Фенилсодержащие n-ацильные производные аминов и аминокислот, способ их получения, фармацевтическая композиция и их применение
Номер патента: 13644
Опубликовано: 30.06.2010
Авторы: Ковалева Виолетта Леонидовна, Небольсин Владимир Евгеньевич, Кромова Татьяна Александровна, Желтухина Галина Александровна




















Формула / Реферат
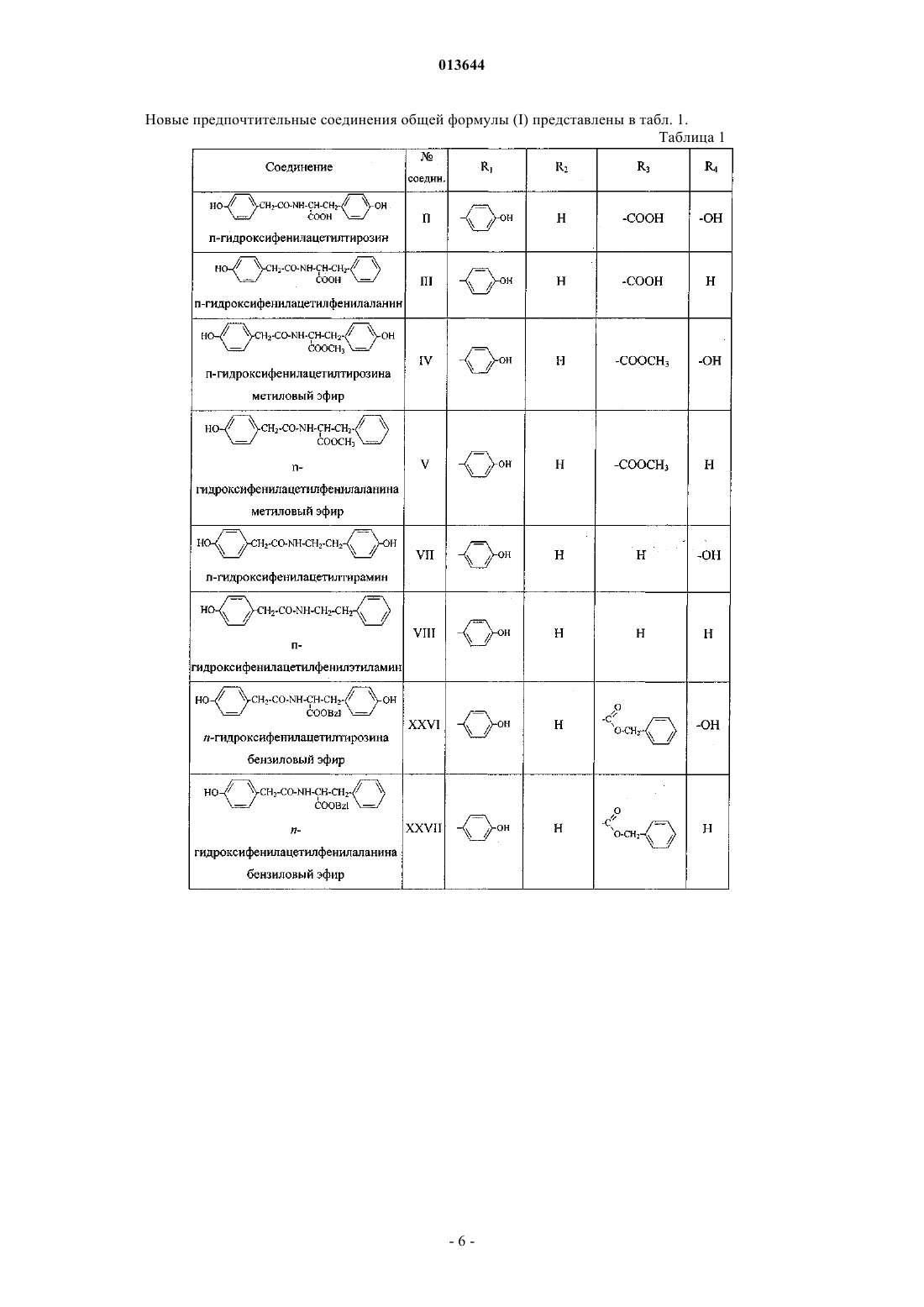
1. Фенилсодержащие N-ацильные производные биогенных аминов общей формулы (I)
![]()
где R1 представляет
![]()
или
![]()
где R5 представляет водород или гидроксильную группу; R2представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН или
-COOR6, где R6 представляет C1-С6-алкил или

где R7 представляет водород или гидроксильную группу, R4представляет водород, гидроксильную группу;
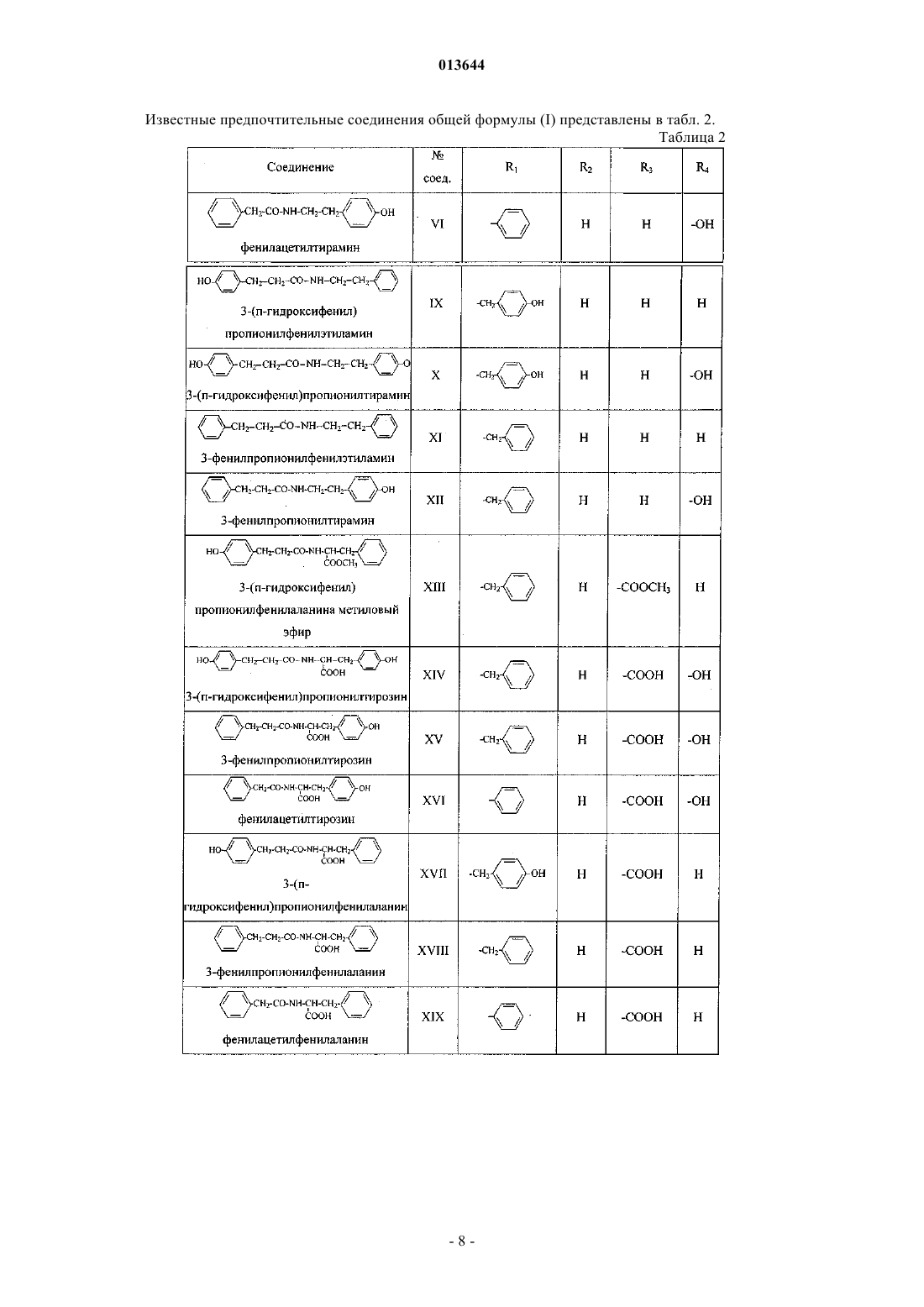
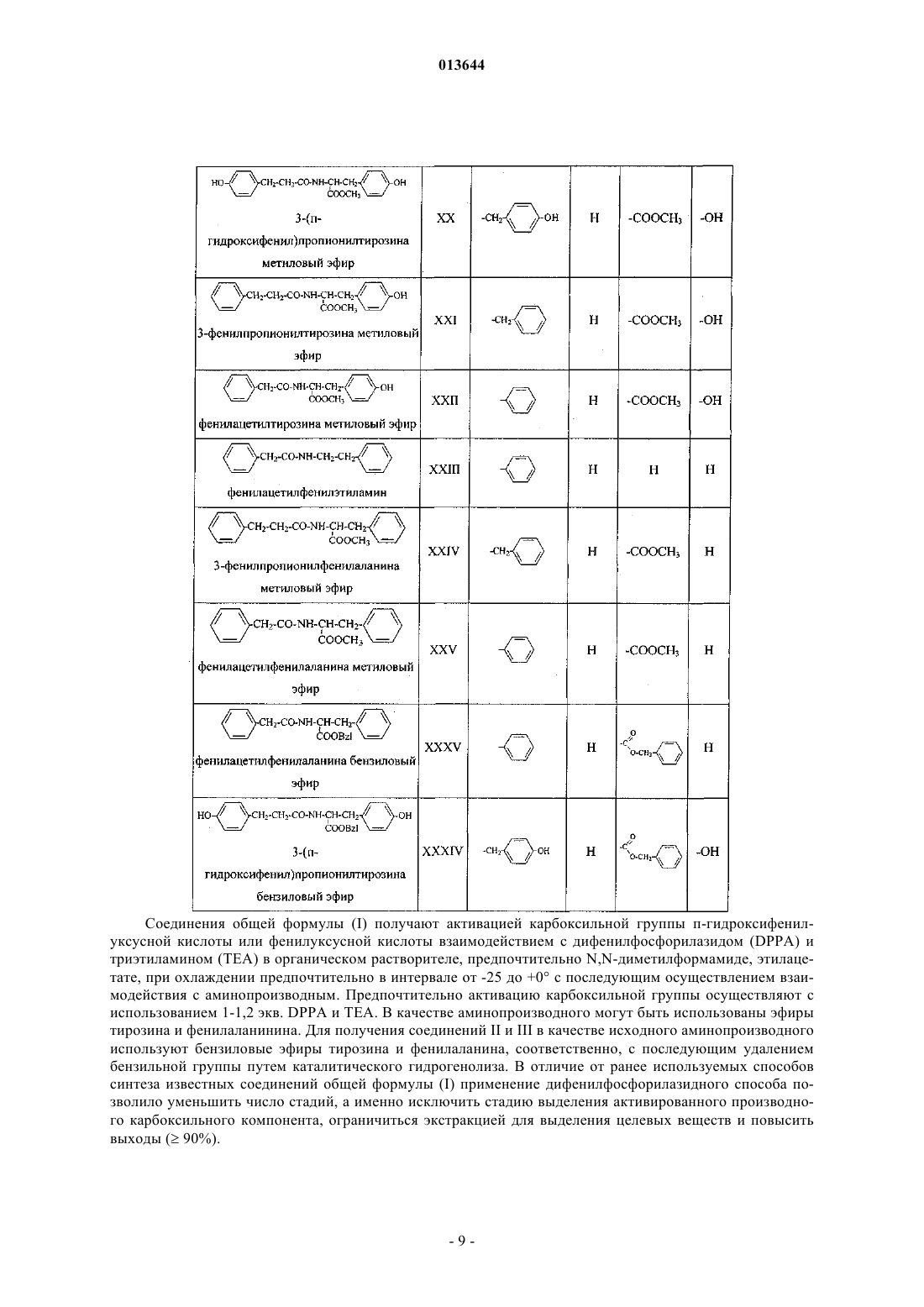
при условии, что соединение общей формулы (I) не является
фенилацетилтирамином,
3-(п-гидроксифенил)пропионилтирамином,
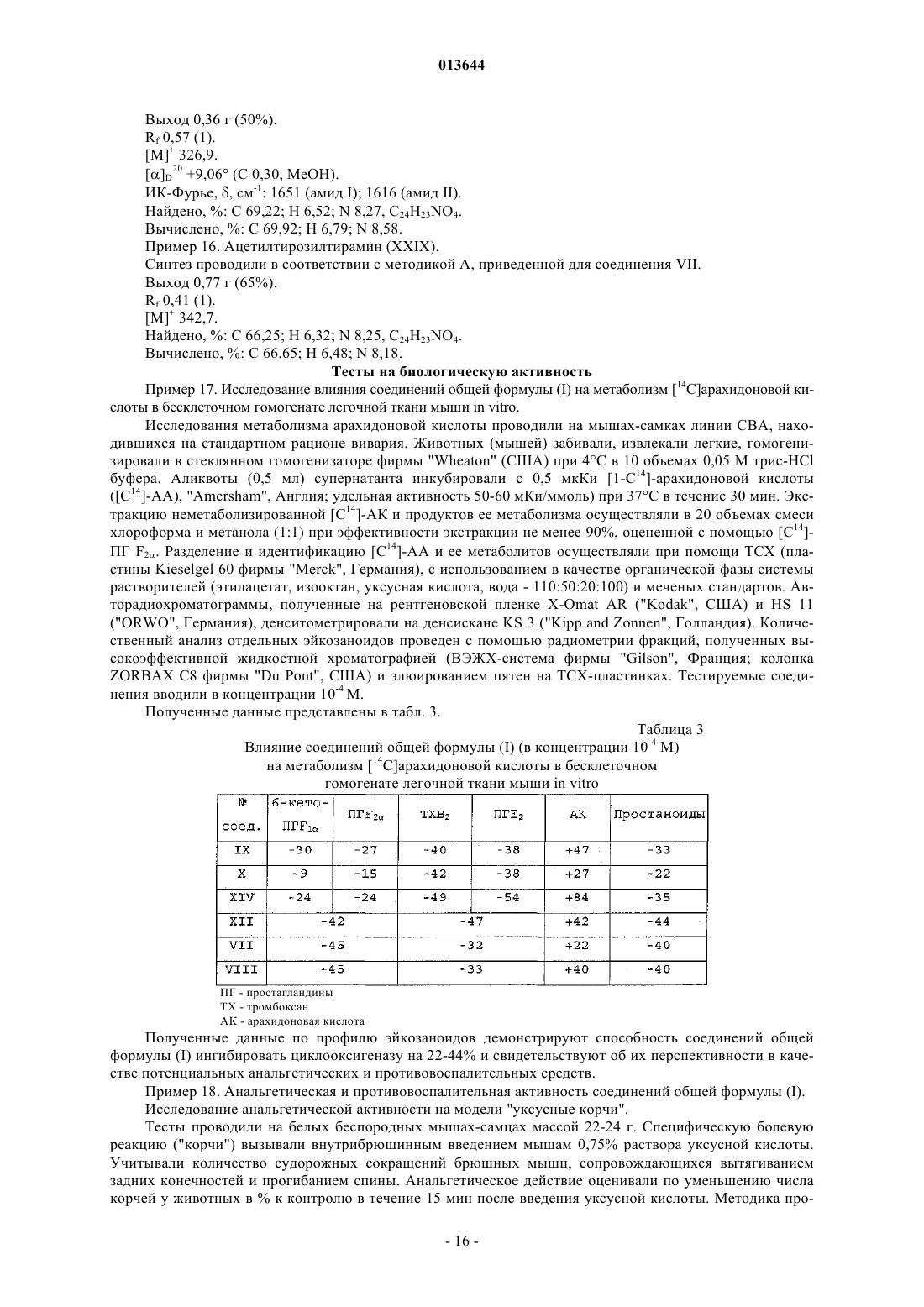
3-фенилпропионилфенилэтиламином,
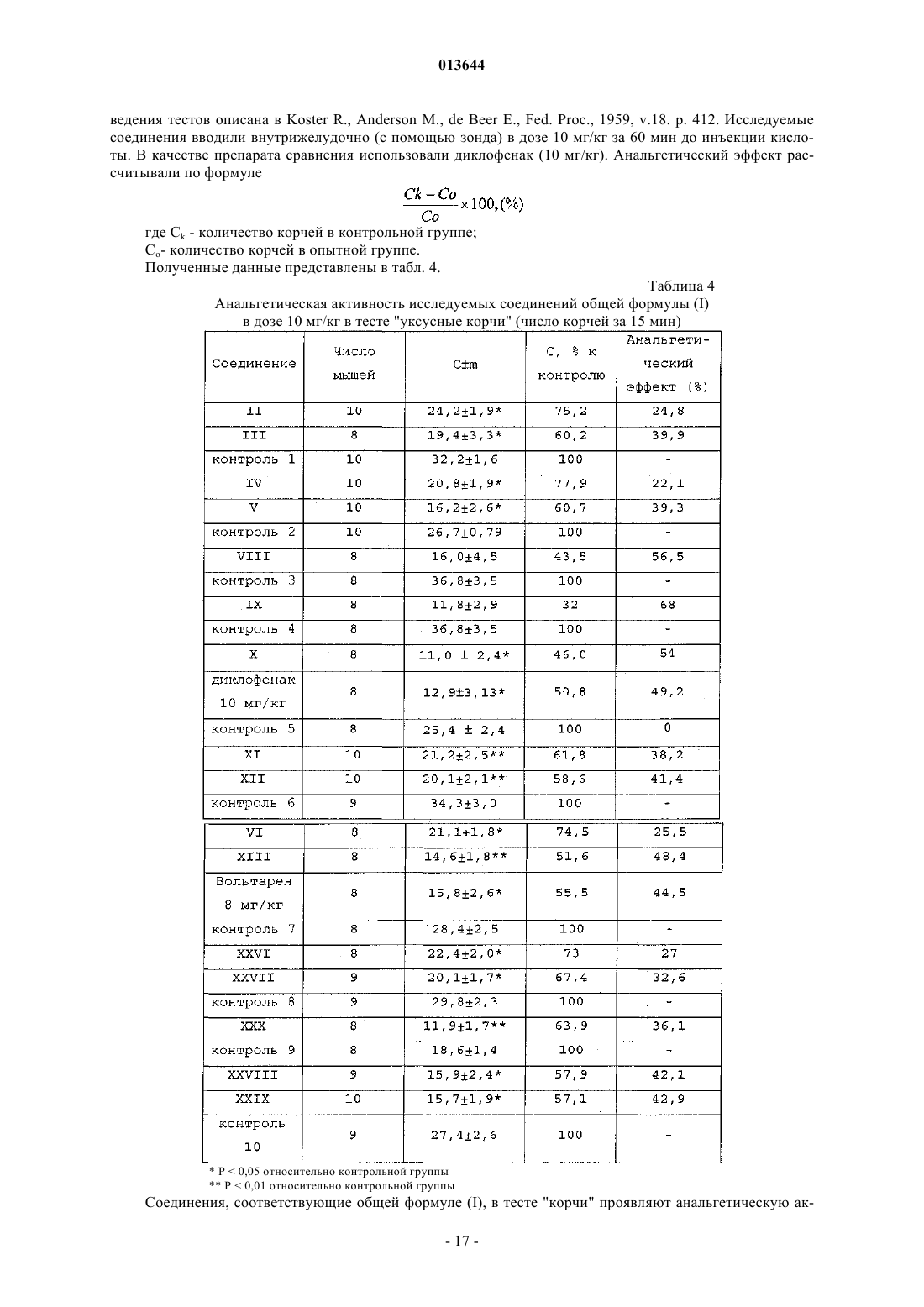
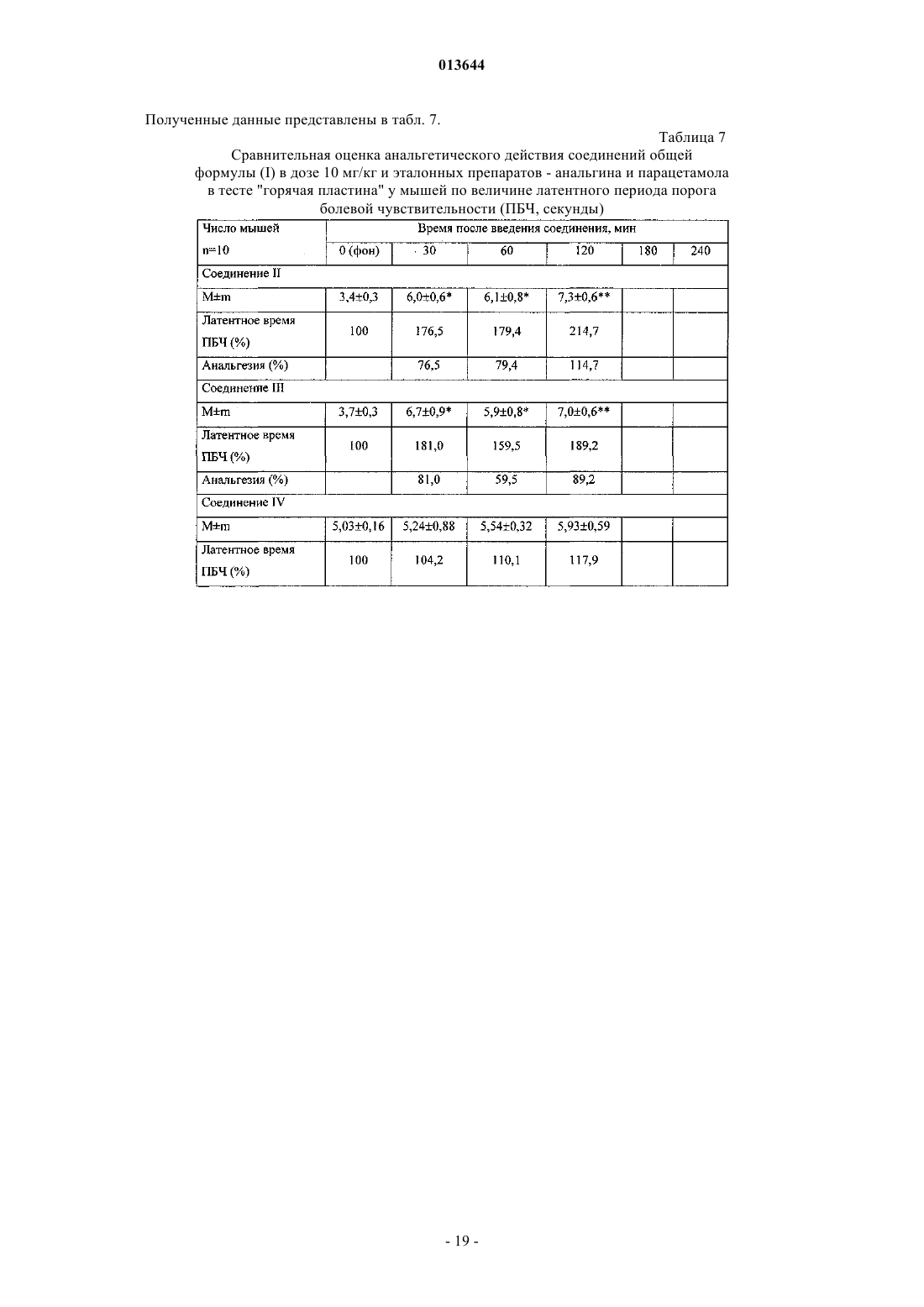
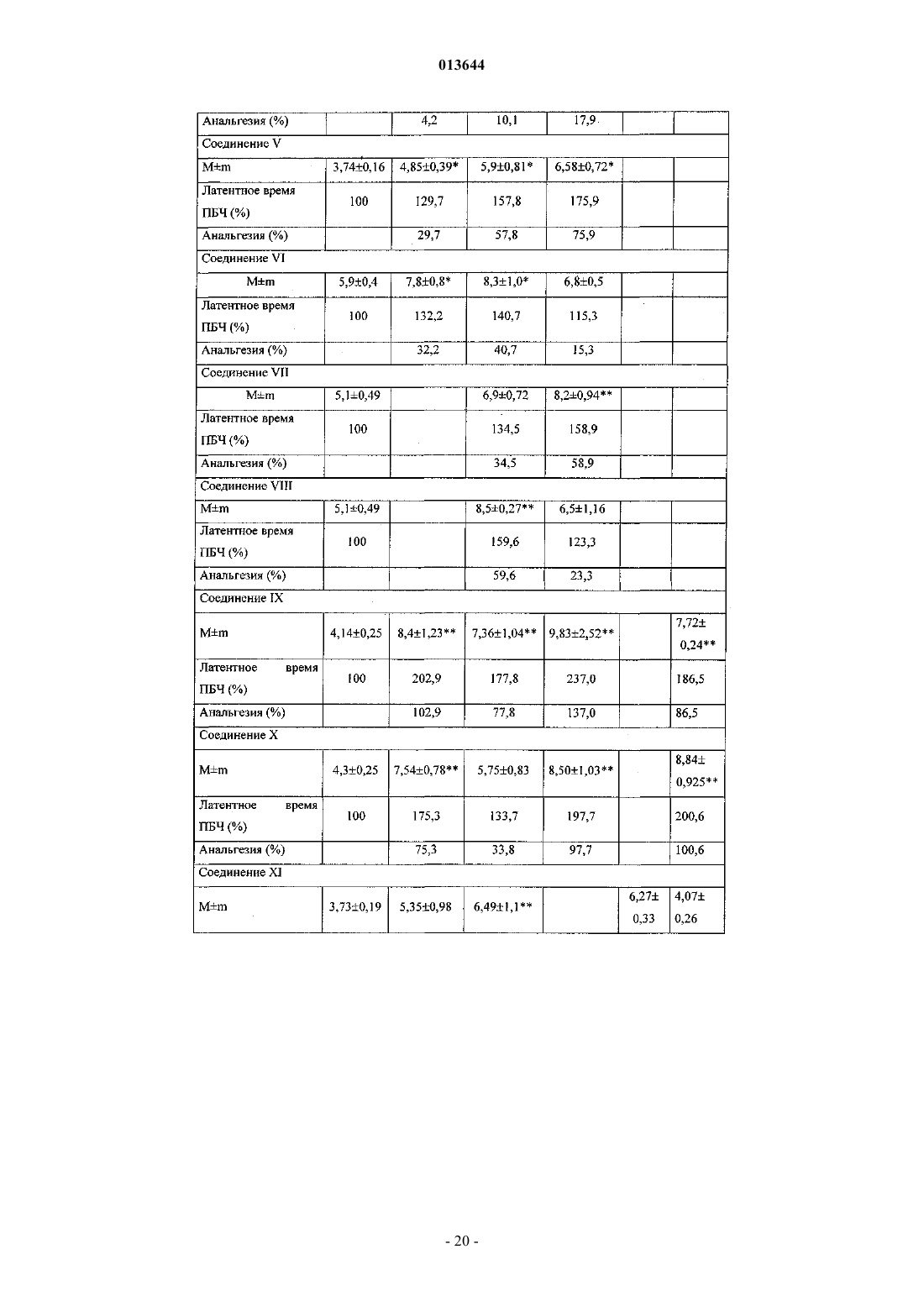
3-фенилпропионилтирамином,
3-(п-гидроксифенил)пропионилфенилаланина метиловым эфиром,
3-(п-гидроксифенил)пропионилтирозином,
3-фенилпропионилтирозином,
