Галогензамещенные ингибиторы протеинкиназы с.
Номер патента: 1450
Опубликовано: 23.04.2001
Авторы: Ву Гуо-Жанг, Джироусек Майкл Роберт, Гоэкджян Питер Г.







Формула / Реферат
1. Соединение формулы

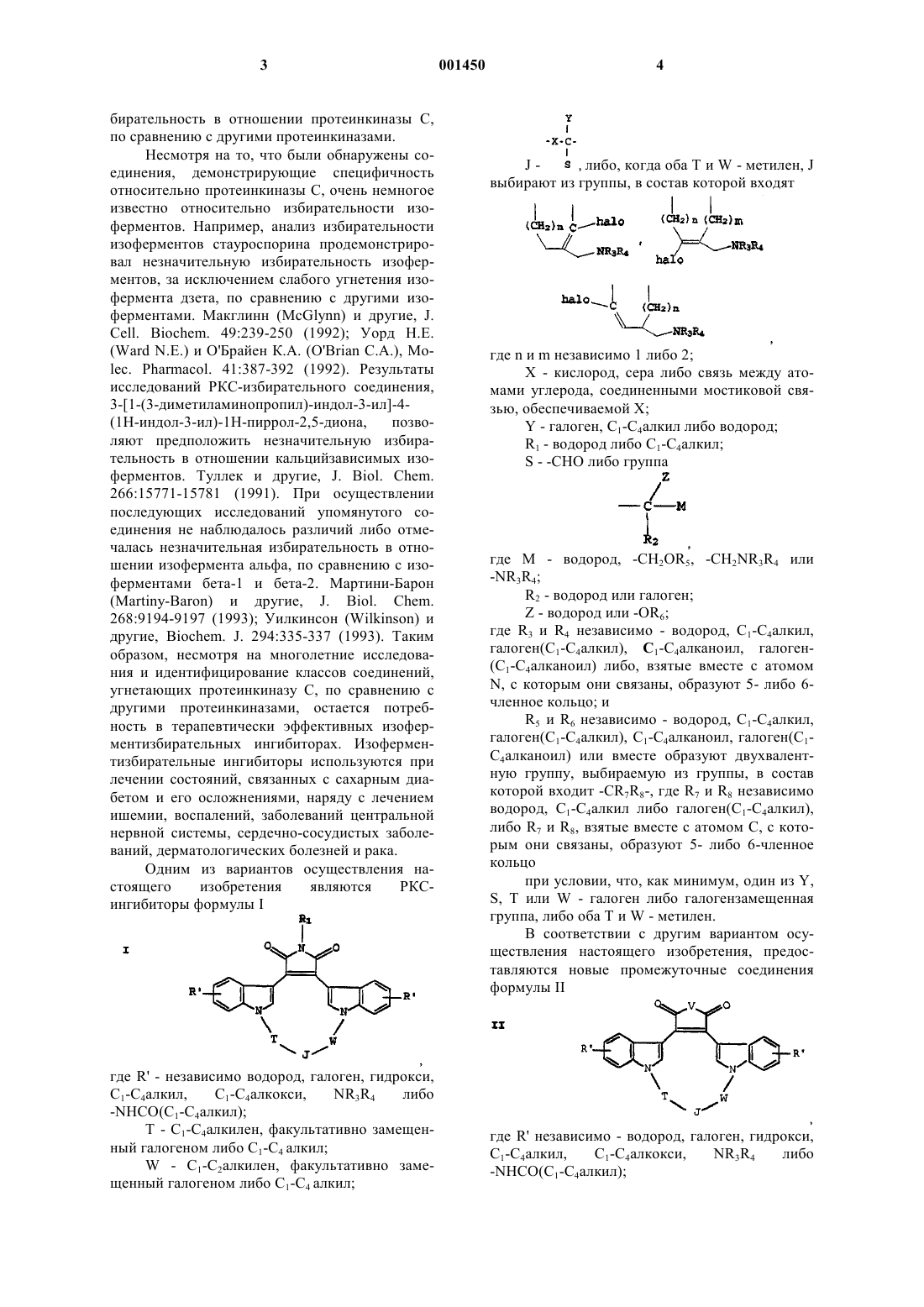
где R' - независимо водород, галоген, гидрокси, C1-C4алкил, C1-C4алкокси, NR3R4 либо -NHCO(C1-C4алкил);
V - -NH- или -NC1-C4алкил-;
Т - C1-C4алкилен, факультативно замещенный галогеном, либо C1-C4 алкил;
W - C1-C2алкилен, факультативно замещенный галогеном, либо C1-C4 алкил;
J -
либо, когда оба Т и W - метилен, J выбирают из группы, в состав которой входят

где n и m - независимо 1 либо 2;
X - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;
Y - галоген, C1-C4алкил либо водород;
S - -СНО либо группа

где М - водород, -CH2OR5, -CH2NR3R4 или -NR3R4;
R2 - водород или галоген; и
Z - водород или -OR6;
где R3 и R4 - независимо водород, C1-C4алкил, галоген(C1-C4алкил), C1-C4алканоил, галоген(C1-C4алканоил) либо R3 и R4, взятые вместе с атомом N, с которым они связаны, образуют 5- либо 6-членное кольцо; и
R5 и R6 - независимо водород, C1-C4алкил, галоген(C1-C4алкил), C1-C4алканоил, галоген(C1-C4алканоил) или вместе образуют двухвалентную группу формулы -CR7R8-, где R7 и R8 независимо водород, C1-C4алкил либо галоген(C1-C4алкил), либо R7 и R8, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6-членное кольцо;
при условии, что, как минимум, один из Y, S, Т или W - галоген либо галогензамещенная группа, либо оба Т и W - метилен.
2. Соединение по п.1, где, как минимум, один из Y, S, Т или W - фтор либо фторзамещенная группа.
3. Соединение формулы

где R1 - C1-C4алкил или водород;
Т - C2-C4алкилен, факультативно замещенный галогеном, либо C1-C4алкил;
W - этилен, факультативно замещенный галогеном, либо C1-C4алкил;
Х - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;
Y - галоген, C1-C4алкил либо водород;
S - -СНО либо группа

где М - водород, -CH2OR5, -CH2NR3R4 или -NR3R4;
R2 - водород или галоген; и
Z - водород или -OR6;
где R3 и R4 - независимо водород, C1-C4алкил, галоген(C1-C4алкил), C1-C4алканоил, галоген(C1-C4алканоил), либо R3 и R4, взятые вместе с атомом N, с которым они связаны, образуют 5- либо 6-членное кольцо; и
R5 и R6 - независимо водород, C1-C4алкил, галоген(C1-C4алкил), C1-C4алканоил, галоген(C1-C4алканоил) или вместе образуют двухвалентную группу формулы -CR7R8-, где R7 и R8 независимо водород, C1-C4алкил либо галоген(C1-C4алкил), либо R7 и R8, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6-членное кольцо
при условии, что, как минимум, один из Y, S, Т или W - галоген либо галогензамещенная группа.
4. Соединение по п.3, где, как минимум, один из Y, S, Т или W - фтор либо фторзамещенная группа.
5. Соединение по п.3, где W - фторзамещенный этилен.
6. Соединение по п.3, где Т - фторзамещенный этилен.
7. Соединение по п.3, где Т - фторзамещенный триметилен.
8. Соединение по п.3, где R1 и Y - водород, X - кислород.
9. Соединение по п.5, где R1 и Y - водород, X - кислород,
S -

10. Соединение по п.9, где Т - этилен.
11. Соединение формулы

где J выбирают из группы, в состав которой входят

где R1 - C1-C4алкил или водород;
n и m - независимо 1 либо 2; и
R3 и R4 - независимо водород, C1-C4алкил, галоген(C1-C4алкил), C1-C4алканоил, галоген(C1-C4алканоил), либо R3 и R4, взятые вместе с атомом N, с которым они связаны, образуют 5- либо 6-членное кольцо.
12. Соединение по п.9, где R1 - водород.
13. Соединение по п.9, где галогеновым заместителем J является фтор.
14. Соединение формулы

где R' - независимо водород, галоген, гидрокси, C1-C4алкил, C1-C4алкокси, NR3R4 либо -NHCO(C1-C4алкил);
V - -O-,
Т - C1-C4алкилен, факультативно замещенный галогеном, либо C1-C4алкил;
W - C1-C2алкилен, факультативно замещенный галогеном, либо C1-C4алкил;
J -

либо, когда оба Т и W - метилен, J выбирают из группы, в состав которой входят

где n и m - независимо 1 либо 2;
Х - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;
Y - галоген, C1-C4алкил либо водород;
S - -CHO либо группа

где М - водород, -CH2OR5, -CH2NR3R4 или -NR3R4;
R2 - водород или галоген; и
Z - водород или -OR6;
где R3 и R4 - независимо, водород, C1-C4алкил, галоген(C1-C4алкил), C1-C4алканоил, галоген(C1-C4алканоил) либо R3 и R4, взятые вместе с атомом N, с которым они связаны, образуют 5- либо 6-членное кольцо; и
R5 и R6 - независимо водород, C1-C4алкил, галоген(C1-C4алкил), C1-C4алканоил, галоген(C1-C4алканоил) или вместе образуют двухвалентную группу формулы -CR7R8-, где R7 и R8 - независимо водород, C1-C4алкил либо галоген(C1-C4алкил), либо R7 и R8, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6-членное кольцо
при условии, чтю, как минимум, один из Y, S, Т или W - галоген либо галогензамещенная группа, либо оба Т и W - метилен.
15. Фармацевтическая композиция, включающая соединение формулы

где R' - независимо водород, галоген, гидрокси, C1-C4алкил, C1-C4алкокси, NR3R4 либо -NHCO(C1-C4алкил);
R1 - C1-C4алкил или водород;
Т - C1-C4алкилен, факультативно замещенный галогеном, либо C1-C4 алкил;
W - C1-C2алкилен, факультативно замещенный галогеном, либо C1-C4 алкил;
J -

либо, когда оба Т и W - метилен, J выбирают из группы, в состав которой входят

где n и m - независимо 1 либо 2;
Х - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;
Y - галоген, C1-C4алкил либо водород;
S - -СНО либо группа

где М - водород, -CH2OR5, -CH2NR3R4 или -NR3R4;
R2 - водород или галоген; и
Z - водород или -OR6;
где R3 и R4 - независимо водород, C1-C4алкил, галоген(C1-C4алкил), C1-C4алканоил, галоген(C1-C4алканоил) либо R3 и R4, взятые вместе с атомом N, с которым они связаны, образуют 5- либо 6-членное кольцо; и
R5 и R6 - независимо водород, C1-C4алкил, галоген(C1-C4алкил), C1-C4алканоил, галоген(C1-C4алканоил) или вместе образуют двухвалентную группу формулы -CR7R8-,
где R7 и R8 - независимо водород, C1-C4алкил либо галоген(C1-C4алкил), либо R7 и R8, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6-членное кольцо,
при условии, что, как минимум, один из Y, S, Т или W - галоген либо галогензамещенная группа, либо оба Т и W - метилен; и
фармацевтически приемлемый наполнитель, носитель либо разбавитель.
16. Фармацевтическая композиция по п.15, где
J -
17. Фармацевтическая композиция по п.16, где S -

X - кислород; и
R1 - водород.
18. Фармацевтическая композиция по п.17, где, как минимум, один из Y, S, Т или W - фтор либо фторзамещенная группа.
19. Фармацевтическая композиция по п.15, где Т и W - метилен, а галогеновым заместителем является фтор.
20. Способ лечения млекопитающего, имеющего заболевание либо состояние, связанное с аномальной активностью протеинкиназы С, причем упомянутый способ включает введение упомянутому млекопитающему фармацевтически эффективного количества соединения формулы

где R' - независимо водород, галоген, гидрокси, C1-C4алкил, C1-C4алкокси, NR3R4 либо -NHCO(C1-C4алкил);
R1 - C1-C4алкил или водород;
Т - C1-C4алкилен, факультативно замещенный галогеном, либо C1-C4 алкил;
W - C1-C2алкилен, факультативно замещенный галогеном, либо C1-C4 алкил;
J -

