Способ получения эзетимиба и промежуточных продуктов, используемых в этом способе
Номер патента: 14331
Опубликовано: 29.10.2010
Авторы: Боди Йожеф, Сёке Каталин, Гати Тамаш, Киш-Бартош Доротья, Темешвари Кристина, Вукич Кристина, Елеш Янош







Формула / Реферат
1. Способ получения эзетимиба формулы I

содержащий следующие стадии:
а) превращение этиленгликолевого эфира 4-(4-фторбензоил)масляной кислоты формулы II в 4-[2-(4-фторфенил)-[1,3]диоксалан-2-ил]масляную кислоту формулы IV через невыделенное промежуточное соединение формулы III
![]()
(b) ацилирование хирального соединения формулы V соединением формулы IV с получением ацилированного производного оксазолидинона формулы VI

где соединение формулы V выбирают из соединений формул Va, Vb, Vc или Vd

и где R1, R2 и R3представляют собой:
в случае Va: R1=Ph, R2=R3=H,
в случае Vb: R1=R2=R3=Ph,
в случае Vc: R1=метил, R2=Ph, R3=H,
в случае Vd: R1=изопропил, R2= R3=Ph,
и где Ph представляет собой фенильную группу.
с) взаимодействие ацилированного производного оксазолидинона формулы VI с защищенным иминосоединением формулы VII и выделение соединения формулы VIII, где R4 представляет собой силильную группу

циклизацию соединения формулы VIII с получением защищенного производного азетидинона общей формулы IX

d) гидролиз кетальной группы соединения формулы IX с получением соединения формулы X

е) энантиоселективное восстановление соединения общей формулы X с получением соединения формулы XI
![]()
где одно из хиральных CBS-оксазаборолидиновых соединений формул XIIa, XIIb, XIIc и XIId выбрано в качестве катализатора и

f) удаление силильной защитной группы в соединении общей формулы XI с получением конечного продукта, эзетимиба формулы I

2. Способ получения соединения формулы VIIIa

включающий изомеризацию соединения формулы VIIIb в присутствии производного Ti(IV)

3. Соединение формулы III, IV

где R представляет собой группу -СН2-СН2-ОН или -Н.
4. Соединение формулы VI

где R1, R2 и R3представляют собой:
(a): R1=Ph,R2=R3=H,
(b): R1=R2 =R3=Ph
(c): R1=метил, R2=Ph, R3=H
(d): R1=изопропил, R2=R3=Ph,
и где Ph представляет собой фенильную группу
5. Соединение формулы VIII

где обозначения R1, R2, R3 независимо представляют собой Va, Vb, Vc или Vd и R4 является силильной защитной группой.
6. Соединение общей формулы IX