фенилацетилтирозином,
3-(п-гидроксифенил)пропионилфенилаланином,
3-фенилпропионилфенилаланином,
фенилацетилфенилаланином,
3-(п-гидроксифенил)пропионилтирозина метиловым эфиром,
3-фенилпропионилтирозина метиловым эфиром,
фенилацетилтирозина метиловым эфиром,
фенилацетилфенилэтиламином,
3-фенилпропионилфенилаланина метиловым эфиром,
фенилацетилфенилаланина метиловым эфиром,
3-(п-гидроксифенил)пропионилтирозина бензиловым эфиром,
3-фенилпропионилтирозина бензиловым эфиром,
фенилацетилтирозина бензиловым эфиром,
тирозилтирозином,
фенилаланилтирозином,
фенилацетилфенилаланина бензиловым эфиром,
или их фармацевтически приемлемые соли.
2. Соединение по п.1, в котором R3 представляет -СООН, -СООСН3.
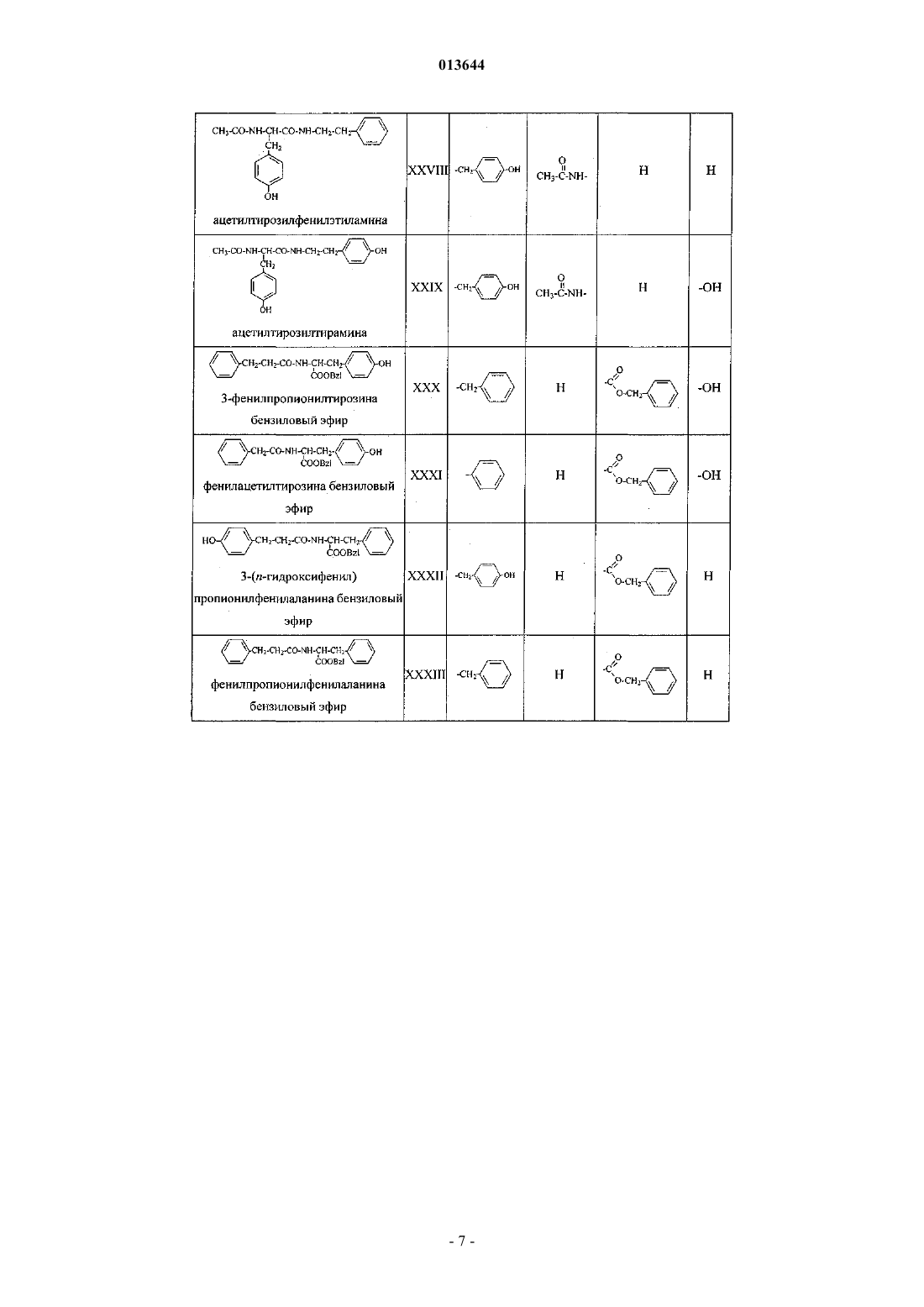
3. Соединение по п.1, выбранное из
п-гидроксифенилацетилтирозина,
п-гидроксифенилацетилфенилаланина,
п-гидроксифенилацетилтирозина метилового эфира,
п-гидроксифенилацетилфенилаланина метилового эфира,
п-гидроксифенилацетилтирозина бензилового эфира,
п-гидроксифенилацетилфенилаланина бензилового эфира,
N-ацетилтирозилфенилэтиламина,
N-ацетилтирозилтирамина,
п-гидроксифенилацетилтирамина,
п-гидроксифенилацетилфенилэтиламина,
3-(п-гидроксифенил)пропионилфенилаланина бензиловым эфиром,
3-фенилпропионилфенилаланина бензиловым эфиром,
или их фармацевтически приемлемых солей.
