3-(имидазолил)пиразоло[3,4-b]пиридины







Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемая соль, гидрат или N-оксид.
2. Соединение по п.1 в форме гидрата.
3. Соединение по п.1 в форме фармацевтически приемлемой соли.
4. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически приемлемый эксципиент или носитель.
5. Способ получения соединения по п.1, включающий контактирование соединения формулы

с имидазолобразующим реагентом.
6. Способ по п.5, где имидазолобразующий реагент выбирают из группы, состоящей из глиоксаля или эквивалента глиоксаля.
7. Способ по п.5, где имидазолобразующий реагент представляет собой глиоксаль и контактирование происходит в присутствии ацетата аммония.
8. Способ получения соединения по п.1, включающий следующие стадии:
(a) контактирование соединения формулы

с этилендиамином с образованием имидазолина и
(b) окисление имидазолина с образованием соединения по п.1.
9. Способ по п.8, где окисление осуществляют реагентом, выбранным из группы, состоящей из KMnO4, MnO2, PhI(OAc)2, реагентов окисления по Сверну и периодинана Десс-Мартина.
10. Способ лечения опосредуемых CCR1 заболеваний или состояний, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по п.1.
11. Способ по п.10, где указанное опосредуемое CCR1 заболевание или состояние представляет собой воспалительное состояние.
12. Способ по п.10, где указанное опосредуемое CCR1 заболевание или состояние представляет собой иммунорегуляторное расстройство.
13. Способ по п.10, где указанное опосредуемое CCR1 заболевание или состояние выбирают из группы, состоящей из ревматоидного артрита, рассеяного склероза, отторжения трансплантата, рестеноза, дерматита, экземы, крапивницы, васкулита, воспалительного заболевания кишечника, пищевой аллергии, астмы, болезни Альцгеймера, болезни Паркинсона, псориаза, красной волчанки, остеоартрита, удара и энцефаломиелита.
14. Способ по п.10, где указанное введение является пероральным, парентеральным, ректальным, трансдермальным, сублингвальным, назальным или местным.
15. Способ по п.10, где указанное соединение вводят в сочетании с противовоспалительным средством, анальгетиком, антипролиферативным средством, ингибитором метаболизма, ингибитором миграции лейкоцитов или иммуномодулятором.
16. Способ по п.10, где указанное соединение представляет собой

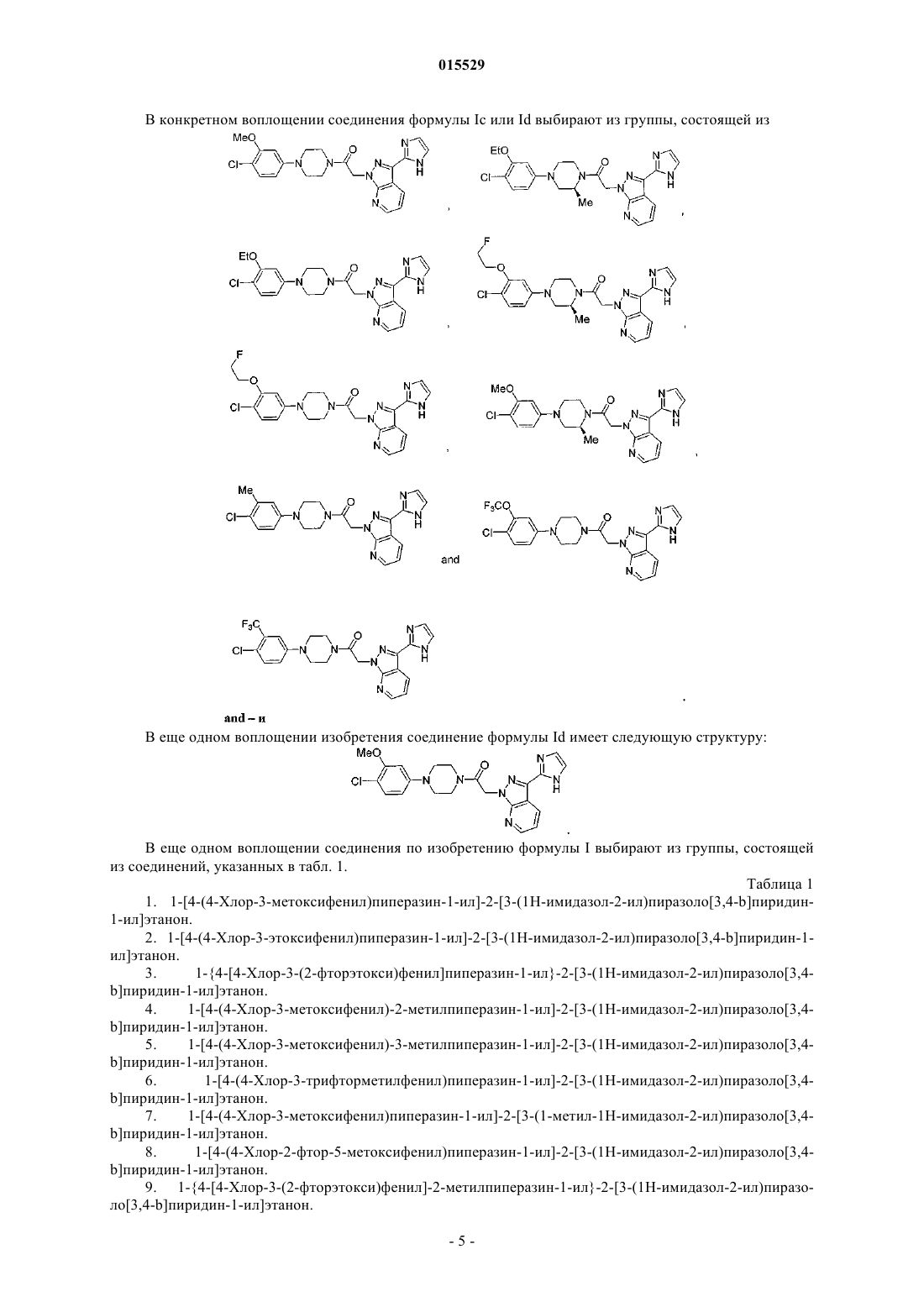
Текст
ОПИСАНИЕ ИЗОБРЕТЕНИЯ К ЕВРАЗИЙСКОМУ ПАТЕНТУ Дата публикации и выдачи патента Описываются соединения, которые действуют как сильные антагонисты рецептора CCR1 и обладают in vivo противовоспалительной активностью. Соединения представляют собой производные 3-имидазолилпиразоло[3,4-b]пиридина и применимы в фармацевтических композициях, способах лечения заболевания, опосредуемого CCR1, и в качестве контроля в анализах для идентификации конкурентных антагонистов CCR1. 015529 Область техники, к которой относится изобретение Настоящее изобретение относится к соединениям, фармацевтическим композициям, содержащим одно или несколько указанных соединений или их фармацевтически приемлемые соли, которые эффективны при ингибировании связывания различных хемокинов, таких как MIP-1, лейкотактин, MPIF-1 иRANTES, с рецептором CCR1. Как антагонисты или модуляторы рецептора CCR1 соединения и композиции применимы при лечении состояний воспалительных и иммунных расстройств и заболеваний. Предшествующий уровень техники Здоровье человека зависит от способности организма обнаруживать и разрушать чужеродные патогены, которые в ином случае могут отбирать жизненные ресурсы у индивидуума и/или вызывать заболевание. Иммунная система, включающая лейкоциты (белые клетки крови (WBC): T- и B-лимфоциты, моноциты, макрофаги, гранулоциты, NK-клетки, тучные клетки, дендритные клетки и иммунопроизводные клетки (например, остеокласты), лимфоидные ткани и лимфатические сосуды, является защитной системой организма. Для борьбы с инфекцией белые клетки крови циркулируют по организму для обнаружения патогенов. Как только патоген обнаружен, природные иммуноциты и, в частности, цитотоксическиеT-клетки собираются в месте инфекции для разрушения патогена. Хемокины действуют как молекулярные знаки для сбора и активации иммуноцитов, таких как лимфоциты, моноциты и гранулоциты, идентифицируя участки, в которых существуют патогены. Несмотря на регуляцию патогенов иммунной системой, может развиваться некоторая неподходящая передача сигналов хемокинами и связываться с провокацией или поддержанием воспалительных расстройств, таких как ревматоидный артрит, рассеянный склероз и других. Например, при ревматоидном артрите нерегулируемое накопление хемокинов в суставах притягивает и активирует инфильтрирующие макрофаги и T-клетки. Активность таких клеток вызывает пролиферацию синовиальных клеток,что ведет, по меньшей мере, частично к воспалению и возможной потере костной и хрящевой ткани (см.DeVries M.E. et al., Semin. Immunol., 11(2): 95-104 (1999. Признаком некоторых демиелинизирующих заболеваний, таких как рассеянный склероз, является хемокинопосредованный рекруитмент моноцитов/макрофагов или T-клеток к центральной нервной системе (см. Kennedy et al., J. Clin. Immunol., 19(5): 273-279 (1999. Рекруитмент хемокинов деструктивных WBC к трансплантатам вовлекается в их последующее отторжение. См. DeVries M.E. et al., цит. выше. Поскольку хемокины играют основные роли в развитии воспаления и лимфоцитов, возможность специфически манипулировать их активностью имеет огромное значение для уменьшения симптомов и прекращения заболеваний, для которых в настоящее время не имеется удовлетворительного лечения. Кроме того, отторжение трансплантата можно минимизировать без генерализованных и осложняющих воздействий дорогостоящих иммунодепрессивных фармацевтических препаратов. Хемокины - группа из более чем 40 небольших пептидов (7-10 кД) - лигируют рецепторы, экспрессированные главным образом на WBC или иммунопроизводных клетках, и передают сигналы через каскады передачи сигналов, соединенные с G-белком, для опосредования их хемоатрактантных и хемостимулирующих функций. Рецепторы могут связываться с несколькими лигандами; например рецепторCCR1 лигирует хемокины RANTES (регулируемые на нормальных экспрессированных Т-клетках активации), MIP-1 (макрофаговый белок зоны воспаления), MPIF-1/CK8 и лейкотактин (среди прочих с меньшей аффинностью). На сегодня известно 24 хемокиновых рецептора. Существующий ряд хемокинов, рецепторы, связывающие различные лиганды, и различные профили рецепторов на иммуноцитах дают возможность для строго регулируемых и специфических иммунных реакций, см. Rossi et al., Ann.Rev. Immunol., 18(1): 217-242 (2000). Активность хемокинов можно регулировать через модуляцию их соответствующих рецепторов, излечивая соответствующие воспалительные и иммунологические заболевания и делая годными трансплантаты органов и тканей. Рецептор CCR1 и его хемокиновые лиганды, например MIP-1, MPIF-1/CK8, лейкотактин иRANTES, представляют собой важные терапевтические мишени (см. Saeki et al., Current PharmaceuticalDesign, 9: 1201-1208 (2003, так как они связаны с ревматоидным артритом, отторжением трансплантата(см. DeVries M.E. et al., цит. выше) и рассеянным склерозом (см. Fischer et al., J. Neuroimmunol., 110(1-2): 195-208 (2000); Izikson et al., J. Exp. Med., 192(7): 1075-1080 (2000) и Rottman et al., Eur. J. Immunol., 30(8): 2372-2377 (2000. Действительно, обнаружены блокирующие функции антитела, модифицированные лиганды хемокиновых рецепторов и низкомолекулярные органические соединения, из которых некоторые, как было успешно показано, предупреждают или лечат некоторые хемокинопосредуемые заболевания (обзор в Rossi et al., цит. выше). Например, на экспериментальной модели ревматоидного артрита развитие заболевания ослабевает, когда вводят лиганд, модифицированный RANTES, блокирующий передачу сигнала (см. Plater-Zyberk et al., Immunol. Lett., 57(1-3): 117-120 (1997. Хотя терапии с блокирующими функции антителами и низкомолекулярными пептидами являются многообещающими, к их недостаткам относятся опасность деградации, весьма короткие периоды полувыведения, когда их вводят,и чрезмерно высокие расходы на разработку и производство, характерные для многих белков. Низкомолекулярные органические соединения предпочтительны, так как они часто имеют более длительные периоды полувыведения in vivo, требуют меньшие дозы для воздействия, их часто можно вводить перо-1 015529 рально и, наконец, они менее дорогостоящие. Некоторые органические антагонисты CCR1 описаны ранее (см. Hesselgesser et al., J. Biol. Chem., 273(25): 15687-15692 (1998); Ng et al., J. Med. Chem., 42(22): 4680-4694 (1999); Liang et al., J. Biol. Chem., 275(25): 19000-19008 (2000); и Liang et al., Eur. J. Parmacol.,389(1): 41-40 (2000. С учетом эффективности, показанной при лечении заболеваний на животных моделях (см. Liang et al., J. Biol. Chem., 275(25): 19000-19008 (2000, продолжается поиск для идентификации других соединений, которые можно использовать при лечении заболеваний, опосредуемых передачей сигналов CCR1. Сущность изобретения Настоящее изобретение относится к соединениям формулы I или их фармацевтически приемлемым солям, гидратам или N-оксидам, где R1 представляет собойC1-4-алкил или C1-4 галогеналкил; m равен целому числу от 0 до 1; R2a, R2c и R2d представляют собой заместители, выбранные, каждый независимо, из группы, состоящей из водорода, галогена, C1-4-алкокси,C1-4-алкила, -O-C1-4-галогеналкила и C1-4-галогеналкила, и R3 представляет собой заместитель, выбранный из группы, состоящей из водорода и C1-4-алкила. Кроме соединений, описанных в данном описании, изобретение также относится к фармацевтическим композициям, содержащим одно или несколько указанных соединений, а также к способам применения указанных соединений, главным образом, для лечения заболеваний, связанных с сингальной активностью CCR1, CCR2 и/или CCR3. Осуществление изобретенияI. Аббревиатуры и определения. Термин "алкил" сам по себе или как часть другого заместителя обозначает, если не указано иное,линейный или разветвленный углеводородный радикал с обозначенным числом атомов углерода (т.е. C1-4 обозначает число атомов углерода от одного до четырех). Примеры алкильных групп включают метил,этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил и втор-бутил. Термин "алкокси" используется в его обычном смысле и относится к алкильным группам, присоединяемым к остальной части молекулы через атом кислорода. Примеры алкоксигрупп включают метокси, этокси, изопропокси и подобные группы. Термины "гало" и "галоген", сами по себе или как части других заместителей, обозначают, если не указано иное, атомы фтора, хлора, брома или йода. Также термины, такие как "галогеналкил", имеют значения, включающие моноалкил и полигалогеналкил. Например, термин "C1-4-галогеналкил" предназначен для обозначения трифторметила, 2,2,2-трифторэтила, 4-хлорбутила, 3-бромпропила и т.п. Термин "защитная группа" относится к группе, за исключением алкильных групп, которая, когда присоединена к реакционноспособной группе в рамках молекулы, уменьшает или предотвращает ее реакционную способность. Примеры защитных групп можно найти в изданиях T.W. Greene andP.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd edition. John WileySons, New York, 1999 и Harrison and Harrison et al., Compendium of Synthetic Organic Methods, vols. 1-8 (John Wiley and Sons,1971-1996), включенных в данное описание в качестве ссылок. Характерные гидроксизащитные группы включают ацильные группы, образующие простые эфиры бензиловую и третиловую группы, образующую простые эфиры тетрагидрофуратильную группу, образующие простые эфиры триалкилсилильные и аллильные группы. Характерные аминозащитные группы включают формильную, ацетильную, трифторацетильную, бензильную, бензилоксикарбонильную (CBZ), трет-бутоксикарбонильную (BOC), триметилсилильную (TMS), 2-триметилсилилэтансульфонильную (SES), тритильную и замещенную тритильную группы, аллилоксикарбонил, 9-флуоренилметилоксикарбонил (FMOC), нитровератрилоксикарбонил(NVOC) и т.п. Термин "реагент аминокислотного сочетания" относится к реагенту, такому как HBTU (гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония) и т.д., который будет взаимодействовать с карбоксильной группой аминокислоты с образованием активированного промежуточного соединения,которое можно использовать для конденсации с широким рядом нуклеофилов, например аминами, спиртами и тиолами, для получения эфирных, тиоэфирных или амидных групп. Термин "фармацевтически приемлемые соли" предназначен для включения солей активных соединений, которые получают с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей, находящихся в соединениях, описанных в данном описании. Когда соединения по настоящему изобретению содержат относительно кислотные функциональные группы, можно получить соли присоединения оснований посредством приведения в контакт нейтральной формы таких соединений с достаточным количеством нужного основания или в чистом виде, или в подходящем инертном растворителе. Примеры солей,образованных с фармацевтически приемлемыми неорганическими основаниями, включают соли алюми-2 015529 ния, аммония, кальция, меди, железа трех- и двухвалентного, лития, магния, марганца, калия, натрия,цинка и т.