Пептидные ингибиторы пути трансдукции сигнала jnk, обладающие способностью проникать в клетку




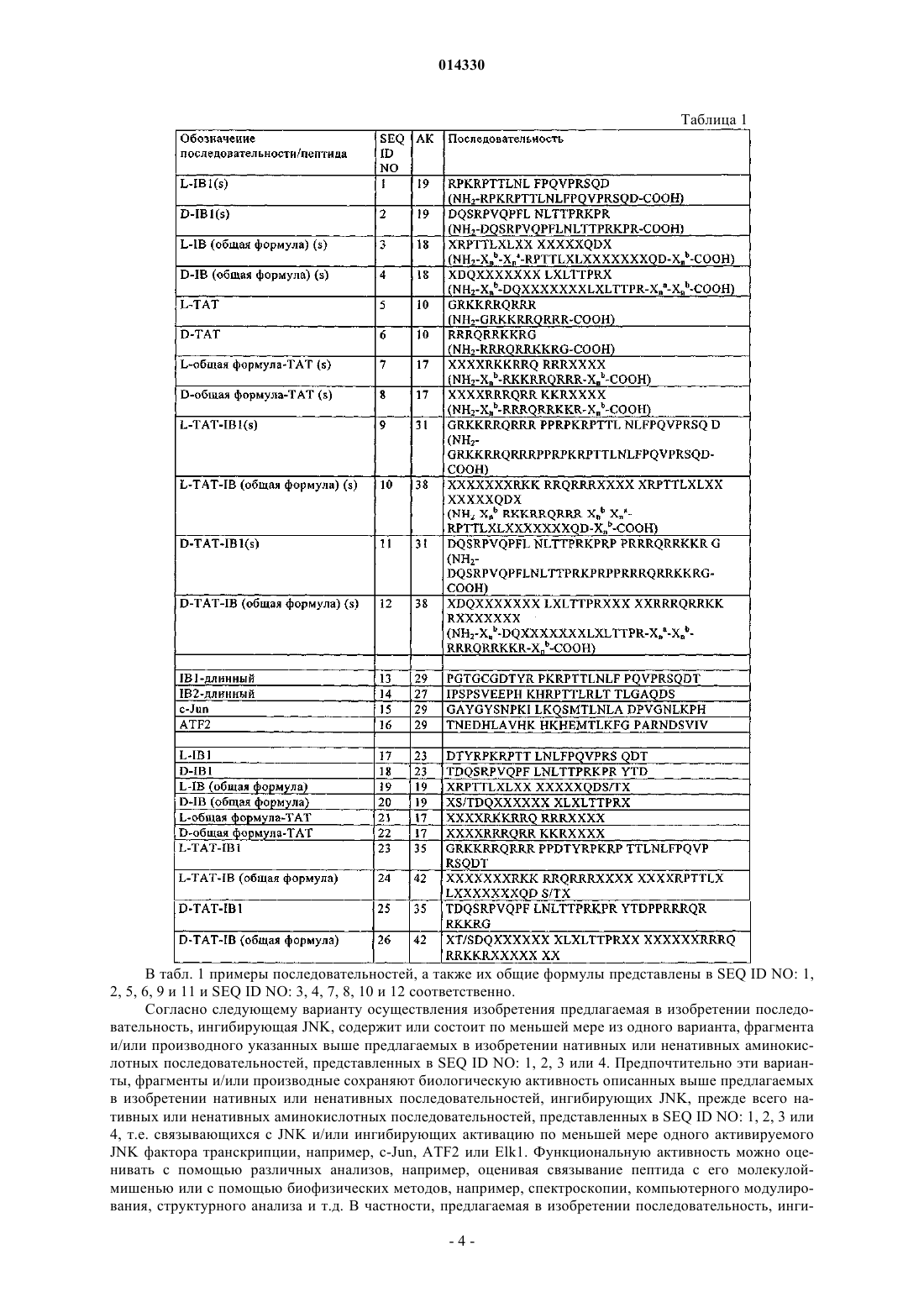

























Формула / Реферат
1. Последовательность, ингибирующая JNK, которая содержит менее 150 аминокислот, где последовательность-ингибитор содержит или состоит по меньшей мере из одной аминокислотной последовательности, представленной в SEQ ID NO: 2 или 4, где последовательность, ингибирующая JNK, не включает последовательности SEQ ID NO: 18, 20, 25 или 26.
2. Последовательность, ингибирующая JNK, по п.1, где последовательность, ингибирующая JNK, содержит от 5 до 150 аминокислотных остатков, более предпочтительно от 10 до 100 аминокислотных остатков, еще более предпочтительно от 10 до 75 аминокислотных остатков и наиболее предпочтительно от 15 до 50 аминокислотных остатков.
3. Последовательность, ингибирующая JNK, по одному из пп.1, 2, где последовательность, ингибирующая JNK, связывается с аминоконцевой киназой c-Jun (JNK).
4. Последовательность, ингибирующая JNK, по одному из пп.1-3, где последовательность, ингибирующая JNK, ингибирует активацию по меньшей мере одного являющегося мишенью для JNK фактора транскрипции, когда последовательность, ингибирующая JNK, присутствует в экспрессирующей JNK клетке.
5. Последовательность, ингибирующая JNK, по одному из пп.1-4, где являющийся мишенью для JNK фактор транскрипции выбран из группы, включающей c-Jun, ATF2 и Elk1.
6. Последовательность, ингибирующая JNK, по одному из пп.1-5, где последовательность, ингибирующая JNK, изменяет действие JNK, когда пептид присутствует в клетке, экспрессирующей JNK.
7. Химерный пептид, содержащий по меньшей мере один первый домен и по меньшей мере один второй домен, которые связаны ковалентной связью, где первый домен содержит транспортирующую последовательность, а второй домен содержит последовательность, ингибирующую JNK по любому из пп.1-6.
8. Пептид по п.7, в котором транспортирующая последовательность содержит аминокислотную последовательность полипептида ТАТ вируса иммунодефицита человека.
9. Пептид по одному из пп.7, 8, в котором транспортирующая последовательность содержит аминокислотную последовательность, представленную в SEQ ID NO: 5, 6, 7 или 8.
10. Пептид по одному из пп.7-9, в котором транспортирующие последовательности усиливают поглощение пептида клеткой.
11. Пептид по одному из пп.7-9, где транспортирующая последовательность обеспечивает ядерную локализацию пептида.
12. Пептид по одному из пп.7-11, в котором последовательность, ингибирующая JNK, включает аминокислотную последовательность, представленную в любой из SEQ ID NO: 11 или 12.
13. Пептид по п.12, где пептид содержит аминокислотную последовательность SEQ ID NO: 11.
14. Антитело, которое иммуноспецифически связывается с последовательностью, ингибирующей JNK, по одному из пп.1-6, или с химерным пептидом по одному из пп.7-13.
15. Фармацевтическая композиция, которая содержит последовательность, ингибирующую JNK, по одному из пп.1-6 или химерный пептид по одному из пп.7-13 и фармацевтически приемлемый носитель.
16. Применение последовательности, ингибирующей JNK, по одному из пп.1-6 или химерного пептида по одному из пп.9-13 для приготовления фармацевтической композиции, предназначенной для лечения патофизиологического состояния, выбранного из группы, включающей злокачественные заболевания легкого, молочной железы, лимфатической системы, желудочно-кишечного тракта и мочеполового пути, а также аденокарциномы, включая злокачественные, такие как различные типы рака ободочной кишки, почечно-клеточная карцинома, рак предстательной железы, немелкоклеточный рак легкого, рак тонкого кишечника и рак пищевода, а также лейкозы и рак, связанный с онкогенной трансформацией Bcr-Abl, псориаза, обыкновенной пузырчатки, синдрома Бехчета, острого респираторного дистресс-синдрома (ARDS), ишемического заболевания сердца, синдрома, связанного с состоянием после диализа, ревматоидного артрита, синдрома приобретенного иммунодефицита, васкулита, септического шока, рестеноза, потери слуха, ушной травмы, ишемии, инсульта, повреждения, связанного с реперфузией, гипоксии, вторичных эффектов, связанных с лечением, провоспалительными цитокинами, сердечной гипертрофии и связанных с артериосклерозом повреждений, патологических состояний, индуцированных ионизирующей радиацией, применяемой при лучевой терапии, ультрафиолетовым светом (УФ-свет), патологических состояний, индуцированных повреждающими ДНК агентами, включая химиотерапевтические лекарственные средства, гипо- и гипертермии, воспалительных, аутогенных воспалительных, иммунных и аутоиммунных заболеваний, дегенеративных заболеваний, миопатий, кардиомиопатий и отторжения трансплантата.
17. Применение по п.16, в котором фармацевтическую композицию следует применять с помощью пути введения, выбранного из группы, включающей внутрибрюшинное, назальное, внутривенное, оральное введение и введение с помощью бляшки.
18. Набор, который содержит последовательность, ингибирующую JNK, по одному из пп.1-6, и/или химерный пептид по одному из пп.7-13, и/или антитело по п.14.
Текст
ПЕПТИДНЫЕ ИНГИБИТОРЫ ПУТИ ТРАНСДУКЦИИ СИГНАЛА JNK, ОБЛАДАЮЩИЕ СПОСОБНОСТЬЮ ПРОНИКАТЬ В КЛЕТКУ В изобретении описаны ингибиторы протеинкиназ и более конкретно ингибиторы протеинкиназы, такой как аминоконцевая киназа c-Jun. Кроме того, в изобретении описаны последовательности ингибиторов JNK, химерные пептиды, нуклеиновые кислоты, кодирующие их, а также фармацевтические композиции, предназначенные для лечения патофизиологических состояний, ассоциированных с передачей сигнала JNK. 014330 Настоящее изобретение относится к ингибиторам протеинкиназ и более конкретно к ингибиторам протеинкиназы, представляющей собой аминоконцевую киназу c-Jun. Кроме того, настоящее изобретение относится к последовательностям ингибиторов JNK, химерным пептидам, нуклеиновым кислотам,кодирующим их, а также фармацевтическим композициям, предназначенным для лечения патофизиологических состояний, ассоциированных с передачей сигнала JNK. Аминоконцевая киназа c-Jun (JNK) является представителем группы активируемых стрессом активируемых митогеном протеин-(МАР)-киназ. Эти киназы принимают участие в контроле роста и дифференцировке клеток и в более общем плане в ответе клеток на стимулы окружающей среды. Путь трансдукции сигнала JNK активируется в ответ на связанный с окружающей средой стресс и при участии нескольких классов рецепторов на клеточной поверхности. Эти рецепторы могут представлять собой рецепторы цитокинов, рецепторы серпентина и рецепторы тирозинкиназ. В клетках млекопитающих JNK принимает участие в биологических процессах, таких как онкогенная трансформация и опосредующие адаптивные ответы на связанный с окружающей средой стресс. JNK ассоциирована с модулирующими иммунными ответами, включая созревание и дифференцировку иммунных клеток, а также участие в запрограммированной гибели клеток, выявленных иммунной системой в качестве мишеней для разрушения. Это уникальное свойство делает передачу сигнала JNK перспективной мишенью при разработке средств, предназначенных для фармакологического вмешательства. С передачей сигнала JNK связаны некоторые неврологические нарушения, прежде всего ишемический удар и болезнь Паркинсона. Одним из путей борьбы с нарушениями, в значительной степени связанными с передачей сигналаJNK, является создание ингибиторов пути передачи сигнала JNK. К таким ингибиторам, уже известным в данной области, относятся, прежде всего, например, ингибиторы, находящиеся перед киназой в пути передачи сигнала (например, СЕР-1347), низкомолекулярные химические ингибиторы JNK (SP600125 иAS601245), которые непосредственно оказывают воздействие на киназную активность, например, в результате конкуренции с АТФ-связывающим доменом протеинкиназы, и пептидные ингибиторы взаимодействия между JNK и ее субстратами (D-JNKI и I-JIP) (см., например, Kuan et al., Current Drug Targets CNSNeurological Disorders, т. 4,1, февраль 2005 г., с. 63-67, (5. Ингибитор СЕР-1347 (KT7515), находящийся перед киназой в пути передачи сигнала, представляет собой полусинтетический ингибитор семейства киназ смешанной линии дифференцировки. СЕР-1347(KT7515) усиливает выживание нейронов в дозах, которые ингибируют активацию аминоконцевых киназc-Jun (JNK) в первичных эмбриональных культурах и дифференцированных РС 12-клетках после удаления из питательной среды и у мышей, обработанных 1-метил-4-фенилтетрагидропиридином (МФТП). Кроме того, СЕР-1347 (KT7515) может увеличивать продолжительность выживания культивируемых куриных эмбриональных ганглиев заднего корешка спинного мозга, симпатических, цилиарных и моторных нейронов (см., например, Borasio et al., Neuroreport. 9 (7), 11 мая 1998 г., с. 1435-1439). Установлено, что низкомолекулярный химический ингибитор SP600125 снижает уровни фосфорилирования c-Jun, защищает допаминергические нейроны от апоптоза и частично восстанавливает уровень допамина при индуцированной МФТП болезни Паркинсона (БП) у мышей линии C57BL/6N (Wanget al., Neurosci Res., 48(2); февраль 2004 г., с. 195-202). Эти результаты свидетельствуют также о том, что путь JNK представляет собой основной медиатор нейротоксического действия МФТП in vivo, и ингибирование JNK может стать новой и эффективной стратегией лечения БП. Другим примером низкомолекулярных химических ингибиторов является вышеуказанный ингибитор JNK AS601245. AS601245 ингибирует путь передачи сигнала JNK и увеличивает выживание клеток после церебральной ишемии. In vivo AS601245 обеспечивает выраженное защитное действие в отношении отложенной потери СА 1-нейронов гиппокампа на модели кратковременной общей ишемии у песчанок. Это действие опосредуется ингибированием JNK и, как следствие, экспрессией и фосфорилированием c-Jun (см., например, Carboni et al., J. Pharmacol. Exp. Ther., 310(1), июль 2004 г., с. 25-32, Epub 26 февраля 2004 г.). К третьему классу ингибиторов пути передачи сигнала JNK относятся указанные выше пептидные ингибиторы взаимодействия между JNK и ее субстратами. В качестве отправной точки для конструирования таких пептидных ингибиторов JNK можно использовать сравнительных анализ первичной структуры встречающихся в естественных условиях белков JNK. Как правило, такие белки содержат JNKсвязывающие домены (JBD) и встречаются в различных инсулинсвязывающих (IB) белках, таких как IB1 или IB2. Примером такого сравнительного анализа первичной структуры последовательностей является,например, сравнительный анализ первичной структуры последовательностей JNK-связывающих доменовIB1 [SEQ ID NO: 13], IB2 [SEQ ID NO: 14], c-Jun [SEQ ID NO: 15] и ATF2 [SEQ ID NO: 16] (см., например, фиг. 1A-B). Такой сравнительный анализ первичной структуры последовательностей выявил частично консервативную состоящую из 8 аминокислот последовательность (см., например, фиг. 1 А). Сравнение JBD, встречающихся в IB1 и IB2, дополнительно подтвердило наличие двух высококонсервативных для двух последовательностей блоков, состоящих из 7 и 3 аминокислот. Последовательности, сконструированные на основе такого сравнительного анализа первичной структуры, описаны, например, в WO 01/27268. В частности, в WO 01/27268 описаны низкомолекулярные слитые пептиды, проникающие в клетку, которые содержат так называемую ответственную за обес-1 014330 печение проникновения в клетки последовательность ТАТ, которую получают из основной обладающей способностью обеспечивать перенос последовательности белка ВИЧ-TAT и состоящей как минимум из 20 аминокислот ингибирующей последовательности IB1. Оба компонента ковалентно связывают друг с другом. Примеры (в настоящее время единственные) ингибиторов пути передачи сигнала MAPK-JNK приведены в WO 01/27268, и они представляют собой, например, L-JNKI1 (JNK - пептид-ингибитор, состоящий из L-аминокислот) или устойчивые к протеазам D-JNKI1-пептиды (JNK - пептид-ингибитор,состоящий из ненативных D-аминокислот). Эти пептиды-ингибиторы JNK (JNKI) являются специфическими для JNK (JNK1, JNK2 и JNK3). В отличие от указанных низкомолекулярных ингибиторов ингибирующие последовательности, описанные в WO 01/27268, например JNKI1, более вероятно, ингибируют взаимосвязь между JNK и ее субстратом. С помощью его обеспечивающей перенос последовательности,выведенной из ТАТ, слитый пептид эффективно транспортируется в клетку. Благодаря новым свойствам,полученным с помощью обеспечивающего перенос компонента, слитые белки активно транспортируются в клетку, где они сохраняют эффективность вплоть до протеолитического расщепления. Однако пептиды, описанные в WO 01/27268, еще легко доступны для фосфорилаз (киназ). Любая аминокислота пептида служит мишенью для киназ и, следовательно, может подвергаться фосфорилированию, что представляет собой важный фактор инактивации таких пептидов. Таким образом, первым объектом настоящего изобретения являются новые ингибирующие последовательности пути передачи сигнала JNK, которые сохраняют функциональные свойства пептидов, описанных в WO 01/27268, но обладают повышенной стабильностью в отношении фосфорилаз (киназ). Кроме того, для получения ингибирующих последовательностей, описанных в WO 01/27268, требуются дорогостоящая стадия выделения и очистки, особенно при получении в крупных масштабах (например, при промышленном получении). Таким образом, вторым объектом настоящего изобретения являются ингибирующие последовательности, которые можно получать с помощью более простого и эффективного с позиций стоимости процесса получения и выделения по сравнению с известным к настоящему времени в данной области. Эти задачи решаются с помощью последовательности, ингибирующей JNK, состоящей менее чем из 150 аминокислот, где ингибирующая JNK последовательность содержит или состоит по меньшей мере из одной аминокислотной последовательности, представленной в SEQ ID NO: 1, 2, 3 или 4, или ее варианта, фрагмента или производного. Предпочтительно предлагаемая в изобретении последовательность, ингибирующая JNK, связывается с JNK и/или ингибирует активацию по меньшей мере одного активируемого JNK фактора транскрипции, например с-Jun, или ATF2 (см., например, SEQ ID NO: 15 и 16 соответственно), или Elk1. Как правило, предлагаемые в настоящем изобретении последовательности, ингибирующие JNK, содержат в целом менее 150 аминокислотных остатков, предпочтительно от 5 до 150 аминокислотных остатков, более предпочтительно от 10 до 100 аминокислотных остатков, еще более предпочтительно от 10 до 75 аминокислотных остатков и наиболее предпочтительно от 15 до 50 аминокислотных остатков. Предлагаемая в изобретении последовательность, ингибирующая JNK, предпочтительно содержит или состоит по меньшей мере из одной аминокислотной последовательности, представленной в SEQ IDNO: 1, 2, 3 или 4, или ее варианта, фрагмента или производного. Более предпочтительно предлагаемая в изобретении последовательность, ингибирующая JNK, может содержать 1, 2, 3, 4 или даже большее количество копий аминокислотной последовательности, представленной в SEQ ID NO: 1, 2, 3 или 4, или ее варианта, фрагмента или производного. При наличии более одной копии предлагаемые в изобретении аминокислотные последовательности, представленные в SEQ ID NO: 1, 2, 3 или 4, или их варианты,фрагменты или производные можно непосредственно связывать друг с другом без какой-либо линкерной последовательности или с помощью линкерной последовательности, которая содержит 1-10, предпочтительно 1-5 аминокислот. Аминокислоты, из которых состоит линкерная последовательность, предпочтительно выбирают из аминокислотных остатков глицина или пролина. Более предпочтительно указанные предлагаемые в изобретении аминокислотные последовательности, представленные в SEQ ID NO: 1, 2, 3 или 4, или ее варианты, фрагменты или производные можно отделять друг от друга с помощью шарнира,состоящего из 2, 3 или большего количества остатков пролина. Предлагаемые в изобретении последовательности, ингибирующие JNK, как они определены выше,могут состоять из L-аминокислот, D-аминокислот или их комбинации. Предпочтительно предлагаемые в изобретении последовательности, ингибирующие JNK, содержат по меньшей мере 1, предпочтительно по меньшей мере 3, более предпочтительно по меньшей мере 6 и еще более предпочтительно по меньшей мере 10 D- и/или L-аминокислот, где D- и/или L-аминокислоты могут входить в состав предлагаемых в изобретении последовательностей, ингибирующих JNK, в виде блока, не в виде блока или в виде другой структуры. Согласно одному из предпочтительных вариантов осуществления изобретения предлагаемые в изобретении последовательности, ингибирующие JNK, могут состоять только из L-аминокислот. Предлагаемые в изобретении последовательности, ингибирующие JNK, могут также содержать или состоять по меньшей мере из одной "нативной последовательности, ингибирующей JNK", которая представлена вSEQ ID NO: 1 или 3. В этом контексте понятие "нативная или нативная(ые) последовательность(и), инги-2 014330 бирующая(ие) JNK" относится к неизмененным предлагаемым в изобретении последовательностям, ингибирующим JNK, представленным в любой из SEQ ID NO: 1 или 3, которые полностью состоят из Lаминокислот. Таким образом, предлагаемая в изобретении последовательность, ингибирующая JNK, может содержать или состоять по меньшей мере из одной (нативной) аминокислотной последовательности NH2Xnb-Xna-RPTTLXLXXXXXXXQD-Xnb-COOH [SEQ ID NO: 3]. В контексте настоящего описания X каждый обозначает аминокислотный остаток, предпочтительно выбранный из любого (нативного) аминокислотного остатка. Xna обозначает один аминокислотный остаток, предпочтительно представляющий собой любой аминокислотный остаток кроме серина или треонина, где n обозначает 0 или 1. Кроме того,Xnb каждый может представлять собой любой аминокислотный остаток, где n обозначает число от 0 до 5,от 5 до 10, от 10 до 15, от 15 до 20, от 20 до 30 или более при условии, что если n обозначает 0 в Xna, тоXnb не должен нести серин или треонин на С-конце, для того чтобы избегать присутствия серина или треонина в этом положении. Предпочтительно Xnb представляет собой состоящий из смежных пептидных остатков фрагмент, выведенный из SEQ ID NO: 1 или 3. Более предпочтительно предлагаемая в изобретении последовательность, ингибирующая JNK, может дополнительно содержать или состоять по меньшей мере из однойNH2RPKRPTTLNLFPQVPRSQD-COOH [SEQ ID NO: 1]. Xna и Xnb обозначают либо D- или L-аминокислоты. Согласно другому предпочтительному варианту осуществления изобретения предлагаемые в изобретении последовательности, ингибирующие JNK, могут частично или полностью состоять из Dаминокислот. Более предпочтительно эти предлагаемые в изобретении последовательности, ингибирующие JNK, состоящие из D-аминокислот, представляют