либо, когда оба Т и W - метилен, J выбирают из группы, в состав которой входят

где n и m - независимо 1 либо 2;
Х - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;
Y - галоген, C1-C4алкил либо водород;
S - -СНО либо группа

где М - водород, -CH2OR5, -CH2NR3R4 или -NR3R4;
R2 - водород или галоген; и
Z - водород или -OR6;
где R3 и R4 - независимо водород, C1-C4алкил, галоген(C1-C4алкил), C1-C4алканоил, галоген(C1-C4алканоил), либо R3 и R4, взятые вместе с атомом N, с которым они связаны, образуют 5- либо 6-членное кольцо; и
R5 и R6 - независимо водород, C1-C4алкил, галоген(C1-C4алкил), C1-C4алканоил, галоген(C1-C4алканоил) или вместе образуют двухвалентную группу формулы -CR7R8-, где R7 и R8 - независимо водород, C1-C4алкил либо галоген(C1-C4алкил), либо R7 и R8, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6-членное кольцо,
при условии, что, как минимум, один из Y, S, Т или W - галоген либо галогензамещенная группа, либо оба Т и W - метилен; и
фармацевтически приемлемый наполнитель, носитель либо разбавитель.
21. Способ по п.20, где J -

22. Способ по п.21, где S -

X - кислород; и R1 - водород.
23. Способ по п.21, где, как минимум, один из Y, S, Т или W - фтор либо фторзамещенная группа.
24. Способ по п.20, где Т и W - метилен, а галогеновым заместителем является фтор.
25. Способ по п.20, где W - фторзамещенный этилен.
26. Способ по п.20, где Т - фторзамещенный этилен.
27. Способ по п.20, где Т - фторзамещенный триметилен.
Текст
1 Протеинкиназа С (РКС) представляет собой семейство близкородственных ферментов,которые функционируют, как серин/треонинкиназы. Протеинкиназа С играет важную роль в передаче сигналов между клетками, экспрессии генов и в контролировании дифференциации и роста клеток. В настоящее время известно, как минимум, десять изоферментов РКС, различающихся по своему распределению в тканях,ферментной специфичности и регуляции. Нишизука И. (Nishizuka Y.) Annu. Rev. Biochem. 58:31-44 (1989); Нишизука И. Science 258:607614 (1992). Изоферменты протеинкиназы С представляют собой одноцепочечный полипептид длиной от 592 до 737 аминокислот. Упомянутые изоферменты включают регуляторный домен и каталитический домен, соединенные линкерным пептидом. Упомянутые регуляторный и каталитический домены могут быть дополнительно подразделены на константные и вариабельные области. Каталитический домен протеинкиназы С весьма подобен каталитическим доменам,имеющимся у других протеинкиназ, в то время как регуляторный домен характерен только для изоферментов РКС. Упомянутые изоферменты РКС демонстрируют 40-80% гомологию на аминокислотном уровне в пределах группы. Гомология отдельного изофермента у различных видов, однако, как правило, превышает 97%. Протеинкиназа С является мембранассоциированным ферментом, который аллостерически регулируется рядом факторов, в том числе, мембранными фосфолипидами, кальцием и некоторыми мембранными липидами, например, диацилглицеринами, которые высвобождаются в ответ на активность фосфолипаз. БеллP.M. (Bell R.M.) и Бернс Д.Дж. (Burns D.J.),J.Biol.Chem. 266:4661-4664 (1991); Нишизука И., Science 258:607-614 (1992). Для полной активации изоферментов протеинкиназы С альфа,бета-1, бета-2 и гамма необходимы мембранный фосфолипид, кальций и диацилглицериновые/форболовые эфиры. Способ активации дельта, эпсилон, эта и тета форм РКС является кальцийнезависимым. Дзета и лямбда формы РКС не зависят ни от кальция, ни от диацилглицерина, и для их активации, как полагают, необходим лишь мембранный фосфолипид. В конкретное болезненное состояние может быть вовлечен только один либо два из упомянутых изоферментов протеинкиназы С. Например, следствием повышенных уровней глюкозы в крови, обнаруживаемых при диабете,является изоферментспецифическое повышение содержания изофермента бета-2 в сосудистых тканях. Иногачи (Inoguchi) и другие, Рrос. Natl.Acad. Sci. USA 89: 11059-11065 (1992). Связанное с диабетом повышение содержания изофермента бета в тромбоцитах человека коррелирует с изменением реакции последних на воздейст 001450 2 вие агонистов. Бастир III И.Дж. (Bastyr III E.J.) и Лу Дж. (Lu J.) Diabetes 42 (Suppl. 1) 97 А (1993). Было показано, что рецепторы витамина D человека избирательно фосфорилируются протеинкиназой С бета. Упомянутое фосфорилирование связывается с изменениями функционирования упомянутого рецептора. Си (Hsieh) и другие, Proc. Natl. Acad. Sci. USA 88:9315-9319(1991); Си и другие, J. Biol. Chem. 268:1511815126 (1993). В дополнение к этому, результаты недавно проведенного исследования показали,что изофермент бета-2 несет ответственность за пролиферацию клеток при эритролейкозе, в то время как изофермент альфа вовлечен в дифференциацию мегакариоцитов в этих же самых клетках. Мюррей (Murray) и другие, J. Biol.Chem. 268:15847-15853 (1993). Убиквитарная природа изоферментов протеинкиназы С и важная роль, осуществляемая ими в физиологии, являются побудительной причиной для получения высокоизбирательных ингибиторов РКС. Учитывая данные, демонстрирующие связь определенных изоферментов с болезненными состояниями, вполне естественно предположить, что ингибирующие соединения,избирательно действующие на один либо два изофермента протеинкиназы С, по сравнению с другими изоферментами РКС и другими протеинкиназами, являются великолепными терапевтическими средствами. Такие соединения,вследствие своей специфичности, должны демонстрировать повышенную эффективность и пониженную токсичность. Стауроспорин, индолокарбазол микробного происхождения, является мощным ингибитором протеинкиназы С, взаимодействующим с каталитическим доменом упомянутого фермента. Тамаоки (Tamaoki) и другие, Biochem. Biophys. Res. Commun. 135:397-402 (1986); Гросс(Gross) и другие, Biochem. Pharmacol. 40:343350 (1990). Однако терапевтическая пригодность упомянутой молекулы и близкородственных соединений ограничивается отсутствием специфичности относительно протеинкиназы С,по сравнению с другими протеинкиназами. Регг А.Т. (Ruegg U.Т.) и Баргесс Г.М. (Burgess G.M.),Trends Pharmacol. Sci. 10:218-220 (1989). Следствием упомянутого отсутствия избирательности является неприемлемая токсичность молекул этого класса. Последние исследования были сосредоточены на бисиндолмалеимидах, соединениях дополнительного класса, связанных со стауроспорином. Дэвис (Davis) и другие, FEBS Lett. 259:61-63 (1989); Твими (Twoemy) и другие,Biochem. Biophys. Res. Commun. 171:1087-1092Med. Chem. 36:21-29 (1993). Некоторые из упомянутых соединений продемонстрировали из 3 бирательность в отношении протеинкиназы С,по сравнению с другими протеинкиназами. Несмотря на то, что были обнаружены соединения, демонстрирующие специфичность относительно протеинкиназы С, очень немногое известно относительно избирательности изоферментов. Например, анализ избирательности изоферментов стауроспорина продемонстрировал незначительную избирательность изоферментов, за исключением слабого угнетения изофермента дзета, по сравнению с другими изоферментами. Макглинн (McGlynn) и другие, J.(Ward N.E.) и О'Брайен К.А. (O'Brian C.A.), Molec. Pharmacol. 41:387-392 (1992). Результаты исследований РКС-избирательного соединения,3-[1-(3-диметиламинопропил)-индол-3-ил]-4(1H-индол-3-ил)-1 Н-пиррол-2,5-диона,позволяют предположить незначительную избирательность в отношении кальцийзависимых изоферментов. Туллек и другие, J. Biol. Chem. 266:15771-15781 (1991). При осуществлении последующих исследований упомянутого соединения не наблюдалось различий либо отмечалась незначительная избирательность в отношении изофермента альфа, по сравнению с изоферментами бета-1 и бета-2. Мартини-Барон(Martiny-Baron) и другие, J. Biol. Chem. 268:9194-9197 (1993); Уилкинсон (Wilkinson) и другие, Biochem. J. 294:335-337 (1993). Таким образом, несмотря на многолетние исследования и идентифицирование классов соединений,угнетающих протеинкиназу С, по сравнению с другими протеинкиназами, остается потребность в терапевтически эффективных изоферментизбирательных ингибиторах. Изоферментизбирательные ингибиторы используются при лечении состояний, связанных с сахарным диабетом и его осложнениями, наряду с лечением ишемии, воспалений, заболеваний центральной нервной системы, сердечно-сосудистых заболеваний, дерматологических болезней и рака. Одним из вариантов осуществления настоящего изобретения являются РКСингибиторы формулы IC1-C4 алкил,-NHCO(C1-C4 алкил); Т - C1-C4 алкилен, факультативно замещенный галогеном либо C1-C4 алкил;W - C1-C2 алкилен, факультативно замещенный галогеном либо C1-C4 алкил;X - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;R5 и R6 независимо - водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) или вместе образуют двухвалентную группу, выбираемую из группы, в состав которой входит -CR7R8-, где R7 и R8 независимо водород, C1-C4 алкил либо галоген(C1-C4 алкил),либо R7 и R8, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6-членное кольцо при условии, что, как минимум, один из Y,S, Т или W - галоген либо галогензамещенная группа, либо оба Т и W - метилен. В соответствии с другим вариантом осуществления настоящего изобретения, предоставляются новые промежуточные соединения формулы IIV - кислород либо Т - C1-C4 алкилен, факультативно замещенный галогеном либо C1-C4 алкил;W - C1-C2 алкилен, факультативно замещенный галогеном либо C1-C4 алкил;X - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;R5 и R6 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) или вместе образуют двухвалентную группу, выбираемую из группы, в состав которой входит -CR7R8-, где R7 и R8 независимо водород, C1-C4 алкил либо галоген(C1-C4 алкил),либо R7 и R8, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6-членное кольцо при условии, что, как минимум, один из Y,S, Т или W - галоген либо галогензамещенная группа, либо оба Т и W - метилен. Одним из аспектов настоящего изобретения является способ получения соединений формулы I, включающий этапы объединения, в концентрации приблизительно от 0,001 молярного до приблизительно 1,5 молярного соединения формулы-NHCO(C1-C4 алкил); и алкилирующего агента в концентрации приблизительно от 0,001 молярного до приблизительно 1,5 молярного формулы где L - отщепляемая группа; Т - C1-C4 алкилен, факультативно замещенный галогеном либо C1-C4 алкил;W - C1-C2 алкилен, факультативно замещенный галогеном либо C1-C4 алкил;X - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;R5 и R6 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) или вместе образуют двухвалентную группу, выбираемую из группы, в состав которой входит -CR7R8-, где R7 - водород или метил и R8 - C1-C4 алкил, галоген(C1-C4 алкил),либо, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6-членное кольцо,и приблизительно от 0,5 до приблизительно 10 эквивалентов Сs2 СО 3 со скоростью приблизительно от 0,1 мл/ч до приблизительно 2,0 мл/ч в полярном апротонном растворителе. В соответствии с одним из вариантов осуществления настоящего изобретения, предоставляется соединение формулы III где R1 - C1-C4 алкил или водород; Т - С 2-С 4 алкилен, факультативно замещенный галогеном либо C1-C4 алкил;W - этилен, факультативно замещенный галогеном либо C1-C4 алкил;X - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;R5 