Новые ингибиторы глутаминилциклазы
Номер патента: 13790
Опубликовано: 30.06.2010
Авторы: Нистрой Андре Иоганнес, Хайзер Ульрих, Демут Ханс-Ульрих, Буххольц Мирко, Шиллинг Штефан


















Формула / Реферат
1. Соединение формулы 1, включая его фармацевтически приемлемые соли, включая все стереоизомеры и полиморфы
![]()
в которой
А обозначает неразветвленную С3алкильную цепь и
В представляет собой группу, выбранную из (VI) или (VII)

где D обозначает замещенный фенил, в котором заместителями являются оксиалкил, тиоалкил, галогенил; или D обозначает дигидробензодиоксин, бензодиоксол, бензодитиол, дигидробензотидин, бензооксатиол или дигидробензооксатин;
X обозначает S, N-CN;
Y обозначает S;
R17 и R18 оба обозначают Н; один из R17 и R18 обозначает Н, а другой обозначает метил; или R17 и R18 могут быть соединены с образованием карбоцикла, имеющего вплоть до 6 кольцевых атомов;
и где понятие алкил обозначает метил, этил, пропил или бутил.
2. Соединение по п.1, в котором В обозначает группу (VI).
3. Соединение по п.2, в котором X обозначает S.
4. Соединение по п.1, в котором В обозначает группу (VII).
5. Соединение по п.4, в котором R17 и R18 оба обозначают Н.
6. Соединение по п.4, в котором один из R17 и R18 обозначает Н, а другой обозначает метил.
7. Соединение по п.4, в котором R17 и R18 соединены с образованием карбоцикла, имеющего вплоть до 6 кольцевых атомов.
8. Соединение формулы 1а, включая его фармацевтически приемлемые соли, включая все стереоизомеры и полиморфы

в котором группа R обозначает метил, трет-бутил, бензил, фенил, 4-фторфенил, 4-хлорфенил, 4-этилфенил, 4-(трифторметил)фенил, 4-(метоксикарбонил)фенил, 4-(метилкарбонил)фенил, 4-(метокси)фенил, бицикло[2.2.1]гепт-5-ен-2-ил, 3,4-(диметокси)фенил, 2,4-(диметокси)фенил, 3,5-(диметокси)фенил, 2-(метоксикарбонил)фенил, 4-(оксазол-5-ил)фенил, 4-(пиразол-1-ил)фенил, 4-изопропилфенил, 4-(пиперидин-1-сульфонил)фенил, 4-(морфолин-4-ил)фенил, 4-цианфенил, 2,3-дигидробензо[1,4]диоксин-6-ил, бензо[1,3]диоксил-5-ил, 3,4,5-(триметокси)фенил, 3-(метокси)фенил, 4-(этокси)фенил, 4-(бензилокси)фенил, 4-(метокси)бензил, 4-йодфенил, 4-бромфенил, 4-метилфенил, нафтилин-1-ил, 4-нитрофенил, н-бутил, циклооктил, фуран-2-илметил, тетрагидрофуран-2-илметил, бензо[1,3]диоксол-5-илметил, 2-(морфолин-4-ил)этил, 4-(метилсульфанил)фенил, 4-(диметиламино)фенил, 4-(трифторметокси)фенил, бензоил или пиридин-4-ил, при условии (д), что исключены следующие соединения:

9. Соединение формулы 1а по п.8, в котором группа R обозначает 3,4-(диметокси)фенил.
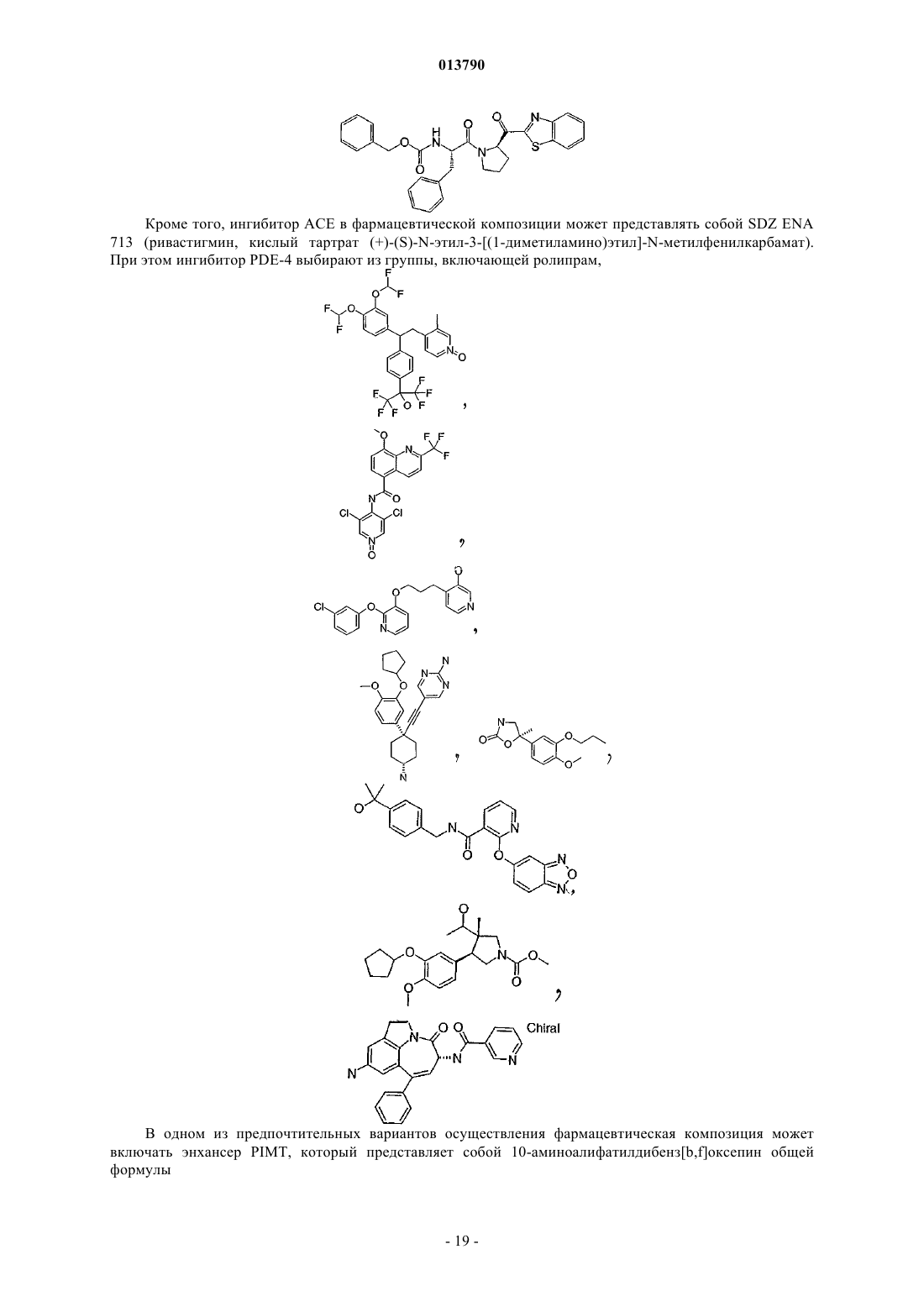
10. Соединение формулы 1б, включая его фармацевтически приемлемые соли, включая все стереоизомеры и полиморфы

в котором группа R2 обозначает метил, трет-бутил, бензил, фенил, 4-фторфенил, 4-этилфенил, 4-(трифторметил)фенил, 4-(метилкарбонил)фенил, 4-(метокси)фенил, 3,4-(диметокси)фенил, 2,4-(диметокси)фенил, 2,3,4-(триметокси)фенил, 3,5-(диметокси)фенил, 2,3-дигидробензо[1,4]диоксин-6-ил, бензо[d][1,3]диоксол-6-ил, 3-(метокси)фенил, 4-(этокси)фенил, 4-(бензилокси)фенил, 4-йодфенил, 4-бромфенил, 4-метилфенил, нафтилин-1-ил, 4-нитрофенил, н-бутил, циклооктил, фуран-2-илметил, тетрагидрофуран-2-илметил, бензо[d][1,3]диоксол-6-илметил, 2-(морфолин-4-ил)этил, 4-(диметиламино)фенил, 4-(метилсульфанил)фенил или тритил.
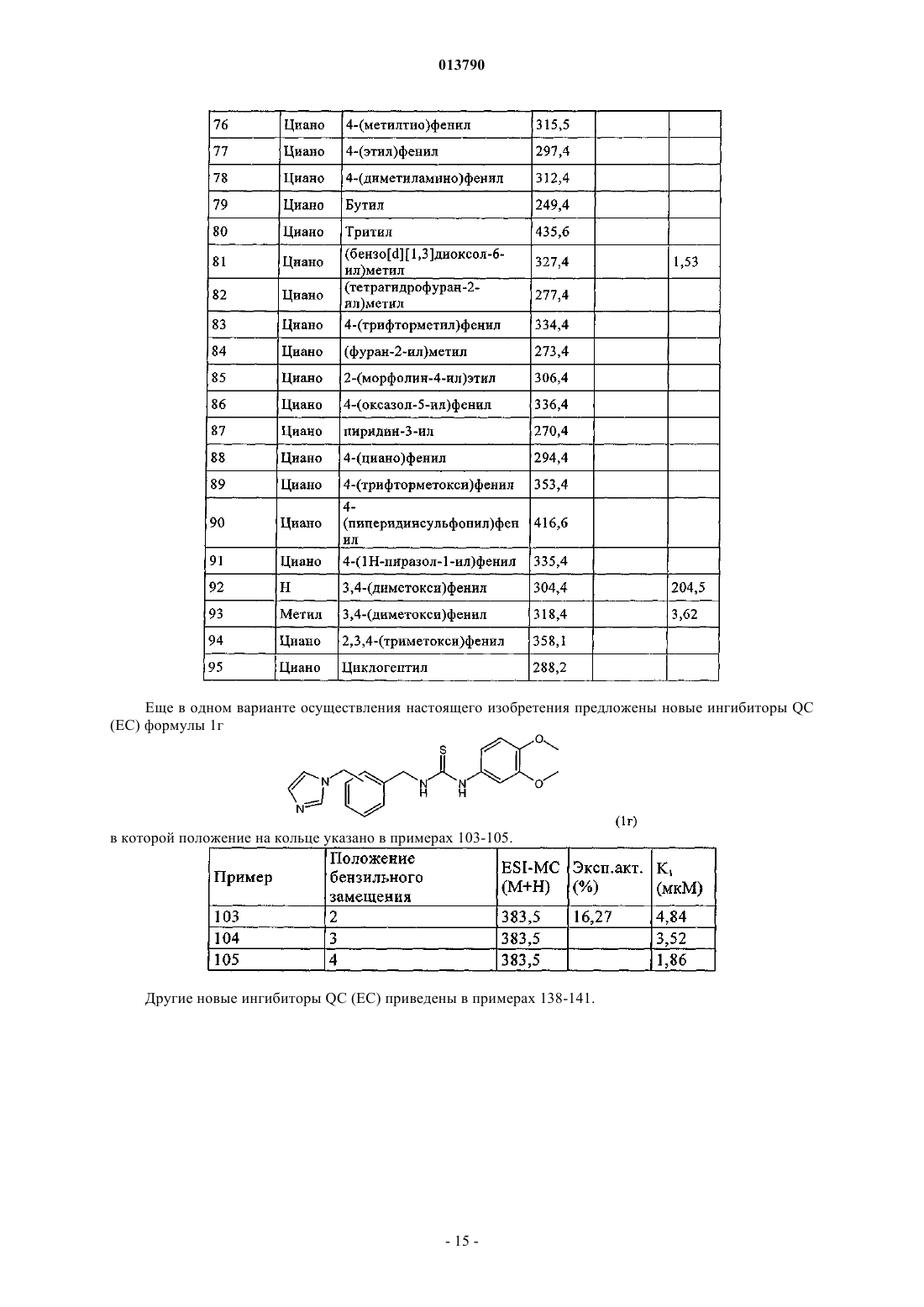
11. Соединение формулы 1г, включая его фармацевтически приемлемые соли, включая все стереоизомеры и полиморфы

в котором группа R7 обозначает 3,4-(диметокси)фенил, а группы R8 и R9обе обозначают Н; или группа R7обозначает 4-(хлор)фенил, а группы R8и R9 вместе с атомом углерода, с которым они связаны, образуют циклобутильное или циклопетильное кольцо; или R7обозначает 4-(метокси)фенил, а группы R8и R9 вместе с атомом углерода, с которым они связаны, образуют циклопропильное или циклогексильное кольцо; или группы R8 и R9вместе с атомом углерода, с которым они связаны, образуют циклопропильное кольцо, а группа R7 обозначает 3,4-(диметокси)фенил.
12. Соединение формулы 1г по п.11, в котором группа R7обозначает 3,4-(диметокси)фенил, а группы R8и R9 обе обозначают Н; или группа R7 обозначает 4-(хлор)фенил, а группы R8 и R9вместе с атомом углерода, с которым они связаны, образуют циклобутильное или циклопентильное кольцо; или R7 обозначает 4-(метокси)фенил, а группы R8и R9 вместе с атомом углерода, с которым они связаны, образуют циклопропильное или циклогексильное кольцо.
13. Соединение формулы 1г по п.11, в котором группа R7обозначает 3,4-(диметокси)фенил, а группы R8и R9 обе обозначают Н.
14. Соединение формулы 1г по п.11, в котором группы R8и R9 вместе с атомом углерода, с которым они связаны, образуют циклопропильное кольцо, а группа R7обозначает 3,4-(диметокси)фенил.
15. Соединение

включая его фармацевтически приемлемые соли и полиморфы.
16. Соединение формулы 1, включая его фармацевтически приемлемые соли, включая все стереоизомеры и полиморфы
![]()
в которой
А обозначает неразветвленную С3алкильную цепь и
В представляет собой группу, выбранную из (VI) или (VII)

