Производные 6н-дибензо[b,e]оксепина в качестве нестероидных антагонистов минералокортикоидных рецепторов
Номер патента: 17668
Опубликовано: 28.02.2013





























Формула / Реферат
1. Соединение формулы

где R1 и R2, каждый независимо, представляет собой водород или фтор;
L представляет собой -(СН2)2-, -СН(СН3)-СН2- или прямую связь;
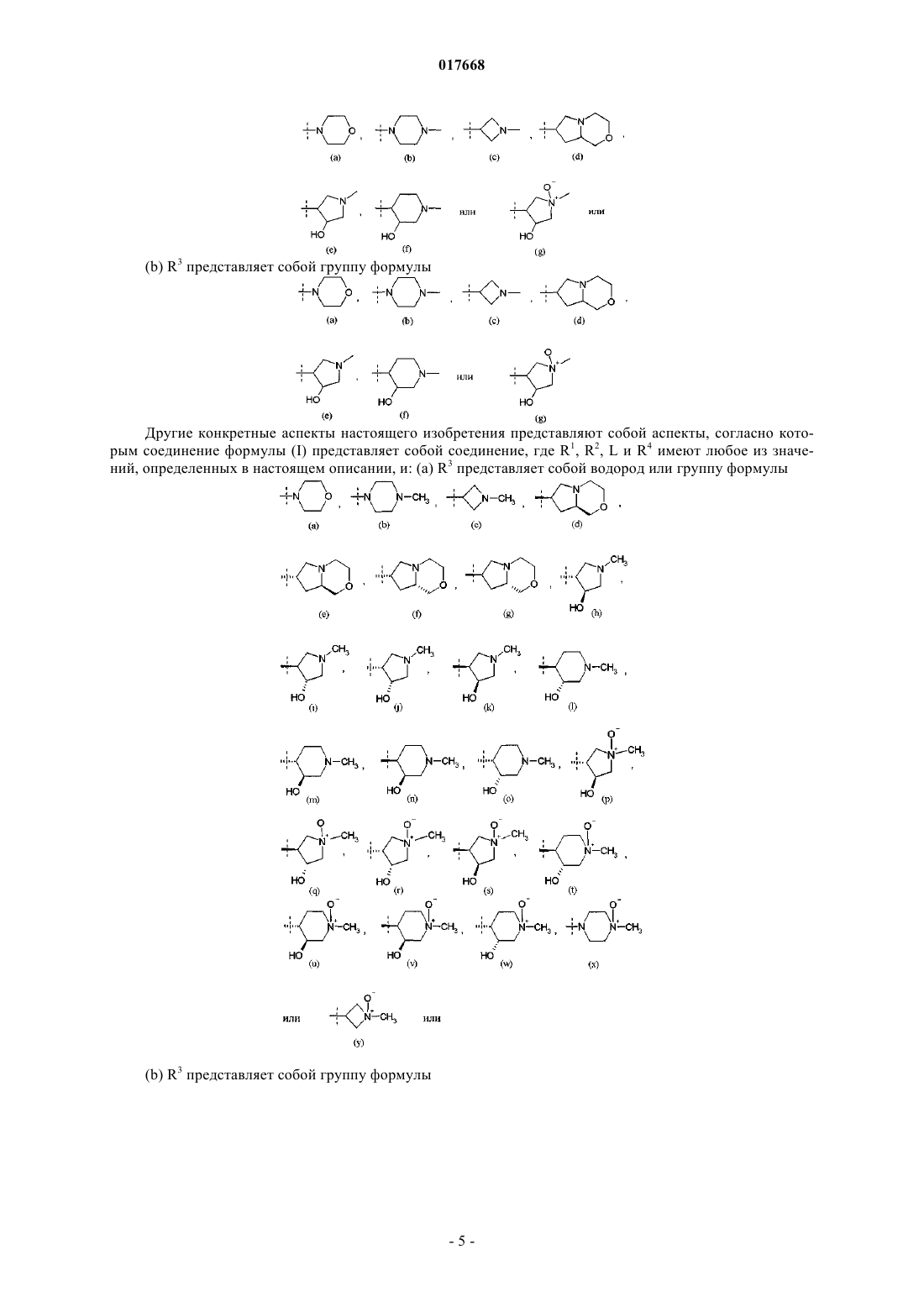
R3 представляет собой водород или группу формулы

R4 представляет собой -CN или -C(O)NH2,
или его фармацевтически приемлемая соль.
2. Соединение или соль по п.1, где R1 представляет собой водород, a R2 представляет собой водород или фтор.
3. Соединение или соль по п.1, где R1 представляет собой водород или фтор, a R2 представляет собой водород.
4. Соединение или соль по п.1, где R1 и R2, каждый независимо, представляет собой водород.
5. Соединение или соль по любому из пп.1-4, где L представляет собой -СН(СН3)-СН2- или прямую связь.
6. Соединение или соль по любому из пп.1-5, где R3 представляет собой группу формулы

7. Соединение или соль по любому из пп.1-6, где R4 представляет собой -CN.
8. Соединение или соль по любому из пп.1-6, где R4 представляет собой -C(O)NH2.
9. Соединение или соль по п.1, где R1 и R2, каждый независимо, представляет собой водород или фтор;
L представляет собой -CH(CH3)-CH2- или прямую связь;
R3 представляет собой водород или группу формулы

R4 представляет собой -CN или -C(O)NH2.
10. Соединение или соль по любому из пп.1-9, выбранные из группы, включающей
(E)-N-(5-((Е)-3-фтор-6H-дибензо[b,е]оксепин-11-илиденметил)-1-(1-метилазетидин-3-ил)-1,3-дигидробензимидазол-2-илиден)цианамид;
(Е)-N-(5-((Е)-(3-фтордибензо[b,е]оксепин-11(6Н)илиден)метил)-1-((3S,4S)-4-гидрокси-1-метилпирролидин-3-ил)-1Н-бензо[d]имидазол-2(3H)-илиден)цианамид;
малеат (Е)-N-(5-((Е)-(3-фтордибензо[b,е]оксепин-11(6Н)-илиден)метил)-1-((3S,4S)-4-гидрокси-1-метилпирролидин-3-ил)-1Н-бензо[d]имидазол-2(3H)-илиден)цианамида;
(E)-N-[5-((Е)-3-фтор-6Н-дибензо[b,е]оксепин-11-илиденметил)-1-((3S,4S)-4-гидрокси-1-метилпирролидин-3-ил)-1,3-дигидробензимидазол-2-илиден]мочевину;
(Е)-N-(5-((Е)-3-фтор-6H-дибензо[b,е]оксепин-11-илиденметил)-1-((R)-1-метил-2-морфолин-4-илэтил)-1,3-дигидробензимидазол-2-илиден)мочевину и
(Е)-N-(5-((Е)-3-фтор-6H-дибензо[b,е]оксепин-11-илиденметил)-1-((7S,8aR)-гексагидропирроло[2,1-c] [1,4]оксазин-7-ил)-1,3-дигидробензимидазол-2-илиден)мочевину.
11. Соединение по п.10, представляющее собой (Е)-N-(5-((Е)-(3-фтордибензо[b,е]оксепин-11(6Н)-илиден)метил)-1-((3S,4S)-4-гидрокси-1-метилпирролидин-3-ил)-1Н-бензо[d]имидазол-2(3H)-илиден) цианамид или его фармацевтически приемлемую соль.
12. Соль по п.10, представляющая собой малеат (Е)-N-(5-((Е)-(3-фтордибензо[b,е]оксепин-11(6Н)-илиден)метил)-1-((3S,4S)-4-гидрокси-1-метилпирролидин-3-ил)-1Н-бензо[d]имидазол-2(3H)-илиден) цианамида.
13. Соединение по п.10, представляющее собой (Е)-N-(5-((Е)-3-фтор-6Н-дибензо[b,е]оксепин-11-илиденметил)-1-((R)-1-метил-2-морфолин-4-илэтил)-1,3-дигидробензимидазол-2-илиден)мочевину или его фармацевтически приемлемую соль.
14. Применение соединения или соли по любому из пп.1-13 в лечении застойной сердечной недостаточности, диабетической нефропатии, хронического заболевания почек, гипертензии, гипокалиемии, миокардиальной аритмии, синдрома Барттера, первичного или вторичного гиперальдостеронизма или синдрома Конна.
15. Применение по п.14 в лечении застойной сердечной недостаточности, гипертенизии, диабетической нефропатии или хронического заболевания почек.
16. Фармацевтическая композиция, содержащая соединение или соль по любому из пп.1-13 в комбинации с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или эксципиентами.
17. Фармацевтическая композиция по п.16, содержащая соединение, представляющее собой (Е)-N-(5-((Е)-3-фтор-6Н-дибензо[b,е]оксепин-11-илиденметил)-1-((R)-1-метил-2-морфолин-4-илэтил)-1,3-дигидробензимидазол-2-илиден)мочевину, или его фармацевтически приемлемую соль в комбинации с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или эксципиентами.
Текст
ПРОИЗВОДНЫЕ 6 Н-ДИБЕНЗО[B,E]ОКСЕПИНА В КАЧЕСТВЕ НЕСТЕРОИДНЫХ АНТАГОНИСТОВ МИНЕРАЛОКОРТИКОИДНЫХ РЕЦЕПТОРОВ Изобретение предлагает соединение формулы (I) Гавардинас Константинос, Джадхав Прабхакар Кондаджи (US) и способы лечения физиологических расстройств, в частности застойной сердечной недостаточности, гипертензии, диабетической нефропатии или хронического заболевания почек. Настоящее изобретение относится к нестероидным антагонистам минералокортикоидных рецепторов.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) 017668 Изобретение относится к трициклическим соединениям, подходящим в качестве терапевтических агентов в лечении физиологических расстройств, реагирующих на антагонисты минералокортикоидных рецепторов, фармацевтическим композициям, содержащим указанные соединения, способам применения указанных соединений для лечения физиологических расстройств у пациентов и промежуточным соединениям и способам, подходящим для синтеза указанных соединений. Альдостерон, основной эндогенный минералокортикоид, регулирует гемодинамический гомеостаз путем промотирования реабсорбции натрия и воды и выведения калия после взаимодействия с минералокортикоидным рецептором (МР). Благодаря роли альдостерона в поддержании электролитного и водного баланса, антагонистов МР использовали для лечения многочисленных физиологических расстройств, включая гипертензию, гипокалиемию, миокардиальную аритмию, синдром Барттера, а также расстройства при первичном или вторичном гиперальдостеронизме, такие как синдром Конна. В последнее время антагонистов МР используют в лечении застойной сердечной недостаточности и острого инфаркта миокарда. Кроме того, антагонисты МР также доказали свою эффективность в доклинических моделях заболевания почек и в комбинации со стандартной терапией для снижения протеинурии у пациентов, страдающих от расстройств почек, таких как хроническое заболевание почек, включая диабетическую нефропатию. Однако существующие антагонисты МР оказывают сопутствующие эффекты, ограничивающие их безопасность и/или эффективность. Например, спиронолактон, сильный антагонист МР, является неселективным и вступает в перекрестную реакцию с другими ядерными гормональными рецепторами (например, андрогенным рецептором (АР), прогестероновым рецептором (ПР) или глюкокортикоидным рецептором (ГР, которые опосредуют другие физиологические процессы. Терапия спиронолактоном была связана с гиперкалиемией, а также гинекомастией, эректильной дисфункцией, пониженным либидо,нерегулярным менструальным циклом, а также желудочным расстройством. Эплеренон, несмотря на то что является селективным в отношении МР относительно других ядерных гормональных рецепторов,также был связан с гиперкалиемией. Таким образом, в данной области техники остается необходимость в разработке альтернатив существующей терапии антагонистами МР. Задачей настоящего изобретения является получение нестероидных лигандов МР, обладающих антагонистической активностью в отношении МР. В предпочтительном варианте реализации задачей является получение нестероидных антагонистов МР, которые связываются с МР с большей аффинностью,чем с АР, ПР и ГР. В более предпочтительном варианте реализации задачей настоящего изобретения является получение нестероидных антагонистов МР, которые связываются с МР с большей аффинностью,чем с АР, ПР и ГР и которые обладают сильной рено-и кардиопротективной активностью. В еще более предпочтительном варианте реализации задачей настоящего изобретения является получение нестероидных антагонистов МР, которые связываются с МР с большей аффинностью, чем с АР, ПР и ГР и которые обладают сильной рено- и кардиопротективной активностью, но сниженной частотой или вероятностью гиперкалиемии. Важный вопрос относительно любого терапевтического агента заключается в том, способен ли указанный агент вызывать пролонгацию интервала QT. Основной механизм, посредством которого терапевтические агенты могут индуцировать пролонгацию интервала QT, заключается в блокировании каналовhERG в сердечной мышце. Блокирование канала hERG препятствует прохождению ионов калия через мембраны клеток сердца, приводя к пролонгации потенциалов действия клеток, что может привести к опасной аритмии сердца. Таким образом, другой предпочтительный вариант реализации настоящего изобретения заключается в получении антагонистов МР, обладающих сниженной частотой или вероятностью блокирования каналов hERG. В данной области техники известны трициклические лиганды МР. Например, в WO 04/052847 иWO 05/066161 описаны трициклические модуляторы стероидных гормональных рецепторов, подходящие для лечения расстройств, чувствительных к модуляции минералокортикоидных рецепторов или глюкокортикоидных рецепторов. Настоящее изобретение относится к обнаружению того факта, что некоторые новые трициклические соединения, определяемые формулой (I) ниже, обладают конкретными характеристиками активности, как подтверждено тестированием in vitro и in vivo, которые свидетельствуют о том, что они полезны в лечении или предупреждении расстройств, реагирующих на терапию антагонистами минералокортикоидных рецепторов. В частности, приведенные в качестве примера соединения формулы (I) представляют собой сильные лиганды МР, которые вызывают антагонизм к минералокортикоидному рецептору. Соответственно, согласно настоящему изобретению предложено соединение формулы (I) где R1 и R2 каждый независимо представляет собой водород или фтор;R3 представляет собой водород или группу формулы:R4 представляет собой -CN или -C(O)NH2,или его фармацевтически приемлемая соль. В другом варианте реализации настоящего изобретения предложен способ лечения или предупреждения застойной сердечной недостаточности, диабетической нефропатии, хронического заболевания почек, гипертензии, гипокалиемии, миокардиальной аритмии, синдрома Барттера, первичного или вторичного гиперальдостеронизма или синдрома Конна, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Согласно более конкретному аспекту настоящего изобретения предложен способ лечения или предупреждения застойной сердечной недостаточности, гипертензии, диабетической нефропатии или хронического заболевания почек. Также согласно настоящему изобретению предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении или предупреждении застойной сердечной недостаточности, диабетической нефропатии, хронического заболевания почек, гипертензии, гипокалиемии,миокардиальной аритмии, синдрома Барттера, первичного или вторичного гиперальдостеронизма или синдрома Конна. Более конкретно, согласно настоящему изобретению предложено соединение формулы(I) или его фармацевтически приемлемая соль для применения в лечении или предупреждении застойной сердечной недостаточности, гипертензии, диабетической нефропатии или хронического заболевания почек. Кроме того, согласно настоящему изобретению предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии. В другом варианте реализации настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или предупреждения застойной сердечной недостаточности, диабетической нефропатии, хронического заболевания почек, гипертензии, гипокалиемии, миокардиальной аритмии, синдрома Барттера, первичного или вторичного гиперальдостеронизма или синдрома Конна. Более конкретно, согласно настоящему изобретению предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или предупреждения застойной сердечной недостаточности, гипертензии, диабетической нефропатии или хронического заболевания почек. Кроме того, согласно настоящему изобретению предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или эксципиентами. Более конкретно, согласно настоящему изобретению предложена фармацевтическая композиция для лечения или предупреждения застойной сердечной недостаточности, гипертензии, диабетической нефропатии или хронического заболевания почек, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с одним или несколькими фармацевтически приемлемыми носителями,разбавителями или эксципиентами. Настоящее изобретение также включает новые промежуточные соединения и способы, подходящие для синтеза соединения формулы (I). Настоящее изобретение также относится к сольватам соединения формулы (I) или фармацевтически приемлемым солям соединений формулы (I). Поэтому, в настоящем описании термин "Формула (I)" или любое конкретное соединение формулы (I) включает в рамках своего значения любой сольват указанного соединения. Примеры фармацевтически приемлемых солей и способы их получения хорошо известны специалистам в данной области техники. См., например, Stahl et al., "Handbook of Pharmaceutical Salts:Pharmaceutical New Chemical Entities," Organic Process Research and Development, 4: 427-435 (2000). Особое внимание уделяется гидрохлоридным, малеатным и мезилатным солям соединений формулы (I).