и R6 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) или вместе образуют двухвалентную группу, выбираемую из группы, в состав которой входит -CR7R8-, где R7 - водород или метил и R8 - C1-C4 алкил, галоген(C1-C4 алкил),либо, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6-членное кольцо при условии, что, как минимум, один из Y,S, Т или W - галоген либо галогензамещенная группа. В другом варианте осуществления настоящего изобретения предоставляется соединение формулы IVR3 и R4 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) либо, взятые вместе с атомом N, с которым они связаны, образуют 5- либо 6 членное кольцо. В соответствии с одним из вариантов осуществления настоящего изобретения предоставляется способ угнетения активности РКС. Упомянутый способ включает введение млекопитающему, нуждающемуся в подобном лечении,фармацевтически эффективного количества соединения формулы I. Настоящим изобретением,кроме того, предоставляется способ избирательного угнетения изоферментов протеинкиназы С бета-1 и бета-2, причем упомянутый способ включает введение млекопитающему, нуждающемуся в подобном лечении, фармацевтически эффективного количества соединения формулы I. Настоящее изобретение дополнительно предоставляет способы лечения состояний, в патологии которых, как было показано, протеинкиназа С играет роль, например, ишемии,воспалений, заболеваний центральной нервной системы, сердечно-сосудистых заболеваний,дерматологических болезней и рака. Упомянутый способ включает введение млекопитающему, нуждающемуся в подобном лечении, фармацевтически эффективного количества соединения формулы I. Настоящее изобретение особо пригодно для лечения диабетических осложнений. Таким образом, настоящим изобретением дополнительно предоставляется способ лечения сахарного диабета, который включает введение млекопитающему, нуждающемуся в подобном лечении, фармацевтически эффективного количества соединения формулы I. Еще одним аспектом настоящего изобретения является фармацевтическая лекарственная форма, включающая соединение формулы I вместе с одним или более фармацевтически приемлемыми наполнителями, носителями либо разбавителями. Подробное описание настоящего изобретения Как отмечалось ранее, настоящее изобретение предоставляет соединения формулы I,которые избирательно угнетают протеинкиназу С. К числу предпочтительных соединений, соответствующих настоящему изобретению, относятся соединения формулы I, в состав состав 9 ляющих -T-J-W- которых входит от 4 до 8 атомов, которые могут быть незамещенными либо замещенными (галогенами либо алкильными группами). В более предпочтительном варианте,в состав составляющих -T-J-W- входит 6 атомов. К числу других предпочтительных соединений, соответствующих настоящему изобретению, относятся соединения формулы I,где R1 - водород,JОдной из предпочтительных групп соединений являются соединения формулы V где R1 - C1-C4 алкил или водород; Т - С 2-С 4 алкилен, факультативно замещенный галогеном либо C1-C4 алкил;W - этилен, факультативно замещенный галогеном либо C1-C4 алкил;X - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;R5 и R6 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) или вместе образуют двухвалентную группу, выбираемую из группы, в состав которой входит -CR7R8-, где R7 - водород или метил и R8 - C1-C4 алкил, галоген(C1-C4 алкил),либо, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6-членное кольцо,и где, как минимум, один из Y, R2, Z, М, Т или W - галоген либо галогензамещенная группа, в более предпочтительном варианте, фтор либо фторзамещенная группа. К числу предпочтительных соединений формулы V относятся соединения, где X - кислород, R1 - водород, Т - факультативно замещенный С 2-С 3 алкилен, W - факультативно замещенный этилен. В предпочтительном варианте, Т и/или W замещаются одной или более фторогруппами. 10 В другом варианте осуществления настоящего изобретения предоставляется соединение формулы VIR3 и R4 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) либо, взятые вместе с атомом N, с которым они связаны, образуют 5- либо 6 членное кольцо. Термин "галоген" обозначает фтор, хлор,бром или йод. Термин "C1-C4 алкил" обозначает циклическую алкильную группу с прямой либо разветвленной цепью, имеющей от одного до четырех атомов углерода, например, метил, этил, nпропил, изопропил, циклопропил, n-бутил, изобутил, втор-бутил, t-бутил и т.п. Подобным же образом термин "С 2-С 4 алкил" обозначает циклическую алкильную группу с прямой либо разветвленной цепью, имеющей от двух до четырех атомов углерода. Галогеналкильной группой является алкильная группа, замещенная одним или более атомами галогена, в предпочтительном варианте, одним-тремя атомами галогена. Одним из примеров галогеналкильной группы является трифторметил. C1-C4 алкокси являетсяC1-C4 алкильной группой, ковалентно связанной через посредство -О-связи. Термин "C1-C4 алкилен" обозначает алкиленовую составляющую формулы -(СН 2)r-, гдеr=1-4. К числу примеров C1-C4 алкилен относятся метилен, этилен, триметилен, тетраметилен и т.п. Подобным же образом "С 2-С 4 алкилен" представляет собой алкиленовую составляющую с прямой цепью, включающей от двух до четырех атомов углерода. Термин "C1-C4 алкенилен" обозначает углеводород с прямой цепью, включающей от одного до четырех атомов углерода и имеющей одну либо более двойных связей, как правило, одну или две двойные связи. Примерами С 2 С 4 алкениленовых групп являются этенилен,пропенилен и 1,3-бутадиенил."C1-C4 алканоил" обозначает ацильный остаток C1-C4 карбоновой кислоты. Примерами C1-C4 алканоильных групп являются ацетил, пропаноил, бутаноил и т.п. Галогеналканоильной группой является алканоильная группа, замещенная одним или более атомами галогена, как правило, одним-тремя атомами галогена. Примером галогеналканоильной группы является трифторацетил. Термин "отщепляемая группа", используемый в настоящем описании, понятен специалистам в этой области техники. В целом, отщепляемой группой является любая группа либо атом, которые повышают степень электрофильности атома, с которым они соединяются для замещения. Предпочтительными отщепляемыми группами являются трифлат, мезилат, тозилат,имидат, хлорид, бромид и иодид. Термин "карбоксильная защитная группа"(Р), используемый в настоящем описании, обозначает одно из эфирных производных группы карбоновой кислоты, которое обычно используется для блокирования либо защиты группы карбоновой кислоты, в то время как реакции осуществляются с другими функциональными группами данного соединения. Типы используемой карбоксильной защитной группы не являются критическими до тех пор, пока дериватизованная карбоновая кислота является устойчивой при условиях последующих реакций и может удаляться в соответствующей точке без нарушения целостности оставшейся части молекулы. В работе Т.В. Грина (T.W. Greene) и П. Вутца (P. Wuts), Protective Groups in OrganicSynthesis, издательство "John Wiley and Sons",Нью-Йорк, Нью-Йорк, 1991, глава 5, приведен перечень традиционно используемых защитных групп. См. также Э. Хезлем (Е. Haslam), Protective Groups in Organic Chemistry, издатель Дж.Г.В. Макоми (J.G.W. McOmie), Plenum Press,Нью-Йорк, Нью-Йорк, 1973. Родственным термином является термин "защищенная карбоксильная группа", который обозначает карбоксильную группу, защищенную карбоксильной защитной группой. Термин "гидроксильная защитная группа"(Р), используемый в настоящем описании, обозначает одно из простых либо сложных эфирных производных гидроксильной группы, которое обычно используется для блокирования либо защиты гидроксильной группы, в то время как реакции осуществляются с другими функциональными группами данного соединения. Типы используемой гидроксильной защитной группы не являются критическими до тех пор,пока дериватизованная гидроксильная группа является устойчивой при условиях последующих реакций и может удаляться в соответствующей точке без нарушения целостности оставшейся части молекулы. В работе Т.В. Грина и П. Вутца, Protective Groups in Organic Synthesis, издательство "John Wiley and Sons", Нью 001450 12 Йорк, Нью-Йорк, 1991, приведен перечень традиционно используемых защитных групп. Предпочтительными гидроксильными защитными группами являются трет-бутилдифенилсилилокси (TBDPS), трет-бутилдиметилсилилокси (TBDMS), трифенилметил (тритил), метокситритил либо алкиловый или ариловый эфир. Родственным термином является термин "защищенная гидроксильная группа", который обозначает гидроксильную группу, защищенную гидроксильной защитной группой. Термин "аминозащитная группа" (Р), используемый в настоящем описании, обозначает заместителей аминогруппы, которые, обычно,используются для блокирования либо защиты функциональной аминогруппы, в то время как реакции осуществляются с другими функциональными группами данного соединения. Типы используемой аминозащитной группы не являются критическими до тех пор, пока дериватизованная аминогруппа является устойчивой при условиях последующих реакций и может удаляться в соответствующей точке без нарушения целостности оставшейся части молекулы. В работе Т.В. Грина и П. Вутца Protective Groups inOrganic Synthesis, глава 7, приведен перечень традиционно используемых защитных групп. См. также Дж.В. Бартон (J.W. Barton), ProtectiveGroups in Organic Chemistry, глава 2. Предпочтительными аминозащитными группами являются t-бутоксикарбонил, фталид, циклический алкил и бензилоксикарбонил. Родственным термином является термин "защищенная аминогруппа", который обозначает аминогруппу, защищенную защитной аминогруппой, как указано. Термин "-NH защитные группы", используемый в настоящем описании, обозначает подкласс защитных аминогрупп, которые обычно используются для блокирования либо защиты функциональной группы -NH, в то время как реакции осуществляются с другими функциональными группами данного соединения. Типы используемой защитной группы не являются критическими до тех пор, пока дериватизованная аминогруппа является устойчивой при условиях последующих реакций и может удаляться в соответствующей точке без нарушения целостности оставшейся части молекулы. В работе Т.В. Грина П. Вутца, Protective Groups in Organic Synthesis, глава 7, с. 362-385 приведен перечень традиционно используемых защитных групп. Предпочтительными -NH защитными группами являются карбамат, амид, алкил- либо арилсульфонамид. Родственный термин "защищенная -NH" обозначает группу, замещенную-NH защитной группой, как определено ранее. Термин "фармацевтически эффективное количество", используемый в настоящем описании, обозначает количество соединения, соответствующего настоящему изобретению, способное ингибировать активность РКС у млеко 13 питающих. Конкретная доза упомянутого соединения, введенная в соответствии с настоящим изобретением, будет, конечно, определяться особыми обстоятельствами, связанными с настоящим случаем, в том числе вводимым соединением, путем введения, конкретным состоянием, которое подвергается лечению и т.