собой ненативные последовательности описанных выше (нативных) последовательностей, ингибирующих JNK, в D-ретроинвертированной форме. Понятие"ретроинвертированные последовательности" относится к изомерам линейной пептидной последовательности, в которой направление последовательности изменено на противоположное и хиральность каждого аминокислотного остатка является инвертированной (см., например, Jameson et al., Nature, 368,1994, с. 744-746; Brady et al., Nature, 368, 1994, с. 692-693). Преимущество объединения D-энантиомеров и обратного синтеза состоит в том, что положения карбонильных и аминогрупп в каждой амидной связи меняются местами, при этом положение групп боковой цепи на каждом альфа-атоме углерода сохраняется. Если специально не указано иное, предполагается, что любая предлагаемая в настоящем изобретенииL-аминокислотная последовательность или пептид могут быть превращены в D-ретроинвертированную последовательность или пептид путем синтеза обратных относительно нативной L-аминокислотной последовательности или пептида последовательности или пептида. Предлагаемые в изобретении D-ретроинвертированные последовательности, как они определены выше, обладают рядом ценных свойств. Например, предлагаемые в изобретении Dретроинвертированные последовательности более эффективно проникают в клетку по сравнению с Lаминокислотными последовательностями, предлагаемыми в настоящем изобретении, при этом предлагаемые в изобретении D-ретроинвертированные последовательности являются более стабильными по сравнению с соответствующими L-аминокислотными последовательностями. Таким образом, предлагаемые в изобретении последовательности, ингибирующие JNK, могут содержать или состоять по меньшей мере из одной D-ретроинвертированной последовательности, соответствующей аминокислотной последовательности NH2-Xnb-DQXXXXXXXLXLTTPR-Xna-Xnb-COOH [SEQID NO: 4]. В контексте настоящего описания X, Xna и Xnb имеют указанные выше значения (предпочтительно обозначают D-аминокислоты), где Xnb предпочтительно обозначает состоящий из смежных остатков фрагмент, полученный из SEQ ID NO: 2 или 4. Более предпочтительно предлагаемые в изобретении последовательности, ингибирующие JNK, могут содержать или состоять по меньшей мере из одной Dретроинвертированной последовательности, соответствующей аминокислотной последовательностиNH2-DQSRPVQPFLNLTTPRKPR-COOH [SEQ ID NO: 2]. Предлагаемые в изобретении последовательности, ингибирующие JNK, как они описаны выше,представлены в табл. 1 (SEQ ID NO: 1-4). В таблице представлены обозначения предлагаемых в изобретении последовательностей, ингибирующих JNK, а также идентификационный номер последовательности, их длина и аминокислотная последовательность. Для сравнения представлены также известные из прототипа последовательности, которые описаны в WO 01/27268 (SEQ ID NO: 17-26). Эти известные из прототипа последовательности не относятся к предлагаемым в изобретении последовательностям, ингибирующим JNK, или предлагаемым в изобретении химерным пептидам, и таким образом, они специально исключены из объема настоящего изобретения посредством письменного отказа от права на них. В табл. 1 примеры последовательностей, а также их общие формулы представлены в SEQ ID NO: 1,2, 5, 6, 9 и 11 и SEQ ID NO: 3, 4, 7, 8, 10 и 12 соответственно. Согласно следующему варианту осуществления изобретения предлагаемая в изобретении последовательность, ингибирующая JNK, содержит или состоит по меньшей мере из одного варианта, фрагмента и/или производного указанных выше предлагаемых в изобретении нативных или ненативных аминокислотных последовательностей, представленных в SEQ ID NO: 1, 2, 3 или 4. Предпочтительно эти варианты, фрагменты и/или производные сохраняют биологическую активность описанных выше предлагаемых в изобретении нативных или ненативных последовательностей, ингибирующих JNK, прежде всего нативных или ненативных аминокислотных последовательностей, представленных в SEQ ID NO: 1, 2, 3 или 4, т.е. связывающихся с JNK и/или ингибирующих активацию по меньшей мере одного активируемогоJNK фактора транскрипции, например, c-Jun, ATF2 или Elk1. Функциональную активность можно оценивать с помощью различных анализов, например, оценивая связывание пептида с его молекулоймишенью или с помощью биофизических методов, например, спектроскопии, компьютерного модулирования, структурного анализа и т.д. В частности, предлагаемая в изобретении последовательность, инги-4 014330 бирующая JNK, или ее варианты, фрагменты и/или производные можно изучать с помощью анализа гидрофильности (см., например, Норр и Woods, Proc. Natl. Acad. Sci. USA 78, 1981, c. 3824-3828), который можно использовать для идентификации гидрофобных и гидрофильных пептидов, что позволяет создавать субстраты для экспериментальных исследований, таких как эксперименты по связыванию, или для синтеза антител. Для идентификации областей (предлагаемой в изобретении) последовательности, ингибирующей JNK, или ее вариантов, фрагментов и/или производных можно осуществлять также анализ вторичной структуры, с помощью которого определяют специфические структурные мотивы (см., например, Chou и Fasman, Biochem 13, 1974, с. 222-223). Для манипуляции, трансляции, предсказания вторичной структуры, определения профилей гидрофильности и гидрофобности, предсказания и создания открытых рамок считывания и определения гомологии последовательности можно использовать компьютерные пакеты программ, существующие в данной области. Можно применять также другие методы структурного анализа, включающие, например, рентгеновскую кристаллографию (см., например, Engstrom, Biochem. Exp. Biol. 11, 1974, с. 7-13), масс-спектроскопию и газовую хроматографию (см., например, Methods in Protein Science, изд-во J. Wiley and Sons, New York, NY, 1997) и компьютерное моделирование (см., например, Computer Graphics and Molecular Modeling, в: Current Communications in Molecular Biology, под ред. Fletterick и Zoller, изд-во Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY,1986). Таким образом, предлагаемая в изобретении последовательность, ингибирующая JNK, может содержать или состоять по меньшей мере из одного варианта (нативных или ненативных) аминокислотных последовательностей, представленных в SEQ ID NO: 1, 2, 3 или 4. В контексте настоящего описания понятие "вариант (нативной или ненативной) аминокислотной последовательности, представленной в SEQID NO: 1, 2, 3 или 4" предпочтительно обозначает последовательность, выведенную из любой из последовательностей, представленных в SEQ ID NO: 1, 2, 3 или 4, где вариант содержит изменения аминокислот в аминокислотных последовательностях, представленных в SEQ ID NO: 1, 2, 3 или 4. Такие изменения, как правило, представляют собой 1-20, предпочтительно 1-10 и более предпочтительно 1-5 замен,добавлений и/или делеций аминокислот в последовательностях, представленных в SEQ ID NO: 1, 2, 3 или 4, где последовательность варианта идентична любой из последовательностей, представленных вSEQ ID NO: 1, 2, 3 или 4, по меньшей мере примерно на 30, 50, 70, 80, 90, 95, 98 или даже 99%. Если предлагаемые в изобретении варианты (нативных или ненативных) аминокислотных последовательностей, представленных в SEQ ID NO: 1, 2, 3 или 4, получают путем замены конкретных аминокислот, такие замены предпочтительно представляют собой консервативные аминокислотные замены. Консервативные аминокислотные замены могут включать синонимические аминокислотные остатки внутри группы, которые обладают в достаточной степени сходными физико-химическими свойствами,такие как замены на остатки из рассматриваемой группы, что должно сохранять биологическую активность молекулы (см., например, Grantham R., Science 185, 1974, с. 862-864). Специалистам в данной области должно быть очевидно, что аминокислоты можно встраивать и/или изымать путем делеции из указанных выше последовательностей без изменения их функции, в частности, если инсерции и/или делеции затрагивают лишь несколько аминокислот, например менее 20 и предпочтительно менее 10, и что недопустимо удалять или заменять аминокислоты, которые имеют решающее значение для функциональной активности. Кроме того, из вариантов, предлагаемых в изобретении, следует исключать замены, которые приводят к появлению дополнительных остатков треонина в положениях аминокислотной последовательности, которые доступны для фосфорилазы, предпочтительно киназы, для того, чтобы избегать инактивации предлагаемой в изобретении последовательности-ингибитора JNK или предлагаемого в изобретении химерного пептида in vivo или in vitro. Предпочтительно синонимические аминокислотные остатки представляют собой остатки, которые отнесены к одной и той же группе и которые, как правило, можно заменять друг на друга путем консервативных аминокислотных замен, такие группы приведены в табл. 2.-5 014330 Таблица 2 Предпочтительные группы синонимических аминокислотных остатков Конкретной формой варианта SEQ ID NO: 1, 2, 3 или 4, предлагаемого в изобретении, является фрагмент предлагаемых в изобретении (нативных или ненативных) аминокислотных последовательностей, представленных в SEQ ID NO: 1, 2, 3 или 4, который, как правило, изменяют путем по меньшей мере одной делеции по сравнению с SEQ ID NO: 1, 2, 3 или 4. Предпочтительно фрагмент содержит по меньшей мере 4 смежных аминокислоты любой из SEQ ID NO: 1, 2, 3 или 4, эта длина, как правило, является достаточной для специфического распознавания эпитопа любой из этих последовательностей. Еще более предпочтительно фрагмент содержит от 4 до 18, от 4 до 15 или наиболее предпочтительно от 4 до 10 смежных аминокислот любой из SEQ ID NO: 1, 2, 3 или 4. Делецию аминокислот можно осуществлять в любом положении SEQ ID NO: 1, 2, 3 или 4, предпочтительно на N- или С-конце. В частности,в пептидах,имеющих последовательность,представленную в(DQSRPVQPFLNLTTPRKPR), можно изымать путем делеции на N- и/или С-конце 1, 2, 3, 4 или 5 аминокислот. Другими словами, предпочтительные укороченные версии SEQ ID NO: 2 могут иметь последовательностьQSRPVQPFLNLTTPRKP,SRPVQPFLNLTTPRKP,SRPVQPFLNLTTPRK,RPVQPFLNLTTPRKP,RPVQPFLNLTTPRK или PVQPFLNLTTP. Чем короче применяемая пептидная последовательность,предлагаемая в изобретении, тем меньше ее неспецифическая клеточная токсичность. В определенных вариантах осуществления изобретения применяемые пептиды должны иметь максимально возможную короткую последовательность, которая еще сохраняет свою биологическую функцию, например, способность ингибировать JNK. Кроме того, предлагаемый в изобретении фрагмент (нативных или ненативных) аминокислотных последовательностей, представленных в SEQ ID NO: 1, 2, 3 или 4, можно обозначать как последовательность, которая идентична любой из последовательностей, представленных в SEQ ID NO: 1, 2, 3 или 4, по меньшей мере примерно на 30, 50, 70, 80, 90, 95, 98 или даже на 99%. Предлагаемые в изобретении последовательности, ингибирующие JNK, могут также содержать или состоять по меньшей мере из одного производного (нативных или ненативных) аминокислотных последовательностей, представленных в SEQ ID NO: 1, 2, 3 или 4. В этом контексте понятие "производное (нативных или ненативных) аминокислотных последовательностей, представленных в SEQ ID NO: 1, 2, 3 или 4", предпочтительно обозначает аминокислотную последовательность, выведенную из любой из последовательностей, представленных в SEQ ID NO: 1, 2, 3 или 4, где производное содержит по меньшей мере одну модифицированную L- или D-аминокислоту (образованную из не встречающихся(ейся) в естественных условиях аминокислот(ы, предпочтительно от 1 до 20, более предпочтительно от 1 до 10 и еще более предпочтительно от 1 до 5 модифицированных L- или D-аминокислот. Производные вариантов или фрагментов также подпадают под объем настоящего изобретения. Любой из представленных в настоящем описании пептидов может иметь концевую модификацию либо на С-, либо на N-конце, либо на обоих концах. С-конец предпочтительно можно модифицировать с помощью амидной модификации, а N-конец можно модифицировать с помощью любой приемлемойNH2-защитной группы, например, путем ацилирования. В этом контексте понятие "модифицированная аминокислота" может означать любую аминокислоту, измененную, например, в результате различного гликозилирования в различных организмах, фосфо-6 014330 рилирования или мечения конкретными аминокислотами. Такую метку, как правило, выбирают из группы меток, включающей:(I) радиоактивные метки, т.е. радиоактивное фосфорилирование или радиоактивную метку, представляющую собой радиоактивную серу, водород, углерод, азот и т.д.;(V) группы для иммобилизации на твердой фазе (например, His-метка, биотин, strep-метка, flagметка, антитела, антиген и т.д.); и(VI) комбинацию меток из двух или большего количество меток, указанных в подпунктах (I)-(V). В контексте вышесказанного под аминокислотной последовательностью, "последовательность которой идентична" по меньшей мере, например, на 95% рассматриваемой аминокислотной последовательности, предлагаемой в настоящем изобретении, понимают последовательность представленной аминокислотной последовательности, которая идентична рассматриваемой последовательности за исключением того, что представленная аминокислотная последовательность может нести вплоть до 5 аминокислотных изменений на каждые 100 аминокислот рассматриваемой аминокислотной последовательности. Другими словами для получения аминокислотной последовательности, последовательность которой по меньшей мере на 95% идентична рассматриваемой аминокислотной последовательности, вплоть до 5%(5 из 100) аминокислотных остатков в представленной последовательности можно встраивать или заменять другими аминокислотами или изымать путем делеции. Для последовательностей, не имеющих точного соответствия друг другу, можно определять "% идентичности" первой последовательности относительно второй последовательности. В целом, метод заключается в том, что эти две подлежащие сравнению последовательности подвергают сравнительному анализу, добиваясь максимальной корреляции между последовательностями. Эта процедура может включать встраивание "брешей" в одну или обе последовательности для повышения эффективности сравнительного анализа первичной структуры последовательностей. Далее % идентичности можно определять по всей длине каждой подлежащей сравнению последовательности (так называемый глобальный сравнительный анализ первичной структуры), что является наиболее приемлемым для последовательностей, имеющих одинаковую или близкую длину, или для более коротких участков определенной длины(так называемый локальный сравнительный анализ первичной структуры), что является наиболее приемлемым для последовательностей, имеющих разную длину. Методы сравнения идентичности и гомологии двух или большего количества последовательностей хорошо известны в данной области. Так, например, для определения % идентичности двух полинуклеотидов и % идентичности и % гомологии двух полипептидных последовательностей можно использовать пакет программ анализа последовательностей группы компьютерной генетики Биотехнологического центра Университета Висконсина (Wisconsin Sequence Analysis Package, версия 9.1. Devereux et al., Nucleic Acids Res. 12, 1984, c. 387-395), например, программы BESTFIT и GAP. В программе BESTFIT использован алгоритм "локальной гомологии" (Smith и Waterman, J. Mol. Biol. 147, 1981, c. 195-197), и она позволяет находить одну характеризующуюся наиболее высоким уровнем сходства область в двух последовательностях. В данной области известны также другие программы, предназначенные для определения идентичности и/или сходства между последовательностями, например, семейство программBLAST (Altschul et al., J. Mol. Biol. 215, 1990, c. 403-410), которые доступны на домашней странице NCBI на сайте в сети Интернет ncbi.nlm.nih.gov и FASTA (Pearson, Methods Enzymol. 183, 1990, c. 63-98; Pearson и Lipman, Proc. Natl. Acad. Sci. USA 85, 1988, c. 2444-2448). Последовательности-ингибиторы JNK, предлагаемые в настоящем изобретении, можно получать или создавать с помощью методов, хорошо известных в данной области, например, путем химического синтеза или генной инженерии, как будет описано ниже. Например, пептид, соответствующий части предлагаемой в изобретении последовательности, ингибирующей JNK, которая включает требуемую область указанной последовательности, ингибирующей JNK, или которая опосредует требуемую активность in vitro или in vivo, можно синтезировать с помощью пептидного синтезатора. Следующим объектом настоящего изобретения является химерный пептид, включающий по меньшей мере один первый домен и по меньшей мере один второй домен, где первый домен содержит транспортирующую последовательность, а второй домен содержит указанную выше предлагаемую в изобретении последовательность, ингибирующую JNK. Как правило, химерные пептиды, предлагаемые в настоящем изобретении, состоят по меньшей мере из 25 аминокислотных остатков, например от 25 до 250 аминокислотных остатков, более предпочтительно от 25 до 200 аминокислотных остатков, еще более предпочтительно от 25 до 150 аминокислотных остатков, от 25 до 100 аминокислотных остатков и наиболее предпочтительно от 25 до 50 аминокислотных остатков. Первый домен предлагаемого в изобретении химерного пептида предпочтительно содержит транспортирующую последовательность, которую, как правило, выбирают из любой последовательности аминокислот, обеспечивающей перенос пептида (в котором она присутствует) к требуемой области клеточ-7 014330 ной локализации. Так, согласно настоящему изобретению транспортирующая последовательность, как правило, обеспечивает перенос пептида через плазматическую мембрану, например, из области, расположенной вне клетки, через плазматическую мембрану в цитоплазму. В другом или дополнительном варианте транспортирующая последовательность может обеспечивать перенос пептида к требуемой области внутри клетки, например ядру, рибосоме, эндоплазматическому ретикулуму (ЭР), лизосоме или пероксисоме, например, путем объединения двух компонентов (например, компонента, ответственного за клеточную проницаемость, и компонента, ответственного за ядерную локализацию) или с помощью одного компонента, который обладает, например, способностью осуществлять транспорт через клеточную мембрану и обеспечивать направленный перенос, например, внутриядерный транспорт. Транспортирующая последовательность может содержать также другой компонент, который обладает способностью связываться с цитоплазматическим компонентом или любым другим компонентом или компартментом клетки (например, с эндоплазматическим ретикулумом, митохондриями, аппаратом с неизвестной функцией (gloom apparatus), лизосомными пузырьками). Таким образом, транспортирующая последовательность первого домена и последовательность, ингибирующая JNK, второго домена может, например, быть локализована в цитоплазме или любом другом компартменте клетки. Это позволяет определять локализацию химерного пептида в клетке после его проникновения. Предпочтительно аминокислотная последовательность транспортирующей последовательности(входящая в состав первого домена химерного пептида, предлагаемого в изобретении) состоит из 5-150 аминокислот, более предпочтительно из 5-100 и наиболее предпочтительно из 5-50, 5-30 или даже 5-15 аминокислот. Более предпочтительно транспортирующая последовательность (входящая в состав первого домена химерного пептида, предлагаемого в изобретении) может представлять собой состоящий из смежных аминокислот фрагмент в первом домене. В другом варианте транспортирующая последовательность в первом домене может быть разделена на два или нескольких фрагментов, где все эти фрагменты представляют собой полную транспортирующую последовательность и могут быть отделены друг от друга 110, предпочтительно 1-5 аминокислотами при условии, что такая транспортирующая последовательность сохраняет свои описанные выше свойства носителя. Аминокислоты, разделяющие фрагменты транспортирующей последовательности, можно, например, выбирать из аминокислотных последовательностей,отличных от аминокислот транспортирующей последовательности. В другом варианте первый домен может содержать транспортирующую последовательность, которая состоит более чем из одного компонента, каждый компонент со своей собственной функцией в отношении транспорта своего "груза", т.