Производные дигидроимидазотиазола
Номер патента: 12374
Опубликовано: 30.10.2009
Авторы: Томас Джерард Хью, Роули Роберт Джон, Смит Дональд, Крулле Томас Мартин, Барба Оскар, Досон Грэм Джон





























Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемая соль,
где R1 представляет собой водород, галогено, С1-6алкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С3-6циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С1-2алкилС3-6циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С1-6алкоксикарбонил, циано, -C=N-OR7, С2-6алкенил, возможно замещенный одним или более атомами галогена или группами гидрокси, в котором группа гидрокси не присоединена непосредственно ни к одному из атомов углерода с двойной связью, С2-6алкинил, возможно замещенный одним или более атомами галогена или группами гидрокси, в котором группа гидрокси не присоединена непосредственно ни к одному из атомов углерода с тройной связью, (CH2)mNR5R6, C1-3алкокси, C1-3алкилтио, C1-3алкоксиС1-3алкил или C1-3алкилтиоС1-3алкил;
R2 представляет собой нафталин-1-ил, нафталин-2-ил, тиено[2,3-b]тиофен-2-ил, хинолин-2-ил, изохинолинил или бензоизотиазол-3-ил;
R2 может быть возможно замещен одной или более группами, выбранными из галогено, циано, гидрокси, NR5R6, CONR5R6 или COOR7, или C1-3алкила, С2-3алкенила, С2-3алкинила, С3-6циклоалкила, С1-3алкокси, С1-3гидроксиалкила, С2-3алкоксиалкила или группы C1-3алкилS(O)n, любая из которых может быть возможно замещена одним или более атомами галогена;
R3 и R4 независимо представляют собой водород или C1-3алкил;
R5 и R6 независимо представляют собой водород или C1-3алкил, либо вместе с азотом, к которому они присоединены, образуют 5- или 6-членную гетероциклильную группу;
R7 представляет собой водород или C1-3алкил;
m равен 1, 2 или 3; и
n равен 0, 1 или 2;
при условии, что данное соединение не является:
а) гидробромидом 3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазола или
б) гидробромидом 3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазола.
2. Соединение по п.1, где R1 представляет собой водород, C1-6алкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С3-6циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси, или C1-2алкилС3-6циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси.
3. Соединение по п.2, где R1 представляет собой C1-6алкил.
4. Соединение по любому из пп.1-3, где R2 представляет собой нафталин-1-ил.
5. Соединение по любому из пп.1-4, где R2 замещен одним или двумя заместителями, выбранными из галогено и C1-3алкила.
6. Соединение по любому из пп.1-5, где R2 представляет собой нафталин-1-ил, который не замещен или замещен галогеном по одному или двум положениям 4, 5 или 7.
7. Соединение по п.6, где R2 представляет собой нафталин-1-ил, замещенный фтором или хлором по одному или двум положениям 4, 5 или 7.
8. Соединение по любому из пп.1-7, где R3 и R4 оба представляют собой водород.
9. Соединение формулы (I), выбранное из
2-метил-3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазола;
2-бром-3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазола;
2-хлор-3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазола;
3-тиено[2,3-b]тиофен-2-ил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(4-метилнафталин-1-ил)-5,6-дигидроимидазо[2,1-b]тиазола;
2-(5,6-дигидроимидазо[2,1-b]тиазол-3-ил)хинолина;
3-(4-фторнафталин-1-ил)-5,6-дигидроимидазо[2,1-b]тиазола;
3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазол-2-карбоновой кислоты этилового эфира;
2-бром-3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазола;
2-метил-3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазола;
3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазол-2-илметанола;
2-этинил-3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(7-хлорнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
1-(3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазол-2-ил)этанола;
3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазол-2-карбонитрила;
2-метилсульфанил-3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазола;
(3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазол-2-ил)метанола;
1-(3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазол-2-ил)этанола;
2-(3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазол-2-ил)пропан-2-ола;
3-(4-фторнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
2-этил-3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазола;
2-изопропил-3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазола;
[3-(7-хлорнафталин-1-ил)-5,6-дигидроимидазо[2,1-b]тиазол-2-ил]метанола;
3-(6-фторнафталин-2-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(6-хлорнафталин-2-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
2-этил-3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(5-хлорнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-нафталин-1-ил-2-пропил-5,6-дигидроимидазо[2,1-b]тиазола;
2-метоксиметил-3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(4-хлорнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
2-циклопропил-3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(5-хлорнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
2-изопропил-3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(4-фторнафталин-1-ил)-2-изопропил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(5-хлорнафталин-1-ил)-2-циклопропил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(8-хлорнафталин-2-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(4,5-дифторнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(4,5-дифторнафталин-1-ил)-2-этил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(5,7-дихлорнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(7-хлорнафталин-1-ил)-2-этил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(7-хлорнафталин-1-ил)-5,6-дигидроимидазо[2,1-b]тиазола;
3-(7-хлорнафталин-1-ил)-2-изопропил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(7-хлорнафталин-1-ил)-2-пропил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(4-хлорнафталин-1-ил)-2-этил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(7-метоксинафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(4-метоксинафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
2-метил-3-(5-метилнафталин-1-ил)-5,6-дигидроимидазо[2,1-b]тиазола;
3-(5-метоксинафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
2-метил-3-(5-трифторметилнафталин-1-ил)-5,6-дигидроимидазо[2,1-b]тиазола;
3-(7-фторнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(5-фторнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
1-(2-метил-5,6-дигидроимидазо[2,1-b]тиазол-3-ил)изохинолина;
3-(6-хлорнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(6-фторнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(6,7-дифторнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(5,7-дифторнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(4-хлор-7-фторнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
2-этил-3-(5-фторнафталин-1-ил)-5,6-дигидроимидазо[2,1-b]тиазола;
3-(5-фторнафталин-2-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
2-метил-3-(7,8-дифторнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(4,5-дифторнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(4-гидроксинафталин-1-ил)-2-метиы-5,6-дигидроимидазо[2,1-b]тиазола;
3-(7-гидроксинафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазола;
3-(5-хлорнафталин-1-ил)-5,6-дигидроимидазо[2,1-b]тиазола;
2-этил-3-(7-фторнафталин-1-ил)-5,6-дигидроимидазо[2,1-b]тиазола и
2-аллил-3-(7-хлорнафталин-1-ил)-5,6-дигидроимидазо[2,1-b]тиазола;
в виде свободного основания или его фармацевтически приемлемой соли.
10. Соединение формулы (I), представляющее собой 3-(5-фторнафталин-1-ил)-2-метил-5,6-дигидроимидазо[2,1-b]тиазол или его фармацевтически приемлемую соль.
11. Фармацевтическая композиция, содержащая соединение по любому из пп.1-10 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
12. Способ лечения заболевания или состояния, в котором играет роль обратный захват норадреналина и возможно также серотонина, включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-10, включая соединения из условий (а) и (б), или его фармацевтически приемлемой соли.
13. Способ лечения заболевания или состояния, в котором играет роль обратный захват норадреналина и возможно также серотонина и в котором желателен 5-НТ1А-агонизм, включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-10, включая соединения из условий (а) и (б), или его фармацевтически приемлемой соли.
14. Способ регулирования потребления пищи и/или насыщения, включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-10, включая соединения из условий (а) и (б), или его фармацевтически приемлемой соли.
15. Способ лечения ожирения, включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-10, включая соединения из условий (а) и (б), или его фармацевтически приемлемой соли.
16. Способ лечения метаболического заболевания, выбранного из диабета II типа, метаболического синдрома (синдрома X), нарушенной толерантности к глюкозе, дислипидемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии, низких уровней HDL (липопротеин высокой плотности) и гипертензии, включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-10, включая соединения из условий (а) и (б), или его фармацевтически приемлемой соли.
17. Способ уменьшения возможности возникновения сердечно-сосудистых побочных эффектов в лечении заболевания или состояния, как оно определено в любом из пп.12-16, включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-10, включая соединения условий из (а) и (б), или его фармацевтически приемлемой соли.
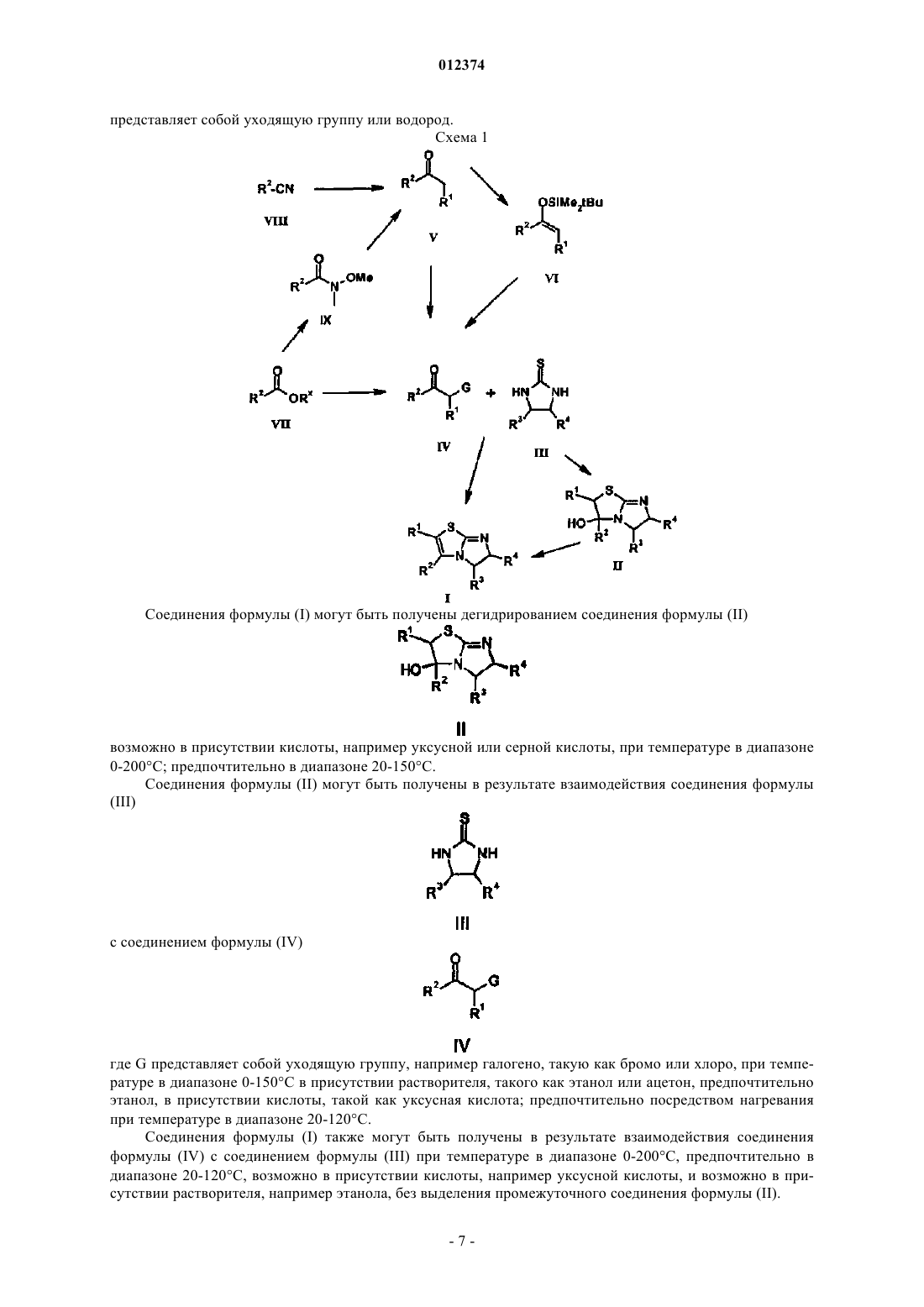
18. Способ получения соединения формулы (I), включающий стадию взаимодействия соединения формулы (III)

с соединением формулы (IV)

где R1-R4 такие, как они определены в п.1, a G представляет собой водород или уходящую группу.
19. Соединение формулы (II)

где R1, R2, R3 и R4 такие, как они определены в п.1.
20. Соединение формулы (X)

где R2, R3 и R4 такие, как они определены в п.1.
