Пептидные производные – ингибиторы слияния при вич-инфекции






Формула / Реферат
1. Изолированный пептид FB005, содержащий последовательность SEQ ID NO:1.
2. Изолированный пептид FB006, содержащий последовательность SEQ ID NO:2.
3. Изолированный пептид FB066, содержащий последовательность SEQ ID NO:7.
4. Изолированный модифицированный пептид, выбранный из группы, состоящей из:
(a) SEQ ID NO:1;
(b) SEQ ID NO:2;
(c) SEQ ID NO:3 и
(d) SEQ ID NO:7,
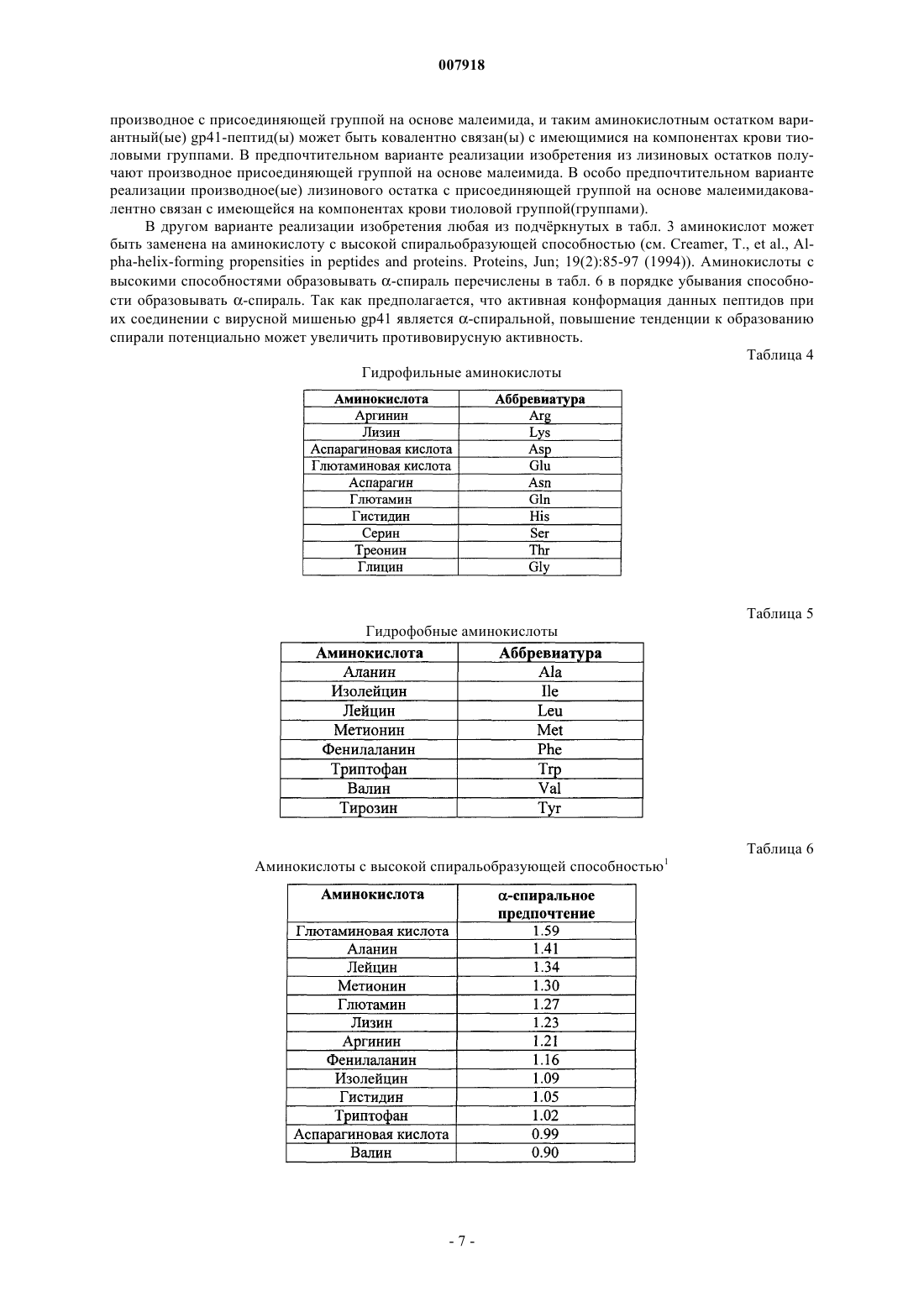
и имеющий по меньшей мере одну замену аминокислотного остатка в предварительно определённом положении пептидной цепи, при этом по меньшей мере один привнесённый аминокислотный остаток является гидрофильным аминокислотным остатком, гидрофобным аминокислотным остатком, аминокислотным остатком со склонностью образовывать альфа-спирали, D-изомером природной L-аминокислоты или не встречающимся в природе аминокислотным остатком.
5. Производное изолированного пептида, которое выбирают из группы, состоящей из:
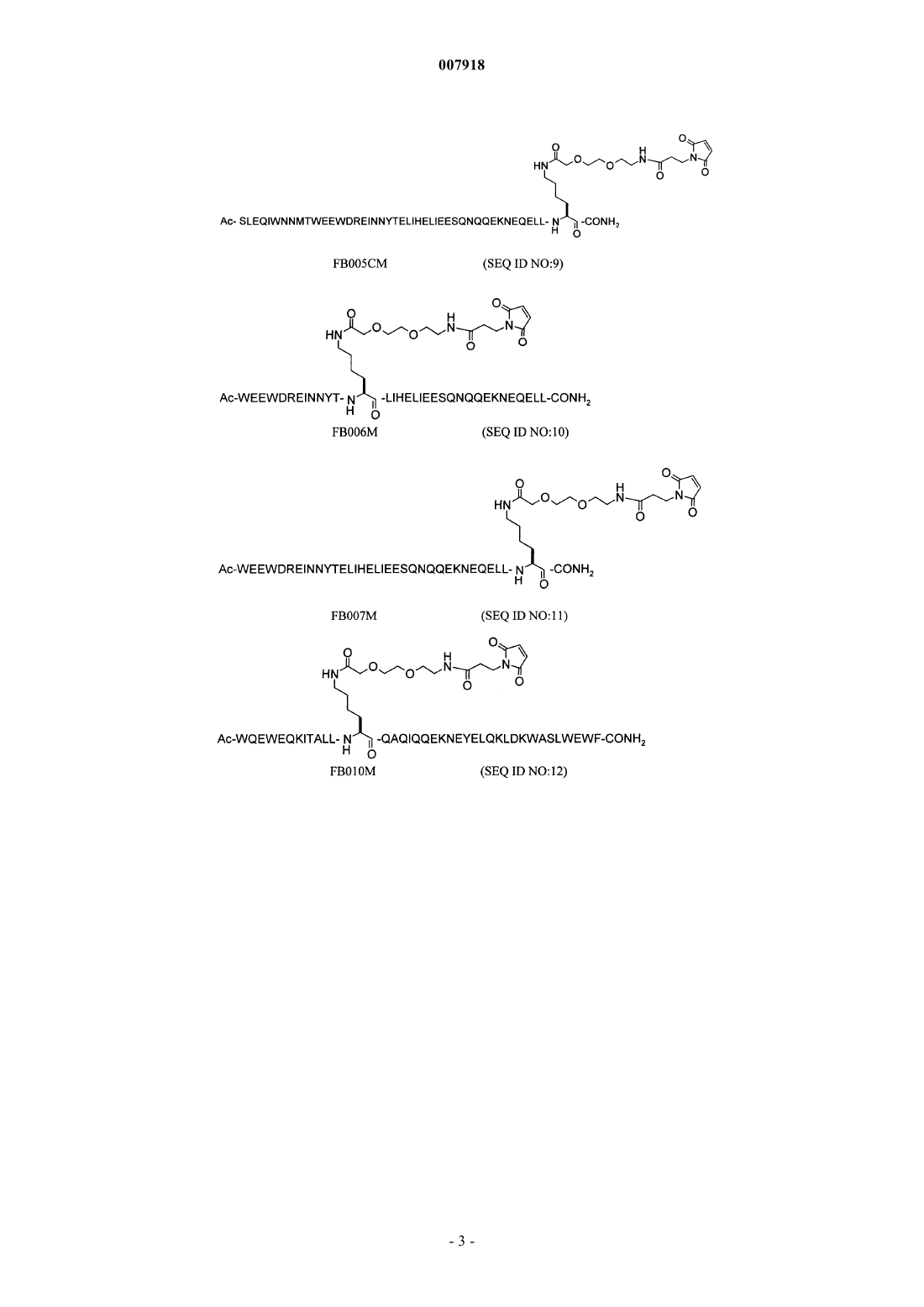
(a) пептида FB005M последовательности SEQ ID NO:8;
(b) пептида FB005CM последовательности SEQ ID NO:9;
(c) пептида FB006M последовательности SEQ ID NO:10;
(d) пептида FB007M последовательности SEQ ID NO:11;
(e) пептида FB010M последовательности SEQ ID NO:12;
(f) пептида FB010KM последовательности SEQ ID NO:13;
(g) пептида FB066M последовательности SEQ ID NO:14 и
(h) пептида FB066KM последовательности SEQ ID NO:15.
6. Производное изолированного пептида, выбранное из группы, состоящей из:
(a) SEQ ID NO:1;
(b) SEQ ID NO:2;
(c) SEQ ID NO:3 и
(d) SEQ ID NO:7,
причем производные заданных остатков аминокислот пептидной цепи получены путём конъюгации указанных заданных аминокислотных остатков с присоединяющими группами.
7. Модифицированный пептид по п.4, отличающийся тем, что производные заданных остатков аминокислот пептидной цепи получены путём конъюгации указанных аминокислотных остатков с присоединяющими группами.
8. Изолированный пептид по п.1, отличающийся тем, что представляет собой производное, полученное путём прикрепления присоединяющей группы к лизину, замещающему остаток глутаминовой кислоты в положении 23 или добавленному с С-конца.
9. Изолированный пептид по п.2, отличающийся тем, что он представляет собой производное, полученное путём прикрепления присоединяющей группы к лизину в положении 13.
10. Изолированный пептид по п.2, отличающийся тем, что он модифицирован путём замены лизина в положении 13 на остаток глутаминовой кислоты, и из такого пептида получено производное путём прикрепления присоединяющей группы к дополнительному лизиновому остатку, добавленному с С-конца.
11. Производное изолированного пептида, содержащее последовательность SEQ ID NO:3, отличающееся тем, что пептид модифицирован путём замены остатка глутаминовой кислоты в положении 13 на лизин и прикрепления присоединяющей группы к лизину, или получено производное пептида путём конъюгации присоединяющей группы с остатком лизина, добавленным с С-конца.
12. Изолированный пептид по п.3, из которого получено производное путём прикрепления присоединяющей группы к лизину в положении 13 или дополнительному лизиновому остатку, добавленному с С-конца.
13. Производное пептида по любому из пп.5-12, в котором присоединяющая группа выбрана из:
(a) малеимидной группы;
(b) сукцинимидильной группы;
(c) гидразиновой группы и
(d) карбонильной группы.
14. Производное пептида по п.13, в котором малеимидная группа связана через 3'-малеимидопропионат с ипсилон-аминогруппой лизина при помощи [2-(2-амино)этоксил]этоксиуксусной кислоты.
15. Фармацевтический состав, содержащий пептид по любому из пп.1-4 или производное пептида по любому из пп.5-12.
16. Конъюгат, содержащий производное пептида по любому пп.5-12, конъюгированное с компонентом крови.
17. Конъюгат по п.16, у которого компонент крови выбран из группы, состоящей из следующих белков:
(a) сывороточный альбумин человека;
(b) трансферрин человека;
(c) ферритин человека;
(d) иммуноглобулины человека;
(e) ферритин человека;
(f) a-2-макроглобулин человека;
(g) тироксинсвязывающий белок человека;
(h) стероидсвязывающие белки человека и
(i) их сочетания.
18. Способ подавления вирусной инфекции в клетках млекопитающего, который включает доставку к указанным клеткам пептида по любому из пп.1-4 или производного пептида по любому из пп.5-12.
19. Способ профилактики вирусной инфекции в клетках млекопитающего, который включает доставку к указанным клеткам пептида по любому из пп.1-4 или производного пептида по любому из пп.5-12.
20. Способ предотвращения репликации вируса в клетках млекопитающего, который включает доставку к указанным клеткам пептида по любому из пп.1-4 или производного пептида по любому из пп.5-12.
21. Способ по любому из пп.18-20, в котором упомянутый пептид доставляют в присутствии упомянутого вируса.
22. Способ по любому из пп.18-20, в котором вирус выбирают из группы, включающей:
(a) вирус иммунодефицита человека (ВИЧ) и
(b) вирус иммунодефицита обезьян (ВИО).
23. Способ по любому из пп.18-20, в котором пептид или производное пептида назначают перорально, местно, внутривенно, внутриартериально, внутримышечно или подкожно.
24. Способ по любому из пп.18-20, в котором пептид или производное пептида назначают совместно с одним или более препаратами для лечения ВИЧ-инфекции.
25. Способ по любому из пп.18-20, в котором указанный один или более дополнительный препарат для лечения ВИЧ-инфекции представляет собой вариантный gp41-пептид.
26. Способ по п.24, в котором дополнительный препарат для лечения ВИЧ-инфекции выбирают из группы, включающей следующие препараты:
(a) AGENERASE;
(b) COMBIVIR;
(c) CRIXIVAN;
(d) EMTRIVA;
(e) EPIVIR;
(f) FORTOVASE;
(g) HIVID;
(h) INVIRASE;
(i) KALETRA;
j) NORVIR;
(k) RESCRIPTOR;
(l) RETROVIR;
(m) REYATAZ;
(n) SUSTIVA;
(o) TRIZIVIR;
(p) VIDEX EC;
(q) VIDEX;
(r) VIRACEPT;
(s) VIRAMUNE;
(t) VIREAD;
(u) ZERIT и
(v) ZIAGEN.
27. Способ по любому из пп.18-20, в котором вирус представляет собой ВИЧ, а клетки млекопитающего представляют собой клетки человека.
28. Способ профилактики или подавления ВИЧ-инфекции, согласно которому назначают производные вариантных gp41-пептидов по любому из пп.5-12 пациенту, чьи клетки подверглись контакту с ВИЧ, при этом указанное производное пептида конъюгируют с компонентом крови указанного пациента, увеличивая таким образом период полураспада пептида в крови указанного пациента.
