Пирролопиримидины как ингибиторы протеинкиназы
Номер патента: 7415
Опубликовано: 27.10.2006
Авторы: Эдлин Крис, Кокс Пол Джозеф, Амендола Шелли, Эдвардз Майкл, Педгрифт Брайан Лесли, Маджид Тахир Надим, Бодуэн Бернар, Алдос Дейвис Джон, Депре Стефани Даниель, Маклей Лэйн Макфарлан, Халли Фрэнк




















Формула / Реферат
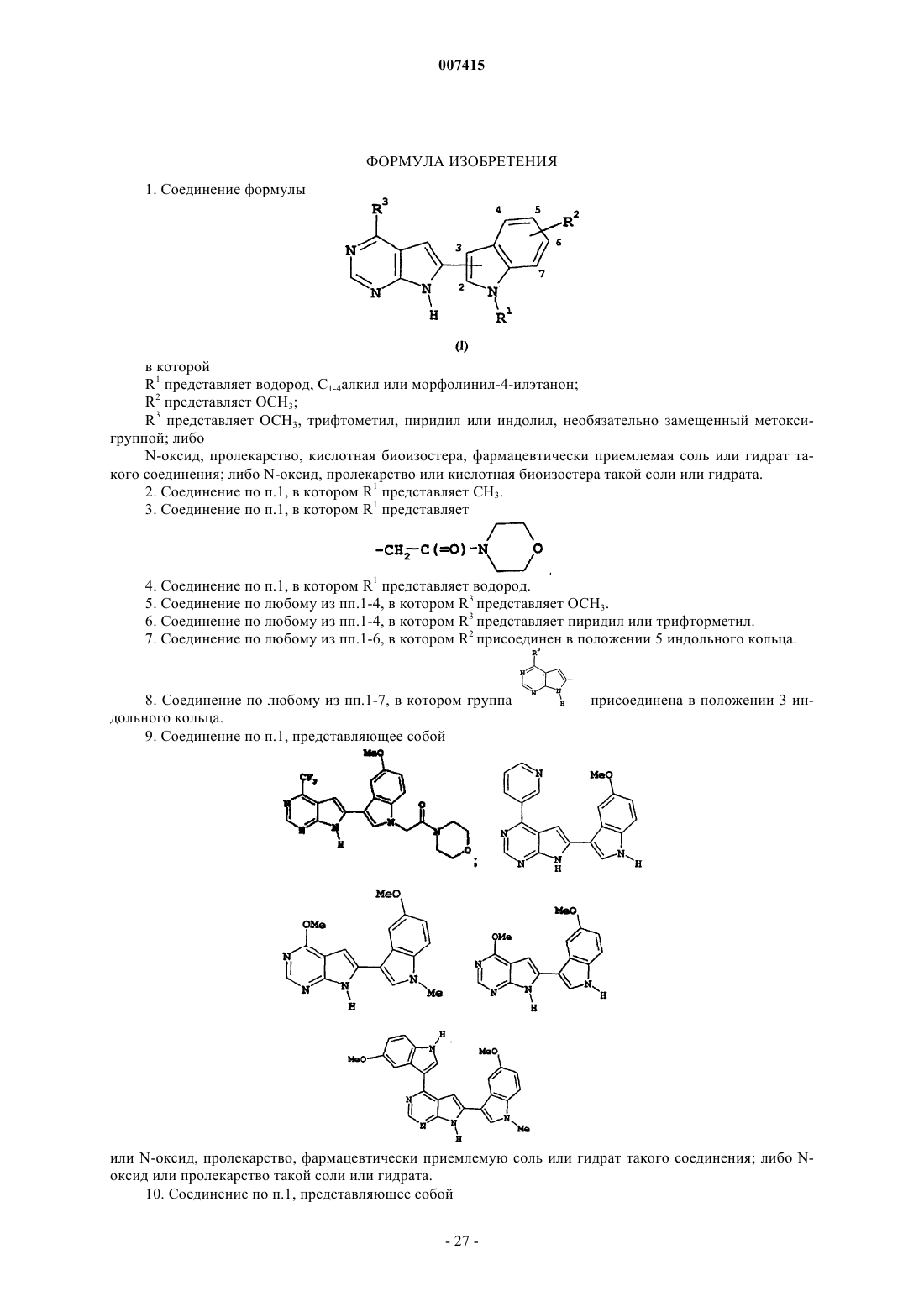
1. Соединение формулы

в которой
R1 представляет водород, С1-4алкил или морфолинил-4-илэтанон;
R2 представляет ОСН3;
R3 представляет ОСН3, трифтометил, пиридил или индолил, необязательно замещенный метоксигруппой; либо
N-оксид, пролекарство, кислотная биоизостера, фармацевтически приемлемая соль или гидрат такого соединения; либо N-оксид, пролекарство или кислотная биоизостера такой соли или гидрата.
2. Соединение по п.1, в котором R1 представляет СН3.
3. Соединение по п.1, в котором R1 представляет

4. Соединение по п.1, в котором R1 представляет водород.
5. Соединение по любому из пп.1-4, в котором R3 представляет ОСН3.
6. Соединение по любому из пп.1-4, в котором R3 представляет пиридил или трифторметил.
7. Соединение по любому из пп.1-6, в котором R2 присоединен в положении 5 индольного кольца.
8. Соединение по любому из пп.1-7, в котором группа  присоединена в положении 3 индольного кольца.
присоединена в положении 3 индольного кольца.
9. Соединение по п.1, представляющее собой

или N-оксид, пролекарство, фармацевтически приемлемую соль или гидрат такого соединения; либо N-оксид или пролекарство такой соли или гидрата.
10. Соединение по п.1, представляющее собой

N-оксид, пролекарство, фармацевтически приемлемую соль или гидрат такого соединения; либо N-оксид или пролекарство такой соли или гидрата.
11. Фармацевтическая композиция, содержащая фармацевтически эффективное количество соединения по любому из пп.1-10, вместе с одним или несколькими фармацевтически приемлемыми носителями или эксципиентами.
12. Способ лечения пациента, подверженного или страдающего от состояний, которые могут быть улучшены в результате введения ингибитора каталитической активности Syk, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11.
13. Способ лечения воспалительного заболевания у нуждающегося в этом пациента, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11.
14. Способ лечения пациента, подверженного или страдающего от состояний, которые могут быть улучшены в результате введения ингибитора каталитической активности FAK, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11.
15. Способ лечения пациента, подверженного или страдающего от состояний, которые могут быть улучшены в результате введения ингибитора каталитической активности KDR, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11.
16. Способ лечения пациента, подверженного или страдающего от состояний, которые могут быть улучшены в результате введения ингибитора каталитической активности Aurora2, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11.
17. Способ лечения рака у нуждающегося в этом пациента, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11.
18. Способ по п.13, в котором воспалительное заболевание представляет собой астму, воспалительный дерматоз, аллергический ринит, аллергический конъюнктивит или воспаление суставов.
19. Способ по п.13, в котором воспалительное заболевание представляет собой астму, псориаз, герпетиформный дерматит, экзему, некротизирующий васкулит, кожный васкулит, буллезное заболевание, аллергический ринит, аллергический конъюнктивит, артрит, ревматоидный артрит, артрит коревой краснухи, псориатический артрит или остеоартрит.
20. Способ лечения хронической обструктивной болезни легких у нуждающегося в этом пациента, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11.
21. Способ по п.17, в котором подвергаемые лечению виды рака включают колоректальный рак, рак предстательной, молочной, щитовидной железы, рак кожи, рак прямой кишки или рак легких.
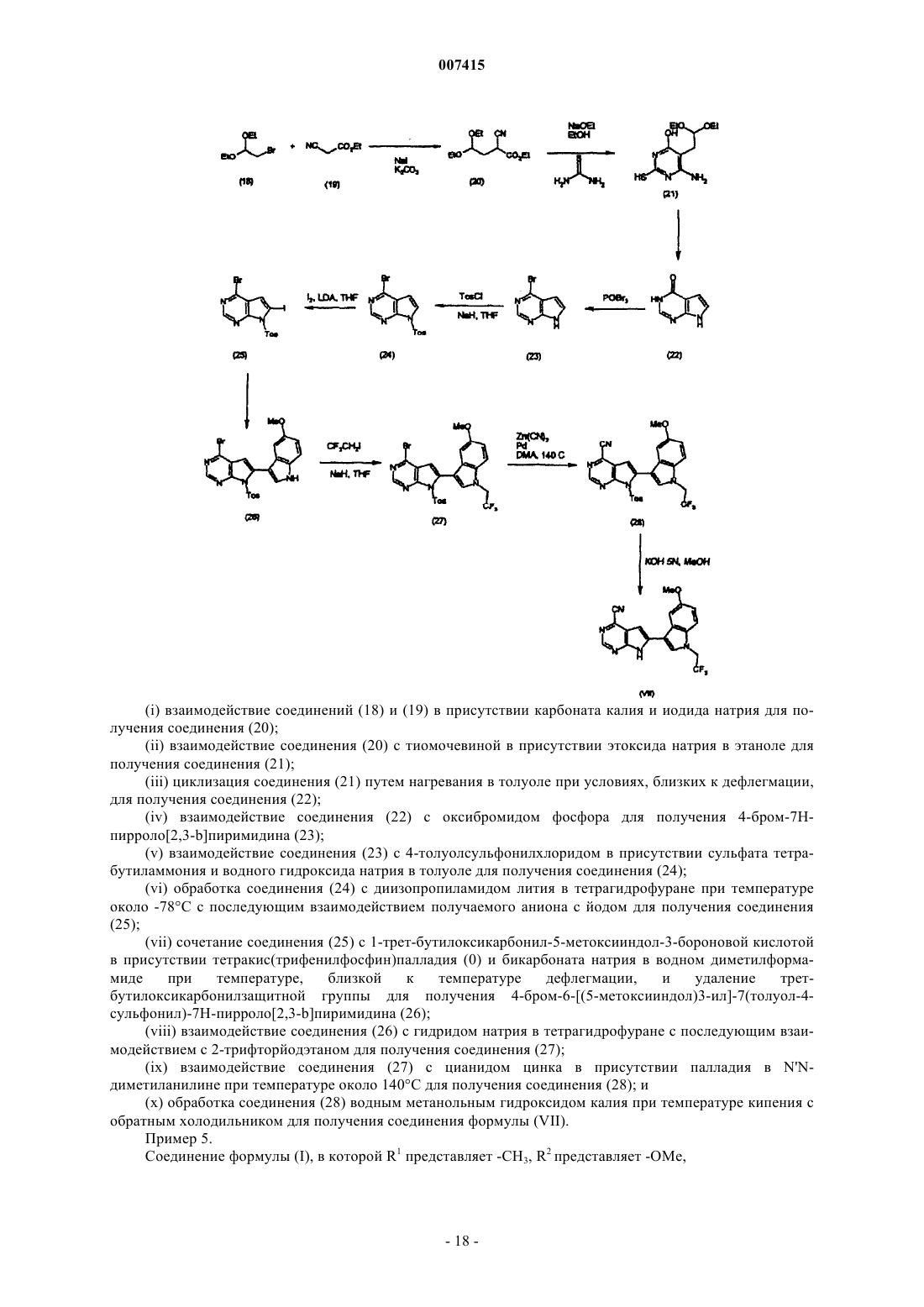
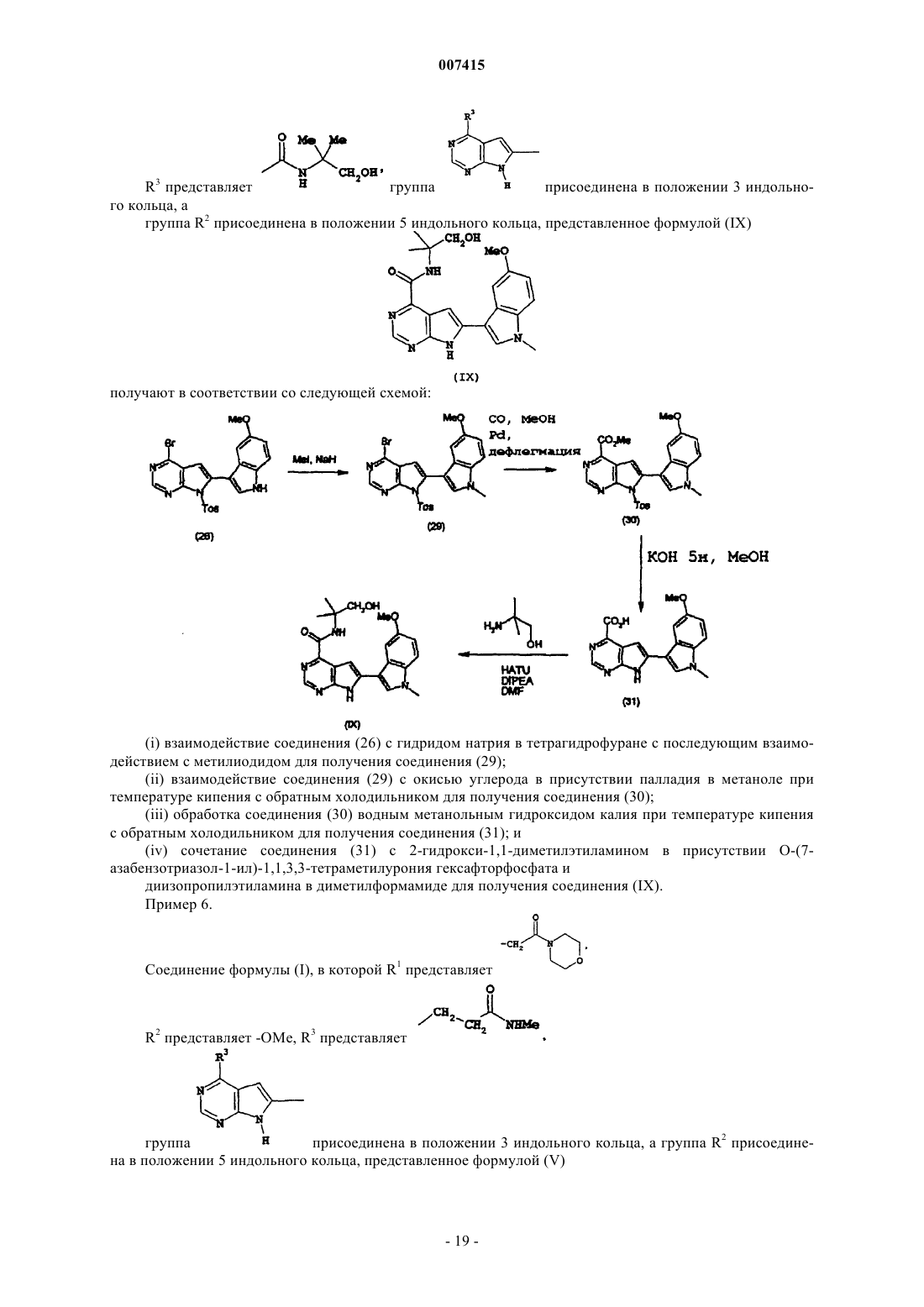
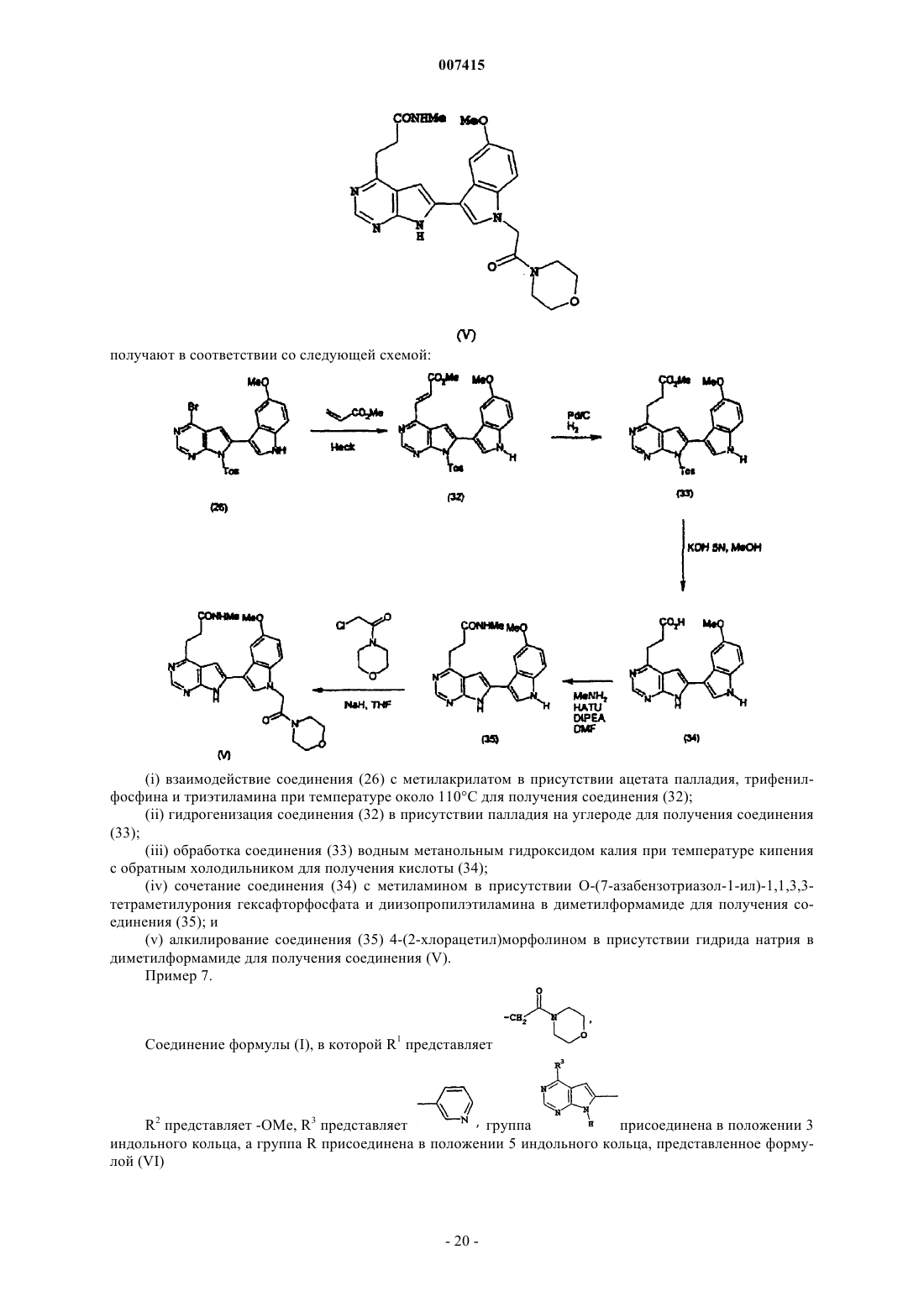
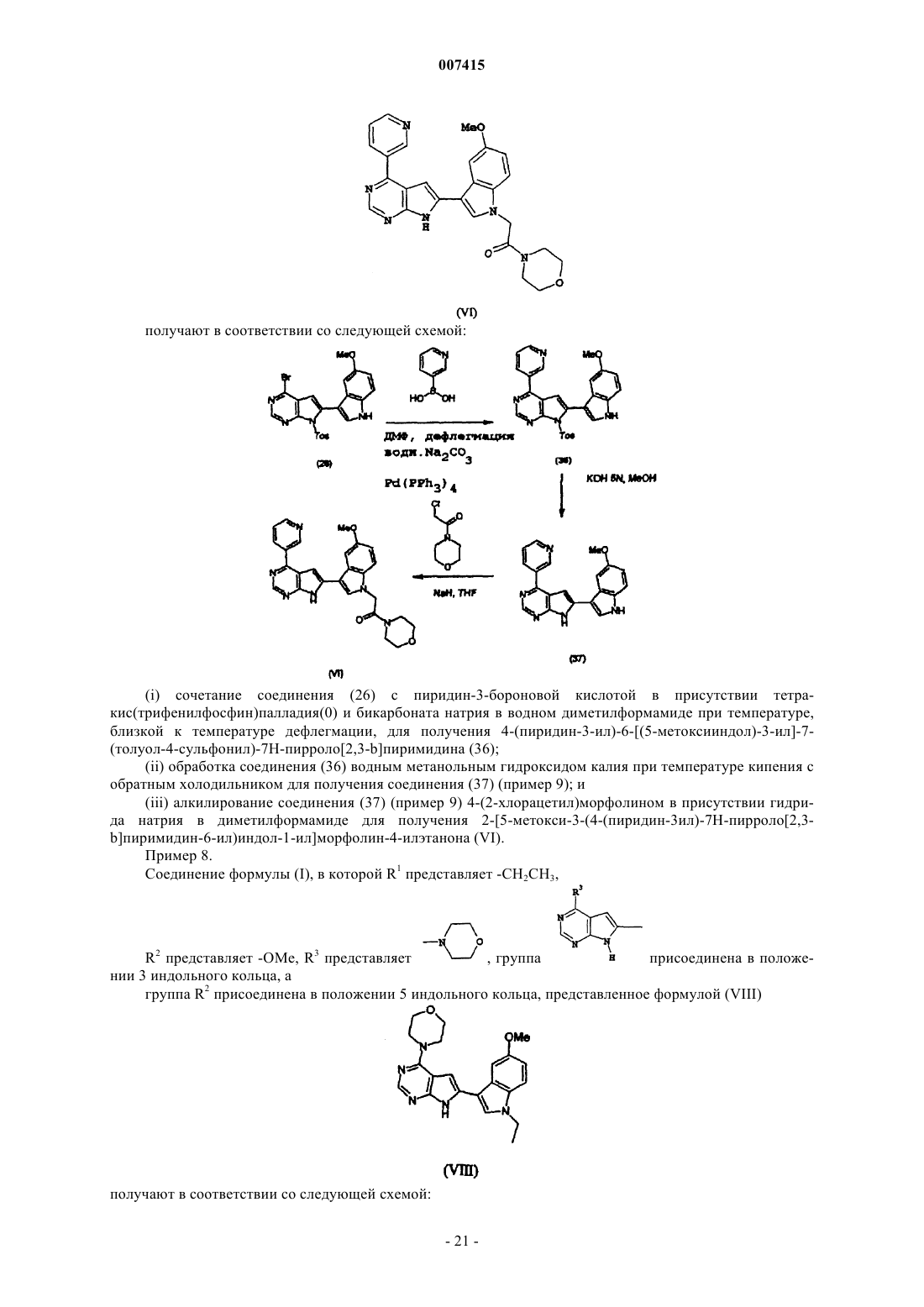
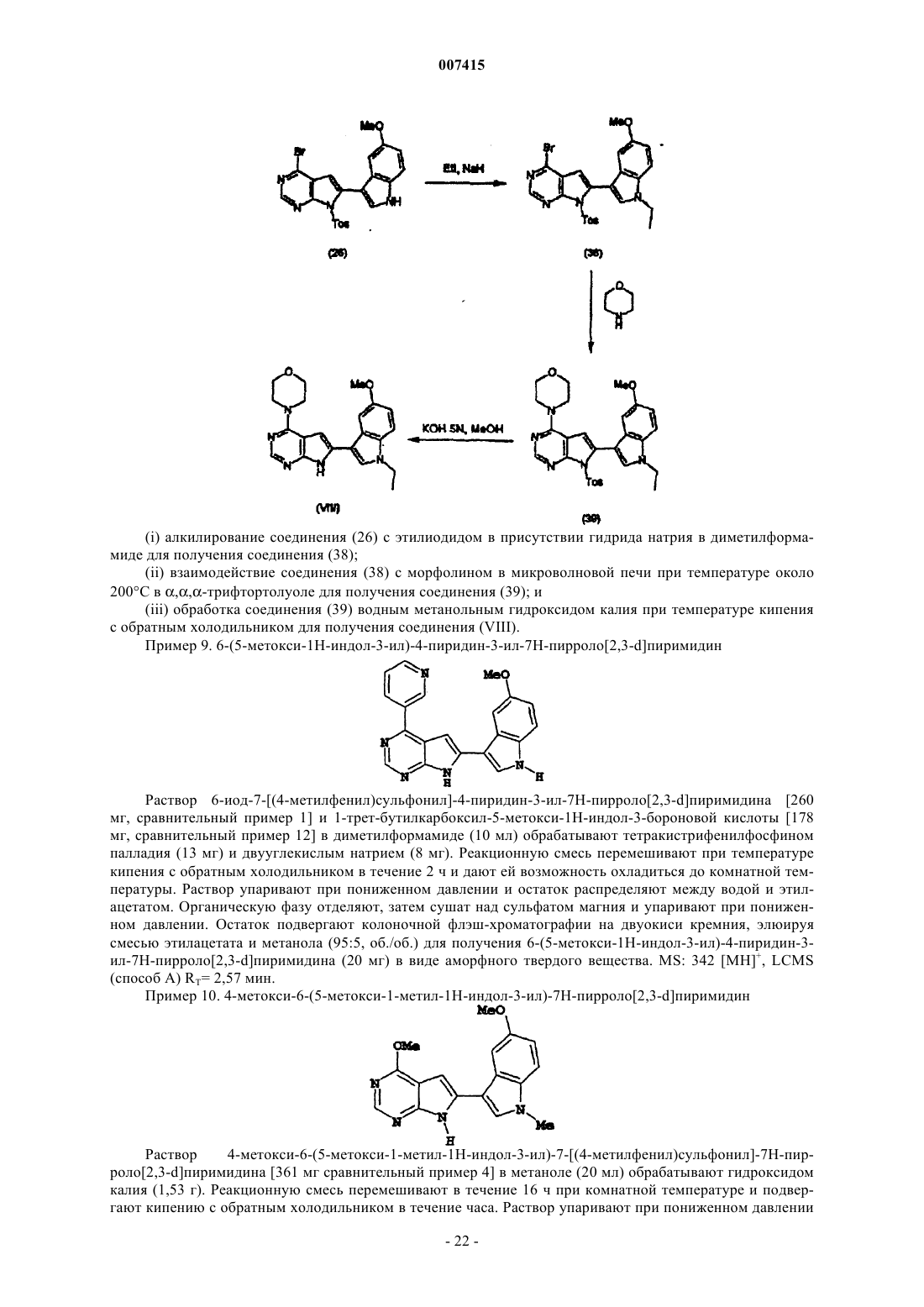
Текст
007415 Данное изобретение относится к замещенным пирролопиримидинам, их получению, фармацевтическим композициям, содержащим указанные соединения, а также к их фармацевтическому применению для лечения болезненных состояний, которые могут быть модулированы ингибированием протеинкиназ. Протеинкиназы участвуют в процессах передачи сигналов, контролирующих активацию, рост и дифференциацию клеток в ответ на внеклеточные медиаторы и на изменения в окружающей среде. В целом, указанные киназы подразделяются на несколько групп: киназы, предпочтительно фосфорилирующие остатки серина и/или треонина, и киназы, предпочтительно фосфорилирующие остатки тирозина [S.K. Hanks and Т. Hunter, FASEB. J., 1995, 9, p. 576-596]. Серин/треонинкиназы включают, например,изоформы протеинкиназы С [А.С. Newton, J. Biol. Chem., 1995, 270, p. 28495-28498] и группу циклинзависимых киназ, таких как cdc2 [J. Pines, Trends in Biochemical Sciences, 1995, 18, p. 195-197]. Тирозинкиназы включают рецепторы фактора роста, соединяющие мембраны, такие как рецептор эпидермального фактора роста [S. Iwashita and M. Kobayashi, Cellular Signalling, 1992, 4, p. 123-132], а также цитозольные безрецепторные киназы, такие как p56tck, p59fYn, ZAP-70 и csk киназы [С. Chan et al., Ann. Rev. Immunol., 1994, 12, p. 555-592]. Излишне высокая активность протеинкиназы наблюдается при многих заболеваниях, возникающих в результате ненормальной клеточной функции. Она может возникнуть прямо или косвенно, например, в результате сбоя правильных контрольных механизмов киназы, вызванного, к примеру, мутацией, сверхэкспрессией или неправильной активацией фермента; либо избыточным или недостаточным продуцированием цитокинов или факторов роста, также принимая участие в трансдукции сигналов в прямом или обратном направлении от киназы. Ожидается, что во всех указанных случаях селективное ингибирование действия киназы может играть положительную роль.Syk представляет собой 72-kDa цитоплазматическую протеинтирозинкиназу, экспрессируемую во многих кроветворных клетках и являющуюся существенным элементом нескольких каскадов, связывающих рецепторы антигена с клеточными ответами. Таким образом, Syk играет центральную роль в сигнализации рецептора IgE с высокой аффинностью, FcRl, в тучных клетках и в антигене рецептора,сигнализирующем в Т- и В-лимфоцитах. Пути трансдукции сигнала в тучных, Т- и В-клетках имеют общие признаки. Лиганд-связывающий домен рецептора не обладает внутренней активностью тирозинкиназы. Однако они взаимодействуют с трансдуцирующими субъединицами, содержащими тирозиновый иммунорецептор на основе мотивов активации (ITAM)[M. Reth, Nature, 1989, 338, p. 383-384]. Указанные мотивы присутствуют как в -, так и -субъединицах FcRl, в -субъединице рецептора Т-клетки (TCR), а также в субъединицах IgG и IgG рецептора В-клетки (BCR). [N.S.van Oers and A. Weiss, Seminars inImmunology, 1995, 7, p. 227-236]. После связывания антигена и мультимеризации остатки ITAM фосфорилируются протеинтирозинкиназами из семейства Src. Syk принадлежит к уникальному классу тирозинкиназ, имеющему двухтандемные домены Src гомологии 2 (SH2) и С-концевой каталитический домен. Указанные домены SH2 с высокой аффинностью связываются с ITAM, и такая SН 2-опосредованная ассоциация Syk с активированным рецептором стимулирует активность Syk киназы и локализует Syk в мембране плазмы. У мышей с дефицитом Syk дегрануляция тучных клеток ингибируется, позволяя высказать предположение о том, что она является важной целью для разработки агентов, стабилизирующих тучные клетки[P.S. Costello, Oncogene, 1996, 13, p. 2595-2605]. Подобные исследования показали важную роль Syc вBCR и TCR сигнализации [A.M. Cheng, Nature, 1995, 378, p. 303-306, (1995), и D.H. Chu et al., Immunological Reviews, 1998, 165, p. 167-180]. Оказывается, что Syk также принимает участие в выживании эозинофилов в ответ на IL-5 и GM-CSF [S. Yousefi et al., J. Exp. Med., 1996, 183, p. 1407-1414]. Несмотря на ключевую роль Syk в тучной клетке, BCR и Т-клеточной сигнализации, имеется мало сведений о механизме передачи Syk эффекторов в прямом направлении. Оказалось, что два адаптерных белка, BLNK(линкер-белок В-клетки, SLP-65) и SLP-76 являются субстратами Syk в В-клетках и тучных клетках, соответственно, и обусловливают раздел Syk с эффекторами в прямом направлении [М. Ishiai et al., Immunity, 1999, 10, p. 117-125, и L.R. Hendricks-Taylor et al., J. Biol. Chem., 1997, 272, p. 1363-1367]. Кроме того, Syk, оказывается, играет важную роль в сигнализирующем пути CD40, который, в свою очередь, играет важную роль в пролиферации В-клеток [М. Faris et al., J. Exp. Med., 1994, 179, p. 1923-1931].Syk также принимает участие в активации тромбоцитов, стимулируемой через низкую аффинностьIgG рецептора (Fc гамма-RIIA) или стимулируемой коллагеном [F. Yanaga et al., Biochem. J., 1995, 311,(Pt.2), p. 471-478]. Фокальная адгезионная киназа (FAK) представляет собой безрецепторную тирозинкиназу, участвующую в интегрин-опосредованных сигнальных трансдукционных путях. FAK солокализуется с интегринами в фокальных контактных сайтах и активации FAK, при этом оказалось, что фосфорилирование ее тирозина во многих видах клеток зависит от интегринов, связывающихся с их внеклеточными лигандами. Результаты нескольких исследований подтверждают гипотезу о том, что ингибиторы FAK могут быть полезны при лечении рака. Например, клетки с дефицитом FAK слабо мигрируют в ответ на хемотактические сигналы и сверхэкспрессия С-концевого домена FAK блокирует клеточное распространение,а также хемотактическую миграцию (Sieg et al., J. Cell Science, 1999, 112, 2677-2691; Richardson A. andParsons Т., Cell, 1997, 97, 221-231); кроме того, опухолевые клетки, обработанные антисмысловыми олигонуклеотидами FAK, теряют способность присоединения и подвергаются апоптозу (Xu et al., CellGrowth Differ., 1996, 4, 413-418). Была описана сверхэкспрессия FAK при раке предстательной железы,молочной железы, щитовидной железы, прямой кишки и легких. Уровень экспрессии FAK напрямую коррелирует с опухолями, демонстрирующими самый агрессивный фенотип. Ангиогенез или образование новых кровеносных сосудов в результате роста из уже существующей сосудистой сети имеет основное значение для эмбрионального развития и органогенеза. Ненормальная повышенная неоваскуляризация наблюдается при ревматоидном артрите, диабетической ретинопатии и во время развития опухоли (Folkman, Nat. Med., 1995, 1, 27-31). Ангиогенез является сложным многоступенчатым процессом, включающим активацию, миграцию, пролиферацию и выживание эндотелиальных клеток. Обширные исследования в области опухолевого ангиогенеза на протяжении последних двух десятилетий выявили ряд терапевтических мишеней, включая киназы, протеазы и интегрины, что привело к открытию многих новых антиангиогенных агентов, включая ингибиторы KDR, некоторые из которых в настоящее время проходят клинические испытания (Jekunen et al., Cancer Treatment Rev., 1997, 23, 263286). Ингибиторы ангиогенеза могут быть использованы в качестве первоочередных, вспомогательных или даже профилактических средств, предупреждающих возникновение или повторный рост злокачественных опухолей. Несколько белков, принимающих участие в сегрегации хромосом и сборке веретен, были идентифицированы в дрожжах и дрозофилах. Разрыв таких белков приводит к неправильной сегрегации хромосом и образованию монополярных или разорванных веретен. К указанным киназам относятся Ipl1 иaurora киназы из S. cerevisiae и дрозофилы, соответственно, которые необходимы для разделения центросом и сегрегации хромосом. Один человеческий гомолог Ipl1 дрожжей был недавно клонирован и охарактеризован различными лабораториями. Такая киназа, названная Aurora2, STK15 или ВТАК, принадлежит к семейству серин/треонинкиназы. Bischoff et al. показали, что Aurora2 является онкогенной и амплифицируется при колоректальном раке человека (EMBO J. , 1998, 17, 3052-3065). Ее присутствие также было подтверждено примерами при раковых заболеваниях, включающих эпителиальные опухоли,таких как рак молочной железы. Данное изобретение предусматривает замещенные пирролопиримидины формулы (I), способные ингибировать одну или несколько протеинканаз, более предпочтительно, FAK, KDR, Syk киназу илиR3 представляет ОСН 3, трифтометил, пиридил или индолил, необязательно