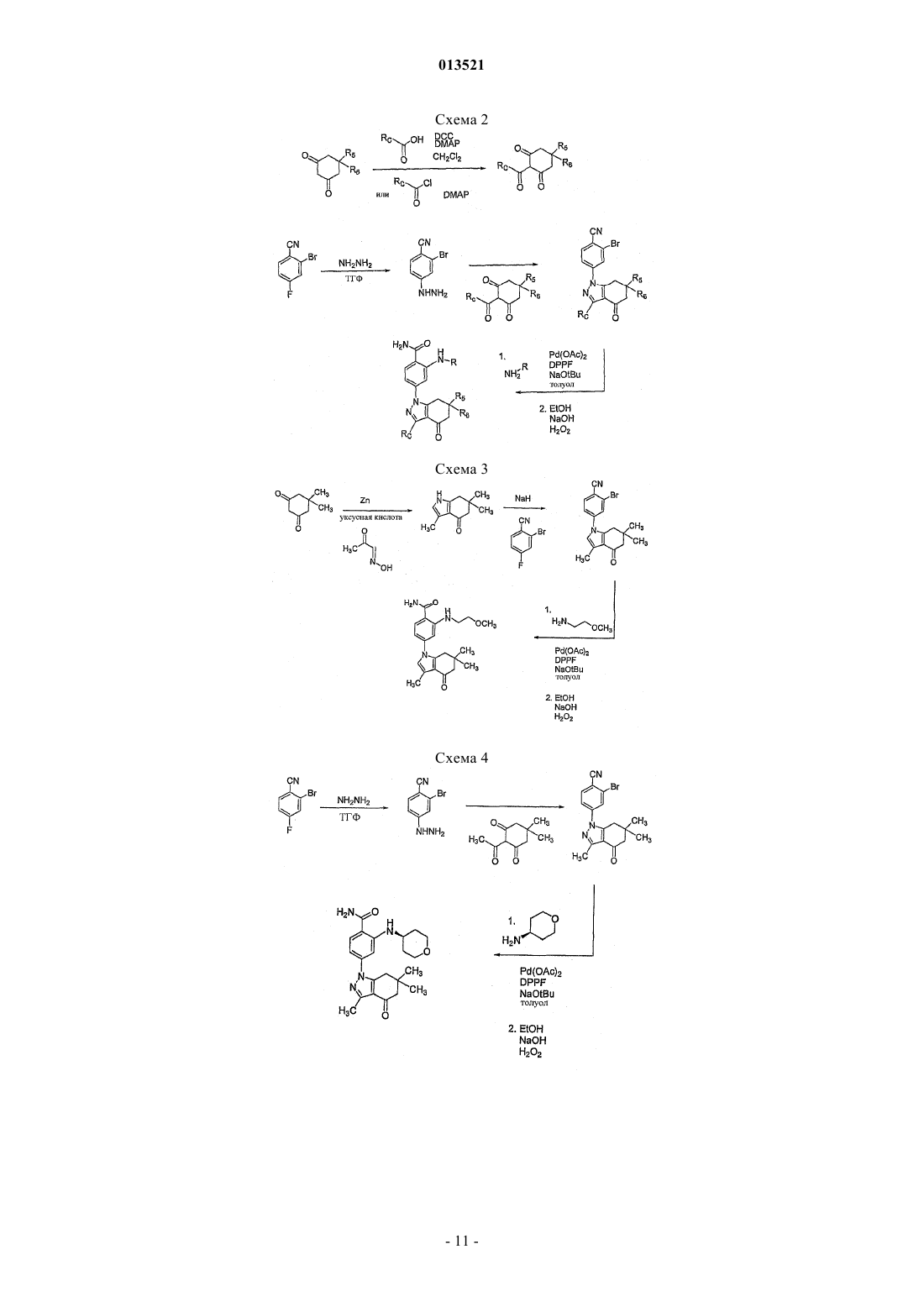
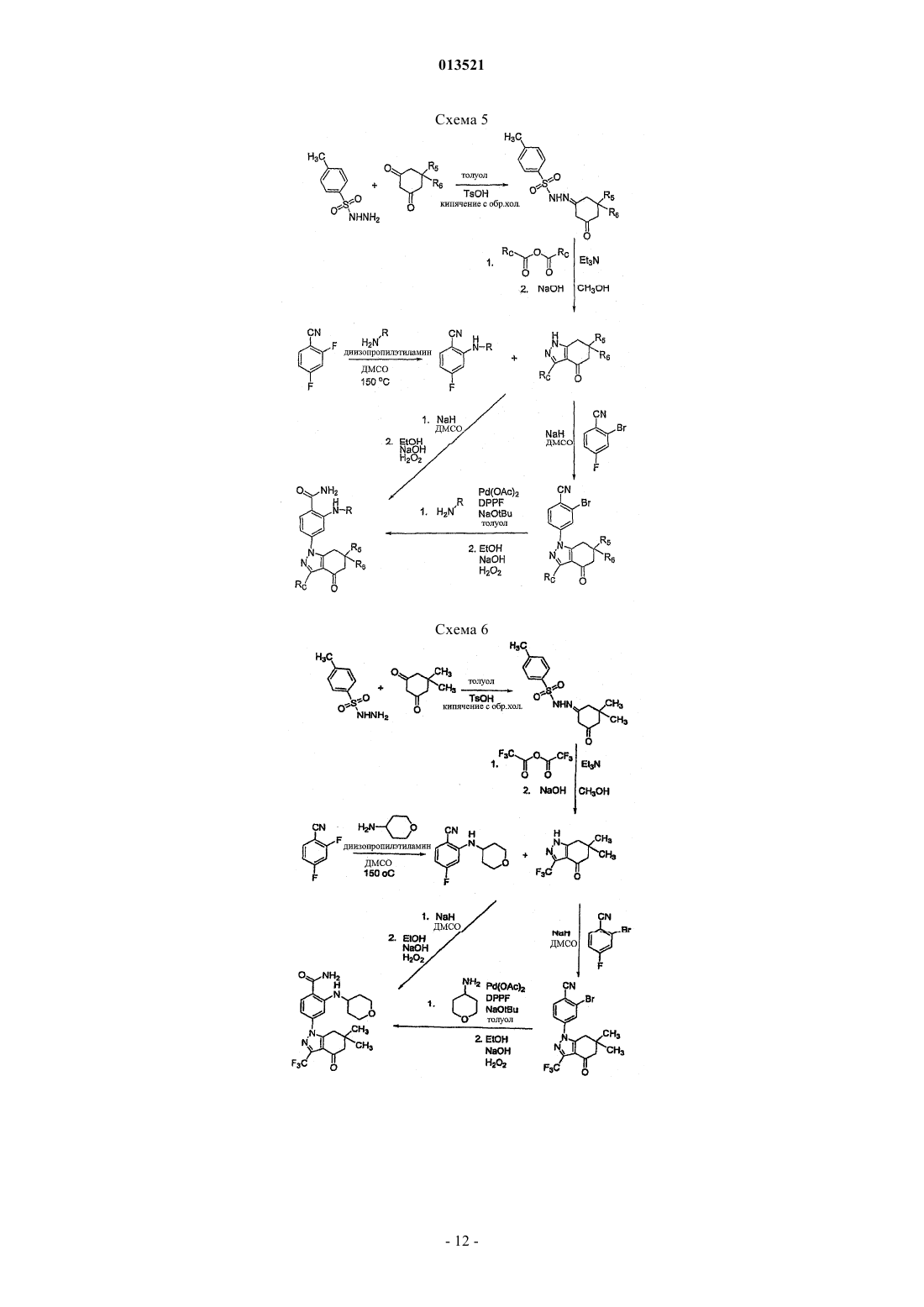
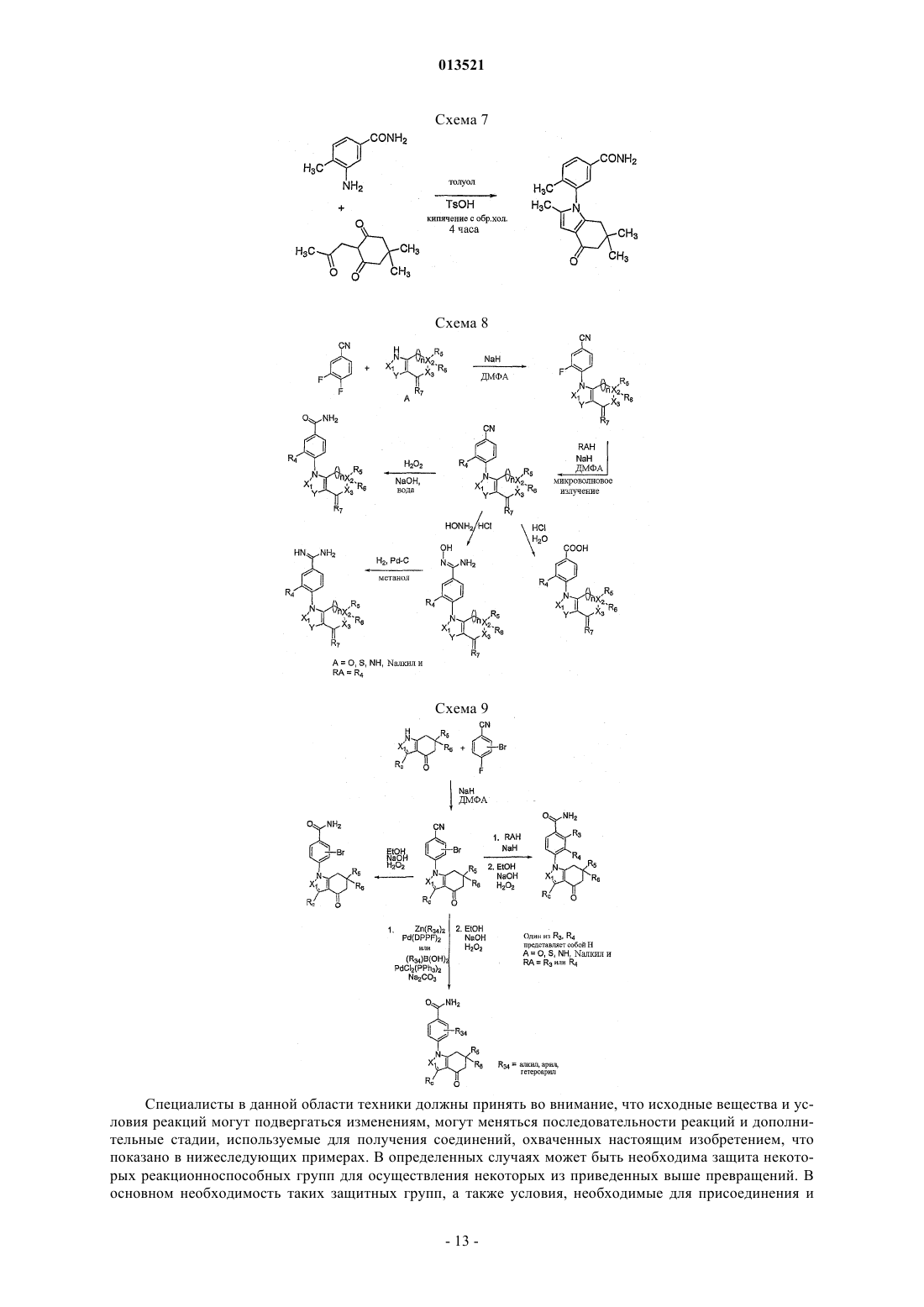
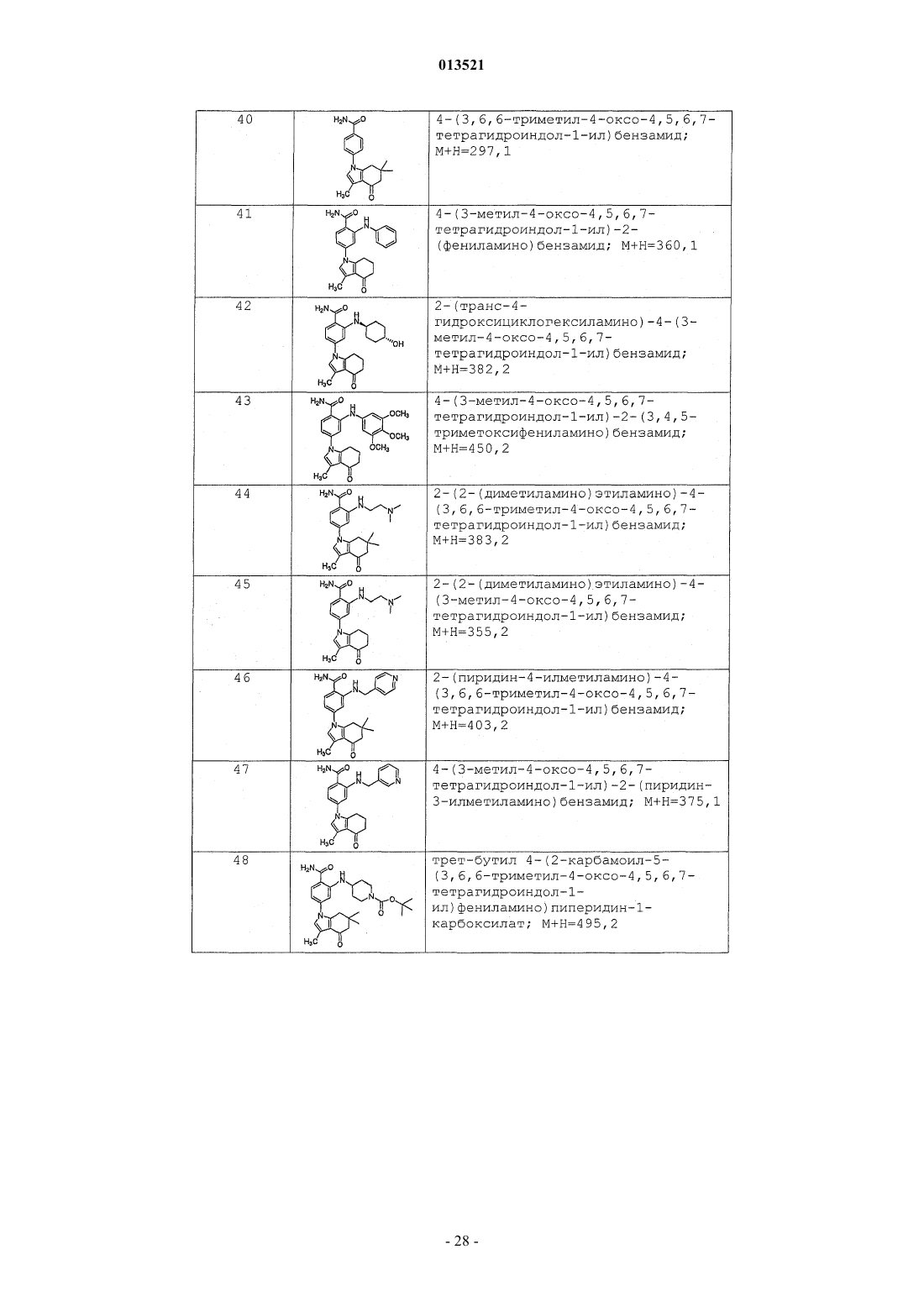
Производные тетрагидроиндазолона
Номер патента: 13521
Опубликовано: 30.06.2010
Авторы: Барта Томас, Хуан Кеннет Хэ, Хинкли Линдзи, Ивз Джерон, Хансон Гуннар Дж., Гэн Лифэн, Вил Джеймс

























Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемая соль,
где R4представляет собой Н;
R3 представляет собой:
(a) галоген или
(b) С1-С15-алкильную группу, причем до шести атомов углерода в указанной алкильной группе необязательно независимо заменены R22, карбонилом, этенилом, этинилом или фрагментом, выбранным из N, О, S, SO2 или SO, при условии, что два атома О, два атома S или атомы О и S не находятся в непосредственном соседстве друг с другом,
где R22 представляет собой:
(i) 5-10-членный гетероарил, содержащий 1-3 гетероатома, выбранных из группы, включающей N, О и S;
(ii) С6-С10-арил,
(iii) насыщенный или ненасыщенный С3-С10-циклоалкил или
(iv) насыщенный или ненасыщенный C2-С10-гетероциклоалкил, содержащий 1-3 гетероатома, выбранных из группы, включающей N, О и S, где каждый арил, гетероарил, насыщенный или ненасыщенный циклоалкил или насыщенный или ненасыщенный гетероциклоалкил независимо необязательно замещен по меньшей мере одной группой, которая независимо представляет собой гидрокси, галоген, амино, циано, карбокси, карбоксамидо, нитро, оксо, -S-(C1-C6)алкил, -SO2-(C1-C6)алкил, -SO2-(С6-С10)арил, -SO-(C1-C6)алкил, -SO-(C6-С10)арил, -SO2NH2, -SO2NH-(C1-C6)алкил, -SO2NH-(С6-С10)арил, (C1-С6)алкокси или моно- или ди(C1-С10)алкиламино; и
каждый из R22 необязательно конденсирован с С6-С10арильной группой, С5-С8 насыщенной циклической группой или С5-С10 гетероциклоалкильной группой, содержащей 1-3 гетероатома, выбранных из группы, включающей N, О и S;
где каждая из групп (b) необязательно замещена по любому доступному положению C1-С10-алкилом, C1-С10-галогеналкилом, С2-С10-алкенилом, С2-С10-алкинилом, гидрокси, карбокси, карбоксамидо, оксо, галогеном, амино, циано, нитро, -SH, -S-(C1-С6)алкилом, -SO2-(C1-C6)алкилом, -SO2NH2, -SO2NH-(C1-C6)алкилом,
-SO2NH-(C6-С10)арилом, -SO-(C1-С6)алкилом, -SO2-(С6-С10)арилом, C1-С6-алкокси, С2-С10-алкенилокси, С2-С10-алкинилокси, моно- или ди(С1-С10)алкиламино, -OC1-С10-алкил-Z или R23,
где Z представляет собой OR0или -N(R30)2,
где каждый из R30 независимо представляет собой -Н или C1-С6-алкил или -N(R30)2 представляет собой пирролидинил, пиперидинил, пиперазинил, азепанил, 1,3- или 1,4-диазепанил или морфолинил, каждый из которых необязательно замещен гидрокси, амино, аминоалкилом, С1-С6-алкилом, моно- или ди(С3-С6)алкиламино, C1-C6-алкокси или галогеном;
R0 представляет собой -Н, C1-С10-алкил, С2-С10-алкенил, С2-С10-алкинил, С6-С10-арил, 5-10-членный гетероарил, содержащий 1-3 гетероатома, выбранных из группы, включающей N, О и S, или C1-C6-ацил;
R23 представляет собой:
(1) 5-10-членный гетероарил, содержащий 1-3 гетероатома, выбранных из группы, включающей N, О и S,
(2) С6-С10-арил,
(3) насыщенный или ненасыщенный C5-С10-циклоалкил или
(4) насыщенный или ненасыщенный С5-С10-гетероциклоалкил, содержащий 1-3 гетероатома, выбранных из группы, включающей N, О и S, и
группы R23 необязательно замещены по меньшей мере одной группой, которая независимо представляет собой гидрокси, оксо, галоген, амино, циано, нитро, -SH, -S-(C1-C6)алкил, -SO2-(C1-С6)алкил, -SO2-арил,
-SO-(C1-C6)алкил, -SO-(С6-С10)арил, -SO2NH2, -SO2NH-(C1-С6)алкил, -SO2NH-(С6-С10)арил, (C1-C6)алкокси или моно- или ди(C1-C10)алкиламино;
R7 представляет собой О, N-OH, N-O-(С0-С6)алкил-R122, где R122 представляет собой C1-С6-алкил или фенил, или N-(C1-С6-алкенокси);
Rc представляет собой водород, галоген, циано, C1-С10-алкил, С2-С10-алкенил, С2-С10-алкинил, C1-С10-галогеналкил, С3-С7-циклоалкил, С3-С7-циклоалкил(C1-С10)алкил;
каждый RQ независимо представляет собой водород, галоген, -N(RCN)2, C1-С6-алкил, C1-С6-галогеналкил или С3-С7-циклоалкил;
каждый из RCN независимо представляет собой -Н, -С1-С10-алкил, -C1-С10-галогеналкил или -С3-С7-циклоалкил,
R21 представляет собой группу формулы

где R1и R2 независимо представляют собой Н, гидрокси или C1-C6-алкил,
Х4 представляет собой О или NOH;
R5 и R6независимо представляют собой Н, C1-С6-алкил или С6-С10-арил, где арил необязательно замещен 1-4 группами, которые независимо представляют собой C1-C6-алкил, C1-C6-алкокси, галоген, гидрокси, амино, моно- или ди(C1-С6)алкиламино, нитро, галоген(C1-С6)алкил, галоген(C1-С6)алкокси или карбоксамид,
где любые два заместителя в соседних положениях в арильном фрагменте совместно с атомами углерода, к которым они присоединены, образуют ненасыщенный С3-С8-циклоалкил или С3-С7-гетероциклоалкил, содержащий 1-3 гетероатома, выбранных из группы, включающей N, О и S; или
R5 и R6совместно с атомом углерода, к которому они присоединены, образуют 3-8-членный цикл и
R9a и R9b независимо представляют собой Н, C1-C6-алкил или моно- или ди(C1-C6)алкиламино(C1-С6)алкил.