где R4является силильной защитной группой.
7. Соединение формулы Ха

8. Соединение формулы XIa

Текст
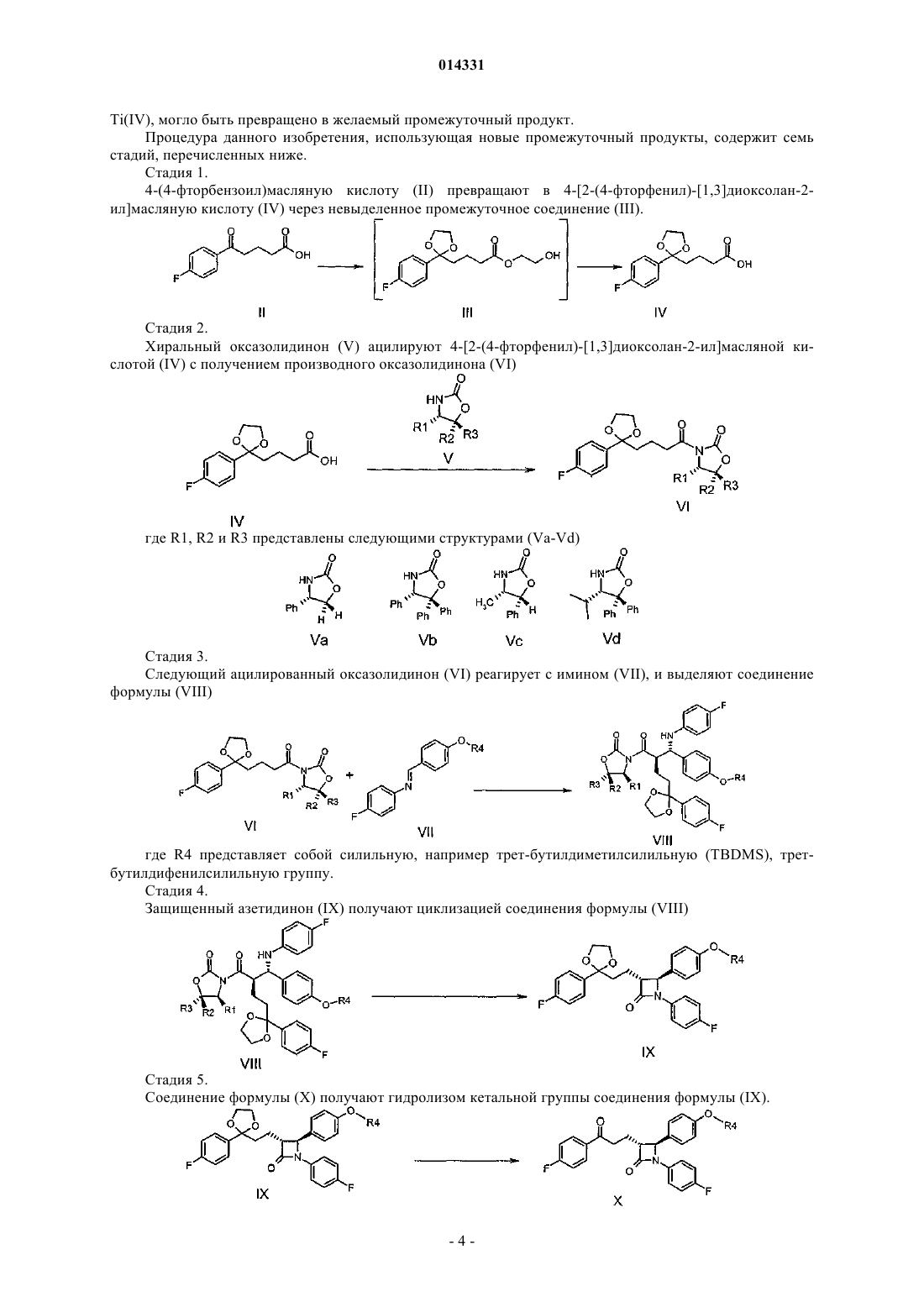
СПОСОБ ПОЛУЧЕНИЯ ЭЗЕТИМИБА И ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ, ИСПОЛЬЗУЕМЫХ В ЭТОМ СПОСОБЕ Настоящее изобретение обеспечивает новый, легко реализуемый в промышленности и рентабельный способ, содержащий только небольшое число стадий и основанный на использовании новых промежуточных продуктов, для получения 1-(4-3(R)-[3(S)-(4-фторфенил)3-гидрокспропил]-4(S)-(4-гидроксифенил)-2-азетидинона (эзетимиба) согласно следующей реакционной схеме: (I), (II), (III), (IV), (V), (VI), (VII), (VIII), (IX), (X), (XI) где соединения общих формул II, IV, VI, VIII, IX, X и XI являются новыми веществами, формула III обозначает невыделенный промежуточный продукт, R1, R2 и R3 представлены соединениями формул VaVd, (Va), (Vb), (Vc), (Vd) и R4 представляет собой синильную, например третбутилдиметилсилильную, трет-бутилдифенилсилильную группу. 014331 Область техники, к которой относится изобретение Данное изобретение относится к новому способу получения эзетимиба, т.е., 1-(4)-3(R)-[3(S)-(4 фторфенил)-3-гидроксипропил]-4(S)-(4-гидроксифенил)-2-азетидинона формулы I. Кроме того, данное изобретение относится к новым промежуточным продуктам, используемым в данном способе. Уровень техники В развитых странах значительная часть смертей вызвана сердечно-сосудистыми нарушениями. Эти заболевания, главным образом, инициируются атеросклеротическими изменениями коронарных артерий. Среди факторов риска, приводящих к развитию болезни, таких как высокое кровяное давление, диабет,курение и т.д. наиболее важным является высокая концентрация холестерина в сыворотке крови. Активные ингредиенты и композиции, уменьшающие концентрацию сывороточного холестерина, являются полезными агентами в лечении и предотвращении атеросклероза. Эзетимиб,т.е.,1-(4)-3(R)-[3(S)-(4-фторфенил)-3-гидроксипропил]-4(S)-(4-гидроксифенил)-2 азетидинон формулы I является активным ингредиентом некоторых современных продаваемых на рынке фармацевтических препаратов, проявляющих значительный гипохолестеринемический эффект, используемых для лечения и предотвращения атеросклероза, описанных в патенте США 5767115 (ScheringCo. U.S.A.) и Европейском патенте 720599. Первые синтетические способы получения эзетимиба и его производных опубликованы в этих описаниях. Согласно одному из описанных в них способов соответствующее транс-производное азетидинона получают в одной стадии по реакции основания [4-(бензилокси)бензилиден]-(4-фторфенил)амина с метил-4-(хлорформил)бутиратом, и после гидролиза и образования хлорангидрида данным 3-[2-(4 бензилоксифенил)-1-(4-фторфенил)-4-оксоазетидин-3-ил]пропионилхлоридом ацилируют(4 фторфенил)цинкхлорид в присутствии тетраксис(трифенилфосфин)палладия. Чистый 1-(4-фторфенил)3(R)-[3-оксо-3-(4-фторфенил)пропил)]-4(S)-(4-бензилоксифенил)-2-азетидинон получают разделением с использованием хиральной ВЭЖХ, и затем конечный продукт эзетимиб получают последующим энантиоселективным восстановлением и каталитическим гидрированием. В этом способе кольцо замещнного азетидинона не образуется энантиоселективным способом, поэтому предпоследний промежуточный продукт очищают способом хиральной колоночной хроматографии. Таким образом по меньшей мере 50% последнего промежуточного продукта теряется, что значительно увеличивает стоимость процедуры. Чтобы избежать использования дорогостоящей хиральной хроматографии, в патенте США 5919672 (Schering Co.) использовали микробиологический и ферментативный способы разделения. Хотя микробиологический способ снижает стоимость разделения рацемата, даже в этом случае выход на стадии разделения не может быть увеличен больше 50%. В Европейском патенте 720599 (Schering Co.) раскрыты способы получения некоторых тризамещнных производных азетидинона, обладающих гипохолестеринемической активностью. Для образования -лактамного кольца описаны одностадийный и двухстадийный способы, и формирование арилгидроксиалкильной боковой цепи проводят несколькими способами. Для синтеза эзетимиба предложен энантиоселективный способ. Сначала азетидиновое кольцо образуется в результате двухстадийного синтеза из метилового эфира 5-оксо-5-S)-2-оксо-4-фенилоксазолидин-3-ил)пентановой кислоты и [4(бензилокси)бензилиден]-(4-фторфенил)амина. Ацилирование проводят при помощи полученного 3[(2S,3R)-2-(4-бензилоксифенил)-1-(4-фторфенил)-4-оксоазетидин-3-ил]пропионилхлорида в присутствии(4-фторфенил)цинкхлорида-тетракис(трифенилфосфин)палладия. Данный промежуточный продукт 1-(4 фторфенил)-3(R)-[3-оксо-3-(4-фторфенил)пропил)]-4(S)-(4-бензилоксифенил)-2-азетидинон очищают колоночной хроматографией, затем после энантиоселективного восстановления оксогруппы и удаления защитной группы получают активный ингредиент. Из предшествующих процедур стратегически отличающийся способ был опубликован в международной заявке на патентWO 97/45406 и в патенте США 5739321 (Schering Co). Согласно этим публикациям энантиоселективное образование транс-замещнного промежуточного продукта азетидинона выполняют взаимодействием 4(S)-гидроксибутиролактона с защищенным имином в присутствии основания, затем 3-(4-фторфенил)-3-гидроксипропильную боковую группу формируют синтезом, состоящим из нескольких стадий, с использованием в качестве исходного вещества вышеупомянутого промежуточного продукта 1-(4-фторфенил)-3(R)-[3-оксо-3-(4-фторфенил)пропил]-4(S)-(4-бензилоксифенил)-2 азетидинона. Бензильную защитную группу удаляют каталитическим гидрированием. Другой реакционный путь раскрыт в патенте США 5856473 (Schering Co.). Согласно описанию(3R,4S)-4-(4-бензилоксифенил)-1-(4-фторфенил)-3-[(Е)-3-(4-фторфенил)аллил]азетидин-2-он, содержащий двойную связь в боковой цепи, получают алкилированием 1-4-(фторфенил)-4(S)-(4-1 014331 бензилоксифенил)-2-азетидинон-4-фторциннамоилбромидом, или энантиоселективным синтезом с использованием в качестве исходного вещества (S)-3-[5-(4-фторфенил)пент-3-еноил]-4-фенилоксазолидин 2-она. Промежуточный продукт 1-(4-фторфенил)-3(R)-[3-оксо-3-(4-фторфенил)пропил]-4(S)-(4 бензилоксифенил)-2-азетидинон получают окислением боковой цепи, после чего конечный продукт эзетимиб получают удалением защитной группы вышеупомянутым энантиоселективным восстановлением. Эти энантиоселективные процедуры обычно используют впоследствии при использовании многостадийных способов синтеза с оптически чистыми производными азетидинона, полученными относительно дорогостоящими энантиоселективными синтетическими способами. Ключевой промежуточный продукт, 1-(4-фторфенил)-3(R)-[3-оксо-3-(4-фторфенил)пропил]-4(S)-(4-бензилоксифенил)-2-азетидинон,очищают только хроматографически, что значительно увеличивает стоимость промышленных способов. В заявках на патентWO 2000/34240 (Schering Со.) иWO 1995/08 532 и в европейском патенте 0720599 (Schering Co.) описан улучшенный и более эффективный энантиоселективный способ синтеза эзетимиба. Согласно данной процедуре сначала 3-[(S)-5-(4-фторфенил)-5-гидроксипентаноил]оксазолидин-2-он получают с 98%-ной чистотой (de, диастереомерный избыток) из соответствующего оксосоединения энантиоселективным восстановлением. 3-[(S)-5-(4-фторфенил)-5 гидроксипентаноил]оксазолидин-2-он и N-(4-гидроксибензилиден)-4-фторанилин силилируют in situ хлортриметилсиланом в одном сосуде. Соответствующий продукт -аминоамид получают обработкой смеси реагентом TiCl4 в присутствии основания с использованием процедуры, хорошо известной в данной области. Авторы обнаружили неожиданную стабильность триметилсилильной группы, являющейся защитной группой для фенольной ОН-группы. Несмотря на стабильность силильной группы промежуточный продукт удалось выделить только с 65% выходом после обработки и дополнительной стадии силилирования. Эзетимиб получают после циклизации -аминоамида и последующего удаления защитных групп. В этой процедуре образование 3(S)-гидроксигруппы осуществляют в начале синтеза относительно дорогостоящим энантиоселективным способом, затем продукт выделяют после дополнительных стадий реакции и процедуры очистки. Стереоселективное образование 3(S)-OH группы является одной из ключевых стадий получения эзетимиба. В каждой из вышеупомянутых процедур используют один из вариантов способов энантиоселективного восстановления, катализируемого CBS-оксазаборолидином, хорошо известный из литературы (Е. J. Corey et al., J.Am.Chem.Soc. 1987, 109, 5551-5553). Достигаемая величина de (диастереомерный избыток) составляет 88-98%, как и должно быть в типичных случаях. Патенты США с номерами 5886171 и 5856473 (Schering Co.) описывают способ энантиоселективного восстановления с использованием CBS-оксазаборолидинового катализатора, в котором защищенный 1-(4-фторфенил)-3(R)-[3-оксо-3-(4-фторфенил)пропил]-4(S)-(4-гидроксифенил)-2-азетидинон превращается в защищенный 1-(4-фторфенил)-3(R)-[3-гидрокси-3-(4-фторфенил)пропил]-4(S)-(4-гидроксифенил)2-азетидинон. Патенты США с номерами 6207822 и 6627757 (Schering Co.) описывают применение подобных восстанавливающих агентов и хиральных катализаторов для превращения 3-[5-(4-фторфенил)-5 оксопентаноил]-4(S)-4-фенил-1,3-оксазолидин-2-она в 3-[5(S)-5-(4-фторфенил)-5-гидроксипентаноил]4(S)-4-фенил-1,3-оксазолидин-2-он. В патенте США 5618707 и в заявке на патентWO 1997/12053 (Schering Co.) описана другая возможность энантиоселективного восстановления, когда предыдущий промежуточный продукт 3-[5-(4 фторфенил)-5-оксопентаноил]-4(S)-4-фенил-1,3-оксазолидин-2-он превращают стереоселективным микробиологическим восстановлением в 3-[5(S)-5-(4-фторфенил)-5-гидроксипентаноил]-4(S)-4-фенил-1,3 оксазолидин-2-он. Достигаемая величина de 95% (диастереомерный избыток) сходна с величиной, полученной с использованием CBS-оксазаборолидинового катализатора. В патенте США 6133001 стереоселективное микробиологическое восстановление описано для превращения 1-(4-фторфенил)-3(R)-[3-оксо-3-(4-фторфенил)пропил]-4(S)-(4-гидроксифенил)-2 азетидинона в 1-(4-фторфенил)-3(R)-[3-гидрокси-3-(4-фторфенил)пропил]-4(S)-(4-гидроксифенил)-2 азетидинон (эзетимиб). Конечный продукт получают в малых количествах и очищают препаративной тонкослойной хроматографией. В международной заявке на патентWO 2005/066120 (Ranbaxy Ltd.) энантиоселективное восстановление оксогруппы 3-[5-(4-фторфенил)-5-оксопентаноил]-4(S)-4-фенил-1,3-оксазолидин-2-она и 1-(4 фторфенил)-3(R)-[3-оксо-3-(4-фторфенил)пропил]-4(S)-(4-гидроксифенил)-2-азетидинона выполняют при помощи (-)-В-хлордиизопинокамфеилборана, достигая селективности, подобной полненной приCBS-восстановлении. Однако интересно, что при восстановлении 20 г 1-(4-фторфенил)-3(R)-[3-оксо-3-(4 фторфенил)пропил]-4(S)-(4-гидроксифенил)-2-азетидинона таким способом после очистки колоночной хроматографией получают только 3 г конечного продукта эзетимиба.