Текст
012374 Предшествующий уровень техники Настоящее изобретение относится к производным дигидроимидазо[2,1-b]тиазола, демонстрирующим 5-НТ 1 А-агонизм в дополнение к ингибированию обратного захвата норадреналина и возможно также к ингибированию обратного захвата 5-НТ, полезным для лечения ожирения, например, в качестве регуляторов потребления пищи и/или насыщения. Ожирение характеризуется избыточной массой жировой ткани по отношению к размерам тела. В клинике жировую массу тела оценивают по индексу массы тела (BMI; масса (кг)/рост (м 2 или окружности талии. Субъекты считаются тучными, когда BMI составляет более 30 и установлены медицинские последствия избыточного веса. С некоторого времени в медицинском мире признано, что увеличенная масса тела, в особенности как результат тучности тела в абдоминальной области, ассоциирована с повышенным риском диабета, гипертензии, болезни сердца и другими многочисленными осложнениями состояния здоровья, такими как артрит, инсульт, заболевание желчного пузыря, проблемы, связанные с мышцами и дыханием, боль в спине и даже некоторые виды рака. Фармакологические подходы к лечению ожирения главным образом касаются снижения жировой массы путем изменения баланса между поглощением и расходованием энергии. Во многих исследованиях четко установлена связь между ожирением и мозговым кровообращением, вовлеченным в регуляцию энергетического гомеостаза. Прямые и опосредованные данные позволяют предположить, что в центральной нервной системе в регуляцию поглощения и расходования энергии вовлечены серотонинергический, дофаминергический, адренергический, холинергический, эндоканнабиноидный, опиоидный и гистаминергический проводящие пути в дополнение ко многим нейропептидным проводящим путям(например, с использованием нейропептида Y и меланокортинов). Кроме того, способностью чувствовать периферические гормоны, вовлеченные в поддержание массы тела и степени ожирения, такие как инсулин и лептин и происходящие из жировой ткани пептиды, обладают гипоталамические центры. Лекарственные средства, имеющие целью патофизиологию, ассоциированную с инсулинзависимым диабетом I типа и инсулиннезависимым диабетом II типа, имеют много сильных побочных эффектов и недостаточно направлены на дислипидемию и гипергликемию у значительной части пациентов. Лечение зачастую фокусируется на том, что конкретный пациент нуждается в диете, физических упражнениях,гипогликемических агентах и инсулине, тем не менее, постоянно существует потребность в новых антидиабетических агентах, конкретно в таких, которые могут иметь лучшую толерантность при еще более незначительных неблагоприятных эффектах. Аналогичным образом, метаболический синдром (синдром X), который характеризуется гипертензией и ассоциированными с ней патологиями, включающими атеросклероз, липидемию, гиперлипидемию и гиперхолестеринемию, ассоциирован с пониженной чувствительность к инсулину, что может приводить к аномальным уровням сахара в крови при его введении. Миокардиальная ишемия и микрососудистое заболевание являются установленными заболеваниями, ассоциированными с неизлечимым или слабо регулируемым метаболическим синдромом. Полагают, что класс соединений, названных как ингибиторы обратного захвата серотонина/норадреналина (SNRI), уменьшают потребление пищи и увеличивают расходование энергии посредством усиления функционирования 5-НТ и норадреналина (NA) в центральной нервной системе. Показано, что сибутрамин +)- и (-)-энантиомеры 1-(4-хлорфенил)-N,N-диметил(2-метилпропил)циклобутанметанамина), являющийся членом этого класса соединений, обеспечивает дозозависимое продолжительное уменьшение массы у тучных пациентов путем усиления естественного насыщения и увеличения расходования энергии посредством стимулирования термогенеза. Наиболее общие побочные эффекты, ассоциированные с терапией сибутрамином, включают головную боль, сухость во рту, запор и бессонницу. Однако эта терапия также ассоциирована с дозозависимыми увеличениями частоты сердечных сокращений и кровяного давления, что лимитирует возможность достижения уменьшения массы и противопоказано пациентам с сердечно-сосудистой историей болезни. Ожидается, что SNRI с дополнительной 5-НТ 1 А-агонистической активностью будет иметь улучшенный профиль в отношении сердечно-сосудистой системы по сравнению с одним только SNRI посредством активации постсинаптических 5-НТ 1 А-рецепторов, ввиду чего уменьшается стимуляция симпатической нервной системы (van den Buuse M.Wegener N., 2005, Eur. J. Pharmacol., Vol. 507(1-3), pp. 187-98; Chamienia A.L.Johns E.J., 1996, Brit. J. Pharmacol., Vol. 118(8), pp. 1891-1898). Кроме этого, 5HT1A-агонисты увеличивают активность норадренергических нейронов в locus coeruleus (голубоватое пятно) (Szabo ST.Blier P., 2001, Eur. J. Neuroscience, Vol. 13, pp. 2077-2087) посредством уменьшения возбуждения 5-НТ-нейронов в области линии сращения через 5-НТ 1 А-ауторецепторную активацию, тем самым переводя тоническое торможение 5-НТ в норадренергическую активность в головном мозгеMontigny СBlier P., 1997, Brit. J. Pharmacol., Vol. 120, pp. 865-875). Продемонстрировано, что постсинаптические 5-НТ 1 А-рецепторы ингибируются в незначительной степени после повторного введения 5 НТ 1 А-агонистов, и это показывает, что не будет наблюдаться никакого уменьшения в эффективности при лечении хронических заболеваний (Anxiety and the Serotonin1A Receptor, Jeremy D. Coplan, Susan I. Wolk-1 012374 16(3), pp. 253-260). В WO 97/02269 и WO 00/71549 раскрыты конденсированные тиазоловые производные, обладающие аффинностью к 5-НТ-рецепторам. В WO 98/41528 раскрыты соединения, демонстрирующие ингибирование обратного захвата моноаминов. В WO 02/26747 раскрыто применение соединений, обладающих двойной 5-HT1Aагонистической/ингибиторной активностью в отношении обратного захвата моноаминов для использования в лечении ожирения. В WO 01/62341 раскрыт способ лечения ожирения, включающий введение ингибитора обратного захвата моноаминов и 5-НТ 1 А-агониста. В WO 01/68653 раскрыты дигидроимидазо[2,1-b]тиазолы и дигидро-5 Н-тиазоло[3,2-а]пиримидины,обладающие двойной 5-НТА-агонистической/ингибиторной активностью в отношении обратного захвата моноаминов для применения в лечении депрессии, ожирения и других расстройств. В Sharpe et al., J. Med. Chem., 1971, 14(10), 977 раскрыты фенацилтиоимидазолины и 3-арил-5,6 дигидроимидазо[2,1-b]тиазолы, в том числе соединения гидробромид 3-нафталин-1-ил-5,6 дигидроимидазо[2,1-b]тиазола и гидробромид 3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазола, обладающие антидепрессивной активностью. Для этих соединений механизм действия не раскрыт или о нем не высказано никакого предположения. В заявке на патент СССР 910637 раскрыты 3-(3-хлор-4-пропоксифенил)-5,6-дигидроимидазо[2,1b]тиазол и 3-(3-хлор-4-бутилоксифенил)-5,6-дигидроимидазо[2,1-b]тиазол, обладающие мутагенной активностью. В патентах США 3671533 и 3806515 раскрыты 5,6-дигидроимидазо[2,1-b]тиазоловые производные - 3-(4-хлорфенил)-2-этил-5,6-дигидроимидазо[2,1-b]тиазол и 3-(4-хлорфенил)-2-метил-5,6 дигидроимидазо[2,1-b]тиазол и некоторые 2,3,5,6-тетрагидроимидазо[2,1-b]тиазоловые производные. Установлено, что 2,3,5,6-тетрагидроимидазо[2,1-b]тиазоловые производные предпочтительны. Установлено, что эти соединения обладают активностью, стимулирующей ЦНС, и их можно применять в качестве антидепрессантов, аноректических и диуретических средств. В патенте США 3715367 раскрыты соединения 3-(3,4-дихлорфенил)-5,6-дигидроимидазо[2,1b]тиазол, 3-(2-гидрокси-5-метилфенил)-5,6-дигидроимидазо[2,1-b]тиазол и 3-(4-аминофенил)-5,6-дигидроимидазо[2,1-b]тиазол, обладающие антидепрессивной активностью. Существует постоянная необходимость в новых агентах против ожирения и антидиабетических агентах, конкретно в таких, которые могут иметь лучшую толерантность при еще более незначительных неблагоприятных эффектах. Сущность изобретения Соединения формулы (I) или их фармацевтически приемлемые соли демонстрируют 5-HT1A-агонизм в дополнение к ингибированию обратного захвата норадреналина и возможно также к ингибированию обратного захвата 5-НТ и полезны для лечения ожирения, например, в качестве регуляторов потребления пищи и/или насыщения. Подробное описание изобретения Настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли,где R1 представляет собой водород, галогено, С 1-6 алкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С 3-6 циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С 1-2 алкилС 3-6 циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С 1-6 алкоксикарбонил, циано, -C=N-OR7, С 2-6 алкенил, возможно замещенный одним или более атомами галогена или группами гидрокси, в котором группа гидрокси не присоединена непосредственно ни к одному из атомов углерода с двойной связью, С 2-6 алкинил,возможно замещенный одним или более атомами галогена или группами гидрокси, в котором группа-2 012374 гидрокси не присоединена непосредственно ни к одному из атомов углерода с тройной связью,(CH2)mNR5R6, C1-3 алкокси, C1-3 алкилтио, C1-3 алкоксиС 1-3 алкил или C1-3 алкилтиоС 1-3 алкил;R2 представляет собой 8-10-членную бициклическую ароматическую группу, возможно содержащую до 3 гетероатомов включительно, выбранных из N и S, или фенил, при условии, что R2 не является бензо[b]тиофеном;R2 может быть возможно замещен одной или более группами, выбранными из галогено, циано, гидрокси, NR5R6, CONR5R6 или COOR7, или C1-3 алкила, С 2-3 алкенила, С 2-3 алкинила, С 3-6 циклоалкила, C1-3 алкокси, C1-3 гидроксиалкила, С 2-3 алкоксиалкила или группы C1-3 алкилS(О)n, любая из которых может быть возможно замещена одним или более атомами галогена; или когда R2 представляет собой фенил,два заместителя на фениле могут соединяться с образованием конденсированного С 5-6 карбоциклического кольца;R3 и R4 независимо представляют собой водород или С 1-3 алкил;R5 и R6 независимо представляют собой водород или C1-3 алкил либо вместе с азотом, к которому они присоединены, образуют 5- или 6-членную гетероциклильную группу;R7 представляет собой водород или C1-3 алкил;n равен 0, 1 или 2; при условии, что данное соединение не является:ai) 3-(3,4-дихлорфенил)-2-фенил-5,6-дигидроимидазо[2,1-b]тиазолом. Предпочтительной группой соединений по изобретению являются соединения формулы (Ia) или их фармацевтически приемлемая соль,-3 012374 где R1 представляет собой водород, галогено, C1-6 алкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С 3-6 циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С 1-2 алкилС 3-6 циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С 1-6 алкоксикарбонил, циано, -C=N-OR7, С 2-6 алкенил, возможно замещенный одним или более атомами галогена или группами гидрокси, в котором группа гидрокси не присоединена непосредственно ни к одному из атомов углерода с двойной связью, С 2-6 алкинил,возможно замещенный одним или более атомами галогена или группами гидрокси, в котором группа гидрокси не присоединена непосредственно ни к одному из атомов углерода с тройной связью,(CH2)mNR5R6, C1-3 алкокси, С 1-3 алкилтио, С 1-3 алкоксиС 1-3 алкил или С 1-3 алкилтиоС 1-3 алкил;R2 представляет собой 8-10-членную бициклическую ароматическую группу, возможно содержащую до 3 гетероатомов включительно, выбранных из N и S, при условии, что R2 не является бензо[b]тиофеном;R2 может быть возможно замещен одной или более группами, выбранными из галогено, циано, гидрокси, NR5R6, CONR5R6 или COOR7, или C1-3 алкила, C1-3 алкокси, C1-3 гидроксиалкила, C2-3 алкоксиалкила или группы C1-3 алкилS(О)n, любая из которых может быть возможно замещена одним или более атомами галогена;R3 и R4 независимо представляют собой водород или C1-3 алкил;R5 и R6 независимо представляют собой водород или C1-3 алкил либо вместе с азотом, к которому они присоединены, образуют 5- или 6-членную гетероциклильную группу;R7 представляет собой водород или C1-3 алкил;n равен 0, 1 или 2; при условии, что данное соединение не является: а) гидробромидом 3-нафталин-1-ил-5,6-дигидроимидазо[2,1-b]тиазола или б) гидробромидом 3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазола. Следующей группой соединений, которые могут быть упомянуты, являются соединения формулы или их фармацевтически приемлемая соль, гдеR1 представляет собой водород, галогено, C1-6 алкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С 3-6 циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси, C1-2 алкилС 3-6 циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси, C1-6 алкоксикарбонил, циано, -C=N-OR7, С 2-6 алкенил, возможно замещенный одним или более атомами галогена или группами гидрокси, в котором гидроксил не присоединен непосредственно ни к одному из атомов углерода с двойной связью, С 2-6 алкинил, возможно замещенный одним или более атомами галогена или группами гидрокси, в котором гидроксил не присоединен непосредственно ни к одному из атомов углерода с тройной связью, (CH2)mNR5R6, C1-3 алкокси,C1-3 алкилтио, C1-3 алкоксиС 1-3 алкил или C1-3 алкилтиоС 1-3 алкил;R2 представляет собой фенил, замещенный одной или более группами, выбранными из галогено,циано, гидрокси, NR5R6, CONR5R6 или COOR7, или C1-3 алкила, С 2-3 алкенила, С 2-3 алкинила, С 3-6 циклоалкила, C1-3 алкокси, C1-3 гидроксиалкила, C2-3 алкоксиалкила или группы C1-3 алкилS(О)n, любая из которых может быть возможно замещена одним или более атомами галогена;R3 и R4 независимо представляют собой водород или C1-3 алкил;R5 и R6 независимо представляют собой водород или C1-3 алкил либо вместе с азотом, к которому они присоединены, образуют 5- или 6-членную гетероциклильную группу;R7 представляет собой водород или C1-3 алкил;n равен 0, 1 или 2; при условии, что данное соединение не является:R1 предпочтительно представляет собой водород, C1-6 алкил, возможно замещенный одним или более атомами галогена или группами гидрокси, С 3-6 циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси, или С 1-2 алкилС 3-6 циклоалкил, возможно замещенный одним или более атомами галогена или группами гидрокси. Более предпочтительно R1 представляет собой С 1-6 алкил, особенно метил. Следующей конкретной группой соединений, которые могут быть упомянуты, являются соединения, где R1 не является водородом. Когда R1 представляет собой C1-6 