Соединения на основе 4-фенил-6-(2,2,2-трифтор-1-фенилэтокси)пиримидина и способы их применения
Номер патента: 18150
Опубликовано: 30.05.2013
Авторы: Ши Чжи-Цай, Чжан Чэньминь, Ван Ин, Туноори Ашок, Цзинь Хайхун, Девасагаярадж Арокиасами






Формула / Реферат
1. Соединение формулы I

и его фармацевтически приемлемая соль и сольват,
где A1 представляет собой 5- или 6-членный гетероцикл, необязательно замещенный одним или более C1-6-алкилом, галогеном, оксо, C(O)RA, ORA, NRBRC или S(O2)RA, причем C1-6-алкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой;
каждый R1 независимо представляет собой галоген, водород, С1-6-алкил, C(O)RA, ORA, NRBRC, S(O2)RA, причем C1-6-алкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой;
R2 независимо представляет собой водород или NRBRC;
R3 представляет собой водород или С1-4-алкил;
R4 представляет собой водород или C1-4-алкил;
каждый RA независимо представляет собой водород или C1-6-алкил, необязательно замещенный одной или более амино, циано, галоген или гидроксигруппой;
каждый RB независимо представляет собой водород или C1-6-алкил, необязательно замещенный одной или более амино, циано, галоген или гидроксигруппой;
каждый RC независимо представляет собой водород или С1-6-алкил, необязательно замещенный одной или более амино, циано, галоген или гидроксигруппой; и
m имеет значение 1-4.
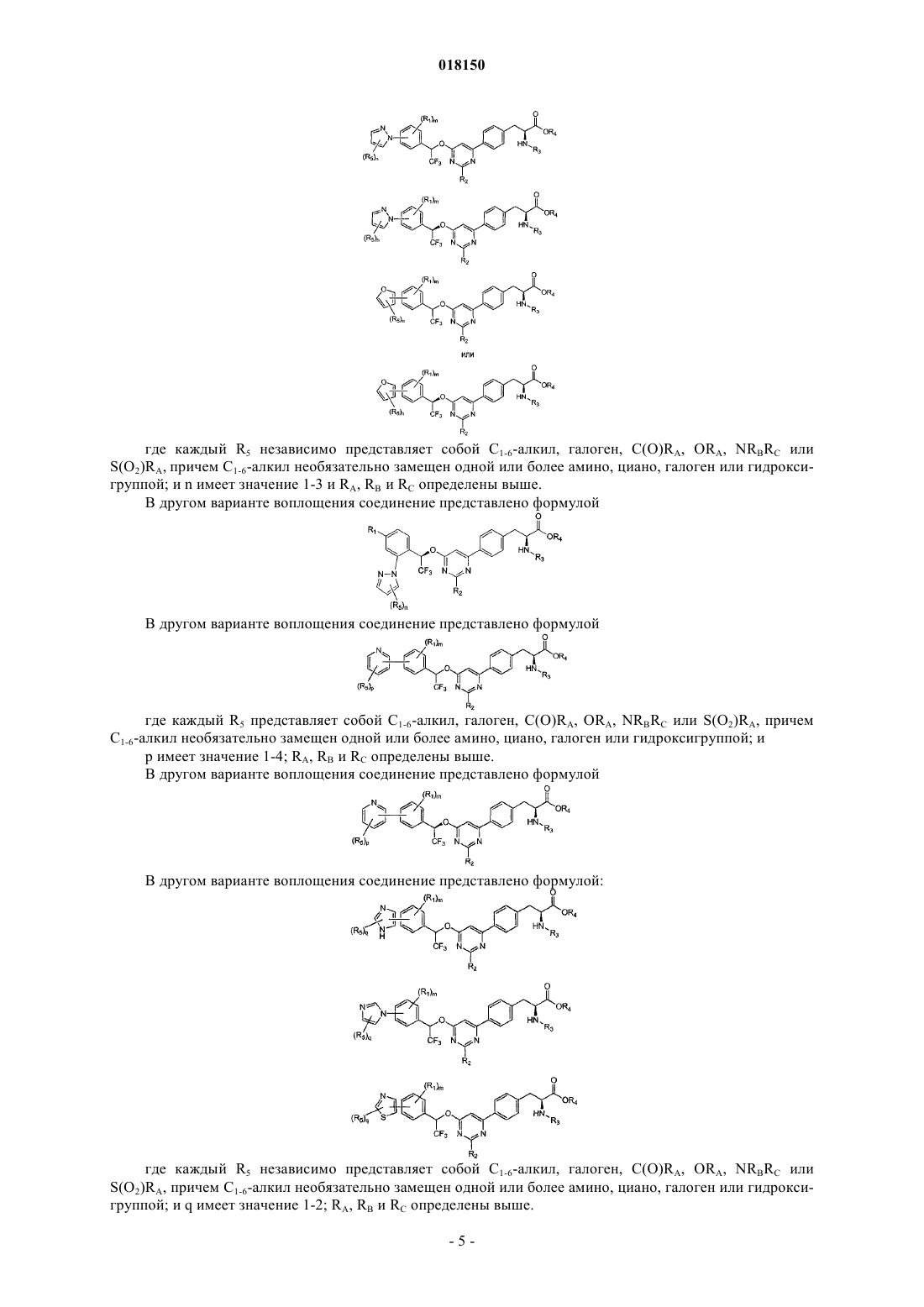
2. Соединение по п.1, которое представлено формулой, выбранной из

где каждый R5 независимо представляет собой С1-6-алкил, галоген, C(O)RA, ORA, NRBRC или S(O2)RA, причем С1-6-алкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой;
n имеет значение 1-3;
RA, RB и RC определены в п.1.
3. Соединение по п.2, которое представлено формулой

4. Соединение по п.1, которое представлено формулой

где каждый R5 представляет собой C1-6-алкил, галоген, C(O)RA, ORa, NRBRC или S(O2)RA, причем C1-6-алкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой;
р имеет значение 1-4;
RA, RB и RC определены в п.1.
5. Соединение по п.4, которое представлено формулой

6. Соединение по п.1, которое представлено формулой, выбранной из

где каждый R5 независимо представляет собой C1-6-алкил, галоген, C(O)RA, ORA, NRBRC или S(O2)RA, причем C1-6-алкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой;
q имеет значение 1-2;
RA, RB и RC определены в п.1.
7. Соединение по п.6, которое представлено формулой, выбранной из


8. Соединение по любому из пп.2-8, где R5 представляет собой C1-4-алкил.
9. Соединение или его фармацевтически приемлемая соль или сольват, где соединение представляет собой
(S)-2-амино-3-(4-{2-амино-6-[2,2,2-трифтор-1-(4-пиридин-4-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-(4-{6-[2,2,2-трифтор-1-(2-пиридин-4-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-(4-{2-амино-6-[2,2,2-трифтор-1-(2-(4-метилтиофен-3-ил)фенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-(4-{2-амино-6-[2,2,2-трифтор-1-(2-(5-метилтиофен-3-ил)фенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-(4-{2-амино-6-[2,2,2-трифтор-1-(4-фуран-3-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-[4-{2-амино-6-{1-[2-(5-диметиламинометилфуран-2-ил)фенил]-2,2,2-трифторэтокси}пиримидин-4-ил}фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{1-[2-(6-цианопиридин-3-ил)фенил]-2,2,2-трифторэтокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-(4-{2-амино-6-[2,2,2-трифтор-1-(2-имидазол-1-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-(4-{6-[2,2,2-трифтор-1-(2-пиразол-1-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[2-(3-трифторметилпиразол-1-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{1-[2-(3,5-диметилпиразол-1-ил)фенил]-2,2,2-трифторэтокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[2-(3-фенилпиразол-1-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[5-метокси-2-(4-метилпиразол-1-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{(R)-2,2,2-трифтор-1-[2-(3-метилпиразол-1-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{l-[4-хлор-2-(3-метилпиразол-1-ил)фенил]-2,2,2-трифторэтокси}пиримидин-4-ил)фенил]пропионовую кислоту;
этиловый эфир (S)-2-амино-3-[4-(2-амино-6-(R-1-[4-хлор-2-(3-метилпиразол-1-ил)фенил]-2,2,2-трифторэтокси}пиримидин-4-ил)фенил]пропионовой кислоты;
(S)-2-амино-3-(4-(2-амино-6-((R)-1-(4-хлор-2-(3-метил-1Н-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропановую кислоту;
(S)-2-амино-3-(4-{2-амино-6-[2,2,2-трифтор-1-(2-тиазол-2-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[2-(пиридин-3-илокси)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[4-(пиридин-3-илокси)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(6-{2,2,2-трифтор-1-[4-(пиридин-3-илокси)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-(4-{2-амино-6-[2,2,2-трифтор-1-(4-тиофен-2-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-(4-{6-[2,2,2-трифтор-1-(4-имидазол-1-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-(4-{2-амино-6-[2,2,2-трифтор-1-(4-[1,2,4]триазол-1-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-(4-{2-амино-6-[2,2,2-трифтор-1-(4-фтор-2-тиофен-3-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[4-фтор-2-(4-метилтиофен-2-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{1-[2-(3,5-диметилизоксазол-4-ил)-4-фторфенил]-2,2,2-трифторэтокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[5-фтор-2-(3-метилпиразол-1-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[5-хлор-2-(3-метилпиразол-1-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[4-(2-оксопирролидин-1-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{(R)-2,2,2-трифтор-1-[5-фтор-2-(3-метилпиразол-1-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[4-(6-метоксипиридин-2-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[2-фтор-4-(5-метоксипиридин-3-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{(S)-2,2,2-трифтор-1-[4-(2-фторпиридин-4-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{(S)-2,2,2-трифтор-1-[4-(5-метоксипиридин-3-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{(S)-2,2,2-трифтор-1-[4-(4-трифторметилпиридин-3-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-(4-{2-амино-6-[(S)-2,2,2-трифтор-1-(4-изоксазол-4-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-(4-{2-амино-6-[2,2,2-трифтор-1-(2-пиримидин-5-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-(4-{2-амино-6-[2,2,2-трифтор-1-(2-тиофен-3-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-[4-(2-амино-6-{2,2,2-трифтор-1-[2-(1-метил-1Н-пиразол-4-ил)фенил]этокси}пиримидин-4-ил)фенил]пропионовую кислоту;
(S)-2-амино-3-(4-{6-[2,2,2-трифтор-1-(2-фуран-3-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(S)-2-амино-3-(4-{6-[2,2,2-трифтор-1-(2-фуран-2-илфенил)этокси]пиримидин-4-ил}фенил)пропионовую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(4-(пиридин-3-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(2-(2-метилпиридин-4-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(2-(4-метилтиофен-3-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-3-(4-(6-(1-(2-(1Н-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)-2-аминопиримидин-4-ил)фенил)-2-аминопропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(4-(фуран-2-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(6-(2,2,2-трифтор-1-(2-(пиридин-3-илокси)фенил)этокси)пиримидин-4-ил)фе-нил)пропановую кислоту;
(2S)-3-(4-(6-(1-(2-(1Н-1,2,4-триазол-1-ил)фенил)-2,2,2-трифторэтокси)-2-аминопиримидин-4-ил)фенил)-2-аминопропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(2-(фуран-3-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(4-(фуран-2-ил)-3-метоксифенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(5-(2,2,2-трифтор-1-(2-(фуран-2-ил)фенил)этокси)пиразин-2-ил)фенил)пропановую кислоту;
(2S)-3-(4-(5-(1-(2-(1Н-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиразин-2-ил)фенил)-2-аминопропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(1-(4,5-диметокси-2-(1Н-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(2-(2-метил-1Н-имидазол-1-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(2-(5-метилтиофен-2-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(1-(2-(5-(диметилкарбамоил)фуран-2-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(4-фтор-2-(тиофен-2-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(6-(2,2,2-трифтор-1-(4-фтор-2-(тиофен-2-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(6-(2,2,2-трифтор-1-(4-фтор-2-(тиофен-3-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(6-(2,2,2-трифтор-1-(4-фтор-2-(4-метилтиофен-2-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(S)-2-амино-3-(4-(2-амино-6-((R)-2,2,2-трифтор-1-(4-(6-фторпиридин-3-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-3-(4-(6-(1-(4-(1Н-имидазол-1-ил)фенил)-2,2,2-трифторэтокси)-2-аминопиримидин-4-ил)фенил)-2-аминопропановую кислоту;
(2S)-2-амино-3-(4-(6-(2,2,2-трифтор-1-(4-(тиофен-2-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(S)-2-амино-3-(4-(2-амино-6-((R)-2,2,2-трифтор-1-(4-(пиримидин-5-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(6-(1-(2-(3,5-диметилизоксазол-4-ил)-4-фторфенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропановую кислоту;
(S)-2-амино-3-(4-(2-амино-6-((R)-2,2,2-трифтор-1-(4-(2-метилпиридин-4-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-3-(4-(6-(1-(4-(1Н-1,2,4-триазол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)-2-аминопропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(4-(пиперидин-1-илметил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(2-фтор-4-(2-метилпиридин-4-ил)фенил)этокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(1-(4-(6-хлорпиридазин-3-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(1-(4-(4-трет-бутилтиазол-2-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропановую кислоту;
(2S)-2-амино-3-(4-(2-амино-6-(2,2,2-трифтор-1-(3'-метокси-3-(3-метил-1Н-пиразол-1-ил)бифенил-4-ил)этокси)пиримидин-4-ил)фенил)пропановую кислоту или
(2S)-2-амино-3-(4-(2-амино-6-(1-(5-хлор-2-(3-метил-1Н-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропановую кислоту.
10. Фармацевтическая композиция в качестве лекарственного средства, включающая соединение по любому из пп.1-9 и фармацевтически приемлемый эксципиент или разбавитель.
11. Композиция по п.10, применяемая в качестве лекарственного средства для лечения, предотвращения или контроля заболевания или расстройства, опосредованного периферическим серотонином.
12. Композиция по п.10, применяемая в качестве лекарственного средства для лечения, предотвращения или контроля заболевания или расстройства, выбранного из группы, состоящей из карциноидного кризиса или карциноидного синдрома, желудочно-кишечного заболевания или расстройства, сердечно-сосудистого или легочного заболевания или расстройства, склеродермии или серотонинового синдрома.
13. Единичная стандартная лекарственная форма, используемая в качестве лекарственного средства, включающая композицию по п.10.
14. Единичная стандартная лекарственная форма по п.13, применяемая в качестве лекарственного средства для лечения, предотвращения или контроля заболевания или расстройства, опосредованного периферическим серотонином.
15. Единичная стандартная лекарственная форма по п.13, применяемая в качестве лекарственного средства для лечения, предотвращения или контроля заболевания или расстройства, выбранного из группы, состоящей из карциноидного кризиса или карциноидного синдрома, желудочно-кишечного заболевания или расстройства, сердечно-сосудистого или легочного заболевания или расстройства, склеродермии или серотонинового синдрома.
16. Способ ингибирования ТРН1 активности in vitro, который включает контактирование ТРН1 с соединением по любому из пп.1-9.
17. Применение соединения по любому из пп.1-9 в качестве лекарственного средства, ингибирующего ТРН1 активность.
18. Применение соединения по любому из пп.1-9 в качестве лекарственного средства для лечения, предотвращения или контроля заболевания или расстройства, опосредованного периферическим серотонином.
19. Применение соединения по любому из пп.1-9 в качестве лекарственного средства для лечения, предотвращения или контроля заболевания или расстройства, выбранного из группы, состоящей из карциноидного кризиса или карциноидного синдрома, желудочно-кишечного заболевания или расстройства, сердечно-сосудистого или легочного заболевания или расстройства, склеродермии или серотонинового синдрома.
