 5,6 г соединения в виде белого порошка. Выход 65%. Точка плавления: 130 С. МН+: 248,1 (время удерживания: 5,45 мин, условие 1). 3.2: гидрохлорид 2-амино-1-этил-N-метил-4-оксо-7-5-[2-оксо-2-(пиридин-2 иламино)этил]пиридин-2-ил-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 19). Работают таким же способом, как описано в примере 1, стадия 1.12, но исходят из 0,32 г (1,3 ммоль) соединения, полученного на стадии 3.1, 0,41 г (1,4 ммоль) соединения, полученного на стадии 1.7, 74 мг(0,06 ммоль) тетракис-(трифенилфосфин)палладия, 14,5 мл насыщенного раствора гидрокарбоната в смеси 1,2-диметоксиэтана/этанола (2/1) (19 мл) и 0,26 мл раствора HCl (1 М) в простом эфире. Получают 0,095 г гидрохлоридной соли продукта в виде порошка желтого цвета. МН+: 458,3 (время удерживания: 6,3 мин, условие 1). Выход 16%. Точка плавления: 268-288 С (разложение). 1(4 мл), последовательно прибавляют 0,09 г (0,95 ммоль) 2-аминопиридина, 0,31 г (2,4 ммоль) диизопропилэтиламина и 0,35 г (1,1 ммоль) TBTU. Реакционную смесь перемешивают 3 ч при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении и остаток обрабатывают этилацетатом (20 мл), затем полученный таким путем органический слой промывают насыщенным раствором хлорида аммония (15 мл) и насыщенным раствором гидрокарбоната (15 мл), затем сушат над Na2SO4,фильтруют и концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией на силикагеле (элюент: дихлорметан/этилацетат, 1% NH4OH) с градиентом этилацетата от 0 до 20%, 1%NH4OH. Получают 0,125 г соединения в виде порошка белого цвета. Выход: 45%. Точка плавления: 131 С. МН+: 292-294 (время удерживания: 6,1 мин, условие 1). 4.2: 2-амино-1-этил-N-метил-4-оксо-7-6-[2-оксо-2-(пиридин-2-иламино)этил]пиридин-3-ил-1,4 дигидро-1,8-нафтиридин-3-карбоксамид (соединение 21). Работают таким же способом, как описано в примере 1, стадия 1.12, но исходят из 0,42 г (1,4 ммоль) соединения, полученного на стадии 4.1, 0,46 г (1,6 ммоль) соединения, полученного на стадии 1.7, 83 мг(0,07 ммоль) тетракис-(трифенилфосфин)палладия, 15 мл насыщенного раствора гидрокарбоната в смеси 1,2-диметоксиэтана/этанола (2/1) (24 мл) и получают 0,03 г продукта в виде порошка белого цвета. Выход: 5%. Точка плавления: 266 С. МН+: 458,1 (время удерживания: 6,5 мин, условие 1). 1(д, 3 Н, 4,6 Гц); 1,33 (т, 3 Н, 6,8 Гц). Пример 5: гидрохлорид 2-амино-7-[4-(2-[(4-циклопропилморфолин-3-ил)метил]амино-2 оксоэтил)фенил]-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 33). 5.1: 4-циклопропилморфолин-3-карбоксамид. К 1,2 г (7,2 ммоль) гидрохлорида морфолин-3-карбоксамида (синтез которого описан в международной заявке WO 2005026156, Hennequin L.F.A. et coll), растворенного в метаноле (36 мл), последовательно прибавляют 2,9 г молекулярного сита 3 , 4,3 г (72 ммоль) уксусной кислоты, 7 г (43,2 ммоль) 2[(1-этоксициклопропил)окси]триметилсилана и 4,4 г (31,7 ммоль) цианборгидрида натрия. Реакционную смесь нагревают при 70 С в течение 3,5 ч, затем охлаждают до комнатной температуры, затем фильтруют. Фильтрат концентрируют при пониженном давлении, затем остаток обрабатывают дихлорметаном (200 мл) и промывают 3 раза водным раствором NaOH (1 н.) (100 мл). Органический слой сушат над Na2SO4, фильтруют, затем концентрируют при пониженном давлении. Получают 0,58 г продукта в виде белого порошка. Выход: 47%. Точка плавления: 116 С. МН+: 171,2 (время удерживания: 1,03 мин, условие 1). 