4. Способ получения соединений общей формулы (I)
![]()
где R1 представляет
![]()
или
![]()
где R5 представляет водород или гидроксильную группу; R2представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН, -COOR6, где R6представляет C1-С6-алкил или
![]()
где R7представляет водород или гидроксильную группу; R4представляет водород, гидроксильную группу;
включающий активацию карбоксильной группы соединения общей формулы
![]()
взаимодействием с дифенилфосфорилазидом и триэтиламином в органическом растворителе при охлаждении с последующим взаимодействием с аминосоединением общей формулы
![]()
где R1-R4 принимают значения, определенные для соединений общей формулы (I).
5. Способ по п.4, в котором используют 1-1,2 экв. дифенилфосфорилазида и триэтиламина.
6. Способ по п.4 или 5, в котором в качестве аминопроизводных используют эфир тирозина или фенилаланина.
7. Способ по любому из пп.4-6, в котором в качестве органического растворителя используют N,N-диметилформамид или этилацетат.
8. Способ по любому из пп.4-6, который осуществляют в интервале температур от -25 до 0°С.
9. Способ получения соединений по п.1 или их фармацевтически приемлемых солей, включающий превращение п-гидроксифенилуксусной кислоты, фенилуксусной кислоты или N-замещенного тирозина общей формулы
![]()
в активированный N-оксисукцинимидный эфир общей формулы
![]()
N,N'-дициклогексилкарбодиимидным методом с последующим взаимодействием активированного N-оксисукцинимидного эфира с аминопроизводным общей формулы
![]()
где R1-R4 принимают значения, определенные для соединений общей формулы (I) в п.1.
10. Способ по п.9, в котором аминопроизводным является эфир тирозина или фенилаланина.
11. Фармацевтическая композиция, обладающая способностью ингибировать циклооксигеназу спазмолитическими, антигипоксическими, антидепрессантными и противопаркинсоническими свойствами, а также способностью потенцировать действие анальгетиков, включающая в качестве активного агента эффективное количество соединения общей формулы (I)
![]()
где R1 представляет
![]()
или
![]()
где R5 представляет водород или гидроксильную группу; R2представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН, -COOR6, где R6представляет C1-С6-алкил или
![]()
где R7представляет водород или гидроксильную группу; R4представляет водород, гидроксильную группу,
или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
12. Фармацевтическая композиция, обладающая анальгетическими, противовоспалительными свойствами при отсутствии ульцерогенности, включающая в качестве активного агента эффективное количество соединения общей формулы (I)
![]()
где R1 представляет
![]()
или
![]()
где R5 представляет водород или гидроксильную группу; R2представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН, -COOR6, где R6представляет C1-С6-алкил или
![]()
где R7представляет водород или гидроксильную группу; R4представляет водород, гидроксильную группу;
при условии, что соединение общей формулы (I) не является
фенилацетилтирамином,
3-(п-гидроксифенил)пропионилтирамином,
3-фенилпропионилфенилэтиламином,
3-фенилпропионилтирамином,
3-фенилпропионилтирозина метиловым эфиром,
или его фармацевтически приемлемые соли, а также в сочетании с другими анальгетиками для потенцирования эффекта.
13. Лекарственное средство, обладающее способностью ингибировать циклооксигеназу спазмолитическими, антигипоксическими, антидепрессантными и противопаркинсоническими свойствами, а также способностью потенцировать действие анальгетиков, включающее соединение общей формулы (I)
![]()
где R1 представляет
![]()
или
![]()
где R5 представляет водород или гидроксильную группу; R2представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН, -COOR6, где R6представляет C1-С6-алкил или
![]()
где R7представляет водород или гидроксильную группу; R4представляет водород, гидроксильную группу;
или его фармацевтически приемлемые соли.
14. Лекарственное средство, обладающее анальгетическими, противовоспалительными свойствами при отсутствии ульцерогенности, включающее соединение общей формулы (I) по п.1
![]()
где R1 представляет
![]()
или
![]()
где R5 представляет водород или гидроксильную группу; R2представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН, -COOR6, где R6представляет C1-C6-алкил или
![]()
где R7представляет водород или гидроксильную группу; R4представляет водород, гидроксильную группу;
при условии, что соединение общей формулы (I) не является
фенилацетилтирамином,
3-(п-гидроксифенил)пропионилтирамином,
3-фенилпропионилфенилэтиламином,
3-фенилпропионилтирамином,
3-фенилпропионилтирозина метиловым эфиром,
или его фармацевтически приемлемые соли, а также сочетание с другими анальгетиками для потенцирования эффекта.
15. Применение соединений общей формулы (I)
![]()
где R1 представляет
![]()
или
![]()
где R5 представляет водород или гидроксильную группу; R2представляет водород или аминогруппу, необязательно замещенную группой СН3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН, -COOR6, где R6представляет C1-С6-алкил или
![]()
где R7представляет водород или гидроксильную группу; R4представляет водород, гидроксильную группу;
или их фармацевтически приемлемых солей для получения лекарственного средства, обладающего способностью ингибировать циклооксигеназу, спазмолитическим, антигипоксическим, антидепрессантным и противопаркинсоническим действием, а также способностью потенцировать действие анальгетиков.
16. Применение соединений общей формулы (I)
![]()
где R1 представляет
![]()
или

где R5 представляет водород или гидроксильную группу; R2представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН, -COOR6, где R6представляет C1-С6-алкил или
![]()
где R7представляет водород или гидроксильную группу; R4представляет водород, гидроксильную группу,
при условии, что соединение общей формулы (I) не является
фенилацетилтирамином,
3-(п-гидроксифенил)пропионилтирамином,
3-фенилпропионилфенилэтиламином,
3-фенилпропионилтирамином,
3-фенилпропионилтирозина метиловым эфиром,
или их фармацевтически приемлемые соли для получения анальгетических, противовоспалительных лекарственных средств, не обладающих ульцерогенностью, а также в сочетании с другими анальгетиками для потенцирования эффекта.
17. Способ лечения болевых синдромов различного генеза, воспалительных и воспалительно-дегенеративных заболеваний суставов и соединительной ткани, костно-мышечной системы, заболеваний, сопровождающихся воспалением, а также послеоперационной боли, посттравматической боли, болевых синдромов гинекологической, неврологической, онкологической, стоматологической природы, ревматоидного артрита, артропатии, болезни Бехтерева, неспецифических спондиллоартритов, подагрического артрита, остеоартроза, внесуставного ревматизма и тромбофлебита, включающий введение млекопитающему эффективного количества соединения общей формулы (I)
![]()
где R1 представляет
![]()
или
![]()
где R5 представляет водород или гидроксильную группу; R2представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН, -COOR6, где R6представляет С1-С6-алкил или
![]()
где R7представляет водород или гидроксильную группу; R4представляет водород, гидроксильную группу;
при условии, что соединение общей формулы (I) не является
фенилацетилтирамином,
3-(п-гидроксифенил)пропионилтирамином,
3-фенилпропионилфенилэтиламином,
3-фенилпропионилтирамином,
3-фенилпропионилтирозина метиловым эфиром,
или его фармацевтически приемлемой соли, а также в сочетании с другими анальгетиками для потенцирования эффекта.
18. Способ лечения заболеваний, сопровождающихся спазмами, депрессией, гипоксией, явлениями Паркинсонизма, а также эмоционально-стрессовых состояний и нарушений, вызываемых спазмами, гипоксией и сопровождающих болезнь Паркинсона, включающий введение млекопитающему эффективного количества соединения общей формулы (I)
![]()
где R1 представляет
![]()
или
![]()
где R5 представляет водород или гидроксильную группу; R2представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН, -COOR6, где R6представляет C1-C6-алкил или
![]()
где R7представляет водород или гидроксильную группу; R4представляет водород, гидроксильную группу,
или его фармацевтически приемлемой соли.
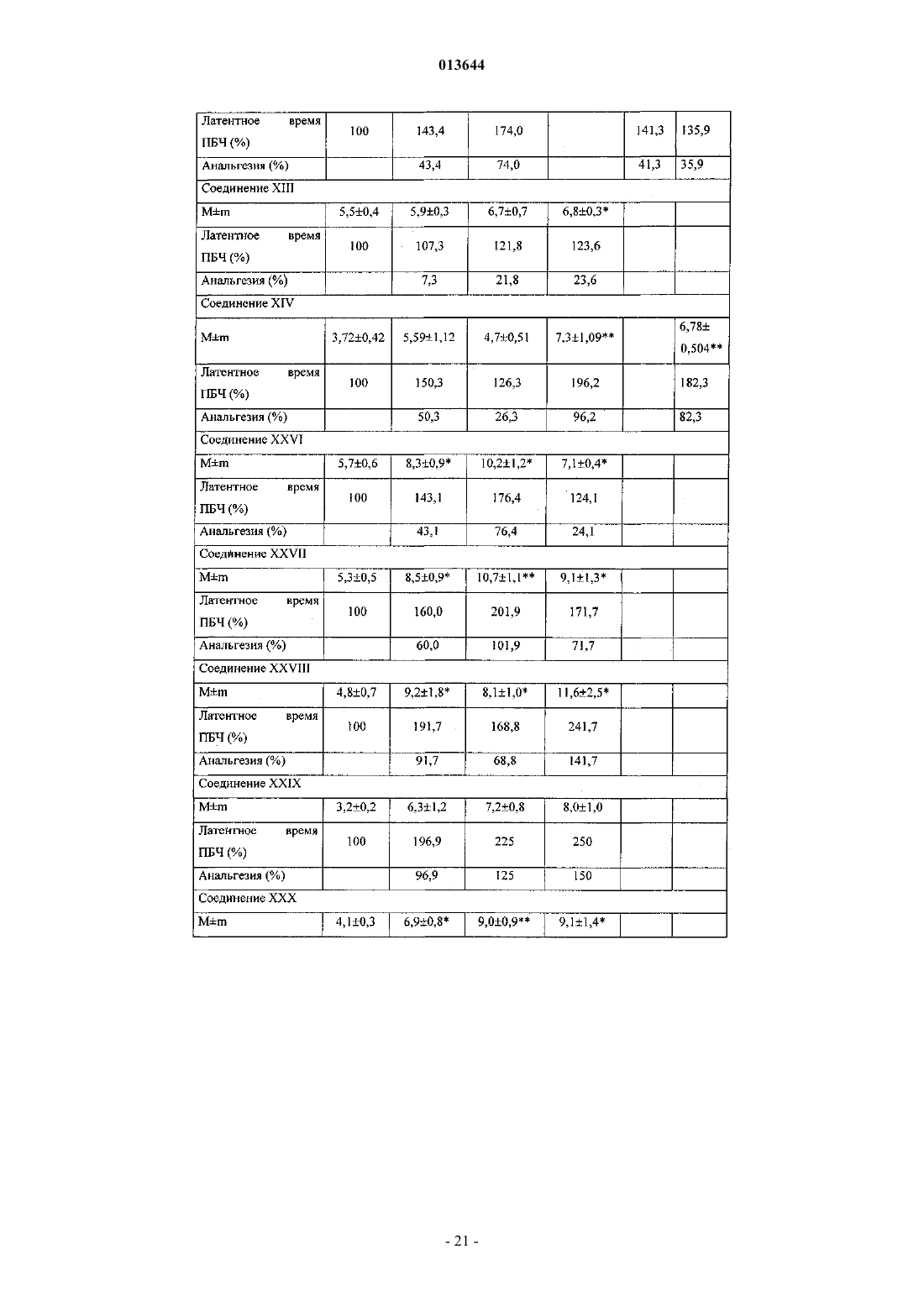
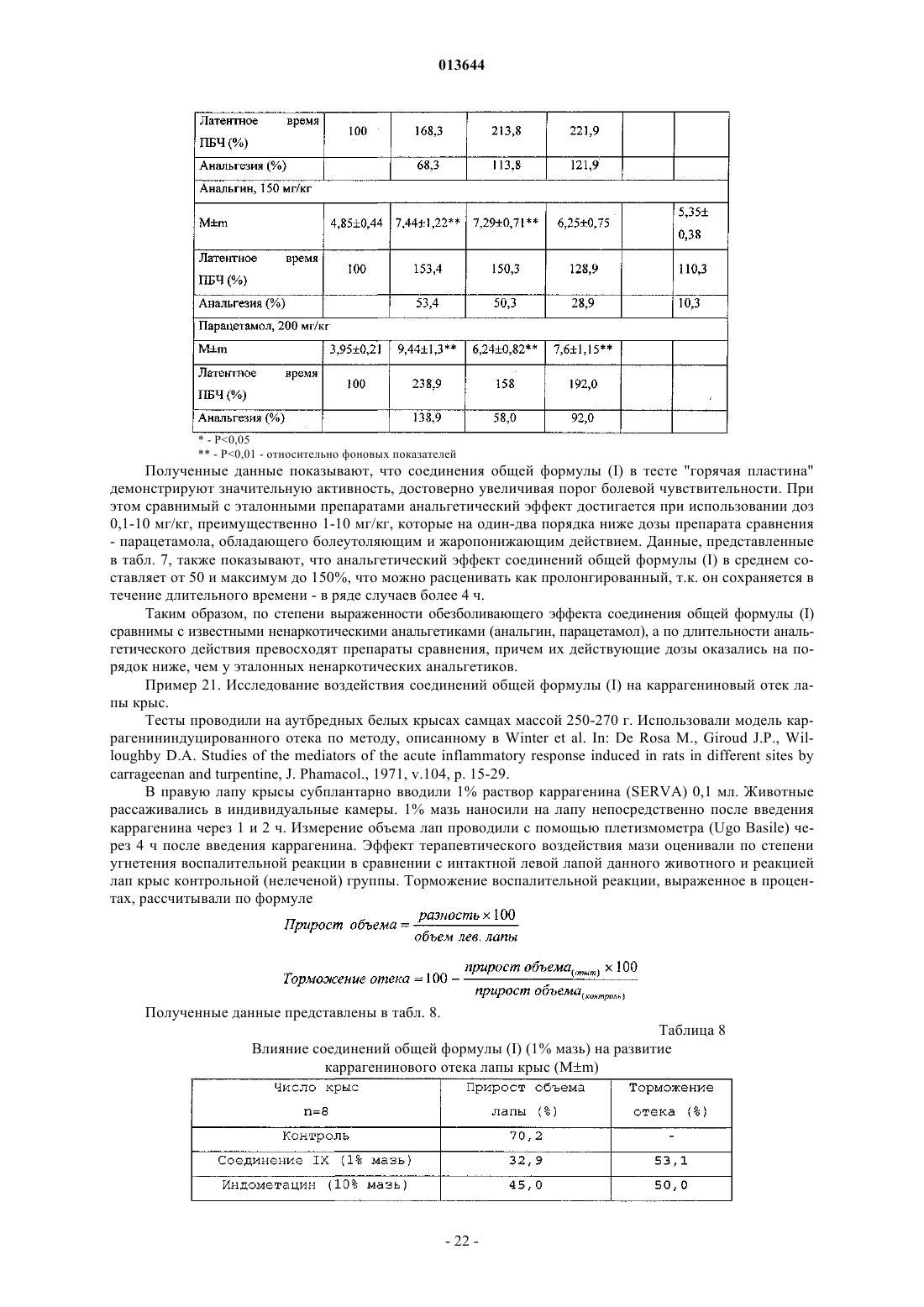
Текст
013644 Настоящее изобретение относится к области биоорганической химии и касается новых соединений - фенилсодержащих N-ацильных производных биогенных аминов, а также способа синтеза новых и известных соединений, их применения в медицине в качестве потенциальных анальгетических, противовоспалительных, спазмолитических и антигипоксических средств, а также средств, обладающих антидепрессантным, противопаркинсоническим действием и способностью потенцировать действие других анальгетиков. Предшествующий уровень техники В публикации международной заявки WO 97/23202 раскрыты фенилсодержащие N-ацильные производные аминов общей формулы (XV) которая, среди прочих, включает 3-(п-гидроксифенил)пропионилфенилэтиламин, 3-(п-гидроксифенил)пропионилтирамин и 3-фенилпропионилфенилэтиламин (соединения IX, X, XI настоящего изобретения соответственно) в качестве промежуточных соединений, а также синтез соединений общей формулы (XV) и их применение в качестве селективных лигандов подтипов NMDA рецепторов, используемых для лечения хронической боли, мигреневой головной боли, а также анестетиков. Однако в указанной публикации не описаны и не охарактеризованы конкретные структуры, соответствующие соединениям X и XI настоящего изобретения и отсутствуют какие-либо данные, подтверждающие заявленный вид активности, а соединение IX в качестве промежуточного соединения и его синтез раскрыты лишь в способе получения других производных аминов. Соединения IX, X и XI настоящего изобретения также описаны в более ранних публикациях, ставших общедоступными до даты приоритета вышеуказанной международной заявки WO 97/23202, для использования по иному назначению. 3-(п-Гидроксифенил)пропионилфенилэтиламин (IX) раскрыт в Jacobson K.А., Kirk K.L. New highperformance liguid chromatographic procedure for the detection and quantification of -phenyletylamine, J.A. Amides, coumarine and other constituents from simsia cronquistii, Phytochem., 1992, p. 1413-1414. В публикации международной заявки WO 97/23202 указана возможность использования соединений общей формулы (XV) для предотвращения некоторых специфических видов боли, таких как мигреневая головная боль, хроническая боль, а также применение их для анестезии, обусловленная способностью данных соединений проявлять действие селективных лигандов подтипов NMDA рецепторов. Однако в WO 97/23202 нет каких-либо данных, подтверждающих заявленную активность указанной группы соединений и, следовательно, возможность применения данных соединений по указанному назначению,на конкретных моделях на животных in vivo, и, таким образом, выводы о возможных фармакологических эффектах основаны исключительно на утверждении о том, что все раскрытые в указанной международной заявке соединения являются селективными лигандами подтипов NMDA-рецепторов. В публикации международной заявки WO 97/23202 описан способ синтеза 3-(п-гидроксифенил)пропионилфенилэтиламина (IX) с использованием 1-гидроксибензотриазола в присутствии N,N'-дициклогексилкарбодиимида (DCC). Не описан способ выделения и очистки данного соединения, из физикохимических констант приведены температура плавления и данные 1 Н-ЯМР-спектроскопии. В статье Jacobson K.А., Kirk K.L. New high-performance liguid chromatographic procedure for the detection and quantification of -phenyletylamine, J. Chromatography, 1987, v.415, p. 124-128 раскрыт синтез 3-(п-гидроксифенил)пропионилфенилэтиламина (IX) с применением модифицированного N-оксисукцинимидного эфира 3-(п-гидроксифенил)пропионовой кислоты. Реакцию проводят в смеси метанол-1 МNa2HPO4, pH 8 (1:1), используя сульфосукцинимидил-3-(п-гидроксифенил)пропионат (сульфатированный реагент Bolton-Hunte). Полученный продукт охарактеризован только температурой плавления. В соответствии с данной статьей полученный 3-(п-гидроксифенил)пропионилфенилэтиламин используют в качестве внутреннего стандарта в электрохимическом детекторе при количественном определении уровня эндогенного фенилэтиламина в биологических жидкостях методом ВЭЖХ. В статье Herbert R.B., Kattah A.E. The biosynthesis of Sceletium alkaloids in Sceletium subvelutinum L.Bolus, Tetrahedron, 1990, v.46,20, p. 7105-7118 описано применение 3-(п-гидроксифенил)пропионилтирамина (X) в качестве промежуточного продукта в синтезе алкалоидов Sceletium subvelutinum, а также его способ синтеза методом DCC. Недостатком данного способа является необходимость применения для очистки целевого продукта колоночной хроматографии, его сравнительно невысокий выход - около 48%. В статье Maldonado E., Hernandez E., Ortega A. Amides, coumarine and other constituents from simsiacronquistii, Phytochem., 1992, p. 1413-1414 описано выделение 3-фенилпропионилфенилэтиламина (XI) из наземной части растений Simsia cronquistii и представлены данные масс-спектрометрии, 1 Н-ЯМР-спек-1 013644 троскопии, а также температура плавления. Данных по биологической активности не приведено. Синтез соединения XI с применением конденсирующего агента 4-(4,6-диметокси-1,3,5-триазин-2 ил)-4-метилморфолина хлорида (DMT-MM) описан в Kunishima М., Kawachi С., Hioki K. et al. Formationwater-soluble condensing agent: DMT-MM, Tetrahedron, 2001, v.57,8, p. 1551-1558. Недостатками данного способа синтеза являются образование побочного продукта и необходимость применения препаративной тонкослойной хроматографии для очистки целевого продукта, что усложняет процесс и должно с неизбежностью приводить к снижению выхода. Несмотря на это, указывается высокий выход продукта(XI), составляющий 98%. Соединение XI было синтезировано с целью изучения применимости нового конденсирующего агента DMT-MM. Синтез