алкил, возможно замещенный одним или более атомами галогена,он может быть фторалкильной группой. Примеры 8-10-членных бициклических ароматических групп, которые могут быть представлены как R2, включают нафталинил, например нафталин-1-ил или нафталин-2-ил, тиенотиофенил, например тиено[2,3-b]тиофен-2-ил, индолил, хинолинил, например хинолин-2-ил, изохинолинил и бензизотиазол,например бензизотиазол-3-ил. Предпочтительно R2 является нафталинилом, особенно нафталин-1-илом. Когда R2 представляет собой замещенную 8-10-членную бициклическую ароматическую группу,например нафталинил, он предпочтительно замещен одним или двумя заместителями, предпочтительно выбранными из галогено, например фторо или хлоро, и C1-3 алкила, например метила. Когда R2 представляет собой нафталин-1-ил, он предпочтительно не замещен или замещен по одному или двум положениям 4, 5 или 7 галогеном, например фтором или хлором. Когда R2 представляет собой фенил, он предпочтительно замещен по положениям 3, 4 и/или 5. Когда R2 представляет собой фенил, он предпочтительно замещен одной или двумя группами, выбранными из галогено и C1-3 алкила, возможно замещенного одним или более атомами галогена. Когда R2 представляет собой фенил, конкретной группой соединений по изобретению, которые могут быть упомянуты, являются соединения, в которых, если R1 представляет собой незамещенный C1-4 алкил с прямой цепью, a R3 и R4 представляют собой водород, R2 не является фенилом, замещенным только одним, двумя или тремя атомами фтора или хлора по положениям 3, 4 и/или 5.R3 и R4 предпочтительно независимо представляют собой водород или метил, более предпочтительно, когда R3 и R4 оба являются водородом. Молекулярная масса соединений формул (I), (Ia) и (Ib) предпочтительно равна менее 800, более предпочтительно менее 600, еще более предпочтительно менее 500. Конкретными соединениями по изобретению, которые могут быть упомянуты, являются соединения, включенные в примеры, и их фармацевтически приемлемые соли. Как использовано в данном описании, если не установлено иное, алкил, равно как и другие группы, имеющие префикс алк, такие как, например, алкенил, алкинил и тому подобное, означает углеродные цепи, которые могут быть линейными или разветвленными, или их комбинации. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил и-5 012374 тому подобное. Алкенил, алкинил и другие подобные термины включают углеродные цепи, имеющие по меньшей мере одну ненасыщенную углерод-углеродную связь. Термин фторалкил включает алкильные группы, замещенные одним или более атомами фтора,например CH2F, CHF2 и CF3. Термины циклоалкил и карбоциклическая группа означают карбоциклы, не содержащие гетероатомы, и включают моноциклические насыщенные карбоциклы. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил и циклогексил. Термин галогено включает атомы фтора, хлора, брома и иода. Термин арил включает фенил и нафтил, в особенности фенил. Термин гетероциклил включает 5- и 6-членные насыщенные кольца, содержащие один или два атома азота. Примеры гетероциклильных групп (колец) включают азетидин, пирролидин, пиперидин и пиперазин. Гетероциклильные группы также могут содержать дополнительные гетероатомы, например морфолин. Соединения, рассмотренные в данном описании, могут содержать один или более асимметрических центров и поэтому могут давать энантиомеры, диастереомеры и оптические изомеры. Настоящее изобретение включает все такие возможные энантиомеры, диастереомеры, равно как и их рацемические смеси,их, по существу, чистые, разделенные энантиомеры, все возможные геометрические изомеры и их фармацевтически приемлемые соли. Приведенное выше соединение формулы (I) показано без определенной стереохимии по некоторым положениям. Настоящее изобретение включает все стереоизомеры соединений формулы (I) и их фармацевтически приемлемые соли. Кроме того, также включены смеси стереоизомеров, равно как и выделенные конкретные стереоизомеры. В ходе синтеза, используемого для получения таких соединений, или при использовании методик рацемизации или эпимеризации, известных специалистам в данной области техники, продукты таких методик могут представлять собой смесь стереоизомеров. Соединения по изобретению также могут демонстрировать атропизомерию; настоящее изобретение включает все атропизомеры формулы (I) и их смеси. Если имеется таутомер соединения формулы (I), настоящее изобретение включает все возможные таутомеры и их фармацевтически приемлемые соли и их смеси, за исключением случаев, когда конкретно отмечено или оговорено иное. Когда соединение формулы (I) и его фармацевтически приемлемые соли существуют в форме сольватов или в полиморфных формах, настоящее изобретение включает любые возможные сольваты и полиморфные формы. Тип растворителя, в котором образуется сольват, не является особенно лимитирующим, если растворитель является фармакологически приемлемым. Например, можно использовать воду,этанол, пропанол, ацетон или тому подобное. Термин фармацевтически приемлемые соли относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот. Когда соединение по настоящему изобретению является кислотным, его соответствующую соль можно удобно получить из фармацевтически приемлемых нетоксичных оснований, в том числе неорганических оснований и органических оснований. Соли, произведенные из таких неорганических оснований, включают соли алюминия, аммония, кальция, меди (двухвалентной и одновалентной), трехвалентного железа, двухвалентного железа, лития, магния, калия, натрия, цинка и тому подобные соли. Особенно предпочтительны соли аммония, кальция, магния, калия и натрия. Соли, произведенные из фармацевтически приемлемых органических нетоксичных оснований,включают соли первичных, вторичных и третичных аминов, равно как и циклических аминов и замещенных аминов, таких как замещенные амины природного происхождения и синтезированные замещенные амины. Другие фармацевтически приемлемые органические нетоксичные основания, из которых могут быть образованы соли, включают аргинин, бетаин, каффеин, холин, N',N'-дибензилэтилендиамин,диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и тому подобное. Когда соединение по настоящему изобретению является основным, его соответствующую соль можно удобно получить из фармацевтически приемлемых нетоксичных кислот, в том числе неорганических и органических кислот. Такие кислоты включают, например, уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую,бромисто-водородную, соляную, изетионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, щавелевую, памовую, пантотеновую, фосфорную, янтарную, серную,винную, п-толуолсульфоновую кислоту и тому подобное. Поскольку соединения формулы (I) предназначены для фармацевтического применения, они предпочтительно предложены, по существу, в чистой форме, например по меньшей мере с чистотой 60%,более приемлема чистота по меньшей мере 75%, особенно чистота по меньшей мере 98% (% представляет собой % мас./мас.). Соединения формулы (I) могут быть получены, как описано ниже и как суммировано на схеме 1, где R1, R2, R3 и R4 определены далее, Rx представляет собой водород или C1-6 алкил, a G-6 012374 представляет собой уходящую группу или водород. Схема 1 Соединения формулы (I) могут быть получены дегидрированием соединения формулы (II) возможно в присутствии кислоты, например уксусной или серной кислоты, при температуре в диапазоне 0-200 С; предпочтительно в диапазоне 20-150 С. Соединения формулы (II) могут быть получены в результате взаимодействия соединения формулы где G представляет собой уходящую группу, например галогено, такую как бромо или хлоро, при температуре в диапазоне 0-150 С в присутствии растворителя, такого как этанол или ацетон, предпочтительно этанол, в присутствии кислоты, такой как уксусная кислота; предпочтительно посредством нагревания при температуре в диапазоне 20-120 С. Соединения формулы (I) также могут быть получены в результате взаимодействия соединения формулы (IV) с соединением формулы (III) при температуре в диапазоне 0-200 С, предпочтительно в диапазоне 20-120 С, возможно в присутствии кислоты, например уксусной кислоты, и возможно в присутствии растворителя, например этанола, без выделения промежуточного соединения формулы (II).-7 012374 Соединения формулы (I) также могут быть получены в результате взаимодействия соединения формулы (III) с соединением формулы (IV), где G представляет собой Н, в присутствии растворителя,например уксусной кислоты, и кислоты, например серной или соляной кислоты, и возможно в присутствии второго дегидрирующего агента, например уксусного ангидрида, при температуре в диапазоне 0200 С, предпочтительно в диапазоне 20-150 С. Согласно следующему аспекту изобретения предложен способ получения соединения формулы (I),включающий стадию взаимодействия соединения формулы (III) где R1-R4 такие, как они определены для формулы (I), a G представляет собой водород или уходящую группу. Соединения формулы (I), в которых R1 представляет собой бромо или хлоро, могут быть получены в результате взаимодействия соединения формулы (I), где R1 представляет собой Н, с галогенирующим агентом, например бромом или тетрахлориодатом бензилтриметиламмония, при температуре в диапазоне 50-150 С, возможно в присутствии растворителя, например дихлорметана, тетрагидрофурана или ацетона. Соединения формулы (I), где R1 представляет собой этоксикарбонил, могут быть получены в результате взаимодействия соединения формулы (IV), где R1 представляет собой этоксикарбонил, a G представляет собой бромо, и соединения формулы (III), описанного выше. Соединения формулы (IV), гдеR1 представляет собой этоксикарбонил, a G представляет собой бромо, могут быть получены из соединений формулы (V), где R1 представляет собой этоксикарбонил, описанными ниже способами галогенирования. Соединения формулы (V), где R1 представляет собой алкоксикарбонил, например этоксикарбонил,могут быть получены из соединения формулы (V), где R1 представляет собой Н, в результате взаимодействия, например, с диэтилкарбонатом в присутствии основания, такого как гидрид натрия. Соединения формулы (III), как правило, имеются в продаже. Соединения формулы (IV), где G представляет собой галогено, могут быть получены в результате взаимодействия соединения формулы (V) с галогенирующим агентом, например бромирующим агентом, таким как трибромид фенилтриметиламмония (РТАТ), бромат натрия, бром или бромид меди(II), в диапазоне 0-200 С в присутствии растворителя, например тетрагидрофурана; предпочтительно в результате нагревания при температуре в диапазоне 20-120 С. Альтернативно, соединения формулы (IV), где G представляет собой бромо, a R2 содержит основные атомы, например R2 представляет собой хинолиновую или изохинолиновую группу, могут быть получены более подходящим способом в результате взаимодействия соединения формулы (V) с трибромидом пиридиния в растворителе, таком как уксусная кислота, при температуре в диапазоне 20-120 С. В дополнение к этому соединения формулы (IV), где G представляет собой галогено, a R2 содержит основные атомы, например R2 представляет собой хинолиновую группу, могут быть получены более подходящим способом в результате взаимодействия соединения формулы (V) с третбутилдиметилсилилтрифлатом и основанием, таким как триэтиламин, в растворителе, таком как дихлорметан, в растворителе, таком как уксусная кислота, при температуре в диапазоне 0-120 С с получением соединения формулы (VI) которое затем можно привести во взаимодействие с галогенирующим агентом, например бромирующим агентом, таким как трибромид фенилтриметиламмония (РТАТ), при температуре в диапазоне 0-200 С в присутствии растворителя, например тетрагидрофурана; предпочтительно в результате нагревания при температуре в диапазоне 20-120 С, с получением соединений формулы (IV) после кислотной обработки при -78 С с использованием смеси минеральных кислот, например бромисто-водородной кислоты и уксусной кислоты. Кроме того, соединения формулы (IV), где G представляет собой бромо, a R1 представляет собой водород, могут быть получены в результате взаимодействия соединения формулы (VII) где Rx представляет собой C1-6 алкил, с дибромметаном и органолитиевым реагентом, таким как метиллитий, в растворителе, таком как тетрагидрофуран, при температуре в диапазоне от -78 С до точки кипения выбранного растворителя, или путем использования дибромметана и основания, такого как диизопропиламин лития (LDA), в растворителе, таком как тетрагидрофуран, при температуре в диапазоне от -78 С до точки кипения выбранного растворителя. Соединения формулы (VII), как правило, имеются в продаже. Соединения формулы (V) могут быть получены непосредственно в результате взаимодействия соединения формулы (VIII) с металлоорганическим реагентом, например соединением формулы R1CH2MgX, где X представляет собой галогено, например хлоро, в присутствии растворителя, например тетрагидрофурана или эфира, при температуре в диапазоне от -50 С до точки кипения выбранного растворителя с последующим гидролизом промежуточной соли имина, возможно в присутствии кислоты, например соляной кислоты. Соединения формулы (VIII) могут быть получены способами, известными специалистам в данной области техники, или они имеются в продаже. В дополнение к этому, соединения формулы (V), где R1 представляет собой Н, также могут быть получены в результате взаимодействия соединения формулы (VII), где Rx представляет собой водород, с дибромметаном и органолитиевым реагентом, таким как метиллитий, в растворителе, таком как тетрагидрофуран, при температуре в диапазоне от -50 С до точки кипения выбранного растворителя. Соединения формулы (V) также могут быть получены в результате взаимодействия соединения формулы (IX) общеизвестного как амид Вейнраба (Weinreb), с металлоорганическим реагентом, например соединением формулы R1CH2MgX, где X представляет собой галогено, например хлоро, в присутствии растворителя,например тетрагидрофурана или эфира, при температуре в диапазоне от -50 С до точки кипения выбранного растворителя с последующим гидролизом промежуточной соли имина, возможно в присутствии кислоты, например соляной кислоты. Кроме этого, соединения формулы (V) также могут быть получены непосредственно из соединений