2. Соединение или соль по п.1,
где R3представляет собой галоген или Z1RZ1,
где Z1представляет собой -O-, -NH-, -S(O)p- или -S(O)2NH-,
где р означает 0, 1 или 2; и
RZ1 представляет собой С1-С14 алкильную группу, в которой до пяти атомов углерода необязательно независимо заменены R22, карбонилом, этенилом, этинилом или фрагментом, выбранным из N, О, S, SO2 или SO, при условии, что два атома О, два атома S или атомы О и S не соседствуют друг с другом непосредственно,
где фрагмент RZ1 необязательно замещен по любому доступному положению C1-С10-алкилом, C1-С10-галогеналкилом, C2-С10-алкенилом, C2-С10-алкинилом, гидрокси, карбокси, карбоксамидо, оксо, галогеном, амино, циано, нитро, -SH, -S-(C1-С6)алкилом, -SO2-(C1-С6)алкилом, -SO2NH2, -SO2NH-(C1-С6)алкилом,
-SO2NH-арилом, -SO-(C1-С6)алкилом, -SO2-арилом, C1-С6-алкокси, C2-С10-алкенилокси, C2-С10-алкинилокси, моно- или ди(C1-С10)алкиламино, -OC1-C10-алкил-Z или R23.
3. Соединение или соль по п.2,
где R7представляет собой О;
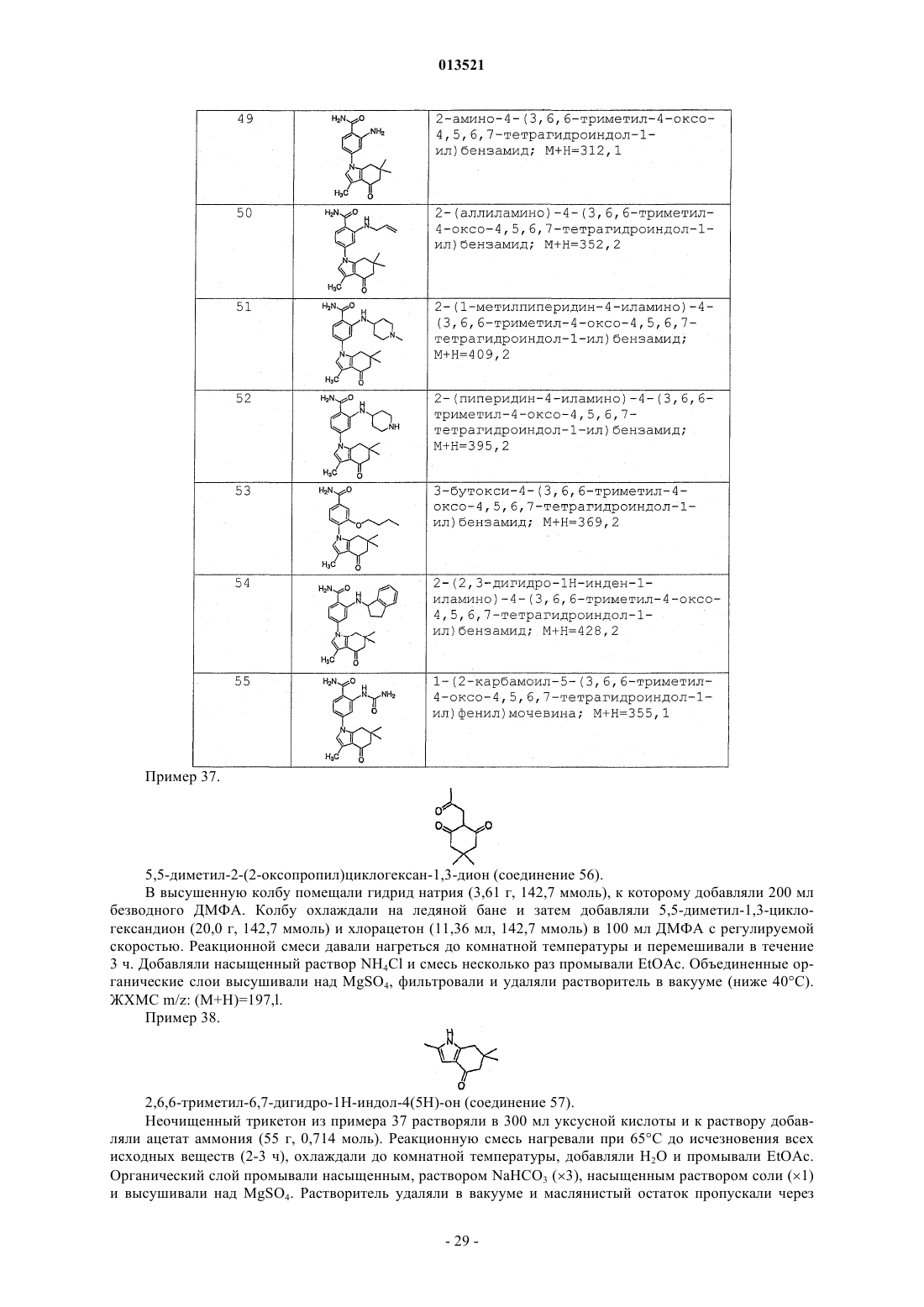
Rc представляет собой -Н, -СН3, этил, циклопропил или -CF3.
4. Соединение или соль по п.1,
где R5и R6 независимо представляют собой Н или C1-С6-алкил.
5. Соединение или соль по п.1,
где каждый RQ независимо представляет собой Н или C1-С6-алкил.
6. Соединение или соль по п.1 формулы

7. Соединение или соль по п.6,
где R3представляет собой галоген или -Z1RZ1,
где Z1представляет собой -O-, -NH-, -S(O)p- или -S(O)2NH-,
где р означает 0, 1 или 2, и
RZ1 представляет собой С1-С14алкильную группу, в которой до пяти атомов углерода необязательно независимо заменены R22, карбонилом, этенилом, этинилом или фрагментом, выбранным из N, О, S, SO2или SO, при условии, что два атома О, два атома S или атомы О и S не соседствуют друг с другом непосредственно,
где фрагмент RZ1 необязательно замещен по любому доступному положению C1-С10-алкилом, C1-С10-галогеналкилом, С2-С10-алкенилом, C2-С10-алкинилом, гидрокси, карбокси, карбоксамидо, оксо, галогеном, амино, циано, нитро, -SH, -S-(C1-С6)алкилом, -SO2-(C1-C6)алкилом, -SO2NH2, -SO2NH-(C1-C6)алкилом,
-SO2NH-арилом, SO2-арилом, -SO-(С1-С6)алкилом, -SO2-арилом, C1-С6алкокси, С2-С10алкенилокси, C2-С10алкинилокси, моно- или ди(C1-С10)алкиламино, -OC1-C10-алкил-Z или R23.
8. Соединение или соль по п.6,
где R3представляет собой галоген или -Z1RZ1,
где Z1представляет собой -О- или -NH- и
RZ1 представляет собой C1-C14 алкильную группу, в которой до пяти атомов углерода необязательно независимо заменены R22, карбонилом, этенилом, этинилом или фрагментом, выбранным из N, О, S, SO2 или SO, при условии, что два атома О, два атома S или атомы О и S не соседствуют друг с другом непосредственно,
где фрагмент RZ1 необязательно замещен по любому доступному положению C1-С10-алкилом, C1-С10-галогеналкилом, C2-С10-алкенилом, C2-С10-алкинилом, гидрокси, карбокси, карбоксамидо, оксо, галогеном, амино, циано, нитро, -SH, -S-(C1-С6)алкилом, -SO2-(C1-C6)алкилом, -SO2NH2, -SO2NH-(C1-C6)алкилом,
-SO2NH-арилом, -SO2-арилом, -SO-(C1-C6)алкилом, -SO2-арилом, C1-C6-алкокси, С2-С10-алкенилокси, С2-С10-алкинилокси, моно- или ди(C1-С10)алкиламино, -OC1-С10-алкил-Z или R23.
9. Соединение, представляющее собой 4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол-1-ил)-2-(транс-4-гидроксициклогексиламино)бензамид, или его фармацевтически приемлемая соль.
10. Соединение, представляющее собой транс-4-({2-(аминокарбонил)-5-[6,6-диметил-4-оксо-3-(трифторметил)-4,5,6,7-тетрагидро-1Н-индазол-1-ил]фенил}амино)циклогексилглицинат, или его фармацевтически приемлемая соль.
11. Фармацевтическая композиция, включающая соединение или соль по п.1 и фармацевтически приемлемый носитель.
12. Применение соединения или соли по п.1 для получения лекарственного средства для лечения рака.
Текст
013521 Перекрестные ссылки на родственные заявки Настоящая заявка претендует на приоритет предварительной заявки 60/656230, поданной 25 февраля 2005, предварительной заявки 60/705715, поданной 4 августа 2005 и предварительной заявки 60/727965, поданной 18 октября 2005. Предпосылки изобретения Область техники, к которой относится изобретение Настоящее изобретение относится к производным бензола, пиридина и пиридазина и, более конкретно, к таким соединениям, которые применимы в лечении и/или профилактике заболеваний и/или состояний, связанных с клеточной пролиферацией, как, например, рака, воспаления и расстройств, связанных с воспалением, а также состояний, связанных с ангиогенезом. Кроме того, соединения по настоящему изобретению применимы в лечении и/или профилактике инфекционных заболеваний, в частности грибковых инфекций. Уровень техники Раковые заболевания отличаются аномальной клеточной пролиферацией. Раковые клетки демонстрируют ряд свойств, которые делают их опасными для их носителя, в число этих свойств, главным образом, входит способность проникать в другие ткани и вызывать врастание капилляров, что гарантирует пролиферирующим раковым клеткам достаточное кровоснабжение. Признаком раковых клеток является их аномальная реакция на управляющие механизмы, которые регулируют деление в нормальных клетках, и продолжение деления до тех пор, пока они окончательно не убивают своего носителя. В нормальных условиях ангиогенез является в высокой степени регулируемым процессом, однако многие заболевания развиваются в результате стойкого нарушения регулирования ангиогенеза. Нарушение регулирования ангиогенеза может либо непосредственно вызывать конкретное заболевание, либо осложнять имеющееся патологическое состояние. Например, неоваскуляризация глаз не только рассматривается в качестве наиболее частой причины появления слепоты, но также считается основной причиной многих глазных болезней. Далее, при некоторых уже имеющихся состояниях, например артрите,вновь образующиеся капиллярные кровеносные сосуды проникают в суставы и разрушают хрящ или же,в случае диабета, новые капилляры, образующиеся в сетчатке, проникают в стекловидное тело, кровоточат и вызывают слепоту. Рост и метастазирование солидных опухолей также находится в зависимости от ангиогенеза (Folkman, J. Cancer Research, 46, 467-473 (1986); Folkman J., Journal of the National CancerInstitute, 82, 4-6 (1989. Например, было показано, что опухоли, которые увеличились до размеров свыше 2 мм, должны иметь собственный источник кровоснабжения, и это достигается за счет начала роста новых капиллярных кровеносных сосудов. Если эти новые кровеносные сосуды стали прорастать в опухоль, появляется возможность участия опухолевых клеток в кровотоке и возникновения метастазов на удаленных участках, таких как печень, легкие или костная ткань (Weidner, N. et al. The New EnglandJournal of Medicine, 324(1), 1-8 (1991. В условиях неуправляемого ангиогенеза способы лечения, разработанные для регулирования, подавления и/или сдерживания ангиогенеза, могли бы привести к ликвидации или облегчению указанных состояний или заболеваний. Воспаление связано с рядом таких расстройств, как боль, головные боли, лихорадка, артрит, астма,бронхит, менструальные спазмы, тендонит, бурсит, псориаз, экзема, ожоги, дерматит, синдром воспаления кишечника, болезнь Крона, гастрит, синдром раздраженного кишечника, язвенный колит, сосудистые заболевания, болезнь Ходжкина, склеродома, ревматический полиартрит, диабет I типа, тяжелая псевдопаралитическая миастения, саркоидоз, нефротический синдром, синдром Бехчета, полимиозит,аллергия, конъюктивит, гингивит, посттравматическое опухание, ишемия миокарда и т.п. Белок теплового шока 90 (HSP-90) является клеточным шапероном, необходимым для активации нескольких эукариотических протеин киназ, включая циклинзависимую киназу CDK-4. Было показано,что гелданамицин, т.е. ингибитор активности HSP-90 по свертыванию белковых цепей, обладает антипролиферативным и противоопухолевым действием.HSP-90 является молекулярным шапероном, который управляет нормальным свертыванием, внутриклеточным размещением и протеолитическим метаболизмом многих ключевых регуляторов роста и выживания клетки. Во время онкогенеза его функции нарушаются, что делает возможным злокачественное перерождение, облегчает быстрые соматические изменения и дает возможность мутантным белкам сохранять или даже усиливать свою деятельность. Ингибирование HSP-90 должно затормозить этот процесс и, следовательно, имеет потенциальное терапевтическое применение (Whitesell L, Lindquist, S.L.Nature Rev. Cancer, 2005, 10, 761-72). Полагают, что ансамициновые антибиотики, например гербимицин А (НА), гелданамицин (GM) и 17-аллиламиногелданамицин (17-AAG), проявляют свое противораковое действие за счет тесного связывания с N-концевым карманом HSP-90 и тем самым дестабилизируют субстраты, которые обычно взаимодействуют с HSP-90 (Stebbins С. et al. Cell., 1997, 89, 239-250). Этот карман является высоко консервативным и обладает слабой гомологией с АТФ-связывающим сайтом ДНК-гиразы (Stebbins С. et al., смотрите выше; Grenert J. P. et al. J. Biol. Chem., 1997, 272, 23843-50). Исследования in vitro и in vivo продемонстрировали, что занятие этого N-концевого кармана ансамицинами и другими ингибиторами изменяет действие HSP-90 и ингибирует свертывание белка. Было-1 013521 показано, что при высоких концентрациях ансамицины и другие ингибиторы HSP-90 предотвращают связывание белковых субстратов с HSP-90 (Scheibel Т.Н. et al. Proc. Natl. Acad. Sci., USA, 1999, 96,1297-302; Schulte T.W. et al. J. Biol. Chem., 1995, 270, 24585-8; Whitesell L. et al. Proc. Natl. Acad. Sci.,USA, 1994, 91, 8324-8328). Кроме того, было продемонстрировано, что ансамицины ингибируют АТФзависимое высвобождение связанных с шапероном белковых субстратов (Schneider C.L. et al. Proc. Natl.Acad. Sci., USA, 1996, 9314536-41; Sepp-Lorenzino et al. J. Biol. Chem., 1995, 270, 16580-16587). В любом случае, субстраты разлагаются в протеасоме в результате убиквитинзависимого процесса (Schneider С.L.HSP-90 одинаковым образом происходит в опухолевых и не трансформированных клетках и, как было показано, особенно сильно влияет на подгруппу регуляторов передачи сигнала, например Raf (Schulte Т.W. et al. Biochem. Biophys., Res. Commun., 1997, 239, 655-9; Schulte T.W. et al., J. Biol. Chem. 1995, 270,24585-8), рецепторы ядерных стероидов (Segnitz В.U. Gehring J. Biol. Chem., 1997, 272, 18694-18701;Smith D.F. et al. Mol. Cell Biol., 1995, 15, 6804-12), v-Src (Whitesell L. et al. Proc. Natl. Acad. Sci., USA,1994, 91, 8324-8328) и некоторые трансмембранные тирозинкиназы (Sepp-Lorenzino L. et al. J. Biol. Chez.,1995, 270, 16580-16587), как, например, рецептор EGF (EGFR) и HER2/Neu (Hartmann F. et al. Int. J. Cancer, 1997, 70, 221-9; Miller P. et al. Cancer Res., 1994, 54, 2724-2730; Mimnaugh E.G. et al. J. Biol. Clzem.,1996, 271, 22796-801; Schnur R. et al. J. Med. Chenu., 1995, 38, 3806-3812), CDK4 и мутантный p53. Erlichman et al. Proc. AACR, 2001, 42, реферат 4474. Вызванное ансамицинами разрушение этих белков ведет к селективному разрыву некоторых регуляторных путей передачи сигнала и приводит к остановке роста на конкретных фазах клеточного цикла (Muise-Heimericks R.C. et al. J. Biol. Chez., 1998, 273, 2986472) и апоптозу и/или дифференцировке клеток, обработанных указанными антибиотиками (Vasilevskaya A. et al.Cancer Res., 1999, 59, 3935-40). Таким образом, ингибиторы HSP-90 являются многообещающими средствами для лечения и/или профилактики многих типов раковых заболеваний и пролиферативных расстройств и, кроме того, перспективны в качестве традиционных антибиотиков. Кроме того, известно, что ингибирование HSP-90 приводит к повышающей регуляции экспрессии шаперона HSP70. Считается, что повышающая регуляция приносит терапевтическую пользу при лечении широкого круга нейродегенеративных расстройств, включая, но не ограничиваясь, перечисленные: болезнь Альцгеймера, болезнь Паркинсона, деменцию с тельцами Леви, боковой амиотрофический склероз(ALS), полиглутаминовую болезнь, болезнь Хантингтона, бульбарную спинальную мышечную атрофию(SBMA) и спинно-мозжечковую атаксию (SCA1-3,7). Следовательно, соединения, описанные в настоящем изобретении, обладают потенциальной терапевтической применимостью для лечения таких нейродегенеративных расстройств (Muchowski, P.J., Wacker J.L., Nat. Rev. Neurosci. 2005, 6, 11-22; Shen H.Y. etal. J. Biol. Chem., 2005, 280, 39962-9). Ингибирование HSP-90 приводит также к противогрибковой активности как при самостоятельном лечении, так и в комбинации со стандартными способами лечения против грибков, как, например, с препаратами класса азолов. Следовательно, соединения, описанные в настоящем изобретении, обладают потенциальной терапевтической применимостью для лечения грибковых инфекций, включая, но не ограничиваясь указанным, опасные для жизни системные грибковые инфекции (Cowen L.E., Lindquist S., Science, 2005, 309, 2185-9). Кроме того, ингибирование HSP-90 обеспечивает антималярийное действие и таким образом ингибиторы этого белка применимы в качестве средств против малярии. Следовательно, в технике продолжает существовать потребность в новых способах лечения рака,воспаления и связанных с воспалением расстройств, а также состояний или заболеваний, связанных с неуправляемым ангиогенезом. Сущность изобретения В широком аспекте настоящее изобретение охватывает соединения показанной ниже формулы (I),фармацевтические композиции, содержащие эти соединения, а также применение таких соединений или композиций для получения лекарственного средства в лечении заболеваний и/или состояний, связанных с клеточной пролиферацией, таких как рак, воспаление, артрит, ангиогенез или подобных им. Изобретение относится к соединениям формулы (I)-2 013521 или их фармацевтически приемлемым солям,где R4 представляет собой Н;(b) С 1-С 15 алкильную группу, причем до шести атомов углерода в указанной алкильной группе необязательно независимо заменены R22, карбонилом, этенилом, этинилом или фрагментом, выбранным изN, О, S, SO2 или SO, при условии, что два атома О, два атома S или атомы О и S не находятся в непосредственном соседстве друг с другом,где R22 представляет собой:(ii) С 6-С 10-арил,(iii) насыщенный или ненасыщенный С 3-С 10-циклоалкил или(iv) насыщенный или ненасыщенный С 2-С 10-гетероциклоалкил, содержащий 1-3 гетероатома, выбранных из группы, включающей N, О и S, где каждый арил, гетероарил, насыщенный или ненасыщенный циклоалкил или насыщенный или ненасыщенный гетероциклоалкил независимо необязательно замещен по меньшей мере одной группой, которая независимо представляет собой гидрокси, галоген, амино, циано, карбокси, карбоксамидо, нитро, оксо, -S-(C1-C6)алкил, -SO2- (C1-C6)алкил, -SO2-(С 6-С 10)арил,-SO-(C1-C6)алкил, -SO-(C6-С 10)арил, -SO2NH2, -SO2NH-(C1-C6)алкил, -SO2NH-(С 6-С 10)арил, (C1-С 6)алкокси или моно- или ди(C1-С 10)алкиламино; и каждый из R22 необязательно конденсирован с С 6-С 10 арильной группой, С 5-С 8 насыщенной циклической группой или С 5-С 10 гетероциклоалкильной группой, содержащей 1-3 гетероатома, выбранных из группы, включающей N, О и S; где каждая из групп (b) необязательно замещена по любому доступному положениюC1-С 10-алкилом, C1-С 10-галогеналкилом, С 2-С 10-алкенилом, C2-С 10-алкинилом, гидрокси, карбокси, карбоксамидо, оксо, галогеном, амино, циано, нитро, -SH, -S-(С 1-С 6)алкилом, -SO2-(C1-C6)алкилом, -SO2NH2,-SO2NH-(C1-C6)алкилом, -SO2NH-(C6-С 10)арилом, -SO-(C1-С 6)алкилом, -SO2-(С 6-С 10)арилом, C1-С 6-алкокси, С 