где D обозначает замещенный фенил, в котором заместителями являются оксиалкил, тиоалкил, галогенил; или D обозначает дигидробензодиоксин, бензодиоксол, бензодитиол, дигидробензотидин, бензооксатиол или дигидробензооксатин;
X обозначает S, N-CN;
Y обозначает S;
R17 и R18 оба обозначают Н; один из R17 и R18 обозначает Н, а другой обозначает метил; или R17 и R18 могут быть соединены с образованием карбоцикла, имеющего вплоть до 6 кольцевых атомов; и
где понятие алкил обозначает метил, этил, пропил или бутил для применения в качестве фармацевтического средства.
17. Фармацевтическая композиция, содержащая по меньшей мере одно соединение по одному из пп.1-16 необязательно в сочетании с терапевтически приемлемым носителем и/или эксципиентом.
18. Фармацевтическая композиция по п.17, предназначенная для парентерального, энтерального или орального введения.
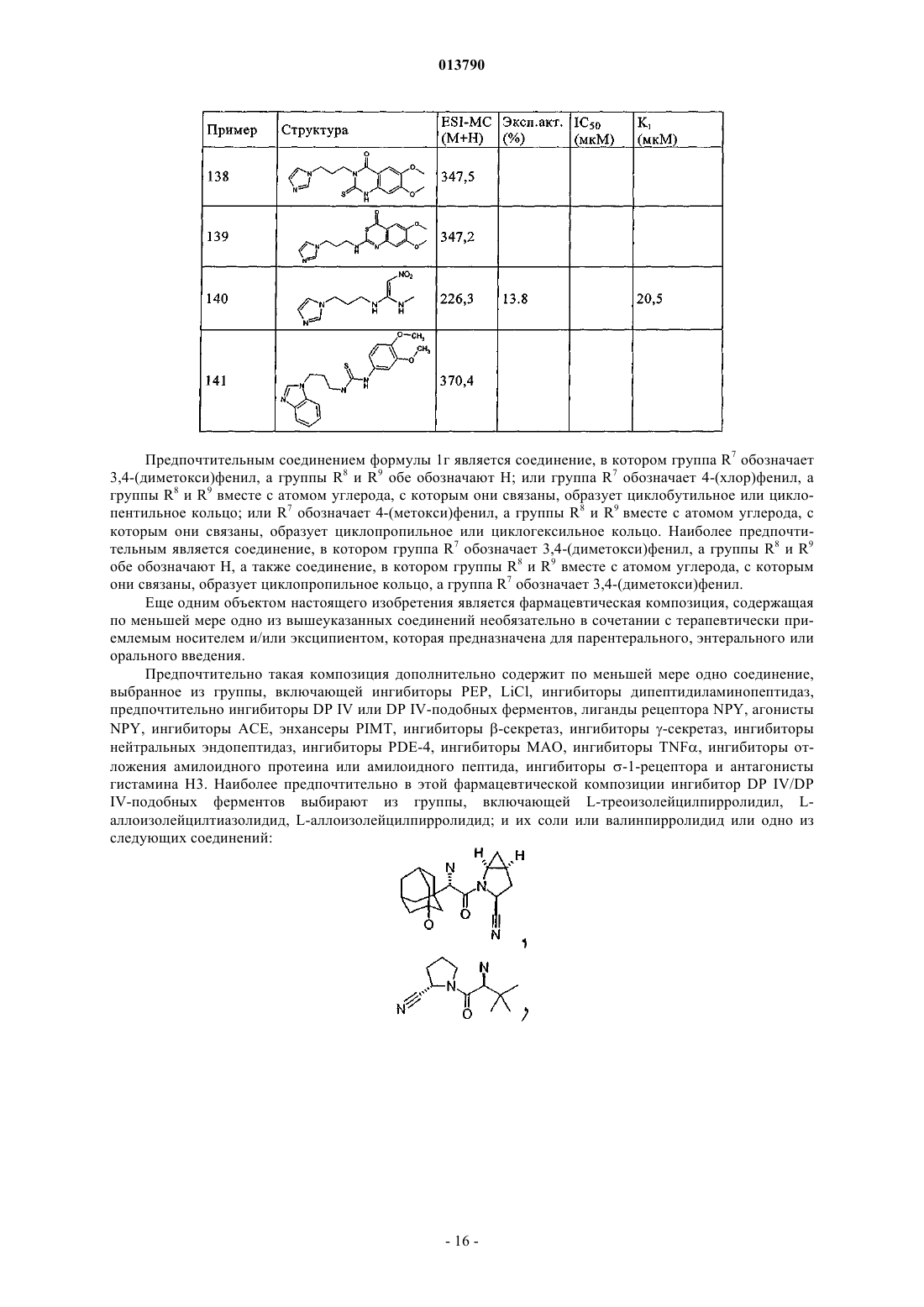
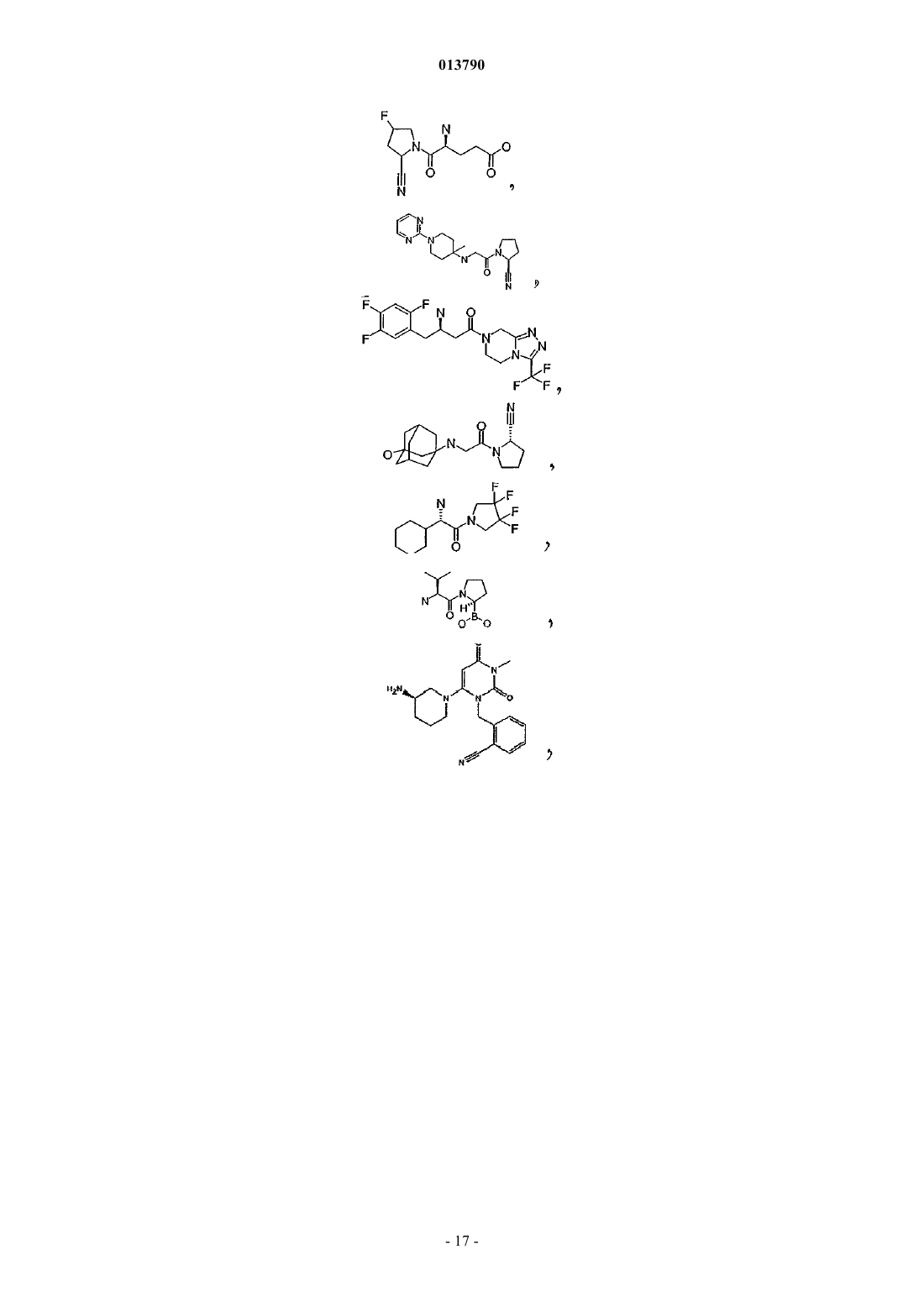
19. Фармацевтическая композиция по п.17 или 18, которая дополнительно содержит по меньшей мере одно соединение, выбранное из группы, включающей ингибиторы PEP, LiCl, ингибиторы дипептидиламинопептидаз, предпочтительно ингибиторы DP IV или DP IV-подобных ферментов, лиганды рецептора NPY, агонисты NPY, ингибиторы АСЕ, энхансеры PIMT, ингибиторы b-секретаз, ингибиторы g-секретаз, ингибиторы нейтральных эндопептидаз, ингибиторы PDE-4, ингибиторы МАО, ингибиторы TNFa, ингибиторы отложения амилоидного протеина или амилоидного пептида, ингибиторы s-1-рецептора и антагонисты гистамина Н3.
20. Фармацевтическая композиция по п.19, в которой ингибитор DP IV/DP IV-подобных ферментов выбирают из группы, включающей L-треоизолейцилпирролидил, L-аллоизолейцилтиазолидид, L-аллоизолейцилпирролидид; и их соли или валинпирролидид или одно из следующих соединений:



21. Фармацевтическая композиция по п.19, в которой антагонист NPY выбирают из 3а,4,5,9b-тетрагидро-1h-бенз[е]индол-2-иламина, (R)-N2-(дифенилацетил)-(R)-N-[1-(4-гидроксифенил)этил]аргинининамида

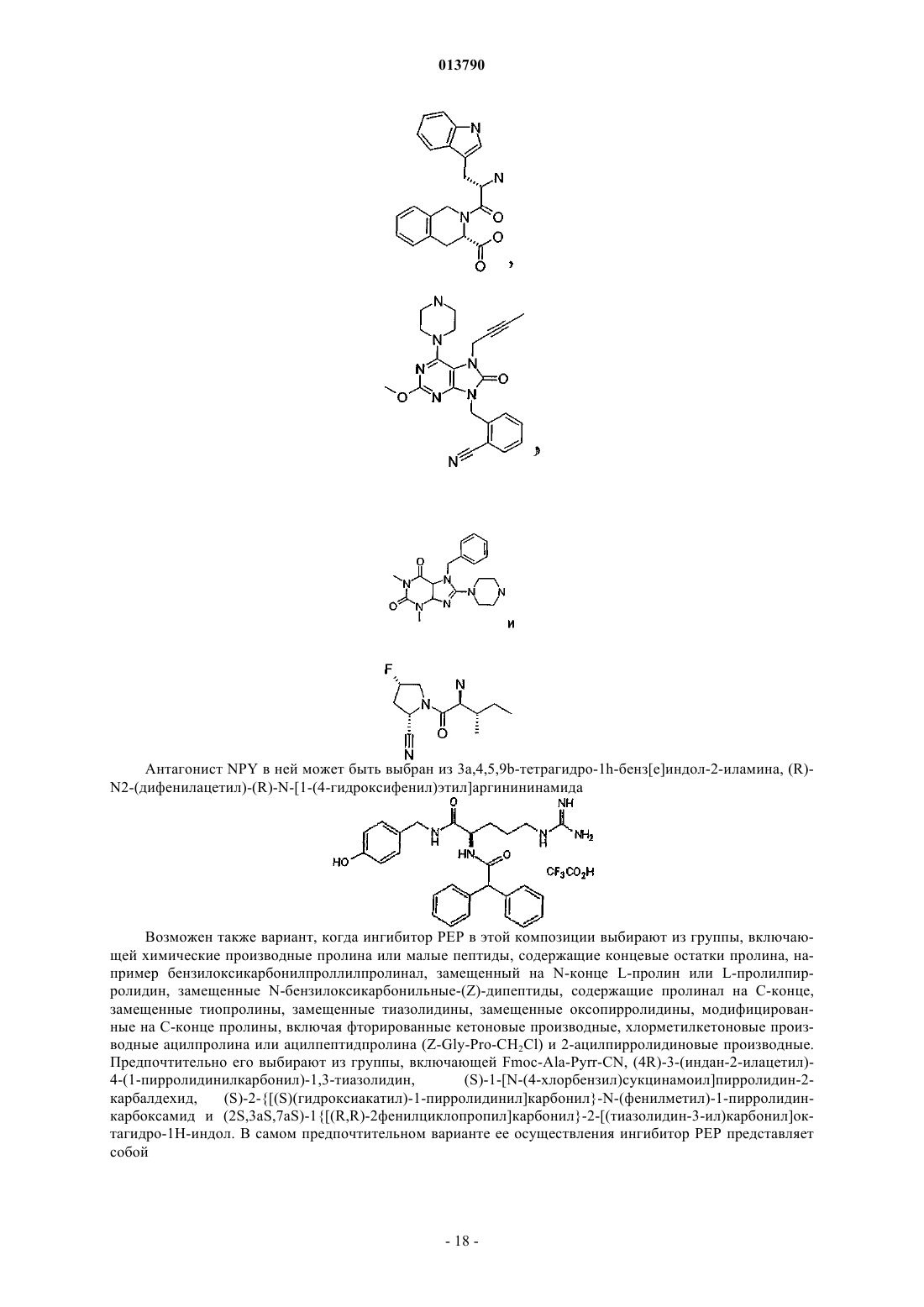
22. Фармацевтическая композиция по п.19, в которой ингибитор PEP выбирают из группы, включающей химические производные пролина или малые пептиды, содержащие концевые остатки пролина, например бензилоксикарбонилпроллилпролинал, замещенный на N-конце L-пролин или L-пролилпирролидин, замещенные N-бензилоксикарбонильные-(Z)-дипептиды, содержащие пролинал на С-конце, замещенные тиопролины, замещенные тиазолидины, замещенные оксопирролидины, модифицированные на С-конце пролины, включая фторированные кетоновые производные, хлорметилкетоновые производные ацилпролина или ацилпептидпролина (Z-Gly-Pro-CH2Cl) и 2-ацилпирролидиновые производные.
23. Фармацевтическая композиция по п.19, в которой ингибитор PEP выбирают из группы, включающей Fmoc-Ala-Pyrr-CN, (4R)-3-(индан-2-илацетил)-4-(1-пирролидинилкарбонил)-1,3-тиазолидин, (S)-1-[N-(4-хлорбензил)сукцинамоил]пирролидин-2-карбалдехид, (S)-2-{[(S)(гидроксиакатил)-1-пирролидинил]карбонил}-N-(фенилметил)-1-пирролидин-карбоксамид и (2S,3aS,7aS)-1{[(R,R)-2-фенилциклопропил]карбонил}-2-[(тиазолидин-3-ил)карбонил]октагидро-1H-индол.
24. Фармацевтическая композиция по п.19, в которой ингибитор PEP представляет собой

25. Фармацевтическая композиция по п.19, в которой ингибитор АСЕ представляет собой SDZ ENA 713 (ривастигмин, кислый тартрат (+)-(S)-N-этил-3-[(1-диметиламино)этил]-N-метилфенилкарбамат.
26. Фармацевтическая композиция по п.19, в которой ингибитор PDE-4 выбирают из группы, включающей ролипрам