-2 014331 Сущность изобретения Данное изобретение обеспечивает новый, легко реализуемый в промышленности и экономичный способ, состоящий всего из небольшого числа стадий, и основанный на использовании новых промежуточных продуктов для полученияl-(4-3(R)-[3(S)-(4-фторфенил)-3-гидроксипропил)]-4(S)-(4 гидроксифенил)-2-азетидинона (эзетимиба) в соответствии со следующей реакционной схемой: где соединения общих формул II, IV, VI, VIII, IX, X и XI являются новыми; формула III представляет собой невыделенный промежуточный продукт;R1, R2 и R3 представлены соединениями формул Va-Vd и R4 представляет собой силильную, например трет-бутилдиметилсилильную, третбутилдифенилсилильную группу. Подробное описание изобретения Принимая во внимание недостатки известных синтетических процедур получения эзетимиба, авторы попытались разработать промышленный безопасный способ получения, содержащий рентабельные технологические стадии, обеспечивающие активный ингредиент с чистотой, удовлетворяющей требованиям фармакологии. Авторы поставили задачу разработать такую стратегию синтеза, которая не содержит утомительных технологических стадий или стадий, требующих экстремальных условий, и в котором промежуточные продукты могут быть получены простыми процедурами с высокой эффективностью и могут быть выделены с высокой чистотой. Для защиты функциональных групп авторы старались использовать такие защитные группы, которые являются стабильными во время синтеза, могут быть введены простыми и дешвыми способами и селективно удалены. Авторы намеревались осуществлять более дорогостоящие технологические стадии в конце синтеза и регенерировать дорогие вспомогательные материалы (например, оптически активные). Во время экспериментов авторы неожиданно обнаружили, что в следующем реакционном пути синтеза с использованием комбинации специальных защитных групп в большинстве случаев получаются такие превосходные промежуточные продукты, которые могут быть легко очищены простыми способами и с высокими эффективностями, благодаря их выдающейся способности кристаллизоваться. Некристаллизующиеся промежуточные продукты могли быть использованы на следующих стадиях без очистки. Была открыта новая реакция, не опубликованная ранее в литературе, где стереоизомерное соединение, образующееся в качестве побочного продукта в реакции типа реакции Манниха, катализируемойTi(IV), могло быть превращено в желаемый промежуточный продукт. Процедура данного изобретения, использующая новые промежуточный продукты, содержит семь стадий, перечисленных ниже. Стадия 1. 4-(4-фторбензоил)масляную кислоту (II) превращают в 4-[2-(4-фторфенил)-[1,3]диоксолан-2 ил]масляную кислоту (IV) через невыделенное промежуточное соединение (III). Стадия 2. Хиральный оксазолидинон (V) ацилируют 4-[2-(4-фторфенил)-[1,3]диоксолан-2-ил]масляной кислотой (IV) с получением производного оксазолидинона (VI) где R1, R2 и R3 представлены следующими структурами (Va-Vd) Стадия 3. Следующий ацилированный оксазолидинон (VI) реагирует с имином (VII), и выделяют соединение формулы (VIII) где R4 представляет собой силильную, например трет-бутилдиметилсилильную (TBDMS), третбутилдифенилсилильную группу. Стадия 4. Защищенный азетидинон (IX) получают циклизацией соединения формулы (VIII) Стадия 5. Соединение формулы (X) получают гидролизом кетальной группы соединения формулы (IX).-4 014331 Стадия 6. Энантиоселективным восстановлением соединения формулы X получают соединение формулы XI Стадия 7. Удалением силильной защитной группы в соединении формулы XI получают конечный продукт,эзетимиб (I) В следующей части все стадии описаны более подробно. Стадия 1. В инертном, безводном растворителе, например в дихлорметане, в присутствии сильной кислоты,например, концентрированной серной кислоты или п-толуолсульфоновой кислоты, преимущественно концентрированной серной кислоты, и связывающего воду вспомогательного материала, например триметил-ортоформиата, 4-(4-фторбензоил)масляная кислота (II) взаимодействует в одной стадии с этиленгликолем при температуре 20-25 С. Реакцию останавливают добавлением основания, например NaHCO3. Растворитель заменяют на спиртовой растворитель, предпочтительно на метанол, и образующийся сложноэфирный промежуточный продукт (III) гидролизуют раствором основания, предпочтительно раствором гидроксида калия. Образующуюся 4-[2-(4-фторфенил)-[1,3]диоксалан-2-ил]масляную кислоту (IV) выделяют после концентрирования реакционной смеси, затем после подкисления слабой кислотой, например винной кислотой, лимонной кислотой, предпочтительно лимонной кислотой, с последующей экстракцией подходящим растворителем, например этилацетатом. Продукт очищают кристаллизацией из неполярного растворителя, например из н-гексана или н-гептана. Стадия 2. Продукт со стадии 1 превращают в смешанный ангидрид в инертном, безводном растворителе, например тетрагидрофуране или дихлорметане, предпочтительно в тетрагидрофуране, с использованием 11,7-кратного молярного количества, предпочтительно 1,05-1,10 эквивалента хлорангидрида, например пивалоилхлорида в присутствии триэтиламина при температуре в диапазоне от -20 до -10 С. Оксазолидинон формулы V, предпочтительно S-(+)-4-фенил-2-оксазолидинон (Va), добавляют в раствор полученного смешанного ангидрида в присутствии подходящего активирующего агента, например хлорида лития (LiCl) или 4-диметиламинопиридина, предпочтительно хлорида лития, затем раствор перемешивают в течение 4-8 ч при температуре в диапазоне от -20 до 25 С. Продукт выделяют экстракцией и очищают кристаллизацией. Стадия 3. Способ А: Продукт стадии 2 взаимодействует с имином формулы VII (где R4 представляет собой, предпочтительно, трет-бутилдиметилсилильную группу) в инертном, безводном растворителе, например дихлорметане, и в атмосфере N2 при температуре в диапазоне от - 40 до -25 С в присутствии TiCl4 и Ti(IV)изопропоксида и в присутствии третичного основания, например диизопропилэтиламина в течение 1-2 ч. Реакцию останавливают добавлением спирта, предпочтительно изопропилового спирта, и продукт (VIII) выделяют экстракцией и после упаривания очищают при его перемешивании с метанолом. Для защиты фенольного гидроксила используют силильную защитную группу, предпочтительно трет-бутилдиметилсилильную группу, которая особенно предпочтительна в сравнении с другими группами силильного типа, способными отщепляться даже в более мягких условиях, и в сочетании с другими защитными группами алкильного и ацильного типа. Поскольку трет-бутилдиметилсилильная защитная группа стабильна в условиях синтеза, нет необходимости повторно силилировать перед выделением промежуточный продукт, который во время обработки частично теряет свою защитную группу. Кроме того, трет-бутилдиметилсилильная защитная группа может быть легко удалена при обработке кислотой в отсутствие побочных реакций. С другой стороны, удаление защитных групп бензильного типа требует или технологически более трудомкого и более опасного каталитического гидрирования или способа удаления более сильной кислотой. К сожалению, карбениевый катион, образующийся во время отщепления защитных групп бензильного и алкильного типа в кислой среде, приводит к образованию значительного количества побочных продуктов алкилирования фенильного кольца. Как хорошо известно из лите-5 014331 ратуры, удаление защитных групп ацильного типа основанием сопровождается значительным количеством побочных реакций (например, раскрытием лактамного цикла). Эксперименты авторов доказали, что в равновесной реакции типа реакции Манниха, катализируемой Ti(IV), кроме ожидаемого R4=трет-бутилдиметилсилильного производного (VIIIa), также образуется изомерный побочный продукт (VIIIb) в значительных количествах. Авторы показали, что с использованием в качестве исходных веществ с (VIIIa) и (VIIIb) в описанных выше условиях реакции образуются те же самые продукты, что и при проведении реакции с использованием в качестве исходных веществ (VIa) и (VIIa). При подходящем выборе параметров эксперимента равновесие может быть сдвинуто в направлении, благоприятствующем образованию (VIIIa), и продукт может быть выделен с выходом 73-78%. Изомер (VIIIb), который может присутствовать в маточном растворе, полученном после фильтрования продукта из метанольной суспензии, может быть превращен в (VIIIa) в условиях Ti (IV)катализируемой реакции типа реакции Манниха. Таким образом, выход реакции может быть значительно повышен. С этой целью метанольный маточный раствор упаривают, растворитель заменяют на подходящий растворитель, например толуол, раствор обесцвечивают силикагелем и затем после фильтрования упаривают. При использовании этой процедуры из этой смеси может быть получено дополнительное количество продукта формулы VIII следующим образом. Осадок после упаривания растворяют в дихлорметане и в присутствии Ti(IV)-изопропоксида и третичного основания, например диизопропилэтиламина, раствор перемешивают в инертной атмосфере,например N2, при температуре в диапазоне от - 40 до -25 С в течение 1-2 ч. Чистый продукт формулы В альтернативной процедуре сначала получают соединение формулы (VIIa) in-situ в дихлорметане в присутствии диизопропилэтиламина (DIPEA) реакцией (Е)-(4-гидроксибензилиден)-(4-фторфенил)амина(VIIb) с трет-бутилдиметилсилилхлоридом (TBDMS-Cl), затем раствор полученного продукта формулыVIIa используют, как описано в способе А. Стадия 4. Продукт со стадии 4 формулы VIII силилируют в подходящем растворителе, например в тетрагидрофуране, толуоле, метил-трет-бутиловом эфире или ацетонитриле, предпочтительно, в ацетонитриле,подходящим силилирующим агентом, например бис(триметилсилил)ацетамидом, при температуре в диапазоне от 20 до 25 С в течение 1-3 ч. Фторсодержащее соединение, предпочтительно тригидрат тетрабутиламмонийфторида добавляют к смеси в каталитическом количестве (0,1-10 мол.%), предпочтительно в количестве 0,5-1 мол.