Антагонисты пути hedgehog и их терапевтические применения
Номер патента: 17918
Опубликовано: 30.04.2013
Авторы: Минетто Джакомо, Перикот Мор Гал.Ла, Томас Рассел Джон, Ферруцци Пьетро, Баккер Анетта Корнелия






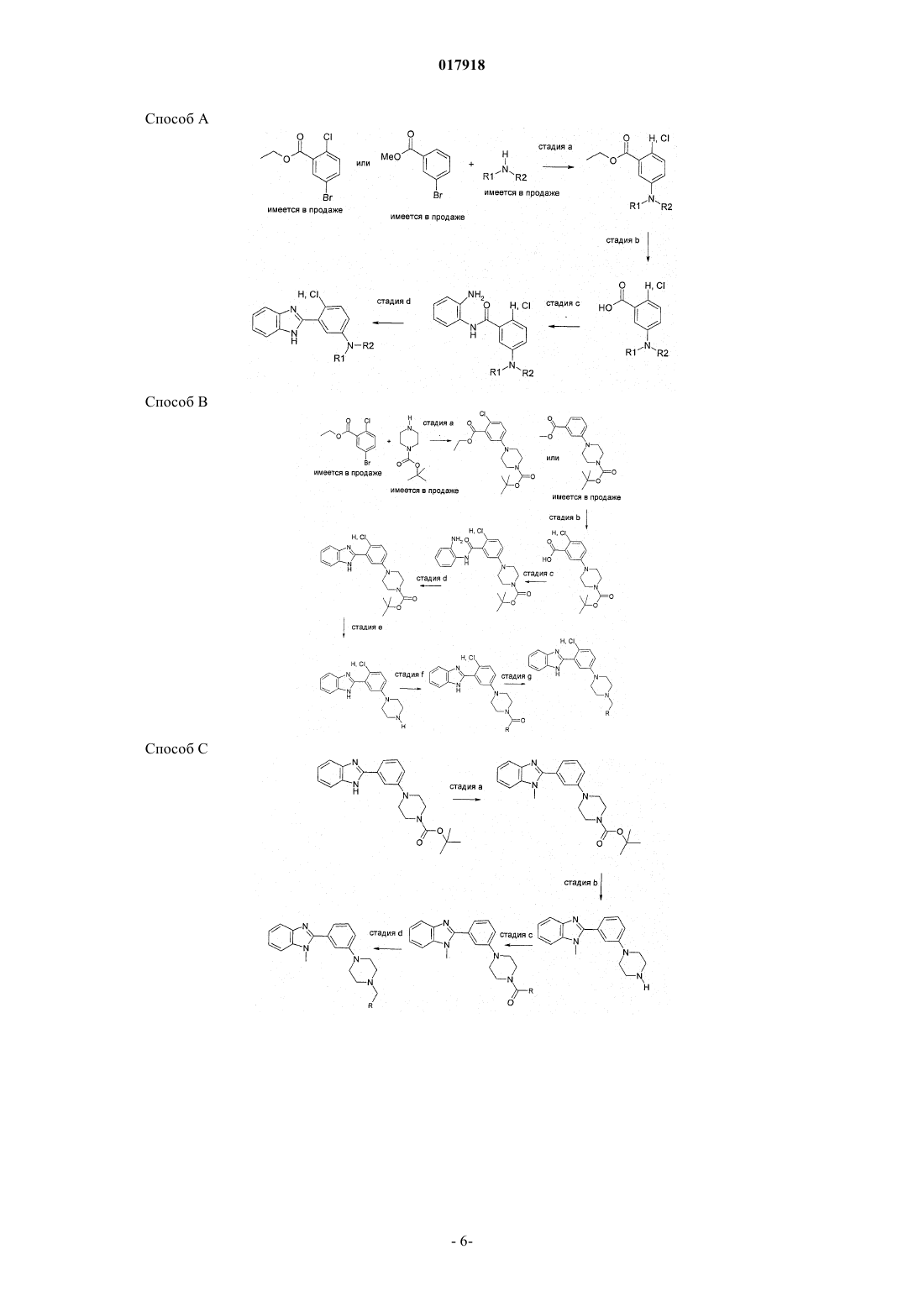
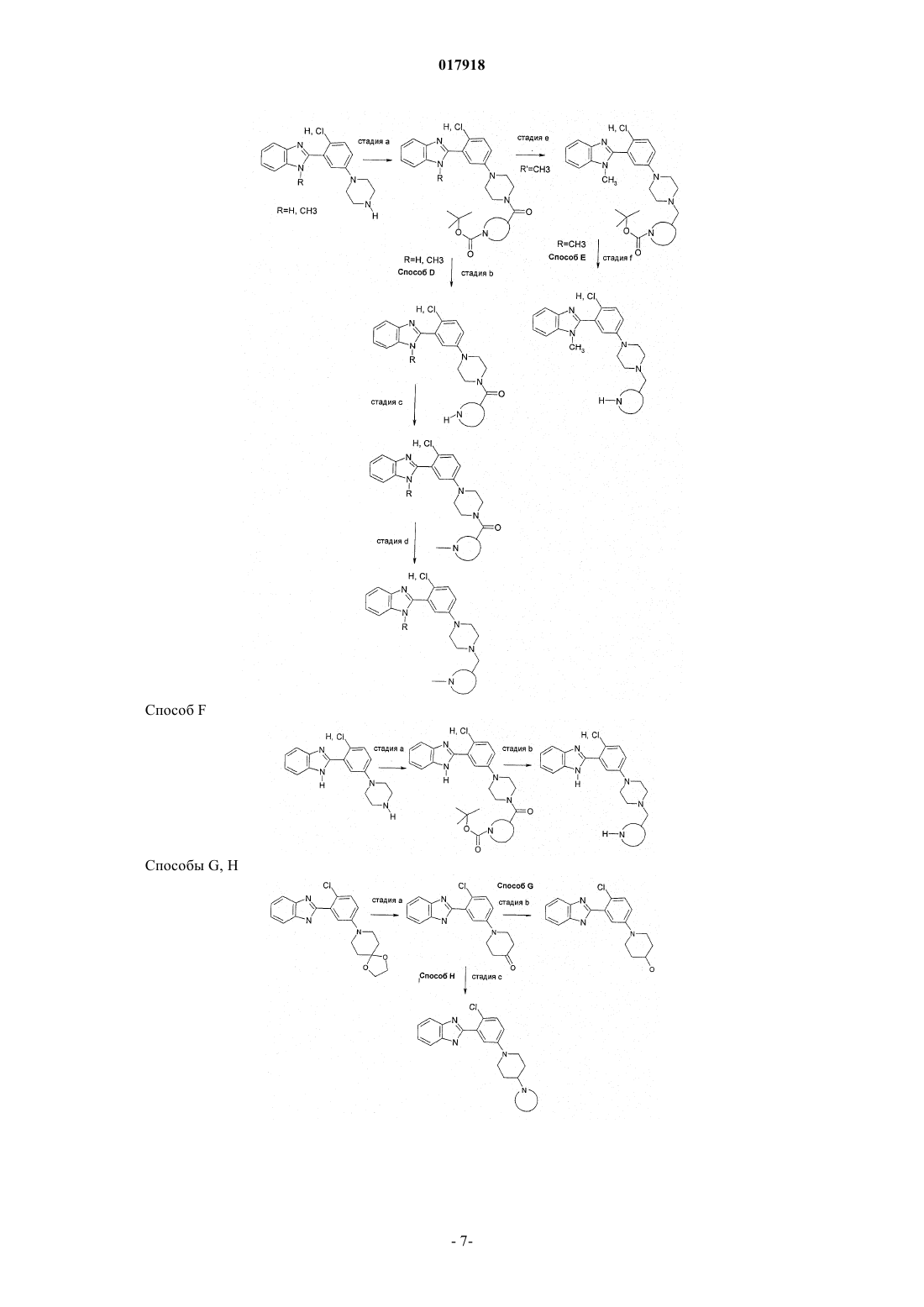
Формула / Реферат
1. Соединения формулы (I)

и их фармацевтически приемлемые соли, где
R1 представляет собой Н; линейную, разветвленную или циклическую (С1-С4)алкильную группу, возможно замещенную одним или более чем одним галогеном;
r равен 0, 1, 2 или 3;
R1' представляет собой, независимо друг от друга, когда r больше 1, галоген;
R2 может представлять собой Н, Cl, F или Br;
X может представлять собой N или СН;
i и j могут быть равны 1, 2 или 3, сумма i+j не может превосходить 5, и когда X представляет собой N, тогда i и j не могут быть равны 1;
R3 может представлять собой Н; линейную, разветвленную или циклическую группу (С1-С6)алкил, оксаалкил, алкилкарбонил, алкилсульфонил, оксаалкилкарбонил, алкоксикарбонил, алкиламинокарбонил, гидрокси, алкокси, возможно замещенную карбамоилом или одним или более чем одним атомом фтора; Ar; Ar-аминокарбонил; линейную или разветвленную группу (С1-С4)алкил, алкиламино, алкилкарбонил, алкоксикарбонил или алкиламинокарбонил, замещенную одним или двумя Ar;

Q является таким, что не образуется прямая связь между двумя атомами азота или между атомом азота и атомом кислорода, и может представлять собой карбонил; аминокарбонил; карбониламино; линейный или разветвленный (С1-С6)алкил, где одна метиленовая группа может быть заменена на О, NRx или карбонил или где две последовательные метиленовые группы могут быть заменены группой карбониламино, аминокарбонил;
Ar представляет собой 5-10-членное ароматическое или гетероароматическое кольцо, возможно замещенное одной или более чем одной группой, независимо выбранной из галогена, гидрокси, меркапто, амино, линейного, разветвленного или циклического (С1-С4)алкила, гидроксиалкила, моно- или диалкиламино, алкокси, алкилкарбонила, алкоксикарбонила, алкилкарбониламино, и таким образом, что два из этих заместителей могут образовывать 5-8-членное кольцо с конденсированной связью с Ar;
Rx может представлять собой Н или линейный, разветвленный или циклический (С1-С4)алкил;
q может быть равен 0 или 1;
k может быть равен 1, 2, 3 или 4;
l, m, n, р, р' и s независимо могут быть равны 1, 2 или 3;
t может быть равен 0, 1 или 2;
суммы l+m, n+p или р'+s+t не могут превосходить 5;
независимо друг от друга Т и T' представляют собой водород; линейную, разветвленную или циклическую цепь (С1-С6)алкил, азаалкил, оксаалкил, возможно замещенную амино, гидрокси, оксо, линейным, разветвленным или циклическим (С1-С3)алкилкарбонилом, алкоксикарбонилом;
Z может представлять собой О или NRy';
Ry и Ry' независимо представляют собой Н; линейную, разветвленную или циклическую группу (С1-С6)алкил, алкилкарбонил или алкоксикарбонил;
у и у' независимо могут быть равны 0, 1, 2 или 3;
Y1 и Y2 независимо представляют собой галоген; гидрокси; амино; линейную или разветвленную группу (С1-C6)алкил, алкиламино, меркаптоалкил, алкокси, алкилтио;
и за исключением 2-[4-(4-метилпиперазин-1-ил)фенил]-1Н-бензоимидазола; 2-[4-(4-пиридин-2-илпиперазин-1-ил)фенил]-1Н-бензоимидазола.
2. Соединения по п.1 формулы (Ia)

где R1, R1', R2, R3, X, Y1, r, i, j и у являются такими, как описано в п.1.
3. Соединения по п.2, где
X представляет собой N;
R3 может представлять собой Н; линейную, разветвленную или циклическую группу (С1-C6)алкил, оксаалкил, алкилкарбонил, алкилсульфонил, оксаалкилкарбонил, алкоксикарбонил или алкиламинокарбонил, возможно замещенную карбамоилом или одним или более чем одним атомом фтора; Ar; Ar-аминокарбонил; линейную или разветвленную группу (С1-С4)алкил, алкиламино, алкилкарбонил, алкоксикарбонил или алкиламинокарбонил, замещенную одним или двумя Ar;

Q является таким, что не образуется прямая связь между двумя атомами азота или между атомом азота и атомом кислорода, и может представлять собой карбонил; аминокарбонил; карбониламино; линейный или разветвленный (С1-C6)алкил, где одна метиленовая группа может быть заменена на О, NRx или карбонил или где две последовательные метиленовые группы могут быть заменены группой карбониламино, аминокарбонил.
4. Соединения по п.2, где X представляет собой СН.
5. Соединения по пп.3 и 4, где R3 представляет собой

q равен 1.
6. Соединения по п.5, где q равен 1 и Q представляет собой карбонил; линейный или разветвленный (С1-C6)алкил, (С1-С5)алкилкарбонил или карбонил(С1-С5)алкил.
7. Соединения по п.4, где q равен 0 и R3 выбран из

8. Фармацевтическая композиция, ингибирующая путь hedgehog, содержащая соединение по пп.1-7 с фармацевтически приемлемым носителем или эксципиентом.
9. Применение соединения по пп.1-7 для изготовления лекарственного средства для лечения или предупреждения остеопороза или рака.
10. Применение по п.9 для лечения рака, выбранного из немелкоклеточной карциномы легкого; мелкоклеточного рака легкого; рака молочной железы; опухолей яичников; опухолей пищеварительного тракта; рака головного мозга; рака предстательной железы; рака поджелудочной железы; базальноклеточной карциномы; злокачественной меланомы; плоскоклеточных карцином; множественной миеломы; лимфомы; форм рака мезенхимы; хронической миелоидной лейкемии; карциномы эндометрия; гепатоклеточной карциномы.
11. Способ лечения заболеваний, состояний или дисфункций, на которые может благоприятно влиять ингибирование пути hedgehog, включающий введение субъекту, нуждающемуся в таком введении, эффективного количества соединения по пп.1-7.
12. Способ по п.11 для лечения остеопороза или рака, в частности немелкоклеточной карциномы легкого; мелкоклеточного рака легкого; рака молочной железы; опухолей яичников; опухолей пищеварительного тракта; рака головного мозга; рака предстательной железы; рака поджелудочной железы; базальноклеточной карциномы; злокачественной меланомы; плоскоклеточных карцином; множественной миеломы; лимфомы; форм рака мезенхимы; хронической миелоидной лейкемии; карциномы эндометрия; гепатоклеточной карциномы.