е. последовательности, ингибирующей JNK, которая находится во втором домене, например, в определенный клеточный компартмент. Транспортирующая последовательность, как она определена выше, может состоять из Lаминокислот, D-аминокислот или их комбинации. Предпочтительно предлагаемые в изобретении транспортирующие последовательности содержат по меньшей мере 1, предпочтительно по меньшей мере 3,более предпочтительно по меньшей мере 6 и еще более предпочтительно по меньшей мере 10 Lаминокислот и/или D-аминокислот, где D- и/или L-аминокислоты могут входить в состав предлагаемых в изобретении последовательностей, ингибирующих JNK, в виде блока или не в виде блока, или в виде другой структуры. Согласно одному из альтернативных вариантов осуществления изобретения транспортирующая последовательность предлагаемого в изобретении химерного пептида может состоять исключительно из Lаминокислот. Более предпочтительно транспортирующая последовательность предлагаемого в изобретении химерного пептида содержит или состоит по меньшей мере из одной "нативной" транспортирующей последовательности, как она определена выше. В этом контексте понятие "нативный" относится к неизмененным транспортирующим последовательностям, полностью состоящим из L-аминокислот. Согласно другому альтернативному варианту осуществления изобретения транспортирующая последовательность предлагаемого в изобретении химерного пептида может состоять исключительно из Dаминокислот. Более предпочтительно транспортирующая последовательность предлагаемого в изобретении химерного пептида может содержать D-ретроинвертированный пептид описанных выше последовательностей. Транспортирующую последовательность первого домена химерного пептида, предлагаемого в изобретении, можно получать из встречающихся в естественных условиях источников или можно получать с помощью методов генной инженерии или химическим синтезом (см., например, Sambrook, J., Fritsch,E.F., Maniatis, Т., Molecular cloning: A laboratory manual. 2-е изд., 1989, изд-во Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.). Источниками транспортирующей последовательности первого домена, которые можно применять,являются, например, нативные белки, такие, например, как белок ТАТ (например, описанный вUS 5804604 и 5674980, каждый из которых включен в настоящее описание в качестве ссылки), VP22(Jackson et al., Proc. Natl. Acad. Sci. USA 89, 1992, с. 10691-10695), транспортирующие последовательности, полученные из представителей Antennapedia (например, последовательность-носитель из Anten-8 014330napedia) или из основных пептидов, например, пептидов, состоящих из 5-15 аминокислот, предпочтительно 10-12 аминокислот и содержащих по меньшей мере 80%, более предпочтительно 85% или даже 90% основных аминокислот, таких, например, как аргинин, лизин и/или гистидин. Кроме того, в качестве указанных в настоящем описании транспортирующих последовательностей применяют варианты, фрагменты и производные одного из нативных белков. Касательно вариантов, фрагментов и производных можно использовать определения, которые указаны выше для последовательностей, ингибирующих JNK. Варианты, фрагменты, а также производные соответствуют изложенным выше определениям последовательностей, ингибирующих JNK. В частности, в контексте транспортирующей последовательности вариант или фрагмент, или производное можно определять как последовательность, идентичная последовательности нативных белков, применяемых в качестве указанных выше транспортирующих последовательностей, по меньшей мере примерно на 30, 50, 70, 80, 90, 95, 98 или даже 99%. В предпочтительном варианте предлагаемого в изобретении химерного пептида транспортирующая последовательность первого домена содержит или состоит из последовательности, полученной из белка ТАТ человеческого вируса иммунодефицита (ВИЧ)1, в частности содержит некоторые или все 86 аминокислот, из которых состоит белок ТАТ. Для предлагаемой в изобретении транспортирующей последовательности можно использовать частичные последовательности полноразмерного белка ТАТ, получая функционально эффективный фрагмент белка ТАТ, т.е. пептид ТАТ, который включает область, опосредующую проникновение и поглощение клетками. Представляет ли такая последовательность функционально эффективный фрагмент белка ТАТ, можно определять с помощью известных методов (см., например, Franked et al., Proc. Natl.Acad. Sci, USA 86, 1989, с. 7397-7401). Так, транспортирующую последовательность первого домена предлагаемого в изобретении химерного пептида можно получать из функционально эффективного фрагмента или части последовательности белка ТАТ, которые содержат менее 86 аминокислот и которые поглощаются клетками и необязательно поглощаются ядрами клеток. Более предпочтительно предполагается, что частичные последовательности (фрагменты) ТАТ, которые можно использовать в качестве носителя для опосредования проникновения химерного пептида через клеточную мембрану, должны содержать основную часть (аминокислоты 48-57 или 49-57) полноразмерного ТАТ. Согласно более предпочтительному варианту осуществления изобретения предлагаемая в изобретении транспортирующая последовательность может содержать или состоять из аминокислотной последовательности, которая содержит остатки ТАТ 48-57 или 49-57 и наиболее предпочтительно общую последовательность ТАТ NH2-Xnb-RKKRRQRRR-Xnb-COOH [SEQ ID NO: 7], где Xnb имеет указанные выше значения. В другом варианте предлагаемая в изобретении транспортирующая последовательность может содержать или состоять из пептида, который содержит, например, аминокислотную последовательностьNH2-GRKKRRQRRR-COOH [SEQ ID NO: 5]. Согласно другому более предпочтительному варианту осуществления изобретения транспортирующая последовательность, предлагаемая в изобретении, может содержать D-ретроинвертированный пептид, который имеет указанные выше последовательности, т.е. D-ретроинвертированную последовательность общей последовательности ТАТ, имеющей последовательность NH2-Xnb-RRRQRRKKR-XnbCOOH [SEQ ID NO: 8]. И в этом случае также Xnb имеет указанные выше значения (предпочтительно обозначает D-аминокислоты). Кроме того, количество остатков "Xnb" в любой из SEQ ID NO: 7-8 не ограничено указанным и может варьироваться, как указано выше. Наиболее предпочтительно предлагаемая в изобретении транспортирующая последовательность может содержать D-ретроинвертированную последовательность NH2-RRRQRRKKRG-COOH [SEQ ID NO: 6]. Согласно другому варианту осуществления изобретения транспортирующая последовательность,предлагаемая в изобретении, может содержать или состоять из вариантов транспортирующих последовательностей, как они определены выше. "Вариант транспортирующей последовательности" предпочтительно обозначает последовательность, полученную из указанной выше транспортирующей последовательности, где вариант имеет модификацию, например, добавление, (внутреннюю) делецию (что приводит к образованию фрагмента) и/или замену по меньшей мере одной аминокислоты, присутствующей в транспортирующей последовательности, как она определена выше. Такая(ие) модификация(и), как правило, представляет(ют) собой 1-20, предпочтительно 1-10 и более предпочтительно 1-5 замен, добавлений и/или делеций аминокислот. Кроме того, для варианта предпочтительно характерна идентичность последовательности транспортирующей последовательности, как она определена выше, более предпочтительно любой из последовательностей, представленных в SEQ ID NO: 5-8, по меньшей мере примерно на 30, 50, 70, 80, 90, 95, 98 или даже 99%. Предпочтительно такая модификация транспортирующей последовательности приводит к получению транспортирующей последовательности с повышенной или пониженной стабильностью. Альтернативно этому варианты транспортирующей последовательности можно создавать для модуляции внутриклеточной локализации химерного пептида, предлагаемого в изобретении. При экзогенном введении указанных выше вариантов их, как правило, создают так, чтобы сохранялась способность транспортирующей последовательности проникать в клетки (т.е. поглощение клеткой варианта транспортирующей последовательности практически такое же, как у нативного белка применяемой транспортирующей по-9 014330 следовательности). Например, изменение основной области, которое, как предполагается, является важным для ядерной локализации (см., например, Dang и Lee, J. Biol. Chem. 264, 1989, с. 18019-18023; Hauber et al., J. Virol. 63, 1989, с. 1181-1187; Ruben et al., J. Virol. 63, 1989, с. 1-8), может приводить к цитоплазматической локализации или частичной цитоплазматической локализации транспортирующей последовательности, и, как следствие, последовательности, ингибирующей JNK, как компонента предлагаемого в изобретении химерного пептида. Помимо вышеуказанного, в вариант можно интродуцировать другие модификации, например, путем связи, например, холестерина или других липидных фрагментов,с транспортирующей последовательностью с получением транспортирующей последовательности, которая обладает повышенной растворимостью в мембране. Любые из вышеуказанных вариантов транспортирующих последовательностей, предлагаемых в изобретении, можно получать с помощью методов, которые, как правило, известны специалистам в данной области (см., например, Sambrook J., Fritsch E.F.,Maniatis Т., Molecular cloning: A laboratory manual. 2-е изд., 1989, изд-во Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.) Второй домен химерного пептида, предлагаемого в изобретении, как правило, содержит предлагаемую в изобретении последовательность, ингибирующую JNK, выбранную из любой из предлагаемых в изобретении последовательностей, ингибирующих JNK, как они определены выше, включая варианты,фрагменты и/или производные этих предлагаемых в изобретении последовательностей, ингибирующихJNK. Оба домена, т.е. первый и второй домены предлагаемого в изобретении химерного пептида, могут быть связаны с получением функциональной единицы. Можно применять любой метод связывания первого и второго домена, известный в данной области. Согласно одному из вариантов осуществления изобретения первый и второй домены химерного пептида, предлагаемого в изобретении, предпочтительно связывают с помощью ковалентной связи. В контексте настоящего описания ковалентная связь может представлять собой, например, пептидную связь, которую можно получать путем экспрессии предлагаемого в изобретении химерного белка в виде слитого белка. Слитые белки в контексте настоящего описания можно получать и применять путями,аналогичными или легко получаемыми на основе стандартных методов рекомбинантной ДНК, которые описаны ниже. Однако оба домена можно также связывать через боковые цепи или их можно связывать с помощью фрагмента, представляющего собой химический линкер. Первый и/или второй домены предлагаемого в изобретении химерного пептида могут присутствовать в предлагаемом в изобретении химерном пептиде в виде одной или нескольких копий. Если оба домена присутствуют в виде одной копии, то первый домен можно связывать либо с N-концом, либо с Сконцом второго домена. При наличии нескольких копий первый и второй домены могут располагаться в любом возможном порядке. Например, первый домен может присутствовать в предлагаемом в изобретении химерном пептиде в виде нескольких копий, например в двух, трех или большем числе копий, которые предпочтительно расположены последовательно, а второй домен может присутствовать в виде одной копии на N- или С-конце последовательности, содержащей первый домен. В альтернативном варианте второй домен может присутствовать в виде нескольких копий, например, в двух, трех или большем числе копий, а первый домен может присутствовать в виде одной копии. В обоих вариантах первый и второй домены могут находиться в любом месте и могут располагаться последовательно. Ниже представлены примеры порядка расположения: например, первый домен - первый домен - первый домен - второй домен; первый домен - первый домен - второй домен - первый домен; первый домен - второй домен - первый домен - первый домен или, например, второй домен - первый домен - первый домен - первый домен. Как должно быть очевидно специалисту в данной области, эти примеры даны только с целью иллюстрации и не ограничивают объем изобретения. Таким образом, число копий и их расположение может варьироваться, как указано ранее. Предпочтительно первый и второй домены можно связывать друг с другом непосредственно без какого-либо линкера. В другом варианте их можно связывать друг с другом через линкерную последовательность, содержащую от 1 до 10, предпочтительно от 1 до 5 аминокислот. Аминокислоты, входящие в линкерную последовательность, предпочтительно выбирают из глицина или пролина в качестве аминокислотных остатков. Более предпочтительно первый и второй домены можно отделять друг от друга с помощью шарнира, состоящего из двух, трех или большего количества остатков пролина, между первым и вторым доменами. Описанный выше химерный пептид, предлагаемый в изобретении, который содержит по меньшей мере один первый и по меньшей мере один второй домен, может состоять из L-аминокислот, Dаминокислот или их комбинации. Кроме того, каждый домен (а также используемые линкеры) могут состоять из L-аминокислот, D-аминокислот или их комбинации (например, D-TAT и L-IB1(s) или L-TAT иD-IB1(s) и т.д.). Предпочтительно предлагаемый в изобретении химерный пептид содержит по меньшей мере 1, предпочтительно по меньшей мере 3, более предпочтительно по меньшей мере 6 и еще более предпочтительно по меньшей мере 10 L-аминокислот и/или D-аминокислот, где D- и/или Lаминокислоты могут располагаться в химерном пептиде, предлагаемом в изобретении, в виде блока или не в виде блока или в виде другой структуры.- 10014330 Согласно конкретному варианту осуществления изобретения химерный пептид, предлагаемый в изобретении, содержит или состоит из L-аминокислотных химерных пептидов, имеющих общую формулу L-TAT-IB-пептида [NH2-Xnb-RKKRRQRRR-Xnb-Xna-RPTTLXLXXXXXXXQD-Xnb-СООН, SEQ ID NO: 10], где X, Xna и Xnb предпочтительно имеют указанные выше значения. Более предпочтительно химерный пептид, предлагаемый в изобретении, содержит или состоит из L-аминокислотного химерного пептида L-TAT-IB1 [NH2-GRKKRRQRRRPPRPKRPTTLNLFPQVPRSQD-COOH, SEQ ID NO: 9]. Согласно другому конкретному варианту осуществления изобретения химерный пептид, предлагаемый в изобретении, содержит или состоит из D-аминокислотных химерных пептидов описанных выше L-аминокислотных химерных пептидов. Примерами D-ретроинвертированных химерных пептидов,предлагаемых в настоящем изобретении, например, являются пептиды D-TAT-IB общей формулы [NH2Xnb-DQXXXXXXXLXLTTPR-Xna-Xnb-RRRQRRKKR-Xnb-СООН, SEQ ID NO: 12]. При этом X, Xna и Xnb предпочтительно имеют указанные выше значения (предпочтительно обозначают D-аминокислоты). Более предпочтительно химерный пептид, предлагаемый в изобретении, содержит или состоит из Dаминокислотных химерных пептидов, описываемых формулой пептида TAT-IB1 [NH2DQSRPVQPFLNLTTPRKPRPPRRRQRRKKRG-COOH, SEQ ID NO: 11]. Однако связывающая область первого и второго домена (вместо РР) может содержать -Xna-Xnb-, которые имеют указанные выше значения. В частности, второй(ые) домен(ы), который(е) имеет(ют) последовательность, представленную вSEQ ID NO: 11, в том числе содержащую -Xna-Xnb- вместо РР, могут иметь делецию на их N- и/или Сконце 1, 2, 3, 4 или 5 аминокислот. В другом предпочтительном варианте осуществления изобретения первый домен, который имеет последовательность, представленную в SEQ ID NO: 11, может иметь делецию на его N- и/или С-конце 1 или 2 аминокислот. Эту(и) делецию(и) можно применять в сочетании с делецией(ями), описанной(ыми) для аминокислотных остатков на концах второго домена. Таким образом, предпочтительными вариантами SEQ ID NO: 11 являются, например, следующие последовательности с делециями второго домена на N- и/или С-конце(ах): Предпочтительными вариантами осуществления изобретения, в которых делеции затрагивают только конец(ы) первого домена, являются следующие последовательности: Примерами последовательностей, полученных в результате комбинаций делеций первого и второго домена, являются: И в этом случае, более короткие пептиды обладаю более низкой (неспецифической) токсичностью для клеток. Однако пептиды должны сохранять их биологическую функцию, т.е. способность проникать через клеточную мембрану (первый домен) и способность ингибировать JNK (второй домен). Первый и второй домены предлагаемого в изобретении химерного пептида, как он определен выше,можно связывать друг с другом с помощью химического или биохимического сшивания, которое осуществляют любым приемлемым путем, известным в данной области, например, создавая пептидную связь между первым и вторым доменами, например, путем экспрессии первого и второго домена в виде слитого белка, или, например, путем перекрестного сшивания первого и второго домена предлагаемого в изобретении химерного пептида. Многие известные методы химического перекрестного сшивания являются неспецифическими, т.