%. Такую реакционную смесь, полученную при циклизации, дополнительно перемешивают в течение 0,5-3 ч, предпочтительно в течение 0,5 ч, затем реакцию останавливают водой и продукт формулы IX выделяют при помощи растворителя алканового типа, например н-гексана. Хиральный вспомогательный материал S-(+)-4-фенил-2-оксазолидинон (Va), после извлечения из ацетонитрильной фазы и его последующего концентрирования экстрагируют дихлорметаном и очищают кри-6 014331 сталлизацией. Стадия 5. Соединение формулы IX, полученное на стадии 4, обрабатывают в инертном растворителе, например, дихлорметане, минеральной глиной кислотного типа, предпочтительно, с монтмориллонитом при температуре в диапазоне от 20 до 25 С в течение 3-6 ч. В этих условиях трет-бутилдиметилсилильная защитная группа стабильна, и кетальная защитная группа может быть селективно удалена. Полученный таким образом продукт формулы X отделяют простым фильтрованием и после упаривания очищают кристаллизацией. Стадия 6. В процедуре авторов энантиоселективное восстановление для образования 3-(S)-гидроксильной группы проводят в конце синтеза. В этом случае особые расходы на дорогой хиральный катализатор являются более низкими. Поскольку асимметрический центр входит в состав оптически чистого индивидуального изомера, очистка конечного продукта упрощается до разделения двух диастереомеров. Соответственно, полученное таким образом соединение формулы X на стадии 5 восстанавливают восстанавливающим агентом боранового типа, таким как боран-диметилсульфид, боран-тетрагидрофуран, борандиэтиланилин или катехин-боран, предпочтительно, смесью боран-диметилсульфида и борантетрагидрофурана в присутствии хирального катализатора CBS-оксазаборолидинового типа, хорошо известного для достижения данной цели, в инертном растворителе, например дихлорметане, в инертной атмосфере, например, в N2, при температуре в диапазоне от -20 до 20 С, предпочтительно от -5 до +5 С. Хиральный CBS-оксазаборолидин (соединения XIIa-XIId), предпочтительно оксазаборолидин (соединение XIIа), используют в качестве катализатора. Продукт выделяют экстракцией и затем вводят в следующую реакцию без очистки. Стадия 7. Полученный таким образом продукт формулы XI нагревают в смеси с разбавленным водным раствором хлористо-водородной или серной кислоты, предпочтительно с раствором серной кислоты и спиртовым растворителем, например метанолом или изопропиловым спиртом, предпочтительно изопропиловым спиртом, при температуре в диапазоне от 50 до 70 С в течение 1-3 ч. Конечный продукт кристаллизуют из реакционной смеси с добавлением воды, затем очищают перекристаллизацией. Преимущества данного изобретения суммированы следующим образом:a) в способе авторов на новом синтетическом пути, основанном на использовании новых соединений, ключевые промежуточный продукты, благодаря их превосходной способности кристаллизоваться могут быть эффективно очищены простыми операциями кристаллизации;b) для защиты фенольной ОН-группы используют группу силильного типа, предпочтительно третичную бутилдиметилсилильную группу, которая является более предпочтительной, чем отщепляющиеся в более мягких условиях, например, в сравнении с группами алкильного и ацильного типа;c) в энантиоселективной катализируемой Ti(IV) реакции типа реакции Манниха соответствующий промежуточный продукт (VIIIa) образуется с высоким выходом (85-90%), однако стереоизомерный побочный продукт не теряется в равновесной реакции, а главным образом превращается в желаемый промежуточный продукт;d) таким образом, большая часть хирального вспомогательного вещества, S-(+)4-фенил-2 оксазолидинона (70% введнного количества) регенерируется простым способом в процессе синтеза; е) в этой процедуре энантиоселективное восстановление с образованием 3-(S)-гидроксильной группы проводят в конце синтеза. Благодаря этому специальные затраты на дорогостоящий хиральный катализатор являются меньшими. Поскольку асимметрический центр входит в состав оптически чистого индивидуального изомера, очистка конечного продукта упрощается до разделения двух диастереомеров. Подводя итоги, можно сказать, что в данном изобретении раскрыта такая новая процедура, которая может быть использована для рентабельного получения эзетимиба в промышленном масштабе. Чистота активного ингредиента, получаемого с использованием этой процедуры, может отвечать современным вс более и более растущим требованиям к качеству фавмацевтически активных ингредиентов. Примеры Следующие примеры являются иллюстративными и не предназначены для ограничения объма заявленного изобретения. Пример 1. Получение 4-[2-(4-фторфенил)-[1,3]диоксалан-2-ил]масляной кислоты (IV) 21,0 г (0,1 моль) 4-(4-фторбензоил)масляной кислоты (II) взвешивают в круглодонной колбе на 500 мл и суспендируют в 210 мл дихлорметана. При непрерывном перемешивании к суспензии по каплям-7 014331 добавляют 28 мл (31,2 г, 0,5 моль) этиленгликоля, 32 мл (31,04 г, 0,3 моль) триметил-о-формиата и 0,5 мл концентрированной серной кислоты. Реакционную смесь перемешивают при 20-25 С в течение 3-6 ч. Реакцию контролируют аналитически тонкослойной хроматографией. Когда кетон заканчивается, о чм судят по данным тонкослойной хроматографии, показывающим исчезновение его пятна, реакцию останавливают добавлением 5 г тврдого NaHCO3. Суспензию перемешивают в течение 0,5 мин, затем растворитель удаляют выпариванием и осадок растворяют в 150 мл метанола. Этот раствор охлаждают на водяной бане со льдом и добавляют при охлаждении 100 мл 10%-ного раствора NaOH. Колбу закрывают и мутную смесь перемешивают при 20-25 С в течение 1 ч. Гидролиз контролируют аналитически тонкослойной хроматографией. Когда сложный эфир заканчивается, о чм судят по данным тонкослойной хроматографии, показывающим исчезновение его пятна, метанол удаляют выпариванием в вакууме и при интенсивном охлаждении на водяной бане со льдом добавляют раствор 350 мл 10% лимонной кислоты для достижения значения кислого рН в диапазоне от 3 до 4. Осажднный продукт экстрагируют 200 мл этилацетата. Водную фазу дважды экстрагируют 50-50 мл этилацетата и затем объединнную органическую фазу промывают до нейтральной реакции 550 мл воды. Этилацетатный раствор сушат над безводным Na2SO4, дегидратирующий агент отфильтровывают, и фильтрат упаривают в вакууме. Осадок после упаривания кристаллизуют добавлением 50 мл н-гексана при 0 С. Кристаллический материал соединения (IV) выделяют фильтрованием и сушат. Выход: 23 г, (90%). Точка плавления: 65-67 С Данные 1 Н ЯМР: (500 МГц, DMSO-d6, 25 С)1,41-1,52 (м, 2 Н), 1,79-1,87 (м, 2 Н), 2,18 (т, J=7,5 Hz,2 Н), 3,63-3,73 (м, 2 Н), 3,91-4,01 (м, 2 Н), 7,13-7,22 (м, 2 Н), 7,37-7,45 (м, 2 Н), 11,97 (шире, 1 Н) м. д. Пример 2. Получение(S)-3-4-[2-(4-фторфенил)-[1,3]диоксалан-2-ил]бутирил-4-фенилоксазолидин-2-она (VIa) 42 г (165 ммоль) соединения формулы IV, продукта примера 1, растворяют в 340 мл безводного тетрагидрофурана и сосуд продувают сухим газообразным N2. Раствор охлаждают до -20 С и добавляют 55 мл (390 ммоль) триэтиламина. Через капельную воронку добавляют смесь 40 мл тетрагидрофурана и 20,2 мл пивалоилхлорида (19,8 г, 164 ммоль) в течение приблизительно 30 мин при температуре в диапазоне от -10 до - 20 С. Смесь, содержащую осадок, перемешивают в течение 2 ч при температуре в диапазоне от -10 до -20 С и затем к ней последовательно добавляют 24,45 г (150 ммоль) тврдого S (+)-4 фенил-2-оксазолидинона (Va) и 7,5 г (177 ммоль) безводного хлорида лития. Затем суспензию перемешивают в течение 4 ч при нагревании до 20-25 С. Реакцию контролируют аналитически тонкослойной хроматографией. Когда пятно S(+)-4-фенил-2 оксазолидинона уменьшается до 3%, реакцию останавливают добавлением 300 мл толуола и 150 мл насыщенного раствора хлорида аммония. Фазы разделяют, затем водную фазу экстрагируют 50 мл толуола. Объединнный толуольный раствор промывают 2150 мл раствором 10% лимонной кислоты, 2150 мл раствором 1 М NaOH и, наконец, 3150 мл воды. Органическую фазу сушат над безводным Na2SO4, дегидратирующий агент отфильтровывают и фильтрат упаривают в вакууме. Осадок кристаллизуют при 0 С в 150 мл изопропилового спирта. Продукт (VIa) сушат в вакууме в присутствии Р 2 О 5. Выход: 55,7 г[]25D =+54,3, (с=1, дихлорметан) Данные 1 Н ЯМР: (500 МГц, DMSO-d6, 25 С)1,42-1,56 (м, 2 Н), 1,76-1,85 (м, 2 Н), 2.80 (дт, J= 17,2,7,5 Hz, 1H), 2,90 (дт, J=17,2, 7,5 Hz, 1H), 3,61-3,71 (м, 2 Н), 3,89-3,99 (м, 2 Н), 4,13 (дд, J = 8,7, 3,6 Hz, 1H),4,71 (т, J=8,7 Hz, 1H), 5,43 (дд, J=8,7, 3,6 Hz, 1H), 7,12-7,19 (м, 2H), 7,23-7,28 (м, 2H), 7,29-7,34 (м, 1H),7,34-7,42 (м, 4 Н) м.д. Пример 3. Получение (S)-3-(R)-2-[(S)-[4-(трет-бутилдиметилсиланилокси)фенил]-(4-фторфениламин)метил]4-[2-(4-фторфенил)-[1,3]диоксалан-2-ил]бутирил-4-фенилоксазолидин-2-она (VIIIa) Получение реагента трихлоридизопропоксида титана реагента 0,95 мл (0,9 г, 3,2 ммоль) изопропоксида Ti(IV) добавляли к раствору 0,99 мл (1,71 г, 9 ммоль) TiCl4, приготовленному в 34 мл дихлорметана при температуре 0 С и в атмосфере N2. Смесь перемешивают в течение 15 мин при 0 С. Данный раствор используют на следующей стадии конденсации. Конденсация (способ А). 4,0 г (10 ммоль) соединения формулы VIa и 6,6 г (20 ммоль) иминосоединения формулы VIIa взвешивают в сосуде на 250 мл, снабжнном магнитной мешалкой, термометром, капельной воронкой и входным отверстием для N2, и растворяют в 50 мл дихлорметана. Смесь охлаждают до -40 С и прибавляют 3,6 мл (20,7 ммоль) DIPEA. Раствор титантрихлоридизопропоксидного реагента постепенно прибавляют из капельной воронки в течение приблизительно 30 мин. Смесь перемешивают в течение 1 ч при температуре в диапазоне от -30 до -40 С, затем реакцию останавливают добавлением 25 мл изопропилового спирта и 50 мл дихлорметана при температуре между -30 и -40 С и после этого реакцию перемешивают дополнительно в течение 30 мин при той же самой температуре. Полученную таким образом оран-8 014331 жевую суспензию медленно выливают в 100 мл тартратного буфера при рН=7, затем после 15-минутного перемешивания фазы разделяют. Водную фазу экстрагируют дополнительным количеством дихлорметана (3x30 мл), затем объединнный дихлорметановый раствор промывают 30 мл воды, сушат безводнымNa2SO4, дегидратирующий агент отфильтровывают и фильтрат упаривают в вакууме. К осадку прибавляют 50 мл метанола, полученную таким образом суспензию перемешивают при 20-25 С в течение 10 мин и затем продукт выделяют фильтрованием. Белое кристаллическое соединение (VIIIa) сушат в вакууме в присутствии Р 2 О 5. Выход: 5,5 г (76%). Конденсация (способ В). 25,8 г (120 ммоль) (Е)-(4-гидроксибензилиден)-(4-фторфенил)амина взвешивают в сосуде на 2 л,снабжнном магнитной мешалкой, термометром, капельной воронкой и входным отверстием для N2, его растворяют в 500 мл дихлорметана, затем добавляют 57,8 мл (332 ммоль) диизопропилэтиламина(DIPEA) при 20-25 С. Добавляют 19,9 г (132 ммоль) трет-бутилдиметилсилилхлорида и раствор перемешивают при 20-25 С в течение 1-2 ч. Гидролиз контролируют аналитически тонкослойной хроматографией. Когда на хроматограмме исчезает пятно исходного вещества, (Е)-(4-гидроксибензилиден)-(4-фторфенил)амина, добавляют 40 г (100 ммоль) соединения (VIa) и смесь охлаждают до температуры в диапазоне от -25 до 30 С. Приблизительно через 30-минутный период через капельную воронку постепенно добавляют раствор 9,5 мл (9 г, 32 ммоль) тетраизопропоксида титана и 9,9 мл (17,1 г, 90 ммоль) тетрахлорида титана (TiCl4) в 340 мл дихлорметана при 0 С. Смесь перемешивают в течение 0,5 ч при температуре от -25 до -30 С, реакцию в смеси останавливают добавлением 250 мл изопропилового спирта и 500 мл дихлорметана при температуре в диапазоне от -30 до 40 С и после этого перемешивают в течение ещ 30 мин при той же самой температуре. Полученную таким образом смесь медленно выливают в 1000 мл тартратного буфера при рН=7, затем после 15-минутного перемешивания фазы разделяют. Водную фазу экстрагируют дополнительным количеством дихлорметана (3250 мл), затем объединнный дихлорметановый раствор промывают 300 мл воды, сушат безводным Na2SO4, дегидратирующий агент отфильтровывают и фильтрат упаривают в вакууме. К осадку прибавляют 500 мл метанола, полученную таким образом суспензию перемешивают при 20-25 С в течение 10 мин, и затем продукт выделяют фильтрованием. Белое кристаллическое соединение (VIIIa) сушат в вакууме в присутствии Р 2 О 5. Выход: 57 г (78%). Точка плавления: 211-213 С[]25D=-0,9, (с=1, дихлорметан) Данные 1 Н ЯМР: (500 МГц, CDCl3, 25 С)0,17 (с, 6 Н), 0,97 (с, 9 Н), 1,22-1,35 (м, 1 Н), 1,66-1,90 (м,3 Н), 3,58-3,77 (м, 2 Н), 3,84-3,96 (м, 2 Н), 4,21 (дд, J = 8,7, 2,9 Hz, 1H), 4,26 (д, J = 9,1 Hz, 1H), 4,46-4,57 (м,1H), 4,66 (т, J = 8,7 Hz, 1H), 5,06 (шир, 1H), 5,44 (дд, J = 8,7, 2,9 Hz, 1H), 6,33-6,41 (м, 2 Н), 6,65-6,78 (м,4 Н), 6,91-6,98 (м 2 Н), 7,02-7,13 (м, 6 Н), 7,13-7,19 (м, 1 Н), 7,25-7,31 (ср, 2 Н) м.