Текст
Гетероциклические соединения формулы (I), модулирующие сигнальный путь hedgehog, их фармацевтическая композиция и их терапевтические применения где все заместители указаны в формуле изобретения. Область изобретения Настоящее изобретение относится к органическим соединениям, их фармацевтическим композициям и их применению в терапии и/или профилактике у млекопитающего, в частности к гетероциклическим соединениям, которые модулируют сигнальный путь hedgehog. Предшествующий уровень техники В результате аутопротеолиза человеческого 45 кДа белка-предшественника Shh получается 20 кДаN-концевой фрагмент, который отвечает за нормальный сигнальный путь hedgehog и 25 кДа С-концевой фрагмент, вовлеченный в аутопроцессирующую активность, в котором N-концевой фрагмент конъюгирован с холестерином (Lee et al. Science, 266, 1528-1537 (1994) и Bumcrot et al. Mol. Cell Biol., 15, 22942303 (1995. Нормально функционирующий сигнальный путь Hedgehog (Hh) определяет картину развития эмбриона путем направления клеточной дифференцировки и пролиферации, о чем впервые сообщили в результате исследований на Drosophilia melanogaster (Nusslein-Vollhard et al. Roux. Arch. Dev. Biol., 193: 267-282 (1984. Клеточные ответы на секретируемый полипептид Hh опосредованы двумя интегральными мембранными белками, Patched (Ptc) и Smoothened (Smo). Hh связывается с трансмембранным белкомPtc, имеющим двенадцать трансмембранных доменов, и таким образом обращает опосредованную Ptc супрессию трансмембранного белка Smo, имеющего семь трансмембранных доменов. Эта активацияSmo затем запускает серию внутриклеточных событий, завершающуюся стабилизацией транскрипционного фактора Cubitus interruptus (Ci) и экспрессией Ci-зависимых генов. Эти события повторяются во время развития млекопитающего и опухолеобразования посредством множественных белковых гомологов, включающих три отдельных члена семейства Hh [Sonic (Shh), Indian (Ihh) и Desert (Dhh)], два белкаPtc (Ptch1 и Ptch2) и три Ci-подобных транскрипционных фактора (Gli1, Gli2 и Gli3). Однако существует один единый гомолог Smo у позвоночных животных, который вовлечен во все формы сигнального путиHh, как показал генетический анализ Drosophila, мышей и полосатого данио (Chen et al. PNAS 99(22): 14071-14076 (2002.Smo инициирует сигнальный каскад, вызывающий активацию транскрипционных факторов Gli и их последующую транслокацию в ядро, что приводит в результате к контролю транскрипции геновмишеней. Путем отрицательной регуляции по механизму обратной связи Gli влияет на транскрипцию Ptc и Hip1 (белок, взаимодействующий с hedgehog 1 (Hip1, который ингибирует путь Hh. Утрата контроля над активацией пути Hh связана с увеличивающимся диапазоном форм рака, включающих формы, поражающие головной мозг, таких как медуллобластома (Romer and Curran, Cancer Res 65(12) 4975-4978(2005 и глиобластома (Bar et al. Stem Cells, 25(10):2524-33 (2007; рак предстательной железы (Sanchezet al. PNAS, 101(34), 12561-12566 (2004; рак поджелудочной железы (Thayer et al. Nature 423 851- 856(2003; немелкоклеточная карцинома легкого (Yuan et al. Oncogene, 26, 1046-1055 (2007); мелкоклеточный рак легкого (Watkins et al. Nature, 422, 313-317 (2003; рак молочной железы (Kubo et al. Cancer Res,64, 6071-6074 (2004; различные опухоли пищеварительного тракта (Berman et al. Nature, 425, 846-851et al. PNAS, 100(8), 4616-4621 (2003; злокачественная меланома (Pons и Quintanilla Clin Trans Oncol.,8(7), 466-474 (2006; плоскоклеточные карциномы (Xuan et al. Mod Pathol., 19(8), 1139-47 (2006; Bклеточные злокачественные новообразования, такие как множественная миелома и лимфомы (Dierks etal. Nat. Med., 13(8), 944-951 (2007); Peacock et al. PNAS, 104(10), 4048-4053 (2007; мезенхимные формы рака, такие как хондросаркома (Tiet et al. Am. J. Pathol., 168(1), 321-330 (2006, светлоклеточная саркома почки (Cutcliffe et al. Clin Cancer Res., 11(22):7986-94 (2005 и рабдомиосаркома (Tostar et al. J. Pathol.,208(1), 17-25 (2006; хроническая миелоидная лейкемия (Sengupta et al. Leukemia, 21(5), 949-955 (2007; карцинома эндометрия (Feng et al. Clin. Cancer Res., 13(5), 1389-1398 (2007); гепатоклеточные карциномы(Huang et al. Carcinogenesis, 27(7), 1334-1340 (2006; опухоли яичников (Chen et al. Cancer Sci., 98(1), 6876 (2007. Также обнаружено, что сигнальный путь Hh регулирует экспрессию белков-транспортеров ABC: белка-1 множественной лекарственной устойчивости (MDR1, АВСВ 1 (белок 1 подсемейства В АТФсвязывающей кассеты), Р-гликопротеин) и (BCRP (белок резистентности рака молочной железы), ABCG2(белок 2 подсемейства G АТФ-связывающей кассеты и что подавление экспрессии генов MDR1 иBCRP путем направленного нокдауна с помощью малых интерферирующих РНК частично обращает вызванную Hh химиорезистентность. Последнее может свидетельствовать о том, что путь Hh может служить мишенью для преодоления MDR (множественная лекарственная устойчивость) и увеличения химиотерапевтического ответа (Sims-Mourtada et al. Oncogene, 26(38), 5674-5679 (2007. Обнаружено, что блокировка сигнального пути sonic hedgehog усиливает антипролиферативное действие ингибиторовEGFR (рецептор эпидермального фактора роста) в клетках рака поджелудочной железы (Hu et al. ActaPharmacol Sin., 28(8), 1224-30 (2007 и клетках рака предстательной железы (Mimeault et al. Int. J. Cancer,118(4), 1022-31 (2006. Также показано, что путь hedgehog ассоциирован с повторным ростом опухоли после химиотерапии и рассматривается в качестве потенциальной мишени для улучшения ответа на излучение (Sims-Mourtadaet al. Clin. Cancer Res., 12(21), 6565-6572 (2006 и циклопамин, представляющий собой антагонист путиhedgehog, увеличивает цитотоксические действия паклитаксела и излучения в клетках рака поджелудочной железы, экспрессирующих Hh (Shafaee et al. Cancer Chemother. Pharmacol., 58(6), 765-70 (2006. Также сообщалось о том, что ингибирование сигнального пути hedgehog может применяться для лечения диапазона заболеваний, связанных с воспалением, гиперплазией эпителиальных клеток, фиброзом ткани или иммунными расстройствами (Lamb et al. EP 1183040). Сообщалось о том, что ингибирование сигнального пути sonic hedgehog уменьшает хроническое отторжение и увеличивает выживаемость аллотрансплантата в модели ортотопической трансплантации тонкой кишки у крыс. Хотя острое отторжение трансплантата можно контролировать при помощи иммуносупрессивных агентов, хроническое отторжение, которое характеризуется артериосклерозом сосудов донорного органа, представляет собой основное препятствие для длительного выживания аллотрансплантата. Выживание трансплантата в модели ортотопической трансплантации тонкой кишки у крыс значительно увеличивалось после обработки антителами к Shh по сравнению с контролем, в качестве которого использовали обработку иммуноглобулином G (116 по сравнению с 77,5 сутками). Отложение коллагена и закупорка сосудов в мезентерии значительно уменьшалось у реципиентов антител к Shh (Chen et al. Transplantation, 83(10), 1351-1357(2007); Lamb et al. EP 1183040B1). Также сообщалось о том, что sFRP-1 представляет собой располагающийся далее по цепи генмишень сигнального пути Hh и что увеличенная экспрессия секретированного связанного с ожогом(Frizzled related) белка-1 (sFRP-1) после активации пути Hh обеспечивает молекулярную связь для ингибирующего действия на сигнальный путь Wnt (He et al. J. Biol. Chem., 281(47), 35598-35602 (2006. Таким образом, модулирование сигнального пути Wnt путем антагонистического действия на путь Hh черезsFRP-1 может представлять собой способ лечения ряда заболеваний, таких как остеопороз (Ai et al. Mol.Cell. Biol., 25(12), 4946-4955 (2005 среди прочих (Luo et al. Laboratory Investigation, 87, 97-103 (2007. Исследованы различные ингибиторы пути Hh, включающие природный продукт циклопамин, который, как полагают, действует путем связывания с гептаспиральной областью Smo. Дополнительно, в течение последних лет сообщали о множестве синтетических низкомолекулярных антагонистов рецептораLubisch et al. раскрывает серии 2-фенилбензимидазолов в качестве ингибиторов PARP, полезных для лечения различных заболеваний, включая рак (WO 2000026192), и в области косметических средств(WO 2001082877). Повторяющая особенность заключается в наличии карбамоильной группировки в 4 позиции бензимидазольного кольца.Arienti et al. (WO 2003032984) и Ameriks et al. (WO 2004093873 и US 2004214857) раскрывают серию 2-фенилбензимидазольных производных в качестве ингибиторов киназы 2 контрольной точки клеточного цикла для лечения рака, дополнительно характеризующихся тем, что положение 5 бензимидазольного кольца всегда замещено карбоксилатной, карбамоильной или сульфамоильной группой.Ohemeng et al. (WO 9911627 и US 5942532) раскрывает серию соединений 5-карбоксиимидамидов 2-фенилбензимидазолов в качестве антибактериальных агентов.Mjalli et al. (WO 2003075921) описывают фармацевтические применения серии 2 фенилбензимидазольных производных.Khaled et al.1 (Bulletin of the Faculty of Pharmacy (Cairo University), 40(1), 7-13 (2002 описывает синтез и антигипертензивную активность 2-фенилбензимидазольных производных, тогда как ДНКсвязывающие свойства некоторых других описаны Kobuta et al. (Nucleic Acids Research Supplement, 2hedgehog для лечения различных форм рака. Подробное описание изобретения В настоящем изобретении предложены соединения формулы (I) где, если позволяет валентность и стабильность, тоR1 представляет собой Н, линейную, разветвленную или циклическую группу (С 1-С 4)алкил, возможно замещенную одним или более чем одним галогеном, разветвленной или линейной (С 1-С 4)алкокси или моно- или ди- линейной, разветвленной или циклической (С 1-С 6)алкиламино группой;R1' представляет собой, независимо друг от друга, когда r больше 1, галоген, линейную, разветвленную или циклическую (С 1-С 4)алкоксигруппу; линейный, разветвленный или циклический (С 1-С 4)алкил,возможно замещенный линейной или разветвленной (С 1-С 4)алкокси, алкиламино или диалкиламино группой;R2 может представлять собой Н, Cl, F или Br;X может представлять собой N или СН;i и j могут быть равны 1, 2 или 3, сумма i+j не может превосходить 5 и когда X представляет собойN, тогда i и j не могут быть равны 1;R3 может представлять собой Н, линейную, разветвленную или циклическую группу (С 1-С 6)алкил,оксаалкил, алкилкарбонил, алкилсульфонил, оксаалкилкарбонил, алкоксикарбонил, алкиламинокарбонил, оксаалкенил, алкенилкарбонил, оксаалкенилкарбонил, алкенилоксикарбонил, алкениламинокарбонил, алкилиден, алкилоксиимино, гидрокси, алкокси, алкенилокси, возможно замещенную карбамоилом или одним или более чем одним атомом фтора; Ar; Ar-аминокарбонил; линейную или разветвленную группу (С 1-С 4)алкил, алкиламино, азаалкил, оксаалкил, алкилкарбонил, алкоксикарбонил или алкиламинокарбонил, замещенную одним или двумя Ar и возможно замещенную одним или более чем одним атомом фтораQ является таким, что не образуется прямая связь между двумя атомами азота или между атомом азота и атомом кислорода и может представлять собой карбонил; аминокарбонил; карбониламино имин;SO2, линейный или разветвленный (С 1-С 6)алкил, возможно замещенный одним или более чем одним атомом фтора, где одна метиленовая группа может быть замещена на О, NRx, карбонил или SO2 или где две последовательные метиленовые группы могут быть замещены группой карбониламино, аминокарбонил, сульфониламино, аминосульфонил;Ar представляет собой 5-10-членное ароматическое или гетероароматическое кольцо, возможно замещенное одной или более чем одной группой, независимо выбранной из галогена, гидрокси, меркапто,амино, циано, нитро, карбамоила, сульфамоила, тригалогенометила, тригалогенометокси, линейного,разветвленного или циклического (C1-С 4)алкила, гидроксиалкила, моно- или диалкиламино, алкокси,алкилкарбонила, алкоксикарбонила, алкилкарбониламино, оксаалкила, азаалкила, и таким образом, что два из этих заместителей могут образовывать 5-8-членное кольцо с конденсированной связью с Ar;Rx может представлять собой Н или линейный, разветвленный или циклический (С 1-С 4)алкил, дигалогеноалкил или тригалогеноалкил;q может быть равен 0 или 1;k может быть равен 1, 2, 3 или 4;l, m, n, р, р' и s независимо могут быть равны 1, 2 или 3;t может быть равен 0, 1 или 2; суммы l+m, n+p или р'+s+t не могут превосходить 5; независимо друг от друга Т и T' представляют собой водород; линейную, разветвленную или циклическую цепь (С 1-С 6)алкил, азаалкил, оксаалкил, алкенил, азаалкенил, оксаалкенил, возможно замещенную галогеном, амино, циано, гидрокси, оксо, линейным, разветвленным или циклическим (С 1 С 3)алкилкарбонилом, алкоксикарбонилом, алкилкарбониламино, алкиламинокарбонилом, карбамоилом,гуанидино, алкенилкарбонилом, оксаалкенилкарбонилом, алкенилоксикарбонилом, алкениламинокарбонилом;Z может представлять собой О, S, SO2, SO, или NRy';Ry и Ry' независимо представляют собой Н; линейную, разветвленную или циклическую группу(С 1-С 6)алкил, алкилкарбонил, алкоксикарбонил или алкиламинокарбонил, возможно замещенную одним или более чем одним атомом фтора; у и у' независимо могут быть равны 0, 1, 2 или 3;Y1 и Y2 независимо представляют собой галоген; гидрокси; амино; циано; нитро; оксо; линейную или разветвленную группу (С 1-С 6)алкил, дигалогеноалкил, азаалкил, оксаалкил, алкилкарбонил, оксаалкилкарбонил, алкоксикарбонил, алкиламинокарбонил, алкилкарбониламино, алкенил, оксаалкенил, азаалкенил, алкенилкарбонил, оксаалкенилкарбонил, алкенилоксикарбонил, алкениламинокарбонил, алкиламино, меркаптоалкил, алкокси, алкилтио, возможно замещенную одним или более чем одним атомом фтора; где две группы Y2 могут образовывать 5-8-членное кольцо со спиро или конденсированной связью. И за исключением 2-[4-(4-метилпиперазин-1-ил)фенил]-1 Н-бензоимидазола; 2-[4-(4-пиридин-2 илпиперазин-1-ил)фенил]-1 Н-бензоимидазола. Предпочтительная группа соединений представляет собой соединения формулы (Ia) Конкретное воплощение формулы (Ia) охватывает те соединения, гдеR3 представляет собой линейную, разветвленную или циклическую группу (С 1-С 6)алкил, оксаалкил,алкилкарбонил, алкилсульфонил, оксаалкилкарбонил, алкоксикарбонил, алкиламинокарбонил, оксаалкенил, алкенилкарбонил, оксаалкенилкарбонил, алкенилоксикарбонил или алкениламинокарбонил, возможно замещенную карбамоилом или одним или более чем одним атомом фтора; Ar; Ar-аминокарбонил; линейную или разветвленную группу (С 1-С 4)алкил, алкиламино, азаалкил, оксаалкил, алкилкарбонил,алкоксикарбонил или алкиламинокарбонил, замещенную одним или двумя Ar и возможно замещенную одним или более чем одним атомом фтора;Q является таким, что не образуется прямая связь между двумя атомами азота или между атомом азота и атомом кислорода, и может представлять собой карбонил; аминокарбонил; карбониламино, имин;SO2; линейный или разветвленный (С 1-С 6)алкил, возможно замещенный одним или более чем одним атомом фтора, где одна метиленовая группа могут быть замещена на О, NRx, карбонил или SO2, или где две последовательные метиленовые группы могут быть замещены группой карбониламино, аминокарбонил, сульфониламино, аминосульфонил. Во втором конкретном воплощении формулы (Ia) X представляет собой СН. В предпочтительном аспекте этого воплощения q равен 0 и R3 выбран из Еще одно предпочтительное воплощение, попадающее под формулу (Ia), далее названное как G1,охватывает соединения, для которых q равен 1, и R3 представляет собой ский (С 1-С 6)алкил, (С 1-С 5)алкилкарбонил или карбонил(С 1-С 5)алкил, возможно замещенный одним или более чем одним атомом фтора, составляют предпочтительное воплощение. Фармакологическая активность репрезентативной группы соединений формулы (I) продемонстрирована с использованием двух анализов in vitro, описанных ниже. В соответствии с еще одним аспектом изобретение, таким образом, относится к способу лечения рака или остеопороза, который включает введение субъекту, предпочтительно человеку, нуждающемуся в таком введении, эффективного количества соединения формулы (I). Типы рака, которые можно лечить с использованием