производных аминокислот тирозина и фенилаланина 3-(п-гидроксифенил)пропионилтирозина, фенилпропионилтирозина, фенилацетилтирозина, фенилпропионилфенилаланина и фенилпропионилтирозина метилового эфира (соединения XIV, XV, XVI, XVIII и XXI настоящего изобретения соответственно) и изучение их ингибирующего действия на нейрон TAN, идентифицированный в ганглии улитки Achatina fulica ferussac, описаны в статьях Takeuchi Н., Ariyoshi Y. Effects of N-betaphenylpropionyl-L-tyrosine and its derivatives on the excitability of an identifiable giant neuron of Achatinaof an identifiable giant neurone of an African giant snail, Br. J. Pharmacol., 1982, v.77, p. 631-639. Описана типичная методика синтеза соединений XIV, XV, XVI, XVIII, XXI методом активированных N-оксисукцинимидных эфиров с использованием в качестве аминопроизводного метилового эфира тирозина, с последующим его омылением (для соединений XIV, XV, XVI, XVIII), но физико-химические константы и выходы для указанных соединений не приведены. Кроме того, синтез фенилацетилтирозина (XV) с высоким выходом (94%) с использованием 1-гидроксибензотриазола и этил-3(3-диметиламино)пропилкарбодиимида, с использованием в качестве исходных соединений этилового эфира тирозина и фенилпропионовой кислоты, с последующим омылением этилового эфира описан в Tangpasuthadol V., Pendharkar S.M., Kohn J. Hydrolytic degradation of tyrosine-derived polycarbonates, a class of new biomaterials.Chemistry, 1974, v.12,3, p. 757-763. Приведены температура плавления и элементный анализ. Синтез был проведен для изучения степени ингибирования соединением XVIII глутаминсинтетазы. Фенилпропионилтирозин метиловый эфир (XXI) упоминается в патенте Японии JP 57193437 (пример 4), где его синтез осуществлен методом активированных N-оксисукцинимидных эфиров. Синтез фенилацетилфенилаланина (XIX), подобный синтезу соединения XVIII, с использованием хлорангидрида фенилуксусной кислоты, раскрыт в Chen H.M., Hsu M.S., Huang L.J. et al. Effect ofN-phenylacetyl L-amino acids on the differentiation of HL-60 cells, Chinese Pharmaceutical Journal, 2001, v.53, 3, p. 157-167. Приведены физико-химические характеристики целевого соединения: температура плавления, данные 1 Н-ЯМР- и ИК-спектроскопии, масс-спектрометрии. Было установлено, что фенилацетилфенилаланин (XIX) является индуктором дифференцировки клеток. 3-(п-Гидроксифенил)пропионилтирозина метиловый эфир (XX) упоминается в публикации международной заявки WO 99/52962, однако методика синтеза и физико-химические характеристики не приведены. Соединение (XX) синтезировано с целью его использования в качестве мономера для получения биоразлагаемых полимеров, совместимых с тканями. Природное соединение, выделенное из симбиотической бактерии Xenorhabdus nematophilus, фенилацетилфенилэтиламин (XXIII), было синтезировано хлорангидридным методом и охарактеризовано физико-химическими данными 1 Н-ЯМР-, 13 С-ЯМР- и ИК-спектроскопии, масс-спектрометрии, температурой плавления в публикации международной заявки WO 01/49656. Исследована противоопухолевая активность соединения XXIII in vitro. Общая формула соединений, раскрытых в публикации международной заявки WO 01/49656, охватывает также и другие соединения настоящего изобретения: п-гидроксифенилацетилтирамин, п-гидроксифенилацетилфенилэтиламин и фенилацетилтирамин (соединения VII, VIII и VI настоящего изобретения соответственно). Однако данная публикация не раскрывает ни конкретных структурных формул указанных соединений, ни методик их синтеза, ни физико-химических констант, ни данных по биологической активности. Фенилпропионилтирамин (XII) упоминается в статье Takeuchi Hiroshi; Tamura Hiroko The effects of(Achatina fulica Ferussac), British Journal of Pharmacology, 1980, v.69,1, p. 29-34, но без описания его синтеза, физико-химических характеристик и назначения. В статье Garrett C.E., Jiang X., Prasad K., Repic О. New observations on peptide bond formation usingCDMT, Tetrahedron Letters, 2002, v.43,23, p. 4161-4165 раскрыт фенилпропионилфенилаланина метиловый эфир (XXIV) и способ его синтеза с применением нового конденсирующего агента 2-хлор-4,6 диметокси-1,3,5-триазина (CDMT) в присутствии N-метилморфолина. Однако не приведено ни физикохимических характеристик указанного соединения, ни данных по его активности, лишь сообщается, что данный способ синтеза имеет преимущества: синтез в одну стадию и выделение продукта путем высаживания водой приводят к хроматографически чистому продукту с высоким выходом 90%. В статье Peric M., Vercek В., Petric А. -Diazoacetophenones as reagents for a mild and selective protection of an amino group, Acta Chimica Slovenica, 1996, v.43,2, p. 163-173 описан синтез фенилацетилтирозина метилового эфира (XXII), промежуточного соединения для синтеза пептидов, конденсацией фенилуксусной кислоты с метиловым эфиром тирозина через образование диазокетона. Для очистки соединения XXII обязательно использовали колоночную хроматографию. Приведены температура плавления,данные 1 Н-ЯМР спектроскопии и элементного анализа. Фенилацетилфенилаланин метиловый эфир (XXV) в соответствии с Votano J.R., Altman J., Wilchek M.disease, Proceedings of the National Academy of Sciences of the United States of America, 1984, v.81,10,p. 3190-3194 был синтезирован методом активированных N-оксисукцинимидных эфиров с последующей очисткой колоночной хроматографией. Физико-химических констант для данного соединения не приведено. В указанной статье соединение XXV является промежуточным в синтезе соединения XIX, которое исследуется в качестве потенциального средства для лечения серповидноклеточного заболевания. Кроме того, известен ферментативный способ синтеза соединения XXV [Didziapetris R., Drabnig В.,Schellenberger V., Jakubke H.D., Svedas V. Penicillin acylase-catalyzed protection and deprotection of aminoblood lipid level-related diseases comprising the same, US 2003199566 описан синтез 3-(п-гидроксифенил)пропионилфенилаланина (XVII) и 3-(п-гидроксифенил)пропионилфенилаланина метилового эфира (XIII) с использованием 1-гидроксибензотриазола и 1-[3-(диметиламино)пропил]-3-этилкарбодиимида гидрохлорида в присутствии триэтиламина. Для получения 3-(п-гидроксифенил)пропионилфенилаланина (XVII) далее проводили омыление соединения (XIII) с выходом целевого продукта 78%. Для обоих соединений приведены данные 13 Н-ЯМР- и 13 С-ЯМР-спектроскопии. Соединения XVII и XIII предлагается использовать для предупреждения и лечения заболеваний, связанных с уровнем липидов в крови. В публикации международной заявки WO 9952962 описан 3-(п-гидроксифенил)пропионилтирозина бензиловый эфир (XXXIV). Приведены температура плавления, данные 1 Н-ЯМР- и 13 С-ЯМР-спектроскопии. Известно, что анальгетическое действие может осуществляться в соответствии с различными механизмами, в частности, путем ингибирования фермента циклооксигеназы в каскаде арахидоновой кислоты[Машковский М.Д. Лекарственные средства. - М.: Новая волна, 2005, с. 163-164]. Наиболее выраженным обезболивающим эффектом среди препаратов, снижающих синтез альгогенов, обладают ненаркотические анальгетики и нестероидные противовоспалительные средства. Ненаркотические анальгетики представлены салицилатами (аспирин), производными пиразолона (амидопирин,анальгин) и парааминофенола (парацетамол). К нестероидным противовоспалительным средствам относятся производные салициловой, уксусной, пропионовой и антраниловой кислот. Ненаркотические анальгетики и нестероидные противовоспалительные средства, наряду с болеутоляющим эффектом, обладают противовоспалительным и жаропонижающим действием [Кукушкин М.Л., Хитров Н.К. Общая патология боли. - М.: Медицина, 2004, 142 с.]. Основным побочным эффектом нестероидных противовоспалительных средств является ульцерогенность. У анальгетиков с другим механизмом действия часто наблюдается проспазматический побочный эффект [Машковский М.Д. Лекарственные средства. - М.: Новая волна, 2005, с.154]. Известны противопаркинсонические свойства нестероидных противовоспалительных препаратов салицилата натрия, индометацина и пироксикама [Кадиева М.Г., Оганесян Э.Т., Мацуева С.Х. Нейротоксины и средства для лечения болезни Паркинсона. III. Средства, опосредовано влияющие на дофаминэргическую систему, Хим.-фарм. журнал, 2005, т.39, 11, с.3-11]. Предполагают, что такая активность, в частности, реализуется опосредовано через простагландины, воздействующие на дофаминовую систему. Известно также, что антисеротониновые препараты оказывают позитивное влияние на дофаминовую систему при болезни Паркинсона, способствуя связыванию рецепторов с дофаминовыми антагонистами [Кадиева М.Г., Оганесян Э.Т., Мацуева С.Х. Нейротоксины и средства для лечения болезни Паркинсона. III. Средства, опосредовано влияющие на дофаминэргическую систему, Хим.-фарм. журнал,2005, т.39, 11, c. 3-11], существуют и другие механизмы действия противопаркинсонических препара-3 013644 тов [Машковский М.Д. Лекарственные средства. - М.: Новая волна, 2005, с.138]. В зависимости от механизма действия антидепрессанты подразделяются на несколько групп, в частности ингибиторы моноаминооксидазы, трициклические антиоксиданты, блокаторы гистаминовых,серотониновых, холецистокининовых, -адренорецепторов [Машковский М.Д. Лекарственные средства.- М.: Новая волна, 2005, с.109]. Так как применение известных антидепрессантов и структурно родственных соединений сопровождается многочисленными серьезными побочными эффектами, то поиск новых более безопасных и эффективных препаратов такого действия является актуальным. Применение соединений настоящего изобретения для купирования депрессивных состояний не было известно. Гипоксия наблюдается при многочисленных патологических состояниях, в том числе нарушениях функций мозга. Антигипоксанты улучшают утилизацию организмом циркулирующего кислорода, повышая устойчивость организма к кислородной недостаточности. Препараты такого действия немногочисленны [Машковский М.Д. Лекарственные средства. - М.: Новая волна, 2005, с. 729]. Многие препараты, в том числе регулирующие деятельность ЦНС, обладают дополнительно антигипоксическими свойствами, повышающими эффективность их действия. Для группы соединений настоящего изобретения антигипоксический эффект ранее описан не был. Целью настоящего изобретения является синтез и применение новых и известных фенилсодержащих N-ацильных производных биогенных аминов и аминокислот в качестве нетоксичных, более эффективных анальгетиков и противовоспалительных средств без побочных эффектов, в частности ульцерогенности и проспазматического действия, обладающих также антигипоксическим, антидепрессантным и противопаркинсоническим действием, а также способностью потенцировать действие других анальгетиков. Краткое описание изобретения Настоящее изобретение относится к новым фенилсодержащим N-ацильным производным аминов общей формулы (I) где R5 представляет водород или гидроксильную группу; R2 представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -COOH,-COOR6, где R6 представляет C1-C6-алкил или где R7 представляет водород или гидроксильную группу, R4 представляет водород, гидроксильную группу; при условии, что соединение общей формулы (I) не является фенилацетилтирамином,3-(п-гидроксифенил)пропионилфенилэтиламином,3-(п-гидроксифенил)пропионилтирамином,3-фенилпропионилфенилэтиламином,3-фенилпропионилтирамином,3-(п-гидроксифенил)пропионилфенилаланина метиловым эфиром,3-(п-гидроксифенил)пропионилтирозином,3-фенилпропионилтирозином,фенилацетилтирозином,3-(п-гидроксифенил)пропионилфенилаланином,3-фенилпропионилфенилаланином,фенилацетилфенилаланином,3-(п-гидроксифенил)пропионилтирозина метиловым эфиром,3-фенилпропионилтирозина метиловым эфиром,фенилацетилтирозина метиловым эфиром,фенилацетилфенилэтиламином,3-фенилпропионилфенилаланина метиловым эфиром,-4 013644 фенилацетилфенилаланина метиловым эфиром,3-(п-гидроксифенил)пропионилтирозина бензиловым эфиром; или их фармацевтически приемлемым солям, обладающим ингибирующей циклооксигеназу активностью, противовоспалительным и анальгетическим действием, спазмолитическим, антигипоксическим,антипаркинсоническим и антидепрессивным действием, а также способностью потенцировать действие других анальгетиков. Настоящее изобретение также относится к применению соединений общей формулы (I) где R5 представляет водород или гидроксильную группу; R2 представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН,-COOR6, где R6 представляет C1-С 6-алкил или где R7 представляет водород или гидроксильную группу; R4 представляет водород, гидроксильную группу; или их фармацевтически приемлемых солей в качестве ингибиторов циклооксигеназы, анальгетических и противовоспалительных, спазмолитических, антигипоксических, антипаркинсонических и антидепрессантных средств, а также средств, способных потенцировать действие других анальгетиков. Далее, настоящее изобретение относится к фармацевтической композиции или средству, обладающему ингибирующей циклооксигеназу активностью, противовоспалительным и анальгетическим действием, а также антидепрессантным, спазмолитическим, антигипоксическим и противопаркинсоническим действием, содержащим эффективное количество соединения общей формулы (I) или его фармацевтически приемлемой соли и необязательно фармацевтически приемлемый носитель. Еще одним объектом изобретения является способ лечения болевых синдромов различного генеза, а также заболеваний, сопровождающихся воспалением, спазмами, гипоксией, депрессией и явлениями Паркинсонизма, включающий введение эффективного количества соединения общей формулы (I) или его фармацевтически приемлемой соли, необязательно в сочетании с другими анальгетиками. Настоящее изобретение также относится к новым способам получения соединений общей формулы(I). Детальное описание изобретения Предпочтительными соединениями формулы (I) являются соединения, в которых R3 представляет-5 013644 Новые предпочтительные соединения общей формулы (I) представлены в табл. 1. Таблица 1-7 013644 Известные предпочтительные соединения общей формулы (I) представлены в табл. 2. Таблица 2 Соединения общей формулы (I) получают активацией карбоксильной группы п-гидроксифенилуксусной кислоты или фенилуксусной кислоты взаимодействием с дифенилфосфорилазидом (DPPA) и триэтиламином (TEA) в органическом растворителе, предпочтительно N,N-диметилформамиде, этилацетате, при охлаждении предпочтительно в интервале от -25 до +0 с последующим осуществлением взаимодействия с аминопроизводным. Предпочтительно активацию карбоксильной группы осуществляют с использованием 1-1,2 экв. DPPA и TEA. В качестве аминопроизводного могут быть использованы эфиры тирозина и фенилаланинина. Для получения соединений II и III в качестве исходного аминопроизводного используют бензиловые эфиры тирозина и фенилаланина, соответственно, с последующим удалением бензильной группы путем каталитического гидрогенолиза. В отличие от ранее используемых способов синтеза известных соединений общей формулы (I) применение дифенилфосфорилазидного способа позволило уменьшить число стадий, а именно исключить стадию выделения активированного производного карбоксильного компонента, ограничиться экстракцией для выделения целевых веществ и повысить выходы ( 90%).