формулы (VII) (где Rx представляет собой Н) в результате взаимодействия сначала с оксалилхлоридом в дихлорметане, содержащем N,N-диметилформамид (DMF), с получением промежуточного хлорангидрида кислоты, который затем далее взаимодействует с металлоорганическим реагентом, например соединением формулы R1CH2MgX, где X представляет собой галогено, например хлоро, в присутствии растворителя, например тетрагидрофурана, в присутствии ацетилацетоната железа(III). Соединения формулы (IX) могут быть получены в результате взаимодействия соединения формулы(VII), где Rx представляет собой водород, и метоксиметиламина в стандартных условиях амидного соче-9 012374 тания, известных специалистам в данной области техники. Дальнейшие подробности получения соединений формулы (I) представлены в примерах. Соединения формулы (I) могут быть получены по отдельности или в виде библиотек соединений,содержащих по меньшей мере 2 соединения, например 5-1000, и более предпочтительно 10-100 соединений формулы (I). Библиотеки соединений могут быть получены с использованием комбинаторного подхода разделить на части и смешать или в результате многочисленных параллельных процедур синтеза с применением либо химии в растворе, либо твердофазной химии с использованием методик, известных специалистам в данной области техники. В процессе синтеза соединений формулы (I) лабильные функциональные группы в промежуточных соединениях, например группы гидрокси, карбокси и амино, можно защитить. Защитные группы можно удалить на любой стадии синтеза соединений формулы (I), или они могут присутствовать в конечном соединении формулы (I). Исчерпывающее обсуждение путей возможной защиты различных функциональных групп и способов расщепления полученных защищенных производных приведено, например, вProtective Groups in Organic Chemistry, T.W. Greene and P.G.M. Wuts, (1991) Wiley-Interscience, New York,2nd edition. Все новые промежуточные производные, которые определены выше, такие как соединения формулы (II), также включены в объем данного изобретения. Другими новыми промежуточными соединениями, включенными в объем данного изобретения, являются соединения формулы (X) где R2, R3 и R4 такие, как они определены ранее. Соединения формулы (X) являются полезными промежуточными соединениями для получения соединений формулы (I), где R1 представляет собой, например,циано или С 1-6 алкил, замещенный гидрокси. Предпочтения, указанные выше для соединений формул (I), (Ia) и (Ib), также применимы к любым промежуточным соединениям, таким как соединения формул (II) и (X). Как указано выше, соединения формулы (I) полезны в качестве ингибиторов обратного захвата норадреналина и также возможно серотонина, например для лечения ожирения. Для такого применения соединения формулы (I) обычно будут введены в форме фармацевтической композиции. Данное изобретение также охватывает фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с фармацевтически приемлемым носителем. Предпочтительно такая композиция содержит фармацевтически приемлемый носитель и нетоксичное терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли. Более того, согласно изобретению также предложена фармацевтическая композиция для лечения заболевания посредством ингибирования обратного захвата норадреналина и возможно также серотонина, например, имеющая резульатом лечение ожирения, содержащая фармацевтически приемлемый носитель и нетоксичное терапевтически эффективное количество соединения формулы (I), в том числе соединений из условий (а) и (b), или его фармацевтически приемлемой соли. Фармацевтические композиции могут возможно содержать другие терапевтические ингредиенты или адъюванты. Эти композиции включают композиции, подходящие для перорального, ректального,местного и парентерального (включая подкожное, внутримышечное и внутривенное) введения, хотя наиболее подходящий путь в любом данном случае будет зависеть от конкретного реципиента и природы и тяжести состояний, для которых собираются вводить активный ингредиент. Для удобства фармацевтические композиции можно представлять в стандартной лекарственной форме и изготавливать любым из способов, хорошо известных в фармацевтической области. На практике соединения формулы (I) или их фармацевтически приемлемые соли можно комбинировать в виде активного ингредиента в однородной смеси с фармацевтическим носителем в соответствии с традиционными методиками фармацевтического производства. Носитель может быть представлен широким разнообразием форм в зависимости от формы препарата, необходимой для введения, например пероральной или парентеральной (включая внутривенную). Так, фармацевтические композиции могут быть представлены в виде дискретных единиц, подходящих для перорального введения, таких как капсулы, облатки или таблетки, содержащие каждая предварительно определенное количество активного ингредиента. Кроме того, данные композиции могут быть представлены в виде порошка, в виде гранул, в виде раствора, в виде суспензии в водной жидкости, в- 10012374 виде не-водной жидкости, в виде эмульсии масло-в-воде или в виде эмульсии вода-в-масляной жидкости. В дополнение к общепринятым лекарственным формам, приведенным выше, соединение формулы (I) или его фармацевтически приемлемую соль также можно вводить с применением средств и/или устройств для доставки с регулируемым высвобождением. Такие композиции можно изготовить любым из способов фармации. В общем случае такие способы включают стадию объединения активного ингредиента с носителем, состоящим из одного или более необходимых ингредиентов. В общем случае такие композиции изготавливают путем однообразного и однородного смешивания активного ингредиента с жидкими носителями или тонкоизмельченными твердыми носителями или ими обоими. Затем продукту придают удобную форму для желаемого представления. Кроме того, соединения формулы (I) или их фармацевтически приемлемые соли могут быть включены в фармацевтические композиции в комбинации с одним или более чем одним другим терапевтически активным соединением. Используемый фармацевтический носитель может быть, например, твердым, жидким или газообразным. Примеры твердых носителей включают лактозу, ультрачистый гипс (terra alba), сахарозу, тальк,желатин, агар, пектин, аравийскую камедь, стеарат магния и стеариновую кислоту. Примерами жидких носителей являются сахарный сироп, кокосовое масло, оливковое масло и вода. Примеры газообразных носителей включают углекислый газ и азот. При изготовлении композиций для пероральной лекарственной формы можно использовать любые удобные фармацевтические среды. Например, воду, гликоли, масла, спирты, корригенты, консерванты,красители и тому подобное можно использовать для создания пероральных жидких композиций, таких как суспензии, эликсиры и растворы; в то время как такие носители, как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие вещества, связующие, разрыхлители и тому подобное, можно использовать для создания пероральных твердых композиций, таких как порошки, капсулы и таблетки. Ввиду легкости их введения таблетки и капсулы являются предпочтительными пероральными лекарственными формами, где применяются твердые фармацевтические носители. Возможно, таблетки могут иметь покрытие, наносимое с использованием стандартных методик в водных или неводных условиях. Таблетка, содержащая композицию по данному изобретению, может быть получена прессованием или формованием, возможно с одним или более чем одним вспомогательным ингредиентом или адъювантом. Прессованные таблетки можно получить прессованием, в подходящей машине, активного ингредиента в свободно текучей форме, такой как порошок или гранулы, возможно смешанного со связующим, смазывающим веществом, инертным разбавителем, поверхностно-активным веществом или диспергирующим агентом. Формованные таблетки можно изготовить формованием в подходящей машине смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Каждая таблетка предпочтительно содержит от приблизительно 0,05 мг до приблизительно 5 г активного ингредиента и каждая облатка или капсула предпочтительно содержит от приблизительно 0,05 мг до приблизительно 5 г активного ингредиента. Например, композиция, предназначенная для перорального введения людям, может содержать от приблизительно 0,5 мг до приблизительно 5 г активного агента, смешанного с подходящим и приемлемым количеством вещества-носителя, которое может варьировать от приблизительно 5 до приблизительно 95% суммарной композиции. Стандартные лекарственные формы в общем случае будут содержать от приблизительно 1 мг до приблизительно 2 г активного ингредиента, в типичном случае 25, 50, 100, 200,300, 400, 500, 600, 800 или 1000 мг. Фармацевтические композиции по настоящему изобретению, подходящие для парентерального введения, можно изготовить в виде растворов или суспензий активных соединений в воде. Туда можно вводить подходящее поверхностно-активное вещество, такое как, например, гидроксипропилцеллюлоза. Дисперсии также можно изготовить в глицерине, жидких полиэтиленгликолях и их смесях в маслах. Кроме того, для предотвращения вредного роста микроорганизмов можно включить консервант. Фармацевтические композиции по настоящему изобретению, подходящие для инъекционного применения, включают стерильные водные растворы или дисперсии. Кроме того, такие композиции могут быть в форме стерильных порошков для быстрого приготовления таких стерильных инъекционных растворов или дисперсий. Во всех случаях конечная инъекционная форма должна быть стерильной и должна представлять собой эффективно текучую среду для облегчения набирания в шприц. Фармацевтические композиции должны быть стабильными в условиях производства и хранения: так, они предпочтительно должны быть защищены от загрязняющего действия таких микроорганизмов, как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, включающую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), растительные масла и их подходящие смеси. Фармацевтические композиции по настоящему изобретению могут находиться в форме, подходящей для местного применения, такой как, например, аэрозоль, крем, мазь, лосьон, присыпка или тому подобное. Кроме того, такие композиции могут быть в форме, подходящей для применения в трансдермальных устройствах. Такие композиции могут быть изготовлены с использованием соединения форму- 11012374 лы (I) или его фармацевтически приемлемой соли традиционными способами изготовления. Например,крем или мазь готовят путем смешивания гидрофильного вещества и воды вместе с соединением в количестве от приблизительно 5 до приблизительно 10 мас.% до получения крема или мази желаемой консистенции. Фармацевтические композиции по данному изобретению могут находиться в форме, подходящей для ректального введения, где носитель представляет собой твердое вещество. Предпочтительно, чтобы из смеси можно было сформировать суппозитории, содержащие стандартную дозу. Подходящие носители включают масло какао и другие вещества, обычно используемые в данной области техники. Для удобства суппозитории можно создавать сначала путем смешивания композиции с размягченным(и) или расплавленным(и) носителем(ями), а затем охлаждения и формования в формах. В дополнение к вышеупомянутым ингредиентам-носителям описанные выше фармацевтические композиции могут включать, как будет целесообразно, один или более дополнительных ингредиентовносителей, таких как разбавители, буферы, корригенты, связующие, поверхностно-активные вещества,загустители, смазывающие вещества, консерванты (включая антиоксиданты) и тому подобное. Кроме того, можно включать другие адъюванты, чтобы сделать композицию изотоничной крови предполагаемого реципиента. Композиции, содержащие соединение формулы (I) или их фармацевтически приемлемые соли, также могут быть получены в концентрированной порошковой или жидкой форме. В общем случае в лечении вышеуказанных состояний полезны уровни дозировок порядка от 0,01 до приблизительно 150 мг/кг массы тела в сутки или, альтернативно, от приблизительно 0,5 мг до приблизительно 7 г на одного пациента в сутки. Например, ожирение можно эффективно лечить путем введения от приблизительно 0,01 до 50 мг соединения на килограмм массы тела в сутки или, альтернативно, от приблизительно 0,5 мг до приблизительно 3,5 г на одного пациента в сутки. Тем не менее очевидно, что определенный уровень доз для любого конкретного пациента будет зависеть от ряда факторов, включая возраст, массу тела, общее состояние здоровья, пол, питание, время введения, путь введения, скорость экскреции, лекарственную комбинацию и тяжесть конкретного заболевания, подвергаемого терапии. Соединения формулы (I), включая соединения из условий (а), (b) и (f)-(ai), можно применять в лечении заболеваний или состояний, в которых играет роль обратный захват норадреналина и возможно также серотонина. 5-НТ 1 А-агонистическая активность, демонстрируемая соединениями формулы (I), означает, что такие соединения будут иметь большую эффективность и меньше побочных эффектов, чем только SNRI, в лечении этих заболеваний или состояний. Таким образом, согласно изобретению также предложен способ лечения заболевания или состояния, в котором играет роль обратный захват норадреналина и возможно также серотонина, включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I),включая соединения из условий (а), (b) и (f)-(ai), или его фармацевтически приемлемой соли. Согласно изобретению также предложен способ лечения заболевания или состояния, в котором играет роль обратный захват норадреналина и возможно также серотонина и в котором желателен 5-НТ 1 Аагонизм, включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I), включая соединения из условий (а), (b) и (f)-(ai), или его фармацевтически приемлемой соли. Заболевания или состояния, в которых играет рольобратный захват норадреналина и возможно также серотонина, включают ожирение. В контексте настоящей заявки полагают, что лечение ожирения охватывает лечение заболеваний или состояний, таких как ожирение и другие расстройства приема пищи, ассоциированные с избыточным потреблением пищи, например путем снижения аппетита и массы тела, поддержания уменьшения массы и предупреждения обратной реакции. Соединения по изобретению также можно применять для лечения других заболеваний, в которых ожирение является фактором, включая метаболические заболевания, такие как диабет II типа, метаболический синдром (синдром X), нарушенная толерантность к глюкозе, дислипидемия, гиперлипидемия,гипертриглицеридемия, гиперхолестеринемия, низкие уровни HDL (липопротеин высокой плотности) и гипертензия. Другие заболевания или состояния, в которых играет роль обратный захват норадреналина и возможно также серотонина, включают таковые, описанные в WO 01/68653, например депрессию, тревогу,психозы (например, шизофрению), позднюю дискинезию, привыкание к употреблению лекарственных средств, злоупотребление лекарственными средствами, когнитивные расстройства, болезнь Альцгеймера,обсессивно-компульсивное поведение, приступы паники, социальные фобии, расстройства приема пищи,такие как булимия, анорексия, прием пищи на ходу и одновременно со спиртными напитками, стресс как содействие в прекращении курения, эпилептические припадки, неврологические расстройства, такие как эпилепсия и/или состояния, в которых имеет место неврологическое поражение, такое как инсульт,травма головного мозга, церебральная ишемия, повреждения головы и кровоизлияние. Другие показания включают недержание мочи при напряжении, невропатическую боль и хроническую боль, ассоциированную с лекарственной терапией или лучевой терапией.