2-С 10-алкенилокси, C2-С 10-алкинилокси, моно- или ди(C1-С 10)алкиламино, -OC1-С 10-алкил-Z или R23,где Z представляет собой OR0 или -N(R30)2,где каждый из R30 независимо представляет собой -Н или C1-С 6-алкил или -N(R30)2 представляет собой пирролидинил, пиперидинил, пиперазинил, азепанил, 1,3- или 1,4-диазепанил или морфолинил, каждый из которых необязательно замещен гидрокси, амино, аминоалкилом, C1-С 6-алкилом, моно- или ди(С 1-С 6)алкиламино, C1-С 6-алкокси или галогеном;(1) 5-10-членный гетероарил, содержащий 1-3 гетероатома, выбранных из группы, включающей N,О и S,(2) С 6-С 10-арил,(3) насыщенный или ненасыщенный С 5-С 10-циклоалкил или(4) насыщенный или ненасыщенный С 5-С 10-гетероциклоалкил, содержащий 1-3 гетероатома, выбранных из группы, включающей N, О и S, и группы R23 необязательно замещены по меньшей мере одной группой, которая независимо представляет собой гидрокси, оксо, галоген, амино, циано, нитро, -SH, -S-(C1-С 6)алкил, -SO2-(C1-С 6)алкил,-SO2-арил, -SO-(C1-C6)алкил, -SO-(С 6-С 10)арил, -SO2NH2, -SO2NH-(C1-С 6)алкил, -SO2NH-(С 6-С 10)арил,(C1-C6)алкокси или моно- или ди(C1-С 10)алкиламино;Rc представляет собой водород, галоген, циано, C1-С 10-алкил, C2-С 10-алкенил, С 2-С 10-алкинил,C1-С 10-галогеналкил, С 3-С 7-циклоалкил, С 3-С 7-циклоалкил(C1-С 10)алкил; каждый RQ независимо представляет собой водород, галоген, -N(RCN)2, С 1-С 6-алкил, C1-С 6 галогеналкил или С 3-С 7-циклоалкил; каждый из RCN независимо представляет собой -Н, -C1-С 10-алкил, -C1-С 10-галогеналкил или -С 3-С 7 циклоалкил,R21 представляет собой группу формулы где R1 и R2 независимо представляют собой Н, гидрокси или C1-C6-алкил; Х 4 представляет собой О или NOH;R5 и R6 независимо представляют собой Н, C1-С 6-алкил или С 6-С 10-арил, где арил необязательно за-3 013521 мещен 1-4 группами, которые независимо представляют собой C1-С 6-алкил, C1-С 6-алкокси, галоген, гидрокси, амино, моно- или ди(C1-С 6)алкиламино, нитро, галоген (C1-С 6)алкил, галоген (C1-С 6)алкокси или карбоксамид,где любые два заместителя в соседних положениях в арильном фрагменте совместно с атомами углерода, к которым они присоединены, образуют ненасыщенный С 3-С 8-циклоалкил или С 3-С 7 гетероциклоалкил, содержащий 1-3 гетероатома, выбранных из группы, включающей N, О и S; илиR9a и R9b независимо представляют собой Н, C1-C6-алкил, или моно- или ди(C1-C6)алкиламино(C1C6)алкил. Также в изобретении разработаны фармацевтические композиции, включающие соединение или фармацевтически приемлемую соль формулы (I) и хотя бы один фармацевтически приемлемый носитель,растворитель, вспомогательное вещество или разбавитель. Настоящее изобретение относится также к применению соединения или соли формулы (I) с целью производства лекарственного средства, предназначенного для применения в лечении рака. Подробное описание изобретения Как отмечено выше, заместитель R3 в формуле (I) представляет собой (а) галоген или (b) алкильную группу, включающую 1-15 атомов углерода. Каждый из атомов углерода алкильной группы, но в общей сложности не более примерно шести, могут быть независимо заменены различными группами, перечисленными выше в связи с формулой (I). Так, например, если алкильная группа представляет собой метил, т.е. алкильную группу с одним атомом углерода, при замене этого атома углерода, например, азотом или серой приведет к образованию группы, которая не будет являться алкильной группой, но вместо этого будет представлять собой амино или тиогруппу соответственно. Аналогично, если заменяемый атом углерода находится в концевом положении рассматриваемой алкильной группы, концевая группа станет другим фрагментом, таким как пиримидинил, амино, фенил или гидрокси. Замена атома углерода на такую группу, как, например, кислород, азот или сера потребует соответствующего изменения количества атомов водорода или других атомов, требуемых для насыщения валентности заменяющего атома. Так, если заменой являются атомы N или О, количество групп, присоединенных к замененному атому, сократится на одну или две для насыщения валентностей азота или кислорода соответственно. Подобные рассуждения должны быть легко понятны специалисту в данной области техники в отношении замены на этенил или этинил. Таким образом, разрешенные в настоящей заявке замены приводят к тому, что термин C1-C15 алкил согласно определению, данному в связи с формулой (I), охватывает такие группы, как, не ограничиваясь перечисленными: амино, гидрокси, фенил, бензил, пропиламиноэтокси, бутоксиэтиламино, пирид-2-илпропил, диэтиламинометил, пентилсульфонил, метилсульфонамидоэтил, 3-[(4-бутилпиримидин-2-ил)этил]фенил, бутокси, диметиламино, 4-(2-(бензиламино)этил)пиридил, бут-2-ениламино, 4-(1-метиламино)пент-3-ен-2 илтио)фенил, 2-N-метилгексанамидо)этокси)метил и 4-3-метокси-4-(4-метил-1 Н-имидазол-2-ил)бут 1-енил)(метил)амино)метил)фенил. Предпочтительные соединения формулы (I) включают те соединения, в которых R3 представляет собой галоген или Z1RZ1,где Z1 представляет собой -O-, -NH-, -S(O)p- или -S(O)2NH-,где р означает 0, 1 или 2; иRZ1 представляет собой C1-C14 алкильную группу, в которой до пяти атомов углерода необязательно независимо заменены R22, карбонилом, этенилом, этинилом или фрагментом, выбранным из N, О, S, SO2 или SO, при условии, что два атома О, два атома S или атомы О и S не соседствуют друг с другом непосредственно,где фрагмент RZ1 необязательно замещен по любому доступному положению C1-C10-алкилом, C1 С 10-галогеналкилом, С 2-С 10-алкенилом, С 2-С 10-алкинилом, гидрокси, карбокси, карбоксамидо, оксо, галогеном, амино, циано, нитро, -SH, -S-(C1-С 6)алкилом, -SO2-(C1-C6)алкилом, -SO2NH2, -SO2NH-(C1C6)алкилом, -SO2NH-арилом, -SO-(C1-C6)алкилом, -SO2-арилом, C1-C6-алкокси, С 2-С 10-алкенилокси, С 2 С 10-алкинилокси, моно- или ди(C1-С 10)алкиламино, -OC1-С 10-алкил-Z или R23. Еще более предпочтительные соединения формулы (I) включают те соединения, в которых R7 представляет собой О;Rc представляет собой -Н, -СН 3, этил, циклопропил или -CF3. Дополнительные предпочтительные соединения формулы (I) включают те соединения, в которыхR5 и R6 независимо представляют собой Н или C1-С 6-алкил. Наиболее предпочтительные соединения формулы (I) включают те соединения, в которых каждыйRQ независимо представляет собой Н или C1-С 6-алкил.-4 013521 Дополнительные предпочтительные соединения формулы (I) включают соединения, отвечающие формуле в которых R3 представляет собой галоген или -Z1RZ1,где Z1 представляет собой -O-, -NH-, -S(O)p- или -S(O)2NH-,где р означает 0, 1 или 2; иRZ1 представляет собой C1-C14 алкильную группу, в которой до пяти атомов углерода необязательно независимо заменены R22, карбонилом, этенилом, этинилом или фрагментом, выбранным из N, О, S, SO2 или SO, при условии, что два атома О, два атома S или атомы О и S не соседствуют друг с другом непосредственно,где фрагмент RZ1 необязательно замещен по любому доступному положению C1-C10-алкилом,C1-С 10-галогеналкилом, С 2-С 10-алкенилом, С 2-С 10-алкинилом, гидрокси, карбокси, карбоксамидо, оксо,галогеном, амино, циано, нитро, -SH, -S-(C1-C6)алкилом, -SO2-(C1-С 6)алкилом, -SO2NH2, -SO2NH-(C1 С 6)алкилом, -SO2NH-арилом, -SO2-арилом, -SO-(C1-C6)алкилом, -SO2-арилом, C1-C6-алкокси, С 2-С 10-алкенилокси, С 2-С 10-алкинилокси, моно- или ди(C1-С 10)алкиламино, -OC1-С 10-алкил-Z или R23. Наиболее предпочтительные соединения вышеуказанной формулы включают те соединения, в которых R3 представляет собой галоген или -Z1RZ1,где Z1 представляет собой -О- или -NH- иRZ1 представляет собой C1-C14 алкильную группу, в которой до пяти атомов углерода необязательно независимо заменены R22, карбонилом, этенилом, этинилом или фрагментом, выбранным из N, О, S, SO2 или SO, при условии, что два атома О, два атома S или атомы О и S не соседствуют друг с другом непосредственно,где фрагмент RZ1 необязательно замещен по любому доступному положению C1-С 10-алкилом,C1-С 10-галогеналкилом, С 2-С 10-алкенилом, С 2-С 10-алкинилом, гидрокси, карбокси, карбоксамидо, оксо,галогеном, амино, циано, нитро, -SH, -S-(C1-С 6)алкилом, -SO2-(C1-C6)алкилом, -SO2NH2, -SO2NH-(C1C6)алкилом, -SO2NH-арилом, SO2-арилом, -SO-(C1-С 6)алкилом, -SO2-арилом, C1-С 6-алкокси, С 2-С 10 алкенилокси, С 2-С 10-алкинилокси, моно- или ди(C1-С 10)алкиламино, -OC1-С 10-алкил-Z или R23. Настоящее изобретение относится к соединению, представляющему собой 4-(6,6-диметил-4-оксо-3 трифторметил-4,5,6,7-тетрагидроиндазол-1-ил)-2-(транс-4-гидроксициклогексиламино)бензамид, или его фармацевтически приемлемая соль. Другим предпочтительным соединением настоящего изобретения является транс-4-(2(аминокарбонил)-5-[6,6-диметил-4-оксо-3-(трифторметил)-4,5,6,7-тетрагидро-1 Н-индазол-1 ил]фениламино)циклогексилглицинат или его фармацевтически приемлемая соль. В другом аспекте изобретение охватывает применение терапевтически эффективного количества соединения формулы (I) или его соли с целью получения лекарственного средства для лечения у пациента рака в случае необходимости такого лечения. В другом аспекте изобретение охватывает упаковку, включающую соединение или соль формулы(I) в контейнере с инструкцией по применению находящегося в упаковке соединения. Определения Термин алкокси относится к алкильной группе с указанным количеством атомов углерода, присоединенных к остальной части молекулы через атом кислорода. Примеры алкоксигрупп включают, например, метокси, этокси, пропокси и изопропокси. В настоящем описании термин алкил включает алкильные группы, содержащие обозначенное количество атомов углерода. Алкильные группы могут быть линейными или разветвленными. Примеры алкилов включают метил, этил, пропил, изопропил, бутил, изо-, втор- и трет-бутил, пентил, гексил,гептил, 3-этилбутил и т.п. Термин алкенил в настоящем описании означает углеводородный остаток с линейной или разветвленной цепью, содержащий от 2 до 10 атомов углерода и содержащий хотя бы одну двойную углерод-углеродную связь, образованную путем удаления двух атомов водорода. Характерные примеры алкенилов включают, не ограничиваясь перечисленным, этенил, 2-пропенил, 2-метил-2-пропенил,3-бутенил, 4-пентенил, 5-гексенил, 2-гептенил, 2-метил-1-гептенил и 3-деценил. Термин алкенокси относится к алкенильной группе, присоединенной к остальному фрагменту мо-5 013521 лекулы через атом кислорода. Термин алкинил в настоящем описании означает углеводородную группу с линейной или разветвленной цепью, содержащую от 2 до 10 атомов углерода и включающую хотя бы одну тройную углерод-углеродную связь. Типовые примеры алкинилов включают, не ограничиваясь перечисленным, ацетиленил, 1-пропинил, 2-пропинил, 3-бутинил, 2-пентинил и 1-бутинил. Термин арил относится к ароматической углеводородной циклической системе, включающей хотя бы один ароматический цикл. Ароматический цикл может быть необязательно конденсирован или другим образом присоединен к другим ароматическим углеводородным циклам или не ароматическим углеводородным циклам. Примеры арильных групп включают, например, фенил, нафтил, 1,2,3,4 тетрагидронафталин и бифенил. Предпочтительные примеры арильных групп включают фенил, нафтил и антраценил. Более предпочтительными арильными группами являются фенил и нафтил. Наиболее предпочтителен фенил. Арильные группы в соединениях по настоящему изобретению могут быть замещены различными группами, указанными в данной заявке. Так, например, любой атом углерода, имеющийся в арильной циклической системе и доступный для замещения, может быть дополнительно связан с одним из ряда заместителей циклической системы, как, например, галогеном, гидрокси, нитро, циано, амино,С 1-С 8-алкилом, C1-C8-алкокси, моно- и ди(С 1-С 8-алкил)амино, С 3-С 10-циклоалкилом, (С 3-С 10-циклоалкил)алкилом, (С 3-С 10-циклоалкил)алкокси, С 2-С 9-гетероциклоалкилом, C1-C8-алкенилом, C1-C8 алкинилом, галоген(С 1-С 8)алкилом, галоген(C1-C8)алкокси, оксо, амино(C1-C8)алкилом, моно- или ди(С 1 С 8-алкил)амино(C1-C8)алкилом, C1-C8-ацилом, C1-C8-ацилокси, C1-C8-сульфонилом,-C1-C8-тио, C1-C8 сульфонамидо, С 1-С 8-аминосульфонилом. Термин карбокси в настоящей заявке означает группу -CO2H. Термин циклоалкил относится к циклическому углеводороду С 3-С 8. Примеры циклоалкилов включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Более предпочтительными являются С 3-С 6 циклоалкильные группы. Циклоалкильные группы по настоящему изобретению могут быть замещены различными группами, указанными в настоящем описании. Так, например, любой атом углерода, имеющийся в циклоалкильной циклической системе и доступный для замещения, может быть дополнительно связан с различными циклическими заместителями, как, например,галогеном, гидрокси, нитро, циано, амино, С 1-С 8-алкилом, C1-C8-алкокси, моно- и ди(C1-C8-алкил)амино,С 3-С 10-циклоалкилом, (С 3-С 10-циклоалкил)алкилом, (С 3-С 10-циклоалкил)алкокси, С 2-С 9-гетероциклоалкилом, C1-C8-алкенилом, C1-C8-алкинилом, галоген(C1-C8)алкилом, галоген(C1-C8)алкокси, оксо, амино(C1-C8)алкилом и моно- или ди(С 1-С 8-алкил)амино(C1-С 8)алкилом. Термины галоген или гало указывают на фтор, хлор, бром и йод. Термин галогеналкокси относится к алкоксигруппе, замещенной одним или несколькими атомами галогенов, где каждый атом галогена независимо представляет собой F, Cl, Br или I. Предпочтительными галогенами являются F и Cl. Предпочтительные галогеналкоксигруппы содержат 1-6 атомов углерода,более предпочтительно 1-4 атома углерода и еще более предпочтительно 1-2 атома углерода. Термин галогеналкокси включает пергалогеналкоксигруппы, такие как OCF3 или OCF2CF3. Предпочтительной галогеналкоксигруппой является трифторметокси. Термин галогеналкил относится к алкильной группе, замещенной одним или несколькими атомами галогенов, где каждый атом галогена независимо представляет собой F, Cl, Br или I. Предпочтительными галогенами являются F и Cl. Предпочтительные галогеналкильные группы содержат 1-6 атомов углерода, более предпочтительно 1-4 атома углерода и еще более предпочтительно 1-2 атома углерода. Термин галогеналкил включает пергалогеналкильные группы, такие как CF3 или CF2CF3. Предпочтительной галогеналкильной группой является трифторметил. Термин гетероциклоалкил относится к циклу или циклической системе, содержащей как минимум один гетероатом, выбранный из азота, кислорода и серы, где указанный гетероатом находится в неароматическом цикле. Гетероциклоалкил необязательно конденсирован или другим образом присоединен к другим гетероциклоалкилам, и/или неароматическим углеводородным циклам, и/или фенильным циклам. Предпочтительные гетероциклоалкильные группы включают от 3 до 7 членов цикла. Более предпочтительно гетероциклоалкильные группы включают 5 или 6 членов цикла. Примеры гетероциклоалкильных групп включают, например, 1,2,3,4-тетрагидроизохинолинил, пиперазинил, морфолинил, пиперидинил, тетрагидрофуранил, пирролидинил, пиридинонил и пиразолидинил. Предпочтительные гетероциклоалкильные группы включают пиперидинил, пиперазинил, морфолинил, пирролидинил, пиридинонил, дигидропирролидинил и пирролидинонил. Гетероциклоалкильные группы по настоящему изобретению могут быть замещены различными группами, указанными в настоящей заявке. Так, например, любой атом углерода, имеющийся в гетероциклоалкильной циклической системе и доступный для замещения, может быть дополнительно связан с различными заместителями, как, например, галогеном, гидрокси, нитро, циано, амино, -C1-C8-алкилом, C1-C8-алкокси, моно- и ди(C1-C8-алкил)амино, С 3-С 10-циклоалкилом, (С 3-С 10-циклоалкил)алкилом, (С 3-С 10-циклоалкил)алкокси, С 2-С 9-гетероциклоалкилом, C1-C8 алкенилом, C1-C8-алкинилом, галоген(C1-С 8)алкилом, галоген(C1-C8)алкокси, оксо, амино(C1-С 8)алкилом,и моно- или ди(С 1-С 8-алкил)амино(C1-C8)алкилом.