замещенный метоксигруппой; и их соответствующие N-оксиды, пролекарства, и их кислотные биоизостеры; а также фармацевтически приемлемые соли и гидраты таких соединений; и их N-оксиды, пролекарства и их кислотные биоизостеры; вместе с одним или несколькими фармацевтически приемлемыми носителями или эксципиентами. В данном описании термин соединения настоящего изобретения и подобные ему выражения означают описанные выше соединения формулы (I) и включают также пролекарства, фармацевтически приемлемые соли и сольваты, к примеру, гидраты, там, где это позволяет контекст. Подобным образом,ссылка на промежуточные соединения, независимо от того, являются ли они сами объектом притязаний,относится также к их солям и сольватам там, где это позволяет контекст. Для уточнения, там, где это позволяет контекст, в описании иногда приводятся конкретные случаи, однако такие случаи являются чисто иллюстративными и не исключают других случаев. Если не указано иначе, то в тексте описания данного изобретения нижеприведенные термины имеют следующие значения: Пациент относится к людям и другим млекопитающим. Кислотная биоизостера означает группу, имеющую химическое и физическое сходство, обеспечивающее явно сходные биологические свойства, с карбоксигруппой (см. Lipinsky, Annual Reports in Medicinal Chemistry, 1986, 21, p.283, "Bioisosterism In Drug Design"; Yun, Hwahak Sekye, 1993, 33, p. 576-579,"Application of Bioisosterism To New Drug Design"; Zhao, Huaxue Tongbao, 1995, p. 34-38, "Bioisosteric Re-2 007415placement and Development Of Lead Compounds In Drug Design"; Graham, Theochem., 1995, 343, p. 105-109,"Theoretical Studies Applied To Drug Design: ab initio Electronic Distributions In Bioisosteres"). Примеры подходящих кислотных биоизостер включают: -C(=O)-NHOH, -С(=O)-СН 2OН, -С(=O)-CH2SH, -С(=O)NH-CN, сульфо, фосфоно, алкилсульфонилкарбамоил, тетразолил, арилсульфонилкарбамоил, гетероарилсульфонилкарбамоил, N-метоксикарбамоил, 3-гидрокси-3-циклобутен-1,2-дион, 3,5-диоксо-1,2,4 оксадиазолидинил или гетероциклические фенолы, такие как 3-гидроксиизооксазолил и 3-гидрокси-1 метилпиразолил. Ацил означает Н-СО- или алкил-СО-группу, в которой алкилгруппа имеет указанные в данном описании значения. Ациламино означает ацил-NH-группу, в которой ацил имеет указанные в данном описании значения. Алкенил означает алифатическую углеводородную группу, содержащую двойную связь углеродуглерод, которая может быть прямой или разветвленной, имеющую от 2 до 15 атомов углерода в цепи. Предпочтительные алкенилгруппы имеют от 2 до 12 атомов углерода в цепи; более предпочтительно - от 2 до 6 атомов углерода (к примеру, от 2 до 4 атомов углерода) в цепи. Разветвленный в данном описании означает, что одна или несколько низших алкилгрупп, таких как метил, этил или пропил, присоединены к линейной цепи; в данном случае - к линейной алкенильной цепи. Низший алкенил означает от 2 до 4 атомов углерода в цепи, которая может быть прямой или разветвленной. Примеры алкенилгрупп включают этенил, пропенил, н-бутенил, изо-бутенил, 3-метилбут-2-енил, н-пентенил, гептенил, октенил,циклогексилбутенил и деценил. Алкенилокси представляет алкенил-O-группу, в которой алкенил имеет указанные в данном описании значения. Примером алкенилоксигрупп является аллилокси. Алкокси представляет алкил-О-группу, в которой алкилгруппа имеет указанные в данном описании значения. Примеры алкоксигрупп включают дифторметокси, метокси, трифторметокси, этокси, нпропокси, изо-пропокси, н-бутокси и гептокси. Алкоксикарбонил представляет алкил-O-СО-группу, в которой алкилгруппа имеет указанные в данном описании значения. Примеры алкоксикарбонилгрупп включают метокси- и этоксикарбонил. Если не указано иначе, алкил означает алифатическую углеводородную группу, которая может иметь прямую или разветвленную цепь, имеющую приблизительно от 1 до 15 атомов углерода в цепи,необязательно замещенных одним или несколькими атомов галогена. Конкретные алкилгруппы имеют от 1 до 6 атомов углерода. Низший алкил как группа или часть низшей алкокси, низшей алкилтио,низшей алкилсульфинил или низшей алкилсульфонилгруппы означает, если не указано иначе, алифатическую углеводородную группу, которая может иметь прямую или разветвленную цепь, имеющую 1 до 4 атомов углерода. Примеры алкилгрупп включают метил, этил, н-пропил, изо-пропил, н-бутил, вторбутил, т-бутил, н-пентил, 3-пентил, гептил, октил, нонил, децил и додецил. Иллюстративные алкилгруппы, замещенные одним или несколькими атомами галогена, включают трифторметил. Алкилен означает алифатический двухвалентный радикал, полученный из алкилгруппы с прямой или разветвленной цепью, в котором алкилгруппа имеет указанные в данном описании значения. Примеры алкиленрадикалов включают метилен, этилен и триметилен. Алкилендиокси означает -О-алкилен-O-группу, в которой алкилен имеет указанные выше значения. Примеры алкилендиоксигрупп включают метилендиокси и этилендиокси. Алкилсульфинил означает алкил-SO-группу, в которой алкилгруппа имеет указанные ранее значения. Предпочтительными алкилсульфинилгруппами являются такие группы, в которых алкилгруппа представляет С 1-4 алкил. Алкилсульфонил означает алкил-SO2-группу, в которой алкилгруппа имеет указанные ранее значения. Предпочтительными алкилсульфонилгруппами являются такие группы, в которых алкилгруппа представляет С 1-4 алкил. Алкилсульфонилкарбамоил означает алкил-SO2-NH-C(=O)-группу, в которой алкилгруппа имеет указанные ранее значения. Предпочтительными алкилсульфонилкарбамоилгруппами являются такие группы, в которых алкилгруппа представляет С 1-4 алкил. Алкилтио означает алкил-S-группу, в которой алкилгруппа имеет указанные ранее значения. Примеры алкилтиогрупп включают метилтио, этилтио, изопропилтио и гептилтио. Алкинил означает алифатическую углеводородную группу, содержащую тройную связь углеродуглерод, которая (группа) может иметь прямую или разветвленную цепь, содержащую от 2 до 15 атомов углерода. Предпочтительные алкинилгруппы имеют от 2 до 12 атомов углерода в цепи, наиболее предпочтительно - от 2 до 6 атомов углерода (к примеру, от 2 до 4 атомов углерода). Примеры алкинилгрупп включают этинил, пропинил, н-бутинил, изобутинил, 3-метилбут-2-инил и н-пентинил. Ароил означает арил-СО-группу, в которой арилгруппа имеет указанные в данном описании значения. Примеры ароилгрупп включают бензоил и 1- и 2-нафтоил. Ароиламино означает ароил-NH-группу, в которой ароил имеет указанные ранее значения. Арил как группа или часть группы означает: (i) необязательно замещенный моноциклический или полициклический ароматический карбоциклический остаток, имеющий от 6 до 14 атомов углерода,-3 007415 такой как фенил или нафтил; либо (ii) необязательно замещенный, частично насыщенный полициклический ароматический карбоциклический остаток, в котором арильная и циклоалкильная или циклоалкенильная группа сконденсированы вместе, образуя циклическую структуру, такую как тетрагидронафтильное, инденильное или инданильное кольцо. За исключением специально оговоренных случаев, арилгруппы могут быть замещены одним или несколькими заместителями арилгрупп, которые могут быть одинаковыми или различными, при этом заместитель арилгруппы включает, например, ацил, ациламино, алкокси, алкоксикарбонил, алкилендиокси, алкилсульфинил, алкилсульфонил, алкилтио, ароил, ароиламино, арил, арилалкилокси, арилалкилоксикарбонил, арилалкилтио, арилокси, арилоксикарбонил,арилсульфинил, арилсульфонил, арилтио, карбокси (или кислотная биоизостера), циано, галоген, гетероароил, гетероарил, гетероарилалкилокси, гетероароиламино, гетероарилокси, гидрокси, нитро, трифторметил, -NY3Y4, -CONY3Y4, -SO2NY3Y4, -NY3-C(=O)алкил, -NY3SO2 алкил или алкил, необязательно замещенный арилом, гетероарилом, гидрокси или -NY3Y4. Арилалкил означает арилалкилгруппу, в которой арильный и алкильный остатки имеют указанные ранее значения. Предпочтительные арилалкилгруппы содержат остаток С 1-4 алкила. Примеры арилалкилгрупп включают бензил, 2-фенетил и нафталинметил. Арилалкилокси означает арилалкил-О-группу, в которой арилалкилгруппы имеет указанные ранее значения. Примеры арилалкилоксигрупп включают бензилокси и 1- или 2-нафталинметокси. Арилалкилоксикарбонил означает арилалкил-O-СО-группу, в которой арилалкилгруппы имеют указанные ранее значения. Примером арилалкилоксикарбонилгруппы является бензилоксикарбонил. Арилалкилтио означает арилалкил-S-группу, в которой арилалкилгруппа имеет указанные ранее значения. Примером арилалкилтиогруппы является бензилтио. Арилокси означает арил-О-группу, в которой арилгруппа имеет указанные ранее значения. Примеры арилоксигрупп включают фенокси и нафтокси, каждая из которых необязательно замещена. Арилоксикарбонил означает арил-О-С(=O)-группу, в которой арилгруппа имеет указанные ранее значения. Примеры арилоксикарбонилгрупп включают феноксикарбонил и нафтоксикарбонил. Арилсульфинил означает арил-SO-группу, в которой арилгруппа имеет указанные ранее значения. Арилсульфонил означает арил-SO2-группу, в которой арилгруппа имеет указанные ранее значения. Арилсульфонилкарбамоил означает арил-SO2-NH-C(=O)-группу, в которой арилгруппа имеет указанные ранее значения. Арилтио означает арил-S-группу, в которой арилгруппа имеет указанные ранее значения. Примеры арилтиогрупп включают фенилтио и нафтилтио. Азагетероарил означает ароматический карбоциклический остаток, имеющий от 5 до 10 кольцевых членов, в котором одним из кольцевых членов является азот, а другие кольцевые члены выбраны из углерода, кислорода, серы и азота. Примеры азагетероарилгрупп включают бензимидазолил, имидазолил, индазолинил, индолил, изохинолинил, пиридил, пиримидинил, пирролил, хинолинил, хиназолинил и тетрагидроиндолизинил. Циклический амин означает 3-8-членную моноциклическую циклоалкильную кольцевую систему, в которой один из кольцевых атомов углерода заменен азотом и которая (i) может также содержать дополнительную гетероатомсодержащую группу, выбранную из О, S, SO2, или NY5 (где Y5 представляет водород, алкил, арил, арилалкил, -C(=O)-R7, -С (O)-OR7 или -SO2R7); и (ii) может быть сконденсирована с дополнительными арильными (к примеру, фенильными), гетероарильными (к примеру, пиридильными),гетероциклоалкильными или циклоалкильными кольцами для получения бициклической или трициклической кольцевой системы. Примеры циклических аминов включают пирролидин, пиперидин, морфолин, пиперазин, индолин, пириндолин, тетрагидрохинолин и тому подобные группы. Циклоалкенил означает неароматическую моноциклическую или полициклическую кольцевую систему, содержащую по меньшей мере одну двойную связь углерод-углерод и имеющую от 3 до 10 атомов углерода. Примеры моноциклических циклоалкенильных колец включают циклопентенил, циклогексенил и циклогептенил. Циклоалкил означает насыщенную моноциклическую или бициклическую кольцевую систему,имеющую от 3 до 10 атомов углерода, необязательно замещенную оксо. Примеры моноциклических циклоалкильных колец включают С 3-8 циклоалкильные кольца, такие как циклопропил, циклопентил, циклогексил и циклогептил. Циклоалкилалкил означает циклоалкилалкилгруппу, в которой циклоалкильные и алкильные остатки имеют ранее указанные значения. Примеры моноциклических циклоалкилалкилгрупп включают циклопропилметил, циклопентилметил, циклогексилметил и циклогептилметил. Галоген означает фтор, хлор, бром или йод. Предпочтительными являются фтор и хлор. Гетероароил означает гетероарил-С(=O)-группу, в которой гетероарилгруппа имеет указанные в данной заявке значения. Примеры гетероарилгрупп включают пиридилкарбонил. Гетероароиламино означает гетероароил-NH-группу, в которой гетероарильный остаток имеет указанные ранее значения.