-2 017668 Соединения согласно настоящему изобретению могут содержать один или несколько хиральных центров и, следовательно, могут существовать во множестве стереоизомерных конфигураций. Вследствие наличия данных хиральных центров соединения согласно настоящему изобретению могут встречаться в виде рацематов, смесей энантиомеров и индивидуальных энантиомеров, а также диастереомеров и смесей диастереомеров. За исключением случаев, которые специально указаны в настоящем описании,все указанные рацематы, энантиомеры и диастереомеры входят в объем настоящего изобретения. Энантиомеры соединений, предложенных согласно настоящему изобретению, могут быть разделены, например, специалистом в данной области техники с использованием стандартных технологий, таких как технологии, описанные J. Jacques, et al., "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, Inc.,1981, а также технологий, приведенных в разделах "Схемы", "Синтезы" и "Примеры" в настоящем описании. Конкретные стереоизомеры и энантиомеры соединений формулы I могут быть получены специалистом в данной области техники с использованием хорошо известных технологий и способов, таких как описаны Eliel and Wilen, "Stereochemistry of Organic Compounds", John WileySons, Inc., 1994, Глава 7;Resolutions", John WileySons, Inc., 1981, а также технологий, приведенных в разделах "Схемы", "Синтезы" и "Примеры" в настоящем описании. Например, конкретные стереоизомеры и энантиомеры могут быть получены путем стереоспецифического синтеза с использованием энантиомерно и геометрически чистых или энантиомерно или геометрически обогащенных исходных веществ. Кроме того, конкретные стереоизомеры и энантиомеры могут быть разделены и выделены с помощью технологий, таких как хроматография на хиральных неподвижных фазах, ферментативное разделение или фракционированная перекристаллизация аддитивных солей, образованных реагентами, используемыми для этой цели. В настоящем описании термины "R" и "S" используются так, как обычно используются в органической химии для обозначения конкретных конфигураций хирального центра. Термины , "R/S" или"RS" относятся к рацемической конфигурации хирального центра. Неполный перечень приоритетов и рассмотрение стереохимии содержится в "Nomenclature of Organic Compounds: Principles and Practice",(J.H. Fletcher, et al., eds., 1974). Как очевидно для специалиста в данной области техники, молекулы, содержащие двойную связь углерод-углерод или углерод-азот, могут существовать в виде геометрических изомеров. Для обозначения конкретных изомеров обычно используют два способа: способ "цис-транс" и способ "Е и Z" в зависимости от того, являются ли группы, присоединенные к каждому из соединенных двойной связью атомов, одинаковыми или разными. Рассмотрение геометрической изомерии и способов названия конкретных изомеров приведено в March, "Advanced Organic Chemistry", John WileySons,1992, глава 4. Все указанные геометрические изомеры, а также смеси индивидуальных изомеров предусмотрены и предложены согласно настоящему изобретению. Соединения согласно настоящему изобретению могут быть получены в виде части фармацевтической композиции. Поэтому, фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с фармацевтически приемлемым носителем, разбавителем или эксципиентом, представляет собой вариант реализации настоящего изобретения, имеющий важное значение. Примеры фармацевтических композиций и способы их получения хорошо известны в данной области техники. См., например, REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY (A. Gennaro, et al., eds., 19th ed., Mack Publishing (1995. Иллюстративные композиции, содержащие соединения формулы (I), включают, например: соединение формулы (I) в суспензии, содержащей 0,5% карбоксиметилцеллюлозы, 0,25% Полисорбата 80 и 2,7% NaCl; или соединение формулы (I) в суспензии, содержащей 1% карбоксиметилцеллюлозы и 0,25% Полисорбата 80; или соединение формулы (I) в суспензии, содержащей 1% карбоксиметилцеллюлозы и 0,25% Полисорбата 80 и 0,05% AntiFoam 1510 в очищенной воде. Однако следует понимать, что предпочтительная композиция согласно настоящему изобретению содержит соединение формулы (I) или его фармацевтически приемлемую соль,полученную в капсуле или таблетке. Соединение формулы (I) или композиция, содержащая соединение формулы (I), могут быть введены любым способом, в результате которого указанное соединение становится биодоступным, включая пероральные и парентеральные способы введения. Для специалиста в данной области техники очевидно, что размер частиц может влиять на растворение in vivo фармацевтического агента, которое в свою очередь может влиять на абсорбцию указанного агента. В настоящем описании термин "размер частиц" относится к диаметру частицы фармацевтического агента, определяемому стандартными технологиями, такими как рассеяние лазерного излучения, лазерная дифракция, рассеяние Ми, седиментационное фракционирование в потоке под действием поля,фотон-корреляционная спектроскопия и т.п. В случае когда фармацевтические агенты обладают плохой растворимостью, небольшие или уменьшенные размеры частиц могут способствовать растворению и таким образом увеличивать абсорбцию указанного агента. Amidon et al., Pharm.Research, 12; 413-420(1995). Способы уменьшения или регулирования размера частиц являются стандартными и включают размол, мокрое измельчение, микронизацию и т.п. Другой способ регулирования размера частиц включа-3 017668 ет приготовление фармацевтического агента в наносуспензии. Конкретный вариант реализации настоящего изобретения включает соединение формулы (I) или фармацевтическую композицию, содержащую соединение формулы (I), при этом указанное соединение обладает размером частиц d90 (т.е. размером,при котором 90% частиц меньше или равны) менее примерно 50 мкм. Более конкретный вариант реализации включает соединение формулы I, обладающее размером частиц d90 менее примерно 10 мкм. В настоящем описании термин "пациент" относится к человеку или млекопитающему кроме человека, такому как собака, кошка, корова, обезьяна, лошадь или овца. Более конкретно, термин "пациент" относится к человеку. В настоящем описании термин "лечение" (или "лечить") включает предотвращение, предупреждение, ограничение, замедление, прекращение или изменение развития или тяжести имеющегося симптома или расстройства. В настоящем описании термин "предупреждение" (или "предупреждать") относится к предотвращению, ограничению или блокированию частоты или появлению симптома или расстройства. Как очевидно для специалиста в данной области техники, физиологические расстройства могут представлять собой "хроническое" состояние или "острый" случай. Таким образом, лечение расстройств предусматривает как острые случаи, так и хронические состояния. В остром случае соединение вводят при появлении симптомов и прекращают при исчезновении симптомов, в то время как хроническое состояние лечат на протяжении всего течения заболевания. В настоящем описании термин "эффективное количество" относится к количеству или дозе соединения формулы (I), которая при введении однократных доз или доз для многократного введения пациенту обеспечивает необходимый эффект у пациента, подвергаемому диагностике или лечению. Эффективное количество может быть легко определено лечащим диагностом, как специалистом в данной области техники с учетом ряда факторов, таких как вид млекопитающего; его размер, возраст и общее состояние здоровья; конкретное рассматриваемое заболевание; степень или тяжесть заболевания; реакция индивидуального пациента; конкретное вводимое соединение; способ введения; характеристики биодоступности вводимого лекарственного средства; выбранный режим дозирования; и одновременное применение каких-либо лекарственных средств. При использовании совместно со способами и применением согласно настоящему изобретению соединения и композиции согласно настоящему изобретению могут быть введены либо отдельно, либо в комбинации со стандартными терапевтическими агентами, используемыми для лечения конкретного заболевания или состояния. В случае, когда соединения или композиции согласно настоящему изобретению применяют как часть комбинации, соединение или композиция, содержащая соединение формулы(I), может быть введена отдельно или как часть состава, содержащего терапевтический агент, с которым его комбинируют. В настоящем описании обозначение "" относится к связи, которая выступает вперед за плоскость страницы, в то время как обозначение "" относится к связи, которая выступает назад за плоскость страницы. Конкретные аспекты настоящего изобретения Следующие перечни определяют несколько групп конкретных конфигураций, заместителей и переменных для соединений формулы (I). Следует понимать, что соединения формулы (I), обладающие указанными конкретными конфигурациями, заместителями или переменными, а также способы, применение и композиции, содержащие данные соединения, представляют собой конкретные аспекты настоящего изобретения. Таким образом, конкретный аспект настоящего изобретения представляет собой аспект, согласно которому соединение формулы (I) представляет собой соединение, где L, R3 и R4 имеют любое из значений, определенных в настоящем описании, и:(a) R1 представляет собой водород, a R2 представляет собой водород или фтор; или(b) R1 представляет собой фтор, a R2 представляет собой водород или фтор; или(c) R1 представляет собой водород или фтор, a R2 представляет собой водород; или(d) R1 представляет собой водород или фтор, a R2 представляет собой фтор; или(e) R1 и R2 каждый независимо представляет собой водород. Дополнительные конкретные аспекты настоящего изобретения представляют собой аспекты, согласно которым соединение формулы (I) представляет собой соединение, где R1, R2, R3 и R4 имеют любое из значений, определенных в настоящем описании, и:(c) L представляет собой прямую связь. Дополнительные конкретные аспекты настоящего изобретения представляют собой аспекты, согласно которым соединение формулы (I) представляет собой соединение, где R1, R2, L и R4 имеют любое из значений, определенных в настоящем описании, и: (a) R3 представляет собой водород или группу формулы(b) R3 представляет собой группу формулы Другие конкретные аспекты настоящего изобретения представляют собой аспекты, согласно которым соединение формулы (I) представляет собой соединение, где R1, R2, L и R4 имеют любое из значений, определенных в настоящем описании, и: (a) R3 представляет собой водород или группу формулы(b) R3 представляет собой группу формулы(с) R3 представляет собой группу формулы Дополнительные конкретные аспекты настоящего изобретения представляют собой аспекты, согласно которым соединение формулы (I) представляет собой соединение, где R1, R2, L и R3 имеют любое из значений, определенных в настоящем описании, и:(b) R4 представляет собой -C(O)NH2,Более конкретный аспект настоящего изобретения представляет собой аспект, согласно которому соединение формулы (I) представляет собой соединение, гдеR1 и R2 каждый независимо представляет собой водород или фтор;L представляет собой -CH(CH3)-CH2- или прямую связь; R3 представляет собой водород или группу формулы:R4 представляет собой -CN или -C(O)NH2,или его фармацевтически приемлемую соль. Еще более конкретный аспект настоящего изобретения представляет собой аспект, согласно которому соединение формулы (I) представляет собой соединение, гдеR1 и R2 каждый независимо представляет собой водород или фтор;-СН(СН 3)-СН 2-, то R3 представляет собой группу формулы: и если L представляет собой прямую связь, то R3 представляет собой группу формулы:R4 представляет собой -CN или -C(О)NH2,-6 017668 или его фармацевтически приемлемую соль. Дополнительные конкретные аспекты настоящего изобретения определены соединениями формулыI(а) и формулы I(b) ниже. Следует понимать, что соединения формулы I(а) и формулы I(b), a также способы и применение указанных соединений и композиции, содержащие указанные соединения, представляют собой дополнительные аспекты настоящего изобретения. Таким образом, конкретный аспект настоящего изобретения определен соединением формулы I(а): где R1 и R2, каждый независимо, представляет собой водород или фтор;L представляет собой -СН(СН 3)-СН 2- или прямую связь; и R3 представляет собой водород или группу формулы: или его фармацевтически приемлемой солью. Еще более конкретный аспект настоящего изобретения представляет собой соединение формулы IR1 и R2, каждый независимо, представляет собой водород или фтор; и-СН(СН 3)-СН 2-, то R3 представляет собой группу формулы: и если L представляет собой прямую связь, то R3 представляет собой группу формулы: или его фармацевтически приемлемую соль. Еще один конкретный аспект настоящего изобретения определен соединением формулы I(b): где R1 и R2 каждый независимо представляет собой водород или фтор;L представляет собой -СН(СН 3)-СН 2- или прямую связь; и R3 представляет собой группу формулы: или его фармацевтически приемлемой солью. Еще более конкретный аспект настоящего изобретения представляет собой соединение формулыI(b), где R1 и R2 каждый независимо представляет собой водород или фтор; и(СН 3)-СН 2-, то R3 представляет собой группу формулы: и если L представляет собой прямую связь, то R3 представляет собой группу формулы: или его фармацевтически приемлемую соль. Кроме того, наиболее конкретный аспект настоящего изобретения определен соединениями формулы (I), приведенными в качестве примера в настоящем описании, главным образом(Е)-N-(5-Е)-3-фтор-6H-дибензо[b,е]оксепин-11-илиденметил)-1-7S,8aR)-гескагидропирроло[2,1c][1,4]оксазин-7-ил)-1,3-дигидробензимидазол-2-илиден)мочевиной. Соединения формулы (I) можно получить посредством различных процедур, известных в данной области техники, а также процедур, описанных в схемах, синтезах и примерах. Однако нижеследующее обсуждение не предполагает каким-либо образом ограничить объем настоящего изобретения. Например,отдельные стадии синтеза для каждого из описанных способов можно комбинировать различным образом, или сочетать со стадиями из различных схем, для получения дополнительных соединений формулы(I). Продукты, полученные на каждой стадии согласно схемам, представленным ниже, можно выделить обычными способами, в том числе, посредством экстракции, осаждения, испарения, хроматографии,фильтрации, растирания в порошок, кристаллизации и т.п. Заместители, если не указано иначе, определены ранее. Реагенты и исходные материалы легко доступны обычному специалисту в данной области техники. Другие необходимые реагенты и исходные материалы можно получить с помощью процедур, которые выбирают из стандартных методов органической и гетероциклической химии, методов, которые аналогичны способам синтеза известных структурно похожих соединений, и процедур, описанных в приведенных ниже синтезах и примерах, в том числе,любых новых процедур. В настоящем документе, следующие термины имеют указанные значения: "МеОН" относится к метанолу; "EtOH" относится к этанолу; "EtOAc" относится к этилацетату; "ДМФ" относится к диметилформамиду; "ДМСО" относится к диметилсульфоксиду; "ТФК" относится к трифторуксусной кислоте; Получение промежуточного соединения формулы (3) можно осуществить согласно реакциям, как изображено на схеме 1. На схеме 1, амин формулы (1) реагирует с 4-бром-1-фтор-2-нитробензолом согласно реакции вытеснения при нуклеофильном ароматическом замещении (SNAr) с получением аминонитрофенилбромида формулы (3). Например, амин (1) может реагировать в инертном растворителе, таком как этилацетат,ТГФ или протоносодержащем растворителе, таком как этанол, при температуре в диапазоне от комнатной до температуры флегмы растворителя. Квалифицированному специалисту понятно, что соединения формулы (1) можно легко получить способами, аналогичными способам, описанным в настоящем документе, или путем применения процедур, которые приняты в данной области техники. Например, метилзамещенный или незамещенный морфолинилэтиламин можно получить путем мезилирования N-трет-бутилкарбоксилэтаноламина с последующей заменой мезилата на морфолин для получения требуемого морфолинилэтиламина. Также является очевидным, что выбор и применение подходящих защитных групп хорошо известно и используется в данной области техники (см., например, Protecting Groups in Organic Synthesis, TheodoraGreene (Wiley-Interscience. Аминовая функциональная группа, такая как группа, присутствующая в азетидиниловой группировке, может быть лишена защиты и впоследствии может еще реагировать с получением дополнительных соединений согласно изобретению. Например, после снятия защиты третбутилового эфира 3-(4-бром-2-нитрофениламино)азетидин-1-карбоновой кислоты в кислотных условиях,азетидин можно алкилировать при восстановительном аминировании, например, с применением формальдегида, с получением N-метилазетидинильного промежуточного соединения. Схема 2 Получение промежуточного соединения формулы (9) можно осуществить согласно реакциям, как изображено на схеме 2. На схеме 2, стадия 1, эфир формулы (4) восстанавливают до спирта формулы (5). Специалисту в данной области техники известно, что существует целый ряд способов восстановления эфиров. Например, эфир формулы (4) обрабатывают борогидридом лития примерно при от 0 до 40 С в инертном растворителе, таком как диэтиловый эфир. Гашение реакции через 10 мин позволяет выделить спиртовой азид формулы (5), который затем восстанавливают до амина на стадии 2. Восстановление азида можно выполнить в условиях каталитического гидрирования, общепринятых в данной области техники, и получить аминоспирт формулы (6). На схеме 2, стадия 3, 4-бром-1-фтор-2-нитробензол подвергают реакции SNAr с амином формулы(6), как описано ранее для схемы 1. На стадии 4, с фениламинопирролидина формулы (7) снимают защиту путем применения кислотных условий, например, используя хлористый водород или трифторуксусную кислоту, и продукт циклизуют на стадии 5 с получением оксезинона формулы (8). Циклизацию выполняют, применяя хлорацетилхлорид в присутствии неорганического основания, такого как гидроксид натрия, поддерживая рН между 10-12. Реакция протекает в смеси ТГФ и воды (примерно 1:1).