п. Соли, образованные с фармацевтически приемлемыми органическими основаниями, включают соли первичных, вторичных и третичных аминов, в том числе замещенных аминов, циклических аминов, аминов, встречающихся в природе, и т.п., таких как аргинин, бетаин, кофеин, холин,N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин,этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперадин, полиамины, прокаин, пурины,теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Когда соединения по настоящему изобретению содержат относительно основные функциональные группы, можно получить соли присоединения кислот посредством приведения в контакт нейтральной формы таких соединений с достаточным количеством нужной кислоты или в чистом виде, или в подходящем инертном растворителе. Примеры фармацевтически приемлемых солей присоединения кислот включают соли, образованные с неорганическими кислотами, такими как хлороводородная, бромоводородная, азотная, угольная, моногидрокарбоновая, фосфорная, моногидрофосфорная, дигидрофосфорная, серная, моногидросерная, йодоводородная или фосфористая кислоты и т.п., а также соли, образованные с относительно нетоксичными органическими кислотами, такими как уксусная, пропионовая, изомасляная, малоновая, бензойная, янтарная, суберовая, фумаровая, миндальная, фталевая, бензолсульфоновая, п-толуолсульфоновая, лимонная, винная, метансульфоновая кислоты и т.п. Также включаются соли аминокислот, такие как аргинат и т.п., и соли органических кислот, подобных глюкуроновой или галактуроновой кислотам, и т.п. (см., например, Berge S.M. et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science, 1977, 66, 1-19). Некоторые специфические соединения по настоящему изобретению содержат как основные, так и кислотные функциональные группы, что позволяет превращать соединения в соли присоединения или оснований,или кислот. Нейтральные формы соединений можно регенерировать приведением в контакт соли с основанием или кислотой и извлечения исходного соединения обычным способом. Исходная форма соединения отличается от различных солевых форм по некоторым физическим свойствам, таким как растворимость в полярных растворителях, но в других отношениях для цели настоящего изобретения соли эквивалентны исходной форме соединения. Кроме солевых форм настоящее изобретение относится к соединениям, которые находятся в форме пролекарства. Пролекарства соединений, описанных в данном описании, представляют собой соединения, которые легко претерпевают химические изменения в физиологических условиях с образованием соединений по настоящему изобретению. Кроме того, пролекарства можно превратить в соединения по настоящему изобретению химическими или биохимическими способами в среде ex vivo. Например, пролекарства могут постепенно превращаться в соединения по настоящему изобретению, когда помещены в резервуар трансдермального пэтча с подходящим ферментом или химическим реагентом. Некоторые соединения по настоящему изобретению могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. Как правило, сольватированные формы эквивалентны несольватированным формам и, как предполагается, входят в объем настоящего изобретения. Некоторые соединения по настоящему изобретению могут существовать в нескольких кристаллических или аморфных формах. Как правило, все физические формы эквивалентны для применений, рассматриваемых в настоящем изобретении, и, как предполагается, входят в объем настоящего изобретения. Некоторые соединения по настоящему изобретению включают асимметричные атомы углерода (оптические центры) или двойные связи; рацематы, диастереомеры, геометрические изомеры, региоизомеры и отдельные изомеры (например, отдельные энантиомеры), как предполагается, все входят в объем настоящего изобретения. Соединения по настоящему изобретению также могут содержать неестественные пропорции атомных изотопов одного или нескольких атомов, которые составляют такие соединения. Например, соединения могут быть меченны радиоактивными изотопами, такими как, например, тритий(3H), йод-125 (125I) или углерод-14 (14C). Предполагается, что все изотопные вариации соединений по настоящему изобретению, являются ли они радиоактивными или нет, включены в объем настоящего изобретения.II. Общие положения. Настоящее изобретение основано на открытии, что соединения формулы I действуют как сильные антагонисты рецептора CCR1. Соединения обладают противовоспалительной активностью in vivo и имеют превосходные фармакокинетические свойства. Соответственно, соединения, описанные в данном описании, применимы в фармацевтических композициях, способах лечения заболеваний, опосредуемыхCCR1, и в качестве контроля в анализах для идентификации конкурентных антагонистов CCR1.III. Соединения. В одном аспекте настоящее изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, гидрату или N-оксиду, где R1 представляет собой C1-4 алкил или C1-4-галогеналкил; m равен целому числу от 0 до 1; R2a, R2c и R2d представляют собой заместители, выбранные, каждый независимо, из группы, состоящей из водорода, галогена, C1-4-алкокси, C1-4 алкила, -O-C1-4-галогеналкила и C1-4-галогеналкила; R3 представляет собой заместитель, выбранный из группы, состоящей из водорода и C1-4-алкила; и R4 представляет собой C1-4-алкил; n равен целому числу от 0 до 2. В одном воплощении R3 представляет собой водород. В другом воплощении R3 представляет собой водород и n равен 0. В другом воплощении R1 в формуле I представляет собой метил, трифторметил или этил и m равен 1. В другом воплощении m равен 0. В еще одном воплощении R2a, R2c и R2d выбраны, каждый независимо, из группы, состоящей из фтора, хлора, брома, йода, метокси, этокси, трифторметокси, метила, этила, трифторметила и 2-фторэтокси. В еще одном воплощении R2a представляет собой водород и R2c и R2d выбраны, каждый независимо, из группы, состоящей из фтора, хлора, брома,йода, метокси, этокси, трифторметокси, метила, этила, трифторметила и 2-фторэтокси. В одном предпочтительном воплощении соединения по изобретению имеют формулы Ia или Ib В одном воплощении R2a и R2c в формуле Ia или Ib выбраны, каждый независимо, из группы, состоящей из фтора, хлора, брома и йода; и R2d выбран из группы, состоящей из метокси, этокси, трифторметокси, метила, этила, трифторметила и 2-фторэтокси. В конкретном воплощении соединения формулы Ia или Ib выбирают из группы, состоящей из В другом предпочтительном воплощении соединения по изобретению имеют формулы Ic или Id-4 015529 В конкретном воплощении соединения формулы Ic или Id выбирают из группы, состоящей из В еще одном воплощении изобретения соединение формулы Id имеет следующую структуру: В еще одном воплощении соединения по изобретению формулы I выбирают из группы, состоящей из соединений, указанных в табл. 1. Таблица 1 1. 1-[4-(4-Хлор-3-метоксифенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин 1-ил]этанон. 2. 1-[4-(4-Хлор-3-этоксифенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1 ил]этанон. 3. 1-4-[4-Хлор-3-(2-фторэтокси)фенил]пиперазин-1-ил-2-[3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]этанон. 4. 1-[4-(4-Хлор-3-метоксифенил)-2-метилпиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]этанон. 5. 1-[4-(4-Хлор-3-метоксифенил)-3-метилпиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]этанон. 6. 1-[4-(4-Хлор-3-трифторметилфенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]этанон. 7. 1-[4-(4-Хлор-3-метоксифенил)пиперазин-1-ил]-2-[3-(1-метил-1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]этанон. 8. 1-[4-(4-Хлор-2-фтор-5-метоксифенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]этанон. 9. 1-4-[4-Хлор-3-(2-фторэтокси)фенил]-2-метилпиперазин-1-ил-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон.-5 015529 10. 1-[4-(4-Хлор-3-метоксифенил)-2-метилпиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]этанон. 11. 1-[4-(4-Хлор-5-этокси-2-фторфенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон. 12. 1-[4-(4-Хлор-3-трифторметоксифенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]этанон. 13. 1-[4-(4-Хлор-2-фтор-5-метоксифенил)-2-метилпиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон. 14. 1-[4-(4-Хлор-3-этоксифенил)-2-метилпиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]этанон. 15. 1-4-[4-Хлор-2-фтор-5-(2-фторэтокси)фенил]пиперазин-1-ил-2-[3-(1 Н-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон. 16. 1-[4-(4-Хлор-3-метоксифенил)-3-метилпиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]этанон. 17. 1-[4-(4-Хлор-3-метилфенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1 ил]этанон. 18. 1-[4-(4-Хлор-2-фтор-5-метоксифенил)-2-метилпиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон. Получение соединений. Приведенные ниже схемы показывают некоторые пути синтеза, которым можно следовать для получения некоторых соединений по настоящему изобретению. Другие пути или модификации путей,представленных ниже, могут быть легко различимы для специалистов и входят в объем изобретения. Схема 1 иллюстрирует синтез 3-имидазолилзамещенных пиразоло[3,4-b]пиридинов. R' представляет собой немешающий заместитель, такой как, например, защитная группа, или карбоксиэфирная группа. Как видно на схеме 1, взаимодействие NH2OH с 3-цианопиразоло[3,4-b]пиридином i будет приводить к гидроксиламидину ii. Восстановление ii с использованием газа водорода и катализатора (например, Pd/C или Pd(OH)2) будет давать амидин iii. Циклизация iii посредством обработки хлорацетальдегидом будет давать имидазол iv. Схема 1 Схема 2 иллюстрирует синтез 3-имидазолилзамещенных пиразоло[3,4-b]пиридинов. R' представляет собой немешающий заместитель, такой как, например, защитная группа, или карбоксиэфирная группа. На схеме 2 взаимодействие 3-цианопиразоло[3,4-b]пиридина с этилендиамином дает циклизованный продукт имидазолин v, который после окисления будет образовывать амидазол vi. Схема 2 Схема 3 иллюстрирует синтез 3-имидазолилзамещенных пиразоло[3,4-b]пиридинов. R' представляет собой немешающий заместитель, такой как, например, защитная группа, или карбоксиэфирная группа,или остаток соединения формулы I (см. также пример 18). Как видно на схеме 3, с использованием про-6 015529 цесса переметаллирования 3-йодпиразоло[3,4-b]пиридин vii можно превратить в 3-формилпиразоло[3,4b]пиридин viii, который после обработки глиоксалем циклизуется с образованием 3-имидазолилпиразоло[3,4-b]пиридина ix. Схема 3 Процедура аминокислотного сочетания, которую можно использовать для получения соединений по изобретению, иллюстрируется схемой 4. На схеме 4 R и R" представляют собой немешающие заместители. Соединения по изобретению можно получить, например, сочетанием карбоксипроизводного 3 имидазолилпиразоло[3,4-b]пиридина x с производным пиперазина xi с использованием любого реагента аминокислотного сочетания (например, HBTU, HATU, pyBOP и т.д.) с образованием 3-имидазолилпиразоло[3,4-b]пиридина (xii) по настоящему изобретению. Схема 4III. Фармацевтические композиции. Кроме соединений, описанных выше, композиции для модуляции активности CCR1, CCR2 и CCR3 у людей и животных типично будут содержать фармацевтический носитель или разбавитель. Предполагается, что термин "композиция", используемый в данном описании, включает продукт,включающий специфические ингредиенты в определенных количествах, а также любой продукт, который является результатом, прямо или косвенно, сочетания специфических ингредиентов в определенных количествах. Термин "фармацевтически приемлемый" означает, что носитель, разбавитель или эксципиенты должны быть совместимы с другими ингредиентами состава и не наносить вред их реципиенту. Фармацевтические композиции для введения соединений по данному изобретению обычно могут находиться в стандартной лекарственной форме и могут быть получены любым из способов, хорошо известных в области фармации и доставки лекарственных средств. Все способы включают стадию приведения активного ингредиента в ассоциацию с носителем, который состоит из одного или нескольких вспомогательных ингредиентов. Как правило, фармацевтические композиции получают посредством равномерного и тщательного введения активного ингредиента в смесь с жидким носителем или тонко измельченным твердым носителем либо тем и другим, и последующим, при необходимости, приданием продукту формы желательного препарата. В фармацевтическую композицию активное целевое соединение включают в количестве, достаточном для получения желательного действия на болезненный процесс или состояние. Фармацевтические композиции, содержащие активный ингредиент, могут находиться в форме,подходящей для перорального применения, например в виде таблеток, пастилок, лепешек, водных или масляный суспензий, диспергируемых порошков или гранул, эмульсий и собственно эмульгируемых композиций, описанных в патенте США 6451339, твердых или мягких капсул, сиропов, эликсиров,растворов, буккального пэтча, перорального геля, жвачки, жевательных таблеток, шипучего порошка или шипучих таблеток. Композиции, предназначенные для перорального применения, можно получить согласно любому способу, известному в технике для получения фармацевтических композиций, и такие композиции могут содержать одно или несколько средств, выбранных из группы, состоящей из подслащивающих агентов, корригентов, красителей, антиоксидантов и консервантов, для того, чтобы получить фармацевтически элегантные и приятные препараты. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые подходят для получения таблеток. Такими эксципиентами могут являться, например, инертные разбавители, такие как целлюлоза, диоксид кремния, оксид алюминия, карбонат кальция, карбонат натрия, глюкоза, маннит, сорбит, лактоза,-7 015529 фосфат кальция или фосфат натрия; вещества, способствующие грануляции и рассыпанию, например кукурузный крахмал или альгиновая кислота; связующие вещества, например PVP, целлюлоза, ПЭГ(PEG), крахмал, желатин или аравийская камедь, и смазывающие вещества, например стеарат магния,стеариновая кислота или тальк. Таблетки могут быть без покрытия или на них может быть нанесено покрытие, энтеросолюбильное или иное, посредством чего обеспечивается пролонгированное действие в течение более длительного периода. Например, можно использовать материал для отсрочки действия,такой как глицерилмоностеарат или глицерилдистеарат. Покрытие на таблетки также можно наносить методами, описанными в патентах США 4256108, 4166452 и 4265874, для формирования осмотических терапевтических таблеток с регулируемым высвобождением. Составы для перорального применения также могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или мягких желатиновых капсул, в которых активный ингредиент смешан с водой или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом. Кроме того, можно получить эмульсии с ингредиентом, несмешивающимся с водой, таким как масла, и стабилизированными поверхностно-активными веществами, такими как моноглицериды, эфиры ПЭГ и т.п. Водные суспензии содержат активные материалы в смеси с эксципиентами, подходящими для получения водных суспензий. Такими эксципиентами являются суспендирующие вещества, например натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая смола и аравийская камедь; диспергирующие или смачивающие вещества могут представлять собой встречающиеся в природе фосфатиды, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными эфирами, образованными жирными кислотами и гекситом, такие как моноолеат полиоксиэтиленсорбитола, или продукты конденсации этиленоксида с неполными эфирами, образованными жирными кислотами и ангидридами гексита, например моноолеат полиэтиленсорбитана. Водные суспензии также могут содержать один или несколько консервантов, например этил- или н-пропил-п-гидроксибензоат, один или несколько красителей, один или несколько корригентов и один или несколько подслащивающих веществ, таких как сахароза или сахарин. Масляные суспензии можно получить суспендированием активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, сезамовом масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Могут быть добавлены подслащивающие вещества, такие как указанные выше, и корригенты для получения приятного перорального препарата. Такие композиции могут сохраняться за счет добавления антиоксиданта, такого как аскорбиновая кислота. Диспергируемые порошки и гранулы, подходящие для получения водной суспензии посредством добавления воды, предоставляют активный ингредиент в смеси с диспергирующим или смачивающим веществом, суспендирующим веществом и одним или несколькими консервантами. Примерами подходящих диспергирующих или смачивающих веществ и суспендирующих веществ являются вещества, уже указанные выше. Также могут присутствовать другие эксципиенты, например подслащивающие вещества, корригенты и красители. Фармацевтические композиции по изобретению также могут находиться в форме эмульсий маслов-воде. Масляная фаза может представлять собой растительное масло, например оливковое масло, или арахисовое масло, или минеральное масло, например жидкий парафин, или их смеси. Подходящими эмульгаторами могут являться встречающиеся в природе смолы, например аравийская камедь или трагакантовая смола, встречающиеся в природе фосфатиды, например, сои, лецитин, и эфиры или неполные эфиры, образованные жирными кислотами и ангидридами гексита, например моноолеат сорбитана, и продукты конденсации указанных неполных эфиров с этиленоксидом, например моноолеат полоксиэтиленсорбитана. Эмульсии также могут содержать подслащивающие вещества и корригенты. Сиропы и эликсиры можно получить с подслащивающими веществами, например глицерином,пропиленгликолем, сорбитом или сахарозой. Такие композиции также могут содержать средства,уменьшающие раздражение, консерванты и корригенты, и красители. Пероральные растворы можно получить в сочетании с, например, циклодекстрином, ПЭГ и поверхностно-активными веществами. Фармацевтические композиции могут находиться в форме стерильной водной или масляной суспензии для инъекции. Такая суспензия может быть получена согласно известному уровню техники с использованием таких подходящих диспергирующих или смачивающих веществ и суспендирующих веществ, которые указаны выше. Стерильный препарат для инъекции также может представлять собой стерильный раствор или суспензию для инъекции в нетоксичном парентерально приемлемом разбавителе или растворителе, например раствор в 1,3-бутандиоле. К числу приемлемых носителей и растворителей,которые можно использовать, относятся вода, раствор Рингера и изотоничный раствор хлорида натрия. Кроме того, в качестве растворителя или суспензионной среды обычно используют стерильные нелету-8 015529 чие масла. С такой целью можно использовать любое мягко действующее нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, при получении препаратов для инъекций находят применение жирные кислоты, такие как олеиновая кислота. Соединения по настоящему изобретению также можно вводить в форме суппозиториев для ректального введения лекарственного средства. Такие композиции можно получить, смешивая лекарственное средство с подходящим не вызывающим раздражение эксципиентом, который является твердым при обычных температурах, но жидким при ректальной температуре, и поэтому будет плавиться в прямой кишке, высвобождая лекарственное средство. Такие материалы включают масло какао и полиэтиленгликоли. Кроме того, соединения можно вводить доставкой в глаза в виде растворов или мазей. Также еще можно осуществлять трансдермальную доставку указанных соединений с помощью пэтчей для ионофореза и подобных средств. Для местного применения используют кремы, мази, гели, растворы или суспензии и т.д., содержащие соединения по настоящему изобретению. Используемый в данном описании термин "местное применение" также включает применение жидкостей для полоскания рта и горла. Соединения по изобретению можно ввести в составы для депонирования в медицинском устройстве, которое может включать различные обычные трансплантаты, стенты, включая стент-трансплантаты,катетеры, баллоны, сетки, или другом устройстве, которое можно разместить или перманентно имплантировать в полость трубчатого органа. Как конкретный пример, желательно иметь устройства и способы,которые могут доставлять соединения по изобретению к участку тела, который лечат методом вмешательства. В примере воплощения ингибирующее средство по данному изобретению можно депонировать в медицинском устройстве, таком как стент, и доставить в место лечения для лечения части организма. Стенты используют как носители для доставки терапевтических средств (т.е. лекарственных средств). Внутрисосудистые стенты, как правило, перманентно имплантируют в коронарные или периферические сосуды. Конструкции стентов включают конструкции, описанные в патентах США 4733655 (Palmaz), 4800882 (Gianturco) или 4886062 (Wiktor). Такие конструкции включают как металлические, так и полимерные стенты, а также саморасширяющиеся и способные расширяться стентыбаллоны. Стенты также можно использовать для доставки лекарственного средства в место контакта с сосудистой сетью, как описано, например, в патенте США 5102417 (Palmaz) и в международных заявкахWO 91/12779 (Medtronic, Inc.) и WO 90/13332 (Cedars-Sanai Medical Center), патенте США 5419760 (Narciso, Jr.) и патенте США 5429634 (Narciso, Jr.). Стенты также используют для доставки вирусов к стенке просвета для генной доставки, как раскрывается в заявке на патент США рег.5833651 (Donovan et al.). Термин "депонирование" означает, что ингибитор (ингибирующий агент) покрыт, абсорбирован,размещен или иным образом внедрен в устройство способами, известными в технике. Например, ингибитор может быть заделан и высвобождается из ("матричный тип") или окружен и высвобождается через("резервуарный тип") полимерные материалы, которые покрывают или включают медицинское устройство. В последнем примере ингибитор может быть захвачен полимерными материалами или присоединен к полимерным материалам с использованием одного или нескольких методов генерации таких материалов, известных в технике. В других составах ингибитор может быть связан с поверхностью медицинского устройства без необходимости наносить покрытие связями, которые можно разорвать, и высвобождается со временем, может быть удален активными механическими или химическими процессами или находиться в перманентно иммобилизованной форме для доставки ингибитора в место имплантации. В одном воплощении ингибитор может быть внедрен с полимерными композициями во время образования биосовместимых покрытий для медицинских устройств, таких как стенты. Покрытия, полученные из таких компонентов, типично однородны и применимы для нанесения на ряд устройств, созданных для имплантации. Полимер может быть или биологически устойчивым или биологически поглощаемым полимером, в зависимости от нужной скорости высвобождения или нужной степени устойчивости полимера, но для такого воплощения предпочтителен биопоглощаемый полимер, так как в отличие от биологически устойчивого полимера он не будет присутствовать длительное время после имплантации и вызывать какую-либо вредную хроническую локальную реакцию. Биопоглощаемые полимеры, которые можно использовать, включают, но не ограничиваются перечисленным, поли-L-молочную кислоту, поликапролактон, полигликолид (PGA), сополимер лактида и гликолида (PLLA/PGA), полигидроксибутират, сополимер гидроксибутирата и валерата, полидиоксанон (PDS), сложный полиортоэфир, полиангидрид, полигликолевую кислоту, поли-D-молочную кислоту, поли-D,L-молочную кислоту, поли(D,L-лактид) (PLA),поли-L-лактид (PLLA), сополимер гликолевой кислоты и триметиленкарбоната (PGA/PTMC), полиэтиленоксид (PEO), сложный полифосфоэфир, полифосфоэфируретан, полиаминокислоты, цианоакрилаты,политриметиленкарбонат, полииминокарбонат, сополимеры простых и сложных эфиров (например,PEO/PLA), полалкиленоксалаты, полифосфазены и биомолекулы, такие как фибрин, фибриноген, целлюлоза, крахмал, коллаген и гиалуроновая кислота, полиэпсилонкапролактон, полиоксимасляная кислота,полиацетали, полидигидропираны, полицианоакрилаты, сшитые или амфипатические блок-сополимеры гидрогелей и другие подходящие биопоглощаемые полимеры, известные в технике. Также можно ис-9 015529 пользовать биологически устойчивые полимеры с относительно слабой хронической реакцией тканей,такие как полиуретаны, силиконы и сложные полиэфиры, и также можно использовать другие полимеры,если они могут растворяться и затвердевать или полимеризоваться на медицинском устройстве, такие как полиолефины, полиизобутилен и сополимеры этилена и альфа-олефинов; акриловые полимеры и сополимеры, полимеры и сополимеры винилгалогенидов, такие как поливинилхлорид; поливинилпирролидон; простые поливиниловые эфиры, такие как поливинилметиловый эфир; поливинилиденгалогениды, такие как поливинилиденфторид и поливинилиденхлорид; полиакрилонитрил, поливинилкетоны; полимеры винилароматических соединений, такие как полистирол, сложные поливиниловые эфиры, такие как поливинилацетат; сополимеры виниловых мономеров друг с другом и олефинами, такие как сополимеры этилена и метилметакрилата, сополимеры акрилонитрила и стирола, смолы ABS и сополимеры этилена и винилацетата; сополимеры пирана; полигидроксипропилметакриламидфенол; полигидроксиэтиласпартамидфенол; полиэтиленоксидполилизин, замещенный пальмитоиловыми остатками; полиамиды, такие как найлон-66 и поликапролактам; алкидные смолы, поликарбонаты; полиоксиметилены; полиимиды; простые полиэфры; эпоксидные смолы, полиуретаны; гидратцеллюлозное волокно; триацетат гидратцеллюлозы; целлюлоза, ацетат целлюлозы, бутират целлюлозы; ацетатбутират целлюлозы; целлофан; нитрат целлюлозы; пропионат целлюлозы; простые эфиры целлюлозы и карбоксиметилцеллюлоза. Полимеры и полупроницаемые полимерные матрицы можно формовать в изделия, имеющие форму, такие как клапаны, стенты, трубки, протезы и т.