или их аналоги

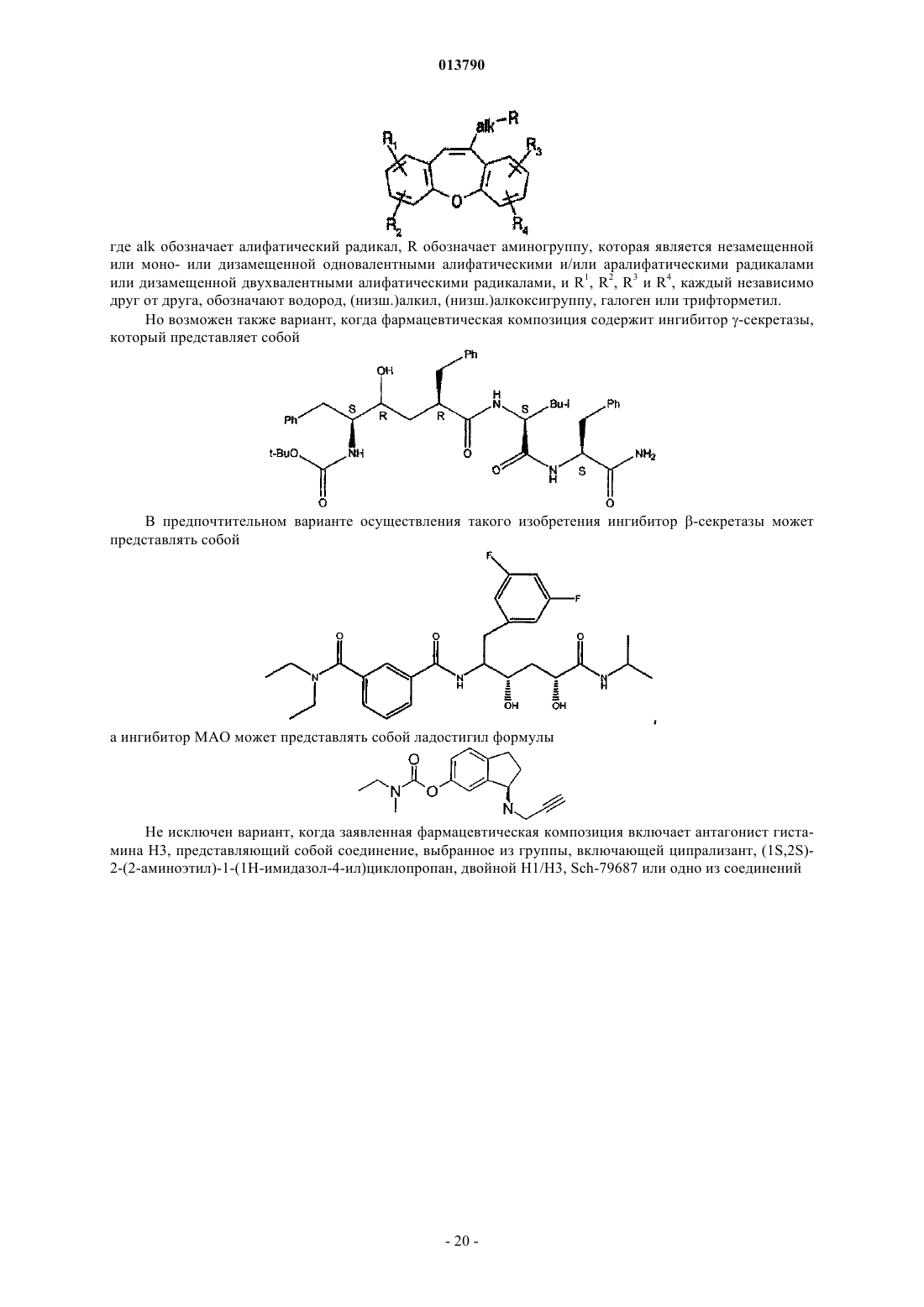
27. Фармацевтическая композиция по п.19, в которой энхансер PIMT представляет собой 10-аминоалифатилдибенз[b,f]оксепин общей формулы

где alk обозначает алифатический радикал, R обозначает аминогруппу, которая является незамещенной или моно- или дизамещенной одновалентными алифатическими и/или аралифатическими радикалами или дизамещенной двухвалентными алифатическими радикалами, и R1, R2, R3и R4, каждый независимо друг от друга, обозначают водород, (низш.)алкил, (низш.)алкоксигруппу, галоген или трифторметил.
28. Фармацевтическая композиция по п.19, в которой ингибитор g-секретазы представляет собой

29. Фармацевтическая композиция по п.19, в которой ингибитор b-секретазы представляет собой

30. Фармацевтическая композиция по п.19, в которой ингибитор МАО представляет собой ладостигил формулы

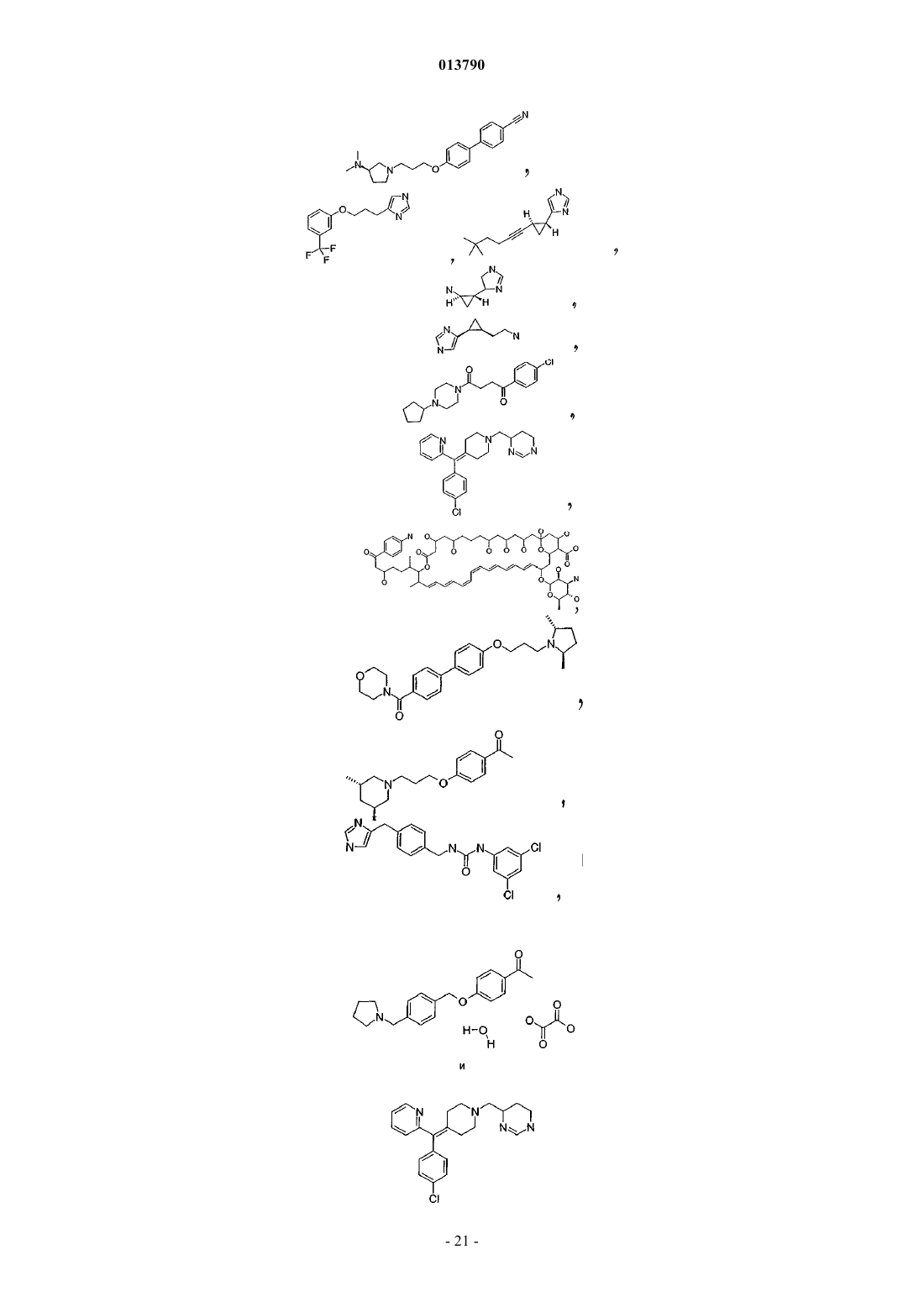
31. Фармацевтическая композиция по п.19, в которой антагонист гистамина Н3 представляет собой соединение, выбранное из группы, включающей ципрализант, (1S,2S)-2-(2-аминоэтил)-1-(1Н-имидазол-4-ил)циклопропан, двойной Н1/Н3, Sch-79687 или одно из соединений