5.2: 1-(4-циклопропилморфолин-3-ил)метанамин, гидрохлорид. В инертной атмосфере к раствору 0,58 г (3,4 ммоль) соединения, полученного на стадии 5.1, в безводном ТГФ, охлажденному до 0 С, прибавляют по каплям 13,6 мл (13,6 ммоль) раствора (1 н.) тетрагид- 21019362 рофуранового комплекса с тригидридом бора в ТГФ. Реакционную смесь нагревают при 70 С в течение 3 ч. Прибавляют 15 мл раствора HCl (1 н.) к реакционной смеси, охлажденной до комнатной температуры, после перемешивания в течение 30 мин водный слой декантируют и экстрагируют 2 раза эфиром(15 мл), затем подщелачивают добавлением раствора (1 н.) едкого натра. Затем водный слой экстрагируют 4 раза (20 мл) этилацетатом и 4 раза (20 мл) дихлорметаном. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток обрабатывают метанолом(4 мл) и добавляют 3,4 мл раствора HCl в эфире, раствор перемешивают 30 мин, затем добавляют 5 мл эфира, образовавшийся твердый продукт выделяют фильтрацией и сушат в вакууме над Р 2 О 5. Получают 0,51 г продукта в виде белого порошка. Выход: 78%. МН+: 157,2 (время удерживания: 0,4 мин, условие 1). 5.3: гидрохлорид 2-амино-7-[4-(2-[(4-циклопропилморфолин-3-ил)метил]амино-2-оксоэтил)фенил]-1-этил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 33). Работают таким же способом, как описано в примере 2, стадия 2.6, но исходят из 0,33 г (0,88 ммоль) соединения, полученного на стадии 2.5, суспензированного в безводном ДМФ (5 мл), добавляют 0,15 г(0,92 ммоль) CDl и смесь перемешивают при комнатной температуре в течение 2 ч 40 мин. Параллельно,в инертной атмосфере к 0,22 г (0,97 ммоль) соединения, полученного на стадии 5.2, растворенного в ДМФ (2 мл), прибавляют 0,11 г (1,1 ммоль) карбоната натрия и смесь перемешивают при КТ в течение 2 ч, затем супернатант отбирают и прибавляют по каплям к полученному выше промежуточному имидазолиду. Смесь нагревают при 80 С в течение 2 ч. Выпаривают ДМФ при пониженном давлении и остаток очищают флэш-хроматографией на силикагеле (элюент: дихлорметан/метанол) с градиентом метанола от 0 до 5%. Получают 0,13 г продукта в виде белого порошка. Выход 28%. К 0,13 г (0,25 ммоль) полученного соединения, растворенного в 2 мл метанола, прибавляют 0,02 г(0,3 ммоль) 35,6% концентрированного раствора HCl. Смесь перемешивают при КТ в течение 1 ч, образовавшийся твердый продукт выделяют фильтрацией, сушат над Р 2 О 5 в вакууме. Получают 0,13 соединения в виде белого порошка. Выход: 92%. Точка плавления: 250 С. МН+: 519,2 (время удерживания: 5,5 мин, условие 1). 1H ЯМР (400 МГц, ДМСО-d6),(м.д.): 11,74 (с, 1 Н); 11,13 (кв, 1 Н, 4,6 Гц); 10,33 (с, 1 Н); 8,53 (д, 1 Н,8,1 Гц); 8,38 (с, 1 Н); 8,16 (д, 2 Н, 8,1 Гц); 8,0 (с, 1 Н); 7,97 (д, 1 Н, 8,1 Гц); 7,45 (д, 2 Н, 8,1 Гц); 4,61 (м, 2 Н); 4,03-3,79 (м, 3 Н); 3,69-3,24 (м, 8 Н); 2,95 (м, 1 Н); 2,81 (с, 3 Н); 1,32 (т, 3 Н, 6,9 Гц); 1,16-0,92 (м, 4 Н). Пример 6: гидрохлорид 2-амино-1-этил-7-4-[2-([(2S,4R)-1-этил-4-фторпирролидин-2 ил]метиламино)-2-оксоэтил]фенил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 26). 6.1: (4R)-1-этил-4-фтор-L-пролинамид. Работают таким же образом, как описано в примере 2, стадия 2.1, но исходят из 0,88 г (5,2 ммоль) 4(R)-фтор-2(S)-пирролинкарбоксамида (синтез которого описан в Bioorg Med Chem 12(23), 2004, 60536061, Fukushima H. Er coll), 0,90 г (5,7 ммоль) этилиодида и 1,53 г (18,3 ммоль) NaHCO3 в безводном ДМФ (17 мл), получают 0,84 г продукта в виде белого порошка. Выход: 100%. Точка плавления: 127 С. МН+: 161,2 (время удерживания: 0,36 мин, условие 1). 6.2: 1-[(2S,4R)-1-этил-4-фторпирролидин-2-ил]метанамин, гидрохлорид. Работают таким же образом, как описано в примере 5, стадия 5.2, но исходят из 0,69 г (4,3 ммоль) соединения, полученного на стадии 6.1, 17,3 мл (17,3 ммоль) раствора (1 М в ТГФ) тетрагидрофуранового комплекса с тригидридом бора в безводном ТГФ (30 мл) и из 4,3 мл раствора HCl (1 М) в эфире, получают 0,33 г продукта в виде желтого масла, используемого без очистки на следующей стадии. Выход: 52%. МН+: 147,3 (время удерживания: 0,36 мин, условие 1). 