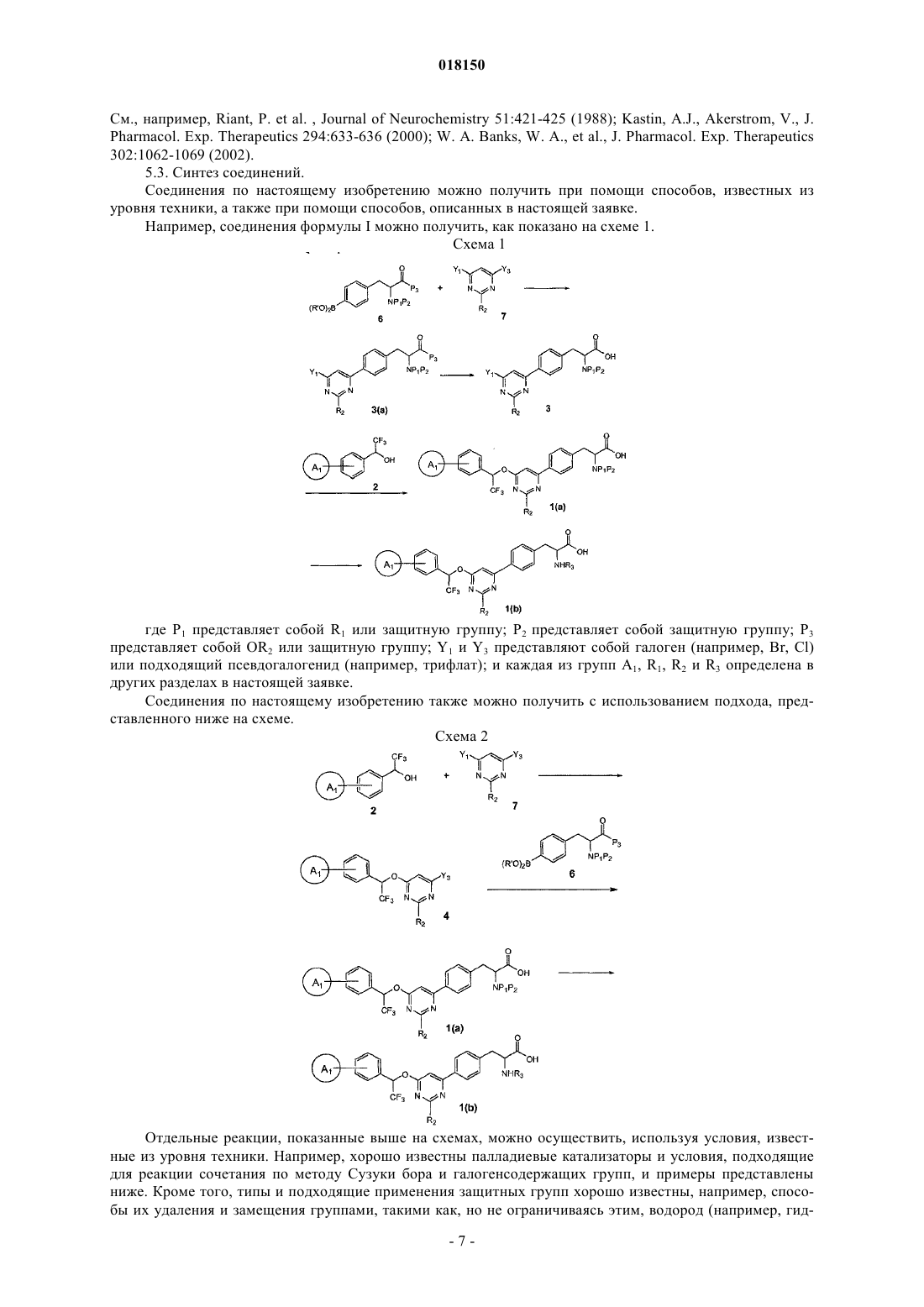
Текст
СОЕДИНЕНИЯ НА ОСНОВЕ 4-ФЕНИЛ-6-(2,2,2-ТРИФТОР-1 ФЕНИЛЭТОКСИ)ПИРИМИДИНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ Раскрыты соединения формулы I, а также включающие их композиции и способы, их использования для лечения, предотвращения и/или контроля заболевания и расстройства. Заявка на патент заявляет приоритет предварительной заявки на патент США 60/874596, поданной 12 декабря 2006 года, включенной в полном объеме в настоящую заявку посредством ссылки. 1. Область, к которой относится изобретение Изобретение относится к соединениям на основе 4-фенил-6-(2,2,2-трифтор-1-фенилэтокси)пиримидина, включающим их композициям и их применению для лечения, предотвращения и контроля при заболеваниях и расстройствах. 2. Предпосылки создания изобретения Нейротрансмиттер серотонин [5-гидрокситриптамин (5-НТ)] участвует в различных аспектах центральной нервной системы, связанных с контролем настроения и регуляцией сна, беспокойством, алкоголизмом, наркотической зависимостью, потреблением пищи и сексуальным поведением. В периферических тканях серотонин, по имеющимся сведениям, вовлечен в регулирование сосудистого тонуса, перистальтику кишечника, первичный гемостаз и иммунный клеточный ответ. Walther, D. J., et al., Science 299:76 (2003). Фермент триптофангидроксилаза (ТРН) катализирует стадию, ограничивающую скорость биосинтеза серотонина. Сообщалось о двух изоформах ТРН: ТРН 1, которая экспрессируется на периферии,преимущественно в желудочно-кишечном (GI) тракте; и ТРН 2, которая экспрессируется в серотонергических нейронах. Id. Изоформа ТРН 1 кодируется геном tph1; TPH2 кодируется геном tph2. Id. Сообщалось о мышах, дефицитных по гену tph1 (мыши с генным "нокаутом"). Как сообщалось, в одном случае мыши экспрессировали нормальные количества серотонина в классических серотонергических отделах головного мозга, но большой недостаток серотонина наблюдался на периферии. Id. В другом случае мыши с "нокаутом" демонстрировали аномальную сердечную активность, которую приписывали недостатку периферического серотонина. Cote, F., et al., PNAS 100(23): 13525-13530 (2003). Поскольку серотонин вовлечен в такое множество биохимических процессов, лекарственные средства, которые воздействуют на серотониновые рецепторы, часто имеют побочные неблагоприятные эффекты. Таким образом, существует необходимость в новых способах лечения заболеваний и расстройств,которые вызваны, опосредованы или связаны с серотонином. 3. Краткое изложение изобретения Изобретение частично направлено на соединения формулы I и его фармацевтически приемлемая соль и сольват,где A1 представляет собой 5- или 6-членный гетероцикл, необязательно замещенный одним или более С 1-6-алкилом, галогеном, оксо, C(O)RA, ORA, NRBRC или S(O2)RA, причем С 1-6-алкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой; каждый R1 независимо представляет собой галоген, водород, С 1-6-алкил, C(O)RA, ORA, NRBRC,S(O2)RA, причем C1-6-лкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой;R2 независимо представляет собой водород или NRBRC;R3 представляет собой водород или С 1-4-алкил;R4 представляет собой водород или C1-4-алкил; каждый RA независимо представляет собой водород или C1-6-алкил, необязательно замещенный одной или более амино, циано, галоген или гидроксигруппой; каждый RB независимо представляет собой водород или C1-6-алкил, необязательно замещенный одной или более амино, циано, галоген или гидроксигруппой; каждый RC независимо представляет собой водород или C1-6-алкил, необязательно замещенный одной или более амино, циано, галоген или гидроксигруппой; иm имеет значение 1-4. Конкретные соединения ингибируют TPH (например, ТРН 1) активность. Настоящее изобретение также относится к фармацевтическим композициям. 4. Краткое описание рисунков Фиг. 1 представляет доза-зависимый эффект соединения по настоящему изобретению на уровни 5 НТ в тонкой кишке у мышей. Соединение вводили перорально в растворе 15% Captisol при 15, 50, 150 и 300 мг/кг. 5. Подробное описание изобретения Изобретение основано, частично, на том открытии, что отключение гена tph1 у мышей значительно снижает уровни GI серотонина, но при этом оказывает незначительный, если вообще таковой имеется,измеримый эффект на центральную нервную систему (ЦНС). Изобретение также основано на открытии соединений, которые ингибируют ТРН (например,ТРН 1). При введении млекопитающим, предпочтительные соединения по настоящему изобретению снижают уровни серотонина, обладают фармакокинетическими и фармакодинамическими свойствами, что делает возможным их практическое применения для лечения, предотвращения и контроля при заболеваниях и расстройствах, и они имеют широкий предел безопасности между фармакологическим эффектом и токсичностью или нежелательными побочными реакциями. 5.1. Определения. Если не указано иное, термин "алкил" означает углеводород с прямой цепью, разветвленный и/или циклический ("циклоалкил"), содержащий от 1 до 20 (например, от 1 до 10 или от 1 до 4) атомов углерода. Алкильные группы, содержащие от 1 до 4 углеродов относят к "низшему алкилу". Примеры алкильных групп включают метил, этил, пропил изопропил, н-бутил, трет-бутил изобутил, пентил, гексил, изогексил, гептил, 4,4-диметилпентил, октил, 2,2,4-триметилпентил, нонил, децил, ундецил и додецил. Циклоалкильные группы могут быть моноциклическими или полициклическими, и примеры включают циклопропил, циклобутил, циклопентил, циклогексил и адамантил. Дополнительные примеры алкильных групп включают линейные, разветвленные и/или циклические части (например, 1-этил-4-метилциклогексил). Термин "алкил" включает насыщенные углеводороды, также как и алкенильные и алкинильные группы. Если не указано иное, фразы "заболевание или расстройство, опосредованное периферическим серотонином" и "заболевание и расстройство, опосредованное периферическим серотонином" означают заболевание и/или расстройство, имеющие один или несколькими симптомов, на тяжесть которых влияют уровни периферического серотонина. Если не указано иное, термин "галоген" и включает фтор, хлор, бром и йод. Если не указано иное, термин "гетероцикл" относится к ароматическому, частично ароматическому или неароматическому моноциклическому или полициклическому кольцу или кольцевой системе, состоящей из углерода, водорода и, по меньшей мере, одного гетероатома (например, N, О или S). Гетероцикл может включать несколько (то есть два или более) колец, конденсированных или связанных вместе. Гетероциклы включают гетероарилы. Примеры включают бензо[1,3]диоксолил, 2,3-дигидробензо[1,4]диоксинил, циннолинил, фуранил, гидантоинил, морфолинил, оксетанил, оксиранил, пиперазинил, пиперидинил, пирролидинонил, пирролидинил, тетрагидрофуранил, тетрагидропиранил, тетрагидропиридинил, тетрагидропиримидинил, тетрагидротиофенил, тетрагидротиопиранил и валеролактамил. Если не указано иное, термин "гетероциклоалкил" или "гетероциклалкил" относится к гетероциклической группе, связанной с алкильной группой. Если не указано иное, термин "гетероциклоалкил" относится к неароматическому гетероциклу. Если не указано иное, термины "контролировать", "контролирующий" и "контроль" охватывают предотвращение рецидива определенного заболевания или расстройства или одного или нескольких его симптомов у пациента, который уже перенес заболевание или расстройство, и/или продление периода времени, в течение которого пациент, который уже перенес заболевание или расстройство, остается в ремиссии. Термины охватывают модуляцию порога, развития и/или длительности заболевания или расстройства, или изменение того, как пациент отвечает на заболевание или расстройство. Если не указано иное, термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных кислот или оснований, включая неорганические кислоты и основания, а также органические кислоты и основания. Подходящие фармацевтически приемлемые основно-аддитивные соли включают соли металлов, образованные из алюминия, кальция, лития, магния,калия, натрия и цинка, или органические соли, образованные из лизина, N,N'-дибензилэтилендиамина,хлорпрокаина, холина, диэтаноламина, этилендиамина, меглумина (N-метилглюкамина) и прокаина. Подходящие нетоксичные кислоты включают неорганические и органические кислоты, такие как уксусная, альгиновая, антраниловая, бензолсульфоновая, бензойная, камфорсульфоновая, лимонная, этенсульфоновая, муравьиная, фумаровая, фуранкарбоновая, галактоуроновая, глюконовая, глюкуроновая,глутаминовая, гликолевая, бромисто-водородная, хлористо-водородная, изетионовая, молочная, малеиновая, яблочная, миндальная, метансульфоновая, муциновая, азотная, памовая, пантотеновая, фенилуксусная, фосфорная, пропионовая, салициловая, стеариновая, янтарная, сульфаминовая, серная, винная кислота и паратолуолсульфоновая кислота. Конкретные нетоксичные кислоты включают хлористоводородную, бромисто-водородную, фосфорную, серную и метансульфоновую кислоты. Примеры конкретных солей, таким образом, включают гидрохлорид и мезилат. Другие являются хорошо известными в данной области. См., например, Remington's Pharmaceutical Sciences, 18th ed. (Mack Publishing, EastonPA: 1990) и Remington: The Science и Practice of Pharmacy, 19th ed. (Mack Publishing, Easton PA: 1995). Если не указано иное, термин "сильный ингибитор ТРН 1" представляет собой соединение, которое имеет значение ТРН 1 IC50 менее чем около 10 мкм. Если не указано иное, термины "предотвращать," "предотвращающий" и "предотвращение" предполагают действие, которое имеет место до того, как пациент начинает страдать определенным заболеванием или расстройством, которое ингибирует или снижает тяжесть заболевания или расстройства или одного или нескольких его симптомов. Термины охватывают профилактику. Если не указано иное, "профилактически эффективное количество" соединения представляет собой количество, достаточное для предотвращения заболевания или состояния или одного или нескольких симптомов, связанных с заболеванием или состоянием, или предотвращение его рецидива. Профилактически эффективное количество соединения представляет собой количество терапевтического средства,отдельно или в комбинации с другими средствами, которое обеспечивает профилактическую пользу в предотвращении заболевания. Термин "профилактически эффективное количество" может охватывать количество, которое улучшает профилактику в целом или повышает профилактическую эффективность другого профилактического средства. Если не указано иное, термин "селективный ингибитор ТРН 1" представляет собой соединение, которое имеет значение ТРН 2IC50, по меньшей мере, приблизительно в 10 раз больше чем его значение ТРН 1IC50. Если не указано иное, термин "стереомерно обогащенная композиция" соединения относится к смеси указанного соединения и его стереоизомера(ов), которая содержит больше указанного соединения,чем его стереоизомера(ов). Например, стереоизомерически обогащенная композиция (S)-бутан-2-ола охватывает смеси (S)-бутан-2-ола и (R)-бутан-2-ола в соотношениях, например, около 60/40, 70/30, 80/20,90/10, 95/5 и 98/2. Если не указано иное, термин "стереоизомерная смесь" охватывает рацемические смеси, а также стереомерно обогащенные смеси (например, R/S=30/70, 35/65, 40/60, 45/55, 55/45, 60/40, 65/35 и 70/30). Если не указано иное, термин "стереомерно чистый" означает композицию, которая содержит один стереоизомер соединения и, по существу, не содержит других стереоизомеров этого соединения. Например, стереомерно чистая композиция соединения, имеющего один стереоцентр, по существу, не содержит противоположного стереоизомера соединения. Стереомерно чистая композиция соединения, имеющего два стереоцентра, по существу, не содержит других диастереомеров соединения. Стереомерно чистая композиция соединения, которое имеет несколько стереоцентров, но которое представлено или названо таким способом, что стереохимии определены не для всех его стереоцентров, по существу, не содержит изомеров соединения, которые имеют разные стереохимии на стереоцентрах, для которых стереохимия определена. Например, "стереомерно чистый 1R)-1,2-дихлорпропил)бензол" относится к 1R)-1,2-дихлорпропил)бензолу, который, по существу, не содержит 1S)-1,2-дихлорпропил)бензола. Типичное стереомерно чистое соединение содержит более чем около 80 мас.% одного стереоизомера соединения и менее чем около 20 мас.% других стереоизомеров соединения, более чем около 90 мас.% одного стереоизомера соединения и менее чем около 10 мас.% других стереоизомеров соединения,более чем около 95 мас.% одного стереоизомера соединения и 5 мас.% других стереоизомеров соединения, более чем около 97 мас.% одного стереоизомера соединения и менее чем около 3 мас.% других стереоизомеров соединения, или более чем около 99 мас.% одного стереоизомера соединения и менее чем около 1 мас.% других стереоизомеров соединения. Если не указано иное, термин "замещенный", когда он используется для описания химической структуры или группы, относится к производному этой структуры или группы, где один или несколько атомов водорода замещены атомом, химической группой или функциональной группой, такой как, но не ограничиваясь этим, спирт, альдегид, алкокси, алканоилокси, алкоксикарбонил, алкенил, алкил (например, метил, этил, пропил, трет-бутил), алкинил, алкилкарбонилокси (-ОС(О)алкил), амид (-С(О)NHалкил- или -алкил-NHC(O)алкил), амидинил(-С(NH)NH-алкил или -C(NR)NH2), амин (первичный, вторичный и третичный, такой как алкиламино, ариламино, арилалкиламино), ароил, арил, арилокси, азо,карбамоил (-NHC(О)O-алкил- или -OC(O)NH-алкил), карбамил (например, CONH2, а также CONH-алкил,CONH-арил и CONH-арилалкил), карбонил, карбоксил, карбоновая кислота, ангидрид карбоновой кислоты, хлорид карбоновой кислоты, циано, сложный эфир, эпоксид, эфир (например, метокси, этокси), гуанидино, галоген, галогеналкил (например, -CCl3, -CF3, -C(CF3)3), гетероалкил, полуацеталь, имин (первичный и вторичный), изоцианат, изотиоцианат, кетон, нитрил, нитро, кислород (то есть, для получения оксогруппы), фосфодиэфир, сульфид, сульфонамидо (например, SO2NH2), сульфон, сульфонил (включая алкилсульфонил, арилсульфонил и арилалкилсульфонил), сульфоксид, тиол (например, сульфгидрил,тиоэфир) и мочевина (-NHCONH-алкил-). Если не указано иное, "терапевтически эффективное количество" соединения представляет собой количество, достаточное для обеспечения терапевтически благоприятного эффекта при лечении или контроле заболевания или состояния, или для отсрочки или минимизирования одного или нескольких симптомов, связанных с заболеванием или состоянием. Терапевтически эффективное количество соединения представляет собой количество терапевтического средства, отдельно или в сочетании с другими терапевтическими средствами, которое обеспечивает терапевтическую пользу при лечении или контроле заболевания или состояния. Термин "терапевтически эффективное количество" может охватывать количество,которое обеспечивает терапевтическое улучшение в целом, уменьшает или помогает избежать симптомы или причины заболевания или состояния или повышает терапевтическую эффективность другого терапевтического средства. Если не указано иное, термин "ТРН 1IC50" представляет собой IC50 соединения для ТРН 1, опреде-3 018150 ленную с использованием анализа ингибирования in vitro, описанного в примерах ниже. Если не указано иное, термин " ТРН 2IC50" представляет собой IC50 соединения для ТРН 2, определенную с использованием анализа ингибирования in vitro, описанного в примерах ниже. Если не указано иное, термины "лечить" "лечащий" и "лечение" предполагают действие, которое происходит в то время, когда пациент страдает от конкретного заболевания или расстройства, которое снижает тяжесть заболевания или расстройства или одного или нескольких его симптомов, или тормозит или замедляет развитие заболевания или расстройства. Если не указано иное, термин "включают" имеет то же значение, что и "включают, но не ограничиваются этим", а термин "включает" имеет то же значение, что и "включает, но не ограничивается этим". Аналогичным образом, термин "такой как" имеет то же значение, что и термин "такой как, но не ограничиваясь этим". Если не указано иное, одно или несколько прилагательных, непосредственно предшествующих ряду существительных, должны быть истолкованы применительно к каждому из существительных. Например, фраза "необязательно замещенный алкил или гетероцикл" имеет то же значение, что и "необязательно замещенный алкил, необязательно замещенный гетероцикл." Следует заметить, что химическая группа, которая образует часть более крупного соединения, может быть описана в настоящей заявке, используя название, обычно соответствующее ей, когда она существует в виде отдельной молекулы, или название, обычно соответствующее ее радикалу. Например, термины "пиридин" и "пиридил" соответствуют тем же значениям, которые использовали для описания группы, присоединенной к другим химическим группам. Таким образом, две фразы "ХОН, где X представляет собой пиридил", и "ХОН, где X представляет собой пиридин", соответствуют тем же значениям и охватывают соединения пиридин-2-ол, пиридин-3-ол и пиридин-4-ол. Также следует заметить, что если стереохимия структуры или части структуры не указана, например, жирной или пунктирной линией, структуру или часть структуры следует интерпретировать как охватывающую все ее стереоизомеры. Аналогичным образом, названия соединений, имеющих один или несколько хиральных центров, которые точно не определяют стереохимию этих центров, охватывают чистые стереоизомеры и их смеси. Более того, предполагается, что любой атом, показанный на рисунке с ненасыщенной валентностью, присоединен к достаточному количеству атомов водорода для соответствия валентностям. Кроме того, химические связи, изображенные одной сплошной линией, параллельной одной пунктирной линии, охватывают как простые, так и двойные (например, ароматические) связи, если допускают валентности. 