6-замещённые циклогексилалкил 2-замещённые хинолиноны и 2-хиноксалиноны в качестве ингибиторов поли (adp-рибоза) полимеразы
Номер патента: 10592
Опубликовано: 30.10.2008
Авторы: Ваутерс Вальтер Баудевейн Леопольд, Ван Дюн Якобус Альфонсус Йозефус, Мабир Доминик Жан-Пьер, Сомерс Мария Викторина Франциска




























Формула / Реферат
1. Соединение формулы (I)

его N-оксидные формы, аддитивные соли и стереохимические изомерные формы, в котором
n представляет собой 0 или 1;
s представляет собой 0 или 1;
X представляет собой -N= или -СН=;
Y представляет собой -N< или -СН<;
Q представляет собой -NH-, -O-, -С(О)-, -СН2-СН2- или -CHR5-,
где R5 является водородом, гидрокси, C1-6алкилом, арилС1-6алкилом, C1-6алкилоксикарбонилом, C1-6 алкилоксиC1-6алкиламино или галогениндазолилом;
R1 является C1-6алкилом;
R2 является водородом или взятый вместе с R3 может образовывать =O;
R3 является водородом, C1-6алкилом или радикалом, выбранным из
| -NR6R7 | (а-1), |
| -O-Н | (а-2), |
| -OR8 | (а-3) или |
| -S-R9 | (а-4), |
где R6 представляет собой ди(C1-6алкил)аминоC1-6алкил или C1-6алкилоксиC1-6алкил; и
R7 является водородом;
R8 является ди(C1-6алкил)аминоC1-6алкилом и
R9 является ди(C1-6алкил)аминоC1-6алкилом;
или R3 представляет собой группу формулы
-(CH2)t-Z- (b-1),
в которой t равно 0, 1 или 2;
Z является гетероциклической кольцевой системой, выбранной из

где каждый R10 независимо представляет собой водород, C1-6алкил, гидрокси, C1-6алкилоксиC1-6алкил, C1-6алкилоксиC1-6алкиламино, морфолино, C1-6алкилимидазолил или пиридинилC1-6алкиламино;
каждый R11 независимо является водородом или гидрокси;
арил является фенилом или замещенным галогеном, C1-6алкилом или C1-6алкилоксифенилом
при условии, что исключен 6-(циклогексил-1Н-имидазол-1-илметил)-3-метил-2(1Н)-хиноксалинон.
2. Соединение по п.1, в котором n равно 0; X является СН; Q является -NH-, -СН2-СН2- или -CHR5-, где R5 является водородом, гидрокси или арилC1-6алкилом; R1 является C1-6алкилом; R2 является водородом; R3 представляет собой водород, гидрокси или группу формулы (b-1); t равно 0; Z представляет собой гетероциклическую кольцевую систему, выбранную из (с-8) или (с-13); каждый R10 независимо представляет собой водород; и арил является фенилом.
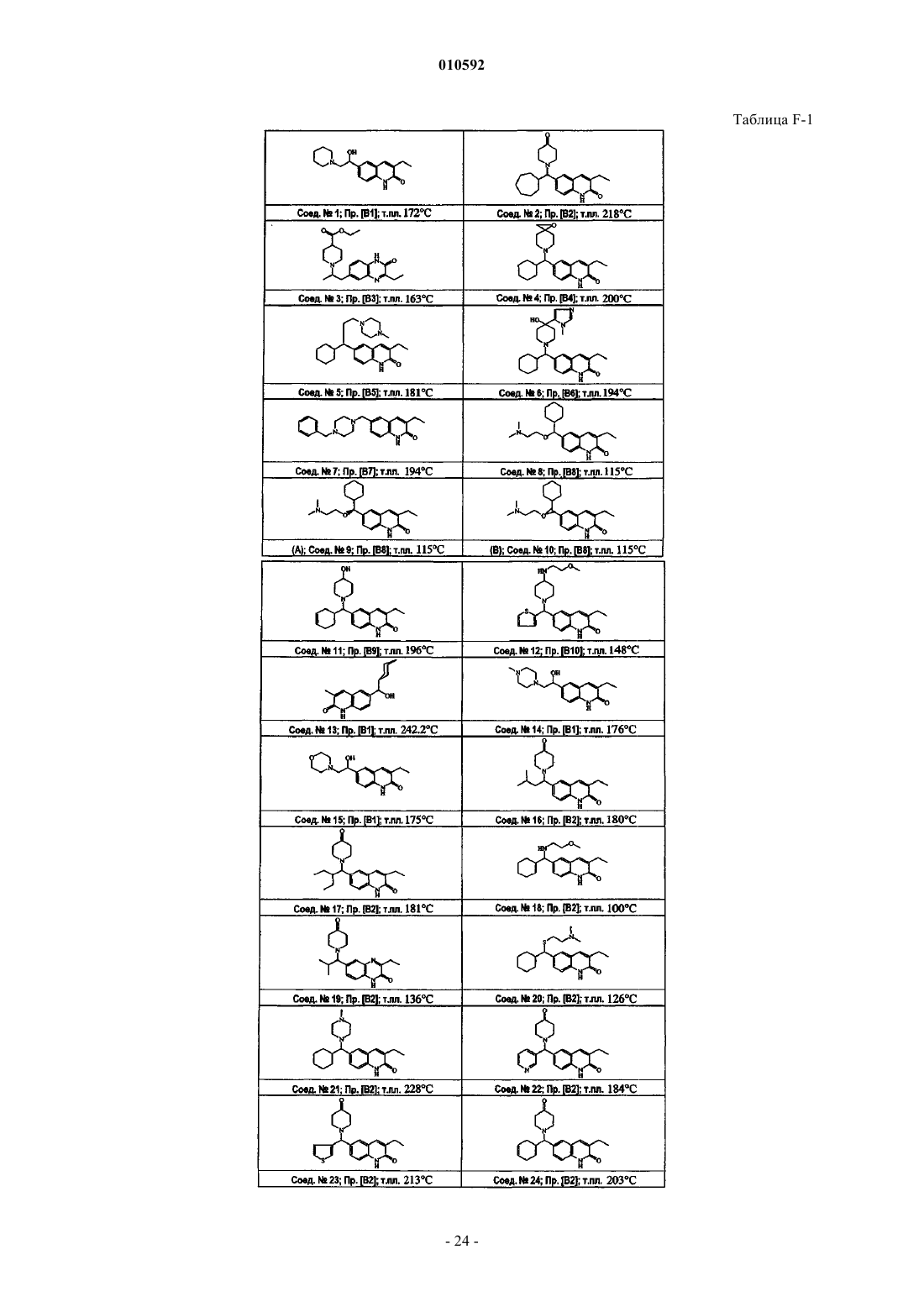
3. Соединение по п.1, в котором соединение выбирают из соединения ь 7, соединения ь 2, соединения ь 1 и соединения ь 11.

4. Применение соединения по любому из пп.1-3 в качестве лекарственного средства.
5. Фармацевтическая композиция, содержащая фармацевтически приемлемые носители и в качестве активного ингредиента терапевтически эффективное количество соединения по пп.1-3.
6. Способ получения фармацевтической композиции по п.5, в котором фармацевтически приемлемые носители и соединение по пп.1-3 тщательно смешаны.
7. Применение соединения для получения лекарственного препарата для лечения PARP-опосредованного нарушения, в котором соединение представляет собой соединение формулы (I)

его N-оксидные формы, аддитивные соли и стереохимические изомерные формы, в котором
n представляет собой 0 или 1;
s представляет собой 0 или 1;
X представляет собой -N= или -СН=;
Y представляет собой -N< или -СН<;
Q представляет собой -NH-, -O-, -С(О)-, -СН2-СН2- или -CHR5-,
где R5 является водородом, гидрокси, C1-6алкилом, арилC1-6алкилом, C1-6алкилоксикарбонилом, C1-6 алкилоксиC1-6алкиламино или галогениндазолилом;
R1 является C1-6алкилом;
R2 является водородом или взятый вместе с R3 может образовывать =O;
R3 является водородом, C1-6алкилом или радикалом, выбранным из

где R6 представляет собой ди(C1-6алкил)аминоC1-6алкил или C1-6алкилоксиC1-6алкил; и
R7 является водородом;
R8 является ди(C1-6алкил)аминоC1-6алкилом; и
R9 является ди(C1-6алкил)аминоC1-6алкилом;
или R3 представляет собой группу формулы
-(CH2)t-Z- (b-1),
в которой t равно 0, 1 или 2;
Z является гетероциклической кольцевой системой, выбранной из

где каждый R10 независимо представляет собой водород, C1-6алкил, гидрокси, C1-6алкилоксиC1-6алкил, C1-6алкилоксиC1-6алкиламино, морфолино, C1-6алкилимидазолил или пиридинилC1-6алкиламино;
каждый R11 независимо является водородом или гидрокси;
арил является фенилом или замещенным галогеном, C1-6алкилом или C1-6алкилоксифенилом
при условии, что исключен 6-(циклогексил-1Н-имидазол-1-илметил)-3-метил-2(1Н)-хиноксалинон.
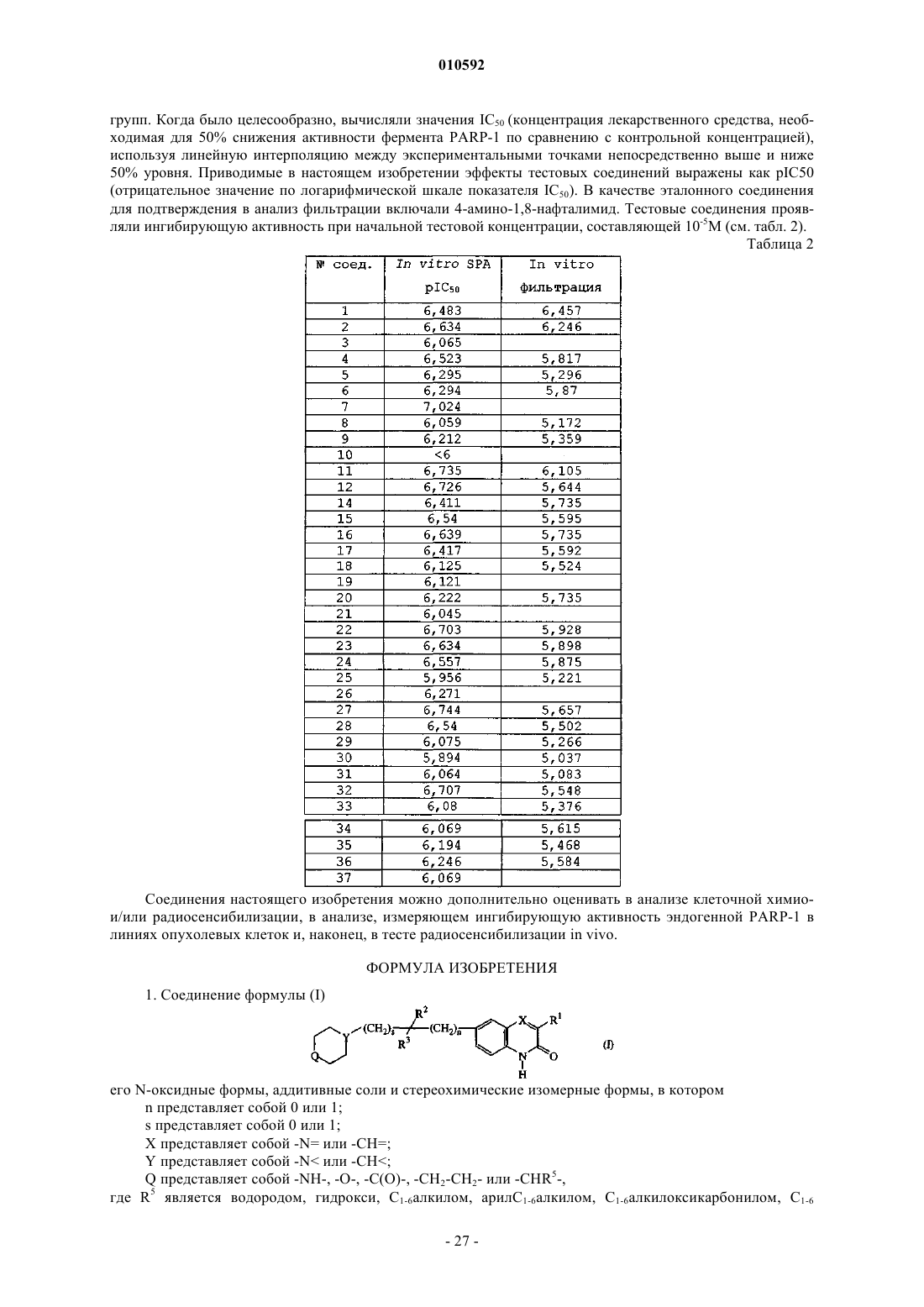
8. Применение по п.7 для получения лекарственного препарата для лечения химиосенсибилизации.
9. Применение по п.7 для получения лекарственного препарата для лечения радиосенсибилизации.
10. Способ получения соединения по п.1, отличающийся тем, что включает гидролиз промежуточных продуктов формулы (VIII), согласно известным в данной области техники способам, путем обработки промежуточных продуктов формулы (VIII) подходящими реагентами, такими как хлорид олова, уксусная кислота и соляная кислота, в присутствии реакционно-инертного растворителя, например тетрагидрофурана

11. Способ получения соединения по п.1, отличающийся тем, что включает циклизацию промежуточных продуктов формулы (X) согласно известным в данной области техники способам циклизации в соединения формулы (I), где X представляет собой СН, указанные как соединения формулы (I-j), предпочтительно в присутствии подходящей кислоты Льюиса, например хлорида алюминия, или в неразбавленном виде, или растворенной в подходящем растворителе, таком как, например, ароматические углеводороды, например в бензоле, хлорбензоле, метилбензоле и т.п.; в галогенированных углеводородах, например в трихлорметане, тетрахлорметане и т.п.; в эфире, например в тетрагидрофуране, 1,4-диоксане и т.п. или в смеси таких растворителей

12. Способ получения соединения по п.1, отличающийся тем, что включает конденсацию подходящего ортобензолдиамина формулы (XI) со сложным эфиром формулы (XII) с получением соединений формулы (I), указанных как соединения формулы (I-а-1), в которых X является N и R2, взятый вместе с R3, образует связь =O, в присутствии карбоновой кислоты, например уксусной кислоты и т.п., неорганической кислоты, такой как, например, соляная кислота, серная кислота, или в присутствии сульфонотющ кислоты, такой как, например, метансульфоновая кислота, бензолсульфоновая кислота, 4-метилбензолсульфоновая кислота и т.п.