такого способа, включают,но не ограничиваются такими типами рака, как немелкоклеточная карцинома легкого; мелкоклеточный рак легкого; рак молочной железы; опухоли яичников; опухоли пищеварительного тракта; формы рака головного мозга, такие как медуллобластома и глиобластома; рак предстательной железы; рак поджелудочной железы; базальноклеточная карцинома; злокачественная меланома; плоскоклеточные карциномы; множественная миелома; лимфомы; мезенхимные формы рака, такие как хондросаркома, светлоклеточная саркома почки и рабдомиосаркома; хроническая миелоидная лейкемия; карцинома эндометрия; гепатоклеточные карциномы. Как правило, соединения формулы (I) могут быть использованы для лечения любого заболевания,состояния или дисфункции, на которые может благоприятно влиять ингибирование пути hedgehog путем связывания соединений с рецептором Smo, включая, но не ограничиваясь, такими заболеваниями, состояниями и дисфункциями как остеопороз и формы рака, выбранные из немелкоклеточной карциномы легкого; мелкоклеточного рака легкого; рака молочной железы; опухолей яичников; опухолей пищеварительного тракта; форм рака головного мозга, таких как медуллобластома и глиобластома; рака предстательной железы; рака поджелудочной железы; базальноклеточной карциномы; злокачественной меланомы; плоскоклеточных карцином; множественной миеломы; лимфом; мезенхимных форм рака, таких как хондросаркома, светлоклеточная саркома почки и рабдомиосаркома; хронической миелоидной лейкемии; карциномы эндометрия; гепатоклеточных карцином. Доза соединений для применения в терапии может варьировать в зависимости, например, от пути введения, природы и тяжести заболевания. Как правило, приемлемое фармакологическое действие у человека может быть достигнуто при суточных дозах, находящихся в диапазоне от 0,01 до 200 мг/кг. В еще одном аспекте изобретение относится к фармацевтической композиции, содержащей одно или более чем одно соединение формулы (I) в составе с фармацевтически приемлемыми носителями и эксципиентами. Фармацевтические композиции могут находиться в форме твердых, полутвердых или жидких препаратов предпочтительно в форме растворов, суспензий, порошков, гранул, таблеток, капсул,сиропов, суппозиториев, аэрозолей или систем с контролируемой доставкой. Композиции могут быть введены множеством путей, включающих пероральный, трансдермальный, подкожный, внутривенный,внутримышечный, ректальный и интраназальный и предпочтительно приготовлены в стандартной лекарственной форме. Лекарственные формы для перорального введения могут содержать от приблизительно 1 до приблизительно 1000 мг соединения по изобретению. Для соединений, которые могут находиться в форме свободных оснований, изобретение также включает соли присоединения кислот, предпочтительно соли с фармацевтически приемлемыми кислотами. Изобретение также включает отдельные изомеры и диастереомеры соединений I или их смеси (например, рацемические смеси). Принципы и способы изготовления фармацевтических композиций описаны, например, в Remington's Pharmaceutical Science, Mack Publishing Company, Easton (PA). Соединения формулы (I), их оптические изомеры или диастереомеры могут быть очищены или разделены в соответствии с хорошо известными методами, которые включают хроматографию с хиральной матрицей и фракционную кристаллизацию, не ограничиваясь этими способами. Синтез соединений и экспериментальные методы Соединения по настоящему изобретению могут быть получены с использованием различных путей синтеза, включающих описанные в нижеприведенных способах A-Z, начиная с имеющихся в продаже соединений. Материалы и методы Все реагенты и растворители были получены из коммерческих источников. Жидкие растворы, чувствительные к воздуху и влаге, переносили посредством шприца. Протекание реакций контролировали при помощи тонкослойной хроматографии (TLC) и/или жидкостной хроматографии-масс-спектрометрии(LC-MS). Все спектры ядерного магнитного резонанса регистрировали с использованием спектрометра VarianMercury Plus 400 МГц, оборудованном широкополосным зондом PFG АТВ. Способы длительностью 10 мин осуществляли с использованием модуля для разделения Waters 2795, оборудованного Waters Micromass ZQ (ES ионизация (путем распыления электронов и WatersMicromass ZQ (ES) или Waters 2487 DAD (детектирование с помощью диодной матрицы) с использованием колонки Supelco Discovery HS C18 5,0 мкм 1021,2 мм. Градиенты осуществляли, используя 0,1% муравьиную кислоту/воду и 0,1% муравьиную кислоту/ацетонитрил с градиентом от 5/95 до 95/5 при указанном времени анализа. Очистки осуществляли с использованием картриджей силикагеля isolute flash Si, с чистотой более 95%. Все анализы методом TLC осуществляли на силикагеле (Merck 60 F254) и пятна выявляли путем визуализации в UV (ультрафиолетовом свете) при 254 нм и окрашивании KMnO4 или нингидрином. Микроволновая обработка: Personal Chemistry, Emrys Optimizer, микроволновой реактор, поглощение устанавливалось до нормального с временем до перемешивания 10 с. Пример 1. (2-(4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]пиперазин-1-ил)этил)диметиламин. 2-Хлор-5-[4-(2-диметиламиноэтил)пиперазин-1-ил]бензойной кислоты этиловый эфир. Способ А, стадия а. Этил 5-бром-2-хлорбензоат (0,80 г, 3,04 ммоль), рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP) (0,19 г, 0,30 ммоль), трис-(дибензилиденацетон)дипалладий(Pd2(dba)3) (0,28 г, 0,30 ммоль) и карбонат цезия (Cs2CO3) (1,39 г, 4,26 ммоль) помещали в сосуд Шленка и продували путем повторяющихся циклов азот/вакуум в течение 30 мин. Затем добавляли безводный толуол (6 мл) и 1-(2-диметиламиноэтил)пиперазин (0,55 мл, 3,65 ммоль). Реакционную смесь перемеши- 12 017918 вали в течение 10 мин при комнатной температуре, нагревали при 85 С в течение ночи и затем охлаждали до комнатной температуры. Раствор разбавляли EtOAc (30 мл), нерастворимый материал отфильтровывали и фильтрат промывали насыщенным рассолом (15 мл). Органическую фазу сушили над Na2SO4,фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали с помощью флэш-хроматографии (с элюцией градиентом: EtOAc, EtOAc:МеОН/1:1, EtOAc:MeOH:NH3 в МеОН (2 М)/1:1:0,2) с получением 0,30 г указанного в заголовке соединения (30%). 1 Н-ЯМР (ядерный магнитный резонанс) (400 МГц, DMSO (диметилсульфоксид:1.28 (3 Н, t), 2.12 (6 Н, s), 2.32-2.42 (4 Н,m), 2.47-2.51 (4 Н, m), 3.11-3.14 (4 Н, m), 4.27 (2 Н, q), 7.06-7.09 (1 Н, m), 7.18-7.19 (1 Н, m), 7.30-7.32 (1 Н,m). 2-Хлор-5-[4-(2-диметиламиноэтил)пиперазин-1-ил]бензойная кислота. Способ А, стадия b. Раствор этилового эфира 2-хлор-5-[4-(2-диметиламиноэтил)пиперазин-1 ил]бензойной кислоты (0,46 г, 1,36 ммоль) в EtOH (6 мл) и NaOH 10% (2 мл) нагревали путем кипячения с обратным холодильником в течение ночи, затем реакционную смесь охлаждали до комнатной температуры и органический растворитель концентрировали путем упаривания при пониженном давлении. Получающийся в результате раствор нейтрализовали путем добавления по каплям 1 н. HCl и воду затем упаривали при пониженном давлении. Получающийся в результате остаток растворяли в EtOH (10 мл),нерастворимое вещество отфильтровывали и фильтрат концентрировали при пониженном давлении при комнатной температуре с получением 0,24 г указанного в заголовке соединения (58%) без дополнительной очистки. 1 Н-ЯМР (400 МГц, DMSO):2.27 (6 Н, s), 2.46-2.55 (8 Н, m), 3.04-3.06 (4 Н, m), 7.76-7.79 (1 Н,m), 6.92-6.93 (1 Н, m), 7.07-7.09 (1 Н, m).N-(2-Аминофенил)-2-хлор-5-[4-(2-диметиламиноэтил)пиперазин-1-ил]бензамид. Способ А, стадия с. К раствору 2-хлор-5-[4-(2-диметиламиноэтил)пиперазин-1-ил]бензойной кислоты (0,40 г, 1,29 ммоль) в безводном DMF (диметилформамид) (5 мл) добавляли 1-(3 диметиламинопропил)-3-этилкарбодиимид гидрохлорид (EDC) (0,25 г, 1,29 ммоль), а затем 1 гидроксибензотриазол гидрат (1-HOBt) (0,11 г, 0,77 ммоль) и диметилпиридин-4-иламин (DMAP) (0,08 г,0,64 ммоль). После перемешивания в течение 3 ч при комнатной температуре добавляли 1,2 фенилендиамин (0,21 г, 1,93 ммоль) и получающуюся в результате смесь перемешивали при комнатной температуре в течение ночи. Раствор упаривали при пониженном давлении, затем разбавляли дихлорметаном (20 мл) и промывали насыщенным раствором Na2CO3 (25 мл). Органический слой сушили надNa2SO4, фильтровали и упаривали при пониженном давлении. Получающееся в результате твердое вещество очищали с помощью флэш-хроматографии (элюент: дихлорметан:MeOH:NH3 в МеОН (2 М)/9:0,5:0,5) с получением 0,18 г (35%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, DMSO):2.13 (6 Н, m), 2.35-2.41 (4 Н, m), 2.48-2.53 (4 Н, m), 3.14-3.18 (4 Н, m), 4.91 (2 Н, br s), 6.54-6.59 (1 Н, m), 6.736.75 (1 Н, m), 6.92-7.00 (2 Н, m), 7.11-7.12 (1 Н, m), 7.23-7.25 (1 Н, m), 7.28-7.33 (1 Н, m), 9.62 (1 Н, br s).(2-4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]пиперазин-1-илэтил)диметиламин. Способ А, стадия d. Раствор N-(2-аминофенил)-2-хлор-5-[4-(2-диметиламиноэтил)пиперазин-1 ил]бензамида (0,35 г, 0,87 ммоль) в уксусной кислоте (15 мл) нагревали в течение 7 ч при 85 С, затем растворитель удаляли при пониженном давлении и остаток растирали с Et2O с получением 0,25 г (75%) указанного в заголовке соединения в виде твердого вещества без дополнительной очистки. 1 Н-ЯМР (400 МГц, DMSO):2.12 (6 Н, s), 2.33-2.42 (4 Н, m), 2.49-2.57 (4 Н, m), 3.16-3.19 (4 Н, m), 7.07 (1 Н, dd), 7.18-7.23(2 Н, m), 7.33 (1 Н, d), 7.38-7.41 (1 Н, m), 7.52 (1 Н, d), 7.65-7.67 (1 Н, m), 12.60 (1 Н, s); m/z 384 (М+Н)+, время удерживания равно 0,68. Пример 2. 4-[3-(1 Н-Бензоимидазол-2-ил)фенил]пиперазин-1-карбоновой кислоты трет-бутиловый эфир. 4-(3-Карбоксифенил)пиперазин-1-карбоновой кислоты трет-бутиловый эфир. Способ В, стадия b. Раствор 4-(3-этоксикарбонилфенил)пиперазин-1-карбоновой кислоты третбутилового эфира (5,00 г, 14,95 ммоль) в EtOH (50 мл) и NaOH 10% (20 мл) кипятили с обратным холодильником в течение 3 ч, затем реакционную смесь охлаждали до комнатной температуры и органический растворитель упаривали при пониженном давлении. Получающийся в результате раствор нейтрализовали путем добавления по каплям 1 н. HCl и водный раствор экстрагировали EtOAc (350 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 3,90 г указанного в заголовке соединения (85%) без дополнительной очистки. 1 Н-ЯМР(400 МГц, DMSO):1.40 (9 Н, s), 3.15-3.18 (4 Н, m), 3.47-3.49 (4 Н, m), 7.27-7.29 (1 Н, m), 7.32-7.37 (1 Н, m),7.42-7.44 (1 Н, m), 7.54 (1H, br s). 4-[3-(2-Аминофенилкарбамоил)фенил]пиперазин-1-карбоновой кислоты трет-бутиловый эфир. Способ В, стадия с. Раствор 4-(3-карбоксифенил)пиперазин-1-карбоновой кислоты трет-бутилового эфира (5,80 г, 18,95 ммоль) и 1,1-карбонилдиимидазола (CDI) (3,07 г, 18,95 ммоль) в ацетонитриле (120 мл) перемешивали в течение 3 ч при комнатной температуре, затем добавляли 1,2-фенилендиамин (2,25 г, 20,85 ммоль). Получающуюся в результате суспензию кипятили с обратным холодильником в течение 3 ч, затем охлаждали до комнатной температуры и растворитель удаляли при пониженном давлении. Получающийся в результате остаток разбавляли дихлорметаном (200 мл), промывали водой (340 мл) и органический слой сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Неочищенный продукт перекристаллизовывали из EtOAc с получением 2,99 г (40%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, DMSO):1.40 (9 Н, m), 3.15-3.17 (4 Н, m), 3.45-3.47 (4 Н, m), 4.85 (2 Н, br(1 Н, m), 7.51 (1 Н, br s), 9.60 (1 Н, br s). 4-[3-(1 Н-Бензоимидазол-2-ил)фенил]пиперазин-1-карбоновой кислоты трет-бутиловый эфир. Способ В, стадия d. Раствор 4-[3-(2-аминофенилкарбамоил)фенил]пиперазин-1-карбоновой кислоты трет-бутилового эфира (1,20 г, 3,03 ммоль) в уксусной кислоте (12 мл) нагревали в течение ночи при 55 С, затем растворитель удаляли при пониженном давлении. Получающееся твердое вещество растирали с Et2O с получением указанного в заголовке соединения в виде белого твердого вещества (0,90 г, 79%) без дополнительной очистки. 1 Н-ЯМР (400 МГц, DMSO):1.41 (9 Н, m), 3.19-3.21 (4 Н, m), 3.48-3.50 (4 Н,m), 7.05-7.08 (1 Н, m), 7.17-7.18 (2 Н, m), 7.35-7.40 (1 Н, m), 7.51-7.73 (4 Н, m), 12.80 (1 Н, br s); m/z 379(М+Н)+; время удерживания равно 2,58. Пример 3. 2-(3-Пиперазин-1-илфенил)-1 Н-бензоимидазол дигидрохлорид. 2-(3-Пиперазин-1-илфенил)-1 Н-бензоимидазол дигидрохлорид. Способ В, стадия е. К раствору 4-[3-(1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбоновой кислоты трет-бутилового эфира (1,80 г, 4,76 ммоль) в МеОН (2 мл) добавляли 2 М HCl в Et2O (8 мл) и получающуюся в результате смесь перемешивали в течение 6 ч при комнатной температуре. Растворитель затем удаляли при пониженном давлении и получающуюся в результате соль растирали с Et2O с получением 1,66 г (100%) указанного в заголовке соединения в виде соли дигидрохлорида. 1 Н-ЯМР (400 МГц,CD3OD):3.34-3.37 (4 Н, m), 3.54-3.57 (4 Н, m), 7.34-7.37 (1 Н, m), 7.52-7.58 (4 Н, m), 7.74-7.77 (3 Н, m); m/z 279 (М+Н)+; время удерживания равно 0,26. Пример 4. 4-[3-(1 Н-Бензоимидазол-2-ил)фенил]пиперазин-1-ил-(1-метилпиперидин-4-ил)метанон. 4-[3-(1 Н-Бензоимидазол-2-ил)фенил]пиперазин-1-ил-(метилпиперидин-4-ил)метанон. Способ В, стадия f. К раствору 1-метилпиперидин-4-карбоновой кислоты гидрохлорида (0,13 г, 0,72 ммоль) и N,N-диизопропилэтиламина (DIPEA) (0,16 мл, 0,72 ммоль) в ацетонитриле (8 мл) добавлялиCDI (0,12 г, 0,72 ммоль) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение 5 ч. Затем добавляли 2-(3-пиперазин-1-илфенил)-1 Н-бензоимидазол дигидрохлорид(0,20 г, 0,57 ммоль) и получающийся в результате раствор нагревали при 65 С в течение ночи. Раствор затем охлаждали до комнатной температуры и растворитель удаляли. Неочищенный продукт перерастворяли в дихлорметане (15 мл) и добавляли насыщенный раствор NaHCO3 (6 мл) при перемешивании. Полученный осадок затем фильтровали, промывали водой (3 мл) и диэтиловым эфиром (5 мл) и сушили при пониженном давлении с получением 0,20 г (87%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, CD3OD):1.80-1.84 (4 Н, m), 2.19-2.26 (2 Н, m), 2.35 (3 Н, s), 2.79-2.83 (1 Н, m), 2.99-3.02 (2 Н, m),3.28-3.33 (4 Н, m), 3.78-3.79 (4 Н, m), 7.10-7.15 (1 Н, m), 7.24-7.27 (2 Н, m), 7.40-7.44 (1 Н, m), 7.55-7.59 (3 Н,m), 7.73-7.74 (1 Н, m); m/z 404 (M+H)+; время удерживания равно 1,10. Пример 5. 2-3-[4-(1-Метилпиперидин-4-илметил)пиперазин-1-ил]фенил-1H-бензоимидазол формиат. 2-3-4-(1-Метилпиперидин-4-ил метил)пиперазин-1-ил]фенил-1 Н-бензоимидазол. Способ В, стадия g. К раствору 4-[3-(1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-ил-(1 метилпиперидин-4-ил)метанона (0,12 г, 0,35 ммоль) в безводном THF (тетрагидрофуран) (4 мл) добавляли боран-метилсульфидный комплекс в THF CH3)2S.BH3) (0,07 мл, 0,74 ммоль) и получающуюся в результате суспензию нагревали при 60 С в течение ночи. Реакционную смесь затем охлаждали до комнатной температуры, растворитель удаляли при пониженном давлении и 1 н. HCl (3 мл) добавляли к остатку,а затем нагревали при 100 С в течение 5 ч. Реакционную смесь затем охлаждали до комнатной температуры, нейтрализовали 10% NaOH и экстрагировали дихлорметаном (310 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали путем препаративной HPLC (высокоэффективная жидкостная хроматография) с получением 0,08 г (69%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, CD3OD):1.55-1.61 (2 Н, m), 2.01-2.06 (3 Н, m), 2.63-2.81 (5 Н, m), 2.94-3.00 (2 Н,m), 3.09-3.11 (4 Н, m), 3.43-3.48 (6 Н, m), 7.08-7.11 (1 Н, m), 7.26-7.28 (2 Н, m), 7.38-7.42 (1 Н, m), 7.57-7.63(3 Н, m), 7.70-7.71 (1 Н, m), 8.36 (3 Н, s); m/z 390 (М+Н)+; время удерживания равно 0,21. Пример 6. 