5.2. Соединения. Изобретение охватывает, inter alia, соединения формулы I и их фармацевтически приемлемые соли и сольваты, где A1 представляет собой 5- или 6-членный гетероцикл, необязательно замещенный одним или более C1-6-алкилом, галогеном, оксо, C(O)RA, ORA,NRBRC или S(O2)RA, причем С 1-6-алкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой; каждый R1 независимо представляет собой галоген, водород, C1-6-алкил, C(O)RA, ORA, NRBRC,S(O2)RA, причем С 1-6-алкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой; R2 независимо представляет собой водород или NRBRC; R3 представляет собой водород илиC1-4-алкил; R4 представляет собой водород или C1-4-алкил; каждый RA независимо представляет собой водород или С 1-6-алкил, необязательно замещенный одной или более амино, циано, галоген или гидроксигруппой; каждый RB независимо представляет собой водород или C1-6-алкил, необязательно замещенный одной или более амино, циано, галоген или гидроксигруппой; каждый RC независимо представляет собой водород или С 1-6-алкил, необязательно замещенный одной или более амино, циано, галоген или гидроксигруппой; и m имеет значение 1-4. В одном варианте воплощения соединение представлено формулой В другом варианте соединение представлено формулой где каждый R5 независимо представляет собой С 1-6-алкил, галоген, C(O)RA, ORA, NRBRC илиS(O2)RA, причем C1-6-алкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой; и n имеет значение 1-3 и RA, RB и RC определены выше. В другом варианте воплощения соединение представлено формулой В другом варианте воплощения соединение представлено формулойC1-6-алкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой; и р имеет значение 1-4; RA, RB и RC определены выше. В другом варианте воплощения соединение представлено формулой В другом варианте воплощения соединение представлено формулой: где каждый R5 независимо представляет собой С 1-6-алкил, галоген, C(O)RA, ORA, NRBRC илиS(O2)RA, причем С 1-6-алкил необязательно замещен одной или более амино, циано, галоген или гидроксигруппой; и q имеет значение 1-2; RA, RB и RC определены выше. В другом варианте воплощения соединение представлено формулой В отношении различных формул, раскрытых в настоящей заявке, в конкретных соединениях по настоящему изобретению A1 является гетероциклом. В некоторых случаях А 1 необязательно замещен одним или несколькими атомами галогена или низшим алкилом. В некоторых случаях R1 представляет собой водород или галоген. В некоторых случаях m имеет значение 1. В некоторых случаях R2 представляет собой водород. В некоторых случаях R3 представляет собой водород или низший алкил. RA представляет собой С 1-6 алкил. В некоторых случаях R4 представляет собой водород или низший алкил. Настоящее изобретение охватывает стереомерно чистые соединения и их стереомерно обогащенные композиции. Стереоизомеры могут быть асимметрично синтезированы или разделены с использованием стандартных технических приемов, таких как хиральные колонки, хиральные агенты разделения или ферментативное разделение. См., например, Jacques, J., et al., Enantiomers, Racemates and ResolutionsResolutions, p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972). Конкретные соединения по настоящему изобретению являются сильными ингибиторами ТРН 1. Отдельные соединения имеют значения ТРН 1 IC50 менее чем около 10, 5, 2,5, 1, 0,75, 0,5, 0,4, 0,3, 0,2, 0,1 или 0,05 мкм. Конкретные соединения являются селективными ингибиторами ТРН 1. Отдельные соединения имеют значения ТРН 1 IC50 примерно в 10, 25, 50, 100, 250, 500 или 1000 раз меньше, чем их значения ТРН 2IC50. Конкретные соединения не ингибируют существенным образом тирозингидроксилазу человека(ТН). Например, отдельные соединения имеют значение IC50 для ТН более чем около 100, 250, 500 или 1000 мкм. Конкретные соединения не ингибируют существенным образом фенилаланингидроксилазу человека (РАН). Например, отдельные соединения имеют значение IC50 для РАН более чем около 100, 250, 500 или 1000 мкм. Конкретные соединения по настоящему изобретению не демонстрируют существенное связывание(например, ингибируют со значением IC50 более чем около 10, 25, 50, 100, 250, 500, 750 или 1000 мкм) с одним или несколькими из следующих: ангиотензин-превращающим ферментом, рецептором эритропоэтина (ЕРО), фактором IX, фактором XI, интегрином (например, 4), изоксазолиновым или изоксазоловым рецептором фибриногена, металлопротеазой, нейтральной эндопептидазой (NEP), фосфатазой (например, тирозинфосфатазой), фосфодиэстеразой (например, PDE-4), полимеразой, PPARy, TNF-, адгезивной молекулой-1 клеток сосудов (VCAM-1) или рецептором витронектина. Способность соединения связываться (например, ингибировать) с любой из этих мишеней можно легко определить, используя способы, известные из уровня техники, как описано в ссылочных документах, приведенных выше. Отдельные соединения по настоящему изобретению не ингибируют клеточную адгезию. При введении млекопитающим (например, мышам, крысам, собакам, обезьянам или людям), некоторые соединения по настоящему изобретению не обладают способностью легкого проникновения через гемато-энцефалический барьер (например, менее чем около 5, 2,5, 2, 1,5, 1, 0,5 или 0,01 процентов соединения в крови проходит в головной мозг). Способность или неспособность соединения проникать через гемато-энцефалический барьер можно определить при помощи способов, известных из уровня техники.Pharmacol. Exp. Therapeutics 294:633-636 (2000); W. A. Banks, W. A., et al., J. Pharmacol. Exp. Therapeutics 302:1062-1069 (2002). 5.3. Синтез соединений. Соединения по настоящему изобретению можно получить при помощи способов, известных из уровня техники, а также при помощи способов, описанных в настоящей заявке. Например, соединения формулы I можно получить, как показано на схеме 1. Схема 1 где P1 представляет собой R1 или защитную группу; Р 2 представляет собой защитную группу; Р 3 представляет собой OR2 или защитную группу; Y1 и Y3 представляют собой галоген (например, Br, Cl) или подходящий псевдогалогенид (например, трифлат); и каждая из групп A1, R1, R2 и R3 определена в других разделах в настоящей заявке. Соединения по настоящему изобретению также можно получить с использованием подхода, представленного ниже на схеме. Схема 2 Отдельные реакции, показанные выше на схемах, можно осуществить, используя условия, известные из уровня техники. Например, хорошо известны палладиевые катализаторы и условия, подходящие для реакции сочетания по методу Сузуки бора и галогенсодержащих групп, и примеры представлены ниже. Кроме того, типы и подходящие применения защитных групп хорошо известны, например, способы их удаления и замещения группами, такими как, но не ограничиваясь этим, водород (например, гид-7 018150 ролиз в кислой или основной среде). Сложноэфирные производные соединений по настоящему изобретению можно легко получить с использованием способов, таких как представленные ниже на схеме 3. Схема 3 Применяя способы, известные из уровня техники, синтетические подходы, показанные выше, можно легко модифицировать для получения широкого ряда соединений. Например, хиральная хроматография и другие методы, известные из уровня техники, можно использовать для разделения стереоизомеров конечного продукта. См., например, Jacques, J., et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen, S. H., et al. Tetrahedron 33:2725 (1977); Eliel, E. L., Stereochemistry of Carbon Compounds (McGraw Hill, NY, 1962); и Wilen, S. H., Tables of Resolving Agents and Optical Resolutions,p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972). Кроме того, как показано выше на некоторых схемах, в синтезе можно использовать хиральные исходные вещества для получения стереомерно обогащенных или чистых продуктов. 5.4. Способы применения. Настоящее изобретение охватывает способ ингибирования ТРН, который включает контактирование ТРН с соединением по настоящему изобретению (то есть соединением, раскрытым в настоящей заявке). В одном конкретном способе ТРН представляет собой ТРН 1. В другом ТРН представляет собой ТРН 2. В одном способе имеет место ингибирование in vitro. В другом имеет место ингибирование invivo. Один вариант воплощения охватывает способ ингибирования ТРН 1 у млекопитающего (например,человека), который включает введение млекопитающему соединения по настоящему изобретению. В одном конкретном способе ТРН 2 существенно не ингибируется. В одном способе соединение не может легко проникать через гемато-энцефалический барьер. В другом соединение является селективным ингибитором ТРН 1. Настоящее изобретение охватывает применение соединений по изобретению для лечения, предотвращения и контроля различных заболеваний и расстройств, опосредованных периферическим серотонином, которые включают ингибирование ТРН 1 активности у пациента, нуждающегося в таком лечении,предотвращении или контроле. В конкретном варианте воплощения ингибирование осуществляют путем введения пациенту терапевтически или профилактически эффективного количества сильного ингибитора ТРН 1. Примеры сильных ингибиторов ТРН 1 раскрыты в настоящей заявке. Конкретные заболевания и расстройства включают карциноидный синдром и желудочно-кишечные заболевания и расстройства. Примеры конкретных заболеваний и расстройств включают боль в брюшной полости (например, связанную с медуллярной карциномой щитовидной железы), беспокойство, карциноидный синдром, глютеновую болезнь, констипацию (например, констипацию ятрогенного характера и идиопатическую констипацию), болезнь Крона, депрессию, диабет, диарею (например, желче-кислотную диарею, энтеротоксин-индуцированную секреторую диарею, диарею ятрогенного характера, идиопатическую диарею (например, идиопатическую секреторную диарею) и диарею путешественников), рвоту,-8 018150 функциональную абдоминальную боль, функциональные аноректальные расстройства, функциональный метеоризм, функциональную диспепсию, функциональные растсройства желчного пузыря, синдром раздраженной толстой кишки (IBS; включая IBD-d, IBS-с и IBS-a), непереносимость лактозы, MEN типа I иII, тошноту, синдром Огилви, панкреатический холерный синдром, недостаточность поджелудочной железы, феохромацитому, склеродермию, соматизацию, сфинктер при расстройствах Одди, язвенный колит и ульцерогенную аденому поджелудочной железы (синдром Золлингера-Эллисона). Дополнительные заболевания и расстройства включают сердечно-сосудистые и легочные заболевания и расстройства, такие как острая и хроническая гипертензия, хроническое обструктивное легочное заболевание (COPD), эмболия сосудов легких (например, бронхостеноз и легочная гипертензия в результате эмболии сосудов легких), легочная гипертензия (например, легочная гипертензия, связанная с портальной гипертензией) и лучевой пневмонит (включая, пневмонит, приводящий к или способствующий легочной гипертензии). Другие заболевания включают мигрень, сопровождающуюся тошнотой и рвотой,респираторный дистресс-синдром взрослых (ARDS), карциноидный кризис, CREST синдром (кальциноз,феномен Рейно, желудочная дисфункция, склеродактилия, телеангиэктазия), серотониновый синдром и субарахноидальное кровоизлияние. В конкретных вариантах настоящего изобретения лечение, контроль и/или предотвращение заболеваний или расстройств достигаются без неблагоприятных эффектов, связанных с изменениями уровней серотонина в центральной нервной системе (ЦНС). Примеры таких неблагоприятных эффектов включают тревожное возбуждение, беспокойство, депрессию и расстройства сна (например, бессонницу и нарушение сна). 5.5. Фармацевтические композиции. Настоящее изобретение охватывает фармацевтические композиции, включающие одно или несколько соединений по настоящему изобретению. Некоторые фармацевтические композиции представляют собой отдельные стандартные лекарственные формы, подходящие для перорального введения, введения через слизистую оболочку (например, назального, сублингвального, вагинального, буккального или ректального), парентерального (например, подкожного, внутривенного, болюсной инъекции, внутримышечного или внутриартериального) или чрескожного введения пациенту. Примеры лекарственных форм включают, но не ограничиваются этим: таблетки; каплеты; капсулы, такие как мягкие эластичные желатиновые капсулы; саше; пастилки; таблетки для рассасывания; дисперсии; суппозитории; мази; припарки (горячие комперессы); пасты; порошки; повязки; кремы; пластыри; растворы; накладки; аэрозоли (например, назальные спреи или ингаляторы); гели; жидкие лекарственные формы, подходящие для перорального введения или введения через слизистую оболочку пациенту, включая суспензии (например, водные или неводные жидкие суспензии, эмульсии масло-в-воде или жидкие эмульсии вода-вмасле), растворы и эликсиры; жидкие лекарственные формы, подходящие для парентерального введения пациенту; и стерильные твердые вещества (например, кристаллические или аморфные твердые вещества), которые можно реструктурировать с получением жидких лекарственных форм, подходящих для парентерального введения пациенту. Композиция должна соответствовать способу введения. Например, пероральное введение соединения, которое может разлагаться в желудке, можно осуществлять с использованием энтеросолюбильного покрытия. Аналогичным образом, композиция может содержать ингредиенты, которые ускоряют доставку активного ингредиента (ингредиентов) к участку их действия. Например, соединения можно вводить в формах, представляющих собой липосомы, для их защиты от разлагающих ферментов, ускорения их транспорта в кровотоке и осуществления их доставки через клеточные мембраны. Аналогичным образом, плохо растворимые соединения можно включать в жидкие лекарственные формы (и лекарственные формы, подходящие для реструктурирования) при помощи солюбилизирующих веществ, эмульгаторов и поверхностно-активных веществ, таких как, но не ограничиваясь этим, циклодекстрины (например, -циклодекстрин, -циклодекстрин, Captisol и Encapsin (см., например, Davisand Brewster, Nat. Rev. Drug Disc. 3:1023-1034 (2004, Labrasol, Labrafil, Labrafac, кремафор и неводные растворители, такие как, но не ограничиваясь этим, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, диметилсульфоксид (ДМСО), биосовместимые масла (например, масло семян хлопчатника, арахисовое масло, кукурузное масло, масло зародышей растений, оливковое масло, касторовое масло и кунжутное масло), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли, сложные эфиры жирных кислот сорбитана и их смеси (например, ДМСО:кукурузное масло). Плохо растворимые соединения также можно включать в суспензии, используя другие методики,известные из уровня техники. Например, наночастицы соединения можно суспендировать в жидкости с получением наносуспензии (см., например, Rabinow, Nature Rev. Drug Disc. 3:785-796 (2004. Формы наночастиц соединений, описанных в настоящей заявке, можно получить при помощи способов, описанных в патентных пуликациях США 2004-0164194, 2004-0195413, 2004-0251332, 2005-0042177 А 1,2005-0031691 А 1 и патентах США 5145684, 5510118, 5518187, 5534270, 5543133, 5662883, 5665331,5718388, 5718919, 5834025, 5862999, 6431478, 6742734, 6745962, каждый из которых в полном объеме включен в настоящую заявку посредством ссылки. В одном варианте воплощения форма наночастиц включает частицы, имеющие средний размер частиц менее чем около 2000 нм, менее чем около 1000 нм или менее чем около 500 нм. Композиция, форма и тип лекарственной формы типично варьируют в зависимости от применения. Например, лекарственная форма, применяемая при лечении острой формы заболевания, может содержать большие количества одного или нескольких активных ингредиентов по сравнению с лекарственной формой, применяемой при лечении хронической формы того же заболевания. Аналогичным образом,лекарственная форма для парентерального введения может содержать меньшие количества одного или нескольких активных ингредиентов по сравнению с лекарственной формой для перорального введения,применяемой для лечения того же самого заболевания. Такие отличия должны быть очевидны для специалистов в данной области. См., например, Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing, Easton PA (1990). 