-4 007415 Гетероарил как группа или часть группы означает: (i) необязательно замещенный ароматический моноциклический или полициклический органический остаток, имеющий приблизительно от 5 до 10 кольцевых членов, в котором один или несколько кольцевых членов представляет(-ют) элемент(-ы), отличные от углерода, например, азот, кислород или сера (примеры таких групп включают бензимидазолил, бензтиазолил, фурил, имидазолил, индолил, индолизинил, изоксазолил, изохинолинил, изотиазолил,оксадиазолил, пиразинил, пиридазинил, пиразолил, пиридил, пиримидинил, пирролил, хиназолинил, хинолинил, 1,3,4-тиадиазолил, тиазолил, тиенил и триазолилгруппы, необязательно замещенные одним или несколькими заместителями арилгрупп, имеющими вышеуказанные значения, за исключением особо оговоренных случаев); (ii) необязательно замещенный, частично насыщенный полициклический гетерокарбоциклический остаток, в котором гетероарил и циклоалкил или циклоалкенилгруппа сконденсированы вместе, образуя циклическую структуру (примеры таких групп включают пиринданилгруппы, необязательно замещенные одним или несколькими заместителями арилгрупп, имеющими вышеуказанные значения, за исключением особо оговоренных случаев). Необязательные заместители включают один или несколько заместителей арилгрупп, имеющих вышеуказанные значения, за исключением особо оговоренных случаев. Гетероарилалкил означает гетероарилалкилгруппу, в которой гетероарильные и алкильные остатки имеют указанные ранее значения. Предпочтительные гетероарилалкилгруппы содержат остаток С 1-4 алкила. Примеры гетероарилалкилгрупп включают пиридилметил. Гетероарилалкилокси означает гетероарилалкил-О-группу, в которой гетероарилалкилгруппа имеет указанные ранее значения. Примеры гетероарилалкилоксигрупп включают необязательно замещенный пиридилметокси. Гетероарилокси означает гетероарил-О-группу, в которой гетероарилгруппа имеет указанные ранее значения. Примеры гетероарилоксигрупп включают необязательно замещенный пиридилокси. Гетероарилсульфонилкарбамоил означает гетероарил-SO2-NH-С(=O)-группу, в которой гетероарилгруппа имеет указанные ранее значения. Гетероциклоалкил означает: (i) циклоалкилгруппу, имеющую от 3 до 7 кольцевых членов и содержащую один или несколько гетероатомов или гетероатомсодержащих групп, выбранных из O, S иNY5, которая может быть необязательно замещена оксо; (ii) частично насыщенный полициклический гетерокарбоциклический остаток, в котором арильное (или гетероарильное) кольцо, каждое из которых необязательно замещено одним или несколькими заместителями арилгрупп, и гетероциклоалкилгруппа сконденсированы вместе, образуя циклическую структуру. (Примеры таких групп включают хроманил, дигидробензофуранил, индолинил и пириндолинилгруппы). Гетероциклоалкилалкил означает гетероциклоалкилалкилгруппу, в которой остатки гетероциклоалкила и алкила имеет указанные ранее значения. Пролекарство означает соединение, которое преобразуется in vivo метаболическими путями (к примеру, гидролиз) в соединение формулы (I), включая его N-оксиды. Например, сложный эфир соединения формулы (I), содержащего гидроксигруппу, может быть превращен гидролизом in vivo в родительскую молекулу. Альтернативно, сложный эфир соединения формулы (I), содержащего карбоксигруппу,может быть превращен гидролизом in vivo в родительскую молекулу. Подходящие сложные эфиры соединений формулы (I), содержащих гидроксигруппу, включают, например, ацетаты, цитраты, лактаты, тартраты, малонаты, оксалаты, салицилаты, пропионаты, сукцинаты,фумараты, малеаты, метилен-бисгидроксинафтоаты, гентисаты, изетионаты, ди-п-толуоилтартраты,метансульфонаты, этансульфонаты, бензолсульфонаты, п-толуолсульфонаты, циклогексилсульфаматы и хинаты. Подходящие сложные эфиры соединений формулы (I), содержащих карбоксигруппу, описаны, например, F.J. Leinweber, Drug Metab. Res., 1987, 18, page 379. Подходящие сложные эфиры соединений формулы (I), содержащих в остатке -L1-Y как карбоксигруппу, так и гидроксигруппу, включают лактоны, образующиеся в результате потери воды между указанными карбокси и гидроксигруппами. Примеры таких лактонов включают капролактоны и бутиролактоны. Особенно полезный класс сложных эфиров соединений формулы (I), содержащих гидроксигруппу,может быть получен из кислотных остатков, выбранных из остатков, описанных Bundgaard et al., J. Med.Chem., 1989, 32, p. 2503-2507, и включающих замещенные (аминометил)бензоаты, например диалкиламинометилбензоаты, в которых две алкилгруппы могут быть соединены вместе и/или прерваны атомом кислорода или необязательно замещенным атомом азота, например, алкилированным атомом азота, более конкретно, (морфолинометил)бензоаты, к примеру, 3- или 4-(морфолинометил)бензоаты, и (4 алкилпиперазин-1-ил)бензоаты, к примеру, 3- или 4-(4-алкилпиперазин-1-ил)бензоаты. Если соединение данного изобретения содержит карбоксигруппу или достаточно кислотную биоизостеру, то могут быть получены аддитивные соли основания, которые являются просто более удобной формой для применения; на практике применение солевой формы по своему существу равносильно применению свободной кислотной формы. Основания, которые могут быть использованы для получения их аддитивных солей, предпочтительно включают такие основания, которые после соединения со свобод-5 007415 ной кислотой приводят к получению фармацевтически приемлемых солей, т.е. солей, катионы которых нетоксичны для пациента в фармацевтических дозах таким образом, что полезное ингибирующее действие, оказываемое свободным основанием, не устраняется побочными действиями, оказываемыми катионами. Фармацевтически приемлемые соли, включающие соли, получаемые из солей щелочных и щелочно-земельных металлов, в объеме данного изобретения включают соли, получаемые из следующих оснований: гидрид натрия, гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид алюминия,гидроксид лития, гидроксид магния, гидроксид цинка, аммиак, этилендиамин, N-метилглюкамин, лизин,аргинин, орнитин, холин, N,N'-дибензилэтилендиамин, хлорпрокаин, диэтаноламин, прокаин, Nбензилфенетиламин, диэтиламин, пиперазин, трис(гидроксиметил)аминометан, гидроксид тетраметиламмония и т.п. Некоторые из соединений настоящего изобретения являются основными, и такие соединения могут быть использованы в виде свободного основания или в виде его фармацевтически приемлемой кислотноаддитивной соли. Кислотно-аддитивные соли являются более удобной формой для применения; на практике применение солевой формы по своему существу равносильно применению свободной основной формы. Кислоты, которые могут быть использованы для получения их аддитивных солей, предпочтительно включают такие кислоты, которые после соединения со свободным основанием приводят к получению фармацевтически приемлемых солей, т.е. солей, анионы которых нетоксичны для пациента в фармацевтических дозах таким образом, что полезное ингибирующее действие, оказываемое свободным основанием, не устраняется побочными действиями, оказываемыми анионами. Несмотря на то, что предпочтительными являются фармацевтически приемлемые соли указанных основных соединений, все кислотноаддитивные соли могут быть использованы как источники свободной основной формы, даже в том случае, если получение конкретной соли per se желательно только в виде промежуточного продукта, как,например, в том случае, когда соль получают только для целей очистки и идентификации, либо когда ее применяют в качестве промежуточного соединения для получения фармацевтически приемлемой соли ионообменным способом. Фармацевтически приемлемые соли в объеме данного изобретения включают соли, получаемые из минеральных и органических кислот, и включают гидрогалоиды, к примеру, гидрохлориды и гидробромиды, сульфаты, фосфаты, нитраты, сульфаматы, ацетаты, цитраты, лактаты, тартраты, малонаты, оксалаты, салицилаты, пропионаты, сукцинаты, фумараты, малеаты, метилен-бис-бетагидроксинафтоаты, гентисаты, изетионаты, ди-п-толуоилтартраты, метансульфонаты, этансульфонаты,бензолсульфонаты, п-толуолсульфонаты, циклогексилсульфаматы и хинаты. Помимо того, что они могут быть полезны как активные соединения сами по себе, соли соединений данного изобретения могут быть использованы для очистки соединений, например, благодаря использованию различной растворимости солей и родительских соединений, побочных продуктов и/или исходных материалов способами, хорошо известными специалистам в данной области техники. Со ссылкой на вышеприведенную формулу (I) следующие группировки являются конкретными и предпочтительными:(ii) -СН 3; Особенно предпочтительными являются соединения формулы (I), в которых R1 представляет водород, -СН 3, или Еще более предпочтительно R1 представляет водород.(iii) необязательно замещенный индолил (к примеру, незамещенный пиридил или необязательно замещенный индолил Особенно предпочтительными являются соединения формулы (I), в которых R3 представляет пиридил или трифторметил.-6 007415 Еще более предпочтительно R3 представляет -ОСН 3. предпочтительно присоединена в положение 3 индольного кольца. Группа Подразумевается, что данное изобретение включает все подходящие комбинации упоминаемых здесь конкретных и предпочтительных группировок. Особенно предпочтительными соединениями данного изобретения являются следующие соединения: и соответствующие N-оксиды, и их пролекарства; а также фармацевтически приемлемые соли и гидраты таких соединений и их N-оксиды и пролекарства. Особенно предпочтительными соединениями данного изобретения являются 4-метокси-6-(5-метокси-1-Н-индол-3-ил)-7 Н-пирроло[2,3-d]пиримидин; и соответствующие N-оксиды, и их пролекарства; а также фармацевтически приемлемые соли и сольваты (к примеру, гидраты) таких соединений и их N-оксиды и пролекарства. Соединения данного изобретения проявляют полезную фармакологическую активность и, соответственно, могут быть введены в фармацевтические композиции и использованы для лечения пациентов,страдающих от определенных медицинских нарушений. Таким образом, настоящее изобретение предусматривает, в соответствии с дальнейшим аспектом, соединения данного изобретения и композиции, содержащие соединения данного изобретения, для применения в терапии. Соединения, входящие в объем настоящего изобретения, в соответствии с тестами, описанными в литературе, а также описываемыми ниже методиками in vitro, блокируют каталитическую активность киназы, при этом предполагается, что полученные результаты тестов коррелируют с фармакологической активностью у людей и других млекопитающих. Таким образом, дальнейший вариант осуществления настоящего изобретения предусматривает соединения данного изобретения, а также композиции, содержащие соединения данного изобретения, для применения при лечении пациента, подверженного или-7 007415 страдающего от состояний, которые могут быть улучшены введением ингибиторов протеинкиназы (к примеру, Syk, FAK, KDR или Aurora2), в частности, ингибитора Syk киназы. Например, соединения настоящего изобретения полезны при лечении воспалительных заболеваний, к примеру, астмы; воспалительных дерматозов (например, псориаз, герпетиформный дерматит, экзема, некротизирующий и кожный васкулит, буллезное заболевание); аллергического ринита и аллергического конъюнктивита; воспаления суставов, включая артрит, ревматоидный артрит и других артритических состояний, таких как ревматоидный спондилит, подагрический артрит, травматический артрит, артрит коревой краснухи, псориатический артрит и остеоартрит. Данные соединения также полезны при лечении хронической обструктивной болезни легких (ХОБЛ), острого синовита, аутоиммунного диабета, аутоиммунного энцефаломиелита, колита, атеросклероза, периферического васкулярного заболевания, сердечно-сосудистого заболевания, рассеянного склероза, рестеноза, миокардита, лимфом В-клеток, системной красной волчанки, болезни трансплантат против хозяина и других заболеваний, связанных с отторжением трансплантата, различных видов рака и опухолей (таких как колоректальный рак, рак предстательной, молочной, щитовидной железы, рак прямой кишки и легких), а также воспалительного заболевания кишечника. Кроме того, данные соединения полезны как опухолевые антиангиогенные агенты. Особым вариантом осуществления терапевтических способов настоящего изобретения является лечение астмы. Другим особым вариантом осуществления терапевтических способов настоящего изобретения является лечение псориаза. Другим особым вариантом осуществления терапевтических способов настоящего изобретения является лечение воспаления суставов. Другим особым вариантом осуществления терапевтических способов настоящего изобретения является лечение воспалительного заболевания кишечника. Другим особым вариантом осуществления терапевтических способов настоящего изобретения является лечение различных видов рака и опухолей. Согласно дальнейшим особенностям данного изобретения предусматривается способ лечения человека или животного, подверженного или страдающего от состояний, которые могут быть улучшены введением ингибиторов протеинкиназы (к примеру, Syk, FAK, KDR или Aurora2), например, вышеописанных состояний, включающий введение пациенту эффективного количества соединения данного изобретения или композиции, содержащей соединение данного изобретения. Эффективное количество означает количество