4-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбоновой кислоты третбутиловый эфир. 4-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбоновой кислоты трет-бутиловый эфир. Способ С, стадия а. Смесь 4-[3-(1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбоновой кислоты трет-бутилового эфира (0,83 г, 2,20 ммоль) (полученного, как описано в способе В) и NaH 60% масла(0,11 г, 4,39 ммоль) в THF (10 мл) перемешивали при комнатной температуре в течение 1 ч. Добавляли метилйодид (0,27 мл, 4,39 ммоль) и получающийся в результате раствор перемешивали в течение ночи. К реакционному раствору добавляли воду (15 мл), затем суспензию нейтрализовали 1 н. HCl и экстрагировали EtOAc (340 мл). Объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. Остаток растирали с Et2O и получающееся твердое вещество фильтровали с получением 0,76 г (88%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, DMSO):1.40(9 Н, m), 3.18-3.20 (4 Н, m), 3.45-3.48 (4 Н, m), 3.85 (3 Н, br s), 7.14-7.15 (1 Н, m), 7.22-7.29 (3 Н, m), 7.33 (1 Н,br s), 7.39-7.42 (1 Н, m), 7.60-7.62 (1 Н, m), 7.65-7.67 (1 Н, m); m/z 393 (М+Н)+; время удерживания равно 2,56. Пример 7. 1-Метил-2-(3-пиперазин-1-илфенил)-1 Н-бензоимидазол дигидрохлорид. 1-Метил-2-(3-пиперазин-1-илфенил)-1 Н-бензоимидазол дигидрохлорид. Способ С, стадия b. К раствору 4-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1 карбоновой кислоты трет-бутилового эфира (1,30 г, 3,32 ммоль) в МеОН (4 мл) добавляли 2 М HCl вEt2O (9 мл) и получающийся в результате раствор перемешивали в течение 6 ч при комнатной температуре. Растворитель затем удаляли при пониженном давлении и получающуюся в результате соль растирали с Et2O с получением 1,21 г (100%) указанного в заголовке соединения в виде соли дигидрохлорида. 1 Н-ЯМР (400 МГц, CD3OD):3.42-3.44 (m, 4H), 3.60-3.63 (m, 4H), 4.13 (s, 3 Н), 7.41-7.48 (m, 2H), 7.537.55 (m, 1H), 7.65-7.73 (m, 3 Н), 7.85-7.88 (m, 1H), 7.97-7.99 (m, 1H); m/z 293 (М+Н)+; время удерживания равно 0,30. Пример 8. Циклопропил-(4-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-ил)метанон. Циклопропил-4-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-ил)метанон. Способ С, стадия с. К раствору циклопропанкарбоновой кислоты (0,06 г, 0,68 ммоль) в ацетонитриле (8 мл) добавляли CDI (0,11 г, 0,68 ммоль) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение 5 ч. Добавляли 1-метил-2-(3-пиперазин-1-илфенил)-1 Н-бензоимидазол дигидрохлорид (0,20 г, 0,55 ммоль) и получающийся в результате раствор нагревали при 65 С в течение ночи. Реакционную смесь затем охлаждали до комнатной температуры, растворитель удаляли при пониженном давлении. Неочищенный продукт перерастворяли в дихлорметане (15 мл) и промывали насыщенным раствором NaHCO3 (23 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии (элюент: циклогексан:EtOAc, градиент от 100% циклогексана до 100% этилацетата) с получением 0,17 г (85%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, CD3OD):0.82-0.84 (2 Н, m), 0.89-0.91 (2 Н, m), 1.992.00 (1 Н, m), 3.24-3.28 (2 Н, m), 3.29-3.35 (2 Н, m), 3.67-3.78 (2 Н, m), 3.85 (3 Н, br s), 3.89-3.95 (2 Н, m), 7.167.22 (1 Н, m), 7.28-7.35 (3 Н, m), 7.44 (1 Н, t), 7.51-7.53 (1 Н, m), 7.66-7.68 (1 Н, m); m/z 361 (М+Н)+; время удерживания равно 1,75. Пример 9. 2-(3-[4-Циклопропилметилпиперазин-1-ил]фенил)-1-метил-1 Н-бензоимидазол формиат. 2-3-[4-Циклопропилметилпиперазин-1-ил]фенил)-1-метил-1 Н-бензоимидазол. Способ С,стадия(CH3)2S.BH3 в THF (0,11 мл, 1,18 ммоль) и получающуюся в результате суспензию нагревали при 65 С в течение ночи. Реакционный раствор затем охлаждали до комнатной температуры, растворитель удаляли при пониженном давлении и к остатку добавляли 1 н. HCl (3 мл), а затем нагревали при 100 С в течение 6 ч. Реакционную смесь охлаждали до комнатной температуры, затем подщелачивали путем добавления по каплям 15% NaOH и экстрагировали EtOAc (310 мл). Объединенные органические слои сушили надNa2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали путем препаративной HPLC с получением 0,05 г (32%) указанного в заголовке соединения. 1 НЯМР (400 МГц, CD3OD):0.42-0.46 (2 Н, m), 0.74-0.78 (2 Н, m), 1.14-1.17 (1 Н, m), 3.06-3.08 (2 Н, m), 3.47m/z 347 (М+Н)+; время удерживания равно 0,65. Пример 10. 3-4-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбонилморфолин-4 карбоновой кислоты трет-бутиловый эфир. 3-4-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбонилморфолин-4-карбоновой кислоты трет-бутиловый эфир. Способы D, Е, стадия а. К раствору морфолин-3,4-дикарбоновой кислоты-4-трет-бутилового эфира(0,16 г, 0,71 ммоль) в ацетонитриле (8 мл) добавляли CDI (0,16 г, 0,71 ммоль) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение ночи. Добавляли 1-метил-2-(3 пиперазин-1-илфенил)-1 Н-бензоимидазол дигидрохлорид (0,20 г, 0,55 ммоль) (полученный, как описано в способе С) и DIPEA (0,19 мл, 1,10 ммоль) и получающийся в результате раствор нагревали при 65 С в течение ночи. Реакционную смесь затем охлаждали до комнатной температуры, растворитель удаляли,неочищенный продукт растворяли в дихлорметане (15 мл) и промывали насыщенным раствором NaHCO3(23 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии (элюент: градиент от циклогексана до циклогексан:EtOAc 2:1) с получением 0,11 г (40%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц,CD3OD):1.44 (9 Н, br s), 3.30-3.89 (16 Н, br m), 4.02-4.18 (2H, m), 7.20-7.26 (2H, m), 7.30-7.37 (3 Н, m),- 15 017918 7.47 (1H, t), 7.55-7.57 (1H, m), 7.67-7.68 (1H, m); m/z 506 (M+H)+; время удерживания равно 2,24. Пример 11. (4-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-ил)морфолин-3-илметанон гидрохлорид.(3 мл) перемешивали при комнатной температуре в течение ночи. Осадок фильтровали, промывали диэтиловым эфиром, твердое вещество растворяли в воде (15 мл) и промывали дихлорметаном (23 мл). Органический слой затем сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 0,09 г (79%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, CD3OD):2.81(3 Н, m), 7.32 (1 Н, t), 7.38-7.40 (1 Н, d), 7.56-7.58 (1 Н, d); m/z 406 (М+Н)+; время удерживания равно 0,52. Пример 12. 4-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-ил-(4-метилморфолин-3 ил)метанон. 4-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-ил-(4-метилморфолин-3-ил)метанон. Способ D, стадия с. К суспензии 4-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1 илморфолин-3-илметанона (0,08 г, 0,20 ммоль) и триацетоксиборгидрида натрия (NaB(ОАс)3 Н) (0,17 г,0,79 ммоль) в дихлорэтане (2 мл) добавляли формальдегид 37% (0,64 мл, 7,90 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, гасили насыщенным раствором Na2CO3(3 мл) и затем экстрагировали дихлорметаном (42 мл). Объединенные органические слои сушили надNa2SO4, фильтровали и концентрировали при пониженном давлении с получением желтого масла (0,08 г,97%). 1 Н-ЯМР (МГц, CD3OD):2.24 (3 Н, s), 2.33-2.39 (1 Н, m), 2.78-2.82 (1 Н, m), 3.25-3.28 (2 Н, m), 3.303.32 (2 Н, m), 3.34-3.37 (1 Н, m), 3.47-3.52 (1 Н, m), 3.62-3.68 (1 Н, m), 3.76-3.85 (4 Н, m), 3.87 (3 Н, s), 3.943.96 (2 Н, m), 7.18-7.24 (2 Н, m), 7.28-7.36 (3 Н, m), 7.44-7.48 (1 Н, m), 7.53-7.55 (1 Н, m), 7.66-7.69 (1 Н, m);m/z 420 (М+Н)+; время удерживания равно 1,12. Пример 13. 1-Метил-2-3-[4-(4-метилморфолин-3-илметил)пиперазин-1-ил]фенил-1 Нбензоимидазол формиат. 1-Метил-2-3-[4-(4-метилморфолин-3-илметил)пиперазин-1-ил]фенил)-1 Н-бензоимидазол. Способ D, стадия d. К раствору 4-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-ил-(4 метилморфолин-3-ил)метанона (0,06 г, 0,16 ммоль) в безводном THF (2 мл) при комнатной температуре в атмосфере азота добавляли алюмогидрид лития (LiAlH4) (0,02 г, 0,47 ммоль). Реакционную смесь нагревали путем кипячения с обратным холодильником в течение 2 ч и с дополнительным количеством LiAlH4(0,01 г, 0,24 ммоль). Через 30 мин раствор охлаждали до комнатной температуры, гасили 10% NaOH (3 мл) и затем экстрагировали дихлорметаном (33 мл). Объединенные органические слои сушили надNa2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество затем очищали путем препаративной HPLC с получением 0,03 г (41%) указанного в заголовке соединения. 1 Н-ЯМРm/z 406 (М+Н)+; время удерживания равно 0,92. Пример 14. 3-4-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-илметилморфолин-4 карбоновой кислоты трет-бутилового эфира формиат. 3-4-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-илметилморфолин-4-карбоновой кислоты трет-бутиловый эфир. Способ Е, стадия е. К раствору 3-4-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1 карбонилморфолин-4-карбоновой кислоты трет-бутилового эфира (0,21 г, 0,42 ммоль) в безводном THF(3 мл) при комнатной температуре в атмосфере азота добавляли алюмогидрид лития (LiAlH4) (0,05 г, 1,27 ммоль). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 2 ч. Раствор затем охлаждали до комнатной температуры, гасили 15% NaOH и затем экстрагировали дихлорметаном(310 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество затем очищали путем препаративной HPLC с получением 0,09 г (43%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, CD3OD):1.48 (9 Н, s), 3.02-3.25 (6 Н,m), 3.39-3.46 (6 Н, m), 3.55-3.59 (1 Н, m), 3.76-3.90 (6 Н, m), 4.15-4.35 (1 Н, m), 7.20-7.24 (2 Н, m), 7.64-7.41(3 Н, m), 7.45-7.49 (1 Н, m), 7.58-7.60 (1 Н, m), 7.69-7.71 (1 Н, m); m/z 492 (М+Н)+; время удерживания равно 1,45. Пример 15. 1-Метил-2-[3-(4-морфолин-3-илметилпиперазин-1-ил)фенил]-1 Н-бензоимидазол. 1-Метил-2-[3-(4-морфолин-3-илметилпиперазин-1-ил)фенил]-1 Н-бензоимидазол. Способ Е, стадия f. К раствору 3-4-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1 илметилморфолин-4-карбоновой кислоты трет-бутилового эфира (0,07 г, 0,15 ммоль) в дихлорметане (1 мл) добавляли 2 М HCl в Et2O (3 мл) и получающийся в результате раствор перемешивали в течение 7 ч при комнатной температуре. Образующийся в результате осадок отфильтровывали, промывали диэтило- 16 017918 вым эфиром и затем очищали путем препаративной HPLC. Фракции подщелачивали K2CO3 для того,чтобы избежать потенциального формилирования, и затем растворитель удаляли при пониженном давлении. Неочищенное вещество затем выделяли с дихлорметаном (15 мл) и органический слой промывали водой (34 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 0,02 г (37%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, DMSO):2.11-2.24 (2 Н, m),2.48-2.58 (3 Н, m), 2.71-2.76 (2 Н, m), 2.83-2.89 (1 Н, m), 2.98-3.06 (1 Н, m), 3.19-3.24 (4 Н, m), 3.28-3.38 (2 Н,m), 3.62-3.71 (2 Н, m), 3.86 (3 Н, s), 7.08-7.12 (1 Н, m), 7.17-7.30 (4 Н, m), 7.35-7.40 (1 Н, m), 7.58-7.67 (2 Н,m). m/z 392 (М+Н)+; время удерживания равно 0,77. Пример 16. (2-4-[3-(1 Н-Бензоимидазол-2-ил)фенил]пиперазин-1-ил-2-оксоэтил)карбаминовой кислоты трет-бутиловый эфир.(2-4-[3-(1 Н-Бензоимидазол-2-ил)фенил]пиперазин-1-ил-2-оксоэтил)карбаминовой кислоты третбутиловый эфир. Способ F, стадия а. К раствору N-(трет-бутоксикарбонил)глицина (0,13 г, 0,74 ммоль) в ацетонитриле (8 мл) добавляли CDI (0,12 г, 0,68 ммоль) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение ночи. Затем добавляли 2-(3-пиперазин-1-илфенил)-1 Нбензоимидазол дигидрохлорид (0,20 г, 0,57 ммоль) (полученный, как описано в способе В) и DIPEA (0,20 мл, 1,14 ммоль) и получающийся в результате раствор нагревали при 65 С в течение ночи. Реакционную смесь затем охлаждали до комнатной температуры, растворитель удаляли, неочищенное вещество выделяли с дихлорметаном (15 мл) и промывали насыщенным раствором NaHCO3 (23 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии (элюент: циклогексан:EtOAc/2:1) с получением 0,14 г (56%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, CD3OD):1.46 (9 Н, s), 3.29-3.36 (4 Н, m), 3.69-3.71 (2 Н, m),3.67-3.80 (2 Н, m), 4.00 (2 Н, s), 7.13-7.16 (1 Н, m), 7.24-7.27 (2 Н, m), 7.42 (1 Н, t), 7.56-7.62 (3 Н, m), 7.73-7.74(1 Н, m); m/z 436 (М+Н)+; время удерживания равно 2,13. Пример 17. 2-4-[3-(1 Н-Бензоимидазол-2-ил)фенил]пиперазин-1-ил]этиламин. 2-4-[3-(1 Н-Бензоимидазол-2-ил)фенил]пиперазин-1-ил]этиламин. Способ F, стадия b. К раствору (2-4-[3-(1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-ил-2 оксоэтил)карбаминовой кислоты трет-бутилового эфира (0,12 г, 0,28 ммоль) в безводном THF (3,50 мл) добавляли (СН 3)2S.ВН 3 в THF (0,06 мл, 0,69 ммоль) и получающуюся в результате суспензию нагревали при 65 С в течение ночи. Реакционную смесь затем охлаждали до комнатной температуры, растворитель удаляли при пониженном давлении и к остатку добавляли 1 н. HCl (3 мл), а затем нагревали при 100 С в течение 6 ч. Реакционную смесь охлаждали до комнатной температуры, затем подщелачивали путем добавления по каплям 15% NaOH, полученное белое твердое вещество фильтровали и промывали водой и диэтиловым эфиром с получением 0,06 г (73%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц,CD3OD):2.60-2.70 (6 Н, m), 2.98-3.01 (2 Н, m), 3.30-3.34 (6 Н, m), 7.10 (1 Н, d), 7.24-7.26 (2 Н, m), 7.39 (1 Н,t), 7.52 (1 Н, d), 7.53-7.60 (2 Н, m), 7.73 (1 Н, br s); m/z 322 (М+Н)+; время удерживания равно 0,22. Пример 7. 1-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]пиперидин-4-ол. 8-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-1,4-диокса-8-азаспиро[4,5]декан. Использовали тот же самый способ, который использовали и в способе А, начиная с 1,4-диокса-8 азаспиро[4,5]декан гидрохлорида. Получали 1,25 г указанного в заголовке соединения (77%, последняя стадия). 1 Н-ЯМР (400 МГц, DMSO):1.70 (4 Н, br s), 3.31-3.34 (4 Н, m), 3.90 (4 Н, br s), 7.09-7.12 (1 Н, m),7.19-7.21 (2 Н, m), 7.37-7.40 (2 Н, m), 7.60 (2 Н, brs), 12.62 (1H, s). 1-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенилпиперидин-4-он. Способы G, H, стадия а. К суспензии 8-[3-(1 Н-бензоимидазол-2-ил)-4-хлорфенил]-1,4-диокса-8 азаспиро[4,5]декана (2,00 г, 5,42 ммоль) в воде (50 мл) по каплям добавляли H2SO4 (2,50 мл) и получающийся в результате раствор перемешивали в течение 72 ч при комнатной температуре. Раствор нейтрализовали Na2CO3 и получающееся масло затвердевало при помещении в воду. Осадок фильтровали и сушили с получением 1,60 г (90%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, DMSO):2.43 (4 Н,t), 3.68 (4 Н, t), 7.22-7.25 (1 Н, m), 7.33-7.36 (2 Н, m), 7.46-7.50 (2 Н, m), 7.69-7.71 (2 Н, m). Пример 18. 1-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]пиперидин-4-ол. 1-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]пиперидин-4-ол. Способ G, стадия b. К суспензии 1-[3-(1 Н-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-4-она (0,05 г, 0,17 ммоль) в метаноле (50 мл) добавляли боргидрид натрия (NaBH4) (0,01 г, 0,17 ммоль), получающуюся в результате смесь перемешивали в течение 1 ч при комнатной температуре и затем гасили несколькими каплями воды. Растворитель удаляли при пониженном давлении, неочищенное вещество растворяли в EtOAc (5 мл) и фильтровали, затем органическую фазу упаривали при пониженном давлении с получением 0,02 г (36%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, DMSO):1.43-1.45 (2 Н,m), 1.78-1.80 (2 Н, m), 2.89-2.94 (2 Н, m), 3.56-3.64 (3 Н, m), 4.68 (1 Н, d), 7.06-7.08 (1 Н, m), 7.18-7.21 (2 Н,m), 7.33-7.40 (2 Н, m), 7.52 (1 Н, d), 7.66 (1 Н, d), 12.59 (1 Н, s); m/z 328 (M+H)+; время удерживания равно 1,55. Пример 19. 2-[5-(4-Азепан-1-илпиперидин-1-ил)-2-хлорфенил]-1 Н-бензоимидазол. 2-[5-(4-Азепан-1-илпиперидин-1-ил)-2-хлорфенил]-1 Н-бензоимидазол. Способ Н, стадия с. К суспензии 1-[3-(1 Н-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-4-она (0,10 г, 0,31 ммоль) в дихлорметане (2 мл) добавляли азепан (0,05 мл, 0,46 ммоль). После перемешивания в течение 30 мин при комнатной температуре добавляли каплю уксусной кислоты и через 1 ч добавляли дополнительную аликвоту азепана (0,05 мл, 0,46 ммоль), а затем триацетоксиборгидрид натрия(NaB(OAc)3H) (0,10 г, 0,46 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем растворитель удаляли при пониженном давлении и неочищенный продукт растворяли в дихлорметане (10 мл). К раствору добавляли PS-изоцианатную смолу (PS-NCO) (Argonaut, загрузка 0,93 ммоль/г), затем смесь встряхивали при комнатной температуре в течение ночи и фильтровали. Растворитель удаляли при пониженном давлении с получением твердого остатка, который промывалиEtOAc и Et2O, а затем сушили с получением желаемого продукта (0,07 г, 58%). 1 Н-ЯМР (400 МГц,DMSO):1.45-1.50 (9 Н, m), 1.74 (2 Н, d), 2.61-2.74 (6 Н, m), 3.31 (2 Н, s), 3.78 (2 Н, d), 7.06-7.08 (1 Н, m),7.20-7.22 (2 Н, m), 7.33-7.38 (2 Н, m), 7.52 (1 Н, d), 7.66 (1 Н, d), 12.60 (1H, s); m/z 409 (М+Н)+; время удерживания равно 1,32. Пример 20. 