е. они не направляют точку сшивания в конкретный сайт транспортного полипептида или транспортируемой макромолекулы. В результате, неспецифические перекрестносшивающие агенты в случае их использования могут "атаковать" функциональные сайты или стерически блокировать активные сайты, приводя к тому, что слитые белки становятся биологически неактивными. Таким образом, предпочтительно применять такие методы введения перекрестных сшивок, которые позволяют осуществлять более специфическое сшивание первого и второго домена. Специфичность сочетания можно повышать путем непосредственного химического сочетания с функциональной группой, встречающейся только один раз или лишь небольшое количество раз в одном- 13014330 или обоих первых и вторых доменах, которые подлежат перекрестному сшиванию. Примером является цистеин, который представляет собой единственную аминокислоту, содержащую тиольную группу, и который во многих белках встречается лишь небольшое количество раз. Также, например, если полипептид не содержит остатков лизина, то перекрестносшивающий реагент, обладающий специфичностью в отношении первичных аминов, будет обладать избирательным действием в отношении аминоконца указанного полипептида. Однако для успешного применения этого подхода с целью повышения специфичности сочетания необходимо, чтобы полипептид имел достаточно редко встречающиеся и реакционноспособные остатки в областях молекулы, которые можно изменять без потери биологической активности молекулы. Остатки цистеина можно заменять, если они встречаются в таких частях последовательности полипептида, где в противном случае их участие в реакции перекрестного сшивания может влиять на биологическую активность. Если остаток цистеина заменяют, то, как правило, желательно минимизировать образующиеся изменения складчатости полипептида. Изменения складчатости полипептида минимальны, когда замена является химически и стерически близкой цистеину. Таким образом, для замены цистеина предпочтительно использовать серин. Как продемонстрировано ниже в примерах, остаток цистеина можно интродуцировать в аминокислотную последовательность полипептида с целью осуществления перекрестного сшивания. При интродукции остатка цистеина предпочтительной является интродукция на амино- или карбоксильный конец или вблизи них. В настоящее время доступны приемлемые методы таких модификаций аминокислотных последовательностей, при которых представляющий интерес полипептид получают путем химического синтеза или посредством экспрессии рекомбинантной ДНК. Сочетание первого и второго доменов можно осуществлять посредством связующего или конъюгирующего агента. Существует несколько агентов для межмолекулярного перекрестного сшивания, которые можно использовать (см., например, Means и Feeney, Chemical Modification of Proteins, изд-во Holden-Day, 1974, c. 39-43). К этим реагентам относятся, например, N-сукцинимидил-3-(2 пиридилдитио)пропионат (СПДП) или N,N'-(1,3-фенилен)бис-малеимид (оба обладают высокой специфичностью в отношении сульфгидрильных групп и образуют необратимые связи); N,N'-этилен-бис(йодацетамид) или другой аналогичный реагент, имеющий 6-11 углеродных метиленовых мостиков (которые обладают относительной специфичностью в отношении сульфгидрильных групп); и 1,5-дифтор 2,4-динитробензол (образующий необратимые связи между аминогруппами и тирозиновыми группами). К другим перекрестносшивающим реагентам, пригодным для этой цели, относятся: n,n'-дифтор-м,м'динитрофенилсульфон, который образует необратимые связи с аминогруппами и фенольными группами; диметиладипимидат (который является специфическим для аминогрупп); фенол-1,4-дисульфонилхлорид(который вступает в реакцию главным образом с аминогруппами); гексаметилендиизоцианат или диизотиоцианат, или азофенил-n-диизоцианат (который вступает в реакцию главным образом с аминогруппами); глутаровый альдегид (который вступает в реакцию с несколькими различными боковыми цепями) и дисдиазобензидин (который вступает в реакцию прежде всего с тирозином и гистидином). Перекрестносшивающие реагенты могут быть гомобифункциональными, т.е. иметь две функциональные группы, участвующие в одной и той же реакции. Предпочтительным гомобифункциональным перекрестносшивающим реагентом является бисмалеимидогексан (БМГ). БМГ содержит две малеимидные функциональные группы, которые специфически взаимодействуют с содержащими сульфгидрил соединениями в мягких условиях (рН 6,5-7,7). Две малеимидные группы соединены углеводородной цепью. Таким образом, БМГ можно применять для необратимого перекрестного сшивания полипептидов,которые содержат остатки цистеина. Перекрестносшивающие реагенты могут быть также гетеробифункциональными. Гетробифункциональные перекрестносшивающие реагенты имеют две различные функциональные группы, например реактивную аминогруппу и реактивную тиольную группу, которые должны перекрестно сшивать два белка, имеющие свободные амины и тиолы соответственно. Примерами гетеробифункциональных перекрестносшивающих агентов являются сукцинимидил-4(N-малеимидометил)циклогексан-1-карбоксилат(СМЦК),сложный м-малеимидобензоил-Nгидроксисукцинимидиловый эфир (МБС) и сукцинимид-4-(n-малеимидофенил)бутират (СМФБ), представляющий собой аналог МБС с удлиненной цепью. Сукцинимидильная группа этих перекрестносшивающих линкеров взаимодействует с первичным амином и реактивная тиольная группа малеимида образует ковалентную связь с тиольной группой остатка цистеина. Перекрестносшивающие реагенты часто обладают низкой растворимостью в воде. К перекрестносшивающему реагенту можно добавлять гидрофильный фрагмент, такой как сульфонатная группа, для повышения его растворимости в воде. В этой связи сульфо-МБС и сульфо-СМЦК являются примерами перекрестносшивающих реагентов, модифицированных с целью повышения растворимости в воде, которые можно применять согласно настоящему изобретению. Многие перекрестносшивающие реагенты приводят к образованию конъюгата, который практически не расщепляется в условиях клеточного окружения. Поэтому некоторые перекрестносшивающие реагенты содержат ковалентную связь, такую как дисульфид, которая расщепляется в условиях клеточ- 14014330 ного окружения. Например, реагент Траута, дитио-бис-(сукцинимидилпропионат) (ДСП) и Nсукцинимидил-3-(2-пиридилдитио)пропионат (СПДП) представляют собой широко известные расщепляемые перекрестносшивающие линкеры. Использование расщепляемого перекрестносшивающего реагента позволяет транспортируемому фрагменту отделяться от транспортного полипептида после введения в клетку-мишень. Для этой цели можно применять также прямую дисульфидную связь. В настоящее время поступают в продажу многочисленные перекрестносшивающие агенты, включая описанные выше. Подробные инструкции по их применению легко можно получить от коммерческих поставщиков. Общие сведения о перекрестном сшивании белков и получении конъюгатов приведены уWong, Chemistry of Protein Conjugation and Cross-Linking, изд-во CRC Press (1991). Химическое перекрестное сшивание можно осуществлять также с использованием спейсерных плечей. Спейсерные плечи обеспечивают внутримолекулярную гибкость или регулируют внутримолекулярные расстояния между конъюгированными фрагментами и тем самым могут помогать сохранять биологическую активность. Спейсерное плечо может представлять собой фрагмент полипептида, который содержит спейсерные аминокислоты, например, пролин. В альтернативном варианте спейсерное плечо может представлять собой часть перекрестносшивающего реагента, такого как "СПДП с длинной цепью"(фирма Pierce Chem. Co., Рокфорд, шт. Иллинойс, каталожный номер 21651 Н). Кроме того, настоящее изобретение относится также к вариантам, фрагментам или производным одного из описанных выше химерных пептидов. Применительно к фрагментам и вариантам, как правило,используют определение, приведенное выше для последовательности, ингибирующей JNK. В частности, в контексте настоящего изобретения понятие "вариант химерного пептида" предпочтительно обозначает последовательность, выведенную из любой из последовательностей, представленных в SEQ ID NO: 9-12, где химерный вариант несет изменения аминокислотных последовательностей предлагаемых в изобретении химерных пептидов, представленных в SEQ ID NO: 9-12. Такие изменения,как правило, представляют собой 1-20, предпочтительно 1-10 и более предпочтительно 1-5 замен, добавлений и/или делеций (приводящих к образованию фрагментов) аминокислот, представленных в SEQ IDNO: 9-12, где последовательность измененного предлагаемого в изобретении химерного пептида идентична любой из последовательностей, представленных в SEQ ID NO: 9, 10, 11 или 12, по меньшей мере примерно на 30, 50, 70, 80 или 95, 98 или даже 99%. Предпочтительно указанные варианты сохраняют биологическую активность первого и второго домена, присущую предлагаемому в изобретении химерному пептиду, т.е. указанную выше транспортирующую активность первого домена и активность второго домена в отношении связывания JNK и/или ингибирования активации по меньшей мере одного активируемого JNK фактора транскрипции. Таким образом, предлагаемый в изобретении химерный пептид представляет собой также фрагменты описанных выше предлагаемых в изобретении химерных пептидов, прежде всего последовательности предлагаемых в изобретении химерных пептидов, которые представлены в SEQ ID NO: 9, 10, 11 или 12. Таким образом, в контексте настоящего изобретения понятие "фрагмент предлагаемого в изобретении химерного пептида" предпочтительно обозначает последовательность, выведенную из любой из последовательностей, представленных в SEQ ID NO: 9, 10, 11 или 12, где фрагмент содержит по меньшей мере 4 смежные аминокислоты любой из последовательностей, представленных в SEQ ID NO: 9, 10, 11 или 12. Указанный фрагмент предпочтительно имеет длину, достаточную для специфического распознавания эпитопа любой из указанных последовательностей и для транспорта последовательности в клетки, ядра или другую предпочтительную область. Еще более предпочтительно фрагмент содержит от 4 до 18, от 4 до 15 или наиболее предпочтительно от 4 до 10 смежных аминокислот любой из последовательностей,представленных в SEQ ID NO: 9, 10, 11 или 12. Фрагменты предлагаемого в изобретении химерного пептида можно определять также как последовательность, идентичную любой из последовательностей,представленных в SEQ ID NO: 9, 10, 11 или 12, по меньшей мере примерно на 30, 50, 70, 80 или 95, 98 или даже 99%. И, наконец, предлагаемый в изобретении химерный пептид представляет собой также производные описанных выше предлагаемых в изобретении химерных пептидов, прежде всего предлагаемых в изобретении химерных пептидов, последовательности которых представлены в SEQ ID NO: 9, 10, 11 или 12. Настоящее изобретение относится также к нуклеотидным последовательностям, которые кодируют предлагаемые в изобретении последовательности, ингибирующие JNK, предлагаемые в изобретении химерные пептиды или их фрагменты, варианты или производные, как они определены выше. Предпочтительную пригодную нуклеиновую кислоту, которая кодирует предлагаемую в изобретении последовательность, ингибирующую JNK, выбирают из нуклеиновой кислоты человеческого IB1 (GenBank, регистрационныйAF074091), нуклеиновой кислоты крысиного IB1 (GenBank, регистрационныйAF 108959) или человеческого IB2 (GenBank, регистрационныйAF218778). Нуклеиновые кислоты, которые кодируют предлагаемые в изобретении последовательности, ингибирующие JNK, или химерные пептиды можно получать с помощью любого метода, известного в данной области (например, с помощью ПЦР-амплификации с использованием синтетических праймеров, которые гибридизуются с 3'- и 5'-концами последовательности, и/или путем клонирования ДНК или геномной библиотеки с использованием олигонуклеотидных последовательностей, специфических для данной- 15014330 генной последовательности). Кроме того, представленные в настоящем описании нуклеотидные последовательности представляют собой последовательности, которые гибридизуются также в строгих условиях с соответствующей цепью, кодирующей (нативную) предлагаемую в изобретении последовательность, ингибирующую JNK,или химерный пептид, как они определены выше. Предпочтительно указанные нуклеотидные последовательности содержат по меньшей мере 6 (смежных) нуклеиновых кислот, которые имеют длину, достаточную для осуществления специфической гибридизации. Более предпочтительно такие нуклеотидные последовательности содержат от 6 до 38, еще более предпочтительно от 6 до 30 и наиболее предпочтительно от 6 до 20 или от 6 до 10 (смежных) нуклеиновых кислот."Строгие условия" зависят от последовательности и отличаются в различных ситуациях. Как правило, строгие условия можно выбирать из условий, включающих применение температуры примерно на 5 С более низкой, чем температура плавления (Тпл) конкретной последовательности при определенной ионной силе и значении рН. Тпл представляет собой температуру (при определенной ионной силе и значения рН), при которой 50% последовательности-мишени гибридизуется с полностью совместимым зондом. Как правило, строгие условия представляют собой условия, при которых концентрация соли составляет по меньшей мере примерно 0,02 М при рН 7 и температура составляет по меньшей мере примерно 60 С. Поскольку другие факторы могут влиять на строгость гибридизации (в том числе среди прочего состав оснований и размер комплементарных цепей), присутствие органических растворителей и уровень ошибочного спаривания оснований, то комбинация параметров является более важной, чем абсолютный уровень любого из них."Очень строгие условия" могут представлять собой, например, следующие условия. Стадия 1: содержащие ДНК фильтры предварительно обрабатывают в течение промежутка времени от 8 ч до всей ночи при 65 С в буфере, содержащем 6SSC, 50 мМ Трис-HCl (рН 7,5), 1 мМ ЭДТК, 0,02% ПВП, 0,02% Фиколла, 0,02% БСА и 500 мкг/мл денатурированной ДНК спермы лосося. Стадия 2: фильтры гибридизуют в течение 48 ч при 65 С в вышеуказанной смеси для предварительной гибридизации, в которую добавляют 100 мг/мл денатурированной ДНК спермы лосося и меченный с помощью 32 Р зонд (5-20106 cpm (импульсы в минуту. Стадия 3: фильтры отмывают в течение 1 ч при 37 С в растворе, содержащем 2SSC, 0,01% ПВП,0,01% Фиколла и 0,01% БСА. Затем осуществляют отмывку в 0,1SSC при 50 С в течение 45 мин. Стадия 4: фильтры оценивают с помощью авторадиографии. Можно использовать другие очень строгие условия, известные в данной области (см., например, Current Protocols in Molecular Biology, под ред. Ausubel et al., изд-во John Wiley and Sons, NY, 1993; и Kriegler, 1990, Gene Transfer and Expression, a"Условия умеренной строгости" могут представлять собой следующие условия. Стадия 1: содержащие ДНК фильтры предварительно обрабатывают в течение 6 ч при 55 С в растворе, содержащем 6SSC, 5 раствор Денхардта, 0,5% ДСН и 100 мг/мл денатурированной ДНК спермы лосося. Стадия 2: фильтры гибридизуют в течение 18-20 ч при 55 С в таком же растворе, в который добавляют меченный с помощью 32 Р зонд (5-20106 cpm). Стадия 3: фильтры отмывают при 37 С в течение 1 ч в растворе, содержащем 2SSC, 0,1% ДСН, затем отмывают дважды в течение 30 мин при 60 С в растворе, содержащем 1SSC и 0,1% ДСН. Стадия 4: фильтры промокают досуха и экспонируют для авторадиографии. Можно использовать другие условия умеренной строгости, известные в данной области (см., например, Current Protocols in Molecular Biology, под ред. Ausubel et al., изд-во John Wiley and Sons, NY,1993; и Kriegler, Gene Transfer and Expression, a Laboratory Manual, изд-во Stockton Press, NY, 1990). И, наконец, "расслабленные условия " могут представлять собой следующее. Стадия 1: содержащие ДНК фильтры предварительно обрабатывают в течение 6 ч при 40 С в растворе, содержащем 35% формамида, 5SSC, 50 мМ Трис-HCl (рН 7,5), 5 мМ ЭДТК, 0,1% ПВП, 0,1% Фиколла, 1% БСА и 500 мкг/мл денатурированной ДНК спермы лосося. Стадия 2: фильтры гибридизуют в течение 18-20 ч при 40 С в таком же растворе, в который добавляют 0,02% ПВП, 0,02% Фиколла, 0,2% БСА, 100 мкг/мл ДНК спермы лосося, 10% мас./об. сульфата декстрана и меченный с помощью 32 Р зонд (5-20106 cpm). Стадия 3: фильтры отмывают в течение 1,5 ч при 55 С в растворе, содержащем 2SSC, 25 мМ ТрисHCl (рН 7,4), 5 мМ ЭДТК и 0,1 % ДСН. Раствор для отмывки заменяют свежим раствором и инкубируют еще в течение 1,5 ч при 60 С. Стадия 4: фильтры промокают досуха и экспонируют для авторадиографии. При необходимости фильтры отмывают в третий раз при 65-68 С и повторно экспонируют на пленку. Другие расслабленные условия, которые можно применять, хорошо известны в данной области (например, представляют собой условия, который применяют для межвидовой гибридизации) (см., например, Current Protocols in Molecular Biology, под ред. Ausubel et al., изд-во John Wiley and Sons, NY, 1993; и Kriegler, Gene Transfer and Expression, a Laboratory Manual, изд-во Stockton Press, NY, 1990).- 16014330 Нуклеотидные последовательности, предлагаемые в настоящем изобретении, можно применять для экспрессии предлагаемых в изобретении пептидов, т.е. предлагаемой в изобретении последовательности,ингибирующей JNK, предлагаемого в изобретении химерного пептида, с целью анализа, характеризации и терапевтического применения; в качестве маркеров для тканей, в которых соответствующие (предлагаемые в изобретении) пептиды предпочтительно экспрессируются (либо конститутивно, либо на определенной стадии дифференцировки или развития ткани, либо в состояниях заболевания). Другие варианты применения нуклеиновых кислот включают, например, применение в качестве маркеров молекулярной массы в основанном на использовании гель-электрофореза в анализе нуклеиновых кислот. Другим вариантом осуществления настоящего изобретения являются также экспрессионные векторы, предназначенные для рекомбинантной экспрессии одной или нескольких предлагаемых в изобретении последовательностей, ингибирующих JNK, и/или химерных пептидов, как они определены выше. Понятие "экспрессионный вектор" в контексте настоящего описания относится либо к кольцевой, либо линейной ДНК или РНК, которая являться либо двухцепочечной, либо одноцепочечной. Он содержит также по меньшей мере одну предлагаемую в изобретении нуклеиновую кислоту для трансформации клетки-хозяина или одноклеточного или многоклеточного организма-хозяина. Предлагаемый в изобретении экспрессионный вектор предпочтительно содержит предлагаемую в изобретении нуклеиновую кислоту, которая кодирует предлагаемую в изобретении последовательность, ингибирующую JNK, или ее фрагмент, или вариант либо предлагаемый в изобретении химерный пептид, или его фрагмент, или вариант. Кроме того, экспрессионный вектор, предлагаемый в настоящем изобретении, предпочтительно содержит соответствующие элементы, предназначенные для поддержания экспрессии, включая различные регуляторные элементы, такие как энхансеры/промоторы вирусов, бактерий, растений, млекопитающих и других эукариотических организмов, которые обеспечивают экспрессию встраиваемого полинуклеотида в клетке-хозяине, такие как изоляторы, связывающие элементы, LCR (например, описанные у Blackwood и Kadonaga, Science 281, 1988, c. 61-63) или области прикрепления к матрице/носителю (например, описанные у Li, Harju и Peterson, Trends Genet. 