29. Способ создания противовирусного конъюгата, который включает смешивание производного вариантного gp41-пептида (или пептидов) с компонентами крови с формированием ковалентных связей между производным вариантного gp41-пептида и компонентами крови.
30. Способ по любому из пп.27, 28, в котором компонент крови выбирают из группы, включающей следующие белки:
(a) сывороточный альбумин человека;
(b) трансферрин человека;
(c) ферритин человека;
(d) иммуноглобулины человека;
(e) ферритин человека;
(f) a-2-макроглобулин человека;
(g) тироксинсвязывающий белок человека;
(h) стероидсвязывающие белки человека и
(i) их сочетания.
31. Способ по п.29, в котором компонент крови представляет собой сывороточный альбумин человека.
32. Способ по любому из пп.27, 28, в котором конъюгация происходит in vivo.
33. Способ по любому из пп.27, 28, в котором конъюгация происходит ex vivo.
34. Способ по п.32, в котором перед конъюгацией с производным пептида компонент(ы) крови разделяют при помощи плазмафореза.
35. Фармацевтический состав, содержащий изолированный пептид по любому из пп.1-14 с фармацевтически приемлемым носителем.
36. Способ генерирования пептидов с противовирусным, виростатическим шыш антифузогенным действием, включающий:
(a) скрининг белков вирулентности вируса для определения в них последовательностей с альфа-спиральобразующими свойствами;
(b) создание изменённого пептида путём модификации или получения производного хотя бы одного аминокислотного остатка указанной последовательности;
(c) синтез указанных изменённых пептидов и
(d) тестирование указанных пептидов для определения их противовирусного, виростатического или антифузогенного действия.
Текст
007918 Область техники Данное изобретение относится к производным С-концевого пептида gp41 вируса иммунодефицита человека (далее "ВИЧ"), являющимся ингибиторами вирусной инфекции и/или проявляющим антифузогенные свойства. В частности, данное изобретение относится к пептидным производным, обладающим ингибирующей активностью по отношению к ВИЧ и вирусу иммунодефицита обезьян (далее "ВИО"),имеющим улучшенную растворимость и увеличенное время действия, которые используют при лечении соответствующих вирусных инфекций. Уровень техники Слияние мембран является обычным явлением в нормальных биологических процессах клетки,кроме того, слияние мембран задействовано в различных патологических состояниях, включая, например, проникновение вируса, окружнного внешней оболочкой, в клетку. Процесс слияния некоторых вирусов с внешней оболочкой с клетками-мишенями происходит при помощи специфических реакций связывания между белками внешней оболочки вируса (envelop) и белками на поверхности клетки, что приводит к конформационным изменениям ассоцированных вирусных белков, что, в свою очередь, обеспечивает слияние вирусной оболочки с мембраной клетки. Одним из вирусов, обладающих внешней оболочкой, является ВИЧ, который относится к семейству ретровирусов, подсемейству лентивирусов. Существует два основных варианта ВИЧ, ВИЧ-1 и ВИЧ-2,для каждого из которых выделено несколько серотипов. Слияние ВИЧ с клетками хозяина осуществляется путм связывания белков оболочки вируса gp120 и gp41 с гликопротеином CD4 и хемокиновым корецептором на поверхности клетки. После связывания gp120 с CD4 и с корецептором (например, CCR5 или CXCR4) на поверхности Т-лимфоцита, происходит встраивание gp41 в мембрану клетки-мишени; после этого спиральные участки N-концевой области gp41 образуют структуры типа скрученной спирали(coiled coil) со спиралями С-концевой области этого же белка, что сближает вирус с клеткой для последующего слияния (Malashkevich, et al., Proc. Natl. Acad. Sci. USA, 1998 Aug 4; 95(16):9134-9). Известно, что пептиды ингибируют или другим способом нарушают процессы, связанные со слиянием мембран, в том числе, например, ингибируют передачу ретровируса в неинфицированные клетки. Было показано, что пептиды участка второго гептадного повтора белка внешней оболочки ВИЧ - gp41, в т.ч. Т 20 (DP178) и С 34 обладают сильной противовирусной активностью по отношению к ВИЧ in vitro(см. Wild, et al., 1994, Proc. Natl. Acad. Sci. USA, 91:9770-4; Chan, et al., 1998, Proc. Natl. Acad. Sci. USA,95:15613-15617). Продемонстрированная противовирусная активность включает в себя ингибирование инфекции CD4+-клеток свободным вирусом и/или ингибирование индуцируемого ВИЧ формирования синцитиев между инфицированными и интактными CD4+-клетками. Предполагается, что ингибирование происходит за счт связывания этих пептидов с участком первого гептадного повтора gp41, что предотвращает образование участками первого и второго гептадного повтора фузогенной шпилечной структуры (hairpin structure). Хотя многие противовирусные или антифузогенные белки, описанные в данной области, проявляют сильную противовирусную и/или антифузогенную активность in vitro, in vivo они имеют короткий период полураспада, главным образом из-за быстрого клиренса из сыворотки и действия пептидаз и протеиназ. Это, в свою очередь, сильно снижает их противовирусную эффективность. Следовательно, существует потребность в создании способа продления периода полураспада пептидов in vivo без значительного нарушения их антифузогенной активности. Один из способов продления периода полураспада пептидов приведн в патенте США 5612034, в котором описан способ ковалентного связывания терапевтического пептида с природным белком, имеющимся в кровотоке. В этом случае к пептиду добавляется химически активная часть, которая способна реагировать с функциональными группами белков крови. После введения модифицированного пептида в кровь он связывается с долгоживущим компонентом крови, формируя долговременное депо этого пептида. Однако из-за того, что молекулярная масса белков крови находится в пределах 50-600 кДа, существуют опасения, что биологическая активность пептидов, связанных таким способом, может быть нарушена вследствие стерического влияния гораздо большего по размеру белка. Попытка увеличить период полураспада известного антифузогенного белка описана в публикации заявки WO 00/69902 (далее "публикация '902") фирмой Conjuchem, Inc. В описании указанной заявкиDP178 модифицируют присоединением 3-малеимидопропионовой кислоты при помощи амидной связи к ипсилон-аминогруппе лизина, который, в свою очередь, пептидной связью соединн с С-концевым PheDP178. Публикация '902 также предлагает аналоги модифицированного DP178, которые представляют собой либо усеченные формы DP178, либо соответствующие фрагменты gp41 других выделенных разновидностей ВИЧ. Публикация '902 не предлагает никаких других подходов к созданию антифузогенных пептидов. Таким образом, остатся потребность в создании способа, который обеспечивает увеличение периода полураспада пептидов in vivo без значительного нарушения их антифузогенной активности. Краткое описание изобретения Данное изобретение относится к пептидным производным gp41 ВИЧ, обладающим противовирусной, виростатической и/или антифузогенной активностью, в том числе, но не только, модифицированными пептидами, приведенными в табл. 1, 2 и 3 и фиг. 1, а также их модифицированные формы и произ-1 007918 водные (далее обобщнно называемые "вариантные gp41 пептиды"). Такие вариантные gp41 пептиды обеспечивают повышенную стабильность in vivo и большую устойчивость к пептидазной или протеазной деградации. В результате вариантные gp41 пептиды сводят к минимуму потребность в более частом, или даже непрерывном введении, которое потребовалось бы при использовании немодифицированных пептидов gp41 ВИЧ. Настоящие пептидные производные, а также производные, полученные согласно способам данного изобретения на основе gp41-подобных последовательностей других вирусов, можно использовать, например, в качестве профилактического средства и/или для лечения инфекций, вызванных некоторыми вирусами, в том числе, но не только, ВИЧ и ВИО. В соответствии с настоящим изобретением ниже описаны пептидные производные, имеющие улучшенную растворимость и противовирусную активность по сравнению с соответствующей немодифицированной пептидной последовательностью gp41 ВИЧ. В особенности данное изобретение касается соединений, формулы которых описаны ниже в табл. 1, 2 и 3 и на чертеже, куда относятся пептидные производные, способные реагировать in vivo или ex vivo с тиоловыми группами компонентов крови с образованием стабильной ковалентной связи. Таблица 1 Пептидные фрагменты gp41 и модифицированные аналоги Данное изобретение описывает новые составы, содержащие пептиды, в которых по сравнению с природным пептидом присутствуют модификации определнных остатков аминокислот (т.е. точечные мутации), которые введены для улучшения активности и растворимости. Вышеуказанные остатки аминокислот представляют собой подчркнутые остатки аминокислот в пептидных последовательностях из табл. 3. Пептиды с модифицированными остатками аминокислот включают в себя, но не ограничиваются пептидами, содержащими с замены природных остатков аминокислот на аминокислотные остатки,имеющие повышенные гидрофильные или гидрофобные свойства. В вариантных gp41-пептидах аминокислотные остатки также могут быть заменены на остатки, имеющие повышенную способность к формированию альфа-спирали. Другие пептиды с модифицированными остатками аминокислот включают в себя, но не ограничиваются пептидами, содержащими производные аминокислотных остатков, в которых связующая группа конъюгирована с заранее определнным аминокислотным остатком, что позволяет получать ковалентное связывание пептидного производного с компонентом крови. В другом своем аспекте данное изобретение относится к фармацевтическим составам, содержащим производные вышеперечисленных формул в комбинации с фармацевтически приемлемым носителем. Такие составы можно использовать для подавления активности ВИЧ (включая ВИЧ-1, ВИЧ-2 и их серотипы) и ВИО. В качестве следующего варианта реализации настоящего изобретения описан способ подавления инфекции, вызванной ВИЧ или ВИО. Способ состоит в назначении субъекту, предпочтительно млекопитающему, ещ более предпочтительно человеку, одного или более вариантных пептидов gp41 в количестве, эффективном для ингибирования вируса, отдельно или в сочетании с фармацевтическим носителем,либо в сочетании с другими противовирусными агентами, включая другие вариантные gp41-пептиды. Особо предпочтительным вариантом реализации данного изобретения является назначение субъекту вирус-ингибирующей дозы хотя бы одного из вариантных gp41-пептидов, отдельно или в сочетании с фармацевтическим носителем, либо в сочетании с другими противовирусными агентами, включая другие вариантные gp41-пептиды. Ещ в одном своем аспекте настоящее изобретение относится к конъюгату, который содержит по меньшей мере один вариантный gp41-пептид, ковалентно связанный с компонентом крови. В одном из вариантов реализации данного изобретения предпочтительные компоненты крови для реакции с соединениями согласно настоящему изобретению включают такие белки, как иммуноглобулины, в т.ч. IgG иIgM, сывороточный альбумин, ферритин, стероидсвязывающие белки, трансферрин, тироксинсвязывающий белок, -2-макроглобулин и т.д., при этом сывороточный альбумин и IgG являются наибо-4 007918 лее предпочтительными вариантами, а сывороточный альбумин является самым предпочтительным вариантом реализации данного изобретения. Ещ в одном своем аспекте настоящее изобретение относится к способу увеличения периода полураспада вариантных gp41-пептидов in vivo, заключающемуся в ковалентном связывании одного или более вариантных gp41-пептидов с компонентом крови. Другой аспект настоящего изобретения относится к способу конструирования, синтеза и тестирования новых пептидов, обладающих противовирусной, виростатической или антифузогенной активностью по отношению к различным вирусам. Способ включает скрининг вирусных белков, участвующих в проникновении вируса в клетку, для определения в них пептидных последовательностей, обладающих свойствами формировать альфа-спирали, и разработке на основе этих пептидов составов, которые можно использовать для лечения заболевания, вызванного этим вирусом. Кроме того, указанный способ также предусматривает проверку пептидов in vitro для подтверждения их противовирусной, виростатической или антифузогенной активности. Краткое описание чертежа На чертеже показаны линейные последовательности различных пептидов, описанных в данном изобретении. Подробное описание изобретения В данном документе термин "получение производного" означает добавление к пептидным последовательностям групп, по которым происходит присоединение. Типичные присоединяющие группы более подробно описаны ниже. В данном документе термин "модификация" означает замену одной аминокислоты в природной пептидной последовательности на другую аминокислоту. Вторая аминокислота может быть выбрана из неограниченной группы гидрофильных аминокислот, гидрофобных аминокислот, аминокислот со спиральобразующими свойствами, не встречающихся в природе аминокислот и D-изомеров природных L-аминокислот. Слияние (фузия) ВИЧ-1 и родственных лентивирусов с клетками-мишенями может быть ингибировано пептидными фрагментами природных белков наружной оболочки вируса, осуществляющих слияние. Эти пептидные фрагменты могут связываться с белками оболочки и блокировать связывание дистальных участков белков оболочки, таким образом, ингибируя конформационные изменения природного белка, которые необходимы для осуществления слияния ВИЧ-1 с клеткой-мишенью. Такие пептиды прерывают инфекционный процесс, необходимый для прогрессирования заболевания, посредством блокирования слияния вируса с клетками. Настоящее изобретение улучшает свойства существующих противовирусных и антифузогенных(блокирующих слияние) пептидов и представляет новые пептидные составы, которые можно использовать для лечения ВИЧ и ВИО. Вирусы, развитие которых могут ингибировать пептиды согласно настоящему изобретению, включают в себя, но не ограничиваются, ретровирусы человека - ВИЧ (в т.