1-(4-4-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбонил[1,4]диазепан-1-ил)этанон. 2-(3-Бромфенил)-1 Н-бензоимидазол. Способы I, J, K, стадия а. О-фенилендиамин (81,8 г, 756,6 ммоль) и щавелевую кислоту (3,40 г, 37,8 ммоль) полностью растворяли в смеси этанол/вода 1:1 (2L), предварительно нагретой до 80 С. Затем к раствору по каплям добавляли 3-бромбензальдегид (44,1 мл, 378,3 ммоль). Реакционную смесь перемешивали в течение ночи при 70 С и оставляли на открытом воздухе на двое суток. Твердое вещество отфильтровывали и растирали с метанолом (150 мл) с получением продукта в виде бледно-желтого твердого вещества (27,50 г). 3,8 г продукта выделяли из маточных растворов. Общий выход 31,30 г (30%). 1 НЯМР (400 МГц, DMSO):7.24 (2 Н, m), 7.54 (2 Н, m), 7.70 (m, 2 Н), 8.19 (1 Н, m), 8.37 (1 Н, t), 13.2 (1 Н, s);m/z 273 (М+Н)+; время удерживания равно 8,60. 2-(3-Бромфенил)-1-дифторметил-1 Н-бензоимидазол. Способы I, J, K, стадия b. В автоклав помещали 2-(3-бромфенил)-1 Н-бензоимидазол (14,10 г, 51,6 ммоль) 50% водн. NaOH (8,3 мл, 154,8 ммоль), ТЕВАС (триэтилбензиламмония хлорид) (0,58 г, 2,5 ммоль) и THF (120 мл). Смесь перемешивали при комнатной температуре в течение 2 ч, затем хлордифторметан продували до полного превращения исходного вещества (2 бар (200 кПа), приблизительно 3 ч). Смесь разбавляли DCM (дихлорметан) (100 мл), органическую фазу сливали и сушили над Na2SO4. Растворитель выпаривали при пониженном давлении с получением темно-красного масла, которое очищали с помощью флэш-хроматографии на диоксиде кремния versaflash (1 г продукта/30 г диоксида кремния)(элюент: циклогексан:AcOEt градиент от циклогексан 95:AcOEt 5 до циклогексан 7:AcOEt 3) с получением 14,30 г указанного в заголовке соединения (86%). 1 Н-ЯМР (400 МГц, DMSO):7.45 (2 Н, m), 7.59(1H, t); 7.75 (1H, m), 7.83 (2 Н, m), 7.95 (1 Н, m), 8.09 (1 Н, s); m/z 323 (М+Н)+; время удерживания равно 8,60. 1-(4-4-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбонил-[1,4]диазепан-1 ил)этанон. Способ J, стадия с. 2-(3-Бромфенил)-1-дифторметил-1 Н-бензоимидазол (0,10 г, 0,31 ммоль), карбонат цезия (0,14 г, 0,43 ммоль), рац-2,2' бис-(дифенилфосфино)-1,1'-бинафтил (BINAP) (0,007 г, 0,01 ммоль) и трис-(дибензилиденацетон)дипалладий(0) (0,004 г) помещали в 7 мл герметизируемый сосуд и продували путем повторяющихся циклов азот/вакуум в течение 10 мин. Добавляли безводный толуол(0,6 мл) и N-ацетилгомопиперазин (0,05 мл, 0,37 ммоль) и реакционную смесь нагревали при 85 С в течение 24 ч при перемешивании. Реакционную смесь охлаждали до комнатной температуры и фильтровали через картридж SCX (2 г), (элюент: DCM/метанол 1:1). Органический слой концентрировали при пониженном давлении и очищали путем препаративной HPLC с получением 58 мг указанного в заголовке соединения (48%). 1 Н-ЯМР (400 МГц, CD3OD):1.95-2.08 (5 Н, m), 3.48-3.55 (2 Н, m), 3.65-3.77 (4 Н, m),3.80-3.84 (2 Н, m), 6.93-6.95 (1 Н, m), 7.04-7.11 (2 Н, m), 7.41-7.67 (4 Н, m), 7.73-7.82 (2 Н, m); m/z 385(М+Н)+; время удерживания равно 3,30. Пример 21. 1-Дифторметил-2-(3-пиперазин-1-илфенил)-1 Н-бензоимидазол гидрохлорид. 4-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбоновой кислоты третбутиловый эфир. Способ K, стадия d. 2-(3-Бромфенил)-1-дифторметил-1 Н-бензоимидазол (1,00 г, 3,10 ммоль), пиперазин-1-карбоновой кислоты трет-бутиловый эфир (0,75 г, 4,02 ммоль) и карбонат цезия (5,04 г, 15,48 ммоль) помещали в сухую круглодонную колбу объемом 250 мл и продували путем повторящихся циклов азот/вакуум в течение 30 мин и добавляли безводный толуол (50 мл). Одновременно ацетат палладия(139 мг, 0,62 ммоль) и рац-2,2' бис-(дифенилфосфино)-1,1'-бинафтил (BINAP) (0,58 г, 0,30 ммоль) помещали в сухую 100 мл круглодонную колбу и продували путем повторяющих циклов азот/вакуум в течение 30 мин. Добавляли безводный толуол (50 мл) и эту суспензию перемешивали в течение 10 мин, а затем добавляли в первую круглодонную колбу. Реакционную смесь кипятили с обратным холодильником в течение ночи в атмосфере азота и охлаждали до комнатной температуры. Смесь фильтровали и нерастворимое вещество промывали EtOAc (320 мл). Органический раствор концентрировали при пониженном давлении и неочищенное вещество очищали с помощью флэш-хроматографии (элюент: циклогексан:AcOEt градиент от 100% циклогексан до циклогексан 5: AcOEt 1) с получением 1,10 г указанного в заголовке соединения (83%). 1 Н-ЯМР (400 МГц, DMSO):1.41 (9 Н, s), 3.19-3.22 (4 Н, m), 3.46-3.48(4 Н, m), 7.08-7.10 (1 Н, m), 7.20-7.23 (2 Н, m), 7.37-7.46 (3 Н, m), 7.71-7.80 (3 Н, m); m/z 429 (М+Н)+; время удерживания равно 4,53. 1-Дифторметил-2-(3-пиперазин-1-илфенил)-1 Н-бензоимидазол. Способ K, стадия е. К раствору 4-[3-(1-дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1 карбоновой кислоты трет-бутилового эфира (1,10 г, 2,57 ммоль) в DCM (2 мл) добавляли 2 М HCl в Et2O(11 мл) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение ночи. Осадок фильтровали и получающуюся в результате соль растирали с Et2O (320 мл) с получением 1,02 г (100%) указанного в заголовке соединения в виде соли гидрохлорида. 1 Н-ЯМР (400 МГц, DMSO):3.21 (4 Н, bs), 3.47-3.50 (4 Н, m), 7.11-7.20 (1 Н, m), 7.25-7.30 (2 Н, m), 7.41-7.51 (3 Н, m), 7.74-8.02 (3 Н, m),9.35 (2 Н, bs); m/z 329 (М+Н)+; время удерживания равно 1,77. Пример 22. 1-4-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-ил-2 метилпропан-1-он. 1-4-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-ил-2-метилпропан-1-он. Способ K, стадия f. CDI (0,05 г, 0,32 ммоль) добавляли к раствору изомасляной кислоты (0,029 г,0,32 ммоль) в ацетонитриле (2,50 мл). Получающийся в результате раствор перемешивали при комнатной температуре в течение 5 ч. Добавляли 1-дифторметил-2-(3-пиперазин-1-илфенил)-1 Н-бензоимидазол(0,10 г, 0,27 ммоль) и N,N-диизопропилэтиламин (DIPEA) (0,05 мл, 0,27 ммоль) и реакционную смесь нагревали при 65 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры, растворитель удаляли и добавляли DCM (3 мл). Органический раствор промывали водой (22 мл) и насыщенным раствором Na2CO3 (22 мл). Органический слой концентрировали при пониженном давлении и неочищенное вещество очищали с помощью флэш-хроматографии (элюент: циклогексан:AcOEt градиент от 100% циклогексана до циклогексан 3: AcOEt 1) с получением 0,067 г указанного в заголовке соединения (61%). 1 Н-ЯМР (400 МГц, CDCl3):1.09 (6 Н, d), 2.77 (1 Н, septuplet), 3.21-3.29 (4 Н, m), 3.64-3.75 (4 Н,m), 7.04-7.07 (2 Н, m), 7.10-7.41 (5 Н, m), 7.70-7.73 (1 Н, m), 7.77-7.81 (1 Н, m); m/z 399 (М+Н)+; время удерживания равно 3,68. Пример 23. 4-4-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1 карбонилпиперидин-1-карбоновой кислоты трет-бутиловый эфир. 4-4-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбонил)пиперидин-1-карбоновой кислоты трет-бутиловый эфир. Способ K, стадия g. CDI (0,053 мг, 0,32 ммоль) добавляли к раствору пиперидин-1,4-дикарбоновой кислоты моно-трет-бутилового эфира (0,074 г, 0,32 ммоль) в ацетонитриле (2,50 мл). Получающийся в результате раствор перемешивали при комнатной температуре в течение 5 ч. Добавляли 1-дифторметил-2-(3-пиперазин-1-илфенил)-1 Н-бензоимидазол (0,10 г, 0,27 ммоль) иN,N-диизопропилэтиламин (DIPEA) (0,05 мл, 0,27 ммоль) и реакционную смесь нагревали при 65 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры, растворитель удаляли и добавляли DCM (3 мл). Органический раствор промывали водой (22 мл) и насыщенным раствором Na2CO3(22 мл). Органический слой концентрировали при пониженном давлении и неочищенное вещество очищали с помощью флэш-хроматографии (элюент: циклогексан:AcOEt градиент от 100% циклогексана до циклогексан 3: AcOEt 1) с получением 0,067 г указанного в заголовке соединения (70%). 1 Н-ЯМР (400 МГц, CDCl3):1.39 (9 Н, s), 1.60-1.74 (4 Н, m), 2.57-2.77 (3 Н, m), 3.21-3.26 (4 Н, m), 3.64-3.74 (4 Н, m), 4.10(2 Н, m), 7.04-7.07 (2 Н, m), 7.11-7.41 (5 Н, m), 7.71-7.73 (1 Н, m), 7.78-7.82 (1 Н, m); m/z 540 (М+Н)+; время удерживания равно 4,25. Пример 24. 4-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-илпиперидин-4 илметанон. 4-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-илпиперидин-4-илметанон. Способ K, стадия h. К раствору 4-4-[3-(1-дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперазин 1-карбонилпиперидин-1-карбоновой кислоты трет-бутилового эфира (0,075 г, 0,14 ммоль) в DCM (0,5 мл) добавляли 2 М HCl в Et2O (2,5 мл) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение 6 ч. Осадок фильтровали и растирали с Et2O (320 мл). Твердое вещество очищали путем препаративной HPLC. Фракции HPLC нейтрализовали K2CO3 и концентрировали. Добавляли DCM (5 мл) и воду (5 мл), органическую фазу отделяли и концентрировали с получением 44 мг указанного в заголовке соединения (72%). 1 Н-ЯМР (400 МГц, CDCl3):1,82-2.15 (4 Н, m), 2.72-2.86 (1 Н, m),3.09-3.34 (6 Н, m), 3.48-3.64 (2 Н, m), 3.63-3.83 (4 Н, m), 7.09-7.15 (2H, m), 7.29-7.30 (2H, m), 7.37-7.47 (3H,m), 7.75-7.78 (1H, m), 7.81-7.85 (1H, m); m/z 440 (M+H)+; время удерживания равно 2,03. Пример 25. 1-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты этиловый эфир. 1-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты этиловый эфир. Способ I, стадия i. Тот же самый способ, как описанный в способе K, стадия d, но начиная с пиперидин-4-карбоновой кислоты этилового эфира вместо пиперазин-1-карбоновой кислоты трет-бутилового эфира. Количественный выход. 1 Н-ЯМР (400 МГц, CD3OD):1.26 (3 Н, t), 1.77-1.87 (2 Н, m), 2.01-2.05(2 Н, m), 2.50-2.57 (1 Н, m), 2.88-2.94 (2 Н, m), 3.76-3.81 (2 Н, m), 4.12-4.17 (2 Н, q), 7.09-7.11 (1 Н, m), 7.247.26 (1 Н, m), 7.29-7.30 (1 Н, m), 7.41-7.81 (6 Н, m); m/z 400 (М+Н)+; время удерживания равно 4,25. Пример 26. 1-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил-[4 метилпиперазин-1-ил)метанон. 1-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты. Способ I, стадия I. Порошкообразный NaOH (0,24 г, 6,02 ммоль) добавляли к раствору 1-[3-(1 дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты этилового эфира (1,20 г,3,01 ммоль) в этанол.вода 3:1 и реакционную смесь нагревали при 80 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры и органический растворитель концентрировали путем упаривания при пониженном давлении. Получающийся в результате раствор нейтрализовали путем добавления по каплям 6 н. HCl. Образовывался бледно-коричневый осадок, и его отфильтровывали и сушили с получением 0,80 г указанного в заголовке соединения (72%). 1 Н-ЯМР (400 МГц, CD3OD):1.83-1.93 (2 Н,m), 2.06-2.11 (2 Н, m), 2.52-2.59 (1 Н, m), 3.14-3.17 (2 Н, m), 3.71-3.76 (2 Н, m), 7.33-7.35 (1 Н, m), 7.40-7.42(2 Н, m), 7.44-7.78 (6 Н, m); m/z 372 (М+Н)+; время удерживания равно 3,25. 1-[3-(1-Дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил-(4-метилпиперазин-1 ил)метанон. Способ I, стадия m. HATU (2-(7-аза-1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат) (0,11 г, 0,30 ммоль) и диизопропилэтиламин (DIPEA) (0,09 мл, 0,54 ммоль) добавляли к суспензии 1-[3-(1-дифторметил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты (0,10 г, 0,27 ммоль) в дихлорметане при комнатной температуре. Реакционную смесь нагревали при 35 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры и промывали насыщенным растворомNa2CO3 (24 мл) и водой (13 мл). Органический слой концентрировали при пониженном давлении и неочищенное вещество очищали с помощью флэш-хроматографии (элюент: циклогексан:AcOEt градиент от циклогексан 1: AcOEt 1 до 100% AcOEt и наконец AcOEt:2,0 н. аммиаком в метаноле 5:1) с получением 0,025 г указанного в заголовке соединения (20%). 1 Н-ЯМР (400 МГц, CD3OD):1.79-1.88 (4 Н, m),2.31 (3 Н, s), 2.39-2.48 (4 Н, m), 2.80-2.92 (3 Н, m), 3.60-3.68 (4 Н, m), 3.85-3.88 (2 Н, m), 7.08-7.10 (1 Н, m),7.20-7.25 (1 Н, m), 7.28-7.29 (1 Н, m), 7.40-7.81 (6 Н, m); m/z 454 (М+Н)+; время удерживания равно 2,00. Пример 27. 4-[4-Фтор-3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-илпирролидин-1 илметанон.(DMAP) (0,03 г, 0,2 ммоль) помещали в 100 мл круглодонную колбу. Добавляли безводный DMF (60 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении, затем неочищенное вещество разбавляли DCM (15 мл) и 0,4 М растворомNa2CO3 (15 мл). Образовывался белый осадок и его отфильтровывали, вновь промывали DCM (10 мл) и сушили с получением 4,96 г указанного в заголовке соединения. Растворы собирали, органическую фазу отделяли от водной фазы и концентрировали при пониженном давлении: образовывался еще осадок, и его отфильтровывали, промывали DCM (3 мл) с получением еще 0,68 г желаемого соединения. Общий выход 81%. 1 Н-ЯМР (400 МГц, DMSO):4.84-5.14 (2 Н, m), 6.54-6.59 (1 Н, m), 6.73-6.76 (1 Н, m), 6.93-6.97(1 Н, m), 7.21-7.24 (1 Н, m), 7.31-7.35 (1 Н, m), 7.72-7.76 (1 Н, m), 9.62-9.64 (1 Н, m); m/z 308/310 (М+Н)+; время удерживания равно 1,87. 2-(5-Бром-2-фторфенил)-1 Н-бензоимидазол. Способы L, М, стадия b. Суспензию N-(2-аминофенил)-5-бром-2-фторбензамида (5,64 г, 18,3 ммоль) в уксусной кислоте (20 мл) нагревали при 60 С. Через 30 мин суспензия превращалась в раствор,и ее оставляли на ночь. Растворитель удаляли при пониженном давлении, и остаток растирали с DCM (10 мл) и отфильтровывали. Осадок промывали DCM (310 мл) и сушили с получением 4,76 г указанного в заголовке соединения (89%). 1 Н-ЯМР (400 МГц, DMSO):7.24-7.27 (2 Н, m), 7.43-7.48 (1 Н, m), 7.63-7.66(2 Н, m), 7.72-7.76 (1 Н, m), 8.33-8.35 (1 Н, m); m/z 290/292 (М+Н)+; время удерживания равно 1,85. 2-(5-Бром-2-фторфенил)-1-метил-1 Н-бензоимидазол. Способ L, стадия с. 60% суспензию NaH в минеральном масле (0,61 г, 25,5 ммоль) добавляли к раствору 2-(5-бром-2-фторфенил)-1 Н-бензоимидазола (3,70 г, 12,76 ммоль) в безводном THF (100 мл), реакционную смесь перемешивали при комнатной температуре в течение 30 мин, затем добавляли метилйо- 20 017918 дид (1,19 мл, 19,1 ммоль). Реакционную смесь оставляли перемешиваться в течение 2 ч, затем гасили водой (20 мл). Раствор концентрировали, затем добавляли AcOEt (30 мл). Органический раствор отделяли, промывали рассолом (10 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 3,80 г желаемого соединения (97%). 1 Н-ЯМР (400 МГц, CD3OD):3.76 (3 Н, s), 7.31-7.42 (3 Н,m), 7.59-7.61 (1 Н, m), 7.70-7.72 (1 Н, m), 7.78-7.82 (1 Н, m), 7.85-7.87 (1 Н, m); m/z 304/306 (М+Н)+; время удерживания равно 1,80. 4-[4-Фтор-3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-илпирролидин-1-илметанон. Способ L, стадия d. Следовали тому же самому способу, как способ, описанный на стадии с способаJ, но начиная с 2-(5-бром-2-фторфенил)-1-метил-1 Н-бензоимидазола и пиперазин-1-ил-пирролидин-1 илметанона. Неочищенную реакционную смесь очищали путем препаративной HPLC с получением 0,06 г указанного в заголовке соединения (44%). 1 Н-ЯМР (400 МГц, CD3OD):1.82-1.89 (4 Н, m), 3.20-3.23(4 Н, m), 3.40-3.42 (4 Н, m), 3.44-3.47 (4 Н, m), 3.77-3.78 (3 Н, m), 7.20-7.22 (1 Н, m), 7.24-7.26 (2 Н, m), 7.317.40 (2 Н, m), 7.57-7.59 (1 Н, m), 7.69-7.71 (1 Н, m); m/z 408 (М+Н)+; время удерживания равно 2,32. Пример 28. 4-[4-Фтор-3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбоновой кислоты трет-бутиловый эфир. 4-[4-Фтор-3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбоновой кислоты третбутиловый эфир. Способ L, стадия е. 2-(5-Бром-2-фторфенил)-1-метил-1 Н-бензоимидазол (2,10 г, 6,9 ммоль), пиперазин-1-карбоновой кислоты трет-бутиловый эфир (1,16 г, 8,3 ммоль), карбонат цезия (3,14 г, 9,6 ммоль) рац-2,2' бис-(дифенилфосфино)-1,1'-бинафтил (BINAP) (0,17 г, 0,28 ммоль) и трис(дибензилиденацетон)дипалладий(0) (0,06 г, 0,07 ммоль) помещали в сосуд Шленка и продували путем повторящихся циклов азот/вакуум в течение 10 мин. Добавляли безводный толуол (13 мл) и реакционную смесь перемешивали при 85 С в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли AcOEt (20 мл) и промывали водой (10 мл). Органический слой сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенную реакционную смесь очищали с помощью флэш-хроматографии (элюент: циклогексан:AcOEt градиент от 100% циклогексан до циклогексан 2:(2 Н, m), 7.56-7.58 (1 Н, m), 7.68-7.71 (1 Н, m); m/z 411 (М+Н)+; время удерживания равно 1,90. Пример 29. 