п. соображениями. Упомянутые соединения могут вводиться различными путями, в том числе перорально, ректально, чрескожно, подкожно,локально, интравенозно, внутримышечно либо интраназально. При всех показаниях типичная дневная доза будет включать приблизительно от 0,01 до приблизительно 20 мг/кг активного соединения, соответствующего настоящему изобретению. Предпочтительная дневная доза будет составлять приблизительно от 0,05 до приблизительно 10 мг/кг в идеальном случае, приблизительно от 0,1 до приблизительно 5 мг/кг. Однако в случае местного введения, типичная доза составляет приблизительно от 1 до приблизительно 500 мкг соединения на см 2 пораженной ткани. В предпочтительном варианте примененное количество соединения будет составлять приблизительно от 30 до приблизительно 300 мкг/см 2, в более предпочтительном варианте приблизительно от 50 до приблизительно 200 мкг/см 2 и в наиболее предпочтительном варианте приблизительно от 60 до приблизительно 100 мкг/см 2. Термин "лечение", используемый в настоящем описании, обозначает оказание помощи и наблюдение за пациентом с целью преодоления заболевания, состояния либо расстройства и включает введение соединения, соответствующего настоящему изобретению, для предотвращения появления симптомов либо осложнений, облегчения симптомов либо осложнений, либо ликвидации заболевания, состояния или расстройства. Термин "изоферментизбирательный" обозначает предпочтительное угнетение одного изофермента протеинкиназы С (либо подгруппы), по сравнению с другими изоферментами. Например, соединение может избирательно угнетать изоферменты бета-1 или бета-2 по сравнению с изоферментами протеинкиназы С альфа, гамма, дельта, эпсилон, дзета и эта. В общем, изоферментизбирательные соединения демонстрируют, как минимум, восьмикратное различие (в предпочтительном варианте, десятикратное различие) в дозировке, необходимой для угнетения одного подтипа, по сравнению с дозировкой, необходимой для подобного же угнетения другого подтипа (например, угнетение изофермента РКС бета-1 или бета-2 по сравнению с изоферментом протеинкиназы С альфа), судя по результатам измерений, проведенных в процессе анализа РКС. Упомянутые соединения демонстрируют это различие в диапазоне угнетения, и величина различия выражается в виде IС 50, т.е. 50% угнетения. Таким об 001450 14 разом, например, бета-1 и бета-2 изоферментизбирательное соединение угнетает изоферменты бета-1 и бета-2 протеинкиназы С в гораздо более низких концентрациях с более низкой токсичностью вследствие минимального характерного для него угнетения других изоферментов РКС. Соединения формулы I, имеющие основную составляющую, например, NR3R4, могут также быть в форме их фармацевтически приемлемых солей, полученных добавлением кислот. К кислотам, традиционно используемым для получения таких солей, относятся неорганические кислоты,например,хлористоводородная, бромисто-водородная, иодистоводородная, серная и фосфорная кислота, а также неорганические кислоты, например, паратолуолсульфоновая, метансульфоновая, щавелевая, парабромфенилсульфоновая, угольная, янтарная, лимонная, бензойная, уксусная кислота,а также родственные неорганические и органические кислоты. Таким образом, к числу упомянутых фармацевтически приемлемых солей относятся сульфат, пиросульфат, бисульфат,сульфит, бисульфит, фосфат, моногидрогенфосфат, дигидрогенфосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутират, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, себацат, фумарат, малеат, 2-бутин-1,4-диоат, 3-гексин-2,5-диоат, бензоат, хлорбензоат, гидроксибензоат, метоксибензоат, фталат, ксилолсульфонат, фенилацетат,фенилпропионат, фенилбутират, цитрат, лактат,гиппурат, -гидроксибутират, гликолят, малеат,тартрат, метансульфонат, пропансульфонат,нафталин-1-сульфонат, нафталин-2-сульфонат,манделат и т.п. соли. Наряду с фармацевтически приемлемыми солями, могут быть использованы другие соли в соответствии с настоящим изобретением. Они могут использоваться в качестве промежуточных веществ при очистке соединений, при получении других солей либо при идентифицировании и определении характеристик соединений либо промежуточных веществ. Фармацевтически приемлемые соли соединений формулы I могут существовать также в виде различных сольватов, например, в воде,метаноле, этаноле, диметилформамиде, этилацетате и т.п. Могут быть получены также смеси таких сольватов. Источником таких сольватов может быть растворитель кристаллизации,являющийся либо обязательным компонентом для растворителя, используемого при приготовлении препарата либо в процессе кристаллизации, либо же добавочным для такого растворителя. Подобные сольваты находятся в пределах объема настоящего изобретения. Могут существовать различные стереоизомерные формы соединений формулы I; например, в состав Т либо W может входить хи 15 ральный атом углерода в замещенной алкиленовой составляющей. Соединения формулы I получают, как правило, в виде рацематов, и они обычно могут использоваться в том же виде,однако, в случае необходимости, с помощью традиционных способов могут быть выделены либо синтезированы отдельные энантиомеры. Подобные рацематы и отдельные энантиомеры,а также их смеси образуют часть настоящего изобретения. Настоящее изобретение охватывает также фармацевтически приемлемые пролекарства соединений формулы I. Пролекарство представляет собой лекарство, которое подверглось химической модификации и может быть биологически пассивным (на участке действия фермента), но которое может быть подвергнуто разложению либо модификации с помощью одного либо нескольких ферментных или других процессов in vivo с получением родительской биоактивной формы. Фармакокинетический профиль упомянутого пролекарства в предпочтительном варианте отличается от родительского,что обеспечивает облегченное поглощение упомянутого пролекарства эпителием слизистой,лучшее образование соли либо повышенную растворимость и/или повышенную системную устойчивость (например, увеличение периода полувыведения из плазмы). Имеется описание традиционных способов отбора и получения подходящих производных пролекарств (например, Г. Бундгард (Н. Bundgaard), Design of Prodrugs, (1985). Подобные химические модификации включают, как правило, следующее: 1) производные эфиров либо амидов, которые могут расщепляться эстеразами либо липазами; 2) пептиды, которые могут распознаваться специфическими либо неспецифическими протеазами; либо 3) производные, которые накапливаются в месте действия посредством отбора на мембране пролекарственной формы; либо модифицированной пролекарственной формы или любого сочетания пп.1-3, см. выше. Синтез определенных бис-индол-Nмалеимидных производных описан у Дэвиса и других, патент США 5057614, раскрытие сущности которого включено в настоящее описание ссылкой. В целом, соединения, соответствующие настоящему изобретению, могут быть получены следующим образом: Галогеновой группой, в соответствии со схемой 1, является в предпочтительном варианте фтор, хлор, бром или йод. Соединением 1 в предпочтительном варианте является 2,3-дихлорN-метилмалеимид. Реакция между соединением 1 и индолом, соединением 2, известна под традиционным названием реакции Гриньяра. Упомянутая реакция протекает в инертном органическом растворителе, например, толуоле, при температуре реакционной среды в пределах от комнатной до температуры перегонки. Наиболее существенный момент заключается в том,что реакция, изображенная на схеме 1, зависит от состояния растворителя. При осуществлении в системе растворителей толуол:тетрагидрофуран:эфир, упомянутая реакция обеспечивает выход соединения 3 в количестве, превышающем 80% и с чистотой, превосходящей 95%. Упомянутый продукт осаждают из реакционной смеси с помощью хлорида аммония, NH4Cl. Образовавшееся промежуточное вещество, соединение 3, может быть выделено стандартными методами. После этого бис-3,4(3'-индолил)-1N-метилпиррол-2,5-дион, соединение 3, посредством щелочного гидролиза может быть превращено в соответствующий ангидрид формулы 4 с помощью методов, известных в данной отрасли техники и описанных Бреннером (Brenner) и другими, Tetrahedron 44:2887-2892 (1988). В предпочтительном варианте соединение 3 реагирует с 5N КОН в этаноле при температуре в пределах от 25 С до температуры перегонки до получения соединения формулы 4 17 Соединения формулы 3, как правило, являются более стойкими, чем соединения формулы 4. Таким образом, предпочтение отдается тому, чтобы соединения 3 реагировали в соответствии со схемой 2 для образования соединений формулы 1. Специалисту в данной области техники, однако, понятно, что соединения формулы 4 могут реагировать также в соответствии со схемой 2. Схема 2 Т, J и W являются теми же самыми, что и определявшиеся ранее. L представляет собой отщепляемую группу, например, хлор, бром,иод, мезил, тозил и т.п. L может также быть гидроксильной группой либо другим предшественником, который может легко превращаться в отщепляемую группу с помощью способов, известных в данной области техники. Например,гидроксильная группа может легко превращаться в сульфоэфир, например, мезил, посредством реагирования упомянутой гидроксильной группы с метансульфонилхлоридом до образования мезилатной отщепляемой группы. Упомянутая реакция, представленная схемой 2, осуществляется любым из известных способом получения N-замещенных индолов. Эта реакция, как правило, включает, приблизительно, эквимолярные количества двух реактивов, хотя могут быть использованы и другие соотношения, особенно такие, в которых алкилирующий реактив используется в избыточном количестве. Упомянутая реакция наилучшим образом осуществляется в полярном апротонном растворителе с использованием соли щелочного металла либо в других подобных условиях алкилирования, которые известны в данной области техники. В случае если отщепляемой группой является бром либо хлор, для ускорения протекания реакции может добавляться каталитическое количество иодидной соли, например, иодида калия. Условия реакции включают следующее: гексаметилдисилазид калия в диметилформамиде либо тетрагидрофуране,гидрид натрия в диметилформамиде. В предпочтительном варианте упомянутая реакция осуществляется при медленном обратном добавлении карбоната цезия в ацетонитриле, диметилформамиде (DMF) либо тетрагидрофуране (THF). Температура реакционной смеси в предпочтительном варианте колеблется приблизительно от комнатной температуры до приблизительно температуры перегонки. Специалисту в данной области техники понятно, что реакция, описанная на схеме 2,может осуществляться с соединениями L-T' и L 001450W, где Т' и W представляют собой защищенную карбоксильную группу, защищенную гидроксильную группу либо защищенную аминогруппу. После алкилирования по схеме 2, Т' иW' могут превращаться в составляющие, способные к взаимодействию с образованием J. Взаимодействие Т' и W' с образованием различных эфирных либо тиоэфирных производных известно в данной области техники и описано,например, Ито (Ito) и другими, Chem. Pharm.Org. Chem. 50:1830 (1985). Специалисту в данной области техники понятно также, что соединение 3 может быть превращено в соединения формулы I посредством двухэтапного синтеза, как описано на схеме 3. Схема 3 Т, J, W, V и L являются теми же самыми,что и определявшиеся ранее. L2 представляет собой защищенную гидроксильную либо иную группу, которая может быть легко