д. Обработка маточного раствора Полученный метанольный маточный раствор упаривают, растворитель заменяют на 200 мл толуола,к толуольному раствору добавляют 10 г силикагеля Si 60, суспензию перемешивают при 20-25 С в течение 15 мин. Силикагель отфильтровывают, промывают толуолом и фильтрат упаривают. Остаток после упаривания растворяют в 100 мл дихлорметана, смесь охлаждают до -30 С и добавляют 7 мл (40 ммоль)DIPEA в атмосфере N2. 2 мл раствора реагента трихлоридизопропоксида титана, приготовленного из (1,9 г, 6,74 ммоль) тетраизопропоксид титана и 1,81 мл (3,12 г, 16,3 ммоль) TiCl4, добавляют через капельную воронку в течение 30-минутного периода. Реакционную смесь перемешивают при температуре между -30 и -40 С, затем чистый продукт формулы VIIIa выделяют таким же способом, как в случае реакции конденсации. Выход: 8,0 г. Объединнный выход: 65 г (89%). Пример 4. Получение(3R,4S)-4-[4-(трет-бутилдиметилсиланилокси)фенил]-1-(4-фторфенил)-3-2-[2-(4 фторфенил)-[1,3]диоксалан-2-ил]этилазетидин-2-она (IX, R4=TBDMS) 20,25 г (28 ммоль) соединения формулы VIIIa суспендируют в 556 мл безводного ацетонитрила при 20-25 С, затем добавляют 13,6 мл (56 ммоль) N,O-бис-(триметилсилил)ацетамида. Реакционную смесь перемешивают при 20-25 С в течение 2 ч, затем добавляют 0,1 г (0,28 ммоль) тригидрата тетрабутиламмонийфторида и дополнительно перемешивают при той же самой температуре. В конце реакции (0,5-1 ч) суспензия становится прозрачным раствором. Реакцию контролируют аналитически тонкослойной хроматографией. Когда исчезает пятно исходного вещества, аминосоединения с линейной цепью (VIIIa),реакционную смесь разбавляют 556 мл воды и 556 мл н-гексана. После разделения фаз водную ацетонитрильную фазу экстрагируют 556 мл н-гексана. Объединнную н-гексановую фазу сушат безводнымNa2SO4, дегидратирующий агент отфильтровывают и фильтрат упаривают в вакууме. Полученное таким образом соединение (IXa) представляет собой масло, которое используют без дальнейшей очистки на следующей стадии реакции. Данные 1 Н ЯМР: (500 МГц, DMSO-d6, 25 С)(м.д.) 0,16 (с, 3 Н), 0,16 (с, 3 Н), 0,92 (с, 9 Н), 1,70-1,82-9 014331 Регенерация S(+)-4-фенил-2-оксазолидинона, возвращнного в качестве побочного продукта из водной ацетонитрильной фазы: Ацетонитрильную водную фазу, полученную, как описано выше, концентрируют до объма приблизительно в 500 мл и продукт, осажднный из оставшегося раствора, экстрагируют дихлорметаном(2100 мл). Объединнный дихлорметановый раствор упаривают, осадок кристаллизуют из смеси этилацетата и н-гексана. Регенерированный S (+)-4-фенил-2-оксазолидинон выделяют фильтрованием. Выход: приблизительно 3,9 г (приблизительно 85%, в расчте на введнный VIIIa). Пример 5. Получение (3R,4S)-4-[4-(трет-бутилдиметилсиланилокси)фенил]-1-(4-фторфенил)-3-[3-(4-фторфенил)-3-оксопропил]азетидин-2-она (X, R4=TBDMS) Приблизительно 17 г соединения, полученного по примеру 4 (IX, R4=TBDMS) (с содержанием по меньшей мере 15,8 г, 28 ммоль), растворяют в 330 мл дихлорметана и прибавляют при 20-25 С 42 г монтмориллонита К 10. Гетерогенную смесь перемешивают при 20-25 С в течение 2-4 ч. Реакцию контролируют аналитически тонкослойной хроматографией. После исчезновения пятна исходного материала на хроматограмме реакционную смесь фильтруют, отфильтрованный монтмориллонит К 10 А сначала промывают 50 мл дихлорметана, а затем 350 мл смесью дихлорметана и метанола (2:1 об./об.). Объединнный фильтрат упаривают, осадок кристаллизуют из смеси этанола и воды при 0 С. Выход: 11,6 г высушенного продукта (80%, объединены стадии 4 и 5.) Точка плавления: 110-112 С[]25D =+4,0, (с=1, дихлорметан) Данные 1 Н ЯМР: (500 МГц, DMSO-d6, 25 С)0,16 (с, 3 Н), 0,17 (с, 3 Н), 0,93 (с, 9 Н), 2,12- 2,23 (м,2 Н), 3,14-3,30 (м, 3 Н), 4,99 (д, J = 2,3 Hz, 1 Н), 6,81-6,88 (м, 2 Н), 7,10-7,18 (м, 2 Н), 7,20-7,27 (м, 2 Н), 7,297,38 (м, 4 Н), 7,99-8,07 (м, 2 Н) м.д. Пример 6. Получение (3R,4S)-4-[4-(трет-бутилдиметилсиланилокси)фенил]-1-(4-фторфенил)-3-[(5)-3-(4-фторфенил)-3-гидроксипропил]азетидин-2-она (XIа) 5,00 г (9,6 ммоль) (3R,4S)-4-[4-(трет-бутилдиметилсиланилокси)фенил]-1-(4-фторфенил)-3-[3-(4 фторфенил)-3-оксопропил]азетидин-2-она растворяют в 9,6 мл не содержащего воду дихлорметана, и затем добавляют 0,5 М толуольный раствор 1,92 мл (0,96 ммоль) (R)-о-толил-CBS-оксазаборолидина. Смесь охлаждают до температуры в диапазоне от 0 до -5 С и при этой температуре в течение 6 ч прибавляют 1,9 мл 1,0 М дихлорметанового раствора боран-диметилсульфида. Реакционную смесь перемешивают при этой температуре, пока по данным тонкослойной хроматографии не исчезнет пятно исходного кетона. Затем добавляют 10 мл метанола, 0,5 мл 5%-ного раствора пероксида водорода и 10 мл 2 М серной кислоты. После перемешивания этой смеси в течение 0,5 ч фазы разделяют. Органическую фазу промывают 50 мл 2 N серной кислоты и затем 50 мл 5 %-ного раствора сульфита. Раствор сушат над безводным сульфатом натрия, фильтруют и упаривают. Выход: 5,05 г бесцветного масла. Диастереомерный избыток: 98% de (хиральная ВЭЖХ) Данные 1 Н ЯМР: (500 МГц, DMSO-d6, 25 С)0,17 (с, 3 Н), 0,18 (с, 3 Н), 0,93 (с, 9 Н), 1,65-1,94 (м,4 Н), 3,07-3,15 (м, 1H), 4,46-4,54 (м, 1H), 4,88 (д, J = 2,3 Hz, 1H), 5,29 (д, J = 4,5 Hz, 1H), 6,83-6,89 (м, 2 Н),7,07-7,17 (м, 4 Н), 7,19-7,25 (м, 2 Н), 7,27-7,34 (м, 4 Н) м.д. Пример 7. Получение(3R,4S)-1-(4-фторфенил)-3-[(S)-3-(4-фторфенил)-3-гидроксипропил]-4-(4-гидроксифенил)азетидин-2-она (I, эзетимиба) 5,0 г (9,6 ммоль) (3R,4S)-4-[4-(трет-бутилдиметилсиланилокси)фенил]-1-(4-фторфенил)-3-[(S)-3-(4 фторфенил)-3-гидроксипропил)азетидин-2-она (XI, R4=TBDMS) растворяют в 35 мл 2-пропанола и добавляют 10 мл 2 М серной кислоты. Раствор нагревают при 60-70 С в течение 1-2 ч и затем ему дают охладиться. Продукт кристаллизуют добавлением деионизованной воды. Кристаллический продукт отфильтровывают и промывают водой до нейтральной реакции. Выход: 3,2 г (81%, объединнные стадии 7 и 8.) Данные 1 Н ЯМР: (500 МГц, DMSO-d6, 25 С)1,65-1,92 (м, 4 Н), 3,05-3,13 (м, 1 Н), 4,46-4,55 (м, 1H),4,81 (д, J = 2,3 Hz, 1H), 5,29 (д, J = 3,7 Hz, 1H), 6,74-6,80 (м, 2 Н), 7,08-7,17 (м, 4 Н), 7,19-7,26 (м, 4 Н), 7,287,35 (м, 2 Н), 9,54 (м, 1H) м.д. Пример 8. Получение (Е)-[4-(трет-бутилдиметилсиланилокси)бензилиден]-(4-фторфенил)амина (VIIIa) 21,5 г (0,1 моль) (Е)-(4-гидроксибензилиден]-(4-фторфенил)амина (VIIIb) растворяют в 125 мл безводного тетрагидрофурана, к раствору добавляют 10,2 г (0,15 моль) имидазола и затем к нему добавляют по каплям 40 мл тетрагидрофуранового раствора 18,8 г (0,125 моль) трет-бутилдиметилсилилхлорида при 20-25 С. Реакционную смесь перемешивают при этой температуре до исчезновения в реакционной смеси, по данным тонкослойной хроматографии, исходного материала. Ожидаемое время реакции составляет 1-2 ч. Реакционную смесь разбавляют 50 мл толуола и выливают е в 100 мл воды. Водную фазу- 10014331 экстрагируют 50 мл толуола и затем объединнную органическую фазу промывают 350 мл воды до нейтральной реакции. Раствор упаривают и продукт кристаллизуют из холодного н-гексана. Выход: 28 г (85%). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения эзетимиба формулы I содержащий следующие стадии: а) превращение этиленгликолевого эфира 4-(4-фторбензоил)масляной кислоты формулы II в 4-[2-(4 фторфенил)-[1,3]диоксалан-2-ил]масляную кислоту формулы IV через невыделенное промежуточное соединение формулы III(b) ацилирование хирального соединения формулы V соединением формулы IV с получением ацилированного производного оксазолидинона формулы VI где соединение формулы V выбирают из соединений формул Va, Vb, Vc или Vd и где R1, R2 и R3 представляют собой: в случае Va: R1=Ph, R2=R3=H,в случае Vb: R1=R2=R3=Ph,в случае Vc: R1=метил, R2=Ph, R3=H,в случае Vd: R1=изопропил, R2=R3=Ph,и где Ph представляет собой фенильную группу. с) взаимодействие ацилированного производного оксазолидинона формулы VI с защищенным иминосоединением формулы VII и выделение соединения формулы VIII, где R4 представляет собой силильную группу циклизацию соединения формулы VIII с получением защищенного производного азетидинона общей формулы IXd) гидролиз кетальной группы соединения формулы IX с получением соединения формулы X е) энантиоселективное восстановление соединения общей формулы X с получением соединения формулы XI где одно из хиральных CBS-оксазаборолидиновых соединений формул XIIa, XIIb, XIIc и XIId выбрано в качестве катализатора иf) удаление силильной защитной группы в соединении общей формулы XI с получением конечного продукта, эзетимиба формулы I 2. Способ получения соединения формулы VIIIa включающий изомеризацию соединения формулы VIIIb в присутствии производного Ti(IV) где R представляет собой группу -СН 2-СН 2-ОН или -Н. 4. Соединение формулы VI(d): R1 =изопропил, R2 =R3=Ph,и где Ph представляет собой фенильную группу 5. Соединение формулы VIII где обозначения R1, R2, R3 независимо представляют собой Va, Vb, Vc или Vd и R4 является силильной защитной группой. 6. Соединение общей формулы IX где R4 является силильной защитной группой. 7. Соединение формулы Ха
МПК / Метки
МПК: C07D 317/30, C07D 405/06, C07D 205/08, C07F 7/02, C07D 413/06
Метки: используемых, промежуточных, получения, продуктов, эзетимиба, способе, этом, способ
Код ссылки
<a href="https://eas.patents.su/14-14331-sposob-polucheniya-ezetimiba-i-promezhutochnyh-produktov-ispolzuemyh-v-etom-sposobe.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения эзетимиба и промежуточных продуктов, используемых в этом способе</a>
Способ получения 7-альфа-карбоксил-9,11-эпоксистероидов и промежуточных продуктов, используемых для их получения, и общий способ эпоксидирования по олефиновым двойным связям