-9 013644 Общая схема синтеза дифенилфосфорилазным методом представлена на схеме 1. Схема 1 Новые соединения II, III, IV, V, VII, VIII, в том числе содержащие фенольные гидроксильные группы, могут быть получены также методом активированных N-оксисукцинимидных эфиров, преимуществом которого является доступность реагентов, водорастворимость выделяющегося N-гидроксисукцинимида, быстрота протекания как реакции получения N-оксисукцинимидных эфиров ацилирующих агентов, так и реакции образования амидной связи, и возможность достижения высоких выходов целевых продуктов (70-80%), несмотря на наличие в них фенольного гидроксила. В соответствии с предлагаемым способом синтез N-оксисукцинимидных эфиров ацилирующих агентов осуществляется превращением п-гидроксифенилуксусной кислоты или фенилуксусной кислоты в активированный N-оксисукцинимидный эфир N,N'-дициклогексилкарбодиимидным методом (DCC-методом) с высоким выходом (около 90%), с последующим образованием амидной связи реакцией N-оксисукцинимидных эфиров с аминопроизводным, также с высокими выходами (70-80%), за короткое время (1-2 ч) и без применения хроматографической очистки целевого продукта. В качестве аминопроизводного могут быть использованы эфиры тирозина и фенилаланинина. Аналогично могут быть получены известные соединения X, XI, XII,XIII, XV, XVII, XIX, XX, XXII, XXIII, XXIV, синтез которых методом активированных N-оксисукцинимидных эфиров не описан в уровне техники. Общая схема синтеза соединений общей формулы (I) методом активированных N-оксисукцинимидных эфиров представлена на схеме 2. Схема 2 Синтез гидроксифенилпропионилтирозина (XIV) может быть осуществлен также методом активированных N-оксисукцинимидных эфиров, причем с целью уменьшения числа стадий может быть использован незащищенный по С-концу тирозин. Кроме того, это позволяет избежать длительного воздействия щелочи, которая была бы необходима для омыления метилового эфира тирозина, что могло бы неблагоприятно отразиться на оптической чистоте получаемого соединения [Шредер Э., Любке К. Пептиды. М.: Мир, 1967, 2 т.; Гросс Э., Майенхофер И. Пептиды. Основные методы образования пептидной связи.- М.: Мир, 1983, с. 422]. Проблема весьма низкой растворимости незащищенного тирозина как в органических растворителях, так и в воде решена путем его перевода в растворимую Na-соль в результате добавления к суспензии тирозина в DMF 2-х эквивалентов 1N раствора NaOH, в результате чего наблюдалось полное растворение аминокислоты. Реакция полученного таким образом раствора аминопроизводного с N-оксисукцинимидным эфиром 3-(п-гидроксифенил)пропионовой кислоты проходит практически полностью и быстро (за 2 ч). После выделения экстракцией без применения хроматографической очистки выход целевого (XIV) продукта составил около 63%. Соединения общей формулы (I) также могут быть получены в виде фармацевтически приемлемых аддитивных солей с нетоксичными кислотами, такими как фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, лимонная кислота, винная кислота и подобные, и солей с основаниями, такими как гидроксид натрия, гидроксид калия, карбонат натрия и подобные. Соединения общей формулы (I) обладают ингибирующей циклооксигеназу активностью и могут быть использованы для лечения болевых синдромов различного генеза, воспалительных и воспалительно-дегенеративных заболеваний суставов и соединительной ткани, а также костно-мышечной системы,других заболеваний, сопровождающихся воспалением, спазмами, гипоксией, для потенцирования других анальгетиков, а также нарушений, обусловленных депрессией и болезнью Паркинсона. В частности, соединения настоящего изобретения могут быть использованы для лечения послеоперационной боли, посттравматической боли, а также болевых синдромов гинекологической, неврологической, онкологической, стоматологической природы, ревматоидного артрита, артропатии, болезни Бехтерева, неспецифических спондиллоартритов, подагрического артрита, остеоартроза, внесуставного ревматизма и тромбофлебита, других заболеваний, сопровождающихся воспалением, спазмами, гипоксией, а также нарушений, обусловленных болезнью Паркинсона, эмоционально-стрессовыми состояниями. Соединения настоящего изобретения вводятся в эффективном количестве, которое обеспечивает желаемый терапевтический результат. Соединения формулы (I) могут быть введены перорально, местно, парентерально, путем ингаляций и ректально в виде стандартных лекарственных форм, содержащих нетоксичные фармацевтически приемлемые носители. Используемый в настоящем описании термин парентеральное введение означает подкожные, внутривенные, внутримышечные или внутригрудные инъекции или вливания.- 10013644 Соединения настоящего изобретения могут быть введены пациенту в дозах, составляющих от 0,1 до 10 мг/кг веса тела в день, предпочтительно в дозах от 0,5 до 5 мг/кг один или более раз в день. При этом следует отметить, что конкретная доза для каждого конкретного пациента будет зависеть от многих факторов, включая активность данного используемого соединения, возраст, вес тела, пол, общее состояние здоровья и режим питания пациента, время и способ введения лекарственного средства,скорость его выведения из организма, конкретно используемую комбинацию лекарственных средств, а также тяжесть заболевания, подвергаемого лечению. Фармацевтические композиции по настоящему изобретению содержат соединение по настоящему изобретению в количестве, эффективном для достижения желаемого результата и могут быть введены в виде стандартных лекарственных форм (например, в твердой, полутвердой или жидкой формах), содержащих соединения настоящего изобретения в качестве активного ингредиента в смеси с носителем или наполнителем, пригодным для внутримышечного, внутривенного, перорального, сублингвального, ингаляционного и интраректального введения. Активный ингредиент может быть включен в композицию вместе с обычно используемыми нетоксичными фармацевтически приемлемыми носителями, пригодными для изготовления растворов, таблеток, пилюль, капсул, драже, суппозиториев, эмульсий, суспензий,мазей, гелей и любых других лекарственных форм. В качестве наполнителей могут быть использованы различные вещества, такие как сахариды, например глюкоза, лактоза или сахароза, маннит или сорбит, производные целлюлозы и/или фосфаты кальция, например трикальций фосфат или кислый фосфат кальция, в качестве связующего компонента могут быть использованы такие, как крахмальная паста, например кукурузный, пшеничный, рисовый,картофельный крахмал, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий карбоксиметилцеллюлоза и/или поливинилпирролидон. При необходимости могут быть использованы разрыхляющие агенты, такие как вышеупомянутые крахмалы и карбоксиметилкрахмал, поперечно-сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия. Могут быть использованы необязательные добавки, такие как агенты, регулирующие текучесть, и смазывающие агенты, такие как диоксид кремния, тальк, стеариновая кислота и ее соли, такие как стеарат магния, или стеарат кальция, и/или пролиленгликоль. Ядро драже обычно покрывают слоем, который устойчив к действию желудочного сока. Для этой цели могут быть использованы концентрированные растворы сахаридов, которые могут необязательно содержать аравийскую камедь, тальк, поливинилпирролидон, полиэтиленгликоль и/или диоксид титана,и подходящие органические растворители или их смеси. В качестве добавок могут быть также использованы стабилизаторы, загустители, красители и отдушки. В качестве мазевой основы могут быть использованы углеводородные мазевые основы, такие как вазелин белый и желтый (Vaselinum album, Vaselinum flavum), вазелиновое масло (Oleum Vaselini), мазь белая и жидкая (Unguentum album, Unguentum flavum), а в качестве добавок для придания более плотной консистенции, такие как твердый парафин и воск; абсорбтивные мазевые основы, такие как гидрофильный вазелин (Vaselinum hydrophylicum), ланолин (Lanolinum), кольдкрем (Unguentum leniens); мазевые основы, смываемые водой, такие как гидрофильная мазь (Unguentum hydrophylum); водорастворимые мазевые основы, такие как полиэтиленгликолевая мазь (Unguentum Glycolis Polyaethyleni), бентонитовые основы и другие. В качестве основы для гелей могут быть использованы метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, оксипропилцеллюлоза, полиэтиленгликоль или полиэтиленоксид, карбопол. В качестве основы для суппозитория могут быть использованы основы, не растворимые в воде, такие как масло какао; основы, растворимые в воде или смешиваемые с водой, такие как желатино-глицериновые или полиэтиленоксидные; комбинированные основы - мыльно-глицериновые. При приготовлении стандартной лекарственной формы количество активного ингредиента, используемого в комбинации с носителем, может варьироваться в зависимости от реципиента, подвергающегося лечению, от конкретного способа введения лекарственного средства. Так, например, при использовании соединений настоящего изобретения в виде растворов для инъекций содержание активного агента в них составляет 0,01-5%. В качестве разбавителей могут быть использованы 0,9% раствор хлорида натрия, дистиллированная вода, раствор новокаина для инъекций, раствор Рингера, раствор глюкозы, специфические добавки для растворения. При введении в организм соединений настоящего изобретения в виде таблеток и суппозиториев их количество составляет 5,0-500 мг на стандартную лекарственную форму. Лекарственные формы настоящего изобретения получают по стандартным методикам, таким как,например, процессы смешивания, гранулирования, формирование драже, растворение и лиофилизация. Следует отметить, что соединения настоящего изобретения проявляют биологическую активность в дозах на два-три порядка ниже по сравнению с известными препаратами, использованными для сравнения, при практически одинаковой эффективности, и для них не выявлено отрицательных побочных действий и не обнаружено противопоказаний к применению. При этом при исследовании токсичности соединений настоящего изобретения в дозе 1000 мкг/кг перорально не зарегистрировали гибели экспери- 11013644 ментальных животных. Детальное описание соединений настоящего изобретения, их получения и исследования фармакологической активности представлено в нижеследующих примерах, предназначенных для иллюстрации предпочтительных вариантов изобретения, и не ограничивающими его объем. Примеры синтеза соединений настоящего изобретения Индивидуальность полученных соединений проверяли методом ТСХ на пластинках "Kieselgel 60 F254"("Merck", Германия) в системе растворителей: хлороформ-метанол 9:1 (1), хлороформ-метанол-этилацетат 6:1:3 (2), хлороформ-метанол-аммиак 6:3:0,5 (3). Хроматограммы проявляли хлортолидиновым реактивом, нингидрином, йодом и по свечению в УФ-свете. 1 Н-ЯМР регистрировали на приборе "АМХ-400 Bruker" (Германия). ИК-Фурье спектры снимали в таблетках KBr на приборе "Magna 750" ("Nicolet" США). Температуру плавления определяли на приборе "Boetius" (Германия). Масс-спектры высокого разрешения получали на времяпролетном масс-спектрометре методом матриксной лазернодесорбционной ионизации с использованием в качестве матрицы 2,5-дигидроксибензойной кислоты на приборах и REFLEX III (Bruker, Германия). Аналитическую обращенно-фазовую ВЭЖХ проводили на приборах хроматограф "Breeze", детектор "Waters" (США), детекция при 214 нм, скорость элюирования 1 мл/мин, в условиях (1): колонка Symmetry 300 C18, 3,9150 мм, 5 мкм, элюция 0,1%-ной водной TFA с градиентом ацетонитрила от 0 до 60% за 18 мин; хроматограф "System Gold" ("Beckman", США), скорость элюирования 0,25 мл/мин, детекция при 220 нм, в условиях (2): колонка "Phenomenex" (США) C18, 2250 мм, 5 мкм, элюция 0,1%-ной TFA с градиентом 0,08% TFA в 100% MeCN от 0 до 100% за 50 мин; хроматограф "Breeze", детектор "Waters" (США), детекция при 214 нм, скорость элюирования 1 мл/мин, в условиях (3): колонка Symmetry 300 С 18, 4,6250 мм, 20 мкм, элюция 0,1%-ной TFA с градиентом 0,09% TFA в смеси 60:40 ацетонитрил-вода от 0 до 100% за 15 мин. Пример 1. п-Гидроксифенилацетилтирамин (VII). Методика А. При перемешивании к раствору 0,40 г (2,63 ммоль) п-гидроксифенилуксусной кислоты в 3,5 млDMF прибавляли 0,35 г (2,63 ммоль) тирамина. Раствор охлаждали до -10 С и прибавляли 0,68 мл(3,16 ммоль) дифенилфосфорилазида и 0,44 мл (3,16 ммоль) триэтиламина. Перемешивали 2 ч при -10 С и оставляли при 20 С на 15 ч. К реакционной массе прибавляли 35 мл воды, экстрагировали 20 мл этилацетата. Этилацетатный слой промывали 10 мл 5% раствора Na2CO3, водой до рН 7, 10 мл 5% раствораHCl, водой до рН 7. Этилацетатный слой сушили над Na2SO4, отфильтровывали Na2SO4, этилацетат удаляли в вакууме. Маслообразный остаток растирали со смесью эфир-гексан (1:1). Образующийся белый осадок отфильтровывали и сушили в вакууме над CaCl2. Выход 0,68 г (95%).