-9 017668 На схеме 2, стадия 6, оксезинон формулы (8) восстанавливают до оксезина формулы (9). Карбонильную функциональную группу восстанавливают путем применения комплекса боран - тетрагидрофуран или комплекса боран - диметилсульфид в инертном растворителе, таком как ТГФ, при температуре в диапазоне примерно от 0 С до температуры флегмы растворителя. Хиральные пирролидины формулы (4) известны в данной области техники и могут быть легко получены из стереоизомеров 3-гидроксипролина. Спирт можно мезилировать и затем вытеснить путем стереохимической инверсии с образованием азида. Альтернативным образом, спирт можно превратить в бромид в условиях Мицунобу (Mitsinobu), применяя тетрабромид углерода, путем стереохимической инверсии с последующей заменой на ион азида для повторной стереохимической инверсии. Трансгидроксипролин можно превратить в цис-конфигурацию путем обработки уксусной кислотой и уксусным ангидридом при 90 С (С. G. Levins и С. Е. Schafmeister J. Org. Chem., 2005, 70, 9002-9008). Схема 3 Получение промежуточного соединения формулы (14 а и b) или (17 а и b) можно осуществить согласно реакциям, как изображено на схеме 3. На схеме 3, стадия 1 а, рацемический транс-гидроксибензиламинпирролидин формулы (10) можно разделить путем применения подходящей энантиомерно чистой кислоты. Например, обработка (+)миндальной кислотой в подходящем растворителе, таком как ацетонитрил, с применением примерно 1% воды, приводит к селективной перекристаллизации соли (S, S)гидроксибензиламин(+)манделата при очень высоком диастереомерном избытке. Затем соль можно нейтрализовать путем применения подходящего основания, такого как водный карбонат калия. На стадии 1b, маточный раствор, полученный в результате вышеуказанной перекристаллизации, теперь обогащенный (R, R)-энантиомером, можно выпарить и обработать (-) миндальной кислотой в растворителе, таком как ацетонитрил, содержащем примерно 2,5% воды, и получить очищенный (R, R)-энантиомер. На стадии 2, воздействие на свободный (S, S) или (R, R) гидроксибензиламин стандартными условиями гидрирования с применением обычного катализатора, такого как палладий на углероде, в обычном растворителе, таком как метанол, н-бутанол или тетрагидрофуран, позволяет получить соответствующий(S, S) или (R, R) гидроксиламин. На схеме 3, стадия 3, реакция SNAr образовавшегося аминопирролидина формулы (12 а или b) с 4 бром-1-фтор-2-нитробензолом обеспечивает образование фениламинопирролидина формулы (13 а или b). Реакцию можно осуществить в присутствии подходящего основания, такого как триэтиламин, в обычном растворителе, таком как тетрагидрофуран или этилацетат. На схеме 3, стадия 4, хиральность гидроксильной группы пирролидина формулы (13 а или b) превращают из (S, S) и (R, R) в (3R, 4S) и (3S, 4R), соответственно. Хиральное превращение осуществляют,применяя реакцию Мицунобу. Квалифицированный специалист знает, что существуют различные условия Мицунобу, применяемые в данной области техники. Например, спирт формулы (15 а или b) растворяют в подходящем безводном растворителе подобно ТГФ, CH2Cl2, толуолу и т.д., и обрабатывают три- 10017668 алкил- или триарилфосфином, таким как Me3P, Bu3P, или Ph3P, и диалкилазодикарбоксилатом, таким какDEAD или DIAD. На стадии 5, 3S, 4R и 4S, SR-хлорацетокси эфир формулы (15 а или b) гидролизуют с получением гидроксипирролидина формулы (16 а или b). Гидролиз осуществляют путем применения неорганического основания, такого как водный гидроксид лития, в растворителе, таком как метанол, при 0-50 С в течение 4-24 ч. На схеме 3, стадия 6, с четырех диастереомерных аминопирролидинов формулы (13 а или b) и формулы (16 а или b) снимают защиту, применяя кислотные условия. Снятие трет-бутоксикарбонильных защитных групп хорошо известно в данной области техники. При предпочтительных условиях применяют инертный растворитель, такой как этилацетат, обработанный безводным хлористым водородом (газ) примерно при 0 С в течение 10 мин. Образовавшийся NH-пирролидин подвергают воздействию условиями восстановительного аминирования и получают N-метилпирролидин формулы (14 а или b) и формулы (17 а или b). Реакцию проводят в инертном растворителе, таком как дихлорметан, ТГФ или, предпочтительно, ацетонитрил, и обрабатывают водным формальдегидом. Реакцию обрабатывают восстановителем, таким как борогидрид натрия, цианоборогидрид натрия или, предпочтительно, триацетоксиборогидрид натрия, примерно при 0-40 С в течение 4-24 ч. Квалифицированному специалисту понятно, что соединения формулы (10) можно легко получить способами, аналогичными способам, описанным в настоящем документе, или путем применения процедур, которые приняты в данной области техники. Например, 3-пирролин можно защитить с помощью дитрет-бутилдикарбоната и затем бромировать в ДМСО/вода с получением транс-3-бром-4 гидроксипирролидина (A. Kamal et. al. TeTpahedron Lett., 2004, 45, 8057-8059; A. Kamal et. al. TeTpahedron Asymmetry, 2006, 17, 2876-2883). При реакции с гидроксидом натрия образуется in situ эпоксид, который реагирует с бензиламином с получением рацемического транс-гидроксибензиламинпирролидина формулы (10). Более конкретно, эпоксид может непосредственно образоваться и выделяться при окислении 3-пирролина посредством МСРВА. Согласно еще одному альтернативному способу синтеза, 2,4 дихлор-2-бутен может реагировать с трет-бутилкарбаматом с образованием 1-Вос-3-пирролина (Y. Tsuzuki, et. al. TeTpahedron Asymmetry, 2001, 12, 2989-2997). Защитную t-Boc-группу можно полностью удалить путем проведения реакции с метиламином вместо трет-бутилкарбамата. Последующее окисление до эпоксида посредством МСРВА и разрыв (связи) с помощью аммиака позволяет получить рацемический транс-4-амино-1-метилпирролидин-3-ол. Он может реагировать с 1-бром-4-фтор-3-нитробензолом с получением соединения формулы (14 аb) в виде смеси энантиомеров. Разделение этой энантиомерной смеси можно осуществить способами, известными в данной области техники, такими как хиральная ВЭЖХ или перекристаллизация с солью энантиомерно чистой карбоновой кислоты, такой как D-(-) или На схеме 4, стадия 1, транс-аминогидроксипиперидин формулы (18) подвергают восстановительному аминированию с бензальдегидом и получают бензиламинопиперидин формулы (19). Квалифицированный специалист понимает, что существуют различные условия, которые подходят для проведения восстановительного аминирования. Например, реакция протекает в растворителе, таком как этанол, в присутствии уксусной кислоты и восстановителя, такого как цианоборогидрид натрия. На схеме 4, стадия 2 а, рацемический транс-гидроксибензиламинпиперидин формулы (19) разделяют путем применения подходящей энантиомерно чистой кислоты. Например, обработка (+)-винной кислотой в подходящем растворителе, таком как ацетонитрил, с применением примерно 5% воды, приводит к селективной перекристаллизации (3R, 4R) гидроксибензиламин(+)тартрата формулы (20 а) при очень высоком диастереомерном избытке. Подобным образом, на стадии 2b, рацемическое вещество перекристаллизовывают посредством (-) винной кислоты в растворителе, таком как ацетонитрил, содержа- 11017668 щем примерно 2,5% воды, и получают очищенный (S, S) энантиомер формулы (20b). На стадии 3, разделенные 3R, 4R и 3S, 4S энантиомеры формулы (20 а) и (20b), соответственно,можно дебензилировать путем каталитического гидрирования с образованием аминогидроксипиперидина формулы (21 а или b). В начале, тартрат промывают раствором карбоната натрия или калия, а затем экстрагируют дихлорметаном для удаления винной кислоты. Затем свободный бензиламин гидрируют в инертном растворителе, таком как этанол или метанол, применяя 5 или 10% палладий на углероде. На схеме 4, стадия 4, аминогидроксипиперидин формулы (21 а или b) превращают в фениламинопиперидин формулы (22 а или b), как показано на схеме 3, стадия 3. На стадии 5, с фениламинопиперидина формулы (22 а или b) снимают защиту и алкилируют путем восстановительного аминирования с образованием N-метилпиперидина формулы (23 а или b). Реакция протекает в смеси растворителей из уксусной кислоты и воды в присутствии формальдегида и восстановителя, такого как цианоборогидрид натрия. 4-Амино-3-гидроксипиперидины формулы (18) известны в данной области техники и могут быть легко получены из 4-пиперидона путем восстановления до 4-гидроксипиперидина, защищенного boc или другой защитной группой. Спирт можно мезилировать, расщеплять до олефина и затем эпоксидировать с получением рацемического трет-бутилового эфира цис-7-окса-3-азабицикло[4.1.0]гептан-3-карбоновой кислоты. Затем эпоксид раскрывают с помощью аниона азида для осуществления транс-региохимии, с последующим восстановлением и получением аминогидроксилпиперидина формулы (18). Схема 5 Получение соединения формулы (30) или (31) можно осуществить согласно реакциям, как изображено на схеме 5. Подходящее соединение формулы (24) представляет собой соединение, где R1 и R2 определены при описании формулы (I) и X = Br или I. Подходящие соединения формулы (30) или (31) представляют собой соединения, в которых R1, R2, L, и R3 определены при описании формулы (I). На схеме 5, стадия 1 и стадия 2, галобензилфениловый эфир формулы (24) подвергают двойной реакции сочетания Хека посредством метил- или этилакрилата с получением винилового эфира формулы(26) с Е региохимией при двойной связи. Реакция происходит в присутствии палладиевого катализатора,такого как тетракис(трифенилфосфин)палладий (0), хлорид палладия или, предпочтительно, ацетат палладия (II). В реакции участвует основание, такое как триэтиламин, карбонат калия, ацетат натрия или четвертичная аммониевая соль, такая как тетра-н-бутиламмоний бромид. Реакция протекает в инертном растворителе, таком как ДМФ или N-метилпирролидин, при температуре примерно от 40 до 130 С в течение 4-48 ч с получением промежуточного соединения формулы (25). Это соединение выделяют и затем циклизуют с образованием винилового эфира формулы (26), по существу, при сходных реакционных условиях при более высокой температуре, такой как 90-150 С. Альтернативным образом, галобензилфениловый эфир формулы (24) можно применять непосредственно с виниловым эфиром формулы (26) в одном сосуде без выделения промежуточного соединения формулы (25) при температуре примерно от 90 до 150 С. На стадии 3 эфир формулы (26) гидролизуют и превращают в винилбромид формулы (27). Гидролиз можно осуществить путем применения неорганического основания, такого как гидроксид калия, натрия или, предпочтительно, лития. Затем образовавшуюся карбоновую кислоту можно обработать уксусной кислотой и NBS при температуре от 40 до 100 С. На схеме 5, стадия 4, винилбромид формулы (27) превращают в винилпинаколборонат формулы(28). Винилбромид реагирует с бис(пинаколато)дибором в присутствии основания, такого как ацетат цезия или калия, в инертном растворителе, таком как ТГФ, 1,2-диметоксиэтан или, предпочтительно 1,4 диоксан. Реакцию проводят при температуре в диапазоне примерно от 40 С до температуры флегмы рас- 12017668 творителя, в присутствии палладиевого катализатора, такого как дихлорид 1,1'-бис(дифенилфосфино) ферроцен палладия (II) (Pd2(dppf)Cl2) или, альтернативным образом, трис(дибензилиденацетон)дипалладий(0) (Pd2(dba)3), в присутствии фосфинового лиганда, такого как трициклогексилфосфин. На стадии 5 винилпинаколборонат формулы (28) реагирует с аминонитробромбензолом в условиях реакции кросс-сочетания Сузуки с получением винилнитрофенильного промежуточного соединения формулы (29). Квалифицированный специалист знает, что существуют различные условия, применимые для стимулирования таких реакций кросс-сочетания. Реакционные условия включают применение подходящего растворителя, такого как диоксан/вода или ТГФ/метанол. Реакцию проводят в присутствии основания, такого как карбонат натрия, метоксид натрия, карбонат калия или ацетат калия. Реакция происходит в присутствии палладиевого катализатора, такого как тетракистрифенилфосфин палладий (0), в инертной атмосфере при температуре примерно 70-120 С в течение примерно 8-24 ч. На схеме 5, стадии 6 и 7, винилнитрофенильное промежуточное соединение формулы (29) восстанавливают до диаминофенильного промежуточного соединения (не показано) при стандартных условиях гидрирования с применением 5 или 10% платины на углероде или 5 или 10% палладия на углероде в подходящем растворителе, таком как тетрагидрофуран, этилацетат или изопропанол. После удаления катализатора диаминофенильное промежуточное соединение можно превратить в цианогуанидин формулы (30), применяя дифенил-N-цианокарбонимидат в диапазоне от комнатной температуры до 100 С. Применяют инертный растворитель, такой как изопропанол, пиридин или ТГФ, с неорганическим основанием, таким как бикарбонат натрия, или без него. Очистку конечного продукта можно осуществить способами, принятыми в данной области техники, такими как хроматография или перекристаллизация. На схеме 5, стадия 8, цианогуанидин формулы (30) гидролизуют в кислотных условиях с получением мочевины формулы (31). Реакция протекает с применением 4 н. хлористо-водородной кислоты в диоксан/вода или в ТФК/вода в течение 4-72 ч в диапазоне от комнатной до температуры примерно 80 С. Квалифицированному специалисту понятно, что соединения формулы (24) можно легко получить способами, аналогичными способам, описанным в настоящем документе, или путем применения процедур, которые приняты в данной области техники. Например, 2-бром-5-фторфенол или 2-бром-5 хлорфенол алкилируют посредством 2-иод- или 2-бромбензилбромида с получением фенилового эфира формулы (24). Соединения формулы (24), где R1 или R2=F, требуемый бензилбромид получают посредством, например, литирования 1-бром-3-фторбензола и реакции с N-метил-N-фенилформамидом для получения альдегида. Последующее восстановление до бензилового спирта и реакция с бромирующим реагентом, таким как фосфорный трибромид, позволяет получить 1-бром-3-фторбензилбромид. Определение биологической активности В настоящем описании "Kd" обозначает равновесную константу диссоциации для комплекса лиганд-рецептор; "Ki" обозначает равновесную константу диссоциации для комплекса лекарственное вещество-рецептор и представляет собой концентрацию лекарственного средства, при которой связана половина всех сайтов