6.3: гидрохлорид 2-амино-1-этил-7-4-[2-([(2S,4R)-1-этил-4-фторпирролидин-2-ил]метиламино)2-оксоэтил]фенил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 26). В инертной атмосфере к суспензии 0,85 г (2,2 ммоль) соединения, полученного на стадии 2.5, в дихлорметане (9 мл), прибавляют 0,45 г (4,4 ммоль) триэтиламина. Смесь охлаждают до 0 С (ледяная баня) и по каплям прибавляют 0,61 г (4,4 ммоль) цианурилфторида, растворенного в (1 мл) дихлорметана. Смесь перемешивают при КТ в течение 3 ч 30 мин, затем разбавляют 30 мл дихлорметана, промывают 20 мл предварительно охлажденного насыщенного раствора NaHCO3, сушат над Na2SO4, фильтруют и концентрируют. Полученный фторангидрид кислоты используют на следующей стадии без очистки. К 0,24 г (1,6 ммоль) соединения, полученного на стадии 6.2, растворенного в безводном ДМФ(4 мл), прибавляют 0,41 г (4,1 ммоль) триэтиламина с последующим добавлением 0,63 г (1,7 ммоль) фторангидрида кислоты, полученного на предыдущей стадии. Смесь перемешивают при КТ в течение ночи,затем выпаривают ДМФ в вакууме, остаток обрабатывают дихлорметаном (20 мл) и органический слой промывают насыщенным раствором NaHCO3, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией на силикагеле (элюент: дихлорметан/метанол) с градиентом метанола от 0 до 3%. Получают 0,075 г соединения в виде белого порошка. Выход 9%. Альфа D=-12 (концентрация 25 мг/мл в МеОН). МН+: 509 (время удерживания: 5,2 мин, условие 1). К 0,07 г (0,13 ммоль) соединения, полученного на предыдущей стадии, растворенного в метаноле(2,5 мл), прибавляют 0,13 мл (0,13 ммоль) раствора HCl (1 М) в эфире. Смесь перемешивают при КТ в течение 1 ч, концентрируют при пониженном давлении. Твердый продукт растирают в дихлорметане(2 мл), фильтруют и сушат в вакууме над Р 2 О 5. Получают 0,05 г соединения в виде порошка светложелтого цвета. Выход: 59%. Точка плавления: 170 С. МН+: 509,3 (время удерживания: 5,2 мин, условие 1). 1H ЯМР (400 МГц, ДМСО-d6),(м.д.): 11,73 (с, 1 Н); 11,12 (м, 1 Н); 10,56 (с, 1 Н); 8,59 (т, 1 Н, 5,6 Гц); 8,54 (д, 1 Н, 8,1 Гц); 8,16 (д, 2 Н, 8,3 Гц); 7,99 (с, 1 Н); 7,96 (д, 1 Н, 8,1 Гц); 7,48 (д, 2 Н, 8,3 Гц); 5,41 (дт, 1 Н,4,9-53,2 Гц); 4,61 (м, 2 Н); 3,9 (м, 1 Н); 3,82-3,44 (м, 6 Н); 3,38 (м, 1 Н); 3,12 (м, 1 Н); 2,82 (м, 3 Н); 2,36 (м,1 Н); 2,02 (м, 1 Н); 1,33 (т, 3 Н, 6,8 Гц); 1,24 (т, 3 Н, 7,9 Гц). Пример 7: гидрохлорид 2-амино-1-этил-7-4-[2-([(2S)-1-этил-4,4-дифторпирролидин-2 ил]метиламино)-2-оксоэтил]фенил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 24). 7.1: 1-этил-4,4-дифтор-L-пролинамид. Работают таким же образом, как описано в примере 2, стадия 2.1, но исходят из 0,2 г (1,1 ммоль) 4,4-дифтор-2 (R)-пирролинкарбоксамида (синтез которого описан в Bioorg Med Chem 12(23), 2004, 60536061, Fukushima H. Er coll), 0,18 г (1,2 ммоль) этилиодида и 0,32 г (3,7 ммоль) NaHCO3 в безводном ДМФ(3,5 мл), и получают 0,18 г продукта в виде белого порошка. Выход: 95%. Точка плавления: 138 С. МН+: 179,2 (время удерживания: 1,25 мин, условие 1). 7.2: гидрохлорид 1-[(2S)-1-этил-4,4-дифторпирролидин-2-ил]метанамин. Работают таким же образом, как описано в примере 5, стадия 5.2, но исходят из 0,18 г (1 ммоль) соединения, полученного на стадии 7.1, 8,2 мл (8,2 ммоль) раствора (1 М в ТГФ) тетрагидрофуранового комплекса с тригидридом бора в безводном ТГФ (12 мл) и из 2 мл раствора HCl (1 М) в эфире, получают 0,2 г продукта в виде порошка серого цвета, используемого без какой-либо очистки. Выход: 100%. МН+: не обнаружено. 