32. Применение фармацевтического соединения по одному из пп.1-16, которое является ингибитором глутаминилциклазы, для приготовления лекарственного средства, предназначенного для лечения нейронных нарушений, прежде всего таких заболеваний, как болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона, хорея Гентингтона, патогенные психические состояния, шизофрения, нарушение всасывания пищи, нарушение сна, нарушение связанной с гомеостазом регуляции энергетического метаболизма, нарушение функции вегетативной нервной системы, нарушение гормонального баланса, нарушение регуляции общей воды организма, гипертензия, лихорадка, нарушение регуляции сна, анорексия, связанные со страхом нарушения, включая депрессию, припадки, включая эпилепсию, синдром отмены лекарственного средства и алкоголизм, нейродегенеративные заболевания, включая когнитивную дисфункцию и деменцию.
33. Применение по п.32 для лечения нейронных нарушений, выбранных из группы, включающей болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона и хорею Гентингтона.
34. Применение по п.32 или 33, в котором ингибирование ингибитором глутаминилциклазы характеризуется значением Ki, составляющим 1 мкМ или менее.
35. Применение по п.32 или 33, в котором ингибирование глутаминилциклазы характеризуется значением Ki, составляющим 0,1 мкМ или менее.
36. Применение по п.32 или 33, в котором ингибирование ингибитором глутаминилциклазы характеризуется значением Ki, составляющим 0,01 мкМ или менее.
37. Применение по одному из пп.32-36, в котором ингибитор глутаминилциклазы имеет молекулярную массу 1000 г/моль или менее.
38. Применение по одному из пп.32-36, в котором ингибитор глутаминилциклазы имеет молекулярную массу 500 г/моль или менее.
39. Применение по одному из пп.32-36, в котором ингибитор глутаминилциклазы имеет молекулярную массу 400 г/моль или менее.
40. Применение по одному из пп.32-36, в котором ингибитор глутаминилциклазы имеет молекулярную массу 350 г/моль или менее.
41. Применение по одному из пп.32-40, в котором ингибитор глутаминилциклазы представляет собой конкурентный ингибитор.
42. Применение по одному из пп.32-41, в котором ингибитор глутаминилциклазы представляет собой конкурентный обратимый ингибитор.
43. Применение по одному из пп.32-42, в котором ингибитор глутаминилциклазы выбирают из соединений по одному из пп.1-16.
44. Применение по п.43, в котором ингибитор глутаминилциклазы вводят млекопитающему необязательно в комбинации с соединением, выбранным из группы, включающей ингибиторы PEP, LiCl, ингибиторы дипептидиламинопептидаз, предпочтительно ингибиторы DP IV или подобных DP IV ферментов, лиганды рецептора NPY, агонисты NPY, ингибиторы АСЕ, энхансеры PIMT, ингибиторы b-секретаз, ингибиторы g-секретаз, ингибиторы нейтральных эндопептидаз, ингибиторы PDE-4, ингибиторы МАО, ингибиторы TNFa, ингибиторы отложения амилоидного протеина или амилоидного пептида, ингибиторы s-1-рецептора и антагонисты гистамина Н3.
45. Способ лечения нейронных нарушений, прежде всего, таких как болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона, хорея Гентингтона, патогенные психические состояния, шизофрения, нарушение всасывания пищи, нарушение сна, нарушение связанной с гомеостазом регуляции энергетического метаболизма, нарушение функции вегетативной нервной системы, нарушение гормонального баланса, нарушение регуляции общей воды организма, гипертензия, лихорадка, нарушение регуляции сна, анорексия, связанные со страхом нарушения, включая депрессию, припадки, включая эпилепсию, синдром отмены лекарственного средства и алкоголизм, нейродегенеративные заболевания, включая когнитивную дисфункцию и деменцию, который заключается в том, что млекопитающему вводят эффективное количество соединения по одному из пп.1-16, которое является ингибитором глутаминилциклазы.
46. Способ по п.45, в котором ингибирование ингибитором глутаминилциклазы характеризуется значением Ki, составляющим 1 мкМ или менее.
47. Способ по п.45, в котором ингибирование глутаминилциклазы характеризуется значением Ki, составляющим 0,1 мкМ или менее.
48. Способ по п.45, в котором ингибирование ингибитором глутаминилциклазы характеризуется значением Ki, составляющим 0,01 мкМ или менее.
49. Способ по одному из пп.45-48, в котором ингибитор глутаминилциклазы имеет молекулярную массу 1000 г/моль или менее.
50. Способ по одному из пп.45-48, в котором ингибитор глутаминилциклазы имеет молекулярную массу 500 г/моль или менее.
51. Способ по одному из пп.45-48, в котором ингибитор глутаминилциклазы имеет молекулярную массу 400 г/моль или менее.
52. Способ по одному из пп.45-48, в котором ингибитор глутаминилциклазы имеет молекулярную массу 350 г/моль или менее.
53. Способ по одному из пп.45-52, в котором ингибитор глутаминилциклазы представляет собой конкурентный ингибитор.
54. Способ по одному из пп.45-53, в котором ингибитор глутаминилциклазы представляет собой конкурентный обратимый ингибитор.
55. Способ по одному из пп.45-54, в котором ингибитор глутаминилциклазы выбирают из соединений по одному из пп.1-16.
56. Способ по п.55, в котором ингибитор глутаминилциклазы вводят млекопитающему необязательно в комбинации с соединением, выбранным из группы, включающей ингибиторы PEP, LiCl, ингибиторы дипептидиламинопептидаз, предпочтительно ингибиторы DP IV или подобных DP IV ферментов, лиганды рецептора NPY, агонисты NPY, ингибиторы АСЕ, энхансеры PIMT, ингибиторы b-секретаз, ингибиторы g-секретаз, ингибиторы нейтральных эндопептидаз, ингибиторы PDE-4, ингибиторы МАО, ингибиторы TNFa, ингибиторы отложения амилоидного протеина или амилоидного пептида, ингибиторы s-1-рецептора и антагонисты гистамина Н3.
57. Применение фармацевтической композиции по одному из пп.17-31 для приготовления лекарственного средства, предназначенного для лечения нейронных нарушений, прежде всего таких заболеваний, как болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона, хорея Гентингтона, патогенные психические состояния, шизофрения, нарушение всасывания пищи, нарушение сна, нарушение связанной с гомеостазом регуляции энергетического метаболизма, нарушение функции вегетативной нервной системы, нарушение гормонального баланса, нарушение регуляции общей воды организма, гипертензия, лихорадка, нарушение регуляции сна, анорексия, связанные со страхом нарушения, включая депрессию, припадки, включая эпилепсию, синдром отмены лекарственного средства и алкоголизм, нейродегенеративные заболевания, включая когнитивную дисфункцию и деменцию.
58. Применение по п.57 для лечения нейронных нарушений, выбранных из группы, включающей болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона и хорею Гентингтона.
59. Применение по п.57 или 58, в котором ингибирование ингибитором глутаминилциклазы характеризуется значением Ki, составляющим 1 мкМ или менее.
60. Применение по п.57 или 58, в котором ингибирование глутаминилциклазы характеризуется значением Ki, составляющим 0,1 мкМ или менее.
61. Применение по п.57 или 58, в котором ингибирование ингибитором глутаминилциклазы характеризуется значением Ki, составляющим 0,01 мкМ или менее.
62. Применение по одному из пп.57-61, в котором ингибитор глутаминилциклазы имеет молекулярную массу 1000 г/моль или менее.
63. Применение по одному из пп.57-61, в котором ингибитор глутаминилциклазы имеет молекулярную массу 500 г/моль или менее.
64. Применение по одному из пп.57-61, в котором ингибитор глутаминилциклазы имеет молекулярную массу 400 г/моль или менее.
65. Применение по одному из пп.57-61, в котором ингибитор глутаминилциклазы имеет молекулярную массу 350 г/моль или менее.
66. Применение по одному из пп.57-65, в котором ингибитор глутаминилциклазы представляет собой конкурентный ингибитор.
67. Применение по одному из пп.57-66, в котором ингибитор глутаминилциклазы представляет собой конкурентный обратимый ингибитор.
68. Применение по одному из пп.57-67, в котором ингибитор глутаминилциклазы выбирают из соединений по одному из пп.1-16.
69. Применение по п.68, в котором ингибитор глутаминилциклазы вводят млекопитающему необязательно в комбинации с соединением, выбранным из группы, включающей ингибиторы PEP, LiCl, ингибиторы дипептидиламинопептидаз, предпочтительно ингибиторы DP IV или подобных DP IV ферментов, лиганды рецептора NPY, агонисты NPY, ингибиторы АСЕ, энхансеры PIMT, ингибиторы b-секретаз, ингибиторы g-секретаз, ингибиторы нейтральных эндопептидаз, ингибиторы PDE-4, ингибиторы МАО, ингибиторы TNFa, ингибиторы отложения амилоидного протеина или амилоидного пептида, ингибиторы s-1-рецептора и антагонисты гистамина Н3.
Текст
013790 Область техники, к которой относится изобретение Настоящее изобретение относится к глутаминилциклазе (QC, КФ 2.3.2.5), которая катализирует внутримолекулярную циклизацию N-концевых остатков глутамина с образованием пироглутаминовой кислоты (5-оксопролин, pGlu) с выделением в свободном состоянии аммиака и внутримолекулярную циклизацию N-концевых остатков глутамата с образованием пироглутаминовой кислоты с выделением в свободном состоянии воды. Предпосылки создания изобретения Глутаминилциклаза (QC, КФ 2.3.2.5) катализирует внутримолекулярную циклизацию N-концевых остатков глутамина с образованием пироглутаминовой кислоты (pGlu) с выделением в свободном состоянии аммиака. QC впервые была выделена Messer из латекса тропического растения Carica papaya в 1963 г. (Messer M. Nature 4874, 1963, с. 1299). Спустя 24 г. соответствующая ферментативная активность была обнаружена в гипофизе животных (Busby W.H.J. и др. J Biol Chem 262, 1987, cc. 8532-8536; FischerGln в pGlu с помощью QC было обнаружено для предшественников TRH (тиреолиберин) и GnRH (гонадолиберин) (Busby W.H.J. и др., J Biol Chem 262, 1987, cc. 8532-8536; Fischer W.H. и Spiess J., Proc NatlAcad Sci USA 84, 1987, cc. 3628-3632). Кроме того, предварительные эксперименты по выявлению локализации QC позволили обнаружить ее совместную локализацию с предполагаемыми продуктами катализа в бычьем гипофизе, что является дополнительным доказательством ее функции в синтезе пептидных гормонов (Bockers Т.М. и др. J. Neuroendocrinol 7, 1995, cc. 445-453). В противоположность этому физиологическая функция растительной QC все еще остается неясной. Предполагается, что фермент из С. papaya играет роль в защите растений от патогенных микроорганизмов (El Moussaoui А. и др. Cell Mol LifeSci 58, 2001, cc. 556-570). В настоящее время на основе сравнительного анализа последовательностей были выявлены предполагаемые QC из других растений (Dahl S.W. и др., Protein Expr Purif 20, 2000, cc. 27-36). Однако физиологическая функция этих ферментов все еще остается неясной. Известные выделенные из растений и животных QC обладают строгой специфичностью в отношении L-глутамина в N-концевом положении субстратов и установлено, что их кинетические характеристики описываются уравнением Михаэлиса-Ментена (Pohl Т. и др., Proc Natl Acad Sci USA 88, 1991, cc. 10059-10063; Consalvo A.P. и др., Anal Biochem 175, 1988, cc. 131-138; Gololobov M.Y. и др., Biol ChemHoppe Seyler 377, 1996, cc. 395-398). Однако сравнение первичной структуры QC из С. papaya и высококонсервативных QC млекопитающих не выявило никакой гомологии последовательностей (Dahl S.W. и др., Protein Expr Purif 20, 2000, cc. 27-36). В то время как растительные QC, вероятно, принадлежат к новому семейству ферментов, (Dahl S.W. и др., Protein Expr Purif 20, 2000, cc. 27-36), установлено, что QC млекопитающих обладают выраженной гомологией последовательностей с бактериальными аминопептидазами (Bateman R.С. и др., Biochemistry 40, 2001, cc. 11246-11250), что позволило сделать заключение о различном эволюционном происхождении QC из растений и животных. В настоящее время установлено, что рекомбинантная человеческая QC, а также активность QC в экстрактах, выделенных из головного мозга, катализирует циклизацию как N-концевого глутаминила,так и глутамата. Более конкретно установлено, что для катализируемого циклазой Glu1-превращения оптимальным является значение рН, близкое к 6,0, а для Gln1-превращения в pGlu-производные оптимальным является значение рН, близкое к 8,0. Поскольку образование пептидов, родственных pGlu-A, можно подавлять путем ингибирования рекомбинантной человеческой QC и QC-активности экстрактов, выделенных из гипофиза свиньи, то фермент QC является мишенью для разработки лекарственных средств,предназначенных для лечения болезни Альцгеймера. В ЕР 02011349.4 описаны полинуклеотиды, кодирующие глутаминилциклазу насекомых, а также кодируемые ими полипептиды. В этой заявке описаны также клетки-хозяева, которые содержат экспрессионные векторы, несущие полинуклеотиды, предлагаемые в изобретении. Выделенные полипептиды и клетки-хозяева, содержащие QC насекомых, применяют в методах скрининга агентов, которые снижают глутаминилциклазную активность. Такие агенты можно применять в качестве пестицидов. Определения Ингибиторы ферментов. Обратимые ингибиторы ферментов включают конкурентные ингибиторы, неконкурентные обратимые ингибиторы, медленно связывающиеся или трудно связывающиеся ингибиторы, аналоги переходного состояния и мультисубстратные аналоги. Для конкурентных ингибиторов характерно:I) нековалентные взаимодействия с ферментом,II) конкуренция с субстратом за активный сайт фермента. Основной механизм действия обратимого ингибитора фермента и определение константы диссоциации можно представить в виде следующей схемы: Образование комплекса фермент-ингибитор [E-I] препятствует связыванию субстрата, в результате не может происходить реакция с образованием обычного физиологического продукта Р. Более высокая концентрация ингибитора [I] приводит к более высокой концентрации [E-I], что обусловливает снижение уровня свободного фермента, с которым субстрат может связываться. Для неконкурентных ингибиторов характерно:I) связывание в сайте, отличном от активного сайта (аллостерический сайт связывания),II) способность вызывать конформационное изменение в ферменте, которое снижает или устраняет каталитическую активность. Для медленно связывающихся или трудно связывающихся ингибиторов характерно:I) они представляют собой конкурентные ингибиторы, для которых характерно медленное достижение равновесия между ингибитором и ферментом,II) значение kon характеризует медленный процесс, вероятно из-за конформационных изменений,которые должны происходить в ферменте или ингибиторе,а) часто представляют собой аналоги переходного состояния,б) обладают эффективностью при концентрации, близкой к концентрации фермента (субнаномолярные значения KD),в) из-за того, что значения koff являются столь низкими, эти типы ингибиторов является практически (почти) необратимыми. Аналоги переходного состояния. Представляют собой конкурентные ингибиторы, которые имитируют переходное состояние фермента, катализирующего реакцию. Происходит ферментный катализ, приводящий к снижению энергии переходного состояния, в результате связывание в переходном состоянии является предпочтительным по сравнению со связыванием с субстратом. Мультисубстратные аналоги. Для реакции, включающей два или большее количество субстратов, можно создавать конкурентный ингибитор или аналог переходного состояния, который имеет структурные характеристики, сходные с характеристиками двух или большего количества субстратов. Необратимые ингибиторы ферментов: обусловливают равновесие между несвязанным ферментом и ингибитором и комплексом фермент-ингибитор (E+IE-I), во всех случаях в соответствии с образованием ковалентной связи (100 ккал/моль), что делает ингибирование необратимым. Агенты, действие которых основано на аффинности. Необратимые ингибиторы, связывающиеся с активным сайтом (мишенью которых является активный сайт) (конкурентные необратимые ингибиторы) распознаются ферментом (обратимое специфическое связывание) с последующим формированием ковалентной связи иI) они обладают структурным сходством с субстратом, переходным состоянием или продуктом, что позволяет осуществлять специфическое взаимодействие между лекарственным средством и ферментоммишенью,II) содержат реактивную функциональную группу (например, нуклеофил, -COCH2Br), что позволяет осуществлять формирование ковалентной связи. Приведенная ниже реакционная схема описывает взаимодействие реагента, связывающегося с активным сайтом, с ферментом-мишенью, где KD обозначает константу диссоциации и kinactivation (инактивация)-2 013790 обозначает скорость образования ковалентной связи. Инактиваторы, действие которых основано на механизме, связанном с ферментом (называемые также ингибиторы-самоубийцы) представляют собой реагенты, связывающиеся с активным сайтом (нереактивные), которые связываются с активным сайтом ферментом, где они трансформируются в реактивную форму (активированную форму) под воздействием каталитической активности фермента. После активации формируется ковалентная связь между ингибитором и ферментом. На приведенной ниже реакционной схеме показан механизм действия инактиватора, действие которого основано на механизме, связанном с ферментом, где KD характеризует комплекс диссоциации, k2 обозначает скорость активации ингибитора после связывания с ферментом, k3 обозначает скорость диссоциации активированного ингибитора, Р из фермента (продукт может еще сохранять реактивность) и k4 обозначает скорость формирования ковалентной связи между активированным ингибитором и ферментом. Инактивация (формирование ковалентной связи k4) должна происходить до диссоциации (k3), в противном случае реактивный ингибитор будет высвобождаться в окружающую среду. Коэффициент распределения, т.е. k3/k4 соотношение высвободившегося продукта и инактивированного продукта должно быть минимизировано для эффективной инактивации системы и получения минимальных нежелательных побочных реакций. Высокое значение коэффициента распределения (преобладающая диссоциация) приводит к неспецифическим реакциям. Неконкурентные ингибиторы ферментов. Исходя из определения неконкурентного ингибитора (ингибитор, который связывается только с ESкомплексами (субстрат-фермент можно вывести следующее уравнение: Для диссоциации субстрата из ES-комплекса характерно значение константы диссоциации, равноеKS, в то время как диссоциация комплекса ESI не происходит (т.е. значение KS равно нулю). Ожидается,что значения Km для ферментов, действие которых удовлетворяет уравнению скорости ферментативной реакции Михаэлиса-Ментена, будет пониженным. Повышение концентрации субстрата приводит к повышению концентрации ESI (из комплекса не может происходить образование продуктов реакции), поэтому не происходит прекращение ингибирования. Предпочтительными согласно настоящему изобретению являются конкурентные ингибиторы ферментов. Наиболее предпочтительными являются обратимые конкурентные ингибиторы ферментов. Понятия "ki" или "Ki" и "KD" относятся к константам связывания, которые характеризуют связывание ингибитора с ферментом и последующее высвобождение из фермента. Другой мерой является значение "IC50", отражающее концентрацию ингибитора, которая при данной концентрации субстрата приводит к 50%-ному ингибированию активности фермента. Понятие "DP IV-ингибитор" или "ингибитор дипептидилпептидазы IV" хорошо известно специалистам в данной области и означает ингибиторы фермента, которые ингибируют каталитическую активность DP IV или подобных DP IV ферментов. Понятие "DP IV-активность" означает каталитическую активность дипептидилпептидазы IV (DPIV) и подобных DP IV ферментов. Эти ферменты представляют собой осуществляющие расщепление в положении после пролина (в меньшей степени после аланина, после серина или после глицина) сериновые протеазы, обнаруженные в различных тканях организма млекопитающего, включая почку, печень и кишечник, и они отщепляют дипептиды от N-концов биологически активных пептидов с высокой специфичностью, когда пролин или аланин представляют собой остатки, примыкающие к N-концевой аминокислоте в их последовательности. Понятие "РЕР-ингибитор" или "ингибитор пролилэндопептидазы" хорошо известно специалистам в данной области и означает ингибиторы фермента, которые ингибируют каталитическую активность пролилэндопептидазы (PEP, пролилолигопептидаза, POP). Понятие "РЕР-активность" означает каталитическую активность эндопротеазы, которая обладает-3 013790 способностью гидролизовать расположенные за пролином связи в пептидах или белках, в которых пролин является аминокислотой в положении 3 или в более удаленном от N-конца положении пептидного или белкового субстрата. Понятие "QC" в контексте настоящего описания обозначает глутаминилциклазу (QC) и QCподобные ферменты. QC и QC-подобные ферменты имеют идентичную или сходную ферментативную активность, обозначенную далее как QC-активность. Следует отметить, что QC-подобные ферменты могут принципиально отличаться от QC по молекулярной структуре. Понятие "QC-активность" в контексте настоящего описания обозначает внутримолекулярную циклизацию N-концевых остатков глутамина с образованием пироглутаминовой кислоты (pGlu) или Nконцевого L-гомоглутамина или Lгомоглутамина с образованием циклического пирогомоглутаминового производного с выделением в свободном состоянии аммиака (см. ниже схемы 1 и 2). Схема 1 Циклизация глутамина с помощью QC Понятие "ЕС" в контексте настоящего описания обозначает побочную активность QC и QCподобных ферментов, таких как глутаматциклаза (ЕС), которая обозначена далее как ЕС-активность. Понятие "ЕС-активность" в контексте настоящего описания обозначает внутримолекулярную циклизацию N-концевых глутаматных остатков с образованием пироглутаминовой кислоты (pGlu) с помощью QC (см. ниже схему 3). Схема 3N-Концевая циклизация незаряженных глутамильных пептидов с помощью QC (ЕС) Понятие "ингибитор QC" или "ингибитор глутаминилциклазы" хорошо известно специалистам в данной области и означает ингибиторы фермента, которые ингибируют каталитическую активность глутаминилциклазы (QC) или глутамилциклазы (ЕС). Эффективность ингибирования QC. С учетом корреляции с ингибированием QC в предпочтительных вариантах осуществления изобретения, заявляемом способе и при медицинском применении используют агент, ингибирование которымQC характеризуется значением Ki 10 мкм или менее, более предпочтительно 1 мкМ или менее, еще более предпочтительно 0,1 мкМ или менее или 0,01 мкМ или менее или наиболее предпочтительно 0,01 мкМ или менее. Фактически, рассматриваются ингибиторы, для которых значения Ki составляют менее микромоля, предпочтительно находятся на наномолярном уровне или еще более предпочтительно на пикомолярном уровне. Таким образом, хотя представленные в настоящем описании активные агенты для-4 013790 удобства обозначены как "ингибиторы QC", должно быть, очевидно, что согласно изобретению такая номенклатура не ограничена каким-либо конкретным механизмом действия. Молекулярная масса ингибиторов QC. В целом, ингибиторы QC, которые используют в заявляемом способе или в медицине, должны представлять собой небольшие молекулы, например, с молекулярной массой 100 г/моль или менее, 500 г/моль или менее, предпочтительно 400 г/моль или менее и еще более предпочтительно 350 г/моль или менее и наиболее предпочтительно 300 г/моль или менее. Понятие "индивидуум" в контексте настоящего описания относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, которого подвергают лечению, наблюдению или экспериментальной обработке. Понятие "терапевтически эффективное количество" в контексте настоящего описания означает количество действующего вещества или фармацевтического агента, вызывающее биологический или медицинский ответ в тканевой системе животного или человека, который требуется получить исследователю,ветеринару или врачу или другому клиницисту, облегчающее симптомы заболевания или нарушения,подлежащего лечению. В контексте настоящего описания понятие "фармацевтически приемлемый" относится к применению как для лечения человека, так и в ветеринарии, например, к "фармацевтически приемлемым" относятся приемлемое для ветеринарии соединение или соединение, приемлемое для медицины и медикосанитарной помощи человеку. В контексте настоящего описания и в формуле изобретения понятие "ацил", если не указано иное,означает С 1-С 12 ацильный остаток, предпочтительно С 1-С 8 ацильный остаток и особенно предпочтительно С 1-С 4 ацильный остаток. Примерами ацила являются указанные ниже алканоильные группы и бензоил."