соединения настоящего изобретения, эффективно ингибирующее каталитическую активность протеинкиназы, такой как Syk, FAK, KDR или Aurora2, и таким образом обеспечивающее желательное терапевтическое действие. Лечение в соответствии с данным описанием включает профилактическое лечение, а также лечение установленных состояний. В объем настоящего изобретения также включены фармацевтические композиции, содержащие, по меньшей мере, одно соединение данного изобретения в сочетании с фармацевтически приемлемым носителем или эксципиентом. Соединения данного изобретения могут быть введены любыми подходящими способами. На практике соединения настоящего изобретения могут быть введены парентерально, локально, ректально, перорально либо ингаляцией, особенно пероральным способом. Композиции данного изобретения могут быть получены известными способами с применением одного или нескольких фармацевтически приемлемых адъювантов или эксципиентов. Адъюванты включают, среди прочего, разбавители, стерильные водные среды и различные нетоксичные органические растворители. Композиции могут иметь вид таблеток, пилюль, гранул, порошков, водных растворов или суспензий, растворов для инъекций, эликсиров или сиропов, и могут содержать один или несколько агентов, выбранных из группы, включающей подсластители, вкусовые вещества, красители или стабилизаторы, для получения фармацевтически приемлемых препаратов. Выбор носителя и содержания в нем активного вещества обычно определяют в соответствии с растворимостью и химическими свойствами активного соединения, конкретным способом введения и мерами предосторожности, соблюдаемыми в фармацевтической практике. Например, для получения таблеток могут быть использованы эксципиенты,такие как лактоза, цитрат натрия, карбонат кальция, фосфат дикальция, и дезинтегрирующие агенты,такие как крахмал, альгиновая кислота и некоторые сложные силикаты в сочетании с лубрикантами, такими как стеарат магния, лаурилсульфат натрия и тальк. Для получения капсул предпочтительно использование лактозы и высокомолекулярных полиэтиленгликолей. Получаемые водные суспензии могут содержать эмульгирующие агенты или агенты, облегчающие суспендирование. Также могут быть использованы разбавители, такие как сахароза, этанол, полиэтиленгликоль, пропиленгликоль, глицерин и хлороформ или их смеси. Для парентерального введения применяют эмульсии, суспензии или растворы продуктов данного изобретения в растительном масле, например кунжутном масле, арахисовом масле или оливковом масле,либо водно-органические растворы, такие как вода и пропиленгликоль, в сложных органических эфирах для инъекций, таких как этилолеат, а также стерильных водных растворах фармацевтически приемлемых-8 007415 солей. Растворы солей продуктов данного изобретения особенно подходят при введении внутримышечных или подкожных инъекций. Водные растворы, также содержащие растворы солей в чистой дистиллированной воде, могут быть использованы для внутривенного введения при условии, что их рН соответствующим образом доведен, что они правильно забуферены и превращены в изотонические с применением достаточного количества глюкозы или хлорида натрия и что они подвергнуты стерилизации нагреванием, облучением или микрофильтрацией. Для местного применения могут быть использованы гели (на основе воды или спирта), кремы или мази, содержащие соединения данного изобретения. Указанные соединения также могут быть введены в гелевую или матричную основу для применения в виде пластыря, обеспечивающего контролируемое высвобождение соединения через трансдермальный барьер. Для введения путем ингаляции соединения данного изобретения могут быть растворены или суспендированы в подходящем носителе для использования в распылителе или аэрозоле, суспензии или растворе, либо могут быть абсорбированы или адсорбированы на подходящем твердом носителе для использования в ингаляторе с сухим порошком. Твердые композиции для ректального введения включают суппозитории, полученные согласно известным способам и содержащие по меньшей мере одно соединение данного изобретения. Процентное содержание активного ингредиента в композициях данного изобретения может быть различным, при этом необходимо, чтобы оно составляло часть, обеспечивающую подходящую дозировку. Очевидно, что приблизительно в одно и то же время могут быть введены несколько единичных дозированных форм. Применяемая доза определяется врачом и зависит от желаемого терапевтического действия, способа введения, продолжительности лечения и состояния пациента. Для взрослых дозы обычно составляют приблизительно от 0,001 до 50, предпочтительно - приблизительно от 0,001 до 5, мг/кг массы тела в сутки при ингаляции; приблизительно от 0,01 до 100, предпочтительно - от 0,1 до 70, более предпочтительно - от 0,5 до 10, мг/кг массы тела в сутки при пероральном введении; и приблизительно от 0,001 до 10, предпочтительно - от 0,01 до 1, мг/кг массы тела в сутки при внутривенном введении. В каждом конкретном случае дозы определяют в соответствии с факторами,присущими подвергаемому лечению пациенту, такими как возраст, масса, общее состояние здоровья и другие характеристики, которые могут повлиять на эффективность медицинского продукта. Соединения данного изобретения могут вводиться с необходимой частотой с целью получения желательного терапевтического эффекта. Некоторые пациенты могут быстро отвечать на более высокую или низкую дозу, при этом для них намного более низкие поддерживающие дозы могут оказаться адекватными. Для других пациентов может понадобиться длительное лечение на уровне от 1 до 4 доз в сутки,в соответствии с физиологическими требованиями каждого конкретного пациента. В целом, активный продукт может вводиться перорально от 1 до 4 раз в сутки. Безусловно, для некоторых пациентов может понадобиться не более одной или двух доз в сутки. Соединения данного изобретения могут быть получены в результате применения или адаптации известных способов, включающих используемые до настоящего времени или описанные в литературе способы, например, способы, описанные R.C. Larock, Comprehensive Organic Transformations, VCHPublishers, 1989. В описываемых ниже взаимодействиях может потребоваться защита реакционноспособных функциональных групп, например, гидрокси, амино, имино, тио или карбоксигрупп, в тех случаях, когда их присутствие желательно в конечном продукте, для того чтобы избежать их нежелательного участия во взаимодействиях. Могут быть использованы известные защитные группы в соответствии со стандартной практикой; примеры приведены в T.W. Greene, P.G.M. Wuts, "Protective Groups in Organic Chemistry",John Wiley and Sons, 1991. Соединения формулы (I), где R1, R2 и R3 имеют вышеуказанные значения, получают взаимодействием соединений формулы (XXVIII): в которой R1 и R2 имеют вышеуказанные значения. Реакция сочетания может быть целесообразно осуществлена, к примеру, в присутствии комплексного металлического катализатора, такого как тетракис(трифенилфосфин)палладий(0) и бикарбонат натрия, в водном диметилформамиде при температуре до температуры дефлегмации. Данную реакцию целесообразно осуществлять с пирролом NH в соединении (XXVIII), защищенном, к примеру, тозилгруппой, и индолом NH в соединении (XXIX), защищенном, к примеру, трет-бутоксикарбонилгруппой. Соединения формулы (I), в которых R2 и R3 имеют вышеуказанные значения, a R1 представляет необязательно замещенный алкил, получают взаимодействием соответствующих соединений формулы (I),в которых R2 и R3 имеют вышеуказанные значения, a R1 представляет водород, с соответствующим галоидалкилом R2-X2, в котором R2 представляет необязательно замещенный алкил, а X2 представляет галоген. Данное взаимодействие особенно подходит для получения соединений формулы (I), в которых R1 представляет морфолиноацетил. Соединения данного изобретения могут быть также получены путем взаимного превращения других соединений данного изобретения. Таким образом, к примеру, соединения формулы (I) , содержащие карбоксигруппу, могут быть получены гидролизом соответствующих сложных эфиров. Гидролиз может быть целесообразно осуществлен путем щелочного гидролиза с применением основания, такого как гидроксид щелочного металла, к примеру, гидроксид лития, или карбонат щелочного металла, такой как карбонат калия, в присутствии смеси водного/органического растворителя с применением органических растворителей, таких как диоксан, тетрагидрофуран или метанол, при температуре приблизительно от температуры окружающей среды до температуры дефлегмации. Гидролиз сложных эфиров может быть также осуществлен в результате кислотного гидролиза с применением неорганической кислоты, такой как хлористо-водородная кислота,в присутствии смеси водного/инертного органического растворителя с применением органических растворителей, таких как диоксан или тетрагидрофуран, при температуре приблизительно от 50 до 80 С. В качестве другого примера, соединения формулы (I), содержащие карбоксигруппу, могут быть получены в результате катализируемого кислотой удаления трет-бутилгруппы соответствующих сложных трет-бутиловых эфиров с применением стандартных реакционных условий, например, взаимодействия с трифторуксусной кислотой при температуре, близкой к комнатной температуре. В качестве другого примера, соединения формулы (I), содержащие карбоксигруппу, могут быть получены в результате гидрогенизации соответствующих сложных бензиловых эфиров. Взаимодействие может быть осуществлено в присутствии формиата аммония и подходящего металлического катализатора, к примеру, палладия, на подложке из инертного носителя, такого как углерод, предпочтительно, в растворителе, таком как метанол или этанол, и при температуре, близкой к температуре дефлегмации. Взаимодействие может быть альтернативно осуществлено в присутствии подходящего металлического катализатора, к примеру, платины или палладия, на подложке из инертного носителя, такого как углерод,предпочтительно, в растворителе, таком как метанол или этанол. В качестве другого примера процесса взаимного превращения, соединения формулы (I), содержащие группу -С (=O)-NY1Y2, могут быть получены путем сочетания соединений формулы (I), содержащих карбоксигруппу, с амином формулы HNY1Y2 для получения амидной связи в результате обычного процесса пептидного связывания, к примеру, сочетания в присутствии O-(7-азабензотриазол-1-ил)-1,1,3,3 тетраметилурония гексафторфосфата и триэтиламина (или диизопропилэтиламина) в тетрагидрофуране(или диметилформамиде) при комнатной температуре. Данная методика особенно применима для получения (i) соединений формулы (I), в которых R3 представляет -C(=O)-NY1Y2 , или (ii) соединений формулы (I), в которых R2 представляет -С(=O)-NY1Y2. Сочетание также может быть осуществлено взаимодействием соединений формулы (I), содержащих карбоксигруппу, с N-оксидом N-(диметиламино) (1 Н-1,2,3-триазоло[4,5-b]пиридин-1-ил)метилен-N-метилметанаминий гексафторфосфата в присутствии подходящего основания, такого как диизопропилэтиламин, в инертном растворителе, таком как диметилформамид, и при температуре, близкой к комнатной температуре, с последующим взаимодействием с амином формулы HNY1Y2 (хлорид аммония может быть использован для получения соединений формулы(I), содержащих группу -C(=O)-NH2). Сочетание также может быть осуществлено взаимодействием соединений формулы (I), содержащих карбоксигруппу, с 2-(1 Н-бензотриазол-1-ил)-1,1,3,3 тетраметилурония гексафторфосфатом в сухом диметилформамиде с последующим взаимодействием с амином формулы HNY1Y2 в присутствии диизопропилэтиламина.