ч. ВИЧ-1 и ВИЧ-2, а также их серотипы) и ВИО. Модифицированные пептиды В соответствии с данным изобретением могут быть получены модифицированные и производные пептиды с антифузогенной активностью по отношению к лентивирусам. Антифузогенные пептиды представляют собой спиральобразующие пептиды на основе природной последовательности белка gp41, которые модифицированы путм замены определнных аминокислот в пептидах. Модифицированные аминокислоты выбраны таким образом, чтобы избежать нарушения взаимодействий, способствующих формированию скрученных спиральных комплексов со спиральными участками белка оболочки вирусаgp41. В одном варианте реализации для модификации выбирают остатки аминокислот, боковые цепи которых находятся в стороне от области образования скрученной спирали. Эти остатки заменяют на альтернативные остатки, которые усилят гидрофобные или гидрофильные свойства пептидов, или из них получают производные для создания реакционных групп, которые обеспечат ковалентное связывание пептидов с циркулирующими в крови белками. Введение гидрофильных остатков в пептидную последовательность повысит растворимость пептида. Введение гидрофобных остатков в пептидную последовательность уменьшит растворимость пептида. В одном из вариантов реализации изобретения модифицированные пептиды включают в себя пептиды, обозначенные как FB005, FB006 и FB066, в особенности производные этих пептидов с присоединяющими группами на основе малеимида, например, 3 малеимидопропионовой кислоты, присоединнной к лизину через [2-(2-аминоэтокси)этокси]уксусную кислоту, или других эквивалентных присоединяющих структур. В другом варианте реализации изобретения аминокислоты в пептидной последовательности заменяют на аминокислоты со способностью формировать альфа-спирали. В качестве альтернативы химические группы могут быть добавлены с амино- и/или карбоксиконцов, например, для того, чтобы увеличить стабильность, реактивность и/или растворимость пептидов. Например, гидрофобные группы, такие как карбобензоксильная, дансильная, ацетильная или тбутилоксикарбонильная группы могут быть присоединены по аминоконцу пептида. Аналогичным образом ацетильная группа или 9-флюоренилметоксикарбонильная группа может быть присоединена по аминоконцу пептида. Дополнительно гидрофобная группа, т-бутилоксикарбонильная или амидная группа-5 007918 может быть добавлена к карбоксиконцу пептида. Аналогичным образом паранитробензилэфирная группа может быть размещена на карбоксиконце пептида. Техника произведения таких модификаций хорошо известна специалистам в данной области. Вышеуказанные пептиды можно синтезировать с изменнной стерической конфигурацией. Например, можно использовать D-изомер вместо обычного L-изомера одного или более аминокислотных остатков. В одном варианте реализации данного изобретения не менее двух аминокислот представляют собой D-изомеры природных L-аминокислот. В другом варианте реализации каждая из природных Lаминокислот в полной пептидной последовательности заменена на D-изомер той же самой кислоты. Изобретение также предусматривает вариант, когда хотя бы один из аминокислотных остатков вариантногоgp41-пептида заменяют на один из хорошо известных не встречающихся в природе аминокислотных остатков. В другом варианте реализации настоящего изобретения в вариантном gp41-пептиде осуществляют любую комбинацию замен на D-изомеры природных L-аминокислот или на аминокислоты, не встречающиеся в природе. Подобными изменениями можно увеличить стабильность, протеиназную резистентность, активность, реактивность и/или растворимость вариантных gp41-пептидов. Не встречающиеся в природе аминокислоты хорошо известны в данной области. Кроме того, способы синтеза пептидов, содержащих D-изомеры природных L-аминокислот или не встречающиеся в природе аминокислоты хорошо известны в данной области (см., например, описания патентов US PatentNos. 5840697 и 6268479, а также Biochemistry (Chap. 4), D. Voet and J.G. Voet, WileySons (1990), которые упомянуты в качестве справки), и также рассматриваются в данном изобретении. В одном из вариантов реализации настоящего изобретения модифицированные пептиды включают в себя пептиды, обозначенные как FB005, FB006 и FB066, в особенности производные этих пептидов,содержащие присоединяющие группы на основе малеимида, например, 3-малеимидопропионовую кислоту, соединнную с лизином посредством [2-(2-аминоэтокси)этокси]уксусной кислоты или другие эквивалентные присоединяющих структур. Изобретение также охватывает вариантные gp41-пептиды, у которых аминокислотные остатки заменены либо на гидрофильные, либо на гидрофобные остатки, что изменяет свойства растворимости этих пептидов. В качестве альтернативы из других аминокислотных остатков вариантных gp41-пептидов получают производные путем присоединения присоединяющей группы на основе малеимида части. В предпочтительном варианте реализации данного исследования подчркнутые аминокислотные остатки нижеследующих вариантных gp41-пептидов (описанных в табл. 3) заменены на гидрофильные или гидрофобные остатки, либо из них получены производные с присоединяющей группой на основе малеимида. В любых других пептидах, охваченных областью настоящего изобретения и имеющих С-концевой лизиновый остаток, этот С-концевой лизиновый остаток может также быть заменн гидрофильными остатками, либо из него получено производное со связующей частью малеимида. Таблица 3 Гидрофильные аминокислоты, на которые можно заменить любые из подчркнутых аминокислотных остатков, перечислены в табл. 4. Гидрофобные аминокислоты, на которые можно заменить любые из подчркнутых аминокислот, перечислены в табл. 5. Дополнительно с использованием любого из подчркнутых аминокислот из табл. 3 можно получить-6 007918 производное с присоединяющей группой на основе малеимида, и таким аминокислотным остатком вариантный(ые) gр 41-пептид(ы) может быть ковалентно связан(ы) с имеющимися на компонентах крови тиоловыми группами. В предпочтительном варианте реализации изобретения из лизиновых остатков получают производное присоединяющей группой на основе малеимида. В особо предпочтительном варианте реализации производное(ые) лизинового остатка с присоединяющей группой на основе малеимидаковалентно связан с имеющейся на компонентах крови тиоловой группой(группами). В другом варианте реализации изобретения любая из подчркнутых в табл. 3 аминокислот может быть заменена на аминокислоту с высокой спиральобразующей способностью (см. Creamer, Т., et al., Alpha-helix-forming propensities in peptides and proteins. Proteins, Jun; 19(2):85-97 (1994. Аминокислоты с высокими способностями образовывать -спираль перечислены в табл. 6 в порядке убывания способности образовывать -спираль. Так как предполагается, что активная конформация данных пептидов при их соединении с вирусной мишенью gp41 является -спиральной, повышение тенденции к образованию спирали потенциально может увеличить противовирусную активность. Таблица 4 Гидрофильные аминокислоты Аминокислоты с высокой спиральобразующей способностью 1 В общем случае пептиды согласно настоящему изобретению являются аналогами С-34, состоящего из пяти гептад одной альфа-спирали скрученного спирального белкового комплекса, при этом предпочтительные аналоги имеют присоединяющие группы на основе малеимида, и их остатки более полярны по сравнению с родительскими остатками, они заменены в 2 из 7 остатков первой гептады, 6 из 7 остатков второй гептады, 3 из 7 остатков третьей гептады и/или 7 из 7 остатков четвртой гептады. В другом варианте реализации пептиды изобретения охватывают вышеперечисленные пептиды, но в них дополнительно включены 10 остатков аминокислот из gp41, добавленных с N-конца пептида С-34. Пептид FB006 основан на пептиде С 34 со вторым и семнадцатым остатком, мутированным в глютамат, а тринадцатым остатком, мутированным в лизин. Места мутации были выбраны с учтом кристаллической структуры комплекса N36/C34. Критерием выбора является то, что эти остатки не задействованы в связывании со спиралями N36. Мутации в глютамат и лизин нацелены на улучшение растворимости и спиральобразующей способности, т.е. тенденции образовывать спираль в водном растворе. Так как предполагается,что активная конформация С 34 является альфа-спиральной, например, в кристаллической структуре комплекса N36/C34, то повышение способности к образованию спирали должно увеличивать биологическую активность. Пептиды FB005, FB006, FB066, FB005M, FB005CM, FB006M и FB007M также имеют такие замены. К вариантным пептидам gp41 относятся пептидные последовательности, приведнные в табл. 1, 2 и 3 на фиг. 1, а также их модифицированные формы и производные. Пептид FB005 основан на пептидеFB006, но дополнительно имеет 10 аминокислотных остатков, расположенных с N-конца и родственных другим вариантным gp41-пептидам. Пептид FB066 основан на пептиде FB006. Он отличается от FB006 тем, что имеет одну аминокислотную замену, в нм лизин в положении 28 заменяют на глютаминовую кислоту. Это оставляет 13-й аминокислотный остаток единственным лизиновым остатком, функционирующим как сайт конъюгации. Такое изменение значительно облегчает синтез аналогов с малеимидными модификациями. Изобретение также относится к производным на основе FB005, FB006, и Т-1249 (см. WO 01/03723), которые конъюгируют с сывороточным альбумином и таким образом увеличивают время жизни ингибиторов. ПептидыFB005M и FB005CM основаны на последовательности FB005; пептиды FB006M и FB007M основаны на последовательности FB006; пептиды FB010M и FB010KM основаны на последовательности Т-1249. Новым также является способ выбора места присоединения пептида к белку крови. Изобретатели обнаружили, что связывание вариантного gp41-пептида с альбумином через внутренний лизиновый остаток пептида делает конъюгат более эффективным по сравнению с присоединением по С-концу. ЗначенияIC50 (значение IC50 - это концентрация препарата, требуемая для достижения угнетения вируса на 50%, а значение ТС 50 - это концентрация препарата, при которой достигается цитотоксичность 50%) для FB006,FB006M и FB007M составляют 1,4; 3,9 и 9,1 нМ соответственно. FB006 является естественным пептидом, FB006M является модифицированным комплексом, имеющим малеимидную связь по 13-му остатку,a FB007M связан через С-конец. Когда FB006M связывается с сывороточным альбумином, его количество, требуемое для достижения противовирусного эффекта, возрастает в 2,8 раза, в то время как связывание с альбумином через С-конец у FB007M вызывает повышение значения IC50 в 6,5 раза. Несмотря на то, что ожидали увеличения периода полураспада пептида при его связывании с молекулой-носителем,концептуально конъюгация с альбумином (белок массой 66 кДа) должна блокировать биологическую активность пептидов в результате стерических помех. Однако, когда изобретатели получили пептидыFB006M, связанные с альбумином, совершенно неожиданно выяснилось, что противовирусная активность комплекса существенно не нарушилась (повышение только в 2,8 раза). Присоединяющие группы в данном изобретении - это химические группы, способные формировать ковалентные связи с функциональными структурами, имеющимися на компоненте крови. Присоединяющие группы обычно стабильны в водной среде. Реактивные функциональные структуры для ковалентного связывания с присоединяющими группами, имеющиеся на компонентах крови, это главным образом аминогруппы, карбоксильные группы и тиоловые группы. В одном варианте реализации изобретения присоединяющие группы включают в себя, но не ограничиваются следующим: реактивные двойные связи, карбоксильные, фосфорильные или удобные ацильные группы, либо в виде эфира, либо смешанного ангидрида, либо имидат; они способны формировать ковалентные связи с функциональными структурами, такими как аминогруппы, гидроксильные группы или тиоловые группы, в определнном-8 007918 месте мобильных белков, в особенности белков крови. Реактивные эфирные присоединяющие группы состоят из фенольных соединений, тиоловых эфиров, алкильных эфиров, фосфатных эфиров и т.п. В особенно предпочтительном варианте реализации изобретения, присоединяющие группы состоят из сукцинимидильных или малеимидных групп. Целью данного изобретения является модифицирование последовательностей пептида gp41 для получения улучшенной биодоступности, большего периода полураспада и лучшего распределения (путм избирательной конъюгации пептида с белковым носителем) пептидов без значительного изменения их противовирусных, виростатических или антифузогенных свойств. Получение производных вариантныхgp41-пептидов, описанное в настоящем документе, позволяет производным пептидам реагировать с группами на компонентах крови (особенно с доступными тиоловыми группами) и формировать стабильные ковалентные связи. Предпочтительные производные вариантных gp41-пептидов разработаны для специфического реагирования с тиоловыми группами на мобильных белках крови. Такая реакция происходит при ковалентном связывании пептида, имеющего малеимидное связующее звено, с тиоловой группой на мобильном белке крови, например сывороточном альбумине или IgG. Таким образом, к одному из вариантов реализации данного изобретения относится модифицированный пептид, ковалентно связанный с белком крови, в т.ч. с мобильным белком крови. К особо предпочтительному варианту реализации изобретения относится ковалентное связывание модифицированного пептида с сывороточным альбумином. Компоненты крови, с которыми происходит ковалентное связывание производных вариантныхgp41-пептидов, могут быть фиксированными или мобильными. Фиксированные компоненты крови - это неподвижные компоненты крови, к которым относятся ткани, мембранные рецепторы, интерстициальные белки, фибриновые белки, коллагены, тромбоциты, эндотелиоциты, эпителиоциты и их связанные с мембранами и мембранные рецепторы, соматические клетки, клетки скелетных и гладких мышц, нейрональные компоненты, остеоциты и остеокласты и все ткани тела, особенно связанные с кровеносной и лимфатической системой. Мобильные компоненты крови - это компоненты крови, у которых нет постоянного местонахождения. Их положение меняется в течение некоторого промежутка времени (обычно не более 5 мин, а чаще всего не более одной минуты). Эти компоненты крови не связаны с мембранами и имеются в крови длительное время в концентрации не менее 0,1 мкг/мл. К мобильным компонентам крови относятся сывороточный альбумин, трансферрин, ферритин и иммуноглобулины, такие как IgM иIgG. Период полураспада мобильных компонентов крови составляет приблизительно не менее 12 ч. В целях данного изобретения в качестве носителя выбирают альбумин, связанный через его свободную тиольную группу. В другом варианте реализации изобретения описан способ генерирования пептидных ингибиторов инфузии с противовирусными, виростатическими или антифузогенными свойствами для профилактики или лечения вирусных, в т.