-6 013521 Термин гетероарил относится к ароматической циклической системе, содержащей хотя бы один гетероатом, выбранный из азота, кислорода или серы. Гетероарильный цикл может быть конденсирован или иным образом присоединен к одному или нескольким гетероарильным циклам, ароматическим или неароматическим углеводородным циклам или гетероциклоалкилам. Примеры гетероарильных групп включают, например, пиридин, фуран, тиенил, 5,6,7,8-тетрагидроизохинолин и пиримидины. Гетероарильные группы в соединениях по настоящему изобретению могут быть замещены различными группами, указанными в настоящей заявке. Так, например, любой атом углерода, имеющийся в гетероарильной циклической системе и доступный для замещения, может быть дополнительно связан с одним из ряда различных заместителей, как, например, галогеном, гидрокси, нитро, циано, амино, C1-C8-алкилом, C1C8-алкокси, моно- и ди(C1-C8-алкил)амино, С 3-С 10-циклоалкилом, (С 3-С 10-циклоалкил)алкилом, (С 3-С 10 циклоалкил)алкокси, С 2-С 9-гетероциклоалкилом, C1-C8-алкенилом, C1-C8-алкинилом, галоген(C1-C8)алкилом, галоген(С 1-С 8)алкокси, оксо, амино(C1-C8)алкилом, и моно- или ди(С 1-С 8-алкил)амино(C1-C8)алкилом. Предпочтительные примеры гетероарильных групп включают тиенил, бензотиенил, пиридил, хинолил, пиразолил, пиримидил, имидазолил, бензимидазолил, фуранил, бензофуранил, дибензофуранил,тиазолил, бензотиазолил, изоксазолил, оксадиазолил, изотиазолил, бензизотиазолил, триазолил, пирролил, индолил, пиразолил и бензопиразолил. Соединения по настоящему изобретению могут содержать один или несколько асимметрических атомов углерода, в результате чего эти соединения могут существовать в различных стереоизомерных формах. Эти соединения могут быть, например, рацематами, хиральными не рацемическими или диастереомерами. В этих ситуациях чистые энантиомеры, т.е. оптически активные формы, могут быть получены асимметрическим синтезом или разделением рацемических смесей. Разделение рацемических смесей может быть достигнуто, например, стандартными способами, такими как кристаллизация в присутствии разделяющего реагента; хроматография с применением, например, хиральной ВЭЖХ-колонки; или получение производных рацемической смеси с разделяющим реагентом для образования диастереомеров,разделение диастереомеров с помощью хроматографии и удаление разделяющего реагента для получения исходного соединения в форме, обогащенной одним из энантиомеров. Любая из указанных выше методик может быть повторена для увеличения энантиомерной чистоты соединения. Если соединения, описанные в настоящей заявке, содержат олефиновые двойные связи или другие центры геометрической асимметрии и если не указано иное, имеется в виду, что соединения включают цис-, транс-, Z- и Е-конфигурации. Аналогично предполагается, что все таутомерные формы также включены в соединения по настоящему изобретению. Фармацевтические композиции Соединения общей формулы (I) могут вводиться перорально, местно, парентерально, с помощью ингаляции или распыления или ректально в виде дозированных лекарственных составов, содержащих обычные нетоксичные фармацевтически приемлемые носители, вспомогательные вещества и основы. Термин парентеральный в настоящей заявке включает методики введения через кожу, подкожных, внутрисосудистых (например, внутривенных), внутримышечных или интратекальных инъекций или инфузий и т.п. Кроме того, предложен фармацевтический состав, включающий соединение общей формулы (I) и фармацевтически приемлемый носитель. Может присутствовать одно или несколько соединений общей формулы (I) в сочетании с одним или несколькими нетоксичными фармацевтически приемлемыми носителями, и/или разбавителями, и/или вспомогательными веществами, а также, если необходимо, другими действующими ингредиентами. Фармацевтические композиции, содержащие соединения общей формулы (I), могут быть получены в форме, подходящей для перорального применения, например в виде таблеток, пастилок, ромбовидных таблеток, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул или сиропов или эликсиров. Композиции, предназначенные для перорального применения, могут быть получены способами, известными в технике для производства фармацевтических композиций, и такие композиции могут содержать одно или несколько средств, выбранных из группы, состоящей из подсластителей, вкусоароматических добавок, красителей и консервантов, для получения отличных с фармацевтической точки зрения препаратов с хорошим вкусом. Таблетки содержат действующий ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, которые подходят для производства таблеток. Эти наполнители могут быть, например, инертными разбавителями, такими как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующими и дезинтегрирующими средствами, например кукурузным крахмалом или альгиновой кислотой; связующими средствами, например крахмалом, желатином или гуммиарабиком, а также средствами для скольжения, например стеаратом магния, стеариновой кислотой или тальком. Таблетки могут не иметь покрытия или могут быть покрытыми по известным методикам. В некоторых случаях такие покрытия могут быть получены по известным методикам с целью замедления разрушения таблетки и всасывания действующих веществ в желудочно-кишечном тракте для обеспечения длительного действия в течение более продолжительного периода. Например, может применяться материал, продлевающий время распада, такой как глицерил моно-7 013521 стеарат или глицерил дистеарат. Составы для перорального применения также могут принимать форму твердых желатиновых капсул, в которых действующий ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или мягких желатиновых капсул, где действующий ингредиент смешан с водной или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом. Составы для перорального применения также могут быть представлены в форме ромбовидных таблеток. Водные суспензии содержат действующие вещества в смеси с наполнителями, подходящими для получения водных суспензий. Такими наполнителями являются суспендирующие средства, например натриевое производное карбоксиметилцеллюлозы, метилцеллюлозы, гидропропилметилцеллюлозы, альгинат натрия, поливинилпирролидон, смола трагаканта и гуммиарабик; диспергирующие или смачивающие средства могут быть природными фосфатидами, например лецитином, или продуктами конденсации алкиленоксидов с жирными кислотами, например пропоксиэтиленстеаратом, или продуктами конденсации оксида этилена с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанолом, или продуктами конденсации оксида этилена с продуктами частичной этерификации, полученными из жирных кислот и гексита, как, например, полиоксиэтилен моноолеатом сорбита, или продуктами конденсации оксида этилена с продуктами частичной этерификации, полученными из жирных кислот и ангидридов гексита, например полиэтиленсорбитан моноолеатом. Водные суспензии также могут содержать один или несколько консервантов, например этил или н-пропил п-гидроксибензоат, или одно или несколько окрашивающих средств, одну или несколько вкусоароматических добавок и один или несколько подсластителей, таких как сахароза или сахарин. Масляные суспензии могут быть получены суспендированием действующих ингредиентов в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле,или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Подсластители и вкусоароматические добавки могут быть добавлены для получения препаратов с приятным вкусом. Сохранность композиций может быть обеспечена добавлением антиоксиданта, как, например, аскорбиновой кислоты. Диспергируемые порошки и гранулы, подходящие для получения водных суспензий при добавлении воды, получают в виде смеси действующего ингредиента с диспергирующим или смачивающим средством, суспендирующим средством и одним или несколькими консервантами. Примеры подходящих диспергирующих или смачивающих средств или суспендирующих средств соответствуют примерам, уже приведенным выше. Также могут присутствовать дополнительные наполнители, например подсластители, вкусоароматические добавки и красящие средства. Фармацевтические композиции по настоящему изобретению также могут иметь форму эмульсий масло в воде. Масляная фаза может быть растительным маслом, минеральным маслом или их смесями. Подходящими эмульгирующими средствами могут быть природные смолы, например гуммиарабик или смола трагаканта, природные фосфатиды, например соевый лецитин, и сложные эфиры или частично этерифицированные производные, полученные из жирных кислот и гексита, ангидридов, например моноолеат сорбитана, а также продукты конденсации упомянутых частично этерифицированных производных с оксидом этилена, например моноолеат полиоксиэтилен сорбитана. Описываемые эмульсии могут также содержать подсластители и вкусоароматические средства. В состав сиропов и эликсиров могут быть включены подсластители, например глицерин, пропиленгликоль, сорбит, глюкоза или сахароза. Кроме того, такие составы могут содержать мягчительное средство, консервант, а также вкусоароматические и красящие средства. Фармацевтические композиции могут быть получены в форме стерильных водных или масляных суспензий, пригодных для инъекций. Эти суспензии могут быть приготовлены в соответствии с известным уровнем техники с применением тех подходящих диспергирующих или смачивающих и суспендирующих средств, которые упоминались выше. Стерильные пригодные для инъекций препараты могут быть также стерильными пригодными для инъекций растворами или суспензиями в нетоксичных приемлемых для парентерального введения разбавителях или растворителях, например в виде раствора в 1,3-бутандиоле. В число приемлемых носителей и растворителей, которые могут применяться в данном случае, входят вода, раствор Рингера и изотонический раствор хлорида натрия. В дополнение, в качестве растворителя и суспендирующей среды обычно применяются стерильные нелетучие масла. С этой целью может применяться любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, при приготовлении пригодных для инъекций препаратов находят применение жирные кислоты, как, например, олеиновая кислота. Соединения общей формулы (I) могут также вводиться в форме суппозиториев, например, с целью ректального введения препарата. Эти композиции могут быть получены смешиванием лекарственного препарата с подходящим не раздражающим наполнителем, который является твердым при обычных температурах, но становится жидким при температуре прямой кишки и, следовательно, будет плавиться в прямой кишке, высвобождая лекарственное средство. Такие вещества включают масло какао и полиэтиленгликоли.-8 013521 Соединения общей формулы (I) могут вводиться парентерально в стерильной среде. Лекарственное средство в зависимости от применяемого носителя и концентрации может быть суспендировано или растворено в носителе. Вспомогательные вещества, такие как местные анестетики, консерванты и буферирующие реагенты, могут быть преимущественно растворены в носителе. В случае заболевания глаз или других внешних тканей, например рта и кожи, составы преимущественно применяются в виде геля, спрея, мази или крема местного действия или в виде суппозитория, содержащего действующие ингредиенты в общем количестве, например, от 0,075 до 30 мас.%, предпочтительно от 0,2 до 30 мас.% и наиболее предпочтительно от 0,4 до 15 мас.%. При включении в состав мази действующие ингредиенты могут применяться в сочетании с основой мази либо парафиновой, либо смешиваемой с водой. С другой стороны, действующие ингредиенты могут быть включены в состав крема в сочетании с основой крема масло в воде. Если желательно, водная фаза основы крема может включать, например, как минимум 30 мас.% многоатомного спирта, такого как пропиленгликоль, бутан-1,3-диол, маннит, сорбит,глицерин, полиэтиленгликоль и их смесей. Желательно, чтобы составы местного применения включали соединение, которое улучшает абсорбцию или проникновение действующего ингредиента через кожу или другие пораженные болезнью области. Примеры таких средств, улучшающих проникновение, включают диметилсульфоксид и его аналоги. Соединения по настоящему изобретению также могут вводиться с помощью средства для трансдермального введения. Предпочтительно местное введение должно осуществляться применением пластыря, либо имеющего резервуар и пористую мембрану, либо являющегося разновидностью твердой матрицы. В любом случае действующее средство непрерывно поступает из резервуара или микрокапсулу через мембрану в липкий слой, проницаемый для действующего средства,который находится в соприкосновении с кожей или слизистой оболочкой реципиента. Если действующее средство поглощается через кожу, в организм реципиента поступает регулируемый и заранее заданный поток действующего средства. В случае микрокапсул, инкапсулирующее средство также может действовать в качестве мембраны. Трансдермальный пластырь может включать соединение в подходящей системе растворителей в сочетании с клейкой системой материалов, как, например, акриловой эмульсией и полиэфирным пластырем. Масляная фаза эмульсий по настоящему изобретению может быть создана известным способом из известных ингредиентов. Хотя эта фаза может включать только эмульгатор, она может включать смесь по крайней мере одного эмульгатора с жиром или маслом или как с жиром, так и с маслом. Предпочтительно в ее состав входит гидрофильный эмульгатор совместно с липофильным эмульгатором, который действует как стабилизатор. Кроме того, предпочтительно включить как масло,так и жир. Эмульгатор (эмульгаторы) вместе со стабилизатором (стабилизаторами) или без него (без них) представляют собой так называемый эмульгированный воск, и этот воск в сочетании с маслом и жиром образует так называемую эмульгирующую основу мази, которая образует диспергированную в масле фазу состава крема. Эмульгаторы и стабилизаторы эмульсии, подходящие для применения в составе по настоящему изобретению, включают, в том числе, Tween 60, Span 80, цетостеариловый спирт, миристиловый спирт, глицерилмоностеарат и лаурилсульфат натрия. Выбор подходящих для составов масел или жиров основан на достижении желаемых косметических свойств, т.к. растворимость действующего соединения в большинстве масел, которые, вероятно, будут использоваться в составах фармацевтических эмульсий, является очень низкой. Следовательно, крем предпочтительно должен быть не жирным, не пачкающим и легко смываемым продуктом с консистенцией, подходящей для того, чтобы избежать утечки из тюбиков или других контейнеров. Могут применяться алкиловые эфиры с линейной или разветвленной цепью, образованные одно- и двухосновными кислотами, такие как диизоадипат, изоцетил,стеарат, диэфир пропиленгиколя и жирных кислот, содержащихся в кокосовом масле, изопропилмиристат, децилолеат, изопропилпальмитат, бутилстеарат, 2-этилгексилпальмитат или смесь сложных эфиров с разветвленной цепью. Указанные вещества могут применяться самостоятельно или в комбинации, в зависимости от требуемых свойств. С другой стороны, могут применяться липиды с высокой температурой плавления, такие как белый мягкий парафин и/или жидкий парафин, или другие минеральные масла. Составы, подходящие для местного введения в глаза, включают также глазные капли, в которых действующие ингредиенты растворены или суспендированы в подходящем носителе, главным образом в водном растворителе для действующих ингредиентов. Противовоспалительные действующие ингредиенты предпочтительно присутствуют в таких составах в концентрации от 0,5 до 20 мас.