п. В одном воплощении изобретения ингибитор по изобретению присоединяют к полимеру или полупроницаемой полимерной матрице, которые формуют в виде стента или стента-трансплантата. Типично полимеры наносят на поверхность имплантируемого устройства центрифугированием, погружением или распылением. Для такой цели также можно использовать другие способы, известные в технике. Методы распыления включают традиционные способы, а также методы микроосаждения с помощью распределителя инжекторного типа. Кроме того, полимер можно осадить на имплантируемое устройство с использованием фотошаблонов для размещения полимера только на определенных частях устройства. Такое покрытие устройства дает равномерный слой на устройстве, который дает возможность улучшенной диффузии различных исследуемых веществ через покрытие на устройстве. В предпочтительных воплощениях изобретения ингибитор вводят в состав для высвобождения из полимерного покрытия в окружающую среду, в которой размещено медицинское устройство. Предпочтительно ингибитор высвобождается регулируемым способом в течение длительного времени (например,месяцев) с использованием по меньшей мере одного из нескольких хорошо известных методов, включающих полимерные носители или слои для регулируемой элюции. Некоторые из таких методов описаны ранее в заявке на патент США 20040243225 A1. Более того, как описано, например, в патенте США 6770729, реагентами и условиями взаимодействия полимерных композиций можно манипулировать таким образом, что можно регулировать высвобождение ингибитора из полимерного покрытия. Например, можно модулировать коэффициент диффузии одного или нескольких полимерных покрытий для регулирования высвобождения ингибитора из полимерного покрытия. В варианте данного положения коэффициент диффузии одного или нескольких полимерных покрытий можно регулировать для модуляции способности исследуемого вещества, которое присутствует в окружающей среде, в которой размещено медицинское устройство (например, исследуемого вещества, которое облегчает распад или гидролиз некоторой части полимера), иметь доступ к одному или нескольким компонентам в полимерной композиции (и, например, посредством этого модулировать высвобождение ингибитора из полимерного покрытия). Еще одно воплощение изобретения включает устройство с несколькими полимерными покрытиями, причем каждое имеет несколько коэффициентов диффузии. В таких воплощениях изобретения высвобождение ингибитора из полимерного покрытия можно модулировать за счет нескольких полимерных покрытий. В еще одном воплощении изобретения высвобождение ингибитора из полимерного покрытия регулируют путем модуляции одного или нескольких свойств полимерной композиции, таких как присутствие одного или нескольких эндогенных или экзогенных соединений, или, с другой стороны, pH полимерной композиции. Например, можно создавать некоторые полимерные композиции для высвобождения ингибитора в ответ на снижение pH полимерной композиции. С другой стороны, можно создавать некоторые полимерные композиции для высвобождения ингибитора в ответ на присутствие пероксида водорода.IV. Способы лечения заболеваний, модулированных CCR1, CCR2 и/или CCR3. В еще одном аспекте настоящее изобретение относится к способам лечения опосредуемых CCR1,CCR2 и/или CCR3 состояний или заболеваний посредством введения субъекту, имеющему такое заболевание или состояние, терапевтически эффективного количества соединения формулы I, описанного выше. "Субъект" в данном описании определяется как термин, включающий животных, таких как млекопитающие, в том числе, но без ограничения, приматы (например, люди), коровы, овцы, козы, лошади, собаки, кошки, кролики, крысы, мыши и т.п.CCR1 предоставляет мишень для препятствия промотирования специфических аспектов функций- 10015529 иммуноцитов, или в более общем смысле, функциям, связанным с экспрессией CCR1 в широком интервале типов клеток у млекопитающего, такого как человек. Соединения, которые ингибируют CCR1, особенно применимы в лечебных целях для модуляции функции моноцитов, макрофагов, лимфоцитов, гранулоцитов, NK-клеток, тучных клеток, дендритных клеток и некоторых иммунопроизводных клеток (например, остеокластов). Соответственно настоящее изобретение относится к соединениям, которые применимы при предупреждении и/или лечении широкого ряда воспалительных и иммунорегуляторных расстройств и заболеваний (см. Sacki et al., Current Pharmaceutical Design, 9: 1201-1208 (2003. Например, соединение по настоящему изобретению, которое ингибирует одну или несколько функций CCR1, можно вводить для ингибирования (т.е. ослабления или предупреждения) воспаления или клеточной инфильтрации, связанной с иммунным расстройством. В результате может быть ингибирован один или несколько воспалительных процессов, таких как эмиграция или инфильтрация лейкоцитов, хемотаксис, экзоцитоз (например, ферментов, гистамина) или высвобождение медиаторов воспаления. Например, инфильтрация моноцитов в область воспаления (например, пораженный сустав при артрите или в ЦНС при MS) можно ингибировать согласно способу по настоящему изобретению. Подобным образом, обсуждаемое соединение, которое промотирует одну или несколько функцийCCR1, вводят для стимуляции (вызывания или усиления) воспалительной реакции, такой как эмиграция лейкоцитов, хемотаксис, экзоцитоз (например, ферментов, гистамина) или высвобождение медиаторов воспаления. Например, можно вызвать приток моноцитов для борьбы с бактериальными инфекциями. Заболевания и состояния, связанные с воспалением, иммунными расстройствами и инфекцией,можно лечить с использованием способа по настоящему изобретению. В предпочтительном воплощении заболевание или состояние является таким заболеванием или состоянием, при котором действия иммуноцитов, таких как моноциты, макрофаги, лимфоциты, гранулоциты, NK-клетки, тучные клетки, дендритные клетки и некоторые иммунопроизводные клетки (например, остеокласты), должны быть ингибированы или промотированы для того, чтобы модулировать воспалительную или аутоиммунную реакцию. В одной группе воплощений заболевания или состояния, в том числе хронические заболевания людей или других видов, можно лечить модуляторами функции CCR1, CCR2 или CCR3. Такие заболевания или состояния включают (1) аллергические заболевания, такие как системная анафилаксия или аллергические реакции, лекарственная аллергия, аллергические реакции на укусы насекомых и пищевые аллергии, (2) воспалительные заболевания кишечника, такие как болезнь Крона, неспецифический язвенный колит, илеит и энтерит, (3) вагинит, (4) псориаз и воспалительные дерматозы, такие как дерматит, экзема,атопический дерматит, аллергический контактный дерматит, крапивница и зуд, (5) васкулит, (6) спондилоартропатии, (7) склеродермия, (8) астма и респираторные аллергические заболевания, такие как астма,аллергическая астма, аллергический ринит, аллергические болезни легких и т.п., (9) аутоиммунные заболевания, такие как фибромиалгия, склеродермия, анкилозирующий спондилит, ювенильный RA, болезнь Стилла, ювенильный полиартрит, ювенильный олигоартрит, ревматоидная полимиалгия, артрит Такуяси,ревматоидный артрит, псориатический артрит, остеоартрит, полиартрит, рассеянный склероз, системная красная волчанка, диабет типа I, диабет типа II, диабет типа I (в самом начале), ретробульбарный неврит,гломерулонефрит и т.п., (10) отторжение трансплантата, в том числе отторжение аллотрансплантата и острая и хроническая болезнь "трансплантат против хозяина", (11) фиброз, например фиброз легких (т.е. идиопатический фиброз легких, интерстициальный фиброз легких), фиброз, связанный с конечной стадией болезни почек, фиброз, вызванный облучением, тубулоинтерстициальный фиброз, субэпителиальный фиброз, склеродермия (прогрессирующий системный склероз), фиброз печени (в том числе фиброз,вызванный алкогольным или вирусным гепатитом), первичный и вторичный цирроз, (12) острое и хроническое воспаление легких (хроническая обструктивная болезнь легких, хронический бронхит, респираторный дистресс-синдром взрослых, респираторный дистресс-синдром младенцев, иммунный комплексный альвеолит) и (13) другие заболевания, при которых следует ингибировать нежелательные воспалительные реакции или иммунные расстройства, такие как сердечно-сосудистое заболевание, включая атеросклероз, сосудистое воспаление вследствие пересадки тканей или во время рестеноза (в том числе рестеноз после ангиопластики и/или установки стента и другие), другие острые и хронические воспалительные состояния, такие как миозит, нейродегенеративные заболевания (например, болезнь Альцгеймера), энцефалит, менингит, гепатит, нефрит, сепсис, саркоидоз, аллергический конъюнктивит, отит, синусит, синовиальное воспаление, вызванное артроскопией, гиперуремия, травма, ишемическое реперфузионное повреждение, назальный полиоз, преэклампсия, ротовой плоский лишай, синдром Гийена-Барре,грануломатозные заболевания, состояния, связанные с продукцией лептина, синдром Бехчета и подагра,и при применениях для заживления ран, (14) иммуноопосредованные пищевые аллергии, такие как чревная болезнь. В другой группе воплощений заболевания или состояния можно лечить модуляторами функцииCCR1. Примеры заболеваний, которые лечат модуляторами функции CCR1, включают онкозаболеванияLymphoma, 2005, 46(7); 967-972), сердечно-сосудистые заболевания, заболевания, при которых играют роль ангиогенез или неоваскуляризация (неопластические заболевания, ретинопатия и дегенерация желтого пятна), инфекционные заболевания (вирусные инфекции, например ВИЧ-инфекция, и бактериаль- 11015529 ные инфекции) и иммунодепрессивные заболевания, такие как состояния при пересадке органов и состояния при пересадке кожи. Термин "состояния при пересадке органов" означает, что включены состояния при пересадке костного мозга и солидных органов (например, почки, печени, легких, сердца, поджелудочной железы или их сочетания). Фармацевтические композиции по данному изобретению также могут ингибировать продукцию металлопротеиназ и цитокинов в местах воспаления или прямо или косвенно (как следствие снижения клеточной инфильтрации), причем таким образом оказывают благоприятное действие при заболеваниях или состояниях, связанных с такими цитокинами. Соединения по настоящему изобретению соответственно применимы при предупреждении и лечении самых разных воспалительных и иммунорегуляторных расстройств и заболеваний. В зависимости от заболевания, от которого лечат, и состояния субъекта соединения по настоящему изобретению можно вводить пероральным, парентеральным (например, внутримышечным, интраперитонеальным, внутривенным, ICV, интрацистеральной инъекции или инфузии, подкожной инъекции или имплантата) путем ингаляцией спреем, назальным, вагинальным, ректальным, сублингвальным или местным путем введения и можно вводить в состав, одни или вместе, подходящих стандартных форм, содержащих обычные нетоксичные фармацевтически приемлемые носители, адъюванты и среды, соответствующие каждому способу введения. При лечении или предупреждении состояний, при которых требуется модуляция, соответствующий уровень дозировки будет составлять, как правило, примерно 0,001-100 мг на 1 кг массы тела пациента в сутки, которые можно вводить в однократной дозе или нескольких дозах. Предпочтительно уровень дозировки будет составлять от примерно 0,01 до примерно 25 мг/кг в сутки, предпочтительнее от примерно 0,05 до примерно 10 мг/кг в сутки. Подходящий уровень дозировки может составлять примерно 0,01-25 мг/кг в сутки, примерно 0,05-10 мг/кг в сутки или примерно 0,1-5 мг/кг в сутки. В пределах указанного интервала дозировка может составлять 0,005-0,05, 0,05-0,5 или 0,5-5 мг/кг в сутки. Для перорального введения композиции предпочтительно предоставляют в форме таблеток, содержащих 1,0-1000 мг активного ингредиента, в частности 1,0, 5,0, 10,0, 15,0, 20,0, 25,0, 50,0, 75,0, 100,0, 150,0,200,0, 250,0, 300,0, 400, 500,0, 600,0, 750,0, 800,0, 900,0 и 1000,0 мг активного ингредиента для симптоматической корректировки дозировки для пациента, которого лечат. Соединения можно вводить по схеме приема 1-4 раза в сутки, предпочтительно один или два раза в сутки. Однако следует представлять, что конкретный уровень дозы и частота дозировки для любого отдельного пациента может изменяться и будет зависеть от ряда факторов, включая активность конкретного используемого соединения, метаболическую устойчивость и длительность действия указанного соединения, возраст, массу тела, наследственные свойства, общее состояние здоровья, пол и питание субъекта, а также способ и время введения, скорость экскреции, сочетание лекарственных средств и тяжесть определенного состояния субъекта, претерпевающего лечение. Заболевания и состояния, связанные с воспалением, иммунным расстройством, инфекцией и раком,можно лечить или предупреждать с помощью соединений, композиций и способов по настоящему изобретению. Соединения и композиции по настоящему изобретению можно сочетать с другими соединениями и композициями, имеющими родственную применимость для предупреждения и лечения состояния или заболевания, представляющего интерес, такого как воспалительные или аутоиммунные расстройства,состояния