5.5.1. Лекарственные формы для перорального введения. Фармацевтические композиции по настоящему изобретению, подходящие для перорального введения, могут быть представлены в виде дискретных лекарственных форм, таких как, но не ограничиваясь этим, таблетки (например, жевательные таблетки), каплеты, капсулы и жидкости (например, сиропы с приданным им приятным вкусом). Такие лекарственные формы содержат предварительно определенные количества активных ингредиентов и могут быть получены способами, используемыми в фармацевтике и хорошо известными специалистам в данной области. См., в основном, Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing, Easton PA (1990). Типичные лекарственные формы для перорального введения получают путем тщательного перемешивания активного ингредиента (ингредиентов) с, по меньшей мере, одним эксципиентом, в соответствии с традиционными методами составления фармацевтических композиций. Эксципиенты могут иметь самые различные формы, в зависимости от формы препарата, желательной для введения. По причине легкости их введения таблетки и капсулы представляют наиболее выгодные стандартные лекарственные формы для перорального введения. Если необходимо, на таблетки может быть нанесено покрытие при помощи стандартных водных или неводных технологий. Такие лекарственные формы можно получить традиционными методами, используемыми в фармацевтике. Главным образом, фармацевтические композиции и лекарственные формы получают путем однородного и тщательного смешивания активных ингредиентов с жидкими носителями, тонкоизмельченными твердыми носителями или и теми и другими, а затем продукту придают желаемую для представления форму, если требуется. Разрыхлители можно включать в твердые лекарственные формы, что способствует быстрой растворимости. Смазывающие вещества также могут быть включены, что облегчает изготовление лекарственных форм(например, таблеток). 5.5.2. Лекарственные формы для парентерального введения. Лекарственные формы для парентерального введения можно вводить пациентам различными путями, включая подкожное, внутривенное (включая болюсную инъекцию), внутримышечное и внутриартериальное введение. Поскольку их введение обычно обходит стороной естественную защиту пациентов от загрязняющих примесей, лекарственные формы для парентерального введения являются особенно стерильными или способными к стерилизации перед введением пациенту. Примеры лекарственных форм для парентерального введения включают растворы, готовые для инъекций, сухие продукты, готовые к растворению или суспендированию в фармацевтически приемлемом носителе для инъекций, суспензии,готовые для инъекций, и эмульсии. Подходящие носители, которые могут быть использованы для получения лекарственных форм для парентерального введения по настоящему изобретению, хорошо известны специалистам в данной области. Примеры включают воду для инъекций USP; водные наполнители, такие как хлорид натрия для инъекций, раствор Рингера для инъекций, раствор декстрозы для инъекций, раствор декстрозы и хлорида натрия для инъекций и лактатсодержащий раствор Рингера для инъекций; водорастворимые наполнители, такие как этиловый спирт, полиэтиленгликоль и полипропиленгликоль; и неводные наполнители,такие как кукурузное масло, хлопковое масло, арахисовое масло, кунжутное масло, этилолеат, изопропилмиристат и бензилбензоат. 6. Примеры 6.1. Получение мышей с прерванным геном tph1. Экзон 3 мышиного гена ТРН 1 удаляли методом генного таргетирования, в основном, как описаноWattler et al., Biotechniques 26(6): 1150-6 (1999). Полученные животные с генным "нокаутом" демонстрировали нормальную активность ТРН в головном мозге, но существенное снижение экспрессии ТРН в кишке. 6.2. Физиологические эффекты прерывания гена tph1. Мышей, гомозиготных (-/-) по прерыванию гена tphl, исследовали вместе с мышами, гетерозиготными (+/-) по прерыванию этого гена, вместе с мышами дикого типа (+/+). В ходе анализа мышей подвергали медицинской обработке, используя комплекс медицинских диагностических процедур, разработанных для оценки основных систем органов субъекта-млекопитающего. Путем исследования гомози- 10018150 готных (-/-) мышей с генным "нокаутом" в желательных количествах и вместе с гетерозиготными (+/-) мышами и мышами дикого типа (+/+) были получены более достоверные и повторяемые данные. Прерывание гена tphl, прежде всего, влияло на изоформу ТРН в желудочно-кишечном тракте(ТРН 1) и оказывало незначительный эффект или не оказывало никакого эффекта на изоформу ТРН в головном мозге (ТРН 2). Прерывание гена не оказывало никаких измеримых побочных эффектов на центральную нервную систему. Это было подтверждено иммунохимией серотонина, показавшей значительное снижение или отсутствие серотонина в желудке, двенадцатиперстной кишке, тощей кишке, подвздошной кишке, слепой кишке и толстой кишке, тогда как уровни серотонина в вставочных нейронах не изменялись. Некоторые мыши, гомозиготные (-/-) по прерыванию гена tphl, демонстрировали уменьшение тромбоза, без существенного усиления кровотечения или других неблагоприятных показаний. 6.3. Методы ВЭЖХ анализа. В некоторых из следующих примеров синтеза представлены данные времени удерживания, полученные методом высокоэффективной жидкостной хроматографии (ВЭЖХ). Различные условия, используемые для получения этих значений времени удерживания, описаны ниже: Способ A: YMC-PACK ODS-A 3,050 мм; Растворитель А=Н 2 О, 0,1% ТФУК; Растворитель В=МеОН, 0,1% ТФУК; В% от 10 до 90% в течение 4 мин; скорость потока=2 мл/мин; длина волны наблюдения=220 и 254 нм. Способ В: YMC-PACK ODS-A 3,050 мм; Растворитель А=90% воды, 10% МеОН с 0,1% ТФУК; Растворитель В=90% МеОН, 10% воды с 0,1% ТФУК; В% от 0 до 100% в течение 4 мин; скорость потока=2 мл/мин; длина волны наблюдения=220 и 254 нм. Способ С: ShimPack VP ODS 4,650 мм; Растворитель А=90% H2O, 10% МеОН, 1%ТФУК; Растворитель В=10% H2O, 90% МеОН, 1%ТФУК; В% от 0 до 100% в течение 2 мин; скорость потока=3,5 мл/мин; длина волны наблюдения=220 и 254 нм. Способ D: Shim VP ODS 4,650 мм; Растворитель А=Н 2 О с 0,1% ТФУК; Растворитель В=МеОН с 0,1% ТФУК; В% от 0 до 100% в течение 4 мин; скорость потока=3 мл/мин; длина волны наблюдения=254 нм. Способ Е: YMC Pack ODS-A 4,633 мм; Растворитель А=Н 2 О, 0,1% ТФУК; Растворитель В=МеОН с 0,1% ТФУК; В% от 10 до 90% в течение 3 мин; скорость потока 2 мл/мин; длина волны наблюдения 220 и 254 нм. Способ F: YMC-Pack ODS-A 3,050 мм; Растворитель А=90% H2O, 10% МеОН, 1% ТФУК; Растворитель В=10% Н 2 О, 90% МеОН, 1% ТФУК; В% от 10 до 90% в течение 4 мин; скорость потока=2 мл/мин длина волны наблюдения=220 и 254 нм. 6.4. Синтез (S)-2-амино-3-(4-2-амино-6-[2,2,2-трифтор-1-(4-пиридин-4-илфенил)этокси]пиримидин-4-илфенил)пропионовой кислоты Тетрабутиламмонийфторид (0,027 мл; 1,0 М раствор в тетрагидрофуране) добавляли к раствору 4 пиридин-4-илбензальдегида (500 мг, 2,73 ммоль) и трифторметилтриметилсилана (TMSCF3) (485 мкл,3,28 ммоль) в 5 мл ТГФ при 0 С. Полученную смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь затем обрабатывали при помощи 5 мл 1 н. раствора HCl и перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали досуха, добавляли 9 мл 1 М водного раствора карбоната натрия, водную фазу экстрагировали при помощи хлороформа (310 мл) и объединенный слой хлороформа промывали водой, сушили над MgSO4. Органический растворитель удаляли в вакууме с получением 360 мг 2,2,2-трифтор-1-(4-пиридин-4 илфенил)этанола, выход: 51%. Смесь 2,2,2-трифтор-1-(4-пиридин-4-илфенил)этанола (100 мг, 0,40 ммоль), 2-амино-4,6 дихлорпиримидина (60 мг, 0,38 ммоль) и карбоната цезия (468 мг, 1,4 4 ммоль) растворяли в 2 мл 1,4 диоксана в 50-мл герметично закрытой трубке. Смесь нагревали при 110 С в течение ночи, затем охлаждали до комнатной температуры; добавляли 10 мл этилацетата и затем фильтровали через целит. Фильтрат концентрировали с получением 120 мг 4-хлор-6-[2,2,2-трифтор-1-(4-пиридин-4-илфенил)этокси]пиримидин-2-иламина, выход: 80%. В микроволновом сосуде 4-хлор-6-[2,2,2-трифтор-1-(4-пиридин-4-илфенил)этокси]пиримидин-2 иламина (30 мг, 0,080 ммоль), 4-бороно-L-фенилаланин (21 мг, 0,098 ммоль) и 1 мл актонитрила и 0,7 мл воды смешивали вместе. Затем к смеси добавляли 0,3 мл 1 н. водного раствора карбоната натрия, с последующим добавлением 5% мол. дихлорбис-(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали при 150 С в течение 5 мин в условиях микроволнового облучения. После ох- 11018150 лаждения реакционную смесь упаривали досуха. Остаток растворяли в 2,5 мл метанола и затем очищали при помощи препаративной ЖХ с получением 6,7 мг (S)-2-амино-3-(4-2-амино-6-[2,2,2-трифтор-1-(4 пиримидин-4-илфенил)этокси]пиримидин-4-илфенил)пропионовой кислоты. 1H ЯМР (400 МГц,CD3OD)(ч/млн) 8,82 (с, 2H), 8,26 (с, 2 Н), 8,02 (д, J=8 Гц, 2R), 7,97 (д, J=8,4 Гц, 2 Н), 7,86 (д, J=8,4 Гц,2 Н), 7,45 (д, J=8 Гц, 2 Н), 6,89 (кв., J=6,8 Гц, 1 Н), 6,81 (д, J=2 Гц, 1 Н), 4,29 (т, J=l,6 Гц, 1 Н), 3,39 (м, 1 Н),3,19 (м, 1 Н). 6.5. Синтез (S)-2-амино-3-(4-6-[2,2,2-трифтор-1-2-пиридин-4-илфенил)этокси]пиримидин-4-ил фенил)пропионовой кислоты Тетрабутиламмонийфторид (0,027 мл; 1,0 М раствор в тетрагидрофуране) добавляли к раствору 2 пиридин-4-илбензальдегида (500 мг, 2,73 ммоль) и трифторметилтриметилсилана (TMSCF3) (485 мкл, 3,2 8 ммоль) в 5 мл ТГФ при 0 С. Полученную смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь затем обрабатывали при помощи 5 мл 1 н. раствора HCl и перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали досуха, добавляли 9 мл 1 М водного раствора карбоната натрия, водную фазу экстрагировали при помощи хлороформа (310 мл) и объединенный органический слой промывали водой, сушили над MgSO4. Органический растворитель выпаривали с получением 300 мг 2,2,2-трифтор-1-(2-пиридин-4-илфенил) этанола, выход: 43%. Смесь 2,2,2-трифтор-1-(2-пиридин-4-илфенил)этанола (100 мг, 0,40 ммоль), 4,6-дихлорпиримидина(54 мг, 0,38 ммоль), карбоната цезия (468 мг, 1,44 ммоль) и 1,4-диоксана (1 мл). Смесь нагревали при 110 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры. Добавляли 10 мл этилацетата и затем смесь фильтровали через целит. Фильтрат концентрировали с получением 110 мг 4-хлор 6-[2,2,2-трифтор-1-(2-пиридин-4-илфенил)этокси]пиримидина, выход: 76%. В микроволновом сосуде вместе смешивали 4-хлор-6-[2,2,2-трифтор-1-(4-пиридин-4-илфенил) этокси]пиримидин (30 мг, 0,082 ммоль), 4-бороно-L-фенилаланин (21 мг, 0,098 ммоль), 1 мл актонитрила и 0,7 мл воды. Затем к смеси добавляли 0,3 мл 1 н. водного раствора карбоната натрия, с последующим добавлением 5% мол. дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали при 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения реакционную смесь упаривали досуха. Остаток растворяли в 2,5 мл метанола и затем очищали при помощи препаративной ЖХ с получением 19 мг (S)-2-амино-3-(4-6-[2,2,2-трифтор-1-(2-пиридин-4 илфенил)этокси]пиримидин-4-илфенил)пропионовой кислоты. 1 Н ЯМР (400 МГц, CD3OD)(ч/млн) 8,94 (д, J=6 Гц, 2 Н), 8,79 (д, J=l,2 Гц, 1 Н), 8,15 (м, 4 Н), 7,84 (т, J=5,2 Гц, 1 Н), 7,62 (м, 3 Н), 7,46 (м, 3 Н),6,66 (кв., J=6,4 Гц, 1 Н), 4,31 (кв., J=6 Гц, 1 Н), 3, 41 (м, 1 Н), 3,26 (м, 1 Н). 6.6. Синтез (S)-2-амино-3-(4-2-амино-6-[2,2,2-трифтор-1-(2-(4-метилтиофен-3-ил)фенил]этокси пиримидин-4-ил)фенил]пропионовой кислоты В микроволновом сосуде смешивали вместе 3-бром-4-метилтиофен (653 мг, 3,69 ммоль), 2 формилфенилбороновую кислоту (500 мг, 3,36 ммоль) и 7 мл актонитрила. К полученному раствору добавляли 6,7 мл 1 н. водного раствора карбоната натрия, с последующим добавлением 5 мол.% дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали при 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения добавляли 50 мл этилацетата,органический слой отделяли, промывали водой, сушили при помощи сульфата натрия. Органический растворитель выпаривали с получением неочищенного продукта, который очищали при помощи колонки(TMSCF3) (228 мкл, 1,54 ммоль) в 5 мл ТГФ. Образовавшуюся смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь затем обрабатывали при помощи 5 мл 1 н. раствора HCl и перемешивали при комнатной температуре в течение ночи. Продукт экстрагировали при помощи этилацетата (350 мл). Органический слой отделяли и сушили над сульфатом натрия. Органический растворитель выпаривали с получением 340 мг 2,2,2-трифтор-1-[2-(4 метилтиофен-3-ил)фенил]этанола, выход 97%. Смесь 2,2,2-трифтор-1-[2-(4-метилтиофен-3-ил)фенил]этанола (100 мг, 0,37 ммоль), 2-амино-4,6 дихлорпиримидина (54 мг, 0,33 ммоль), карбоната цезия (481 мг, 1,48 ммоль) и 1,4-диоксана (1 мл) нагревали при 110 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры; добавляли 10 мл этилацетата. Смесь затем фильтровали через целит, фильтрат концентрировали с получением 100 мг 4-хлор-6-2,2,2-трифтор-1-[2-(4-метилтиофен-3-ил)фенил]этоксипиримидин-2-иламина, выход: 76%. В микроволновом сосуде смешивали 4-хлор-6-2,2,2-трифтор-1-[2-(4-метилтиофен-3-ил)фенил] этоксипиримидин-2-иламин (30 мг, 0,075 ммоль), 4-бороно-L-фенилаланин (19 мг, 0,09 ммоль), 1 мл актонитрила и 0,7 мл воды. К смеси добавляли 0,3 мл 1 н. водного раствора карбоната натрия, с последующим добавлением 5 мол.% дихлорбис-(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали при 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения реакционную смесь упаривали досуха. Остаток растворяли в 2,5 мл метанола и затем очищали при помощи препаративной ВЭЖХ с получением 15,1 мг (S)-2-амино-3-(4-2-амино-6-[2,2,2-трифтор-1(2-(4-метилтиофен-3-ил)фенил]этоксипиримидин-4-илфенил)пропионовой кислоты. 1 Н ЯМР (400 МГц,CD3OD):(ч/млн) 7,94 (д, J=8 Гц, 2 Н), 7,80 (с, 1 Н), 7,50 (м, 5 Н), 7,25 (м, 2 Н), 7,03 (с, 1H), 6,94 (с, 1 Н),4,31 (т, J=5,6 Гц, 1 Н), 3,48 (м, 1 Н), 3,26 (м, 1 Н), 1,98 (с, 3 Н). 6.7. Синтез (S)-2-амино-3-(4-2-амино-6-[2,2,2-трифтор-1-(2-(5-метилтиофен-3-ил)фенил]этокси пиримидин-4-ил)фенил]пропионовой кислоты В микроволновом сосуде смешивали 4-бром-2-метилтиофен (653 мг, 3,69 ммоль), 2 формилфенилбороновую кислоту (500 мг, 3,36 ммоль) и 7 мл актонитрила. К полученному раствору добавляли 6,7 мл 1 н. водного раствора карбоната натрия, с последующим добавлением 5 мол.% дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали при 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения добавляли 50 мл этилацетата,органический слой отделяли, промывали водой, сушили при помощи сульфата натрия, органический растворитель выпаривали и остаток очищали при помощи ISCO с получением 550 мг 2-(5-метилтиофен-3 ил)бензальдегида, выход 81%. Тетрабутиламмонийфторид (0,028 мл; 1,0 М раствор в тетрагидрофуране) добавляли к раствору 2(5-метилтиофен-3-ил)бензальдегида (550 мг, 1,29 ммоль) и трифторметилтриметилсилана (TMSCF3) (483 мкл, 3,27 ммоль) в 10 мл ТГФ при 0 С. Образовавшуюся смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь затем обрабатывали при помощи 10 мл 1 н. раствора HCl и перемешивали при комнатной температуре в течение ночи. Продукт экстрагировали при помощи этилацетата (350 мл). Органический слой отделяли и сушили над сульфатом натрия. Органический растворитель выпаривали с получением 650 мг 2,2,2-трифтор-1-[2-(5 метилтиофен-3-ил)фенил]этанола, выход: 87%. Смесь 2,2,2-трифтор-1-[2-(5-метилтиофен-3-ил)фенил]этанола (100 мг, 0,37 ммоль), 2-амино-4,6 дихлорпиримидина (54 мг, 0,33 ммоль), карбоната цезия (481 мг, 1,48 ммоль) и 1,4-диоксана (2 мл) нагревали при 110 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры; добавляли 10 мл этилацетата. Смесь затем фильтровали через целит, фильтрат концентрировали с получением 90 мг 4-хлор-6-2,2,2-трифтор-1-[2-(5-метилтиофен-3-ил)фенил]этоксипиримидин-2-иламина, выход: 68%. В микроволновом сосуде смешивали 4-хлор-6-2,2,2-трифтор-1-[2-(5-метилтиофен-3-ил)фенил] этоксипиримидин-2-иламин (30 мг, 0,075 ммоль), 4-бороно-L-фенилаланин (19 мг, 0,09 ммоль), 1 мл актонитрила и 0,7 мл воды. К смеси добавляли 0,3 мл 1 н. водного раствора карбоната натрия, с последующим добавлением 5 мол.% дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали при 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения реакционную смесь упаривали досуха. Остаток растворяли в 2,5 мл метанола и затем очищали при помощи препаративной ЖХ с получением 10,1 мг (S)-2-амино-3-(4-2-амино-6-[2,2,2-трифтор-1-(2(5-метилтиофен-3-ил)фенил)этокси]пиримидин-4-илфенил)пропионовой кислоты. 1 Н ЯМР (400 МГц,CD3OD):(ч/млн) 7,83 (д, J=8,4 Гц, 2 Н), 7,63 (д, J=7,2 Гц, 1 Н), 7,34 (м, 4 Н), 7,26 (м, 1 Н), 7,12 (д, J=l,2 Гц,1 Н), 6,92 (кв., J=6,8 Гц, 1H), 6,82 (д, J=l,2 Гц, 1 Н), 6,64 (с, 1H), 4,21 (т, J=5,6 Гц, 1 Н), 3,29 (м, 1 Н), 3,20 (м,1 Н), 2,47 (с, 3 Н). 6.8. Синтез (S)-2-амино-3-(4-2-амино-6-[2,2,2-трифтор-1-(4-фуран-3-илфенил)этокси]пиримидин-4 илфенил)пропионовой кислоты В микроволновом сосуде смешивали 3-бромфуран (590 мг, 4,02 ммоль), 4-формилфенилбороновую кислоту (600 мг, 4,02 ммоль) и 7 мл актонитрила. К смеси затем добавляли 8 мл 1 н. водного раствора карбоната натрия, с последующим добавлением 5 мол.% дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали при 150 С в течение 7 мин в условиях микроволнового облучения. После охлаждения добавляли 50 мл этилацетата, органический слой отделяли, промывали водой, сушили над сульфатом натрия. Органический растворитель выпаривали с получением неочищенного продукта, который очищали при помощи ISCO, с получением 410 мг 4-фуран-3-илбензальдегида, выход: 60%. Тетрабутиламмонийфторид (0,024 мл; 1,0 М раствор в тетрагидрофуране) добавляли к раствору 4 фуран-3-илбензальдегида (410 мг, 2,38 ммоль) и трифторметилтриметилсилана (TMSCF3) (423 мкл, 2,86 ммоль) в 5 мл ТГФ при 0 С. Образовавшуюся смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь затем обрабатывали при помощи 5 мл 1 н. раствора HCl и перемешивали при комнатной температуре в течение ночи. Продукт экстрагировали при помощи этилацетата (350 мл). Органический слой отделяли и сушили над сульфатом натрия. Органический растворитель выпаривали с получением 480 мг 2,2,2-трифтор-1-(4-фуран-3-илфенил)этанола, выход: 83%. Смесь 2,2,2-трифтор-1-(4-фуран-3-илфенил)этанола (100 мг, 0,4 ммоль), 2-амино-4,6-дихлорпиримидина (60 мг, 0,36 ммоль), карбоната цезия (468 мг, 1,44 ммоль) и 1,4-диоксана (1 мл) нагревали при 110 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры; добавляли 10 мл этилацетата. Смесь затем фильтровали через целит, фильтрат концентрировали с получением 110 мг 4-хлор 6-[2,2,2-трифтор-1-(4-фуран-3-илфенил)этокси]пиримидин-2-иламина, выход: 72%. В микроволновом сосуде смешивали 4-хлор-6-[2,2,2-трифтор-1-(4-фуран-3-илфенил)этокси]пиримидин-2-иламин (30 мг, 0,081 ммоль), 4-бороно-L-фенилаланин (20 мг, 0,098 ммоль), 1 мл актонитрила и 0,7 мл воды. К смеси затем добавляли 0,3 мл 1 н. водного раствора карбоната натрия, с последующим добавлением 5 мол.% дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали при 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения реакционную смесь упаривали досуха. Остаток растворяли в 2,5 мл метанола и затем очищали при помощи препаративной ЖХ с получением 7,2 мг (S)-2-амино-3-(4-2-амино-6-[2,2,2-трифтор-1-(4-фуран-3 илфенил)этокси]пиримидин-4-илфенил)пропионовой кислоты. 1 Н ЯМР (400 МГц, CD3OD):(ч/млн) 7,96 (м, 3 Н), 7,61 (м, 5 Н), 6,81 (с, 1 Н), 6,77 (д, J=6,8 Гц, 1 Н), 6,74 (д, J=4,8 Гц, 1 Н), 4,27 (кв., J=5,6 Гц, 1 Н),3,36 (м, 1 Н), 3,21 (м, 1 Н). 6.9. Синтез (S)-2-амино-3-[4-2-амино-6-1-[2-(5-диметиламинометилфуран-2-ил)фенил]-2,2,2 трифторэтоксипиримидин-4-ил)фенил]пропионовой кислоты(DCE). Затем добавляли 0,2 мл НОАс. Смесь перемешивали при комнатной температуре в течение ночи,с последующим добавлением 15 мл DCE. Органическую фазу промывали водой, сушили над сульфатом натрия. Растворитель удаляли при помощи роторного испарителя с получением 400 мг (5-бромфуран-2 илметил)диметиламина, выход: 97%. В микроволновом сосуде смешивали (5-бромфуран-2-илметил)диметиламин (385 мг, 1,88 ммоль), 2 формилфенилбороновую кислоту (288 мг, 1,93 ммоль) и 3,7 мл актонитрила. Затем к смеси добавляли 3,7 мл 1 н. водного раствора карбоната натрия, с последующим добавлением 5 мол.% дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали при 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения добавляли 20 мл 1 н. раствораHCl. Смесь экстрагировали при помощи этилацетата (310 мл) и этилацетатный слой отбрасывали. К водной фазе добавляли 1 н. раствор NaOH, чтобы довести уровень рН до 10, затем экстрагировали при помощи этилацетата (320 мл). Объединенный органический слой промывали водой и сушили над сульфатом натрия. Растворитель выпаривали с получением 300 мг 2-(4-диметиламинометилциклопента-1,3 диенил)бензальдегида, выход: 69%. Тетрабутиламмонийфторид (0,013 мл; 1,0 М раствор в тетрагидрофуране) добавляли к раствору 2(4-диметиламинометилциклопента-1,3-диенил)бензальдегида (287 мг, 1,25 ммоль) и трифторметилтриметилсилана (TMSCF3) (222 мкл, 1,5 ммоль) в 5 мл ТГФ при 0 С. Образовавшуюся смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь затем обрабатывали при помощи 5 мл 1 н. раствора НСl и перемешивали при комнатной температуре в течение ночи. Продукт экстрагировали при помощи этилацетата (350 мл). Органический слой отделяли и сушили над сульфатом натрия. Органический растворитель выпаривали с получением 250 мг 1-[2-(5 диметиламинометилфуран-2-ил)фенил]2,2,2-трифторэтанола, выход 66%. Смесь 1-[2-(5-диметиламинометилфуран-2-ил)фенил]-2,2,2-трифторэтанола (225 мг, 0,75 ммоль), 2 амино-4,6-дихлорпиримидина (111 мг, 0,67 ммоль), карбоната цезия (978 мг, 3,01 ммоль) и 1,4-диоксана(3 мл) нагревали при 110 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры; добавляли 10 мл зтилацетата. Смесь затем фильтровали через целит, фильтрат концентрировали с получением 110 мг 4-хлор-6-1-[2-(5-диметиламинометилфуран-2-ил)фенил]2,2,2-трифторэтоксипиримидин-2-иламина, выход 87%. В микроволновом сосуде смешивали 4-хлор-6-1-[2-(5-диметиламинометилфуран-2-ил)фенил]2,2,2 трифторэтоксипиримидин-2-иламин (37 мг, 0,087 ммоль), 4-бороно-L-фенилаланин (22 мг, 0,10 ммоль),1 мл актонитрила и 0,7 мл воды. Затем добавляли 0,3 мл 1 н. водного раствора карбоната натрия, с последующим добавлением 5 мол.% дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали при 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения реакционную смесь упаривали досуха. Остаток растворяли в 2,5 мл метанола и затем очищали при помощи препаративной ЖХ с получением 16 мг (S)-2-амино-3-[4-2-амино-6-1-[2-(5 диметиламинометилфуран-2-ил)фенил]-2,2,2-трифторэтоксипиримидин-4-илфенил]пропионовой кислоты. 1 Н ЯМР (400 МГц, CD3OD):7,88 (д, J=8,4 Гц, 2 Н), 7,71 (д, J=7,6 Гц, 1 Н), 7,62 (д, J=7,6 Гц, 1 Н),7,42 (м, 2 Н), 7,40 (д, J=l,6 Гц, 2 Н), 7,34 (д, J=8,4 Гц, 1 Н), 6,89 (кв., J=3,6 Гц, 2 Н), 666 (с, 1 Н), 4,54 (с, 2 Н),4,20 (кв., J=6 Гц, 1 Н), 3,3 (м, 1 Н), 3,14 (м, 1 Н), 2,84 (с, 6 Н). 6.10. Синтез (S)-2-амино-3-[4-(2-амино-6-1-[2-(6-цианопиридин-3-ил)фенил]-2,2,2-трифторэтокси пиримидин-4-ил)фенил]пропионовой кислоты В микроволновом сосуде смешивали 5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)пиридин-2 карбонитрил (279 мг, 1,51 ммоль), 2-бромбензальдегид (230 мг, 1 ммоль) и 2 мл актонитрила. Затем добавляли 2 мл 1 н. водного раствора карбоната натрия с последующим добавлением 5 мол.% дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали при 100 С в течение 10 мин в условиях микроволнового облучения. После охлаждения добавляли 50 мл этилацетата,органический слой отделяли, промывали водой и сушили над сульфатом натрия. Органический растворитель выпаривали с получением неочищенного продукта, который очищали при помощи ISCO, с получением 150 мг 5-(2-формилфенил)пиридин-2-карбонитрила, выход 72%. Тетрабутиламмонийфторид (5,3 мкл, 1,0 М раствор в тетрагидрофуране) добавляли к раствору 5-(2 формилфенил)пиридин-2-карбонитрила (110 мг, 0,53 ммоль) и трифторметилтриметилсилана (120 мкл,0,81 ммоль) в 5 мл ТГФ при 0 С. Образовавшуюся смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь затем обрабатывали при помощи 5 мл 1 н. раствора HCl и перемешивали при комнатной температуре в течение ночи. Продукт экстрагировали при помощи этилацетата (350 мл). Органический слой отделяли и сушили над сульфатом натрия. Органический растворитель выпаривали с получением 140 мг 5-[2-(2,2,2-трифтор-1 гидроксиэтил)фенил]пиридин-2-карбонитрила, выход 95%. Смесь 5-[2-(2,2,2-трифтор-1-гидроксиэтил)фенил]пиридин-2-карбонитрила (46 мг, 0,165 ммоль),(S)-3-[4-(2-амино-6-хлорпиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовой кислоты (59 мг, 0,15 ммоль), карбоната цезия (195 мг, 0,6 ммоль) и 1,4-диоксана (1 мл) нагревали при 110 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры, затем вливали в 5 мл воды. Добавляли 1 н. раствор НСl, чтобы довести уровень рН до 4,5, водную фазу экстрагировали при помощи этилацетата(310 мл). Объединенный органический слой промывали насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали с получением 80 мг неочищенного (S)-3-[4-(2-амино-6-1[2-(6-цианопиридин-3-ил)фенил]-2,2,2-трифторэтоксипиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовой кислоты, выход 84%. 80 мг (S)-3-[4-(2-амино-6-1-[2-(6-цианопиридин-3-ил)фенил]-2,2,2-трифторэтоксипиримидин-4 ил)фенил]-2-трет-бутоксикарбониламинопропионовой кислоты растворяли в растворе 30% трифторуксусной кислоты в дихлорметане (5 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель выпаривали и остаток очищали при помощи препаративной ВЭЖХ с получением 12,6 мг К 2-имидазол-1-илбензальдегиду (0,344 г, 2 ммоль) в ТГФ (8 мл) добавляли трифторметилтриметилсилан (0,341 г, 2,4 ммоль). Реакционную смесь охлаждали до 0-5 С (баня с ледяной водой) и добавляли тетра-н-бутиламмонийфторид (0,035 мл, 0,035 ммоль, 1 М в ТГФ). Ледяную баню удаляли и смесь перемешивали при комнатной температуре в течение 6 ч. Добавляли 2 н. раствор HCl (5 мл) и реакционную смесь дополнительно перемешивали в течение 3 ч при комнатной температуре. Растворитель удаляли на роторном испарителе при пониженном давлении. Неочищенный остаток растворяли в ДХМ (30 мл),промывали водой (20 мл), насыщенным солевым раствором (20 мл) и сушили при помощи сульфата натрия. Растворитель удаляли с получением неочищенного 2,2,2-трифтор-1-(2-имидазол-1-илфенил)этанола (0,45 г, 93%), который непосредственно использовали на следующей стадии. 2-Амино-4,6-дихлорпиримидин (0,107 г, 0,65 ммоль), 2,2,2-трифтор-1-(2-имидазол-1-илфенил)этанол (0,157 г, 0,65 ммоль) и NaH (0,03 г, 0,78 ммоль) добавляли к безводному ТГФ (10 мл) в атмосфере азота. Реакционную смесь перемешивали при 40-45 С в течение 6 ч и затем охлаждали до комнатной температуры и гасили водой (0,2 мл). Реакционную смесь концентрировали с получением неочищенного 4-хлор-6-[2,2,2-трифтор-1-(2-имидазол-1-илфенил)этокси]пиримидин-2-иламина (0,24 г, чистота 90% по данным ЖХМС), который непосредственно использовали на следующей стадии. Вышеуказанный неочищенный промежуточный продукт (0,24 г), L-пара-боронофенилаланин (0,140 г, 0,67 ммоль), карбонат натрия (0,14 г, 1,32 ммоль) и дихлорбис(трифенилфосфин)палладий(II) (15 мг,0,021 ммоль) растворяли в смеси MeCN (2,0 мл) и Н 2 О (2,0 мл) в микроволновом сосуде. Реакционную смесь герметично закрывали и перемешивали в микроволновом реакторе при 150 С в течение 6 мин. Смесь фильтровали и фильтрат концентрировали. Остаток растворяли в МеОН и Н 2 О (1:1) и очищали при помощи препаративной ВЭЖХ, используя в качестве системы растворителей МеОН/Н 2 О/ТФУК, с получением (S)-2-амино-3-(4-[2-амино-6-[2,2,2-трифтор-1-(2-имидазол-1-илфенил)этокси]пиримидин-4 ил]фенилпропионовой кислоты в виде соли ТФУК. ЖХМС: М+1=499. 1 Н ЯМР (400 МГц, CD3OD): К 2-пиразол-1-илбензальдегиду (0,344 г, 2 ммоль) в ТГФ (8 мл) добавляли трифторметилтриметилсилан (0,341 г, 2,4 ммоль). Смесь охлаждали до 0-5 С (баня с ледяной водой) и добавляли тетра-нбутиламмонийфторид (0,035 мл, 0,035 ммоль, 1 М в ТГФ). Ледяную баню удаляли и смесь перемешивали при комнатной температуре в течение 6 ч. Добавляли 2 н. раствор HCl (5 мл) и реакционную смесь дополнительно перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли на роторном испарителе при пониженном давлении. Остаток растворяли в ДХМ (30 мл), промывали водой (20 мл), насыщенным солевым раствором (20 мл) и сушили над сульфатом натрия. Растворитель удаляли в вакууме с получением неочищенного 2, 2,2-трифтор-1-(2-пиразол-1-илфенил)этанола (0,45 г, 93%) который непосредственно использовали в следующем эксперименте. 4,6-Дихлорпиримидин (0,082 г, 0,55 ммоль), 2,2,2-трифтор-1-(2-пиразол-1-илфенил)этанол (0,121 г,0,50 ммоль), NaH (0,03 г, 0,78 ммоль) добавляли к безводному ТГФ (10 мл) в атмосфере азота. Реакционную смесь перемешивали при 40-45 С в течение 6 ч и затем охлаждали до комнатной температуры и гасили водой (0,2 мл). Реакционную смесь концентрировали с получением неочищенного 4-хлор-6-[2,2,2 трифтор-1-(2-пиразол-1-илфенил)этокси]пиримидина (0,20 г, чистота 90% по данным ЖХМС), который непосредственно использовали на следующей стадии. Неочищенный промежуточный продукт (0,20 г), L-параборонофенилаланин (0,105 г, 0,50 ммоль),карбонат натрия (0,105 г, 1 ммоль), дихлорбис(трифенилфосфин)палладий(II) (15 мг, 0,021 ммоль) растворяли в смеси MeCN (2,0 мл) и Н 2 О (2,0 мл) в микроволновом сосуде. Сосуд герметично закрывали и реакционную смесь нагревали в микроволновом реакторе при 150 С в течение 6 мин. Смесь фильтровали и фильтрат концентрировали. Остаток растворяли в МеОН и Н 2 О (1:1) и затем очищали при помощи препаративной ВЭЖХ, используя в качестве системы растворителей МеОН/Н 2 О/ТФУК, с получением 2,2,2-Трифтор-1-(2-йодфенил)этанол (0,331 г, 1,1 ммоль), 3-трифторметилпиразол (0,136 г, 1,0 ммоль), CuI (0,019 г, 0,1 ммоль), K2CO3 (0,290 г, 2,1 ммоль), (1R,2R)-N,N'-диметилциклогексан-1,2 диамин (0,028 г, 0,2 ммоль) и толуол (10 мл) объединяли в 20-мл трубке высокого давления. Смесь нагревали при 130 С (температура масляной бани) в течение 12 ч. Реакционную смесь разбавляли этилацетатом и промывали Н 2 О (220 мл), насыщенным солевым раствором и сушили при помощи сульфата натрия. Удаление растворителя давало неочищенный продукт, который очищали при помощи колоночной хроматографии ISCO, используя 5-10% этилацетат в гексане в качестве растворителя, с получением 140 мг 2,2,2-трифтор-1-[2-(3-трифторметилпиразол-1-ил)фенил]этанола. 2-Амино-4,6-дихлорпиримидин (0,074 г, 0,45 ммоль), 2,2,2-трифтор-1-[2-(3-трифторметилпиразол 1-ил)фенил]этанол (0,140 г, 0,45 ммоль) и NaH (0,022 г, 0,59 ммоль) добавляли к безводному ТГФ (10 мл) в атмосфере азота. Реакционную смесь перемешивали при 40-45 С в течение 6 ч и затем охлаждали до комнатной температуры и гасили водой (0,2 мл). Реакционную смесь концентрировали с получением неочищенного 4-хлор-6-[2,2,2-трифтор-1-[2-(3-трифторметилпиразол-1-ил)фенил]этокси]пиримидин-2 иламина (0,21 г, чистота 90% по данным ЖХМС), который непосредственно использовали на следующей стадии. Неочищенный промежуточный продукт (0,21 г), L-параборонофенилаланин (0,1 г, 0,48 ммоль), карбонат натрия (0,1 г, 0,94 ммоль) и дихлорбис(трифенилфосфин)палладий(II) (15 мг, 0,021 ммоль) растворяли в смеси MeCN (2,0 мл) и Н 2 О (2,0 мл) в микроволновом сосуде. Сосуд герметично закрывали и реакционную смесь нагревали в микроволновом реакторе при 150 С в течение 6 мин. Реакционную смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта, который растворяли в МеОН и Н 2 О (1:1) и очищали при помощи препаративной ВЭЖХ, используя в качестве системы растворителей MeOH/H2O/ТФУК, с получением (S)-2-амино-3-[4-(2-амино-6-[2,2,2-трифтор-1-(2-(3-трифторметилпиразол-1-илфенил)этокси]пиримидин-4-ил]фенилпропионовой кислоты в виде соли ТФУК. ЖХМС: М+1=567. 1 Н ЯМР (400 МГц, CD3OD):(ч/млн) 3,2 (м, 1 Н), 3,35 (м, 1 Н), 4,30 (т, 1 Н), 6,80 (с,1 Н), 6,85 (м, 1 Н), 6,98 (д, 1 Н), 7,45 (д, 2 Н), 7,59 (м, 1 Н), 7,68 (м, 2 Н), 7,88 (м, 1 Н), 7,95 (д, 2 Н), 8,20 (1 Н). 6.14. Синтез (3)-2-амино-3-[4-(2-амино-6-1-[2-(3,5-диметилпиразол-1-ил)фенил]-2,2,2-трифторэтоксипиримидин-4-ил)фенил]пропионовой кислоты 2,2,2-Трифтор-1-(2-йодфенил)этанол (0,331 г, 1,1 ммоль), 3,5-диметилпиразол (0,096 г, 1,0 ммоль),CuI (0,019 г, 0,1 ммоль), K2CO3 (0,290 г, 2,1 ммоль), (1R,2R)-N,N'-диметилциклогексан-1,2-диамин (0,028 г, 0,2 ммоль) и толуол (10 мл) объединяли в 20-мл трубке высокого давления и смесь нагревали при 130 С (температура масляной бани) в течение 12 ч. Смесь разбавляли этилацетатом и промывали Н 2 О(220 мл), насыщенным солевым раствором и сушили над сульфатом натрия. Удаление растворителя давало неочищенный продукт, который очищали при помощи колоночной хроматографии ISCO, используя 5-10% этилацетат в гексане в качестве растворителя, с получением 1-[2-(3,5-диметилпиразол-1 ил)фенил]-2,2,2-трифторэтанола (120 мг). 