%, преимущественно от 0,5 до 10 мас.% и конкретно примерно 1,5 мас.%. Для терапевтических целей действующие соединения по настоящему изобретению обычно комбинируют с одним или несколькими вспомогательными веществами, которые подходят для указанного пути введения. При пероральном введении соединения могут быть смешаны с лактозой, сахарозой, порошком крахмала, сложными эфирами целлюлозы и жирных кислот, алкиловыми эфирами целлюлозы, тальком, стеариновой кислотой, стеаратом магния, оксидом магния, натриевыми и кальциевыми солями фосфорной и серной кислот, желатином, гуммиарабиком, альгинатом натрия, поливинилпирролидоном и/или поливиниловым спиртом и затем таблетированы или инкапсулированы для удобства введения. Такие капсулы или таблетки могут содержать состав для регулируемого высвобождения, что может быть достигнуто при диспергировании действующего соединения в гидроксипропилметилцеллюлозе. Составы для парентерального введения могут быть в форме-9 013521 водных или неводных изотонических стерильных растворов или суспензий для инъекций. Эти растворы и суспензии могут быть получены из стерильных порошков или гранул, содержащих один или несколько носителей или разбавителей из числа упомянутых для применения в составах для перорального введения. Соединения могут быть растворены в воде, полиэтиленгликоле, пропиленгликоле, этаноле, кукурузном масле, хлопковом масле, арахисовом масле, кунжутном масле, бензиловом спирте, растворе хлорида натрия и/или различных буферных растворах. Другие вспомогательные вещества и способы введения широко и хорошо известны в фармацевтической технике. При лечении указанных выше состояний применимы уровни дозировки порядка от примерно 0,1 до примерно 140 мг на килограмм веса тела в день (от примерно 0,5 мг до примерно 7 г на одного пациента в день). Количество действующего ингредиента, которое может быть объединено с носителями, для получения одной единицы дозированной лекарственной формы будет меняться в зависимости от подвергаемого лечению субъекта и конкретного способа введения. Дозированные лекарственные формы в основном должны содержать от примерно 1 до примерно 500 мг действующего ингредиента. Дневная доза может вводиться в виде одной-четырех доз в течение дня. В случае заболеваний кожи может оказаться предпочтительным применять местное нанесение препаратов соединений по настоящему изобретению на пораженную область два-четыре раза в день. Однако следует понимать, что конкретный уровень дозировки для любого отдельно взятого пациента будет зависеть от ряда факторов, включая активность конкретного применяемого соединения, возраст, массу тела, общее состояние здоровья, пол, рацион питания, время введения, путь введения, скорость выведения, комбинацию лекарств и тяжесть конкретного заболевания, которое подвергается лечению. Для введения животным, кроме человека, композиция также может быть добавлена в пищу животного или питьевую воду. Может быть удобным готовить состав пищи и питьевой воды животного таким образом, чтобы животное получало композиции по настоящему изобретению вместе с пищей в терапевтически достаточном количестве. Может быть удобным включать композиции в заранее приготовленную смесь для добавления к пище или питьевой воде. Предпочтительно животные, кроме человека, включают одомашненных животных. Соединения по настоящему изобретению могут вводиться пациенту самостоятельно или в комбинации хотя бы с одним дополнительным терапевтическим средством или способом лечения, например радиационной терапией, при необходимости такого лечения. Дополнительное терапевтическое средство или способ лечения могут применяться одновременно, отдельно или последовательно по отношению к введению соединения по настоящему изобретению. Упомянутые дополнительные терапевтические средства включают, не ограничиваясь перечисленными, противораковые средства, противовоспалительные средства и т.п. Соединения по настоящему изобретению могут быть получены по известным химическим реакциям и методикам. Типовые способы синтеза соединений по настоящему изобретению представлены ниже. Следует понимать, что природа заместителей, наличие которых необходимо в желаемом целевом соединении, часто определяет предпочтительный способ синтеза. В раскрытых способах все изменяемые группы являются теми, которые указаны в общем описании, если они конкретно не определены ниже. Способы получения Общая методика. Типовые методики синтеза, предназначенные для получения соединений по настоящему изобретению, изображены ниже на следующих схемах. Если не указано иное, символы X1, X2, Х 3, n, R5, R6, R7, RC,R11 и Y заключают в себе определения, данные в связи с описанием формулы (I). Схема 1 Специалисты в данной области техники должны принять во внимание, что исходные вещества и условия реакций могут подвергаться изменениям, могут меняться последовательности реакций и дополнительные стадии, используемые для получения соединений, охваченных настоящим изобретением, что показано в нижеследующих примерах. В определенных случаях может быть необходима защита некоторых реакционноспособных групп для осуществления некоторых из приведенных выше превращений. В основном необходимость таких защитных групп, а также условия, необходимые для присоединения и- 13013521 удаления этих групп, должны быть ясны специалисту в области органического синтеза. Содержание всех статей и ссылок, упомянутых в настоящей заявке, в том числе патентов, включено в настоящую заявку во всей полноте с помощью ссылок. Примеры. Получение соединений по настоящему изобретению далее проиллюстрировано следующими примерами, которые не следует истолковывать, как ограничивающие суть и объем изобретения конкретными методиками и соединениями, описанными в этих примерах. Во всех случаях, если не указано иное,колоночную хроматографию проводили с использованием силикагелевой неподвижной фазы. Пример 1. 3,6,6-триметил-1,5,6,7-тетрагидроиндол-4-он (соединение 1). К раствору антиоксима пировиноградного альдегида-1 (10 г, 1 экв.) и 5,5-диметил-1,3-циклогександиона (16,1 г, 1 экв.) в смеси НОАс-Н 2 О (7:3, 200 мл) медленно с охлаждением на водяной бане при комнатной температуре добавляли порошок цинка (14,95 г, 2 экв.). Затем смесь кипятили с обратным холодильником в течение ночи, концентрировали до сухого состояния, распределяли между насыщенным раствором соли (300 мл) и дихлорметаном (300 мл). Значение рН доводили приблизительно до 6 добавлением насыщенного водного раствора NaHCO3, затем экстрагировали смесь дихлорметаном (3200 мл). Органические слои объединяли, высушивали над Na2SO4, фильтровали, концентрировали. Неочищенный продукт очищали флэш-хроматографией, элюируя 5% этилацетатом в дихлорметане. Объединенные органические фракции концентрировали, растирали со смесью эфир-гексан (2:1) в течение 1 ч, затем фильтровали, промывали гексаном, получая чистое соединение, указанное в заглавии примера (9 г, выход 45%) в виде твердого вещества. ЖХМС m/z: (M+H)=178,1. Пример 2. 2-бром-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндол-1-ил)бензонитрил (соединение 2). Соединение примера 1 (9,8 г, 55,3 ммоль) и 2-бром-4-фторбензонитрил (13,27 г, 66,4 ммоль) растворяли в безводном диметилформамиде (ДМФА, 300 мл). К полученному раствору добавляли гидрид натрия (95%, 2,79 г, 111 ммоль) и реакционную смесь перемешивали при 55 С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли воду. Осаждалось твердое вещество желтоватокоричневого цвета, которое отделяли фильтрованием, промывали водой и эфиром и затем высушивали в вакууме (16,5 г, 84%). ЖХМС m/z: (М+Н)=358,1. Пример 3.(соединение 3). В микроволновый сосуд "Personal Chemistry" загружали соединение примера 2 [2-бром-4-(3,6,6 триметил-4-оксо-4,5,6,7-тетрагидроиндол-1-ил)бензонитрил (1,072 г, 3,0 ммоль), транс-4-аминоциклогексанол (1,382 г, 12,0 ммоль), ацетат палладия(II) (33,7 мг, 5 мол.%), 1,1'-бис(дифенилфосфино)ферроцен (DPPE) (166,3 мг, 10 мол.%) и трет-бутоксид натрия (576,7 мг, 6,0 ммоль). К полученной смеси добавляли толуол (20 мл) и реакционную смесь нагревали микроволновым излучением до 115 С в течение 15 мин. Реакционному сосуду давали остыть, затем полученную суспензию фильтровали и фильтрат упаривали. Остаток очищали флэш-хроматографией. Промежуточный продукт гидролизовали растворением в 25% диметилсульфоксиде/этаноле, добавляя 0,5 мл 1 н. раствора гидроксида натрия и 0,5 мл 30% водного раствора пероксида водорода, с последующим перемешиванием при комнатной температуре в течение 4 ч. После завершения реакции, судя по данным ТСХ, смесь ДМСО/этанол разбавляли водой и экстрагировали этилацетатом (3). Объединенные органические слои промывали насыщенным раствором соли, (2), высушивали над Na2SO4 и упаривали. Соединение очищали колоночной хроматографией, элюируя смесью EtOAc-MeOH и получая 575 мг (выход 47%) соединения, указанного в заглавии примера в виде порошка белого цвета. ЖХМС m/z: (M+H)=410,3.(соединение 4). В микроволновый сосуд "Personal Chemistry" загружали соединение примера 2 (2,858 г, 8,0 ммоль),4-аминотетрагидропиран (3,236 г, 32,0 ммоль), ацетат палладия(II) (89,8 мг, 5 мол.%), 1,1'-бис(дифенилфосфино)ферроцен (443,6 мг, 10 мол.%) и трет-бутоксид натрия (1,538 г, 16,0 ммоль). Реагенты суспендировали в толуоле (40 мл) и нагревали микроволновым излучением до температуры 115 С в течение 15 мин. Реакционному сосуду давали остыть, затем фильтровали полученную суспензию и фильтрат концентрировали. После очистки промежуточно полученного нитрила флэш-хроматографией нитрил растворяли в 25% диметилсульфоксиде/этаноле и добавляли 2 мл 1 н. раствора гидроксида натрия и 2 мл 30% водного раствора пероксида водорода, после чего перемешивали при комнатной температуре в течение 16 ч. Затем смесь ДМСО/этанол разбавляли водой и экстрагировали этилацетатом (3). Объединенные органические фазы промывали насыщенным раствором соли (2), высушивали надNa2SO4 и упаривали. Остаток очищали колоночной хроматографией (EtOAc/MeOH), получая 1,132 г(36%) указанного в заглавии примера соединения в виде порошка не совсем белого цвета. ЖХМС m/z: 2-(2-метоксиэтиламино)-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндол-1-ил)бензамид (соединение 5). В микроволновый сосуд "Personal Chemistry" загружали соединение примера 2 (107,1 мг,0,3 ммоль), 2-метоксиэтиламин (91,3 мг, 1,2 ммоль), ацетат палладия(II) (3,4 мг, 5 мол.%), 1,1'-бис(дифенилфосфино)ферроцен (16,6 мг, 10 мол.%) и трет-бутоксид натрия (57,7 мг, 0,6 ммоль). Реагенты суспендировали в толуоле (2 мл) и нагревали микроволновым излучением до температуры 115 С в течение пятнадцати минут. Реакционному сосуду давали остыть, затем фильтровали суспензию и упаривали фильтрат. После очистки неочищенного промежуточного нитрила флэш-хроматографией проводили гидролиз, растворяя остаток в 25% диметилсульфоксиде/этаноле, добавляя 5 капель 1 н. гидроксида натрия и 5 капель 30% водного раствора пероксида водорода, с последующим перемешиванием при комнатной температуре в течение 3 ч. Затем смесь ДМСО/этанол разбавляли водой и экстрагировали CH2Cl2 (3). Объединенные органические фазы промывали насыщенным раствором соли (2), высушивали над Na2SO4 и концентрировали. Вещество очищали колоночной хроматографией (гексан/EtOAc) , получая соединение, указанное в заглавии примера (94,4 мг, выход 85%) в виде порошка не совсем белого цвета. ЖХМС m/z:(соединение 6). В микроволновый сосуд "Personal Chemistry" загружали соединение примера 2 (107,1 мг, 0,3 ммоль),3,4,5-триметоксианилин (219,9 мг, 1,2 ммоль), ацетат палладия(II) (3,4 мг, 5 мол.%), 1,1'-бис(дифенилфосфино)ферроцен (16,6 мг, 10 мол.%) и трет-бутоксид натрия (57,7 мг, 0,6 ммоль). Реагенты суспендировали в толуоле (2 мл) и нагревали микроволновым излучением до температуры 115 С в течение пятнадцати минут. Реакционному сосуду давали остыть, затем фильтровали суспензию и упаривали фильтрат. Остаток очищали флэш-хроматографией и проводили гидролиз, растворяя остаток в 25% диметилсульфоксиде/этаноле, добавляя 5 капель 1 н. гидроксида натрия и 5 капель 30% водного раствора пероксида- 15013521 водорода, с последующим перемешиванием при комнатной температуре в течение 3 ч. Затем смесь ДМСО/этанол разбавляли водой и экстрагировали EtOAc (3). Объединенные органические фазы промывали насыщенным раствором соли (2), высушивали над Na2SO4 и концентрировали. Вещество очищали колоночной хроматографией (гексан/EtOAc), получая соединение, указанное в заглавии примера 28,4 мг (выход 20%) в виде порошка желтого цвета. ЖХМС m/z: (M+H)=478,3. Пример 7.(соединение 7). В микроволновый сосуд "Personal Chemistry" загружали соединение примера 2 (107,1 мг, 0,3 ммоль),3-(аминометил)пиридин (129,8 мг, 1,2 ммоль), ацетат палладия(II) (3,4 мг, 5 мол.%), 1,1'-бис(дифенилфосфино)ферроцен (16,6 мг, 10 мол.%) и трет-бутоксид натрия (57,7 мг, 0,6 ммоль). Реагенты суспендировали в толуоле (2 мл) и нагревали микроволновым излучением до температуры 125 С в течение пятнадцати минут. Неочищенный промежуточный нитрил очищали флэш-хроматографией (гексан/EtOAc). Проводили гидролиз, растворяя остаток в 25% диметилсульфоксиде/этаноле, добавляя 4 капли 1 н. гидроксида натрия и 4 капли 30% водного раствора пероксида водорода, с последующим перемешиванием при комнатной температуре в течение 15 мин. Затем смесь ДМСО/этанол разбавляли водой и экстрагировали EtOAc (3). Объединенные органические фазы промывали насыщенным раствором соли (2),высушивали над Na2SO4 и концентрировали. Вещество очищали колоночной хроматографией(гексан/EtOAc), получая 50 мг (41%) соединения, указанного в заглавии примера, в виде порошка желтого цвета. ЖХМС m/z: (М+Н)=403. Пример 8. 2-(4-оксоциклогексиламино)-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндол-1-ил)бензамид (соединение 8). Соединение примера 3 [2-(транс-4-гидроксициклогексиламино)-4-(3,6,6-триметил-4-оксо-4,5,6,7 терагидроиндазол-1-ил)бензамид] (150 мг, 0,366 ммоль) и перйодинан Дресса-Мартина (0,366 ммоль) растворяли в безводном CH2Cl2 и перемешивали при комнатной температуре в течение 1 ч. Концентрировали реакционную смесь и выделяли указанное в заглавии примера соединение в виде твердого вещества белого цвета (36,2 мг, выход 24%) после очистки колоночной хроматографией с элюированием смесью EtOAc-MeOH. ЖХМС m/z: (М+Н)=408,3. Пример 9. 2-[транс-4-(2-морфолин-4-илэтокси)циклогексиламино]-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндол-1-ил)бензамид (соединение 9). В микроволновый реакционный сосуд загружали соединение примера 3 [2-(транс-4-гидроксициклогексиламино)-4-(3,6,6-триметил-4-оксо-4,5,6,7-терагидроиндазол-1-ил)бензамид] (100 мг, 0,244 ммоль), гидрохлорид 4-(2-хлорэтил)морфолина (45,4 мг, 0,244 ммоль), гидрид натрия (17,3 мг,0,732 ммоль) и каталитическое количество йодида калия. После суспендирования в ДМФА реакционную смесь нагревали до 180 С в микроволновой печи "Personal Chemistry" в течение 8 мин. Затем разбавляли раствор водой и экстрагировали EtOAc (2). Органические слои экстрагировали 2 н. HCl (2). Водные экстракты подщелачивали водным раствором гидроксида натрия и экстрагировали EtOAc (3). Объединенные органические экстракты высушивали над Na2SO4, концентрировали и очищали колоночной хроматографией (EtOAc/MeOH), получая указанное в заглавии соединение в виде твердого вещества белого цвета. ЖХМС m/z: (M+H)=523,9. 