ч. и ретровирусных, инфекций. В соответствии с этим способом идентифицируют вирусные белки, принимающие участие во внедрении вируса в клетку и/или имеющие фузогенную активность. После этого аминокислотные последовательности этих вирусных белков скринируются на спиральформирующие регионы, которые, как предполагается, участвуют во взаимодействии белокбелок. Специалисты в данной области могут использовать компьютерные алгоритмы скрининга на альфа-спиральформирующие участки в белковых последовательностях. Компьютерные алгоритмы, которые можно использовать для определения альфа-спиральформирующих участков включают в себя, но не ограничиваются индексами спирального предпочтения Гарние-Робсона (Garnier-Robson) и Чау-Фасмана(Chou-Fasman), имеющиемися в таких программных пакетах, как DNASTAR. Пептиды, являющиеся производными альфа-спиральформирующих участков вирусных белков, могут быть созданы в соответствии со способами, описанными выше, путм замены определнных аминокислотных остатков на аминокислотные остатки, которые будут увеличивать гидрофильность, гидрофобность или способность формировать альфа-спираль у пептидной последовательности. В качестве альтернативы в пептидах данного изобретения можно использовать замены на D-изомеры вместо природных L-аминокислот или не встречающиеся в природе аминокислоты. В одном варианте реализации данного изобретения не менее двух аминокислотных замен представляют собой D-изомеры природных L-аминокислот. В другом варианте реализации вся пептидная последовательность состоит из D-изомеров природных L-аминокислот. Подобные изменения призваны увеличить стабильность, протеиназную резистентность, активность, реактивность и/или растворимость пептидов согласно настоящему изобретению. Производные формы этих пептидов можно использовать в качестве лечебного средства с увеличенным периодом полураспада благодаря их конъюгации с компонентами крови, такими как сывороточный альбумин. Ожидается, что пептидные последовательности, имеющие D-изомеры природных Lаминокислот, будут проявлять повышенную резистентность к действию протеиназ, пропорциональную количеству D-изомеров природных L-аминокислот в пептидной последовательности, независимо от того,конъюгированы пептиды с компонентами крови или нет. Данный способ изобретения подробно рассматривает тестирование пептидных составов in vitro на противовирусные, виростатические или антифузогенные свойства. Например, специалист в данной области сможет модифицировать принципы из примера 9 данного документа, чтобы создать тестовую систему для скрининга на противовирусную активность. Неограничительным образом специалист данной области может использовать или модифициро-9 007918 вать принципы из примера 9 для проверки эффектов противовирусных пептидов в присутствии вируса,специфичного к определнному типу клеток, например моноциту периферической крови (РВМС), чтобы определить значения IC50 и ТС 50. После инфицирования клеток определенного типа как в присутствии,так и в отсутствие ингибитора (с соответствующими контролями), производят инкубацию упомянутых клеток и измеряют титры вируса и значения IC50 и TC50. Вирусы, к которым применим способ согласно настоящему изобретению, включают, но не ограничивают, ретровирусами человека, в т.ч. ВИЧ-1 и ВИЧ-2, Т-лимфотропными вирусами человека (HTLV-I и HTLV-II) и прочими ретровирусами, в т.ч. вирус лейкоза коров, вирус саркомы кошек, вирус лейкоза кошек, вирус иммунодефицита обезьян (ВИО), вирус саркомы обезьян, лейкоза обезьян и вирус прогрессирующей пневмонии овец. Противовирусные, виростатические или антифузогенные пептиды также могут ингибировать неретровирусные вирусы, в том числе, но не только, респираторно-синцитиальный вирус человека(RSV), вирус собачьей чумы, вирус Нью-касльской болезни, вирус парагриппа человека(HPV), вирус гриппа, вирус кори, вирусы Эпштейна-Барра, вирусы гепатита В и вирусы МасонаПфайзера обезьян. Пептиды согласно настоящему изобретению также могут ингибировать вирусы без внешней оболочки (envelop), в том числе, но не только, пикорнавирусы (например, вирусы полиомиелита), вирус гепатита А, энтеровирусы, ЕСНО-вирусы, вирусы Коксаки, паповавирусы (например, папалломавирус), парвовирусы, аденовирусы и реовирусы. Синтез пептидов Производные вариантных gp41-пептидов можно синтезировать стандартными способами твердофазной химии пептидов, хорошо известными любому специалисту в данной области. Например, пептиды можно синтезировать способами твердофазной химии по процедурам, описанным Стюардом (Steward etal. in Solid Phase Peptide Synthesis, 2nd Ed., Pierce Chemical Company, Rockford, Ill., (1984 с использованием синтезатора Rainin PTI Symphony. В качестве альтернативы могут быть синтезированы фрагменты пептидов, которые впоследствии комбинируют или соединяют друг с другом в растворе, с образованием последовательности gp41-пептида (конденсация сегментов описана, например, в US Patent 6281331 (описания обоих способов упомянуты в данном документе в качестве ссылки. Для твердофазного синтеза пептидов общие сведения о многочисленных способах можно найти в следующих источниках: Stewart etal. in "Solid Phase Peptide Synthesis", W. H. Freeman Co. (San Francisco), 1963 и Meienhofer, Hormonal Proteins and Peptides, 1973, 246. Для получения информации о классическом способе синтеза в растворе, см.,например, Schroder et al. in "The Peptides", vol. 1, Acacemic Press (New York). В общем случае такие способы подразумевают последовательное добавление одной или более аминокислот или должным образом защищенных аминокислот к растущей пептидной цепи на полимере. Обычно либо амино-, либо карбоксильную группу первой аминокислоты защищают подходящей защитной группой. После этого защищенную аминокислоту и/или ее производную либо присоединяют к инертной тврдой основе, либо использует в растворе путм добавления следующей по порядку аминокислоты, имеющей должным образом защищенную комплементарную группу (амино- или карбоксильную), в условиях, необходимых для формирования амидной связи. Затем защитную группу удаляют с только что присоединнного аминокислотного остатка, добавляют следующую кислоту (соответствующим образом защищенную) и так далее. После того, как все аминокислоты присоединены в нужной последовательности, оставшиеся защитные группы (или тврдую основу) отсоединяют последовательно или одновременно для получения конечного пептида. Простой модификацией данной общей процедуры к растущей цепи можно добавлять более одной аминокислоты одновременно, например, путм спаривания (в условиях, не рацемизирующих хиральные центры) защищенного трипептида с должным образом защищенным дипептидом для получения после снятия защиты пентапептида. Во время синтеза имеющегося пептидного производного могут потребоваться защитные группы. Эти защитные группы общеизвестны в области пептидного синтеза и могут в общем случае быть описаны как химические группы, способные защищать пептидное производное от реагирования с другими функциональными группами. Различные защитные группы имеются в продаже, и их примеры могут быть найдены в патенте US Patent 5493007, упомянутом в данном документе в качестве справки. Типичные примеры подходящих защитных групп включают в себя ацетил, флюоренилметилоксикарбонил (FMOC), т-бутилоксикарбонил (ВОС), бензилоксикарбонил(CBZ) и т.д. В табл. 7 описаны трхбуквенные и однобуквенные сокращения природных аминокислот. Особо предпочтительным способом получения вариантных gp41-пептидов является твердофазный способ синтеза пептидов, при котором аминокислоту с -N-конца защищают кислото- или основаниечувствительной группой. Такие защитные группы должны быть стабильны в условиях формирования пептидной связи, но в то же время легко отделяемы без разрушения растущей пептидной цепочки или рацемизации какого-либо хирального центра в этой цепи. Примеры N-концевых защитных групп и карбоксизащитных групп приведены в публикации Greene, "Protective Groups In Organic Synthesis" (JohnWileySons, New York, pp. 152-186 (1981, приведенной в данном документе в качестве ссылки. Примеры N-концевых защитных групп включают в себя, но не ограничиваются, следующие: группы низшего алканоила, например, ацетил ("Ас"), пропионил, пивалоил, т-бутилацетил и т.п.; к другим ацильным группам относятся 2-хлорацетил, 2-бромацетил, трифторацетил, трихлорацетил, фталил, о-нитрофеноксиацетил, -хлорбутирил, бензоил, 4-хлорбензоил, 4-бромбензоил, 4-нитробензоил и т.п.; сульфонильные группы, такие как бензенесульфонил, п-толуенесуольфонил, о-нитрофенилсульфонил, 2,2,5,7,8 пентаметилхроман-6-сульфонил (pmc) и т.п.; карбаматобразующие группы, такие как т-амилоксикарбонил, бензилоксикарбонил, п-хлоробензилоксикарбонил, п-метоксибензилоксикарбонил, п-нитробензилоксикарбонил,2-нитробензилоксикарбонил,п-бромобензилоксикарбонил,3,4-диметоксибензилоксикарбонил, 3,5-диметоксибензилоксикарбонил, 2,4-диметоксибензилоксикарбонил, 4-етоксибензилоксикарбонил, 2-нитро-4,5-диметоксибензилоксикарбонил, 3,4,5-триметоксибензилоксикарбонил,1-(п-бифенилил)-1-метилетоксикарбонил, ,-диметил-3,5-диметоксибензилоксикарбонил, бензгидрилоксикарбонил, т-бутилоксикарбонил (boc), диизопропилметоксикарбонил, изопропилоксикарбонил,этоксикарбонил, метоксикарбонил, аллиллоксикарбонил (Aloc), 2,2,2-трихлорэтоксикарбонил, феноксикарбонил, 4-нитрофеноксикарбонил, флуоренил-9-метоксикарбонил, изоборнилоксикарбонил, циклопентилоксикарбонил, адамантилоксикарбонил, циклогексилоксикарбонил, фенилтиокарбонил и т.п.; арилалкильные группы, такие как бензил, бифенилизопропилоксикарбонил, трифенилметил, бензилоксиметил,9-флуоренилметоксикарбонил (Fmoc) и т.п., а также силильные группы, такие как триметилсилил и т.п. Предпочтительные -N-защитные группы следующие: о-нитрофенилсульфенил, 9-флуоренилметоксикарбонил, т-бутилоксикарбонил (boc), изоборнилоксикарбонил, 3,5-диметоксибензилоксикарбонил, т-амилоксикарбонил, 2-циано-т-бутилоксикарбонил и подобные ему, предпочтительным из них является 9-флюоренилметилоксикарбонил (Fmoc), при этом к предпочтительным N-защитным группам боковых цепей относятся 2,2,5,7,8-пентаметилхроман-6-сульфонил (pmc), нитро, птолуенсульфонил, 4-метоксибензинсульфонил, Cbz, Воc и адамантилоксикарбонил для таких аминокислот боковых цепей, как лизин и аргинин; Aloc для лизина; бензил, о-бромобензилоксикарбонил, 2,6 дихлоробензил, изопропил, т-бутил (t-Bu), циклогексил, сиклопентил и ацетил (Ас) для тирозина; тбутил, бензил и тетрагидропиранил для серина; тритил, бензил, Cbz, п-толуенсульфонил и 2,4 динитрофенил для гистидина; формил для триптофана; бензил и т-бутил для аспарагиновой кислоты и глютаминовой кислоты; трифенилметил (тритил) для цистеина.- 11007918 К карбоксизащитной группе обычно относят эфирную или амидную группу, защищающие карбоксильную кислоту. Такие карбоксизащитные группы хорошо известны специалистом в данной области и широко используются при защите карбоксильных групп при производстве пенициллинов и цефалоспоринов, что описано в патентах U.S. Patent Nos. 3840556 и 3719667, упомянутых в данном документе в качестве ссылки. К представителям карбоксизащитных групп относятся без ограничения: С 1-С 8 низшие алкилы; арилалкилы, например фенетил или бензил и их производные, такие как алкоксибензильная или нитробензильная группы; арилалкенилы, например, фенилетенил; арилы и их производные, например, 5 инданил; диалкиламиноалкилы, например, диметиламиноэтил; алканоилоксиалкильные группы, например ацетоксиметил, бутироксиметил, валериоксиметил, изобутирилоксиметил, изовалериоксиметил, 1(пропионилокси)-1-этил, 1-(пивалоилоксил)-1-этил, 1-метил-1-(пропионилокси)-1-этил, пивалоилоксиметил, пропионилоксиметил; циклоалканоилоксиалкильные группы, например циклопропилкарбонилоксиметил,циклобутилкарбонилоксиметил,циклопентилкарбонилоксиметил,циклогексилкарбонилоксиметил; арилоксиалкилы, например бензилоксиметил, бензоилоксиэтил; арилалкилкарбонилоксиалкилы, например бензилкарбонилоксиметил, 2-бензилкарбонилоксиэтил; алкоксикарбонилалкилы или циклоалкилоксикарбонилалкилы, например метоксикарбонилметил, циклогексилоксикарбонилметил, 1-метоксикарбонил-1-этил; алкоксикарбонилоксиалкилы или циклоалкоксикарбонилоксиалкилы,например метоксикарбонилоксиметил, т-бутилоксикарбонилоксиметил, 1-этоксикарбонилокси-1-этил, 1 циклогексилоксикарбонилокси-1-этил; арилоксикарбонилоксиалкилы, например 2-(феноксикарбонилокси)этил, 2-(5-инданилоксикарбонилокси)этил; алкоксиалкилкарбонилоксиалкилы, например 2-(1 метокси-2-метилпропан-2-оилокси)этил; арилалкилоксикарбонилоксиалкилы, например 2-(бензилоксикарбонилокси)этил; арилалкенилоксикарбонилоксиалкилы, например 2-(3-фенилпропен-2-илоксикарбонилокси)этил; алкоксикарбониламиноалкилы, например, т-бутилоксикарбониламинометил; алкиламинокарбониламиноалкилы, например метиламинокарбониламиноэтил; алканоиламиноалкилы, напримен,ацетиламинометил; гетероциклические карбонилоксиалкилы,например 4-метилпиперазинилкарбонилоксиметил; диалкиламинокарбонилалкилы, например диметиламинокарбонилметил, диэтиламинокарбонилметил; (5-(низший алкил)-2-оксо-1,3-диоксолен-4-ил)алкилы, например (5-т-бутил-2-оксо 1,3-диоксолен-4-ил)метил; (5-фенил-2-оксо-1,3-диоксолен-4-ил)алкилы, например (5-фенил-2-оксо-1,3 диоксолен-4-ил)метил. Представителями амидных карбоксизащитных групп являются, в частности, аминокарбонильные и низшеалкиламинокарбонильные группы. Из вышеперечисленных карбоксизащитных групп предпочитаемыми являются низшеалкильные,циклоалкильные или арилалкильные эфиры, например метиловый эфир, этиловый эфир, пропиловый эфир, изопропиловый эфир, бутиловый эфир, секбутиловый эфир, изобутиловый эфир, амиловый эфир,изоамиловый эфир, октиловый эфир, циклогексиловый эфир, фенилэтиловый эфир и т.