Пептиды" выбирают из группы от дипептидов до декапептидов, предпочтительными являются дипептиды, трипептиды и пентапетиды. Аминокислоты, из которых состоят "пептиды", можно выбирать из перечисленных ниже. В контексте настоящего описания и в формуле изобретения, если не указано иное, понятие "алкил" обозначает С 1-С 12 алкильную группу, предпочтительно C1-С 6 алкильную группу. Алкильные группы могут быть с прямой цепью или разветвленными. Пригодными алкильными группами являются, например,метил, этил, пропил (например, н-пропил и изопропил), (н-бутил, трет-бутил и втор-бутил), пентил, гексил, гептил (например, н-гептил) и октил (например, н-октил). Понятие "алк", например, в понятии "алкокси", и понятие "алкан", например, в понятии "алканоил", следует интерпретировать в соответствии с понятием "алкил". Примерами алкоксигрупп являются метоксигруппа, этоксигруппа, бутоксигруппа (например, н-бутоксигруппа), гептилоксигруппа (например, н-гептилоксигруппа) и октилоксигруппа (например, н-октилоксигруппа). Примерами алканоила (т.е. ацильных групп) являются этаноил (т.е. ацетил),пропионил и бутирил. Понятие "алкенил", если не указано иное, обозначает С 2-С 12 алкенильную группу, предпочтительно С 2-С 6 алкенильную группу, которая содержит по меньшей мере одну двойную связь в любом требуемом положении. Алкенильные группы могут быть с прямой или разветвленной цепью. Примерами алкенильных групп являются этенил, пропенил и бутенил. Понятие "алкинил", если не указано иное, обозначает С 2-С 12 алкинильную группу, предпочтительно С 2-С 6 алкинильную группу, которая содержит по меньшей мере одну тройную связь в любом требуемом положении. Алкинильные группы могут быть с прямой или разветвленной цепью. Примерами алкенильных групп являются этинил, пропинил и бутинил. Понятие "циклоалкил", если не указано иное, обозначает С 3-С 12 циклоалкильную группу, предпочтительно С 3-С 8 циклоалкильную группу. Примерами циклоалкильных групп являются циклопропил, циклобутил, циклопропил, циклогексил, циклогептил и циклооктил. Циклоалкильные группы могут быть разветвленными, в этом случае количество атомов углерода обозначает общее количество атомов во фрагменте. Понятие "гетероциклический", если не указано иное, обозначает циклоалкильный фрагмент, в котором один или несколько (например, 1, 2 или 3) кольцевых атома замещены гетероатомами, выбранными из N, S или О. Примерами гетероциклических групп, содержащих один гетероатом, являются пирролидин, тетрагидрофуран и пиперидин. Такие группы необязательно могут быть замещены, например, алкилом, оксогруппой или гидроксилом. Конкретными примерами гетероциклической группы являются замещенные или незамещенные оксираногруппа, азиридиногруппа, оксациклопропил, азациклопропил, тиираногруппа, оксетаногруппа,тиетаногруппа, пирролидиногруппа, тетрагидрофураногруппа, тиоланогруппа, 1,1-диоксотиоланогруппа,1,3-диоксоланогруппа, тиазолидиногруппа, имидазолидиногруппа, оксазолидиногруппа, пиразолидиногруппа, тетрагидропираногруппа, пиперидиногруппа, уротропиногруппа, пиперазиногруппа, Nметилпиперазиногруппа, (2-(N-метил)-N'-пиперазинил)этил, (4N-(2'-гидроксиэтил)-1N-пиперазинил), (2(4N-(2'-гидроксиэтил)-1N-пиперазинил)этилокси), морфолиногруппа, 2-(N-морфолино)этильная группа,а также лактамы, лактоны, циклические имиды и циклические ангидриды. Понятие "карбоциклический", если не указано иное, обозначает карбоциклическую группу, содер-5 013790 жащую от 3 до 12 атомов углерода, более предпочтительно от 3 до 8 атомов углерода. В контексте настоящего описания понятие карбоциклическая группа относится к группе, отличной от арила или циклоалкила, которая содержит по меньшей мере одно кольцо, состоящее из атомов углерода, и не содержащее гетероатомов. Примерами карбоциклических групп являются соединенные мостиками кольцевые системы (например, бицикло[2.2.1]гептенил) и частично ненасыщенные кольцевые системы. Понятие "арил", если не указано иное, обозначает С 6-С 12 арильную группу, предпочтительно С 6 С 8 арильную группу. Арильные группы могут содержать по меньшей мере одно ароматическое кольцо(например, одно, два или три кольца), но могут также содержать частично или полностью ненасыщенные кольца. Примером арильной группы с одним ароматическим кольцом является фенил. Представителем ароматических групп с двумя ароматическими кольцами является нафтил. Примерами арильных групп,которые содержат частично или полностью ненасыщенные кольца, являются пентален и инден. Как указано ниже, арильные группы необязательно могут быть замещенными. Другими примерами арильных групп являются 4-фторфенильная, 3-фторфенильная, пентафторфенильная, 4-гидроксифенильная, 3 нитрофенильная, 4-(трифторметил)фенильная, 4-анилинильная, 2-бифенилильная, 3-бифенилильная, 4 бифенилильная, инденильная, 1-нафтильная или 2-нафтильная, 1-антраценильная, 2-антраценильная, 3 антраценильная группы. Примерами алкиларила являются фенилметил-(бензил) и фенилэтил, 2-фенилэт-1-ил, паратолилметил, паратолилэтил, метатолилметил, метатолилэтил, ортотолилметил, ортотолилэтил, 2-(4 этилфенил)эт-1-ил, 2,3-диметилфенилметил, 2,4-диметилфенилметил, 2,5-диметилфенилметил, 2,6 диметилфенилметил, 3,4-диметилфенилметил, 3,5-диметилфенилметил, 2,4,6-триметилфенилметил, 2,3 диметилфенилэтил, 2,4-диметилфенилэтил, 2,5-диметилфенилэтил, 2,6-диметилфенилэтил, 3,4 диметилфенилэтил, 3,5-диметилфенилэтил, 2,4,6-триметилфенилэтил, бензгидрил (т.е. дифенилметил),бензгидрил (т.е. дифенилэтил), тритил (т.е. трифенилметил), тритил (т.е. трифенилэтил), а-стирил, bстирил, кумил, 2-этилфенилметил, 3-этилфенилметил, 4-этилфенилметил, 2-этилфенилэтил, 3 этилфенилэтил, 4-этилфенилэтил, 2-фторбензил, 1-метил-2-фторфен-6-илметил, 1-метил-2-фторфен-4 илметил-, 1-метил-2-фторфен-6-илэтил, 1-метил-2-фторфен-4-илэтил, 1 Н-инденилметил, 2 Н-инденилметил, 1 Н-инденилэтил, 2 Н-инденилэтил, инданилметил, индан-1-он-2-илметил, индан-1-он-2-илэтил,тетралинилметил, тетралинилэтил, флуоренилметил, флуоренилэтил, (3-фенил)циклопент-1-ил, дигидронафталинилметил, дигидронафталинилэтил или (4-циклогексил)фенилметил, (4-циклогексил)фенилэтил. Понятие "гетероарил", если не указано иное, обозначает арильный фрагмент, в котором один или несколько (например 1, 2, 3 или 4, предпочтительно 1, 2 или 3) кольцевых атома замещены гетероатомами, выбранными из N, S и О, или 5-членное ароматическое кольцо, содержащее один или несколько (например, 1, 2, 3 или 4, предпочтительно 1, 2 или 3) кольцевых атома, выбранных из N, S и О. Как указано ниже, гетероарильные группы необязательно могут быть замещены. Примерами гетероарильных групп являются пиридин (например, 2, 3 или 4-пиридин), пиримидин, хинолин, пиррол, фуран, тиофен, оксазол,пиразол, бензодиоксолан, бензодиоксан, бензотиофен, бензодиоксепин и тиазолил, 1-имидазолил, 2 имидазолил, 4-имидазолил, 3-фенил-1-пирролил, изоксазолил, изотиазолил, 3-пиразолил, 1,2,3 триазолил, 1,2,4-триазолил, тетразолил, пиридазинил, пиразинил, индазолил, 6-индолил, бензимидазолил, изохинолинил, пуринил, карбазолинил, акридинил и 2,3'-бифурил. Примерами алкилгетероарила являются пиридинилметил, N-метилпиррол-2-метил-N-метилпиррол 2-этил, N-метилпиррол-3-метил, N-метилпиррол-3-этил, 2-метилпиррол-1-метил, 2-метилпиррол-1-этил,3-метилпиррол-1-метил, 3-метилпиррол-1-этил, 4-пиридинометил, 4-пиридиноэтил, 2-(тиазол-2-ил)этил,тетрагидроизохинолинилметил, тетрагидроизохинолинилэтил, 2-этилиндол-1-метил, 2-этилиндол-1-этил,3-этилиндол-1-метил, 3-этилиндол-1-этил, 4-метилпиридин-2-метил, 4-метилпиридин-2-илэтил, 4 метилпиридин-3-метил, 4-метилпиридин-3-этил. Вышеуказанные арильные и гетероарильные группы при необходимости необязательно могут быть замещены. Понятие "замещение" или "замещенный" обозначает замещение одной или несколькими (например,1, 2 или 3, предпочтительно 1 или 2) одновалентными или мультивалентными функциональными группами. Пригодными замещающими группами являются алкил, циклоалкил, арил (например, фенил), гетероарил (например, фурил), карбоциклическая группа, гетероциклическая группа, алкоксигруппа, циклоалкоксигруппа, арилоксигруппа, гетероарилоксигруппа, карбоциксиоксигруппа, гетерцикликоксигруппа,алкенилоксигруппа, алкинилоксигруппа, алкенил, алкинил, ацил, алканоил, алкоксиалканоил, алкоксиалкил, гетероарилалкил, арилалкил, арилалкилоксигруппа, гетероарилалкилоксигруппа, нитрогруппа, -Sалкил (например, метилтио)галогруппа (например, содержащая фтор, хлор, бром и йод группа), цианогруппа, гидроксил, -SO2 алкил, -SO2 арил, -SO2 гетероарил, -SO2 циклоалкил-SO2 гетероциклическая группа,-СО 2 Н, -СО 2 алкил, -NH2, -NHалкил, -N(алкил)2 (например, диметиламино), -СО-N(алкил)2 и -СОNH(алкил). Алкильные группы, включая производные, такие как алкоксигруппа, а также алкенильная, алкинильная и циклоалкильная группы, необязательно могут быть замещены галогеном, например замещены фтором. Например, к замещенным галогеном алкильным группам относится трифторметил, а к замещенным галогеном алкоксигруппам относится трифторметоксигруппа. Понятие "галоген" обозначает фтор(F), хлор (Cl), бром (Br) и йод (I) соответственно. В качестве аминокислот, которые можно применять согласно настоящему изобретению, могут служить L- и D-аминокислоты, N-алкилированные аминокислоты, N-метиламинокислоты, азааминокислоты; алло- и трео-формы Ile и Thr, которые могут представлять собой, например, -, - или аминокислоты, причем предпочтительными являются -аминокислоты. Примерами аминокислот являются аспарагиновая кислота (Asp), глутаминовая кислота (Glu), аргинин (Arg), лизин (Lys), гистидин(Nle), цистеиновая кислота (Суа) и метионинсульфоксид (MSO), ацетил-Lys, модифицированные аминокислоты, такие как фосфорилсерин (Ser(P, бензилсерин (Ser(Bzl и фосфорилтирозин (Tyr(P, 2 аминомасляная кислота (Abu), аминоэтилцистеин (Cmc), дегидроаланин (Dha), дегидроамино-2-масляная кислота (Dhb), карбоксиглутаминовая кислота (Gla), гомосерин (Hse), гидроксилизин (Hyl), цисгидроксипролин (cisHyp), транс-гидроксипролин (transHyp), изовалин (Iva), пироглутаминовая кислота"Пептиды" выбирают из группы от дипептидов до декапептидов, предпочтительными являются дипептиды, трипептиды и пентапептиды. Аминокислоты, из которых состоят "пептиды", можно выбирать из перечисленных выше. Понятие "азааминокислота" обозначает аминокислоту, в которой хиральная -СН-группа заменена атомом азота, в то время как "азапептид" обозначает пептид, в котором хиральная -СН-группа одного или нескольких аминокислотных остатков в пептидной цепи заменена атомом азота. Другие аминокислотные замены, кодируемые генетическим кодом, могут также присутствовать в пептидных соединениях, предлагаемых в изобретении, и их можно классифицировать согласно указанной общей схеме. Протеиногенными аминокислотами являются встречающиеся в естественных условиях-аминокислоты, полученные из белков. Непротеиногенными аминокислотами являются все другие аминокислоты, которые не являются "строительными" блоками обычных встречающихся в естественных условиях белков. Понятие "пептидные миметики" само по себе является известным специалисту в данной области. Предпочтительно оно относится к соединениям, которые имеют вторичную структуру типа пептида и-7 013790 необязательно дополнительные структурные характеристики; их механизм действия в основном аналогичен или идентичен механизму действия нативного пептида; однако их активность (например, в качестве антагониста или ингибитора) может быть изменена по сравнению с нативным пептидом, прежде всего в отношении рецепторов или ферментов. Кроме того, они могут имитировать действие нативного пептида (агонист). Примерами пептидных миметиков являются миметики-носители, непептидные миметики,пептоиды, нуклеиновые кислоты пептида, олигопирролиноны, винилогпептиды и олигокарбаматы. Определение таких пептидных миметиков см. в Lexikon der Chemie, изд-во Spektrum Akademischer Verlag,Heidelberg, Berlin, 1999. Обладающие свойствами миметиков структуры применяют с целью повышения активности, повышения избирательности для снижения побочных действий, защиты соединения от ферментативного расщепления с целью пролонгирования действия. Стереоизомеры. Под объем настоящего изобретения подпадают все возможные стереоизомеры соединений, предлагаемых в изобретении. Если соединения, предлагаемые в изобретении, имеют по меньшей мере один хиральный центр, то они могут существовать в виде энантиомеров. Если соединения имеют два или более хиральных центра,то они могут, кроме того, существовать в виде диастереоизомеров. Следует иметь в виду, что все такие изомеры и их смеси подпадают под объем настоящего изобретения. Получение и выделение стереоизомеров. Если способы получения соединений, предлагаемых в изобретении, приводят к получению смеси стереоизомеров, эти изомеры можно разделять с помощью общепринятых методов, таких как препаративная хроматография. Соединения можно получать в рацемической форме или в виде индивидуальных энантиомеров с помощью либо энантиомерного синтеза, либо путем разделения. Соединения, например,можно разделять на компоненты-энантиомеры с помощью стандартных методов, таких как образование диастереомерных пар посредством солеобразования с использованием оптически активной кислоты, такой как (-)-ди-паратолуол-d-винная кислота и/или (+)-ди-паратолуол-1-винная кислота с последующей фракционированной кристаллизацией и регенерацией свободного основания. Соединения можно разделять также путем образования диастереомерных сложных эфиров или амидов с последующим хроматографическим разделением и удалением хиральных вспомогательных веществ. В альтернативном варианте соединения можно разделять с помощью хиральной ЖХВР-колонки. Фармацевтически приемлемые соли. Принимая во внимание близость соединений в свободной форме соединений в форме солей или сольватов, подразумевается, что когда в контексте настоящего описания упоминается соединение, то при этом подразумевается также соответствующая соль или сольват, при условии, что возможно их образование в конкретных условиях. Соли и сольваты соединений формулы (1) и их физиологически функциональные производные,пригодные для применения в медицине, представляют собой соединения, в которых противоион или связанный растворитель являются фармацевтически приемлемыми. Однако под объем настоящего изобретения подпадают соли и сольваты, содержащие неприемлемые с фармацевтической точки зрения противоионы и связанные растворители, например, в качестве промежуточных продуктов при получении других соединений и их фармацевтически приемлемых солей и сольватов. Пригодными солями, которые можно применять согласно изобретению, являются соли как с органическими, так и неорганическими кислотами или основаниями. К фармацевтически приемлемым кислотно-аддитивным солям относятся соли, образованные с такими кислотами, как соляная, бромистоводородная, серная, азотная, лимонная, винная, фосфорная, молочная, пировиноградная, уксусная, трифторуксусная, трифенилуксусная, сульфамовая, сульфаниловая, янтарная, щавелевая, фумаровая, малеиновая, яблочная, миндальная, глутамовая, аспарагиновая, оксоуксусная, метансульфоновая, этансульфоновая, арилсульфоновая (например, паратолуолсульфоновая, бензолсульфоновая, нафталинсульфоновая или нафталиндисульфоновая), салициловая, глутаровая, глюконовая, трикарбаллиловая, коричная, замещенная коричная (например, коричная кислота, замещенная фенилом, метилом, метоксигруппой или галогеном, включая 4-метил- и 4-метоксикоричную кислоту), аскорбиновая, олеиновая, нафтойная, гидроксинафтойная (например, 1- или 3-гидрокси-2-нафтойная), нафталинакриловая (например, нафталин-2 акриловая), бензойная, 4-метоксибензойная, 2- или 4-гидроксибензойная, 4-хлорбензойная, 4 фенилбензойная, бензолакриловая (например, 1,4-бензолдиакриловая), изетионовые кислоты, перхлорная, пропионовая, гликолевая, гидроксиэтансульфоновая, памовая, циклогексансульфамовая, салициловая, сахариновая и трифторуксусная кислота. К фармацевтически приемлемым солям с основаниями относятся аммониевые соли, соли щелочных металлов, таких как натрий и калий, щелочно-земельных металлов, таких как кальций и магний, и соли органических оснований, таких как дициклогексиламин и Nметил-D-глукамин. Подразумевается, что все соединения, предлагаемые в настоящем изобретении, в форме фармацевтически приемлемых кислотно-аддитивных солей подпадают под объем настоящего изобретения. Примерами сольватов являются гидраты.-8 013790 Полиморфные кристаллические формы. Кроме того, некоторые кристаллические формы соединений могут существовать в виде полиморфов, и они подпадают под объем настоящего изобретения. Некоторые соединения могут образовывать также сольваты с водой (т.е. гидраты) или с обычными органическими растворителями, и подразумевается, что такие сольваты подпадают под объем настоящего изобретения. Соединения, включая их соли,можно получать также в форме их гидратов или в форме, включающей другие растворители, применяемые для кристаллизации. Пролекарства. Кроме того, под объем настоящего изобретения подпадают пролекарства соединений, предлагаемых в настоящем изобретении. Как правило, такие пролекарства представляют собой функциональные производные соединений, которые легко превращаются in vivo в требуемое обладающее терапевтической активностью соединение. Таким образом, в этих случаях подразумевается, что в способах лечения, предлагаемых в настоящем изобретении, понятие введение включает лечение различных указанных нарушений с помощью вариантов пролекарства одного или нескольких заявленных соединений, которые превращаются в указанное выше соединение in vivo после введения индивидууму. Стандартные процедуры отбора и получения пригодных производных, представляющих собой пролекарства, описаны, например,в "Design of Prodrugs", под ред. Н. Bundgaard, изд-во Elsevier, 1985 и заявках на патент DE 19828113, DE 19828114, WO 99/67228 и WO 99/67279, которые полностью включены в настоящее описание в качестве ссылки. Защитные группы. В процессе получения соединений, предлагаемых в настоящем изобретении, может оказаться необходимым и/или целесообразным защищать чувствительные или реактивные группы на рассматриваемых молекулах. Это можно осуществлять с помощью стандартных защитных групп, таких, которые описаны в Protective Groups in Organic Chemistry, под ред. J.F.W. McOmie, изд-во Plenum Press, 1973; и T.W.GreeneP.G.M. Wuts, Protective Groups in Organic Synthesis, изд-во John WileySons, 1991, которые полностью включены в настоящее описание в качестве ссылки. Защитные группы можно удалять на соответствующей последующей стадии с помощью методов, известных в данной области. В контексте настоящего описания подразумевается, что понятие "композиция" включает продукт,содержащий заявленные соединения в терапевтически эффективных количествах, а также любой продукт, который образуется непосредственно или опосредованно из комбинаций заявленных соединений. Носители и добавки для галеновых препаративных форм. Для жидких препаратов для орального введения, таких, например, как суспензии, элексиры и растворы, пригодные носители и добавки предпочтительно могут представлять собой воду, гликоли, масла,спирты, корригенты, консерванты, красители и т.п.; для твердых препаратов для орального введения,таких, например, как порошки, капсулы, желатиновые капсулы и таблетки, пригодными носителями и добавками являются крахмалы, сахара, разбавители, агенты, способствующие грануляции, замасливатели, связующие вещества, диспергирующие агенты и т.п. Носители, которые можно добавлять к смеси, содержат необходимые и инертные фармацевтические эксципиенты, включая (но не ограничиваясь ими) пригодные связующие вещества, суспендирующие агенты, замасливатели, корригенты, подслащивающие вещества, консерванты, покрытия, разрыхлители, красители и окрашивающие вещества. В качестве пригодных полимеров, которые можно применять в качестве носителей доставляемого к мишени лекарственного средства, можно применять поливинилпирролидон, пирановый сополимер, полигидроксипропилметакриламидофенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидеполилизин, замещенный пальмитоильным фрагментом. Кроме того, соединения, предлагаемые в настоящем изобретении, можно соединять с классом биоразложимых полимеров, применяемых для достижения контролируемого высвобождения лекарственного средства, таких, например, как полиактовая кислота,полиэпсилонкапролактон, полигидроксимасляная кислота, сложные полиортоэфиры, полиацетали, полидигидропираны, полицианакрилаты и сшитые или амфипатические блок-сополимеры гидрогелей. Пригодными связующими веществами являются (но не ограничиваясь ими) крахмал, желатин, натуральные сахара, такие как глюкоза или беталактоза, сахаристые вещества из кукурузы, натуральные и синтетические камеди, такие как аравийская камедь, трагакант или олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. В качестве разрыхлителей можно применять (без ограничения) крахмал, метилцеллюлозу, агар,бентонит, ксантановую камедь и т.п. Пептидные последовательности. Упомянутые и применяемые согласно настоящему описанию пептиды имеют следующие последовательности:- 10013790 Краткое изложение сущности изобретения В настоящем изобретении предложены соединения, которые могут действовать в качестве ингибиторов глутаминилциклазы (QC, КФ 2.3.2.5). Настоящее изобретение относится к новым физиологическим субстратам QC в организме млекопитающих, выбранным из группы, включающей [Glu3]амилоидный -протеин (3-40/42), [Gln3]амилоидный-протеин (3-40/42), гастрин, нейротензин, FPP, CCL 2, CCL 7, CCL 8, CCL 16, CCL 18, фракталкин,орексин A, [Gln3]глюкагон(3-29) и [Gln5]субстанция Р(5-11). Соединения, предлагаемые в настоящем изобретении, и фармацевтические композиции, содержащие по меньшей мере одно соединение, предлагаемое в настоящем изобретении, можно применять для лечения состояний, которые можно лечить путем модуляции активности QC. Путем введения млекопитающему ингибиторов активности QC (ЕС) можно предупреждать или облегчать или лечить нейронные нарушения (болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона,хорея Гентингтона, патогенные психические состояния, шизофрения, нарушение всасывания пищи, нарушение сна, нарушение связанной с гомеостазом регуляции энергетического метаболизма, нарушение функции вегетативной нервной системы, нарушение гормонального баланса, нарушение регуляции общей воды организма, гипертензия, лихорадка, нарушение регуляции сна, анорексия, связанные со страхом нарушения, включая депрессию, припадки, включая эпилепсию, синдром отмены лекарственного средства и алкоголизм, нейродегенеративные заболевания, включая когнитивную дисфункцию и деменцию). Кроме того, путем введения млекопитающему соединения, предлагаемого в настоящем изобретении, можно подавлять пролиферацию миелоидных клеток-предшественников. Путем введения ингибитора QC, предлагаемого в настоящем изобретении, можно также подавлять мужскую фертильность. Предпочтительным вариантом осуществления настоящего изобретения является применение ингибиторов активности QC (ЕС) в сочетании с другими агентами, прежде всего для лечения нейронных нарушений. Подробное описание изобретения В настоящем изобретении предложены новые ингибиторы QC (ЕС) формулы 1 где D обозначает замещенный фенил, в котором заместителями являются оксиалкил, тиоалкил, галогенил; или D обозначает дигидробензодиоксин, бензодиоксол, бензодитиол, дигидробензотидин, бензооксатиол или дигидробензооксатин; X обозначает S, N-CN; Y обозначает S; R17 и R18 оба обозначают Н; один из R17 и R18 обозначает Н, а другой обозначает метил; или R17 и R18 могут быть соединены с образованием карбоцикла, имеющего вплоть до 6 кольцевых атомов, и где понятие алкил означает метил, этил,пропил или бутил. Во втором варианте осуществления изобретения группа В представляет собой группу формулы(VI). В еще одном варианте осуществления изобретения В представляет собой группу формулы (VII). Предпочтительно X обозначает S. В одном из вариантов осуществления изобретения R17 и R18 оба обозначают Н, а в другом - один из R17 и R18 обозначает Н, а другой обозначает метил. Предпочтительными являются соединения, в которых R17 и R18 соединены с образованием карбоцикла, имеющего вплоть до 6 кольцевых атомов. В настоящем изобретении предложены также ингибиторы QC формулы 1(а)-(г), включая его фармацевтически приемлемые соли, включая все стереоизомеры и полиморфы. Еще в одном варианте осуществления настоящего изобретения предложены новые ингибиторы QC В следующем варианте осуществления настоящего изобретения предложены новые ингибиторы QC Еще в одном варианте осуществления настоящего изобретения предложены новые ингибиторы QC в которой положение на кольце указано в примерах 103-105. Другие новые ингибиторы QC (ЕС) приведены в примерах 138-141. Предпочтительным соединением формулы 1 г является соединение, в котором группа R7 обозначает 3,4-(диметокси)фенил, а группы R8 и R9 обе обозначают Н; или группа R7 обозначает 4-(хлор)фенил, а группы R8 и R9 вместе с атомом углерода, с которым они связаны, образует циклобутильное или циклопентильное кольцо; или R7 обозначает 4-(метокси)фенил, а группы R8 и R9 вместе с атомом углерода, с которым они связаны, образует циклопропильное или циклогексильное кольцо. Наиболее предпочтительным является соединение, в котором группа R7 обозначает 3,4-(диметокси)фенил, а группы R8 и R9 обе обозначают Н, а также соединение, в котором группы R8 и R9 вместе с атомом углерода, с которым они связаны, образует циклопропильное кольцо, а группа R7 обозначает 3,4-(диметокси)фенил. Еще одним объектом настоящего изобретения является фармацевтическая композиция, содержащая по меньшей мере одно из вышеуказанных соединений необязательно в сочетании с терапевтически приемлемым носителем и/или эксципиентом, которая предназначена для парентерального, энтерального или орального введения. Предпочтительно такая композиция дополнительно содержит по меньшей мере одно соединение,выбранное из группы, включающей ингибиторы PEP, LiCl, ингибиторы дипептидиламинопептидаз,предпочтительно ингибиторы DP IV или DP IV-подобных ферментов, лиганды рецептора NPY, агонистыNPY, ингибиторы АСЕ, энхансеры PIMT, ингибиторы -секретаз, ингибиторы -секретаз, ингибиторы нейтральных эндопептидаз, ингибиторы PDE-4, ингибиторы МАО, ингибиторы TNF, ингибиторы отложения амилоидного протеина или амилоидного пептида, ингибиторы -1-рецептора и антагонисты гистамина Н 3. Наиболее предпочтительно в этой фармацевтической композиции ингибитор DP IV/DPIV-подобных ферментов выбирают из группы, включающей L-треоизолейцилпирролидил, Lаллоизолейцилтиазолидид, L-аллоизолейцилпирролидид; и их соли или валинпирролидид или одно из следующих соединений: Антагонист NPY в ней может быть выбран из 3 а,4,5,9b-тетрагидро-1h-бенз[е]индол-2-иламина, (R)N2-(дифенилацетил)-(R)-N-[1-(4-гидроксифенил)этил]аргинининамида Возможен также вариант, когда ингибитор PEP в этой композиции выбирают из группы, включающей химические производные пролина или малые пептиды, содержащие концевые остатки пролина, например бензилоксикарбонилпроллилпролинал, замещенный на N-конце L-пролин или L-пролилпирролидин, замещенные N-бензилоксикарбонильные-(Z)-дипептиды, содержащие пролинал на С-конце,замещенные тиопролины, замещенные тиазолидины, замещенные оксопирролидины, модифицированные на С-конце пролины, включая фторированные кетоновые производные, хлорметилкетоновые производные ацилпролина или ацилпептидпролина (Z-Gly-Pro-CH2Cl) и 2-ацилпирролидиновые производные. Предпочтительно его выбирают из группы, включающей Fmoc-Ala-Pyrr-CN, (4R)-3-(индан-2-илацетил)4-(1-пирролидинилкарбонил)-1,3-тиазолидин,(S)-1-[N-(4-хлорбензил)сукцинамоил]пирролидин-2 карбалдехид,(S)-2-[(S)(гидроксиакатил)-1-пирролидинил]карбонил-N-(фенилметил)-1-пирролидинкарбоксамид и (2S,3aS,7aS)-1[(R,R)-2 фенилциклопропил]карбонил-2-[(тиазолидин-3-ил)карбонил]октагидро-1H-индол. В самом предпочтительном варианте ее осуществления ингибитор PEP представляет собой Кроме того, ингибитор АСЕ в фармацевтической композиции может представлять собой SDZ ENA 713 (ривастигмин, кислый тартрат (+)-(S)-N-этил-3-[(1-диметиламино)этил]-N-метилфенилкарбамат). При этом ингибитор PDE-4 выбирают из группы, включающей ролипрам, В одном из предпочтительных вариантов осуществления фармацевтическая композиция может включать энхансер PIMT, который представляет собой 10-аминоалифатилдибенз[b,f]оксепин общей формулы где alk обозначает алифатический радикал, R обозначает аминогруппу, которая является незамещенной или моно- или дизамещенной одновалентными алифатическими и/или аралифатическими радикалами или дизамещенной двухвалентными алифатическими радикалами, и R1, R2, R3 и R4, каждый независимо друг от друга, обозначают водород, (низш.)