Номер патента: 9176
Опубликовано: 28.12.2007
Авторы: Виецзорек Джозеф, Ванцанелла Фортунато, Лю Синь, Баес Хулио А., Муссиарьелло Дженнаро, Нг Джон С., Поццо Марк Дж., Лосан Джон П., Кунда Сэстри А., Син Юань-Лун Л, Ван Пин Т., Андерсон Деннис К., Летендре Лео Дж., Эрб Дернхард
МПК: C07J 31/00, C07J 1/00, C07J 21/00...
Метки: олефиновым, 7-альфа-карбоксил-9,11-эпоксистероидов, эпоксидирования, способ, двойным, промежуточных, связям, продуктов, получения, используемых, общий
Формула / Реферат:
1. Способ получения соединения формулы в которой -A-A- обозначает группу -CHR4-CHR5- или -CR4=CR5-, R3, R4 и R5 независимо друг от друга выбирают из группы, включающей водород, галоген, гидроксил, C1-C7алкил, C1-C7алкоксигрушгу, гидроксиалкил, алкоксиалкил, гидроксикарбонил, цианогруппу, арилоксигруппу, R1 обозначает a-ориентированный C1-C7алкоксикарбонильный или гидроксикарбонильный радикал, -B-B- обозначает группу -CHR6-CHR7- либо a- или...
Способ получения иопамидола и новые промежуточные соединения, получаемые в этом способе