связывания при равновесии; "Kb" обозначает равновесную константу диссоциации для комплекса антагонист-рецептор; "IC50" обозначает концентрацию агента, обеспечивающую 50% максимально возможной ингибирующей реакции этого агента, или, альтернативно, концентрацию агента, обеспечивающую 50%-ное замещение лиганда, связывающегося с рецептором, "ЕС 50" Обозначает концентрацию агента, обеспечивающую 50% максимально возможной реакции агента, и "ED50" обозначает дозу вводимого терапевтического агента, обеспечивающую 50%-ную максимальную реакцию этого агента. Анализ связывания ядерного рецептора стероидных гормонов Лизаты клеток линии мезонефроса человека НЕК 293 со сверхэкспрессией MR человека (минералокортикоидный рецептор (МР, GR человека (глюкокортикоидный рецептор (ГР, AR человека (андрогенный рецептор (АР или PR человека (прогестероновый рецептор (ПР использовали для анализа конкурентного связывания рецептора с лигандом для определения значений Ki. В общих чертах, анализы конкурентного связывания стероидного рецептора проводят в буферном растворе, содержащем 20 мМ буфера HEPES (рН 7,6), 0,2 мМ EDTA, 75 мМ NaCl, 1,5 мМ МgС 12, 20% глицерина, 20 мМ молибдата натрия, 0,2 мМ DTT (дитиотреитол), 20 мкг/мл апротинина и 20 мкг/мл лейпептина (буфер для анализа). Обычно анализы по связыванию для стероидного рецептора включают радиоактивно меченые лиганды, например, 0,25 нМ [3 Н]-альдостерона для связывания MR, 0,3 нМ [3 Н]дексаметазона для связывания GR, 0,36 нМ [3 Н]-метилтриенолона для связывания AR и 0,29 нМ [3 Н]метилтриенолона для связывания PR и также 20 мкг 293-MR лизата, 20 мкг 293-GR лизата, 22 мкг 293AR лизата или 40 мкг 293-PR лизата на лунку. Обычно анализ проводят на 96-луночном формате планшета. Конкурирующие тестовые вещества добавляют в различных концентрациях приблизительно от 0,01 нМ до 10 мкМ. Неспецифичное связывание определяют в присутствии 500 нМ альдостерона для связывания MR, 500 нМ дексаметазона для GR связывания или 500 нМ метилтриенолона для AR и PR связывания. Реакционные смеси (140 мкл) для связывания инкубируют при 4 С в течение ночи, затем добавляют 70 мкл холодного буфера на древесном угле-декстране (содержащий на 50 мл буфера для анализа, 0,75 г древесного угля и 0,25 г декстрана) в каждую реакционную смесь. Планшеты перемешивают в течение 8 мин на орбитальном шейкере при 4 С. Затем планшеты центрифугируют при 3000 об/мин- 13017668 при 4 С в течение 10 мин. Аликвоту 120 мкл реакционной смеси для связывания затем переносят в другой 96-луночный планшет и в каждую лунку добавляют 175 мкл сцинтилляционной жидкости WallacOptiphase Hisafe 3. Планшеты запечатывают и интенсивно встряхивают на орбитальном шейкере. После 2-часовой инкубации планшеты считывают на счетчике Wallac Microbeta. Данные используют для расчета значения IC50 и процента ингибирования при 10 мкМ. Kd [3H]альдостерона для MR связывания, [3 Н]-дексаметазона для GR связывания, [3 Н]-метилтриенолона для AR связывания или [3 Н]-метилтриенолона для PR связывания определяют с помощью связывания при насыщении. Величины IC50 для соединений переводят в Ki с помощью уравнения Ченга-Прусова. Согласно протоколу, по существу описанному выше, соединения примеров 1-50 в анализе по связыванию MR имеют Ki 10 нМ. Предпочтительно, соединения согласно настоящему изобретению в анализе по связыванию MR имеют Ki 5 нМ, и более предпочтительно 1 нМ. В частности, соединения согласно примерам 1, 8 и 23 в анализе по связыванию MR имеют Ki приблизительно 0,79 нМ, 0,08 нМ и 0,23 нМ, соответственно (представленные величины являются средними геометрическими при n=2), что указывает на то, что соединения в объеме настоящего изобретения являются сильными лигандами MR человека. Функциональные анализы модулирования ядерного рецептора стероидных гормонов Альдостерон проявляет физиологическое действие посредством взаимодействия с минералокортикоидным рецептором. После цитоплазматического связывания альдостерона с MR комплекс лиганда и рецептора перемещается к ядру клетки, где он связывается с элементами ответа на гормон на ДНК для инициации экспрессии генов-мишеней. Для того чтобы продемонстрировать способность соединений согласно настоящему изобретению модулировать активность рецепторов стероидных гормонов (т.е. проявлять агонизирующее, частично агонизирующее, частично антагонизирующее или антагонизирующее действие), проводятся биоанализы, в которых определяют функциональное модулирование экспрессии генов-мишеней в клетках, которые временно трансфицировали генетической конструкцией, состоящей из белка ядерного рецептора и гена-репортера элемента гормонального ответа. Растворители, реагенты и лиганды, которые применяют в функциональных анализах, являются коммерчески доступными или могут быть приготовлены специалистом в данной области техники. А. Панель скрининга ядерного гормонального рецептора. Линию клеток мезонефроса человека НЕК 293 трансфицировали с рецептором стероидного гормона и плазмидами гена-репортера с помощью подходящего агента для трансфекции, например, Fugene. Вкратце, репортерную плазмиду, которая содержит две копии ARE пробазина и промотор ТК (тимидинкиназа) в направлении ап-стрим (против хода транскрипции) к люциферазной репортерной кДНК,трансфицируют в клетки НЕК 293 с помощью плазмиды, постоянно экспрессирующей андрогенный рецептор человека (AR), с использованием вирусного промотора CMV (цитомегаловирус). Репортерную плазмиду, которая содержит две копии GRE и промотор ТК в направлении ап-стрим (против хода транскрипции) к люциферазной репортерной кДНК, трансфицируют с помощью плазмиды, постоянно экспрессирующей глюкокортикоидный рецептор человека (GR), минералокортикоидный рецептор человека(MR) либо прогестероновый рецептор человека (PR), с помощью вирусного промотора CMV. Клетки трансфицируют в Т 150 см колбах в среде DMEM с 5% адсорбированной на угле эмбриональной бычьей сывороткой (FBS). После инкубации в течение ночи трансфицированные клетки трипсинизировали, высевали на 96-луночные планшеты в среду DMEM, содержащую 5% адсорбированной на угле FBS, инкубировали в течение 4 ч, затем исследовали тестируемые соединения в различных концентрациях от 0,01 нМ до 10 мкМ. В модели антагонистического действия анализов в среду добавляли в небольших концентрациях агонист каждого из соответствующих рецепторов (0,08 нМ альдостерона для MR, 0,25 нМ дексаметазона для GR, 0,66 нМ метилтриенолона для AR и 0,08 нМ промегестона для PR). Спустя 24 ч инкубации с тестовыми соединениями, клетки лизировали, и с помощью стандартных методов определяли активность люциферазы. Для определения значений ЕС 50 данные наносили на четырехпараметровую логистическую кривую. Процентную эффективность (соединения с реакциями максимального насыщения) или процент максимальной стимуляции (соединения с максимальными реакциями, которые не являются насыщенными) определяли относительно максимальной стимуляции, полученной со следующими эталонными агонистами: 30 нМ альдостерона для анализа MR, 100 нМ метилтриенолона для анализа AR, 30 нМ промегестона для анализа PR и с 100 нМ дексаметазона для анализа GR. Значения IC50 определяли аналогично с помощью данных, полученных в ходе анализа модели антагонистического действия. В антагонистической модели процентное ингибирование определяли посредством сравнения активности тестовых соединений в присутствии низких концентраций агонистов (0,08 нМ альдостерона для MR, 0,25 нМ дексаметазона для GR, 0,66 нМ метилтриенолона для AR и 0,08 нМ промегестона для PR) с ответом, полученным в присутствии тех же низких концентраций агониста в отсутствии тестовых соединений. В. Анализ конкурентных антагонистов hMR. Клетки мезонефроса человека HEK293 трансфицировали MR человека с помощью аналогичных реагентов для трансфекции, плазмид, промоторов, репортерных конструкций, буферов и процедур, опи- 14017668 санных выше для скрининга панели ядерных гормональных рецепторов. Трансфицированные клетки обрабатывали трипсином, высевали в 96-луночные планшеты в среду DMEM, содержащую 5% адсорбированной на угле FBS, инкубировали в течение 4 ч и затем добавляли альдостерон в различных концентрациях (10 разведений) (приблизительно от 0,001 нМ до 0,03 мкМ). Способность альдостерона проявлять агонизирующее действие к hMR определяли в присутствии или отсутствии заданных концентраций тестового соединения и отслеживали с помощью измерения активности люциферазы с помощью стандартных методов. Kb тестового соединения можно определить с помощью анализа Шилдса, представляющем графическую зависимость log (отношение доз - 1) от log концентрации антагониста с помощью уравнения: Log (DR-1) = Log [антагонист] - Log Kb, где отношение доз (DR) представляет собой отношение ЕС 50 альдостерона в присутствии тестового соединения к ЕС 50 альдостерона в отсутствии тестового соединения. Согласно протоколу, по существу, изложенному выше, приведенные в качестве примера соединения согласно настоящему изобретению в анализе конкурентных антагонистов MR имеют Kb 200 нМ. Предпочтительно, соединения согласно настоящему изобретению в анализе конкурентных антагонистовMR имеют Kb 50 нМ и более предпочтительно 10 нМ. В частности, соединения согласно примерам 1,8, и 23 в анализе конкурентных антагонистов MR имеют Kb приблизительно 1,2, 1,1 и 2,5 нМ, соответственно (величины являются средним геометрическим при n=2), что свидетельствует о том, что соединения в объеме настоящего изобретения являются сильными антагонистами MR человека.In vivo модель болезни почек, опосредованной альдостероном Самцов крыс Sprague Dawley (240-280 г) с одной удаленной почкой в течение одной недели индивидуально содержали на ad lib воде и диете для грызунов 5001. После акклиматизации собирали базовые(исходные) 24-часовые образцы мочи и анализировали на содержание общего белка и креатинина. Животных распределяли по исследовательским группам по массе тела и исходному содержанию белка мочи. С помощью методики tail-clip собирали исходную сыворотку и определяли азот мочевины крови(BUN), содержание креатинина и электролиты. После забора исходных образцов всех крыс, кроме контрольной группы, переводили на диету, включающую 6% солей, и питьевую воду с 0,3% KCl на протяжении всего периода исследования. Контрольных животных содержали на диете 5001 и питьевой воде на протяжении периода исследования, без введения им альдостерона. Введение животным, не входящим в контрольную группу (например, группе тестируемого соединения и группе, получавшей только носитель), в течение 28 дней 2,5 мкл/ч d-альдостерона в 0,01% DMSO при 0,75 мкг/ч проводили подкожно при помощи имплантированных мини-насосов Alza при анестезии изофлураном. Тестовое соединение в носителе, включающем 1% карбоксиметилцеллюлозу (CMC)/0,25% полисорбата 80 или только носитель затем вводили раз в сутки с помощью принудительного перорального введения (10 мл/кг), начиная со дня после введения альдостерона. Повторные образцы мочи собирали после 2 и 4 недель введения соединения или только носителя и проводили анализ на содержание общего белка мочи и креатинина. В конце исследования забирали фармакокинетические образцы, соответсвующие 8 промежуткам времени(0,5, 1, 2, 3, 6, 8, 12 и 24 ч). Также удаляли сердце и почки и фиксировали в 10% забуференном формалине для окрашевания гематоксилином и эозином (НЕ) и трехцветного окрашивания по Массону для определения структурных нарушений в сердечной и почечной ткани. С помощью сердечной пункции в конце исследования забирали сыворотку для дополнительного определения сывороточного BUN, креатинина и электролитов. Согласно протоколам, по существу описанным выше, соединения примеров 1, 8 и 23, при введении 10 мг/кг/сутки в течение 14 суток, снижают выделение почками белка по сравнению с животными, которым вводили носитель примерно на 49, 83 и 64 мг/сутки соответственно (значение представляют среднее из n=8 для каждого соединения), что свидетельствует о том, что соединения в рамках настоящего изобретения проявляют in vivo ренопротективное действие. Для того чтобы показать, что соединение понижало вероятность или частоту гиперкалемии, можно использовать следующую модель.In vivo анализ модуляции электролитов Самцов крыс Sprague Dawley (240-280 г) андреналэктомировали (проводили адренэктомию) и содержали на диете для грызунов 5001 и питьевом 1% растворе NaCl в течение 6 дней после операции. Затем животных подвергали голоданию на протяжении ночи, а питьевую воду с 1%-ным содержанием солей заменяли питьевой водой ad lib. Утром голодающих животных распределяли по группам на основании массы тела натощак. Контрольным животным (т.е., тем, которым не давали ни альдостерон, ни тестовое соединение) вводили 10 мл/кг носителя тестового соединения, содержащего 0,5% СМС/0,25% полисорбата 80/2,7% NaCl с помощью перорального принудительного введения и 1 мл/кг носителя альдостерона (0,01% DMSO/вода) подкожно. Животным, которые получали носитель, вводили тот же носитель тестового соединения посредством перорального принудительного кормления и альдостерон 3 мкг/кг подкожно. Тестовые соединения суспендировали в носителе карбоксиметилцеллюлоза/NaCl. Опытной группе животных давали тестовое соединение, суспендированное в носителе карбоксиметилцеллюлоза/NaCl, и альдостерон 3 мкг/кг подкожно. Непосредственно после введения дозы животных- 15017668 помещали в метаболическую камеру с ad lib доступом к питьевой воде. Спустя 5 ч после введения дозировки собирали образцы мочи и проводили анализ на выделение электролитов. Данные представлены в виде логарифмического соотношения экскреции Na/K. Для того чтобы определить, может ли соединение вызывать повышение соотношения Na/K в моче (показатель повышенной концентрации калия в сыворотке), можно протестировать соединения при различных дозировках. Предполагается, что специалист в данной области техники без дополнительных исследований может использовать предшествующие описания для практического применения настоящего изобретения в полном объеме. Представленные ниже синтезы и примеры приводятся с целью описания изобретения в подробностях и представляют типичный синтез соединений формулы (I). Однако они не ограничивают настоящее изобретение никоим образом. Реагенты и исходные материалы являются коммерчески доступными или могут быть синтезированы специалистом в данной области техники. Специалисты в данной области техники способны быстро определить приемлемые допустимые отклонения от процедур, описанных в примерах. Названия веществ согласно настоящему изобретению обычно приведены согласноChemDraw Ultra версия 10.0. Синтез 1. 1-Бром-4-фтор-2-(2-йодбензилокси)бензол. Смесь 2-йодбензилбромида (0,29 мол, 90 г), 2-бром-5-фторфенола (0,29 мол, 57,9 г) и карбоната калия (0,46 мол, 63 г) в N,N-диметилформамиде (750 мл) перемешивают при комнатной температуре в течение 16 ч. Добавляют воду (1 л), перемешивают полученную смесь в течение одного часа, отфильтровывают твердые вещества, промывают водой и сушат в вакуумном сушильном шкафу (20 мм. рт.ст./60 С), получая названное соединение (121 г, 100%). 