- 10007415 В качестве другого примера процесса взаимного превращения соединения формулы (I), содержащих группу -СН 2 ОН, могут быть получены в результате восстановления соответствующих соединений формулы (I), содержащих группу -СНО или -CO2R7 (где R7 представляет низший алкил). К примеру, восстановление может быть целесообразно осуществлено взаимодействием с литийалюминий гидридом в инертном растворителе, таком как тетрагидрофуран, и при температуре приблизительно от комнатной температуры до температуры дефлегмации. В качестве другого примера процесса взаимного превращения, соединения формулы (I), в которыхR2 представляет гидрокси, могут быть получены взаимодействием соответствующих соединений формулы (I), в которых R1 представляет метокси, с кислотой Льюиса, такой как трибромид бора, в инертном растворителе, таком как дихлорметан, и при температуре приблизительно от 0 С до комнатной температуры. В качестве другого примера процесса взаимного превращения, соединения формулы (I), в которыхR2 представляет -OR4 (где R4 представляет необязательно замещенный алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил или гетероциклоалкилалкил), могут быть получены алкилированием соответствующих соединений формулы (I), в которых R2 представляет гидрокси, с соединениями формулыR4-X3 (XXX) 4 в которой R имеет указанные выше значения, а X3 представляет галоген, предпочтительно, атом брома,или тозилгруппу, с применением стандартных условий алкилирования. Алкилирование, к примеру, может быть осуществлено в присутствии основания, такого как карбонат щелочного металла (к примеру,карбонат калия или карбонат цезия), алкоксид щелочного металла (к примеру, третичный бутоксид калия) или гидрид щелочного металла (к примеру, гидрид натрия), в диметилформамиде или диметилсульфоксиде при температуре приблизительно от 0 до 100 С. В качестве другого примера процесса взаимного превращения, соединения формулы (I), в которыхR1 представляет алкил, алкенил, циклоалкил, гетероциклоалкил или алкил, замещенный -C(=O)NY1Y2,-OR7, -C(=O)-OR7, -NY1Y2, могут быть получены алкилированием соответствующих соединений формулы (Iа), в которых R1 представляет водород, с соответствующим галоидом формулы (XXXI)-C(=O)NY1Y2, -OR7, -C(=O)-OR7, -NY1Y2, a X4 представляет галоген, предпочтительно бром, с применением стандартных условий алкилирования, к примеру, описанных выше. В качестве другого примера процесса взаимного превращения, соединения формулы (I), содержащие сульфоксидные связи, могут быть получены окислением соответствующих соединений формулы (I),содержащих -S-связи. К примеру, окисление может быть целесообразно осуществлено при помощи взаимодействия с пероксикислотой, к примеру, 3-хлорпербензойной кислотой, предпочтительно в инертном растворителе, к примеру, дихлорметане, предпочтительно при комнатной или близкой к ней температуре, либо, альтернативно, при помощи вторичного кислого пероксомоносульфата калия в среде, такой как водный метанол, забуференный до приблизительно рН 5, при температуре приблизительно от 0 С до комнатной температуры. Данный способ является предпочтительным для соединений, содержащих кислотно-лабильную группу. В качестве другого примера процесса взаимного превращения, соединения формулы (I), содержащие сульфоновые связи, могут быть получены окислением соответствующих соединений формулы (I),содержащих -S- или сульфоксидные связи. К примеру, окисление может быть целесообразно осуществлено взаимодействием с пероксикислотой, к примеру, 3-хлорпербензойной кислотой, предпочтительно в инертном растворителе, к примеру, дихлорметане, предпочтительно при комнатной или близкой к ней температуре. В качестве другого примера процесса взаимного превращения, соединения формулы (I), содержащие цианогруппу, могут быть получены взаимодействием соответствующих соединений формулы (I),содержащих группу -C(=O)-NH2, с пентахлоридом фосфора в присутствии триэтиламина. Взаимодействие может быть целесообразно осуществлено в инертном растворителе, таком как тетрагидрофуран, при температуре, близкой к температуре дефлегмации. В качестве другого примера процесса взаимного превращения, соединения формулы (I), содержащие группу -C(=O)-NH2, могут быть получены взаимодействием соответствующих соединений формулы(I), содержащих цианогруппу, с перекисью водорода в присутствии гидроксида натрия. Взаимодействие может быть целесообразно осуществлено в метаноле при температуре, близкой к комнатной температуре. В качестве другого примера процесса взаимного превращения соединения формулы (I), в которыхR3 представляет -NY1Y2 (где Y1 и Y2 имеют вышеуказанные значения), могут быть получены взаимодействием соответствующих соединений формулы (I), в которых R3 представляет галоген (к примеру, хлор),с амином формулы HNY1Y2 (где Y1 и Y2 имеют указанные выше значения). В качестве другого примера процесса взаимного превращения, соединения формулы (I), в которыхR3 представляет циано, могут быть получены взаимодействием соединений формулы (I), в которых X1- 11007415 представляет галоген, предпочтительно, хлор, с цианидом цинка в присутствии порошка цинка, комплекса [1'1-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) и дихлорметана (каталитическое количество) и N,N-диметилацетамида при температуре приблизительно до 150 С. В качестве другого примера процесса взаимного превращения, соединения формулы (I), содержащие группу -C(=O)-OR5 (в которой R5 имеет вышеуказанные значения), могут быть получены взаимодействием соответствующих соединений формулы (I), содержащих группу -С(=O)-ОН, со спиртами формулы R5-OH. К примеру, если R5 представляет трет-бутил, то взаимодействие может быть целесообразно осуществлено в присутствии 1,1'-карбонилдиимидазола и 1,8-диазабицикло[5.4.0]ундец-7-ена при температуре, близкой к комнатной температуре. Необходимо принимать во внимание, что соединения настоящего изобретения могут содержать асимметричные центры. Такие асимметричные центры могут независимо друг от друга быть в R- или Sконфигурации. Специалистам в данной области техники понятно, что определенные соединения данного изобретения могут также иметь геометрическую изомерию. Также подразумевается, что настоящее изобретение включает отдельные геометрические изомеры и стереоизомеры, а также их смеси, включая рацемические смеси, соединений вышеприведенной формулы (I). Такие изомеры могут быть выделены из их смесей в результате применения или адаптации известных способов, например, способов хроматографии и перекристаллизации, либо они могут быть получены отдельно из соответствующих изомеров их промежуточных соединений. Согласно дальнейшим особенностям настоящего изобретения аддитивные соли кислоты соединений данного изобретения могут быть получены взаимодействием свободного основания с соответствующей кислотой в результате применения или модификации известных способов. Например, кислотноаддитивные соли соединений данного изобретения могут быть получены либо путем растворения свободного основания в воде или водном спиртовом растворе, или ином подходящем растворителе, содержащем соответствующую кислоту, и выделения соли в результате упаривания раствора, либо путем взаимодействия свободного основания и кислоты в органическом растворителе, в данном случае соль выделяется непосредственно или может быть получена в результате концентрации раствора. Кислотно-аддитивные соли соединений данного изобретения могут быть получены из солей в результате применения или адаптации известных способов. Например, родительские соединения данного изобретения могут быть получены из их кислотно-аддитивных солей в результате обработки щелочью, к примеру, водным раствором бикарбоната натрия или водным раствором аммиака. Соединения данного изобретения могут быть получены из их основно-аддитивных солей в результате применения или модификации известных способов. Например, родительские соединения данного изобретения могут быть получены из их основно-аддитивных солей в результате обработки кислотой, к примеру, хлористоводородной кислотой. Соединения настоящего изобретения могут быть целесообразно получены или образованы в ходе процесса данного изобретения в виде сольватов (к примеру, гидратов). Гидраты соединений настоящего изобретения могут быть целесообразно получены перекристаллизацией из смеси водного/органического растворителя с применением органических растворителей, таких как диоксан, тетрагидрофуран или метанол. Согласно дальнейшим особенностям настоящего изобретения основно-аддитивные соли соединений данного изобретения могут быть получены взаимодействием свободной кислоты с соответствующим основанием в результате применения или модификации известных способов. Например, основноаддитивные соли соединений данного изобретения могут быть получены либо путем растворения свободной кислоты в воде или водном спиртовом растворе, или ином подходящем растворителе, содержащем соответствующее основание, и выделения соли в результате упаривания раствора, либо путем взаимодействия свободной кислоты и основания в органическом растворителе, в данном случае соль выделяется непосредственно или может быть получена в результате концентрирования раствора. Исходные материалы и промежуточные соединения могут быть получены в результате применения или адаптации известных способов, например способов, описанных в сравнительных примерах или их очевидных химических эквивалентах. Промежуточные соединения формулы (XXVIII), в которых R3 имеет вышеуказанные значения, X1 представляет йод,а пиррол NH защищен тозилгруппой, могут быть получены в соответствии со схемой 1.(i) взаимодействием соединений формулы (XXVII) с пара-толуолсульфонилхлоридом в присутствии водного гидроксида натрия и сульфата тетрабутиламмония в инертном растворителе, таком как толуол, при комнатной температуре;(ii) последующей обработкой полученного соединения формулы (XXVIII) бутиллитием в тетрагидрофуране при температуре около -78 С;(iii) взаимодействием полученного аниона с йодом. Промежуточные соединения формулы (XXVIII), в которой R3 представляет гетероарил, могут быть получены взаимодействием соединений формулы (XXVIII), в которой R3 представляет галоген, к примеру, хлор, с бораном формулы R3BEt2, в которой R3 представляет гетероарил. Взаимодействие может быть целесообразно осуществлено в присутствии тетракис(трифенилфосфин)палладия(0) и карбоната калия в тетрагидрофуране при температуре до температуры дефлегмации. Данное взаимодействие особенно подходит для получения соединений формулы (XXVIII), в которой R3 представляет пиридил. Промежуточные соединения формулы (XXVIII), в которой R3 представляет гетероарил, могут быть также получены взаимодействием соединений формулы (XXVIII), в которой R3 представляет галоген, к примеру хлор, с гетероарилбороновыми кислотами формулы R3B(OH)2 в присутствии тетракис (трифенилфосфин)палладия(0) и водного бикарбоната натрия в диметилформамиде при температуре до температуры дефлегмации. Данное взаимодействие особенно подходит для получения соединений формулы(XXVIII), в которой R3 необязательно представляет замещенный индолил. Промежуточные соединения формулы (XXVIII), в которой R3 представляет OR4, где R4 имеет вышеуказанные значения, могут быть получены взаимодействием соединений формулы (XXVIII), в которой R3 представляет галоген, к примеру хлор, с соединениями формулы R4ONa (получены взаимодействием спирта формулы R4OH с натрием) при температуре около 65 С. Данное взаимодействие особенно подходит для получения соединений формулы (XXVIII), в которой R3 представляет ОМе. Далее настоящее изобретение проиллюстрировано примерами, однако оно не ограничивается нижеприведенными иллюстративными и сравнительными примерами. Условия для осуществления высокоэффективной жидкостной хроматографии - масс-спектрометрии(LC-MS) с целью определения времени удерживания (RT) следующие: Способ А: колонка Hypersil BDS С-18 (4,6 мм х 50 мм), работающая с обращенной фазой в условиях градиентного элюирования смесями (А) воды, содержащей 0,05% трифторуксусной кислоты и (В) ацетонитрила, содержащего 0,05% трифторуксусную кислоту, в качестве градиента подвижной фазы: (0,00 мин 100%А:0%В; линейный градиент до 100% в течение 2 мин; затем удержание до 3,5 мин); скорость потока 1 мл/мин с приблизительно 0,25 мл/мин расщеплением на масс-спектрометре; объем впрыскивания 10 мкл; длина волны детектора Hewlett Packard Model HP1100 Series UV 200 нм; определение рассеивания света при испарении (ELS) - температура 46 С, давление азота 4 бара. Способ В: модель инжектора Gilson 251 с применением колонки Hypersil HyPURITY С-18-5 мк (4,6 мм х 50 мм), работающей в условиях градиентного элюирования смесями (А) воды, содержащей 0,05% трифторуксусной кислоты и (В) ацетонитрила, содержащего 0,05% трифторуксусную кислоту, в качестве градиента подвижной фазы: (0,00 мин 95%А:5%В; линейный градиент до 95%В в течение 4 мин; затем до 5%В в течение 4,5 мин; затем удержание до 6 мин); объем впрыскивания 5 мкл и скорость потока 1 мл/мин до УФ детектора (DAD) с последующим приблизительно 0,100 мл/мин расщеплением на массспектрометре (положительный электроспрей) с остатком - до детектора ELS. СПОСОБ С: прибор для определения микромассы, модель LCT, соединенный с прибором моделиHP 1100. Избыток соединения определяют, применяя HP фотодиодный антенно-решетчатый детектор,модель G1315A, в диапазоне