7.3: гидрохлорид 2-амино-1-этил-7-4-[2-([(2S)-1-этил-4,4-дифторпирролидин-2-ил]метиламино)2-оксоэтил]фенил-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 24). В инертной атмосфере к 0,32 г (0,85 ммоль) соединения, полученного на стадии 2.5, суспензированного в безводном ДМФ (4 мл), последовательно прибавляют 0,37 мл (3,8 ммоль) диизопропилэтиламина,затем раствор в ДМФ (2 мл) 0,20 г (0,85 ммоль) соединения, полученного на стадии 7.2, в присутствии 0,3 мл (1,7 ммоль) диизопропилэтиламина, к полученной реакционной смеси, охлажденной на ледяной бане, добавляют 0,31 г (0,9 ммоль) TBTU. Реакционную смесь перемешивают 6 ч при 70 С, затем концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией на силикагеле (элюент: дихлорметан/этилацетат, 1% NH4OH) с градиентом этилацетата от 0 до 100%, 1% NH4OH. Получают 0,087 г соединения в виде порошка розоватого цвета. К 0,087 г (0,17 ммоль) полученного на предыдущей стадии соединения, растворенного в 2 мл метанола, прибавляют 0,17 мл (0,17 ммоль) раствора HCl (1 М в эфире). Смесь перемешивают при КТ в течение 1 ч., затем прибавляют 5 мл эфира и образовавшийся твердый продукт выделяют фильтрованием,сушат над Р 2 О 5 в вакууме. Получают 0,065 г продукта в виде порошка розоватого цвета. Выход: 70%. Точка плавления: 266 С. МН+: 527,3 (время удерживания: 6,04 мин, условие 1). 1(с, 3 Н); 2,75 (м, 1 Н); 2,43 (м, 1 Н); 1,32 (т, 3 Н, 6,8 Гц); 1,21 (т, 3 Н, 7,1 Гц). Пример 8: гидрохлорид 2-амино-1-этил-7-[4-(2-[(4-этилморфолин-3-ил)метил]амино-2 оксоэтил)фенил]-N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 22). 8.1: 4-этилморфолин-3-карбоксамид. Работают таким же образом, как описано в примере 2, стадия 2.1, но исходят из 0,2 г (1,1 ммоль) морфолин-3-карбоксамида (синтез которого описан в международной заявке WO 2005026156, HennequinL.F.A. et coll), из 0,18 г (1,2 ммоль) этилиодида и 0,32 г (3,7 ммоль) NaHCO3 в безводном ДМФ (3,5 мл), и получают 0,18 г продукта в виде белого порошка. Выход: 95%. Точка плавления: 127 С. МН+: 159,3 (время удерживания: 0,35 мин, условие 1). 8.2: 1-(4-этилморфолин-3-ил)метанамин. В инертной атмосфере к 0,06 г (0,38 ммоль) соединения, полученного на стадии 8.1, растворенного в безводном ТГФ, прибавляют 0,76 мл (0,76 ммоль) раствора (1 М) в безводном ТГФ литийалюминийгидрида, затем реакционную смесь нагревают при 70 С в течение 5 ч. К смеси, охлажденной до КТ, последовательно прибавляют 28 мкл воды, 28 мкл едкого натра (1 н.), 84 мкл воды для образованием "лепешки". Смесь интенсивно перемешивают в течение 30 мин, затем осадок удаляют фильтрацией. Фильтрат концентрируют при пониженном давлении с образованием 0,06 г продукта в виде бесцветного масла,которое используют без очистки на следующей стадии. Выход: 100%. 1 3 Н); 1,53 (с, 2 Н); 1,1 (т, 3 Н, 7,5 Гц). 8.3: гидрохлорид 2-амино-1-этил-7-[4-(2-[(4-этилморфолин-3-ил)метил]амино-2-оксоэтил)фенил]N-метил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамида. Работают таким же образом, как описано в примере 7, стадия 7.3, но исходят из 0,14 г (0,37 ммоль) соединения, полученного на стадии 2.5, 0,05 г (0,37 ммоль) соединения, полученного на стадии 8.2, 0,13 гH ЯМР (400 МГц, ДМСО-d6),(м.д.): 11,73 (с, 1 Н); 11,12 (кв, 1 Н, 4,6 Гц); 11,03 + 10,76 (2 с, 1 Н); 8,52 (д, 2 Н, 8,1 Гц); 8,15 (д, 2 Н, 8,1 Гц); 7,98 (с шир., 1 Н); 7,95 (д, 1 Н, 8,1 Гц); 7,47 (д, 2 Н, 8,1 Гц); 4,61 (м,2 Н); 3,94 (м, 2 Н); 3,81 (м, 1 Н); 3,58 (м, 3 Н); 3,38 (м, 4 Н); 3,18 (м, 3 Н); 2,81 (д,3 Н, 4,6 Гц); 1,28 (м, 6 Н). Пример 9: 1-этил-7-[5-(2-[(1-этилпирролидин-2-ил)метил]амино-2-оксоэтил)тиофен-2-ил]-N,2 диметил-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 52). 