п., либо алканоилоксиалкилы, циклоалканоилоксиалкилы, арилоксиалкилы или арилалкилкарбонилоксиалкильные эфиры. Предпочитаемыми амидными карбоксизащитными группами являются низшеалкиламинокарбонильные группы. В способе твердофазного синтеза пептида -С-концевую аминокислоту присоединяют к подходящей тврдой основе или смоле. К тврдым основам, пригодным для вышеупомянутого способа синтеза,относятся такие материалы, которые инертны по отношению к реагентам и условиям реакции пошаговой конденсации-депротекции, а также нерастворимы в используемой среде. Предпочтительной тврдой основой для синтеза -С-концевых карбоксильных пептидов является 4-гидроксиметилфеноксиацетил-4'метилбензилгидриламиновая смола (НМР-смола). Предпочтительной тврдой основой для -С-концевых амидных пептидов является Fmoc-защищнная смола Ramage, производимая и продаваемая фирмойBachem Inc., California. В предпочтительном способе синтеза связываемый лизин защищают Aloc. После завершения синтеза Aloc отделяют при помощи Pd(Ph3)4, когда пептид вс ещ находится на смоле, после чего можно соединить связующую (линкерную) молекулу и малеимидную группу. В частном случае связующим звеном является [2-(2-амино)этоксил]этоксиуксусная кислота, а малеимидной группой - 3'-малеимидопропионовая кислота. После произведнной модификации Fmoc-группы удаляют и пептид отделяют от смолы. По завершении твердофазного синтеза пептид отделяют от смолы и снимают защиту, что осуществляется одной или несколькими последовательными операциями. Отделение пептида и снятие защиты производят одновременно общепринятым способом - обработкой связанного со смолой полипептида отщепляющим реагентом, состоящим из тиоанизола, триизопропил силана, фенола и трифторуксусной кислоты. В том случае, когда на -С-конце пептида находится алкиламид, смолу отделяют путм аминолизиса при помощи алкиламина. Другим решением является отделение пептида путм трансэстерификации, например, при помощи метанола, за которой следует аминолизис, либо путм прямой трансамидации. На этой стадии защищенный пептид очищают либо сразу используют на следующем этапе синтеза. Удаление групп, защищающих боковые цепи, осуществляется при помощи отщепляющего реагента,описанного выше. Пептид с полностью снятой защитой очищают последовательными хроматографиче- 12007918 скими обработками любого из следующих типов: ионный обмен на слабощелочной смоле (ацетатная форма); гидрофобная адсорбционная хроматография на неизменнном полистерин-дивинилбензине (например, Amberlite XAD); силикагелевая адсорбционная хроматография; ионообменная хроматография на карбоксиметилцеллюлозе; тонкослойная хроматография, например, на приборе Sephadex G-25, LH-20 либо противоположное распределение; высокоэффективная жидкостная хроматография (ВЭЖХ), особенно ВЭЖХ с обратной фазой или с наполнением колонки октил- либо фенил/гексилсилилсиликасвязанной фазой. Опытный специалист сможет определить предпочтительные хроматографические этапы или последовательности, требуемые для получения нужной степени очистки вариантных gp41 пептидов. В качестве альтернативы фрагменты пептидов синтезируют (в т.ч. с присоединением малеимидных групп) в тврдой фазе, а конечное производное пептида получают соединением этих фрагментов в растворе. Молекулярные массы этих пептидов можно определить, используя массовую спектроскопию Electrospray или массовую спектроскопию MALDI-TOF. Терапевтическое применение модифицированных пептидов Вариантные gp41-пептиды, в т.ч. вещества, приведнные в табл. 1, 2 и 3 и на фиг. 1, ингибируют вирусную инфекцию в клетках, например, путм ингибирования клеточно-клеточного слияния или свободной вирусной инфекции. Пути проникновения инфекции могут задействовать мембранное слияние,что происходит в случае с покрытыми внешней оболочкой или инкапсулированными вирусами, или другой вид слияния, в котором участвуют вирусные и клеточные структуры, например клеточные рецепторы. Вариантные gp41-пептиды назначают in vivo, так, что конъюгация с компонентами крови происходила in vivo, либо предварительно конъюгируют с компонентами крови ex vivo, после чего конъюгированное производное вводят in vivo. В другом варианте реализации изобретения используют плазмаферез для выделения нужных компонентов крови из образца, забранного у больного, которые затем конъюгируют с пептидами изобретения и обратно вводятся больному. Тиоловые группы реже встречаются in vivo на белках плазмы, чем, например, аминогруппы. Поэтому малеимидмодифицированные вариантныеgp41-пептиды будут ковалентно связываться с меньшим количеством белков. Например, у альбумина(самого распространнного белка крови) имеется только одна тиоловая группа. Таким образом, конъюгат модифицированного gp41-пептида с альбумином будет стремиться составить молярное соотношениеgp41-пептида к альбумину 1:1. Кроме альбумина имеются молекулы IgG (класс II) с тремя тиоловыми группами. Так как IgG и сывороточный альбумин составляют большую часть растворимых белков крови,то они же имеют и большую часть тиоловых групп крови, доступных для ковалентного связывания с вариантными gp41-пептидами. Кроме того, даже среди белков крови, имеющих свободный тиол, в т.ч. и IgG, специфическое связывание с малеимидом приводит преимущественному образованию конъюгатов gp41-пeптид-мaлeимидaльбyмин, вызванному уникальными характеристиками самого альбумина. Единственная тиоловая группа альбумина, высоко консервативная у данного вида животных, расположена у аминокислотного остатка 34 (Cys34). Недавно было продемонстрировано, что остаток Cys34 альбумина имеет повышенную реактивность свободных тиолов по сравнению с другими белками, имеющими свободный тиол. Частично это связано с очень низким значением pK остатка Cys34 альбумина, равным 5,5. Это гораздо меньше, чем типичное значение pK для цистеина, которое обычно равно 8. Благодаря такому низкому значению pK остаток Cys34 на альбумине при нормальных условиях находится преимущественно в ионизированном виде, что значительно повышает его реактивность. Кроме такого низкого значения pK остатка Cys34 альбумина имеется и другой фактор, который увеличивает реактивность Cys34 в данном положении, это гидрофобный карман, расположенный близко к поверхности одной петли участка V альбумина. Такое расположение делает Cys34 легко доступным для лигандов всех типов, оно является важным фактором биологической роли Cys34 как ловушки для свободных радикалов и свободных тиолов. Эти свойства делают Cys34 высоко реактивным по отношению к gp41-пептидам, имеющим малеимидные связующие звенья, и увеличение скорости реакции может быть до 1000 раз больше по сравнению со скоростями реакций вариантных gp41-пептидов, имеющих малеимидные связующие звенья, с другими белками, имеющими свободный тиол. Другое преимущество конъюгатов gp41-пептид-малеимид-альбумин - это воспроизводимость, связанная с соотношением связи пептида и альбумина 1:1, и возникновением е только на месте Cys34. Другие техники связывания, например глютаральдегид, DCC (N,N'-дициклогексилкарбодиимид), EDC (этилена дихлорид) и другие химические активации, например, свободных аминов, не имеют такой селективности. Например, альбумин содержит 52 лизиновых остатка, из них 25-30 расположены на поверхности альбумина и поэтому доступны для конъюгации. Активация этих лизиновых остатков или модифицирование вариантных gp41-пептидов для связывания с этими лизиновыми остатками приводит к образованию гетерогенной популяции конъюгатов. Даже если для связывания малеимидных gp41-пептидов с альбумином используется молярное соотношение 1:1, на выходе аминное производное альбумина будет состоять из большого количества видов конъюгатов, содержащих 0, 1, 2 или более gp41-пептидов на мо- 13007918 лекулу альбумина, и в каждом из них пептид будет случайным образом присоединн к одному или нескольким из 25-30 доступных лизиновых отрезков. При множестве возможных комбинаций охарактеризовывание точного состава и свойств каждой партии конъюгата становится затруднительным, а точное воспроизведение партий практически невозможным, что сделает такие конъюгаты менее удобными как лечебное средство. Конъюгация через лизиновые остатки может иметь преимущество доставки большего количества лечебного агента на каждой молекуле альбумина, но недавние исследования показали, что предпочтительным соотношением лечебного агента к альбумину является 1:1. В статье Stehle, et al., "TheLoading Rate Determines Tumor Targeting properties of Methotrexate-Albumin Conjugates in Rats," AntiCancer Drugs, Vol. 8, pp. 677-685 (1988), полностью включнной в данный документ в качестве ссылки,авторы сообщают, что соотношение 1:1 противоракового препарата метотрексат с альбумином в их конъюгате через глютаральдегид дало самые обещающие результаты. Такие конъюгаты имели преимущество при их захвате опухолевыми клетками, при этом конъюгаты, несущие молекулы метотрексата в соотношении от 5:1 до 20:1, имели изменнные профили ВЭЖХ и быстро захватывались печенью in vivo. Было заявлено, что при таких высоких соотношениях конформационные изменения альбумина уменьшают его эффективность как переносчика лекарственных средств. При контролируемом назначении вариантных gp41-пептидов in vivo можно контролировать специфичное связывание альбумина и IgG in vivo. При типичном назначении 80-90% назначаемых производных вариантных gp41-пептидов свяжется с альбумином, и менее 5% свяжется с IgG. Происходит также связывание со свободными тиолами, например глютатионом, в следовых количествах. Такая специфичность связывания предпочтительна при использовании in vivo, так как она позволяет точно вычислять предполагаемый период полураспада вариантных gp41-пептидов. Помимо получения контролированного специфичного связывания in vivo, производные вариантныеgp41-пептиды дают специфичное связывание с сывороточным альбумином и IgG ex vivo. К такому связыванию ex vivo относятся добавление вариантных gp41-пептидов с малеимидными связующими звеньями к крови, сыворотке или физиологическому раствору, содержащему альбумин и/или IgG. После конъюгации вариантных gp41-пептидов ex vivo кровь, сыворотка или физиологический раствор вводят обратно в кровь больного для лечения in vivo либо лиофилизируют. Вариантные gp41-пептиды можно использовать как одни, так и в комбинации для оптимизирования их лечебного действия. В другом варианте реализации изобретения вариантные gp41-пептиды назначают совместно с одним или более противовирусными препаратами для лечения ВИЧ-инфекции. Дополнительные противовирусные препараты для лечения ВИЧ-инфекции, которые назначают совместно с вариантными gp41-пептидами, включают в себя, но не ограничиваются следующими: AGENERASE (amprenavir; GlaxoSmithKline); COMBIVIR (lamivudine, zidovudine; GlaxoSmithKline); CRIXIVAN (indinavir,IDV, MK-639; Merck); EMTRIVA (FTC, emtricitabine; Gilead Sciences); EPIVIR (lamivudine, 3TC; GlaxoSmithKline); FORTOVASE (saquinavir; Hoffmann-La Roche); HIVID (Zalcitabine, ddC, dideoxycytidine;GlaxoSmithKline). В дополнительном варианте реализации изобретения вариантные gp41-пептиды назначают совместно с одним или более дополнительных препаратов для лечения ВИЧ-инфекции и связанных с ней заболеваний. Эти дополнительные препараты, которые назначают совместно с вариантными gp41-пептидами,включают в себя, но не ограничиваются следующими: TRIMETREXATE GLUCURONATE (для лечения пневмонии, вызванной Pneumocystis carinii); GANCICLOVIR (для лечения цитомегаловирусного ретинита); PENTAMIDINE - аэрозольная форма (для лечения пневмонии, вызванной Pneumocystis carinii);avium); VISTIDE (для лечения рецидивирующего цитомегаловирусного ретинита); SEROSTIM (для лечения похудания, связанного со СПИДом). Вариантные gp41-пептиды, в том числе, но не только, пептиды, приведнные в табл. 1, 2 и 3, а также на фиг. 1, назначают совместно с одним или несколькими дополнительными вариантными gp41 пептидами, приведнными в табл. 1, 2 и 3, а также на фиг. 1. В другом варианте реализации изобретения вариантные gp41-пептиды, в том числе, но не только, пептиды, приведнные в табл. 1, 2 и 3, а также на фиг. 1, назначают совместно с пептидами Т-20 или Т-1249. Вариантные gp41-пептиды назначают в физиологически приемлемой среде, например деионизированной воде, фосфатном буферном растворе (PBS), солевом физиологическом растворе, водном растворе этанола или другого спирта, плазме, белковых растворах, маннитоле, водном растворе глюкозы, спирте,- 14007918 растительном масле и т.п. Фармацевтический состав, содержащий вариантные gp41-пептиды, предпочтительным образом назначают совместно с фармацевтически приемлемым носителем. Другие доплнительные компоненты включают в себя буферы, в которых рН среды обычно поддерживается в пределах от 5 до 10 с концентрацией буфера от 50 до 250 мМ; соль, концентрация которой обычно варьирует от 5 до 500 мМ; физиологически приемлемые стабилизаторы и т.п. Для удобства хранения и транспортировки составы могут быть лиофилизированы. Вариантные пептиды назначают перорально, парентерально, например внутривенно (в/в), внутриартериально (в/а), внутримышечно (в/м), подкожно (п/к) или другими способами. В подходящих случаях препарат можно вводить путм инфузии. В некоторых случаях, при которых реакция функциональной группы протекает относительно медленно, назначение производят оральным, назальным, ректальным,трансдермальным или аэрозольным путм, при этом свойства конъюгата должны позволять ему проникать в сосудистую систему. Обычно используют одна инъекция, хотя при желании можно производить более одной инъекции. Пептидное производное вводят любым удобным инструментом, в т.ч. шприцом,троакаром, катетером и т.п. Конкретный режим назначения будет варьировать в зависимости от количества назначаемого препарата, от того, назначается ли он болюсно или продолжительно и т.