2-Амино-4,6-дихлорпиримидин (0,074 г, 0,45 ммоль), 1-[2-(3,5-диметилпиразол-1-ил)фенил]-2,2,2 трифторэтанол (0,120 г, 0,45 ммоль), NaH (0,022 г, 0,59 ммоль) добавляли к безводному ТГФ (10 мл) в атмосфере азота. Реакционную смесь перемешивали при 40-45 С в течение 6 ч и затем охлаждали до комнатной температуры и гасили водой (0,2 мл). Реакционную смесь концентрировали с получением неочищенного 4-хлор-6-1-[2-(3,5-диметилпиразол-1-ил)фенил]-2,2,2-трифторэтокси]пиримидин-2-иламина (0,195 г, чистота 90% по данным ЖХМС), который непосредственно использовали на следующей стадии. Неочищенный промежуточный продукт (0,195 г), L-параборонофенилаланин (0,10 г, 0,48 ммоль),- 17018150 карбонат натрия (0,10 г, 0,95 ммоль) и дихлорбис(трифенилфосфин)палладий(II) (15 мг, 0,021 ммоль) растворяли в смеси MeCN (2,0 мл) и Н 2 О (2,0 мл) в микроволновом сосуде. Сосуд герметично закрывали и реакционную смесь нагревали в микроволновом реакторе при 150 С в течение 6 мин. Смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта, который растворяли в МеОН иH2O (1:1) и очищали при помощи препаративной ВЭЖХ, используя в качестве системы растворителей МеОН/Н 2 О/ТФУК, с получением (S)-2-амино-3-[4-(2-амино-6-[1-(2-(3,5-диметилпиразол-1-ил)фенил]2,2,2-трифторэтоксипиримидин-4-ил)фенил]пропионовой кислоты в виде соли ТФУК. ЖХМС: М+1=527. 1 Н ЯМР (400 МГц, CD3OD):(ч/млн) 4,32 (т, 1 Н), 3,39 (м, 1 Н), 3,25 (м, 1 Н), 2,30 (с, 3 Н), 2,10(0,019 г, 0,1 ммоль), K2CO3 (0,290 г, 2,1 ммоль), (1R,2R)-N,N'-диметилциклогексан-1,2-диамин (0,028 г,0,2 ммоль) и толуол (10 мл) переносили в 20-мл трубку высокого давления и смесь нагревали при 130 С(температура масляной бани) в течение 12 ч. Смесь разбавляли этилацетатом и промывали H2O (220 мл), насыщенным солевым раствором и сушили над сульфатом натрия. Удаление растворителя давало неочищенный продукт, который очищали при помощи колоночной хроматографии ISCO, используя 510% зтилацетат в гексане в качестве растворителя, с получением 2,2,2-трифтор-1-[2-(3-фенилпиразол-1 ил)фенил]этанола (75 мг). 2-Амино-4,6-дихлорпиримидин (0,041 г, 0,25 ммоль), 2,2,2-трифтор-1-[2-(3-фенилпиразол-1 ил)фенил]этанол (0,070 г, 0,22 ммоль) и NaH (0,012 г, 0,31 ммоль) добавляли к безводному ТГФ (7 мл) в атмосфере азота. Реакционную смесь перемешивали при 40-45 С в течение 6 ч и затем охлаждали до комнатной температуры и гасили водой (0,04 мл). Реакционную смесь концентрировали с получением неочищенного 4-хлор-6-2,2,2-трифтор-1-[2-(3-фениллиразол-1-ил)фенил]этокси]пиримидин-2-иламина(0,110 г, чистота 90% по данным ЖХМС), который непосредственно использовали на следующей стадии. Неочищенный промежуточный продукт (0,110 г), L-параборонофенилаланин (0,050 г, 0,24 ммоль),карбонат натрия (0,050 г, 0,48 ммоль) и дихлорбис(трифенилфосфин)палладий(II) (8 мг, 0,010 ммоль) растворяли в смеси MeCN (2,0 мл) и Н 2 О (2,0 мл) в микроволновом сосуде. Сосуд герметично закрывали и реакционную смесь нагревали в микроволновом реакторе при 150 С в течение 6 мин. Смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта, который растворяли в МеОН и Н 2 О (1:1) и очищали при помощи препаративной ВЭЖХ, используя в качестве системы растворителей МеОН/Н 2 О/ТФУК, с получением (S)-2-амино-3-[4-(2-амино-6-2,2,2-трифтор-1-[2-(3-фенилпиразол-1-ил) фенил]этоксипиримидин-4-ил)фенил]пропионовой кислоты в виде соли ТФУК. ЖХМС: М+1=575. 1 Н ЯМР (400 МГц, CD3OD):(ч/млн) 3,20 (м, 1 Н), 3,38 (м, 1 Н), 4,30 (т, 1 Н), 6,80 (с, 1 Н), 7,00 (с, 1 Н), 7,307,48 (м, 7 Н), 7,62 (м, 3 Н), 7,90 (м, 4 Н), 8,10 (с, 1 Н). 6.16. Синтез (S)-2-амино-3-[4-(2-амино-6-2,2,2-трифтор-1-[5-метокси-2-(4-метилпиразол-1-ил)фенил]этоксипиримидин-4-ил)фенил]пропионовой кислоты 1-(2-Бром-5-метоксифенил)-2,2,2-трифторэтанол (0,570 г, 2,0 ммоль), 4-метилпиразол (0,164 г, 2,0 ммоль), CuI (0,057 г, 0,3 ммоль), K2CO3 (0,580 г, 4,2 ммоль), (1R,2R)-N,N'-диметилциклогексан-1,2 диамин (0,071 г, 0,5 ммоль) и толуол (10 мл) объединяли в 20-мл трубке высокого давления и смесь нагревали при 130 С (температура масляной бани) в течение 12 ч. Смесь разбавляли этилацетатом и промывали Н 2 О (220 мл), насыщенным солевым раствором и сушили над сульфатом натрия. Удаление растворителя давало неочищенный продукт, который очищали при помощи колоночной хроматографии метанола в ДХМ (50 мл). Смесь фильтровали. Фильтрат концентрировали с получением неочищенного продукта, который переносили в 20% раствор ТФУК в ДХМ (30 мл) и перемешивали в течение 30 мин при комнатной температуре. Анализ ЖХМС показал завершение реакции с требуемым продуктом. Реакционную смесь концентрировали с получением неочищенного продукта, который растворяли в МеОН и Н 2 О (1:1) и очищали при помощи препаративной ВЭЖХ, используя в качестве системы растворителей МеОН/Н 2 О/ТФУК, с получением (S)-2-амино-3-[4-(2-амино-6-2,2,2-трифтор-1-[5-метокси-2-(4-метилпиразол-1-ил)фенил]этокси]пиримидин-4-ил)фенил]пропионовой кислоты. ЖХМС: М+1=543. 1H ЯМР (400 МГц, CD3OD):(ч/млн) 2,20 (с, 3 Н), 3,22 (м, 1 Н), 3,40 (м, 1 Н), 3,84 (с, 3 Н), 4,35 (т, 1 Н), 6,84 (с, 1 Н), 6,98(0,456 г, 2,4 ммоль), K2CO3 (2,07 г, 15 ммоль), (1R,2R)-N,N'-диметилциклогексан-1,2-диамин (0,170 г, 1,2 ммоль) и толуол (10 мл) объединяли в 20-мл трубке высокого давления и смесь нагревали при 130 С(температура масляной бани) в течение 12 ч. Реакционную смесь разбавляли этилацетатом и промывали Н 2 О (220 мл), насыщенным солевым раствором и сушили над сульфатом натрия. Удаление растворителя давало неочищенный продукт, который очищали при помощи колоночной хроматографии ISCO, используя 5-10% зтилацетата в гексане в качестве растворителя, с получением R-2,2,2-трифтор-1-[2-(3 метилпиразол-1-ил)фенил]этанола (1,8 г). 2-Амино-4,6-дихлорпиримидин (1,2 г, 7,4 ммоль), R-2,2,2-трифтор-1-[2-(3-метилпиразол-1-ил) фенил]этанол (1,8 г, 7,03 ммоль) и NaH (0,380 г, 10 ммоль) добавляли к безводному ТГФ (40 мл) в атмосфере азота. Реакционную смесь перемешивали при 40-45 С в течение 6 ч и затем охлаждали до комнатной температуры и гасили водой (0,1 мл). Реакционную смесь концентрировали с получением 4-хлор-6-R2,2,2-трифтор-1-[2-(3-метилпиразол-1-ил)фенил]этоксипиримидин-2-иламина (3,0 г, чистота 90% по данным ЖХМС), который непосредственно использовали на следующей стадии. Неочищенный промежуточный продукт (0,750 г), L-параборонофенилаланин (0,420 г, 2,0 ммоль),карбонат натрия (0,430 г, 4,0 ммоль) и дихлорбис(трифенилфосфин)палладий (II) (30 мг, 0,043 ммоль) растворяли в смеси MeCN (7,0 мл) и Н 2 О (7,0 мл) в микроволновом сосуде. Сосуд герметично закрывали и реакционную смесь нагревали в микроволновом реакторе при 150 С в течение 7 мин. Смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта, который растворяли в МеОН иH2O (1:1) и очищали при помощи препаративной ВЭЖХ, используя в качестве системы растворителей МеОН/Н 2 О/ТФУК, с получением (S)-2-амино-3-[4-(2-aMHHO-6-R-2,2,2-трифтор-1-[2-(3-метилпиразол 1-ил)фенил]этоксипиримидин-4-ил)фенил]пропионовой кислоты в виде соли ТФУК. ЖХМС: М+1=514. 1 Н ЯМР (400 МГц, CD3OD):(ч/млн) 2,40 (с, 3 Н), 3,30 (м, 1 Н), 3,42 (м, 1 Н), 4,38 (т, 1 Н), 6,21 (с, 1 Н), 7,02 1-(4-Хлор-2-йодфенил)-2,2,2-трифторэтанол (0,840 г, 2,5 ммоль), 3-метилпиразол (0,230 г, 2,8 ммоль), CuI (0,190 г, 1,0 ммоль), K2CO3 (0,863 г, 6,25 ммоль), (1R,2R)-N,N'-диметилциклогексан-1,2 диамин (0,071 г, 0,5 ммоль) и толуол (10 мл) объединяли в 20-мл трубке высокого давления и смесь нагревали при 130 С (температура масляной бани) в течение 12 ч. Смесь разбавляли этилацетатом и промывали H2O (220 мл), насыщенным солевым раствором и сушили над сульфатом натрия. Удаление растворителя давало неочищенный продукт, который очищали при помощи колоночной хроматографииISCO, используя 5-10% этилацетат в гексане в качестве растворителя, с получением 1-[4-хлор-2-(3 метилпиразол-1-ил)фенил]-2,2,2-трифторэтанола (240 мг). 1-[4-Хлор-2-(3-метилпиразол-1-ил)фенил]-2,2,2-трифторэтанол (0,120 г, 0,41 ммоль), (S)-3-[4-(2 амино-6-хлорпиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовую кислоту (0,176 г, 0,45 ммоль), 1,4-диоксан (4 мл) и CS2CO3 (0,533 г, 1,64 ммоль) объединяли в герметично закрытой трубке объемом 20 мл и смесь нагревали при 100 С в течение 12 ч. Смесь концентрировали. К остатку добавляли 10% метанол в ДХМ (50 мл) и смесь фильтровали. Фильтрат концентрировали с получением неочи- 19018150 щенного продукта, который переносили в смесь ТГФ/3 н. НСl (30 мл/15 мл) и полученную смесь перемешивали при 40-45 С в течение 12 ч. Анализ ЖХМС показал завершение реакции с требуемым продуктом. Смесь концентрировали с получением неочищенного продукта, который растворяли в МеОН и H2O(1:1) и очищали при помощи препаративной ВЭЖХ, используя в качестве системы растворителей МеОН/Н 2 О/ТФУК, с получением (S)-2-амино-3-[4-(2-амино-6-1-[4-хлор-2-(3-метилпиразол-1-ил)фенил]2,2,2-трифторэтоксипиримидин-4-ил)фенил]пропионовой кислоты в виде соли ТФУК. ЖХМС: М+1=547. 1H ЯМР (400 МГц, CD3OD):(ч/млн) 2,30 (с, 3 Н), 3,10-3,30 (м, 2 Н), 4,20 (т, 1 Н), 6,32 (д, 1 Н),6,74 (с, 1 Н), 7,0 (кв., 1 Н), 7,38 (д, 2 Н), 7,50 (м, 2 Н), 7,72 (м, 1 Н), 7,90 (м, 3 Н). 6.19. Синтез этилового эфира (S)-2-амино-3-[4-(2-амино-6-R-1-[4-хлор-2-(3-метилпиразол-1-ил) фенил]-2,2,2-трифторэтоксипиримидин-4-ил)фенил]пропионовой кислоты Указанное в заголовке соединение получали постадийно, как описано ниже: Стадия 1. Синтез 1-(2-бром-4-хлорфенил)-2,2,2-трифторэтанона. В 2-горлую круглодонную колбу объемом 500 мл, содержащую безводный метанол (300 мл), добавляли тионилхлорид (29,2 мл, 400 ммоль) по каплям при 0-5 С (баня с ледяной водой) в течение 10 мин. Баню с ледяной водой удаляли и добавляли 2-бром-4-хлорбензойную кислоту (25 г, 106 ммоль). Смесь нагревали до слабого кипения с обратным холодильником в течение 12 ч. Ход реакции отслеживали при помощи ТСХ и ЖХМС. После завершения реакции реакционную смесь концентрировали. Неочищенный продукт растворяли в дихлорметане (ДХМ, 250 мл), промывали водой (50 мл), насыщенным водным раствором NaHCO3 (50 мл), насыщенным солевым раствором (50 мл), сушили над сульфатом натрия и концентрировали с получением метилового эфира 2-бром-4-хлорбензойной кислоты (26 г, 99%), который непосредственно использовали на следующей стадии. Метиловый эфир 2-бром-4-хлорбензойной кислоты (12,4 г, 50 ммоль) в толуоле (200 мл) охлаждали до -70 С и добавляли трифторметилтриметилсилан (13 мл, 70 ммоль). Тетрабутиламмонийфторид (1 М,2,5 мл) добавляли по каплям и смесь оставляли нагреваться до комнатной температуры в течение 4 ч,после этого ее перемешивали в течение 10 ч при комнатной температуре. Реакционную смесь концентрировали с получением неочищенного [1-(2-бром-4-хлорфенил)-2,2,2-трифтор-1-метоксиэтокси]триметилсилана. Неочищенный промежуточный продукт растворяли в метаноле (100 мл) и добавляли 6 н. раствор HCl (100 мл). Смесь выдерживали при 45-50 С в течение 12 ч. Метанол удаляли и неочищенное вещество экстрагировали дихлорметаном (200 мл). Объединенный ДХМ слой промывали водой (50 мл),NaHCO3 (50 мл), насыщенным солевым раствором (50 мл) и сушили над сульфатом натрия. Удаление растворителя давало неочищенный продукт, который очищали при помощи колоночной хроматографииISCO, используя 1-2% этилацетат в гексане в качестве растворителя, с получением 1-(2-бром-4 хлорфенил)-2,2,2-трифторэтанона (10 г, 70%). 1H ЯМР (300 МГц, CDCl3):(ч/млн) 7,50 (д, 1 Н), 7,65 (д,1 Н), 7,80 (с, 1 Н). Стадия 2. Синтез R-1-(2-бром-4-хлорфенил)-2,2,2-трифторэтанола. К катехолборану (1 М в ТГФ 280 мл, 280 ммоль) в 2-литровой 3-горлой круглодонной колбе в атмосфере азота добавляли S-2-метил-CBS оксазаборолидин (1,16 г, 28 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 20 мин. Реакционную смесь охлаждали до -78 С (баня сухой лед/ацетон) и по каплям добавляли 1-(2-бром-4-хлорфенил)-2,2,2-трифторэтанон (40 г, 139 ммоль) в ТГФ (400 мл) в течение 2 ч. Реакционную смесь оставляли нагреваться до -36 С и перемешивали при этой температуре в течение 24 ч и далее перемешивали при -32 С еще в течение 24 ч. Добавляли 3 н. раствор NaOH (250 мл) и охлаждающую баню заменяли баней лед-вода. Затем по каплям добавляли 30% раствор пероксида водорода в воде (250 мл) в течение 30 мин. Баню с ледяной водой удаляли и смесь перемешивали при комнатной температуре в течение 4 ч. Органический слой отделяли, концентрировали и повторно растворяли в простом эфире (200 мл). Водный слой экстрагировали простым эфиром (2200 мл). Объединенные органические слои промывали 1 н. водным раствором NaOH (4100 мл), насыщенным солевым раствором и сушили над сульфатом натрия. Удаление растворителя давало неочищенный продукт, который очищали при помощи колоночной хроматографии, используя от 2 до 5% этилацетата в гексане в качестве растворителя, с получением желаемого спирта 36,2 г (90%, 95% э.и.). Спирт (36,2 г) кристаллизовали из гексана (80 мл) с получением R-1-(2-бром-4-хлорфенил)-2,2,2-трифторэтанола 28,2 гR-1-(2-бром-4-хлорфенил)-2,2,2-трифторэтанол (15,65 г, 54,06 ммоль), 3-метилпиразол (5,33 г, 65 ммоль), CuI (2,06 г, 10,8 ммоль), K2CO3 (15,7 г, 113,5 ммоль), (1R,2R)-N,N'-диметилциклогексан-1,2 диамин (1,54 г, 10,8 ммоль) и толуол (80 мл) объединяли в трубке высокого давления объемом 250 мл и нагревали до 130 С (температура масляной бани) в течение 12 ч. Реакционную смесь разбавляли этил- 20018150 ацетатом и промывали Н 2 О (4100 мл), насыщенным солевым раствором и сушили над сульфатом натрия. Удаление растворителя давало неочищенный продукт, который очищали при помощи колоночной хроматографии ISCO, используя 5-10)% этилацетат в гексане в качестве растворителя, с получением R-1[4-хлор-2-(3-метилпиразол-1-ил)фенил]-2,2,2-трифторэтанола (13,5 г; 86%). 1H ЯМР (400 МГц, CDCl3):(ч/млн) 2,30 (с, 3 Н), 4,90 (м, 1 Н), 6,20 (с, 1 Н), 6,84 (д, 1 Н), 7,20 (с, 1 Н), 7,30 (д, 1 Н), 7,50 (д, 1 Н). Стадия 4. Синтез этилового эфира (S)-2-амино-3-[4-(2-амино-6-R-1-[4-хлор-2-(3-метилпиразол-1 ил)фенил]-2,2,2-трифторэтоксипиримидин-4-ил)фенилпропионовой кислоты.R-1-[4-хлор-2-(3-метилпиразол-1-ил)фенил]-2,2,2-трифторэтанол (17,78 г, 61,17 ммоль), (S)-3-[4-(2 амино-6-хлорпиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовую кислоту (20,03 г, 51 ммоль), 1,4-диоксан (250 мл) и Cs2CO3 (79,5 г, 244 ммоль) объединяли в 3-горлой круглодонной колбе объемом 500 мл и нагревали до 100 С (температура масляной бани) в течение 12-24 ч. Ход реакции отслеживали при помощи ЖХМС. После завершения реакции смесь охлаждали до 60 С и добавляли воду(250 мл) и ТГФ (400 мл). Органический слой отделяли и промывали насыщенным солевым раствором(150 мл). Растворитель удаляли с получением неочищенного ВОС-защищенного продукта, который переносили в ТГФ (400 мл), 3 н. раствор HCl (200 мл). Смесь нагревали при 35-40 С в течение 12 ч. ТГФ удаляли в вакууме. Оставшийся водный слой экстрагировали при помощи изопропилацетата (2100 мл) и концентрировали отдельно для извлечения непрореагировавшего спирта (3,5 г). Следы оставшегося органического растворителя удаляли из водной фракции под вакуумом. В 1-л химический стакан, снабженный терморегулятором и рН-метром, добавляли Н 3 РО 4 (40 мл,85% раствор в воде) и воду (300 мл) затем 50% раствор NaOH в воде, чтобы довести уровень рН до 6,15. Температуру повышали до 58 С и вышеуказанный кислотный водный раствор добавляли по каплям в буфер с одновременным добавлением 50% раствора NaOH в воде, таким образом, чтобы поддержать уровень рН в пределах 6,1-6,3. После завершения добавления осажденное твердое вещество фильтровали и промывали горячей водой (50-60 С) (2200 мл) и сушили с получением неочищенной (S)-2-амино-3-[4(2-амино-6-R)-1-[4-хлор-2-(3-метилпиразол-1-ил)фенил]-2,2,2-трифторэтокси)пиримидин-4-ил)фенил] пропионовой кислоты (26,8 г; 95%). ЖХМС и ВЭЖХ анализ показал соединение с чистотой около 9697%. К безводному этанолу (400 мл) по каплям добавляли SOCl2 (22 мл, 306 ммоль) при 0-5 С. Добавляли неочищенную кислоту (26,8 г) из вышеописанной реакции. Баню с ледяной водой удаляли и реакционную смесь нагревали при 40-45 С в течение 6-12 ч. После завершения реакции этанол удаляли в вакууме. К остатку добавляли ледяную воду (300 мл) и экстрагировали изопропилацетатом (2100 мл). Водный раствор нейтрализовали при помощи насыщенного раствора Na2CO3 до доведения рН до 6,5. Раствор экстрагировали этилацетатом (2300 мл). Объединенный этилацетатный слой промывали насыщенным солевым раствором и концентрировали с получением 24 г неочищенного сложного эфира(ВЭЖХ чистота 96-97%). Неочищенный сложный эфир затем очищали при помощи колоночной хроматографии ISCO, используя 5% этанола в ДХМ в качестве растворителя, с получением этилового эфира(S)-2-амино-3-[4-(2-амино-6-R)-1-[4-хлор-2-(3-метилпиразол-1-ил)фенил]-2,2,2-трифторэтокси)пиримидин-4-ил)фенил]пропионовой кислоты (20,5 г; 70%; чистота по данным ВЭЖХ 98%). ЖХМС М+1=575. 