-С-С-); 1226 (вал., -С-О, фенольный). Найдено, %: С 70,57; Н 6,43; N 5,50, C16H17NO3. Вычислено, %: С 70,83; Н 6,32; N 5,16. ВЭЖХ в условиях (1): индивидуальный пик, время удерживания 8,71 мин. Методика Б. К раствору 0,70 г (4,60 ммоль) п-гидроксифенилуксусной кислоты в 17 мл этилацетата при перемешивании прибавляли 0,53 г (4,60 ммоль) N-гидроксисукцинимида, раствор охлаждали до 0 С и прибавляли 0,95 г (4,60 ммоль) N,N'-дициклогексилкарбодиимида (DCC). Перемешивали 2 ч при 0 С и оставили на 20 ч при 4 С. Осадок N,N'-дициклогексилмочевины (DCU) отфильтровали. Растворитель удалили в вакууме. Маслообразный остаток растирали с гексаном. Образовавшийся белый твердый осадок отфильтровывали, промывали гексаном и сушили в вакууме над CaCl2. Получили 1,08 г (94,6%). Rf 0,58 (1). При перемешивании к раствору 0,30 г (1,2 ммоль) N-оксисукцинимидного эфира п-гидроксифенилуксусной кислоты в 8 мл N,N-диметилформамида (DMF) прибавляли 0,16 г (1,2 ммоль) тирамина. Реакционную смесь перемешивали 2 ч при 20 С, оставляли при 4 С на 20 ч. DMF удаляли в вакууме. Маслообразный остаток растирали с водой. Образующийся белый осадок отфильтровывали, промывали водой. Выход 0,26 г (80%).- 12013644 Найдено, %: С 71,05; Н 6,10; N 5,25, C16H17NO3. Вычислено, %: С 70,83; Н 6,32; N 5,16. Пример 2. п-Гидроксифенилацетилфенилэтиламин (VIII). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,57 г (90,5%).[М]+ 255,5. 1 Н-ЯМР DMSO-d6, , м.д.: 2,68 (т, J=8 Гц, 2H, -СН 2-РЕА), 3,22-3,26 (м, -СН 2-PEA), 3,36 (с, 2 Н,СН 2-(OH-PhAc, 6,66 (д, J=4 Гц, 2 Н, м-СН-аром. OH-PhAc), 7,00 (д, J=4 Гц, 2 Н, м-СН-аром. OH-PhAc),7,14-7,28 (м, 5 Н, аром.-СН-PEA), 8,0 (уш. с, 1 Н, NH-PEA), 9,20 (с, 1 Н, ОН-(OH-PhAc. ИК-Фурье, см-1: 3332 (вал. ОН); 3087 (вал., =С-Н, аром.); 1626 (амид I); 1558 (амид II); 1515 (аром. -C-C); 1249 (вал., -С-О, фенольный). Найдено, %: С 75,57; Н 6,80; N 5,77, C16H17NO2. Вычислено, %: С 75,27; Н 6,71; N 5,49. ВЭЖХ в условиях (1): индивидуальный пик, время удерживания 11,17 мин. Синтез проводили в соответствии с методикой Б, приведенной для соединения VII. Выход 0,50 г (79,4%).[М]+ 255,7. Найдено, %: С 75,17; Н 6,87; N 5,75, C16H17NO2. Вычислено, %: С 75,27; Н 6,71; N 5,49. Пример 3. 3-(п-Гидроксифенил)пропионилтирамин (X). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,41 г (95%).Rf 0,38 (1). Тпл=174-176. 1 Н-ЯМР, DMSO-d6, , м.д.: 2,26 (т, J=8 Гц, 2 Н, -СН 2-(НО-PhPr, 2,53 (т, J=6 Гц, 2 Н, -CH2-Tyra),2,67 (т, J=8 Гц, 2 Н, -СН 2-(HO-PhPr, 3,16 (т, J=6 Гц, 2 Н, -СН 2-Tyra), 6,62 (д, J=7 Гц, 2 Н, м-CH-BzlTyra), 6,65 (д, J=7 Гц, 2 Н, м-CH-Bzl-(НО-PhPr, 6,92-6,96 (м, 4 Н, o-CH-Bzl-Tyra и o-CH-Bzl-(HO-PhPr,7,79 (с, 1 Н, NH-Tyra), 9,09 (уш. с, 2 Н, ОН-Tyra и ОН-(НО-PhPr. ИК-Фурье, см-1: 3249 (вал. ОН), 1621 (амид I), 1515 (аром.), 1541 (амид II). Найдено, %: С 71,56; Н 6,78; N 4,97. Вычислено, %: С 71,56; Н 6,71; N 4,91, C17H19NO3. ВЭЖХ в условиях (2): индивидуальный пик, время удерживания 25,62 мин. Синтез проводили в соответствии с методикой Б, приведенной для соединения VII. Выход 0,37 г (85%).[M]+ 285,3. Пример 4. 3-Фенилпропионилфенилэтиламин (XI). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,26 г (97%).(т, J=8 Гц, 2 Н, -CH2-PhPro), 3,24 (т, J=6 Гц, 2 Н, -СН 2-РЕА), 7,25-7,30 (м, 10 Н, СН-аром.), 7,89 (уш. с,1 Н, NH-PEA). ИК-Фурье, см-1: 1637 (амид I), 1546 (амид II). Найдено, %: С 80,24; Н 7,61; N 5,54. Вычислено, %: С 80,60; Н 7,56; N 5,53, C17H19NO3. ВЭЖХ в условиях (2): индивидуальный пик, время удерживания 37,86 мин. Синтез проводили в соответствии с методикой Б, приведенной для соединения VII. Выход 0,20 г (77%).Rf 0,80 (1). Найдено, %: С 80,39; Н 7,53; N 5,30. Вычислено, %: С 80,60; Н 7,56; N 5,53, C17H19NO3. Пример 5. 3-(п-Гидроксифенил)пропионилфенилэтиламин (IX). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,20 г (90%).[M]+ 269, 6. 1 Н-ЯМР, CDCl3, , м.д.: 2,39 (т, J=7 Гц, 2 Н, -СН 2-(НО-PhPr, 2,73 (м, 2 Н, -СН 2-РЕА), 2,86 (т, J=7 Гц, 2 Н, -СН 2-(НО-PhPr, 3,48 (м, 2 Н, -СН 2-РЕА), 6,75 (м, 2 Н, м-CH-аром. НО-PhPr), 7,03 (м, 2H, o-СНаром. HO-PhPr), 7,09 (м, 2 Н, o-СН-аром. PEA), 7,3 (м, 3 Н, м,п-СН-аром, PEA). ИК-Фурье, см-1: 3263 (вал. ОН); 1618 (амид I); 1537 (амид II). Найдено, %: С 75,57; Н 6,93; N 5,09, C17H19NO2. Вычислено, %: С 75,81; Н 7,11; N 5,20. ВЭЖХ в условиях (3): индивидуальный пик, время удерживания 14,77 мин. Пример 6. п-Гидроксифенилацетилтирозина метиловый эфир (IV). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,17 г (39%).[]D25 +12,22 (C 0,36; МеОН). 1 Н-ЯМР, DMSO-d6, , м.д.: 2,78 (дд, 1 Н, СН 2-Tyr), 2,9 (дд, 1 Н, СН 2-Tyr), 3,25-3,45 (м, 2 Н,CH2-HOPhAc), 4,3-4,4 (м, 1 Н, -СН-Tyr), 3,6 (с, 3 Н, ОСН 3 Tyr), 6,55-7,1 (м, 8 Н, аром. Н), 8,25 (д, 1 Н,NH-Tyr). ИК-Фурье, , см-1: 1649 (амид I); 1515 (амид II); 1263 (амид III). Найдено, %: С 65,75; Н 5,75; N 4,23. Вычислено, %: С 65,64; Н 5,81; N 4,25. ВЭЖХ в условиях (3), индивидуальный пик, время удерживания 7,25 мин. Пример 7. п-Гидроксифенилацетилфенилаланина метиловый эфир (V). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,40 г (39%), масло.[]D20 +35,05 (С 0,19, этилацетат). 1 Н-ЯМР, DMSO-d6, , м.д.: 2,9 (дд, 1 Н, CH2-Phe), 3,05 (дд, 1 Н, CH2-Phe), 3,25-3,4 (м, 2 Н,CH2-HOPhAc), 3,6 (с, 3 Н, ОСН 3 Phe), 4,4-4,5 (м, 1 Н, -CH-Phe), 6,55-6,95 (м, 4H, аром. Н HOPhAc),7,1-7,3 (м, 5 Н, аром. Н Phe), 8,3 (д, 1 Н, NH-Phe), 9,2 (с, 1 Н, OH-Ar HOPhAc). ИК-Фурье, , см-1: 1663 (амид I); 1515 (амид II); 1263 (амид III). Найдено, %: С 69,08; Н 6,05; N 4,45. Вычислено, %: С 68,99; Н 6,11; N 4,47. ВЭЖХ в условиях (3), индивидуальный пик, время удерживания 8,57 мин. Пример 8. Фенилацетилтирамин (VI). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,35 г (37,6%).[М+1]+ 256,2. 1 Н-ЯМР, DMSO-d6, , м.д.: 2,6 (т, 2 Н, -СН 2-ТА), 3,2 (кв, 2 Н, -СН 2-ТА), 3,4 (с, 2 Н, CH2-PhAc), 6,67,0 (м, 4 Н, аром. Н ТА), 7,15-7,3 (м, 5 Н, аром. Н PhAc), 8,0 (т, 1 Н, NH-TA), 9,1 (с, 1 Н, ОН-ТА). ИК-Фурье, , см-1: 1646 (амид I); 1516 (амид II); 1264 (амид III). Найдено, %: С 75,37; Н 6,69; N 5,45. Вычислено, %: С 75,27; Н 6,71; N 5,49. ВЭЖХ в условиях (3), индивидуальный пик, время удерживания 8,06 мин. Пример 9. 3-(п-Гидроксифенил)пропионилфенилаланина метиловый эфир (XIII). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,37 г (38%), масло.- 14013644 Пример 10. Бензиловый эфир п-гидроксифенилацетилтирозина (XXVI). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,59 г (55,7%).[]D20-9,18 (С 0,20, МеОН). ИК-Фурье, , см-1: 1649 (амид I); 1515 (амид II); 1737 (вал С=О слож.эф). Найдено, %: С 71,05; Н 5,70; N 3,43. Вычислено, %: С 71,10; Н 5,72; N 3,45. Пример 11. п-Гидроксифенилацетилтирозин (II). К раствору 0,59 г (1,47 ммоль) бензилового эфира п-гидроксифенилацетилтирозина в 10 мл метанола прибавляли 0,20 г 10%-го палладия на угле и при интенсивном перемешивании гидрировали в токе водорода в течение 1,5 ч. Катализатор отфильтровывали. Растворитель из фильтрата удаляли в вакууме. Маслообразный остаток растирали со смесью эфир-гексан (1:1). Образующийся белый осадок отфильтровывали и сушили в вакууме над CaCl2 и Р 2 О 5. Получали 0,32 г (68%). Выход 37%.[]D20 +28,03 (С 0,31, MeOH). 1 Н-ЯМР, DMSO-d6, , м.д.: 2,75 (дд, 1 Н, СН 2-Tyr), 2,9 (дд, 1 Н, СН 2-Tyr), 3,2-3,4 (м, 2 Н,CH2-HOPhAc), 4,3-4,4 (м, 1 Н, -СН-Tyr), 6,55-7,1 (м, 8 Н, аром. Н), 8,05 (д, 1 Н, NH-Tyr). ИК-Фурье, , см-1: 1614 (амид I); 1516 (амид II); 1254 (амид III). Найдено, %: С 64,65; Н 5,41; N 4,37, C17H17NO5. Вычислено, %: С 64,75; Н 5,43; N 4,44. ВЭЖХ в условиях (1), индивидуальный пик, время удерживания 6,33 мин. Пример 12. Бензиловый эфир п-гидроксифенилацетилфенилаланина (XXVII). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,76 г (74%).[]D20 -19,47 (С 0,19, МеОН). ИК-Фурье, , см-1: 1649 (амид I); 1515 (амид II); 1740 (вал С=О слож.эф). Найдено, %: С 74,12; Н 5,92; N 3,57. Вычислено, %: С 74,02; Н 5,95; N 3,60. Пример 13. п-Гидроксифенилацетилфенилаланин (III). К раствору 0,65 г (1,67 ммоль) бензилового эфира п-гидроксифенилацетилфенилаланина в 10 мл метанола прибавляли 0,30 г 10%-го палладия на угле и при интенсивном перемешивании гидрировали в токе водорода в течение 1,5 ч. Катализатор отфильтровывали. Растворитель из фильтрата удаляли в вакууме. Маслообразный остаток растирали с гексаном. Образующийся белый осадок отфильтровывали и сушили в вакууме над CaCl2 и Р 2 О 5. Получали 0,27 г (53%). Выход 39,2%.H-ЯМР, DMSO-d6, , м.д.: 2,85 (дд, 1 Н, CH2-Phe), 3,1 (дд, 1 Н, CH2-Phe), 3,2-3,35 (м, 2 Н,CH2-HOPhAc), 4,4-4,5 (м, 1 Н, -CH-Phe), 6,55-6,95 (м, 4 Н, аром. Н HOPhAc), 7,1-7,3 (м, 5 Н, аром. Н Phe),8,15 (д, 1 Н, NH-Phe). ИК-Фурье, , см-1: 1611 (амид I); 1512 (амид II). Найдено, %: С 68,30; Н 5,68; N 4,65. Вычислено, %: С 68,21; Н 5,72; N 4,68. ВЭЖХ в условиях (3), индивидуальный пик, время удерживания 7,59 мин. Пример 14. Бензиловый эфир 3-фенилпропионилтирозина (XXX). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,94 г (70%).[]D20 -11,93 (С 0,18, МеОН). Найдено, %: С 74,22; Н 6,92; N 3,57, C24H23NO4. Вычислено, %: G 74,42; Н 6,25; N 3,47. Пример 15. Ацетилтирозилфенилэтиламин (XXVIII). Синтез проводили в соответствии с методикой А, приведенной для соединения VII.[]D20 +9,06 (С 0,30, МеОН). ИК-Фурье, , см-1: 1651 (амид I); 1616 (амид II). Найдено, %: С 69,22; Н 6,52; N 8,27, C24H23NO4. Вычислено, %: С 69,92; Н 6,79; N 8,58. Пример 16. Ацетилтирозилтирамин (XXIX). Синтез проводили в соответствии с методикой А, приведенной для соединения VII. Выход 0,77 г (65%).[М]+ 342,7. Найдено, %: С 66,25; Н 6,32; N 8,25, C24H23NO4. Вычислено, %: С 66,65; Н 6,48; N 8,18. Тесты на биологическую активность Пример 17. Исследование влияния соединений общей формулы (I) на метаболизм [14 С]арахидоновой кислоты в бесклеточном гомогенате легочной ткани мыши in vitro. Исследования метаболизма арахидоновой кислоты проводили на мышах-самках линии СВА, находившихся на стандартном рационе вивария. Животных (мышей) забивали, извлекали легкие, гомогенизировали в стеклянном гомогенизаторе фирмы "Wheaton" (США) при 4 С в 10 объемах 0,05 М трис-HCl буфера. Аликвоты (0,5 мл) супернатанта инкубировали с 0,5 мкКи [1-С 14]-арахидоновой кислоты([С 14]-АА), "Amersham", Англия; удельная активность 50-60 мКи/ммоль) при 37 С в течение 30 мин. Экстракцию неметаболизированной [С 14]-АК и продуктов ее метаболизма осуществляли в 20 объемах смеси хлороформа и метанола (1:1) при эффективности экстракции не менее 90%, оцененной с помощью [С 14]ПГ F2. Разделение и идентификацию [С 14]-АА и ее метаболитов осуществляли при помощи ТСХ (пластины Kieselgel 60 фирмы "Merck", Германия), с использованием в качестве органической фазы системы растворителей (этилацетат, изооктан, уксусная кислота, вода - 110:50:20:100) и меченых стандартов. Авторадиохроматограммы, полученные на рентгеновской пленке X-Omat AR ("Kodak", США) и HS 11("ORWO", Германия), денситометрировали на денсискане KS 3 ("Kipp and Zonnen", Голландия). Количественный анализ отдельных эйкозаноидов проведен с помощью радиометрии фракций, полученных высокоэффективной жидкостной хроматографией (ВЭЖХ-система фирмы "Gilson", Франция; колонкаZORBAX С 8 фирмы "Du Pont", США) и элюированием пятен на ТСХ-пластинках. Тестируемые соединения вводили в концентрации 10-4 М. Полученные данные представлены в табл. 3. Таблица 3 Влияние соединений общей формулы (I) (в концентрации 10-4 M) на метаболизм [14 С]арахидоновой кислоты в бесклеточном гомогенате легочной ткани мыши in vitro Полученные данные по профилю эйкозаноидов демонстрируют способность соединений общей формулы (I) ингибировать циклооксигеназу на 22-44% и свидетельствуют об их перспективности в качестве потенциальных анальгетических и противовоспалительных средств. Пример 18. Анальгетическая и противовоспалительная активность соединений общей формулы (I). Исследование анальгетической активности на модели "уксусные корчи". Тесты проводили на белых беспородных мышах-самцах массой 22-24 г. Специфическую болевую реакцию ("корчи") вызывали внутрибрюшинным введением мышам 0,75% раствора уксусной кислоты. Учитывали количество судорожных сокращений брюшных мышц, сопровождающихся вытягиванием задних конечностей и прогибанием спины. Анальгетическое действие оценивали по уменьшению числа корчей у животных в % к контролю в течение 15 мин после введения уксусной кислоты. Методика про- 16013644 ведения тестов описана в Koster R., Anderson M., de Beer E., Fed. Proc., 1959, v.18. p. 412. Исследуемые соединения вводили внутрижелудочно (с помощью зонда) в дозе 10 мг/кг за 60 мин до инъекции кислоты. В качестве препарата сравнения использовали диклофенак (10 мг/кг). Анальгетический эффект рассчитывали по формулеCo- количество корчей в опытной группе. Полученные данные представлены в табл. 4. Таблица 4 Анальгетическая активность исследуемых соединений общей формулы (I) в дозе 10 мг/кг в тесте "уксусные корчи" (число корчей за 15 мин) Р 0,05 относительно контрольной группыР 0,01 относительно контрольной группы Соединения, соответствующие общей формуле (I), в тесте "корчи" проявляют анальгетическую ак- 17013644 тивность, близкую к диклофенаку и вольтарену - препаратам сравнения (табл. 4), при этом анальгетический эффект большинства соединений составляет от 38 до 68%. Пример 19. Влияние соединений общей формулы (I) на анальгетическое действие трамала и анальгина на модели "уксусные корчи". Исследования проводили в соответствии с методикой, приведенной в примере 18. Таблица 5 Влияние исследуемых соединений общей формулы (I) в дозе 10 мг/кг на анальгетическую активность трамала (10 мг/кг)- статистически значимо относительно контрольной группы, р 0,05- статистически значимо относительно трамала, р 0,05 