п. Предпочтительно производить интраваскулярное назначение, при этом место введения не имеет принципиального значения, но желательно, чтобы в этом месте имелся быстрый кровоток, например, внутривенно - в периферическую или центральную вену. Другие пути назначения могут оказаться полезными там, где назначение связано с техникой медленного высвобождения или защитным матриксом. Преследуемой целью является эффективное распределения вариантных gp41-пептидов в крови для того, чтобы они могли прореагировать с компонентами крови. Количество вводимого конъюгата будет сильно варьировать,приблизительно в пределах от 1 до 500 мг. Общее количество, введнное внутрисосудисто, будет обычно в пределах 0,5-50 мг/кг массы тела, чаще всего от 0,5 до 10 мг/кг. Связывание с долгоживущими компонентами крови, такими как иммуноглобулины, сывороточный альбумин, эритроциты и тромбоциты, дат несколько преимуществ. Действие вариантных gp41-пептидов продлевается на дни и даже недели. За этот период времени требуется всего одно назначение препарата. Достигается большая специфичность, так как активное вещество будет, в основном, связано с большими молекулами, и вероятность попадания его внутрь клеток и нарушения физиологических процессов станет низкой. Формирование ковалентной связи с компонентом крови может быть осуществлено in vivo или exvivo. Для формирования ковалентной связи ex vivo производные вариантные gp41-пептиды добавляют к сыворотке крови или физиологическому раствору, содержащему очищенные компоненты крови, например, человеческий сывороточной альбумин или IgG, для того, чтобы между производным и компонентом крови образовалась ковалентная связь. В предпочтительном варианте реализации вариантные gp41 пептиды реагируют с человеческим сывороточным альбумином в солевом физиологическом растворе. После формирования конъюгата он может быть введн больному или лиофилизирован. Активность и/или наличие вариантных gp41-пептидов в крови млекопитающего можно отслеживать. Производя заборы крови от хозяина в различные моменты времени, можно определить, связались ли вариантные gp41-пептиды с долгоживущими компонентами крови в количестве, достаточном для их терапевтической активности, после чего определять уровень вариантных gp41-пептидов в крови. При желании можно также определить, с каким из компонентов крови ковалентно связались gp41-пептиды. Мониторинг можно также осуществлять при помощи специфических анализов на активность вариантных gp41-пептидов, антитела против вариантных gp41-пептидов или HPLC-MS (ВЭЖХ (высокоэффективная жидкостная хроматография)-масс-спектрография). Вариантные gp41-пептиды назначают больным в соответствии со способами, описанными в данном документе, или другими способами, известными в данной области. К больным, для которых рассматривается лечение, относятся больные, инфицированные одним из приведнных в данном документе вирусов, в особенности ВИЧ-1 и ВИЧ-2. Эффективные лечебные дозы вариантных gp41-пептидов могут быть определены по процедурам, хорошо известным специалистам, при этом должны учитываться соображения о потенциальной токсичности данных вариантных gp41-пептидов. Вариантные gp41-пептиды также можно назначать профилактически ранее не инфицированным лицам. Такое назначение может быть особо показано в случаях, когда лицо было подвергнуто высокому риску попадания вируса, который, например, может возникнуть при контакте с инфицированным лицом и высоком риске передачи вируса. Это может быть особенно показано тогда, когда лечения для вируса не известно, например, как в случае с ВИЧ. В качестве примера, не ограничивающего другие применения,можно сказать, что профилактическое назначение gp41-пептида было бы показано в ситуации, когда работник здравоохранения подвергся попаданию на него крови от ВИЧ-инфицированного индивида, или другой ситуации, когда пациент занимался деятельностью, связанной с высоким риском попадания ВИЧ. Другие применения вариантных gp41-пептидов включают в себя назначение их лицам, являющимся носителями вируса, например ВИЧ, для предотвращения передачи вируса от инфицированного лица к неинфицированному. Такое использование также включает в себя предотвращение передачи от матери ребнку при кормлении грудью или других повседневных контактах, или передачи во время сексуальных- 15007918 контактов. В другом варианте реализации изобретения вариантные gp41-пептиды, в том числе, но не только те, что перечислены в табл. 1, 2, 3 и на фиг. 1, назначают совместно с одним или несколькими дополнительными пептидами, приведнными в табл. 1, 2 и 3 и на фиг. 1, Т-20, Т-1249 или другими препаратами для лечения ВИЧ-инфекции. Такое назначение производят для предотвращения репликации ВИЧ (в т.ч. ВИЧ-1, ВИЧ-2 и их серотипы) и частиц ВИО в организме больного. Местное использование Вариантные gp41-пептиды, приведнные в табл. 1, 2 и 3 на фиг. 1, используют отдельно или в виде состава, содержащего или состоящего, в основном, из эффективной концентрации пептида и фармацевтически приемлемого носителя. Эффективная концентрация может быть установлена путм наблюдения за подавлением вирусной инфекции при назначении агента (агентов). Составы, предлагаемые данным изобретением, включают в себя микробицидное, виростатическое и антифузогенное применение как invitro, так и in vivo, особенно для интравагинального и интраректального использования. Для этих целей модифицированный пептид может быть включн в лекарственную форму с любой подходящей основой,при условии, что антифузогенная активность модифицированного пептида не нарушается этой основой. Таким образом, составы могут быть в форме кремов, гелей, пенок, лосьонов, мазей, таблеток, растворов или спреев. Формообразующая основа или растворитель может быть водным или неводным, например,спиртовым или масляным, или их смесью, а также может дополнительно содержать другие сурфактанты,эмульгаторы, любриканты, стабилизаторы, красители, отдушки, противомикробные агенты либо в качестве активных компонентов, либо в качестве консервантов, кислоты или основания для регуляции рН. Предпочтительное значение рН находится в пределах от 4 до 5. Для приготовления составов используют общепринятые способы. Предпочтительно, чтобы фармацевтически приемлемый носитель или формообразующая основа для топически (местно) применяемых составов была в виде жидкости, желе или пенки, содержащей вещество данного изобретения. Вещество может быть включено в состав: (а) мазей и желе, (b) вводимых предметов (суппозитории, губки и т.п.), (с) пенок, (d) спринцеваний и (е) очищающих жидкостей. Состав лучше всего вводить во влагалище женщины или прямую кишку мужчины или женщины в момент, совпадающий по времени, а лучше предшествующий сексуальному контакту, но можно также использовать на других слизистых оболочках. Составы можно использовать для лечения и предупреждения заболеваний, передающихся половым путм, в т.ч. и ВИЧ-инфекции. Предпочтительно использовать такой способ назначения, который обеспечил бы прямой контакт пептидсодержащего состава данного изобретения с возбудителями заболеваний, передающихся половым путм. При топическом назначении фармацевтически приемлемый носитель может дополнительно содержать органические растворители, эмульгаторы, желатинизаторы, увлажнители, стабилизаторы, другие сурфактанты, размягчители, консерванты, агенты задержки высвобождения, а также небольшие количества увлажнителей, секвестраторов, красителей, отдушек и других компонентов, обычно используемых в фармацевтических составах, предназначенных для местного использования. Что касается предметов, относящихся к настоящему изобретению, составами изобретения можно импрегнировать адсорбирующие материалы, например, губки, или ими можно покрывать поверхность тврдых материалов, например,презервативов, диафрагм или медицинских перчаток с целью доставки составов к вагинальному или другому потенциально инфицируемому эпителию, желательно до или во время полового акта. Другие предметы и системы доставки подобного типа будут известны специалистам в данной области. На данный момент предпочтительными предметами являются презервативы, которые покрываются слоем модифицированных пептидов путм их распыления на поверхности презервативов, или пропитывания пептидами презервативов в процессе их производства, способом, известным специалистам. К предпочтительным составам для нанесения на поверхность относится силикон, который обеспечивает скольжение и замедленное высвобождение модифицированных пептидов. Биоадгезивные полимеры можно также использовать для продления времени высвобождения из конкретных местных или других медицинских форм. Тврдые лекарственные формы для местного применения включают в себя суппозитории, порошки,таблетки и гранулы. В твердых лекарственных формах к составу может быть добавлен как минимум один инертный разбавитель, например, сахароза, лактоза или крахмал, они также могут содержать любриканты, буферы и другие компоненты, хорошо известные специалистам в данном деле. Конкретные значения доз модифицированных пептидов в составах или предметах данного изобретения могут варьировать для достижения таких уровней их концентрации на месте передачи жидкостей при половом контакте, которые позволят достичь требуемого лечебного или профилактического эффекта для конкретного пептида и способа назначения. Соответственно, выбранные дозировочные уровни будут зависеть от природы и места инфекции, требуемого терапевтического эффекта, пути назначения, требуемой длительности лечения и других факторов. В общем случае предпочтительная доза модифицированных пептидов данного изобретения будет варьировать в пределах от 0,01 до 2,0 вес.%. Предпочтительной местной вагинальной формой является крем или суппозиторий, описанные выше, содержащие от 0,01 до 2,0 вес.% состава, согласно настоящему изобретению. Во время лечения, обычно два раза в день, примерно от 1 до 5 мл такого состава вводятся интравагинально, желательно высоко во влагалищное отверстие, либо в прямую кишку. Большие количества обычно не используются во избежание подтеканий.- 16007918 Способы и составы данного изобретения можно использовать для профилактики и лечения широкого спектра инфекций, вызванных патогенными микроорганизмами. Примеры Для удобства и более полного понимания данного изобретения ниже приведено несколько примеров. Однако широта охвата данного изобретения не ограничивается конкретными вариантами реализации, раскрытыми в этих примерах, которые служат только иллюстративным целям. Общие положения Если об этом не сказано отдельно, синтез каждого вариантного gp41-пептида производили по процедуре автоматического твердофазного синтеза на пептидном синтезаторе Symphony с ручным вмешательством во время получения производного. Синтез производили на Fmoc-защищнной амидной связующей смоле Ramage с Fmoc-защищнными аминокислотами. Присоединение производили с использованием О-бензотриазол-1-ил-N,N,N',N'тетраметилюроний гексафлюорофосфата (HBTU) в качестве активатора в растворе N,N-диметилформамида (DMF) и диизопропилэтиламина (DIEА) в качестве основания. Защитная группа Fmoc удалялась при помощи 20% пиперидина/DMF. Все аминокислоты, использованные в синтезе, обладают Lстереохимией. Во время синтеза для проведения реакций использовали стеклянную посуду. Пример 1. Синтез FB005. Шаг 1. Пример описывает тврдофазный синтез пептида в количествах порядка 1 ммоль. К смоле последовательно добавляли следующие защищенные аминокислоты: Fmoc-Leu-OH, Fmoc-Leu-OH, FmocGlu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Lys(Boc)-OH, FmocGlu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, FmocSer(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Glu(tBu)-OH,Fmoc-His-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Glu-OH, Fmoc-Thr(tBu)-OH, Fmoc-Tyr(tBu)-OH, FmocAsn(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Ile-OH, Fmoc-Glu(tBu)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Asp(tBu)OH, Fmoc-Trp(Boc)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu-OH, Ртос-Тrp(Вос)-ОН, Fmoc-Thr-OH, Fmoc-MetOH, Fmoc-Asn-OH, Fmoc-Asn-OH, Fmoc-Тrp-ОН, Fmoc-Ile-OH, Fmoc-Gln-OH, Fmoc-Glu-OH, Fmoc-LeuOH, Fmoc-Ser-OH. Они были растворены в N,N-диметилформамиде (DMF) и, в соответствии с процедурой, активированы при помощи О-бензотриазол-1-ил-N,N,N',N'-тетраметилюроний гексафлюорофосфата(HBTU) и диизопропилэтиламина (DIEA). Удаление защитных групп Fmoc осуществляли при помощи 20% (об.) раствора пиперидина в N,N-диметилформамиде (DMF) в течение 20 мин (шаг 1). Аминогруппа последней аминокислоты была ацетилирована с использованием уксусной кислоты, активированной при помощи O-бензотриазол-1-ил-N,N,N',N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEA). Шаг 2. Пептид отделяли от смолы при помощи 85% TFA/5% триизопропилсилана (TIPS)/5% тиоанизола и 5% фенола, после того как его осадили при помощи Et2O температуры сухого льда (шаг 2). Пример 2. Синтез FB005M. Шаг 1. Пример описывает тврдофазный синтез пептида в количествах порядка 1 ммоль. К смоле последовательно добавляли следующие защищенные аминокислоты: Fmoc-Leu-OH, Fmoc-Leu-OH, FmocGlu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Lys(Boc)-OH, FmocGlu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, FmocSer(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Glu(tBu)-OH,Fmoc-His-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Lys(Aloc)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Tyr(tBu)-OH,Fmoc-Asn(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Ile-OH, Fmoc-Glu(tBu)-OH, Fmoc-Arg(Pbf)-OH, FmocAsp(tBu)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu-OH, Fmoc-Trp(Boc)-OH, Fmoc-Thr-OH,Fmoc-Met-OH, Fmoc-Asn-OH, Fmoc-Asn-OH, Fmoc-Trp-OH, Fmoc-Ile-OH, Fmoc-Gln-OH, Fmoc-Glu-OH,Fmoc-Leu-OH, Fmoc-Ser-OH. Они были растворены в N,N-диметилформамиде (DMF) и в соответствии с процедурой активированы при помощи О-бензотриазол-1-ил-N,N,N',N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEA). Удаление защитных групп Fmoc осуществляли при помощи 20% (об.) раствора пиперидина в N,N-диметилформамиде (DMF) в течение 20 мин (шаг 1). Аминогруппа последней аминокислоты была ацетилирована с использованием уксусной кислоты, активированной при помощи О-бензотриазол-1-ил-N,N,N',N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEА). Шаг 2. Селективное снятие защиты с группы Lys (Aloc) производили вручную и осуществляли путм обработки смолы раствором 3-эквивалентного Pd(PPh3)4, растворнного в 5 мл С 6 Н 6 СНСl3 (1:1): 2,5% (об.) NMM: 5% (об.) АсОН в течение 2 ч (шаг 2). После этого смолу промывали СНСl3 (65 мл),20% АсОН в DCM (65 мл), DCM (65 мл) и DMF (65 мл). Шаг 3. После этого синтез опять переводили в автоматический режим и добавляли Fmoc-AEEA-OH и малеимидопропионовую кислоту (шаг 3). Между каждым присоединением смолу промывали 3 разаN,N-диметилформамидом (DMF) и 3 раза изопропанолом (iPrOH). Шаг 4. Пептид отделяли от смолы при помощи 85% TFA/5% триизопропилсилана (TIS)/5% тиоанизола и 5% фенола, после того как его осаждали при помощи Et2O температуры сухого льда (шаг 4).