2-Диметиламино-1-4-[4-фтор-3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1 илэтанон. 2-(2-Фтор-5-пиперазин-1-илфенил)-1-метил-1 Н-бензоимидазол. Способ L, стадия f. К раствору 4-[4-фтор-3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1 карбоновой кислоты трет-бутилового эфира (0,86 г, 1,71 ммоль) в DCM (2,0 мл) добавляли 2,0 М HCl вEt2O (10 мл) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение ночи. Осадок отфильтровывали и растирали с Et2O (315 мл) с получением указанного в заголовке соединения (количественный выход). 1 Н-ЯМР (400 МГц, DMSO):3.22-3.25 (4 Н, m), 3.44-3.47 (4 Н, m),3.94 (3 Н, s), 7.43-7.53 (3 Н, m), 7.60-7.67 (2 Н, m), 7.87-7.89 (1 Н, m), 8.01-8.03 (1 Н, m), 9.42 (2 Н, m); m/z 311(М+Н)+; время удерживания равно раздвоенный пик. 2-Диметиламино-1-(4-[4-фтор-3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-ил)этанон. Способ L, стадия g. Тот же самый способ, как используемый в способе L-стадия h. 1 Н-ЯМР (400 МГц, CD3OD):2.30 (6 Н, s), 3.18-3.25 (6 Н, m), 3.74-3.78 (7 Н, m), 7.21-7.28 (3 Н, m), 7.31-7.40 (2 Н, m),7.57-7.59 (1 Н, m), 7.69-7.71 (1 Н, m); m/z 396 (М+Н)+; время удерживания равно раздвоенный пик. Пример 30. (S)-2-4-[4-Фтор-3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбонилпирролидин-1-карбоновой кислоты трет-бутиловый эфир.(S)-2-4-[4-Фтор-3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперазин-1-карбонилпирролидин-1 карбоновой кислоты трет-бутиловый эфир. Способ L, стадия h. CDI (0,05 г, 0,31 ммоль) добавляли к раствору N-Boc-L-пролина (0,07 г, 0,31 ммоль) в ацетонитриле (2,50 мл). Получающийся в результате раствор перемешивали при комнатной температуре в течение 6 ч. Добавляли 2-(2-фтор-5-пиперазин-1-илфенил)-1-метил-1 Н-бензоимидазол (0,10 г, 0,26 ммоль) иN,N-диизопропилэтиламин (DIPEA) (0,09 мл, 0,52 ммоль) и реакционную смесь нагревали при 65 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры, растворитель удаляли при пониженном давлении и добавляли DCM (3 мл). Органический раствор промывали водой (22 мл) и насыщенным раствором Na2CO3 (22 мл). Органический слой концентрировали при пониженном давлении и неочищенное вещество очищали с помощью флэш-хроматографии (элюент: циклогексан:AcOEt градиент от циклогексан 2: AcOEt 1 до 100% AcOEt) с получением 0,067 г указанного в заголовке соединения(1 Н, m), 7.69-7.71 (1 Н, m); m/z 508 (M+H)+; время удерживания равно 2,63.(S)-2-4-[4-фтор-3-(1-метил-1 Н-бензоимидазол-2 ил)фенил]пиперазин-1-карбонилпирролидин-1-карбоновой кислоты трет-бутилового эфира (0,05 г, 0,10 ммоль) в DCM (0,5 мл) добавляли 2 М HCl в Et2O (2,0 мл) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении, затем добавляли воду (4,0 мл) и смесь вновь концентрировали с получением 0,04 г указанного в заголовке соединения в виде соли хлоргидрата (86%). 1 Н-ЯМР (400 МГц, CD3OD):1.852.04 (4 Н, m), 2.42-2.51 (1 Н, m), 3.60-3.81 (4 Н, m), 3.98-3.99 (3 Н, m), 3.23-3.36 (5 Н, m), 4.60-4.69 (1 Н, m),7.32-7.44 (3 Н, m), 7.62-7.68 (2 Н, m), 7.77-7.80 (1 Н, m), 7.89-7.93 (1 Н, m); m/z 408 (М+Н)+; время удерживания равно раздвоенный пик. Пример 32. 1-4-[3-(1 Н-Бензоимидазол-2-ил)-4-фторфенил]пиперазин-1-ил-2-диметиламиноэтанон. 2-(5-Бром-2-фторфенил)бензоимидазол-1-карбоновой кислоты трет-бутиловый эфир. Способ М, стадия I. 60% дисперсию NaH в минеральном масле (0,70 г, 17,34 ммоль) добавляли к раствору 2-(5-бром-2-фторфенил)-1 Н-бензоимидазола (3,87 г, 13,3 ммоль) в безводном THF (140 мл) в атмосфере азота при комнатной температуре. Через 10 мин добавляли раствор ди-трет-бутилдикарбоната(0,30 г, 13,3 ммоль) в безводном THF (50 мл) и реакционную смесь оставляли на 30 ч. Добавляли воду(40 мл) и реакционную смесь концентрировали при пониженном давлении. Добавляли AcOEt (30 мл) и органический слой отделяли. Водный раствор промывали AcOEt (210 мл). Органические слои собирали,сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенную реакционную смесь очищали с помощью флэш-хроматографии (элюент: циклогексан:AcOEt градиент от 100% циклогексана до циклогексан 10: AcOEt 1) до 4,7 г указанного в заголовке соединения (90%). 1 Н-ЯМР (400 МГц,CD3OD):1.44 (9 Н, s), 7.23-7.27 (1 Н, m), 7.41-7.50 (2 Н, m), 7.73-7.77 (2 Н, m), 7.84-7.86 (1 Н, m), 8.10-8.12(1 Н, m); m/z 392 (М+Н)+; время удерживания равно 2,88. 2-[5-(4-трет-Бутоксикарбонилпиперазин-1-ил)-2-фторфенил]бензоимидазол-1-карбоновой кислоты трет-бутиловый эфир/ Способ М, стадия m. 2-(5-Бром-2-фторфенил)бензоимидазол-1-карбоновой кислоты трет-бутиловый эфир (2,52 г, 6,5 ммоль), пиперазин-1-карбоновой кислоты трет-бутиловый эфир (1,56 г, 8,40 ммоль) и карбонат цезия (10,50 г, 32,20 ммоль) помещали в сухую 500 мл круглодонную колбу в атмосфере азота и добавляли безводный толуол (140 мл). Одновременно ацетат палладия (0,29 г, 1,30 ммоль) и BINAP(1,20 г, 1,93 ммоль) помещали в сухую 250 мл круглодонную колбу в атмосфере азота и добавляли безводный толуол (140 мл). Через 10 мин суспензию, содержащую BINAP и ацетат палладия, добавляли в круглодонную 500-мл колбу. Реакционную смесь нагревали при 85 С в течение ночи в атмосфере азота и охлаждали до комнатной температуры. Смесь фильтровали и нерастворимое вещество промывали EtOAc(320 мл). Органический раствор концентрировали при пониженном давлении и неочищенное вещество очищали с помощью флэш-хроматографии (элюент: циклогексан:AcOEt градиент от 100% циклогексана до циклогексан 4: AcOEt 1) с получением 2,48 г указанного в заголовке соединения (76%). 1 Н-ЯМР (400 МГц, DMSO): 1.36 (9 Н, s), 3.08-3.10 (4 Н, m), 3.43-3.46 (4 Н, m), 7.12-7.18 (1 Н, m), 7.20-7.25 (2 Н, m), 7.387.47 (2 Н, m), 7.76-7.78 (1 Н, m), 7.97-7.99 (1 Н, m); m/z 497 (М+Н)+; время удерживания равно 2,93. 2-(2-Фтор-5-пиперазин-1-илфенил)-1 Н-бензоимидазол. Способ М,стадияn. К раствору 2-[5-(4-трет-бутоксикарбонилпиперазин-1-ил)-2 фторфенил]бензоимидазол-1-карбоновой кислоты трет-бутилового эфира (0,45 г, 0,91 ммоль) в DCM (1,0 мл) добавляли 2 М HCl в Et2O (8,0 мл) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение ночи. Добавляли еще 2 М HCl в Et2O (5,0 мл) и реакционную смесь оставляли перемешиваться на 24 ч. Осадок отфильтровывали и растирали Et2O (315 мл) с получением 0,21 г указанного в заголовке соединения (55%). 1 Н-ЯМР (400 МГц, DMSO):3.26 (4 Н, bs), 3.49-3.51 (4 Н, m),7.36-7.40 (1 Н, m), 7.45-7.53 (3 Н, m), 7.80-7.84 (2 Н, m), 7.91-7.93 (1 Н, m), 9.28 (2 Н, bs); m/z 297 (М+Н)+; время удерживания равно раздвоенный пик. 1-4-[3-(1 Н-Бензоимидазол-2-ил)-4-фторфенил]пиперазин-1-ил-2-диметиламиноэтанон. Способ М, стадия о. CDI (0,052 г, 0,32 ммоль) добавляли к раствору N,N-диметилглицина (0,033 г,0,32 ммоль) в ацетонитриле (2,50 мл). Получающийся в результате раствор перемешивали при комнатной температуре в течение 5 ч. Затем добавляли 2-(2-фтор-5-пиперазин-1-илфенил)-1 Н-бензоимидазол (0,10 г, 0,27 ммоль) и N,N-диизопропилэтиламин (DIPEA) (0,11 мл, 0,6 ммоль) и реакционную смесь нагревали при 65 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли при пониженном давлении. DCM (2 мл) добавляли к неочищенному веществу, органический раствор промывали водой (24 мл) и насыщенным раствором Na2CO3 (24 мл) и концентрировали. Неочищенную реакционную смесь очищали с помощью флэш-хроматографии (элюент: MeOH:AcOEt градиент от 100% AcOEt до МеОН 0,6: AcOEt 4) с получением 0,049 г указанного в заголовке соединения (43%). Н-ЯМР (400 МГц, CD3OD):2.33 (6 Н, s), 3.22-3.29 (4 Н, m), 3.29 (2 Н, s), 3.77-3.79 (4 Н, m), 7.16-7.24 (2 Н,m), 7.26-7.31 (2 Н, m), 7.59-7.70 (2 Н, bp), 7.75-7.77 (1 Н, m); m/z 382 (М+Н)+; время удерживания равно раздвоенный пик. Пример 33. 4-4-[3-(1 Н-Бензоимидазол-2-ил)-4-фторфенил]пиперазин-1-илпиперидин-4 илметанон. 4-4-[3-(1 Н-Бензоимидазол-2-ил)-4-фторфенил]пиперазин-1-карбонилпиперидин-1-карбоновой кислоты трет-бутиловый эфир. Способ М, стадия р. CDI (0,047 г, 0,29 ммоль) добавляли к раствору пиперидин-1,4-дикарбоновой кислоты моно-трет-бутилового эфира (0,07 г, 0,29 ммоль) в ацетонитриле (2,50 мл). Получающийся в результате раствор перемешивали при комнатной температуре в течение 5 ч. Добавляли 2-(2-фтор-5 пиперазин-1-илфенил)-1 Н-бензоимидазол (0,10 г, 0,27 ммоль) и DIPEA (0,05 мл, 0,26 ммоль) и реакционную смесь нагревали при 65 С в течение ночи. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли при пониженном давлении. DCM (2 мл) добавляли к неочищенному веществу, органический раствор промывали водой (24 мл) и насыщенным раствором Na2CO3 (24 мл) и концентрировали. Неочищенную реакционную смесь очищали с помощью флэш-хроматографии (элюент: циклогексан:AcOEt градиент от 100% циклогексана до 100% AcOEt) с получением 0,08 г указанного в заголовке соединения (60%). 1 Н-ЯМР (400 МГц, CD3OD):1.45 (9 Н, s), 1.55-1.66 (2 Н, m), 1.72-1.76 (2 Н,m), 2.82-3.00 (3 Н, m), 3.21-3.31 (4 Н, m), 3.77-3.83 (4 Н, m), 4.07-4.17 (2 Н, m), 7.17-7.24 (2 Н, m), 7.26-7.31(2 Н, m), 7.55-7.72 (2 Н, m), 7.75-7.78 (1 Н, m). 4-4-[3-(1 Н-Бензоимидазол-2-ил)-4-фторфенил]пиперазин-1-илпиперидин-4-илметанон. Способ М, стадия q. К раствору 4-4-[3-(1 Н-бензоимидазол-2-ил)-4-фторфенил]пиперазин-1 карбонилпиперидин-1-карбоновой кислоты трет-бутилового эфира (0,08 г, 0,16 ммоль) в DCM (1 мл) добавляли 2 М HCl в Et2O (3,0 мл) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение 3 суток. Реакционную смесь концентрировали при пониженном давлении,добавляли воду (8 мл) и раствор концентрировали при пониженном давлении. Твердое вещество очищали путем препаративной HPLC. Фракции после HPLC нейтрализовали K2CO3 и концентрировали. Добавляли DCM (5 мл) и воду (5 мл), органическую фазу отделяли и концентрировали с получением 44 мг указанного в заголовке соединения (72%). 1 Н-ЯМР (400 МГц, CD3OD):1.70-1.80 (4 Н, m), 2.73-2.80 (2 Н, m),2.91-2.98 (1 Н, m), 3.13-3.17 (2 Н, m), 3.20-3.28 (4 Н, m), 7.15-7.32 (2 Н, m), 7.26-7.31 (2 Н, m), 7.62-7.65 (2 Н,m), 7.75-7.77 (1 Н, m); m/z 408 (М+Н)+; время удерживания равно раздвоенный пик. Пример 34. 2-Этокси-N-1-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-илацетамид. 2-(3-Бромфенил)-1 Н-бензимидазол. Способ N, стадия а. О-фенилендиамин (81,8 г, 756,6 ммоль) и щавелевую кислоту (3,4 г, 37,8 ммоль) полностью растворяли в EtOH-H2O /1:1 (2 л), предварительно нагретой до 80 С. Затем к раствору по каплям добавляли 3-бромбензальдегид (44,10 мл, 378,30 ммоль). Реакционную смесь перемешивали в течение ночи при 70 С на открытом воздухе. На следующий день твердое вещество отфильтровывали и растирали с МеОН (150 мл) с получением продукта в виде бледно-желтого твердого вещества (27,50 г). Из маточных растворов выделяли 3,8 г. Общий выход 31,30 г (30%). 1 Н-ЯМР (400 МГц, DMSO):7.24(2 Н, m), 7.54 (2 Н, m), 7.70 (m, 2 Н), 8.19 (1 Н, m), 8.37 (1 Н, t), 13.2 (1 Н, s); m/z 273 (M+H)+; время удерживания равно 8,60. 2-(3-Бромфенил)-1-метил-1 Н-бензимидазол. Способ N, стадия b. 2-(3-Бромфенил)-1 Н-бензимидазол (7,8 г, 28,6 ммоль) полностью ратворяли в безводном THF (300 мл), затем к прозрачному желтому раствору порциями добавляли NaH 60 мас./мас.%(1,49 г, 37,2 ммоль). Светло-коричневую суспензию перемешивали в течение 1 ч при к.т., затем по каплям добавляли CH3I (2,5 мл, 40,0 ммоль). Реакционную смесь перемешивали при к.т. в течение ночи. Реакционную смесь гасили H2O (300 мл) и экстрагировали EtOAc (2450 мл). Органические экстракты сушили над MgSO4, фильтровали и упаривали с получением соединения в виде желто-коричневого твердого вещества (7,4 г, 70%). 1 Н-ЯМР (400 МГц, DMSO):3.90 (3 Н, s), 7.30 (2 Н, m), 7.55 (1 Н, t), 7.64 (1 Н, d),7.70 (1 Н, d), 7.77 (1 Н, m), 7.88 (1 Н, m), 8.05 (1 Н, m); m/z=287 [M+H]+, время удерживания равно 7,70.(1-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-илкарбаминовой кислоты третбутиловый эфир. Способ N, стадия с. В четырехгорлую круглодонную колбу, высушенную в атмосфере Ar, помещали BINAP (5,85 г, 9,4 ммоль) и Pd(OAc)2 (1,41 г, 6,3 ммоль). Смесь перемешивали в течение 10 мин в потоке Ar, затем добавляли 300 мл безводного толуола. В то же время в еще одну четырехгорлую круглодонную колбу, высушенную в атмосфере Ar, помещали 2-(3-бромфенил)-1-метил-1 Н-бензимидазол,предварительно растворенный в 300 мл безводного толуола, (9,0 г, 31,3 ммоль), 4-N-ВОСаминопиперидин (8,18 г, 40,7 ммоль) и Cs2CO3 (50,99 г, 156,5 ммоль). Смесь перемешивали в течение приблизительно 10 мин, затем содержимое первой колбы добавляли во вторую. Реакционную смесь перемешивали в течение ночи при температуре дефлегмации. После охлаждения до комнатной температуры суспензию фильтровали через слой целита в фильтровальном тигле Гуча. Прозрачный раствор упаривали при пониженном давлении и остаток растирали в МТВЕ (метил-трет-бутиловом эфире) с получени- 23 017918 ем 7,1 г (55%) чистого продукта в виде не совсем белого твердого вещества. 1 Н-ЯМР (DMSO): 1.39 (s,9 Н), 1.49 (m, 2 Н), 1.82 (m, 2 Н), 2.83 (m, 2 Н), 3.44 (bs, 1 Н), 3.75 (m, 2 Н), 3.87 (s, 3 Н), 6.86 (d, 1 Н), 7.11-7.67(m, 8 Н); m/z=407 [М+Н]+; время удерживания равно 8,02. 1-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-иламин гидрохлорид. Способ N, стадия d. К смеси 1-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4 илкарбаминовой кислоты трет-бутилового эфира (2,00 г, 4,93 ммоль) в дихлорметане (2 мл) и метанола(1 мл) добавляли 2 М HCl в Et2O (25 мл) и получающуюся в результате смесь перемешивали в течение ночи при комнатной температуре. Твердое вещество отфильтровывали, сушили в вакууме, перерастворяли в воде (50 мл) и промывали дихлорметаном (310 мл). Воду удаляли при пониженном давлении с получением 1,48 г (89%) указанного в заголовке соединения в виде соли гидрохлорида. 1 Н-ЯМР (400 МГц,DMSO):1.67-1.77 (2 Н, m), 2.01-2.04 (2 Н, m), 2.95-3.00 (2 Н, m), 3.19-3.26 (1 Н, m), 3.89-3.95 (2 Н, m), 4.05[M+H]+; время удерживания равно 0,17. 2-Этокси-N-1-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-илацетамид. Способ N, стадия е. К раствору этоксиуксусной кислоты (0,05 мл, 0,49 ммоль) в ацетонитриле (3 мл) добавляли CDI в виде концентрированного раствора (0,80 г, 0,49 ммоль) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение 3 ч. Добавляли 1-[3-(1-метил-1 Нбензоимидазол-2-ил)фенил]пиперидин-4-иламин гидрохлорид (0,13 г, 0,41 ммоль) и DIPEA (0,07 мл,0,41 ммоль) и получающийся в результате раствор нагревали при 70 С в течение ночи. Реакционную смесь затем охлаждали до комнатной температуры, растворитель удаляли при пониженном давлении. Неочищенный продукт перерастворяли в дихлорметане (4 мл) и промывали насыщенным растворомNa2CO3 (2 мл) и водой (2 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии (элюент: дихлорметан:МеОН, градиент от 100% дихлорметана до 95% дихлорметан:5% МеОН) с получением 0,70 г (44%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, CDCl3):1.17 (3 Н, t), 1.51-1.61 (2 Н, m), 1.96-2.00(2 Н, m), 2.89-2.95 (2 Н, m), 3.50 (2 Н, q), 3.67-3.71 (2 Н, m), 3.81 (3 Н, s), 3.86 (2 Н, s), 3.91-4.01 (1 Н, m), 6.446.46 (1 Н, m), 6.99-7.01 (1 Н, m), 7.05-7.07 (1 Н, m), 7.23-7.35 (5 Н, m), 7.77-7.80 (1 Н, m); m/z=393 [M+H]+; время удерживания равно 1,67. Пример 35. 4-Фтор-1'-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]-[1,4']бипиперидинил. 2-(3-Бромфенил)-1-метил-1 Н-бензимидазол. Способы О, Оа, стадия а. 2-(3-Бромфенил)-1 Н-бензимидазол (описанный в способе N, стадия а) (7,8 г, 28,6 ммоль) полностью растворяли в безводном THF (300 мл), затем NaH 60 мас./мас.% (1,49 г, 37,2 ммоль) порциями добавляли к прозрачному желтому раствору. Светло-коричневую суспензию перемешивали при комнатной температуре в течение 1 ч, затем по каплям добавляли CH3I (2,5 мл, 40,0 ммоль). Реакционную смесь перемешивали при к.т. в течение ночи. Реакционную смесь гасили H2O (300 мл) и экстрагировали EtOAc (2450 мл). Органические экстракты сушили над MgSO4, фильтровали и упаривали с получением 7,4 г указанного в заголовке соединения в виде желто-коричневого твердого вещества(1 Н, m), 7.88 (1 Н, m), 8.05 (1 Н, m); m/z 287 (М+Н)+; время удерживания равно 7,70. 