15, 1999, c. 403-408). В некоторых вариантах осуществления изобретения регуляторные элементы являются гетерологичными (т.е. ненативный промотор гена). В других вариантах необходимые сигналы транскрипции и трансляции могут обеспечиваться нативным для генов и/или фланкирующих областей промотором. Понятие "промотор" в контексте настоящего описания относится к области ДНК, функциями которой является контроль транскрипции одной или нескольких предлагаемых в изобретении нуклеотидных последовательностей, который структурно идентифицируют по присутствию сайта связывания ДНКзависимой РНК-полимеразы и других последовательностей ДНК, которые связаны с регуляторной промоторной функцией. Функциональный усиливающий экспрессию фрагмент промотора представляет собой укороченную или процессированную промоторную последовательность, которая сохраняет активность в качестве промотора. Активность промотора можно оценивать с помощью любого анализа, известного в данной области (см., например, Wood, de Wet, Dewji и DeLuca, Biochem Biophys. Res. Commun. 124, 1984, c. 592-596; Seliger и McElroy, Arch. Biochem. Biophys. 88, 1960, c. 136-141) или набора,поступающего в продажу под названием Promega. Понятие "энхансерная область" в контексте настоящего описания касательно предлагаемого в изобретении экспрессионного вектора, как правило, относится к области ДНК, функцией которой является повышение транскрипции одного или нескольких генов. Более конкретно понятие "энхансер" в контексте настоящего описания означает регуляторный элемент ДНК, который усиливает, увеличивает, усовершенствует или улучшает экспрессию гена вне зависимости от его локализации и ориентации относительно гена, подлежащего экспрессии, и который может усиливать, увеличивать, совершенствовать или улучшать экспрессию более чем одного промотора. В качестве промоторных/энхансерных последовательностей, как они определены выше касательно предлагаемого в изобретении экспрессионного вектора, можно применять регуляторные последовательности растений, животных, насекомых или грибов. Например, можно использовать промоторные/энхансерные элементы из дрожжей и других грибов (например, промотор GAL4, промотор алкогольдегидрогеназы, промотор фосфоглицеринкиназы, промотор щелочной фосфатазы). В другом или дополнительном варианте они могут включать контролирующие транскрипцию области животного происхождения, например, (I) контролирующую область гена инсулина из панкреатических -клеток (см., например, Hanahan et al., Nature 315, 1985, с. 115-122); (II) контролирующую область гена иммуноглобулина,активную в лимфоидных клетках (см., например, Grosschedl et al., Cell 38, 1984, с. 647-658); (III) контролирующую область гена альбумина, активную в печени (см., например, Pinckert et al., Genes and Dev 1,1987, с. 268-276); (IV) контролирующую область гена основного белка миелина, активную в олигодендроцитах головного мозга (см., например, Readhead et al., 1 Cell 48, 1987, с. 703-712); и (V) контролирующую область гена гонадотропин-рилизинг гормона, активную в гипоталамусе (см., например, Mason etal., Science 234, 1986, с. 1372-1378) и т.п. Кроме того, предлагаемый в изобретении экспрессионный вектор может содержать маркер амплификации. Указанный маркер амплификации можно выбирать из группы, включающей, например, адено- 17014330 зиндеаминазу (ADA), дигидрофолатредуктазу (DHFR), ген, обусловливающий устойчивость ко многим лекарствам (MDR), орнитиндекарбоксилазу (ODC) и ген, обусловливающий устойчивость к N(фосфонацетил)-L-аспартату (CAD). Амплификация генов, кодирующих вышеуказанные белки, т.е. представляющие интерес белки (ПИБ), и/или слитого белка, предлагаемого в изобретении, позволяет повышать уровень экспрессии этих белков при интеграции вектора в клетку (Kaufman et al., Mol. CellBiol. 5, 1985, с. 1750-1759). Примерами экспрессионных векторов или их производных, которые можно применять в настоящем изобретении, являются, в частности, например, вирусы человека или животных (например, вирус коровьей оспы или аденовирус); вирусы насекомых (например, бакуловирусы); векторы на основе дрожжей; векторы на основе бактериофагов (например, фага лямбда); плазмидные векторы и космидные векторы. В настоящем изобретении предложены также различные системы хозяин-вектор, которые можно применять для экспрессии предлагаемых в изобретении нуклеиновых кислот, которые кодируют последовательности пептидов, как они определены выше. К ним относятся (но не ограничиваясь ими): (I) системы на основе клеток млекопитающих, которые заражают вирусом коровьей оспы, аденовирусом и т.п.;(II) системы на основе клеток насекомых, зараженных бакуловирусом и т.п.; (III) дрожжи, содержащие векторы на основе дрожжей, или (IV) бактерии, трансформированные бактериофагом, ДНК, плазмидной ДНК или космидной ДНК. В зависимости от применяемой системы хозяин-вектор можно использовать любые из многочисленных пригодных транскрипционных и трансляционных элементов. Предпочтительно можно выбирать линию клеток-хозяев, пригодную для такой системы хозяинвектор, которая модулирует экспрессию встроенных представляющих интерес последовательностей или модифицирует или процессирует экспрессируемые пептиды, кодируемые последовательностями, конкретным требуемым образом. Кроме того, экспрессию в выбранной линии-хозяине под контролем определенных промоторов можно усиливать в присутствии определенных индукторов; облегчая тем самым контроль экспрессии созданного с помощью генной инженерии пептида. Кроме того, в различных клетках-хозяевах экспрессируемый пептид может подвергаться характерным и специфическим механизмам трансляционного и посттрансляционного процессинга и модификации (например, гликозилированию,фосфорилированию и т.п.). Таким образом, можно выбирать соответствующие линии клеток или систем хозяев для гарантии достижения требуемой модификации и процессинга чужеродного пептида. Например, экспрессию пептида в бактериальной системе можно использовать для получения негликозилированного корового пептида; в то время как экспрессия в клетках млекопитающих обеспечивает "нативное" гликозилирование гетерологичного пептида. Настоящее изобретение относится также к антителам к предлагаемым в изобретении последовательностям, ингибирующим JNK, и/или предлагаемым в изобретении химерным пептидам. Кроме того,предложены эффективные способы получения антител, специфических в отношении последовательностей, ингибирующих JNK, предлагаемых в настоящем изобретении, или предлагаемых в изобретении химерных пептидов, которые содержат такую ингибирующую последовательность. Согласно изобретению последовательности, ингибирующие JNK, и/или предлагаемые в изобретении химерные пептиды, а также их фрагменты, варианты или производные можно использовать в качестве иммуногенов для производства антител, которые иммуноспецифически связываются с указанными пептидными компонентами. Такие антитела включают, например, поликлональные, моноклональные,химерные, одноцепочечные антитела, Fab-фрагменты и экспрессионные библиотеки Fab-фрагментов. Конкретным вариантом осуществления настоящего изобретения являются антитела к предлагаемым в изобретении химерным пептидам или к последовательностям, ингибирующим JNK, как они определены выше. В данной области известны различные методики, которые можно применять для получения указанных предлагаемых в изобретении антител. Например, различных животных-хозяев можно иммунизировать для получения поликлональных антител путем инъекции любого предлагаемого в изобретении химерного пептида или последовательности, ингибирующей JNK, как они определены выше. При этом в терапии можно использовать различные адъюванты, повышая тем самым иммунологический ответ, к которым относятся (но не ограничиваясь ими) адъювант Фрейнда (полный и неполный), минеральные гели (например, гидроксид алюминия), поверхностно-активные вещества (например, лизолецитин, плюроновые полиолы, полианионы, пептиды,масляные эмульсии, динитрофенол и т.д.), CpG, полимеры, Плюроники и применяемые для человека адъюванта, такие как бацилла Кальмета-Герена и Corynebacterium parvum. Для получения моноклональных антител к предлагаемому в изобретении химерному пептиду или последовательности, ингибирующей JNK, как они определены выше, можно использовать любые методики, которые обеспечивают получение молекул антител в непрерывной культуре клеточной линии. Такие методики представляют собой (но не ограничиваясь ими) метод на основе гибридом (см. Kohler иMilstein, Nature 256, 1975, с. 495-497); метод на основе триом; метод на основе В-клеточных гибридом(см. Kozbor et al., Immunol Today 4, 1983, с. 72) и метод на основе EBV-(вирус Эпштейна-Барра)гибридомы, которые предназначены для получения человеческих моноклональных антител (см. Cole etal., 1985. В: Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, Inc., c. 77-96). Человеческие моноклональные антитела можно применять для воплощения на практике настоящего изобретения и их мож- 18014330 но получать с помощью человеческих гибридом (см. Cote et al., Proc. Natl. Acad. Sci. USA 80, 1983, c. 2026-2030) или путем трансформации человеческих В-клеток вирусом Эпштейна-Барра in vitro (см. Coleet al., Monoclonal Antibodies and Cancer Therapy, изд-во Alan R. Liss, Inc. 1985, c. 77-96). Согласно изобретению методики можно адаптировать для получения одноцепочечных антител,специфических в отношении предлагаемых в изобретении последовательностей, ингибирующих JNK,и/или предлагаемых в изобретении химерных пептидов (см., например, US 4946778). Кроме того, можно адаптировать методы конструирования экспрессионных библиотек Fab-фрагментов (см., например, Huseet al., Science 246, 1989, с. 1275-1281), позволяющие быстро и эффективно идентифицировать моноклональные Fab-фрагменты с требуемой специфичностью в отношении указанных предлагаемых в изобретении последовательностей, ингибирующих JNK, и/или предлагаемых в изобретении химерных пептидов, как они определены выше. Антитела из организмов кроме человека можно "гуманизировать" с помощью методик, хорошо известных в данной области (см., например, US 5225539). Фрагменты антител,содержащие идиотипы к последовательностям, ингибирующим JNK, и/или предлагаемому в изобретении химерному пептиду, которые можно получать с помощью известных методик, включают, например, (I)F(ab')2-фрагмент, получаемый путем расщепления пепсином молекулы антитела; (II) Fab-фрагмент, создаваемый восстановлением пептидных мостиков F(ab')2-фрагмента; (III) Fab-фрагмент, создаваемый обработкой молекулы антитела папаином и восстановителем, и (IV) Fv-фрагменты. В одном из вариантов осуществления настоящего изобретения используют методы скрининга предлагаемых в изобретении антител, обладающие требуемой специфичностью, к которым относятся (но не ограничиваясь ими) твердофазный иммуноферментный анализ (ELISA) и другие иммунологические анализы, известные в данной области. В конкретном варианте осуществления изобретения селекция антител, специфических в отношении конкретного эпитопа предлагаемой в изобретении последовательности,ингибирующей JNK, и/или предлагаемого в изобретении химерного пептида (например, их фрагмента,состоящего, как правило, из 5-20, предпочтительно 8-18 и наиболее предпочтительно 8-11 аминокислот),облегчается созданием гибридом, которые связываются с фрагментом предлагаемой в изобретении последовательности, ингибирующей JNK, и/или предлагаемого в изобретении химерного пептида, которые несут указанный эпитоп. Под объем изобретения подпадают также указанные антитела, специфические в отношении эпитопа, как они описаны выше. Предлагаемые в изобретении антитела можно применять в методах, известных в данной области,для локализации и количественной оценки предлагаемой в изобретении последовательности, ингибирующей JNK (и/или соответственно химерного пептида, предлагаемого в изобретении), например, для оценки уровней пептида в соответствующих физиологических образцах, для применения в диагностических методах или для применения для визуализации пептида и т.п. Предлагаемые в изобретении последовательности, ингибирующие JNK, химерные пептиды и/или нуклеиновые кислоты, предлагаемые в изобретении, можно приготавливать в виде фармацевтических композиций, которые также подпадают под объем изобретения. Эти композиции могут содержать помимо одной из указанных субстанций фармацевтически приемлемый эксципиент, носитель, буфер, стабилизатор или другие материалы, хорошо известные специалистам в данной области. Такие материалы должны быть нетоксичными и не должны влиять на эффективность действующего вещества. Точная природа носителя или другого материала может зависеть от пути введения, например орального, внутривенного, кожного или подкожного, назального, внутримышечного, внутрибрюшинного пути или нанесения с помощью бляшки. Фармацевтические композиции для орального введения могут иметь форму таблетки, капсулы, порошка или жидкости. Таблетка может включать твердый носитель, такой как желатин или адъювант. Жидкие фармацевтические композиции, как правило, включают жидкий носитель, такой как вода, вазелин, животные или растительные масла, минеральное масло или синтетическое масло. В их состав могут входить физиологический соляной раствор, декстроза или другой раствор сахарида или гликоли, такие как этиленгликоль, пропиленгликоль или полиэтиленгликоль. Для внутривенной, кожной или подкожной инъекции или инъекции в область поражения действующее вещество должно находиться в форме приемлемого для парентерального введения водного раствора, который не содержит пирогены и имеет приемлемые значение рН, изотоничность и стабильность. Специалисты в данной области легко могут приготавливать приемлемые растворы с использованием,например, изотонических наполнителей, таких как хлорид натрия для инъекций, раствор Рингера для инъекций, лактированный раствор Рингера для инъекций. При необходимости можно включать консерванты, стабилизаторы, буферы, антиоксиданты и/или другие добавки. Вне зависимости от того, применяют ли полипептид, пептид или молекулу нуклеиновой кислоты, другое фармацевтически приемлемое соединение, предлагаемое в настоящем изобретении, которое вводят индивидууму, введение предпочтительно осуществлять в "профилактически эффективном количестве" или в "терапевтически эффективном количестве" (в качестве одного из возможных вариантов), которое является достаточным для оказания благоприятного действия на индивидуума. Фактическое вводимое количество, скорость и временные особенности введения должны зависеть от природы и серьезности состояния, подлежащего лечению. Решение о схеме лечение, например, касающееся доз и т.д., находится в компетенции обычных- 19014330 практикующих врачей и других лечащих врачей и, как правило, учитывает нарушение, подлежащее лечению, состояние каждого пациента, место введения и метод введения и другие факторы, известные практикующим специалистам в данной области. Примеры методов и упомянутых протоколов приведены в справочнике Remington's Pharmaceutical Sciences, 16-ое изд., под ред. Osol A., 1980. В другом варианте прицельную терапию можно использовать для введения предлагаемых в изобретении последовательностей, ингибирующих JNK, химерных пептидов и нуклеиновых кислот, предлагаемых в изобретении, более специфично в определенный тип клеток с помощью обеспечивающих направленный перенос систем, таких как антитело (против клетки-мишени) или специфические для клетки лиганды. Антитела против клетки-мишени, как правило, являются специфическими для поверхностных клеточных белков клеток, ассоциированных с любым из указанных ниже заболеваний. Например, эти антитела могут представлять собой антитела против антител клеточной поверхности, таких, например,как ассоциированные с поверхностью В-клеток белки, такие как белок DR ГКГ класса II, CD 18 (бетацепь LFA-1), CD45RO, CD40 или Bgp95, или белков клеточной поверхности, выбранных, например, изCD2, CD3, CD4, CD5, CD7, CD8, CD9, CD10, CD13, CD16, CD19, CD20, CD21, CD22, CD23, CD24,CD25, CD30, CD33, CD34, CD38, CD39, CD4, CD43, CD45, CD52, CD56, CD68, CD71, CD138 и т.д. Обеспечивающие направленный перенос конструкции, как правило, можно получать путем ковалентного связывания предлагаемых в изобретении последовательностей, ингибирующих JNK, химерных пептидов и нуклеиновых кислот с антителом, специфическим для белка клеточной поверхности, или путем связывания со специфическим для клетки лигандом. Белки, например, можно связывать с таким антителом или присоединять к нему с помощью пептидной связи или путем химического сочетания, перекрестного сшивания и т.