- 12012374 Согласно изобретению также предложен способ регулирования потребления пищи и/или насыщения, включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I), включая соединения условий из (а), (b) и (f)-(ai), или его фармацевтически приемлемой соли. Согласно изобретению также предложен способ лечения ожирения, включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I), включая соединения из условий (а), (b) и (f)-(ai), или его фармацевтически приемлемой соли. Согласно изобретению также предложен способ уменьшения возможности возникновения сердечно-сосудистых побочных эффектов при лечении заболевания или состояния, которое определено выше,включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I), включая соединения из условий (а), (b) и (f)-(ai), или его фармацевтически приемлемой соли. Согласно изобретению также предложен способ лечения метаболического заболевания, выбранного из диабета II типа, метаболического синдрома (синдрома X), нарушенной толерантности к глюкозе, дислипидемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии, низких уровней HDL и гипертензии, включающий стадию введения субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I), включая соединения из условий (а), (b) и (f)-(ai), или его фармацевтически приемлемой соли. Согласно изобретению также предложено применение соединения формулы (I), включая соединения из условий (а), (b) и (f)-(ai), или его фармацевтически приемлемой соли в лечении состояния, которое определено выше. Согласно изобретению также предложено применение соединения формулы (I), включая соединения из условий (а), (b) и (f)-(ai), или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения состояния, которое определено выше. В способах по данному изобретению термин лечение включает как терапевтическое, так и профилактическое лечение. Соединения формулы (I) или их фармацевтически приемлемые соли могут быть введены сами по себе или в комбинации с одним или более чем одним другим терапевтически активным соединением. Другие терапевтически активные соединения могут представлять собой соединения для лечения того же заболевания или состояния, что и соединения формулы (I), либо другого заболевания или состояния. Терапевтически активные соединения можно вводить одновременно, последовательно или по отдельности. Соединения формулы (I) можно вводить с другими активными соединениями для лечения ожирения и/или диабета, например инсулином и аналогами инсулина, ингибиторами липазы желудка, ингибиторами панкреатической липазы, сульфонилмочевинами и их аналогами, бигуанидами, 2-агонистами,глитазонами, агонистами PPAR (-рецепторы, активируемые пролифератором пероксисом), агонистамиRXR (ретеноидные Х-рецепторы), ингибиторами окисления жирных кислот, ингибиторами глюкозидазы, -агонистами, ингибиторами фосфодиэстеразы, понижающими содержание липидов агентами, ингибиторами гликоген-фосфорилазы, антагонистами МСН-1 (меланин-концентрирующий гормон 1) и антагонистами СВ-1 (каннабиноидные рецепторы-1), амилиновыми антагонистами, ингибиторами липоксигеназы, аналогами сомостатина, активаторами глюкокиназы, глюкагоновыми антагонистами,агонистами инсулинового сигналинга, ингибиторами РТР 1 В (protein tyrosine phosphatase 1 В; протеинтирозинфосфатаза 1 В), ингибиторами глюконеогенеза, антилиполитическими агентами, ингибиторамиGSK (киназа гликогенсинтазы), агонистами галанинового рецептора, аноректическими агентами, агонистами рецепторов CCK (холецистокинин), лептином, серотонинергическими/дофаминергическими лекарственными средствами против ожирения, антагонистами CRF (corticotrophin-releasing factor; кортикотропин-рилизинг-фактор), CRF-связывающими белками, тиреомиметическими соединениями, ингибиторами альдозоредуктазы, антагонистами глюкокортикоидных рецепторов, ингибиторами NHE-1 (Na-Hexchange-1; натрий-водородный обмен типа 1) или ингибиторами сорбитолдегидрогеназы. При применении в комбинированной терапии соединения формулы (I) предпочтительно вводят в комбинации с другими не затрагивающими центральную нервную систему подходами к ожирению, например, их вводят с орлистатом (Xenical) или с агонистом GPR119 (член семейства G-белоксвязывающих рецепторов (GPCR (GPR119 идентифицирован как SNORF25 в WO 00/50562, в которой раскрыты как рецепторы человека, так и рецепторы крысы, и в патенте США 6468756, в котором кроме того раскрыт рецептор мыши), если необходимо периферическое действие. Все публикации, включая патенты и заявку на патент, приведенные в данном описании, но ими не ограничиваясь, включены в данное описание посредством ссылки, как будто показано, что каждая отдельная публикация конкретно и отдельно включена в данное описание посредством ссылки в полном объеме. Теперь изобретение будет описано со ссылкой на следующие далее примеры, которые представлены для иллюстративных целей и не предназначены для ограничения объема настоящего изобретения.- 13012374 Примеры Материалы и методы. Колоночную хроматографию проводили на SiO2 (40-63 меш), если не указано иное. Данные LCMS(жидкостная хроматография с масс-спектрометрией) получали следующим образом: колонка Atlantis, 3 мкм C18 (3,020,0 мм; скорость потока = 0,85 мл/мин) с элюированием раствором Н 2 О-CH3CN, содержащим 0,1%-ную НСО 2 Н, в течение 6 мин, с детектированием в ультрафиолете (УФ) при 220 нм. Информация о градиенте: 0,0-0,3 мин: 100% Н 2 О; 0,3-4,25 мин: изменение до 10% Н 2 О-90% CH3CN; 4,25-4,4 мин: изменение до 100% CH3CN; 4,4-4,9 мин: выдерживание при 100% CH3CN; 4,9-5,0 мин: возвращение к 100% Н 2 О; 5,0-6,0 мин: выдерживание при 100% Н 2 О. Масс-спектры получали с использованием источника ионизации электрораспылением как в режиме положительно заряженных ионов (ES+), так и в режиме отрицательно заряженных ионов (ES-). Спектры ЯМР получали при 27 С на спектрометре VarianMercury 400, действующем при 400 МГц, или на спектрометре Bruker AMX2 500, действующем на 500 МГц. Сокращения и акронимы. Ас: ацетил; AIBN: 2,2'-азобисизобутиронитрил; DCM: дихлорметан; DIPEA: N,N'-диизопропилэтиламин; DMF: N,N-диметилформамид; DMSO: диметилсульфоксид; EDCI: 1-этил-3-(3'-диметиламинопропил)карбодиимид; Et: этил; HOBt: 1-гидроксибензотриазол; Me: метил; iPr: изопропил; РТАТ: трибромид фенилтриметиламмония; RT: время удерживания; кт: комнатная температура; TFA: трифторуксусная кислота; THF: тетрагидрофуран. Подготовительный пример 1. 2-Бром-1-нафталин-1-илэтанон.(1,10 г; 2,92 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь фильтровали и твердое вещество промывали THF (220 мл). Фильтрат концентрировали в вакууме и остаток распределяли между водой (30 мл) и DCM (230 мл). Объединенные органические фазы сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле, элюируя смесью EtOAс:гексаны (1:9), что позволило получить указанное в заголовке соединение. H (CDCl3): 4.56 2-Нафтонитрил (3 г; 19,6 ммоль) растворяли в Et2O (20 мл) с последующим добавлением хлорида этилмагния (2,0 М в Et2O; 9,8 мл; 19,6 ммоль) и реакционную смесь нагревали до температуры дефлегмации в течение 5 ч, затем перемешивали при комнатной температуре в течение 16 ч. Реакцию гасили 2 н.HCl (20 мл) и водой (20 мл), затем экстрагировали в DCM (340 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексаны (1:9), что позволило получить указанное в заголовке соединение. Н (CDCl3): 1.30 1-Нафталин-2-илпропан-1-он (подготовительный пример 2; 3,5 г; 19,0 ммоль) растворяли в THF (50 мл) с последующим добавлением РТАТ (7,1 г; 19,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Твердое вещество отфильтровывали, фильтрат концентрировали в вакууме и очищали на силикагеле, элюируя смесью EtOAc:гексаны (1:9), что позволило получить указанное в заголовке соединение. Н (CDCl3): 1.98 (3 Н, d), 5.47 (1 Н, q), 7.57 (1 Н, m), 7.63 (1 Н, m), 7.91 (2 Н,m), 7.99 (1 Н, d), 8.08 (1H,dd), 8.57 (1 Н, s). Подготовительный пример 4. Метоксиметиламид тиено[2,3-b]тиофен-2-карбоновой кислоты.- 14012374 Тиено[2,3-b]тиофен-2-карбоновую кислоту (1 г; 5,43 ммоль), гидрохлорид N,О-диметилгидроксиламина (0,53 г; 5,43 ммоль) и HOBt (0,73 г; 5,43 ммоль) растворяли в DMF (20 мл) и DIPEA (2,9 мл; 16,8 ммоль). Через 5 мин добавляли EDCI (1,35 г; 7,1 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 24 ч. Растворитель удаляли в вакууме и остаток распределяли между водой (30 мл) и EtOAc (330 мл). Объединенные органические фракции промывали 1 н. NaOH (220 мл), 1 н. HCl (220 мл), соляным раствором (20 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексаны (2:3), что позволило получить указанное в заголовке соединение. Н (CDCl3): 3.40 (3 Н, s), 3.82 (3 Н, s), 7.28 (1 Н, d), 7.39 (1 Н, d),8.13 (1 Н, s). Подготовительный пример 5. 1-Тиено[2,3-b]тиофен-2-илэтанон. Метоксиметиламид тиено[2,3-b]тиофен-2-карбоновой кислоты (подготовительный пример 4; 0,958 г; 4,21 ммоль) растворяли в THF (20 мл) в атмосфере аргона и охлаждали до 0 С. По каплям добавляли бромид метилмагния (1,4 М в смеси толуол:ТНР; 6,3 мл; 8,85 ммоль) и реакционную смесь перемешивали при 0 С в течение 2 ч, затем при комнатной температуре в течение 16 ч. Реакцию гасили 5%-ным HCl в МеОН, затем растворитель удаляли в вакууме. Остаток распределяли между 10%-ным растворомNaHCO3 (50 мл) и EtOAc (340 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле, элюируя смесью EtOAc:гексаны (1:3), что позволило получить указанное в заголовке соединение. Н (CDCl3): 2.60 (3 Н, s), 7.28 (1 Н, d), 7.41 (1 Н, d), 7.83THF (20 мл) с последующим добавлением РТАТ (825 мг; 2,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, выпавшее в осадок твердое вещество отфильтровывали и промывали THF (220 мл). Фильтрат концентрировали в вакууме и остаток распределяли между водой(30 мл) и EtOAc (330 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле, элюируя смесью EtOAc:гексаны (1:4), что позволило получить указанное в заголовке соединение. Н (CDCl3): 4.39 (2 Н, s), 7.32 (1 Н, d), 7.45 (1 Н, d), 7.96 (1 Н, s). Подготовительный пример 7. 2-Бром-1-(4-метилнафталин-1-ил)этанон. Смесь 4-метил-1-ацетонафтона (0,5 г; 2,7 ммоль) и РТАТ (1,1 г; 3,0 ммоль) в безводном THF (20 мл) перемешивали при комнатной температуре в атмосфере аргона в течение 3 ч. Реакционную смесь фильтровали и несколько раз промывали THF. Фильтрат концентрировали в вакууме и остаток распределяли между водой (30 мл) и EtOAc (330 мл). Объединенную органическую фазу сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексан (1:10),что позволило получить указанное в заголовке соединение. H (CDCl3): 2.77 (3 Н, s), 4.57 (2 Н, s), 7.37 (1 Н,d), 7.63 (2 Н, m), 7.86 (1 Н, d), 8.07 (1 Н, d), 8.71 (1 Н, d). Подготовительный пример 8. Метоксиметиламид хинолин-2-карбоновой кислоты.DMF (55 мл) добавляли DIPEA (10,5 мл; 61,3 ммоль). Полученный раствор перемешивали в течение 12 ч при комнатной температуре перед тем, как произвести распределение реакционной смеси между EtOAc(100 мл) и смесью вода:рассол (200 мл; 1:1). Слои разделяли и водную фазу экстрагировали EtOAc (350 мл). После промывания разбавленным раствором NaOH (1 М, 50 мл) и рассолом (50 мл) объединенные- 15012374 органические экстракты сушили (MgSO4), фильтровали и концентрировали в вакууме. Очисткой остатка флэш-хроматографией на силикагеле (элюент: EtOAc) получали указанное в заголовке соединение. НTHF при 0 С в атмосфере аргона. После перемешивания на холоду в течение 2 ч реакционную смесь добавляли к концентрированному раствору NH4Cl (400 мл). Экстракцией EtOAc (4100 мл) с последующим промыванием объединенных экстрактов соляным раствором (150 мл), высушиванием (MgSO4), концентрированием в вакууме и очисткой остатка флэш-хроматографией на силикагеле (элюент, гексан:EtOAc(2:1 получали указанное в заголовке соединение. Способ В: метиллитий (1,6 М раствор в диэтиловом эфире; 10 мл) добавляли к раствору хинолин-2 карбоновой кислоты (1,40 г; 8,1 ммоль) в THF (40 мл) при 0 С в атмосфере аргона. Через 2 ч при энергичном перемешивании последовательно добавляли хлорметилсилан (10 мл, 79 ммоль) и затем через 10 мин разбавленную соляную кислоту (1 М, 30 мл). Водный слой отделяли, дополнительно разбавляли водой (200 мл) и нейтрализовали твердым NaHCO3. После обработки и очистки аналогично способу А получали указанное в заголовке соединение. Н (DMSO): 2.80 (3 Н, s), 7.78 (1 Н, m), 7.90 (1 Н, m), 8.07 (1 Н,d), 8.81 (1 Н, d), 8.20 (1 Н, d), 8.57 (1 Н, d); m/z (ES+) = 172,10 [M+H]+; RT = 3,34 мин. Подготовительный пример 10. 