1H ЯМР (400 МГц, ДМСО-d6)5,11 (с, 2 Н),6,81 (т, 1 Н), 7,13 (т, 1 Н), 7,19 (дд, 1 Н), 7,46 (т, 1 Н), 7,59 (д, 1 Н), 7,62 (т, 1 Н), 7,93 (д, 1 Н). Синтез 2. Этиловый эфир 3-[2-(2-бром-5-фторфеноксиметил)фенил]акриловой кислоты. К смеси 1-бром-4-фтор-2-(2-йодбензилокси)бензола (0,29 мол, 117,4 г), ацетата натрия (0,44 мол,36,1 г, 1,5 эквив.), бромида тетра-н-бутиламмония (0,29 мол, 90,3 г), ацетата палладия (II) (8 ммол, 1,8 г,3 мол%) и N-метилпирролидинона (900 мл) добавляют по каплям при 55-60 С раствор этилакрилата(0,32 мол, 34,3 мл) в N-метилпирролидиноне (200 мл). Реакционную смесь охлаждают до комнатной температуры и обрабатывают водой (2 л) и метил-трет-бутиловым эфиром (2 л). Пропускают через диатомитовую землю, добавляют этилацетат (1 л), разделяют слои и промывают водой (2 л). Органическую часть сушат над безводным сульфатом натрия, фильтруют и концентрируют. Полученное твердое вещество суспензируют в гексанах (1 л), замораживают в холодильнике в течение 2 ч, фильтруют и промывают холодными гексанами (500 мл). Сушат в вакуумном сушильном шкафу (50 С/20 мм. рт.ст.), получая названное соединение в виде бледно-желтого твердого вещества (104,4 г, 95%). ЖХ-МС m/z 381,0 [М+Н]+. Синтез 3. Этиловый эфир (Е)-(3-фтор-6 Н-дибензо [b,е]оксепин-11-илиден)уксусной кислоты. Смесь этилового эфира 3-[2-(2-бром-5-фторфеноксиметил)фенил]акриловой кислоты (0,25 мол, 94 г), ацетата натрия (0,37 мол, 30 г, 1,5 эквив.), бромида тетра-н-бутиламмония (0,25 мол, 81 г) и ацетата палладия (II) (7 ммол, 1,7 г, 3 мол.%) в N-метилпирролидинона (850 мл) нагревают при 100-110 С в течение 6 ч. Охлаждают до комнатной температуры, разбавляют водой (1 л), фильтруют через диатомитовую землю и промывают этилацетатомом (2 л). Фильтрат переносят в делительную воронку, добавляют воду(500 мл) и разделяют слои. Органический слой промывают водой (21,5 л), сушат над безводным сульфатом натрия, фильтруют через слой силикагеля, промывают этилацетатом (1,5 л) и концентрируют досуха. К оставшемуся твердому веществу добавляют гексаны (1 л), замораживают в холодильнике в течение 2 ч, фильтруют, промывают гексанами (500 мл) и сушат при 50 С/20 мм. рт.ст., получая названное соединение (64,3 г, 87%). ЖХ-МС m/z 299,0 [М+Н]+. Синтез 4. (Е)-11-Бромметилен-3-фтор-6,11-дигидродибензо[b,е]оксепин. К суспензии этилового эфира (3-фтор-6H-дибензо[b,е]оксепин-11-илиден)уксусной кислоты (0,23 мол, 69,5 г) в изопропаноле (725 мл) добавляют раствор гидроксида лития (0,53 мол, 12,0 г) в воде (125 мл) и нагревают до 70 С в течение 4 ч. Смесь оставляют охлаждаться 40 С и затем обрабатывают ледяной уксусной кислотой (0,44 мол, 25 мл). После перемешивания в течение 15 мин добавляют Nбромсукцинимид (0,25 мол, 44 г). Смесь пузырится, температура поднимается до 45 С, и спустя несколько минут выпадает твердое вещество. Смесь перемешивают при 40-45 С в течение одного часа и охлаждают до комнатной температуры. Добавляют бисульфит натрия (4,5 г) в воде (150 мл), насыщенный водный раствор бикарбоната натрия (150 мл) и воду (450 мл). Полученную суспензию фильтруют и промывают холодной 1:1 смесью изопропанол/вода (300 мл). Твердое вещество сушат при 60 С/20 мм рт.ст. в течение ночи, получая названное соединение (65,8 г, 93%). 1 Н ЯМР (400 МГц, ДМСО-d6)4,9-5,4 К перемешиваемой смеси (Е)-11-(бромметилен)-3-фтор-6,11-дигидродибензо[b,е]оксепина (49 ммол, 15 г) и бис(пинаколято)дибора (64 ммол, 16 г) в 1,4-диоксане (250 мл) добавляют ацетат калия (150 ммол, 15 г). Смесь продувают азотом, добавляют аддукт дихлор[1,1'-бис(дифенилфосфино)ферроцен] палладия(II) и дихлорметана (2,46 ммол, 1,80 г) и нагревают при 65 С в течение ночи. Охлаждают до комнатной температуры, фильтруют через диатомитовую землю, промывают этилацетатом и фильтрат концентрируют в вакууме. Добавляют метанол (200 мл) и смесь перемешивают в течение одного часа на роторном испарителе в отсутствие вакуума, в результате чего образуется коричневое твердое вещество. Темно-коричневые кристаллы собирают фильтрованием и сушат в вакууме в течение ночи, получая названное соединение (7,28 г, 42%). Фильтрат концентрируют и очищают посредством колоночной хроматографии, элюируя смесью от 0 до 16% этилацетата в гексанах, получая названное соединение в виде желтого твердого вещества (3,46 г, 20%). Общий выход реакции составляет 10,7 г (62%). 1 Н ЯМР (300 МГц, CDCl3)7,36 (дд, J=8,8, 6,8 Гц, 1 Н), 7,32-7,27 (м, 4 Н), 6,64-6,57 (м, 1 Н), 6,48 (дд, J=10,3, 2,6 Гц,1 Н), 5,98 (с, 1 Н), 5,20 (ушир.с, 1 Н), 1,15 (с, 12 Н). Синтез 6. 1-Бром-3-фторбензальдегид. К раствору диизопропиламина (2,27 мол, 320 мл) в безводным тетрагидрофуране (800 мл), находящемуся в 5-литровой колбе, при 0 С добавляют по каплям в течение 1,5 ч 1,6 М бутиллитий (1,16 л, 1,86 мол) в гексанах. Полученный желтый раствор перемешивают при 0 С в течение 30 мин. В отдельной 12 л колбе растворяют 1-бром-3-фторбензол (203 мл, 1,86 мол) в безводным тетрагидрофуране (650 мл) и охлаждают до -78 С. Заранее приготовленный раствор LDA переносят в капельную воронку через трубку и добавляют по каплям к раствору 1-бром-3-фторбензола в течение 2 ч, так чтобы температура не поднималась выше -70 С. Полученную суспензию перемешивают при -78 С в течение 30 мин. Затем по каплям при -78 С в течение одного часа добавляют раствор N-метил-N-фенилформамида (230 мл, 1,86 моль,1,00 эквив.) в безводном тетрагидрофуране (1,15 л), поддерживая температуру ниже -70 С. Реакцию перемешивают на холоде в течение 2 ч и медленно оставляют достигать комнатной температуры в течение ночи. Реакционную смесь разбавляют метил-трет-бутиловым эфиром (3 л), тушат 1 М хлористоводородной кислотой (4 л) и энергично перемешивают в течение 3 ч. Слои разделяют и водный слой экстрагируют метил-трет-бутиловым эфиром (1 л). Объединенные органические фазы промывают 1 М хлористо-водородной кислотой (21 л), водой (1 л), солевым раствором (1 л), сушат над сульфатом магния,фильтруют и концентрируют до получения оранжевого масла, которое медленно затвердевает с образованием названного соединения (394 г, 105%). 1 Н ЯМР (CDCl3)10,37 (с, 1 Н), 7,50 (д, 1 Н), 7,44 (м, 1 Н),7,17 (т, 1 Н). Синтез 7. 1-Бром-3-фторбензиловый спирт. К 1-бром-3-фторбензальдегиду (1,60 кг, 7,91 мол) в метаноле (14 л) при 0 С порциями в течение 30 мин добавляют боргидрид натрия (284 г, 7,93 мол), контролируя выделение тепла и газа. Спустя 15 мин,тушат водой (500 мл) и концентрируют, получая желтое масло. Масло растворяют в этилацетате (6 л) и промывают водой (3 л). Водный слой экстрагируют этилацетатом (1 л). Объединенные органические фазы промывают солевым раствором (2 л), сушат над сульфатом натрия, фильтруют и концентрируют, получая названное соединение в виде оранжевого масла (1614 г, 100%). 1H ЯМР (CDCl3)7,40 (д, 1 Н), 7,18(м, 1 Н), 7,07 (т, 1 Н), 4,86 (с, 2 Н). Синтез 8. 1-Бром-3-фторбензилбромид. К раствору 1-бром-3-фторбензилового спирта (1,61 кг, 7,85 мол) в хлороформе (14 л) за один раз при 0 С добавляют пиридин (770 мл, 9,52 мол) (небольшое выделение тепла) и перемешивают при 0 С в течение 5 мин. Добавляют по каплям трибромид фосфора (900 мл, 9,49), поддерживая внутреннюю температуру ниже 20 С, и перемешивают полученный раствор в течение ночи, позволяя ему нагреться до комнатной температуры. Реакционную смесь охлаждают до 0 С и медленно гасят ледяной водой (2 л). Переносят в 50-литровую колбу, слои разделяют и водный слой затем экстрагируют хлороформом (1 л). Объединенные органические фазы промывают 5% серной кислотой (2 л), насыщенным водным раствором бикарбоната натрия (2 л), солевым раствором (2 л), сушат сульфатом магния, фильтруют и концентрируют, получая названное соединение в виде желтого масла (1753 г, 83%). 1 Н ЯМР (CDCl3)7,43 (д,1 Н), 7,21 (м, 1 Н), 7,09 (т, 1 Н), 4,68 (с, 2 Н). Синтез 9. 1-Бром-2-(2-бром-6-фторбензилокси)-4-фторбензол. К раствору 1-бром-3-фторбензилбромида (1,75 кг, 6,52 мол) и 2-бром-5-фторфенола (730 мл, 16,56 мол, 1,01 экв.) в N,N-диметилформамиде (14 л) при 0 С добавляют за один раз карбонат калия (1,36 кг,9,80 мол), и перемешивают полученную суспензию на холоду в течение одного часа и затем при комнатной температуре в течение ночи. Реакционную смесь переносят в 50-литровую колбу, добавляют по каплям в течение 30 мин воду (14 л) и периодически добавляя лед с целью контроля выделения тепла кри- 17017668 сталлизации. Перемешивают при комнатной температуре в течение одного часа, собирают беловатое твердое вещество фильтрованием, промывают водой и сушат в течение ночи при 50 С, получая названное соединение (2,22 кг, 90%). 1H ЯМР (CDCl3)7,51 (м, 2 Н), 7,30 (м, 1 Н), 7,14 (т, 1 Н), 6,89 (дд, 1 Н),6,67 (м, 1 Н), 5,26 (д, 2 Н). Синтез 10. Этиловый эфир (Е)-(3,7-дифтор-6H-дибензо[b,е]оксепин-11-илиден)уксусной кислоты. Смесь 1-бром-2-(2-бром-6-фторбензилокси)-4-фторбензола (886 ммол, 335 г), этилакрилата (931 ммол, 101 мл, 1,05 экв. ), Pd(ОАс)2 (22,2 ммол, 4,97 г), бромида тетрабутиламмония (886 ммол, 286 г) и ацетата натрия (4,40 мол, 363,5 г) в N-метилпирролидиноне (3,3 л) дегазируют, пять раз откачивая газы и вновь заполняя колбу азотом. Смесь нагревают до 120 С в течение 2 ч. Охлаждают до 35 С и добавляют воду (5,5 л) в течение 40 мин. Осадок отфильтровывают, промывают водой (2500 мл) и сушат на воздухе в течение 30 мин. Повторно суспензируют в изопропаноле (1 л) в течение 2 ч и охлаждают на льду в течение одного часа. Темно-серое твердое вещество собирают фильтрованием, промывают холодным изопропанолом (5100 мл) и сушат при 50 С в вакууме, получая названное соединение (191 г, 68%). Синтез 11. (Е)-11-Бромметилен-3,7-дифтор-6,11-дигидродибензо[b,е]оксепин. Перемешиваемую суспензию этилового эфира (Е)-(3,7-дифтор-6H-дибензо[b,е]оксепин-11 илиден)уксусной кислоты (601 ммол, 190 г) в метаноле (1,9 л) и 5 н. гидроксид натрия (1,20 мол, 240 мл) нагревают до 50 С в течение 2 ч. Метанол удаляют в вакууме и повторно суспензируют полученную черную суспензию в воде (3 л). В течение 45 мин добавляют 5 н. хлористо-водородную кислоту (250 мл),перемешивают в течение 2,5 ч, фильтруют, и остаток промывают водой (4200 мл). Серое твердое вещество сушат в вакууме при 60 С в течение ночи и при 80 С в течение 6 ч. Твердое вещество (еще влажное,содержащее приблизительно 95 г воды) и ацетат лития (60,1 ммол, 4 г, 0,1 экв.) растворяют в ацетонитриле (1,7 л) и перемешивают в течение 15 мин. Добавляют за один раз N-бромсукцинимид (661 ммол,118 г) и перемешивают в течение 2,5 ч. Добавляют 0,25 М тиосульфат натрия (400 мл), и затем насыщенный водный раствор бикарбоната натрия (400 мл) и воду (900 мл). Смесь перемешивают в течение одного часа при комнатной температуре и затем перемешивают в течение 2 ч на ледяной бане. Смесь фильтруют и промывают холодным ацетонитрилом (300 мл). Сушат в течение ночи в вакууме при 50 С, получая названное соединение в виде светло-бежевого твердого вещества (173,3 г, 89%). 1H ЯМР (ДМСО-d6)5,25 (с, 2 Н), 6,67 (дд, 1 Н), 6,81 (дт, 1 Н), 7,20 (с, 1 Н), 7,22 (д, 1 Н), 7,28 (т, 1 Н), 7,36 (дд, 1 Н), 7,48 (м, 1 Н). Синтез 12. (Е)-2-3,7-дифтордибензо[b,е]оксепин-11(6 Н)-илиден)метил)-4,4,5,5-тетраметил-1,3,2 диоксаборолан. К смеси (Е)-11-(бромметилен)-3,7-дифтор-6,11-дигидродибензо[b,е]оксепина (46,4 ммол, 15,0 г) и бис(пинаколято)дибора (50,3 ммол, 15,3 г) в 1,4-диоксане (150 мл) добавляют ацетат калия (74,3 ммол,7,29 г), трициклогексилфосфин (6,03 ммол, 1,69 г) и Pd2(dba)3 (2,32 ммол, 2,13 г). Продувают барботажем азота, затем перемешивают при нагревании до 65 С в течение ночи. После охлаждения до комнатной температуры смесь фильтруют через слой диатомитовой земли, промывают слой этилацетатом и растворитель удаляют, получая коричневое масло. Масло разбавляют метанолом (180 мл) и встряхивают полученную смесь на роторном испарителев отсутствие вакуума в течение 3 ч, в результате чего образуется белый осадок. Осадок собирают вакуумным фильтрованием, получая названное соединение (10,8 г, 63%) в виде белого твердого вещества. 1 Н ЯМР (300 МГц, CDCl3)7,34 (дд, J=8,8, 6,7 Гц, 1 Н), 7,24-7,18 (м,1 Н), 7,08-7,01 (м, 2 Н), 6,65-6,59 (м, 1 Н), 6,50 (дд, J=10,3, 2,6 Гц, 1 Н), 5,99 (с, 1 Н), 5,31 (д, J=0,9 Гц, 2 Н),1,16 (с, 12 Н). Синтез 13. (Е)-11-Бромметилен-3,8-дифтор-6,11-дигидродибензо[b,е]оксепин. Названное соединение синтезируют в соответствии с процедурами, описанными в синтезах 9, 10 и 11, используя в качестве исходных материалов 1-бром-2-бромметил-4-фторбензол и 2-бром-5 фторфенол. Синтез 14. (Е)-2-3,8-дифтордибензо[b,е]оксепин-11(6 Н)-илиден)метил)-4,4,5,5-тетраметил-1,3,2 диоксаборолан. К смеси (Е)-11-(бромметилен)-3,8-дифтор-6,11-дигидродибензо[b,е]оксепина (90,0 ммол, 29,0 г) и бис(пинаколято)дибора (117 ммол, 29,5 г) в 1,4-диоксане (295 мл) добавляют ацетат калия (144 ммол,14,1 г), трициклогексилфосфин (11,7 ммол, 3,28 г) и Pd2(dba)3 (4,14 ммол, 3,80 г). Продувают барботажем азота через смесь, затем нагревают при 65 С в течение ночи. После охлаждения до комнатной температуры фильтруют через слой диатомитовой земли и промывают этилацетатом. Фильтрат концентрируют в вакууме, получая коричневое масло. Масло разбавляют метанолом (180 мл) и встряхивают полученную смесь в течение 1,5 ч на роторном испарителе в отсутствие вакуума, в результате чего образуется коричневый осадок. Осадок собирают вакуумным фильтрованием, получая названное соединение в виде коричневого твердого вещества (24,2 г, 73%). 1H ЯМР (300 МГц, CDCl3)7,35 (дд, J=8,8, 6,7 Гц, 1 Н), 7,297,24 (м, 1 Н), 7,05-6,93 (м, 2 Н), 6,65-6,59 (м, 1 Н), 6,49 (дд, J=10,2, 2,6 Гц, 1 Н), 5,99 (с, 1 Н), 5,15 (ушир.с,2 Н), 1,16 (с, 12 Н). Синтез 15. 2-трет-Бутоксикарбониламинопропиловый эфир (R)-метансульфоновой кислоты. Метансульфонилхлорид (0,128 мол, 14,7 г, 1,5 эквив.) добавляют по каплям к раствору (R)-(+)-2 трет-бутоксикарбониламино-1-пропанола (0,085 мол, 15,0 г) и триэтиламина (0,12 мол, 17,3 г, 2,0 эквив.)- 18017668 в дихлорметане (150 мл) при 0 С в атмосфере азота. По завершении добавления реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 ч. Реакционную смесь разбавляют дихлорметаном, промывают 0,1 н. хлористо-водородной кислотой и насыщенным раствором бикарбоната натрия. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме, получая названное соединение (20,8 г, 96%) в виде белого твердого вещества. 1 Н ЯМР (400 МГц,CDCl3)1,24 (д, 3H), 1,43 (с, 9 Н), 3,03 (с, 3H), 3,97 (ушир.с, 1 Н), 4,14 (дд, 1 Н), 4,21 (ушир.с, 1 Н), 4,64(ушир.с, 1 Н, Boc-NH). Синтез 16. трет-Бутиловый эфир (R)-(1-метил-2-морфолин-4-илэтил)карбаминовой кислоты. Смесь 2-трет-бутоксикарбониламинопропилового эфира (R)-метансульфоновой кислоты (0,083 мол,20 г) и морфолина (0,83 мол, 72,2 г) в ацетонитриле (210 мл) нагревают до 65 С в течение 16 ч. Реакционную смесь концентрируют, остаток разбавляют водой, экстрагируют дихлорметаном, объединяют органические слои и промывают их водой. Органический слой сушат над безводным сульфатом натрия,фильтруют и концентрируют в вакууме, получая названное соединение (12 г, 62%) в виде желтого плотного масла. 1 Н ЯМР (400 МГц, CDCl3)1,13 (д, 3H), 1,43 (с, 9 Н), 2,21 (дд, 1 Н), 2,26-2,32 (м, 1 Н), 2,352,39 (м, 2 Н), 2,48-2,55 (м, 2 Н), 3,63-3,70 (м, 5 Н), 4,68 (ушир.