д. Затем можно осуществлять прицельную терапию путем введения пациенту обеспечивающей направленный перенос конструкции в фармацевтически эффективном количестве с помощью указанных ниже путей введения, например внутрибрюшинного, назального, внутривенного, орального,или с помощью бляшек. Предпочтительно предлагаемые в изобретении последовательности, ингибирующие JNK, химерные пептиды или нуклеиновые кислоты, предлагаемые в изобретении, присоединенные к антителам против клеток-мишеней или специфическим для клеток лигандам, как они определены выше, можно высвобождать in vitro или in vivo, например, в результате гидролиза ковалентной связи, под действием пептидаз или с помощью любого другого приемлемого метода. В другом варианте, если последовательности, ингибирующие JNK, химерные пептиды или нуклеиновые кислоты, предлагаемые в изобретении, присоединяют к небольшому специфическому для клетки лиганду, то можно не осуществлять высвобождение лиганда. При нахождении на клеточной поверхности предлагаемые в изобретении химерные пептиды могут проникать в клетку благодаря активности входящей в их состав транспортирующей последовательности. Направленный перенос может требоваться по разным причинам; например,если предлагаемые в изобретении последовательности, ингибирующие JNK, химерные пептиды и нуклеиновые кислоты, предлагаемые в изобретении, обладают неприемлемой токсичностью, или если существуют другие показания к применению слишком высокой дозы. Вместо непосредственного введения предлагаемых в изобретении последовательностей, ингибирующих JNK, и/или химерных пептидов, предлагаемых в изобретении, их можно получать в клеткахмишениях путем экспрессии кодирующего гена, интродуцированного в клетки, например, путем введения вирусного вектора. Вирусный вектор, как правило, кодирует предлагаемые в изобретении последовательности, ингибирующие JNK, и/или химерные пептиды, предлагаемые в изобретении. Можно обеспечивать направленный перенос вектора к специфическим клеткам, подлежащим лечению. Кроме того,вектор может содержать регуляторные элементы, которые "включаются" более или менее избирательно клетками-мишенями при определенной регуляции. Эта методика является вариантом VDEPT-метода (virus-directed enzyme prodrug therapy) (опосредуемая вирусом терапия с использованием пролекарства на основе фермента), в которой применяют зрелые белки вместо их предшественников. В другом варианте предлагаемые в изобретении последовательности, ингибирующие JNK, и/или химерные пептиды можно вводить в форме предшественников с помощью антитела или вируса. Предлагаемые в изобретении последовательности, ингибирующие JNK, и/или химерные пептиды можно затем превращать в активную форму с помощью активирующего агента, который продуцируется в клетках,подлежащих лечению, или направленно переносится к ним. Такой тип лечения иногда называют ADEPT(antibody-directed enzyme prodrug therapy) (опосредуемая антителом терапия с использованием пролекарства на основе фермента) или VDEPT (опосредуемая вирусом терапия с использованием пролекарства на основе фермента); в первом случае обеспечивают направленный перенос активирующего агента в клетки путем конъюгации со специфическим для клетки антителом, а в последнем случае получают активирующий агент, например последовательность, ингибирующую JNK, или химерный пептид, в векторе путем экспрессии кодирующей ДНК в вирусном векторе (см., например, ЕР-А-415731 и WO 90/07936). Настоящее изобретение относится также к применению предлагаемых в изобретении последовательностей, ингибирующих JNK, предлагаемых в изобретении химерных пептидов и/или предлагаемых в изобретении нуклеотидных последовательностей для приготовления фармацевтических композиций,например, как они определены выше, предназначенных для предупреждения и/или лечения связанных с пролиферацией клеток нарушений, ассоциированных с активацией JNK у индивидуума ("ассоциирован- 20014330 ное с JNK нарушение"). Как правило, такая фармацевтическая композиция, применяемая согласно настоящему изобретению, содержит в качестве активного компонента, например: (I) любую(ые) одну или несколько предлагаемую(ых) в изобретении последовательность(ей), ингибирующую(их) JNK, и/или предлагаемые в изобретении химерные пептиды и/или их варианты, фрагменты или производные; и/или(II) нуклеиновые кислоты, кодирующие предлагаемую в изобретении последовательность, ингибирующую JNK, и/или предлагаемый в изобретении химерный пептид и/или их варианты или фрагменты,и/или (III) клетки, содержащие любую(ые) одну или несколько предлагаемую(ых) в изобретении последовательность(ей), ингибирующую(их) JNK, и/или предлагаемые в изобретении химерные пептиды и/или их варианты, фрагменты или производные, и/или (IV) клетки, трансфектированные вектором и/или нуклеиновыми кислотами, которые кодируют предлагаемую в изобретении последовательность, ингибирующую JNK, и/или предлагаемый в изобретении химерный пептид и/или их варианты или фрагменты. Предупреждение и/или лечение согласно настоящему изобретению, как правило, предусматривает введение предлагаемой в изобретении фармацевтической композиции, как она определена выше. Понятие "модулировать" относится к подавлению экспрессии JNK в случае ее сверхэкспрессии. Оно относится также к подавлению фосфорилирования c-jun, ATF2 или NFAT4, например, с помощью пептида, представленного в любой одной или в нескольких SEQ ID NO: 1-4 и/или 9-12, в качестве конкурентного ингибитора встречающихся в естественных условиях в клетке сайтов связывания c-jun, ATF2 и NFAT4. Понятие "модулировать" относится также к подавлению гетеро- и гомомерных комплексов факторов транскрипции c-jun, ATF2 или NFAT4 и родственных партнеров, таких, например, как АР-1-комплекс,включающий c-jun, AFT2 и c-fos. Когда связанное с пролиферацией клеток заболевание ассоциировано со сверхэкспрессией JNK, такие супрессорные последовательности, ингибирующие JNK, можно интродуцировать в клетку. В некоторых случаях понятие "модулировать" может включать повышение уровня экспрессии JNK, например, при использовании специфического для IB-пептида антитела, которое блокирует связывание IB-пептида с JNK, предупреждая тем самым ингибирование JNK родственным IB пептидом. Профилактику и/или лечение индивидуума с помощью предлагаемой в изобретении фармацевтической композиции, как она описана выше, можно, как правило, осуществлять путем введения (in vivo) индивидууму фармацевтической композиции в определенном ("терапевтически эффективном") количестве, где индивидуум может представлять собой, например, любое млекопитающее, например, человека,примата, мышь, крысу, собаку, кошку, корову, лошадь или свинью. Понятие "терапевтически эффективный" означает, что количество активного компонента фармацевтической композиции является достаточным для облегчения ассоциированного с JNK нарушения. Понятие "связанное с пролиферацией клеток нарушение" или "ассоциированное с JNK нарушение" в контексте настоящего описания, как правило, означает популяции злокачественных, а также доброкачественных клеток in vivo и in vitro, которые часто отличаются морфологически и функционально от клеток окружающей ткани и которые, как правило, характеризуются аномальными уровнями JNK. Понятие "аномальный уровень JNK" подразумевает повышенный или пониженный уровень JNK в какой-либо области индивидуума, подлежащего лечению, относительно уровня, присутствующего в аналогичной непораженной области организма пациента, у которого отсутствует нарушение. Например, предлагаемые в изобретении фармацевтические композиции можно применять для предупреждения и/или лечения злокачественных заболеваний систем различных органов, в которых часто выявляется активация JNK, например легкого, молочной железы, лимфатической системы, желудочнокишечного тракта и мочеполового пути, а также аденокарцином, включая злокачественные, такие как большинство видов рака ободочной кишки, почечно-клеточная карцинома, рак предстательной железы,немелкоклеточный рак легкого, рак тонкого кишечника и рак пищевода. Нарушения включают также лейкозы, заболевания или патофизиологические состояния, ассоциированные с онкогенной трансформацией, а также виды рака, связанные с онкогенной трансформацией Bcr-Abl, для чего точно требуется активация JNK. Предлагаемые в изобретении фармацевтические композиции можно применять также для предупреждения и/или лечения доброкачественных или иммунологических связанных с пролиферацией клеток заболеваний, таких как псориаз, обыкновенная пузырчатка, синдром Бехчета, острый респираторный дистресс-синдром (ARDS), ишемическое заболевание сердца, синдром, связанный с состоянием после диализа, ревматоидный артрит, синдром приобретенного иммунодефицита, васкулит, септический шок и другие типы острого воспаления и липидный гистоцитоз. Наиболее предпочтительными являются иммунопатологические нарушения.В сущности, любое нарушение, которое по этиологии связано с киназной активностью JNK, рассматривается как чувствительное к предупреждению или лечению, например, нарушения или патофизиологические состояния, ассоциированные с активацией JNK в клетке или клетках,как указано выше, например, рестеноз, потеря слуха, ушная травма, ишемия, "удар", и/или нарушения или патофизиологические состояния, ассоциированные с созреванием и дифференцировкой иммуноцитов, повреждения, связанные с реперфузией, гипоксия, связанные с апоптозом заболевания (например,встречающиеся при вирусных инфекциях (например, СПИД), аутоиммунные заболевания, нейродегенератиные нарушения (например, "удар", травма головного мозга, повреждение спинного мозг, амиотро- 21014330 фический боковой склероз (ALS), болезнь Гентингтона, болезнь Альцгеймера и болезнь Паркинсона),сердечно-сосудистое заболевание, остеопороз и старение), ответ на вызывающие стресс стимулы, и вторичные эффекты, связанные с лечением, например, провоспалительными цитокинами. Предлагаемые в изобретении фармацевтические композиции можно применять также для лечения или предупреждения воздействий, связанных с диабетом или нарушающим клетки стрессом, например, при патологических состояниях, индуцированных артериальной гипертензией, включая сердечную гипертрофию и связанные с артериосклерозом повреждения, и при бифуракции кровеносных сосудов и т.п., связанных с ионизирующей радиацией, применяемой при лучевой терапии, ультрафиолетовым светом (УФ-свет), связанных со свободными радикалами, повреждающими ДНК агентами, включая химиотерапевтические лекарственные средства, связанные с повреждениями при ишемии/реперфузии, связанными с гипоксией; и/или гипо- и гипертермией. И, наконец, касательно вышеуказанных заболеваний, нарушений или патофизиологических состояний предлагаемую в изобретении фармацевтическую композицию можно применять для ингибирования экспрессии генов, экспрессия которых повышается в присутствии активного полипептида JNK. Эти гены и генные продукты, как правило, включают, например, провоспалительные цитокины. Указанные цитокины обнаружены при всех формах воспалительных, аутогенных воспалительных,иммунных и аутоиммунных заболеваний, дегенеративных заболеваний, при миопатиях, кардиомиопатиях и отторжении трансплантата. Предлагаемые в изобретении последовательности, ингибирующие JNK, предлагаемые в изобретении химерные пептиды или предлагаемые в изобретении нуклеотидные последовательности можно применять также в любой ситуации, при которой требуется ингибирование активности JNK, поскольку JNK и все их изоформы принимают участие в развитии и создании патологических состояний, или в их пути. Такое применение может включать использование in vitro, ex vivo и in vivo. Таким образом, предлагаемые в изобретении нуклеиновые кислоты, как они определены выше,можно применять в конкретных вариантах осуществления настоящего изобретения для модуляции активируемых JNK путей передачи сигналов с использованием генной терапии, предпочтительно для лечения одного из состояний, заболеваний и/или нарушений, как они определены выше. В этом контексте понятие "генная терапия" относится к терапии, которую осуществляют путем введения индивидууму специфической предлагаемой в изобретении нуклеиновой кислоты, например, в виде фармацевтической композиции, как она определена выше, где предлагаемая(ые) в изобретении нуклеиновая(ые) кислота(ы) кодирует(ю) исключительно L-аминокислоты. В этом варианте осуществления настоящего изобретения нуклеиновая кислота продуцирует кодируемый(ые) пептид(ы), который(ые) затем служит(ат) для проявления терапевтического действия путем модуляции функции заболевания или нарушения. Любой из относящихся к генной терапии методов, доступных в данной области, можно применять для воплощения на практике настоящего изобретения (см., например, Goldspiel et al., Clin Pharm 12, 1993, с. 488-505). В предпочтительном варианте осуществления изобретения предлагаемая в изобретении нуклеиновая кислота, которую применяют для генной терапии, является частью экспрессионного вектора, который экспрессирует один или несколько предлагаемых в изобретении родственных IB-пептидов, т.е. предлагаемую в изобретении последовательность, ингибирующую JNK, и/или предлагаемый в изобретении химерный пептид или их фрагменты или производные, в приемлемом хозяине. В конкретном варианте осуществления изобретения указанный экспрессионный вектор несет промотор, функционально связанный с кодирующей(ими) областью(ями) последовательности, ингибирующей JNK. Промотор может представлять собой указанный выше промотор, например индуцибельный или конститутивный промотор, и необязательно тканеспецифический промотор. В другом конкретном варианте осуществления изобретения для генной терапии используют предлагаемую в изобретении молекулу нуклеиновой кислоты, в которой кодирующие последовательности предлагаемой в изобретении молекулы нуклеиновой кислоты (и любых других требуемых ее последовательностей) фланкированы областями, которые усиливают гомологичную рекомбинацию в требуемом сайте в геноме, что требуется для внутрихромосомной экспрессии нуклеиновых кислот (см., например,Koller и Smithies, Proc. Natl. Acad. Sci. USA 86, 1989, c. 8932-8935). Введение предлагаемой в изобретении нуклеиновой кислоты пациенту при применении генной терапии может быть либо непосредственным (т.е. пациента непосредственно обрабатывают нуклеиновой кислотой или вектором, содержащим нуклеиновую кислоту), либо косвенным (т.е. клетки сначала трансформируют нуклеиновой кислотой in vitro, затем трансплантируют пациенту). Эти два подхода называют соответственно как генная терапия in vivo или ex vivo. В конкретном варианте осуществления настоящего изобретения нуклеиновую кислоту вводят непосредственно in vivo, после чего происходит ее экспрессия с образованием кодируемого продукта. Для этой цели можно использовать любой из многочисленных методов, известных в данной области, в том числе, например, конструирование нуклеиновой кислоты в виде части соответствующего экспрессионного вектора нуклеиновой кислоты и введение его так, чтобы он стал внутриклеточным (например, путем заражения с использованием дефектного или ослабленного ретровирусного или другого вирусного вектора; см. US 4980286); непосредственную инъекцию "оголенной" ДНК; применение бомбардировки микрочастицами (например, "GeneGun" (генная пушка); Biolistic, фирма DuPont); нанесение нуклеиновых кислот с помощью липидов; применение ассо- 22014330 циированных с клеточной поверхностью рецепторов/трансфектирующих агентов; капсулирование в липосомах, микрочастицах или микрокапсулах; введение вместе с пептидом, для которого известно, что он обладает способностью проникать в ядро; или их введение вместе с лигандом, предрасположенным к опосредуемому рецептором эндоцитозу (см., например, Wu и Wu, J. Biol. Chem., 262, 1987, с. 4429-4432),который можно применять для "мечения" клеточный типов, которые специфически экспрессируют представляющие интерес рецепторы и т.д. Другой подход генной терапии, который можно применять для воплощения на практике настоящего изобретения, включает перенос гена в клетки in vitro в культуре ткани с помощью таких методов, как электропорация, липофекция, опосредуемая фосфатом кальция трансфекция, вирусное заражение или т.п. Как правило, метод переноса включает сопутствующий перенос в клетки селектируемого маркера. Затем клетки помещают под давление отбора (например, такого фактора, как устойчивость к антибиотикам) так, чтобы облегчать выделение клеток, которые получили и экспрессируют перенесенный ген. Затем эти клетки вводят пациенту. В конкретном варианте осуществления изобретения перед введением invivo полученной рекомбинантной клетки нуклеиновую кислоту интродуцируют в клетку с помощью любого метода, известного в данной области, включая, например, трансфекцию, электропорацию, микроинъекцию, заражение вирусным вектором или вектором на основе бактериофага, содержащим представляющие интерес нуклеотидные последовательности, слияние клеток, опосредуемый хромосомой перенос гена, опосредуемый микроячейкой перенос гена, слияние сферопластов и аналогичные методы, которые гарантируют, что при переносе не будет нарушено развитие и физиологические функции клетокреципиентов (см., например, Loeffler и Behr, Meth Enzymol, 217, 1993, с. 599-618). Выбранная методика должна обеспечивать стабильный перенос нуклеиновой кислоты в клетку, такой, чтобы при этом нуклеиновая кислота экспрессировалась клеткой. Предпочтительно перенесенная нуклеиновая кислота передается по наследству и может экспрессироваться в потомстве клетки. В предпочтительных вариантах осуществления настоящего изобретения образовавшиеся рекомбинантные клетки можно вводить пациенту с помощью различных методов, известных в данной области,включая, например, инъекцию эпителиальных клеток (например, подкожно), обработку пациента рекомбинантными кожными клетками в качестве кожного трансплантата и внутривенную инъекцию рекомбинантных клеток крови (например, гематопоэтических стволовых клеток или клеток-предшественников). Общее количество клеток, которое следует применять, зависит от требуемого действия, состояния пациента и т.п. и его может определять специалист в данной области. Клетки, в которые можно интродуцировать нуклеиновую кислоту при осуществлении генной терапии, относятся к любому требуемому доступному типу клеток, и они могут быть ксеногенными, гетерогенными, сингенными или автогенными. Типы клеток включают (но не ограничиваясь ими) дифференцированные клетки, такие как эпителиальные клетки, эндотелиальные клетки, кератиноциты, фибробласты, мышечные клетки, гепатоциты и кровяные клетки, или различные стволовые клетки или клетки-предшественники, в частности, эмбриональные клетки сердечной мышцы, стволовые клетки печени (опубликованная международная заявка на патентWO 94/08598), нервные стволовые клетки (Stemple и Anderson, Cell 71, 1992, с. 973-985), гематопоэтические стволовые клетки или клетки-предшественники, например, получаемые из костного мозга, клетки крови пуповины, периферической крови, печени эмбриона и т.п. В предпочтительном варианте осуществления изобретения применяемые для генной терапии клетки являются аутологичными для пациента. Согласно другому варианту осуществления изобретения предлагаемые в изобретении последовательности, ингибирующие JNK, предлагаемые в изобретении химерные пептиды, предлагаемые в изобретении нуклеотидные последовательности или антитела к предлагаемым в изобретении последовательностям, ингибирующим JNK, или к предлагаемым в изобретении химерным пептидам можно применять для анализов (in vitro) (например, имуноанализов) для выявления, прогнозирования, диагностики или мониторинга указанных выше различных состояний, заболеваний и/или нарушений или мониторинга их лечения. Иммуноанализ можно осуществлять с помощью метода, предусматривающего контакт образца, полученного из организма пациента, с антителом к предлагаемой в изобретении последовательности, ингибирующей JNK, предлагаемому в изобретении химерному пептиду или предлагаемой в изобретении нуклеотидной последовательности в условиях, при которых может происходить иммуноспецифическое связывание, и последующее выявление или оценку количества любого характеризующегося иммуноспецифическим связыванием антитела. В конкретном варианте осуществления изобретения антитело, специфическое в отношении предлагаемой в изобретении последовательности, ингибирующейJNK, предлагаемого в изобретении химерного пептида или предлагаемой в изобретении нуклеотидной последовательности, можно применять для анализа образца ткани или сыворотки из организма пациента в отношении присутствия JNK или последовательности, ингибирующей JNK; при этом аномальный уровень JNK свидетельствует о наличии болезненного состояния. Иммуноанализы, которые можно использовать, включают (но не ограничиваясь ими) конкурентные или неконкурентные системы анализов, основанные на применении таких методик, как Вестерн-блоттинг, радиоиммуноанализы (РИА), твердофазный иммуноферментный анализ (ELISA), "сэндвич"-иммуноанализы, анализы на основе иммунопреципитации, реакции с использованием преципитина, реакции, основанные на диффузии преципитина в геле, анализы иммунодиффузии, анализы агглютинации, флуоресцентные иммуноанализы, анализы фикса- 23014330 ции комплемента, иммунорадиометрические анализы и иммуноанализы на белке и т.