2-[2-Бром-1-(изопропилдиметилсиланилокси)винил]хинолин. Триэтиламин (0,40 мл; 2,85 ммоль) и трет-бутилдиметилсилилхлорид (TBDMS-хлорид) (0,35 мл; 1,52 ммоль) добавляли к раствору 1-хинолин-2-илэтанона (подготовительный пример 9; 240 мг; 1,40 ммоль) в безводном DCM (10 мл) при 0 С в атмосфере аргона. Через 1 ч добавляли РТАТ (535 мг; 1,42 ммоль), ледяную баню убирали и полученную смесь перемешивали в течение дополнительных 2 ч. После распределения между EtOAc (100 мл) и смесью разбавленного раствора Na2S2O3 (10%-ный; 50 мл) и раствора NaHCO3 (50 мл) следовала дополнительная экстракция водной фазы с использованием EtOAc (350 мл). После промывания рассолом (50 мл) объединенные органические экстракты сушили (MgSO4),фильтровали и концентрировали в вакууме. Очисткой остатка флэш-хроматографией на силикагеле Способ А: суспензию трибромида пиридиния (420 мг; 1,31 ммоль) в АсОН (10 мл) обрабатывали бромисто-водородной кислотой (30%-ная в АсОН; 0,26 мл) и затем перемешивали в течение 30 мин при комнатной температуре. Добавляли раствор 1-хинолин-2-ил-этанона (подготовительный пример 9; 200 мг; 1,08 ммоль) и смесь перемешивали в течение 12 ч при комнатной температуре. После удаления растворителя остаток распределяли между EtOAc (100 мл) и концентрированным раствором NaHCO3 (100 мл). Слои разделяли и водную фазу экстрагировали EtOAc (350 мл). Промывкой объединенных экстрактов рассолом (100 мл), высушиванием (MgSO4) и концентрированием в вакууме с последующей перекристаллизацией из гексана получали указанное в заголовке соединение. Способ В: бромисто-водородную кислоту (30%-ная в АсОН; 0,46 мл) добавляли к раствору 2-[2 бром-1-(изопропилдиметилсиланилокси)винил]хинолина (подготовительный пример 10; 420 мг; 1,15 ммоль) в THF (20 мл) при -78 С в атмосфере аргона. Баню IPA (изопропиловый спирт)/сухой лед убирали и смесь перемешивали в течение 12 ч при комнатной температуре. После обработки и очистки аналогично способу А получали указанное в заголовке соединение. Н (DMSO): 5.24 (2 Н, s), 7.82 (1 Н, m), 7.94 4-Фтор-1-ацетонафтон (500 мг; 2,66 ммоль) растворяли в THF (20 мл) с последующим добавлением РТАТ (1,1 г; 2,92 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч,затем выпавшее в осадок твердое вещество отфильтровывали и промывали THF (220 мл). Фильтрат концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексаны(1:9), что позволило получить указанное в заголовке соединение. Н (CDCl3): 4.55 (2 Н, s), 7.19 (1 Н, m),7.64 (1 Н, m), 7.72 (1 Н, m), 7.99 (1 Н, m), 8.19 (1 Н, d), 8.79 (1 Н, d). Подготовительный пример 13. Этиловый эфир 3-нафталин-2-ил-3-оксопропионовой кислоты. Метил-п-нафтилкетон (5 г; 29,4 ммоль) растворяли в диэтилкарбонате (50 мл) в атмосфере аргона и порциями в течение 10 мин добавляли гидрид натрия (60%-ный в минеральном масле; 2,35 г; 58,8 ммоль). Реакционную смесь нагревали до 100 С в течение 4 ч, затем перемешивали при комнатной температуре в течение 16 ч. Растворитель удаляли в вакууме и остаток распределяли между АсОН (5 мл) в воде (200 мл) и Et2O (3100 мл). Объединенные органические фракции промывали рассолом (50 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексаны (от 1:9 до 1:4), что позволило получить указанное в заголовке соединение. H Этиловый эфир 3-нафталин-2-ил-3-оксопропионовой кислоты (подготовительный пример 13; 6,33 г; 26,1 ммоль) растворяли в THF (100 мл) и охлаждали до 0 С. Добавляли РТАТ (9,82 г; 26,1 ммоль) и реакционную смесь перемешивали при 0 С в течение 2 ч, затем при комнатной температуре в течение 16 ч. Выпавшее в осадок твердое вещество отфильтровывали и промывали THF (230 мл). Фильтрат концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексаны (1:9),что позволило получить указанное в заголовке соединение. H (CDCl3): 1.25 (3 Н, t), 4.30 (2 Н, q), 5.84 (1 Н,s), 7.58 (1 Н, m), 7.64 (1 Н, m), 7.90 (2 Н, m), 7.97 (1 Н, d), 8.02 (1 Н, dd), 8.53 (1 Н, s). Подготовительный пример 15. 1-Нафталин-1-илпропанон. 1-Цианонафталин (4,6 г; 30 ммоль) растворяли в Et2O (15 мл) с последующим добавлением хлорида этилмагния (2,0 М в Et2O; 15 мл) и реакционную смесь нагревали до температуры дефлегмации в течение 17 ч. Реакцию гасили разбавленной HCl (2 н., 20 мл) и смесь дополнительно нагревали в течение 1 ч перед добавлением воды (20 мл), смесь разделяли и экстрагировали DCM (340 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью Et2O:изогексан (1:19), что позволило получить указанное в заголовке соединение. Н (CDCl3): 1.27 (3 Н, t), 3.12 (2 Н, q), 7.58 (3 Н, m), 7.90 (2 Н, m), 8.02 (1 Н, d), 8.61 (1 Н, d); m/z (ES+) = 184,09 [M+H]+; RT = 2,37 мин. Подготовительный пример 16. 2-Бром-1-нафталин-1-илпропанон. К перемешиваемому раствору 1-нафталин-1-илпропанона (подготовительный пример 15; 3,0 г; 16,3 ммоль) в THF (20 мл) добавляли РТАТ (6,3 г; 16,3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь фильтровали и твердое вещество промывалиTHF (220 мл). Фильтрат концентрировали в вакууме и остаток распределяли между EtOAc (50 мл) и насыщенным раствором бикарбоната натрия (50 мл). Объединенные органические фазы сушили (MgSO4) и концентрировали в вакууме, что позволило получить указанное в заголовке соединение. Н (CDCl3): 2.0 К раствору хлорида этилмагния (2,0 М в Et2O; 3,6 мл; 7,28 ммоль) в THF (20 мл), охлажденному до 0 С, порциями в течение 10 мин добавляли гидробромид 2-бром-3-нафталин-1-ил-5,6 дигидроимидазо[2,1-b]тиазола (пример 4; 1 г; 2,43 ммоль) и реакционную смесь перемешивали при 0 С в течение 2 ч. В течение 5 мин добавляли DMF (0,75 мл; 9,71 ммоль) и реакционную смесь перемешивали при 0 С в течение 30 мин, затем при комнатной температуре в течение 16 ч. Реакцию гасили насыщенным раствором NH4Cl (40 мл) и водой (20 мл), затем экстрагировали в EtOAc (340 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью MeOH:DCM (1:19), что позволило получить указанное в заголовке соединение. Н (CDCl3): 3.55 (2 Н, m), 4.31 (2 Н, t), 7.63 (4 Н, m), 7.79 (1 Н, m), 7.99 (1 Н, m), 8.07 (1 Н, m), 9.13 (1 Н,s). Подготовительный пример 18. 1-(7-Хлорнафталин-1-ил)пропан-1-он.DCM (50 мл) при -10 С. После перемешивания в течение 20 мин смесь охлаждали до -78 С и по каплям добавляли пропионилхлорид (5,5 мл; 63,3 ммоль). Полученную суспензию выдерживали при -78 С в течение 4 ч перед добавлением ее к разбавленной HCl (0,3 М; 300 мл). Слои разделяли и водный слой дополнительно экстрагировали EtOAc (2100 мл). Экстракты объединяли, промывали рассолом (100 мл) и сушили (MgSO4). Концентрированием с последующей очисткой остатка флэш-хроматографией на силикагеле (элюент, гексан:EtOAc (10:1 получали указанное в заголовке соединение. Н (DMSO): 1.16 (3 Н,t), 3.15 (2 Н, q), 7.62-7.67 (2 Н, m), 8.07 (1 Н, d), 8.19-8.21 (2 Н, m), 8.62 (1 Н, s); m/z (ES+) = 219,01 [М+Н]+; РТАТ (2,21 г; 5,88 ммоль) добавляли к раствору 1-(7-хлорнафталин-1-ил)пропан-1-она (подготовительный пример 18; 1,28 г; 5,85 ммоль) в THF (50 мл). По мере перемешивания при комнатной температуре в течение 2 ч ярко-оранжевый раствор полностью обесцвечивался и образовывался осадок. Смесь фильтровали через целит, фильтрат разбавляли EtOAc (300 мл) и промывали разбавленным растворомNa2S2O3 (10%-ный; 100 мл) и рассолом (100 мл). Высушиванием (MgSO4) и концентрированием в вакууме с последующей перекристаллизацией из EtOH получали указанное в заголовке соединение. Н Гидробромид 2-бром-3-нафталин-2-ил-5,6-дигидроимидазо[2,1-b]тиазола (пример 11; 4,0 г; 9,7 ммоль) суспендировали в THF (50 мл) в атмосфере аргона и охлаждали до 0 С. Добавляли хлорид этилмагния (2,0 М в Et2O; 14,6 мл; 29,1 ммоль) и реакционную смесь перемешивали при 0 С в течение 1 ч,затем при комнатной температуре в течение 1 ч. Реакционную смесь охлаждали до 0 С и добавляли DMF(3,0 мл; 38,8 ммоль). Реакционную смесь перемешивали при 0 С в течение 1 ч затем при комнатной температуре в течение 16 ч. Реакцию гасили насыщенным раствором NH4Cl (50 мл) и водой (25 мл), затем экстрагировали в EtOAc (350 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью MeOH:DCM (1:19), что позволило получить указанное в заголовке соединение. H (CDCl3): 3.87 (2 Н, m), 4.35 (2 Н, m), 7.54 (1 Н,dd), 7.64 (2 Н, m), 7.94 (2 Н, m), 8.02 (2 Н, m), 9.36 (1 Н, s). Подготовительный пример 21. 1-(4-Фторнафталин-1-ил)пропан-1-он. 1-Циано-4-фторнафталин (1 г; 5,8 ммоль) растворяли в Et2O (10 мл) с последующим добавлением хлорида этилмагния (2,0 М в Et2O; 2,9 мл; 5,8 ммоль) и реакционную смесь нагревали до температуры дефлегмации в течение 16 ч. Реакцию гасили 2 н. HCl (20 мл), затем экстрагировали в DCM (340 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексаны (1:9), что позволило получить указанное в заголовке соединение. Н (CDCl3): 1.29 (3 Н, t), 3.08 (2 Н, q), 7.16 (1 Н, m), 7.60 (1 Н, m), 7.66 (1 Н, m), 7.89 (1 Н,m), 8.16 (1 Н, d), 8.74 (1 Н, d). Подготовительный пример 22. 2-Бром-1-(4-фторнафталин-1-ил)пропан-1-он. 1-(4-Фторнафталин-1-ил)пропан-1-он (подготовительный пример 21; 3,30 г; 16,3 ммоль) растворяли в THF (50 мл) и охлаждали до 0 С. Добавляли РТАТ (6,14 г; 16,3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Твердое вещество отфильтровывали и промывалиTHF (220 мл). Фильтрат концентрировали в вакууме и остаток распределяли между водой (30 мл) иEtOAc (250 мл). Объединенные органические фракции промывали рассолом (320 мл), сушили(MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя DCM, что позволило получить указанное в заголовке соединение. H (CDCl3): 1.98 (3 Н, d), 5.36 (1 Н, q), 7.18 (1 Н, m),7.63 (1 Н, m), 7.70 (1 Н, m), 7.92 (1 Н, m), 8.18 (1 Н, d), 8.59 (1 Н, d). Подготовительный пример 23. 1-Нафталин-1-илбутан-1-он. 1-Цианонафталин (1 г; 6,5 ммоль) растворяли в THF (20 мл) в атмосфере аргона и охлаждали до 0 С. Добавляли хлорид пропилмагния (2,0 М в Et2O; 9,8 мл; 19,6 ммоль) и реакционную смесь перемешивали при 0 С в течение 1 ч, затем при комнатной температуре в течение 16 ч. Реакцию гасили 1 н. HCl (40 мл) и экстрагировали в DCM (330 мл). Объединенные органические фракции сушили (MgSO4), концен- 19012374 трировали в вакууме и очищали на силикагеле, элюируя смесью EtOAc:гексаны (1:4), что позволило получить указанное в заголовке соединение. Н (CDCl3) 1.05 (3 Н, t), 1.84 (2 Н, m), 3.04 (2 Н, t), 7.48-7.71 (3 Н,m), 7.87 (2 Н, m), 7.98 (1 Н, d), 8.57 (1 Н, d). Подготовительный пример 24. 2-Бром-1-нафталин-1-илбутан-1-он. 1-Нафталин-1-илбутан-1-он (подготовительный пример 23, 0,99 г, 5,0 ммоль) растворяли в THF (20 мл) и охлаждали до 0 С. Добавляли РТАТ (1,88 г, 5,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Выпавшее в осадок твердое вещество отфильтровывали и промывали THF (210 мл). Фильтрат концентрировали в вакууме и остаток распределяли между водой (40 мл) и EtOAc (240 мл). Объединенные органические фракции промывали рассолом (20 мл), сушили (MgSO4),концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексаны(19), что позволило получить указанное в заголовке соединение. Н (CDCl3) 1.15 (3 Н, t), 2.20 (1 Н, m), 2.33 1-Цианонафталин (1 г, 6,5 ммоль) растворяли в THF (20 мл) в атмосфере аргона и охлаждали до 0 С. Добавляли бромид изобутилмагния (2,0 М в Et2O, 9,8 мл, 19,6 ммоль) и реакционную смесь перемешивали при 0 С в течение 1 ч, затем при комнатной температуре в течение 16 ч. Реакцию гасили 1 н. HCl(30 мл), затем экстрагировали в DCM (330 мл). Объединенные органические фракции сушили (MgSO4),концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексаны(1 9), что позволило получить указанное в заголовке соединение. H (CDCl3) 1.04 (6 Н, d), 2.35 (1 Н, m),2.95 (2 Н, d), 7.48-7.62 (3 Н, m), 7.83 (1 Н, d), 7.89 (1 Н, d), 7.98 (1 Н, d), 8.56 (1H, d). Подготовительный пример 26. 2-Бром-3-метил-1-нафталин-1-илбутан-1-он.THF (10 мл) и охлаждали до 0 С. Добавляли РТАТ (0,64 г; 1,7 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 72 ч. Твердое вещество отфильтровывали и промывали THF(220 мл). Фильтрат концентрировали в вакууме и остаток распределяли между водой (40 мл) и EtOAc(240 мл). Объединенные органические фракции промывали рассолом (220 мл), сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексаны(1:9), что позволило получить указанное в заголовке соединение. Н (CDCl3): 1.13 (3 Н, d), 1.24 (3 Н, d),2.53 (1 Н, m), 5.03 (1 Н, d), 7.52 (1 Н, m), 7.57 (1 Н, m), 7.63 (1 Н, m), 7.89 (2 Н, m), 8.03 (1 Н, d), 8.46 (1 Н, d). Подготовительный пример 27. Метоксиметиламид 5-хлорнафталин-1-карбоновой кислоты. К раствору 5-хлор-1-нафтойной кислоты (1,12 г; 5,4 ммоль) в DMF (10 мл) добавляли N,Oдиметилгидроксиламин (0,53 г; 5,4 ммоль), HOBt (0,73 г; 5,4 ммоль) и DIPEA (2,9 мл; 17,0 ммоль). Через 5 мин добавляли EDCI (1,35 г; 7,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение 96 ч. Растворитель удаляли в вакууме и масло распределяли между водой (100 мл) и EtOAc (3100 мл). Объединенную органическую фазу сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексан (1:1), что по- 20012374 зволило получить указанное в заголовке соединение. Н (CD3OD): 3.42 (6 Н, br s), 7.51 (1 Н, m), 7.62 (1 Н,d), 7.68 (2 Н, m), 7.79 (1 Н, d), 8.37 (1 Н, d). Подготовительный пример 28. 1-(5-Хлорнафталин-1-ил)пропан-1-он. Перемешиваемый раствор метоксиметиламида 5-хлорнафталин-1-карбоновой кислоты (подготовительный пример 27; 0,89 г; 3,6 ммоль) и THF (50 мл) охлаждали до 0 С в атмосфере аргона. По каплям добавляли бромид этилмагния (2,0 М в Et2O; 1,2 мл; 3,6 ммоль) и перемешивали при комнатной температуре в течение 24 ч. Добавляли дополнительное количество бромида этилмагния (1,2 мл; 3,6 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение следующих 24 ч. Реакционную смесь подкисляли концентрированной HCl (5 мл) в метаноле (40 мл) и концентрировали в вакууме. Остаток распределяли между водой (100 мл) и EtOAc (3100 мл). Объединенную органическую фазу сушили(MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью Смесь 1-(5-хлорнафталин-1-ил)пропан-1-она (подготовительный пример 28; 0,13 г; 0,6 ммоль) и РТАТ (0,25 г; 0,66 ммоль) в безводном THF (10 мл) перемешивали при комнатной температуре в атмосфере аргона в течение 48 ч. Реакционную смесь фильтровали и несколько раз промывали THF. Фильтрат концентрировали в вакууме, получая масло, которое распределяли между водой (100 мл) и EtOAc(3100 мл). Объединенную органическую фазу сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексан (1:19) и получая указанное в заголовке соединение. Н (CDCl3): 1.99 (3 Н, d), 5.35 (1 Н, q), 7.53 (1 Н, m), 7.65 (2 Н, m), 7.89 (1 Н, d), 8.31 (1 Н, d), 8.53 К раствору 6-фтор-2-нафтойной кислоты (1,13 г; 5,9 ммоль) в DMF (10 мл) добавляли N,Oдиметилгидроксиламин (0,58 г; 5,9 ммоль), HOBt (0,80 г; 5,9 ммоль) и DIPEA (3,2 мл; 18,2 ммоль). Через 5 мин добавляли EDCI (1,47 г; 7,7 ммоль) и реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение 24 ч. Растворитель удаляли в вакууме и остаток распределяли междуEtOAc (350 мл) и водой (50 мл). Объединенную органическую фазу сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексан (1:1) и получая указанное в заголовке соединение. Н (CDCl3): 3.42 (3 Н, s), 3.57 (3 Н, s), 7.31 (1 Н, m), 7.48 (1 Н, dd), 7.80 (2 Н,s), 7.91 (1 Н, m), 8.24 (1 Н, s). Подготовительный пример 31. 