- 17007918 Пример 3. Синтез FB005CM. Шаг 1. Пример описывает тврдофазный пептидный синтез вещества в масштабе 1 ммоль. К смоле последовательно добавляли следующие защищенные аминокислоты: Fmoc-Lys(Aloc)-OH, Fmoc-Leu-OH,Fmoc-Leu-OH, Fmoc-Glu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, FmocLys(Boc)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Asn(Trt)-OH, FmocGln(Trt)-OH, Fmoc-Ser(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Ile-OH, Fmoc-Leu-OH,Fmoc-Glu(tBu)-OH, Fmoc-His-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Glu-OH, Fmoc-Thr(tBu)-OH, FmocTyr(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Ile-OH, Fmoc-Glu(tBu)-OH, Fmoc-Arg(Pbf)-OH,Fmoc-Asp(tBu)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu-OH, Fmoc-Trp(Boc)-OH, Fmoc-ThrOH, Fmoc-Met-OH, Fmoc-Asn-OH, Fmoc-Asn-OH, Fmoc-Trp-OH, Fmoc-Ile-OH, Fmoc-Gln-OH, Fmoc-GluOH, Fmoc-Leu-OH, Fmoc-Ser-OH. Они были растворены в N,N-диметилформамиде (DMF) и в соответствии с процедурой активированы при помощи О-бензотриазол-1-ил-N,N,N',N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEA). Удаление защитных групп Fmoc осуществляли при помощи 20% (об.) раствора пиперидина в N,N-диметилформамиде (DMF) в течение 20 мин (шаг 1). Аминогруппа последней аминокислоты была ацетилирована с использованием уксусной кислоты, активированной при помощи О-бензотриазол-1-ил-N,N,N',N'-тетраметилюроний гексафлюорофосфата(HBTU) и диизопропилэтиламина (DIEА). Шаг 2. Селективное снятие защиты с группы Lys (Aloc) производили вручную и осуществляли путм обработки смолы раствором 3-эквивалентного Pd(PPh3)4, растворнного в 5 мл С 6 Н 6 СНСl3 (1:1) : 2,5% (об.) NMM : 5% (об.) АсОН в течение 2 ч (Шаг 2). После этого смолу промывали СНСl3 (65 мл),20% АсОН в DCM (65 мл), DCM (65 мл) и DMF (65 мл). Шаг 3. После этого синтез опять переводили в автоматический режим и добавляли Fmoc-AEEA-OH и малеимидопропионовую кислоту (шаг 3). Между каждым присоединением смолу промывалиь 3 разаN,N-диметилформамидом (DMF) и 3 раза изопропанолом (iPrOH). Шаг 4. Пептид отделяли от смолы при помощи 85% TFA/5% TIS/5% тиоанизола и 5% фенола, после того как его осаждали при помощи Et2O температуры сухого льда (шаг 4). Пример 4. Синтез FB006. Шаг 1. Пример описывает тврдофазный пептидный синтез вещества в масштабе 1 ммоль. К смоле последовательно добавляли следующие защищенные аминокислоты: Fmoc-Leu-OH, Fmoc-Leu-OH, FmocGlu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Lys(Boc)-OH, FmocGlu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, FmocSer(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Glu(tBu)-OH,Fmoc-His-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Lys(Boc)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Tyr(tBu)-OH,Fmoc-Asn(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Ile-OH, Fmoc-Glu(tBu)-OH, Fmoc-Arg(Pbf)-OH, FmocAsp(tBu)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Trp(Boc)-OH. Они были растворены в N,N-диметилформамиде (DMF) и, в соответствии с процедурой, активированы при помощи О-бензотриазол-1-ил-N,N,N',N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEА). Удаление защитных групп Fmoc осуществляли при помощи 20% (об.) раствора пиперидина в N,N-диметилформамиде (DMF) в течение 20 мин (шаг 1). Аминогруппа последней аминокислоты была ацетилирована с использованием уксусной кислоты, активированной при помощи О-бензотриазол-1-илN,N,N'N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEA). Шаг 4. Пептид отделяли от смолы при помощи 85% TFA/5% TIS/5% тиоанизола и 5% фенола, после того как его осаждали при помощи Et2O температуры сухого льда (шаг 4). Пример 5. Синтез FB006M. Шаг 1. Пример описывает твердофазный пептидный синтез вещества в объеме 1 ммоль. К смоле последовательно добавляли следующие защищенные аминокислоты: Fmoc-Leu-OH, Fmoc-Leu-OH, FmocGlu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Lys(Boc)-OH, FmocGlu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, FmocSer(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Glu(tBu)-OH,Fmoc-His-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Lys(Aloc)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Tyr(tBu)-OH,Fmoc-Asn(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Ile-OH, Fmoc-Glu(tBu)-OH, Fmoc-Arg(Pbf)-OH, FmocAsp(tBu)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Trp(Boc)-OH. Они были растворены в N,N-диметилформамиде (DMF) и в соответствии с процедурой активированы при помощи О-бензотриазол-1-ил-N,N,N'N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEА). Удаление защитных групп Fmoc осуществляли при помощи 20% (об.) раствора пиперидина вN,N-диметилформамиде (DMF) в течение 20 мин (шаг 1). Аминогруппа последней аминокислоты была ацетилирована с использованием уксусной кислоты, активированной при помощи О-бензотриазол-1-илN,N,N',N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEА). Шаг 2. Селективное снятие защиты с группы Lys (Aloc) производили вручную и осуществляли путм обработки смолы раствором 3-эквивалентного Pd(PPh3)4, растворнного в 5 мл С 6 Н 6 СНСl3 (1:1): 2,5% (об.) NMM: 5% (об.) АсОН в течение 2 ч (шаг 2). После этого смолу промывали СНСl3 (65 мл),- 18007918 20% АсОН в DCM (65 мл), DCM (65 мл) и DMF (65 мл). Шаг 3. После этого синтез опять переводили в автоматический режим и добавляли Fmoc-AEEA-OH и малеимидопропионовую кислоту (шаг 3). Между каждым присоединением смолу промывали 3 разаN,N-диметилформамидом (DMF) и 3 раза изопропанолом (iPrOH). Шаг 4. Пептид отделяли от смолы при помощи 85% TFA/5% TIS/5% тиоанизола и 5% фенола, после того как его осаждали при помощи Et2O температуры сухого льда (шаг 4). Пример 6. Синтез FB007M. Шаг 1. Пример описывает тврдофазный пептидный синтез вещества в масштабе 1 ммоль. К смоле последовательно добавляли следующие защищенные аминокислоты: Fmoc-Lys(Aloc)-OH, Fmoc-Leu-OH,Fmoc-Leu-OH, Fmoc-Glu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, FmocLys(Boc)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Asn(Trt)-OH, FmocGln(Trt)-OH, Fmoc-Ser(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Ile-OH, Fmoc-Leu-OH,Fmoc-Glu(tBu)-OH, Fmoc-His-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Glu(tBu)-OH, Fmoc-Thr(tBu)-OH,Fmoc-Tyr(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Ile-OH, Fmoc-Glu(tBu)-OH, FmocArg(Pbf)-OH, Fmoc-Asp(tBu)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu(tBu)-OH, FmocTrp(Boc)-OH. Они были растворены в N,N-диметилформамиде (DMF) и в соответствии с процедурой активированы при помощи O-бензотриазол-1-ил-N,N,N',N'-тетраметилюроний гексафлюорофосфата(HBTU) и диизопропилэтиламина (DIEA). Удаление защитных групп Fmoc осуществлялось при помощи 20% (об.) раствора пиперидина в N,N-диметилформамиде (DMF) в течение 20 мин (шаг 1). Аминогруппа последней аминокислоты была ацетилирована с использованием уксусной кислоты, активированной при помощи O-бензотриазол-1-ил-N,N,N',N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEА). Шаг 2. Селективное снятие защиты с группы Lys (Aloc) производили вручную и осуществляли путм обработки смолы раствором 3-эквивалентного Pd(PPh3)4, растворнного в 5 мл С 6 Н 6 CHCl3 (1:1): 2,5% (об.) NMM : 5% (об.) АсОН в течение 2 ч (шаг 2). После этого смолу промывали CHCl3 (65 мл),20% АсОН в DCM (65 мл), DCM (65 мл) и DMF (65 мл). Шаг 3. После этого синтез опять переводили в автоматический режим и добавляли Fmoc-AEEA-OH и малеимидопропионовую кислоту (шаг 3). Между каждым присоединением смолу промывали 3 разаN,N-диметилформамидом (DMF) и 3 раза изопропанолом (iPrOH). Шаг 4. Пептид отделяли от смолы при помощи 85% TFA/5% TIS/5% тиоанизола и 5% фенола, после того как его осаждали при помощи Et2O температуры сухого льда (шаг 4). Пример 7. Синтез FB010M. Шаг 1. Пример описывает тврдофазный пептидный синтез вещества в масштабе 1 ммоль. К смоле последовательно добавляли следующие защищенные аминокислоты: Fmoc-Phe-OH, Fmoc-Trp(Boc)-OH,Fmoc-Glu(tBu)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ser(tBu)-OH, Fmoc-Ala-OH, Fmoc-Trp(Boc)OH, Fmoc-Lys(Boc)-OH, Fmoc-Asp(tBu)-OH, Fmoc-Leu-OH, Fmoc-Lys(Boc)-OH, Fmoc-Gln(Trt)-OH, FmocLeu-OH, Fmoc-Glu(tBu)-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Lys(Boc)OH, Fmoc-Glu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Gln(Trt)-OH, FmocAla-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Aloc)-OH, Fmoc-Leu-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, FmocThr(tBu)-OH, Fmoc-Ile-OH, Fmoc-Lys(Boc)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Trp(Boc)OH, Fmoc-Glu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Trp(Boc)-OH. Они были растворены в N,Nдиметилформамиде (DMF) и в соответствии с процедурой активированы при помощи O-бензотриазол-1 ил-N,N,N',N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEA). Удаление защитных групп Fmoc осуществляли при помощи 20% (об.) раствора пиперидина в N,Nдиметилформамиде (DMF) в течение 20 мин (шаг 1). Аминогруппа последней аминокислоты была ацетилирована с использованием уксусной кислоты, активированной при помощи O-бензотриазол-1-илN,N,N',N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEA). Шаг 2. Селективное снятие защиты с группы Lys (Aloc) производили вручную и осуществляли путм обработки смолы раствором 3-эквивалентного Pd(PPh3)4, растворнного в 5 мл С 6 Н 6 CHCl3 (1:1): 2,5% (об.) NMM: 5% (об.) АсОН в течение 2 ч (шаг 2). После этого смолу промывали CHCl3 (65 мл),20% АсОН в DCM (65 мл), DCM (65 мл) и DMF (65 мл). Шаг 3. После этого синтез опять переводили в автоматический режим и добавляли Fmoc-AEEA-OH и малеимидопропионовую кислоту (шаг 3). Между каждым присоединением смолу промывали 3 разаN,N-диметилформамидом (DMF) и 3 раза изопропанолом (iPrOH). Шаг 4. Пептид отделяли от смолы при помощи 85% TFA/5% TIS/5% тиоанизола и 5% фенола, после того как его осаждали при помощи Et2O температуры сухого льда (шаг 4). Пример 8. Синтез FB010KM. Шаг 1. Пример описывает твердофазный пептидный синтез вещества в масштабе 1 ммоль. К смоле последовательно добавляли следующие защищенные аминокислоты: Fmoc-Lys(Aloc)-OH, Fmoc-Phe-OH,Fmoc-Trp(Boc)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ser(tBu)-OH, Fmoc-AlaOH, Fmoc-Trp(Boc)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Asp(tBu)-OH, Fmoc-Leu-OH, Fmoc-Lys(Boc)-OH,- 19007918Fmoc-Gln(Trt)-OH, Fmoc-Leu-OH, Fmoc-Glu(tBu)-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Glu(tBu)-OH, FmocAsn(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH,Fmoc-Gln(Trt)-OH, Fmoc-Ala-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Ile-OH, Fmoc-Leu-OH,Fmoc-Ala-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-Lys(Boc)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(tBu)OH, Fmoc-Trp(Boc)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Trp(Вос)-ОН. Они были растворены в N,N-диметилформамиде (DMF) и в соответствии с процедурой активированы при помощи Oбензотриазол-1-ил-N,N,N',N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина(DIEA). Удаление защитных групп Fmoc осуществляли при помощи 20% (об.) раствора пиперидина вN,N-диметилформамиде (DMF) в течение 20 мин (шаг 1). Аминогруппа последней аминокислоты была ацетилирована с использованием уксусной кислоты, активированной при помощи О-бензотриазол-1-илN,N,N',N'-тетраметилюроний гексафлюорофосфата (HBTU) и диизопропилэтиламина (DIEА). Шаг 2. Селективное снятие защиты с группы Lys (Aloc) производили вручную и осуществляли путм обработки смолы раствором 3-эквивалентного Pd(PPh3)4, растворнного в 5 мл С 6 Н 6 CHCl3 (1:1): 2,5% (об.) NMM : 5% (об.) АсОН в течение 2 ч (шаг 2). После этого смолу промывали CHCl3 (65 мл),20% АсОН в DCM (65 мл), DCM (65 мл) и DMF (65 мл). Шаг 3. После этого синтез опять переводили в автоматический режим и добавляли Fmoc-AEEA-OH и малеимидопропионовую кислоту (шаг 3). Между каждым присоединением смолу промывали 3 разаN,N-диметилформамидом (DMF) и 3 раза изопропанолом (iPrOH). Шаг 4. Пептид отделяли от смолы при помощи 85% TFA/5% TIS/5% тиоанизола и 5% фенола, после того как его осаждали при помощи Et2O температуры сухого льда (шаг 4). Пример 9. Ингибирование вируса модифицированными пептидами. Противовирусную активность и цитотоксичность FB005, FB006, FB006M, FB007M, FB010KM иFM010M проверяли по отношению к ВИЧ-1IIIB на свежих культурах человеческих РВМС (моноциты периферической крови). Четыре модифицированных пептида FB006M, FB007M, FB010M, и FB010KM конъюгировали с человеческим сывороточным альбумином (HSA) путм их смешения перед противовирусным тестом. Результаты описаны в табл. 8 ниже, в которой значение IC50 - это концентрация препарата, требуемая для достижения угнетения вируса на 50%, а значение ТС 50 - это концентрация препарата,при которой достигается цитотоксичность 50%. Тестирование воздействия на ВИЧ на культуре клеток Заранее оттитрованные порции ВИЧ-1IIIB вынимали из морозильника (-80 С) и быстро размораживали в биологически безопасном шкафу до комнатной температуры непосредственно перед использованием. Свежие человеческие РВМС изолировали от скринированных доноров, серонегативных на ВИЧ иHBV (вирус гепатита В) (Interstate Blood Bank, Inc.; Memphis, TN). Клетки осаждали/промывали 2-3 раза низкоcкоростным центрифугированием и повторным суспендированием в PBS для удаления примесей тромбоцитов. После этого лейкофорезированную кровь разбавляли фосфатным буферным раствором,Dulbecco (PBS) в соотношении 1:1 и наносили слоем поверх 14 мл среды для отделения лимфоцитов в 50-миллилитровую пробирку для центрифугирования, а затем отцентрифугировали в течение 30 мин при 600 g. Затем отслоенные РВМС осторожно аспирировали с полученной поверхности и отмывали 2 раза низкоскоростным центрифугированием в PBS. После последнего отмывания клетки подсчитывали путм исключения трипанового синего и ещ раз суспендировали (1107 клеток/мл) в RPMI 1640 с добавлением 15% фетальной бычьей сывороткой (FBS), 2 мМ L-глютамина, 4 мкг/мл фитогемагглютинина (РНА-Р,Sigma). После этого клетки инкубировали в течение 48-72 ч при 37 С. После инкубации РВМС центрифугировали и ещ раз суспендировали в RPMI 1640 с 15% FBS, 2 мМ L-глютамина, 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина, 10 мкг/мл гентамицина и 20 Ед/мл рекомбинантного человеческого IL2 (RD Systems, Inc.). РВМС поддерживали в этой среде в концентрации 1-2106 клеток/мл со сменой среды два раза в неделю до тех пор, пока их не использовали в анализе согласно протоколу. Клетки поддерживали в культуре не более 2 недель, после чего их не использовали, как слишком старые для анализа. Культура была бедна моноцитами вследствие их прилипания к сосуду с культурой. Для стандартного РВМС-анализа отбирали стимулированные РНА-Р клетки как минимум от двух нормальных доноров, разбавляли в свежей