9.1: (5-хлортиофен-2-ил)уксусная кислота. К 19,7 г (158,8 ммоль) FAMSO прибавляют 1,2 г (29,8 ммоль) порошкообразного гидроксида натрия, смесь нагревают при 70 С в течение 30 мин, затем прибавляют 3 г (19,8 ммоль) 5-хлор-2 тиофенкарбоксальдегида и смесь снова нагревают при 70 С в течение одной ночи. При комнатной температуре прибавляют 850 мл воды, затем водный слой экстрагируют этилацетатом (3 раза по 430 мл). Объединенные органические слои промывают водным насыщенным раствором NaCl, сушат над Na2SO4,фильтруют и концентрируют при пониженном давлении. Получают 6,7 г продукта в виде масла каштанового цвета, используемого без очистки на следующей стадии. МН+: 253 (время удерживания: 7,6 мин,условие 1). К 6,7 г соединения, полученного на предыдущей стадии, растворенного в этаноле (44 мл), прибавляют 33 мл (132 ммоль) раствора HCl (4 М) в диоксане и смесь нагревают при 80 С в течение 2 ч 15 мин. Смесь охлаждают до КТ, разбавляют этилацетатом (400 мл) и промывают насыщенным водным раствором NaHCO3 (350 мл), затем NaCl (200 мл), органический слой сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией на колонке с силикагелем(элюент: гептан/дихлорметан) с градиентом дихлорметана от 0 до 50%. Получают 1,93 г продукта в виде масле каштанового цвета. Выход: 32%. МН+: 205,1 (время удерживания: 5,33 мин, условие 1). К 1,93 г (6,6 ммоль) соединения, полученного на предыдущей стадии, растворенного в растворяющей смеси (ТГФ/метанол/вода 1/1/1) (33 мл), прибавляют 0,97 г (23,1 ммоль) моногидратного гидроксида лития. Смесь нагревают при 70 С в течение 2 ч, охлаждают до КТ и подкисляют до рН 1 добавлением 23 мл водного раствора HCl (1 н.), затем экстрагируют дихлорметаном (2 раза по 30 мл). Объединенные органические слои промывают насыщенным водным раствором NaCl, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Получают 1,44 г продукта в виде порошка каштанового цвета и используемого без очистки на следующей стадии. МН+: 176 (время удерживания: 12,3 мин, условие GCCl/CH4+). 9.2: 2-(5-хлортиофен-2-ил)-N-[(1-этилпирролидин-2-ил)метил]ацетамид. Работают таким же образом, как описано в примере 4, стадия 4.1, но исходят из 1,77 г (10 ммоль) соединения, полученного на предыдущей стадии, 5,4 г (40 ммоль) 1-этилпирролидинметиламина, 6,4 г(20 ммоль) TBTU и 2,58 г (20 ммоль) диизопропилэтиламина в безводном ДМФ (100 мл). Получают 1,62 г продукта в виде белого порошка. Выход: 56%. Точка плавления: МН+: 287,1 (время удерживания: 4,1 мин, условие 1). 9.3: 1-этил-7-[5-(2-[(1-этилпирролидин-2-ил)метил]амино-2-оксоэтил)тиофен-2-ил]-N,2-диметил 4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид (соединение 52). Работают таким же образом, как описано в примере 1, стадия 1.12, но исходят из 1 г (3,5 ммоль) соединения, полученного на стадии 9.2, 1,3 г (4,5 ммоль) соединения, полученного на стадии 1.7, 0,24 г (0,2 ммоль) тетракис-(трифенилфосфин)палладия и 14 мл насыщенного раствора NaHCO3 в смеси ДМЭ/EtOH 2/1 (35 мл). Получают 0,05 г продукта, полученного в виде белого порошка. Выход 2%. Точка плавления: 250 С. МН+: 497 (время удерживания: 5,3 мин, условие 1). 1H ЯМР (400 МГц, ДМСО-d6),(м.д.): 11,7 (с, 1 Н); 11,1 (кв, 1 Н, 4,5 Гц); 8,43 (д, 1 Н, 8,3 Гц); 8,0 (м,1 Н); 7,95 (с шир., 1 Н); 7,82 (д, 1 Н, 8,3 Гц); 7,80 (д, 1 Н, 3,7 Гц); 7,0 (д, 1 Н, 3,7 Гц); 4,5 (м, 2 Н); 3,72 (с, 2 Н); 3,27 (м, 1 Н); 3,01 (м, 1 Н); 2,89 (м, 1 Н); 2,8 (т, 3 Н, 4,5 Гц); 2,76 (м, 1 Н); 2,42 (м, 1 Н); 2,18 (м, 1 Н); 2,07 (м,1 Н); 1,76 (м, 1 Н); 1,61 (м, 2 Н); 1,48 (м, 1 Н); 1,3 (т, 3 Н, 6,9 Гц); 1,01 (т, 3 Н, 7,2 Гц). Пример 10: гидрохлорид 2-амино-1-этил-N-метил-4-оксо-7-4-[2-(пиридин-2-иламино)этил]фенил 1,4-дигидро-1,8-нафтиридин-3-карбоксамида (соединение 64). 