8-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]-1,4-диокса-8-азаспиро[4,5]декан. Способы О, Оа, стадия b. В 4-горлую круглодонную колбу, оборудованную магнитной мешалкой и высушенную в атмосфере аргона, помещали BINAP (3,7 г, 5,9 ммоль, 0,3 экв.) и Pd(OAc)2 (0,9 г, 3,9 ммоль, 0,2 экв.) и оставляли в потоке аргона на 15 мин. В то же время в 4-горлой круглодонной колбе,оборудованной магнитной мешалкой, обратным холодильником и высушенной в атмосфере аргона, 2-(3 бромфенил)-1-метил-1 Н-бензимидазол (5,6 г, 19,5 ммоль, 1 экв.) растворяли в безводном толуоле (300 мл), затем к раствору добавляли 1,4-диокса-8-азаспиро[4,5]декан (3,3 мл, 25,4 ммоль, 1,3 экв.), Cs2CO3(31,8 г, 97,5 ммоль, 5 экв.) и суспензию катализатора. Реакционную смесь перемешивали в течение ночи при температуре дефлегмации, контролируя превращение при помощи LC-MS. Суспензию оставляли охлаждаться до к.т., затем фильтровали через слой целита в фильтровальном тигле Гуча. Растворитель упаривали с получением 16,0 г красно-коричневого масла, которое очищали при помощи автоматической флэш-хроматографии на диоксиде кремния versaflash (1 г продукта/30 г диоксида кремния) путем элюции из Су:МТВЕ=85:15 до Су:МТВЕ=15:85, с получением 2,3 г чистого продукта в виде желтого масла(34%). 1 Н-ЯМР (400 МГц, DMSO):1.70-1.73 (4 Н, m), 3.34-3.37 (4 Н, m), 3.84 (3 Н, s), 3.90 (4 Н, m), 7.117.39 (6 Н, m), 7.57-7.58 (1 Н, m), 7.64-7.65 (1 Н, m); m/z 350 (М+Н)+; время удерживания равно 6,73. 1-[3-(1-Метил-1H-бензо[d]имидазол-2-ил)фенил]пиперидин-4-он. Способы О,Оа,стадия с. 8-[3-(1-Метил-1 Н-бензоимидазол-2-ил)фенил]-1,4-диокса-8 азаспиро[4,5]декан (2,30 г, 6,60 ммоль) растворяли в 120 мл THF. Раствор охлаждали при 0 С на NaClледяной бане, затем добавляли 6 М HCl (18 мл, 108 ммоль). Ход реакции контролировали при помощиHPLC/MS и реакционную смесь перемешивали при к.т. до полного превращения. Раствор концентрировали при пониженном давлении, затем его нейтрализовали 2 М NaOH (80 мл) и, наконец, экстрагировали- 24 017918 получением 2 г чистого продукта в виде бледно-желтого твердого вещества (92%). 1 Н-ЯМР (400 МГц,DMSO):2.46 (4 Н, t), 3.70 (4H, t), 3.88 (3H, s), 7.20-76 (8 Н, m); m/z 306 (М+Н)+; время удерживания равно 5,90. 2-(5-Бром-2-хлорфенил)-1H-бензимидазол. Способы О, Оа, стадия d. В одногорлую круглодонную колбу, оборудованную магнитной мешалкой, помещали 5-бром-2-хлорбензойную кислоту (70,0 г, 297,3 ммоль), о-фенилендиамин (64,3 г, 594,6 ммоль) и метансульфоновую кислоту (140 мл) и нагревали до 170 С для плавления твердых веществ. Систему перемешивали в течение 5 ч при этой температуре, затем оставляли для того, чтобы температура системы достигла к.т. Голубое твердое вещество обрабатывали NaOH 35% (200 мл) с получением фиолетовой суспензии (рН 5), которую фильтровали и промывали 0,5 М NaOH (2 л) и Н 2 О (2 л). Продукт сушили в вакууме (60 С) с получением 61,6 г чистого фиолетового твердого вещества (67%); m/z 307/309(М+Н)+; время удерживания равно 8,73. трет-Бутил 2-(5-бром-2-хлорфенил)-1 Н-бензимидазол-1-карбоксилат. Способы О, Оа, стадия е. В трехгорлой круглодонной колбе, оборудованной магнитной мешалкой,2-(5-бром-2-хлорфенил)-1H-бензимидазол (30,7 г, 99,8 ммоль) суспендировали в THF (1 л). Затем добавляли 50% NaOH (72,0 г, 598 ммоль). Суспензию оставляли при к.т. на 1 ч при перемешивании. (ВОС)2 О(37,0 г, 169,7 ммоль) растворяли в THF (200 мл) и добавляли к реакционной смеси. Реакционную смесь оставляли перемешиваться в течение ночи. Растворитель упаривали при пониженном давлении. Получающийся остаток разбавляли водой (500 мл), фильтровали и сушили в вакууме (60 С) с получением 39,8 г коричневого твердого вещества (98%); m/z 407/409 (М+Н)+; время удерживания равно 10,14. 2-[2-Хлор-5-(1,4-диокса-8-азаспиро[4,5]дек-8-ил)фенил]бензоимидазол-1-карбоновой кислоты третбутиловый эфир. Способы О, Оа, стадия f. В 4-горлую круглодонную колбу, оборудованную магнитной мешалкой и высушенную в атмосфере аргона, помещали BINAP (13,8 г, 22,1 ммоль) и Pd(OAc)2 (3,3 г, 14,7 ммоль) и оставляли в потоке аргона на 15 мин. В то же время в 4-горлой круглодонной колбе, оборудованной магнитной мешалкой, обратным холодильником и высушенной в атмосфере аргона трет-бутил 2-(5-бром-2 хлорфенил)-1 Н-бензимидазол-1-карбоксилат (30,0 г, 73,6 ммоль) растворяли в безводном толуоле (1 л),затем к раствору добавляли 1,4-диокса-8-азаспиро[4,5]декан (12,3 мл, 95,7 ммоль), Cs2CO3 (119,9 г, 368,0 ммоль) и суспензию катализатора. Реакционную смесь перемешивали в течение 30 ч при 80 С, контролируя превращение при помощи LC-MS. Суспензию оставляли охлаждаться до к.т., затем фильтровали через слой целита в фильтровальном тигле Гуча. Растворитель упаривали с получением 64,5 г коричневого масла, которое использовали на следующей стадии без какой-либо дополнительной очистки; m/z 471/473 (М+Н)+; время удерживания равно 9,70. 1-[3-(1H-Бензо[d]имидазол-2-ил)-4-хлорфенил]пиперидин-4-он. Способы О, Оа, стадия g. В 4-горлой круглодонной колбе, оборудованной магнитной мешалкой, 2[2-хлор-5-(1,4-диокса-8-азаспиро[4,5]дек-8-ил)фенил]бензоимидазол-1-карбоновой кислоты третбутиловый эфир (59,5 г, 93,9 ммоль, 1 экв.) растворяли в THF (250 мл), затем добавляли 4 М HCl (1,2 л,4695 ммоль). Систему оставляли перемешиваться при к.т. в течение ночи. Коричневую суспензию фильтровали, твердое вещество промывали холодной водой (500 мл), это твердое вещество представляет собой катализатор с предыдущей стадии. Маточные растворы концентрировали при пониженном давлении для осаждения другого твердого вещества, которое отфильтровывали. Кислотную водную фазу (1,7 л) охлаждали до 0 С при помощи бани лед-NaCl и подщелачивали 4 М NaOH (1,8 л). Твердое вещество фильтровали, промывали водой (600 мл) и сушили в вакууме (60 С) с получением чистых 22,8 г продукта в виде светло-серого твердого вещества (75%). 1 Н-ЯМР (400 МГц, DMSO):2.45 (4 Н, t), 3.70 (4 Н, t),7.17-7.64 (7 Н, m), 12.85 (1 Н, bs); m/z 426/428 (М+Н)+; время удерживания равно 6,97. 4-Фтор-1'-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]-[1,4']бипиперидинил. Способ О, стадия h. Триацетоксиборгидрид натрия (0,10 г, 0,49 ммоль) добавляли к смеси 1-[3-(1 метил-1 Н-бензоимидазол-2-ил)фенил]пиперидин-4-она (0,1 г, 0,33 ммоль) и 4-фторпиперидин гидрохлорида (0,07 г, 0,49 ммоль) в дихлорэтане (2,50 мл). Реакционную смесь оставляли при комнатной температуре на 18 ч. Реакционную смесь разбавляли DCM (4 мл) и промывали насыщенным раствором Na2CO3(4 мл). Органический слой концентрировали и неочищенное вещество фильтровали через картридж SCX(2 г) (элюент: сначала 10 мл DCM:MeOH 1:1, затем 10 мл 2,0 н. раствор аммиака в МеОН). Раствор аммиака концентрировали и неочищенное вещество очищали с помощью флэш-хроматографии (элюент:(М+Н)+; время удерживания равно раздвоенный пик.(0,5 г) нагревали до 40 С в метаноле (5 мл) в течение 16 ч. Добавляли цианоборгидрид натрия (0,025 г,0,4 ммоль) и хлорид цинка (0,027 г, 0,2 ммоль) и реакционную смесь нагревали в течение еще 16 ч, реакционную смесь упаривали и остаток распределялся между 1 н. гидроксидом натрия (2 мл) и дихлорметаном (4 мл). Органический слой отделяли, упаривали и очищали путем препаративной HPLC. 1 Н-ЯМР(400 МГц, CD3OD):1.56 (1 Н, bs), 1.84-1.98 (7 Н, m), 2.20-2.23 (2 Н, m), 2.89-2.94 (2 Н, m), 3.04 (2 Н, bs),3.36-3.43 (1 Н, m), 3.56 (2 Н, bs), 3.94 (3 Н, s), 4.02-4.05 (2 Н, m), 7.23-7.26 (2 Н, m), 7.37-7.45 (3 Н, m), 7.477.51 (1 Н, m), 7.64-7.66 (1 Н, m), 7.71-7-73 (1 Н, m); m/z 375 (М+Н)+; время удерживания равно раздвоенный пик. Пример 37. 2-[3-(3-Фтор-4-морфолин-4-илпиперидин-1-ил)фенил]-1-метил-1 Н-бензоимидазол. 4-Триметилсиланилокси-3,6-дигидро-2 Н-пиридин-1-карбоновой кислоты трет-бутиловый эфир. Способ Р, стадия а. Хлортриметилсилан (1,52 мл, 12,05 ммоль) и безводный триэтиламин (3,42 мл,24,6 ммоль) добавляли к перемешиваемому раствору 4-оксопиперидин-1-карбоновой кислоты третбутиловому эфиру (2,0 г, 10,04 ммоль) в безводном DMF (20 мл). Реакционную смесь нагревали при 80 С в течение 16 ч, затем охлаждали до комнатной температуры и разбавляли н-гексаном (120 мл). Органический раствор промывали охлажденным насыщенным раствором NaHCO3 (60 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 2,56 г указанного в заголовке соединения(94%). 1 Н-ЯМР (400 МГц, DMSO):0.00 (9 Н, s), 1.23 (9 Н, s), 1.83-1.87 (2 Н, m), 3.24-3.27 (2 Н, m), 3.603.61 (2 Н, m), 4.63 (1 Н, bs); m/z 272 (М+Н)+; время удерживания равно 2,78. 3-Фтор-4-оксопиперидин-1-карбоновой кислоты трет-бутиловый эфир. Способ Р, стадия b. Selectfluor (5,52 г, 15,59 ммоль) добавляли к перемешиваемому раствору 4 триметилсиланилокси-3,6-дигидро-2 Н-пиридин-1-карбоновой кислоты трет-бутилового эфира (3,84 г,14,17 ммоль) в безводном ацетонитриле (150 мл) при комнатной температуре в атмосфере азота. Через 75 мин реакционную смесь разбавляли AcOEt (500 мл) и промывали раствором NaCl (300 мл) и затем рассолом (300 мл). Органический слой сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенную реакционную смесь очищали с помощью флэш-хроматографии через нейтральный оксид алюминия (элюент: циклогексан:AcOEt:MeOH градиент от циклогексан: AcOEt 1:1 до циклогексан:(2 мл) в атмосфере азота. Через 10 мин порциями добавляли триацетоксиборгидрид натрия (2,35 г, 11,13 ммоль) и реакционную смесь оставляли на 3 ч. К реакционной смеси добавляли насыщенный растворNa2CO3 (20 мл). Органическую фазу отделяли и водный раствор экстрагировали DCM (310 мл). Органические слои собирали, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенную реакционную смесь очищали с помощью флэш-хроматографии (элюент: циклогексан:AcOEt:2,0 н. раствор аммиака в МеОН, градиент от циклогексан: AcOEt 2:1 до 100% AcOEt и наконец AcOEt:2,0 н. раствор аммиака в МеОН 10:1) с получением 1,28 г указанного в заголовке соединения (57%). 1 Н-ЯМР(400 МГц, CD3OD):1.45 (9 Н, s), 1.74-1.80 (2 Н, m), 2.38-2.51 (1 Н, m), 2.65-2.68 (4 Н, m), 2.71-3.04 (2 Н,m), 3.65-3.72 (4 Н, m), 4.18-4.21 (1 Н, m), 4.37-4.38 (1 Н, m), 4.91-5.03 (1 Н, m); m/z 289 (М+Н)+; время удерживания равно раздвоенный пик. 4-(3-Фторпиперидин-4-ил)морфолин. Способ Р, стадия d. 2,0 н. HCl в Et2O (15 мл) добавляли к раствору 3-фтор-4-морфолин-4 илпиперидин-1-карбоновой кислоты трет-бутилового эфира (1,28 г, 4,44 ммоль) в DCM (2 мл) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение 4 суток. Осадок отфильтровывали и растирали с Et2O (25 мл). Добавляли насыщенный раствор Na2CO3 (10 мл) и получающийся в результате раствор концентрировали при пониженном давлении. Добавляли EtOH (15 мл) и осадок отфильтровывали. Фильтрат концентрировали при пониженном давлении с получением 0,71 г указанного в заголовке соединения (85%). 1 Н-ЯМР (400 МГц, CD3OD):1.84-1.94 (2 Н, m), 2.47-2.60 (1 Н,m), 2.66-2.68 (4 Н, m), 2.78-2.86 (1 Н, m), 2.88-3.02 (1 Н, m), 3.23-3.28 (1 Н, m), 3.36-3.44 (1 Н, m), 3.68-3.71(4 Н, m), 5.01-5.14 (1 Н, m); m/z 189 (М+Н)+; время удерживания равно 0,17. 2-[3-(3-Фтор-4-морфолин-4-илпиперидин-1-ил)фенил]-1-метил-1 Н-бензоимидазол. Способ Р, стадия е. 4-(3-Фторпиперидин-4-ил)морфолин (0,085 г, 0,45 ммоль), 2-(3-бромфенил)-1 метил-1 Н-бензоимидазол (описанный в способе N, стадии а, b) (0,10 г, 0,35 ммоль) и карбонат цезия(10,50 г, 32,2 ммоль) помещали в 7-мл сухой закрытый герметизируемый сосуд в атмосфере азота и добавляли безводный толуол (0,4 мл). Одновременно ацетат палладия (0,016 г, 0,07 ммоль) и BINAP (0,065 г, 0,10 ммоль) помещали в другой 7-мл сухой закрытый герметизируемый сосуд в атмосфере азота и раз- 26 017918 бавляли безводным толуолом (0,8 мл). Приблизительно через 20 мин получающуюся в результате суспензию добавляли в сосуд, содержащий исходные вещества. Реакционную смесь нагревали при 85 С в течение 24 ч, затем охлаждали до комнатной температуры и фильтровали через SCX (элюент: сначала 10 мл DCM.MeOH 1:1, затем 10 мл 2,0 н. раствора аммиака в МеОН). Раствор аммиака концентрировали при пониженном давлении и неочищенное вещество очищали с помощью флэш-хроматографии (элюент:MeOH:AcOEt градиент от 100% AcOEt до MeOH:AcOEt 1:4) с получением 0,08 г указанного в заголовке соединения (58%). 1 Н-ЯМР (400 МГц, CD3OD):1.91-1.95 (1 Н, m), 2.01-2.11 (1 Н, m), 2.41-2.54 (1 Н, m),2.67-2.75 (4 Н, m), 2.84-3.04 (2 Н, m), 3.68-3.76 (4 Н, m), 3.88 (3 Н, s), 3.92-3.98 (1 Н, m), 4.09-4.16 (1 Н, m),5.06-5.19 (1 Н, m); m/z 395 (М+Н)+; время удерживания равно 0,27. Пример 38. (4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепан-1-ил)-(1-метилпиперидин-3 илметанон. 4-(4-Хлор-3-этоксикарбонилфенил)-[1,4]диазепан-1-карбоновой кислоты трет-бутиловый эфир. Способ Q, стадия а. [1,4]-Диазепан-1-карбоновой кислоты трет-бутиловый эфир (11,5 г, 0,057 ммоль), 5-бром-2-хлорбензойной кислоты этиловый эфир (12,5 г, 0,047 ммоль), Pd(OAc)2 (0,21 г, 1 ммоль), рац-2,2' бис-(дифенилфосфино)-1,1'-бинафтил (BINAP) (0,88 г, 1,4 ммоль) и карбонат цезия (21,5 г, 66 ммоль) суспендировали в толуоле (200 мл). Реакционную смесь кипятили с обратным холодильником в течение 16 ч, затем охлаждали до 20 С. Реакционную смесь выливали в воду (200 мл) и экстрагировали AcOEt (3150 мл). Объединенные экстракты промывали рассолом, сушили над MgSO4 и концентрировали при пониженном давлении с получением коричневого масла, которое очищали с помощью флэш-хроматографии (элюент: н-гексан:AcOEt градиент от н-гексан:AcOEt 4:1 до н-гексан:AcOEt 1:1) с получением 17,2 г указанного в заголовке соединения (96%). 1 Н-ЯМР (400 МГц, CDCl3):1.37-1.48 (12 Н,m), 1.96-2.02 (2 Н, m), 3.22-3.35 (2 Н, m), 3.50-3.64 (6 Н, m), 4.39 (2 Н, q), 6.76-6.78 (1 Н, m), 7.10-7.11 (1 Н,m), 7.23-7.26 (1 Н, m); m/z 383 (М+Н)+; время удерживания равно 7,70. 2-(2-Хлор-5-[1,4]диазепан-1-илфенил)-1 Н-бензоимидазол. Способ Q, стадия b. К раствору триметилалюминия (2,0 М раствор в толуоле) (90 мл, 180 ммоль) в толуоле (300 мл), охлажденном до 0 С, в атмосфере азота порциями добавляли бензол-1,2-диамин (6,5 г,60 ммоль). Раствор перемешивали в течение 30 мин при 0 С, затем при 15-20 С до прекращения выделения метана. 4-(4-Хлор-3-этоксикарбонилфенил)-[1,4]диазепан-1-карбоновой кислоты трет-бутиловый эфир (15,2 г, 40 ммоль) добавляли одной порцией к получающемуся в результате раствору. Реакционную смесь нагревали до температуры дефлегмации в течение 24 ч, затем охлаждали до 0 С. Осторожно добавляли воду (70 мл), а затем МеОН (700 мл). Осажденное твердое вещество фильтровали и фильтрат концентрировали при пониженном давлении. Осадок промывали горячим DCM:MeOH 1:1 (400 мл), а затем AcOEt (200 мл). Раствор концентрировали при пониженном давлении. Два экстракта комбинировали и очищали с помощью флэш-хроматографии (элюент: AcOEt:MeOH:NH3, градиент от 100% AcOEt доAcOEt:MeOH:NH3 73:25:2) с получением 9,3 г смеси 2-(2-хлор-5-[1,4]диазепан-1-илфенил)-1 Нбензоимидазола и N-(2-аминофенил)-2-хлордиазепан-1-илбензамида; m/z 345 (М+Н)+; время удерживания равно 3,07; m/z 327 (М+Н)+; время удерживания равно 4,1. 2-(2-Хлор-5-[1,4]диазепан-1-илфенил)-1 Н-бензоимидазол. Способ Q, стадия с. Смесь 3:1 2-(2-хлор-5-[1,4]диазепан-1-илфенил)-1 Н-бензоимидазола и N-(2 аминофенил)-2-хлордиазепан-1-илбензамида (10,1 г) растворяли в уксусной кислоте (120 мл) и нагревали при 55 С в течение 16 ч. Реакционную смесь охлаждали до 20 С и концентрировали при пониженном давлении. Получающееся в результате коричневое масло растирали с Et2O и сушили. Соль ацетат растворяли в 10% MeOH/DCM (100 мл) и с 0,1 М водным раствором NaHCO3 (200 мл). Образовывался осадок, и его перемешивали в течение 2 ч, затем фильтровали и сушили при 60 С с получением 7,92 г указанного в заголовке соединения (61% после двух стадий). 1 Н-ЯМР (400 МГц, DMSO):2.11-2.17 (2 Н, m),3.07-3.08 (2 Н, m), 3.26 (2 Н, bs), 3.60-3.63 (2 Н, m), 3.82-3.84 (2 Н, m), 7.12-7.15, (1 Н, m), 7.40-7.41 (1 Н, m),7.54-7.56 (1 Н, m), 7.60-7.64 (2 Н, m), 7.88-7.92 (2 Н, m), 9.40 (2 Н, bs); m/z 327 (М+Н)+; время удерживания равно 4,1. 