алкил, (низш.)алкоксигруппу, галоген или трифторметил. Но возможен также вариант, когда фармацевтическая композиция содержит ингибитор -секретазы,который представляет собой В предпочтительном варианте осуществления такого изобретения ингибитор -секретазы может представлять собой а ингибитор МАО может представлять собой ладостигил формулы Не исключен вариант, когда заявленная фармацевтическая композиция включает антагонист гистамина Н 3, представляющий собой соединение, выбранное из группы, включающей ципрализант, (1S,2S)2-(2-аминоэтил)-1-(1 Н-имидазол-4-ил)циклопропан, двойной Н 1/Н 3, Sch-79687 или одно из соединений- 21013790 Все вышеуказанные фармацевтические соединения, которые являются ингибиторами глутаминилциклазы, можно применять для приготовления лекарственного средства, предназначенного для лечения нейронных нарушений, прежде всего таких заболеваний, как болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона, хорея Гентингтона, патогенные психические состояния, шизофрения, нарушение всасывания пищи, нарушение сна, нарушение связанной с гомеостазом регуляции энергетического метаболизма, нарушение функции вегетативной нервной системы, нарушение гормонального баланса, нарушение регуляции общей воды организма, гипертензия, лихорадка, нарушение регуляции сна, анорексия,связанные со страхом нарушения, включая депрессию, припадки, включая эпилепсию, синдром отмены лекарственного средства и алкоголизм, нейродегенеративные заболевания, включая когнитивную дисфункцию и деменцию. Более предпочтительно - для лечения нейронных нарушений, выбранных из группы, включающей болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона и хорею Гентингтона. Причем предпочтительным является вариант применения, при котором ингибирование ингибитором глутаминилциклазы характеризуется значением Ki, составляющим 1 мкМ или менее, наиболее предпочтительным - значением Ki, составляющим 0,1 мкМ или менее, еще более предпочтительны значениемKi, составляющим 0,01 мкМ или менее. В одном из вариантов осуществления заявленного применения ингибитор глутаминилциклазы имеет молекулярную массу 1000 г/моль или менее, 500 г/моль или менее, 400 г/моль или менее и 350 г/моль или менее. Предпочтительным применением является применение, когда ингибитор глутаминилциклазы представляет собой конкурентный ингибитор, конкурентный обратимый ингибитор. В самом предпочтительном варианте осуществления данного изобретения ингибитор глутаминилциклазы вводят млекопитающему необязательно в комбинации с соединением, выбранным из группы,включающей ингибиторы PEP, LiCl, ингибиторы дипептидиламинопептидаз, предпочтительно ингибиторы DP IV или подобных DP IV ферментов, лиганды рецептора NPY, агонисты NPY, ингибиторы АСЕ,энхансеры PIMT, ингибиторы -секретаз, ингибиторы -секретаз, ингибиторы нейтральных эндопептидаз, ингибиторы PDE-4, ингибиторы МАО, ингибиторы TNF, ингибиторы отложения амилоидного протеина или амилоидного пептида, ингибиторы -1-рецептора и антагонисты гистамина Н 3. Еще одним объектом заявленного изобретения является способ лечения нейронных нарушений,прежде всего, таких как болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона, хорея Гентингтона,патогенные психические состояния, шизофрения, нарушение всасывания пищи, нарушение сна, нарушение связанной с гомеостазом регуляции энергетического метаболизма, нарушение функции вегетативной нервной системы, нарушение гормонального баланса, нарушение регуляции общей воды организма, гипертензия, лихорадка, нарушение регуляции сна, анорексия, связанные со страхом нарушения, включая депрессию, припадки, включая эпилепсию, синдром отмены лекарственного средства и алкоголизм, нейродегенеративные заболевания, включая когнитивную дисфункцию и деменцию, который заключается в том, что млекопитающему вводят эффективное количество выше описанных ингибиторов глутаминилциклазы. В одном из предпочтительных вариантов осуществления этого способа ингибирование ингибитором глутаминилциклазы характеризуется значением Ki, составляющим 1 мкМ или менее или значениемKi, составляющим 0,1 мкМ или менее, или значением Ki, составляющим 0,01 мкМ или менее. Еще один вариант осуществления этого изобретения предполагает, что ингибитор глутаминилциклазы имеет молекулярную массу 1000 г/моль или менее, или молекулярную массу 500 г/моль или менее,или молекулярную массу 400 г/моль или менее, или молекулярную массу 350 г/моль или менее. Используемый в способе ингибитор глутаминилциклазы представляет собой конкурентный ингибитор или конкурентно обратимый ингибитор. Возможно введение ингибитора глутаминилциклазы млекопитающему необязательно в комбинации с соединением, выбранным из группы, включающей ингибиторы PEP, LiCl, ингибиторы дипептидиламинопептидаз, предпочтительно ингибиторы DP IV или подобных DP IV ферментов, лиганды рецептораNPY, агонисты NPY, ингибиторы АСЕ, энхансеры PIMT, ингибиторы -секретаз, ингибиторы -секретаз,ингибиторы нейтральных эндопептидаз, ингибиторы PDE-4, ингибиторы МАО, ингибиторы TNF, ингибиторы отложения амилоидного протеина или амилоидного пептида, ингибиторы -1-рецептора и антагонисты гистамина Н 3. Выше описанная композиция может применяться для приготовления лекарственного средства,предназначенного для лечения нейронных нарушений, прежде всего таких заболеваний, как болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона, хорея Гентингтона, патогенные психические состояния, шизофрения, нарушение всасывания пищи, нарушение сна, нарушение связанной с гомеостазом регуляции энергетического метаболизма, нарушение функции вегетативной нервной системы, нарушение гормонального баланса, нарушение регуляции общей воды организма, гипертензия, лихорадка, нарушение регуляции сна, анорексия, связанные со страхом нарушения, включая депрессию, припадки, включая эпилепсию, синдром отмены лекарственного средства и алкоголизм, нейродегенеративные заболевания,включая когнитивную дисфункцию и деменцию.- 22013790 В одном из предпочтительных вариантов осуществления этого изобретения ее можно применять для лечения нейронных нарушений, выбранных из группы, включающей болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона и хорею Гентингтона. Возможно применение, при котором ингибирование ингибитором глутаминилциклазы характеризуется значением Ki, составляющим 1 мкМ или менее, или значением Ki, составляющим 0,1 мкМ или менее, или значением Ki, составляющим 0,01 мкМ или менее. Применяемый ингибитор глутаминилциклазы может иметь молекулярную массу 1000 г/моль или менее, или 500 г/моль или менее, или 400 г/моль или менее, или 350 г/моль или менее. Как и в случае способа, применяемый ингибитор глутаминилциклазы может быть конкурентным ингибитором или конкурентно обратимым ингибитором. В предпочтительном варианте осуществления изобретения применение предусматривает, что ингибитор глутаминилциклазы вводят млекопитающему необязательно в комбинации с соединением, выбранным из группы, включающей ингибиторы PEP, LiCl, ингибиторы дипептидиламинопептидаз, предпочтительно ингибиторы DP IV или подобных DP IV ферментов, лиганды рецептора NPY, агонистыNPY, ингибиторы АСЕ, энхансеры PIMT, ингибиторы -секретаз, ингибиторы -секретаз, ингибиторы нейтральных эндопептидаз, ингибиторы PDE-4, ингибиторы МАО, ингибиторы TNF, ингибиторы отложения амилоидного протеина или амилоидного пептида, ингибиторы -1-рецептора и антагонисты гистамина Н 3. Примеры синтеза Схема синтеза 1 Синтез соединений из примеров 1-53, 96-102, 136-137 Реагенты и условия: (a) NaH, ДМФ, 4 ч, КТ.; (б) 8 ч, 100 С; (в) H2N-NH2, EtOH, 8 ч, дефлегмация,затем 4 н. HCl, 6 ч, дефлегмация, (г) R3-NCO, EtOH, 6 ч, дефлегмация, (д) 3,4-диметоксифенилизотиоцианат. Схема синтеза 2 Синтез соединений из примеров 54-95 Реагенты и условия: (a) R-NCS, EtOH, 6 ч, дефлегмация, (б) ВРКД (водорастворимый карбодиимид), 1 Н-имидазол-1-пропангимин, ДМФ, 2 ч, КТ. Схема синтеза 3 Синтез соединений из примеров 103-105- 23013790 Реагенты и условия: (a) NaH, ДМФ, КТ, 3 ч; (б) LiAlH4, диоксан, дефлегмация, 1 ч; (в) R-NCS,EtOH, дефлегмация 6 ч. Схема синтеза 4 Синтез соединений из примеров 106-109 Реагенты и условия: (а) EtOH, 2 ч, дефлегмация. Схема синтеза 5 Синтез соединений из примеров 110-112 Реагенты и условия: (а) 1H-имидазол-1-пропанамин, триэтиламин, толуол, 12 ч, дефлегмация. Схема синтеза 6 Синтез соединений из примеров 113-132 Реагенты и условия: (а) CAIBE, 1 Н-имидазол-1-пропанамин, диоксан, 0 С, 12 ч; (б) реагент Лавессона, EtOH, дефлегмация, 8 ч. Схема синтеза 7 Синтез соединений из примеров 133-135 Реагенты и условия: (а) хлорид 1 Н-имидазол-1-пропановой кислоты, CH2Cl2, - 10 С, 1 ч; (б) реагент Лавессона, диоксан, дефлегмация, 8 ч. Схема синтеза 8 Синтез соединений из примера 138 Реагенты и условия: (a) EtOH, дефлегмация, 8 ч. Схема синтеза 9 Синтез соединений из примера 139- 24013790 Схема синтеза 11 Синтез соединений из примера 141ESI-Масс-спектры получали с помощью спектрометра SCIEX API 365 (фирма Perkin Elmer). Результаты измерений 1 Н-ЯМР (500 МГц) регистрировали с помощью устройства BRUKER АС 500 с использованием ДМСО-D6 в качестве растворителя. Химические сдвиги выражали в виде ч./млн относительно обработки тетраметилсиланом. Схемы расщепления обозначали следующим образом: s (синглет), d (дублет), dd (двойной дублет), t (триплет), m (мультиплет) и br (широкий сигнал). Подробное описание синтеза Примеры 1-12 и 14-53. 1 Н-Имидазол-1-пропанамин подвергали взаимодействию с соответствующим изотиоцианатом в этаноле при температуре дефлегмации в течение 8 ч. Затем растворитель удаляли и оставшееся масло растворяли в метиленхлориде. Органический слой промывали дважды насыщенным раствором NaHCO3,а затем NaHSO4 и соляным раствором, сушили и упаривали. Оставшийся твердый продукт перекристаллизовывали из этилацетата, получая конкретную тимочевину, выход 80-98%. Пример 13. 1-(3-(1 Н-Имидазол-1-ил)пропил)-3-(3,4-диметоксифенил)тиомочевина. 4,0 ммоль 3,4-диметоксифенилизотиоцианата и 4,0 ммоль 3-(1H-имидазол-1-ил)алкил-1-амина растворяли в 10 мл абсолютного этанола. После перемешивания в течение 2 ч при температуре дефлегмации растворитель выпаривали и образовавшийся твердый продукт перекристаллизовывали из этанола. Выход: 0,66 г (51,3%); tпл.: 160,0-161,0 С. 1H-ЯМР 1,8-2,0 (m, 2 Н), 3,4-3,5 (m, 2 Н), 3,75 (s, 6 Н), 3,9-4,0 (m, 2 Н), 6,7-6,8 (m, 1 Н), 6,9 (br m, 2 Н),6,95 (s, 1 Н), 7,15 (s, 1 Н), 7,55 (br s, 1H), 7,6 (s, 1H), 9,3 (s, 1H); МС m/z 321,2 (М+Н), 253,3 (M-C3H3N2). Примеры 96-102. 1 Н-Имидазол-1-пропанамин подвергали взаимодействию с соответствующим изоцианатом в этаноле при температуре дефлегмации в течение 8 ч. Затем растворитель удаляли и оставшееся масло растворяли в метиленхлориде. Органический слой промывали дважды насыщенным раствором NaHCO3, а затем NaHSO4 и соляным раствором, сушили и упаривали. Оставшийся твердый продукт перекристаллизовывали из этилацетата, получая конкретную мочевину, выход 85-90%. Примеры 136, 137. 1 Н-Имидазол-1-алкиламины получали с помощью известных из литературы методов из бромалкилфталимидов и соли имидазолия с последующим гидролизом. Образовавшиеся продукты трансформировали в тиомочевины согласно методу, изложенному в примерах 1-53, выход 88% (пример 136) и 95% (пример 137). Примеры 54-95. Во всех примерах из соответствующих тиомочевин путем взаимодействия с водорастворимым карбодиимидом (ВРКД) и 1 Н-имидазол-1-пропанамидом в безводном диметилформамиде в течение 24 ч при комнатной температуре (КТ) получали тризамещенные гуанидины, выход 40-87%. Примеры 103-105. Имидазол подвергали взаимодействию с соответствующим брометилфенилцианидом в ДМФ, используя 1 экв. NaH, в течение 3 ч при КТ, получая 1 Н-имидазол-1-метилфенилцианиды. Растворитель удаляли и образовавшееся масло повторно растворяли в диоксане. Цианиды превращали в соответствующие амины, используя 1 экв. LiAlH4. После добавления насыщенного раствора KHSO4 диоксан выпаривали и водный слой экстрагировали CHCl3. Органический слой концентрировали в вакууме и амин превращали в вакууме и амин превращали в соответствующие тиомочевины с помощью методов, изложенных в примерах 1-53, выход 78% (пример 103) и 65% (пример 104) и 81% (пример 105). Примеры 106-109. Используя в качестве исходных продуктов соответствующие метансульфонат-2-метилпропилфталимиды, амины синтезировали согласно методам, описанным для аминов в примерах 136-137. Образовавшиеся продукты трансформировали в тиомочевины с помощью методов, изложенных в примерах 1- 25013790 53, общие выходы продуктов из примеров 106-109 составляли 25-30%. Примеры 110-112. 1 Н-Имидазол-1-пропанамин подвергали взаимодействию с соответствующим 2-хлорбензо[d]тиазолом в толуоле в течение 24 ч при температуре 130 С. После удаления растворителя и перекристаллизации из метанола получали продукты из примеров 110-112, выход 55-65%. Примеры 113-118, 120-124 и 126-132. 1 Н-Имидазол-1-пропанамин подвергали взаимодействию с соответствующей 2-фенилуксусной кислотой в безводном диоксане путем добавления эквивалента CAIBE и N-метилморфолина при температуре 0 С. После этого смеси давали нагреться в течение 2 ч до КТ и смесь перемешивали в течение 12 ч. После удаления растворителя образовавшееся масло повторно растворяли в метиленхлориде и органический слой промывали водным раствором NaHCO3 и водой, сушили и растворитель выпаривали. Оставшееся масло растворяли в диоксане, добавляя реагент Лавессона. После перемешивания в течение 12 ч добавляли насыщенный раствор NaHCO3. Диоксан выпаривали и водный слой экстрагировали этилацетатом. Органический слой отделяли, сушили и растворитель выпаривали. Оставшийся твердый продукт кристаллизовали из этилацетата/простого эфира, получая продукты из примеров 113-118, 120-124 и 126132, общие выходы 62-85%. Пример 119. N-(3-(1 Н-Имидазол-1-ил)пропил)-2-(3,4-диметоксифенил)этантиоамид. Смесь, содержащую 4,0 ммоль триэтиламина и 4,0 ммоль 3-(1 Н-имидазол-1-ил)алкил-1-амина в 20 мл диоксана, добавляли по каплям при перемешивании к охлажденному на льду раствору, содержащему 4,0 ммоль 2-(3,4-диметоксифенил)ацетилхлорида в 30 мл диоксана. Смеси давали нагреться до КТ и затем перемешивали в течение 1 ч. После удаления растворителя при пониженном давлении остаток повторно растворяли в 50 мл дихлорметана. Органический слой промывали с помощью 30 мл насыщенного водного раствора NaHCO3 и воды. Органический раствор сушили, фильтровали и растворитель удаляли при пониженном давлении. После повторного растворения в 50 мл безводного диоксана добавляли 2,2 ммоль реагента Лавессона и смесь нагревали до 90 С и перемешивали в течение 8 ч. Растворитель удаляли при пониженном давлении и остаток повторно растворяли в 50 мл дихлорметана. Органический слой промывали трижды насыщенным водным раствором NaHCO3, а затем трижды водой, сушили, фильтровали и затем органический растворитель удаляли. Соединение очищали хроматографией, используя устройство для хроматографии с центрифугой (фирма Harrison Research Ltd.) с применением силикагелевых пластинок с толщиной слоя 2 мм и градиент CHCl3/MeOH в качестве системы для элюирования. Выход: 0,14 г (10,6%); tпл.: 148,0-150,0 С. 1 Н ЯМР 2,0-2,15 (bm, 2 Н), 3,4-3,5 (m, 2 Н), 3,7 (s, 6H), 6,75-6,8 (m, 2 Н), 4,1-4,2 (m, 2 Н), 6,8-6,9 (m,2 Н), 6,95-7,0 (m, 1 Н), 7,4 (s, 1H), 7,75-7,85 (br m, 1H), 8,6 (s, 1H), 10,2 (s, 1H); МС m/z 320,2 (М+Н), 252,2(M-C3H3N2). Пример 125. N-(3-(1 Н-Имидазол-1-ил)пропил)-1-(3,4-диметоксифенил)циклопропанкарботиоамид. 11,06 ммоль 3,4-диметоксифенилацетонитрила, 34,8 ммоль 2-бром-1-хлорэтанола и 1,16 ммоль гидрохлорида триэтилбензиламмония растворяли в 10 мл водного раствора KOH (60%). Смесь переносили в баню для обработки ультразвуком и интенсивно перемешивали в течение 3 ч при комнатной температуре. Образовавшуюся суспензию разводили 40 мл воды и экстрагировали трижды 20 мл дихлорметана. Объединенные органические слои промывали водным раствором соляной кислоты (1 н.), сушили надNa2SO4 и растворитель удаляли при пониженном давлении. Оставшееся масло очищали экспрессхроматографией на силикагеле, используя в качестве системы для элюирования этилацетат/гептан, получая 0,81 г (34,4%) 1-(3,4-диметоксифенил)циклопропанкарбонитрила. 3,9 ммоль 1-(3,4-диметоксифенил) циклопропанкарбонитрила и 11,2 ммоль KOH суспендировали в 80 мл этиленгликоля. Смесь перемешивали в течение 12 ч при температуре дефлегмации. Затем добавляли 80 мл воды и водный слой экстрагировали дважды простым эфиром. После доведения значения рН до 4-5 с помощью HCl (1 н.) водный слой экстрагировали трижды простым эфиром, затем объединенные органические слои сушили над Na2SO4 и растворитель удаляли, получая 0,81 г (93,5%) 1-(3,4-диметоксифенил)циклопропанкарбоновой кислоты. 3,44 ммоль 1-(3,4-диметоксифенил)циклопропанкарбоновой кислоты, 3,5 ммоль N-метилморфолина и 3,5 ммоль изобутилхлормиата растворяли в безводном тетрагидрофуране, перемешивали в течение 15 мин при -15 С. Затем добавляли 3,5 ммоль 3-(1 Н-имидазол-1-ил)алкил-1-амина и смеси давали нагреться до 0 С и перемешивали в течение 12 ч. Растворитель удаляли при пониженном давлении и оставшееся масло повторно растворяли в хлороформе. Затем органический слой промывали дважды насыщенным водным раствором NaHCO3, затем сушили над Na2SO4 и растворитель удаляли. Очистку осуществляли с помощью хроматографии с центрифугой на устройстве типа Chromatotron (фирма Harrison ResearchLtd.), применяя силикагелевые пластинки с толщиной слоя 2 мм и градиент CHCl3/МеОН в качестве системы для элюирования, получая 0,671 г (59,3%) N-(3-(1H-имидазол-1-ил)пропил)-1-(3,4-диметоксифенил)циклопропанкарбоксамида. После повторного растворения в 30 мл безводного диоксана добавляли 1,43 ммоль реагента Лавессона и смесь нагревали до 90 С и перемешивали в течение 8 ч. Растворитель удаляли при пониженном давлении, остатку давали раствориться в 50 мл дихлорметана. Органический слой промывали трижды- 26013790 насыщенным водным раствором NaHCO3, а затем трижды водой, сушили, фильтровали и затем органический растворитель удаляли. Соединение очищали с помощью устройства для хроматографии с центрифугой (фирма Harrison Research Ltd.), применяя силикагелевые пластинки с толщиной слоя 2 мм и градиент CHCl3/МеОН в качестве системы для элюирования. Выход: 0,33 г (46,2%); tпл.: 127,0-127,5 С. 1 Н ЯМР 1,1-1,2 (t, 2H), 1,55-1,6 (t, 2H), 2,0-2,1 (m, 2H), 3,5-3,6 (m, 2 Н), 3,7-3,8 (s, 6 Н), 4,1-4,2 (t,2 Н), 6,8-6,9 (m, 3 Н), 7,65 (s, 1 Н), 7,75 (s, 1H), 8,8 (m, 1 Н), 9,05 (s, 1 Н); МС m/z 346,0 (М+Н), 278,2 (MC3H3N2), 177,1 (М-C6H8N3S). Примеры 133-135. Смесь, содержащую 1 экв. триэтиламина и 3,4-диметоксианилина в диоксане, добавляли при перемешивании к раствору соответствующего -бромалкилхлорангидрида при температуре 0 С. Раствору давали нагреться до КТ и перемешивали в течение 2 ч. Растворитель выпаривали и оставшееся масло повторно растворяли в дихлорметане. Органический слой промывали водой, сушили, фильтровали и растворитель удаляли при пониженном давлении. Имидазол и гидрид натрия суспендировали и смесь перемешивали в инертных условиях при КТ в течение 3 ч. Добавляли -бром-N-(3,4-диметоксифенил)алкиламид и смесь нагревали до 100 С и перемешивали в течение 8 ч. Затем растворитель выпаривали, добавляли горячий толуол и раствор фильтровали. Затем растворитель удаляли при пониженном давлении. Трансформацию в тиоамиды осуществляли согласно методам, описанным в примерах 113-132, с помощью реагента Лавессона, получая соединения,указанные в примерах 133-135, общие выходы которых составляли 13-20%. Для других указанных в примерах соединений, которые синтезировали с помощью описанных выше общих схем синтеза, получены следующие аналитические данные. Пример 1. 1-(3-(1 Н-Имидазол-1-ил)пропил)-3-метилтиомочевина. tпл.: 122-122,5 С. 1H ЯМР 1,85-1,95 (m, 2 Н), 2,8 (s, 3 Н), 3,2-3,5 (br d, 2H), 3,8-3,9 (m, 2 Н), 6,85 (d, 1H), 7,15 (d, 1H),7,3-7,5 (br d, 2H), 7,65 (s, 1H); МС m/z 199,1 (М+Н), 221,3 (M+Na), 131,0 (M-C3H3N2). Пример 2. 1-(3-(1 Н-Имидазол-1-ил)пропил)-3-трет-бутилтиомочевина. tпл.: 147,0-147,5 С. 1 Н ЯМР 1,3-1,4 (s, 9 Н), 1,85-1,95 (m, 2H), 3,5 (t, 2H), 3,8 (t, 2H), 6,85 (d, 1H), 7,15 (d, 1H), 7,3-7,5 (br[Gln5]субстанция Р(5-11). Их дополнительное описание см. в табл. 1. Соединения или комбинации, предлагаемые в настоящем изобретении, и фармацевтические композиции, содержащие по меньшей мере один ингибитор QC (ЕС), можно применять для лечения состояний, которые можно лечить путем модуляции активности QC.- 29013790 Таблица 1 Аминокислотные последовательности физиологически активных пептидов, несущих N-концевой остаток глутамина, который как известно циклизуется с образованием в конце концов pGlu
МПК / Метки
МПК: C07D 405/12, C07D 403/12, A61K 38/06, C07D 235/06, A61P 25/28, C07D 417/12, C07D 233/61, A61K 31/4164, A61P 25/14
Метки: новые, ингибиторы, глутаминилциклазы
Код ссылки
<a href="https://eas.patents.su/30-13790-novye-ingibitory-glutaminilciklazy.html" rel="bookmark" title="База патентов Евразийского Союза">Новые ингибиторы глутаминилциклазы</a>
Новые ингибиторы химазы