и заболевания, включая воспалительную болезнь кишечника, ревматоидный артрит, остеоартрит, псориатический артрит, полиартрит, рассеянный склероз, аллергические заболевания, псориаз, атопический дерматит и астму, и патологии, указанные выше. Например, при лечении или предупреждении воспаления или аутоиммунной реакции или, например, артрита, связанного с потерей костной массы, соединения и композиции по настоящему изобретению можно использовать в сочетании с противовоспалительным или анальгезирующим средством, таким как опиатный агонист, ингибитор липоксигеназы, такой как ингибитор 5-липоксигеназы, ингибитор циклооксигеназы, такой как ингибитор циклооксигеназы-2, ингибитор интерлейкина, такой как ингибитор интерлейкина-1, антагонист NMDA, ингибитор оксида азота или ингибитор синтеза оксида азота, нестероидное противовоспалительное средство или цитокинподавляющее противовоспалительное средство,например, с таким соединением, как ацетаминофен, аспирин, кодеин, фентанил, ибупрофен, индометацин, кеторолак, морфин, напроксен, фенацетин, пироксикам, стероидный аналгетик, суфентанил, сунлиндак, тенидап и подобные средства. Подобным образом, соединения и композиции по настоящему изобретению можно вводить с аналгетиками, указанными выше; потенцирующим средством, таким как кофеин, антагонистом H2 (например, ранитидином), симетиконом, гидроксидом алюминия или магния; противоотечным средством, таким как фенилэфрин, фенилпропаноламин, псевдоэфедрин, оксиметазолин, эфинерфрин, нафазолин, ксилометазолин, пропилекседрин или леводезоксиэфедрин; противокашлевым средством, таким как кодеин, гидрокодон, карамифен, карбетапентан или декстрометорфан; диуретиком и седативным или неседативным антигистаминным средством. Подобным образом соединения и композиции по настоящему изобретению можно использовать в сочетании с другими лекарственными средствами, которые используют при лечении, предупреждении,- 12015529 подавлении или ослаблении симптомов заболеваний или состояний, при которых применимы соединения и композиции по настоящему изобретению. Такие другие лекарственные средства можно вводить способом и в количестве, обычно используемых для них, одновременно или последовательно с соединением и композицией по настоящему изобретению. Когда соединение или композицию по настоящему изобретению используют одновременно с одним или несколькими другими лекарственными средствами,предпочтительна фармацевтическая композиция, содержащая такие другие лекарственные средства в дополнение к соединению или композиции по настоящему изобретению. Соответственно фармацевтические композиции по настоящему изобретению включают композиции, которые также содержат один или несколько других активных ингредиентов или терапевтических средств, кроме соединения или композиции по настоящему изобретению. Примеры других терапевтических средств, которые можно сочетать с соединением или композицией по настоящему изобретению или вводить отдельно или в одних и тех же фармацевтических композициях, включают, но не ограничиваются перечисленным, (a) антагонистыVLA-4, (b) кортикостероиды, такие как беклометазон, метилпреднизолон, бетаметазон, преднизон, преднизолон, дексаметазон, флутиказон, гидрокортизон, будезонид, триамцинолон, салметерол, салбутанол,форметерол; (c) иммунодепрессанты, такие как циклоспорин (циклоспорин A, сандиммун, неорал),такролирнус (FK-506, програф), рапамицин (сиролимус, рапамин) и другие иммунодепрессанты типаFK-506, и микофенолат, например микофенолат мофетил (CellCept); (d) антигистаминные средства (антагонисты H1-гистамина), такие как бромфенирамин, хлорфенирамин, дексхлорфенирамин, трипролидин, клемастин, дифенгидрамин, дифенилпиралин, трипеленнамин, гидроксизин, метдилазин, прометазин, тримепразин, азатадин, ципрогептадин, антазолин, фенирамин, пириламин, астемизол, терфенадин,лоратадин, цетиризин, фексофенадин, дескарбоэтоксилоратадин и подобные средства; (e) нестероидные противоастматические средства (например, тербуталин, метапротеренол, фенотерол, изоэтарин, альбутерол, битолтерол и пирбутерол), теофиллин, кромолиннатрий, атропин, бромид ипратропия, антагонисты лейкотриена (например, зафмлукаст, монтелукаст, пранлукаст, иралукаст, побилукаст и SKB-106203),ингибиторы биосинтеза лейкотриена (зилейтон, BAY-1005); (f) нестероидные противовоспалительные средства (NSAID), такие как производные пропионовой кислоты (например, альминопрофен, беноксапрофен, буклоксовая кислота, карпрофен, фенбуфен, фенопрофен, флупрофен, флубипрофен, ибупрофен,индопрофен, кетопрофен, рниропрофен, напроксен, оксапрозин, пирпрофен, пранопрофен, супрофен,тиапрофеновая кислота и тиоксапрофен), производные уксусной кислоты (например, индометацин, ацеметацин, альклофенак, клиданак, диклофенак, фенклофенак, фенклозовая кислота, фентиазак, фурофенак, изоксепак, окспинак, сулиндак, тиопинак, толметин, зидометацин и зомепирак), производные фенаминовой кислоты (например, флуфенаминовая кислота, меклофенаминовая кислота, мелфенаминовая кислота, нифлумовая кислота и толфенаминовая кислота), производные дифенилкарбоновой кислоты(например, дифлунизал и флуфинезал), оксикамы (например, изоксикам, пироксикам, судоксикам и теноксикам), салицилаты (например, ацетилсалициловая кислота и сульфазалазин) и пиразолоны (например, апазон, безпиперилон, фепразон, мофебутазон, оксифенбутазон и фенилбутазон); (g) ингибиторы циклооксигеназы-2 (COX-2), такие как целекоксиб (целебрекс) и рофекоксиб (Vioxx); (h) ингибиторы фосфодиэстеразы типа IV (PDE IV); (i) соединения золота, такие как ауранафин и ауротиоглюкоза, (j) этанерцепт (энбрел), (k) терапевтические антитела, такие как ортоклон (ОКТ 3), даклизумаб (зенапакс), базиликсимаб (симулект) и инфликсимаб (ремикад), (l) другие антагонисты хемокиновых рецепторов, в особенности CCR5, CXCR2, CXCR3, CCR2, CCR3, CCR4, CCR7, CX3CR1 и CXCR6; (m) смазывающие или смягчающие вещества, такие как петролатум и ланолин, (n) кератолитические средства (например, тазаротен), (o) производные витамина D3, например кальципотреин или кальципотриол(довонекс), (p) PUVA, (q) антралин (дритрокрем), (r) этретинат (тегизон) и изотретиноин, (s) терапевтические средства против рассеяного склероза, такие как интерферон -1 (бетазерон), интерферон(например, преднизолон) и циклофосфамид, (t) DMARDS, такие как метотрексат, (u) другие соединения,такие как 5-аминосалициловая кислота и ее пролекарства; гидроксихлороквин; D-пеницилламин; антиметаболиты, такие как азатиоприн, 6-меркатопурин и метотрексат; ингибиторы синтеза ДНК, такие как гидроксимочевина, и средства, разрушающие микротрубочки, такие как колхицин. Массовое отношение соединения по настоящему изобретению к другому активному ингредиенту может изменяться и будет зависеть от эффективной дозы каждого ингредиента. Как правило, будет использоваться эффективная доза каждого. Так, например, когда соединение по настоящему изобретению объединяют с NSAID, массовое отношение соединения по настоящему изобретению к NSAID, как правило, будет колебаться от примерно 1000:1 до примерно 1:1000, предпочтительно от примерно 200:1 до примерно 1:200. Сочетания соединения по настоящему изобретению и других активных ингредиентов, как правило, также будут находиться в указанном интервале, но в каждом случае следует использовать эффективную дозу каждого ингредиента.V. Примеры. Приведенные далее примеры предназначены для пояснения, но не для ограничения заявляемого изобретения.- 13015529 Используемые ниже реагенты и растворители можно получить из коммерческих источников, таких как Aldrich Chemical Co. (Milwaukee, Wisconsin, USA). 1H-ЯМР регистрируют на ЯМР-спектрометре Varian Mercury, 400 МГц. Значимые пики получают относительно ТМС и вносят в таблицу в порядке мультиплетность (с - синглет, д - дублет, т - триплет, кв - квартет, м - мультиплет) и число протонов. Результаты масс-спектрометрии приводятся в виде отношения массы к заряду с последующим относительным содержанием каждого иона (в скобках). В таблицах приводится одна величина m/е для M+H (или, как указано, M-H) иона, содержащего наиболее обычные атомные изотопы. Изотопные картины соответствуют предполагаемой формуле во всех случаях. Масс-спектрометрический анализ с ионизацией электронным распылением (ESI) проводят на масс-спектрометре с электронным распылением HewlettPackard MSD с использованием HP 1100 ВЭЖХ, снабженного колонкой Agilent Zorbax SB-C18,2,150 мм, 5 мкм, для доставки образца. Обычно анализируемое вещество растворяют в метаноле в концентрации 0,1 мг/мл и 1 мкл вводят с растворителем для доставки в масс-спектрометр, который сканирован от 100 до 1500 Да. Все соединения можно анализировать при положительном типе ESI с использованием смеси ацетонитрил/вода с 1% муравьиной кислоты в качестве растворителя для доставки. Соединения, представленные ниже, также можно анализировать при отрицательном типе ESI с использованием в качестве системы для доставки 2 мМ раствора NH4OAc в смеси ацетонитрил/вода. В примерах и в описании изобретения используются следующие аббревиатуры: ВЭЖХ - высокоэффективная жидкостная хроматография; ДМФА - диметилформамид; ТФУК трифторуксусная кислота; ТГФ - тетрагидрофуран; EtOAc - этилацетат; BOC2O - ди-трет-бутилдикарбонат или BOC-ангидрид; DIPEA - диизопропилэтиламин; HBTU - гексафторфосфат O-(бензотриазол-1 ил)-N,N,N',N'-тетраметилурония; dpff - 1,1'-бис-(дифенилфосфино)ферроцен; Pd2(dba)3 - трис-(дибензилиденацетон)дипалладий(0); DMP - периодинан Десс-Мартина; Me - метил; Et - этил; DCM - дихлорметан. Соединения в объеме данного изобретения можно синтезировать так, как описано ниже, с использованием различных реакций, известных специалистам в данной области техники. Специалисты в данной области техники легко выяснят, что можно использовать альтернативные способы для синтеза целевых соединений данного изобретения, и что подходы, описанные в данном документе, не являются исчерпывающими, но обеспечивают широко приемлемые и практичные пути к соединениям, представляющим интерес. Некоторые молекулы, заявляемые в данной заявке на патент, могут существовать в различных энантиомерных и диастереомерных формах, и заявляются все такие варианты указанных соединений. В данном тексте приводится подробное описание экспериментальных процедур, используемых для синтеза основных соединений, приводящих к соединениям, которые описаны физическими показателями, идентифицирующими их, а также изображениями структур, относящихся к ним. Специалистам в данной области техники также известно, что во время выполнения стандартных процедур органической химии часто используют кислоты и основания. Иногда во время экспериментальных процедур, описанных в данной заявке, получают соли исходных соединений, если соединения обладают необходимой присущей им кислотностью или основностью. Пример 1. Синтез 1-[4-(4-хлор-5-этокси-2-фторфенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2 ил)пиразоло[3,4-b]пиридин-1-ил]этанона Стадия 1. Смесь 3-йод-7-азаиндазола (25,50 г) и K2CO3 (41,4 г) в ДМФА (200 мл) нагревают до 85C и постепенно добавляют трет-бутилхлорацетат (14,3 мл). Смесь перемешивают при указанной температуре в течение 1 ч (час), охлаждают до комнатной температуры и затем добавляют воду (300 мл). Фильтрация реакционной смеси дает трет-бутиловый эфир (3-йодпиразоло[3,4-b]пиридин-1-ил)уксусной ки- 14015529 слоты. Стадия 2. В 250-мл колбу загружают трет-бутиловый эфир (3-йодпиразоло[3,4-b]пиридин-1 ил)уксусной кислоты (15,0 г), PdCl2(dppf) (3,0 г), Zn(CN)2 (4,96 г), ДМФА (200 мл) и H2O (14 мл). Колбу,содержащую полученную суспензию, обезгаживают и снова заполняют азотом в течение 5 мин, а затем к реакционной смеси добавляют Pd2(dba)3 (3,85 г). Реакционную смесь греют в атмосфере N2 при 90C в течение 16 ч, охлаждают до комнатной температуры, разбавляют H2O (800 мл) и фильтруют. Собранное твердое вещество промывают толуолом (10 мл) и получают трет-бутиловый эфир (3-цианопиразоло[3,4b]пиридин-1-ил)уксусной кислоты в виде желтого твердого вещества. Стадия 3. Смесь трет-бутилового эфира (3-цианопиразоло[3,4-b]пиридин-1-ил)уксусной кислоты,гидрохлорида гидроксиламина (8,28 г) и Et3N (22,6 мл) в EtOH (120 мл) греют в течение ночи в атмосфере N2 при 65C. Полученную смесь охлаждают до комнатной температуры, фильтруют, собранное твердое вещество промывают H2O (100 мл) и Et2O (50 мл 2) и получают трет-бутиловый эфир [3-(Nгидроксикарбамимидоил)пиразоло[3,4-b]пиридин-1-ил)уксусной кислоты. Стадия 4. трет-Бутиловый эфир [3-(N-гидроксикарбамимидоил)пиразоло[3,4-b]пиридин-1 ил]уксусной кислоты (6,17 г) загружают в 100-мл флакон с AcOH (45 мл) и Ac2O (4,3 мл). Полученную смесь перемешивают при комнатной температуре в течение 1 ч, и за это время исходная суспензия становится прозрачным раствором. К полученному раствору добавляют Pd/C (10%, 900 мг), перемешивают в атмосфере H2 из баллона и полученную смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь фильтруют через слой целита, промытый DCM/MeOH. Выпаривание растворителя дает трет-бутиловый эфир (3-амидинопиразоло[3,4-b]пиридин-1-ил)уксусной кислоты, который используют без