10.1: N-[2-(4-бромфенил)этил]пиридин-2-амин. В герметизируемом сосуде смесь 1,9 г (20,4 ммоль) 2-аминопиридина и 6,1 г (30,6 ммоль) 4 бромфенилэтиламина, растворенного в бутаноле (10 мл), нагревают при 230 С в течение 1 ч в микроволновой печи. Смесь, охлажденную до комнатной температуры, выливают в воду (50 мл) и экстрагируют этилацетатом (3 раза по 100 мл), объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Остаток очищают флэш-хроматографией на колонке с силикагелем (элюент: дихлорметан/этилацетат) с градиентом этилацетата от 0 до 50%. Получают 1,4 г продукта в виде порошка белого цвета. Выход: 24%. Точка плавления: 71 С. МН+: 278,1 (время удерживания: 3,71 мин, условие 1). 10.2: гидрохлорид 2-амино-1-этил-N-метил-4-оксо-7-4-[2-(пиридин-2-иламино)этил]фенил-1,4 дигидро-1,8-нафтиридин-3-карбоксамида (соединение 64). Работают таким же образом, как описано в примере 1, стадия 1.12, но исходят из 0,3 г (1,1 ммоль) соединения, полученного на стадии 10.1, 0,345 г (1,2 ммоль) соединения, полученного на стадии 1.7,0,06 г (0,05 ммоль) тетракис-(трифенилфосфин)палладия и 4,7 мл насыщенного раствора NaHCO3 в смеси ДМЭ/EtOH 2/1 (15 мл). 0,12 г полученного продукта обрабатывают метанолом (3 мл) и добавляют 0,03 мл 36% концентрированной HCl. Получают 0,130 г продукта в виде белого порошка. Выход: 25%. Точка плавления: 242 С. МН+: 497 (время удерживания: 5,33 мин, условие 1). 1H ЯМР (400 МГц, ДМСО-d6),(м.д.): 13,7 (с, 1 Н); 11,73 (м, 1 Н); 11,12 (кв, 1 Н, 4,5 Гц); 8,9 (с, 1 Н); 8,52 (д, 1 Н, 8,1 Гц); 8,17 (д, 2 Н, 8,3 Гц); 8,02 (с шир., 1 Н); 7,96 (д, 1 Н, 8,1 Гц); 7,89 (м, 1 Н); 7,51 (д, 2 Н, 8,3 Гц); 7,08 (д, 1 Н, 9,1 Гц); 6,84 (т шир., 1 Н, 6,7 Гц); 4,6 (м, 2 Н); 3,69 (м, 2 Н); 3,01 (т, 2 Н, 7,2 Гц); 2,81 (д, 3 Н,4,5 Гц); 1,32 (т, 3 Н, 6,9 Гц). Соединения 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 20, 23, 25, 28, 30, 31, 45, 46, 47, 51,52, 56, 62, 63 и 64 получены согласно варианту синтеза, описанному в примере 1. Соединения 35, 36, 37,38, 40, 41, 42, 44, 48, 49, 50, 53, 54 и 55 получены согласно варианту синтеза, описанному в примере 2. Соединения 29, 34 и 43 получены согласно варианту, описанному в примере 3. Приложенные ниже таблицы иллюстрируют химическую структуру и физические свойства некоторых соединений согласно изобретению. Табл. 1 иллюстрирует соединения формулы (Ia), соответствующие соединениям формулы (I), в которой U означает карбонильную группу и R6=-(CH2)n-L. Табл. 2 иллюстрирует соединения формулы (I), соответствующие соединениям формулы (I), в которых U означает карбонильную группу, Y, V и W означают группу -СН-, Z означает атом углерода, R7 означает атом водорода и R6=-(CH2)n-L. Табл. 3 иллюстрирует соединения формулы (Ic), соответствующие соединениям формулы (I), в которых U означает группу -СН 2-, Z, Y, V и W означают группу -СН-, R7 означает атом водорода иR6=-(CH2)n-L. В таблицах: в колонке "соль" символ "-" означает соединение в форме свободного основания, а "HCl" означает соединение в форме гидрохлорида, а число в скобках указывает на отношение (кислота: основание);Me, Et, n-Pr, i-Pr, n-Bu и i-Bu означают соответственно группы метильную, этильную, нпропильную, изопропильную, н-бутильную и изобутильную;Ph и Bn означают соответственно группы фенильную и бензильную. Таблица 1
МПК / Метки
МПК: A61P 35/00, C07D 471/04, A61K 31/4375
Метки: способ, применение, получения, пиридино-пиридинонов, терапии, производные
Код ссылки
<a href="https://eas.patents.su/30-19362-proizvodnye-piridino-piridinonov-sposob-ih-polucheniya-i-primenenie-v-terapii.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиридино-пиридинонов, способ их получения и применение в терапии</a>
Производные 6-гетероарилпиридоиндолона, способ их получения и применение в терапии

Номер патента: 14873
Опубликовано: 28.02.2011
Авторы: Бурри Бернар, Вермют Камилль Жорж, Чиапетти Паола, Жегам Самир, Дерок Жан-Мари, Мюно Иветт, Казелла Пьер
МПК: A61P 35/00, A61K 31/4375, C07D 471/04...