1-(6-Фторнафталин-2-ил)пропан-1-он. Хлорид этилмагния (2,0 М в Et2O; 5,5 мл; 11 ммоль) добавляли по каплям к раствору метоксиметиламида 6-фторнафталин-2-карбоновой кислоты (подготовительный пример 30; 0,18 г; 5,0 ммоль) в THF(50 мл) при 0 С в атмосфере аргона. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 24 ч. Реакционную смесь подкисляли концентрированной HCl (5 мл) в метаноле (40 мл) и концентрировали в вакууме, полученное твердое вещество распределяли между EtOAc (3100 мл)- 21012374 и водой (100 мл). Объединенную органическую фазу сушили (MgSO4), концентрировали в вакууме, что позволило получить указанное в заголовке соединение. Н (CDCl3): 1.30 (3 Н, t), 3.14 (2 Н, q), 7.33 (1 Н, td),7.50 (1 Н, dd), 7.84 (1 Н, d), 7.97 (1H, m), 8.08 (1H, d), 8.48 (1H, s). Подготовительный пример 32. 2-Бром-1-(6-фторнафталин-2-ил)пропан-1-он. Смесь 1-(6-фторнафталин-2-ил)пропан-1-она (подготовительный пример 31; 0,84 г; 4,0 ммоль) и РТАТ (1,72 г; 4,6 ммоль) в безводном THF (10 мл) оставляли перемешиваться при комнатной температуре в течение 24 ч. Реакционную смесь фильтровали и несколько раз промывали THF. Фильтрат концентрировали в вакууме, что позволило получить указанное в заголовке соединение. Н (CDCl3): 1.98 (3 Н, d),5.44 (1 Н, q), 7.36 (1 Н, td), 7.51 (1 Н, dd), 7.88 (1 Н, d), 8.00 (1 Н, m), 8.10 (1 Н, d), 8.57 (1 Н, s). Подготовительный пример 33. 6-Хлорнафталин-2-карбоновая кислота. Перемешиваемый раствор 6-амино-2-нафтойной кислоты (3,14 г; 17,0 ммоль) в воде (10 мл) и концентрированной HCl (20 мл) охлаждали до 0 С и по каплям добавляли раствор нитрита натрия (1,17 г; 17,0 ммоль) в воде (10 мл). Через 1 ч порциями добавляли раствор хлорида меди(I) (3,36 г; 34,0 ммоль) в концентрированной HCl (10 мл) при 0 С. Реакционную смесь разбавляли водой (400 мл) и перемешивали в течение 30 мин. Выпавшее в осадок твердое вещество собирали фильтрованием, несколько раз промывали водой и сушили, что позволило получить указанное в заголовке соединение. Н (DMSO): 7.63 (1 Н,dd), 8.03 (2 Н, m), 8.17 (2 Н, m), 8.64 (1 Н, s). Подготовительный пример 34. Метоксиметиламид 6-хлорнафталин-2-карбоновой кислоты.(4,3 мл; 25 ммоль). Через 5 мин добавляли EDCI (2,0 г; 10,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение 24 ч. Растворитель удаляли в вакууме и остаток распределяли между EtOAc (3100 мл) и водой (100 мл). Объединенную органическую фазу сушили(MgSO4) и концентрировали в вакууме. Остаток растворяли в EtOAc (100 мл) и промывали HCl (2 М, 50 мл). Органический слой промывали гидроксидом натрия (2 М, 50 мл), сушили (MgSO4) и концентрировали в вакууме, что позволило получить указанное в заголовке соединение. Н (CDCl3): 3.42 (3 Н, s), 3.56 Перемешиваемый раствор метоксиметиламида 6-хлорнафталин-2-карбоновой кислоты (подготовительный пример 34; 1,84 г; 7,4 ммоль) и THF (50 мл) охлаждали до 0 С в атмосфере аргона. По каплям добавляли хлорид этилмагния (2M в Et2O; 7,7 мл; 15,4 ммоль) и перемешивали при комнатной температуре в течение 24 ч. Реакционную смесь подкисляли концентрированной HCl (5 мл) в метаноле (40 мл) и концентрировали в вакууме. Остаток распределяли между EtOAc (3100 мл) и бикарбонатом натрия (100 мл). Объединенную органическую фазу сушили (MgSO4) и концентрировали в вакууме, что позволило получить указанное в заголовке соединение. Н (CDCl3): 1.30 (3 Н, t), 3.14 (2 Н, q), 7.51 (1 Н, dd), 7.82 (1 Н,d), 7.90 (2 Н, m), 8.08 (1H, dd), 8.46 (1H, s). Подготовительный пример 36. 2-Бром-1-(6-хлорнафталин-2-ил)пропан-1-он.- 22012374 РТАТ (2,64 г; 7,0 ммоль) в безводном THF (40 мл) перемешивали при комнатной температуре в атмосфере аргона в течение 24 ч. Реакционную смесь фильтровали и твердое вещество несколько раз промывалиTHF. Фильтрат затем концентрировали в вакууме. Остаток очищали хроматографией на силикагеле,элюируя смесью EtOAc:гексан (8:92), что позволило получить указанное в заголовке соединение. H Хлорид пропилмагния (2,0 М в Et2O; 3,3 мл; 6,5 ммоль) добавляли по каплям к перемешиваемому раствору 2-нафтонитрила (1,0 г; 6,5 ммоль) в Et2O (20 мл) при 0 С в атмосфере аргона. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 48 ч. Реакционную смесь подкисляли HCl (2 М, 15 мл) и концентрировали в вакууме, что позволило получить указанное в заголовке соединение. Н (CDCl3): 1.06 (3 Н, t), 1.85 (2 Н, m), 3.09 (2 Н, t), 7.61 (2 Н, m), 7.90 (2 Н, m), 7.98 (1 Н, d), 8.05 Смесь 1-нафталин-2-илбутан-1-она (подготовительный пример 37; 1,26 г; 6,4 ммоль) и РТАТ (2,65 г; 7,0 ммоль) в безводном THF (20 мл) перемешивали при комнатной температуре в атмосфере аргона в течение 48 ч. Реакционную смесь фильтровали и твердое вещество несколько раз промывали THF. Фильтрат концентрировали в вакууме, получая твердое вещество, которое очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексан (1:19), что позволило получить указанное в заголовке соединение. H (CDCl3): 1.14 (3 Н, t), 2.23 (1 Н, m), 2.30 (1 Н, m), 5.25 (1 Н, t), 7.61 (2 Н, m), 7.92 (2 Н, m), 8.00 (1 Н,d), 8.08 (1H, dd), 8.56 (1H, s). Подготовительный пример 39. Бенз[b]тиофен-2,3-дион. Оксалилхлорид (3,8 мл; 43,0 ммоль) по каплям добавляли к перемешиваемому раствору бензолтиола (3 г; 27,0 ммоль) в Et2O (20 мл) и смесь нагревали при температуре дефлегмации в течение 1,5 ч. Раствор охлаждали до комнатной температуры, затем концентрировали в вакууме. Остаток растворяли вDCM (50 мл) и охлаждали до 0 С. К реакционной смеси порциями добавляли хлорид алюминия (2,3 г; 32,0 ммоль) и раствор нагревали при температуре дефлегмации в течение 1 ч. Раствор охлаждали до комнатной температуры и выливали на лед. Органический слой отделяли и последовательно промывали водным бикарбонатом натрия (100 мл), водой (100 мл) и рассолом (100 мл). Органический слой сушили(MgSO4) и концентрировали в вакууме. При добавлении гексана образовывался осадок, который собирали фильтрованием, что позволило получить указанное в заголовке соединение. Н (CDCl3): 7.38 (1 Н, t),7.43 (1 Н, d), 7.70 (1 Н, t), 7.84 (1 Н, d). Подготовительный пример 40. Амид бензо[d]изотиазол-3-карбоновой кислоты. К перемешиваемому раствору бензо[b]тиофен-2,3-диона (подготовительный пример 39; 0,73 г; 4,4 ммоль) в гидроксиде аммония (28%-ный в Н 2 О; 14 мл) по каплям в течение 5 мин добавляли перекись водорода (30%-ная (об./об.); 1,4 мл) и перемешивали в течение 96 ч. Осадок собирали фильтрованием,что позволило получить указанное в заголовке соединение. Н (CDCl3): 5.62 (1 Н, br s) 7.27 (1 Н, br s), 7.57- 23012374 растворяли в оксилхлориде фосфора (10 мл) при 0 С, нагревали до комнатной температуры, затем нагревали при температуре дефлегмации в течение 3 ч. Растворитель удаляли в вакууме и остаток распределяли между ледяной водой (100 мл) и EtOAc (3100 мл). Объединенную органическую фазу сушили(MgSO4) и концентрировали в вакууме, что позволило получить указанное в заголовке соединение. H Бромид этилмагния (3,0 М в Et2O; 0,83 мл; 2,5 ммоль) добавляли по каплям к перемешиваемому раствору бензо[d]изотиазол-3-карбонитрила (подготовительный пример 41; 0,39 г; 2,5 ммоль) в Et2O (20 мл) при 0 С и перемешивали в течение 24 ч в атмосфере аргона. Реакционную смесь подкисляли HCl (2 М; 15 мл) и концентрировали в вакууме; полученное твердое вещество распределяли между водой (100 мл) иEtOAc (3100 мл). Объединенную органическую фазу сушили (MgSO4) и концентрировали в вакууме,что позволило получить указанное в заголовке соединение. Н (CDCl3): 1.29 (3 Н, t), 3.33 (2 Н, q), 7.56 (2 Н,m), 7.98 (1 Н, d), 8.88 (1 Н, d). Подготовительный пример 43. 1-Бензо[d]изотиазол-3-ил-2-бромпропан-1-он. Смесь 1-бензо[d]изотиазол-3-илпропан-1-она (подготовительный пример 42; 0,35 г; 1,8 ммоль) и РТАТ (0,74 г; 2,0 ммоль) в безводном THF (10 мл) перемешивали при комнатной температуре в атмосфере аргона в течение 72 ч. Реакционную смесь фильтровали и несколько раз промывали THF. Фильтрат концентрировали в вакууме, получая масло, которое распределяли между EtOAc (3100 мл), водой (100 мл) и рассолом (50 мл). Объединенную органическую фазу сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, элюируя смесью EtOAc:гексан (1:19), что позволило получить указанное в заголовке соединение. H (CDCl3): 1.97 (3 Н, d), 5.99 (1 Н, q), 7.60 (2 Н, m), 8.00 (1 Н, dd), 8.85 К перемешиваемому раствору 1-(3,4-дихлорфенил)пропан-1-она (1,0 г; 4,924 ммоль) в THF (30 мл) добавляли РТАТ (1,94 г; 5,170 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь фильтровали и твердое вещество промывали THF (220 мл). Фильтрат концентрировали в вакууме и остаток распределяли между насыщенным раствором NaHCO3 (30 мл) иDCM (230 мл). Объединенные органические фазы сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле, элюируя смесью этилацетат:гексаны (1:4), что позволило получить указанное в заголовке соединение в виде бледно-желтого масла. Н (CDCl3): 8.20 (1 Н, s), 7.90 (1 Н, d), 7.60 Диизопропилэтиламин (7,3 мл; 0,042 моль) добавляли к раствору 4-бром-3-метилбензойной кислоты (3 г; 0,014 моль), N,O-диметилгидроксиламина гидрохлорида (1,36 г; 0,014 моль), EDCI (4,01 г; 0,021 моль) и HOBt (1,94 г; 0,01395 моль) в DMF (20 мл) при комнатной температуре. Через 24 ч растворитель удаляли в вакууме и остаток очищали на диоксиде кремния с элюированием DCM, получая указанное в заголовке соединение, m/z (ES+) = 257,93 [М+Н]+; RT = 3,31 мин. Подготовительный пример 46. 1-(4-Бром-3-метилфенил)этанон.(подготовительный пример 45; 1,53 г; 5,93 ммоль) в THF при -70 С. После нагревания до комнатной температуры в течение ночи растворитель частично удаляли и остаток распределяли между дихлорметаном (50 мл) и насыщенным хлоридом аммония (50 мл). Органический слой сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали на силикагеле с элюированием смесью гексан:DCM 1-(4-Бром-3-метилфенил)этанон (подготовительный пример 46; 0,58 г; 2,732 ммоль) и РТАТ (1,027 г; 2,732 ммоль) совместно добавляли в THF (30 мл) при комнатной температуре. После выдерживания реакционной смеси в течение ночи твердое вещество отфильтровывали и фильтрат концентрировали,получая коричневое масло. Очисткой на силикагеле с элюированием смесью гексан:EtOAc (25:1) получали указанное в заголовке соединение, m/z (ES+) = 292,97 [М+Н]+; RT = 2,60 мин. Подготовительный пример 48. Метоксиметиламид 5-хлорнафталин-1-карбоновой кислоты. 5-Хлор-1-нафтойную кислоту (1,12 г; 5,4 ммоль), гидрохлорид N,O-диметилгидроксиламина (0,53 г; 5,4 ммоль) и HOBt (0,73 г; 5,4 ммоль) растворяли в DMF (15 мл) и DIPEA (2,9 мл; 17,0 ммоль). Через 5 мин добавляли EDCI (1,35 г; 7 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 96 ч. Растворитель удаляли в вакууме и остаток распределяли между водой (100 мл) и EtOAc(3100 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле с элюированием смесью EtOAc:гексаны (1:1), что позволило получить указанное в заголовке соединение. H (CDCl3): 3.41 (6 Н, br s), 7.44 (1H, t), 7.61 (3H, m), 7.83 (1H, d), 8.37 К перемешиваемому раствору 1-хлорнафталина (10 г; 61,5 ммоль) в DCM (150 мл) при -10 С порциями в течение 5 мин добавляли хлорид алюминия (24,6 г; 184,5 ммоль) и реакционную смесь охлаждали до -78 С. По каплям в течение 10 мин добавляли пропионилхлорид (10,7 мл; 123,0 ммоль) и реакционную смесь перемешивали при -78 С в течение 16 ч. Реакционную смесь нагревали до 0 С и гасили 1 МHCl с последующим добавлением воды (200 мл). Органический слой отделяли и водный слой экстрагировали в DCM (2125 мл). Объединенные органические фракции промывали 1 М раствором NaOH (250 мл), рассолом (50 мл), сушили (MgSO4) и концентрировали в вакууме, что позволило получить указанное в заголовке соединение. H (CDCl3): 1.28 (3 Н, t), 3.04 (2 Н, q), 7.58 (1 Н, d), 7.64 (2 Н, m), 7.73 (1 Н, d), 8.34 Раствор 5-хлор-1-нафтойной кислоты (2,08 г; 10,1 ммоль) в тионилхлориде (10 мл) кипятили с обратным холодильником в течение 3 ч, затем охлаждали до комнатной температуры и концентрировали в вакууме. К остатку при 0 С осторожно добавляли концентрированный раствор аммиака (10 мл), реакционную смесь нагревали до комнатной температуры и перемешивали в течение 30 мин. Реакционную смесь разбавляли водой (50 мл) и твердое вещество отфильтровывали, промывали водой и сушили под вакуумом. Твердое вещество растворяли в оксихлориде фосфора (10 мл) и нагревали до температуры дефлегмации в течение 4 ч, затем охлаждали до комнатной температуры и медленно выливали в теплую воду (50 мл). Выпавшее в осадок твердое вещество отфильтровывали, промывали водой (220 мл) и сушили под вакуумом, что позволило получить указанное в заголовке соединение. Н (CDCl3): 7.64 (2 Н, m),7.73 (1 Н, m), 8.00 (1 Н, m), 8.20 (1 Н, d), 8.56 (1H,d). Подготовительный пример 51. Метиловый эфир циклопропилуксусной кислоты. К раствору циклопропилуксусной кислоты (2 г; 20,0 ммоль) в МеОН (10 мл) добавляли концентрированную H2SO4 (5 капель) и реакционную смесь нагревали до температуры дефлегмации в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Остаток распределяли между насыщенным раствором NaHCO3 (30 мл) и Et2O (230 мл). Объединенные органические фракции промывали рассолом (220 мл), сушили (MgSO4) и концентрировали в вакууме, что позволило получить указанное в заголовке соединение. H (CDCl3): 0.15 (2 Н, m), 0.54 (2 Н, m), 1.04 (1 Н, m),2.21 (2 Н, d), 3.68 (3 Н, s). Подготовительный пример 52. Метиловый эфир 2-циклопропил-3-нафталин-1-ил-3-оксопропионовой кислоты. н-Бутиллитий (2,5 М; 8,3 мл; 20,9 ммоль) добавляли к раствору диизопропиламина (2,9 мл; 20,9 ммоль) в THF (20 мл) при 0 С в атмосфере аргона. Реакционную смесь перемешивали при 0 С в течение 1 ч и затем охлаждали до -78 С. По каплям добавляли раствор метилового эфира циклопропилуксусной кислоты (подготовительный пример 51; 1,19 г; 10,4 ммоль) в THF (15 мл) и реакционную смесь перемешивали при -78 С в течение 20 мин. Затем добавляли раствор 1-нафтальдегида (1,56 мл; 11.5 ммоль) вTHF (15 мл) и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 ч. Добавляли воду (50 мл) и реакционную смесь экстрагировали в Et2O (350 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле с элюированием смесью EtOAc:гексаны (1:9). Элюированный продукт растворяли в DCM (50 мл) и добавляли периодинан Десса-Мартина (3,30 г; 7,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли DCM (50 мл) и добавляли воду (100 мл). Осадок отфильтровывали и слои разделяли. Водный слой экстрагировали в DCM (250 мл), объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле, элюируя смесью EtOAc:гексаны (1:9), что позволило получить указанное в заголовке соединение. Н (CDCl3): 0.17 (1 Н, m), 0.40 (1 Н, m), 0.63 (1 Н, m), 0.73 (1 Н, m), 1.58 (1 Н, m), 3.60 (1 Н, d), 3.72 (3 Н,s), 7.50 (1 Н, m), 7.56 (1 Н, m), 7.61 (1 Н, m), 7.82 (1 Н, d), 7.89 (1 Н, d), 8.00 (1 Н, d), 8.55 (1H, d). Подготовительный пример 53. 