длины волн 200-600 нм и детектор рассеивания света при испарении Sedex,модель 65. Данные масс-спектра получают в диапазоне от 180 до 800. Данные анализируют, применяя компьютерную программу Micromass MassLynx. Разделение осуществляют на колонке Hypersil BDS C18 (50 х 4,6 мм) с размером частиц 3 мкм, элюируемой линейным градиентом от 5 до 90% ацетонитрила, содержащего 0,05% (об./об.) трифторуксусной кислоты в воде, содержащей 0,05% (об./об.) трифторуксусной кислоты в течение 3,5 мин со скоростью потока 1 мл/мин. Общая продолжительность работы, включая уравновешивание колонки, составляет 7 мин. получают в соответствии со следующей схемой:(i) обработка 7 Н-пирроло[2,3-b]пиримидина (1) 3-хлорпербензойной кислотой в дихлорметане при температуре около 0 С для получения 7 Н-пирроло[2,3-b]пиримидин-N-оксида (2);(ii) взаимодействие соединения (2) с оксибромидом фосфора при температуре около 50 С для получения 4-бром-7 Н-пирроло[2,3-b]пиримидина (3);(iii) взаимодействие соединения (3) с 4-толуолсульфонилхлоридом в присутствии сульфата тетрабутиламмония и водного гидроксида натрия в толуоле для получения 4-бром-7 Н-пирроло[2,3-b]пиримидина (4);(iv) взаимодействие соединения (4) с трифторметилтриметилсиланом в присутствии фторида калия и иодида меди (I) в диметилформамиде при температуре около 60 С для получения 7-(толуол-4 сульфонил)-4-трифторметил-7 Н-пирроло[2,3-b]пиримидина (5);(v) обработка соединения (5) диизопропиламидом лития в тетрагидрофуране при температуре около-78 С с последующим взаимодействием получаемого аниона с йодом для получения 6-иод-7-(толуол-4 сульфонил)-4-трифлуорометил-7 Н-пирроло[2,3-b]пиримидина (6);(vi) сочетание соединения (6) с 1-трет-бутилоксикарбонил-5-метокси-1 Н-индол-3-бороновой кислотой в присутствии тетракис(трифенилфосфин)палладия(0) и бикарбоната натрия в водном диметилформамиде при температуре, близкой к температуре дефлегмации, и удаление третбутилоксикарбонилзащитной группы с последующей обработкой метилиодидом в присутствии гидрида натрия в тетрагидрофуране для получения 6-(5-метокси-1 Н-индол-3-ил)-7-(толуол-4-сульфонил)-4 трифторметил-7 Н-пирроло[2,3-b]пиримидина (7);(vii) удаление тозилзащитной группы в соединении (7) обработкой гидроксидом калия в метаноле для получения 6-(5-метокси-1 Н-индол-3-ил)-4-трифторметил-7 Н-пирроло[2,3-b]пиримидина (8); и(viii) алкилирование соединения (8) 4-(2-хлорацетил)морфолином в присутствии гидрида натрия в диметилформамиде для получения 2-[5-метокси-3-(4-трифторметил-7 Н-пирроло[2,3-b]пиримидин-6 ил)индол-1-ил]-1-морфолин-4-илэтанона (II). Пример 2. (2-Гидрокси-1,1-диметилэтил) амид 1-метил-3-(7 Н-пирроло[2,3-b]пиримидин-6-ил)-1 Ниндол-5-карбоновой кислоты Соединение формулы (I), в которой R1 представляет -СН 3,получают в соответствии со следующей схемой:(i) взаимодействие соединения (9) с 4-толуолсульфонилхлоридом в присутствии сульфата тетрабутиламмония и водного гидроксида натрия в толуоле для получения соединения (10);(ii) обработка соединения (10) диизопропиламидом лития в тетрагидрофуране при температуре около -78 С с последующим взаимодействием получаемого аниона с йодом для получения соединения (11);(12) в присутствии тетракис(трифенилфосфин)палладия(0) и бикарбоната натрия в водном диметилформамиде при температуре, близкой к температуре дефлегмации, и удаление трет-бутилоксикарбонилзащитной группы с последующей обработкой метилиодидом в присутствии гидрида натрия в тетрагидрофуране для получения 6-[(1-метил-5-карбометоксииндол)-3-ил]-7 Н-пирроло[2,3-b]пиримидина (13);(iv) обработка соединения (13) водным метанольным гидроксидом калия при температуре кипения с обратным холодильником для получения 6-[(1-метил-5-карбоксииндол)-3-ил]-7 Н-пирроло[2,3-b]пиримидина(v) сочетание соединения (14) с 2-гидрокси-1,1-диметилэтиламином в присутствии О-(7 азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфата и диизопропилэтиламина в диметилформамиде для получения (2-гидрокси-1,1-диметилэтил)амида 1-метил-3-(7 Н-пирроло[2,3-b]пиримидин 6-ил)-1 Н-индол-5-карбоновой кислоты (III). Пример 3. 2-[5-метокси-3-(7 Н-пирроло[2,3-b]пиримидин-6-ил)индол-1-ил]-1-морфолин-4-илэтанон получают в соответствии со следующей схемой(15) в присутствии тетракис(трифенилфосфин)палладия(0) и бикарбоната натрия в водном диметилформамиде при температуре,близкой к температуре дефлегмации, и удаление трет-бутилоксикарбонилзащитной группы для получения 6-[(5-метоксииндол)-3-ил]-7-(толуол-4-сульфонил)-7 Н-пирроло[2,3-b]пиримидина (16);(ii) обработка соединения (16) водным метанольным гидроксидом калия при температуре кипения с обратным холодильником для получения 6-[(5-метоксииндол)-3-ил]-7 Н-пирроло[2,3-b]пиримидина (17); и(iii) взаимодействие соединения (17) с гидридом натрия в диметилформамиде с последующим взаимодействием с морфолинамидом 2-бромуксусной кислоты для получения 2-[5-метокси-3-(7 Нпирроло[2,3-b]пиримидин-6-ил)индол-1-ил]-1-морфолин-4-ил-этанона (IV). Пример 4. Соединение формулы (I), в которой R1 представляет -CH2CF3, R2 представляет -ОМе, R3 представ присоединена в положении 3 индольного кольца, а группа R2 присоединеляет -CN, группа на в положении 5 индольного кольца, представленное формулой (VII) получают в соответствии со следующей схемой:(i) взаимодействие соединений (18) и (19) в присутствии карбоната калия и иодида натрия для получения соединения (20);(ii) взаимодействие соединения (20) с тиомочевиной в присутствии этоксида натрия в этаноле для получения соединения (21);(iv) взаимодействие соединения (22) с оксибромидом фосфора для получения 4-бром-7 Нпирроло[2,3-b]пиримидина (23);(v) взаимодействие соединения (23) с 4-толуолсульфонилхлоридом в присутствии сульфата тетрабутиламмония и водного гидроксида натрия в толуоле для получения соединения (24);(vi) обработка соединения (24) с диизопропиламидом лития в тетрагидрофуране при температуре около -78 С с последующим взаимодействием получаемого аниона с йодом для получения соединения(vii) сочетание соединения (25) с 1-трет-бутилоксикарбонил-5-метоксииндол-3-бороновой кислотой в присутствии тетракис(трифенилфосфин)палладия (0) и бикарбоната натрия в водном диметилформамиде при температуре,близкой к температуре дефлегмации,и удаление третбутилоксикарбонилзащитной группы для получения 4-бром-6-[(5-метоксииндол)3-ил]-7(толуол-4 сульфонил)-7 Н-пирроло[2,3-b]пиримидина (26);(viii) взаимодействие соединения (26) с гидридом натрия в тетрагидрофуране с последующим взаимодействием с 2-трифторйодэтаном для получения соединения (27);(ix) взаимодействие соединения (27) с цианидом цинка в присутствии палладия в N'Nдиметиланилине при температуре около 140 С для получения соединения (28); и(х) обработка соединения (28) водным метанольным гидроксидом калия при температуре кипения с обратным холодильником для получения соединения формулы (VII). Пример 5. Соединение формулы (I), в которой R1 представляет -СН 3, R2 представляет -ОМе,R3 представляет группа присоединена в положении 3 индольного кольца, а группа R2 присоединена в положении 5 индольного кольца, представленное формулой (IX) получают в соответствии со следующей схемой:(i) взаимодействие соединения (26) с гидридом натрия в тетрагидрофуране с последующим взаимодействием с метилиодидом для получения соединения (29);(ii) взаимодействие соединения (29) с окисью углерода в присутствии палладия в метаноле при температуре кипения с обратным холодильником для получения соединения (30);(iii) обработка соединения (30) водным метанольным гидроксидом калия при температуре кипения с обратным холодильником для получения соединения (31); и(iv) сочетание соединения (31) с 2-гидрокси-1,1-диметилэтиламином в присутствии О-(7 азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфата и диизопропилэтиламина в диметилформамиде для получения соединения (IX). Пример 6. получают в соответствии со следующей схемой:(i) взаимодействие соединения (26) с метилакрилатом в присутствии ацетата палладия, трифенилфосфина и триэтиламина при температуре около 110 С для получения соединения (32);(ii) гидрогенизация соединения (32) в присутствии палладия на углероде для получения соединения(iii) обработка соединения (33) водным метанольным гидроксидом калия при температуре кипения с обратным холодильником для получения кислоты (34);(iv) сочетание соединения (34) с метиламином в присутствии О-(7-азабензотриазол-1-ил)-1,1,3,3 тетраметилурония гексафторфосфата и диизопропилэтиламина в диметилформамиде для получения соединения (35); и(v) алкилирование соединения (35) 4-(2-хлорацетил)морфолином в присутствии гидрида натрия в диметилформамиде для получения соединения (V). Пример 7. получают в соответствии со следующей схемой:(i) сочетание соединения (26) с пиридин-3-бороновой кислотой в присутствии тетракис(трифенилфосфин)палладия(0) и бикарбоната натрия в водном диметилформамиде при температуре,близкой к температуре дефлегмации, для получения 4-(пиридин-3-ил)-6-[(5-метоксииндол)-3-ил]-7(толуол-4-сульфонил)-7 Н-пирроло[2,3-b]пиримидина (36);(ii) обработка соединения (36) водным метанольным гидроксидом калия при температуре кипения с обратным холодильником для получения соединения (37) (пример 9); и(iii) алкилирование соединения (37) (пример 9) 4-(2-хлорацетил)морфолином в присутствии гидрида натрия в диметилформамиде для получения 2-[5-метокси-3-(4-(пиридин-3 ил)-7 Н-пирроло[2,3b]пиримидин-6-ил)индол-1-ил]морфолин-4-илэтанона (VI). Пример 8. Соединение формулы (I), в которой R1 представляет -СН 2 СН 3,получают в соответствии со следующей схемой:(i) алкилирование соединения (26) с этилиодидом в присутствии гидрида натрия в диметилформамиде для получения соединения (38);(ii) взаимодействие соединения (38) с морфолином в микроволновой печи при температуре около 200 С в -трифтортолуоле для получения соединения (39); и(iii) обработка соединения (39) водным метанольным гидроксидом калия при температуре кипения с обратным холодильником для получения соединения (VIII). Пример 9. 6-(5-метокси-1 Н-индол-3-ил)-4-пиридин-3-ил-7 Н-пирроло[2,3-d]пиримидин Раствор 6-иод-7-[(4-метилфенил)сульфонил]-4-пиридин-3-ил-7 Н-пирроло[2,3-d]пиримидина [260 мг, сравнительный пример 1] и 1-трет-бутилкарбоксил-5-метокси-1H-индол-3-бороновой кислоты [178 мг, сравнительный пример 12] в диметилформамиде (10 мл) обрабатывают тетракистрифенилфосфином палладия (13 мг) и двууглекислым натрием (8 мг). Реакционную смесь перемешивают при температуре кипения с обратным холодильником в течение 2 ч и дают ей возможность охладиться до комнатной температуры. Раствор упаривают при пониженном давлении и остаток распределяют между водой и этилацетатом. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток подвергают колоночной флэш-хроматографии на двуокиси кремния, элюируя смесью этилацетата и метанола (95:5, об./об.) для получения 6-(5-метокси-1 Н-индол-3-ил)-4-пиридин-3 ил-7 Н-пирроло[2,3-d]пиримидина (20 мг) в виде аморфного твердого вещества. MS: 342 [МН]+, LCMS Раствор 4-метокси-6-(5-метокси-1-метил-1 Н-индол-3-ил)-7-[(4-метилфенил)сульфонил]-7 Н-пирроло[2,3-d]пиримидина [361 мг сравнительный пример 4] в метаноле (20 мл) обрабатывают гидроксидом калия (1,53 г). Реакционную смесь перемешивают в течение 16 ч при комнатной температуре и подвергают кипению с обратным холодильником в течение часа. Раствор упаривают при пониженном давлении- 22007415 и остаток распределяют между водой и этилацетатом. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток растирают с простым диэтиловым эфиром, получая 4-метокси-6-(5-метокси-1-метил-1 Н-индол-3-ил)-7 Н-пирроло[2,3-d]пиримидин (155 мг) в виде твердого вещества, т.пл. = 184C. MS: 309 [МН]+. Пример 11. 4-метокси-6-(5-метокси-1 Н-индол-3-ил)-7 Н-пирроло[2,3-d]пиримидин Раствор 4-метокси-6-(5-метокси-1 Н-индол-3-ил)-7-[(4-метилфенил)сульфонил]-7 Н-пирроло[2,3-d]пиримидина [448 мг, сравнительный пример 5] в метаноле (15 мл) обрабатывают гидроксидом калия (1,96 г). Реакционную смесь перемешивают в течение 2 ч при комнатной температуре и растворитель выпаривают при пониженном давлении. Остаток распределяют между водой и этилацетатом. Органическую фазу отделяют,затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток подвергают колоночной флэш-хроматографии на двуокиси кремния, элюируя смесью этилацетата и циклогексана (80:20, об./об.) для получения 4-метокси-6-(5-метокси-1 Н-индол-3-ил)-7 Н-пирроло[2,3-d]пиримидина (320 мг) в виде желтого твердого вещества, т.пл.260 С. MS: 295 [МН]+. Пример 12. 