Метки: производные, применение, 6-гетероарилпиридоиндолона, терапии, получения, способ
Формула / Реферат:
1. Соединение формулы (I)в которойR1 обозначает атом водорода или (С1-С4)алкильную группу, CN, CF3 или CHF2;R2 обозначает атом водорода или (С1-С4)алкильную группу;R3 обозначает фенил, не замещенный или замещенный одним или несколькими заместителями, выбираемыми независимо из атома галогена, (С1-С4)алкильной группы, (С1-С4)алкоксильной группы;R4 обозначает гетероциклический радикал, выбираемый изR5 обозначает атом водорода, атом галогена,...
Производные азакарболинов, способ их получения и применение в терапии

Номер патента: 18945
Опубликовано: 29.11.2013
Авторы: Бедель Оливье, Арендт Кристофер, Муркрофт Нейл, Гуйон Тьерри, Ли Жунхуа, Левит Михаил, Бабэн Дидье, Миньяни Серж, Папэн Давид
МПК: A61P 35/00, C07D 471/14, A61K 31/437...
Метки: азакарболинов, способ, применение, получения, терапии, производные
Формула / Реферат:
1. Соединения общей формулы (I)в которой R3, R4 могут независимо один от другого обозначать:1) Н;2) F;3) Cl;4) Br;5) I;6) OR2a;7) NR1aR1b;8) COR2a;9) CO2R2a;10) CO(NR1aR1b);11) С1-C10-алкил, линейный, или разветвленный, или циклический (С3-С7), необязательно моно-, или ди-, или тризамещенный R2a, R2b, R2c;12) С2-С6-алкенил, линейный или разветвленный, необязательно моно-, или ди-, или тризамещенный R2a, R2b, R2c;13) арил, выбранный из фенила,...
Производные диоксан-2-алкилкарбамата, способ их получения и применение в терапии

Номер патента: 8218
Опубликовано: 27.04.2007
Авторы: Медеско Флоренс, Ли Адриен Так, Даргазанли Джихад, Хорнер Кристиан, Баз Мишель, Абуабделла Ахмед
МПК: A61P 25/08, C07D 319/06, A61K 31/335...
Метки: производные, получения, применение, терапии, способ, диоксан-2-алкилкарбамата
Формула / Реферат:
1. Соединение, соответствующее формуле (I) в которой R1 представляет собой фенильную или нафталинильную группу, возможно замещенную одним(ой) или более атомами галогена или группами гидрокси, циано, нитро, (С1-С3)алкил, (С1-С3)алкокси, трифторметил, трифторметокси, бензилокси, (С3-С6)циклоалкил-O- или (С3-С6)циклоалкил(С1-С3)алкокси; R2 представляет собой либо группу общей формулы CHR3CONHR4, в которой R3 представляет собой атом водорода или...