Текст
010592 Область техники, к которой относится изобретение Настоящее изобретение относится к ингибиторам поли(ADP-рибоза)полимеразы (PARP) и обеспечивает соединения и композиции, содержащие раскрываемые соединения. Кроме того, настоящее изобретение относится к способам применения раскрываемых ингибиторов PARP, например, в качестве лекарственного препарата. Уровень техники изобретения Ядерный фермент поли(ADP-рибоза)полимераза-1 (PARP-1) является членом семейства PARP ферментов, состоящего из PARP-1 и нескольких недавно идентифицированных новых поли(ADPрибозилирующих) ферментов. PARP также упоминается как поли(аденозин-5'-дифосфорибоза)полимераза или PARS, представляющая собой поли(ADP-рибоза)синтетазу.PARP-1 представляет собой главный белок ядра массой в 116 кДа, состоящий из трех доменов: Nконцевого связывающего ДНК домена, содержащего два цинковых пальца, аутомодифицирующего домена и С-концевого каталитического домена. Этот белок имеется практически у всех эукариот. Фермент синтезирует поли(ADP-рибозу), представляющую собой разветвленный полимер, который может состоять из более чем 200 единиц ADP-рибозы. Белковые акцепторы поли(ADP-рибозы) прямо или опосредованно участвуют в поддержании целостности ДНК. Они включают в себя гистоны, топоизомеразы, ДНКи РНК-полимеразы, ДНК-лигазы и Са 2+- и Mg2+-зависимые эндонуклеазы. Высокий уровень экспрессии белка PARP выявляется во многих тканях, особенно значительно в клетках иммунной системы, сердца,мозга и клетках зародышевой линии (germ-line). При нормальных физиологических условиях активностьPARP является минимальной. Однако повреждение ДНК вызывает немедленную, до 500-кратного повышения, активацию PARP. Среди многих функций, свойственных PARP, и особенно PARP-1, ее главной ролью является способствовать репарации ДНК путем ADP-рибозилирования и, таким образом, координировать количество белков репарации ДНК. В результате активации PARP уровни NAD+ значительно снижаются. Расширенная активация PARP приводит к тяжелому истощению NAD+ в клетках, подвергшихся массивному повреждению ДНК. Короткий период полужизни поли(ADP-рибозы) приводит к ускоренному метаболизму. Образовавшаяся поли(ADP-рибоза) быстро разрушается существенно активной поли(ADPрибоза)гликогидролазой (PARG), вместе с фосфодиэстеразой и белком (ADP-рибоза)лиазой. PARP иPARG образуют цикл, который превращает большое количество NAD+ в ADP-рибозу. Гиперстимуляция PARP менее чем за один час может вызвать снижение NAD+ и АТР до уровня, составляющего менее чем 20% от нормального. Такой сценарий представляет наибольшую вредность во время ишемии, когда выработка клеточной энергии уже подвергается значительному риску из-за лишения кислорода. Последующая выработка свободных радикалов во время реперфузии считается основной причиной повреждения тканей. Частично снижение уровня АТР, которое во многих органах является обычным во время ишемии и реперфузии, может быть связано с истощением запасов NAD+ вследствие метаболизма поли(ADP-рибозы). Таким образом, ожидается, что ингибирование PARP или PARG сохранит уровень энергии клетки и, таким образом, потенцирует выживание ишемизированных тканей после инсульта. Синтез поли(ADP-рибозы) также участвует в индуцированной экспрессии множества генов, существенных для воспалительной реакции. Ингибиторы PARP подавляют продукцию индуцируемой формы синтетазы окиси азота (iNOS) в макрофагах, селектина Р-типа и молекул межклеточной адгезии-1(ICAM-1) в эндотелиальных клетках. Такая активность лежит в основе сильного противовоспалительного эффекта, проявляемого ингибиторами PARP. Ингибирование PARP способно уменьшить некроз путем предотвращения перемещения нейтрофилов и инфильтрации ими поврежденных тканей. Активация PARP осуществляется поврежденными фрагментами ДНК и после активации она становится катализатором присоединения до 100 единиц ADP-рибозы к различным ядерным белкам, включая гистоны и непосредственно PARP. Во время основных клеточных стрессов высокая активация PARP может быстро привести к повреждению клетки или ее смерти вследствие истощения энергетических запасов. Поскольку для восстановления каждой молекулы NAD+ расходуются четыре молекулы АТР, расширенная активация PARP истощает NAD+, и попытки ресинтеза NAD+ также могут привести к истощению АТР. Имеются сообщения о том, что активация PARP играет ключевую роль в индукции нейротоксичности NMDA- (N-метил-О-аспартата) и NO-. Это было показано на кортикальных культурах и срезах гиппокампа, где выявлена непосредственная корреляция предотвращения токсичности с возможностью ингибирования PARP. Таким образом, была признана потенциальная роль ингибиторов PARP при лечении нейродегенеративных болезней и травм головы, несмотря на то, что точный механизм действия еще не объяснен. Аналогично было показано, что единственные инъекции ингибиторов PARP уменьшали размер инфаркта, вызванного ишемией и реперфузией сердечной или скелетной мышцы у кроликов. В этих исследованиях единственная инъекция 3-аминобензамида (10 мг/кг), как за одну минуту до окклюзии, так и за одну минуту до реперфузии, вызывала сходные уменьшения размера инфаркта миокарда (32-42%), в то время как другой ингибитор PARP, 1,5-дигидроксиизохинолин (1 мг/кг), имел сопоставимую степень-1 010592 снижения размера инфаркта (38-48%). Эти результаты делают логичным предположение, что ингибиторы PARP могут восстанавливать сердечную мышцу, подвергшуюся ишемии, или ткани скелетных мышц после реперфузионной травмы. Активация PARP может также использоваться в качестве показателя размеров повреждения вследствие нейротоксического поражения, являющегося причиной воздействия любого из следующих индукторов подобно глутамату (путем возбуждения NMDA рецептора), промежуточных продуктов активного кислорода, амилоидного -протеина, N-метил-4-фенил-1,2,3,6-тетрагидропиридина (МРТР) или его активного метаболита N-метил-4-фенилпиридина (МРР+), которые задействуются при патологических состояниях, таких как инсульт, болезнь Альцгеймера и болезнь Паркинсона. Другое исследование продолжило изучение роли активации PARP в зернистых клетках мозжечка in vitro и в нейротоксичности МРТР. Наиболее часто в результате инсульта или других нейродегенеративных процессов наблюдается чрезмерное воздействие на нервную ткань глутамата, являющегося доминирующим нейромедиатором центральной нервной системы и действующего на N-метил-D-аспартат (NMDA) рецепторы и другие подтипы рецепторов. Кислород лишает нейроны способности в больших количествах высвобождать глутамат во время ишемического повреждения мозга, как, например, во время инсульта или сердечного приступа. В свою очередь, это избыточное высвобождение глутамата вызывает гиперстимуляцию (эксайтотоксичность) N-метил-D-аспартатных (NMDA), AMPA-, каинатных и MGR-рецепторов, которые открывают ионные каналы и пропускают неконтролируемый поток ионов (например, Са 2+ и Na+ внутрь клетки и K+ из клетки), что приводит к гиперстимуляции нейронов. Эти гиперстимулированные нейроны выделяют большее количество глутамата, образуя петлю обратной связи или эффект домино, который в конечном счете приводит к повреждению или гибели клетки посредством выработки протеаз, липаз и свободных радикалов. Чрезмерная активация глутаматных рецепторов органически связана с различными неврологическими заболеваниями и состояниями, включая эпилепсию, инсульт, болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз (ALS), болезнь Хантингтона, шизофрению, хроническую боль, ишемию и нарушение иннервации вследствие гипоксии, гипогликемии, ишемии, травмы и инсульта нервной системы. Воздействие глутамата и стимуляция также задействованы в качестве причины навязчивых состояний, в особенности лекарственной зависимости. Доказательства того, что антагонисты глутаматных рецепторов (т.е. соединения, которые блокируют связывание глутамата или возбуждение его рецептора) препятствуют повреждению нервной ткани вследствие сосудистого инсульта, включают в себя данные, полученные на многих видах животных, а также на кортикальных культурах мозга, в которые вводили глутамат или NMDA. Было доказано, что попытки предотвращения эксайтотоксичности путем блокирования NMDA, АМРА, каинатных и MGR-рецепторов вызывают проблемы, так как каждый рецептор имеет множество участков для связывания с глутаматом и, следовательно, остается трудным выявление эффективного сочетания антагонистов или универсального антагониста, предотвращающего связывание всех рецепторов с глутаматом и позволяющего проверить эту теорию. Кроме того, многие из композиций, которые эффективно блокируют рецепторы, также являются токсичными для животных. Таким образом, в настоящее время не известно никакого эффективного лечения расстройств, связанных с глутаматом. Например, глутаматное возбуждение NMDA-рецепторов активирует фермент нейронную синтетазу окиси азота (nNOS), приводя к образованию окиси азота (NO), также являющейся медиатором нейротоксичности. Нейротоксичность NMDA можно предотвратить введением ингибиторов синтетазы окиси азота (NOS) или посредством направленного генетического разрушения nNOS in vitro. Другим применением ингибиторов PARP является лечение периферических повреждений нервов и возникающего в результате этих повреждений синдрома патологической боли, известного как нейропатическая боль, такая как вызванная повреждением от хронического сужения (CCI) общего седалищного нерва, при которой происходит транссинаптическая альтерация дорсального рога спинного мозга, характеризующаяся гиперхроматозом цитоплазмы и кариоплазмы (так называемые "темными" нейронами). Также доказано, что ингибиторы PARP являются полезными для лечения воспалительных заболеваний кишечника, таких как колит. В частности, у крыс вызывали колит внутрипросветным введением гаптена тринитробензолсульфоновой кислоты в 50% этаноле. Опытные крысы получали 3 аминобензамид, специфический ингибитор активности PARP. Ингибирование активности PARP уменьшало воспалительную реакцию и восстанавливало морфологию и энергетическое состояние дистального отдела толстой кишки. Дополнительно доказано предположение, что ингибиторы PARP являются полезными для лечения артрита. Дополнительно выявлено, что ингибиторы PARP являются полезными для лечения диабета. Показано, что ингибиторы PARP полезны для лечения эндотоксинового шока или септического шока. Также ингибиторы PARP применялись для увеличения продолжительности жизни и пролиферативной способности клеток, включая лечение болезней, таких как старение кожи, болезнь Альцгеймера, атеросклероз, остеоартрит, остеопороз, мышечную дистрофию, дегенеративные болезни скелетных мышц, с вовлечением репликационного старения, возрастную мышечную дегенерацию, иммунное старение,СПИД и другие дегенеративные болезни иммунной системы; и альтерацию экспрессии гена старения клеток. Также известно, что ингибиторы PARP, такие как 3-аминобензамид, влияют на общую репарацию-2 010592 ДНК, например, после воздействия перекиси водорода или ионизирующего излучения. Хорошо установлена центральная роль PARP в репарации ДНК с разрывами нитей, в особенности,когда разрывы вызваны непосредственно ионизирующим излучением или, косвенно, после ферментативной репарации поражений ДНК, вызванных метилирующими агентами, ингибиторами Iтопоизомеразы и другими химиотерапевтическими веществами, такими как цисплатин и блеомицин. Роль PARP в репарации и выживании клетки после индуцированного повреждения ДНК была показана в ряде исследований с использованием "нокаутных" мышей, трансдоминантных моделей ингибирования(гиперэкспрессии ДНК-связывающих доменов), антисмысловых и низкомолекулярных ингибиторов. Ингибирование ферментативной активности PARP должно приводить к повышению чувствительности опухолевых клеток к повреждающему ДНК лечению. Сообщается, что ингибиторы PARP являются эффективными для радиосенсибилизации (гипоксических) опухолевых клеток и эффективными для предотвращения регенерации опухолевых клеток от потенциально летальных и сублетальных повреждений ДНК после лучевой терапии, возможно с помощью их способности предотвращать воссоединение разрыва нитей ДНК и вызывать повреждение ДНК некоторых сигнальных путей. Ингибиторы PARP использовались для лечения рака. Кроме того, в патенте U.S. Patent No 5177075 рассматриваются некоторые изохинолины, используемые для повышения летального действия ионизирующего излучения или химиотерапевтических средств на опухолевые клетки. В статье Weltin et al., "Effect of 6-(5-Phenanthridinone, an Inhibitor of Poly(ADP-ribose)Polymerase, on Cultured Tumor Cells", Oncol.Res., 6:9, 399-403 (1994) рассматривается ингибирование активности PARP, сниженная пролиферация опухолевых клеток и заметное синергическое действие при совместном воздействии на клетки опухоли алкилирующих средств. Всесторонний обзор ситуации в данной области техники был недавно опубликован авторами Li иZhang в IDrugs 2001, 4(7): 804-812. Остается потребность в эффективных и мощных ингибиторах PARP и более конкретно в ингибиторах PARP-1, которые обладают минимальными побочными эффектами. Настоящее изобретение относится к соединениям, композициям и способам ингибирования активности PARP для лечения рака и/или предотвращения повреждения клеток, тканей и/или органов по причине повреждения или смерти клеток вследствие, например, некроза или апоптоза. Соединения и композиции настоящего изобретения являются особенно полезными для повышения эффективности химиотерапии и лучевой терапии, где первичным эффектом лечения является вызывание повреждения ДНК в клетках-мишенях. Предшествующий уровень техники Патент ЕР 371564, опубликованный 6 июня 1990 г., раскрывает (1 Н-азол-1-илметил)замещенные производные хинолина, хиназолина или хиноксалина. Описанные соединения подавляют выведение ретиноевых кислот из плазмы. Более конкретно, раскрывается 6-(циклогексил-1 Н-имидазол-1-илметил)-3 метил-2(1 Н)-хиноксалинон (соединение А). Описание изобретения Настоящее изобретение относится к соединениям формулы (I)X представляет собой -N= или -CR4=, где R4 является водородом, или взятый вместе с R1 могут образовывать двухвалентный радикал формулы -СН=СН-СН=СН-;R2 является водородом или взятый вместе с R3 могут образовывать =O;R7 является водородом или C1-6 алкилом;R9 является ди(C1-6 алкил)аминоC1-6 алкилом; или R3 представляет собой группу формулыZ является гетероциклической кольцевой системой, выбранной из где каждый R10 независимо представляет собой водород, C1-6 алкил, аминокарбонил, гидрокси,C1-6 алкилоксиC1-6 алкил, C1-6 алкилоксиC1-6 алкиламино, ди(фенилC2-6 алкенил),пиперидинилC1-6 алкил, C3-10 циклоалкил, C3-10 циклоалкилC1-6 алкил, арилокси(гидрокси)C1-6 алкил, галогениндазолил, арилC1-6 алкил, арилC1-6 алкенил, морфолино, C1-6 алкилимидазолил или пиридинилC1-6 алкиламино; каждый R1 независимо является водородом, гидрокси, пиперидинилом или арилом; арил является фенилом или замещенным галогеном, C1-6 алкилом или C1-6 алкилоксифенилом при условии, что исключен 6-(циклогексил-1 Н-имидазол-1-илметил)-3-метил-2(1 Н)-хиноксалинон. Всегда, если гетероциклическая кольцевая система Z содержит функциональные группы -CH2-,-СН= или -NH-, заместители R10 и R11 или остальная часть молекулы могут присоединяться к атому углерода или азота, и в этом случае замещаются один или оба атома водорода. Соединения формулы (I) могут также существовать в таутомерных формах. Несмотря на то, что такие формы явно не обозначены в вышеописанной формуле, они также входят в объем настоящего изобретения. Ниже объясняется ряд терминов, используемых в настоящем изобретении в предшествующих определениях и в дальнейшем. Эти термины иногда используются сами по себе или в составных терминах. Галоген относится к группе фтора, хлора, брома и йода; C1-6 алкил обозначает прямую и разветвленную цепь насыщенных углеводородных радикалов, имеющих от 1 до 6 атомов углерода, таких как, например, метил, этил, пропил, бутил, пентил, гексил, 1-метилэтил, 2-метилпропил, 2-метилбутил, 2 метилпентил и т.п.; C1-6 алкандиил обозначает двухвалентную прямую и разветвленную цепь насыщенных углеводородных радикалов, имеющих от 1 до 6 атомов углерода, таких как, например, метилен, 1,2 этандиил, 1,3-пропандиил 1,4-бутандиил, 1,5-пентандиил, 1,6-гександиил и их разветвленные изомеры,такие как 2-метилпентандиил, 3-метилпентандиил, 2,2-диметилбутандиил, 2,3-диметилбутандиил и т.п.; тригалогенметил обозначает метил, содержащий три идентичных или различных галогеновых заместителя, например трифторметил; C2-6 алкенил обозначает прямую и разветвленную цепь углеводородных ра-4 010592 дикалов, имеющих одну двойную связь и имеющих от 2 до 6 атомов углерода, таких как, например, этенил, 2-пропенил, 3-бутенил, 2-пентенил, 3-пентенил, 3-метил-2-бутенил и т.п.; и C3-10 циклоалкил включает в себя циклические углеводородные группы, имеющие от 3 до 10 атомов углерода, такие как циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогептил, циклооктил и т.п. Термин "аддитивная соль" включает в себя соли, которые соединения формулы (I) способны образовывать с органическими или неорганическими основаниями, такими как амины, основания щелочных металлов и основания щелочно-земельных металлов, или основания четвертичного аммония, или с органическими или неорганическими кислотами, такими как минеральные кислоты, сульфоновые кислоты,карбоновые кислоты или фосфорсодержащие кислоты. Термин "аддитивная соль" дополнительно включает в себя фармацевтически приемлемые соли,комплексы металлов и сольваты и их соли, которые способны образовывать соединения формулы (I). Термин "фармацевтически приемлемые соли" означает фармацевтически приемлемые аддитивные соли кислоты или основания. Предполагается, что вышеупомянутые фармацевтически приемлемые аддитивные соли кислоты или основания содержат терапевтически активные нетоксичные кислотные и нетоксичные основные формы аддитивной соли, которые способны образовывать соединения формулы(I). Соединения формулы (I), которые имеют свойства оснований, могут превращаться в их фармацевтически приемлемые кислотно-аддитивные соли путем обработки указанной формы основания подходящей кислотой. Подходящие кислоты содержат, например, неорганические кислоты, такие как галогеноводородные кислоты, например хлористо-водородная или бромисто-водородная кислота; серная; азотная; фосфорная и т.п. кислоты; или органические кислоты, такие как, например, уксусная, пропионовая, гидроксиуксусная, молочная, пировиноградная, щавелевая, малоновая, янтарная (т.е. бутандикислота), малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и т.п. кислоты. Соединения формулы (I), которые имеют кислотные свойства, можно превращать в их фармацевтически приемлемые основные аддитивные соли, обрабатывая указанную кислую форму подходящим органическим или неорганическим основанием. Подходящие формы основных солей содержат, например, соли аммония, соли щелочных и щелочно-земельных металлов, например соли лития, натрия, калия, магния,кальция и т.п., соли с органическими основаниями, например бензатиновая, N-метил-D-глюкаминовая,гидрабаминовая соли, и соли с аминокислотами, такими как, например, аргинин, лизин и т.п. Термин кислая или основная аддитивная соль также включает гидраты и растворимые дополнительные формы, которые соединения формулы (I) способны образовывать. Примерами таких форм являются, например, гидраты, алкоголяты и т.п. Термин "комплексные соединения металлов" обозначает комплекс, образованный между соединением формулы (I) и одной или более органической или неорганической солью или солями металлов. Примеры указанных органических или неорганических солей включают галогениды, нитраты, сульфаты, фосфаты,ацетаты, трифторацетаты, трихлорацетаты, пропионаты, тартраты, сульфонаты, например метилсульсульфонаты, 4-метилфенилсульфонаты, салицилаты, бензоаты и т.п. металлов второй основной группы периодической системы, например соли магния или кальция, третьей или четвертой основной группы, например алюминия, олова, свинца, а также от первой до восьмой групп переходных металлов периодической системы, таких как, например, хрома, марганца, железа, кобальта, никеля, меди, цинка и т.