дополнительной очистки. Стадия 5. трет-Бутиловый эфир (3-амидинопиразоло[3,4-b]пиридин-1-ил)уксусной кислоты, полученный выше, загружают в 100-мл флакон с хлорацетилальдегидом (5,72 мл), диоксаном (50 мл) и K2CO3(12,42 г). Полученную смесь перемешивают при 80C в течение 4 ч и добавляют еще хлорацетилальдегид(5,72 мл) и K2CO3 (12,42 г). Смесь перемешивают еще в течение 1 ч при 80C и перемешивают еще при 120C в течение 1 ч, охлаждают до комнатной температуры, разбавляют дихлорметаном (DCM), промывают рассолом, сушат (Na2SO4), фильтруют и упаривают в вакууме. Очистка флэш-хроматографией дает трет-бутиловый эфир 2-(3-(1H-имидазол-2-ил)-1H-пиразоло[3,4-b]пиридин-1-ил)уксусной кислоты в виде коричневого масла. Стадия 6. трет-Бутиловый эфир 2-(3-(1H-имидазол-2-ил)-1H-пиразоло[3,4-b]пиридин-1-ил)уксусной кислоты (977 мг) растворяют в трифторуксусной кислоте (ТФУК) (10 мл) и перемешивают при комнатной температуре в течение 1 ч. Смесь упаривают в вакууме и получают 2-(3-(1H-имидазол-2-ил)-1Hпиразоло[3,4-b]пиридин-1-ил)уксусную кислоту в виде коричневого масла, которое используют без дополнительной очистки. Стадия 7. (Протокол A - процедура сочетания с HBTU). Раствор [3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]уксусной кислоты (0,30 М, 0,40 мл, 0,12 ммоль) переносят во флакон. Во флакон добавляют дигидрохлорид 1-(4-хлор-5-этокси-2-фторфенил)пиперазина (48 мг, 0,14 ммоль), гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (55 мг, 0,14 ммоль) и изо-Pr2NEt (0,30 мл) и смесь перемешивают при температуре окружающей среды. Через 30 мин анализ ЖХ/МС показывает образование нужного продукта и полное израсходование исходной карбоновой кислоты. Смесь разбавляютEtOAc, промывают водой (1) и рассолом (1), сушат над Na2SO4 и упаривают. Остаток очищают хроматографией на силикагеле (1-8% MeOH в CH2Cl2), и получают 1-[4-(4-хлор-5-этокси-2-фторфенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон в виде желтоватокоричневого твердого вещества. 1 Названное в заголовке соединение получают, следуя протоколу A. В качестве компонентов сочетания используют дигидрохлорид 1-[4-хлор-2-фтор-5-(2-фторэтокси)фенил]пиперазина и [3-(1H-имидазол 2-ил)пиразоло[3,4-b]пиридин-1-ил]уксусную кислоту. Сырой продукт реакции очищают хроматографией на силикагеле (2-3,5% MeOH в CH2Cl2) и получают 1-4-[4-хлор-2-фтор-5-(2-фторэтокси)фе- 15015529 нил]пиперазин-1-ил-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон в виде желтоватокоричневого твердого вещества. 1 Названное в заголовке соединение получают, следуя протоколу A. В качестве компонентов сочетания используют дигидрохлорид 1-(4-хлор-3-этоксифенил)пиперазина и [3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]уксусную кислоту. Сырой продукт реакции очищают хроматографией на силикагеле (4-15% MeOH в CH2Cl2) и получают 1-[4-(4-хлор-3-этоксифенил)пиперазин-1-ил]-2-[3-(1Hимидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон в виде желтовато-коричневого твердого вещества Названное в заголовке соединение получают, следуя протоколу A. В качестве компонентов сочетания используют дигидрохлорид 1-(4-хлор-2-фтор-5-метоксифенил)пиперазина и [3-(1H-имидазол-2 ил)пиразоло[3,4-b]пиридин-1-ил]уксусную кислоту. Сырой продукт реакции очищают хроматографией на силикагеле (1-10% MeOH в CH2Cl2) и получают 1-[4-(4-хлор-2-фтор-5-метоксифенил)пиперазин-1 ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон в виде желтовато-коричневого твердого вещества (27 мг). 1 Н NMR (CDCl3, 400 MHz)8.79 (dd, 0.6H), 8.67 (dd, 0.4H), 8.57-8.55 (m, 1H), 7.31-7.20 (m, 2H),7.12-7.06 (m, 2H), 6.49-6.45 (m, 1H), 5.45 (s, 0.6H), 5.43 (s, 1.4H), 3.86 (s, 0.9H), 3.85 (s, 2.1H), 3.81-3.75 (m,4H), 3.15-3.08 (m, 4H); LC/MS m/z (M+H)+ 470.4. Пример 5. Синтез 1-[(S)-4-(4-хлор-3-метоксифенил)-2-метилпиперазин-1-ил]-2-[3-(1H-имидазол-2 ил)пиразоло[3,4-b]пиридин-1-ил]этанона Названное в заголовке соединение получают, следуя протоколу A. В качестве компонентов сочетания используют дигидрохлорид (S)-1-(4-хлор-3-метоксифенил)-3-метилпиперазина и [3-(1H-имидазол-2 ил)пиразоло[3,4-b]пиридин-1-ил]уксусную кислоту. Сырой продукт реакции очищают хроматографией на силикагеле (1-8% MeOH в CH2Cl2) и получают 1-[(S)-4-(4-хлор-3-метоксифенил)-2-метилпиперазин-1 ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон в виде желтовато-коричневого твердого вещества. 1 Н NMR (CDCl3, 400 MHz)8.79 (dd, 0.6H), 8.66 (dd, 0.4H), 8.57-8.55 (m, 1H), 7.29-7.19 (m, 4H),6.44-6.39 (m, 2H), 5.42 (br. s, 2H), 4.83 (br. s, 0.3H), 4.49 (br. s, 0.3H), 4.30 (br. s, 0.3H), 3.89 (s, 1.2H), 3.88 Названное в заголовке соединение получают, следуя протоколу A. В качестве компонентов сочетания используют дигидрохлорид (S)-1-(4-хлор-2-фтор-5-метоксифенил)-3-метилпиперазина и [3-(1Hимидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]уксусную кислоту. Сырой продукт реакции очищают хроматографией на силикагеле (2-3% MeOH в CH2Cl2) и получают 1-[(S)-4-(4-хлор-2-фтор-5-метоксифенил)2-метилпиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон в виде желтоватокоричневого твердого вещества (29 мг). 1 Названное в заголовке соединение получают, следуя протоколу A. В качестве компонентов сочетания используют (R)-1-(4-хлор-3-метоксифенил)-2-метилпиперазин и [3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]уксусную кислоту. Сырой продукт реакции очищают хроматографией на силикагеле Названное в заголовке соединение получают, следуя протоколу A. В качестве компонентов сочетания используют (S)-1-(4-хлор-3-метоксифенил)-2-метилпиперазин и [3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]уксусную кислоту. Сырой продукт реакции очищают хроматографией на силикагеле Названное в заголовке соединение получают, следуя протоколу A. В качестве компонентов сочета- 17015529 ния используют (R)-1-(4-хлор-3-метоксифенил)-2-метилпиперазин и [3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]уксусную кислоту. Сырой продукт реакции очищают хроматографией на силикагеле Названное в заголовке соединение получают, следуя протоколу A. Во флакон, содержащий дигидрохлорид (S)-1-(4-хлор-3-этоксифенил)-3-метилпиперазина (70 мг, 0,21 ммоль), добавляют 2-(3-(1Hимидазол-2-ил)-1H-пиразоло[3,4-b]пиридин-1-ил)уксусную кислоту (51 мг, 0,21 ммоль), HBTU (81 мг,0,21 ммоль), ДМФА (0,7 мл) и DIPEA (0,15 мл, 0,87 ммоль). Реакционную смесь выдерживают при 30C в течение 24 ч. Раствор разбавляют EtOAc (30 мл) и промывают 1 N HCl (210 мл) и насыщ. водн. раствором NaCl (210 мл). Органическую фазу сушат над MgSO4 и концентрируют в вакууме. Полученный остаток очищают препаративной ВЭЖХ (градиент 2095 MeCN-H2O с 0,1% ТФК), чистые фракции лиофилизуют и получают указанное соединение (11 мг, выход 11%). MC (ES), предполагается [M+H]+ 480,2, найдено 480,5. 1(51 мг, 0,21 ммоль), HBTU (83 мг, 0,22 ммоль), ДМФА (0,7 мл) и DIPEA (0,20 мл, 1,2 ммоль). Реакционную смесь выдерживают при 20C в течение 24 ч. Раствор разбавляют EtOAc (30 мл) и промывают 1 NHCl (210 мл) и насыщ. водн. раствором NaCl (210 мл). Органическую фазу сушат над MgSO4 и концентрируют в вакууме. Полученный остаток очищают препаративной ВЭЖХ (градиент 2095 MeCNH2O с 0,1% ТФК), чистые фракции лиофилизуют и получают указанное соединение (20 мг, выход 20%).(80 мг, 0,23 ммоль), добавляют 2-(3-(1H-имидазол-2-ил)-1H-пиразоло[3,4-b]пиридин-1-ил)уксусную кислоту (51 мг, 0,21 ммоль), HBTU (81 мг, 0,21 ммоль), ДМФА (0,7 мл) и DIPEA (0,2 мл, 1,2 ммоль). Реак- 18015529 ционную смесь выдерживают при 30C в течение 24 ч. Раствор разбавляют EtOAc (30 мл) и промывают 1 N HCl (210 мл) и насыщ. водн. раствором NaCl (210 мл). Органическую фазу сушат над MgSO4 и концентрируют в вакууме. Полученный остаток очищают препаративной ВЭЖХ (градиент 2095MeCN-H2O с 0,1% ТФУК), чистые фракции лиофилизуют и получают указанное соединение (14 мг, выход 13%). MC (ES), предполагается [M+H]+ 498,2, найдено 498,4. 1 Н NMR (CDCl3, 400 MHz)9.95 (br s, 1H), 8.79 (dd, J=1.6, 8.2, 1H), 8.55 (dd, J=1.6, 4.4, 1H), 7.197.26 (m, 4H), 6.44-6.50 (m, 2H), 5.40 (br s, 2H), 4.77 (dt, J=4.2, 47.2, 2H), 4.26 (dt, J=4.2, 27.2, 2H), 2.78-4.42 Во флакон загружают 2-(3-(1H-имидазол-2-ил)-1H-пиразоло[3,4-b]пиридин-1-ил)уксусную кислоту(142 мг, 0,50 ммоль), безводный ДМФА (2,0 мл) и DIPEA (0,5 мл). Флакон закрывают крышкой, нагревают до 45C и содержимое перемешивают в течение ночи. На следующий день летучие вещества удаляют в вакууме, и разделение препаративной ВЭЖХ (обращенная фаза, градиент ацетонитрил-вода) дает 1-[4-(4-хлор-3-метилфенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон. MC (ES), [M+H]+ найдено 436,4. Пример 14. Синтез 1-[4-(4-хлор-3-трифторметилфенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2 ил)пиразоло[3,4-b]пиридин-1-ил]этанона Во флакон загружают 2-(3-(1H-имидазол-2-ил)-1H-пиразоло[3,4-b]пиридин-1-ил)уксусную кислоту(55 мг, 0,226 ммоль), HBTU (125 мг, 0,33 ммоль), дигидрохлорид 1-(4-хлор-3-трифторметилфенил)пиперазина (170 мг, 0,50 ммоль), безводный ДМФА (2,0 мл) и DIPEA (0,5 мл). Флакон закрывают крышкой, нагревают до 45C и содержимое перемешивают в течение ночи. На следующий день летучие вещества удаляют в вакууме и разделение препаративной ВЭЖХ (обращенная фаза, градиент ацетонитрил-вода) дает 1-[4-(4-хлор-3-трифторметилфенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон. MC (ES), [M+H]+ найдено 490,4. Пример 15. Синтез 1-[4-(4-хлор-3-трифторметоксифенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2 ил)пиразоло[3,4-b]пиридин-1-ил]этанона Во флакон загружают 2-(3-(1H-имидазол-2-ил)-1H-пиразоло[3,4-b]пиридин-1-ил)уксусную кислоту(55 мг, 0,226 ммоль), HBTU (125 мг, 0,33 ммоль), дигидрохлорид 1-(4-хлор-3-трифторметоксифенил)пиперазина (177 мг, 0,50 ммоль), безводный ДМФА (2,0 мл) и DIPEA (0,4 мл). Флакон закрывают крышкой, нагревают до 45C и содержимое перемешивают в течение ночи. На следующий день летучие вещества удаляют в вакууме и разделение препаративной ВЭЖХ (обращенная фаза, градиент ацетонитрил-вода) дает 1-[4-(4-хлор-3-трифторметоксифенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон. MC (ES), [M+H]+найдено 506,4. Названное в заголовке соединение получают из 1-[4-(4-хлор-3-метоксифенил)пиперазин-1-ил]-2-[3 йодпиразоло[3,4-b]пиридин-1-ил]этанона (см. заявку на патент США, per.11/474132, опубликована как US 20070010524, включена в данное описание в качестве ссылки) согласно процедуре, схожей с описанной на стадиях 2-5 в примере синтеза 1. 1 Н NMR (CDCl3, 400 MHz)10.22 (br, 1H), 8.82 (dd, 1H), 8.56 (dd, 1H), 7.20-7.30 (m, 3H), 7.11 (s,1H), 6.47 (d, 1H), 6.42 (dd, 1H), 5.44 (s, 2H), 3.88 (s, 3H), 3.80 (m, 4H), 3.19 (m, 4H); MS (ES) M+H expect 452.2. Пример 17. Синтез 1-[4-(4-хлор-3-метоксифенил)пиперазин-1-ил]-2-[3-(1-метил-1H-имидазол-2 ил)пиразоло[3,4-b]пиридин-1-ил]этанона К раствору 1-[4-(4-хлор-3-метоксифенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4b]пиридин-1-ил]этанона (50 мг, 0,11 ммоль, 1 экв.) в ТГФ добавляют 60% гидрид натрия (5,7 мг,0,14 ммоль, 1,3 экв.) и перемешивают в течение 1 ч, а затем добавляют йодметан (25,8 мг, 0,16 ммоль,1,5 экв.). Через 2 ч ЖХМС показывает, что главный пик соответствует желаемому продукту, и препаративная ВЭЖХ (обращенная фаза, градиент ацетонитрил-вода) дает 1-[4-(4-хлор-3-метоксифенил)пиперазин-1-ил]-2-[3-(1-метил-1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон. MC (ES), [M+H]+ найдено 465,2. В трехгорлую 5-л колбу Мортона, снабженную механической мешалкой, переходником для газа,нагревательным кожухом и термометром, загружают рац-BINAP (4,24 г, 0,005 экв.) и Pd2(dba)3 (3,20 г,0,0025 экв.). Колбу откачивают и заполняют азотом. Добавляют толуол (100 мл) через канюлю. Смесь перемешивают при комнатной температуре в течение 15 мин и получают пурпурный раствор. Затем добавляют толуол (2,0 л). Добавляют в один прием 2-хлор-5-броманизол (300,3 г, 1,356 моль, 1 экв.). Добавляют в один прием Boc-пиперазин (252,4 г, 1 экв.). Добавляют в один прием трет-бутоксид натрия(183,0 г, 1,4 экв.). Колбу откачивают и заполняют азотом. Затем смесь нагревают до внутренней температуры 60C. Получают гетерогенную светло-оранжевую взвесь. Через 1 ч смесь становится гомогенным коричневым раствором. Еще через 15 ч смесь охлаждают до комнатной температуры. К смеси при перемешивании добавляют EtOAc (2,0 л). Твердое вещество отфильтровывают. Фильтрат промывают EtOAc(100 мл). Объединенный фильтрат промывают 10% водн. раствором K2CO3 (11 л), водой (11 л) и сушат над MgSO4. Растворитель удаляют в вакууме и получают продукт реакции оранжевое твердое вещество В 4-л стакан, снабженный мешалкой, загружают трет-бутил-4-(4-хлор-3-метоксифенил)пиперазин 1-карбоксилат (500 г, 1,53 моль, 1, экв.) и MeOH (1,50 л). При перемешивании при комнатной температуре в течение 5 мин добавляют конц. 37% HCl (500 мл, 4 экв.). Внутренняя температура поднимается до 40C и раствор становится густым с выпадением осадка. Через 15 мин смесь нагревают на плитке до внутренней температуры 60C (начинается вспенивание, когда смесь нагревается до приблизительно 50C.) После 2 ч выдержки при 60C раствор охлаждают до комнатной температуры и затем до 5C в холодильнике. Продукт реакции собирают фильтрацией в две партии. Каждую партию красно- 21015529 коричневого фильтрата промывают EtOAc (2500 мл) и получают светло-желтое твердое вещество. Две партии объединяют и получают продукт (391, 3 г). Фильтрат концентрируют до объема 300 мл в вакууме и обрабатывают горячим (50C) MeOH (500 мл). Смесь охлаждают до 5C в холодильнике в течение 24 ч. Полученное выпавшее в осадок вещество собирают фильтрацией и промывают EtOAc (2200 мл), получают еще 44,3 г продукта (всего 435,6 г, выход 95%). Стадия 3. 