среде до конечной концентрации 1106 клеток/мл и наносили на внутренние ячейки 96-луночного планшета по 50 мкл в ячейку (5104 клеток в ячейку). В каждом планшете имелись ячейки контроля вирус/клетка (клетки плюс вирус), экспериментальные ячейки (препарат плюс клетки плюс вирус) и клетки контроля вещества (препарат плюс среда без клеток, что необходимо для MTS-мониторинга цитотоксичности). Так как ВИЧ-1 не является цитопатичным для РВМС,то можно использовать одну и ту же тестовую систему для измерения как противовирусной активности,так и цитотоксичности. Разбавленные исследуемые препараты готовили в двойной концентрации в пробирках микротитрования и в соответствующие ячейки по стандартной процедуре наносили по 100 мкл каждой концентрации. В каждую тест-ячейку вводили по 50 мкл определнной концентрации вирусной культуры (конечная MOI0,1). После инфицирования культуры РВМС содержали при 7 С, 5% СО 2, после чего образцы надосадочной жидкости без клеток забирали на анализ активности обратной транс- 20007918 криптазы и/или содержания р 24 ВИЧ. После отбора образцов надосадочной жидкости цитотоксичность препарата измеряли путм добавления в планеты MTS и определения клеточной жизнеспособности. Кроме того, лунки изучали микроскопически и отмечали и любые отклонения от нормы. Дополнительный анализ цитотоксичности Для определения цитотоксичности препаратов в более высоких концентрациях по сравнению с теми, что использовались при оценке анти-ВИЧ активности, провели дополнительное тестирование. По существу, это исследование было аналогичным описаному выше для оценки анти-ВИЧ активности, только в этом случае в лунки вирус не добавляли (вместо этого добавляли питательную среду без вируса) и высшая концентрация для теста была увеличена до 25 мкМ. После инкубации планшеты изучали на жизнеспособность клеток при помощи MTS, как это было описано выше. Таблица 8 Несмотря на то, что новое изобретение довольно подробно описано при помощи иллюстрации и примера для лучшего понимания, очевидно, что в пределах поданной заявки на изобретение можно производить определнные изменения и модификации. Предполагается, что модификации вышеописанных моделей реализации изобретения, которые представляются очевидными для специалистов из области медицины, иммунологии, вирусологии, фармакологии, белкового синтеза и модификации и/или связанных с ними областей, охватываются нижеследующей формулой изобретения. Все публикации и описания патентов, упомянутые в данном документе, указывают на уровень мастерства специалистов в той области, к которым относится данное изобретение. Все эти публикации и описания патентов включены в качестве ссылки в том виде, в каком каждая отдельная публикация или описание патента было бы специально отдельно указано для включения в документ в качестве ссылки. Некоторые пептиды и производные из них, которые полезны при профилактике и/или лечении вирусной инфекции, в особенности ВИЧ-инфекции, были описаны в патентной заявке США US Provisional PatentApplication60/412797, поданной в 24 сентября 2002 г.; е содержание (в т.ч. все последовательности,описанные в ней) приведено в данном документе полностью в качестве ссылки. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Изолированный пептид FB005, содержащий последовательность SEQ ID NO:1. 2. Изолированный пептид FB006, содержащий последовательность SEQ ID NO:2. 3. Изолированный пептид FB066, содержащий последовательность SEQ ID NO:7. 4. Изолированный модифицированный пептид, выбранный из группы, состоящей из:(d) SEQ ID NO:7,и имеющий по меньшей мере одну замену аминокислотного остатка в предварительно определнном положении пептидной цепи, при этом по меньшей мере один привнеснный аминокислотный остаток является гидрофильным аминокислотным остатком, гидрофобным аминокислотным остатком, аминокислотным остатком со склонностью образовывать альфа-спирали, D-изомером природной Lаминокислоты или не встречающимся в природе аминокислотным остатком. 5. Производное изолированного пептида, которое выбирают из группы, состоящей из:(d) SEQ ID NO:7,причем производные заданных остатков аминокислот пептидной цепи получены путм конъюгации указанных заданных аминокислотных остатков с присоединяющими группами. 7. Модифицированный пептид по п.4, отличающийся тем, что производные заданных остатков аминокислот пептидной цепи получены путм конъюгации указанных аминокислотных остатков с присоединяющими группами. 8. Изолированный пептид по п.1, отличающийся тем, что представляет собой производное, полученное путм прикрепления присоединяющей группы к лизину, замещающему остаток глутаминовой кислоты в положении 23 или добавленному с С-конца. 9. Изолированный пептид по п.2, отличающийся тем, что он представляет собой производное, полученное путм прикрепления присоединяющей группы к лизину в положении 13. 10. Изолированный пептид по п.2, отличающийся тем, что он модифицирован путм замены лизина в положении 13 на остаток глутаминовой кислоты, и из такого пептида получено производное путм прикрепления присоединяющей группы к дополнительному лизиновому остатку, добавленному с Сконца. 11. Производное изолированного пептида, содержащее последовательность SEQ ID NO:3, отличающееся тем, что пептид модифицирован путм замены остатка глутаминовой кислоты в положении 13 на лизин и прикрепления присоединяющей группы к лизину, или получено производное пептида путм конъюгации присоединяющей группы с остатком лизина, добавленным с С-конца. 12. Изолированный пептид по п.3, из которого получено производное путм прикрепления присоединяющей группы к лизину в положении 13 или дополнительному лизиновому остатку, добавленному с С-конца. 13. Производное пептида по любому из пп.5-12, в котором присоединяющая группа выбрана из:(d) карбонильной группы. 14. Производное пептида по п.13, в котором малеимидная группа связана через 3'малеимидопропионат с ипсилон-аминогруппой лизина при помощи [2-(2-амино)этоксил]этоксиуксусной кислоты. 15. Фармацевтический состав, содержащий пептид по любому из пп.1-4 или производное пептида по любому из пп.5-12. 16. Конъюгат, содержащий производное пептида по любому пп.5-12, конъюгированное с компонентом крови. 17. Конъюгат по п.16, у которого компонент крови выбран из группы, состоящей из следующих белков:(h) стероидсвязывающие белки человека и(i) их сочетания. 18. Способ подавления вирусной инфекции в клетках млекопитающего, который включает доставку к указанным клеткам пептида по любому из пп.1-4 или производного пептида по любому из пп.5-12. 19. Способ профилактики вирусной инфекции в клетках млекопитающего, который включает доставку к указанным клеткам пептида по любому из пп.1-4 или производного пептида по любому из пп.512. 20. Способ предотвращения репликации вируса в клетках млекопитающего, который включает доставку к указанным клеткам пептида по любому из пп.1-4 или производного пептида по любому из пп.512. 21. Способ по любому из пп.18-20, в котором упомянутый пептид доставляют в присутствии упомянутого вируса. 22. Способ по любому из пп.18-20, в котором вирус выбирают из группы, включающей:(a) вирус иммунодефицита человека (ВИЧ) и(b) вирус иммунодефицита обезьян (ВИО). 23. Способ по любому из пп.18-20, в котором пептид или производное пептида назначают перорально, местно, внутривенно, внутриартериально, внутримышечно или подкожно. 24. Способ по любому из пп.18-20, в котором пептид или производное пептида назначают совместно с одним или более препаратами для лечения ВИЧ-инфекции. 25. Способ по любому из пп.18-20, в котором указанный один или более дополнительный препарат для лечения ВИЧ-инфекции представляет собой вариантный gp41-пептид. 26. Способ по п.24, в котором дополнительный препарат для лечения ВИЧ-инфекции выбирают из группы, включающей следующие препараты:(v) ZIAGEN. 27. Способ по любому из пп.18-20, в котором вирус представляет собой ВИЧ, а клетки млекопитающего представляют собой клетки человека. 28. Способ профилактики или подавления ВИЧ-инфекции, согласно которому назначают производные вариантных gp41-пептидов по любому из пп.5-12 пациенту, чьи клетки подверглись контакту с ВИЧ,при этом указанное производное пептида конъюгируют с компонентом крови указанного пациента, увеличивая таким образом период полураспада пептида в крови указанного пациента. 29. Способ создания противовирусного конъюгата, который включает смешивание производного вариантного gp41-пептида (или пептидов) с компонентами крови с формированием ковалентных связей между производным вариантного gp41-пептида и компонентами крови. 30. Способ по любому из пп.27, 28, в котором компонент крови выбирают из группы, включающей следующие белки:(h) стероидсвязывающие белки человека и(i) их сочетания. 31. Способ по п.29, в котором компонент крови представляет собой сывороточный альбумин человека. 32. Способ по любому из пп.27, 28, в котором конъюгация происходит in vivo. 33. Способ по любому из пп.27, 28, в котором конъюгация происходит ex vivo. 34. Способ по п.32, в котором перед конъюгацией с производным пептида компонент(ы) крови разделяют при помощи плазмафореза. 35. Фармацевтический состав, содержащий изолированный пептид по любому из пп.1-14 с фармацевтически приемлемым носителем. 36. Способ генерирования пептидов с противовирусным, виростатическим или антифузогенным действием, включающий:(a) скрининг белков вирулентности вируса для определения в них последовательностей с альфа- 23007918 спиральобразующими свойствами;(b) создание изменнного пептида путм модификации или получения производного хотя бы одного аминокислотного остатка указанной последовательности;(c) синтез указанных изменнных пептидов и(d) тестирование указанных пептидов для определения их противовирусного, виростатического или антифузогенного действия.
МПК / Метки
МПК: A61K 39/21, C07K 7/00
Метки: вич-инфекции, слияния, производные, ингибиторы, пептидные
Код ссылки
<a href="https://eas.patents.su/25-7918-peptidnye-proizvodnye-ingibitory-sliyaniya-pri-vich-infekcii.html" rel="bookmark" title="База патентов Евразийского Союза">Пептидные производные – ингибиторы слияния при вич-инфекции</a>
Пептидные ингибиторы вируса гепатита с

Номер патента: 4765
Опубликовано: 26.08.2004
Авторы: Байли Мюррей Д., Пупар Марк-Андре, Гиро Элиза, Ллина-Брюне Монсе, Тсантризо Иула С., Гудреу Натали, Ранкур Жан, Камерон Даль
МПК: C07K 14/81, A61P 31/14, A61K 38/55...
Метки: вируса, пептидные, ингибиторы, гепатита
Формула / Реферат:
1. Рацематы, диастериоизомеры и оптические изомеры соединения формулы (I) , в которой a равно 0 или 1, b обозначает 0 или 1, Y обозначает H или C1-C6алкил, B обозначает H, ацильное производное формулы R7-C(O)- или сульфонил формулы R7-SO2, где R7 обозначает C1-C10алкил, необязательно замещенный карбоксилом, C1-C6аланоилокси- или C1-C6алкоксигруппой, C6- или C10арил или C7-C16аралкил, необязательно замещенный C1-C6алкилом, гидрокси- или...
Ингибиторы гиразы и их применение для лечения бактериальной инфекции

Номер патента: 5680
Опубликовано: 28.04.2005
Авторы: Стамос Дин, Ронкин Стивен, Бадиа Майкл, Чарифсон Пол, Грийо Анн-Лор, Трюдо Мартэн
МПК: C07D 417/04, A61K 31/4155, A61P 31/04...
Метки: лечения, бактериальной, инфекции, гиразы, ингибиторы, применение
Формула / Реферат:
1. Способ лечения бактериальной инфекции у млекопитающего, в случае необходимости такого лечения, включающий стадию введения указанному млекопитающему терапевтически эффективного количества соединения формулы или его фармацевтически приемлемой соли, где R1 обозначает необязательно замещеннную группу, выбранную из C1-6 алифатической группы, -C (R4)2(CH2)nNRCOR, -C(R4)2(CH2)nNRCO2(C1-6 алифатической группы), -CO2(C1-6 алифатической группы),...
Производные пиразола для лечения инфекции вируса иммунодефицита человека (вич)

Номер патента: 7184
Опубликовано: 25.08.2006
Авторы: Селби Мэттью Данкан, Джоунз Лин Хауард, Маубрей Чарлз Эрик, Стаппл Пол Энтони, Прайс Дейвис Энтони
МПК: A61K 31/4155, A61K 31/4162, A61K 31/415...
Метки: пиразола, иммунодефицита, инфекции, человека, производные, вируса, лечения, вич
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное, где либо R1 представляет собой Н, С1-С6алкил, С3-С7циклоалкил, фенил, бензил, галогено, -CN, -OR7, -CO2R10, -CONR5R10, R8 или R9, причем указанные С1-С6алкил, С3-С7циклоалкил, фенил и бензил возможно замещены галогено, -CN, -OR10, S(O)xR10, -CO2R10, -CONR5R10, -OCONR5R10, -NR5CO2R10, -NR10R11, -NR5COR10, -SO2NR5R10,-NR5CONR5R10, -NR5SO2R10 или R10; и R2...
Производные &beta-d-нуклеозида в качестве лекарственного средства для лечения инфекции вируса гепатита c у хозяина

Номер патента: 7178
Опубликовано: 25.08.2006
Авторы: Соммадосси Жан-Пьер, Лаколла Пауло
МПК: A61K 31/7076, A61K 31/7068, A61P 31/14...
Метки: вируса, beta-d-нуклеозида, производные, средства, инфекции, качестве, хозяина, лечения, гепатита, лекарственного
Формула / Реферат:
1. Применение эффективного против вируса количества производного b-D-нуклеозида структуры или его фармацевтически приемлемой соли или пролекарства, необязательно, в фармацевтически приемлемом носителе или разбавителе при получении лекарственного средства для лечения инфекции вируса гепатита С у хозяина. 2. Применение эффективного против вируса количества производного b-D-нуклеозида структуры или его фармацевтически приемлемой соли или...
Циклические пептидные антигрибковые агенты

Номер патента: 464
Опубликовано: 26.08.1999
Авторы: Тернер Уильям В.Мл., Васудеван Венкатрагхаван, Родригез Майкл Дж., Борромео Питер С., Джемисон Джеймс Э.
МПК: A61K 38/12, C07K 7/56
Метки: циклические, антигрибковые, агенты, пептидные
Формула / Реферат:
1. Соединение формулы I: где R' обозначает водород, метил или NH2C(O)CH2-; R" и R'" обозначают, независимо, метил или водород; Rx1, Rx2, Ry1, Ry2, Ry3, Ry4 обозначают, независимо, гидрокси или водород; R0 обозначает группу формулы: R1 обозначает С1-С6 алкил, C1-C6 алкокси, фенил, п-галогенфенил, п-нитрофенил, фенокси, бензил, п-галогенбензил или п-нитробензил; I) R2 обозначает группу формулы где A) R3 обозначает...
Предыдущий патент: Строительство большепролетных зданий с самораскреплением из составных несущих стеновых панелей и перекрытий
Следующий патент: Средство, обладающее противовирусной активностью, и способ его получения
Случайный патент: Способ заваривания напитка и картридж, содержащий заварочный материал