п. Вышеприведенный термин стереохимические изомерные формы соединений формулы (I) обозначает все возможные соединения, составленные из тех же атомов, соединенных той же последовательностью связей, но имеющие различные возможные у соединений формулы (I) трехмерные структуры,которые не являются взаимозаменяемыми. Если не упомянуто или обозначено иначе, химическое обозначение соединения охватывает смесь всех возможных стереохимических изомерных форм, которые могут иметь указанные соединения. Указанная смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. В фармакологии общепринято, что один энантиомер может иметь более высокую фармакологическую активность по сравнению с другим. Все стереохимические изомерные формы соединений формулы (I) как в форме чистых соединений, так и в смеси друг с другом, входят в объем настоящего изобретения. Предполагается, что N-оксидные формы соединений формулы (I) содержат такие соединения формулы (I), в которых один или несколько атомов азота окислены до так называемого N-оксида, в частности N-оксиды, у которых была N-окислена одна или более из следующих групп: пиперидин-, пиперазинили пиридазинилазотная. Предполагается, что всегда при использовании далее в настоящем изобретении термин "соединения формулы (I)" обозначает включение в него также N-оксидных форм, фармацевтически приемлемых кислых или основных аддитивных солей и всех форм стереоизомеров. Соединения, описанные в ЕР 371564, тормозят выделение из плазмы ретиноевых кислот. Было неожиданно обнаружено, что соединения настоящего изобретения проявляют ингибирующую PARP активность. Первая группа вызывающих интерес соединений состоит из соединений формулы (I), в которых од-5 010592 но или более следующих значений выбраны изc) R3 является водородом, C1-6 алкилом, радикалом, выбранным из (а-1), (а-2), (а-3) или (а-4) или из группы формулы (b-1);h) Z представляет собой гетероциклическую кольцевую систему, выбранную из (с-1), (с-5), (с-6), (с 8), (с-10), (с-12) или (с-13);i) каждый R10 независимо представляет собой водород, C1-6 алкил, гидрокси, C1-6 алкилоксиC1-6 алкил,C1-6 алкилоксиC1-6 алкиламино, морфолино, C1-6 алкилимидазолил или пиридинилC1-6 алкиламино;j) каждый R11 независимо представляет собой водород или гидрокси; иk) арил является фенилом. Вторая группа вызывающих интерес соединений состоит из соединений формулы (I), в которых одно или более следующих значений представляет:f) R3 представляет собой водород, гидрокси или группу формулы (b-1);h) Z представляет собой гетероциклическую кольцевую систему, выбранную из (с-8) или (с-13);i) каждый R10 независимо представляет собой водород;j) арил представляет собой фенил. Третья группа вызывающих интерес соединений состоит из соединений формулы (I), из первой группы интересных соединений или из второй группы интересных соединений, в которых Z является гетероциклической кольцевой системой, отличной от гетероциклической кольцевой системы формулы(с-2) или (с-4). Группа предпочтительных соединений состоит из соединений формулы (I), в которых X представляет собой -N= или -СН=; R1 является C1-6 алкилом; R3 является водородом, C1-6 алкилом, радикалом, выбранным из (а-1), (а-2), (а-3) или (а-4), или из группы формулы (b-1); R6 является ди(C1-6 алкил)аминоC1-6 алкилом или C1-6 алкилоксиC1-6 алкилом; R7 является водородом; R8 является ди(C1-6 алкил)аминоC1-6 алкилом; t равно 0 или 2; Z представляет собой гетероциклическую кольцевую систему, выбранную из (с-1),(с-5), (с-6), (с-8), (с-10), (с-12) или (с-13); каждый R10 независимо является водородом, C1-6 алкилом, гидрокси, C1-6 алкилоксиC1-6 алкилом, C1-6 алкилоксиC1-6 алкиламино, морфолино, C1-6 алкилимидазолилом или пиридинилC1-6 алкиламино; каждый R1 независимо является водородом или гидрокси и арил представляет собой фенил. Дополнительная группа предпочтительных соединений состоит из соединений формулы (I), в которых n равно 0; X представляет собой СН; Q представляет собой -NH-, -СН 2-СН 2- или -CHR5-, где R5 является водородом, гидрокси или арилC1-6 алкилом; R1 является C1-6 алкилом; R2 является водородом; R3 является водородом, гидрокси или группой формулы (b-1); t равно 0; Z представляет собой гетероциклическую кольцевую систему, выбранную из (с-8) или (с-13); каждый R10 независимо является водородом; и арил представляет собой фенил. Еще одна дополнительная группа предпочтительных соединений состоит из соединений формулы(I), из группы предпочтительных соединений или из дополнительной группы предпочтительных соединений, в которых Z представляет собой гетероциклическую кольцевую систему, отличную от гетероциклической кольцевой системы формулы (с-2) или (с-4). Наиболее предпочтительными соединениями являются соединение 7, соединение 2, соединение 1 и соединение 11. Соединения формулы (I) можно получить согласно основным способам, описанным в патенте ЕР 371564. Ряд таких способов получения будет описан более подробно далее в настоящем изобретении. Другие способы получения конечных соединений формулы (I) описаны в примерах. Соединения формулы (I), упомянутые в настоящем изобретении как соединения формулы (I-b), в которых R2 представляет собой водород и R3 представляет собой -NR7-CHO, где R7 является водородом или метилом, можно получить, исходя из соединений формулы (I), упомянутых в настоящем изобретении как соединения формулы (I-а), в которых R2, взятый вместе с R3, образует связь =O, в присутствии муравьиной кислоты и формамида или метилформамида, обозначенных здесь как промежуточные продукты формулы (II). Соединения формулы (I), упомянутые в настоящем изобретении как соединения формулы (1-c), в которых R3 представляет собой гидрокси, можно получить, превращая кетоновую функциональную группу соединений формулы (I-а) в гидроксигруппу подходящим восстановителем, например боргидридом натрия, в подходящем растворителе, например метаноле и тетрагидрофуране. Соединения формулы (I-а) можно получить, превращая соединения формулы (I-c), упомянутые в настоящем изобретении как соединения формулы (I-с-1), в которых R2 представляет собой водород, в присутствии подходящего окислителя, такого как трехокись хрома, и кислоты, такой как серная кислота,в подходящем растворителе, таком как 2-пропанон. Промежуточные продукты формулы (IV), в которой W представляет собой подходящую уходящую группу, такую как, например, хлор, бром, метансульфонилокси или бензолсульфонилокси, можно получить из соединений формулы (I-с-1), обрабатывая указанные соединения подходящим реактивом, например метансульфонилоксихлоридом или бензолсульфонилоксихлоридом, или галогенирующим реагентом, таким как, например, POCl3 или SOCl2. Соединения формулы (I), обозначенные как соединения формулы (I), упомянутые в настоящем изобретении как соединения формулы (I-h), в которых Rb определен в R6 и Rc определен в R7, или Rb и Rc,взятые вместе с азотом, к которому они присоединены, образуют подходящую гетероциклическую кольцевую систему, определенную в Z, можно получить взаимодействием промежуточных продуктов формулы (IV) с промежуточными продуктами формулы (V). Реакцию можно проводить в реакционно-7 010592 инертном растворителе, таком как диметилформамид или ацетонитрил, и, необязательно, в присутствии подходящего основания, такого как, например, карбонат натрия, карбонат калия или триэтиламин. Соединения формулы (I) также можно превращать друг в друга с помощью реакций, известных специалистам в данной области техники, или перегруппировками функциональных групп. Выше было приведено описание ряда таких перегруппировок. Другими примерами являются гидролиз эфиров карбоновой кислоты до соответствующей карбоновой кислоты или спирта; гидролиз амидов до соответствующей карбоновой кислоты или аминов; гидролиз нитрилов до соответствующих амидов; аминогруппы в имидазоле или фениле можно заменять водородом с помощью реакций диазотирования, известных специалистам в данной области техники, и последующей заменой диазогруппы водородом; спирты можно превращать в сложные эфиры и простые эфиры; первичные амины можно превращать во вторичные или третичные амины; двойные связи можно гидрировать до соответствующей одинарной связи; йодные радикалы в фенильной группе можно превращать в сложноэфирную группу введением окиси углерода в присутствии подходящего палладиевого катализатора. Следовательно, соединения формулы (I), (I-a), (I-b), (I-с), (I-c-1), (I-h), (I-i), (I-j) и (I-k) могут необязательно быть объектом одного или более следующих преобразований в любом желательном порядке:(i) преобразование соединения формулы (I) в различные соединения формулы (I);(ii) преобразование соединения формулы (I) в его соответствующую приемлемую соль или N-оксид;(iii) преобразование фармацевтически приемлемой соли или N-оксида соединения формулы (I) в исходное соединение формулы (I);(iv) получение стереохимических изомерных форм соединения формулы (I) или его фармацевтически приемлемой соли или N-оксида. Промежуточные продукты формулы (VII), в которой Rd и Re являются подходящими радикалами или взятые вместе с углеродом, к которому они присоединены, образуют подходящую гетероциклическую кольцевую систему, определенную в Z, можно получить гидролизом промежуточных продуктов формулы (VI), в которой R3 представляет собой группу формулы (b-1) или радикал, упомянутый в настоящем изобретении как Rg, формулы (а-1), где s не равно 0, согласно способам, известным в данной области техники, таким как перемешивание промежуточного продукта (VI) в водном растворе кислоты в присутствии реакционно-инертного растворителя, например тетрагидрофурана. Подходящей кислотой является, например, соляная кислота. Соединения формулы (I), упоминаемые в настоящем изобретении как соединения формулы (I-k), в которой R2 является водородом и Rg является обозначенным выше радикалом, можно получить, исходя из промежуточных продуктов формулы (VII), селективным гидрированием указанного промежуточного продукта с подходящим восстановителем, таким как, например, с катализатором из благородных металлов, таким как платина на угле, палладий на угле и т.п., и подходящим восстановителем, таким как водород, в подходящем растворителе, таком как метанол. Соединения формулы (I) можно получить путем гидролиза промежуточных продуктов формулы(VIII), согласно способам, известным в данной области техники, путем обработки промежуточных продуктов формулы (VIII) подходящими реактивами, такими как хлорид олова, уксусная кислота и соляная кислота, в присутствии реакционно-инертного растворителя, например тетрагидрофурана. Соединения формулы (I) можно получить, исходя из N-оксида формулы (IX), путем преобразования промежуточных продуктов формулы (IX) в присутствии подходящего реактива, такого как карбонат натрия или уксусный ангидрид и, когда это целесообразно, в растворителе, таком как дихлорметан. Соединения формулы (I), упомянутые в настоящем изобретении как соединения формулы (I-j), гдеX представляет собой СН, можно также получать циклизацией промежуточного продукта формулы (X). Реакция циклизации промежуточных продуктов формулы (X) может быть проведена согласно способам циклизации, известным в данной области техники. Реакцию предпочтительно проводят в присутствии подходящей кислоты Льюиса (Lewis Acid), например хлорида алюминия, как в неразбавленном виде, так и растворенной в подходящем растворителе, таком как, например, ароматический углеводород, например бензол, хлорбензол, метилбензол и т.п.; галогенированные углеводороды, например трихлорметан, тетрахлорметан и т.п.; эфир, например тетрагидрофуран, 1,4-диоксан и т.п.; или смеси таких растворителей. Скорость реакции можно увеличить некоторым повышением температуры, предпочтительно от 70 до 100 С, и перемешиванием. Соединения формулы (I), упомянутые в настоящем изобретении как соединения формулы (I-i), гдеX представляет собой N, можно получить конденсацией подходящего ортобензолдиамина формулы (XI) со сложным эфиром формулы (XII), где Rh представляет собой C1-6 алкил. Конденсацию замещенного ортодиамина формулы (XI) и сложного эфира формулы (XII) можно проводить в присутствии карбоновой кислоты, например уксусной кислоты и т.п., неорганической кислоты, такой как, например, соляная кислота, серная кислота, или сульфоновой кислоты, такой как, например, метансульфоновая кислота,бензолсульфоновая кислота, 4-метилбензолсульфоновая кислота и т.п. Скорость реакции можно увеличить некоторым подходящим повышением температуры, и в некоторых случаях реакцию можно проводить даже при температуре кипения с обратным холодильником реакционной смеси. Выделяющуюся во время конденсации воду можно удалять из смеси азеотропной дистилляцией, дистилляцией и подобными способами. Промежуточные продукты формулы (XI) можно получить реакцией восстановления азота в амин,исходя из промежуточного продукта формулы (XIII), в присутствии металлического катализатора, такого как никель Ренея (Raney Nickel), и подходящего восстановителя, такого как водород, в подходящем растворителе, таком как метанол. Промежуточные продукты формулы (XIII) можно получить гидролизом промежуточных продуктов формулы (XIV), согласно способам, известным в данной области техники, таким как перемешивание промежуточного продукта (XIV) в водном растворе кислоты в присутствии реакционно-инертного растворителя, например тетрагидрофурана. Подходящей кислотой является, например, соляная кислота. Промежуточные продукты формулы (X) удобно получать путем взаимодействия анилина формулы Промежуточные продукты формулы (VIII), где n равно О, R2 является водородом или гидрокси, и в случае, когда R является водородом, тогда R3, упомянутый в настоящем изобретении в качестве промежуточного продукта формулы (VIII-а), представляет собой гидрокси, можно получить путем обработки промежуточного продукта формулы (XVII), где W представляет собой галоген, литийорганическим реагентом, таким как, например, н-бутиллитий, в реакционно-инертном растворителе, например тетрагидрофуране, и последующим взаимодействием указанного промежуточного продукта с промежуточным продуктом формулы (XVIII), где R1 является водородом или радикалом, определенным в R3. Настоящее изобретение также относится к соединению приведенной выше формулы (I) для применения в качестве лекарственного средства. Соединения настоящего изобретения обладают свойствами ингибировать PARP, что очевидно из нижеприведенной экспериментальной части настоящего изобретения. Настоящее изобретение также относится к применению соединения для получения лекарственного средства для лечения любой из описанных в настоящем изобретении болезни и нарушения у животного,в котором указанные соединения являются соединением формулы (I) его N-оксидными формами, фармацевтически приемлемыми аддитивными солями и стереохимическими изомерными формами, в которомX представляет собой -N= или -CR4=, где R является водородом, или взятый вместе с R1 могут образовывать двухвалентный радикал формулы -СН=СН-СН=СН-;R2 представляет собой водород, или взятый вместе с R3 могут образовывать связь =O;R7 является водородом или C1-6 алкилом;R9 представляет собой ди(C1-6 алкил)аминоC1-6 алкил; или R3 представляет собой группу формулыZ представляет собой гетероциклическую кольцевую систему, выбранную из где каждый R10 независимо представляет собой водород, C1-6 алкил, аминокарбонил, гидрокси,C1-6 алкоксиC1-6 алкил, C1-6 алкоксиC1-6 алкиламино, ди(фенилC2-6 алкенил),пиперидинилC1-6 алкил, C3-10 циклоалкил, C3-10 циклоалкилC1-6 алкил, арилокси(гидрокси)C1-6 алкил, галогениндазолил, арилC1-6 алкил, арилC2-6 алкенил, морфолино, C1-6 алкилимидазолил или пиридинилC1-6 алкиламино; каждый R11 независимо представляет собой водород, гидрокси, пиперидинил или арил; арил представляет собой фенил или замещенный галогеном, C1-6 алкилом или C1-6 алкилокси фенил. Ввиду свойств соединений настоящего изобретения связываться с PARP, они могут использоваться в качестве эталонных соединений или соединений, содержащих меченые атомы, когда один из атомов в молекуле может быть замещен, например, радиоактивным изотопом. Для получения фармацевтических композиций настоящего изобретения эффективное количество конкретного соединения, в форме основной или кислотно-аддитивной соли в качестве активного ингредиента, тщательно смешивают с фармацевтически приемлемым носителем, который может иметь широко разнообразные формы в зависимости от желательной для введения формы препарата. Желательно,чтобы эти фармацевтические композиции представляли собой одноразовую дозировку, предпочтительно подходящую для введения перорально, ректально, подкожно или для парентеральной инъекции. Например, для получения композиций в форме для перорального применения можно использовать любую из обычных фармацевтических сред, таких как, например, вода, гликоли, масла, спирты и т.п., в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или можно использовать твердые носители, такие как крахмалы, сахара, каолин, скользящие вещества, связующие вещества, дезинтегрирующие вещества и т.п., в случае порошков, пилюль, капсул и таблеток. Благодаря легкости введения таблетки и капсулы представляют собой наиболее предпочтительную единицу дозы пероральной формы, и в этом случае, безусловно, используют твердые фармацевтические носители. Носитель для парентеральных композиций будет обычно содержать стерильную воду, по меньшей мере, в значительной степени, хотя можно включать в него другие компоненты, например, способствующие растворимости. Можно получать растворы для инъекций, в которых носитель содержит, например, физиологический раствор, раствор глюкозы или смесь раствора глюкозы и физиологического раствора. Также можно получать суспензии для инъекций, и в этом случае можно использовать подходящие жидкие носители, суспендирующие вещества и т.п. В композициях, подходящих для чрескожного введения, носитель необязательно содержит вещество, повышающее пенетрацию, и/или подходящее смачивающее вещество,необязательно объединенное с подходящими добавками любого характера в незначительных пропорциях, и эти добавки не оказывают значительного вредного воздействия на кожу. Указанные добавки могут облегчать применение на коже и/или могут быть полезны для получения желательных композиций. Эти композиции можно вводить различными способами, например, как трансдермальный пластырь, как точечное нанесение, как мазь. Особенно предпочтительно составлять вышеупомянутые фармацевтические композиции в единице дозы лекарственной формы для облегчения введения и однократности дозировки. Термин единица дозы лекарственной формы, используемый в описании настоящего изобретения и в- 11010592 формуле изобретения, относится к физически дискретным единицам, подходящим для одноразовых дозировок, и каждая единица содержит заданное количество активного ингредиента, рассчитанного, чтобы вызывать желательный терапевтический эффект, в сочетании с установленным фармацевтическим носителем. Примерами таких единиц дозы лекарственной формы являются таблетки (включая таблетки с насечкой или таблетки, покрытые оболочкой), капсулы, пилюли, пакетики с порошком, пластинки, растворы или суспензии для инъекций, дозы по чайной ложке, по столовой ложке и т.п. и их раздельное многоразовое применение. Соединения настоящего изобретения могут лечить или предотвращать повреждение ткани, вызванное повреждением или смертью клетки вследствие некроза или апоптоза; может улучшать состояние нервной ткани или сердечно-сосудистой системы после повреждения, включающего в себя следующее: очаговую ишемию, инфаркт миокарда и реперфузионную травму; могут лечить различные болезни и состояния, вызванные или усугубленные активностью PARP; могут расширять или повышать продолжительность жизни или пролиферативную способность клеток; могут вызывать альтерацию экспрессии гена старения клеток; могут действовать на радиочувствительность и/или химиочувствительность клеток. В целом, ингибирование активности PARP сберегает клетки от потери энергии, в случае нервных клеток предотвращая необратимую деполяризацию нейронов, и, таким образом, обеспечивает нейропротективное действие. По нижеприведенным причинам настоящее изобретение дополнительно относится к способу введения терапевтически эффективного количества вышеопределенных соединений в количестве, достаточном для ингибирования активности PARP, для лечения или предотвращения повреждения ткани, вызванного повреждением или смертью клетки вследствие некроза или апоптоза, для влияния на нейронную активность, не опосредованную NMDA токсичностью, для влияния на нейронную активность, опосредованную NMDA токсичностью, для лечения повреждения нервной ткани, вызванного ишемией и реперфузионной травмой, неврологических нарушений и нейродегенеративных заболеваний; для предотвращения или лечения сосудистого инсульта; для лечения или предотвращения сердечно-сосудистых нарушений; для лечения других состояний и/или нарушений, таких как возрастная мышечная дегенерация,СПИД и другие иммунодегенеративные болезни, воспаление, подагра, артрит, атеросклероз, кахексия,рак, дегенеративные болезни скелетных мышц, вовлекающие репликационное старение, диабет, травмы головы, воспалительные нарушения кишечника (такие как колит и болезнь Крона), мышечная дистрофия, остеоартрит, остеопороз, хроническая и/или острая боль (такая как нейропатическая боль), почечная недостаточность, ишемия сетчатки, септический шок (такой как эндотоксиновый удар), и старение кожи,для увеличения продолжительности жизни и пролиферативной способности клеток; для альтерации экспрессии гена старения клеток; для химиосенсибилизации и/или радиосенсибилизации (гипоксических) клеток опухоли. Настоящее изобретение также относится к лечению болезней и состояний у животных,которое включает введение указанному животному терапевтически эффективного количества вышеопределенных соединений. В частности, настоящее изобретение относится к способу лечения, предотвращения или торможения неврологического нарушения у животного, который включает введение указанному животному терапевтически эффективного количества вышеопределенных соединений. Неврологические нарушения выбирают из группы, состоящей из периферической нейропатии, вызванной физической травмой или состоянием болезни, травматического повреждения мозга, физического повреждения спинного мозга,инсульта, связанного с повреждением мозга, очаговой ишемии, общей ишемии, реперфузионной травмы,демиелинизирующей болезни и неврологического нарушения, относящегося к нейродегенерации. Настоящее изобретение также рассматривает применение соединений формулы (I) для ингибирования активности PARP для лечения, предотвращения или торможения повреждения ткани, вызванного повреждением или смертью клетки вследствие некроза или апоптоза, для лечения, предотвращения или торможения неврологического нарушения у животного. Термин "предотвращение нейродегенерации" включает в себя способность предотвращать нейродегенерацию у больных с наличием впервые диагностированного нейродегенеративного заболевания, или предотвращать риск развития нового дегенеративного заболевания, и способность предотвращать дополнительную нейродегенерацию у пациентов, которые уже больны или имеют симптомы нейродегенеративного заболевания. Термин "лечение", используемый в настоящем изобретении, охватывает любое лечение болезни и/или состояния у животного, в частности, у человека, и включает в себя: (i) предотвращение появления болезни и/или состояния у объекта, у которого может быть предрасположенность к болезни и/или состоянию, но еще не диагностировано его наличие; (ii) торможение болезни и/или состояния, то есть подавление его развития; (iii) ослабление болезни и/или состояния, то есть появление регресса болезни и/или состояния. Термин "радиосенсибилизатор", используемый в настоящем изобретении, обозначает молекулу,предпочтительно низкого молекулярного веса, которая вводится животным в терапевтически эффективных количествах для увеличения чувствительности клеток к ионизирующему излучению и/или для содействия лечению болезней, которые излечимы ионизирующим излучением. Болезни, которые поддают- 12010592 ся лечению ионизирующим излучением, включают в себя неопластические болезни, доброкачественные и злокачественные опухоли и опухолевые клетки. Также в настоящем изобретении рассматривается лечение ионизирующим излучением других болезней, не перечисленных в настоящем изобретении. Термин "химиосенсибилизатор", используемый в настоящем изобретении, обозначает молекулу предпочтительно низкого молекулярного веса, которая вводится животным в терапевтически эффективных количествах для увеличения чувствительности клеток к химиотерапии и/или для содействия лечению болезней, которые излечимы химиотерапией. Болезни, которые являются излечимыми химиотерапией, включают в себя неопластические болезни, доброкачественные и злокачественные опухоли и опухолевые клетки. Также в настоящем изобретении рассматривается химиотерапевтическое лечение других болезней, не перечисленных в настоящем изобретении. Соединения, композиции и способы согласно настоящему изобретению являются особенно полезными для лечения или предотвращения повреждения тканей по причине