превращена в отщепляемую группу с помощью способов, известных в данной области техники. Упомянутым взаимодействием между соединением 3 или 4 и соединением 6 является алкилирование, как обсуждалось ранее. Моноалкилированное промежуточное вещество 7 деблокируется, и L2 превращается в отщепляемую группу. Например, если гидроксильная группа защищается tбутилдиметилсилилом (TBDMS), последний избирательно удаляется с помощью кислотного метанола. Образовавшаяся свободная гидроксильная группа после этого превращается в отщепляемую группу, например, алкилгалогенид, в предпочтительном варианте, алкилиодид либо бромид (CBr4 в трифенилфосфине) либо сульфонат (мезила хлорид в триэтиламине). После этого получают макролид посредством алкилирования при медленном обратном добавлении к раствору основания, например, гексаметилдисилазида калия либо гидрида натрия, но, в предпочтительном варианте, Cs2CO3, в полярном апротонном растворителе, например, ацетонитриле, DMF, THF при температуре в пределах от температуры окружающей среды до температуры перегонки. Выход соединений формулы I может быть значительно повышен при условии осуществления алкилирования при медленном обратном добавлении к Сs2 СО 3 в полярном апротонном 19 растворителе. Медленное обратное добавление включает объединение смеси соединения и алкилирующего агента (схема 2) либо упомянутого соединения (схема 3) с упомянутым основанием со скоростью в пределах приблизительно от 0,1 мл/ч приблизительно до 2,0 мл/ч. Концентрация каждого реактива в упомянутой смеси составляет приблизительно от 1,5 моль приблизительно до 0,001 моль. При осуществлении упомянутого с использованием моноалкилированного соединения (схема 3) упомянутая концентрация составляет приблизительно от 3 моль приблизительно до 0,001 моль. Следствием медленного добавления является концентрация реактивов в реакционном сосуде приблизительно от 0,01 моль приблизительно до 1,5 моль. Специалисту в данной области техники понятно, что при более высокой скорости добавления в упомянутой реакции может быть использована более низкая концентрация реактивов. Подобным же образом при более медленной скорости добавления в упомянутой реакции может быть использована более высокая концентрация реактивов. В предпочтительном варианте упомянутое соединение добавляется со скоростью приблизительно 0,14 мл/ч при 0,37 мольной концентрации упомянутого соединения и алкилирующего агента(с. 28). В предпочтительном варианте Сs2 СО 3 добавляется в избыточном количестве, в наиболее предпочтительном варианте в соотношенииCs2CO3 к алкилирующему агенту 4:1. Предпочтительными полярными апротонными растворителями являются ацетонитрил,диметилформамид (DMF), ацетон, диметилсульфоксид (DMSO), диоксан, диэтиленгликольметиловый эфир (диглим), тетрагидрофуран (THF) либо другие полярные апротонные растворители, в которых растворяются упомянутые реактивы. Реакция осуществляется при температурах в пределах приблизительно от 0 С до температуры перегонки. Специалисту в данной области техники понятно, что соотношение смеси упомянутого соединения и алкилирующего агента не является критическим. Предпочтение, однако, отдается тому, чтобы упомянутые реактивы смешивались во взаимном соотношении, составляющем от 0,5 до 3 эквивалентов. В наиболее предпочтительном варианте упомянутые реактивы смешиваются в соотношении 1:1.II В случае если V - N-СН 3, соединение превращается в соответствующий ангидрид (V O) посредством щелочного гидролиза. Щелочной гидролиз включает реагирование упомянутого соединения с основанием (например, гидроксидом натрия либо гидроксидом калия) в C1C4 спирте (в предпочтительном варианте этаноле), DMSO/воде, диоксане/воде либо ацетонитриле/воде при температуре в пределах приблизительно от 25 С до в предпочтительном варианте приблизительно температуры перегонки. Концентрация упомянутых реактивов не является критической. 20 Упомянутый ангидрид (V - O) превращается в малеимид формулы I посредством аммонолиза. Аммонолиз включает реагирование упомянутого ангидрида с избытком гексаметилдисилазана либо аммониевой соли (ацетатом,бромидом либо хлоридом аммония) и C1C4 спиртом (в предпочтительном варианте метанолом) в полярном апротонном растворителе,например DMF, при комнатной температуре. В предпочтительном варианте гексаметилдисилазан либо аммониевая соль реагируют при соотношении, превышающем приблизительно 5:1 эквивалентов ангидрида. На схеме 4 показано получение промежуточного вещества, пригодного в соответствии с настоящим изобретением для получения соединений формулы III, где X - О. Схема 4 Представленные далее примеры и препараты предназначены для дополнительной иллюстрации настоящего изобретения, а не для его ограничения. Пример 1. Синтез бисфторированных шестичленных мостиковых бисиндолилмалеимидов. Соединение формулы VII получают следующим образом: Соединение 8 а превращают в соединение формулы VII посредством осуществления этапов, представленных на схеме 4. Соединение формулы VIII Данное соединение, где R' - Н либо F, превращают в соединение формулы IX посредством осуществления этапов, представленных на схеме 4. Пример 2. Синтез С 2-монофторированных семичленных мостиковых бисиндолилмалеимидов. Соединение формулы X Данное соединение превращают в соединение формулы X посредством осуществления этапов, представленных на схеме 4. Пример 3. Синтез С 6-монофторированных семичленных мостиковых бисиндолилмалеимидов. Соединение формулы XI Данное соединение превращают в соединение формулы VIII посредством осуществления этапов, представленных на схеме 4. Соединение формулы IX 24 Пример 6. Синтез С 4-производных с боковой цепью фторированных шестичленных мостиковых бисиндолилмалеимидов. Соединение формулы XIV Данное соединение превращают в соединение формулы XI посредством осуществления этапов, представленных на схеме 4. Пример 4. Синтез С 6-дифторированных семичленных мостиковых бисиндолилмалеимидов. Соединение формулы XII Данное соединение превращают в соединение формулы XII посредством осуществления этапов, представленных на схеме 4. Пример 5. Синтез фторированных шестичленных мостиковых тиобисиндолилмалеимидов. Соединение формулы XIII Данное соединение превращают в соединение формулы XIV посредством осуществления этапов, представленных на схеме 4. Пример 7. Синтез фторалкенилированных шестичленных мостиковых бисиндолилмалеимидов. Соединение формулы XV Данное соединение превращают в соединение формулы XV посредством осуществления этапов, представленных на схеме 4. Пример 8. Синтез фторалкенилированных семичленных мостиковых бисиндолилмалеимидов. Соединение формулы XVI Данное соединение превращают в соединение формулы XIII посредством осуществления этапов, представленных на схеме 4. Данное соединение превращают в соединение формулы XVI посредством осуществления этапов, представленных на схеме 4. Данное соединение превращают в соединение формулы XVII посредством осуществления этапов, представленных на схеме 4. Соединение формулы XVIII Пример 10. Синтез С 3-производных фторированных шестичленных мостиковых бисиндолилмалеимидов. Данное соединение превращают в соединение формулы XVIII посредством осуществления этапов, представленных на схеме 4. Пример 9. Синтез С 2-метилированных фторированных шестичленных мостиковых бисиндолилмалеимидов. Соединение формулы XIX Пример 11. Синтез С 3-производных фторированных шестичленных мостиковых бисиндолилмалеимидов. Пример 12. Синтез С 3-производных фторированных шестичленных мостиковых бисиндолилмалеимидов. В следующих примерах и препаратах, температура плавления, спектры ядерного магнитного резонанса, масс-спектры, жидкостная хроматография на силикагеле, N,N-диметилформамид, палладий на древесном угле, тетрагидрофуран и этилацетат сокращенно обозначаются следующим образом: М. Pt., NMR, MS,HPLC, DMF, Pd/C, THF и EtOAc, соответственно. Термины "NMR" и "MS" обозначают, что спектр соответствует необходимой структуре. Пример 13. Получение бисиндолилмалеимида следующей формулы: Этил(3S,4R)-2-фтор-3-гидрокси-4,5-Оизопропилиденпентаноат. В круглодонную колбу, в которой перемешивали 50 мл безводного THF и 6,1 мл (0,03 моль) гексаметилдисилазана (HMDS) при 0 С,добавляли 12 мл (0,03 моль) 2,5 М раствора бутиллития в гексане. Полученную смесь выдерживали до возврата ее температуры до уровня 28 комнатной и перемешивали в течение 10 мин. После этого упомянутую смесь охлаждали до-85 С. К указанной смеси с максимальной скоростью прикапывали смесь 1,80 г (0,010 моль) гексаметилфосфортриамида (НМРА) и 1,00 мл(0,010 моль) этилфторацетата, не позволяя температуре подниматься выше -85 С. Через 5 мин дополнительно добавляли 880 мг (0,00677 моль) 2,3-О-изопропилиден D-глицеральдегида (8). Упомянутую смесь перемешивали в течение 10 мин, после чего течение реакции прерывали при-85 С добавлением 5 мл охлажденного насыщенного раствора хлорида аммония. После нагревания до комнатной температуры упомянутую смесь разбавляли 80 мл СН 2 Сl2 и промывали водой (60 мл 3). Слой СН 2 Сl2 отделяли и сушили над Na2SO4. После выпаривания летучих веществ in vacuо образовавшийся остаток вносили в короткую колонку с силикагелем. Упомянутую колонку промывали СН 2 Сl2, затем 30% СН 3 СN/ССl4 (либо этилацетатом) с получением неочищенного продукта. Упомянутый неочищенный продукт хроматографировали на колонке с силикагелем,используя в качестве элюента 15% (в объемном отношении) СН 3 СN/ССl4 с получением двух фракций с частичным разделением двух диастереомеров: фракция 1, 177 мг (9 а, 34%, 9b, 66%, по данным 1HNMR) фракция 2, 377 мг (9 а, 62%, 9b, 38%). Общий выход 34,7%, где 9 а - 52,7%, 9b - 47,3%. Этил (3S,4R) 3-аллилокси-2-фтор-4,5-Оизопропилиденпентаноат. Спирт 9, 327 мг (1,39 ммоль, представляющий собой смесь 9 а, 62% и 9b, 38%), выпаривали с толуолом и 327 мг (1,39 ммоль) растворяли в 6 мл циклогексана. В атмосфере N2 с перемешиванием добавляли аллилтрихлорацетимидат (423 мкл, 561 мг, 2,78 ммоль), после чего в течение 20 мин 5 мкл порциями добавляли трифторметансульфокислоту (20 мкл). Сразу же начал образовываться маслянистый осадок темно-коричневого цвета. Упомянутую реакционную смесь перемешивали при комнатной температуре в течение 60 ч. Образовавшийся осадок белого цвета фильтровали и дважды промывали циклогексаном. Образовавшийся фильтрат выпаривали до получения маслянистой жидкости. В результате хроматографирования упомянутой жидкости на силикагелевой колонке с использованием 10% (в объемном отношении) этилацетата-гексана в качестве элюента, было получено две фракции. Первая из упомянутых фракций содержала, главным обра 29 зом, 10 а, вторая упомянутая фракция включала 10b. Упомянутые фракции очищали на силикагелевой колонке, используя в качестве элюента 25% (в объемном отношении) смесь гексана/СН 2 Сl2, с получением 10 а, 133 мг (34,8%) и 10b, 83 мг (21,7%). Общий выход составлял 56,5%. 