д. В альтернативном варианте можно осуществлять анализы (in vitro) путем введения предлагаемых в изобретении последовательностей, ингибирующих JNK, предлагаемых в изобретении химерных полипептидов, предлагаемых в изобретении нуклеотидных последовательностей или антител к предлагаемым в изобретении последовательностям, ингибирующим JNK, или предлагаемым в изобретении химерным пептидам в клеткимишени, которые, как правило, выбирают, например, из культивируемых клеток животных, человеческих клеток или микроорганизмов, и оценивая ответ клетки с помощью биофизических методов, как правило, известных специалистам в данной области. Клетки-мишени, которые обычно применяют для этой цели, могут представлять собой культивируемые клетки (in vitro) или клетки in vivo, т.е. клетки, из которых состоят органы или ткани живых животных или людей, или микроорганизмов, присутствующих в живых животных или людях. Настоящее изобретение относится также к наборам для диагностического или терапевтического применения, в которые входит один или несколько контейнеров, содержащих предлагаемые в изобретении последовательности, ингибирующие JNK, предлагаемые в изобретении химерные пептиды, предлагаемые в изобретении нуклеотидные последовательности и/или антитела к предлагаемым в изобретении последовательностям, ингибирующим JNK, или к предлагаемым в изобретении химерным пептидам,например, антитело к последовательности, ингибирующей JNK, и необязательно меченый связывающийся с антителом компонент. Метка, которую включают в антитело, может представлять собой (но не ограничиваясь ими) хемилюминесцентный, ферментный, флуоресцентный, колориметрический или радиоактивный фрагмент. Другим конкретным вариантом осуществления изобретения являются также наборы для применения в диагностике, включающие один или несколько контейнеров, которые содержат нуклеиновые кислоты, кодирующие или в другом варианте комплементарные предлагаемой в изобретении последовательности, ингибирующей JNK, и/или предлагаемому в изобретении химерному пептиду, необязательно меченый связывающийся с этими нуклеиновыми кислотами компонент. В следующем конкретном варианте осуществления изобретения в состав набора может входить в один или несколько контейнеров пара олигонуклеотидных праймеров (например, каждый длиной 6-30 нуклеотидов), которые могут действовать в качестве праймеров амплификации для полимеразной цепной реакции (ПЦР; см.,например, Innis et al., PCR Protocols, изд-во Academic Press, Inc., San Diego, CA, 1990), лигазной цепной реакции, циклической реакции с зондом и т.п. или при применении других методов, известных в данной области, в которых применяют предлагаемые в изобретении нуклеиновые кислоты. Набор необязательно может включать также предварительно определенное количество очищенной предлагаемой в изобретении последовательности, ингибирующей JNK, предлагаемого в изобретении химерного пептида или кодирующих их нуклеиновых кислот, для применения в качестве диагностического агента, стандарта или контроля в анализах. Объем настоящего изобретения не ограничен приведенными конкретными вариантами его осуществления. Так, различные модификации изобретения помимо представленных в настоящем описании,должны стать очевидны специалистам в данной области после ознакомления с приведенным выше описанием и прилагаемыми чертежами. Такие модификации подпадают под объем прилагаемой формулы изобретения. Различные публикации, процитированные в настоящем описании, включены в него в качестве ссылки во всей их полноте. Если не указано иное, то все технические и научные понятия, использованные в настоящем описании, имеют обычные значения, известные обычному специалисту в области, к которой относится настоящее изобретение. Хотя для воплощения на практике или оценки настоящего изобретения можно применять методы и материалы, аналогичные или эквивалентные представленным в настоящем описании, приемлемые методы и материалы будут описаны ниже. Все публикации, заявки на патент и другие упомянутые в настоящем описании ссылки включены в него в качестве ссылок во всей их полноте. В случае расхождения следует опираться на настоящее описание, включая определения. Кроме того, материалы, методы и примеры приведены только с целью иллюстрации и не направлены на ограничение объема изобретения. Другие особенности и преимущества изобретения станут очевидными после ознакомления с приведенным подробным описанием и формулой изобретения. Описание чертежей На чертежах показано на фиг. 1 А-В - диаграмма, на которой представлены результаты сравнительного анализа первичной структуры областей консервативных доменов JBD указанных факторов транскрипции. Последовательности, ингибирующие JNK, были идентифицированы при оценке структуры этих сравниваемых последовательностей. Результаты сравнительного анализа первичной структуры в качестве примера представлены на фиг. 1 А-1B. На фиг. 1 А показана область наиболее высокой гомологии между JBD, присутствующими в IB1, IB2, c-Jun и ATF2. На панели Б с целью сравнения представлены аминокислотные последовательности JBD L-IB1(s) и L-IB1. Полностью консервативные остатки обозначены звездочками, а остатки с заменой на Ala в векторе GFP-JBD23Mut обозначены незакрашенными окружностями. На фиг. 1B показаны аминокислотные последовательности химерных белков, включающие последовательность, ингиби- 24014330 рующую JNK, и транспортирующую последовательность. В этом примере представленную транспортирующую последовательность выводят из белка ТАТ вируса иммунодефицита человека (ВИЧ), а последовательность, ингибирующую JNK, выводят из полипептида IB1(s). Человеческая, мышиная и крысиная последовательности на панелях Б и В являются идентичными; на фиг. 2 - диаграмма, на которой представлены последовательности общих слитых пептидов TATIB, выведенных из человеческих, мышиных и крысиных последовательностей; на фиг. 3 - результаты оценки нейрозащитного действия от очаговой церебральной ишемии, полученные на перманентной МСАО-модели. Определение эффективности защитного действия осуществляли при использовании различных доз (см. фиг. 3). Как видно из фиг. 3, дозы, составляющие по меньшей мере 11, 3, 0,3 и 0,03 мг/кг, обеспечивали защиту головного мозга. Наиболее высокий уровень защиты обнаружен при использовании дозы 0,03 мг/кг; на фиг. 4 - результаты оценки нейрозащитного действия предлагаемого в изобретении химерного пептида, последовательность которого представлена в SEQ ID NO: 11, после i.v.-введения в отношении очаговой церебральной ишемии на кратковременной МСАО-модели. После создания ишемии у взрослых мышей умерщвляли через 48 ч после реперфузии. Получали серийные криостатические срезы и оценивали объем инфаркта. Как видно из фиг. 4, предлагаемый в изобретении химерный пептид обеспечивал эффективное нейрозащитное действие; на фиг. 5 - результаты анализа на культуре нейронов по оценке высвобождения лактатдегидрогеназы (ЛДГ) после стимуляции с помощью N-метил-D-аспартата (NMDA). Результаты четко демонстрируют нейрозащитное действие предлагаемого в изобретении химерного D-JNKI1-пептида (SEQ ID NO: 11),поскольку дегенеративные изменения, обусловленные обработкой NMDA, полностью ингибировались,что видно по отсутствию значительного превышения высвобождения ЛДГ по сравнению с контролем; на фиг. 6 - результаты ингибирования эндогенной JNK-активности в клетках линии HepG2 при использовании предлагаемых в изобретении слитых пептидов, последовательности которых представлены в SEQ ID NO: 9 и 11, при применении метода анализа "в одной лунке". Как видно на фиг. 6, в частности на панели г на фиг. 6, D-TAT-IB1(s), последовательность которого представлена в SEQ ID NO: 11 (сокращенно обозначен как D-JNKI) эффективно ингибирует активность JNK, даже лучше, чем L-TATIB1(s), последовательность которого представлена в SEQ ID NO: 9 (сокращенно обозначен как L-JNKI); на фиг. 7 - защитное действие D-TAT-IB1(s) в отношении перманентной потери слуха. Представлены данные об изменении в уровне слухового порога (уровень звукового давления в дБ) на морских свинках после шумовой травмы (120 дБ при 6 кГц в течение 30 мин) при максимальной действующей частоте 8 кГц при оценке в течение 20 мин (временный сдвиг порога, TTS, окраска серым цветом) и в течение 15 дней после шумовой обработки (перманентный сдвиг порога, PTS). Морским свинкам наносили D-TATIB1(s), в виде геля на основе гиалуроновой кислоты, на мембрану круглого окна улитки (внутреннего уха) либо за 30 мин до, либо через 30 мин или 4 ч после шумовой травмы; в качестве контроля использовали необработанные уши. TTS оценивали через 20 мин после шумовой травмы, a PTS (окраска черным цветом), соответствующий перманентной потере слуха, определяли через 15 дней. Как видно из представленных данных, D-TAT-IB1(s) не только обеспечивал заметную защиту от потери слуха, вызванного шумовой травмой, при превентивном применении до шумовой обработки, но также и в зависимости от времени при применении после травмы. PTS в обработанных ушах был значительно ниже при применении D-TAT-IB1(s) через 30 мин и 4 ч после травмы по сравнению с необработанными контрольными ушами. Примеры Пример 1. Идентификация последовательностей, ингибирующих JNK. Аминокислотные последовательности, важные для эффективного взаимодействия с JNK, идентифицировали путем сравнительного анализа последовательностей известных JBD. Сравнение последовательностей JBD, присутствующих в IB1 [SEQ ID NO: 13], IB2 [SEQ ID NO: 14], c-Jun [SEQ ID NO: 15] иATF2 [SEQ ID NO: 16], выявило состоящую из 8 аминокислот последовательность с определенным уровнем консервативности (фиг. 1A). Поскольку JBD из IB1 и IB2 примерно в 100 раз превышают по эффективности c-Jun или ATF2 в отношении связывания JNK (Dickens et al. Science 277, 1997, с. 693),логично предположить, что консервативные для IB1 и IB2 остатки могут быть важны с позиций обеспечения максимального связывания. Сравнение JBD, присутствующих в IB1 и IB2, позволило выявить два блока, состоящие из 7 и 3 аминокислот, которые являются высококонсервативными для двух последовательностей. Эти два блока входят в последовательность пептида, состоящего из 19 аминокислот, в L-IB1(s)[SEQ ID NO: 1], и показаны также с целью сравнения в состоящей из 23 аминокислот (ак) пептидной последовательности, выведенной из IB1 [SEQ ID NO: 17]. Эти последовательности показаны на фиг. 1 Б,пунктирной линией в L-IB1-последовательности обозначена брешь в последовательности, введенная для выравнивания консервативных остатков при сравнении с L-IB1(s). Пример 2. Получение слитых белков, содержащих ингибитор JNK. Предлагаемые в изобретении слитые белки-ингибиторы JNK, последовательность которых представлена в SEQ ID NO: 9, синтезировали путем ковалентного связывания С-конца SEQ ID NO: 1 с N- 25014330 концом состоящего из 10 аминокислот пептида-носителя, выведенного из HIV-TAT4g 57 (Vives et al., J.Biol. Chem. 272, 1997, с. 16010), который представлен в SEQ ID NO: 5, через линкер, состоящий из двух остатков пролина. Этот линкер применяли в связи с тем, что он обеспечивает максимальную гибкость и предупреждает нежелательные изменения вторичной структуры. Получали также основные конструкции,которые обозначали как L-IB1(s) (SEQ ID NO: 1) и L-TAT [SEQ ID NO: 5] соответственно. Полностью-D-ретроинвертированные пептиды, представленные в SEQ ID NO: 11, синтезировали аналогичным образом. Получали также основные конструкции, которые обозначали как D-IB1(s) [SEQID NO: 2] и D-TAT [SEQ ID NO: 6] соответственно. Все предлагаемые в изобретении D- и L-слитые пептиды, представленные в SEQ ID NO: 9, 10, 11 и 12, получали путем классического Fmock-синтеза и дополнительно анализировали с помощью массспектрометрии. Их окончательно очищали с помощью ЖХВР. Для выяснения действия пролинового линкера получали два типа ТАТ-пептидов, один, дополненный двумя остатками пролина, а второй без них. Добавление двух остатков пролина не приводило к заметной модификации проникновения или локализации ТАТ-пептида внутри клетки. Общие пептиды, в которых обнаружены консервативные аминокислотные остатки, представлены на фиг. 2. Пример 3. Ингибирование гибели клеток с помощью JBD19. Изучали воздействие состоящей из 19 ак JBD-последовательности, присутствующей в IB1(s), на биологическую активность JNK. Состоящую из 19 ак последовательность связывали через N-конец с зеленым флуоресцентным белком (конструкция GFP JBD19) и оценивали воздействие этой конструкции на апоптоз панкреатических -клеток, индуцированный IL-1. Ранее установлено, что такой механизм апоптоза блокируется трансфекцией JBD1-280, при этом не происходит защита специфическими ингибиторамиERK1/2 или p38 (см. Ammendrup et al., выше). Синтезировали олигонуклеотиды, соответствующие JBD19 и содержащие области, которые кодируют консервативную состоящую из 19 аминокислот последовательность, а также последовательность,несущую мутацию в полностью консервативных областях, и непосредственно встраивали в сайты EcoRI и SalI вектора pEGFP-N1, который кодирует зеленый флуоресцентный белок (GFP) (фирма Clontech). Продуцирующие инсулин клетки ТС-3 культивировали в среде RPMI 1640, дополненной 10% фетальной телячьей сыворотки, 100 мкг/мл стрептомицина, 100 ед./мл пенициллина и 2 мМ глутамином. Продуцирующие инсулин клетки ТС-3 трансфектировали указанными векторами и IL-1 (10 нг/мл) и добавляли в среду для культуры клеток. Подсчитывали количество находящиеся в состоянии апоптоза клеток через 48 ч после добавления IL-1 с помощью инвертного флуоресцентного микроскопа. Находящиеся в состоянии апоптоза клетки отличались от здоровых клеток характерным "вспучиванием" цитоплазмы и их подсчитывали через 2 дня.GFP представляет собой экспрессионный вектор зеленого флуоресцентного белка, который применяют в качестве контроля; JBD19 представляет собой вектор, экспрессирующий химерный GFP, связанный с состоящей из 19 ак последовательностью, выведенной из JBD IB1; JBD19Mut представляет собой такой же вектор, что и GFP-JBD19, но в котором JBD несет мутации 4 консервативных остатков, показанных на фиг. 1 Б; и JBD1-280 представляет собой вектор GFP, связанный с полным JBD (ак 1-280). Экспрессионная конструкция GFP-JBD19 препятствовала индуцируемому IL-1 апоптозу панкреатических-клеток с такой же эффективностью, что и полный JBD1-280. Применяемые в качестве дополнительных контролей последовательности, несущие мутации полностью консервативных остатков IB1(s), обладали существенно меньшей способностью препятствовать апоптозу. Пример 4. Импорт в клетку пептидов TAT-IB1(s). Оценивали способность L- и D-энантиомерных форм ТАТ и предлагаемых в изобретении пептидовTAT-IB1(s) ("TAT-IB-пептиды") проникать в клетки. L-ТАТ, D-TAT, предлагаемые в изобретении LTAT-IB1(s)- и предлагаемые в изобретении D-TAT-IB1(s)-пептиды [SEQ ID NO: 5, 6, 9 и 12 соответственно] метили, добавляя на N-конец остаток глицина, конъюгированный с флуоресцеином. Меченые пептиды (1 мкМ) добавляли в культуры клеток ТС-3, которые поддерживали в условиях, описанных в примере 3. В предварительно определенные моменты времени клетки промывали ЗФР и фиксировали в течение 5 мин в охлажденном на льду метаноле-ацетоне (1:1) перед оценкой с помощью флуоресцентного микроскопа. Меченный флуоресциеном БСА (1 мкМ, 12 моль/моль БСА) применяли в качестве контроля. Полученные результаты свидетельствуют о том, что все указанные выше меченные флуоресцеином пептиды эффективно и быстро (менее чем в течение 5 мин) проникали в клетки при их добавлении в культуральную среду. И, наоборот, меченный флуоресцеином бычий сывороточный альбумин (1 мкМ БСА, 12 молей флуоресцеина/моль БСА) не проникал в клетки. Изучение в зависимости от времени показало, что интенсивность флуоресцентного сигнала при использовании L-энантиомерных форм пептидов снижалась на 70% через 24 ч. Через 48 ч обнаружен небольшой сигнал или отсутствие сигнала. В противоположность этому, D-TAT и предлагаемые в изобретении D-TAT-IB1(s) обладали очень высокой стабильностью в клетках. Флуоресцентные сигналы этих полностью-D-ретроинвертированных пептидов оказались еще очень- 26014330 сильными спустя 1 неделю, и сигнал лишь слегка снижался ко 2 неделе после обработки. Пример 5. In vitro ингибирование фосфорилирования c-JUN, ATF2 и Elk1. В опытах in vitro оценивали воздействие пептидов на JNK-опосредуемое фосфорилирование их факторов транскрипции-мишеней. Рекомбинантные и не активированные JNK1, JNK2 и JNK3 получали с помощью набора для транскрипции и трансляции лизатов кроличьих ретикулоцитов (TRANSCRIPTIONAND TRANSLATION rabbit reticulocyte lysate kit) (фирма Promega) и применяли для твердофазных анализов киназ c-Jun, ATF2 и Elk1 либо индивидуально, либо после слияния с глутатион-S-трансферазой(GST) в качестве субстратов. Осуществляли опыты по оценке зависимости ответа от дозы, смешивая предлагаемые в изобретении пептиды L-TAT или L-TAT-IB1(s) (0-25 мкМ) с рекомбинантными киназами JNK1, JNK2 или JNK3 в реакционном буфере (20 мМ Трис-ацетат, 1 мМ ЭГТК (этиленгликолевая тетраацетиловая кислота), 10 мМ n-нитрофенилфосфат (пНФФ), 5 мМ пирофосфат натрия, 10 мМ nглицерофосфат, 1 мМ дитиотреитол) в течение 20 мин. Затем инициировали киназные реакции, добавляя 10 мМ MgCl2 и 5 пКи 33 РдАТФ и 1 мкг либо GST-Jun (ак 1-89), либо GST-AFT2 (ак 1-96), либо GSTELK1 (ак 307-428). Слитые с GST белки получали от фирмы Stratagene (Ла-Джолла, шт. Калифорния). В смесь добавляли 10 мкл покрытых глутатионом агарозных гранул. Продукты реакции затем разделяли с помощью ДСН-ПААГ, в которых применяли денатурирующий 10% полиакриламидный гель. Гели сушили и затем экспонировали на рентгеновскую пленку (фирма Kodak). Практически полное ингибирование фосфорилирования с-Jim, ATF2 и Elk1 с помощью JNK обнаружено при использовании предлагаемых в изобретении пептидов ТАТ-IB(s) в дозах порядка 2,5 мкМ. Однако важным исключением было отсутствие ингибирования пептидом TAT-IB(s) фосфорилирования Elk1 с помощью JNK3. В целом, для заявляемого в изобретении пептида TAT-IB1(s) обнаружена очень высокая эффективность в отношении ингибирования фосфорилирования семейством JNK их факторов транскрипции-мишеней. Способность D-TAT, предлагаемого в изобретении пептида D-TAT-IB1(s) и предлагаемого в изобретении пептида L-TAT-IB1(s) (изучены дозы 0-250 мкМ) ингибировать фосфорилирование GST-Jun (ак 173) рекомбинантными JNK1, JNK2 и JNK3 анализировали с помощью описанного выше метода. В целом,пептид D-TAT-IB1(s) снижал опосредуемое JNK фосфорилирование c-Jun, но примерно в 10-20 раз менее эффективно по сравнению с пептидом L-TAT-IB1(s). Пример 6. Ингибирование фосфорилирования c-JUN активированными JNK. Воздействие пептидов L-TAT или предлагаемого в изобретении L-TAT-IB1(s) на JNK, которые активировали вызывающими стресс стимулами, оценивали, используя GST-Jun для оценки выделения JNK из облученных УФ-светом клеток линии HeLa или обработанных IL-1 клеток линии РТС. РТС-клетки культивировали согласно описанному выше методу. HeLa-клетки культивировали в среде DMEM, дополненной 10% фетальной телячьей сыворотки, 100 мкг/мл стрептомицина, 100 ед./мл пенициллина и 2 мМ глутамином. За 1 ч до получения клеточного экстракта РТС-клетки активировали IL-1, как описано выше, а HeLa-клетки активировали УФ-светом (20 Дж/м 2). Клеточные экстракты получали из контрольных, облученных УФ-светом HeLa-клеток и обработанных IL-1 ТС-3-клеток путем расщепления клеточных культур в лизирующем буфере (20 мМ Трис-ацетат, 1 мМ ЭГТК, 1% Triton Х-100, 10 мМ nнитрофенилфосфат, 5 мМ пирофосфат натрия, 10 мМ n-глицерофосфат, 1 мМ дитиотреитол). Клеточный дебрис удаляли центрифугированием в течение 5 мин при 15000 об/мин на роторе типа SS-34 Beckman. 