4-(5-метокси-1 Н-индол-3-ил)-6-(5-метокси-1-метил-1 Н-индол-3-ил)-7 Н-пирроло[2,3-d]пиримидин Раствор 4-(5-метокси-1-[(4-метилфенил)сульфонил]-1H-индол-3-ил)-6-(5-метокси-1-метил-1Hиндол-3-ил)-7-[(4-метилфенил)сульфонил]-7H-пирроло[2,3-d]пиримидина [93 мг, сравнительный пример 9] в метаноле (5 мл) обрабатывают гидроксидом калия (249 мг). Реакционную смесь перемешивают в течение 16 ч при комнатной температуре. Раствор упаривают при пониженном давлении и остаток распределяют между этилацетатом и водой. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток очищают при помощи ВЭЖХ, получая 4-(5 метокси-1H-индол-3-ил)-6-(5-метокси-1-метил-1H-индол-3-ил)-7H-пирроло[2,3-d]пиримидин (9 мг) в виде смолы. MS: 424 [МН]+. LCMS (Способ В) RT = 3,15 мин. Сравнительный пример 1. 6-Йод-7-[(4-метилфенил)сульфонил]-4-пиридин-3-ил-7 Н-пирроло[2,3-d]пиримидин К раствору 7-[(4-метилфенил)сульфонил]-4-пиридин-3-ил-7 Н-пирроло[2,3-d]пиримидина [1 г, сравнительный пример 2] в тетрагидрофуране (20 мл) при -78 С по каплям добавляют раствор бутиллития в гексане (2 мл, 1,6 М) в инертной атмосфере. Раствор перемешивают при указанной температуре в течение 1,5 ч и к нему добавляют йод (796 мг). Реакционную смесь перемешивают при температуре -78 С в течение еще 1 ч и дают ей возможность нагреться до комнатной температуры. Реакционную смесь распределяют между этилацетатом и водным раствором сульфита натрия. Органическую фазу отделяют,затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток подвергают колоночной флэш-хроматографии на двуокиси кремния, элюируя градиентом этилацетата и циклогексана(50:50, до 100, об./об.) для получения указанного в заголовке соединения (260 мг) в виде твердого аморфного вещества. MS: 477 [МН]+. LCMS (Способ В) RT = 3,26 мин. Сравнительный пример 2. 7-[(4-Метилфенил)сульфонил]-4-пиридин-3-ил-7 Н-пирроло[2,3-d]пиримидин Раствор 4-хлор-7-[(4-метилфенил)сульфонил]-7 Н-пирроло[2,3-d]пиримидина [4 г, сравнительный пример 3] и диэтил-3-пиридилборана (2,1 г) в тетрагидрофуране (180 мл) обрабатывают тетракистрифенилфосфином палладия (0,65 г) и карбоната калия (3,59 г). Раствор перемешивают при температуре кипения с обратным холодильником в течение 24 ч и упаривают при пониженном давлении. Остаток распределяют между этилацетатом и рассолом. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток дважды подвергают колоночной флэшхроматографии на двуокиси кремния, элюируя смесью этилацетата и метанола (90:10, об./об.), а также- 23007415 смесью этилацетата и циклогексана (50:50, об./об.) для получения указанного в заголовке соединения(2,5 мг) в виде твердого аморфного вещества. MS: 351 [МН]+. LCMS (Способ В) RT = 3,05 мин. Сравнительный пример 3. 4-Хлор-7-[(4-метилфенил)сульфонил]-7 Н-пирроло[2,3-d]пиримидин Раствор 4-хлор-7 Н-пирроло[2,3-d]пиримидина (ссылка: Gerster, John F.; Hinshaw, Barbara С; Robins,Roland K.; Townsend, Leroy B. Study of electrophylic substitution in the pyrrolo[2,3-d]pyrimidine ring. J. Heterocycl. Chem. (1969), -(2), 207-213) (20 г) и пара-толуолсульфонилхлорида (28,6 г) в толуоле (1 л) обрабатывают раствором гидроксида натрия (50 г) в воде (800 мл), а также сульфатом тетрабутиламмония(462 мг). Раствор энергично перемешивают при комнатной температуре в течение 2 ч и распределяют между этилацетатом и рассолом. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток подвергают колоночной флэш-хроматографии на двуокиси кремния, элюируя градиентом этилацетата и циклогексана (от 50:50 до 80:20, об./об.) для получения указанного в заголовке соединения (2,5 г) в виде твердого вещества, т.пл. = 143C. LCMS (Способ В)RT = 2,78 мин. Сравнительный пример 4. 4-Метокси-6-(5-метокси-1-метил-1 Н-индол-3-ил)-7-[(4-метилфенил)сульфонил]-7 Н-пирроло[2,3-d]пиримидин К раствору 4-метокси-6-(5-метокси-1 Н-индол-3-ил)-7-[(4-метилфенил)сульфонил]-7 Н-пирроло[2,3d]пиримидина [448 г, сравнительный пример 5] в диметилформамиде (20 мл) добавляют гидрид натрия (44 мг, 60% дисперсия в масле) и метилиодид (156 мг) в инертной атмосфере. Раствор перемешивают в течение 1 ч при комнатной температуре и растворитель выпаривают при пониженном давлении. Остаток распределяют между водой и этилацетатом. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток подвергают колоночной флэш-хроматографии на двуокиси кремния,элюируя смесью этилацетата и циклогексана (30:70, об./об.) для получения указанного в заголовке соединения (260 мг) в виде твердого аморфного вещества. MS: 464 [МН]+. LCMS (Способ В) RT = 4,39 мин. Сравнительный пример 5. 4-Метокси-6-(5-метокси-1 Н-индол-3-ил)-7-[(4-метилфенил)сульфонил]7 Н-пирроло[2,3-d]пиримидин Раствор 6-йод-4-метокси-7-[(4-метилфенил)сульфонил]-7 Н-пирроло[2,3-d]пиримидина [1,98 г,сравнительный пример 6] и 1-трет-бутилкарбоксил-5-метокси-1H-индол-3-бороновой кислоты [1,26 г,сравнительный пример 12] в диметилформамиде (40 мл) последовательно обрабатывают насыщенным водным раствором бикарбоната натрия (10 мл) и тетракистрифенилфосфином палладия (165 мг). Реакционную смесь перемешивают при температуре кипения с обратным холодильником в течение 3 ч и растворитель выпаривают при пониженном давлении. Остаток распределяют между этилацетатом и водой. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток подвергают колоночной флэш-хроматографии на двуокиси кремния, элюируя смесью этилацетата и циклогексана (50:50, об./об.) для получения указанного в заголовке соединения (1,8 г) в виде серого твердого вещества, т.пл. = 131 С. MS: 450 [МН]+. Сравнительный пример 6. 6-Йод-4-метокси-7-[(4-метилфенил)сульфонил]-7 Н-пирроло[2,3-d]пиримидин К раствору 4-метокси-7-[(4-метилфенил)сульфонил]-7 Н-пирроло[2,3-d]пиримидина [2,23 г, сравнительный пример 7] в тетрагидрофуране (35 мл) при температуре -78 С по каплям добавляют раствор бутиллития в гексане (5 мл, 1,6 М) в инертной атмосфере. Раствор перемешивают при температуре -70 С в течение часа и добавляют йод (2,05 г). Реакционную смесь перемешивают при температуре -70 С в течение еще 1 ч, дают ей возможность нагреться до комнатной температуры и распределяют между этилацетатом и водным раствором сульфита натрия. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении, получая указанное в заголовке соединение (2,64 г) в виде твердого аморфного вещества. MS: 430 [МН]+. LCMS (Способ В) RT = 4,15 мин. Сравнительный пример 7. 4-Метокси-7-[(4-метилфенил)сульфонил]-7 Н-пирроло[2,3-d]пиримидин Раствор 4-хлор-7 Н-пирроло[2,3-d]пиримидина [1,2 г, сравнительный пример 8] и паратолуолсульфонилхлорида (1,77 г) в толуоле (60 мл) обрабатывают раствором гидроксида натрия (3,2 г) в воде (30 мл), а также сульфатом тетрабутиламмония (27 мг). Раствор энергично перемешивают, при комнатной температуре в течение 4 ч и распределяют между этилацетатом и рассолом. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток подвергают колоночной флэш-хроматографии на двуокиси кремния, элюируя градиентом этилацетата и циклогексана (от 50:50 до 80:20, об./об.) для получения указанного в заголовке соединения (2,23 г) в виде аморфного твердого вещества. MS: 304 [МН]+. LCMS (Способ В) RT = 3,88 мин. Сравнительный пример 8. 4-Метокси-7 Н-пирроло[2,3-d]пиримидин К раствору метоксида натрия, полученному в результате постепенного добавления натрия (2 г) в метанол (100 мл) в инертной атмосфере, добавляют 4-хлор-7 Н-пирроло[2,3-d]пиримидин (ссылка: Gerster, John F.; Hinshaw, Barbara С; Robins, Roland K.; Townsend, Leroy B. Study of electrophylic substitutionin the pyrrolo[2,3-d]pyrimidine ring. J. Heterocycl. Chem. (1969), -(2), 207-13.) (3,5 г). Раствор перемешивают при температуре 65 С в течение 16 ч, а затем распределяют между этилацетатом и рассолом. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток подвергают колоночной флэш-хроматографии на двуокиси кремния, элюируя смесью этилацетата- 24007415 и циклогексана (50:50, об./об.) для получения указанного в заголовке соединения (1,2 г) в виде аморфного твердого вещества. MS: 150 [МН]+. LCMS (Способ В) RT = 2,39 мин. Сравнительный пример 9. 4-(5-Метокси-1-[(4-метилфенил)сульфонил]-1H-индол-3-ил)-6-(5 метокси-1-метил-1H-индол-3-ил)-7-[(4-метилфенил)сульфонил]-7H-пирроло[2,3-d]пиримидин К раствору 4-(5-метокси-1-[(4-метилфенил)сульфонил]-1H-индол-3-ил)-6-(5-метокси-1H-индол-3 ил)-7-[(4-метилфенил)сульфонил]-7H-пирроло[2,3-d]пиримидина [270 мг, сравнительный пример 10] в диметилформамиде (10 мл) добавляют гидрид натрия (10 мг, 60% дисперсия в масле) и метилиодид(0,025 мл) в инертной атмосфере. Раствор перемешивают в течение 16 ч при комнатной температуре, и растворитель выпаривают при пониженном давлении. Остаток распределяют между водой и этилацетатом. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток подвергают колоночной флэш-хроматографии на двуокиси кремния, элюируя смесью этилацетата и циклогексана (50:50, об./об.) для получения указанного в заголовке соединения (93 мг) в виде твердого аморфного вещества. MS: 732 [МН]+. LCMS (Способ В) RT = 4,68 мин. Сравнительный пример 10. 4-(5-Метокси-1-[(4-метилфенил)сульфонил]-1H-индол-3-ил)-6-(5 метокси-1H-индол-3-ил)-7-[(4-метилфенил)сульфонил]-7H-пирроло[2,3-d]пиримидин Раствор 4-хлор-6-йод-7-[(4-метилфенил)сульфонил]-7H-пирроло[2,3-d]пиримидина [1,72 г, сравнительный пример 11] и 1-трет-бутилкарбоксил-5-метокси-1H-индол-3-бороновой кислоты [1,26 г, сравнительный пример 12] в диметилформамиде (36,5 мл) последовательно обрабатывают насыщенным водным раствором бикарбоната натрия (9,1 мл) и тетракистрифенилфосфином палладия (0,3 г). Реакционную смесь перемешивают при температуре кипения с обратным холодильником в течение 2 ч, и растворитель выпаривают при пониженном давлении. Остаток распределяют между этилацетатом и водой. Органическую фазу отделяют, затем сушат над сульфатом магния и упаривают при пониженном давлении. Остаток подвергают колоночной флэш-хроматографии на двуокиси кремния, элюируя смесью этилацетата и циклогексана (30:70, об./об.) для получения указанного в заголовке соединения (270 мг) в виде смолы. MS: 718 [МН]+. LCMS (способ В) RT = 4,44 мин. Сравнительный пример 11. 4-Хлор-6-йод-7-[(4-метилфенил)сульфонил]-7H-пирроло[2,3-d]пиримидин К раствору 4-хлор-7-[(4-метилфенил)сульфонил]-7 Н-пирроло[2,3-d]пиримидина [5,4 г, сравнительный пример 3] в тетрагидрофуране (96 мл) при температуре -78 С по каплям добавляют раствор бутиллития в гексане (12,1 мл, 1,6 М) в инертной атмосфере. Раствор перемешивают при температуре -78 С в течение 3 ч и добавляют йод (8,9 г). Реакционную смесь перемешивают при температуре -78 С в течение 2 ч и дают ей возможность нагреться до комнатной температуры. Реакционную смесь распределяют между этилацетатом и водным раствором сульфита натрия, сушат над сульфатом магния, а растворитель выпаривают при пониженном давлении. Остаток подвергают колоночной флэш-хроматографии на двуокиси кремния, элюируя градиентом этилацетата и циклогексана (50:50, до 100, об./об.) для получения указанного в заголовке соединения (1,52 г) в виде аморфного твердого вещества. MS: 434 [МН]+. LCMS(Способ В) RT = 4,26 мин. Сравнительный пример 12. 1-трет-бутилкарбоксил-5-метокси-1 Н-индол-3-бороновая кислота Перемешиваемый раствор сложного трет-бутилового эфира 3-бром-5-метоксииндол-1-карбоновой кислоты [50 г, сравнительный пример 13] в тетрагидрофуране (800 мл), в атмосфере азота обрабатывают трибутилборатом (49,5 мл), затем охлаждают до температуры -100 С, после чего обрабатывают раствором н-бутиллития в гексанах (94 мл, 2,5 М), поддерживая температуру ниже -90 С. Закончив добавление,смеси дают возможность медленно нагреться до комнатной температуры в течение часа, а затем гасят ее,добавляя лед (10 г). Органическую фазу удаляют при пониженном давлении, а остаток распределяют между этилацетатом (500 мл) и водой (400 мл). Органический слой сушат над сульфатом магния, а затем упаривают, получая указанное в заголовке соединение в виде твердого вещества кремового цвета (28 г).MS: 314 [MH+Na]+. LCMS (Способ С) RT = 4,07 мин. Сравнительный пример 13. Сложный трет-бутиловый эфир 3-бром-5-метоксииндол-1-карбоновой кислоты К раствору 5-метоксииндола (10 г) в сухом диметилформамиде (150 мл) при температуре окружающей среды по каплям добавляют бром (4 мл), убедившись в том, что температура не поднимается выше приблизительно 30 С. Смесь сразу же обрабатывают триэтиламином (28 мл) и 4 диметиламинопиридином (0,5 г), а затем раствором ди-трет-бутилдикарбоната (18 г) в сухом диметилформамиде (80 мл), продолжая перемешивание еще в течение 4 ч. Реакционную смесь упаривают, а остаток распределяют между этилацетатом (250 мл) и водой (200 мл). Водный слой экстрагируют этилацетатом (100 мл). Соединенные органические фазы промывают водой (100 мл), затем рассолом (100 мл), после чего сушат над сульфатом магния и упаривают. Остаток подвергают колоночной флэшхроматографии на двуокиси кремния, элюируя смесью пентана и этилацетата (19/1, об./об.) для получения указанного в заголовке соединения (23,4 г) в виде бесцветного твердого вещества, т.