2-Хлор-1-(4-(4-хлор-3-метоксифенил)пиперазин-1-ил)этанон В 3-л колбу, снабженную механической мешалкой, загружают дигидрохлорид 1-(4-хлор-3 метоксифенил)пиперазина (220 г, 0,73 моль, 1 экв.), CH2Cl2 (1000 мл) и воду (1000 мл). Двухфазную смесь охлаждают до 5C на ледяной бане и к раствору при энергичном перемешивании добавляют K2CO3(506 г, 5 экв.) по частям для минимизации вспенивания. Из капельной воронки добавляют по каплям раствор хлорацетилхлорида (124,4 г, 1,5 экв.) в CH2Cl2 (100 мл), поддерживая внутреннюю температуру ниже 8C. Через 1 ч слои разделяют. Водную фазу экстрагируют CH2Cl2 (2300 мл) и объединенные органические слои сушат над смесью Na2SO4/K2CO3 3:1 (добавление K2CO3 способствует фазе раствора стать прозрачной). После фильтрации фильтрат концентрируют в вакууме, остаток сушат в течение 16 ч в вакууме и получают продукт реакции в виде не совсем белого твердого вещества (410 г, выход 92%). Стадия 6. 7-Азаиндазол-3-карбоксальдегид В 5-л 3-горлую колбу, снабженную цифровым термометром, 1-л капельной воронкой и механической мешалкой (всю стеклянную посуду перед использованием высушивают в печи и охлаждают на воздухе в течение 30 мин), загружают 3-йод-7-азаиндазол (196,0 г, 0,80 моль) и 1 л безводного ТГФ (из бутыли SureSeal от Aldrich, используют, как он есть). Твердые вещества полностью растворяются в ТГФ при комнатной температуре с образованием темно-коричневого раствора. Затем колбу охлаждают до-5C на бане со смесью лед/NaCl и при умеренном перемешивании и добавляют по каплям 1 М раствор о-толилмагнийхлорида (в ТГФ, 880 мл, 1,1 экв.), сохраняя внутреннюю температуру от -5 до -3C (после добавления 820 мл раствора о-толилмагнийхлорида температура более не поднимается). Весь процесс добавления занимает 2 ч 25 мин. По окончании добавления смесь представляет собой гомогенный темнокоричневый раствор. Еще через 1 ч добавляют по каплям раствор изопропилмагнийхлорида (2 М в ТГФ, 480 мл, 1,2 экв.),сохраняя внутреннюю температуру 4C. Через 25 мин и после добавления примерно 200 мл раствора изопропилмагнийхлорида начинает образовываться коричневый осадок. После добавления, в общем,380 мл раствора изопропилмагнийхлорида смесь снова становится гомогенной. Весь процесс добавления осуществляют за 45 мин. Еще через 1 ч 25 мин берут небольшое количество образца и гасят D2O. Анализа ЖХМС данного образца показывает полный обмен на йод-Mg. Затем добавляют по каплям 1-формилпиперидин (120 мл, 1,3 экв.), сохраняя внутреннюю температуру где-то 2C. После добавления примерно 30 мл 1-формилпиперидина внутренняя температура более не поднимается и оставшийся 1-формилпиперидин добавляют относительно быстро. Весь процесс добавления занимает 20 мин. По окончании добавления смесь все еще представляет собой темный однородный раствор, и ее постепенно нагревают до комнатной температуры и умеренно перемешивают в течение 18 ч. Смесь снова охлаждают до 0C на бане со смесью лед/NaCl и реакцию гасят, постепенно добавляя смесь насыщенный раствор NH4Cl (750 мл)/концентрированная HCl (250 мл), сохраняя внутреннюю температуру 35C. По завершении добавления перемешивание продолжают в течение 1 ч, и появляется желтый осадок. Смесь фильтруют и твердое вещество промывают ТГФ (100 мл). Собранный фильтрат переносят в делительную воронку и доводят pH водного слоя до 5-6, добавляя NaHCO3 (примерно 5 г). ТГФ-слой отделяют и промывают насыщ. раствором NaCl (2100 мл). Объединенные водные слои(включая промывную жидкость с NaCl и погашенный водный слой) экстрагируют EtOAc (3250 мл). Объединенные органические слои сушат (Na2SO4), фильтруют, упаривают в вакууме (температура бани 30C) и получают коричневатое твердое вещество. Полученное твердое вещество растирают в Et2O(600 мл) и отфильтровывают. Собранное твердое вещество промывают Et2O (2100 мл) и получают 7- 22015529 азаиндазол-3-карбоксальдегид в виде желтоватого твердого вещества (86,6 г, 73%). Стадия 4. 1-[4-(4-Хлор-5-этокси-2-фторфенил)пиперазин-1-ил]-2-[3-формилпиразоло[3,4-b]пиридин-1-ил]этанон(162,5 г, 2 экв.) в ДМФА (0,5 л) в 5-л колбе нагревают до 85C (процесс нагревания занимает примерно 1,5 ч). К реакционной смеси небольшими порциями добавляют 2-хлор-1-(4-(4-хлор-3 метоксифенил)пиперазин-1-ил)этанон (175 г, 1 экв.). Весь процесс добавления занимает примерно 30 мин. Затем смесь перемешивают при 85C в течение 30 мин, и ЖХМС подтверждает, что реакция завершена. После охлаждения до комнатной температуры смесь переносят в 4-л колбу с 2 л льда. Реакционную колбу ополаскивают небольшим количеством ацетона (30 мл), который также переносят в смесь ДМФА/лед в 4-л колбе. В осадок выпадает много коричневатого твердого вещества. После того как лед полностью растопится, смесь фильтруют. Собранное твердое вещество промывают водой (1 л), перемешивают и затем промывают водой (1 л), чтобы избавиться от остаточного ДМФА. Собранное твердое вещество, содержащееся в воде, в таком виде растворяют в CH2Cl2 (4 л) и смесь переносят в 5-л делительную воронку. Нижний слой CH2Cl2 отделяют, а верхний водн. слой промывают CH2Cl2 (2100 мл). Объединенные слои CH2Cl2 сушат (Na2SO4), фильтруют, упаривают в вакууме и получают 1-[4-(4-хлор-5 этокси-2-фторфенил)пиперазин-1-ил]-2-[3-формилпиразоло[3,4-b]пиридин-1-ил]этанон в виде коричневатого твердого вещества (236,4 г, 97%), которое используют без дополнительной очистки. Стадия 5. 1-[4-(4-Хлор-5-этокси-2-фторфенил)пиперазин-1-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон 1-[4-(4-Хлор-5-этокси-2-фторфенил)пиперазин-1-ил]-2-[3-формилпипиразоло[3,4-b]пиридин-1 ил]этанон (300 г, 723 ммоль, 1 экв.), тример дигидрата глиоксаля (60,6 г, 0,4 экв.) и ацетат аммония(222,9 г, 4 экв.) суспендируют в смеси ТГФ (720 мл) и MeOH (720 мл) в 5-л круглодонной колбе, снабженной стержнем для магнитной мешалки и вводом для азота. Добавляют уксусную кислоту (84 мл,2 экв.) и смесь греют на масляной бане при 45C (при нагревании твердые вещества растворяются). Через 12 ч анализ ЖХМС показывает полное израсходование исходного альдегида и образование нужного продукта (ЖХ/МС m/z (M+H)+ 452,1). MeOH/ТГФ удаляют выпариванием на роторном испарителе. Остаток растворяют в смеси 10% MeOH в CH2Cl2 (са. 1,5 л) и смесь энергично встряхивают с водным раствором карбоната калия (са. 210 г карбоната калия в са. 1,5 л воды, pH водного слоя 8-9). Слои разделяют и водный слой экстрагируют смесью 10% MeOH в CH2Cl2 (2100 мл). Объединенные органические слои концентрируют и получают коричневое маслянистое твердое вещество. Сырой продукт реакции суспендируют в смеси 10% MeOH в EtOAc (са. 1 л). Добавляют безводный Na2SO4 (са. 60 г) и силикагель(са. 100 г) и взвесь осторожно нагревают феном для растворения сырого продукта. Взвесь переносят в 2-л воронку с пористым стеклянным фильтром, содержащую силикагель (са. 100 г, предварительно уравновешен 10% MeOH в EtOAc), и продукт элюируют через слой силикагеля 10% MeOH в EtOAc (са. 6 л) и 1% Et3N, 10% MeOH в EtOAc (са. 10 л) (примечание: неполное растворение продукта и/или выпадение продукта в осадок в присутствии силикагеля затрудняют фильтрацию). Растворители удаляют выпариванием на роторном испарителе. Остаток растирают в MeCN (1300 мл), сушат (в высоком вакууме после роторного выпаривания) и получают 1-[4-(4-хлор-5-этокси-2-фторфенил)пиперазин-1-ил]-2-[3-(1Hимидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанон в виде не совсем белого твердого вещества (190 г,58%, чистота по ЖХ/МС 98%). Стадия 7. Гидрохлорид 2-(3-(1H-имидазол-2-ил)-1H-пиразоло[3,4-b]пиридин-1-ил)-1-(4-(4-хлор-3 метоксифенил)пиперазин-1-ил)этанона- 23015529 В 2-л колбу с магнитной мешалкой загружают исходное вещество (5,1 г, 11,28 ммоль) и EtOAc(900 мл). Полученную суспензию греют до образования прозрачного раствора и охлаждают до комнатной температуры при умеренном перемешивании. К полученному раствору при комнатной температуре в течение 5 мин добавляют по каплям раствор HCl в Et2O (2 М, 6,2 мл, 12,42 ммоль). По завершении добавления полученную суспензию перемешивают при комнатной температуре еще в течение 1 ч. Твердое вещество собирают фильтрацией, промывают Et2O (150 мл 2), сушат в вакууме и получают 5,4 г не совсем белого порошка. В 250-мл колбу с магнитной мешалкой загружают полученный выше порошок(5,4 г), ацетон (100 мл) и деионизованную воду (16 мл). Полученную суспензию греют до образования прозрачного раствора и перемешивают для охлаждения. Когда раствор становится мутным (появляются мелкие кристаллы), к суспензии медленно, в течение 20 мин добавляют ацетон (540 мл). Полученную суспензию греют при 50C и перемешивают в течение 2 ч. Фильтрация в горячем состоянии, промывка горячим ацетоном (50 мл 2) и сушка в вакууме дают 3,3 г (60%) продукта в виде не совсем белого твердого вещества, т.пл. 164-165C. В поляризационном микроскопе кристаллы имеют вид призм. Пример 19 В 2000-мл колбу загружают 1-[4-(4-хлор-3-метоксифенил)пиперазин-1-ил]-2-(3-иодпиразоло[3,4b]пиридин-1-ил)этанон (см. заявку на патент США, рег.11/474132, опубликована как US 20070010524,40 г, 78,1 ммоль), dppf (3,86 г, 6,96 ммоль) Zn(CN)2 (9,6 г, 81,6 ммоль), ДМФА (360 мл) и H2O (20 мл). Полученную суспензию обезгаживают с использованием N2 в течение 5 мин, и затем добавляют Pd2(dba)3(4,24 г, 4,64 ммоль). Реакционную смесь греют в атмосфере N2 при 90C в течение 2 ч (контроль ТСХ и ЖХ-MC). После охлаждения до комнатной температуры смесь разбавляют EtOAc (1500 мл), фильтруют для удаления выпавшего в осадок вещества, промывают H2O (1000 мл 2), насыщенным раствором ЭДТК 4Na (800 мл 2) и рассолом и сушат над Na2SO4. После выпаривания растворителя добавляют эфир(150 мл) и перемешивают в течение 2 ч. Полученное твердое вещество отфильтровывают и получают нужный продукт (30 г, 93%) в виде светло-желтого порошка. Перекристаллизация из кипящего CH3CN В 250-мл колбу загружают 1-(2-(4-(4-хлор-3-метоксифенил)пиперазин-1-ил)-2-оксоэтил)-1H-пиразоло[3,4-b]пиридин-3-карбонитрил (15,3 г, 37,2 ммоль) и EtOH (40 мл, 1 М). При охлаждении на ледя- 24015529 ной бане и перемешивании добавляют AcOH (6,75 мл, 112 ммоль), а затем этилендиамин (25 мл,372 ммоль). Полученную смесь греют при 120C (баня) в атмосфере N2 (наблюдаемая смесь начинает закипать) в течение 1,5 ч. TCX и ЖХ-МС показывают исчезновение исходного вещества и образование имидазолина. После охлаждения до комнатной температуры смесь разбавляют DCM (700 мл) и промывают H2O (350 мл). Слой H2O снова экстрагируют DCM (150 мл), объединенный органический слой промывают рассолом (350 мл) и сушат над MgSO4. После выпаривания растворителя при пониженном давлении остаток суспендируют в горячем EtOAc (80 мл). После охлаждения до комнатной температуры твердое вещество собирают фильтрацией, промывают EtOAc (30 мл) и получают названное в заголовке соединение в виде белого порошка (16 г, 95%), который используют непосредственно на следующей стадии; т.пл. 133-135C; Rt=1,369 мин; MC(ES), M+H предполагается 454,2, найдено 454,4. Стадия 3. 2-(3-1H-Имидазол-2-ил)-1H-пиразоло[3.4-b]пиридин-1-ил)-1-(4-(4-хлор-3-метоксифенил)пиперазин-1-ил)этанон К полученному выше имидазолину (12,3 г, 27,1 ммоль) в 500-мл колбе добавляют безводный ДМСО (108 мл, -0,25 М). При перемешивании добавляют по частям DMP (17,2 г, 40,6 ммоль). Полученную смесь перемешивают при 45C в атмосфере N2 в течение 2 ч (контроль ТСХ и ЖХ-МС). После охлаждения до комнатной температуры реакцию гасят насыщ. раствором Na2S2O3 (100 мл) (ледяная баня),затем добавляют 3 N раствор NaOH (100 мл) (pH 12-13) и H2O (300 мл) и смесь экстрагируют DCM(600 + 300 мл). Объединенный органический слой промывают насыщ. раствором NaHCO3 (300 мл) и рассолом(300 мл) и сушат (MgSO4, 120 г). После выпаривания органического растворителя остаток - желтое твердое вещество (11 г) растворяют в горячем CH3CN (20 мл). После охлаждения до комнатной температуры полученное твердое вещество собирают фильтрацией и получают 6,7 г (55%) названного в заголовке соединение в виде светлых желтовато-коричневых кристаллов; т.пл. 149-152C; Rt=1,309 мин; MC(ES),M+H предполагается 452,2, найдено 452,4. Маточную жидкость концентрируют и получают еще 0,6 г(общий выход извлеченного вещества 60%). Пример 20. Приведенный далее пример иллюстрирует неожиданные превосходные фармакокинетические свойства соединения по изобретению в сравнении с подобным соединением, описанным ранее (публикацияUS2007/0010524 А 1). В целях сравнения в табл. 2A приводятся фармакокинетические профили соединения по изобретению (т.е. соединения В, см. также 1.016 в табл. 2B) и двух соединений, описанных в публикации US2007/0010524 А 1 (т.