100 мкг экстрактов инкубировали в течение 1 ч при комнатной температуре с 1 мкг GST-Jun (аминокислоты 1-89) и 10 мкл покрытых глутатионом агарозных гранул (фирма Sigma). После четырех отмывок буфером для расщепления гранулы ресуспендировали в этом же буфере, дополненном пептидами L-TAT или предлагаемым в изобретении пептидом L-TAT-IB1(s) (25 мкМ), в течение 20 мин. Затем киназные реакции инициировали, добавляя 10 мМ MgCl2 и 5 пКи 33 РдАТФ, и инкубировали в течение 30 мин при 30 С. Продукты реакции затем разделяли с помощью ДСН-ПААГ, в которых применяли денатурирующий 10%-ный полиакриламидный гель. Гели сушили и затем экспонировали на рентгеновскую пленку(фирма Kodak). В этих экспериментах установлено, что предлагаемые в изобретении пептиды TAT-IB(s) эффективно предупреждали фосфорилирование c-Jun активированными JNK. Пример 7. In vivo ингибирование фосфорилирования c-JUN предлагаемыми в изобретении пептидами TAT-IB(s). Для выяснения вопроса о том, могут ли предлагаемые в изобретении обладающие способностью проникать в клетку пептиды блокировать передачу сигнала JNK in vivo, при создании изобретения использовали гетерологичную систему GAL4. HeLa-клетки, которые культивировали с помощью описанного выше метода, совместно трансфектировали репортерным вектором 5GAL-LUC в сочетании с экспрессионной конструкцией GAL-Jun (фирма Stratagene), которая содержит домен активации c-Jun (аминокислоты 1-89), сшитый с ДНК-связывающим доменом GAL4. Для активации JNK использовали совместную трансфекцию векторами, экспрессирующими находящиеся непосредственно в обратном направлении киназы MKK4 и MKK7 (см., Whitmarsh et al., Science 285, 1999, с. 1573). В целом, метод состоял в следующем: 3105 клеток трансфектировали плазмидами в 3,5-сантиметровых чашках с использованиемDOTAP (фирма Boehringer Mannheim) согласно инструкциям производителя. Для экспериментов с ис- 27014330 пользованием GAL-Jun 20 мкг плазмиды трансфектировали 1 мкг репортерной плазмиды pFR-Luc (фирма Stratagene) и 0,5 мкг экспрессионной плазмиды либо MKK4, либо MKK7. Через 3 ч после трансфекции клеточные среды заменяли и добавляли пептиды ТАТ и TAT-IB1(s) (1 мкМ). Люциферазную активность оценивали через 16 ч с помощью системы "Dual Reporter System" (фирмы Promega) после стандартизации содержания белка. Добавление пептида TAT-IB1(s) блокировало активацию c-Jun после опосредуемой MKK4 и MKK7 активации JNK. Поскольку в клетках линии HeLa происходит экспрессия изоформ JNK1 и JNK2, но отсутствует экспрессия JNK3, при создании изобретения клетки трансфектировали JNK3. И в этом случае пептид TAT-IB(s) ингибировал опосредуемую JNK2 активацию c-Jun. Пример 8. Ингибирование пептидами ТАТ-IB индуцируемой IL-1 гибели панкреатических клеток. При создании изобретения анализировали воздействие предлагаемых в изобретении пептидов LTAT-IB(s) на развитие апоптоза -клеток, вызванного обработкой IL-1. Культуры ТС-3-клеток инкубировали в течение 30 мин с 1 мкМ предлагаемыми в изобретении пептидами L-TAT-IB1(s) с последующим введением 10 нг/мл IL-1. Второе введение пептида (1 мкМ) осуществляли через 24 ч. Количество находящихся в состоянии апоптоза клеток подсчитывали после инкубации в течение 2 дней с IL-1 с использованием для окрашивания ядер йодида пропидия (клетки, окрашенные в красный цвет, представляют собой погибшие клетки) и Hoechst 33342 (клетки, окрашенные в синий цвет, представляют собой клетки с неповрежденной плазматической мембраной). Добавление предлагаемых в изобретении пептидов TAT-IB(s) ингибировало индуцированный IL-1 апоптоз TC-3-клеток, культивируемых в присутствии IL-1 в течение 2 дней. Продолжительное ингибирование индуцируемой IL-1 гибели клеток оценивали путем обработкиTC-3-клеток аналогично описанному выше, за исключением того, что инкубацию клеток пептидами иIL-1 осуществляли в течение 12 дней. Каждый день вносили дополнительную порцию пептидов (1 мкМ) и каждые два дня вносили дополнительную порцию IL-1 (10 нг/мл). Предлагаемые в изобретении пептиды TAT-IB1(s) в этих условиях обладали выраженным защитным действием от апоптоза. Эти эксперименты в сравнении друг с другом четко демонстрируют, что предлагаемые в изобретении пептидыTAT-IB(s) представляют собой биологически активные молекулы, обладающие способностью предупреждать воздействия передачи сигнала JNK на гибель клеток. Пример 9. Синтез предлагаемых в изобретении полностью-D-ретроинвертированных пептидовIB(s). Пептиды, предлагаемые в изобретении, могут представлять собой пептиды, состоящие из полностью-D-аминокислот, которые подвергали обращенному синтезу для предупреждения естественного протеолиза (т.е. полностью-D-ретроинвертированные пептиды). Полностью-D-ретроинвертированный пептид, предлагаемый в изобретении, представляет собой пептид с аналогичными нативному пептиду функциями, в котором боковые группы входящих в его состав аминокислот должны соответствовать по линеаризованной структуре нативному пептиду, но сохранять устойчивый к протеазам каркас. Ретроинвертированные пептиды, предлагаемые в изобретении, синтезировали аналогично с помощью D-аминокислот путем присоединения аминокислот в пептидной цепи так, чтобы последовательность аминокислот в ретроинвертированном пептидном аналоге была точно противоположна последовательности выбранного пептида, который служит в качестве модели. Например, если встречающийся в естественных условиях белок ТАТ (состоящий из L-аминокислот) имеет последовательностьGRKKRRQRRR [SEQ ID NO: 5], то ретроинвертированный пепидный аналог этого пептида (состоящий из D-аминокислот) должен иметь последовательность RRRQRRKKRG [SEQ ID NO: 6]. Процедуры синтеза цепи D-аминокислот для получения ретроинвертированных пептидов известны в данной областиGuichard et al., J. Med. Chem. 39, 1996, с. 2030-2039). В частности, ретропептиды можно получать с помощью классического F-mock-синтеза и затем анализировать с помощью масс-спектрометриии. Их окончательно очищают с помощью ЖХВР. Поскольку особенностью нативных пептидов является их чувствительность к расщеплению встречающимися в естественных условиях протеазами и им свойственна иммуногенность, то гетеробивалентые или гетеромультивалентые соединения, предлагаемые в настоящем изобретении, следует получать таким образом, чтобы они представляли собой "ретроинвертированный изомер" требуемого пептида. Таким образом, защита пептида от естественного протеолиза должна повышать эффективность гетеробивалентного или гетеромультивалентного соединения как в результате удлинения времени полужизни,так и снижения степени иммунного ответа, направленного на активное разрушение пептидов. Пример 10. Продолжительная биологическая активность предлагаемых в изобретении полностьюD-ретроинвертированных пептидов IB(s). Данные о продолжительной биологической активности, предсказанной для предлагаемого в изобретении содержащего ретроинвертированный пептид D-TAT-IB(s) гетероконъюгата при сравнении с нативным L-аминокислотным аналогом, что обусловлено защитой предлагаемого в изобретении пептидаD-TAT-IB(s) от расщепления нативными протеазами, представлены на фиг. 5.- 28014330 Анализировали ингибирование индуцируемой IL-1 гибели панкреатических -клеток предлагаемым в изобретении пептидом D-TAT-IB1(s). Клетки линии ТС-3 инкубировали согласно описанному выше методу в течение 30 мин с однократным добавлением указанного пептида (1 мкМ), после чего добавляли IL-1 (10 нг/мл). Затем после инкубации в течение 2 дней с IL-1 подсчитывали количество клеток, находящихся на стадии апоптоза, с помощью окрашивания ядер йодидом пропидия и Hoechst 33342. В каждом эксперименте осуществляли подсчет минимум 1000 клеток. Представлены среднеквадратичные отклонения средних значений (СКО) при n=5. Установлено, что пептид D-TAT-IB1 снижал индуцированный IL-1 апоптоз в такой же степени, что и пептиды L-TAT-IB. Анализировали также продолжительное ингибирование индуцируемой IL-1 гибели клеток с помощью пептида D-TAT-IB1. Клетки линии ТС-3 инкубировали с помощью указанного выше метода в течение 30 мин с однократным добавлением указанных пептидов (1 мкМ), после чего добавляли IL-1(10 нг/мл), а затем добавляли цитокин каждые два дня. Затем после инкубации в течение 15 дней с IL-1 подсчитывали количество клеток, находящихся на стадии апоптоза, с помощью окрашивания ядер йодидом пропидия и Hoechst 33342. Следует отметить, что однократное добавление пептида TAT-IB1 не обеспечивает продолжительную защиту. В каждом эксперименте осуществляли подсчет минимум 1000 клеток. В результате установлено, что предлагаемый в изобретении D-TAT-IB1(s), в отличие от не предлагаемого в изобретении L-TAT-IB1(s), обладал способностью обеспечивать продолжительную защиту(15 дней). Пример 11. Ингибирование пептидами TAT-IB(s) индуцированной облучением гибели панкреатических -клеток.JNK активировали также ионизирующим излучением. Для решения вопроса о том, могут ли предлагаемые в изобретении пептиды TAT-IB(s) обеспечивать защиту от индуцируемого облучением повреждения JNK, клетки линии "WiDr" облучали (30 Гр) в присутствии D-TAT, предлагаемого в изобретении,или без него, L-TAT-IB1(s) или предлагаемыми в изобретении пептидами D-TAT-IB1(s) (1 мкМ вносили за 30 мин до облучения). Контрольные клетки (CTRL) не облучали. Клетки анализировали через 48 ч с помощью окрашивания ИП (йодид пропидия) и Hoechst 3342 согласно описанному выше методу. Представлены среднеквадратичные отклонения средних значений (СКО) при n=3. Предлагаемые в изобретении пептиды L-TAT-IB1(s) и D-TAT-IB1(s) оба обладали способностью предупреждать индуцируемый облучением апоптоз этой человеческой линии клеток рака ободочной кишки. Пример 12. Радиационная защита от ионизирующего излучения с помощью предлагаемых в изобретении пептидов TAT-IB(s). Для выявления способности предлагаемых в изобретении пептидов ТАТ-IB(s) осуществлять радиационную защиту мышей линии С 57 В 1/6 (возрастом 2-3 месяца) облучали рентгеновскими лучами (Rлучи) с помощью устройства Phillips RT 250 в дозе 0,74 Гр/мин (17 мА, 0,5 мм Cu-фильтр). За 30 мин до облучения животным вводили внутрибрюшинно (i.p.) следующие пептиды: либо ТАТ, либо предлагаемые в изобретении L-TAT-IB1(s), либо предлагаемые в изобретении D-TAT-IB1(s) (30 мкл 1 мМ раствора). В целом, метод состоял в следующем: для облучения мышей их помещали в небольшие пластиковые боксы, при этом голова оставалась вне бокса. Перед облучением животных клали на спину и фиксировали шею с помощью небольшой пластмассовой трубки для поддержания головы в правильном положении. Тело защищали свинцом. Перед облучением мышей содержали на стандартном пеллетированном корме для мышей, однако после облучения мышей содержали на полужидком корме, который заменяли каждый день. Затем оценивали реакцию слизистой оболочки губ в двух независимых анализах, используя балльную систему, разработанную Parkins с соавторами (Parkins et al., RadiotherapyOncology, 1, 1983, с. 165173), в которой оценивали статус эритемы, а также наличие отека, шелушения и экссудата. Кроме того,животных взвешивали перед каждой оценки статуса эритемы/отека. Результаты этих экспериментов свидетельствуют о том, что предлагаемые в изобретении пептидыTAT-IB(s) могут обеспечивать защиту от связанных с ионизирующим излучением потери веса и эритемы/отека. Пример 13. Подавление факторов транскрипции JNK с помощью предлагаемых в изобретении пептидов L-TAT-IB1(s). Анализы по удерживанию в геле осуществляли с использованием несущего двойную метку зонда АР-1 (5'-CGC TTG ATG AGT CAG CCG GAA-3' (SEQ ID NO: 27. Ядерные экстракты клеток линииHeLa обрабатывали в течение 1 ч 5 нг/мл TNF- или соответственно не обрабатывали. ТАТ и предлагаемые в изобретении пептиды L-TAT-IB1(s) добавляли за 30 мин до TNF-. Выявлена только часть геля,несущая специфический комплекс АР-1-ДНК (что продемонстрировано с помощью конкурентных экспериментов с немечеными специфичными и неспецифичными конкурентами). Предлагаемые в изобретении пептиды L-TAT-IB1(s) снижают образование получаемого посредством связывания АР-1 и ДНК комплекса в присутствии TNF-. Пример 14. Оценка нейрозащитного действия от очаговой церебральной ишемии на перманентной
МПК / Метки
МПК: C07K 14/47
Метки: пути, пептидные, обладающие, ингибиторы, сигнала, клетку, способностью, проникать, трансдукции
Код ссылки
<a href="https://eas.patents.su/30-14330-peptidnye-ingibitory-puti-transdukcii-signala-jnk-obladayushhie-sposobnostyu-pronikat-v-kletku.html" rel="bookmark" title="База патентов Евразийского Союза">Пептидные ингибиторы пути трансдукции сигнала jnk, обладающие способностью проникать в клетку</a>
Конъюгаты специфичных к иммунным клеткам макролидных соединений и противовоспалительных соединений с улучшенной способностью к целевому поступлению в клетку при противовоспалительной терапии

Номер патента: 7061
Опубликовано: 30.06.2006
Авторы: Месич Милан, Маркович Стрибор, Комач Марияна, Мерчеп Младен, Томаскович Линда, Хрвачич Бошка
МПК: A61P 5/44, C07J 43/00, A61K 31/58...
Метки: соединений, поступлению, терапии, клеткам, целевому, способностью, улучшенной, противовоспалительных, макролидных, конъюгаты, специфичных, противовоспалительной, иммунным, клетку
Формула / Реферат:
1. Соединение формулы I где М обозначает макролидную субъединицу, обладающую способностью накапливаться в клетках воспаления, выбранную из группы, включающей где R1 обозначает водород или метильную группу, R2 и R3 оба обозначают водород или вместе образуют связь или R2 обозначает аминогруппу, описываемую субструктурой -NR'R", где R' и R" могут быть независимо друг от друга водородом или любой алкильной или циклоалкильной группой, содержащей...
Рекомбинантные штаммы bcg, обладающие повышенной способностью к высвобождению из эндосомы

Номер патента: 12434
Опубликовано: 30.10.2009
Авторы: Сэдофф Джералд С., Хоун Дэвид Майкл, Сунь Ронггай
МПК: A01N 63/00, A61K 31/4745, A61K 48/00...
Метки: эндосомы, повышенной, обладающие, рекомбинантные, высвобождению, штаммы, способностью
Формула / Реферат:
1. Микобактерия (Mycobacterium), генетически сконструированная так, что она включает экспрессируемый и секретируемый функциональный эндосомолитический белок, который является активным при рН 6-8, где указанным экспрессируемым и секретируемым функциональным эндосомолитическим белком является перфринголизин О из Clostridium или его мутант. 2. Микобактерия по п.1, где аминокислотная последовательность указанного экспрессируемого и секретируемого...
Композиции растительных экстрактов, обладающие иммуностимулирующей способностью, способы их получения и применения

Номер патента: 3750
Опубликовано: 28.08.2003
Автор: Шехадех Ахмад Абдаллах
МПК: A61P 37/04, A61K 35/78
Метки: обладающие, растительных, способы, композиции, иммуностимулирующей, экстрактов, применения, получения, способностью
Формула / Реферат:
1. Композиция растительных экстрактов, обладающая иммуностимулирующей активностью, содержащая экстракт АРУМА. 2. Композиция растительных экстрактов по п.1, содержащая также экстракт ГРАНАТА. 3. Композиция растительных экстрактов по п.1, содержащая также экстракт ГИБИСКУСА. 4. Композиция растительных экстрактов по п.1, содержащая также экстракт ЧАЯ. 5. Композиция растительных экстрактов по п.2, содержащая также экстракт ГИБИСКУСА. 6. Композиция...
Пептидные производные – ингибиторы слияния при вич-инфекции

Номер патента: 7918
Опубликовано: 27.02.2007
МПК: A61K 39/21, C07K 7/00
Метки: производные, ингибиторы, вич-инфекции, слияния, пептидные
Формула / Реферат:
1. Изолированный пептид FB005, содержащий последовательность SEQ ID NO:1. 2. Изолированный пептид FB006, содержащий последовательность SEQ ID NO:2. 3. Изолированный пептид FB066, содержащий последовательность SEQ ID NO:7. 4. Изолированный модифицированный пептид, выбранный из группы, состоящей из: (a) SEQ ID NO:1; (b) SEQ ID NO:2; (c) SEQ ID NO:3 и (d) SEQ ID NO:7, и имеющий по меньшей мере одну замену аминокислотного остатка в предварительно...
Пептидные ингибиторы вируса гепатита с

Номер патента: 4765
Опубликовано: 26.08.2004
Авторы: Ллина-Брюне Монсе, Пупар Марк-Андре, Камерон Даль, Ранкур Жан, Гудреу Натали, Байли Мюррей Д., Гиро Элиза, Тсантризо Иула С.
МПК: C07K 14/81, A61P 31/14, A61K 38/55...
Метки: пептидные, ингибиторы, гепатита, вируса
Формула / Реферат:
1. Рацематы, диастериоизомеры и оптические изомеры соединения формулы (I) , в которой a равно 0 или 1, b обозначает 0 или 1, Y обозначает H или C1-C6алкил, B обозначает H, ацильное производное формулы R7-C(O)- или сульфонил формулы R7-SO2, где R7 обозначает C1-C10алкил, необязательно замещенный карбоксилом, C1-C6аланоилокси- или C1-C6алкоксигруппой, C6- или C10арил или C7-C16аралкил, необязательно замещенный C1-C6алкилом, гидрокси- или...
Предыдущий патент: Пиридинаминосульфонил-замещенные бензамиды в качестве ингибиторов цитохрома р450 3а4 (cyp3a4)
Следующий патент: Способ получения эзетимиба и промежуточных продуктов, используемых в этом способе
Случайный патент: Способ получения фенильных гетероциклов, пригодных в качестве ингибиторов цог-2