Согласно данным табл. 5 анальгетический эффект комбинации соединения IX с трамалом достоверно значимо сильнее, чем эффект соединения IX и трамала в отдельности (6,4 2,0 против 24,03,4 и 17,52,3 соответственно). Таблица 6 Влияние соединений общей формулы (I) в дозе 10 мг/кг на анальгетическое действие анальгина (50 мг/кг) в тесте уксусные корчи- статистически значимо относительно контрольной группы, Р 0,05- статистически значимо относительно анальгина, р 0,05 Соединение IX также усиливает обезболивающее действие анальгина (табл. 6). Таким образом, соединение IX в дозе 10 мг/кг при внутрижелудочном введении статистически значимо усиливает анальгетическое действие трамала и потенцирует анальгетическое действие анальгина. Пример 20. Исследование анальгетической активности на модели "горячая пластина". Анальгетическое действие соединений, соответствующих общей формуле (I), изучали на модели"горячая пластина" по методике, представленной в Woolfe G., McDonald A.D. The evaluation of the analgesic action of pethidine hydrochloride (Demerol), J. Pharmacol. Exp. Ther., 1944, v.80, p. 300-307. Тесты проводили на белых беспородных мышах-самцах массой 22-24 г. Животных поодиночке помещали на горячую пластину (фирмы "Ugo Basile"), температура которой оставалась постоянной и равнялась 55 С. Регистрировали время первых проявлений болевой реакции: облизывание лап, подпрыгивание до введения вещества (фоновые показатели) и через 0,5, 1, 2, 3 и 4 ч после введения вещества. Вещества вводили внутрижелудочно (с помощью зонда). Навеску вещества тщательно размешивали в 0,1 мл Твин 80 до получения раствора, затем добавляли физиологический раствор до объема 0,5 мл. Вычисляли среднее латентное время порога болевой чувствительности (ПБЧ, секунды) в каждой группе. Полученные результаты выражали в % от фоновых значений. Анальгетический эффект (%) рассчитывали по формуле А-100% = X,где А - фоновый показатель;X - анальгетический эффект, (%) А = (время через 0,5-4 ч 100%):фоновое время. В качестве препаратов сравнения использовали: анальгин (150 мг/кг, парацетамол (200 мг/кг), кеторол (10 мг/кг).- 18013644 Полученные данные представлены в табл. 7. Таблица 7 Сравнительная оценка анальгетического действия соединений общей формулы (I) в дозе 10 мг/кг и эталонных препаратов - анальгина и парацетамола в тесте "горячая пластина" у мышей по величине латентного периода порога болевой чувствительности (ПБЧ, секунды) Полученные данные показывают, что соединения общей формулы (I) в тесте "горячая пластина" демонстрируют значительную активность, достоверно увеличивая порог болевой чувствительности. При этом сравнимый с эталонными препаратами анальгетический эффект достигается при использовании доз 0,1-10 мг/кг, преимущественно 1-10 мг/кг, которые на один-два порядка ниже дозы препарата сравнения- парацетамола, обладающего болеутоляющим и жаропонижающим действием. Данные, представленные в табл. 7, также показывают, что анальгетический эффект соединений общей формулы (I) в среднем составляет от 50 и максимум до 150%, что можно расценивать как пролонгированный, т.к. он сохраняется в течение длительного времени - в ряде случаев более 4 ч. Таким образом, по степени выраженности обезболивающего эффекта соединения общей формулы (I) сравнимы с известными ненаркотическими анальгетиками (анальгин, парацетамол), а по длительности анальгетического действия превосходят препараты сравнения, причем их действующие дозы оказались на порядок ниже, чем у эталонных ненаркотических анальгетиков. Пример 21. Исследование воздействия соединений общей формулы (I) на каррагениновый отек лапы крыс. Тесты проводили на аутбредных белых крысах самцах массой 250-270 г. Использовали модель каррагенининдуцированного отека по методу, описанному в Winter et al. In: De Rosa M., Giroud J.P., Willoughby D.A. Studies of the mediators of the acute inflammatory response induced in rats in different sites bycarrageenan and turpentine, J. Phamacol., 1971, v.104, p. 15-29. В правую лапу крысы субплантарно вводили 1% раствор каррагенина (SERVA) 0,1 мл. Животные рассаживались в индивидуальные камеры. 1% мазь наносили на лапу непосредственно после введения каррагенина через 1 и 2 ч. Измерение объема лап проводили с помощью плетизмометра (Ugo Basile) через 4 ч после введения каррагенина. Эффект терапевтического воздействия мази оценивали по степени угнетения воспалительной реакции в сравнении с интактной левой лапой данного животного и реакцией лап крыс контрольной (нелеченой) группы. Торможение воспалительной реакции, выраженное в процентах, рассчитывали по формуле Полученные данные представлены в табл. 8. Таблица 8 Влияние соединений общей формулы (I) (1% мазь) на развитие каррагенинового отека лапы крыс (Мm)- 22013644 Результаты, представленные в табл. 8, демонстрируют выраженную локальную противовоспалительную активность соединений общей формулы (I), сопоставимую с активностью эталонного препарата из группы НПВС - индометацина, при этом действующая доза соединения на порядок ниже, чем у препарата сравнения. Пример 22. Исследование ульцерогенного действия соединений общей формулы (I). Тесты проводили на белых аутбредных крысах самках массой 300-320 г. Тестируемые соединения вводили в дозе 30 мк/кг, однократно, внутрижелудочно крысам, лишенным пищи на 24 ч до опыта. Животным в контрольной группе вводили дистилированную воду в том же объеме. Через 24 ч животных забивали, извлекали желудки. В пустой желудок вводили 2% раствор формалина и помещали его в стакан с формалином. Через 30 мин вскрывали желудок по большой кривизне, расправляли на предметном столике, фиксировали и промывали дистилированной водой. С помощью лупы МБС-9 (8-кр. ув.) измеряли длину и ширину дефектов слизистой желудка и подсчитывали общую площадь в мм 2 (1 деление линейки лупы = 0,1 мм). Ульцерогенный эффект вещества оценивали по площади язвенного поражения слизистой желудка по методике, представленной в Руководстве по экспериментальному (доклиническому) изучению новых фармакологических веществ. - М.: Ремедиум, 2000, 398 с. Полученные данные представлены в табл. 9. Таблица 9 Сравнительное изучение влияния соединений общей формулы (I) и индометацина в дозе 30 мг/кг на слизистую оболочку желудка крыс (Mm) Полученные данные показывают, что при внутрижелудочном введении соединений общей формулы (I) в дозе 30 мг/кг язвенное поражение слизистой оболочки желудка крыс отсутствует. Пример 23. Исследование спазмолитического действия соединений общей формулы (I). Модель серотонининдуцированной контрактуры гладких мышц рога матки [Blattner R., ClassenH.G., Dehnert H. et al. Experiments on isolated smooth muscle preparation, Ed. J.M. Barnden a. R.Colson,1980] создавали на самках крыс Вистар массой 300-350 г. Подготовленный гладкомышечный препарат(ГМП) помещали в термостатируемую камеру (+37 С), содержащую раствор Тироде с пониженным содержанием кальция с целью предупреждения спонтанной сократительной активности гладкомышечного препарата. Сокращение рога матки регистрировали с помощью механотрона 6 М 2 Б, соединенного с самописцем КПС-4, исходная нагрузка на объект составляла 0,5-0,7 г. Контрактуру ГМП вызывали введением в инкубационную среду 0,1 мл серотонина (Sigma) в концентрации 10-5 М. Через 30-60 с после введения медиатора регистрировали максимальную амплитуду сокращения рога матки. Соединение IX вносили в камеру кумулятивно на высоте контрактуры гладкой мышцы в концентрации 10-4 М в объеме 0,1 мл на высоте амплитуды сокращений или в условиях инкубации (в том же диапазоне концентраций) в течение 15 мин. Эффект исследуемого соединения IX оценивали по количеству сокращений и снижению величины амплитуды. Результаты представлены в табл. 10. Таблица 10 Влияние соединений общей формулы (I) на серотонининдуцированную контрактуру ГМП рога матки крысы При добавлении соединения IX на высоте серотонининдуцированной контрактуры ГМП рога матки происходило урежение частоты сокращений (в течение 5 мин) гладкомышечного препарата рога матки крысы: с 9 сокращений в контроле до 5 сокращений в опыте (табл. 10). В условиях преинкубации соединения IX также наблюдалось урежение сокращений в ответ на серотонин (4 сокращения за 5 мин) с последующей полной блокадой контрактуры ГМП (табл. 10).- 23013644 Таким образом, в условиях in vitro соединение IX проявляет спазмолитическое действие (на высоте сокращения ГМП, вызванного серотонином) и тормозит развитие контрактуры ГМП при превентивном введении. Можно предположить, что соединение IX является блокатором серотониновых рецепторов и таким образом оказывает антидепрессивное действие. Пример 24. Исследование антигипоксического действия соединений общей формулы (I). Для воспроизведения острой кислородной недостаточности использовали модель гипоксии с гилеркапнией в гермообъеме [Лукьянова Л.Д., Гацура В.В., Пастушенков Л.В. Методические рекомендации по экспериментальному изучению препаратов, предлагаемых для клинического изучения в качестве антигипоксических средств. - М., 1960, с. 1-19.]. Мышей-самцов массой 27-29 г поодиночно сажали в стеклянные банки объемом 260 мл, которые герметично закрывали. По мере потребления кислорода животным его концентрация в сосуде снижается, что приводит к гибели животных. Регистрировали длительность жизни мышей в минутах. Полученные данные представлены в табл. 11. Таблица 11 Влияние соединений общей формулы (I) на длительность жизни мышей на модели гипоксической гипоксии с гиперкапнией- Р 0,05; статистически значимо относительно контрольной группы Соединение IX в дозе 50 мг/кг достоверно увеличивает длительность жизни мышей, находящихся в состоянии гипоксической гипоксии на 27,3%. Результаты эксперимента свидетельствуют, что соединение IX проявляет антигипоксическое действие. Пример 25. Исследование антидепрессивного действия соединений общей формулы (I) в тесте поведенческого отчаяния (влияние на длительность иммобилизации). Тест поведенческого отчаяния по Porsolt [Porsolt R.D., Bertin A. and Jalfre M. Behavioral Despair inmise: A primary-Screening Test for Antidepressants, Arch int. Pharmfcodyn., 1977, 229, p. 327-336] является прогностическим тестом для препаратов с антидепрессивным действием. Стрессовое состояние вызывают у мышей (масса 27-30 г) форсированным плаванием. Животных помещают в цилиндр (высота - 25 см, диаметр - 10 см), заполненный на 1/3 водой при температуре 21-23 С. Животные самостоятельно не могут выбраться из цилиндра. После короткого времени активности у животных наступает так называемая поведенческая депрессия, характеризующаяся зависанием животных, иммобилизацией, время которой можно фиксировать. Эксперимент проводится в два дня. В первый день животных помещают в цилиндр на 15 мин (претест). После выемки из воды животных обсушивают и вводят исследуемые препараты. Через 24 ч опять вводят препараты и через 1 ч животных помещают в цилиндр на 6 мин. Впервые 2 мин животные активно плавают; в последующие 4 мин развивается поведенческая депрессия, проявляющаяся в иммобилизации (зависании), которая фиксируется в течение 4 мин и измеряется в секундах. Соединение IX и препарат сравнения антидепрессант флуоксетин вводили внутрь в дозе 50 мг/кг. Полученные данные представлены в табл. 12. Таблица 12 Влияние соединения IX и флуоксетина на время иммобилизации в тесте поведенческого отчаяния- Р 0,05- Р 0,01 - статистически значимо относительно контрольной группы В тесте поведенческого отчаяния соединение IX так же, как и антидепрессант - флуоксетин, статистически значимо уменьшает время иммобилизации у мышей.- 24013644 Таким образом, в спектре действия соединения IX обнаружен фармакологический эффект (уменьшение время иммобилизации в тесте поведенческого отчаяния по Porsolt), характерный для препаратов группы антидепрессантов. Примеры стандартных лекарственных форм Пример 26. А. Таблетированная форма. Таблетированную форму получают, используя приведенные ниже ингредиенты Компоненты смешивают и прессуют для образования таблеток весом 300 мг каждая. Б. Суппозитории. Пример состава суппозитория При необходимости возможно изготовление ректальных, вагинальных и уретральных суппозиториев с соответствующими наполнителями. Таким образом, настоящее изобретение относится к новым соединениям общей формулы (I), простым и препаративным способам синтеза новых и известных соединений и их применению в качестве нестероидных противовоспалительных средств, ингибиторов циклооксигеназы, обладающих противовоспалительным и преимущественным анальгетическим действием, не проявляющих побочного ульцерогенного эффекта. Результаты фармакологических исследований свидетельствуют о том, что заявленные соединения обладают уникальной способностью оказывать лечебный эффект при экстремальных воздействиях: эмоциональном стрессе, болевом синдроме, гипоксии, воспалении, спазмах, а также купировать нарушения,обусловленные болезнью Паркинсона, а также потенцировать действие других анальгетиков. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Фенилсодержащие N-ацильные производные биогенных аминов общей формулы (I) где R5 представляет водород или гидроксильную группу; R2 представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН или где R7 представляет водород или гидроксильную группу, R4 представляет водород, гидроксильную группу; при условии, что соединение общей формулы (I) не является фенилацетилтирамином,3-(п-гидроксифенил)пропионилтирамином,3-фенилпропионилфенилэтиламином,3-фенилпропионилтирамином,3-(п-гидроксифенил)пропионилфенилаланина метиловым эфиром,3-(п-гидроксифенил)пропионилтирозином,3-фенилпропионилтирозином,фенилацетилтирозином,3-(п-гидроксифенил)пропионилфенилаланином,3-фенилпропионилфенилаланином,фенилацетилфенилаланином,3-(п-гидроксифенил)пропионилтирозина метиловым эфиром,3-фенилпропионилтирозина метиловым эфиром,фенилацетилтирозина метиловым эфиром,фенилацетилфенилэтиламином,3-фенилпропионилфенилаланина метиловым эфиром,фенилацетилфенилаланина метиловым эфиром,3-(п-гидроксифенил)пропионилтирозина бензиловым эфиром,3-фенилпропионилтирозина бензиловым эфиром,фенилацетилтирозина бензиловым эфиром,тирозилтирозином,фенилаланилтирозином,фенилацетилфенилаланина бензиловым эфиром,или их фармацевтически приемлемые соли. 2. Соединение по п.1, в котором R3 представляет -СООН, -СООСН 3. 