2-Циклопропил-1-нафталин-1-илэтанон. Суспензию метилового эфира циклопропил-3-нафталин-1-ил-3-оксопропионовой кислоты (подготовительный пример 52; 0,61 г; 2,3 ммоль) и NaCl (0,13 г; 2,3 ммоль) в DMSO (15 мл) и воде (0,5 мл) нагревали до 160 С в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры и распреде- 26012374 ляли между водой (50 мл) и EtOAc (340 мл). Объединенные органические фракции сушили (MgSO4),концентрировали в вакууме и хроматографировали на силикагеле, элюируя смесью EtOAc:гексаны (1:9),что позволило получить указанное в заголовке соединение. Н (CDCl3): 0.23 (2 Н, m), 0.61 (2 Н, m), 1.19 Указанное в заголовке соединение получали в соответствии со способом Robert F. O'Malley et al., J. Раствор 1-бром-5-хлорнафталина (подготовительный пример 54; 1 г; 4,14 ммоль) в THF (20 мл) охлаждали до -78 С в атмосфере аргона. По каплям добавляли н-бутиллитий (2,5 М; 1,8 мл; 4,55 ммоль) и реакционную смесь перемешивали при -78 С в течение 1 ч. Добавляли DMF (0,96 мл; 12,4 ммоль) и реакционную смесь перемешивали при -78 С в течение 2 ч, затем нагревали до комнатной температуры и гасили 1 М HCl (30 мл). Реакционную смесь экстрагировали в EtOAc (330 мл), объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле, элюируя смесью EtOAc:гексаны (1:9), что позволило получить указанное в заголовке соединение. Н (CDCl3): 7.60 (1 Н, m), 7.70 (1 Н, d), 7.75 (1 Н, m), 8.05 (1 Н, d), 8.61 (1 Н, d), 9.22 (1 Н, d), 10.41 (1 Н, s). Подготовительный пример 56. Метиловый эфир 3-(5-хлорнафталин-1-ил)-2-циклопропил-3 оксопропионовой кислоты. Указанное в заголовке соединение получали, используя способ, идентичный описанному в подготовительном примере 52, используя в качестве исходного соединения 5-хлорнафталин-1-карбальдегид Указанное в заголовке соединение получали, используя способ, идентичный описанному в подготовительном примере 53, используя в качестве исходного соединения метиловый эфир 3-(5-хлорнафталин 1-ил)-2-циклопропил-3-оксопропионовой кислоты (подготовительный пример 56). Н (CDCl3): 0.22 (2 Н,m), 0.60 (2 Н, m), 1.16 (1 Н, m), 2.95 (2 Н, d), 7.49 (1 Н, m), 7.62 (2 Н, m), 7.83 (1 Н, m), 8.47 (2 Н, m). Подготовительный пример 58. Бромид (3,3-диэтокси-1-метилпропил)трифенилфосфония.- 27012374 Указанное в заголовке соединение получали в соответствии со способом Henri Brunner et al., Synthesis, 1997, 79-86. Подготовительный пример 59. 1-Хлор-7-метилнафталин.(2,5 М; 27 мл; 67,7 ммоль) и реакционную смесь перемешивали при -78 С в течение 1,5 ч. Добавляли раствор 2-хлорбензальдегида (6,9 мл; 61,6 ммоль) в THF (10 мл), реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 ч. Добавляли EtOH (20 мл) и триэтиламин (2 мл) и реакционную смесь распределяли между водой (100 мл) и Et2O (3100 мл). Объединенные органические фракции сушили (MgSO4) и концентрировали в вакууме. Остаток растворяли в ледяной уксусной кислоте(100 мл) и 48%-ном HBr в воде (100 мл) и нагревали до 100 С в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и распределяли между водой (150 мл) и Et2O (3100 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле, элюируя гексанами, что позволило получить указанное в заголовке соединение. Н (CDCl3): 2.59 К кипящему с обратным холодильником раствору N-бромсукцинимида (1,20 г; 6,7 ммоль) и AIBN(84 мг; 0,5 ммоль) в CCl4 (25 мл) добавляли раствор 1-хлор-7-метилнафталина (подготовительный пример 59; 1,13 г; 6,4 ммоль) в CCl4 (25 мл). Реакционную смесь кипятили с обратным холодильником в течение 3 ч, затем охлаждали до комнатной температуры и твердое вещество отфильтровывали и отбрасывали. Фильтрат концентрировали в вакууме и растворяли в CHCl3 (50 мл). Добавляли гексаметилентетрамин (1,08 г; 7,7 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 3 ч, затем перемешивали при комнатной температуре в течение 72 ч. Реакционную смесь концентрировали в вакууме и остаток растворяли в смеси уксусная кислота:вода (1:1,40 мл) и концентрированной HCl (10 мл). Реакционную смесь кипятили с обратным холодильником в течение 3 ч, затем охлаждали до комнатной температуры и распределяли между водой (100 мл) и EtOAc (3100 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле, элюируя смесью EtOAc:гексаны (1:9), что позволило получить указанное в заголовке соединение. Н (CDCl3): 7.56 Раствор 8-хлорнафталин-2-карбальдегида (подготовительный пример 60; 117 мг; 0,61 ммоль) и гидроксиламина гидрохлорида (51 мг; 0,74 ммоль) в муравьиной кислоте (5 мл) нагревали до температуры дефлегмации в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры и распределяли между водой (30 мл) и EtOAc (330 мл). Объединенные органические фракции сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле, элюируя смесью EtOAc:гексаны (1:9), что позволило получить указанное в заголовке соединение. H (CDCl3): 7.57 (1 Н, m), 7.70 (2 Н, m), 7.83 (1 Н,d), 7.98 (1 Н, d), 8.69 (1 Н, s). Подготовительный пример 62. Гидрохлорид 3-диметиламино-1-нафталин-1-илпропан-1-она. К раствору 1-ацетонафтона (10 г; 59 ммоль), параформальдегида (2,3 г; 78 ммоль) и диметиламина гидрохлорида (6,36 г; 78 ммоль) в этаноле (20 мл) добавляли концентрированную HCl (0,5 мл). Реакционную смесь нагревали до температуры дефлегмации в течение 10 ч, затем охлаждали до комнатной- 28012374 температуры и концентрировали в вакууме. Остаток растирали с Et2O (20 мл), что позволило получить указанное в заголовке соединение, m/z (ES+) = 228 [M+H]+; RT = 2,22 мин. Подготовительный пример 63. 1-Нафталин-1-илпропенон. Гидрохлорид 3-диметиламино-1-нафталин-1-илпропан-1-она (подготовительный пример 62; 15,47 г; 59 ммоль) растворяли в воде (400 мл) и подщелачивали до рН 9, используя насыщенный раствор Na2CO3. Свободное основание экстрагировали в EtOAc (3150 мл), объединенные органические фракции сушили(MgSO4) и концентрировали в вакууме. Остаток растворяли в МеОН (25 мл), охлаждали до 0 С и добавляли йодметан (8,4 мл; 135 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Полученное твердое вещество отфильтровывали и промывали Et2O (220 мл). Твердое вещество энергично перемешивали в смеси Et2O (100 мл) и насыщенного раствора NaHCO3 (100 мл) в течение 16 ч. Слои разделяли и водный слой экстрагировали в EtOAc (2100 мл). Объединенные органические фракции промывали 1 М HCl (100 мл), рассолом (150 мл), сушили (MgSO4) и концентрировали в вакууме, что позволило получить указанное в заголовке соединение. Н (CDCl3): 6.06 (1 Н, d), 6.28(3,35 г; 31 ммоль) растворяли в DCM (40 мл) и к раствору добавляли концентрированную H2SO4 (4 капли). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, затем разбавлялиDCM (40 мл) и промывали насыщенным раствором NaHCO3 (50 мл) и водой (50 мл). Органический слой сушили (MgSO4), концентрировали в вакууме и хроматографировали на силикагеле, элюируя смесьюEtOAc:гексаны (1:9). Продукт дополнительно хроматографировали на силикагеле, элюируя DCM, что позволило получить указанное в заголовке соединение. Н (CDCl3): 3.37 (2 Н, t), 3.99 (2 Н, t), 4.57 (2 Н, s),7.32 (4 Н, m), 7.56 (4 Н, m), 7.90 (2 Н, m), 8.00 (1 Н, d), 8.63 (1H, d). Подготовительный пример 65. 3-Бензилокси-2-бром-1-нафталин-1-илпропан-1-он. 3-Бензилокси-1-нафталин-1-илпропан-1-он (подготовительный пример 64; 2,89 г; 9,9 ммоль) и РТАТ (4,12 г; 10,9 ммоль) растворяли в THF (40 мл) и перемешивали при комнатной температуре в течение 72 ч. Осадок отфильтровывали, промывали THF (10 мл) и отбрасывали. Фильтрат концентрировали в вакууме и хроматографировали на силикагеле, элюируя смесью EtOAc:гексаны (1:19), что позволило получить указанное в заголовке соединение. Н (DMSO): 3.95 (1 Н, dd), 4.20 (1 Н, dd), 4.60 (2 Н, d), 5.96(1 Н, t), 7.31 (4 Н, m), 7.65 (4 Н, m), 8.07 (1 Н, d), 8.22 (2 Н, m), 8.38 (1 Н, d). Подготовительные примеры 66-70. Методику, описанную в подготовительном примере 15, использовали для получения соединений из подготовительных примеров 66-70 из соответствующего нитрила и реактива Гриньяра. Подготовительные примеры 71-79. Методику, описанную в подготовительном примере 1, использовали для получения соединений из подготовительных примеров 71-79 из соответствующего кетона.
МПК / Метки
МПК: C07D 513/04, A61K 31/429
Метки: производные, дигидроимидазотиазола
Код ссылки
<a href="https://eas.patents.su/30-12374-proizvodnye-digidroimidazotiazola.html" rel="bookmark" title="База патентов Евразийского Союза">Производные дигидроимидазотиазола</a>
Производные 2,3-бензодиазепина и фармацевтические композиции, содержащие эти производные в качестве активного ингредиента

Номер патента: 5867
Опубликовано: 30.06.2005
Авторы: Грефф Зольтан, Сабо Геза, Леваи Дьердь, Раткаи Зольтан, Гиглер Габор, Мартонне Марко Бернадетт, Шимиг Дьюла, Линг Иштван, Вег Миклош, Баркоци Йожеф, Харшинг Ласло Габор, Сенаши Габор
МПК: A61K 31/551, C07D 243/02, A61P 25/00...
Метки: содержащие, фармацевтические, производные, 2,3-бензодиазепина, композиции, ингредиента, активного, качестве, эти
Формула / Реферат:
1. Производное 2,3-бензодиазепина формулы I где X - водород, хлор или метоксигруппа, Y - водород или галоген, Z - метил или хлор, R - C1-4 алкил или группа формулы -NR1R2, где R1 и R2, независимо, представляют собой водород, C1-4 алкил, C1-4 алкоксил или C3-6 циклоалкил, и его фармацевтически приемлемые соли с кислотами. 2. Производное 2,3-бензодиазепина по п.1, где X - хлор, Y - водород, хлор или бром, R - C1-4 алкил, Z - определен в п.1, и...
Производные бензимидазола, их получение и терапевтическое применение

Номер патента: 5950
Опубликовано: 25.08.2005
Авторы: Болкениус Франк, Ван Дорсселер Вивиан, Барт Франсис, Бишон Даниэль
МПК: C07D 471/06, A61P 9/00, A61K 31/4375...
Метки: производные, получение, применение, бензимидазола, терапевтическое
Формула / Реферат:
1. Соединения, соответствующие формуле (I) в которой R1 представляет собой атом водорода, группу C1-C4алкил, атом галогена, нитрогруппу или группу C1-C4алкокси, R2 и R2' независимо друг от друга представляют собой атом водорода или группу C1-C4алкил, X представляет собой атом азота или атом углерода, n равно 1 или 2, m равно 1 или 2 и, когда X представляет собой атом азота, R3 представляет собой атом водорода или группу C1-C4алкил, давая...
Производные 2н-пиридазин-3-она, фармацевтические композиции, содержащие эти производные, и способ получения активного ингредиента

Номер патента: 6246
Опубликовано: 27.10.2005
Авторы: Шимиг Дьюла, Эдьед Андраш, Паллаги Каталин, Сенаши Габор, Вельман Янош, Гачальи Иштван, Шмидт Эва, Леваи Дьёрди, Рацне Байногель Юдит, Баркоци Йожеф, Котаи Надь Петер, Миклошне Ковач Анико
МПК: A61K 31/50, C07D 413/14
Метки: ингредиента, 2н-пиридазин-3-она, получения, композиции, способ, производные, активного, фармацевтические, содержащие, эти
Формула / Реферат:
1. Производное 2H-пиридазин-3-она формулы где R означает атом водорода или C1-4-алкильную группу, X и Y независимо представляют собой атом водорода, атом галогена или группу формулы при условии, что один из X и Y всегда представляет собой группу формулы II, а другой радикал представляет собой атом водорода или атом галогена, где в формуле II n имеет значение 1 или 2, и его фармацевтически приемлемые соли присоединения кислот. 2. Производное...
Производные 5н, 10н-имидазо{1,2-а}индено {1,2-е} пиразин-4-она, способы их получения, содержащее их лекарственное средство и производные инданона в качестве промежуточных продуктов

Номер патента: 218
Опубликовано: 24.12.1998
Авторы: Рибей Ив, Алу Жан-Клод, Манфр Франко, Жимоне Патрик, Одьо Франсуа, Женевуа-Борелла Ариель, Миньяни Серж, Арди Жан-Клод, Немесе Патрик, Дамур Доминик, Барро Мишель
МПК: C07F 9/6561, A61K 31/495, C07D 487/04...
Метки: продуктов, средство, производные, качестве, способы, пиразин-4-она, 1,2-е, промежуточных, инданона, 10н-имидазо{1,2-а}индено, получения, лекарственное, содержащее
Формула / Реферат:
1. Производные 5Н,10H-имидазо[1,2-а] индено[1,2-е]пиразин-4-она формулы (I) в которой R означает атом водорода или карбоксил, алкоксикарбонил, -CO-NR4R5, -РО3H2 или -СН2ОН; R1 означает радикалы -aлк-NH2, -алк-NН-СО-R3, -алк-СООR4, -aлк-CO-NR5R6 или -CO-NH-R7; R3 означает алкил, фенил, фенилалкил, циклоалкил или -NR6R8; R4 означает атом водорода или алкильный радикал; R5 означает атом водорода, алкил, фенил, циклоалкил или...
Производные бензоксазина в качестве модуляторов 5-нт-6, способ их получения, фармацевтические композиции, содержащие эти производные, и их применение

Номер патента: 9982
Опубликовано: 28.04.2008
Авторы: Чжао Шухай, Кларк Робин Дуглас, Бергер Якоб
МПК: A61K 31/536, C07D 265/36, A61P 25/18...
Метки: применение, 5-нт-6, содержащие, эти, композиции, фармацевтические, получения, качестве, бензоксазина, способ, модуляторов, производные
Формула / Реферат:
1. Соединение формулы его фармацевтически приемлемая соль или пролекарство, где m означает целое число от 0 до 3; n означает 2 или 3; р означает 2; Y означает -S(O2)-; Z1 означает N; R1 и R2 означают водород; R3 означает фенил или нафтил, которые необязательно замещены галогеном, алкилом, алкокси, гидрокси, циано, пиперазинилом, метилсульфониламино, метилсульфонилом, аминокарбонилом, или означает хинолинил, изохинолинил, тиофенил,...
Предыдущий патент: Способы производства творога и сыра, творог, сыр и молочный продукт
Следующий патент: Фунгицидная комбинация биологически активных веществ
Случайный патент: Самонесущая шина, имеющая дополнительный усилитель боковины