Производные 2-карбамид-4-фенилтиазола, способ их получения и их применение в терапии
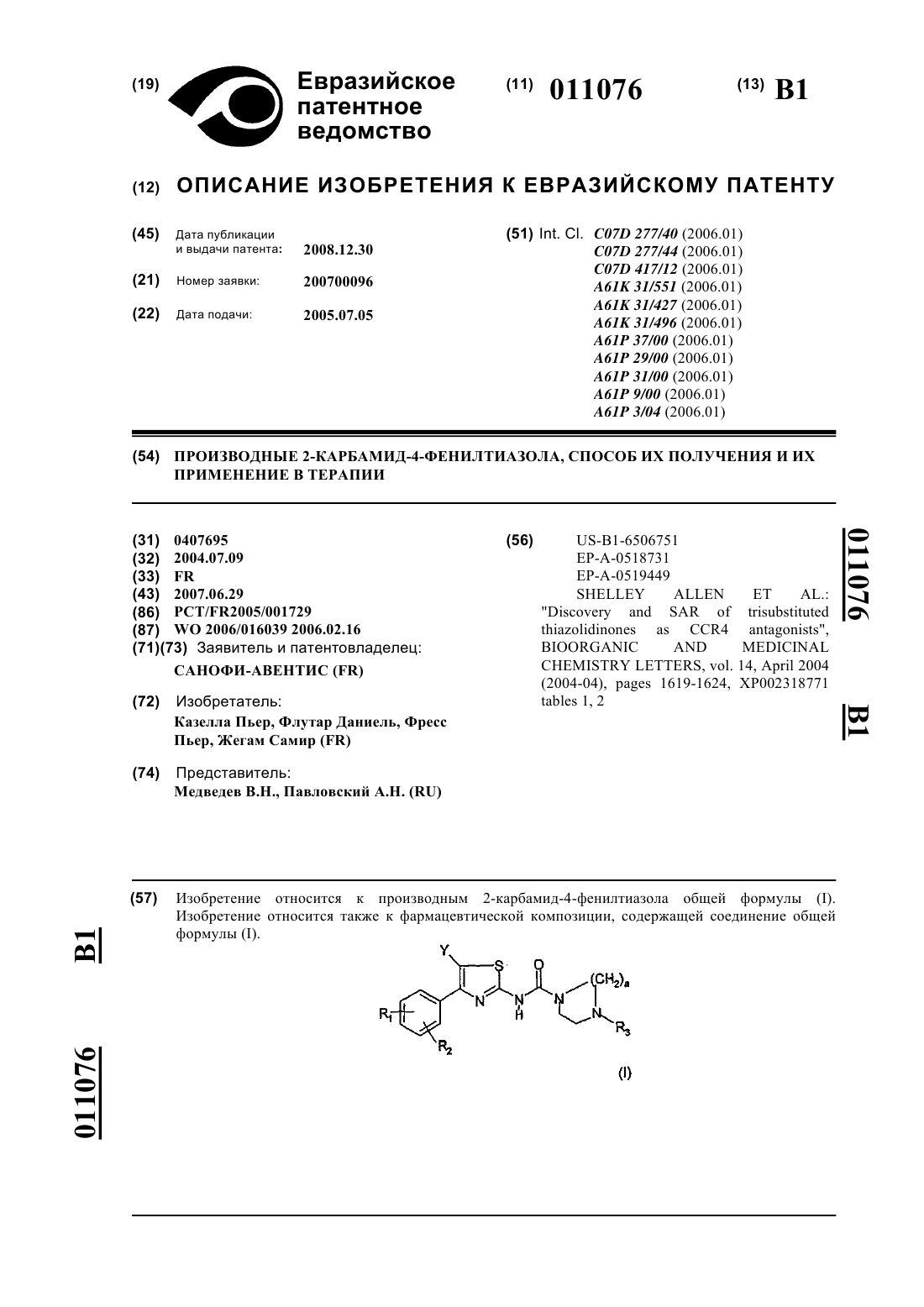
Номер патента: 11076
Опубликовано: 30.12.2008
Авторы: Флутар Даниель, Фресс Пьер, Жегам Самир, Казелла Пьер
МПК: A61K 31/496, A61K 31/427, A61K 31/551...
Метки: производные, 2-карбамид-4-фенилтиазола, получения, способ, применение, терапии
Формула / Реферат:
1. Соединение общей формулы (I) в которой (i) R1 выбран из -О-(C1-C8)алкила; (ii) R2 выбран из группы, состоящей (C1-C8)алкила, перфтор(C1-C4)алкила, (С3-C6)циклоалкила и -О-(C1-C8)алкила; (iii) Y обозначает атом водорода или атом галогена; (iv) R3 обозначает: (a1) группу формулы -(CH2)р-А, в которой р обозначает 0, 1, 2, 3 или 4, и, если р обозначает 2, 3 или 4, то А обозначает группу формулы или -NR4R5, в которой R6 выбран из группы,...
Производные пиперидинил- и пиперазинил- алкилкарбаматов, способы их получения и применение их в терапии

Номер патента: 9468
Опубликовано: 28.12.2007
Авторы: Бюрнье Филипп, Пюэш Фредерик, Женесс Жан, Хорнар Кристиан, Абуабделла Ахмед
МПК: A61K 31/435, A61K 31/436, A61K 31/44...
Метки: применение, пиперазинил, пиперидинил, терапии, производные, алкилкарбаматов, способы, получения
Формула / Реферат:
1. Производные пиперидинил- и пеперазинилалкилкарбаматов, соответствующие формуле (I) где А представляет собой атом азота или группу CR2, в которой R2 представляет собой атом водорода или фтора либо гидроксильную, циано, трифторметильную, C1-6алкильную или C1-6алкоксигруппу; n представляет собой целое число, равное 2 или 3, и m представляет собой целое число, равное 2, когда А представляет собой атом азота; n представляет собой целое число,...
Предыдущий патент: Способ получения замещенных пиримидиновых производных
Следующий патент: Устройство для проведения реакции синтеза углеводородного соединения и способ его эксплуатации
Случайный патент: Способ получения геополимеров