смерти или повреждения клетки вследствие некроза или апоптоза. Соединения настоящего изобретения могут быть "противоопухолевыми средствами", этот термин также охватывает "средства, препятствующие росту опухолевых клеток" и "антибластомные средства". Например, способы настоящего изобретения являются полезными для лечения рака и химиосенсибилизации и/или радиосенсибилизации опухолевых клеток при раковых заболеваниях, таких как АСТН (адренокортикотропный гормон)-продуцирующие опухоли, острый лимфоцитарный лейкоз, острый нелимфоцитарный лейкоз, рак коры надпочечников, рак мочевого пузыря, рак мозга, рак молочной железы, рак шейки матки, хронический лимфоцитарный лейкоз, хронический миелоцитарный лейкоз, колоректальный рак, Т-клеточная лимфома кожи, рак эндометрия, рак пищевода, саркома Юинга, рак желчного пузыря, волосатоклеточный лейкоз, рак головы и шеи, Ходжкинская лимфома, саркома Капоши, рак почки,рак печени, рак легкого (мелкоклеточный и/или немелкоклеточный), злокачественный перитонеальный выпот, злокачественный плевральный выпот, меланома, мезотелиома, множественная миеломная болезнь, нейробластома, неходжкинская лимфома, остеогенная саркома, рак яичников, рак из зародышевых клеток (герминома), рак простаты, рак поджелудочной железы, рак полового члена, ретинобластома, рак кожи, саркома мягких тканей, плоскоклеточные карциномы, рак желудка, рак яичек, рак щитовидной железы, трофобластические опухоли, рак матки, рак влагалища, рак вульвы и опухоль Вильма. В этой связи соединения настоящего изобретения могут использоваться как "радиосенсибилизаторы" и/или "химиосенсибилизаторы". Известно, что радиосенсибилизаторы повышают чувствительность злокачественных клеток к повреждающему воздействию ионизирующего излучения. По литературным данным было предложено несколько механизмов для модели действия радиосенсибилизаторов, включающих в себя: радиосенсибилизаторы гипоксической клетки (например, соединения 2-нитроимидазола и соединения диоксида бензотриазина), имитирование кислорода или альтернативного поведения подобно биовосстановителям при гипоксии; радиосенсибилизаторы негипоксической клетки (например, галогенированные пиримидины) могут быть аналогами оснований ДНК и избирательно внедряться в ДНК раковых клеток и, таким образом, вызывать радиационно-индуцированное разрушение молекул ДНК и/или препятствовать нормальным механизмам репарации ДНК; и были предположены различные другие потенциальные механизмы действия радиосенсибилизаторов в лечении болезни. В настоящее время во многих протоколах лечения рака используют радиосенсибилизаторы в сочетании с рентгеновским излучением. Примеры рентгенактивируемых радиосенсибилизаторов включают в себя, но не ограничиваются перечисленным, следующее: метронидазол, мисонидазол, десметилмисонидазол, пимонидазол, этанидазол, ниморазол, митомицин С, RSU 1069, SR 4233, ЕO9, RB 6145, никотинамид, 5-бромдезоксиуридин (BUdR), 5 йодезоксиуридин (IUdR), бромдезоксицитидин, фтордезоксиуридин (FudR), гидроксимочевина, цисплатин и их терапевтически эффективные аналоги и производные. Фотодинамическая терапия (PDT) раковых заболеваний использует видимый свет в качестве радиоактиватора сенсибилизирующего средства. Примеры фотодинамических радиосенсибилизаторов включают в себя, но не ограничиваются перечисленным, следующее: производные гематопорфирина, фотофрин, производные бензопорфирина, этиопорфирин олова, феоборбид-а, бактериохлорофилл-а, нафталоцианины, фталоцианины, фталоцианин цинка и их терапевтически эффективные аналоги и производные. Радиосенсибилизаторы можно вводить совместно с терапевтически эффективным количеством одного или нескольких других соединений, включая, но не ограничиваясь перечисленным: соединения,которые способствуют внедрению радиосенсибилизаторов в клетки-мишени; соединения, которые регулируют прохождение терапевтических средств, питательных веществ и/или кислорода в клетки-мишени; химиотерапевтические средства, которые воздействуют на опухоль совместно или без дополнительного облучения; или другие терапевтически эффективные соединения для лечения рака или другой болезни. Примеры дополнительных терапевтических средств, которые можно использовать в сочетании с радиосенсибилизаторами, включают в себя, но не ограничиваются перечисленным: 5-фторурацил, лейковорин,5'-амино-5'-дезокситимидин, кислород, карбоген, переливание эритроцитарной массы, перфторуглероды- 13010592 использовать в сочетании с радиосенсибилизаторами, включают в себя, но не ограничиваются перечисленным: адриамицин, камптотецин, карбоплатин, цисплатин, даунорубицин, доцетаксел, доксирубицин,интерферон (альфа, бета, гамма), интерлейкин 2, иринотекан, паклитаксел, топотекан и их терапевтически эффективные аналоги и производные. Химиосенсибилизаторы можно вводить в сочетании с терапевтически эффективным количеством одного или нескольких других соединений, включая, но не ограничиваясь перечисленным: соединения,которые способствуют внедрению химиосенсибилизаторов в клетки-мишени; соединения, которые регулируют прохождение терапевтических средств, питательных веществ и/или кислорода в клетки-мишени; химиотерапевтические средства, которые воздействуют на опухоль, или другие терапевтически эффективные соединения для лечения рака или другой болезни. Примеры дополнительных терапевтических средств, которые могут использоваться в сочетании с химиосенсибилизаторами, включают в себя, но не ограничиваются перечисленным: метилирующие средства, ингибиторы I-топоизомеразы и другие химиотерапевтические средства, такие как цисплатин и блеомицин. Соединения формулы (I) можно также использовать для выявления или идентификации PARP рецепторов и более конкретно PARP-1 рецепторов. С этой целью соединения формулы (I) могут быть мечеными. Указанная метка может быть выбрана из группы, состоящей из радиоизотопа, спиновой метки,антигенной метки, группы ферментных флуоресцентных маркеров или хемолюминесцентной группы. Из представленных в настоящем изобретении результатов тестов специалисты в данной области техники могут легко определить эффективное количество. В целом рассматривается, что эффективным может быть количество, составляющее от 0,01 до 100 мг/кг массы тела и, в частности, от 0,05 до 10 мг/кг массы тела. Подходящим может быть введение необходимой дозы в виде двух, трех, четырех или более субдоз с подходящими интервалами в течение дня. Указанные субдозы могут быть составлены как единицы дозы лекарственной формы, например, содержащие от 0,5 до 500 мг и, в частности, от 1 до 200 мг активного ингредиента в единице дозы лекарственной формы. Следующие примеры поясняют настоящее изобретение. Экспериментальная часть Далее в настоящем изобретении "BuLi" обозначает бутиллитий, "DCM" обозначает дихлорметан,"DlPE" обозначает диизопропиловый эфир, "ДМФА" обозначает N,N-диметилформамид, "ДМСО" обозначает диметилсульфоксид, "EtOAc" обозначает этилацетат, "EtOH" обозначает этанол, "МЕК" обозначает метилэтилкетон, "МеОН" обозначает метанол и "ТГФ" обозначает тетрагидрофуран. А. Получение промежуточных продуктов. Пример А 1. а). Получение промежуточного продукта 1 и промежуточного продукта 2. Хлорид алюминия (0,6928 моль) порциями добавляли к раствору хлорацетилхлорида (0,5196 моль) в DCM (50,2 мл), поддерживая температуру ниже 30 С. Добавляли 3-этил-2(1 Н)-хинолинон (0,1732 моль), поддерживая температуру ниже 30 С. Смесь перемешивали и нагревали при температуре кипения с обратным холодильником в течение 15 ч, охлаждали и вливали в ледяную воду. Осадок отфильтровывали, промывали водой и переносили в DCM. Органический раствор перемешивали и фильтровали. Осадок сушили с получением 33,5 г промежуточного продукта 1. Фильтрат подвергали экстракции. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель досуха с получением 20,46 г промежуточного продукта 2.b). Получение промежуточного продукта 3. Пиперидин (0,24 моль) добавляли по капле при комнатной температуре к раствору промежуточного продукта 1 и промежуточного продукта 2 в ДМФА (300 мл). Смесь перемешивали в течение 5 мин, вливали в воду и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель досуха. Остаток (39,14 г) очищали колоночной хроматографией на силикагеле(20-45 мкм) (элюент: DCM/MeOH/NH4OH 96/4/0,2). Очищенные фракции собирали и выпаривали растворитель. Часть (3,7 г) остатка (13,8 г) кристаллизовали из 2-пропанона. Осадок отфильтровывали, промывали диэтиловым эфиром и сушили с получением 3 г промежуточного продукта 3 с температурой плавления 190 С. Пример А 2. а). Получение промежуточного продукта 4. По капле добавляли nBuLi 1,6M в гексане (0,0764 моль) при температуре -60 С в токе N2 к смеси 6 бром-3-этил-2-метоксихинолина (0,0694 моль) в ТГФ (185 мл). Смесь перемешивали при температуре-60 С в течение 1 ч и затем добавляли по капле при -60 С к смеси N-метокси-N-метилциклогептанкарбоксамида (0,0694 моль) в диэтиловом эфире (100 мл). Смесь перемешивали при -60 С в течение 1 ч,затем доводили до температуры 0 С, вливали в насыщенный раствор NH4Cl и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Продукт использовали без дополнительной очистки, получая 21,61 г (количественно) промежуточного продукта 4.b). Получение промежуточного продукта 5. Смесь промежуточного продукта 4 (0,0694 моль) в 3N соляной кислоте (317 мл) и ТГФ (159 мл) перемешивали и нагревали при температуре кипения с обратным холодильником в течение ночи. Смесь выливали на лед, превращали в основание концентрированным раствором NH4OH и экстрагировалиEtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Продукт использовали без дополнительной очистки, получая 17,59 г (85%) промежуточного продукта 5. с). Получение промежуточного продукта 6. Гидроборат натрия (0,0296 моль) добавляли порциями при температуре 0 С в токе N2 к смеси промежуточного продукта 5 (0,0591 моль) в МеОН (176 мл). Смесь выливали на лед и экстрагировали DCM. Осадок отфильтровывали и сушили. Продукт использовали без дополнительной очистки, получая 6,38 гd). Получение промежуточного продукта 7. Промежуточный продукт 6 (0,0213 моль) добавляли порциями при температуре 0 С к тионилхлориду (32 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали досуха. Продукт использовали без дополнительной очистки, получая 6,77 г (количественно) промежуточного продукта 7. Пример A3. а). Получение промежуточного продукта 8. Смесь N-метокси-N-метил-4-нитробензолацетамида (0,534 моль) в МеОН (1200 мл) гидрировали при комнатной температуре и давлении 3 бар в течение 1 ч с никелем Ренея (60 г) в качестве катализатора. После поглощения H2 (3 экв.) катализатор отфильтровывали и выпаривали фильтрат с получением 102 г (98%) промежуточного продукта 8.b). Получение промежуточного продукта 9. Ацетилангидрид (1,36 моль) добавляли по капле при комнатной температуре к смеси промежуточного продукта 8 (0,525 моль) в DCM (100 мл). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и смесь экстрагировали DCM. Органический слой отделяли, сушили (MgSO4),фильтровали и выпаривали растворитель. Остаток (151,6 г) очищали колоночной хроматографией на силикагеле (20-45 мкм) (элюент: DCM/MeOH/NH4OH 95/5/0,1). Очищенные фракции собирали и выпаривали растворитель с получением 32 г (26%) промежуточного продукта 9. с). Получение промежуточного продукта 10. Метиллитий 1,6 М (74 мл) добавляли при температуре 0 С в токе N2 к смеси промежуточного продукта 9 (0,059 моль) в ТГФ (210 мл). Смесь перемешивали при 0 С в течение 90 мин, вливали в воду и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (20-45 мкм) (элюент:DCM/MeOH/NH4OH 96/4/0,1). Желательные фракции собирали и выпаривали растворитель с получением 7 г (33%) промежуточного продукта 10.d). Получение промежуточного продукта 11. Дымящую азотную кислоту (5,6 мл) добавляли по капле при температуре ниже 30 С к смеси промежуточного продукта 10 (0,037 моль) в ацетилангидриде (100 мл). Смесь перемешивали при температуре ниже 30 С в течение 1 ч, вливали в ледяную воду, превращали в основание концентрированным раствором NH4OH и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (15-35 мкм)(элюент: DCM/MeOH 99/1). Желательные фракции собирали и растворитель выпаривали с получением 4 г промежуточного продукта 11. е). Получение промежуточного продукта 12. Гидроборат натрия (0,0187 моль) добавляли порциями при температуре 5 С к смеси промежуточного продукта 11 (0,017 моль) в МеОН (50 мл). Смесь перемешивали при 5 С, гидролизовали водой и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель с получением 4,2 г (количественно) промежуточного продукта 12.f). Получение промежуточного продукта 13. Смесь промежуточного продукта 12 (0,0176 моль) в 2N гидроксиде натрия (65 мл), ТГФ (25 мл) иEtOH (25 мл) перемешивали при комнатной температуре в течение 15 ч, вливали в воду и экстрагировалиg). Получение промежуточного продукта 14. Триэтиламин (0,03 моль) добавляли при комнатной температуре к смеси промежуточного продукта 13 (0,015 моль) в DCM (40 мл). Метансулфонилхлорид (0,015 моль) добавляли при температуре 0 С в токе N2. Смесь перемешивали при 0 С в течение 1 ч и при комнатной температуре в течение 3 ч. Растворитель выпаривали при комнатной температуре. Продукт использовали без дополнительной очистки,получая (количественно) промежуточный продукт 14.h). Получение промежуточного продукта 15. Смесь промежуточного продукта 14 (0,015 моль), этил 4-пиперидинкарбоксилата (0,045 моль) и карбоната калия (0,045 моль) в ацетонитриле (100 мл) перемешивали и нагревали при температуре кипения с обратным холодильником в течение 15 ч, вливали в воду и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (15-35 мкм) (элюент: DCM/MeOH/NH4OH 97/3/0,1). Очищенные фракции собирали и выпаривали растворитель с получением 0,7 г (14%) промежуточного продукта 15.i). Получение промежуточного продукта 16. Смесь промежуточного продукта 15 (0,002 моль) в МеОН (50 мл) гидрировали при комнатной температуре под давлением 3 бар в течение 1 ч с никелем Ренея (0,7 г) в качестве катализатора. После поглощения H2 (3 экв.) катализатор фильтровали через целит, промывали МеОН и небольшим количествомDCM и выпаривали фильтрат с получением 0,65 г (количественно) промежуточного продукта 16. Пример А 4. Получение промежуточного продукта 17. 1,6 М nBuLi в гексане (0,148 моль) добавляли по капле при температуре -60 С в токе N2 к смеси 6 бром-3-этил-2-метоксихинолина (0,114 моль) в ТГФ (500 мл), и смесь перемешивали при -60 С в течение 1 ч. Циклогексанкарбоксальдегид (0,137 моль) в ТГФ (100 мл) добавляли по капле при температуре-60 С, перемешивали при -60 С в течение 2 ч и затем перемешивали дополнительно при температуре-40 С в течение 1 ч. Смесь вливали в насыщенный NH4Cl и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Продукт использовали без дополнительной очистки, получая 34,13 г (количественно) промежуточного продукта 17.b). Получение промежуточного продукта 18. Смесь промежуточного продукта 17 (0,114 моль) в 3N соляной кислоте (250 мл) и ТГФ (250 мл) перемешивали и нагревали при температуре кипения с обратным холодильником в течение 24 ч. Смесь охлаждали и добавляли DCM (200 мл). Осадок отфильтровывали, промывали водой и сушили. Продукт использовали без дополнительной очистки, получая 12,5 г (39%) промежуточного продукта 18. с). Получение промежуточного продукта 19. Промежуточный продукт 18 (0,035 моль) порциями добавляли при температуре 0 С к тионилхлориду (56,23 мл). Смесь перемешивали при комнатной температуре в течение 3 ч и выпаривали растворитель. Остаток кристаллизовали из диэтилового эфира. Осадок отфильтровывали, промывали несколько раз диэтиловым эфиром и повторно кристаллизовали из воды и DCM. Смесь перемешивали в течение 15 ч. Осадок отфильтровывали и сушили с получением 7,9 г (75%) промежуточного продукта 19.d). Получение промежуточного продукта 20.(250 мл) перемешивали при комнатной температуре в токе N2 в течение 10 мин. Раствор промежуточного продукта 19 (0,0434 моль) в ДМФА (50 мл) медленно добавляли и смесь перемешивали при комнатной температуре в течение 1 ч и затем при температуре 70 С в течение 1 ч. Смесь охлаждали до комнатной температуры и вливали в воду (1500 мл). Осадок отфильтровывали, промывали несколько раз холодной водой и сушили. Продукт использовали на следующей стадии реакции без дополнительной очистки. Часть (3 г) остатка (13,8 г) очищали колоночной хроматографией на силикагеле (15-40 мкм) (элюент:DCM/MeOH/NH4OH 99/1/0,1). Желательные фракции собирали и выпаривали растворитель. Остаток (2,9 г) кристаллизовали из MEK/DIPE, фильтровали и сушили с получением 2,85 г промежуточного продукта 20. Пример А 5. а). Получение промежуточного продукта 21. 1,6 М nBuLi в гексане (0,0516 моль) добавляли по капле при температуре -60 С в токе N2 к смеси 6 бром-3-этил-2-метоксихинолина (0,043 моль) в ТГФ (115 мл). Смесь перемешивали при -60 С в течение 1 ч. Смесь 1-циклогексил-3-(4-метил-1-пиперазинил)-1-пропанона (0,043 моль) в ТГФ (103 мл) добавляли по капле при температуре -60 С. Смесь перемешивали при -60 С в течение 1 ч, доводили до температуры 0 С, вливали в насыщенный раствор NH4Cl и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (20-45 мкм) (элюент: DCM/MeOH/NH4OH 97/3/0,2 и 90/10/0,2). Очищенные фракции собирали и выпаривали растворитель с получением 3,7 г (20%) промежуточного продукта 21.b). Получение промежуточного продукта 22. Смесь промежуточного продукта 21 (0,00841 моль), хлорида олова(II) (0,0336 моль) и 12N соляной кислоты (0,121 моль) в уксусной кислоте (36 мл) перемешивали при температуре 80 С в течение 16 ч. Смесь выливали на лед, превращали в основание концентрированным раствором NH4OH и экстрагировали DCM. Органический слой отделяли, сушили, фильтровали и выпаривали растворитель. Продукт использовали без дополнительной очистки, получая 2,45 г (74%) промежуточного продукта 22. Пример А 6. а). Получение промежуточного продукта 23. Фосфорилхлорид (110,9 мл) добавляли по капле при температуре 5 С к ДМФА (81,5 мл). Смесь перемешивали до полного растворения. Добавляли этиловый эфир 4-[(1-оксобутил)амино]бензойной кислоты (0,34 моль). Смесь перемешивали при температуре 100 С в течение 15 ч, затем охлаждали до комнатной температуры и вливали в ледяную воду. Осадок отфильтровывали, промывали водой и сушили с получением 42,35 г (47%) промежуточного продукта 23.b). Получение промежуточного продукта 24. Смесь промежуточного продукта 23 (0,1606 моль) в 30% растворе метанолята натрия в МеОН(152,8 мл) и МеОН (400 мл) перемешивали и нагревали при температуре кипения с обратным холодильником в течение 15 ч, затем охлаждали и вливали в ледяную воду. Осадок отфильтровывали, промывали- 18010592 водой и вливали в DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель досуха, получая 31,64 г (85%) промежуточного продукта 24. с). Получение промежуточного продукта 25. Алюмотетрагидрид лития (0,1288 моль) порциями добавляли при температуре 0 С в токе N2 к раствору промежуточного продукта 24 (0,1288 моль) в ТГФ (263 мл). Смесь перемешивали в течение 30 мин,вливали в ледяную воду и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель досуха с получением 27,4 г (98%) промежуточного продукта 25.d). Получение промежуточного продукта 26. Метансульфонилхлорид (0,104 моль) добавляли по капле при температуре 0 С в токе N2 к смеси промежуточного продукта 25 (0,069 моль) и триэтиламина (0,207 моль) в DCM (120 мл). Смесь перемешивали при 0 С в течение 4 ч. Растворитель выпаривали досуха (без нагревания). Продукт использовали без дополнительной очистки, получая 20,4 г промежуточного продукта 26. е). Получение промежуточного продукта 27. Смесь промежуточного продукта 26 (0,0691 моль), 1-(фенилметил)пиперазина (0,0829 моль) и карбоната калия (0,145 моль) в ацетонитриле (150 мл) перемешивали и нагревали при температуре кипения с обратным холодильником в течение 12 ч. Растворитель выпаривали досуха. Остаток переносили в DCM и воду. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (24,6 г) очищали колоночной хроматографией на силикагеле (20-45 мкм) (элюент:DCM/MeOH/NH4OH 98/2/0,1). Очищенные фракции собирали и выпаривали растворитель. Остаток (2,7 г) кристаллизовали из DIPE. Осадок фильтровали и сушили с получением 1,6 г промежуточного продукта 27 с температурой плавления 78 С. Пример А 7. а). Получение промежуточного продукта 28. 1,6 М nBuLi в гексане (0,224 моль) добавляли при температуре -78 С в токе N2 к раствору 6-бром-3 этил-2-метоксихинолина (0,188 моль) в ТГФ (500 мл). Смесь перемешивали при -78 С в течение 1 ч. Смесь 3-циклогексен-1-карбоксальдегида (0,182 моль) в ТГФ (500 мл) добавляли по капле при температуре -78 С. Смесь перемешивали при -78 С в течение 2 ч, затем доводили до температуры 0 С, гидролизовали и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (58,9 г) очищали колоночной хроматографией на силикагеле (20-45 мкм)(элюент: DCM/EtOAc 96/4). Очищенные фракции собирали и выпаривали растворитель с получением 40,5 г (72%) промежуточного продукта 28.b). Получение промежуточного продукта 29. Смесь промежуточного продукта 28 (0,131 моль) в 3N соляной кислоте (400 мл) и ТГФ (400 мл) перемешивали при температуре 60 С в течение ночи и затем превращали в основание твердым карбонатом калия. Осадок отфильтровывали, промывали DCM и сушили. Фильтрат экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток кристаллизовали из DCM. Осадок отфильтровывали и сушили. Часть (1,5 г) остатка (16,5 г) переносили в МеОН. Смесь перемешивали в течение ночи. Осадок отфильтровывали и сушили с получением 0,72 г промежуточного продукта 29 с температурой плавления 212 С. с). Получение промежуточного продукта 30. Промежуточный продукт 29 (0,053 моль) при температуре 0 С медленно добавляли к тионилхлориду (150 мл). Смесь перемешивали при комнатной температуре в течение 4 ч. Растворитель выпаривали досуха. Остаток несколько раз переносили в DCM. Растворитель выпаривали. Продукт использовали без дополнительной очистки, получая 16 г (количественно) промежуточного продукта 30.d). Получение промежуточного продукта 31.(200 мл) перемешивали при комнатной температуре в токе N2 в течение 10 мин. Медленно добавляли раствор промежуточного продукта 30 (0,053 моль) в ДМФА (200 мл). Смесь перемешивали при комнатной температуре в течение 1 ч и затем вливали в воду. Осадок отфильтровывали, промывали несколько раз водой и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (19,2 г) очищали колоночной хроматографией на силикагеле (15-40 мкм)(элюент: DCM/MeOH/NH4OH 96/4/0,2). Очищенные фракции собирали и выпаривали растворитель с получением 11,4 г (59%) промежуточного продукта 31. Пример А 8. а). Получение промежуточного продукта 32. 