3-4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепан-1-карбонилпиперидин-1-карбоновой кислоты трет-бутиловый эфир. Способ Q, стадия d. 2-(2-Хлор-5-[1,4]диазепан-1-илфенил)-1 Н-бензоимидазол (0,25 г, 0,76 ммоль),HOBt (0,12 г, 0,86 ммоль), NMM (N-метилморфолин) (0,5 мл, 3,9 ммоль), пиперидин-1,3-дикарбоновой кислоты 1-трет-бутиловый эфир (0,17 г, 0,76 ммоль) и EDC (0,22 г, 1,16 ммоль) растворяли в DCM (8,0 мл) и перемешивали при 20 С в течение 16 ч. Добавляли воду (6,0 мл). Органический слой отделяли,концентрировали при пониженном давлении и неочищенное вещество очищали с помощью флэшхроматографии (элюент: 100% AcOEt) с получением 0,25 г указанного в заголовке соединения (100%). 1 Н-ЯМР (400 МГц, DMSO):1.28-1.51 (11 Н, m), 1.56-1.89 (3 Н, m), 2.01 (1 Н, m,), 2.60-2.71 (3 Н, m), 3.413.76 (7 Н, m), 3.91-4.03 (3 Н, m), 6.90-6.95 (1 Н, m), 7.20-7.30 (3 Н, m), 7.34-7.37 (1 Н, m), 7.63 (2 Н, bs); m/z 536 (М+Н)+; время удерживания равно 1,85. карбонилпиперидин-1-карбоновой кислоты трет-бутиловый эфир (0,21 г, 0,39 ммоль) растворяли в изопропаноле (4 мл) и добавляли ацетилхлорид (0,17 мл, 2,4 ммоль). Реакционную смесь нагревали при 40 С в течение 16 ч, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении и сушили при 60 С с получением 0,19 г указанного в заголовке соединения в виде соли дихлоргидрата (94%). 1 Н-ЯМР (400 МГц, CD3OD):1.56-2.07 (6 Н, m), 3.04-3.25 (5 Н, m), 3.42-3.54 (1 Н, m), 3.593.87 (6 Н, m), 3.91-4.08 (1 Н, m), 7.14-7.20 (1 Н, m), 7.25-7.31 (1 Н, m), 7.50-7.56 (1 Н, m), 7.66-7.71 (2 Н, m),7.87-7.92 (2 Н, m); m/z 438 (М+Н)+; время удерживания равно раздвоенный пик. 4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепан-1-ил-1-метилпиперидин-3-илметанон. Способ Q, стадия f. 4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепан-1-илпиперидин-3 илметанон (0,18 г, 0,34 ммоль) суспендировали в DCM (8 мл). Добавляли триэтиламин (0,5 мл 3,5 ммоль) с получением раствора, а затем АсОН (0,5 мл) и формальдегид (37% водный раствор) (0,1 мл). Добавляли триацетоксиборгидрид натрия (0,14 г, 0,63 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 24 ч и затем при 40 С в течение 4 суток. Реакционную смесь концентрировали до 4 мл, затем вновь добавляли формальдегид (0,1 мл), триэтиламин (0,5 мл, 3,5 ммоль), АсОН (0,5 мл) и триацетоксиборгидрид натрия (0,14 г, 0,63 ммоль) и реакционную смесь нагревали в микроволновой печи при 100 С в течение 20 мин. Реакционную смесь гасили 2,0 н. раствором NaOH (4 мл) и перемешивали в течение 15 мин. Органический слой отделяли и концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии (элюент: н-гексан:AcOEt:MeOH:NH3, градиент от нгексан:AcOEt 1:1 до AcOEt:MeOH:NH3 80:18:2) с получением 0,07 г указанного в заголовке соединения,которое дополнительно очищали путем препаративной HPLC с получением 0,02 г указанного в заголовке соединения (13%). 1 Н-ЯМР (400 МГц, CD3OD):1.37-1.94 (4 Н, m), 1.98-2.10 (2 Н, m), 2.56-2.62 (3 Н, m),2.68-2.80 (2 Н, m), 2.98-3.06 (2 Н, m), 3.44-4.04 (9 Н, m), 6.90-6.98 (1 Н, m), 7.14-7.21 (1 Н, m), 7.28-7.40 (3 Н,m), 7.63-7.67 (2 Н, m); m/z 452 (М+Н)+; время удерживания равно раздвоенный пик. Пример 39. 4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепан-1-карбоновой кислоты фениламид. 4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепан-1-карбоновой кислоты фениламид. Способ R. К раствору 2-(2-хлор-5-[1,4]диазепам-1-илфенил)-1 Н-бензоимидазола (способ Q, стадии а, b, с) (0,10 г, 0,30 ммоль) в дихлорметане (4 мл) добавляли фенилизоцианат (0,03 мл, 0,30 ммоль) и получающуюся в результате суспензию перемешивали при комнатной температуре в течение 16 ч. Осадок фильтровали, затем очищали с помощью флэш-хроматографии (элюент: EtOAc:гексан, 1:4) с получением 0,06 г (45%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, DMSO):1.89-1.95 (2 Н, m), 3.393.42 (2 Н, m), 3.56-3.59 (2 Н, m), 3.64 (4 Н, bs), 6.88-6.93 (2 Н, m), 7.16-7.24 (5 Н, m), 7.33-7.35 (1 Н, m), 7.387.41 (2 Н, m), 7.52-7.54 (1 Н, m), 7.67-7.69 (1 Н, m), 12.56 (1 Н, s); m/z=446 [M+H]+; время удерживания равно 5,87. Пример 40. 4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепан-1-карбоновой кислоты (2 диметиламиноэтил)амид. 4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепан-1-карбоновой кислоты(2 диметиламиноэтил)амид. Способ S. К раствору N,N-диметилэтан-1,2-диамина (0,85 г, 0,96 ммоль) и триэтиламина (0,50 мл,3,84 ммоль) в диоксане по каплям добавляли изопропилхлорформиат (0,10 мл, 0,96 ммоль) и после перемешивания в течение 1 ч при комнатной температуре добавляли 2-(2-хлор-5-[1,4]диазепам-1-илфенил)1 Н-бензоимидазол (способ Q, стадии а, b, с) (0,21 г, 0,64 ммоль). Получающуюся в результате смесь нагревали в течение 1 ч при 150 С в микроволновом реакторе. Растворитель затем удаляли при пониженном давлении, неочищенное вещество распределялось между дихлорметаном (8 мл) и водой (2 мл), органический слой отделяли и загружали на колонку диоксида кремния для очистки (элюент:(1 Н, m), 3.12-3.15 (1 Н, m), 3.41-3.46 (4 Н, m), 3.65-3.74 (6 Н, m), 6.97-7.00 (1 Н, m), 7.21-7.22 (1 Н, m), 7.387.41 (3 Н, m), 7.69-7.72 (2 Н, m); m/z=441 [M+H]+; время удерживания равно 4,12. Пример 41. 1-4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепам-1-ил)-2 диметиламиноэтанон. 1-4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепам-1-ил-2-диметиламиноэтанон. Способ Т, стадия а. Раствор диметилглицина (0,78 г, 0,76 ммоль), N,N-диизопропилэтиламина (0,20 мл, 1,14 ммоль) и о-бензотриазол-N,N,N',N'-тетраметил-уроний-гексафторфосфата (HBTU) (0,32 г, 0,84 ммоль) в дихлорметане (20 мл) перемешивали при комнатной температуре в течение 15 мин, затем добавляли 2-(2-хлор-5-[1,4]диазепам-1-илфенил)-1 Н-бензоимидазол (способ Q, стадии а, b, с) (0,25 г, 0,76 ммоль). После перемешивания в течение 16 ч при комнатной температуре растворитель удаляли и неочищенное вещество очищали с помощью флэш-хроматографии (элюент: градиент отm), 7.33-7.37 (1 Н, m), 7.64 (2 Н, bs); m/z=412 [M+H]+; время удерживания равно 4,24. Пример 42. (2-4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепан-1-илэтил)диметиламин.(2-4-[3-(1 Н-Бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепан-1-илэтил)диметиламин. Способ Т, стадия b. К раствору 1-4-[3-(1 Н-бензоимидазол-2-ил)-4-хлорфенил]-[1,4]диазепам-1 ил-2-диметиламино-этанона (0,13 г, 0,31 ммоль) в тетрагидрофуране (4 мл) добавляли 1 М LiAlH4 (0,31 мл, 0,31 ммоль) и получающиеся в результате смеси перемешивали при комнатной температуре в течение 72 ч, затем гасили водой (20 мл) и 2 н. NaOH (20 мл). Реакционную смесь затем пропускали через короткий слой (Na2SO4), растворитель удаляли при пониженном давлении и неочищенное вещество очищали путем препаративной HPLC с получением 0,03 г (25%) указанного в заголовке соединения. 1 НЯМР (400 МГц, CD3OD):1.98-2.04 (2 Н, m), 2.29 (6 Н, s), 2.53-2.56 (2 Н, m), 2.66-2.70 (4 Н, m), 2.85-2.88(2 Н, m), 3.55-3.58 (2 Н, m), 3.61-3.63 (2 Н, m), 6.86-6.89 (1 Н, m), 7.16-7.17 (1 Н, m), 7.26-7.30 (2 Н, m), 7.327.35 (1 Н, m), 7.63 (2 Н, bs); m/z=398 [M+H]+; время удерживания равно 4,53. Пример 43.Ar, помещали BINAP (5,85 г, 9,4 ммоль) и Pd(OAc)2 (1,41 г, 6,3 ммоль). Смесь перемешивали в течение 10 мин в потоке Ar, затем добавляли 300 мл безводного толуола. В то же время в еще одну четырехгорлую круглодонную колбу, высушенную в атмосфере Ar, помещали 2-(3-бромфенил)-1-метил-1 Нбензимидазол (описанный в способе О, Оа, стадия а), предварительно растворенный в 300 мл безводного толуола (9,0 г, 31,3 ммоль), 1-ВОС-гомопиперазин (8,0 мл, 40,7 ммоль) и Cs2CO3 (50,99 г, 156,5 ммоль). Смесь перемешивали в течение приблизительно 10 мин, затем содержимое первой колбы добавляли во вторую колбу. Реакционную смесь перемешивали в течение ночи при температуре дефлегмации. После охлаждения до комнатной температуры суспензию фильтровали через слой целита в фильтровальном тигле Гуча. Прозрачный раствор упаривали при пониженном давлении и остаток (25,0 г) очищали путем автоматической флэш-хроматографии на диоксиде кремния versaflash (1 г продукта/30 г диоксида кремния) путем элюции от Су:EtOAc=9:1 до Су:EtOAc=1:9, с получением чистого продукта в виде коричневой пены (5,2 г, 41%). 1 Н-ЯМР (d6 DMSO): 1.26 (d, 9H), 1.84(m, 2H), 3.25(m, 2H), 3.61(m, 4H), 3.86(s, 3 Н),6.91 (dd, 1H), 7.00(t, 1H), 7.10(s, 1H), 7.27(m, 3 Н), 7.59(d, 1H), 7.67(d, 1 Н); m/z=407 [M+H]+; время удерживания равно 8,20 мин. Соль дигидрохлорид 2-(3-[1,4]диазепан-1-ил фенил)-1-метил-1 Н-бензоимидазола. Способы U, V, W, Ab, стадия b. К смеси 4-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил][1,4]диазепан-1-карбоновой кислоты трет-бутилового эфира (2,20 г, 5,61 ммоль) в метаноле (3 мл) добавляли 2 М HCl в Et2O (10 мл) и получающуюся в результате смесь перемешивали в течение 6 ч при комнатной температуре. Растворитель затем удаляли при пониженном давлении и получающуюся в результате соль растирали с Et2O, фильтровали при пониженном давлении и сушили в вакууме с получением 1,60 г (93%) указанного в заголовке соединения в виде соли хлорида. 1 Н-ЯМР (400 МГц, DMSO):2.132.19 (2 Н, m), 3.07-3.08 (2 Н, m), 3.24 (2 Н, bs), 3.61-3.64 (2 Н, m), 3.84-3.87 (2 Н, m), 4.06 (3 Н, s), 7.14-7.22(2 Н, m), 7.33 (1 Н, bs), 7.49-7.53 (1 Н, m), 7.62-7.68 (2 Н, m), 7.86-7.90 (1 Н, m), 8.04-8.08 (1 Н, m), 9.54 (2 Н,bs); m/z=307[M+H]+; время удерживания равно раздвоенный пик.N,N-Диметил-2-4-[3-(1-метил-1 Н-бензоимидазол-2-ил)фенил]-[1,4]диазепан-1-илацетамид. Способ U, стадия с. Смесь 2-(3-[1,4]диазепан-1-илфенил)-1-метил-1 Н-бензоимидазола (0,30 г, 0,79 ммоль), 2-хлор-N,N-диметилацетамида (0,14 г, 1,18 ммоль) и триэтиламина (0,44 мл, 3,20 ммоль) в диоксане (8 мл) нагревали в течение 16 ч при 80 С. Реакционную смесь затем охлаждали до комнатной температуры, осадок отфильтровывали и органическую фазу упаривали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии (элюент: от EtOAc 100% до EtOAc:MeOH:NH3 89:10:1) с получением 0,11 г (34%) указанного в заголовке соединения. 1 Н-ЯМР (400 МГц, CD3OD):1.93-2.04 (2 Н,m), 2.15 (3 Н, s), 2.21 (3 Н, s), 3.07-3.21 (2 Н, m), 3.45-3.51 (1 Н, m), 3.54-3.57 (1 Н, m), 3.63-3.70 (3 Н, m), 3.763.84 (3 Н, m), 6.90-6.95 (1 Н, m), 7.20-7.31 (3 Н, m), 7.33-7.37 (1 Н, m), 7.64 (2 Н, bs); m/z=392 [M+H]+; время удерживания равно 4,87 мин. Пример 44. 2-[3-(4-Метансульфонил-[1,4]диазепан-1-ил)фенил]-1-метил-1 Н-бензоимидазол. 2-[3-(4-Метансульфонил-[1,4]диазепан-1-ил)фенил]-1-метил-1 Н-бензоимидазол. Способ V, стадия d. К раствору 2-(3-[1,4]диазепан-1-илфенил)-1-метил-1 Н-бензоимидазола (0,18 г,0,46 ммоль) и триэтиламина (0,32 мл, 2,30 ммоль) в дихлорметане (4 мл) добавляли метансульфонилхлорид (0,04 мл, 0,55 ммоль). После перемешивания в течение 16 ч при комнатной температуре добавляли воду (1 мл), органический слой сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Получающееся в результате неочищенное вещество очищали с помощью флэш-хроматографии
МПК / Метки
МПК: C07D 413/12, C07D 403/12, C07D 401/12, C07D 235/18, A61P 35/00, C07D 405/12, A61K 31/4184, A61P 3/14
Метки: терапевтические, применения, hedgehog, антагонисты, пути
Код ссылки
<a href="https://eas.patents.su/30-17918-antagonisty-puti-hedgehog-i-ih-terapevticheskie-primeneniya.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты пути hedgehog и их терапевтические применения</a>
Соединения 2-(2-галоген-4-аминофенил)пиридиновых ингибиторов передачи сигналов белком hedgehog (варианты), способ их получения, композиция и способы лечения рака и ингибирований ангиогенеза и сигнального пути hedgehog в клетках на их основе

Номер патента: 17262
Опубликовано: 30.11.2012
Авторы: Сатерлин Даниэл, Стэнли Марк, Сэвидж Скотт, Кастанедо Джорджетт, Малески Кимберли, Гунцнер Джанет, Дайна Майкл, Рейнолдс Марк, Ван Шумэй, Бао Лян, Лалонд Ребекка
МПК: C07D 213/40, C07D 213/38, C07D 213/56...
Метки: композиция, способы, пути, передачи, клетках, варианты, получения, ингибирований, способ, ангиогенеза, сигнального, основе, лечения, ингибиторов, сигналов, рака, 2-(2-галоген-4-аминофенил)пиридиновых, соединения, hedgehog, белком
Формула / Реферат:
1. Соединения 2-(2-галоген-4-аминофенил)пиридиновых ингибиторов передачи сигналов белком hedgehog, охватываемые общей структурной формулойгде R1 выбран из группы, содержащей алкил, карбоцикл или гетероцикл, каждый из которых может содержать заместители, выбранные из группы, включающей гидроксил, галоген, амино, карбоксил, амидино, гуанидино, карбонил, нитро, циано, ацил, алкил, галоалкил, сульфонил, сульфинил, алкокси, алкилтио, карбамоил,...
Тетразамещенные пиридазины в качестве антагонистов пути hedgehog

Номер патента: 17386
Опубликовано: 28.12.2012
Авторы: Клэй Джулия Мари, Коэн Джеффри Дэниел, Сэлл Дэниел Джон, Лобб Карен Линн, Хипскинд Филип Артур, Томпсон Майкл Ли, Бастиан Джоли Энн
МПК: C07D 401/14, C07D 401/04, A61K 31/501...
Метки: пиридазины, hedgehog, пути, антагонистов, качестве, тетразамещенные
Формула / Реферат:
1. Соединение следующей формулы:или фармацевтически приемлемая соль указанного соединения, гдеX представляет собой C-R1 или N;R1 представляет собой водород, фтор или циано;R2 представляет собойпиперидинил или гем-ди-F-замещенный циклогексил;R3 представляет собой метил или трифторметил;R4 представляет собой пирролидинил, морфолинил, пиридил, амино или диметиламино;R5 представляет собой трифторметил или метилсульфонил;R6 представляет собой водород...
Бензил- и пиридинилпроизводные в качестве модуляторов пути передачи сигнала hedgehog

Номер патента: 16564
Опубликовано: 30.05.2012
Авторы: Льямас Луис, Джейн Риши Кумар, Дай Миао, Келлехер Джозеф III, Карки Раджеш, Пойкерт Штефан, Юсуфф Наим, Миллер-Мослин Карен, Макюан Майкл А., Хэ Фэн, Ли Джон, Перес Лоренс Блас
МПК: C07D 237/28, A61K 31/4545, A61K 31/502...
Метки: пиридинилпроизводные, бензил, пути, модуляторов, сигнала, качестве, передачи, hedgehog
Формула / Реферат:
1. Соединение формулы Iи его фармацевтически приемлемые соли, гдеR1 обозначает фенил или het, которые могут быть незамещенными или замещенными;R2 обозначает het, в котором по меньшей мере одним гетероатомом является N и который может быть незамещенным или замещенным;L обозначает C1-С6-алкил;X обозначает N;Y обозначает связь, СН2;R3 обозначает арил или het, который является замещенным;Z обозначает Н, C1-С6-алкил, оксогруппу, C(O)OR6; иm равно 0,...
Полиглутаминовые кислоты, функционализированные катионными и гидрофобными группами, и их применения, в частности терапевтические применения

Номер патента: 16911
Опубликовано: 30.08.2012
Авторы: Шан Йу-Пинг, Брейн Оливье, Бонне Гонне Сесиль
МПК: A61K 47/34, C08G 69/48, C08G 69/10...
Метки: терапевтические, гидрофобными, кислоты, катионными, функционализированные, частности, полиглутаминовые, применения, группами
Формула / Реферат:
1. Полиаминокислота, содержащая остатки глутаминовой кислоты, где некоторые остатки глутаминовой кислоты несут боковую катионную группу, которая, если она может быть депротонирована, имеет pKa, большее или равное 7, причем указанные катионные группы являются одинаковыми или различными и соответствуют следующей формуле:в которой X представляет собой O, NH;Y представляет собой независимо Н или CH3;Z представляет собой хлорид, сульфат, фосфат или...
Терапевтические применения вариантов хемокинов

Номер патента: 9414
Опубликовано: 28.12.2007
Авторы: Праудфут Аманда, Шо Джеффри, Джонсон Зо
МПК: A61K 38/19, A61P 29/00, A61P 31/00...
Метки: хемокинов, применения, вариантов, терапевтические
Формула / Реферат:
1. Применение полипептида, содержащего SEQ ID NO:2, для изготовления лекарственного средства для лечения аутоиммунных, воспалительных или инфекционных заболеваний. 2. Применение по п.1, где этот полипептид дополнительно содержит изолейцин в положении 64 SEQ ID NO:2 и таким образом содержит SEQ ID NO:4. 3. Применение по п.1 или 2, дополнительно отличающееся тем, что указанный полипептид не содержит мутацию в положении 9, 10 или 13 в...
Предыдущий патент: Применение производных азабициклогексана
Следующий патент: Производные пиразин-2-карбоксамида для лечения заболеваний, которые поддаются лечению путем блокирования эпителиальных натриевых каналов
Случайный патент: Лифтовое устройство