Номер патента: 5922
Опубликовано: 25.08.2005
Авторы: Анелли Пьер Лучио, Броккетта Марино, Лукс Джованна, Каппеллетти Энрико
МПК: C07C 237/46
Метки: соединения, промежуточные, способе, способ, иопамидола, этом, получения, получаемые, новые
Формула / Реферат:
1. Способ получения (S)-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-5-[(2-гидрокси-1-оксопропил)амино]-2,4,6-трииодо-1,3-бензолдикарбоксамида формулы (I) исходя из соединения формулы (II) причем указанный способ включает а) введение соединения формулы (II) в реакцию с подходящим защитным агентом с образованием соединения формулы (III) где -R - группа формулы A или B где R1 - атом водорода, C1-C4 линейная или разветвленная алкильная группа или...
Способ получения r-альфа-пропил-пиперониламина и его аналогов, промежуточные продукты, используемые в этом способе.

Номер патента: 1207
Опубликовано: 25.12.2000
Авторы: Анзалоне Луиджи, Ли Хью-Йин, Вальтермир Роберт Юджин
МПК: C07D 317/58
Метки: способе, r-альфа-пропил-пиперониламина, способ, используемые, этом, продукты, промежуточные, получения, аналогов
Формула / Реферат:
1. Способ получения R-a-пропил-пиперониламина формулы I: или его стереоизомера, или соли, включающий: (а) взаимодействие соединения формулы II: с хиральным метилбензиламином с получением соединения формулы III: или его стереоизомера, где R выбирают из Н, ОН и ОСН3; (b) гидрирование полученного соединения формулы III или его стереоизомера, или соли в присутствии Ra-Ni (Ni Ренея) с получением соединения формулы IV: или его...
Способ получения промежуточных продуктов