1,6 М nBuLi в гексане (0,154 моль) добавляли по капле при температуре -60 С в токе N2 к смеси 6 бром-3-этил-2-метоксихинолина (0,118 моль) в ТГФ (314 мл), и смесь перемешивали при -60 С в течение 1 ч. 2-Тиофенкарбоксальдегид (0,142 моль) в ТГФ (100 мл) добавляли по капле при температуре -60 С. Смесь перемешивали при -60 С в течение 2 ч, затем при -40 С в течение 1 ч, вливали в насыщенный раствор NH4Cl и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Продукт использовали без дополнительной очистки, получая 35,37 г (количественно) промежуточного продукта 32.b). Получение промежуточного продукта 33. Смесь промежуточного продукта 32 (0,118 моль) в 3N соляной кислоте (426 мл) и ТГФ (274 мл) перемешивали при температуре 70 С в течение 6 ч. Смесь наливали на лед, превращали в основание концентрированным раствором NH4OH и экстрагировали EtOAc. Органический слой отделяли, сушили(MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (20-45 мкм) (элюент: DCM/MeOH 98/2). Очищенные фракции собирали и выпаривали растворитель с получением 9,3 г (28%) промежуточного продукта 33. с). Получение промежуточного продукта 34. Промежуточный продукт 33 (0,0322 моль) порциями добавляли при температуре 0 С к тионилхлориду (46 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали досуха. Продукт использовали без дополнительной очистки, получая 9,78 г (количественно) промежуточного продукта 34.d). Получение промежуточного продукта 35. Карбонат калия (0,161 моль) добавляли к смеси гидрохлорида 4,4-пиперидиндиола (0,0483 моль) в ацетонитриле (74 мл). Смесь перемешивали в токе N2 в течение 15 мин. Смесь промежуточного продукта 34 (0,0322 моль) в ацетонитриле (98 мл) добавляли при комнатной температуре. Смесь перемешивали при температуре 60 С в течение ночи, затем вливали в воду и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 97/3/0,1). Очищенные фракции собирали и выпаривали растворитель с получением 0,46 г (4%) промежуточного продукта 35. В. Получение окончательной композиции. Пример В 1. Получение соединения 1. Гидроборат натрия (0,0318 моль) добавляли при температуре 0 С в токе N2 к раствору промежуточного продукта 3 (0,0245 моль) в МеОН (80 мл). Смесь перемешивали в течение 30 мин. Добавляли воду(10 мл). Выпаривали органический растворитель. Водный концентрат переносили в DCM и воду и смесь экстрагировали. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель досуха. Часть (3 г) остатка (7,5 г) кристаллизовали из 2-пропанона и небольшого количества МеОН. Осадок отфильтровывали и сушили с получением 2,69 г соединения 1 с температурой плавления 172 С. Пример В 2. Получение соединения 2. Карбонат калия (0,107 моль) добавляли к смеси гидрохлорида 4,4-пиперидиндиола (0,032 моль) в ацетонитриле (49 мл). Смесь перемешивали в токе N2 в течение 15 мин. Добавляли смесь промежуточного продукта 7 (0,0213 моль) в ацетонитриле (68 мл). Смесь перемешивали при температуре 60 С в течение 3 ч, затем вливали в воду и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4),фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле(15-40 мкм) (элюент: DCM/MeOH/NH4OH 98,5/1,5/0,1). Очищенные фракции собирали и выпаривали растворитель. Остаток кристаллизовали из диэтилового эфира. Осадок отфильтровывали и сушили с получением 4,16 г (51%) соединения 2 с температурой плавления 218 С. Пример В 3. Получение соединения 3. Смесь промежуточного продукта 16 (0,002 моль) и этил 2-оксобутаноата (0,004 моль) в EtOH (15 мл) перемешивали и нагревали при температуре кипения с обратным холодильником в течение 2,5 ч,вливали в воду и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (0,9 г) очищали колоночной хроматографией на силикагеле (15-40 мкм) (элюент: циклогексан/2-пропанол/NH4OH 85/15/1). Очищенные фракции собирали и растворитель выпаривали. Остаток кристаллизовали из диэтилового эфира. Осадок отфильтровывали и сушили с получением 0,054 г соединения 3 с температурой плавления 163 С. Пример В 4. Получение соединения 4. Смесь гидрида натрия (0,42 г) в ТГФ (10,5 мл) перемешивали при комнатной температуре в течение 10 мин. Затем ТГФ декантировали от ДМСО (32 мл), затем добавляли триметилсульфоксония йодид(0,013 моль). Смесь перемешивали при комнатной температуре в течение 1 ч. Медленно добавляли промежуточный продукт 20 (0,0114 моль). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и смесь экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток кристаллизовали из смеси 2-пропанон/диэтиловый эфир. Осадок отфильтровывали и сушили. Остаток перекристаллизовывали из смеси 2-пропанон/диэтиловый эфир. Осадок отфильтровывали и сушили с получением 1,54 г (36%) соединения 4 с температурой плавления 200 С. Пример В 5. Получение соединения 5. Смесь промежуточного продукта 22 (0,00623 моль) в МеОН (25 мл) гидрировали при комнатной температуре под давлениям 3 бара в течение 8 ч с 10% Pd/C (0,25 г) в качестве катализатора. После поглощения H2 (1 экв.) катализатор фильтровали через целит и фильтрат выпаривали. Остаток переносили в воду и концентрированный раствор NH4OH и смесь экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 94/6/0,3). Очищенные фракции собирали и растворитель выпаривали. Остаток кристаллизовали из 2-пропанона. Осадок отфильтровывали и сушили с получением 1,07 г (43%) соединения 5 с температурой плавления 181 С. Пример В 6. Получение соединения 6. 1,6 М nBuLi в гексане (21,32 мл) добавляли по капле при температуре -70 С в токе N2 к смеси 1 метил-1 Н-имидазола (0,0341 моль) в ТГФ (28 мл). Смесь перемешивали при -70 С в течение 30 мин. Добавляли хлоротриэтилсилан (0,0341 моль). Смесь доводили до комнатной температуры. Добавляли по капле 1,6 М nBuLi в гексане (21,32 мл) при температуре -70 С. Смесь перемешивали при -70 С в течение 1 ч и затем доводили до температуры -15 С. Смесь промежуточного продукта 20 (0,0136 моль) в ТГФ (50 мл) добавляли по капле при температуре -70 С. Смеси давали нагреться до комнатной температуры в течение ночи, затем вливали в насыщенный раствор NH4Cl и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (20-45 мкм) (элюент: DCM/MeOH/NH4OH 94/6/0,5). Очищенные фракции собирали и растворитель выпаривали. Остаток кристаллизовали из 2-пропанона. Осадок фильтровали и сушили с получением 5,05 г (83%) соединения 6 с температурой плавления 194 С. Пример В 7. Получение соединения 7. Смесь промежуточного продукта 27 (0,00319 моль) в 6N соляной кислоте (70 мл) перемешивали при температуре 80 С в течение 30 мин, вливали в воду (50 мл) и твердый карбонат калия. Смесь перемешивали в течение 10 мин. Осадок отфильтровывали, промывали водой и сушили с получением 0,9 г Смесь промежуточного продукта 19 (0,0164 моль) в 2-(диметиламино)этаноле (50 мл) перемешивали и нагревали при температуре кипения с обратным холодильником в течение 2 ч. Смесь вливали в воду и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток (16 г) очищали колоночной хроматографией на силикагеле (15-40 мкм) (элюент:DCM/MeOH/NH4OH 94/6/0,5). Очищенные фракции собирали. Смесь оставляли для кристаллизации в течение нескольких дней (окончательная преципитация). Осадок отфильтровывали, переносили в петролейный эфир, фильтровали и сушили с получением 2,8 г (48%) соединения 8 с температурой плавления 122 С. Соединение 8 (0,02244 моль) отделяли от его энантиомеров колоночной хроматографией (элюент: гексан/2-пропанол 88/12; колонка: CHIRALPAK AD). Очищенные фракции собирали и растворитель выпаривали. Остаток кристаллизовали из гексана и петролейного эфира. Осадок отфильтровывали и сушили с получением 2,2 г соединения 9 с температурой плавления 115 С и 2,02 г соединения 10 с температурой плавления 115 С. Пример В 9. Получение соединения 11. Цианотригидроборат натрия (0,02 моль) порциями при температуре 0 С в токе N2 добавляли к раствору промежуточного продукта 31 (0,02 моль) и 2-метоксиэтанамина (0,024 моль) в МеОН (100 мл). Смесь перемешивали при комнатной температуре в течение 12 ч. Добавляли воду и смесь экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток (9,7 г) очищали колоночной хроматографией на силикагеле (20-45 мкм) (элюент:DCM/MeOH/NH4OH 93/7/0,5). Очищенные фракции собирали и растворитель выпаривали. Остаток кристаллизовали из 2-пропанона и диэтилового эфира. Осадок отфильтровывали и сушили с получением 0,63 г соединения 11 с температурой плавления 196 С. Пример В 10. Получение соединения 12. Цианотригидроборат натрия (0,00126 моль) при температуре 0 С добавляли к смеси промежуточного продукта 35 (0,00126 моль) и 2-метоксиэтанамина (0,00151 моль) в МеОН (10 мл). Смесь перемешивали при комнатной температуре в течение ночи, затем выливали на лед и экстрагировали DCM. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 90/10/0,1). Очищенные фракции собирали и растворитель выпаривали. Остаток кристаллизовали из МЕК и диэтилового эфира. Осадок отфильтровывали и сушили с получением 0,22 г (41%) соединения 12. В табл. F-1 перечислены соединения, полученные согласно одному из вышеописанных примеров. Фармакологический пример. Ингибирующая активность PARP-1 в анализе сцинтилляционных зазоров (SPA) in vitro. Соединения настоящего изобретения тестировали в анализе in vitro согласно методике SPA (запатентованной Amersham Pharmacia Biotech). В принципе, анализ основан на хорошо отработанной методике SPA для обнаружения поли(АТРрибозил)ирования биотинилированных белков-мишеней, т.е. гистонов. Это рибозилирование индуцируется использованием фермента PARP-1, активированного ДНК с разрывами (nicked ДНК), и [3 Н]никотинамидаденин-динуклеотидом ([3H]-NAD+) в качестве АТР-рибозил донора. В качестве индуктора ферментной активности PARP-1 получали ДНК с разрывами. Для этого 25 мг ДНК (поставщик: Sigma) растворяли в 25 мл буфера ДНК (10 мМ Трис-HCl, рН 7,4; 0,5 мг/мл Альбумина бычьей сыворотки (BSA); 5 мМ MgCl2H2O и 1 мМ KCl), к которому добавляли 50 мкл раствора ДНК (1 мг/мл в 0,15 М NaCl). После инкубации в течение 90 мин при температуре 37 С реакцию завершали добавлением 1,45 г NaCl, с последующей дополнительной инкубацией при 58 С в течение 15 мин. Реакционную смесь охлаждали на льду и проводили диализ при 4 С в течение 1,5 и 2 ч соответственно против 1,5 л 0,2 М KCl и дважды против 1,5 л 0,01 М KCl в течение 1,5 и 2 ч соответственно. Смесь была аликвотной, и ее сохраняли при температуре -20 С. Гистоны (1 мг/мл, тип II-A, поставщик Sigma) биотинилировали с использованием набора для биотинилирования Amersham и сохраняли аликвотными при температуре -20 С. Шарики основного раствора SPA в 100 мг/мл поли(винилтолуола) (PVT) (поставщикAmersham) делали в фосфатно-буферном растворе (ФБР). Готовили основной раствор [3H]-NAD+ добавлением 120 мкл [3H]-NAD+ (0,1 мКи/мл, поставщик NEN) к 6 мл инкубационного буфера (50 мМTris/HCl, рН 8; 0,2 мМ дитиотреитола (ДТТ); 4 мМ MgCl2). Раствор 4 мМ NAD+ (поставщик: Roche) готовили в инкубационном буфере (из 100 мМ основного раствора в воде, хранящегося при температуре- 25010592 используемой последовательности белка фермента PARP-1, включающую в себя ссылки на литературные данные, можно найти в базе данных Swiss-Prot под номером первичного запроса P09874. Биотинилированные гистоны и PVT-SPA шарики смешивали и предварительно инкубировали в течение 30 мин при комнатной температуре. Фермент PARP-1 (концентрация зависела от серии) смешивали с ДНК с разрывами и смесь предварительно инкубировали в течение 30 мин при температуре 4 С. Равные части этих растворов гистонов/PVT-SPA шариков и фермента PARP-1/ДНК с разрывами смешивали и на одну лунку добавляли 75 мкл этой смеси вместе с 1 мкл соединения в ДМСО и 25 мкл [3H]-NAD+ в 96 луночном микротитрационном планшете. Конечные концентрации в инкубационной смеси составляли для биотинилированных гистонов 2 мкг/мл, для PVT-SPA шариков 2 мг/мл, для ДНК с разрывами 2 мкг/мл и от 5 до 10 мкг/мл для фермента PARP-1. После инкубации указанной смеси в течение 15 мин при комнатной температуре реакцию завершали добавлением 100 мкл 4 мМ NAD+ в инкубационном буфере (конечная концентрация 2 мМ) и планшеты перемешивали. Шарики оставляли для седиментации в течение по меньшей мере 15 мин и планшеты переносили в счетчик TopCountNXT (Packard) для сцинтилляционного подсчета, показатели обозначались в числе импульсов в минуту (cpm). Для каждого эксперимента параллельно проводили исследования в контрольных группах (содержащих фермент PARP-1 и ДМСО без соединения), слепую инкубацию (содержащую ДМСО, но не содержащую фермент PARP-1 или соединение) и пробы (содержащие фермент PARP-1 и соединение, растворенные в ДМСО). Все тестовые соединения растворяли в ДМСО и, в конечном итоге,дополнительно разбавляли ДМСО. В первом случае соединения тестировали при концентрации 10-6 М. Когда соединения показывали активность при 10-6 М, делали кривую дозовой зависимости, с которой тестировали соединения при концентрациях, составляющих от 10-5 до 10-8 М. Из каждого теста вычитали пустое значение как из контрольных, так и опытных значений. Контрольные пробы представляли максимальную активность фермента PARP-1. Для каждой пробы значение cpm выражали как процент от среднего значения cpm контрольных групп. Когда было целесообразно, вычисляли значения IC50 (концентрация лекарственного средства, необходимая для 50% снижения активности фермента PARP-1 по сравнению с контрольной концентрацией), используя линейную интерполяцию между экспериментальными точками непосредственно выше и ниже 50% уровня. Приводимые в настоящем изобретении эффекты тестовых соединений выражены как pIC50 (отрицательное значение по логарифмической шкале показателя IC50). В качестве эталонного соединения для подтверждения, в анализ SPA включали 4-амино-1,8 нафталимид. Тестовые соединения проявляли ингибирующую активность при начальной тестовой концентрации, составляющей 10-6 М (см. табл. 2). Ингибирующая активность PARP-1 в анализе фильтрации in vitro. Соединения настоящего изобретения тестировали in vitro в анализе фильтрации, оценивающем активность PARP-1 (вызванной в присутствии ДНК с разрывами) посредством ее гистонной поли(ADPрибозил)ирующей активности с использованием [32P]-NAD в качестве ADP-рибозил донора. Радиоактивные рибозилированные гистоны преципитировали трихлоруксусной кислотой (ТСА) в 96-луночном фильтрационном планшете и измеряли захват [32 Р], используя сцинтилляционный счетчик. Готовили смесь гистонов (основной раствор 5 мг/мл в Н 2 О), NAD+ (основной раствор 100 мМ вH2O), и [32P]-NAD+ в инкубационном буфере (50 мМ Tris/HCl, рН 8; 0,2 мМ ДТТ; 4 мМ MgCl2). Также готовили смесь фермента PARP-1 (от 5 до 10 мкг/мл) и ДНК с разрывами. ДНК с разрывами готовили согласно описанию в анализе in vitro SPA ингибирующей активности PARP-1. На лунку добавляли 75 мкл смеси фермент PARP-1/ДНК вместе с 1 мкл соединения в ДМСО и 25 мкл смеси гистоны-NAD+/[32P]-NAD+ В 96-луночный фильтрационный планшет (0,45 мкм, поставщик Millipore). Конечные концентрации в инкубационной смеси составляли 2 мкг/мл для гистонов, 0,1 мМ для NAD+, 200 мкМ (0,5 мкКи) для [32P]-NAD+ и 2 мкг/мл для ДНК с разрывами. Планшеты инкубировали в течение 15 мин при комнатной температуре и реакцию завершали добавлением 10 мкл ледяной 100% ТСА, с последующим добавлением 10 мкл ледяного раствора BSA (1% в Н 2 О). Белковую фракцию оставляли для преципитации в течение 10 мин при температуре 4 С и планшеты подвергали вакуумной фильтрации. Затем планшеты промывали с использованием на каждую лунку 1 мл 10% ледяной ТСА, 1 мл 5% ледяной ТСА и 1 мл 5% ТСА при комнатной температуре. В конце в каждую лунку добавляли 100 мкл сцинтилляционного раствора (Microscint 40, Packard) и планшеты переносили в счетчик TopCountNXT (поставщик Packard) для сцинтилляционного подсчета, показатели обозначались в числе импульсов в минуту (cpm). Для каждого эксперимента параллельно проводили исследования в контрольных группах (содержащих ферментPARP-1 и ДМСО без соединения), слепую инкубацию (содержащую ДМСО, но не содержащую ферментPARP-1 или соединение) и пробы (содержащие фермент PARP-1 и соединение, растворенные в ДМСО). Все тестовые соединения растворяли в ДМСО и, в конечном итоге, дополнительно разбавляли ДМСО. В первом случае соединения тестировали при концентрации 10-5 М. Когда соединения показывали активность при 10-5 М, делали кривую дозовой зависимости, с которой тестировали соединения при концентрациях, составляющих от 10-5 до 10-8 М. Из каждого теста вычитали пустое значение как из контрольных, так и опытных значений. Контрольные пробы представляли максимальную активность ферментаPARP-1. Для каждой пробы значение cpm выражали как процент от среднего значения cpm контрольных- 26010592 групп. Когда было целесообразно, вычисляли значения IC50 (концентрация лекарственного средства, необходимая для 50% снижения активности фермента PARP-1 по сравнению с контрольной концентрацией),используя линейную интерполяцию между экспериментальными точками непосредственно выше и ниже 50% уровня. Приводимые в настоящем изобретении эффекты тестовых соединений выражены как pIC50(отрицательное значение по логарифмической шкале показателя IC50). В качестве эталонного соединения для подтверждения в анализ фильтрации включали 4-амино-1,8-нафталимид. Тестовые соединения проявляли ингибирующую активность при начальной тестовой концентрации, составляющей 10-5 М (см. табл. 2). Таблица 2 Соединения настоящего изобретения можно дополнительно оценивать в анализе клеточной химиои/или радиосенсибилизации, в анализе, измеряющем ингибирующую активность эндогенной PARP-1 в линиях опухолевых клеток и, наконец, в тесте радиосенсибилизации in vivo. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I)R2 является водородом или взятый вместе с R3 может образовывать =O;R9 является ди(C1-6 алкил)аминоC1-6 алкилом; или R3 представляет собой группу формулыZ является гетероциклической кольцевой системой, выбранной из где каждый R10 независимо представляет собой водород, C1-6 алкил, гидрокси, C1-6 алкилоксиC1-6 алкил,C1-6 алкилоксиC1-6 алкиламино, морфолино, C1-6 алкилимидазолил или пиридинилC1-6 алкиламино; каждый R11 независимо является водородом или гидрокси; арил является фенилом или замещенным галогеном, C1-6 алкилом или C1-6 алкилоксифенилом при условии, что исключен 6-(циклогексил-1 Н-имидазол-1-илметил)-3-метил-2(1 Н)-хиноксалинон. 2. Соединение по п.1, в котором n равно 0; X является СН; Q является -NH-, -СН 2-СН 2- или -CHR5-,где R5 является водородом, гидрокси или арилC1-6 алкилом; R1 является C1-6 алкилом; R2 является водородом; R3 представляет собой водород, гидрокси или группу формулы (b-1); t равно 0; Z представляет собой гетероциклическую кольцевую систему, выбранную из (с-8) или (с-13); каждый R10 независимо представляет собой водород; и арил является фенилом. 3. Соединение по п.1, в котором соединение выбирают из соединения 7, соединения 2, соединения 1 и соединения 11. 4. Применение соединения по любому из пп.1-3 в качестве лекарственного средства. 5. Фармацевтическая композиция, содержащая фармацевтически приемлемые носители и в качестве активного ингредиента терапевтически эффективное количество соединения по пп.1-3. 6. Способ получения фармацевтической композиции по п.5, в котором фармацевтически приемлемые носители и соединение по пп.1-3 тщательно смешаны. 7. Применение соединения для получения лекарственного препарата для лечения PARPопосредованного нарушения, в котором соединение представляет собой соединение формулы (I)R2 является водородом или взятый вместе с R3 может образовывать =O;R9 является ди(C1-6 алкил)аминоC1-6 алкилом; или R3 представляет собой группу формулыZ является гетероциклической кольцевой системой, выбранной из где каждый R10 независимо представляет собой водород, C1-6 алкил, гидрокси, C1-6 алкилоксиC1-6 алкил,C1-6 алкилоксиC1-6 алкиламино, морфолино, C1-6 алкилимидазолил или пиридинилC1-6 алкиламино; каждый R11 независимо является водородом или гидрокси; арил является фенилом или замещенным галогеном, C1-6 алкилом или C1-6 алкилоксифенилом при условии, что исключен 6-(циклогексил-1 Н-имидазол-1-илметил)-3-метил-2(1 Н)-хиноксалинон. 8. Применение по п.7 для получения лекарственного препарата для лечения химиосенсибилизации. 9. Применение по п.7 для получения лекарственного препарата для лечения радиосенсибилизации. 10. Способ получения соединения по п.1, отличающийся тем, что включает гидролиз промежуточных продуктов формулы (VIII), согласно известным в данной области техники способам, путем обработки промежуточных продуктов формулы (VIII) подходящими реагентами, такими как хлорид олова, уксусная кислота и соляная кислота, в присутствии реакционно-инертного растворителя, например тетрагидрофурана- 29010592 11. Способ получения соединения по п.1, отличающийся тем, что включает циклизацию промежуточных продуктов формулы (X) согласно известным в данной области техники способам циклизации в соединения формулы (I), где X представляет собой СН, указанные как соединения формулы (I-j), предпочтительно в присутствии подходящей кислоты Льюиса, например хлорида алюминия, или в неразбавленном виде, или растворенной в подходящем растворителе, таком как, например, ароматические углеводороды, например в бензоле, хлорбензоле, метилбензоле и т.п.; в галогенированных углеводородах,например в трихлорметане, тетрахлорметане и т.п.; в эфире, например в тетрагидрофуране, 1,4-диоксане и т.п. или в смеси таких растворителей 12. Способ получения соединения по п.1, отличающийся тем, что включает конденсацию подходящего ортобензолдиамина формулы (XI) со сложным эфиром формулы (XII) с получением соединений формулы (I), указанных как соединения формулы (I-а-1), в которых X является N и R2, взятый вместе сR3, образует связь =O, в присутствии карбоновой кислоты, например уксусной кислоты и т.п., неорганической кислоты, такой как, например, соляная кислота, серная кислота, или в присутствии сульфоновой кислоты, такой как, например, метансульфоновая кислота, бензолсульфоновая кислота, 4 метилбензолсульфоновая кислота и т.п.
МПК / Метки
МПК: C07D 241/44, C07D 401/06
Метки: 6-замещённые, качестве, полимеразы, 2-замещённые, хинолиноны, поли, ингибиторов, 2-хиноксалиноны, циклогексилалкил, adp-рибоза
Код ссылки
<a href="https://eas.patents.su/30-10592-6-zameshhyonnye-ciklogeksilalkil-2-zameshhyonnye-hinolinony-i-2-hinoksalinony-v-kachestve-ingibitorov-poli-adp-riboza-polimerazy.html" rel="bookmark" title="База патентов Евразийского Союза">6-замещённые циклогексилалкил 2-замещённые хинолиноны и 2-хиноксалиноны в качестве ингибиторов поли (adp-рибоза) полимеразы</a>
6-алкенил и 6-фенилалкил замещённые 2-хинолиноны и 2-хиноксалиноны в качестве ингибиторов поли(адф-рибоза) полимеразы