1 Этиловый эфир (S)-2-амино-3-(4-(2-амино-6-R)-1-(4-хлор-2-(3-метилпиразол-1-ил)фенил)-2,2,2 трифторэтокси)пиримидин-4-ил)фенил)пропионовой кислоты (22,2 г, 38,6 ммоль) растворяли в ТГФ (220 мл) и воде (50 мл). Добавляли моногидрат гидроксида лития (5,56 г, 132 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 ч. ТГФ удаляли и к остатку добавляли воду (100 мл) для получения прозрачного раствора. В 1-л химический стакан, снабженный терморегулятором и рН-метром, добавляли Н 3 РО 4 (40 мл,85% в воде), воду (300 мл) и 50% раствор NaOH в воде до доведения уровня рН до 6,15. Температуру повышали до 58 С и в буфер по каплям добавляли водный раствор Li-соли соединения с одновременным добавлением 3 н. раствора НСl, так, чтобы поддержать уровень рН в пределах от 6,1 до 6,2. После завершения добавления осажденное твердое вещество фильтровали и промывали горячей водой (50-60 С)(2200 мл) и сушили с получением (S)-2-амино-3-(4-(2-амино-6-R)-1-(4-хлор-2-(3-метилпиразол-1 ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропионовой кислоты (19,39 г; 92%). ЖХМС и ВЭЖХ анализы показывали чистоту соединения около 98-99%. ЖХМС М+1=547. 1 Н ЯМР (400 МГц,- 21018150 В микроволновой реактор объемом 40 мл, добавляли 1,04 г 2-формилфенилбороновой кислоты (6,9 ммоль), 1,14 г 2-бромтиазола (6,9 ммоль), 240 мг бистрифенилфосфиндихлорида палладия (Pd(PPh3)2Cl2,0,34 ммоль). Затем к смеси добавляли 13,8 мл 1M Na2CO3 (13,8 ммоль) и 10 мл CH3CN. Реактор герметично закрывали и реакционную смесь подвергали микроволновому облучению при 160 С в течение 5 мин. Анализ ЖХМС показал завершение реакции с желаемым продуктом. Реакционную смесь затем выливали в делительную воронку. Затем для экстракции добавляли 200 мл метиленхлорида и 100 мл воды. Слой метиленхлорида сушили над MgSO4. Удаление растворителя давало неочищенный продукт, который очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат(5/1 до 2/1), с получением чистого 2-тиазол-2-ил-бензальдегида (0,5 г, выход: 38%). В круглодонную колбу объемом 50 мл добавляли 184 мг 2-тиазол-2-ил-бензальдегида (0,97 ммоль) и 10 мл безводного тетрагидрофурана (ТГФ). Затем к раствору добавляли 145,4 мг трифторметилтриметилсилана (1,02 ммоль) и 20 мкл раствора 1M трет-бутиламмонийфторида в ТГФ (0,02 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, затем добавляли 10 мл 1 н. раствора HCl и реакционную смесь перемешивали при комнатной температуре в течение 15 мин. ТГФ удаляли в вакууме и смесь экстрагировали при помощи метиленхлорида (350 мл). Объединенный слой CH2Cl2 сушили надMgSO4. Удаление растворителя давало 262 мг неочищенного продукта, чистота которого составляла около 95%, и который использовали на следующей стадии без дополнительной очистки. 2,2,2-Трифтор-1-(2-тиазол-2-илфенил)этанол (260 мг, 1 ммоль), (S)-3-[4-(2-амино-6-хлорпиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовую кислоту (390 мг, 1 ммоль), карбонат цезия(1,3 г, 4 ммоль) и 10 мл 1,4-диоксана смешивали вместе в герметично закрытой трубке объемом 50 мл. Реакционную смесь нагревали при 100 С в течение 3 дней. Добавляли воду (20 мл) и затем медленно добавляли 1 н. водный раствор HCl до доведения рН до 4, затем 1,4-диоксан удаляли в вакууме и полученную смесь экстрагировали при помощи метиленхлорида (350 мл). Объединенный слой метиленхлорида сушили над MgSO4. Удаление растворителя давало неочищенный продукт, который использовали в реакции на следующей стадии без дополнительной очистки. Вышеуказанный неочищенный продукт растворяли в 5 мл метиленхлорида и добавляли 0,4 мл трифторуксусной кислоты. Смесь перемешивали при комнатной температуре в течение ночи. Трифторуксусную кислоту затем удаляли в вакууме с получением неочищенного продукта, который очищали при помощи препаративной ВЭЖХ, с получением 63 мг чистого продукта. ВЭЖХ; YMC Pack ODS-A 350 мм, 7 мкм; Растворитель А=вода с 0,1% ТФУК; Растворитель В=метанол с 0,1% ТФУК. Растворитель В от 10 до 90% в течение 4 мин; Скорость потока=2 мл/мин; Время удерживания=3 мин. ВЭЖХ чистота=100%. ЖХМС: М+1=515,9. 1H ЯМР (400 МГц, CD3OD):8,06 ч/млн (2 Н, м); 7,92 (2 Н, д, J=8 Гц); 7,84 (1 Н, м); 7,81 (1 Н, м); 7,77 (1 Н, д, J=4 Гц); 7,57 (2 Н, м); 7,45 (2 Н, д, J=8 Гц); 6,84 (1 Н, с); 4,30 (2 Н,дд, J=8 Гц); 3,38 (2 Н, дд, J=12,2 Гц); 3,23 (2 Н, дд, J=12,8 Гц). 6.22. Синтез (S)-2-амино-3-[4-(2-амино-6-2,2,2-трифтор-1-[2-(пиридин-3-илокси)фенил]этокси пиримидин-4-ил)фенил]пропионовой кислоты; (S)-2-амино-3-[4-(2-амино-6-2,2,2-трифтор-1-[4-(пиридин-3-илокси)фенил]этоксипиримидин-4-ил)фенил]пропионовой кислоты; (S)-2-амино-3-[4-(6-2,2,2 трифтор-1-[4-(пиридин-3-илокси)фенил]этоксипиримидин-4-ил)фенил]пропионовой кислоты; (S)-2 амино-3-(4-2-амино-6-[2,2,2-трифтор-1-(4-тиофен-2-илфенил)этокси]пиримидин-4-илфенил)пропионовой кислоты; (S)-2-амино-3-(4-6-[2,2,2-трифтор-1-(4-имидазол-1-илфенил)этокси]пиримидин-4-илфенил)пропионовой кислоты; и (S)-2-амино-3-(4-2-амино-6-[2,2,2-трифтор-1-(4-[1,2,4]триазол-1-илфенил) этокси]пиримидин-4-илфенил)пропионовой кислоты В этом способе тетра-н-бутиламмонийфторид (0,05 экв.) добавляли к смеси замещенного бензальдегида (1 экв.) и трифторметилтриметилсилана (1,2 экв.) в ТГФ при 0 С. Затем температуре давали подняться до комнатной температуры. Смесь перемешивали при комнатной температуре в течение 5 ч, затем разбавляли этилацетатом, промывали водой, насыщенным солевым раствором и сушили при помощиMgSO4. Растворитель удаляли при пониженном давлении с получением трифторированного спирта в виде неочищенного продукта, который использовали на следующей стадии без дополнительной очистки. Полученный выше спирт (1 экв.) растворяли в безводном 1,4-диоксане. Добавляли гидрид натрия(60% в минеральном масле, 1,2 экв.) одной порцией и смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли 2-амино-4,6-дихлорпиримидин (1 экв.) и полученную смесь перемешивали при 80 С в течение 2 ч. Растворитель удаляли и остаток суспендировали в этилацетате, который промывали водой, сушили над MgSO4 и затем концентрировали с получением требуемого монохлоридного продукта, который использовали на следующей стадии без дополнительной очистки. Вышеуказанный неочищенный продукт (1 экв.) добавляли в микроволновой сосуд объемом 5 мл,содержащий 4-бороно-L-фенилаланин (1 экв.), Na2CO3 (2 экв.), ацетонитрил (2 мл), воду (2 мл) и дихлорбис(трифенилфосфин)палладий (0,05 экв.). Сосуд закрывали крышкой и смесь нагревали при 150 С в течение 5 мин в условиях микроволнового облучения. Смесь охлаждали, фильтровали через шприцевой фильтр и затем разделяли при помощи препаративной ВЭЖХ с обращенной фазой, используя колонкуYMC-Pack ODS 10030 мм в.д. (система растворителей МеОН/Н 2 О/ТФУК). Чистые фракции объединяли и концентрировали в вакууме. Продукт затем суспендировали в 5 мл воды, замораживали и лиофилизировали с получением продукта в виде соли трифторуксусной кислоты (ТФУК). В этом способе бромзамещенный бензилальдегид (1 экв. ) добавляли в микроволновой сосуд объемом 20 мл, который содержал ароматическую гетероциклическую бороновую кислоту (1 экв.), Na2CO3 (2 экв.), смесь ацетонитрил (8 мл)/вода (8 мл) и дихлорбис(трифенилфосфин)палладий (0,05 экв.). Сосуд закрывали крышкой и перемешивали при 150 С в течение 6 мин в условиях микроволнового облучения. Реакционную смесь охлаждали, фильтровали через шприцевой фильтр и затем разбавляли этилацетатом. Смесь промывали водой. Затем добавляли силикагель для образования пробки и очищали при помощи хроматографии с элюированием гексаном и этилацетатом. Полученные выше альдегиды затем подвергали реакциям, аналогичным описанным в примере 22. Смесь метилового эфира 2-бром-5-фторбензойной кислоты (1 г, 4,292 ммоль), NaBH4 (0,423 г,11,159 ммоль) и LiCl (0,474 г, 11,159 ммоль) в смеси ТГФ/EtOH (20 мл/10 мл) перемешивали при комнатной температуре в течение ночи. Добавляли водный раствор HCl (10 мл, 2 н.) и перемешивали в течение около 10 мин. Затем органический растворитель удаляли под низким вакуумом. Остаток разбавляли водой и экстрагировали при помощи этилацетата. Органический слой промывали водным раствором NaHCO3 (10%), водой и насыщенным солевым раствором и затем сушили (MgSO4) и концентрировали с получением 852 мг (96,8% выход неочищенного продукта) неочищенного продукта, (2-бром-5 фторфенил)метанола, в виде белого твердого вещества, которое использовали без дополнительной очистки. К раствору (2-бром-5-фторфенил)метанола (0,852 г, 4,156 ммоль) в ДХМ (15 мл) добавляли MnO2(4,254 г, 85%, 41,56 ммоль). Смесь перемешивали при комнатной температуре в течение двух дней и затем фильтровали и промывали при помощи ДХМ. Фильтрат концентрировали с получением 777 мг 2 бром-5-фторбензальдегида (92% выход). Свежеполученный альдегид (0,777 г, 3,828 ммоль) затем растворяли в безводном ТГФ (10 мл) и охлаждали до 0 С. Добавляли трифторметилтриметилсилан (1,13 мл,7,656 ммоль) и затем тетрабутиламмонийфторид (0,020 г, 0,076 ммоль). Затем температуре давали подняться до комнатной температуры. Смесь перемешивали в течение 5 ч при комнатной температуре, затем разбавляли этилацетатом, промывали водой, насыщенным солевым раствором и сушили при помощиMgSO4. Растворитель удаляли при пониженном давлении с получением (2-бром-5-фторфенил)-2,2,2 трифторэтанола, 1,1 г (90% чистота) в виде неочищенного продукта, который использовали на следующей стадии без дополнительной очистки. 2-Бром-5-фторфенил-2,2,2-трифторэтанол (0,990 г, 3,263 ммоль, 90%), 3-метилпиразол (0,476 г,4,895 ммоль), CuI (0,367 г, 1,632 ммоль), K2CO3 (1,334 г, 8,158 ммоль), (1R,2R)-N,N'-диметилциклогексан-1,2-диамин (0,110 г, 0,653 ммоль) и толуол (10 мл) объединяли в микроволновом сосуде объемом 20 мл, который затем герметично закрывали и нагревали при 180 С в течение 40 мин. Смесь фильтровали и промывали при помощи этилацетата. Фильтрат промывали водой 3 раза и затем добавляли силикагель для образования пробки. Соединение очищали при помощи колоночной хроматографииISCO, используя 5-10% этилацетат в гексане в качестве растворителя, с получением 1-(5-фтор-2-(3 метилпиразол-1-ил)фенил)-2,2,2-трифторэтанола 75 мг. 1H ЯМР (400 МГц, CDCl3) : 2,29 (с, 3 Н), 4,90 (м,1 Н), 6,21 (д, 1 Н), 7,07-7,11 (м, 1 Н), 7,19-7,22 (м, 1 Н), 7,29-7,32 (м, 1 Н), 7,51 (д, 1 Н). Полученный выше спирт (0,075 г, 0,273 ммоль) растворяли в безводном 1,4-диоксане (3 мл). Одной порцией добавляли гидрид натрия (0,013 г, 0,328 ммоль, 60% в минеральном масле) и смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли 2-амино-4,6-дихлорпиримидин (0,045 г,0,273 ммоль). Смесь перемешивали при 80 С около 2 ч. Растворитель удаляли и остаток суспендировали в этилацетате, который промывали водой, сушили над MgSO4 и затем концентрировали с получением желаемого монохлоридного продукта 100 мг (0,249 ммоль), который добавляли в микроволновой сосуд объемом 5 мл, содержащий 4-бороно-L-фенилаланин (0,052 г, 0,249 ммоль), Na2CO3 (0,053 г, 0,498 ммоль), смесь ацетонитрил (2 мл)/вода (2 мл) и дихлорбис(трифенилфосфин)палладий (5 мг, 0,007 ммоль). Сосуд закрывали крышкой и перемешивали при 150 С в течение 5 мин в условиях микроволнового облучения. Реакционную смесь охлаждали, фильтровали через шприцевой фильтр и затем разделяли при помощи препаративной ВЭЖХ с обращенной фазой, используя колонку YMC-Pack ODS 10030 мм в.д. (система растворителей МеОН/Н 2 О/ТФУК). Чистые фракции концентрировали в вакууме. Продукт затем суспендировали в 5 мл воды, замораживали и лиофилизировали с получением (S)-2-амино-3[4-(2-амино-6-R)-1-[5-фтор-2-(3-метилпиразол-1-ил)фенил]-2,2,2-трифторэтоксипиримидин-4-ил)фенилпропионовой кислоты, 37 мг в виде трифтор-соли. 1H ЯМР (400 МГц, CD3OD) : 2,29 (с, 3 Н), 3,083,30 (м, 2 Н), 4,19 (кв., 1 Н), 6,32 (д, 1 Н), 6,82 (с, 1 Н), 6,85 (м, 1 Н), 7,26 (м, 1 Н), 7,33 (д, 2 Н), 7,42 (м, 2 Н),7,75 (д, 1 Н), 7,87 (д, 2 Н). 6.25. Синтез (S)-2-амино-3-[4-(2-амино-62,2,2-трифтор-1-[5-хлор-2-(3-метилпиразол-1-ил)фенил] этоксипиримидин-4-ил)фенил]пропионовой кислоты Указанные в заголовке соединения получали из R-1-[5-хлор-2-(3-метилпиразол-1-ил)фенил]-2,2,2 трифторэтанола, который получали, используя тот же способ, как описано выше для R-1-[4-хлор-2-(3 метилпиразол-1-ил)фенил]-2,2,2-трифторэтанола. В частности, R-1-[5-хлор-2-(3-метилпиразол-1-ил)фенил]-2,2,2-трифторэтанол (0,959 г, 3,318 ммоль) растворяли в безводном 1,4-диоксане (8 мл). Одной порцией добавляли гидрид натрия (0,159 г, 3,982 ммоль, 60% в минеральном масле) и смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли 2-амино-4,6-дихлорпиримидин (0,544 г, 3,318 ммоль). Смесь перемешивали при 80 С около 2 ч. Растворитель удаляли и остаток суспендировали в этилацетате, который промывали водой,сушили над MgSO4 и затем концентрировали с получением желаемого монохлоридного продукта 1,38 г,который использовали непосредственно без дополнительной очистки. Полученный выше монохлорид(0,460 г, 1,104 ммоль) добавляли в микроволновой сосуд объемом 20 мл, который содержал 4-бороно-Lфенилаланин (0,277 г, 1,325 ммоль), Na2CO3 (0,234 г, 2,208 ммоль), смесь ацетонитрил (8 мл)/вода (8 мл) и дихлорбис(трифенилфосфин)палладий (0,039 г, 0,055 ммоль). Сосуд закрывали крышкой и смесь перемешивали при 150 С в течение 10 мин в условиях микроволнового облучения. Смесь охлаждали, фильтровали через шприцевой фильтр и затем разделяли при помощи препаративной ВЭЖХ с обращенной фазой, используя колонку YMC-Pack ODS 10030 мм в.д. (система растворителей МеОН/Н 2 О/ТФУК). Чистые фракции концентрировали в вакууме. Продукт затем суспендировали в 5 мл воды, замораживали и лиофилизировали с получением 580 мг (S)-2-амино-3-[4-(2-амино-6-(R-1-[5-хлор-2-(3-метилпиразол-1 ил)фенил]-2,2,2-трифторэтоксипиримидин-4-ил)фенилпропионовой кислоты. 1 Н ЯМР (400 МГц,CD3OD):2,40 (с, 3 Н), 3,29-3,46 (м, 2 Н), 4,38 (кв., 1 Н), 6,45 (д, 1 Н), 7,09 (с, 1 Н), 7,24 (м, 1 Н), 7,53-7,70 (м,4 Н), 7,82 (с, 1 Н), 7,90 (д, 1 Н), 7,97 (д, 2 Н). 6.26. Синтез (S)-2-амино-3-[4-(2-амино-6-2,2,2-трифтор-1-[4-(2-оксопирролидин-1-ил)фенил]этоксипиримидин-4-ил)фенил]пропионовой кислоты 4-(2-Оксопирролидин-1-ил)бензальдегид (500 мг, 2,64 ммоль) в ТГФ (20 мл) охлаждали до 0 С и добавляли трифторметилтриметилсилан (375 мг, 2,64 ммоль). По каплям добавляли тетрабутиламмонийфторид (1 М, 0,1 мл) и смесь оставляли нагреваться до комнатной температуры в течение 1 ч и перемешивали далее в течение ночи при комнатной температуре. После завершения реакции добавляли 3H раствор HCl (5 мл) и реакционную смесь перемешивали в течение 2 ч. Смесь концентрировали. Добавляли воду (20 мл) и смесь экстрагировали при помощи EtOAc (220 мл) и промывали при помощи NaHCO3(20 мл), затем насыщенным солевым раствором (20 мл) и сушили над сульфатом натрия и концентрировали с получением 590 мг требуемого продукта, который использовали на следующей стадии без дополнительной очистки (выход 86%). Раствор 4,6-дихлорпиримидин-2-иламина (700 мг, 2,69 ммоль), NaH (194 мг, 8,07 ммоль, 60%) и 1(4-(2,2,2-трифтор-1-гидроксиэтил)фенил)пирролидин-2-она (441 мг, 2,69 ммоль) в безводном ТГФ (10 мл) перемешивали при комнатной температуре в течение ночи. После завершения реакции ТГФ удаляли при пониженном давлении. Добавляли воду (10 мл), охлаждая при этом смесь до 0 С. Смесь затем экстрагировали дихлорметаном (240 мл). Объединенный органический раствор сушили над Na2SO4. Удаление растворителя давало 498 мг желаемого продукта с чистотой 92%, который использовали на следующей стадии без дополнительной очистки (выход 498 мг, 48%). В реакционный сосуд Эмри (20 мл) для микроволнового нагрева загружали 1-(4-(2-амино-6 хлорпиримидин-4-илокси)-2,2,2-трифторэтил)фенил)пирролидин-2-он (200 мг, 0,51 ммоль), 4-бороно-Lфенилаланин (108 мг, 0,51 ммоль) и 5 мл ацетонитрила. К полученному раствору добавляли 5 мл водного раствора карбоната натрия (1M), и затем 5% мол. дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали до 160 С в течение 7 мин в условиях микроволнового облучения. После охлаждения реакционную смесь упаривали досуха. Остаток растворяли в 4 мл метанола и очищали при помощи препаративной ЖХ с получением 153 мг продукта (выход 58%). 1H ЯМР (400 МГц,CD3OD):(ч/млн) 2,1 (м, 2 Н), 2,5 (т, 2 Н), 3,05-3,4 (м, 2 Н), 3,85 (т, 2 Н), 4,2 (м, 1 Н), 6,6 (м, 1 Н), 6,75 (с,- 26018150R-1-(2-Бром-5-фторфенил)-2,2,2-трифторэтанол (4,0 г, 14,65 ммоль), 3-метилпиразол (1,56 г, 19,04 ммоль), CuI (0,557 г, 2,93 ммоль), K2CO3 (4,25 г, 30,76 ммоль), (1R,2R)-N,N'-диметилциклогексан-1,2 диамин (0,416 г, 2,93 ммоль) и толуол (15 мл) переносили в герметично закрытую трубку объемом 50 мл и полученную смесь нагревали при 130 С (температура масляной бани) в течение 2 дней. Смесь разбавляли этилацетатом и промывали при помощи Н 2 О (430 мл), насыщенным солевым раствором и сушили над сульфатом натрия. Удаление растворителя давало неочищенный продукт, который очищали при помощи колоночной хроматографии ISCO, используя 5-10% этилацетат в гексане в качестве растворителя,с получением 1,75 г R-2,2,2-трифтор-1-[5-фтор-2-(3-метилпиразол-1-ил)фенил]этанола (выход: 44%). 