30 тельно перемешивали в течение 1 ч. Летучие вещества удаляли in vacuo. Полученный остаток растворяли в метаноле и добавляли этилацетат для замещения метанола посредством совместного выпаривания. Осадок белого цвета отфильтровывали и полученный фильтрат выпаривали. Остаток хроматографировали на силикагелевой колонке с использованием этилацетата. После удаления следовых количеств неизвестных компонентов было получено 13 мгN2 с перемешиванием в течение 20 мин прикапывали раствор DIBAL-H-толуола (0,68 мл, 1,29 М, 0,877 ммоль). Образовавшийся раствор дополнительно перемешивали в течение 1,5 ч при-75 С. После этого упомянутый раствор выдерживали до нагревания до -5 С и прохождение реакции приостанавливали добавлением 4 мл холодного этилацетата. После перемешивания в течение 10 мин добавляли 2 г влажного Na2SO4. Упомянутую смесь перемешивали при -5 С в течение 2 ч. Твердое вещество отфильтровывали и дважды промывали этилацетатом. Полученный фильтрат выпаривали под пониженным давлением. Полученный остаток хроматографировали на силикагелевой колонке, используя 30% (в объемном отношении) смесь этилацетата-гексана в качестве элюента с получением чистого 11 а, 32 мг (63,4%). 11b получали таким же путем из 10b. Диол (12):(0,099 ммоль), растворяли в 3 мл СН 3 ОН и 1 мл СН 2 Сl2. При температуре -78 С упомянутую смесь барботировали О 3 до тех пор, пока раствор не становился светло-синим. После этого упомянутый раствор в течение 5 мин барботировали аргоном. Добавляли каплю диметилсульфида и полученную смесь перемешивали в течение 10 мин. Добавляли 30 мг (0,794 ммоль,8 экв.) борогидрида натрия и перемешивали в течение 5 мин. Полученную реакционную смесь выдерживали до возвращения ее температуры до уровня температуры окружающей среды,после чего дополнительно перемешивали в течение 1 ч. Добавив 3 капли насыщенного водного раствора NH4Cl, упомянутую смесь дополни Упомянутый диол (12 а), 12 мг (0,050 ммоль), растворяли в 5 мл безводного этилового эфира и охлаждали до 0 С в атмосфере N2. Вначале с перемешиванием добавляли 35 мкл (0,250 ммоль, 5 экв.) Et3N, затем 20 мкл (0,25 ммоль, 5 экв.) мезилхлорида. Образовавшуюся смесь перемешивали при 0 С в течение 5 ч. Осадок отфильтровывали и промывали этиловым эфиром. Полученный фильтрат промывали водой (2 х),рассолом (2 х) и сушили над Na2SO4. После выпаривания растворителей in vacuo было получено масло 13 а желтоватого цвета, 9 мг (43,3%). Образовавшийся осадок растворяли в воде и экстрагировали этилацетатом. Этилацетатный слой дважды промывали водным растворомNаНСО 3 и сушили над Na2SO4. После выпаривания растворителя in vacuo было получено 11 мг (55,3%) 13 а. В целом, было получено 20 мг 13 а, 100% выход. Димезилат 13 а, 20 мг (0,0507 ммоль) и бисиндолмалеимид, 17,3 мг (0,0507 ммоль) смешивали, растворяли в 2,5 мл безводного DMF(высушенного на молекулярных ситах) и с помощью шприц-насоса добавляли к суспензии карбоната цезия (66 мг, 0,203 ммоль) в 3 мл безводного DMF при 50 С в атмосфере N2 в течение 40 ч. Полученную реакционную смесь дополнительно перемешивали при 50 С в течение 10 ч. Упомянутую смесь выпаривали in vacuo для удаления летучих веществ. Полученный остаток растворяли в СНСl3 и промывали насыщенным водным раствором NаНСО 3 и рассолом. Слой хлороформа отделяли и сушили надNa2SO4. После выпаривания твердый осадок красного цвета хроматографировали на силикагелевой колонке с элюированием 10% (в объемном отношении) раствором ацетона/СНСl3. Вначале было элюировано необходимое соединение 14 а, 7 мг (25,4%), затем один из исходных материалов, бисиндолилмалеимид, 13 мг (75%). Пример 14. Получение бисиндолилмалеимида следующей формулы: 32 изомер 22'а был получен из смеси 22' посредством хроматографирования на силикагелевой колонке, элюированной СНСl3. Соединения 21 а и 21b являются стереоизомерами, 21 а находится в конформации 2R; соединение 21b находится в конформации 2S. Номенклатура хирального центра изменяется,когда упомянутый эфир восстанавливается до спирта (т.е. соединение 24 а становится 2S). В круглодонную колбу, в которой находилось 40 мл безводного THF и 5,0 мл (0,0255 моль) гексаметилдисилазана, с перемешиванием добавляли 9,0 мл (0,0225 моль) 2,5 М раствора бутиллития в гексане; реакция протекала в ледяной ванне в атмосфере N2. После перемешивания при комнатной температуре в течение 10 мин упомянутую реакционную смесь охлаждали до -78 С и в течение 5 мин к ней прикапывали 3,6 г (0,020 моль) НМРА и 2,0 мл (0,020 моль) этилфторацетата. После дополнительного перемешивания в течение 5 мин быстро добавляли 760 мг (4,46 ммоль) циклогексилиденглицеральдегида (21, способ получения см. JOC,1992, 57, с. 648, и JOC, 1995, 60, с. 585-587, дистиллированного при 60 С/0,5 мм Hg). Полученную смесь дополнительно перемешивали в течение 10 мин, после чего реакцию останавливали при -78 С добавлением 5 мл холодного насыщенного раствора хлорида аммония. После нагревания до комнатной температуры упомянутую смесь разбавляли 60 мл гексана. Гексановый слой отделяли. Оставшийся слой экстрагировали 30 мл гексана. Гексановые растворы смешивали, промывали насыщенным раствором хлорида аммония (100 мл 3) и сушили надNa2SO4. После выпаривания при пониженном давлении полученный остаток хроматографировали на силикагелевой колонке с использованием 30% смеси этилацетата-гексана до получения основного продукта 22', 740 мг (47,6%), который представлял собой смесь двух изомеров: основного изомера 22'а (74%) и второстепенного изомера 22'b (26%), по данным 1HNMR. Водный смыв экстрагировали СНСl3. После выпаривания и хроматографирования из слоя хлороформа было получено 92 мг (7,5%) 21, который также является смесью двух изомеров. Чистый Упомянутый исходный материал 22', 740 мг (2,1 ммоль) растворяли в 60 мл СН 3 ОН. Добавляли 1,2 г лимонной кислоты. Упомянутую смесь перемешивали при температуре окружающей среды в течение 4 ч. Упомянутый растворитель удаляли на роторном испарителе. Остаток растворяли в этилацетате (100 мл),промывали насыщенным водным растворомNaHCO3 (100 мл 2) и водой. Слой этилацетата сушили над Na2SO4 и выпаривали с получением 22, 600 мг (100%), представляющего собой смесь изомера 22 а (69%) и 22b (31%), по данным 1HNMR. Чистый 22 а получали посредством гидролиза 22'а при тех же самых условиях со 100% выходом. Спирт 22, 655 мг (2,37 ммоль), смесь изомера 21 а (69%) и 21b (31%) растворяли в 30 мл циклогексана и добавляли 1,50 мл (9,8 ммоль) аллилтрихлорацетимидата. После этого в течение 30 мин прикалывали 100 мкл СF3SО 3 Н. Полученную реакционную смесь перемешивали при температуре окружающей среды в атмосфере N2 в течение 46 ч. По данным тонкослойного хроматографирования (TLC), в упомянутой смеси оставалось приблизительно 20% исходного материала. Дополнительно добавляли 60 мкл СF3SО 3 Н и упомянутую реакционную смесь дополнительно перемешивали в течение 24 ч. Осадок отфильтровывали и промывали циклогексаном. Полученный фильтрат выпаривали и остаток хроматографировали на силикагелевой колонке с использованием 10% смеси этилацетата-гексана в качестве элюента, получив 23 а,511 мг (68,1%) и 23b, 160 мг (21,3%). Чистый 23 а также получали из чистого 21 а, используя те же самые условия, что и указанные выше, для сравнения. 34 качестве элюента до получения 25 а, 232 мг(77,5%). Упомянутый диол 25b получали посредством той же самой вышеуказанной процедуры. Упомянутую реакцию начинали 230 мг 24b; было получено 158 мг 25b с 67,7% выходом. Упомянутый эфир 23 а, 635 мг (2,00 ммоль) дважды выпаривали с толуолом и растворяли в 10 мл безводного THF. Упомянутый эфир прикапывали к суспензии 200 мг (5,27 ммоль, 2,5 экв.) LiAlH4 в 40 мл безводного THF при -78 С в атмосфере N2 с перемешиванием. После добавления указанного образца, упомянутую реакционную смесь перемешивали в течение 20 мин. Упомянутую смесь нагревали до температуры 0 С и перемешивали при температуре 0 С в течение 20 мин. После этого добавляли 5 мл этилацетата. После перемешивания в течение 5 мин добавляли 4 г влажного сульфата натрия. Упомянутую смесь перемешивали в течение 30 мин. Твердое вещество отфильтровывали и дважды промывали этилацетатом. Фильтрат выпаривали на роторном испарителе. Полученный остаток хроматографировали на силикагелевой колонке с использованием 30% смеси этилацетата/гексана в качестве элюента. После удаления некоторого количества исходного материала 23 а, 19 мг, элюировали основной продукт, 24 а,413 мг (75,0%), затем некоторое количество 24b,30 мг (5,5%). Соединение 24b (44 мг) было получено посредством восстановления 23b (112 мг) с использованием раствора DIBAL-H в THF посредством процедуры, подобной вышеприведенной,с 45% выходом. Упомянутый спирт 24 а, 295 мг (1,08 ммоль), растворяли в 20 мл смеси растворителей СН 3 ОН/СН 2 Сl2 (1:1). Полученный раствор охлаждали до -78 С и барботировали озоном до начала появления голубого цвета. После этого упомянутый раствор барботировали аргоном для удаления избытка О 3. Добавляли несколько капель СН 3SСН 3 и полученный раствор перемешивали в течение 5 мин. После этого при-78 С добавляли 245 мг (6,48 ммоль) NaBH4. После перемешивания в течение 5 мин упомянутую реакционную смесь выдерживали до возвращения ее температуры до температуры окружающей среды и перемешивали в течение 1 ч. Летучие вещества удаляли in vacuo. Полученный остаток хроматографировали на силикагелевой колонке с использованием этилацетата в Диол 25 а, 195 мг (0,70 ммоль) растворяли в 50 мл диэтилового эфира. Добавляли триэтиламин, 583 мкл (4,2 ммоль), затем 342 мкл (4,2 ммоль) метансульфонилхлорида. Упомянутую смесь перемешивали при температуре окружающей среды в атмосфере N2 в течение 3 ч. Для растворения осадка добавляли 50 мл воды. Слой эфира отделяли и промывали водой (50 мл 2). После высушивания над безводнымNa2SO4 эфир выпаривали при пониженном давлении до получения жидкости 26 а желтоватого цвета, 308 мг (100%).N2, с помощью шприц-насоса в течение 48 ч прикапывали 10,0 мл раствора 26 а, 257 мг (0,59 моль), и бисиндолилмалеимида, 202 мг (0,59 ммоль) в DMF. После дополнительного перемешивания при температуре 50 С в течение 24 ч, упомянутую реакционную смесь разбавляли 100 мл СНСl3 и промывали рассолом (50 мл 2),затем водой (50 мл 2). Слой хлороформа сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Полученный остаток растворяли в СНСl3 и хроматографировали на силикагелевой колонке с использованием 5% смеси ацетона/СНСl3 в качестве элюента. Первым элюированным компонентом был необходимый продукт 27 а, 185 мг. Упомянутый продукт перекристаллизовывали из смеси СНСl3/СН 3 ОН с получением 130 мг (37,7%) и фильтрата. Соединение 27b было получено таким же способом, что и описанный перед тем, за исключением того, что мезилат и бисиндолилмалеимид с помощью шприц-насоса вводили в 35 течение 80 ч. Упомянутую реакцию начинали 97 мг диола 25b, получив 64 мг 27b с общим выходом, составлявшим 26,1%. Упомянутый исходный материал 27 а, 50 мг (0,099 ммоль), растворяли в 50 мл метанола. К упомянутому раствору добавляли 1 мл воды и 200 мг моногидрата р-толуолсульфокислоты(1,05 ммоль). Упомянутую реакционную смесь перемешивали при 50 С в течение 4 ч. Указанную смесь выпаривали на роторном испарителе и полученный остаток хроматографировали на силикагелевой колонке с использованием 5% смеси СН 3 ОН/СНСl3 в качестве элюента. Основной компонент 28 а перекристаллизовывали из смеси ацетона/СН 3 ОН с получением кристаллического вещества 28 а, 23 мг, и фильтрата,который содержал 18 мг 28 а (95,0%). 