Номер патента: 9875
Опубликовано: 28.04.2008
Авторы: Ваутерс Вальтер Баудевейн Леопольд, Сомерс Мария Викторина Франциска, Мабир Доминик Жан-Пьер, Гийемон Жером Эмиль Жорж, Ван Дюн Якобус Альфонсус Йозефус
МПК: A61K 31/498, A61K 31/4704, A61P 1/04...
Метки: 2-хинолиноны, 6-фенилалкил, поли(адф-рибоза, 6-алкенил, 2-хиноксалиноны, полимеразы, замещённые, ингибиторов, качестве
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, соли присоединения и стереохимически изомерные формы, где n равно 0, 1 или 2; X является N или CR7, где R7 является водородом или взятый вместе с R1 может образовывать двухвалентный радикал формулы -СН=СН-СН=СН-; R1 является C1-6алкилом или тиофенилом; R2 является водородом, гидрокси, C1-6алкилом, С3-6алкинилом или взятый вместе с R3 может образовывать =O; R3 является радикалом, выбранным из...
6-замещённые 2-хинолиноны и 2-хиноксалиноны как ингибиторы поли(адф-рибоза)полимеразы

Номер патента: 10488
Опубликовано: 30.10.2008
Авторы: Гийемон Жером Эмиль Жорж, Мабир Доминик Жан-Пьер, Ван Дюн Якобус Альфонсус Йозефус, Ваутерс Вальтер Баудевейн Леопольд, Сомерс Мария Викторина Франциска
МПК: A61P 43/00, A61K 31/498, C07D 241/44...
Метки: 6-замещённые, 2-хиноксалиноны, ингибиторы, 2-хинолиноны, поли(адф-рибоза)полимеразы
Формула / Реферат:
1. Соединения формулы (I) их N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где n равно 0 или 1; X представляет собой N или CR5, где R5 означает водород; R1 представляет собой C1-6-алкил; R2 представляет собой водород или гидроксигруппу или, взятый вместе с R3 или R4, может образовывать =0; R3 представляет собой радикал, выбранный из (а-1), (а-2) или (а-3) или группу формулы (b-1), то есть -Z-;...
Производные нуклеозидов в качестве ингибиторов рнк-зависимой рнк вирусной полимеразы