пл. 111-112 С.- 25007415 Методика тестов in vitro для Syk Ингибирующее действие соединений на Syk киназу Ингибирующее действие соединений на Syk киназу определяют, применяя флуоресцентный анализ с разрешением во времени. Каталитический домен Syk киназы (остатки A340-N635) экспрессируют в виде слитого белка в дрожжевых клетках и очищают до гомогенного состояния. Активность киназы определяют в 50 мМ Трис-HCl буфера, рН 7,0, содержащем 50 мМ NaCl, 5 мМ MgCl2, 5 мМ MnCl2, 1 мкМ аденозинтрифосфата и 10 мкМ синтетического пептида БиотинАланин)3-DEEDYEIPP-NH2. Ферментативные реакции прекращают, добавляя буфер, содержащий 0,4 М KF, 133 мМ ЭДТА, рН 7,0, содержащей конъюгат стрептавидина-XL665 и моноклональное фосфоспецифическое антитело,конъюгированное с криптатом европия (Eu-K). Описание обоих флуорофоров, XL-665 и Eu-K, приведено в G. Mathis et al., Anticancer Research, 1997, 17, p. 3011-3014. Специфический долговременный сигналXL-665, получаемый только при фосфорилировании синтетического пептида Syk, измеряют на аппарате для прочтения планшетов LJL Biosystems Analyst AD. Ингибирование активности syk соединениями данного изобретения выражают в виде процентной величины ингибирования контрольной активности,проявляемой при отсутствии исследуемых соединений. Конкретные предпочтительные соединения данного изобретения ингибируют активность syk с IC50 в интервале от 100 микромолей до 100 наномолей. Особенно предпочтительные соединения данного изобретения ингибируют активность syk с IC50 в интервале от 1 микромолей до 100 наномолей. 2. Антиген-индуцированная дегрануляция клеток базофильного лейкоза у крыс (RBL) 2.1 Клеточная культура, мечение клеток RBL-2H3 и осуществление исследования Клетки RBL-2H3 содержат в колбах Т 75 при температуре 37 С и 5% СО 2 и пересеивают каждые 3-4 дня. Для сбора клеток используют 5 мл трипсина-ЭДТА, промывая колбу один раз, затем в каждую колбу добавляют по 5 мл трипсина и инкубируют при комнатной температуре в течение 2 мин. Клетки переносят в пробирку с 14-мл средой, центрифугируют со скоростью 1100 об./мин при комнатной температуре в течение 5 мин и вновь подвергают суспендированию до 2 х 105/мл. Клетки сенсибилизируют, добавляя к каждым 10 мл клеток 1 мкл DNP-специфического IgE. В каждую лунку плоскодонного 96 луночного планшета (40000 клеток/лунка) добавляют по 200 мкл клеток, после чего планшет инкубируют в течение ночи при температуре 37 С и 5% СО 2. На следующий день соединения получают в 100% ДМСО при 10 мМ. Затем каждое соединение разбавляют 1:100 в буфере для анализов, а затем вновь разбавляют в 1% ДМСО-буфере для анализов для получения конечной концентрации, составляющей 0,03-30 мкМ. В каждую лунку добавляют по 80 мкл буфера для анализов, а затем по 10 мкл разбавленного соединения, после чего в течение 5 мин осуществляют инкубирование. В каждую лунку добавляют по 10 мкл DNP-HSA (100 нг/мл) и инкубируют при температуре 37 С (без СО 2) в течение 30 мин. В качестве одного вида контроля к ряду лунок добавляют только 1% ДМСО (без соединения) для определения общего уровня высвобождения. В качестве другого вида контроля к другому ряду лунок вместо DNP-HSA добавляют буфер для определения величины фона анализа. Через 30 мин надосадочные жидкости переносят в новый 96-луночный планшет. В каждую лунку планшета для исследований добавляют по 50 мкл надосадочной жидкости. В каждую лунку добавляют по 100 мкл раствора субстрата и инкубируют при температуре 37 С в течение 90 мин. С целью прекращения реакции добавляют 50 мкл 0,4 М раствора глицина и планшет прочитывают при 405 нм на аппарате для прочтения планшетов Molecular Devices(i) Подсчитывают среднее SD (стандартное отклонение) каждого набора лунок из трех повторов.(ii) Максимальный ответ означает положительные контрольные лунки, содержащие антиген (100 нг/мл) без соединения.(iii) Минимальный ответ означает контрольные лунки, содержащие буфер (без антигена) и не содержащие соединения.(iv) Применяя данные величины в качестве максимума (100%) и минимума (0%), соответственно,получают экспериментальные данные для определения процентной величины максимального ответа(v) Строят кривую зависимости ответа от дозы и подсчитывают IC50 соединения, используя компьютерную программу Prism GraphPad и нелинейный регрессионный анализ наименьших квадратов. Соединения данного изобретения ингибируют антиген-индуцируемую дегрануляцию клеток базофильного лейкоза крыс (RBL) с EC50 в интервале от 100 до 1 мкмол.N-оксид, пролекарство, кислотная биоизостера, фармацевтически приемлемая соль или гидрат такого соединения; либо N-оксид, пролекарство или кислотная биоизостера такой соли или гидрата. 2. Соединение по п.1, в котором R1 представляет СН 3. 3. Соединение по п.1, в котором R1 представляет 4. Соединение по п.1, в котором R1 представляет водород. 5. Соединение по любому из пп.1-4, в котором R3 представляет ОСН 3. 6. Соединение по любому из пп.1-4, в котором R3 представляет пиридил или трифторметил. 7. Соединение по любому из пп.1-6, в котором R2 присоединен в положении 5 индольного кольца. 8. Соединение по любому из пп.1-7, в котором группа дольного кольца. 9. Соединение по п.1, представляющее собой присоединена в положении 3 ин или N-оксид, пролекарство, фармацевтически приемлемую соль или гидрат такого соединения; либо Nоксид или пролекарство такой соли или гидрата. 10. Соединение по п.1, представляющее собойN-оксид, пролекарство, фармацевтически приемлемую соль или гидрат такого соединения; либо Nоксид или пролекарство такой соли или гидрата. 11. Фармацевтическая композиция, содержащая фармацевтически эффективное количество соединения по любому из пп.1-10, вместе с одним или несколькими фармацевтически приемлемыми носителями или эксципиентами. 12. Способ лечения пациента, подверженного или страдающего от состояний, которые могут быть улучшены в результате введения ингибитора каталитической активности Syk, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11. 13. Способ лечения воспалительного заболевания у нуждающегося в этом пациента, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11. 14. Способ лечения пациента, подверженного или страдающего от состояний, которые могут быть улучшены в результате введения ингибитора каталитической активности FAK, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11. 15. Способ лечения пациента, подверженного или страдающего от состояний, которые могут быть улучшены в результате введения ингибитора каталитической активности KDR, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11. 16. Способ лечения пациента, подверженного или страдающего от состояний, которые могут быть улучшены в результате введения ингибитора каталитической активности Aurora2, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11. 17. Способ лечения рака у нуждающегося в этом пациента, включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11. 18. Способ по п.13, в котором воспалительное заболевание представляет собой астму, воспалительный дерматоз, аллергический ринит, аллергический конъюнктивит или воспаление суставов. 19. Способ по п.13, в котором воспалительное заболевание представляет собой астму, псориаз, герпетиформный дерматит, экзему, некротизирующий васкулит, кожный васкулит, буллезное заболевание,аллергический ринит, аллергический конъюнктивит, артрит, ревматоидный артрит, артрит коревой краснухи, псориатический артрит или остеоартрит. 20. Способ лечения хронической обструктивной болезни легких у нуждающегося в этом пациента,включающий введение указанному пациенту фармацевтически эффективного количества соединения по любому из пп.1-10, либо фармацевтически эффективного количества композиции по п.11. 21. Способ по п.17, в котором подвергаемые лечению виды рака включают колоректальный рак, рак предстательной, молочной, щитовидной железы, рак кожи, рак прямой кишки или рак легких.
МПК / Метки
МПК: C07D 239/00, A61P 29/00, C07D 487/04, C07D 209/00, A61K 31/505
Метки: протеинкиназы, ингибиторы, пирролопиримидины
Код ссылки
<a href="https://eas.patents.su/29-7415-pirrolopirimidiny-kak-ingibitory-proteinkinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Пирролопиримидины как ингибиторы протеинкиназы</a>
Ингибиторы протеинкиназы с.

Номер патента: 598
Опубликовано: 29.12.1999
Авторы: Макдональд Джон Х., Рито Кристофер Дж., Джироусек Майкл Р., Хит Уильям Ф.
МПК: C07D 471/22, A61K 47/22
Метки: протеинкиназы, ингибиторы
Формула / Реферат:
1. Соединение формулы где W является О, -S- или NH, R1 представляет собой независимо водород, галоген, С1-С4-алкил, гидрокси, С1-С4-алкокси, галогеналкил, нитро, -NH(С1-С4-алкил), N(С1-С4-алкил)2 или -NНСО(С1-С4-алкил), R2 является водородом, СН3СО-, NН2 или гидроксигруппой, Z является -(СН2)р- или (СН2)р-O-(СН2)p-, R6 является -NH(CF3) или -N(CF3)(СН3), m независимо является 0, 1, 2 или 3, р независимо является 0, 1 или 2 или...
Галогензамещенные ингибиторы протеинкиназы с.

Номер патента: 1450
Опубликовано: 23.04.2001
Авторы: Гоэкджян Питер Г., Джироусек Майкл Роберт, Ву Гуо-Жанг
МПК: C07D 498/22, A61K 47/16, A61P 9/10...
Метки: ингибиторы, галогензамещенные, протеинкиназы
Формула / Реферат:
1. Соединение формулы где R' - независимо водород, галоген, гидрокси, C1-C4алкил, C1-C4алкокси, NR3R4 либо -NHCO(C1-C4алкил); V - -NH- или -NC1-C4алкил-; Т - C1-C4алкилен, факультативно замещенный галогеном, либо C1-C4 алкил; W - C1-C2алкилен, факультативно замещенный галогеном, либо C1-C4 алкил; J - либо, когда оба Т и W - метилен, J выбирают из группы, в состав которой входят где n и m - независимо 1 либо 2; X - кислород, сера...
Пирролопиримидины и фармацевтические композиции, включающие эти соединения.

Номер патента: 1428
Опубликовано: 26.02.2001
Авторы: Фрай Йорг, Брилл Карл-Дитер Вольфганг, Болд Гидо, Тракслер Питер
МПК: A61K 31/519, A61P 35/00, C07D 471/14...
Метки: эти, соединения, включающие, пирролопиримидины, композиции, фармацевтические
Формула / Реферат:
1. Производное 7Н-пирроло[2,3-d] пиримидина формулы I в которой q - 0 или 1, n - от 1 до 3, если q=0, или от 0 до 3, если q=1, R - галоген, С1-С7алкил, гидрокси, С1-С7алканоилокси, C1-С7алкокси, карбокси, С1-С7алкоксикарбонил, карбамоил, N-С1-С7алкилкарбамоил, N,N-ди-С1-С7алкилкарбамоил, циано, амино, С1-С7алканоиламино, С1-С7алкиламино, N,N-ди-С1-С7алкиламино или трифторметил, причем, если в молекуле присутствуют несколько радикалов R, они...
Ингибитор протеинкиназы с.

Номер патента: 967
Опубликовано: 28.08.2000
Авторы: Инджел Гари Л., Фарид Наджи А., Фол Маргарет М., Виннероски Леонард Л., Ричардсон Лори А., Джираусек Майкл Р.
МПК: A61P 5/48, C07D 413/14, A61K 31/40...
Метки: ингибитор, протеинкиназы
Формула / Реферат:
1. Соль формулы и ее сольваты. 2. Соль по п.1 формулы и ее сольваты. 3. Соль по п.1 или 2, которая по существу является кристаллической. 4. Соль по п.3, которая представляет собой моногидрат метансульфоната (S)-13-[(диметиламино)метил]-10,11,14,15-тетрагидро-4,9:16,21-диметено-1Н,13Н-дибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3(2Н)-диона. 5. Соль по любому одному из пп.1-4, отличающаяся тем, что имеет общее...
Замещенные, азотсодержащие гетероциклы в качестве ингибиторов протеинкиназы р38

Номер патента: 2855
Опубликовано: 31.10.2002
Авторы: Уилсон Кейт П., Бемис Гай В., Харрингтон Эдмунд Мартин, Галулло Винсент П., Кочран Джон Е., Даффи Джон Патрик, Мёрко Марк А., Салитуро Франческо Джеральд, Су Майкл
МПК: A61K 31/505, A61P 25/28, C07D 471/04...
Метки: замещенные, ингибиторов, гетероциклы, качестве, протеинкиназы, азотсодержащие
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль, где каждый из Q1 и Q2 независимо выбран из систем 5-6-членных ароматических карбоциклических или гетероциклических колец или систем 8-10-членных бициклических колец, состоящих из ароматических карбоциклических колец, ароматических гетероциклических колец или сочетания ароматического карбоциклического кольца и ароматического гетероциклического кольца; где Q1 замещен 1-4...