3-бром-4-цианофенилгидразин (соединение 10). В чистой сухой 250-мл круглодонной колбе 2-бром-4-фторбензонитрил (25,34 г) растворяли в тетрагидрофуране (50 мл) в атмосфере N2. К полученному раствору медленно добавляли безводный гидразин (50 мл). Цвет раствора изменялся из желтого в красно-оранжевый. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 16 ч. Из раствора осаждалось бело-желтое твердое кристаллическое вещество. Затем смесь разбавляли ТГФ (50 мл) для растворения выпавших твердых веществ. После этого органический слой промывали насыщенным раствором бикарбоната натрия до тех пор, пока рН органического слоя не достигал приблизительно 8,5. Органический слой отделяли и удаляли растворитель при пониженном давлении, получая белое твердое вещество. Это вещество помещали в воронку с пористым стеклянным фильтром, промывали 1,5 л воды и затем диэтиловым эфиром (приблизительно 200 мл). Затем эфир объединяли с белым твердым веществом и высушивали при пониженном давлении. Указанное в заглавии соединение выделяли в виде рыхлого белого или не совсем белого твердого вещества (23,43 г, выход 87,2%). ЖХМС m/z: рассчитанная масса=212,05; наблюдавшаяся=252,98 2-бром-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил (соединение 11). В чистом сухом 20-мл микроволновом реакционном сосуде соединение примера 10 (2,49 г) смешивали с 2-ацетил-5,5-диметил-1,3-циклогександионом (2,14 г). Содержимое сосуда растворяли в смеси этанолуксусная кислота (12 мл, 3:1). Сосуд плотно закрывали и перемешивали на приборе для встряхивания. Затем сосуд помещали в микроволновый реактор и нагревали до 150 С в течение 15 мин. После этого сосуд охлаждали и помещали в холодильник на 1 ч. Затем охлажденный раствор разбавляли водой(8 мл) и выливали на пористый стеклянный фильтр. Оранжевое твердое вещество промывали Н 2 О(100 мл) и затем этанолом (25 мл). Затем это твердое вещество высушивали при пониженном давлении. Указанное в заглавии примера соединение получали в виде кристаллического твердого вещества светлооранжевого цвета (3,7463 г, выход 88,85%). ЖХМС m/z: (М+Н)=358,1. Пример 12.NaOt-Bu (54 мг, 0,56 ммоль) добавляли в 2-мл микроволновый сосуд. Добавляли толуол (0,5 мл) и 4-аминотетрагидропиран (56 мкл, 0,56 ммоль), сосуд вакуумировали и заполняли N2. Реакционную смесь нагревали при 120 С в течение 15 мин (микроволновая печь). Реакционную смесь фильтровали и твердый остаток промывали метиленхлоридом. Продукт очищали с применением флэш-хроматографии,элюируя гексаном и этилацетатом. Продукт выделяли в виде твердого вещества не совсем белого цвета(88 мг, 83%), ЖХМС m/z: (М+Н)=379,3. К полученному пиразолу (88 мг, 0,23 ммоль) в 2-мл микроволновом сосуде добавляли этанол (0,8 мл), ДМСО (0,2 мл), NaOH (5 н., 93 мкл, 2 мол.экв.) и Н 2 О 2 (0,1 мл,30% раствор в Н 2 О). Реакционную смесь нагревали при 100 С в течение 10 мин. Продукт выделяли промыванием полученной смеси H2O и этилацетатом. Растворитель удаляли в вакууме, получая указанное в заглавии примера соединение в виде твердого вещества желтого цвета (88 мг, 100%), ЖХМС m/z: 2-(тетрагидротиопиран-4-иламино)-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензамид (соединение 13). Тетрагидротиопиран-4-он (10,0 г, 86,0 ммоль) и NH4OAc (60 г, 10 мол.экв.) растворяли в 200 мл смеси MeOH/H2O (1:1). К этой смеси добавляли NaCNBH3 (10,8 г, 172,0 ммоль) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Метанол удаляли в вакууме,продукт экстрагировали из водного слоя этилацетатом (3100 мл) и промывали насыщенным раствором соли. Органический слой промывали 10% водным раствором HCl (150 мл) и затем подщелачивали (до рН 11) 10% раствором NaOH. После этого основной раствор промывали этилацетатом и высушивали надMgSO4, получая 4-аминотетрагидротиопиран, который использовали без дальнейшей очистки ЖХМСm/z: (М+Н)+=118,2. Соединение 11 (1,4 г, 3,9 ммоль), Pd(OAc)2 (44,7 мг, 5 мол.%), DPPF (229 мг,10 мол.%) и NaOt-Bu (790 мг, 7,8 ммоль) добавляли в 20-мл микроволновый сосуд. В этот же сосуд добавляли толуол (12 мл) и 4-аминотетрагитиопиран (550 мг, 1,2 мол.экв.), сосуд вакуумировали и заполняли N2. Реакционную смесь нагревали при 130 С в течение 20 мин (микроволновая печь). Реакционную смесь фильтровали и твердый остаток промывали CH2Cl2. Продукт очищали, применяя флэшхроматографию, элюируя гексаном и этилацетатом. Продукт, т.е. 2-(тетрагидротиопиран-4-иламино)-4(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил, выделяли в виде твердого вещества не совсем белого цвета (350 мг), ЖХМС m/z: (М+Н)+=395,3. Продукт предыдущей стадии (830 мг, 2,1 ммоль) растворяли в этаноле (12 мл) и ДМСО (3 мл), к которым был добавлен NaOH (5 н., 841 мкл, 2 мол.экв.). Реакционную смесь нагревали при 70 С в течение ночи. Реакционную смесь промывали H2O и EtOAc. Продукт очищали флэш-хроматографией, элюируя гексаном и этилацетатом. Указанное в заглавии соединение получали в виде твердого вещества не совсем белого цвета (490 мг), ЖХМС m/z: (М+Н)+=413,2. Пример 14. 2-(1-оксогексагидро-1-тетрагидротиопиран-4-иламино)-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензамид (соединение 14). 2-(тетрагидротиопиран-4-иламино)-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензамид растворяли в этаноле, к которому был добавлен Н 2 О 2 (несколько капель 30% раствора в Н 2 О). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 30 мин. Продукт выделяли экстракцией с использованием EtOAc и H2O. Указанное в заглавии соединение получали в виде твердого вещества не совсем белого цвета (20 мг), ЖХМС m/z: (M+H)+=429,2. Пример 15. 2-(1,1-диоксогексагидро-1-тетрагидротиопиран-4-иламино)-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензамид (соединение 15). 2-(тетрагидротиопиран-4-иламино)-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензамид (25 мг, 0,063 ммоль) растворяли в смеси этанола (1 мл) и ДМСО (0,25 мл), к которой были добавлены NaOH (5 н., 25 мкл, 2 мол.экв.) и Н 2 О 2 (избыток, 30% раствор в Н 2 О). Реакционную смесь нагревали при 70 С в течение ночи. Реакционную смесь промывали H2O и EtOAc. Продукт очищали, применяя флэш-хроматографию, элюируя дихлорметаном и метанолом. Указанное в заглавии примера соединение получали в виде твердого вещества не совсем белого цвета (20 мг), ЖХМС m/z: (M+H)+=445,2.(9,3 г, 49,9 ммоль, 1,0 экв.) и п-толуолсульфоновой кислоты (100 мг, 0,53 ммоль, 0,01 экв.) в 300 мл толуола нагревали до кипения с обратным холодильником. Через 30 мин реакционную смесь охлаждали и к ней добавляли 50 мл толуола. Полученную смесь повторно нагревали до кипения с обратным холодильником. Через 1 ч реакционную смесь охлаждали до комнатной температуры. Твердые вещества отделяли фильтрованием, три раза промывали эфиром и высушивали в вакууме, получая 3,3-диметил-5-(п-толилсульфонилгидразоно)циклогексанон (14,26 г, 93%) в виде твердого вещества светло-желтого цвета,ЖХ/МС (m/z): [М+Н]+=309,1. К раствору/суспензии 3,3-диметил-5-(п-толилсульфонилгидразоно)циклогексанона (4,0 г, 12,97 ммоль,1,0 экв.) в 72 мл тетрагидрофурана и 24 мл триэтиламина добавляли ангидрид трифторуксусной кислоты(1,8 мл, 12,97 ммоль, 1,0 экв.). Темно-красную реакционную смесь нагревали при 55 С. Через 15 мин реакционная смесь превращалась в гомогенную. Через 2 ч реакционную смесь охлаждали до комнатной температуры. Добавляли метанол (16 мл) и смесь 1:1 вода-1 М водный раствор гидроксида натрия(16 мл). После перемешивания в течение 3 ч реакционную смесь разбавляли 50 мл насыщенного водного раствора хлорида аммония и экстрагировали этилацетатом (3 50 мл). Объединенные органические слои промывали насыщенным раствором соли, высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток фильтровали через пробку силикагеля, элюируя этилацетатом. Фильтрат концентрировали в вакууме и остаток обрабатывали эфиром. Твердые вещества отделяли фильтрованием и промывали эфиром. Фильтрат концентрировали в вакууме и образовавшийся остаток обрабатывали эфиром. Твердые вещества отделяли фильтрованием, промывали эфиром и объединяли с ранее полученным твердым продуктом, получая 6,6-диметил-3-трифторметил-1,5,6,7-тетрагидроиндазол-4-он (1,24 г, 41%) в виде твердого вещества красновато-оранжевого цвета, ЖХ/МС (m/z): [M+H]+=233,1. Пример 17. 2-бром-4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил (соединение 17). Гидрид натрия (168 мг, 7,02 ммоль, 1,0 экв.) добавляли к раствору 6,6-диметил-3-трифторметил 1,5,6,7-тетрагидроиндазол-4-она (1,63 г, 7,02 ммоль, 1,0 экв.) в 35 мл безводного диметилсульфоксида. Через 15 мин добавляли твердый 2-бром-4-фторбензонитрил (2,25 г, 11,23 ммоль, 1,6 экв.). Реакционную смесь нагревали при 45 С. Через 23 ч реакционную смесь охлаждали до комнатной температуры и гасили 10 мл насыщенного водного раствора хлорида аммония. Смесь разбавляли водой и экстрагировали этилацетатом (4). Объединенные органические слои промывали насыщенным раствором соли, высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали на колонкеBiotage (SiO2, гексан-этилацетат), получая 2-бром-4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил (1,83 г, 63%) в виде порошка не совсем белого цвета, ЖХ/МС:(64 мг, 5 мол.%), DPPF (328 мг, 10 мол.%) и NaOt-Bu (1,13 мг, 11,2 ммоль) добавляли в 20-мл микроволновый сосуд. В этот же сосуд добавляли толуол (15 мл) и 1-амино-3-циклопентен (11,2 ммоль), сосуд вакуумировали и заполняли N2. Реакционную смесь нагревали при 120 С в течение 15 мин. Реакционную смесь фильтровали и твердый остаток промывали CH2Cl2. Продукт очищали с применением флэш- 19013521 хроматографии, элюируя гексаном и этилацетатом. Получали продукт в виде твердого вещества не совсем белого цвета (1,35 г), ЖХМС m/z: (М+Н)+=361,2. Продукт предыдущей стадии (2,71 г, 7,5 ммоль) растворяли в этаноле (20 мл) и ДМСО (5 мл) и добавляли NaOH (5 н., 2,51 мл, 2 мол.экв.) и Н 2 О 2 (3,0 мл, 30% раствор в Н 2 О). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь промывали Н 2 О и экстрагировалиEtOTAc. Продукт очищали с применением флэш-хроматографии, элюируя гексаном и этилацетатом. Получали указанное в заглавии примера соединение в виде твердого вещества не совсем белого цвета(500 мг, 1,4 0 ммоль, 1,0 экв.), 3,4,5-триметоксианилина (513 мг, 2,80 ммоль, 2,0 экв.), ацетата палладия(II) (16 мг, 0,07 ммоль, 0,05 экв) и 1,1'-бис(дифенилфосфино)ферроцена (78 мг, 0,14 ммоль, 0,10 экв.) в 3,75 мл толуола. Реакционный сосуд закрывали пробкой и реакционную смесь 20 мин нагревали до 120 С при микроволновом облучении. Отдельные реакционные смеси разбавляли этилацетатом, объединяли и распределяли с водой. Слои разделяли и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором соли, высушивали над сульфатом натрия,фильтровали и концентрировали в вакууме. Остаток очищали на колонке Biotage System (SiO2, гексан-этилацетат) получая 4-(6,6-диметил-4 оксо-3-метил-4,5,6,7-тетрагидроиндазол-1-ил)-2-(3,4,5-триметоксианилино)бензонитрил (1,59 г, 82%) в виде твердого вещества желтовато-коричневого цвета, ЖХ/МС (m/z): [М+Н]+=461,9. К смеси 4-(6,6-диметил-4-оксо-3-метил-4,5,6,7-тетрагидроиндазол-1-ил)-2-(3,4,5-триметоксианилино)бензонитрила (1,56 г, 3,39 ммоль, 1,0 экв.) и 17 мл смеси 4:1 этанол-диметилсульфоксид добавляли 2 мл 1 М водного гидроксида натрия и 2 мл 30% пероксида водорода. Через 3 ч реакционную смесь разбавляли водой и экстрагировали этилацетатом (3). Объединенные органические слои промывали насыщенным раствором соли, высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме, получая указанное в заглавии соединение, т.е. 4-(6,6-диметил-4-оксо-3-метил-4,5,6,7-терагидроиндазол-1-ил)-2-(3,4,5-триметоксианилино)бензамид(1,61 г, 99%) в виде твердого вещества желтовато-коричневого цвета, ЖХ/МС (m/z): [М+Н]+=479,3. Пример 20. Соль метансульфоновой кислоты и транс-4-[2-карбамоил-5-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндол-1-ил)фениламино]циклогексилового эфира аминоуксусной кислоты (соединение 20). 2-(транс-4-гидроксициклогексиламино)-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндол-1 ил)бензамид (750,0 мг, 1,832 ммоль), N-(трет-бутоксикарбонил)глицин (641,8 мг, 3,664 ммоль, 2,0 экв.),гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (702,4 мг, 3,664 ммоль, 2,0 экв.) и каталитическое количество 4-диметиламинопиридина растворяли в 20 мл CH2Cl2 и перемешивали при комнатной температуре в течение шестнадцати часов. Растворитель удаляли в вакууме и остаток очищали колоночной хроматографией (элюируя EtOAc), получая 780,5 мг (выход 75,2%) пены вещества бледножелтого цвета, защищенного Вос. Продукт предыдущей стадии растворяли в 15 мл CH2Cl2 и охлаждали до 0 С на ледяной бане. Затем добавляли 15 мл трифторуксусной кислоты и реакционную смесь перемешивали при 0 С в течение 5 и 30 мин при комнатной температуре. Трифторуксусную кислоту и CH2Cl2 удаляли в вакууме и остаток растворяли в воде. Водный раствор подщелачивали добавлением насыщенного водного раствора бикарбоната натрия. Водную суспензию экстрагировали EtOAc (3) и объединенные органические слои промывали водой и высушивали над Na2SO4. Удаление растворителя в вакууме приводило к получению 569,7 мг (выход 66,6%, включая стадии сочетания и снятия защиты) пены не совсем белого цвета, кото- 20013521 рая по данным ЖХ/МС представляла собой чистый продукт (m/z: (M+H)=467,3). Полученное свободное основание было затем превращено в мезилатную соль растворением в CH2Cl2 и добавлением по каплям одного эквивалента метансульфоновой кислоты с последующим перемешиванием при комнатной температуре в течение 1 ч. Удаление растворителя в вакууме позволило получить 680,7 мг указанного в заглавии примера соединения в виде порошка не совсем белого цвета. Пример 21. Соль метансульфоновой кислоты и транс-4-[2-карбамоил-5-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндол-1-ил)фениламино]циклогексилового эфира диметиламиноуксусной кислоты (соединение 21). Применяли методику примера 20, заменяя N-(трет-бутоксикарбонил)глицин диметиламиноуксусной кислотой и получая указанное в заглавии примера соединение, ЖХМС m/z: (M+H)=495,2. Пример 22. 2-[3-(2-диметиламиноэтокси)-4-метоксифениламино]-4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7 тетрагидроиндазол-1-ил)бензамид (соединение 22). К раствору 2-метокси-5-нитрофенола (10 г), гидрохлорида (2-хлорэтил)диметиламина (9,37 г) в диметилформамиде (150 мл) медленно, при комнатной температуре и в атмосфере N2 добавляли NaH(2,98 г). Затем смесь перемешивали при 140 С, нагревая на масляной бане в течение 5 ч. Реакционную смесь концентрировали до сухого состояния, растворяли остаток в 2 М HCl (200 мл) и промывали этилацетатом (2100 мл). Водный слой подщелачивали K2CO3, экстрагировали дихлорметаном (3200 мл) и этилацетатом (2100 мл), высушивали над Na2SO4, фильтровали и концентрировали, получая [2-(2 метокси-5-нитрофенокси)этил]диметиламин (9,17 г, выход 65%). ЖХМС (m/z) M+H: 241,1. Продукт предыдущей стадии (10,53 г) и 10% Pd/C (1 г) смешивали с EtOH (200 мл) и гидрировали в течение ночи при давлении 50 фунтов/кв.