3. Соединение по п.1, выбранное из п-гидроксифенилацетилтирозина,п-гидроксифенилацетилфенилаланина,п-гидроксифенилацетилтирозина метилового эфира,п-гидроксифенилацетилфенилаланина метилового эфира,п-гидроксифенилацетилтирозина бензилового эфира,п-гидроксифенилацетилфенилаланина бензилового эфира,N-ацетилтирозилфенилэтиламина,N-ацетилтирозилтирамина,п-гидроксифенилацетилтирамина,п-гидроксифенилацетилфенилэтиламина,3-(п-гидроксифенил)пропионилфенилаланина бензиловым эфиром,3-фенилпропионилфенилаланина бензиловым эфиром,или их фармацевтически приемлемых солей. 4. Способ получения соединений общей формулы (I) где R5 представляет водород или гидроксильную группу; R2 представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН,-COOR6, где R6 представляет C1-С 6-алкил или где R7 представляет водород или гидроксильную группу; R4 представляет водород, гидроксильную группу; включающий активацию карбоксильной группы соединения общей формулы взаимодействием с дифенилфосфорилазидом и триэтиламином в органическом растворителе при охлаждении с последующим взаимодействием с аминосоединением общей формулы где R1-R4 принимают значения, определенные для соединений общей формулы (I). 5. Способ по п.4, в котором используют 1-1,2 экв. дифенилфосфорилазида и триэтиламина. 6. Способ по п.4 или 5, в котором в качестве аминопроизводных используют эфир тирозина или фенилаланина. 7. Способ по любому из пп.4-6, в котором в качестве органического растворителя используют N,Nдиметилформамид или этилацетат. 8. Способ по любому из пп.4-6, который осуществляют в интервале температур от -25 до 0 С. 9. Способ получения соединений по п.1 или их фармацевтически приемлемых солей, включающий превращение п-гидроксифенилуксусной кислоты, фенилуксусной кислоты или N-замещенного тирозина общей формулы в активированный N-оксисукцинимидный эфир общей формулы где R1-R4 принимают значения, определенные для соединений общей формулы (I) в п.1. 10. Способ по п.9, в котором аминопроизводным является эфир тирозина или фенилаланина. 11. Фармацевтическая композиция, обладающая способностью ингибировать циклооксигеназу спазмолитическими, антигипоксическими, антидепрессантными и противопаркинсоническими свойствами, а также способностью потенцировать действие анальгетиков, включающая в качестве активного агента эффективное количество соединения общей формулы (I) где R5 представляет водород или гидроксильную группу; R2 представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН,-COOR6, где R6 представляет C1-С 6-алкил или где R7 представляет водород или гидроксильную группу; R4 представляет водород, гидроксильную группу,или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель. 12. Фармацевтическая композиция, обладающая анальгетическими, противовоспалительными свойствами при отсутствии ульцерогенности, включающая в качестве активного агента эффективное количество соединения общей формулы (I) где R5 представляет водород или гидроксильную группу; R2 представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН,-COOR6, где R6 представляет C1-С 6-алкил или где R7 представляет водород или гидроксильную группу; R4 представляет водород, гидроксильную группу; при условии, что соединение общей формулы (I) не является фенилацетилтирамином,3-(п-гидроксифенил)пропионилтирамином,3-фенилпропионилфенилэтиламином,3-фенилпропионилтирамином,3-фенилпропионилтирозина метиловым эфиром,или его фармацевтически приемлемые соли, а также в сочетании с другими анальгетиками для потенцирования эффекта. 13. Лекарственное средство, обладающее способностью ингибировать циклооксигеназу спазмолитическими, антигипоксическими, антидепрессантными и противопаркинсоническими свойствами, а также способностью потенцировать действие анальгетиков, включающее соединение общей формулы (I) где R5 представляет водород или гидроксильную группу; R2 представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН,-COOR6, где R6 представляет C1-С 6-алкил или где R7 представляет водород или гидроксильную группу; R4 представляет водород, гидроксильную группу; или его фармацевтически приемлемые соли. 14. Лекарственное средство, обладающее анальгетическими, противовоспалительными свойствами при отсутствии ульцерогенности, включающее соединение общей формулы (I) по п.1 где R5 представляет водород или гидроксильную группу; R2 представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН,-COOR6, где R6 представляет C1-C6-алкил или где R7 представляет водород или гидроксильную группу; R4 представляет водород, гидроксильную группу; при условии, что соединение общей формулы (I) не является фенилацетилтирамином,3-(п-гидроксифенил)пропионилтирамином,3-фенилпропионилфенилэтиламином,3-фенилпропионилтирамином,3-фенилпропионилтирозина метиловым эфиром,или его фармацевтически приемлемые соли, а также сочетание с другими анальгетиками для потенцирования эффекта. 15. Применение соединений общей формулы (I) где R5 представляет водород или гидроксильную группу; R2 представляет водород или аминогруппу, необязательно замещенную группой СН 3 (CH2)mCO-, где m=0-4; R3 представляет водород, -СООН,-COOR6, где R6 представляет C1-С 6-алкил или где R7 представляет водород или гидроксильную группу; R4 представляет водород, гидроксильную группу; или их фармацевтически приемлемых солей для получения лекарственного средства, обладающего способностью ингибировать циклооксигеназу, спазмолитическим, антигипоксическим, антидепрессантным и противопаркинсоническим действием, а также способностью потенцировать действие анальгетиков. 16. Применение соединений общей формулы (I) где R5 представляет водород или гидроксильную группу; R2 представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН,-COOR6, где R6 представляет C1-С 6-алкил или где R7 представляет водород или гидроксильную группу; R4 представляет водород, гидроксильную группу,при условии, что соединение общей формулы (I) не является фенилацетилтирамином,3-(п-гидроксифенил)пропионилтирамином,3-фенилпропионилфенилэтиламином,3-фенилпропионилтирамином,3-фенилпропионилтирозина метиловым эфиром,или их фармацевтически приемлемые соли для получения анальгетических, противовоспалительных лекарственных средств, не обладающих ульцерогенностью, а также в сочетании с другими анальгетиками для потенцирования эффекта. 17. Способ лечения болевых синдромов различного генеза, воспалительных и воспалительнодегенеративных заболеваний суставов и соединительной ткани, костно-мышечной системы, заболеваний,сопровождающихся воспалением, а также послеоперационной боли, посттравматической боли, болевых синдромов гинекологической, неврологической, онкологической, стоматологической природы, ревматоидного артрита, артропатии, болезни Бехтерева, неспецифических спондиллоартритов, подагрического артрита, остеоартроза, внесуставного ревматизма и тромбофлебита, включающий введение млекопитающему эффективного количества соединения общей формулы (I) где R5 представляет водород или гидроксильную группу; R2 представляет водород или аминогруппу, необязательно замещенную группой CH3(CH2)mCO-, где m=0-4; R3 представляет водород, -СООН,-COOR6, где R6 представляет С 1-С 6-алкил или где R7 представляет водород или гидроксильную группу; R4 представляет водород, гидроксильную группу; при условии, что соединение общей формулы (I) не является фенилацетилтирамином,3-(п-гидроксифенил)пропионилтирамином,3-фенилпропионилфенилэтиламином,3-фенилпропионилтирамином,3-фенилпропионилтирозина метиловым эфиром,или его фармацевтически приемлемой соли, а также в сочетании с другими анальгетиками для потенцирования эффекта. 18. Способ лечения заболеваний, сопровождающихся спазмами, депрессией, гипоксией, явлениями Паркинсонизма, а также эмоционально-стрессовых состояний и нарушений, вызываемых спазмами, гипоксией и сопровождающих болезнь Паркинсона, включающий введение млекопитающему эффективного количества соединения общей формулы (I)
МПК / Метки
МПК: C07K 5/065, A61P 29/00, C07C 237/20, A61K 38/05, A61K 31/165, C07C 233/51, C07C 237/22, A61P 25/16
Метки: производные, фармацевтическая, применение, композиция, аминов, аминокислот, получения, способ, n-ацильные, фенилсодержащие
Код ссылки
<a href="https://eas.patents.su/30-13644-fenilsoderzhashhie-n-acilnye-proizvodnye-aminov-i-aminokislot-sposob-ih-polucheniya-farmacevticheskaya-kompoziciya-i-ih-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Фенилсодержащие n-ацильные производные аминов и аминокислот, способ их получения, фармацевтическая композиция и их применение</a>
N-ацильные производные аминокислот, способ их получения, фармацевтическая композиция и применение в качестве противоаллергических, противовоспалительных и гиполипидемических средств

Номер патента: 13057
Опубликовано: 26.02.2010
Авторы: Кромова Татьяна Александровна, Желтухина Галина Александровна, Небольсин Владимир Евгеньевич, Ковалева Виолетта Леонидовна
МПК: A61P 11/00, A61K 31/4172, A61K 31/405...
Метки: фармацевтическая, средств, противовоспалительных, композиция, применение, производные, качестве, способ, аминокислот, получения, n-ацильные, противоаллергических, гиполипидемических
Формула / Реферат:
1. N-Ацильные производные аминокислот общей формулы (I)где n равно 2 или 3 иR1 представляетR2=Н, -СН3, -С2Н5,и их фармацевтически приемлемые соли,при условии, что соединение общей формулы (I) не является Na-сукцинил-L-гистидином, сукцинил-L-триптофаном, сукцинил-D-триптофаном и сукцинил-D,L-триптофаном и его дикалиевой солью, Na-сукцинил-L-триптофаном метиловым эфиром, Na-глутарил-L-гистидином метиловым эфиром, Na-глутарил-L-триптофаном...
Диарильные производные, способы их получения, применение, фармацевтическая композиция и способ лечения

Номер патента: 3641
Опубликовано: 28.08.2003
Авторы: Доналдсон Келли Хорн, Ширер Барри Джордж, Юлинг Дейвид Эдуард
МПК: C07C 255/53, A61P 9/10, A61K 31/455...
Метки: производные, диарильные, способ, композиция, способы, лечения, фармацевтическая, применение, получения
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемое производное где R1 представляет собой феноксиметил или фенил, замещенный хлором, фтором или бромом, метилом или трифторметилом; R2 представляет собой водород или C1-6алкил; X представляет собой кислород или -NH; R3 представляет собой циано, тетразол-5-ил или -CO2R7, где R7 представляет собой водород или C1-6алкил; R4 и R5 независимо представляют собой водород, метил,...
Производные бензоконденсированных гетероциклических оксимов, способ их получения, содержащие их фармацевтическая композиция и комбинация и их применение

Номер патента: 12437
Опубликовано: 30.10.2009
Авторы: Кторза Ален, Кэньяр Даньель-Анри, Леклерк Вероник, Лебеге Никола, Карато Паскаль, Юртеван Орели, Бертоло Паскаль, Даке Катрин, Л'эльгуаль'ш Жан-Марсьяль
МПК: A61P 3/06, A61K 31/5415, A61K 31/538...
Метки: применение, фармацевтическая, комбинация, гетероциклических, получения, способ, оксимов, бензоконденсированных, композиция, содержащие, производные
Формула / Реферат:
1. Соединения формулы (I) в которой X представляет собой атом кислорода или атом серы, А представляет собой (С1-С6)алкиленовую цепь, в которой СН2 группа необязательно может быть заменена атомом кислорода, R1 представляет собой арильную группу, a R2 представляет собой атом водорода либо линейную или разветвленную (С1-С6)алкильную группу, R3 и R4 каждый представляет собой атом водорода, В представляет собой линейную или разветвленную...
Пирролидинил-, пиперидинил- или гомопиперидинилзамещенные производные, способ их получения, их применение в медицине, содержащая их фармацевтическая композиция и способ ее получения

Номер патента: 4648
Опубликовано: 24.06.2004
Авторы: Вирсхюэрен Вим Гастон, Вигеринк Пит Том Берт Поль, Де Брюин Марсель Франс Леопольд, Ван Эмелен Кристоф
МПК: C07D 405/14, A61K 31/505, A61P 9/00...
Метки: содержащая, производные, применение, гомопиперидинилзамещенные, способ, композиция, пирролидинил, фармацевтическая, медицине, пиперидинил, получения
Формула / Реферат:
1. Соединение формулы (I) его стереохимически изомерная форма или его фармацевтически приемлемая кислотно-аддитивная соль, в которых R1, R2 и R3 каждый представляет водород; -Z1-Z2- представляет двухвалентный радикал формулы -O-CH(R4)-CR2-CH2- (a-4), R4 представляет водород; Alk представляет C1-6алкандиил, необязательно замещенный гидроксигруппой; представляет двухвалентный радикал формулы в которой m равно 0 или 1; R6...
Производные 4-(3-трифторметилпиридин-5-ил)пиперазина, фармацевтическая композиция, способы их получения и применение.

Номер патента: 7543
Опубликовано: 27.10.2006
Авторы: Рингберг Эрик, Нильссон Бьерн
МПК: A61P 13/02, A61K 31/444, A61K 31/496...
Метки: получения, фармацевтическая, производные, композиция, 4-(3-трифторметилпиридин-5-ил)пиперазина, способы, применение
Формула / Реферат:
1. Соединение формулы (I) где R1 выбран из Н, C1-4-алкила, 2-гидроксиэтила, 2-цианоэтила, тетрагидропиран-2-ила и азотозащитной группы; R2 и R3 каждый независимо представляет Н или СН3; R4 выбран из O-R5, NH-R5 или S-R5, где R5 выбран из арила, арил-С1-6-алкила, арилокси-С2-6-алкила, гетероарила, гетероарил-С1-6-алкила, гетероарилокси-С2-6-алкила, С3-6-циклоалкила, С3-6-циклоалкил-С1-4-алкила, C1-6-алкила, 2-тетрагидрофурила,...
Предыдущий патент: Фунгицидная смесь на базе протиоконазола с тиаметоксамом
Следующий патент: Твердая фармацевтическая композиция с пролонгированным высвобождением 1-(2,3,4-триметоксибензил)пиперазина и способ ее получения
Случайный патент: Съемная опалубка