Номер патента: 11745
Опубликовано: 30.06.2009
Авторы: Марьянофф Брюс Э., Де Гаравилла Лоуренс, Пауэлл Юджин, Греко Майкл Н., Хокинз Майкл Дж.
МПК: C07F 9/32, A61K 31/662, C07F 9/30...
Метки: ингибиторы, новые, химазы
Формула / Реферат:
1. Соединение формулы (I) где R1 выбран из группы, состоящей из водорода и С1-4алкила; выбрано из группы, состоящей из арила, гетероарила, бензоконденсированного гетероциклила, циклопропила, n равно 0 и один из R2 или R3 представляет собой фенил и бензоконденсированный циклоалкил, и кольцо А необязательно замещено R2 и R3; R2 представляет собой 1-2 заместителя, независимо выбранных из группы, состоящей из C1-6алкила, C2-6алкенила,...
Новые ингибиторы протеазы hiv

Номер патента: 11276
Опубликовано: 27.02.2009
Авторы: Крал Владимир, Покорна Яна, Козисек Милан, Плесек Яромир, Лепсик Мартин, Краусслих Ханс-Георг, Прейдова Яна, Бодем Йохен, Конвалинка Ян, Сиглер Петр, Грюнер Богумир
МПК: A61P 31/18, A61K 31/69, C07F 5/05...
Метки: новые, протеазы, ингибиторы
Формула / Реферат:
1. Применение соединений, содержащих кластеры, выбранные из группы, включающей бораны, и/или карбораны, и/или металкарбораны, имеющие от 6 до 12 атомов бора в каждом кластере, где заряд каждого индивидуального кластера борана, карборана или металкарборана равен 0, -1 или -2; число кластеров борана, карборана или металкарборана в молекуле составляет от 1 до 9 и данные карборановые кластеры в металкарборановых фрагментах координированы с атомом...
Новые ингибиторы гистондеацетилазы