е. соединений A и C). Как видно из таблицы, соединение B имеет превосходные фармакокинетические свойства. Конкретнее, соединение по изобретению B является более выгодным,так как обнаруживает существенное повышение перорального всасывания (измеренное в % пероральной биодоступности) и величин Смакс и AUC по сравнению со схожими по строению соединениями A и C. Протокол ФК (РК) при испытаниях на крысах. При фармакокинетических исследованиях каждое соединение дают четырем самцам крысы Sprague-Dawley, не подвергавшихся никакому воздействию. Двое животных получают однократную дозу соединения (в виде состава с 31,6% пропиленгликоля/31,6% N,N-диметилацетамида/36,8% EtOH) внутривенно (i.v.) в 1 мг/кг, двое животных получают пероральную дозу (р.о.) соединения (в виде состава с 1% НРМС в воде) в 50 мг/кг. В установленные моменты времени (до 24 ч) берут образцы крови после дачи каждой дозы и анализируют соответствующие концентрации соединения в плазме с использованием метода ЖХ-МС/МС. Строят кривые зависимости концентрации в плазме от времени и определяют соответствующие фармакокинетические параметры с использованием некомпартментального анализа. Величины Смакс (максимальная концентрация в плазме) и AUC (площадь под кривой), указанные в таблице, вычисляют на основании кривых зависимости концентрации в плазме от времени после перорального приема дозы. Величины F (пероральная биодоступность) представляют собой отношения площади под кривой после перорального приема дозы (нормализованные к 1 мг/кг) и площади под кривой после iv дозы. Пример 21. Данный пример иллюстрирует оценку биологической активности, связанной с представляющими интерес соединениями по изобретению. Материалы и методы. А. Клетки. 1. Клетки, экспрессирующие CCR1. А. Клетки ТНР-1. Клетки ТНР-1 получают от ATCC (TIB-202) и культивируют в виде суспензии в среде RPMI-1640 с добавлением 2 мМ L-глутамина, 1,5 г/л бикарбоната натрия, 4,5 г/л глюкозы, 10 мМ HEPES, 1 мМ пирувата натрия, 0,05% 2-меркаптоэтанола и 10% FBS. Клетки выращивают в атмосфере 5% CO2/95% воздуха, 100% влажности при 37C и пересевают дважды в неделю при 1:5 (клетки культивируют в интервале плотности 2105-2106 клеток/мл) и собирают при 1106 клеток/мл. Клетки ТНР-1 экспрессируют CCR1 и могут использоваться в анализах связывания и функциональных анализах CCR1. 2. Хемотаксические анализы. Хемотаксические анализы выполняют с использованием поликарбонатных фильтров с диаметром пор 5 мкм, покрытых поливинилпирролидоном, в 96-луночных хемотаксических планшетах (Neuroprobe;Gaitherdburg, MD) с использованием буфера для хемотаксиса (сбалансированный солевой раствор Хэнкса (HBSS) и 1% FBS). Хемокиновые лиганды CCR1 (т.е. MIP-1, CCL15/лейкотактин; системы RD; Миннеаполис, MN) используют для оценки опосредуемого соединением ингибирования миграции, опосредуемой CCR1. Другие хемокины (т.е. SDF-1; системы RD; Миннеаполис, MN) используют в качестве контроля специфичности. В нижний планшет загружают 29 мкл хемокина (т.е. 0,1 нМ CCL15/лейкотактина) и различные количества соединения; верхний планшет содержит 100000 клеток ТНР-1 или моноцитов в 20 мкл. Планшеты инкубируют 1-2 ч при 37C и определяют число клеток в нижнем планшете или прямым подсчетом клеток в пяти мощных зонах на лунку или анализом CyQuant (Molecular Probes) метод с флуоресцентным красителем, которым измеряют содержание нуклеиновой кислоты, и наблюдением под микроскопом.B. Идентификация ингибиторов CCR1. Одной из основных функций хемокинов является их способность опосредовать миграцию клеток,экспрессирующих хемокиновые рецепторы, таких как белые клетки крови. Для того чтобы подтвердить,что соединение, представляющее интерес, ингибирует не только специфическое связывание CCR1 и передачу сигнала (по меньшей мере, как определяют анализы мобилизации кальция), но также опосредуемую CCR1 миграцию, можно использовать хемотаксический анализ. Клетки миеломоноцитарного лейкоза ТНР-1, которые схожи с моноцитами, а также свежеизвлеченные моноциты используют в качестве мишеней для хемоатракции хемокиновыми лигандами CCR1 (т.е. MIP-1, CCL15/лейкотактином). Клетки помещают в верхний компартмент луночного миграционного микропланшета, в то время как MIP-1(или другой потенциальный хемокиновый лиганд CCR1) и представляющее интерес соединение в возрастающих концентрациях загружают в нижний планшет. В отсутствие ингибитора клетки будут мигрировать в нижний планшет в ответ на хемокиновый агонист; если соединение ингибирует функцию CCR1,тогда большинство клеток будут оставаться в верхнем планшете. Для того чтобы подтвердить аффинность представляющего интерес соединения в отношении CCR1, а также подтвердить его способность ингибировать опосредуемую CCR1 миграцию клеток, в данном хемотаксическом анализе ингибирующую активность титруют в интервале концентраций соединения от 110-10 до 110-4 М. В данном анализе- 26015529 количество соединения изменяется, в то время как число клеток и концентрации агониста хемокина выдерживают постоянными. После инкубации хемотаксических планшетов в течение 1-2 ч при 37C количественно определяют отвечающие клетки в нижнем планшете, помечая их анализом CyQuant (MolecularProbes) - методом с флуоресцентным красителем, которым измеряют содержание нуклеиновой кислоты,и определяя Spectrafluor Plus (Tecan). Используют компьютерную программу Prism от GraphPad, Inc. (SanDiego, Ca) для вычисления величин IC50 - величины IC50 представляют собой такие концентрации соединения, которые требуются для ингибирования числа клеток, отвечающих на агонист CCR1, на 50%. 1. Эффективность in vivo.a. Кроличья модель деструктивного воспаления сустава. Исследования с LPS на кроликах проводят, по существу, так, как описано в Podolin et al., J. Immunol, 169(11): 6435-6444 (2002). Самок новозеландских кроликов (массой приблизительно 2 кг) обрабатывают интраартикулярно в оба коленных сустава LPS (10 нг). Представляющее интерес соединение, например 1.016 (в композиции в 1% метоцеле), или среду (1% метоцель) дают перорально в дозе объемом 5 мг/кг два раза (за 2 ч до интраартикулярной инъекции LPS и через 4 ч после интраартикулярной инъекции LPS). Через шестнадцать часов после инъекции LPS коленные суставы промывают и подсчитывают клетки. Благоприятное действие лечения определяют по уменьшению числа клеток в зоне воспаления,собравшихся в воспаленной синовиальной жидкости коленных суставов. Обработка соединениями, представляющими интерес, приводит к значительному уменьшению числа клеток, собравшихся в зоне воспаления.b. Оценка соединений, представляющих интерес, на крысиной модели артрита, вызванного коллагеном. Проводят 17-дневное исследование развивающегося коллагенового артрита типа II для оценки действия соединения, представляющего интерес, на вызванное артритом опухание голеностопного сустава. Коллагеновый артрит у крыс является экспериментальной моделью полиартрита, которую широко используют для преклинического испытания многочисленных противоартритных средств (см. Trentham etal., Arthritis Rheum., 42: 498-506 (1999. Признаками данной модели являются явное начало и развитие сильного легко определяемого полиартритного воспаления, заметная деструкция хряща в связи с образованием паннуса и от слабой до умеренной резорбция кости и пролиферация периостальной кости. Самкам кроликов Lewis (массой приблизительно 0,2 кг) дают наркоз изофлураном и инъецируют им неполный адъювант Фрейнда, содержащий 2 мг/мл бычьего коллагена типа II, в хвост и два участка в боку в дни 0 и 6 такого 17-дневного исследования. Соединение, представляющее интерес, вводят подкожно от 0 дня до 17 в эффективной дозе. Проводят измерения циркулем диаметра голеностопного сустава и уменьшение опухания сустава принимают за меру эффективности. В табл. 2B (приведенной ниже) приводятся структуры и активность характерных соединений, описанных в данном описании. Активность приводится как следующая из хемотаксического анализа, описанного выше: + для IC50100 нМ;для IC50100 нМ. или его фармацевтически приемлемая соль, гидрат или N-оксид. 2. Соединение по п.1 в форме гидрата. 3. Соединение по п.1 в форме фармацевтически приемлемой соли. 4. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически приемлемый эксципиент или носитель. 5. Способ получения соединения по п.1, включающий контактирование соединения формулы с имидазолобразующим реагентом. 6. Способ по п.5, где имидазолобразующий реагент выбирают из группы, состоящей из глиоксаля или эквивалента глиоксаля.
МПК / Метки
МПК: C07D 401/00
Метки: 3-(имидазолил)пиразоло[3,4-b]пиридины
Код ссылки
<a href="https://eas.patents.su/30-15529-3-imidazolilpirazolo34-bpiridiny.html" rel="bookmark" title="База патентов Евразийского Союза">3-(имидазолил)пиразоло[3,4-b]пиридины</a>
Композиции для ингаляции, содержащие трициклические 5,6-дигидро-9н-пиразоло (3,4-с)-1,2,4-триазоло (4,3-α) пиридины

Номер патента: 6742
Опубликовано: 28.04.2006
Авторы: Шеферд Майкл Тревор, Хамфри Майкл Джон, Миллер Пол Роберт
МПК: A61K 31/437, A61K 9/14, A61K 9/72...
Метки: ингаляции, 3,4-с)-1,2,4-триазоло, 4,3-&alpha, содержащие, 5,6-дигидро-9н-пиразоло, пиридины, композиции, трициклические
Формула / Реферат:
1. Препаративная форма для ингаляции, включающая твердые частицы 9-циклопентил-5,6-дигидро-7-этил-3-(тиен-2-ил)-9Н-пиразоло[3,4-с]-1,2,4-триазоло[4,3-a]пиридина или его фармацевтически приемлемой соли в качестве активного соединения, при этом указанные частицы имеют диаметр менее 20 мкм. 2. Препаративная форма для ингаляции по п.1, в которой указанные твердые частицы имеют следующее распределение по размерам: 90% менее чем 10 мкм в диаметре и...
Терапевтические пиразоло[3,4-b]пиридины и индазолы

Номер патента: 12245
Опубликовано: 28.08.2009
Авторы: Юен По-Вай, Щелкун Роберт М.
МПК: A61K 31/4162, A61K 31/416, A61P 25/00...
Метки: индазолы, терапевтические, пиразоло[3,4-b]пиридины
Формула / Реферат:
1. Соединение формулы I где R2 представляет собой фенил, возможно замещенный заместителями в количестве от одного до трех, независимо выбранными из галогено, метила, этила, CF3, метокси, CH2F, CHF2 и СН2ОН; R3 выбран из группы: 3-пирролидинила, 4-пиперидинила, 3-пиперидинила и 2-морфолинила; и R4, R5, R6 и R7 представляют собой Н или три из R4, R5, R6 и R7 представляют собой Н, а один из R4, R5, R6 и R7 выбран из группы: галогено, метокси и...
Пиразоло [3,4 - d] пиримидины с противоконвульсивным, противоаллергическим / противоастматическим действием

Номер патента: 2768
Опубликовано: 29.08.2002
Авторы: Менцер Манфред, Арнольд Томас, Унферферт Клаус, Тобер Кристине, Ланкау Ханс-Йоахим, Росток Ангелика
МПК: C07D 487/04, A61K 31/519, A61P 25/08...
Метки: пиримидины, противоастматическим, противоконвульсивным, противоаллергическим, действием, пиразоло
Формула / Реферат:
1. Соединения общей формулы 1 или их таутомеры, где Х обозначает кислород или серу и Y обозначает галоген, С1-С4-алкил, С1-С4-алкокси, трифторметил или трифторметокси, m равно 1 или 2, за исключением 2-бензил-4,5-дигидро-4-оксо-пиразоло[3,4d]пиримидина. 2. Соединения общей формулы 1, представляющие собой 2-(2-фторбензил)-пиразоло[3,4-d]пиримидин-4(5Н)-он, 2-(2-хлорбензил)-пиразоло[3,4-d]пиримидин-4(5Н)-он, ...
Способ получения пиразоло [4,3-d] пиримидин-7-онов и их промежуточные продукты

Номер патента: 3774
Опубликовано: 28.08.2003
Авторы: Данн Питер Джеймс, Леветт Филип Чарльз
МПК: C07D 487/04
Метки: пиразоло, способ, получения, пиримидин-7-онов, 4,3-d, продукты, промежуточные
Формула / Реферат:
1. Способ получения соединения формул (IA) и (IB) включающий реакцию соединения формулы (IIA) и (IIB) соответственно в присутствии -OR, где R, в случае образования (IA), представляет CH2CH3 и R, в случае образования соединения (IB), представляет CH2CH2CH3, где X представляет уходящую группу при условии, что X не является этокси 2. Способ по п.1, где X выбирают из группы, состоящей из арилсульфонилокси, C1-C4алкилсульфонилокси, нитро- или...
Производные пиразоло [4,3-d]пиримидина

Номер патента: 4982
Опубликовано: 28.10.2004
Авторы: Матайас Джон Пол, Харрис Лоренс Джеймс, Негри Джоанна Териза, Стрит Стивен Дерек Алберт, Вуд Альберт Шо, Леветт Филип Чарльз, Баннейдж Марк Эдвард, Девриз Кит Майкл
МПК: A61P 15/10, C07D 487/04, A61K 31/519...
Метки: производные, 4,3-d]пиримидина, пиразоло
Формула / Реферат:
1. Соединение формулы I или его фармацевтически или ветеринарно приемлемая соль, или фармацевтически или ветеринарно приемлемый сольват, или пролекарство, где R1 представляет собой C1-C6алкил или C3-C6алкенил, C3-C6циклоалкил или C4-C6циклоалкенил, где указанная алкильная группа может быть разветвленной или прямоцепочечной и где, когда R1 представляет собой C1-C3алкил, тогда указанная алкильная группа замещена, и где когда R1 представляет...
Предыдущий патент: Получение полимерных конъюгатов соединений, применяемых в терапии, сельском хозяйстве и в качестве пищевых добавок
Следующий патент: Наружное высокомощное светодиодное осветительное оборудование
Случайный патент: Способ разработки пластов соляных залежей малой мощности подземным выщелачиванием