дюйм. Реакционную смесь фильтровали через целитовую пробку и осадок на фильтре промывали МеОН. Фильтрат концентрировали до сухого состояния, получая 3-(2-диметиламиноэтокси)-4-метоксифениламина (9,05 г, выход 98%). ЖХМС (m/z) M+H: 211,2. 2-бром-4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил (500 мг,1 экв.), 3-(2-диметиламиноэтокси)-4-метоксифениламин (306 мг, 1,2 экв.), Pd(OAc)2 (14 мг), DPPF (66 мг) и NaOt-Bu (214 мг, 2 экв.) смешивали в толуоле (5 мл) и нагревали микроволновым излучением до 140 С в течение 25 мин. Затем добавляли 10 мл смеси EtOH(4)/H2O(1) с последующим добавлением NaOH(700 мг, 2 экв.) и H2O2 (1 мл). Смесь нагревали микроволновым излучением до 100 С в течение 15 мин,затем концентрировали до сухого состояния и очищали флэш-хроматографией, элюируя 20% EtOH в дихлорметане и получая указанное в заглавии соединение (480 мг, выход двух стадий 71%). ЖХМС(14 мг, 0,06 ммоль, 0,05 экв.) и DPPF (67 мг, 0,12 ммоль, 0,10 экв.) в 3,75 мл толуола. Реакционный сосуд закрывали пробкой и реакционную смесь нагревали микроволновым излучением 20 мин при 120 С. Реакционную смесь разбавляли водой и этилацетатом и объединяли. Слои разделяли и водный слой экстра- 21013521 гировали этилацетатом (3), объединенные органические слои промывали насыщенным раствором соли,высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали на системе Biotage (SiO2, гексан-этилацетат), получая 4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол-1-ил)-2-(тетрагидропиран-4-иламино)бензонитрил (772 мг, 49%) в виде твердого вещества желтовато-коричневого цвета, ЖХ/МС (m/z): [М+Н]+=433,7. Дальнейшее элюирование этилацетатом привело к получению 4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол-1-ил)-2-(тетрагидропиран-4-иламино)бензамида (713 мг, 44%) в виде твердого желтого вещества, ЖХ/МС (m/z):[М+Н]+=451,2. К раствору/суспензии 4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол-1-ил)-2-(тетрагидропиран-4-иламино)бензонитрила (772 мг, 1,79 ммоль, 1,0 экв.) в 10 мл смеси этанолдиметилсульфоксид 4:1 добавляли 1 мл 1 М водного раствора гидроксида натрия и 1 мл 30% раствора пероксида водорода. Через 30 мин реакционную смесь разбавляли водой и экстрагировали этилацетатом(4). Объединенные органические слои промывали насыщенным раствором соли, высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме, получая указанное в заглавии примера соединение, т.е. 4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол-1-ил)-2-(тетрагидропиран-4 иламино)бензамид (788 мг, 98%) в виде твердого вещества желто-коричневого цвета, ЖХ/МС (m/z): 3-дифторметил-6,6-диметил-1,5,6,7-тетрагидроиндазол-4-он (соединение 24). Смесь 5,5-диметилциклогексан-1,3-диона (10 г), п-толуолсульфонилгидразина (13,3 г), п-толуолсульфоновой кислоты (140 мг) в толуоле (450 мл) кипятили с обратным холодильником в течение 0,5 ч. Затем добавляли 100 мл толуола и смесь кипятили с обратным холодильником еще 1 ч. Смесь охлаждали до комнатной температуры, фильтровали, твердый остаток промывали эфиром (3200 мл) и полностью высушивали, получая гидразон в виде твердого вещества (20,4 г, 92,7%). ЖХМС М+Н: 309,1 (мол.масса: 308). Полученное соединение применялось в следующей стадии без дальнейшей очистки. Продукт предыдущей стадии (8,86 г) растворяли в смеси тетрагидрофурана (100 мл) и триэтиламина (30 мл), помещали в атмосферу N2 и медленно при перемешивании добавляли ангидрид дифторуксусной кислоты (5 г), затем полученную смесь нагревали до 55 С в течение ночи. Смесь охлаждали до комнатной температуры и добавляли МеОН (35,5 мл) с последующим добавлением 35,5 мл смеси 1:1 Н 2 О и 1 н. NaOH. Эту смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь разбавляли насыщенным водным раствором NH4Cl (120 мл), экстрагировали этилацетатом (4150 мл), высушивали над Na2SO4, фильтровали, концентрировали, очищали колоночной хроматографией, элюируя 1:1 смесью этилацетата и гексана, получая 3-дифторметил-6,6-диметил-1,5,6,7 тетрагидроиндазол-4-он (3,12 г, 50,6%). ЖХМС М+Н: 215,1 (мол.масса: 214). Пример 25. 2-бром-4-(3-дифторметил-6,6-диметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил (соединение 25) NaH (350 мг) добавляли к раствору 3-дифторметил-6,6-диметил-1,5,6,7-тетрагидроиндазол-4 она (3,12 г) в ДМСО (75 мл) при комнатной температуре. После 20-минутного перемешивания добавляли 2-бром-4-фторбензонитрил (4,67 г) и перемешивали при 45 С в течение ночи. Реакционную смесь разбавляли насыщенным водным раствором NH4Cl (100 мл), Н 2 О (100 мл). Смесь экстрагировали этилацетатом (4150 мл), высушивали над Na2SO4, фильтровали, концентрировали, очищали колоночной хроматографией, элюируя смесью 1:2 этилацетат/гексан. Концентрат желаемых фракций превращали в суспензию в эфире, перемешивали в течение 2 ч,фильтровали, промывали гексаном, получая чистый твердый 2-бром-4-(6,6-диметил-4-оксо-3 дифторметил-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил (2,82 г, 49,2%). ЖХМС m/z: (M+H)=395,65(146 мг, 1,52 ммоль, 2,0 экв.) добавляли к перемешиваемому раствору/суспензии 2-бром-4-(6,6-диметил 4-оксо-3-дифторметил-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрила (300 мг, 0,76 ммоль, 1,0 экв.), ацетата палладия(II) (8,5 мг, 0,04 моль, 0,05 экв.) и 1,1'-бис(дифенилфосфино)ферроцена (42 мг, 0,08 ммоль,0,1 экв.) в толуоле (2,25 мл). Реакционный сосуд закрывали пробкой и реакционную смесь нагревали в течение 20 мин при 120 С под действием микроволнового излучения. Реакционную смесь разбавляли водой и этилацетатом и объединяли. Слои разделяли и водный слой экстрагировали этилацетатом (3). Объединенные органические слои промывали насыщенным раствором соли, высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали на системе Biotage (SiO2, гексанэтилацетат), получая 4-(6,6-диметил-4-оксо-3-дифторметил-4,5,6,7-тетрагидроиндазол-1-ил)-2-(тетрагидропиран-4-иламино)бензонитрил (897 мг, выход 65%) в виде твердого вещества желтовато-коричневого цвета,ЖХ/МС (m/z):[M+H]+ 415,2. Дальнейшее элюирование этилацетатом позволило получить соединение,указанное в заглавии примера, т.е. 4-(6,6-диметил-4-оксо-3-дифторметил-4,5,6,7-тетрагидроиндазол-1 ил)-2-(тетрагидропиран-4-иламино)бензамид (131 мг, 9%) в виде твердого вещества желтого цвета,ЖХ/МС (m/z):[М+Н]+ 433,3. К раствору/суспензии 4-(6,6-диметил-4-оксо-3-дифторметил-4,5,6,7-тетрагидроиндазол-1-ил)-2(тетрагидропиран-4-иламино)бензонитрила (897 мг, 2,16 ммоль, 1,0 экв.) в 10,8 мл смеси 4:1 этанолдиметилсульфоксид добавляли 1 мл 1 М водного раствора гидроксида натрия и 1 мл 30% раствора пероксида водорода. Через 30 мин реакционную смесь разбавляли водой и экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором поваренной соли, высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме, получая соединение, указанное в заглавии примера, т.е. 4-(6,6-диметил-4-оксо-3-дифторметил-4,5,6,7-тетрагидроиндазол-1-ил)-2-(тетрагидропиран-4-иламино)бензамид (869 мг, 93%) в виде твердого вещества желтовато-коричневого цвета, ЖХ/МС 2-[3-(2-диметиламиноэтокси)-4-метоксифениламино]-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензамид (соединение 27). 2-бром-4-(6,6-диметил-4-оксо-3-метил-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил (100 мг, 0,28 ммоль),Pd(OAc)2 (3,1 мг, 5 мол.%), DPPF (15,5 мг, 10 мол.%) и NaOt-Bu (52 мг, 0,56 ммоль) добавляли в микроволновый сосуд. В этот же сосуд добавляли толуол (0,5 мл) и 3-(2-диметиламиноэтокси)-4-метоксифениламин (118 мг, 2 мол.экв.), сосуд вакуумировали и заполняли N2. Реакционную смесь нагревали при 130 С в течение 30 мин (микроволновое излучение). Реакционную смесь фильтровали и твердый остаток промывали CH2Cl2. Полученный нитрил очищали с применением флэш-хроматографии, элюируя CH2Cl2 и метанолом. Нитрил (32 мг, 0,66 ммоль) растворяли в этаноле (0,8 мл) и ДМСО (0,2 мл), к которым были добавлены NaOH (5 н., 26 мкл, 2 экв.) и H2O2 (избыток, 30% раствор в H2O). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь промывали H2O и экстрагировали EtOAc. Продукт очищали с применением флэш-хроматографии, элюируя CH2Cl2 и метанолом. Указанное в заглавии соединение получали в виде твердого вещества желтого цвета (10 мг, 30%),ЖХ/МС (m/z): М+Н=506,5.(500,6 мг, 5,0 ммоль) и 4-диметиламинопиридин (916,3 мг, 7,5 ммоль, 1,5 экв.) растворяли в 10 мл CH2Cl2 и охлаждали до 0 С на ледяной бане. По каплям в течение 2 мин к раствору циклогександиона добавляли раствор N,N'-дициклогексилкарбодиимида (1,238 г, 16,0 ммоль, 1,2 экв.) в 5 мл CH2Cl2. После этого реакционную смесь перемешивали при 0 С в течение 10 мин и при комнатной температуре в течение 16 ч. Затем реакционную смесь фильтровали и очищали фильтрат колоночной хроматографией (градиент гексан-30% EtOAc/гексан), получая 1,022 г (выход 92%) 2-(2-циклопропилацетил)-5,5-диметилциклогексан 1,3-диона в виде масла бледно-желтого цвета. 2-(2-циклопропилацетил)-5,5-диметилциклогексан-1,3-дион (111,1 мг, 0,5 ммоль) и гидрохлорид 4-цианофенилгидразина (84,8 мг, 0,5 ммоль, 1,0 экв.) суспендировали в 4 мл смеси 3:1 EtOH:AcOH и подвергали действию микроволнового излучения при 100 С в течение 10 мин. Растворитель удаляли в вакууме и образовавшийся осадок очищали колоночной хроматографией, получая 81,8 мг (выход 51%) 4-(3-циклопропилметил-6,6-диметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрила в виде порошка не совсем белого цвета. После растворения полученного нитрила в 10 мл смеси 4:1 EtOH-ДМСО добавляли приблизительно 0,1 мл 1 н. NaOH (водн.) и 0,1 мл 30% Н 2 О 2 (водн.) и раствор перемешивали в течение 1,5 ч. Реакционную смесь выливали в насыщенный раствор соли, экстрагировали этилацетатом (3) и объединенные органические фазы промывали насыщенным раствором соли (2). Полученные органические экстракты высушивали над Na2SO4 и концентрировали в вакууме, получая 73,2 мг (выход 43%) 4-(3-циклопропилметил 6,6-диметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензамида в виде твердого вещества не совсем белого цвета. ЖХМС m/z: (М+Н)=339,4. Пример 29. Соль метансульфоновой кислоты и 2-[2-(2-диметиламиноэтокси)пиридин-4-иламино]-4-(3,6,6 триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензамида (соединение 29). 2-хлор-4-нитропиридин (5,3 г, 33,43 ммоль) и N,N-диметилэтаноламина (8 мл) запаивали в пробирку высокого давления и перемешивали при 110 С в течение 24 ч. После этого реакционную смесь концентрировали и высушивали при пониженном давлении для удаления избытка N,N-диметилэтаноламина. Полученный остаток желтого цвета разбавляли 1 н. HCl и остаток желтого цвета, который идентифицировали, как 2-хлор-4-нитропиридин отделяли фильтрованием. Кислотный водный раствор промывалиEtOAc (2), подщелачивали добавлением 3 н. раствора NaOH и экстрагировали EtOAc (4). Объединенные органические фазы высушивали над Na2SO4 и удаляли растворитель в вакууме, получая 3,55 г (выход 50,3%) N,N-диметил-N'-(4-нитропиридин-2-ил)этан-1,2-диамина в виде твердого вещества оранжевого цвета. Этот продукт (3,55 г, 16,807 ммоль) растворяли в 60 мл этанола. Добавляли примерно пять капель концентрированной HCl и затем Pd/C (10 мас.%) на кончике шпателя. Реакционный сосуд продували N2,заполняли Н 2, вакуумировали три раза и заполняли Н 2 до давления 60 фунтов/кв.дюйм. После встряхивания при комнатной температуре в течение 3 ч полноту восстановления нитрогруппы проверяли ЖХ/МС. Реакционную смесь фильтровали через целит и удаляли растворитель в вакууме. Затем остаток растворяли в EtOAc, промывали насыщенным водным раствором NaHCO3, водой и насыщенным раствором соли и высушивали над Na2SO4. После удаления растворителя в вакууме получали 2,95 г (выход двух стадий 48,7%) 2-(2-диметиламиноэтокси)пиридин-4-иламина в виде воскообразного твердого вещества красного цвета. 2-Бром-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил (358,2 мг, 1,0 ммоль), 2(2-диметиламиноэтокси) пиридин-4-иламин (362,5 мг, 2,0 ммоль, 2,0 экв.), ацетат палладия(II) (11,2 мг,5 мол.%), 1,1'-бис(дифенилфосфино)ферроцен (55,4 мг, 10 мол.%) и трет-бутоксид натрия (192,2 мг,2,0 ммоль, 2,0 экв.) суспендировали в 4 мл толуола. Реакционную смесь нагревали действием микровол- 24013521 нового излучения при 120 С в течение 20 мин. После охлаждения реакционной смеси растворитель удаляли в вакууме и остаток разбавляли водой. Водную суспензию экстрагировали EtOAc3 и объединенные органические экстракты промывали насыщенным раствором соли, высушивали над Na2SO4 и концентрировали при пониженном давлении. Промежуточно образовавшийся нитрил и желаемый амид собирали, объединяли после очистки колоночной хроматографией (EtOAc/MeOH) и растворяли в 20 мл смеси 4:1 EtOH/ДМСО. Добавляли 5 капель 1 н. NaOH и пять капель 30% Н 2 О 2 и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Раствор разбавляли водой, экстрагировали EtOAc(3), объединенные органические экстракты промывали насыщенным раствором соли (2), высушивали над Na2SO4 и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (EtOAc/MeOH), получая 347,3 мг (выход 72,9%) пены розового цвета. Эту пену растворяли в CH2Cl2 и обрабатывали одним эквивалентом метансульфоновой кислоты, перемешивая в течение 1 ч при комнатной температуре. Смесь упаривали при пониженном давлении, получая соединение, указанное в заглавии примера. ЖХМС m/z: (M+H)=477,3. Пример 30. 2-(2,3-дигидроксипропиламино)-4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол 1-ил)бензамид (соединение 30). Смесь 2,4-дифторбензонитрила (1,39 г, 10 ммоль) 3-аминопропан-1,2-диола (0,911 г, 10 ммоль) и этилдиизопропиламина (1,74 мл) в ДМСО (10 мл) нагревали микроволновым излучением при 200 С в течение 7 мин. После этого реакционную смесь выливали в насыщенный водный раствор NH4Cl (100 мл),экстрагировали EtOAc (3100 мл), высушивали над Na2SO4, фильтровали и концентрировали. Неочищенную смесь очищали флэш-хроматографией на силикагеле (EtOAc-гексан 1:1), получая 2-(2,3 дигидроксипропиламино)-4-фторбензонитрил (0,87 г, выход 41,4%). ЖХМС m/z: (М+Н)=211. К смеси 6,6-диметил-3-трифторметил-1,5,6,7-тетрагидроиндазол-4-она (0,961 г, 4,14 ммоль, 1 экв.),NaH (99 мг, 4,14 ммоль, 1 экв.) и диметилацетамида (20 мл) медленно добавляли 2-(2,3-дигидроксипропиламино)-4-фторбензонитрил (0,87 г, 4,14 ммоль, 1 экв.). После этого реакционную смесь перемешивали при 150 С в течение ночи, охлаждали, выливали в насыщенный водный раствор NH4Cl (100 мл),экстрагировали EtOAc (3150 мл), высушивали над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле, элюируя EtOAc и получая 2-(2,3 дигидроксипропиламино)-4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил (1,7 г, выход 97%). ЖХМС (m/z): (М+Н)=423. 2-(2,3-дигидроксипропиламино)-4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол 1-ил)бензонитрил (1,7 г, 4 ммоль, 1 экв.), NaOH (806 мг, 20 ммоль, 5 экв.) и Н 2 О 2 (3 мл) растворяли в смеси EtOH-вода (4:1) (40 мл). Смесь нагревали микроволновым излучением до 120 С в течение 15 мин,затем концентрировали до сухого состояния и очищали флэш-хроматографией, элюируя 10% МеОН в 4-(6,6-диметил-4-оксо-3-трифторметил-4,5,6,7-тетрагидроиндазол-1-ил)-2-(тетрагидротиопиран-4 иламино)бензамид (соединение 31). 1-(2-бром-4-цианофен-4-ил)-3-трифторметил-6,6-диметилтетрагидроиндазал-4-он (313 мг, 0,76 ммоль),Pd(OAc)2 (8,7 мг, 5 мол.%), DPPF (44,5 мг, 10 мол.%) и NaOt-Bu (153 мг, 1,52 ммоль) добавляли в микроволновый сосуд. Туда же добавляли толуол (1 мл) и 4-аминотетрагидротиопиран (116 мг, 1,3 мол.