Номер патента: 7272
Опубликовано: 25.08.2006
Авторы: Тен Холтэ Петер, Дяткин Алексей Борисович, Пилатт Изабелль Ноэлль Констанс, Ван Брандт Свен Францискус Анна, Ру Брюно, Вердонк Марк Густаф Селин, Анжибо Патрик Рене, Мерпул Ливен
МПК: A61P 35/00, C07D 207/14, A61K 31/4545...
Метки: гистондеацетилазы, ингибиторы, новые
Формула / Реферат:
1. Соединение формулы (I) где n равен 0, 1, 2 или 3 и, когда n равен 0, тогда имеется в виду непосредственная связь; t равен 0, 1, 2, 3 или 4 и, когда t равен 0, тогда имеется в виду непосредственная связь; каждый Q представляет собой азот или каждый X представляет собой азот или каждый Y представляет собой азот или каждый Z представляет собой азот или R1 представляет собой -C(O)NH(OH); R2 представляет собой водород, галоген, гидрокси,...
Карбониламинопроизводные как новые ингибиторы гистондеацетилазы

Номер патента: 7908
Опубликовано: 27.02.2007
Авторы: Ван Брандт Свен Францискус Анна, Вердонк Марк Густаф Селин, Ван Эмелен Кристоф, Бакс Лео Якобус Йозеф
МПК: A61K 31/4468, A61K 31/4439, A61K 31/444...
Метки: ингибиторы, новые, карбониламинопроизводные, гистондеацетилазы
Формула / Реферат:
1. Соединение формулы (I) где n равен 1; m равен 0 или 1, и когда m равен 0, тогда имеется в виду непосредственная связь; t равен 0, 1, или 2 и когда t равен 0, тогда имеется в виду непосредственная связь; каждый Q представляет собой азот или каждый X представляет собой азот или каждый Y представляет собой азот или R1 представляет собой -C(O)NH(OH); R2 представляет собой водород, C1-6-алкил; -L- представляет собой непосредственную связь;...
Пиперазинил-, пиперидинил- и морфолинилпроизводные как новые ингибиторы гистондеацетилазы

Номер патента: 7270
Опубликовано: 25.08.2006
Автор: Ван Эмелен Кристоф
МПК: A61K 31/506, C07D 401/04, A61P 35/00...
Метки: гистондеацетилазы, новые, пиперидинил, морфолинилпроизводные, пиперазинил, ингибиторы
Формула / Реферат:
1. Соединение формулы (I) и его фармацевтически приемлемые аддитивные соли где t равен 0 или 1 и, когда t равен 0, тогда имеется в виду непосредственная связь; каждый Q представляет собой азот или ; каждый Х представляет собой азот или ; каждый Y представляет собой азот или ; каждый Z представляет собой -NH-, -О- или -СН2-; R1 представляет собой -C(O)NH(OH); R2 представляет собой водород, гидрокси, C1-6-алкил или арил-С1-6-алкил; -L-...