с, 1 Н, -NH). Синтез 17. Гидрохлорид (R)-1-метил-2-морфолин-4-илэтиламина. К раствору трет-бутилового эфира (R)-(1-метил-2-морфолин-4-илэтил)карбаминовой кислоты (0,05 мол, 12 г) в сухом дихлорметане (75 мл) при 0 С добавляют 1,6 М раствор хлористого водорода в диоксане (120 мл). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 ч. Осадок отфильтровывают и сушат в вакууме, получая названное соединение (9,2 г, 86%) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО-d6)1,29 (д, 3H), 3,10 (ушир.с, 2 Н), 3,38 (ушир.с, 4 Н), 3,79(ушир.с, 5 Н). Синтез 18. (R)-(4-Бром-2-нитрофенил)-(1-метил-2-морфолин-4-илэтил)амин. Смесь (R)-1-метил-2-морфолин-4-илэтиламингидрохлорида (0,04 мол, 8,7 г), 5-бром-2 фторнитробензола (0,048 мол, 10,5 г), триэтиламина (0,20 мол, 20,2 г) и диметиламинопиридина (0,0004 мол, 0,048 г) кипятят с обратным холодильником в этилацетате (220 мл) в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом и промывают водой и солевым раствором. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют. Очищают на колонке с силикагелем, используя в качестве элюента 10% этилацетат в гексанах, получая названное соединение (12,8 г, 95%) в виде оранжевого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3)1,29 (д, 3H), 2,47-2,51 (м, 5 Н), 2,53-2,58 (м, 1 Н), 3,64-3,69 (м, 4 Н), 3,72-3,78 (м, 1 Н), 6,78 (д, 1 Н), 7,46 (дд,1 Н), 8,30 (д, 1 Н), 8,33 (д, 1 Н). Промежуточные соединения, указанные в приведенной ниже таблице, синтезируют в соответствии с процедурами, описанными в синтезах 15, 16, 17, и 18, используя в качестве исходных материалов Синтез 20. (4-Бром-2-нитрофенил)-(4-метилпиперазин-1-ил)амин. Смесь 1-амино-4-метилпиперазина (0,217 мол, 25,0 г), 5-бром-2-фторнитробензола (0,239 мол, 52,6 г) и триэтиламина (0,46 мол, 46 г) в этилацетате (900 мл) нагревают до кипения в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом и промывают водой и солевым раствором. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют. Очищают на колонке с силикагелем, используя в качестве элюента 5% метанол в дихлорметане,получая названное соединение (43,2 г, 63%). ES-MS m/z 315 [М+1]+. Синтез 21. трет-Бутиловый эфир 3-(4-бром-2-нитрофениламино)азетидин-1-карбоновой кислоты. 5-Бром-2-фторнитробензол (28,8 ммол, 3,55 мл), трет-бутиловый эфир 3-аминоазетидин-1 карбоновой кислоты (28,8 ммол, 4,96 г) и триэтиламин (35,9 ммол, 5,00 мл) растворяют в этилацетате(100 мл) и кипятят с обратным холодильником в атмосфере азота в течение 48 ч. Охлаждают до комнатной температуры. Дважды промывают 0,5 М хлористо-водородной кислотой, сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме, получая ярко-оранжевое твердое вещество (11,04 г, 100%). ЖХ-МС m/z (79Br/81Br) 394,0/396,0 [М+Н]+. Синтез 22. Гидрохлорид азетидин-3-ил-(4-бром-2-нитрофенил)амина. Раствор трет-бутилового эфира 3-(4-бром-2-нитрофениламино)азетидин-1-карбоновой кислоты(29,66 ммол, 11,04 г) в 4,0 М растворе хлористого водорода в диоксане (40 мл) перемешивают при комнатной температуре в атмосфере азота в течение ночи. Из раствора выпадает твердое вещество. Разбавляют эфиром (200 мл), твердые вещества отфильтровывают, промывают большим количеством эфира и сушат (8,54 г, 93%). 1 Н ЯМР (400 МГц, CD3OD): 4,12 (дд, 2 Н), 4,45 (дд, 2 Н), 4,72 (м, 1 Н), 6,76 (д, 1 Н),7,62 (дд, 1 Н), 8,30 (д, 1 Н), ЖХ-МС m/z (79Br/81Br) 272,0/274,0 [М+Н]+. Синтез 23. (4-Бром-2-нитрофенил)-(1-метилазетидин-3-ил)амин. К смеси гидрохлорида азетидин-3-ил-(4-бром-2-нитрофенил)амина (27,7 ммол, 8,54 г) в ацетонитриле (100 мл) и 37% водном формальдегиде (83,0 ммол, 6,28 мл, 3,00 эквив.) при 0 С медленно добавляют триацетоксиборгидрид натрия (88,6 ммол, 18,8 г). Перемешивают при комнатной температуре в атмосфере азота в течение ночи. Летучие вещества удаляют под уменьшенным давлением. Разбавляют этилацетатом, дважды промывают 10% водным раствором бикарбоната натрия, сушат над безводным сульфатом натрия, фильтруют и концентрируют под уменьшенным давлением, получая 8,5 г оранжевого твердого вещества. Очищают на колонке со 120 г силикагеля, элюируя 5% метанолом в дихлорметане,получая названное соединение (5,92 г, 75%). ЖХ-МС m/z (79Br/81Br) 286,0/288,0 [М+Н] + . Синтез 24. 1-трет-Бутиловый эфир 2-метиловый эфир (2S, 4R)-4-гидроксипирролидин-1,2 дикарбоновой кислоты. Тионилхлорид (1,07 мол, 128,2 г) добавляют по каплям к раствору (2S, 4R)-4-гидроксипирролидин 2-карбоновой кислоты (0,70 мол, 93,0 г) в сухом метаноле (2 л) при 0 С в атмосфере азота. По завершении добавления реакционную смесь нагревают до комнатной температуры и перемешивают в течение 6 ч. Реакционную смесь концентрируют при пониженном давлении, получая соответствующий гидрохлорид метилового эфира. К раствору гидрохлорида метилового эфира в сухом дихлорметане (2 л) при 0 С добавляют триэтиламин (1,56 мол, 157,5 г) и перемешивают в течение 30 мин. Затем последовательно добавляют N,N-диметиламинопиридин (0,10 мол, 13 г) и ди-трет-бутилдикарбонат (0,85 мол, 185,7 г). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 18 ч. Реакционную смесь гасят водой, отделяют органический слой и промывают водой и насыщенным раствором бикарбоната натрия. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая названное соединение (160 г, 91%) в виде вязкого масла. ES-MS m/z 246,1 [М+1]+. Синтез 25. Гидрохлорид (2R, 4R)-4-гидроксипирролидин-2-карбоновой кислоты. К смеси уксусного ангидрида (408 г) и уксусной кислоты (1,2 л) при 50 С за один раз добавляют транс-4-гидрокси-L-пролин (0,36 мол, 94 г). Реакционную смесь нагревают в течение 5,5 ч до 90 С и затем концентрируют. Остаток растворяют в 2 н. хлористо-водородной кислоте и кипятят с обратным холодильником в течение 3 ч. Реакционную смесь охлаждают до комнатной температуры, фильтруют через диатомитовую землю, и концентрируют в вакууме до образования белых игл. Кристаллы отфильтровывают, промывают эфиром и сушат в вакууме, получая названное соединение (90,0 г, 75%). []D20 + 10,0(с=1,0 в метаноле). 1H ЯМР (400 МГц, D2O),2,34-2,39 (м, 1 Н), 2,45-2,53 (м, 1 Н), 3,38 (дд, 1 Н), 3,45 (д,1 Н), 4,50 (дд, 1 Н), 4,58 (ушир.с, 1 Н). Синтез 26. 1-трет-Бутиловый эфир 2-метиловый эфир (2R,4R)-4-гидроксипирролидин-1,2 дикарбоновой кислоты. Тионилхлорид (1,81 мол, 213,8 г, 1,5 экв.) добавляют по каплям при 0 С в атмосфере азота к раствору гидрохлорида (2R,4R)-4-гидроксипирролидин-2-карбоновой кислоты (1,19 мол, 200 г) в сухом метаноле (2 л). По завершении добавления реакционную смесь нагревают до комнатной температуры и перемешивают в течение 6 ч. Реакционную смесь концентрируют при пониженном давлении, получая соответствующий гидрохлорид метилового эфира. К раствору гидрохлорида метилового эфира в сухом дихлорметане (2 л) при 0 С добавляют триэтиламин (265,9 г, 2,63 мол) и перемешивают в течение 30 мин. Затем последовательно добавляют N,N-диметиламинопиридин (0,18 мол, 21,9 г) и ди-третбутилдикарбонат (1,43 мол, 313,5 г). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 18 ч. Реакционную смесь гасят водой, отделяют органический слой и промывают водой и раствором NaHCO3. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме, получая названное соединение (260 г, 88%) в виде плотного масла. 1H ЯМР(400 МГц, CDCl3)1,43 (с, 9 Н), 1,46 (с, 9 Н), 2,05-2,10 (м, 2 Н), 2,26-2,35 (м, 2 Н), 3,48-3,56 (м, 2 Н), 3,583,61 (м, 1 Н), 3,64-3,70 (м, 2 Н), 3,77 (с, 3H), 3,79 (с, 3H), 4,27-4,29 (м, 1 Н), 4,34-4,38 (м, 2 Н). Синтез 27. 1-трет-Бутиловый эфир 2-метиловый эфир (2S, 4R)-4-метансульфонилоксипирролидин 1,2-дикарбоновой кислоты. Метансульфонилхлорид (1,56 мол, 179 г) добавляют по каплям при 0 С в атмосфере азота к раствору 1-трет-бутилового эфира 2-метилового эфира (2S, 4R)-4-гидроксипирролидин-1,2-дикарбоновой кислоты (0,65 мол, 160 г) в пиридине (600 мл). По завершении добавления реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 ч. Реакционную смесь разбавляют дихлорметаном, промывают 0,1 н. раствором хлористо-водородной кислоты и насыщенным раствором бикарбоната натрия. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме, получая названное соединение (185 г, 91%) в виде плотного масла. ES-MS m/z 324,1 [М+1]+, 224,0[М-Вос]+. Синтез 28. 1-трет-Бутиловый эфир 2-метиловый эфир (2R, 4R)-4-метансульфонилоксипирролидин 1,2-дикарбоновой кислоты. Названное соединение синтезируют в соответствии с процедурой синтеза 27 из 1-трет-бутилового эфира 2-метилового эфира (2R, 4R)-4-гидроксипирролидин-1,2-дикарбоновой кислоты. ES-MS m/z 324,1(0,88 мол, 292 г) в сухом дихлорметане (1,44 л) при 0 С. По завершении добавления реакционную смесь нагревают до комнатной температуры и перемешивают в течение 4 ч. Добавляют этанол (1,44 л) и перемешивают еще в течение 2 ч. К реакционной смеси добавляют диэтиловый эфир (1,44 л), отфильтровывают осадок трифенилфосфиноксида и фильтрат концентрируют при пониженном давлении. Очищают посредством колоночной хроматографии, используя в качестве элюента 5% этилацетат в гексанах, получая названное соединение (155 г, 85%) в виде плотного масла. ES-MS m/z 308 [М+1]+. Синтез 30. 1-трет-Бутиловый эфир 2-метиловый эфир (2R, 4S)-4-бромпирролидин-1,2-дикарбоновой кислоты. Названное соединение синтезируют в соответствии с процедурой синтеза 29 из 1-трет-бутилового эфира 2-метилового эфира (2R,4R)-4-гидроксипирролидин-1,2-дикарбоновой кислоты. 1H ЯМР (400 МГц, CDCl3)1,45-1,47 (с, 9 Н), 2,38-2,45 (м, 1 Н), 2,54-2,59 (м, 1 Н), 3,72-3,74 (с, 3H), 3,84-3,87 (м, 1 Н),3,92-3,94 (м, 1 Н), 4,44-4,54 (м, 2 Н). Синтез 31. 1-трет-Бутиловый эфир 2-метиловый эфир (2S,4S)-4-азидопирролидин-1,2-дикарбоновой кислоты. Смесь 1-трет-бутилового эфира 2-метилового эфира (2S,4R)-4-метансульфонилоксипирролидин 1,2-дикарбоновой кислоты (0,60 мол, 185 г) и азида натрия (1,20 мол, 78 г) в N,N-диметилформамид (1 л) нагревают до 80 С в течение 16 ч. Реакционную смесь разбавляют водой и экстрагируют этилацетатом. Объединенные органические слои промывают 0,5 н. хлористо-водородной кислотой, насыщенным раствором бикарбоната натрия и солевым раствором. Сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме, получая названное соединение (140 г, 88%) в виде плотного желтого масла.ES-MS m/z 271,2 [М+1]+, 171,0 [М-Вос]+. Промежуточные соединения, указанные в приведенной ниже таблице, синтезируют в соответствии с процедурами, описанными в синтезе 31, используя в качестве исходных материалов подходящий бром или метилсульфонилпирролидин. 65 С в течение 16 ч Синтез 35. трет-Бутиловый эфир (2S,4S)-4-азидо-2-гидроксиметилпирролидин-1-карбоновой кислоты. Боргидрид лития (0,16 мол, 3,6 г) добавляют к раствору 1-трет-бутилового эфира 2-метилового эфира (2S,4S)-4-азидопирролидин-1,2-дикарбоновой кислоты (0,16 мол, 40 г) в безводном диэтиловом эфире (400 мл) при температуре от -10 до -20 С в атмосфере азота. По завершении добавления реакционную смесь перемешивают в течение 30 мин при той же температуре. Реакционную смесь гасят насыщенным раствором бикарбоната натрия при -70 С и оставляют медленно нагреваться до комнатной темпера- 21017668 туры. Органический слой отделяют и водный слой экстрагируют дихлорметаном. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме, получая названное соединение (32 г, сырое) в виде плотного желтого масла. 1H ЯМР (400 МГц, CDCl3)1,46 (с, 9 Н), 2,32-2,35(м, 1 Н), 3,30 (дд, 1 Н), 3,65-3,79 (м, 4 Н), 4,07-4,29 (м, 2 Н). Промежуточные соединения, указанные в приведенной ниже таблице, синтезируют в соответствии с процедурами, описанными в синтезе 35, используя в качестве исходных материалов подходящий метиловый эфир пирролидин-2-карбоновой кислоты. трет-Бутиловый эфир (2S,4S)-4-азидо-2-гидроксиметилпирролидин-1-карбоновой кислоты (32 г) растворяют в метаноле (150 мл). Добавляют 10% палладий на угле (3,2 г) и гидрируют (1 атм) при комнатной температуре в течение 16 ч. Катализатор отфильтровывают через слой диатомитовой земли и концентрируют в вакууме, получая названное соединение (28 г, сырое) в виде плотного желтого масла. Промежуточные соединения, указанные в приведенной ниже таблице, синтезируют в соответствии с процедурами, описанными в синтезе 39, используя в качестве исходных материалов подходящие азидопирролидины.(0,14 мол, 31 г), 5-бром-2-фторнитробензола (0,29 мол, 64,0 г) и триэтиламина (0,58 мол, 59 г) в этилацетате (320 мл) нагревают до кипения с обратным холодильником в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом, и промывают водой и солевым раствором. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая названное соединение (42 г, сырое) в виде плотного темно-оранжевого масла. Синтез 44. Гидрохлорид (2S, 4S)-[4-(4-бром-2-нитрофениламино)пирролидин-2-ил]метанола. При комнатной температуре 4,0 М раствор хлористого водорода в диоксане (950 мл) медленно добавляют к раствору трет-бутилового эфира (2S,4S)-4-(4-бром-2-нитрофениламино)-2-гидроксиметилпирролидин-1-карбоновой кислоты (0,22 мол, 95 г) в сухом дихлорметане (475 мл) и перемешивают в течение 2 ч. Выпавшее твердое вещество отфильтровывают и сушат в вакууме, получая названное соединение (70 г, 87%) в виде оранжевого твердого вещества. 1H ЯМР (400 МГц, CD3OD)1,98-2,05 (м,1 Н), 2,71-2,79 (м, 1 Н), 3,36 (дд, 1 Н), 3,58-3,63 (м, 1 Н), 3,76 (дд, 1 Н), 3,88-3,95 (м, 2 Н), 4,55-4,58 (м, 1 Н),7,03 (д, 1 Н), 7,66 (дд, 1 Н), 8,30 (д, 1 Н). Промежуточные соединения, указанные в приведенной ниже таблице, синтезируют в соответствии с процедурами, описанными в синтезах 43 и 44 используя в качестве исходных материалов подходящие аминопирролидины. Хлорацетилхлорид (0,49 мол, 55,7 г) добавляют по каплям при комнатной температуре к раствору гидрохлорида (2S,4S)-[4-(4-бром-2-нитрофениламино)пирролидин-2-ил]метанола (0,22 мол, 79 г) в (1:1) смеси тетрагидрофуран/вода (1180 мл). Поддерживают рН реакци в диапазоне 10-12 при помощи непрерывного добавления 4 М раствора гидроксида натрия. По окончании добавления хлорацетилхлорида, реакционную смесь перемешивают в течение 4 ч при комнатной температуре. Тетрагидрофуран удаляют при пониженном давлении и водный слой экстрагируют дихлорметаном. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая названное соединение (50 г, 62%) в виде оранжевого твердого вещества. 1H ЯМР (400 МГц, CDCl3)1,59-1,67 (м, 1 Н), 2,58-2,64 (м, 1 Н), 3,40(0,028 мол, 10 г) в тетрагидрофуране (150 мл) при температуре от 0 до -5 С в атмосфере азота добавляют по каплям ВН 3THF (1,0 М в тетрагидрофуране, 0,056 мол, 56 мл). По окончании добавления реакцион- 23017668 ную смесь кипятят с обратным холодильником в течение 3 ч. Реакционную смесь охлаждают до 0 С, гасят 1 н. хлористо-водородной кислотой и затем добавляют 1 н. водный раствор гидроксида натрия. Тетрагидрофуран удаляют при пониженном давлении и водный слой экстрагируют дихлорметаном. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток растворяют в метаноле (160 мл), добавляют 4 н. раствор хлористого водорода в диоксане (160 мл) и реакционную смесь нагревают в течение 4 ч при 80 С. Охлаждают до комнатной температуры и рН доводят до 10-12 добавлением 4 н. водного раствора гидроксида натрия. Метанол удаляют при пониженном давлении, и водный слой экстрагируют дихлорметаном. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая названное соединение в виде кристаллического, желтого твердого вещества (8,0 г, 83%). 1 Н ЯМР (400 МГц, CDCl3)1,32-1,38 (м, 1 Н), 2,19-2,22 (м, 1 Н), 2,34 (дт, 1 Н), 2,42-2,47(м, 1 Н), 2,54-2,58 (м, 1 Н), 2,92 (д, 1 Н), 3,07 (д, 1 Н), 3,34 (т, 1 Н), 3,61 (дт, 1 Н), 3,85 (дд, 1 Н), 3,97 (дд, 1 Н),4,01-4,07 (м, 1 Н), 6,67 (д, 1 Н), 7,48 (дд, 1 Н), 8,17 (д, 1 Н), 8,35 (д, 1 Н). Промежуточные соединения, указанные в приведенной ниже таблице, синтезируют в соответствии с процедурами, описанными в синтезах 48 и 49, используя в качестве исходных материалов подходящие фениламинопирролидины. Восстановление тетрагидропирроло[2,1-c][1,4]оксазин-4-она выполняют комплексом боран-тетрагидрофуран или комплексом боран-диметилсульфид, например, как описано в синтезах 50 и 51. Синтез 53. трет-Бутил-2,5-дигидро-1H-пиррол-1-карбоксилат. Раствор ди-трет-бутилдикарбоната (0,69 мол, 151,0 г) в метиленхлориде (200 мл) добавляют по каплям к раствору 3-пирролина (40,0 г, 0,57 мол) в метиленхлориде (400 мл) в течение 1,5 ч при 0 С и перемешивают при комнатной температуре в течение 10 ч. Растворитель удаляют и сырой продукт используют на следующей стадии в синтезе 47 (95,0 г, 98%). Синтез 54. Рацемический трет-бутиловый эфир транс-3-бром-4-гидроксипирролидин-1-карбоновой кислоты. К перемешиваемой смеси трет-бутил-2,5-дигидро-1H-пиррол-1-карбоксилата (0,29 мол, 50,0 г) в диметилсульфоксиде (360 мл) и воде (18 мл) постепенно в течение 15 мин при 0 С добавляют Nбромсукцинимид (0,325 мол, 58,0 г). После перемешивания при комнатной температуре в течение 2 ч добавляют воду (500 мл) и экстрагируют этилацетатом. Органический слой промывают солевым раствором, сушат над сульфатом натрия и концентрируют в вакууме, получая названное соединение в виде светло-коричневого масла (75,0 г, 95%). 1 Н ЯМР (500 МГц, CDCl3)4,43-4,53 (м, 1 Н), 4,12-4,20 (м, 1H),3,96-4,11 (м, 2H), 3,68-3,94 (м, 2 Н), 3,32-3,48 (м, 1 Н), 1,47 (с, 9 Н). Синтез 55. Рацемический трет-бутиловый эфир транс-3-бензиламино-4-гидроксипирролидин-1 карбоновой кислоты. Смесь сырого рацемического трет-бутилового эфира транс-3-бром-4-гидроксипирролидин-1 карбоновой кислоты (75,0 г) и водного 1 н. гидроксида натрия (400 мл) перемешивают при комнатной температуре в течение 2 ч. Добавляют бензиламин (0,86 мол, 80,0 г), перемешивают при 65 С в течение- 24017668 4,5 ч и затем охлаждают до 0 С. Полученный осадок собирают фильтрованием, промывают водой и изопропиловым эфиром и сушат, получая названное соединение в виде белого твердого вещества (50,0 г,63%). 1H ЯМР (300 МГц, CDCl3)7,21-7,38 (м, 5 Н), 4,06-4,15 (м, 1 Н), 3,84 (д, J=5,0 Гц, 2 Н), 3,57-3,75 (м,2 Н), 3,10-3,35 (м, 3H), 1,65 (ушир.с, 2 Н), 1,46 (с, 9 Н). Синтез 56. трет-Бутиловый эфир (3S,4S)-3-бензиламино-4-гидроксипирролидин-1-карбоновой кислоты, соль (+)-миндальной кислоты. Смесь рацемического трет-бутилового эфира транс-3-бензиламино-4-гидроксипирролидин-1 карбоновой кислоты (0,343 мол, 100,0 г) и (+)-миндальной кислоты (0,378 мол, 57,83 г) в ацетонитриле (1 л) и воде (10 мл) нагревают до 70 С в течение одного часа и затем охлаждают до комнатной температуры в течение 4 ч. Полученный кристаллический осадок собирают фильтрованием, промывают ацетонитрилом и перекристаллизовывают из ацетонитрила/воды (20:1), получая названное соединение в виде белого кристаллического твердого вещества (62,0 г, 40%). 1H ЯМР (500 МГц, CDCl3)7,19-7,44 (м, 8 Н), 5,05(ушир.с, 2 Н), 4,97 (с, 1 Н), 3,95-4,02 (м, 1 Н), 3,73 (с, 2 Н), 3,28-3,50 (м, 4 Н), 2,95-3,18 (м, 3H), 1,39 (с, 9 Н); т. пл, 185-188, []23D +50,0 (с 0,50, МеОН). Синтез 57. трет-Бутиловый эфир (3S,4S)-3-бензиламино-4-гидроксипирролидин-1-карбоновой кислоты. К соли (+)-миндальной кислоты трет-бутилового эфира (3S,4S)-3-бензиламино-4-гидроксипирролидин-1-карбоновой кислоты (60,0 г) добавляют 3% водный карбонат калия (300 мл), и экстрагируют свободный амин этилацетатом (3250 мл). Объединенный органический слой промывают солевым раствором и сушат над безводным сульфатом натрия. Растворитель испаряют при пониженном давлении, получая (S,S)-энантиомер в виде белого твердого вещества (34,0 г, 65%). APCI MS m/z 293 [М+Н]+; т. пл. 6970 С, []23D+18,0 (с, 0,50, МеОН). Синтез 58. трет-Бутиловый эфир (3S,4S)-3-амино-4-гидроксипирролидин-1-карбоновой кислоты. К раствору трет-бутилового эфира (3S,4S)-3-бензиламино-4-гидроксипирролидин-1-карбоновой кислоты (0,054 мол, 16,0 г) в н-бутаноле (225 мл) добавляют 10% Pd/C (5,0 г), и гидрируют смесь при 50 psi(3,45105 Па) в течение 10 ч. Реакционную смесь фильтруют через слой диатомитовой земли и промывают этанолом. Фильтрат выпаривают, получая названное соединение в виде белого твердого вещества(11,10 г, 99%). APCI MS m/z 203 [М+Н]+; []23D+7,8 (с, 0,50, МеОН). Синтез 59. (трет-Бутиловый эфир (3S, 4S)-3-(4-бром-2-нитрофениламино)-4-гидроксипирролидин 1-карбоновой кислоты. К раствору 1-бром-4-фтор-3-нитробензола (0,125 мол, 26,0 г) в этилацетате (300 мл) добавляют трет-бутиловый эфир (3S, 4S)-3-бензиламино-4-гидроксипирролидин-1-карбоновой кислоты (0,113 мол,23,0 г) и триэтиламин (0,339 мол, 35,0 г), кипятят с обратным холодильником в течение 14 ч, охлаждают до комнатной температуры и промывают водой и солевым раствором. Сушат над безводным сульфатом натрия, растворитель испаряют и очищают при помощи колоночной флэш-хроматографии, получая названное соединение в виде желтого твердого вещества (43,0 г, 90%). APCI MS m/z 403 [M+H]+. Синтез 60. трет-Бутиловый эфир (3R, 4R)-3-бензиламино-4-гидроксипирролидин-1-карбоновой кислоты, соль (-)-миндальной кислоты. Рацемический трет-бутиловый эфир транс-3-бензиламино-4-гидроксипирролидин-1-карбоновой кислоты (0,085 мол, 25,0 г) и (-)-миндальную кислоту (0,094 мол, 14,45 г, 1,1 экв.) в ацетонитриле (200 мл) и воде (5 мл) перемешивают при 70 С в течение одного часа. Охлаждают до комнатной температуры в течение 4 ч. Полученный кристаллический осадок собирают фильтрованием, промывают ацетонитрилом и перекристаллизовывают из ацетонитрила/воды (20:1), получая соль (-)-миндальной кислоты в виде белого кристаллического твердого вещества (34,0 г, 88%). 1 Н ЯМР (500 МГц, CDCl3)7,19-7,44 (м, 8 Н); 5,05 (ушир.с, 2 Н), 4,97 (с, 1 Н), 3,95-4,02 (м, 1 Н), 3,73 (с, 2 Н), 3,28-3,50 (м, 4 Н), 2,95-3,18 (м, 3H), 1,39 (с,9 Н); []23D -58,0 (с 0,50, МеОН). Синтез 61. трет-Бутиловый эфир (3R,4R)-3-(4-бром-2-нитрофениламино)-4-гидроксипирролидин-1 карбоновой кислоты. Названное соединение синтезируют в соответствии с процедурами, описанными в синтезах 58, 59 и 60, используя в качестве исходного материала соль (-)-миндальной кислоты трет-бутилового эфира(2,50 ммол, 1,0 г), хлоруксусной кислоты (3,0 ммол, 300 мг) и трифенилфосфина (3,0 ммол, 850 мг) в тетрагидрофуране (40 мл) добавляют диэтилазодикарбоксилат (3,0 ммол, 600 мг) и перемешивают при комнатной температуре в течение 12 ч. Растворитель удаляют, к остатку добавляют этилацетат (50 мл) и промывают водой и солевым раствором. Сушат над безводным сульфатом натрия и испаряют. Остаток очищают посредством колоночной хроматографии, элюируя 20% этилацетатом в метиленхлориде, получая названное соединение в виде желтого твердого вещества (1,10 г, 98%). 1 Н ЯМР (500 МГц, CDCl3)8,35 (с, 1 Н), 8,20 (м, 1 Н), 7,60 (м, 1 Н), 6,82 (м, 1 Н), 5,55 (д, J=6,8 Гц, 1 Н), 4,32 (м, 1 Н), 4,13 (ушир.д, J=8,2- 25017668 Гц, 2 Н), 3,90 (м, 1 Н), 3,79 (м, 1 Н), 3,74 (дд, J=4,2, 10,4 Гц, 1 Н), 3,25 (т, J=7,20 Гц, 1 Н), 1,41 (с, 9 Н). Синтез 63. (3S,4R)-трет-Бутил-3-(4-бром-2-нитрофениламино)-4-гидроксипирролидин-1-карбоксилат. К раствору (3S,4R)-трет-бутил-3-(4-бром-2-нитрофениламино)-4-(2-хлорацетокси)пирролидин-1 карбоксилата (2,20 ммол, 1,0 г) в метаноле добавляют 2,0 М водный гидроксид лития (5 мл) и перемешивают при комнатной температуре в течение 3,5 ч. Растворитель удаляют, разбавляют остаток этилацетатом (50 мл) и промывают водой и солевым раствором. Сушат над безводным сульфатом натрия, фильтруют и растворитель испаряют. Очищают посредством колоночной хроматографии, элюируя 50% этилацетат в метиленхлориде, получая названное соединение в виде желтого твердого вещества (900 мг,98%). APCI MS m/z 462 [М+Н]+. Синтез 64. (3R,4S)-трет-Бутил-3-(4-бром-2-нитрофениламино)-4-гидроксипирролидин-1-карбоксилат. Названное соединение синтезируют в соответствии с процедурами, описанными в cинтезах 62 и 63,используя в качестве исходного материала (3R,4R)-трет-бутил-3-(4-бром-2-нитрофениламино)-4 гидроксипирролидин-1-карбоксилат. Синтез 65. (3S,4S)-4-(4-Бром-2-нитрофениламино)пирролидин-3-ол, гидрохлорид. В суспензию трет-бутилового эфира (3S,4S)-3-(4-бром-2-нитрофениламино)-4-гидроксипирролидин-1-карбоновой кислоты (0,106 мол, 43,0 г) в этилацетате (500 мл) при 0 С барботируют безводный газообразный хлористый водород в течение 10 мин и перемешивают при комнатной температуре в течение одного часа. Растворитель удаляют и полученное твердое вещество сушат в вакууме, получая названное соединение в виде желтого твердого вещества (35,0 г, 98%). APCI MS m/z 302 [М+Н]+. Синтез 66. (3S,4S)-4-(4-Бром-2-нитрофениламино)-1-метилпирролидин-3-ол К гидрохлориду (3S,4S)-4-(4-Бром-2-нитрофениламино)пирролидин-3-ола (0,104 мол, 35,0 г) в ацетонитриле добавляют при 0 С триацетоксиборгидрид натрия (0,37 мол, 78,0 г). К этой смеси медленно добавляют в течение 10 мин 37% водный формальдегид (30 мл) и перемешивают при комнатной температуре в течение 10 ч. Добавляют насыщенный водный раствор бикарбоната натрия и перемешивают в течение одного часа. Экстрагируют этилацетатом, и промывают водой и солевым раствором. Сушат над безводным сульфатом натрия, фильтруют, и растворитель испаряют, получая названное соединение в виде желтого твердого вещества (34,0 г, 98%). APCI MS m/z 302 [М+Н]+; []23D+58,6 (с, 0,50, МеОН). Промежуточные соединения, указанные в приведенной ниже таблице, синтезируют в соответствии с процедурами, описанными в синтезах 65 и 66, в которых подходящий трет-бутил-3-(4-бром-2 нитрофениламино)-4-гидроксипирролидин-1-карбоксилат обрабатывают безводным газообразным HCl в течение 10-30 мин и затем перемешивают в течение 1-4 ч. Полученный пирролидин, с которого снята защита, обрабатывают формальдегидом в течение времени, составляющего от 15 мин до 10 ч. Синтез 70. трет-Бутиловый эфир 4-гидроксипиперидин-1-карбоновой кислоты. Боргидрид натрия (0,82 мол, 31,3 г) добавляют порциями при 0 С к раствору 1-(третбутоксикарбонил)-4-пиперидона (0,74 мол, 150 г) и триэтиламина (1,5 мол, 151,6 г) в дихлорметане (1 л). По завершении добавления реакционную смесь нагревают до комнатной температуры и перемешивают в- 26017668 течение 4 ч. Реакционную смесь гасят насыщенным раствором хлорида аммония и удаляют этанол при пониженном давлении. Остаток растворяют в воде и экстрагируют дихлорметаном. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая названное соединение(150 г, 99%) в виде вязкого твердого вещества. 1H ЯМР (400 МГц, CDCl3)1,45 (с, 9 Н), 1,53 (д, 2 Н), 1,831,87 (м, 2 Н), 3,02 (дт, 2 Н), 3,8-3,85 (м, 3 Н). Синтез 71. трет-Бутиловый эфир 4-метансульфонилоксипиперидин-1-карбоновой кислоты. Метансульфонилхлорид (1,12 мол, 128,8 г) добавляют по каплям при 0 С к раствору третбутилового эфира 4-гидроксипиперидин-1-карбоновой кислоты (0,74 мол, 150 г) и триэтиламина (1,5 мол, 151,6 г) в дихлорметане (1 л). По завершении добавления реакционную смесь нагревают до комнатной температуры и перемешивают в течение 16 ч. Реакционную смесь разбавляют дихлорметаном, промывают насыщенным раствором бикарбоната натрия и водой. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая названное соединение (192 г, 92%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, CDCl3)1,45 (с, 9 Н), 1,78-1,85 (м, 2 Н), 1,93-1,98 (м,2 Н), 3,03 (с, 3H), 3,26-3,32 (м, 2 Н), 3,68-3,71 (м, 2 Н), 4,85-4,90 (м, 1 Н). Синтез 72. трет-Бутиловый эфир 3,6-дигидро-2 Н-пиридин-1-карбоновой кислоты. Раствор трет-бутилового эфира 4-метансульфонилоксипиперидин-1-карбоновой кислоты (0,69 мол,193 г) в DBU (400 мл) нагревают при 80 С в течение 16 ч. Реакционную смесь разбавляют водой, экстрагируют диэтиловым эфиром и органический слой промывают 1 н. хлористо-водородной кислотой и насыщенным раствором бикарбоната натрия. Органический слой сушат над безводным сульфатом натрия,фильтруют и концентрируют, получая названное соединение (116 г, 92%) в виде коричневого масла. 1H ЯМР (400 МГц, CDCl3)1,46 (с, 9 Н), 2,12 (ушир.с, 2 Н), 3,47 (т, 2 Н), 3,87 (с, 2 Н), 5,65 (ушир.с, 1 Н), 5,81(ушир.с, 1 Н). Синтез 73. Рацемический трет-бутиловый эфир цис-7-окса-3-азабицикло[4.1.0]гептан-3-карбоновой кислоты. Раствор м-хлорпероксибензойной кислоты (0,77 мол, 133,1 г) в дихлорметане (200 мл) добавляют по каплям при 0 С к раствору трет-бутилового эфира 3,6-дигидро-2 Н-пиридин-1-карбоновой кислоты(0,64 мол, 117,8 г) в дихлорметане (1 л). По завершении добавления реакционную смесь нагревают до комнатной температуры и перемешивают в течение 16 ч. Реакционную смесь разбавляют дихлорметаном и промывают 4 н. раствором гидроксида натрия, водой и солевым раствором. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая названное соединение (112 г,87%) в виде светло-желтого масла. 1H ЯМР (400 МГц, CDCl3)1,44 (с, 9 Н), 1,87-1,93 (м с, 1 Н), 2,04(ушир.с, 1 Н), 3,11 (ушир.с, 1 Н), 3,20 (ушир.с, 1 Н), 3,28 (с, 1 Н), 3,44 (ушир.с, 1 Н), 3,68 (ушир.с, 1 Н), 3,813,92 (м, 1 Н). ES-MS m/z 204 [М+Н]+. Синтез 74. Рацемический трет-бутиловый эфир транс-4-азидо-3-гидроксипиперидин-1-карбоновой кислоты. Смесь рацемического трет-бутилового эфира транс-7-окса-3-азабицикло[4.1.0]гептан-3-карбоновой кислоты (0,56 мол, 112 г), азида натрия (1,12 мол, 7 3,08 г), хлорида аммония (0,56 мол, 30,07 г) и метанола/воды (3:1) (1 л) нагревают до 65 С в течение 16 ч. Реакционную смесь разбавляют водой, экстрагируют дихлорметаном и органический слой промывают насыщенным раствором бикарбоната натрия и солевым раствором. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают на колонке с силикагелем, используя в качестве элюента 6% этилацетат в гексанах, получая названное соединение (42,3 г, 42%) в виде светло-желтого масла. 1H ЯМР (400 МГц,CDCl3)1,48 (с, 9 Н), 1,84 (ушир.с, 1 Н), 1,97-2,01 (м, 1 Н), 2,74-2,82 (м, 1 Н), 2,88 (ушир.с, 1 Н), 3,37-3,40(0,23 мол, 56,6 г) растворяют в метаноле (500 мл), добавляют 10% палладий на угле (12,0 г) и гидрируют(1 атм) при комнатной температуре в течение 16 ч. Катализатор отфильтровывают через слой диатомитовой земли и фильтрат концентрируют, получая названное соединение (53,7 г, 99%) в виде плотного светло-желтого масла. Синтез 76. Рацемический трет-бутиловый эфир транс-4-бензиламино-3-гидроксипиперидин-1 карболовой кислоты. К раствору рацемического трет-бутилового эфира транс-4-амино-3-гидроксипиперидин-1-карбоновой кислоты (0,24 мол, 53,7 г) и уксусной кислоты (92 мл) в этаноле (400 мл) добавляют бензальдегид(0,24 мол, 26,3 г), и реакционную смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют цианоборгидрид натрия (0,49 мол, 31,2 г) и реакционную смесь перемешивают в течение 3 ч. Реакционную смесь гасят раствором бикарбоната натрия и экстрагируют дихлорметаном. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток растворяют в минимальном количестве дихлорметана и добавляют 0,1 н. водную хлористо-водородную кислоту для поддержания рН в диапазоне 4-5. Органический слой отбрасывают. Кислотный водный слой три раза про- 27017668 мывают дихлорметаном и отбрасывают органический слой. рН водного слоя доводят до 10-12 и экстрагируют дихлорметаном. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая названное соединение (35,7 г, 46%) в виде светло-желтого масла. 1H ЯМР (400 МГц, CDCl3)1,35-1,40 (м, 2 Н), 1,45 (с, 9 Н), 2,05 (ушир.с, 1 Н), 2,43 (т, 1 Н), 2,57 (т, 1 Н), 2,69 (ушир.с,1 Н), 3,27 (ушир.с, 1 Н), 3,70 (д, 1 Н), 3,94 (д, 1 Н), 4,15 (ушир.с, 1 Н), 4,26 (д, 1 Н), 7,23-7,29 (м, 1 Н), 7,307,32 (м, 2 Н), 7,33-7,36 (м, 2 Н). Синтез 77. трет-Бутиловый эфир (3R,4R)-4-бензиламино-3-гидрокси-4-пиперидин-1-карбоновой кислоты, соль L-(+)-винной кислоты. Смесь рацемического трет-бутилового эфира транс-4-бензиламино-3-гидроксипиперидин-1-карбоновой кислоты (0,11 мол, 35,0 г) и L-(+)-винной кислоты (0,12 мол, 18,9 г, 1,2 экв.) и ацетонитрила/воды (20:1) (175 мл) нагревают при 75 С в течение 4 ч. Реакционную смесь концентрируют при пониженном давлении, получая соль винной кислоты (53,0 г). Остаток три раза кристаллизуют из ацетонитрила. Кристаллы отфильтровывают и сушат в вакууме, получая названное соединение (18,3 г, 67%). ВЭЖХ (Колонка: Chiralcel OD-H (250 4,6 мм); система растворителей: изопропанол/0,1% триэтиламин в гексанах (8:92); скорость потока: 0,800 мл/мин; длина волны: 258 нм): 99,9% ее. tR 9,502 мин. tR другого энантиомера(3S,4S)-3-гидрокси-4 фенэтилпиперидин-1-карбоновой кислоты) 8,714 мин. Синтез 78. трет-Бутиловый эфир (3R,4R)-4-амино-3-гидроксипиперидин-1-карбоновой кислоты. Соль L-(+)-винной кислоты трет-бутилового эфира (3R, 4R)-4-бензил-3-гидрокси-4-фенэтилпиперидин-1-карбоновой кислоты (0,04 мол, 18,3 г) растворяют в 4% растворе карбоната калия (500 мл) и перемешивают в течение 30 мин. Свободный амин экстрагируют дихлорметаном. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая свободный амин (11,3 г, 92%). Амин растворяют в метаноле (110 мл), добавляют 10% палладий на угле (2,5 г) и гидрируют (1 атм) при комнатной температуре в течение 16 ч. Катализатор отфильтровывают через слой диатомитовой земли и концентрируют фильтрат, получая названное соединение (7,6 г, 95%) в виде плотного светло-желтого масла. []26,1 +2,141 (с 1,0, метанол). 1 Н ЯМР (400 МГц, ДМСО-d6)1,08 (дкв, 1 Н), 1,38 (с, 9 Н), 1,67 (дд,1 Н), 1,80 (ушир.с, 1 Н), 2,37-2,45 (м, 2 Н), 2,67 (ушир.с, 1 Н), 2,90 (ушир.с, 1 Н), 3,79 (ушир.с, 1 Н), 3,88(ушир.с, 1 Н), 5,0 (ушир.с, 1 Н). ES-MS m/z 217 [М+Н]+. Синтез 79. трет-Бутиловый эфир (3R, 4R)-4-(4-бром-2-нитрофениламино)-3-гидроксипиперидин-1 карбоновой кислоты. Смесь трет-бутилового эфира (3R,4R)-4-амино-3-гидроксипиперидин-1-карбоновой кислоты (0,02 мол, 6,0 г), 5-бром-2-фторнитробензола (0,03 мол, 6,71 г) и триэтиламина (0,058 мол, 5,88 мл) в этилацетате (180 мл) нагревают до кипения в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом и промывают водой и солевым раствором. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая названное соединение (12,35 г,сырое). 1H ЯМР (400 МГц, CDCl3)1,46 9 Н), 1,50-1,51 (м, 1 Н), 2,08-2,12 (м, 1 Н), 2,93 (дд, 1 Н), 3,01(дд, 1 Н), 8,08 (д, 1 Н), 8,30 (д, 1 Н). ES-MS m/z 316 [М-Вос]+. Синтез 80. Гидрохлоридная соль (3R,4R)-4-(4-бром-2-нитрофениламино)пиперидин-3-ола. 4,0 М раствор хлористого водорода в диоксане (70 мл) медленно добавляют при комнатной температуре к раствору трет-бутилового эфира (3R, 4R)-4-(4-бром-2-нитрофениламино)-3-гидроксипиперидин 1-карбоновой кислоты (0,027 мол, 11,5 г) в сухом дихлорметане (40 мл) и перемешивают в течение 16 ч. Осадок отфильтровывают и сушат в вакууме, получая названное соединение (8,68 г, 89%) в виде желтого твердого вещества. Н ЯМР (400 МГц, CD3OD)1,81-1,86 (м, 1 Н), 2,39-2,44 (м, 1 Н), 3,02-3,07 (м, 1 Н),3,18-3,26 (м, 1 Н), 3,34-3,40 (м, 1 Н), 3,45 (дд, 1 Н), 3,87-3,91 (т, 2 Н), 7,17 (д, 1 Н), 7,61 (дд, 1 Н), 8,28 (д, 1 Н). Синтез 81. (3R,4R)-4-(4-Бром-2-нитрофениламино)-1-метилпиперидин-3-ол. К раствору гидрохлоридной соли (3R, 4R)-4-(4-бром-2-нитрофениламино)пиперидин-3-ола (0,0,024 мол, 8,68 г) и уксусной кислоты (10,2 мл) в воде (42 мл) добавляют формальдегид (20,5 мл, 37-41% водный раствор) и реакционную смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют цианоборгидрид натрия (0,073 мол, 4,6 г) и реакционную смесь перемешивают в течение 3 ч. Реакционную смесь гасят раствором бикарбоната натрия и экстрагируют дихлорметаном. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая названное соединение(7,8 г, 97%). 1 Н ЯМР (400 МГц, CDCl3)1,63-1,71 (м, 1 Н), 2,16-2,21 (м, 2 Н), 2,33 (с, 3H), 2,35-2,38 (м,1 Н), 2,60 (ушир.с, 1 Н), 2,80 (д, 1 Н), 3,54-3,56 (м, 1 Н), 3,75-3,79 (м, 1 Н), 6,92 (д, 1 Н), 7,48 (дд, 1 Н), 8,14 (д,1 Н), 8,30 (д, 1 Н). Синтез 82. трет-Бутиловый эфир (3S,4S)-4-бензиламино-3-гидрокси-4-пиперидин-1-карбоновой кислоты, D-(-)-винной кислоты соль. Концентрируют маточный раствор, полученный в синтезе 77, получая обогащенную энантиомером соль L-(+)-винной кислоты трет-бутилового эфира (3S,4S)-3-гидрокси-4-фенэтилпиперидин-1 карбоновой кислоты (0,0377 мол, 17,2 г). Добавляют 4% водный раствор карбоната калия (500 мл) и перемешивают в течение 30 мин. Свободный амин экстрагируют дихлорметаном. Органический слой су- 28017668 шат над безводным сульфатом натрия, фильтруют и концентрируют, получая свободный амин (11,0 г). Нагревают смесь трет-бутилового эфира 4-бензиламино-3-гидроксипиперидин-1-карбоновой кислоты(100 мл) при 75 С в течение 4 ч. Реакционную смесь концентрируют при пониженном давлении, получая сырую соль винной кислоты. Остаток три раза кристаллизуют из ацетонитрила. Кристаллы отфильтровывают и сушат в вакууме, получая названное соединение (14,8 г, 90%). ВЭЖХ (Колонка: Chiralcel OD-H(250 4,6 мм); система растворителей: изопропанол/0,1% триэтиламин в гексанах (8:92); скорость потока: 0,800 мл/минут; длина волны: 258 нм): 96,4% ее. tR 8,714 мин, tR другого энантиомера (соли L-(+)винной кислоты трет-бутилового эфира (3R,4R)-3-гидрокси-4-фенэтилпиперидин-1-карбоновой кислоты) 9,502 мин. Синтез 83. трет-Бутиловый эфир (3S,4S)-4-амино-3-гидроксипиперидин-1-карбоновой кислоты. Соль D-(-)-винной кислоты трет-бутилового эфира (3S,4S)-4-бензиламино-3-гидрокси-4-фенэтилпиперидин-1-карбоновой кислоты (0,036 мол, 16,4 г) растворяют в 4% водном растворе карбоната калия(500 мл) и перемешивают в течение 30 мин. Экстрагируют дихлорметаном, органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая свободный амин (11,0 г, 99%). Амин растворяют в метаноле (110 мл), добавляют 10% палладий на угле (5,5 г) и гидрируют (50 psi = 3,45105 Па) при комнатной температуре в течение 2 ч. Катализатор отфильтровывают через диатомитовую землю и концентрируют, получая названное соединение (7,7 г, 99%) в виде плотного, светложелтого масла. Синтез 84. трет-Бутиловый эфир (3S,4S)-4-(4-бром-2-нитрофениламино)-3-гидроксипиперидин-1 карбоновой кислоты. Смесь трет-бутилового эфира (35,4S)-4-амино-3-гидроксипиперидин-1-карбоновой кислоты (0,035 мол, 7,5 г), 5-бром-2-фторнитробензола (0,038 мол, 8,48 г) и триэтиламина (0,073 мол, 7,44 г) в этилацетате (250 мл) нагревают до кипения в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом и промывают водой и солевым раствором. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая названное соединение (16,16 г,сырое). Синтез 85. Гидрохлорид (3S,4S)-4-(4-бром-2-нитрофениламино)пиперидин-3-ола. 4 М раствор хлористого водорода в диоксане (90 мл) медленно добавляют при комнатной температуре к раствору трет-бутилового эфира (3S,4S)-4-(4-бром-2-нитрофениламино)-3-гидроксипиперидин-1 карбоновой кислоты (0,035 мол, 14,6 г) в сухом дихлорметане (50 мл) и перемешивают в течение 16 ч. Осадок отфильтровывают и сушат в вакууме, получая названное соединение (9,84 г, 80%) в виде желтого твердого вещества. Синтез 86. (35,4S)-4-(4-Бром-2-нитрофениламино)-1-метилпиперидин-3-ол. Формальдегид (24 мл, 37-41% водный раствор) добавляют к раствору гидрохлорида (3S,4S)-4-(4 бром-2-нитрофениламино)пиперидин-3-ола (0,028 мол, 9,8 г) и уксусной кислоты (12 мл) в воде (46 мл),и реакционную смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют цианоборгидрид натрия (0,083 мол, 5,26 г) и реакционную смесь перемешивают в течение 3 ч. Реакционную смесь гасят раствором бикарбоната натрия и экстрагируют дихлорметаном. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая названное соединение (8,4 г,92%). 1 Н ЯМР (400 МГц, CDCl3)1,63-1,71, (м, 1 Н 2,16-2,21 (м, 2 Н), 2,33 (с, 3H), 2,35-2,38 (м, 1 Н), 2,60(0,037 мол, 13,7 г), трифенилфосфина (0,007 мол, 2,02 г) и ацетата калия (0,074 мол, 7,29 г) в диоксане/воде (3:1) (450 мл) три раза дегазируют азотом. К реакционной смеси добавляют ацетат палладия (II)(0,001 мол) и снова дегазируют три раза азотом. Полученную реакционную смесь нагревают при 85 С в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют водой, экстрагируют этилацетатом, обединяют органический слой, промывают водой и солевым раствором. Органическую фазу сушат над безводным сульфатом натрия, фильтруют и концентрируют. Очищают на колонке с силикагелем, используя в качестве элюента 7% этилацетат в гексанах, получая названное соединение (9,1 г, 48%) в виде оранжевого твердого вещества. 1H ЯМР (400 МГц, CDCl3),1,26 (д, 3H), 2,44-2,57 (м, 6 Н),- 29
МПК / Метки
МПК: C07D 498/04, A61P 5/40, A61K 31/4184, C07D 405/14, C07D 405/06
Метки: минералокортикоидных, нестероидных, антагонистов, рецепторов, производные, качестве, 6н-дибензо[b,e]оксепина
Код ссылки
<a href="https://eas.patents.su/30-17668-proizvodnye-6n-dibenzobeoksepina-v-kachestve-nesteroidnyh-antagonistov-mineralokortikoidnyh-receptorov.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 6н-дибензо[b,e]оксепина в качестве нестероидных антагонистов минералокортикоидных рецепторов</a>
Производные фенилпиразола в качестве нестероидных лигандов глюкокортикоидных рецепторов

Номер патента: 15540
Опубликовано: 31.08.2011
Авторы: Ко Диана Мэри, Макдональд Саймон Джон Фосет, Барнетт Хизер Анна, Килинг Стивен Филип, Кэмпбелл Айан Бакстер, Маклэй Айэйн Макфарлэйн, Инглис Грэхам Джордж Адам, Вулвен Джеймс Майкл, Джонс Хэйдн Теренс, Вейнгартен Гордон Гэд, Купер Энтони Уилльям Джеймс, Скоун Филип Алан
МПК: A61K 31/415, C07D 231/38
Метки: производные, лигандов, фенилпиразола, рецепторов, нестероидных, качестве, глюкокортикоидных
Формула / Реферат:
1. Соединение формулы (I)где R1выбран из водорода, метила, этила и 2-фторэтила;каждый из R2 и R3независимо выбран из брома, хлора, фтора, -CHF2, -CF3 и -OCHF2или R2 представляет собой -SO2CH3, и R3 представляет собой водород;n означает целое число, выбранное из 0, 1 и 2,когда n равно 1, X выбран из хлора и фтора, икогда n равно 2, каждый X представляет собой фтор;или его соль или сольват.2. Соединение по п.1, где R1 выбран из водорода и этила.3....
Азабициклические, азатрициклические и азаспироциклические производные аминоциклогексана в качестве антагонистов рецепторов nmda, 5ht и нейронных никотиновых рецепторов

Номер патента: 7098
Опубликовано: 30.06.2006
Авторы: Калвиньш Ивар, Хенрих Маркус, Парсонс Кристофер Грахам Рафаэль, Каусс Валерьянс, Гольд Маркус, Йиргенсонс Айгарс, Ванейевс Максимс, Даныш Войцех
МПК: A61K 31/40, A61P 25/18, A61P 25/16...
Метки: качестве, антагонистов, азатрициклические, производные, аминоциклогексана, азаспироциклические, нейронных, азабициклические, рецепторов, никотиновых, nmda
Формула / Реферат:
1. Соединения формулы (1) где R и R1-R5, каждый независимо, выбран из C1-6-алкильных групп, С2-6-алкенильных групп, С2-6-алкинильных групп, С6-12-арил-С1-4-алкильных групп, необязательно замещенных С6-12-арильных групп и в случае R и R2-R5 - из атомов водорода, с условием, что по меньшей мере один из R2 и R3 и по меньшей мере один из R4 и R5 не является водородом, или R и R1 вместе представляют С3-5-алкиленовую или алкениленовую группу, причем...
О-замещенные производные дибензилмочевины в качестве антагонистов рецепторов trpv1

Номер патента: 15152
Опубликовано: 30.06.2011
Авторы: Павани Мария Джованна, Тревизани Марчелло, Бореа Пьер Андреа, Джеппетти Пьеранджело, Баральди Пьер Джованни, Фруттароло Франческа
МПК: C07C 275/24, A61K 31/17, A61P 29/00...
Метки: trpv1, производные, рецепторов, качестве, дибензилмочевины, о-замещенные, антагонистов
Формула / Реферат:
1. Соединения формулы (I)где R выбран из галогена, алкила, алкокси, арила и гетероарила;R1 выбран из 2-гидроксиэтила, 2,3-дигидроксипропила, 3-гидроксипропила, 2,2-дигидроксиэтила, 3,3-дигидроксипропила, 1,3-диоксоланэтила, 1,3-диоксанметила, 1,3-диоксоланметила, 1,3-диоксанэтила, 3-фтор-2-гидроксипропила, 3-карбокси-2-гидроксипропила, 3-хлор-2-гидроксипропила, 2-гидроксипропила, 2-гидроксипропен-2-ила, морфолиноэтила, пиперазиноэтила,...
Производные бензамидов в качестве антагонистов брадикининовых рецепторов

Номер патента: 14498
Опубликовано: 30.12.2010
Авторы: Шмидт Эва, Ваго Иштван, Кешеру Дьёрдь, Сентирмай Эва, Беке Дьюла, Хорнок Каталин, Бозо Эва, Ваштаг Моника, Фаркаш Шандор
МПК: A61K 31/18, C07D 207/08, A61P 29/00...
Метки: рецепторов, качестве, производные, бензамидов, антагонистов, брадикининовых
Формула / Реферат:
1. Фенилсульфамоил бензамидные производные антагонистов брадикининовых В1-рецепторов формулы (I)где R1представляет атом водорода или С1-С4-алкильную группу;R2 выбирается из(1) атома водорода;(2) C1-C6 линейной или разветвленной алкильной группы;(3) -(CH2)n-NH2;(4) -(СН2)n-ОН;(5) -(CH2)n-CO-NH2;(6) -(CH2)n-COORc;(7) бензила, необязательно замещенного одной или более гидроксигруппой или атомом галогена; илиR1, R2и атом углерода, к которому они оба...
Производные бензамидопиперидина в качестве антагонистов рецепторов вещества р

Номер патента: 5409
Опубликовано: 24.02.2005
Авторы: Хамфри Джон Майкл, Чэппи Томас Аллен, Винсент Лоренс Альберт, Соболов-Джейнс Сьюзн Бет, Хуанг Дзианхуа, О'нэйлл Брайан Томас, Нэйджел Артур Адам, Арнольд Эрик Платт
МПК: A61K 31/5395, C07D 401/14, A61P 1/00...
Метки: производные, рецепторов, антагонистов, качестве, вещества, бензамидопиперидина
Формула / Реферат:
1. Соединение формулы где Q обозначает C=S или C=O; A обозначает CH, CH2, C(C1-C6)алкил, CH(C1-C6)алкил, C(CF3) или CH(CF3), при условии, что, когда B присутствует, A должен представлять либо CH, C(C1-C6)алкил, либо C(CF3); B отсутствует или обозначает метилен или этилен; Y обозначает N и Z обозначает CH или Y обозначает CH и Z обозначает N; G обозначает NH(CH2)q, S(CH2)q или O(CH2)q, где q равно 0 или 1; при условии, что, когда q равно 0,...
Предыдущий патент: Способ и устройство для определения границы раздела по меньшей мере двух текучих сред
Следующий патент: 1,2,4-оксадиазольные соединения для лечения аутоиммунных заболеваний
Случайный патент: Напитки и пищевые продукты, устойчивые к индуцированным светом изменениям вкуса, способы их производства и композиции, обеспечивающие такую устойчивость