Номер патента: 13842
Опубликовано: 30.08.2010
Авторы: Ван Линьсуа, Оудом Франки Ли, Долбир Кристин Андерсон
МПК: C07C 17/093, C07C 25/02
Метки: продуктов, способ, промежуточных, получения
Формула / Реферат:
1. Способ получения соединения формулы Iв которой R0, все независимо друг от друга, обозначают галоген, C1-С6алкил, С2-С6алкенил, С2-С6алкинил, C1-С6галогеналкил, циано-С1-С6алкил, С2-С6галогеналкенил, циано-С2-С6алкенил, С2-С6галогеналкинил, циано-С2-С6алкинил, гидроксигруппу, гидрокси-С1-С6алкил, C1-С6алкоксигруппу, нитрогруппу, аминогруппу, C1-С6алкиламиногруппу, ди(С1-С6алкил)аминогруппу, С1-С6алкилкарбониламиногруппу,...
Способ получения промежуточных продуктов, применяемых при синтезе производных фенилгидразина, обладающих инсектицидной активностью

Номер патента: 4940
Опубликовано: 28.10.2004
Автор: Дзин Хайхонг
МПК: C07C 201/08
Метки: продуктов, обладающих, фенилгидразина, промежуточных, получения, применяемых, синтезе, способ, производных, активностью, инсектицидной
Формула / Реферат:
1. Способ получения соединения, имеющего структурную формулу где X представляет a) фенил, фенил-C1-C4алкокси; фенокси или бензил; либо b) один заместитель из группы a) и один или несколько заместителей, выбранных из C1-C4алкокси, гидроксила, галогена, C1-C6алкила и C1-C6алкилтио; либо c) вместе с фенилом, к которому он присоединен, образует полициклический конденсированный гетероцикл, включающий следующие стадии: A) выбор в качестве...
Предыдущий патент: Пептидные ингибиторы пути трансдукции сигнала jnk, обладающие способностью проникать в клетку
Следующий патент: Селекция клеток-хозяев, экспрессирующих белок на высоких уровнях
Случайный патент: Способ и система для измельчения и очистки пластиковых отходов и способ получения топлива из переработанных отходов