экв.),сосуд вакуумировали и заполняли N2. Реакционную смесь нагревали при 130 С в течение 20 мин (микроволновое излучение). Реакционную смесь фильтровали и твердый остаток промывали CH2Cl2. Продукт очищали с применением флэш-хроматографии, элюируя гексаном и EtOAc. Продукт выделяли в виде твердого вещества не совсем белого цвета (23 мг, 7%), ЖХМС (М+Н)+=449,4. Полученный пиразол (23 мг, 0,05 ммоль) растворяли в этаноле (0,8 мл) и ДМСО (0,2 мл), к которым добавляли NaOH (5 н., 21 мкл, 2 мол.экв.), и реакционную смесь нагревали до 70 С в течение 12 ч. Реакционную смесь промывали водой и EtOAc. Продукт очищали с применением флэш-хроматографии,- 25013521 элюируя гексаном и EtOAc. Указанное в заглавии соединение получали в виде твердого вещества не совсем белого цвета (16 мг, 67%), ЖХ/МС (m/z): (М+Н)=467,2. Пример 32. 4-(3-дифторметил-6,6-диметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)-2-(тетрагидротиопиран-4 иламино)бензамид (соединение 32). 2-бром-4-(3-дифторметил-6,6-диметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил) бензамид (300 мг,0,76 ммоль), Pd(OAc)2 (8,7 мг, 5 мол.%), DPPF (44,5 мг, 10 мол.%) и NaOt-Bu (153 мг, 1,52 ммоль) добавляли в микроволновый сосуд. Туда же добавляли толуол (1 мл) и 4-аминотетрагидротиопиран (116 мг,1,3 мол.экв.), сосуд вакуумировали и заполняли N2. Реакционную смесь нагревали при 120 С в течение 15 мин (микроволновое излучение). Реакционную смесь фильтровали и твердый остаток промывалиCH2Cl2. Продукт очищали с применением флэш-хроматографии, элюируя гексаном и EtOAc. Продукт выделяли в виде твердого вещества не совсем белого цвета (70 мг, 21%). Полученный на предыдущей стадии пиразол (70 мг, 0,16 ммоль) растворяли в этаноле (0,8 мл) и ДМСО (0,2 мл), к которым добавляли NaOH (5 н., 65 мкл, 2 мол.экв.) и реакционную смесь нагревали до 70 С в течение 12 ч. Реакционную смесь промывали H2O и EtOAc. Продукт очищали с применением флэш-хроматографии, элюируя гексаном и EtOAc. Указанное в заглавии соединение получали в виде твердого вещества не совсем белого цвета (40 мг, выход 56%), ЖХ/МС (m/z): (М+Н)=449,2. Пример 33. 2-[(тетрагидрофуран-2-илметил)амино]-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1 ил)бензамид (соединение 33). 2-бром-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил (1,0 ммоль, 358 мг),ацетат палладия(II) (0,13 ммоль, 30 мг), 1,1'-бис(дифенилфосфино)ферроцен (0,1 ммоль, 56 мг), третбутоксид натрия (2 ммоль, 192 мг), толуол (2 мл) и 2-(аминометил)тетрагидрофуран (3 ммоль, 0,31 мл) смешивали в микроволновой пробирке, перемешивали в течение непродолжительного времени и нагревали в микроволновом устройстве Personal Chemistry при высоком поглощении до температуры 110 С в течение 900 с. После охлаждения реакционную смесь растворяли в этилацетате (200 мл) и промывали водой (50 мл). Органическую фазу высушивали над сульфатом магния, фильтровали и концентрировали. Остаток очищали хроматографией, получая желаемый промежуточный нитрил в виде твердого вещества желтовато-коричневого цвета (305 мг, выход 81%). К полученному описанным способом нитрилу (0,79 ммоль, 300 мг) добавляли ДМСО (6 капель) и этанол (4 мл). Реакционный сосуд погружали в масляную баню с температурой 50 С, добавляли KOH(5,8 ммоль, 325 мг) и затем 30% пероксид водорода (приблизительно 10 ммоль, 1 мл). Через 40 мин реакционную смесь растворяли в этилацетате (100 мл) и промывали водой (50 мл). Органический слой высушивали над сульфатом магния, фильтровали и концентрировали. Очистка остатка хроматографией позволила получить указанное в заглавии примера соединение в виде твердого вещества белого цвета- 26013521 времени и нагревали в микроволновом устройстве Personal Chemistry при высоком поглощении до температуры 110 С в течение 900 с. После охлаждения реакционную смесь растворяли в этилацетате(200 мл) и промывали водой (25 мл). Водный слой экстрагировали дополнительным количеством этилацетата (200 мл) и объединенные органические фазы высушивали над сульфатом магния, фильтровали и концентрировали. Остаток очищали хроматографией, получая желаемый нитрил в виде твердого вещества желтовато-коричневого цвета (258 мг, выход 61%). К полученному описанным способом нитрилу (0,60 ммоль, 250 мг) добавляли ДМСО (0,1 мл) и этанол (4 мл). Реакционный сосуд погружали в масляную баню с температурой 50 С, добавляли KOH(4,5 ммоль, 255 мг) и затем 30% пероксид водорода (приблизительно 10 ммоль, 1 мл). Через 30 мин реакционную смесь растворяли в этилацетате (100 мл) и промывали водой (50 мл). Водный слой экстрагировали дополнительной порцией этилацетата (100 мл). Объединенные органические слои высушивали над сульфатом магния, фильтровали и концентрировали. Очистка остатка хроматографией позволила получить указанный в заглавии примера бензамид в виде твердого вещества белого цвета (187 мг, выход 71%). ЖХМС (m/z): М+Н=438,2. Пример 35. 2-(2,2,2-трифторэтиламино)-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензамид (соединение 35). 2-бром-4-(3,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрил (2,0 моль, 714 мг),ацетат палладия(II) (0,13 ммоль, 60 мг), 1,1'-бис(дифенилфосфино)ферроцен (0,2 ммоль, 112 мг), третбутоксид натрия (4 ммоль, 384 мг), толуол (4 мл) и 2,2,2-трифторэтиламин (8 ммоль, 0,63 мл) смешивали в 2-5-мл микроволновой пробирке, перемешивали в течение непродолжительного времени и нагревали в микроволновом устройстве Personal Chemistry при высоком поглощении до температуры 110 С в течение 900 с. После охлаждения реакционную смесь растворяли в этилацетате (200 мл) и промывали водой(25 мл). Органическую фазу высушивали над сульфатом магния, фильтровали и концентрировали. Остаток хроматографировали, получая смесь ожидавшегося 2-(2,2,2-трифторэтиламино)-4-(3,6,6-триметил-4 оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензонитрила (346 мг, 50%) в виде твердого вещества желтоватокоричневого цвета и желаемого конечного продукта, т.е. 2-(2,2,2-трифторэтиламино)-4-(3,6,6-триметил 4-оксо-4,5,6,7-тетрагидроиндазол-1-ил)бензамида в виде твердого вещества желтовато-коричневого цвета (77 мг, 10%). ЖХМС (m/z): М+Н=395,1. Пример 36. Следующие соединения получали в основном согласно методикам, приведенным ранее на схемах 1-4 и описанным в приведенных выше примерах. 5,5-диметил-2-(2-оксопропил)циклогексан-1,3-дион (соединение 56). В высушенную колбу помещали гидрид натрия (3,61 г, 142,7 ммоль), к которому добавляли 200 мл безводного ДМФА. Колбу охлаждали на ледяной бане и затем добавляли 5,5-диметил-1,3-циклогександион (20,0 г, 142,7 ммоль) и хлорацетон (11,36 мл, 142,7 ммоль) в 100 мл ДМФА с регулируемой скоростью. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 3 ч. Добавляли насыщенный раствор NH4Cl и смесь несколько раз промывали EtOAc. Объединенные органические слои высушивали над MgSO4, фильтровали и удаляли растворитель в вакууме (ниже 40 С). ЖХМС m/z: (M+H)=197,l. Пример 38. 2,6,6-триметил-6,7-дигидро-1 Н-индол-4(5 Н)-он (соединение 57). Неочищенный трикетон из примера 37 растворяли в 300 мл уксусной кислоты и к раствору добавляли ацетат аммония (55 г, 0,714 моль). Реакционную смесь нагревали при 65 С до исчезновения всех исходных веществ (2-3 ч), охлаждали до комнатной температуры, добавляли Н 2 О и промывали EtOAc. Органический слой промывали насыщенным, раствором NaHCO3 (3), насыщенным раствором соли (1) и высушивали над MgSO4. Растворитель удаляли в вакууме и маслянистый остаток пропускали через- 29013521 пробку оксида кремния. Собирали соответствующие фракции и удаляли растворитель. Полученное твердое вещество промывали EtOAc и гексаном (если добавлено слишком много гексана, вещество вновь выпадало из раствора, после добавления EtOAc становилось вязким. Чтобы избежать этого, достаточно было просто добавить немного EtOAc). Твердое вещество фильтровали и промывали гексаном. Дополнительное количество твердого вещества может быть выделено при удалении растворителя и повторения описанной процедуры. ЖХМС m/z: (М+Н)=178,1. Было собрано приблизительно 5 г желтоватокоричневого твердого вещества, которое было идентифицировано как 2,6,6-триметилтетрагидроиндол-4 он. Пример 39.(1,69 г, 8,5 ммоль) растворяли в безводном ДМФА (50 мл). К этому раствору добавляли NaH (95%,408 мг, 17,0 ммоль) и перемешивали при 50 С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли Н 2 О. Продукт выпадал из раствора, его отфильтровывали и высушивали в вакууме (2,4 г, 79%). (100% чистота по данным ЖХМС М+Н=357). Пример 40. 3-фтор-4-(2,6,6-триметил-4-оксо-4,5,6,7-тетрагидро-1 Н-индол-1-ил)бензонитрил (соединение 59). Пиррол(2,6,6-триметилтетрагидроиндол-4-он) (1,0 г, 5,6 ммоль) и 3,4-дифторбензонитрил (785 мг,5,6 ммоль) растворяли в безводном ДМФА (20 мл). К этому раствору добавляли NaH (95%, 270 мг,11,2 ммоль) и перемешивали при 50 С в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры и промывали Н 2 О и EtOAc. Органический слой высушивали над MgSO4. Колоночная хроматография на силикагеле с элюированием смесью EtOAc-гексан (1:1) приводила к получению продукта в виде твердого вещества желтого цвета (100% чистота по данным ЖХМС М+Н=397,1). Пример 41. 3-бутокси-4-(2,6,6-триметил-4-оксо-4,5,6,7-тетрагидро-1 Н-индол-1-ил)бензамид (соединение 60). 3-фтор-4-(2,6,6-триметил-4-оксо-4,5,6,7-тетрагидроиндол-1-ил)бензонитрил (148 мг, 0,5 ммоль) и 1-бутанол (185,3 мг, 2,5 ммоль) растворяли в безводном ДМФА (1,5 мл). К этой смеси добавляли NaH(24,0 мг, 1 ммоль). После продувания реакционного сосуда (микроволновый сосуд "Personal Chemistry") азотом, реакционный сосуд запаивали и нагревали до 100 С в течение 300 с в микроволновой печи EmyrsOptimizer. После охлаждения реакционную смесь разбавляли H2O и экстрагировали EtOAc (2). Объединенные органические экстракты промывали насыщенным раствором соли (2), высушивали над Na2SO4 и концентрировали до сухого состояния. Затем желтый остаток (3-бутокси-4-(2,6,6-триметил-4-оксо 4,5,6,7-тетрагидроиндол-1-ил)бензонитрил) растворяли в EtOH (3 мл) и обрабатывали 1 мл 1 н. водного раствора NaOH и каталитическим количеством Н 2 О 2. После нагревания этого раствора при 65 С в течение 3 ч происходило количественное превращение нитрила в амид. Колоночная хроматография на оксиде кремния при элюировании EtOAc приводила к получению продукта в виде твердого вещества белого цвета (119,9 мг, 65%). (100% чистота по данным ЖХ/МС М+Н=369,2).
МПК / Метки
МПК: A61P 35/00, C07D 209/14, C07D 231/56, A61K 31/416
Метки: тетрагидроиндазолона, производные
Код ссылки
<a href="https://eas.patents.su/30-13521-proizvodnye-tetragidroindazolona.html" rel="bookmark" title="База патентов Евразийского Союза">Производные тетрагидроиндазолона</a>
Производные 2,3-бензодиазепина и фармацевтические композиции, содержащие эти производные в качестве активного ингредиента

Номер патента: 5867
Опубликовано: 30.06.2005
Авторы: Линг Иштван, Сабо Геза, Харшинг Ласло Габор, Баркоци Йожеф, Леваи Дьердь, Раткаи Зольтан, Грефф Зольтан, Сенаши Габор, Шимиг Дьюла, Мартонне Марко Бернадетт, Вег Миклош, Гиглер Габор
МПК: C07D 243/02, A61K 31/551, A61P 25/00...
Метки: качестве, производные, эти, 2,3-бензодиазепина, ингредиента, содержащие, фармацевтические, композиции, активного
Формула / Реферат:
1. Производное 2,3-бензодиазепина формулы I где X - водород, хлор или метоксигруппа, Y - водород или галоген, Z - метил или хлор, R - C1-4 алкил или группа формулы -NR1R2, где R1 и R2, независимо, представляют собой водород, C1-4 алкил, C1-4 алкоксил или C3-6 циклоалкил, и его фармацевтически приемлемые соли с кислотами. 2. Производное 2,3-бензодиазепина по п.1, где X - хлор, Y - водород, хлор или бром, R - C1-4 алкил, Z - определен в п.1, и...
Производные 3-деоксидесмикозина

Номер патента: 4059
Опубликовано: 25.12.2003
Авторы: Павлович Дражен, Лопотар Невенка, Джерек Марко, Наранджя Амалия
МПК: A61K 31/70, A61P 31/04, C07H 17/08...
Метки: 3-деоксидесмикозина, производные
Формула / Реферат:
1. Производные 3-деоксидесмикозина формулы I в которой R представляет COH или CH(OCH3)2, R1 и R2 означают H или ацетил, R3 означает H или OH, R4 означает N(CH3)2 или N-O(CH3)2, A представляет карбонильную группу (C=O) или CH, линия --- представляет простую или двойную связь, линия ... представляет или двойную или простую связь и линия представляет двойную или простую связь, при условии, что когда A представляет CH, в положении 2, 3...
Фармацевтические композиции, содержащие производные 3-аминоазетидина, новые производные и способ их получения

Номер патента: 4649
Опубликовано: 24.06.2004
Авторы: Иттэнжер Огюстэн, Филош Брюно, Букерель Жан, Ашар Даниель, Майерс Майкл, Бушар Эрве, Гризони Серж
МПК: C07D 205/04, A61P 25/00, A61K 31/397...
Метки: композиции, получения, фармацевтические, новые, способ, содержащие, производные, 3-аминоазетидина
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы в которой R1 обозначает радикал -NHCOR4 или -N(R5)-Y-R6, Y обозначает CO или SO2, R2 и R3, одинаковые или разные, обозначают либо ароматический радикал, выбранный из фенила, нафтила и инденила, которые могут быть незамещенными или замещенными одним или несколькими заместителями: галоген, алкил, алкокси, формил, гидрокси, трифторметил, трифторметокси,...
Производные 5н, 10н-имидазо{1,2-а}индено {1,2-е} пиразин-4-она, способы их получения, содержащее их лекарственное средство и производные инданона в качестве промежуточных продуктов

Номер патента: 218
Опубликовано: 24.12.1998
Авторы: Барро Мишель, Рибей Ив, Дамур Доминик, Алу Жан-Клод, Женевуа-Борелла Ариель, Миньяни Серж, Одьо Франсуа, Немесе Патрик, Жимоне Патрик, Манфр Франко, Арди Жан-Клод
МПК: C07F 9/6561, A61K 31/495, C07D 487/04...
Метки: промежуточных, пиразин-4-она, качестве, инданона, 10н-имидазо{1,2-а}индено, лекарственное, способы, производные, средство, содержащее, получения, продуктов, 1,2-е
Формула / Реферат:
1. Производные 5Н,10H-имидазо[1,2-а] индено[1,2-е]пиразин-4-она формулы (I) в которой R означает атом водорода или карбоксил, алкоксикарбонил, -CO-NR4R5, -РО3H2 или -СН2ОН; R1 означает радикалы -aлк-NH2, -алк-NН-СО-R3, -алк-СООR4, -aлк-CO-NR5R6 или -CO-NH-R7; R3 означает алкил, фенил, фенилалкил, циклоалкил или -NR6R8; R4 означает атом водорода или алкильный радикал; R5 означает атом водорода, алкил, фенил, циклоалкил или...
Производные 2н-пиридазин-3-она, фармацевтические композиции, содержащие эти производные, и способ получения активного ингредиента

Номер патента: 6246
Опубликовано: 27.10.2005
Авторы: Эдьед Андраш, Сенаши Габор, Котаи Надь Петер, Гачальи Иштван, Шмидт Эва, Рацне Байногель Юдит, Баркоци Йожеф, Вельман Янош, Шимиг Дьюла, Паллаги Каталин, Миклошне Ковач Анико, Леваи Дьёрди
МПК: C07D 413/14, A61K 31/50
Метки: фармацевтические, способ, производные, содержащие, эти, активного, ингредиента, 2н-пиридазин-3-она, получения, композиции
Формула / Реферат:
1. Производное 2H-пиридазин-3-она формулы где R означает атом водорода или C1-4-алкильную группу, X и Y независимо представляют собой атом водорода, атом галогена или группу формулы при условии, что один из X и Y всегда представляет собой группу формулы II, а другой радикал представляет собой атом водорода или атом галогена, где в формуле II n имеет значение 1 или 2, и его фармацевтически приемлемые соли присоединения кислот. 2. Производное...
Предыдущий патент: Ингибиторы протеазы вич
Следующий патент: Алкинилпирролопиримидины и их применение в качестве ингибиторов hsp90
Случайный патент: Трактор с устройством автоматического управления направлением движения