36 выпаривали для уменьшения объема раствора наполовину, после чего смешивали с 10 мл С 2 Н 5 ОН и 2 мл насыщенного водного раствора натрийтартрата калия. Полученную смесь перемешивали в течение 5 ч. Смесь выпаривали и растворяли 30 мл СН 2 Сl2. Слой СН 2 Сl2 отделяли, трижды промывали водой и сушили надNa2SO4. После выпаривания полученный остаток хроматографировали на силикагелевой колонке с использованием 10% раствора этилацетата в СН 2 Сl2 до получения соединения 30'а, 7 мг (14,1%). Основной компонент элюировали этилацетатом с получением необходимого продукта 30 а, 41 мг (79,3%). Соединение 30b было получено из соответствующего диола 28b (с 63,4% выходом) с помощью такой же самой процедуры, что и описанная перед тем, за исключением того, что в качестве растворителя для расщепления упомянутого диола была использована смесь Диол 28 а, 55 мг (0,109 ммоль) растворяли в 20 мл смеси ацетона-НОАс (1:1). К упомянутому раствору добавляли 100 мг раствораNaIO43 Н 2 О (0,373 ммоль) в 2,5 мл воды. Упомянутую смесь перемешивали при температуре окружающей среды в течение 2,5 ч. Упомянутую смесь выпаривали на роторном испарителе для сокращения объема упомянутой смеси наполовину с последующим разбавлением 30 мл СH2Cl2. После этого упомянутую смесь дважды промывали водой, водным раствором NаНСО 3 и снова водой. Слой СН 2 Сl2 отделяли, сушили надNa2SO4 и выпаривали до сухости, получая неочищенный альдегид 29 а. Упомянутый неочищенный альдегид 29 а растворяли в 14 мл СН 2 Сl2 и охлаждали до-78 С. Борогидрид натрия, 30 мг (0,79 ммоль),растворяли в 6 мл спирта с улучшенными технологическими свойствами и добавляли к упомянутому раствору. Полученную смесь перемешивали в атмосфере N2 в течение 40 мин. Реакцию останавливали добавлением 500 мкл холодного СН 3 СНО, после чего реакционную смесь выдерживали до возвращения ее температуры до уровня комнатной. Упомянутую смесь Упомянутый спирт 30 а, 16 мг (0,0338 ммоль), растворяли в 10 мл СH2Cl2. К полученному раствору добавляли 70 мкл (0,52 ммоль) триэтиламина и 28 мкл (0,34 ммоль) метансульфонилхлорида. Полученную реакционную смесь перемешивали при температуре окружающей среды в атмосфере N2 в течение 3 ч. Упомянутую реакционную смесь переносили на делительную воронку и трижды промывали водой. После выпаривания получили неочищенный 37 31 а, 23 мг. При TLC было получено всего одно пятно. Упомянутый неочищенный мезилат 31 а, 23 мг (0,0338 ммоль), вносили в 25 мл круглодонную колбу и добавляли 12 мл С 2 Н 5 ОН. Упомянутую колбу охлаждали посредством помещения на баню со смесью сухого льда/ацетона. В упомянутую колбу с помощью канюли вносили 1,2 г HN(CH3)2 и 3 мл воды. После этого упомянутую колбу герметически закрывали тефлоновой пробкой и обвязывали медной проволокой. Содержимое упомянутой колбы перемешивали при 100 С в течение 8 ч. После охлаждения колбы до комнатной температуры, летучие вещества удаляли на роторном испарителе. Полученный остаток растворяли в 20 мл этилацетата и промывали водным раствором NaHCO3 (20 мл 2) и водой (20 мл 2). После удаления растворителя получили 24 мг неочищенного продукта 32 а, который перекристаллизовывали из метанола с получением чистого 32 а. Упомянутый имид 32 а, 24 мг (0,048 ммоль) растворяли в смеси 3 мл С 2 Н 5 ОН и 3 мл 5N КОН. Полученную смесь перемешивали при 80 С в течение 24 ч. Этанол удаляли на роторном испарителе и упомянутую водную суспензию охлаждали до 0 С и подкисляли 5N НСl. Появлялся осадок фиолетового цвета. После перемешивания в течение 10 мин упомянутую водную смесь нейтрализовали разбавленным КОН и экстрагировали этилацетатом. Слой этилацетата дважды промывали водным раствором NaHCO3 и водой. После высушивания над К 2 СО 3 и выпаривания из упомянутого раствора получали 24 мг неочищенного 33 а. Упомянутый ангидрид 33 а, 24 мг, растворяли в 5 мл безводного DMF. К упомянутому раствору добавляли 250 мкл (1,19 ммоль) 1,1,1,3,3,3-гексаметилдисилазана, затем 25 мкл СН 3 ОН (0,62 ммоль). Полученную смесь перемешивали при температуре окружающей среды в атмосфере N2 в течение 38 ч. Летучие вещества удаляли in vacuo. Остаток растворяли в 6 мл смеси СН 3 СN-1N НСl (2:1) и перемешивали при температуре окружающей среды в течение 1 ч. Упомянутый органический растворитель удаля 001450 38 ли, водную суспензию нейтрализовали 1N КОН и экстрагировали этилацетатом. Слой этилацетата дважды промывали 0,1N КОН и водой и выпаривали до сухости. Остаток разделяли на силикагелевой колонке, элюировавшейся этилацетатом. Второй элюированной полосой являлся необходимый продукт 14 а, который перекристаллизовывали из смеси СH2Cl2-гексана с получением 5 мг продукта (общий выход из 30 а,20%). Пример 15. Альтернативная схема получения соединений 34 (а и b) из соединений 30 (а и Спирт 30 а, 50 мг (0,106 ммоль) смешивали с 20 мл C2H5OH-5N КОН (1:1). Полученную смесь перемешивали при 70 С в атмосфере N2 в течение 20 ч. После этого упомянутую смесь охлаждали до 0 С и подкисляли 5N НСl. Сразу же образовывался осадок красного цвета. Добавляли метиленхлорид, 40 мл. Органический слой отделяли, промывали водой (30 мл 4) и сушили над Na2SO4. После выпаривания остаток хроматографировали на силикагелевой колонке с использованием 5% раствора этилацетата в СН 2 Сl2 до получения 35 а, 33 мг (68%). Упомянутый ангидрид 35 а, 54 мг (0,117 ммоль) растворяли в 5 мл безводного DMF. Добавляли(1,1,1,3,3,3-гексаметилдисилазан), 500 мкл (2,36 ммоль), и метанол, 48 мкл (2,36 ммоль). Упомянутую смесь перемешивали в атмосфере N2 при температуре окружающей среды в течение 36 ч. Летучие вещества удаляли in vacuo и остаток перемешивали с 10 мл СН 3 СN и 5 мл 1N НСl в течение 1 ч. После этого упомянутый раствор концентрировали и экстрагировали с помощью СH2Cl2. Слой метиленхлорида промывали водой, рассолом, сушили над Na2SO4 и выпаривали. Остаток разде 39 ляли на силикагелевой колонке с использованием CH2Cl2-CH3CN (9:1), получая первый компонент, который был исходным материалом (20 мг, 37,0%), затем необходимый продукт 36 а, 31 мг (57,4%). Соединение 36b, 3,4 мг (39%) получали из соответствующего спирта 30b, 9 мг (0,019 ммоль) посредством той же самой процедуры,что и описанная перед тем, за исключением того, что, по сравнению с получением 36 а, было использовано гораздо большее избыточное количество HMDS (250 мкл, 1,18 ммоль) и метанола (24 мкл, 1,18 ммоль). Аминирование Соединение 36 а, 31 мг (0,068 ммоль) растворяли в 15 мл безводного THF. К упомянутому раствору в атмосфере азота добавляли 240 мкл (1,56 ммоль) триэтиламина и 84 мкл (1,02 ммоль) метансульфонилхлорида. Полученную смесь перемешивали при температуре окружающей среды в течение 3 ч. Летучие вещества удаляли in vacuo. Остаток растворяли в 30 мл СН 2 Сl2, дважды промывали 1N HCl и рассолом,сушили над Na2SO4 и выпаривали. Остаток растворяли в 6 мл дистиллированного THF и 1 мл 40% диметиламина в воде. Колбу герметически закрывали тефлоновой пробкой и перемешивали при 50 С в течение 24 ч. Упомянутую смесь охлаждали до 0 С и выпаривали для удаления летучих веществ. Остаток очищали на силикагелевой колонке с использованием 0-10% Et3N в этилацетате, получая необходимое соединение 34 а, 13,2 мг (40,2%). Соединение 34b (1,9 мг) было получено из 36b (3,4 мг) с помощью той же самой процедуры, что и для получения 34 а с 52% выходом. Пример 16. Синтез дитиокарбаматного производного. 20 мг соединения 38 (0,04 ммоль, 1 экв.) вносили в высушенную в печи 25 мл круглодонную колбу, снабженную мешалкой, мембраной и баллоном N2. С помощью канюли в упомянутую колбу вносили 10 мкл THF, 4,45 мг триэтиламина (0,044 ммоль, 1,1 экв.), затем с помощью шприца 3,8 мг дисульфида углерода(0,05 ммоль, 1,2 экв.). Упомянутый раствор красного цвета перемешивали в течение приблизительно 15 мин, после чего в него с помощью шприца добавляли 7,1 мг метилиодида(0,05 ммоль, 1,2 экв.). Упомянутую реакционную смесь перемешивали при комнатной температуре в течение ночи. Упомянутый прозрачный раствор красного цвета замутился приблизительно через два часа. 40 Данные TLC (10% МеОН в СH2Cl2) показали полное исчезновение исходного материала. Упомянутую реакционную смесь переносили в делительную воронку с EtOAc и промывали 40 мл Н 2 О, затем 40 мл рассола. Упомянутый органический слой собирали и пропускали черезMgSO4 в воронке из спеченного стекла для просушивания. Упомянутый растворитель удаляли с получением твердого вещества пурпурного цвета (соединение 39). Образец анализировали средствами инфракрасной спектроскопии/массспектрометрии (IS/MS). С помощью IS/MS пик МН+ был обнаружен при 559, общий выход составлял 23 мг. Превращение дитиокарбамата в трифторметильную группу 23 мг (0,041 ммоль, 1 экв.) дитиокарбамата 39 растворяли в безводном СН 2 Сl2 в сухой 25 мл круглодонной колбе, снабженной мешалкой,мембраной и баллоном N2. Упомянутый раствор охлаждали на ледяной бане в течение приблизительно 15 мин. К упомянутому раствору быстро добавляли 0,047 г 1,3-дибром 5,5 диметилгидантоина (0,164 ммоль, 4 экв.) в виде твердого вещества, затем с помощью шприца 0,06 г дигидрогентрифторида тетрабутиламмония (0,205 ммоль, 5 экв.). (Цвет упомянутого раствора изменился с красновато-пурпурного на оранжево-коричневый). Упомянутую реакционную смесь перемешивали при 0 С в течение 1,5 ч. После этого упомянутую смесь переносили в делительную воронку, заполненную 30 мл Н 2 О. Слой СН 2 Сl2 вновь промывали 25 мл Н 2 О, собирали и сушили над MgSO4. Упомянутый растворитель удаляли с получением масла темнокоричневого/оранжевого цвета. Упомянутый продукт очищали с помощью силикагеля, начиная с СН 2 Сl2, как подвижной фазы, с постепенным добавлением метанола. Было получено три отдельных пятна; третий образец собирали и упомянутый растворитель удаляли с получением 16,5 мг (75%) продукта 40. Пример 17. Ацилирование амина соединения А. 41 7 мг (0,0154 ммоль) соединения А растворяли в 1 мл сухого СН 2 Сl2. Полученный раствор охлаждали до 0 С в атмосфере N2 и перемешивали с добавлением 3,7 мкл (0,046 ммоль, 3 экв.) пиридина, затем 2,06 мкл (0,018 ммоль, 1,2 экв.) трифторуксусного ангидрида (компания Aldrich). Упомянутую реакционную смесь перемешивали при 0 С с 14 ч 17 мин до 16 ч 20 мин(TLC1) и продолжали до 19 ч 00 мин (TLC2). Развития реакции не наблюдалось. Добавляли следующую порцию пиридина (3,7 мкл) и трифторуксусного ангидрида (2,6 мкл). После перемешивания в течение 4 ч данные TLC продемонстрировали незначительное развитие реакции. Упомянутую реакционную смесь выдерживали до нагревания до комнатной температуры и перемешивали в течение 3 ч. И после этого данные TLC продемонстрировали незначительное развитие реакции. Добавили третью порцию пиридина (3,7 мкл) и трифторуксусного ангидрида (2,0 мл); упомянутую реакционную смесь перемешивали в течение 2 ч; данные TLC продемонстрировали улучшение превращения. Упомянутую реакцию останавливали и летучие вещества выпаривали при пониженном давлении. Остаток растворяли в СНСl3, дважды промывали насыщенным водным растворомNaHCO3 и водой, и слой СНСl3 выпаривали. Остаток хроматографировали на силикагелевой колонке с элюированием 10% раствором ацетона-СНСl3 (в объемном отношении); было получено два чистых компонента, 208.1 и 208.2. Исходный материал, оставшийся в колонке, элюировали 10% раствором СН 3 ОН-СНСl3, в состав которого входило 2% Et3N, помеченного как 208.3. 