Номер патента: 7491
Опубликовано: 27.10.2006
Авторы: Прхавк Мариджа, Маккосс Малкольм, Олсен Дэвид Б., Пракаш Тхазха П., Бхат Балкришен, Кэрролл Стивен С., Бхат Неелима, Кук Филлип Дэн, Элдруп Энн Б., Сонг Кванлай
МПК: A61K 31/7064, C07H 19/14, A61P 31/14...
Метки: производные, качестве, полимеразы, рнк, рнк-зависимой, нуклеозидов, вирусной, ингибиторов
Формула / Реферат:
1. Соединение структурной формулы II или его фармацевтически приемлемая соль; где R1 представляет метил, фторметил, гидроксиметил, дифторметил, трифторметил или аминометил; R2 представляет гидрокси, фтор или метокси; R3 представляет водород, фтор, гидрокси, амино или метокси; R5 представляет водород или Р3О9Н4; R8 представляет водород или амино; R9 представляет водород, циано, метил, галоген или CONH2 и R10 и R11, каждый независимо,...
2-амино-6-(2-замещенные-4-фенокси)-замещенные-пиридины в качестве ингибиторов синтазы окиси азота

Номер патента: 3834
Опубликовано: 30.10.2003
Авторы: Лауэ Джон Адамс III, Волкманн Роберт Альфред, Новаковски Йоланта
МПК: A61K 31/4418, A61P 25/00, C07D 213/73...
Метки: синтазы, окиси, ингибиторов, азота, качестве, 2-амино-6-(2-замещенные-4-фенокси)-замещенные-пиридины
Формула / Реферат:
1. Соединение формулы где R1 и R2 выбраны независимо из группы, включающей водород, галоид, гидрокси, (C1-C6)алкокси, (C1-C7)алкил, (C2-C6)алкенил, (C2-C10)алкоксиалкил; G выбирают из группы, включающей водород, (C1-C6)алкил, (C1-C6)алкокси(C1-C3)алкил, аминокарбонил-(C1-C3)алкил-, (C1-C3)алкиламинокарбонил-(C1-C3)алкил-, ди-[(C1-C3)алкил]аминокарбонил-(C1-C3)алкил и N(R3)(R4)(C0-C4)алкил-, где R3 и R4 независимо выбирают из группы, включающей...
Триамид-замещённые индолы, бензофураны и бензотиофены в качестве ингибиторов микросомального белка, переносящего триглицериды, (мтр) и/или ингибиторов секреции аполипопротеина в (аро в)