1H ЯМР (400 МГц, CDCl3):(ч/млн) 2,35 (с, 3 Н), 5,0 (м, 1 Н), 6,3 (с, 1 Н), 7,1 (м, 1 Н), 7,20 (с, 1 Н), 7,35 (д, 1 Н),7,50 (с, 1 Н). Раствор 4,6-дихлорпиримидин-2-иламина (938 мг, 5,72 ммоль), NaH (188 мг, 1,5 экв., 8,17 ммоль,60%) и R-2,2,2-трифтор-1-[5-фтор-2-(3-метилпиразол-1-ил)фенил]этанола (1,5 г, 1 экв. 5,45 ммоль) в безводном ТГФ (10 мл) перемешивали при комнатной температуре при 50 С в течение ночи. После завершения реакции ТГФ удаляли при пониженном давлении. Добавляли воду (10 мл) для гашения реакции. Смесь затем экстрагировали дихлорметаном (240 мл). Объединенный органический раствор сушили надNa2SO4. Удаление растворителя давало желаемый продукт с чистотой 92%, который использовали на следующей стадии без очистки (выход: 85%). В реакционный сосуд Эмри (20 мл) для микроволнового нагрева загружали хлор-6-R-2,2,2-трифтор 1-(5-фтор-2-(3-метилпиразол-1-ил)фенил)этокси)пиримидин-2-иламин (2,18 г, 5,45 ммоль), 4-бороно-Lфенилаланин (1,13 г, 5,45 ммоль), карбонат натрия (1 М 10,90 мл, 2 экв.). К полученному раствору затем добавляли 5 мол.% дихлорбис(трифенилфосфин)палладия(II) (191 мг, 0,27 ммоль) и 5 мл ацетонитрила и 5 мл Н 2 О. Реакционный сосуд герметично закрывали и смесь нагревали при 160 С в течение 10 мин в условиях микроволнового облучения. После охлаждения реакционную смесь упаривали досуха. Остаток растворяли в Н 2 О (10 мл) и экстрагировали эфиром. Эфирный слой отбрасывали. Затем большую часть воды водной фазы удаляли в вакууме с последующим добавлением 10 мл метанола. Неочищенный продукт очищали при помощи препаративной ВЭЖХ с получением 1,163 г (выход 75%) продукта. 1 Н ЯМР Тетрабутиламмонийфторид (TBAF) (0,1 мл 1 М раствора в ТГФ) добавляли к раствору 4-(6 метоксипиридин-2-ил)бензальдегида (213 мг, 1 ммоль) и трифторметилтриметилсилана (0,2 мл, 1,2 ммоль) в 10 мл ТГФ при 0 С. Смесь нагревали до комнатной температуры и перемешивали в течение 4 ч. Реакционную смесь затем обрабатывали 12 мл 1 М раствора НСl и перемешивали в течение ночи. Продукт экстрагировали при помощи этилацетата (320 мл). Органический слой отделяли и сушили над сульфатом натрия. Органический растворитель выпаривали с получением 0,25 г 1-[4-(6-метоксипиридин 2-ил)фенил]-2,2,2-трифторэтанола, который непосредственно использовали на следующей стадии без очистки, выход: 90%.Cs2CO3 (375 мг, 1 ммоль) добавляли к раствору 1-[4-(6-метоксипиридин-2-ил)фенил]-2,2,2 трифторэтанола (67 мг, 0,2 ммоль) в 10 мл безводного 1,4-диоксана. Смесь перемешивали в течение 5 мин, затем добавляли (S)-3-[4-(2-амино-6-хлорпиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовую кислоту (78 мг, 0,2 ммоль) и смесь нагревали при 110 С в течение ночи. После охлаждения добавляли 5 мл воды и для экстракции продукта использовали этилацетат (20 мл). Органический слой сушили над сульфатом натрия. Растворитель удаляли при помощи роторного испарителя с получением 112 мг (S)-3-[4-(2-амино-6-2,2,2-трифтор-1-[4-(6-метоксипиридин-2-ил)фенил]этоксипиримидин-4- 27018150 ил)фенил]-2-трет-бутоксикарбониламинопропионовой кислоты (выход: 88%). Полученный выше продукт (112 мг) добавляли в 5 мл раствора 30% ТФУК/ДХМ. После завершения реакции растворитель выпаривали с получением неочищенного продукта, который очищали при помощи препаративной ВЭЖХ, с получением 5 мг (S)-2-амино-3-[4-(2-амино-6-2,2,2-трифтор-1-[4-(6-метоксипиридин-2-ил)фенил]этоксипиримидин-4-ил)фенил]пропионовой кислоты. 1 Н ЯМР (300 МГц, CD3OD):TBAF (0,1 мл) добавляли к раствору 4-бром-2-фторбензальдегида (2,03 г, 10 ммоль) и TMSCF3 (20 мл, 12 ммоль) в 10 мл ТГФ при 0 С. Образовавшуюся смесь нагревали до комнатной температуры и перемешивали в течение 4 ч. Реакционную смесь затем обрабатывали при помощи 12 мл 3M раствора HCl и перемешивали в течение ночи. Продукт экстрагировали при помощи этилацетата (320 мл). Органический слой отделяли и сушили над сульфатом натрия. Органический растворитель выпаривали с получением 2,4 г 1-(4-бром-2-фторфенил)-2,2,2-трифторэтанола (выход: 90%).Cs2CO3 (8,45 г, 26 ммоль) добавляли к раствору 1-(4-бром-2-фторфенил)-2,2,2-трифторэтанола (1,4 г, 5,2 ммоль) в 10 мл безводного 1,4-диоксана, смесь перемешивали в течение 5 мин, затем добавляли (S)3-[4-(2-амино-6-хлорпиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовую кислоту (2,0 г,5 ммоль) и полученную смесь нагревали при 110 С в течение ночи. После охлаждения добавляли 5 мл воды и для экстракции продукта использовали этилацетат (20 мл). Органический слой сушили над сульфатом натрия. Растворитель удаляли при помощи роторного испарителя с получением 2,6 г (S)-3-(4-2 амино-6-[1-(4-бром-2-фторфенил)-2,2,2-трифторэтокси]пиримидин-4-илфенил)-2-трет-бутоксикарбониламинопропионовой кислоты (выход: 82%). В микроволновой сосуд (2 мл) загружали (S)-3-(4-2-амино-6-[1-(4-бром-2-фторфенил)-2,2,2 трифторэтокси]пиримидин-4-илфенил)-2-трет-бутоксикарбониламинопропионовую кислоту (130 мг, 0,2 ммоль), 3-метокси-5-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиридин (70 мг, 0,3 ммоль) 1 мл ацетонитрила и 0,7 мл воды. К этой смеси добавляли 0,4 мл водного раствора карбоната натрия (1 М), с последующим добавлением 14 мг (5 мол.%) дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали до 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения реакционную смесь упаривали досуха, остаток растворяли в 2,5 мл метанола и очищали при помощи препаративной ВЭЖХ с получением 51 мг (S)-3-[4-(2-амино-6-2,2,2-трифтор-1-[2-фтор-4(5-метоксипиридин-3-ил)фенил]этоксипиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовой кислоты. Полученный выше продукт (51 мг) растворяли в 5 мл раствора 30% ТФУК/ДХМ. Смесь перемешивали при комнатной температуре в течение ночи. Удаление растворителя давало неочищенный продукт,который очищали при помощи препаративной ВЭЖХ с получением 17 мг (S)-2-амино-3-[4-(2-амино-62,2,2-трифтор-1-[2-фтор-4-(5-метоксипиридин-3-ил)фенил]зтоксипиримидин-4-ил)фенил]пропионовой кислоты. 1 Н ЯМР (300 МГц, CD3OD):(ч/млн) 8,73 (с, 1 Н), 8,56 (с, 1 Н), 8,25 (с, 1 Н), 7,94 (д, J=8,2 Гц,2 Н), 7,77 (м, 3 Н), 7,55 (д, J=8,4 Гц, 2 Н), 7,16 (м, 1 Н), 7,00 (с, 1 Н), 4,35 (т, 1 Н), 4,09 (с, 3 Н), 3,4 (м, 2 Н). 6.30. СинтезCs2CO3 (16,25 г, 50 ммоль) добавляли к раствору (S)-1-(4-бромфенил)-2,2,2-трифторэтанола (2,55 г,11,0 ммоль) в 10 мл безводного 1,4-диоксана и смесь перемешивали в течение 5 мин, после чего добавляли (S)-3-[4-(2-амино-6-хлорпиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовую кислоту(3,92 г, 10 ммоль). Полученную смесь нагревали при 110 С в течение ночи. После охлаждения добавляли 5 мл воды и для экстракции продукта использовали этилацетат (20 мл). Органический слой сушили над сульфатом натрия. Растворитель удаляли при помощи роторного испарителя с получением 5,2 г (S)-3-(42-амино-6-[(S)-1-(4-бромфенил)-2,2,2-трифторэтокси]пиримидин-4-илфенил)-2-трет-бутоксикарбониламинопропионовой кислоты (выход: 82%). В микроволновой сосуд (2 мл) загружали (S)-3-(4-2-амино-6-[(S)-1-(4-бромфенил)-2,2,2-трифторэтокси]пиримидин-4-илфенил)-2-трет-бутоксикарбониламинопропионовую кислоту (139 мг, 0,23 ммоль), 2-фторпиридин-4-бороновую кислоту (40 мг, 0,27 ммоль) 1 мл ацетонитрила и 0,7 мл воды. К этой смеси добавляли 0,4 мл водного раствора карбоната натрия (1 М), с последующим добавлением 14 мг (5 мол.%) дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали до 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения реакционную смесь упаривали досуха и остаток растворяли в 2,5 мл метанола. Продукт очищали при помощи препаративной ВЭЖХ с получением 70 мг (S)-3-[4-(2-амино-6-(S)-2,2,2-трифтор-1-[4-(2-фторпиридин 4-ил)фенил]этоксипиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовой кислоты. Полученный выше продукт (70 мг) растворяли в 5 мл 30% раствора ТФУК в ДХМ. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Удаление растворителя давало неочищенный продукт, который очищали при помощи препаративной ВЭЖХ с получением 52 мг (S)-2-амино 3-[4-(2-амино-6-(S)-2,2,2-трифтор-1-[4-(2-фторпиридин-4-ил)фенил]этоксипиримидин-4-ил)фенил] пропионовой кислоты. 1 Н ЯМР (300 МГц, CD3OD):(ч/млн) 8,17 (д, J=5,7 Гц, 1 Н), 7,85 (д, J=8,4 Гц, 2 Н),7,77 (д, J=6,9 Гц, 2 Н), 7,67 (д, J=8,2 Гц, 2 Н), 7,53 (м, 1 Н), 7,38 (д, J=8,4 Гц, 2 Н), 7,30 (с, 1 Н), 6,76 (м, 2 Н),4,21 (т, 1 Н), 3,2 (м, 2 Н). 6.31. Синтез (S)-2-амино-3-[4-(2-амино-6-(S)-2,2,2-трифтор-1-[4-(5-метоксипиридин-3-ил)фенил] этоксипиримидин-4-ил)фенил]пропионовой кислоты В микроволновой сосуд (2 мл) загружали (S)-3-(4-2-амино-6-[(S)-1-(4-бромфенил)-2,2,2 трифторэтокси]пиримидин-4-илфенил)-2-трет-бутоксикарбониламинопропионовую кислоту (139 мг,0,23 ммоль), 3-метокси-5-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиридин (69 мг, 0,27 ммоль), 1 мл ацетонитрила и 0,7 мл воды. К этой смеси добавляли 0,4 мл водного раствора карбоната натрия (1 М),с последующим добавлением 14 мг дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали до 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения реакционную смесь упаривали досуха, остаток растворяли в 2,5 мл метанола и очищали при помощи препаративной ВЭЖХ с получением 60 мг (S)-3-[4-(2-амино-6-(S)-2,2,2-трифтор-1-[4-(5-метоксипиридин-3-ил)фенил]этоксипиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовой кислоты. Полученный выше продукт (60 мг) растворяли в 5 мл 30% раствора ТФУК в ДХМ. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Удаление растворителя давало неочищенный продукт, который очищали при помощи препаративной ВЭЖХ с получением 48 мг (S)-2-амино 3-[4-(2-амино-6-(S)-2,2,2-трифтор-1-[4-(5-метоксипиридин-3-ил)фенил]этоксипиримидин-4-ил)фенил] пропионовой кислоты. 1 Н ЯМР (300 МГц, CD3OD):(ч/млн): 8,54 (д, J=l,5 Гц, 1 Н), 8,37 (д, J=2,7 Гц, 1 Н),8,03 (дд, J=2,7 Гц, 1,5 Гц, 1 Н), 7,84 (д, J=8,2 Гц, 2 Н), 7,78 (д, J=8,4 Гц, 2 Н), 7,70 (д, J=8,4 Гц, 2 Н), 7,41 (д,J=8,4 Гц, 2 Н), 6,81 (с, 1 Н), 6,75 (м, 1 Н), 4,22 (т, 1 Н), 3,95 (т, 3 Н), 3,25 (м, 2 Н). 6.32. Синтез В микроволновой сосуд (2 мл) загружали (S)-3-(4-2-амино-6-[(S)-1-(4-бромфенил)-2,2,2-трифторэтокси]пиримидин-4-илфенил)-2-трет-бутоксикарбониламинопропионовую кислоту (139 мг, 0,23 ммоль), 4-трифторметилпиридин-3-бороновую кислоту (61 мг, 0,3 ммоль), 1 мл ацетонитрила и 0,7 мл воды. К этой смеси добавляли 0,4 мл водного раствора карбоната натрия (1 М), с последующим добавлением 14 мг дихлорбис(трифенилфосфин)палладия(II). Реакционный сосуд герметично закрывали и нагревали до 150 С в течение 5 мин в условиях микроволнового облучения. После охлаждения реакционную смесь упаривали досуха, остаток растворяли в 2,5 мл метанола и очищали при помощи препаративной ВЭЖХ с получением 20 мг (S)-3-[4-(2-амино-6-(S)-2,2,2-трифтор-1-[4-(4-трифторметил-пиридин-3 ил)фенил]этоксипиримидин-4-ил)фенил]-2-трет-бутоксикарбониламинопропионовой кислоты. Полученный выше продукт (20 мг) растворяли в 5 мл 30% раствора ТФУК в ДХМ. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Удаление растворителя давало неочищенный продукт, который очищали при помощи препаративной ВЭЖХ с получением 10 мг (S)-2-амино 3-[4-(2-амино-6-(S)-2,2,2-трифтор-1-[4-(4-трифторметилпиридин-3-ил)фенил]этоксипиримидин-4- 29
МПК / Метки
МПК: A61P 9/00, C07D 405/12, A61P 11/00, C07D 409/12, C07D 413/12, C07D 403/12, A61P 1/00, C07D 417/12, C07D 401/12
Метки: способы, 4-фенил-6-(2,2,2-трифтор-1-фенилэтокси)пиримидина, основе, соединения, применения
Код ссылки
<a href="https://eas.patents.su/30-18150-soedineniya-na-osnove-4-fenil-6-222-triftor-1-feniletoksipirimidina-i-sposoby-ih-primeneniya.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения на основе 4-фенил-6-(2,2,2-трифтор-1-фенилэтокси)пиримидина и способы их применения</a>
Твердые формы (s)-этил-2-амино-3-(4-(2-амино-6-((r)-1-(4-хлор-2-(3-метил-1h-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноата и способы их применения

Номер патента: 17275
Опубликовано: 30.11.2012
Авторы: Чжан Хаймин, Беднарз Марк С., Перлберг Анетт, Де Поль Сьюзан, Канамарлапуди Раманаиах К.
МПК: A61P 1/00, A61P 25/00, A61K 31/506...
Метки: формы, s)-этил-2-амино-3-(4-(2-амино-6-((r)-1-(4-хлор-2-(3-метил-1h-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноата, способы, твердые, применения
Формула / Реферат:
1. Кристаллический (S)-этил-2-амино-3-(4-(2-амино-6-((R)-1-(4-хлор-2-(3-метил-1Н-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноат или его фармацевтически приемлемая соль.2. Соединение по п.1, которое представляет собой кристаллический (S)-этил-2-амино-3-(4-(2-амино-6-((R)-1-(4-хлор-2-(3-метил-1Н-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноат.3. Соединение по п.2, которое имеет температуру плавления,...
Производные пиримидина, применимые в качестве ингибиторов pi3 киназы, и способы их применения

Номер патента: 18083
Опубликовано: 30.05.2013
Авторы: Волива Чарлз, Берджер Маттью, Пун Даниэль, Визманн Марион, Нг Саймон, Кнапп Марк, Барсанти Пол, Пекки Сабина, Чжан Яньчэнь, Фрейзьер Келли, Верхаген Джоелл, Фантл Уэнди, Бартулис Сара, Пик Тереса, Смит Аарон, Иванович Эдуин, Синь Сяхуа, Уагман Аллан, Пфистер Кейт, Луи Алисия, Хендриксон Томас, Ни Чжицзи, Меритт Ханне, Аталлах Гордана
МПК: C07D 401/04, A61K 31/506, A61P 35/00...
Метки: применения, киназы, качестве, применимые, производные, ингибиторов, способы, пиримидина
Формула / Реферат:
1. Соединение формулы Iили его стереоизомер, таутомер или фармацевтически приемлемая соль, в которойW выбран из СН и N;R1 выбран из группы, включающей:(1) замещенный и незамещенный гетероциклил, представляющий собой 3- или 4-членное кольцо, содержащее гетероатом, выбранный из азота, кислорода и серы, или 5- или 6-членное кольцо, содержащее от 1 до 3 гетероатомов, выбранных из группы, состоящей из азота, кислорода или серы, где любое из указанных...
Производные пиперидина, композиции на их основе, способы их применения

Номер патента: 14686
Опубликовано: 30.12.2010
Авторы: Моллейре Луи-Пьер, Кассайре Джером, Питтерна Томас, Майенфиш Петер, Корси Камилла, Седербаум Фредрик
МПК: C07D 211/68, A01N 43/40, C07D 211/06...
Метки: композиции, способы, производные, основе, пиперидина, применения
Формула / Реферат:
1. Способ борьбы с насекомыми, клещами, нематодами или моллюсками или их уничтожения, который включает нанесение на вредителей, на очаг вредителей или на растение, подверженное нашествию вредителей, инсектицидно, акарицидно, нематоцидно или моллюскоцидно эффективного количества соединения формулы Iв которой Y обозначает ординарную связь, С=O или C=S;кольцопредставляет собой бензол, пиридин, пиримидин, пиразин, пиридазин, триазин, пиррол,...
N-арилдиазаспирациклические соединения и способы их получения и применения

Номер патента: 9789
Опубликовано: 28.04.2008
Авторы: Бхатти Балвиндер С., Миллер Крэйг Х., Шмитт Джеффри Д.
МПК: A61K 31/40, C07D 453/02, A61K 31/435...
Метки: соединения, n-арилдиазаспирациклические, способы, получения, применения
Формула / Реферат:
1. Соединение, представленное следующей формулой 1 а также его фармацевтически приемлемые соли, где u, v, w и x, каждый независимо равен 0, 1, 2 или 3, причем значения выбираются так, что соединения выбираются из 1,7-диазаспиро[4,4]нонанов, 2,7-диазаспиро[4,4]нонанов, 1,6-диазаспиро[3,5]нонанов, 2,6-диазаспиро[4,5]нонанов, 1,7-диазаспиро[4,5]деканов и 1,8-диазаспиро[5,5]ундеканов, R представляет собой водород, С1-С8алкил, ацил, алкоксикарбонил...
Арилвинилазациклоалкановые соединения и способы их получения и применения

Номер патента: 10870
Опубликовано: 30.12.2008
Авторы: Капе Марк, Шмитт Джеффри Дэниел, Шев Мишель, Женевуа-Борелла Ариель, Далл Гари Морис
МПК: A61K 31/505, A61K 31/506, A61K 31/44...
Метки: соединения, способы, получения, применения, арилвинилазациклоалкановые
Формула / Реферат:
1. Арилвинилазациклоалкановые соединения формулы где волнистая линия представляет различные возможные геометрии (Е или Z) двойной связи; X представляет азот; R1 представляет водород или галоген; R3 представляет водород; m равно 1-3; n равно 1-3; и их изомеры, смеси, энантиомеры, диастереомеры, таутомеры и фармацевтически приемлемые соли. 2. Соединение по п.1, где R1 представляет водород. 3. Соединение по п.1, где n=1. 4. Соединение по п.1, где...
Предыдущий патент: Винилиндазолильные соединения
Следующий патент: Способ и система для улучшения производительности или эффективности скважинного насоса
Случайный патент: Получение частиц фосфата или полифосфата алюминия