Номер патента: 7008
Опубликовано: 30.06.2006
Авторы: Ли Дзин, Ченг Хенгмиао, Блайз Алан Элвуд, Бертинато Питер, Хуатан Хип, Бронк Брайан Скотт, Мейсон Клайв Филип
МПК: A61K 31/343, A61K 31/404, A61K 31/4439...
Метки: аро, ингибиторов, триглицериды, переносящего, микросомального, бензофураны, белка, качестве, индолы, бензотиофены, триамид-замещённые, аполипопротеина, секреции, мтр
Формула / Реферат:
1. Соединение формулы 1b или его фармацевтически приемлемая соль, где R1 представляет собой заместитель в 5 или 6 положении формулы 1b и имеет структуру или, когда R7 представляет собой фенил, пиридил, фенил-Z1-или пиридил-Z1-, необязательно замещенные от одного до пяти, независимо, выбранными R12, то R1 представляет собой (C1-С6)алкил, (С3-С8)циклоалкил, (С5-С10)бициклоалкил, -(CRaRb)tO(C1-C6 алкил), -(СRaRb)tS(C1-C6 алкил), -(CRaRb)rC(O)R15,...
Предыдущий патент: Способ получения антидетонационной добавки к углеводородному топливу
Следующий патент: Применение антипрогестинов для индукции апоптоза клетки
Случайный патент: Термопластичный композитный материал и способ получения декоративного изделия на его основе