208.11 мг Rf=0,67 208.23 мг Rf=0,45 208.34 мг Rf=0 1HNMR в CDCl3 Файл 208.2: GZW.013 (Отдел 3): -N-CH3,3,10 частей на миллион. Файл 208.3: GZW.014 (Отдел 3): -N-CH3,2,40 частей на миллион. Сигнал метальной группы в CDCl3 демонстрирует существенный слабопольный химический сдвиг от 2,40 частей на миллион до 3,10 частей на миллион. Следовательно, необходимый продукт является продуктом 2 (соединение 41). Упомянутый эксперимент был повторен,как описано далее. Собранный выделенный материал 208.1,5 мг (0,011 ммоль), дважды подвергали совместному выпариванию с толуолом и растворяли в 2 мл сухого СН 2 Сl2 (над молекулярным ситом). Добавляли 40 мкл пиридина (0,497 ммоль, 45 экв.) при 0 С, затем 10 мкл (0,071 ммоль, 6,5 экв.) (СF3 СО)2 О). Упомянутую смесь перемешивали в атмосфере N2 в течение 2 ч. ДанныеTLC1 подтвердили завершение реакции. Летучие вещества выпаривали in vacuo, остаток про 001450 42 пускали через капиллярную силикагелевую колонку, используя 10% смесь ацетона-СНСl3 в качестве элюента, с получением 5 мг продукта,помеченого как 209-2. Пример 18. Получение бисиндолилмалеимида следующей формулы: Соединение 24 а, 140 мг (0,511 ммоль) дважды выпаривали с толуолом и растворяли в 5,0 мл безводного THF свежей перегонки. Раствор охлаждали до 0 С (ледяная баня) и перемешивали в атмосфере N2. К упомянутому раствору с помощью шприца добавляли 5,0 мл (5,0 ммоль) 1,0 М раствора ВН 3 THF в THF. Образовавшуюся смесь выдерживали до медленного нагревания до комнатной температуры и перемешивали в атмосфере N2 в течение 15 ч. Упомянутую смесь охлаждали на ледяной бане и добавляли 10 мл 10% NaOH, затем 10 мл 50% Н 2 О 2. Образовавшуюся мутную смесь белого цвета перемешивали при комнатной температуре в течение 5 ч. Упомянутую смесь выпаривали на роторном испарителе для удаления THF и остаток разбавляли 50 мл воды. Упомянутую смесь экстрагировали этилацетатом (40 мл 3). Слой этилацетата промывали рассолом (50 мл) и сушили над Na2SO4. После выпаривания остаток отделяли на силикагелевой колонке (1 см 12,5 см, отгонка под вакуумом), используя толуол(20 мл), 30-100% этилацетата в гексане (145 мл). Судя по результатам идентификации, четвертым компонентом был необходимый продукт 42 а, 69 мг (46%). Соединение 49 было синтезировано с помощью представленных далее этапов и той же самой общей процедуры, что и подробно обсуждавшаяся в примере 14. 44 Пример 19. Тест ингибирования протеинкиназы С in vitro. Реакционная смесь: 10 мкл Са 2 (концентрированный раствор 9,4 мМ) 55 мкл липидов (PS (фосфолипиды) 5 мкг/лунку, DG (диглицидиловый эфир) 0,6 мкг/лунку) либо HEPES (ГЭПЭС-буфер) 5 мкл соединения, либо DMSO (диметилсульфоксид) (Начальная концентрация экспериментальных соединений 5000 нМ) 10 мкл субстрата основного миелинового белка (МBР) (МBР 3 мг/мл, партия 451-026) 10 мкл АТР (АТФ) (300 мкМ АТР, 0,25 микроКюри/лунку АТ 32 Р и 10 мМ MgCl2) 10 мкл фермента (РКС 1:80 в ГЭПЭСбуфере, II 1:30 в ГЭПЭС-буфере) концентрированный раствор ГЭПЭС-буфера 100 мМ, рН 7,5 Общий объем реакционной смеси составлял 100 мкл; упомянутую реакционную смесь инкубировали в течение 10 мин при 30 С. Реакцию останавливали добавлением 100 мкл 25% холодной трихлоруксусной кислоты (ТСА). Добавляли 25 мкл раствора бычьего сывороточного альбумина (BSA) (1 мг/мл) и 200 мкл упомянутой реакционной смеси переносили на 96 луночный стекловолоконный фильтровальный планшет (компания Millipore, каталожныйMAFCNOB50). Супернатант фильтровали и трижды промывали 10% ТСА. Нижнюю часть фильтровального планшета и установленный носитель удаляли. Добавляли 100 мкл меткиMicroscint-20 с радиоактивным изотопом (компания Packard, каталожный 6013621) и пробы переносили в счетное устройство Packard. Получение липидов Липиды (компания Avanti Polar Lipids) вносили в боросиликатную стеклянную культуральную пробирку (25150 мм). Упомянутые липиды сушили в атмосфере азота до испарения хлороформа. Упомянутые липиды ресуспендировали в ГЭПЭС-буфере, в течение приблизительно 30 с подвергали обработке ультразвуком,после чего тщательно перемешивали на вихревой мешалке. Липиды до использования в упомянутом тесте хранили на тающем льду. Результаты Значения IС 50 для каждого из представленных далее соединений определяли с помощью упомянутого ранее теста in vitro. Пять концентраций каждого соединения испытывали против РКС альфа (РКС ) и РКС бета II (РКС II). Упомянутые концентрации экспериментальных соединений составляли 5000 нМ, 500 нМ, 50 нМ, 5 нМ, 1 нМ и ноль (без соединения,только DMSO). Образец без соединения использовали в этом тесте для определения 100% активности фермента РКС. С помощью упомянутых концентраций по кривым угнетения определяли значения IС 50. Соединения, упомянутые в настоящем описании, будучи ингибиторами протеинкиназы С, пригодны для лечения состояний, в патологии которых упомянутая протеинкиназа С играет определенную роль. К числу таких состояний, признанных в данной области техники,относятся: сахарный диабет и вызванные им осложнения, ишемия, воспаления, заболевания 47 центральной нервной системы, сердечнососудистые заболевания, болезнь Альцгеймера,дерматологические заболевания и рак. Было показано, что ингибиторы протеинкиназы С блокируют воспалительные реакции,например окислительный "взрыв" нейтрофилов,отрицательную негативную модуляцию антигенных маркеров хелперных Т-лимфоцитов и форболиндуцированный отек лап. Твими Б. и другие, Biochem. Biophys. Res. Commun. 171:1087-1092(Mulqueen M.J.) и другие, Agents Actions 37:8589 (1992). Соответственно соединения, соответствующие настоящему изобретению, являющиеся ингибиторами РКС, пригодны для лечения воспалительных процессов. Активность протеинкиназы С играет центральную роль в функционировании центральной нервной системы. Хуанг К.П. (Huang K.P.)Trends Neurosci. 12:425-432 (1989). Наряду с этим, было показано, что ингибиторы протеинкиназы С предупреждают повреждения, которые наблюдаются при очаговых и централизованных ишемических повреждениях головного мозга и отеке головного мозга. Хара X. (Наrа Н.) и другие, J. Cereb. Blood Flow Metab. 10:646-653Res. 594:290-294 (1992). Недавно было установлено, что протеинкиназа С вовлечена в болезнь Альцгеймера. Шимохама С. (Shimohama S.) и другие, Neurology 43:1407-1413 (1993). Соответствующим образом соединения, соответствующие настоящему изобретению, пригодны для лечения болезни Альцгеймера и ишемического повреждения головного мозга. Активность протеинкиназы С давно связывают с ростом клеток, стимуляцией опухолей и раком. Ротенберг С.А. (Rotenberg S.A.) и Вайнштейн И.Б. (Weinstein I.B.) Biochem. Mol.(Ahmad) и другие, Molecular Pharmacology 43:858-862 (1993). Известно, что ингибиторы протеинкиназы С эффективно предупреждают рост опухолей у животных. Мейер Т. (Меуеr Т.) и другие, Int. J. Cancer 43:851-856 (1989); Акинагака С. (Akinagaka S.) и другие, Cancer Res. 51:4888-4892 (1991). Соединения, соответствующие настоящему изобретению, кроме того,действуют как универсальные агенты реверсии лекарственных препаратов (MDR), что обеспечивает эффективность упомянутых соединений,в случае их совместного введения с другими химиотерапевтическими веществами. Активность протеинкиназы С также играет важную роль при сердечно-сосудистых заболеваниях. Было показано, что следствием повышенной активности протеинкиназы С в сосудистой сети является повышенное сужение кровеносных сосудов и повышенная артериальная гипертензия. Известный ингибитор протеинкиназы С предотвращает упомянутое повышение. Билдер Г.Е. (Bilder G.E.) и другие, J. Pharmacol.Exp. Ther. 252:526-530 (1990). Поскольку ингибиторы протеинкиназы С демонстрируют угнетение окислительного "взрыва" нейтрофилов,они также оказываются пригодными для лечения ишемии сердечно-сосудистой системы и улучшения сердечной деятельности после ишемии. Мид Р.Е. (Muid R.E.) и другие, FEBS Lett. 293:169-172 (1990); Соноки X. (Sonoki H.) и другие, Kokyu-To Junkan 37:669-674 (1989). Исследовали также роль протеинкиназы С в функционировании тромбоцитов. Было показано, что повышенные уровни протеинкиназы С коррелируют с усиленной реакцией на агонистов. Бастир III Э.Дж. и Лу (Lu), J. Diabetes 42: (Приложение 1) 97 А (1993). РКС вовлечена в биохимический путь при модуляции капиллярной проницаемости, обусловленной факторами активности тромбоцитов. Кобаяши (Kobayashi) и другие, Amer. Phys. Soc. H1214-H1220 (1994). Было показано, что сильнодействующие ингибиторы протеинкиназы С влияют на индуцированную агонистами агрегацию тромбоцитов. Туллек Д. и другие, J. Biol. Chem. 266:15771-15781 (1991). Ингибиторы протеинкиназы С блокируют также индуцированную агонистами пролиферацию клеток гладкой мускулатуры. Мацумото X. (Matsumoto Н.) и Сасаки И. (Sasaki Y.), Biochem.Biophys. Res. Commun. 158:105-109 (1989). Таким образом, соединения, соответствующие настоящему изобретению, пригодны для лечения сердечно-сосудистых заболеваний, атеросклероза и рестеноза. Аномальная активность протеинкиназы С связана также с кожными заболеваниями, например, псориазом. Горн Ф. (Horn F.) и другие,J. Invest. Dermatol. 88:220-222 (1987); Рейно Ф.(Raynaud F.) и Эвен-Брион Д. (Evain-Brion D.),Br. J. Dermatol. 124:542-546 (1991). Псориаз характеризуется аномальной пролиферацией кератиноцитов. Бьшо показано, что известные ингибиторы протеинкиназы С угнетают пролиферацию кератиноцитов таким образом, который напоминает их действие, как ингибиторов РКС. Хегеманн Л. (Hegemann L.) и другие, Arch.(Bollag W.B.) и другие, J. Invest. Dermatol. 100:240-246 (1993). Соответственно, упомянутые соединения, как ингибиторы РКС, пригодны для лечения псориаза. Протеинкиназа С связана с несколькими различными аспектами диабета. Чрезмерная активность протеинкиназы С связана с нарушением передачи инсулиновых сигналов и, следовательно, с невосприимчивостью к инсулину,наблюдаемой при диабете типа II. Карасик A.(Karasik А.) и другие, J. Biol. Chem. 265:1022610231 (1990); Чен К.С. (Chen K.S.) и другие,Trans. Assoc. Am. Physicians 104:206-212 (1991); Чин Дж.Е. (Chin J.E.) и другие, J. Biol. Chem. 268:6338-6347 (1993). Наряду с этим, результаты исследований продемонстрировали явно выраженное усиление активности протеинкиназыInvest. 87:31-38 (1991); Тесфамарим Б. (Tesfamariam В.) и другие, J. Clin. Invest. 87:16431648 (1991). Лекарственные формы, включающие соединения формулы I, в предпочтительном варианте, готовятся перед введением. Таким образом, еще одним вариантом осуществления настоящего изобретения является фармацевтическая лекарственная форма, включающая соединение формулы I и один или более фармацевтически приемлемых носителей, разбавителей или наполнителей. Фармацевтическая лекарственная форма,соответствующая настоящему изобретению,готовится известными способами с использованием хорошо известных и легко доступных ингредиентов. При изготовлении композиций, соответствующих настоящему изобретению, активный ингредиент будет, как правило, смешиваться с носителем либо разбавляться носителем или заключаться в носитель, который может быть в форме капсулы, саше, емкости из бумаги и т.п. В случае использования носителя в качестве разбавителя, им может быть твердый, полутвердый либо жидкий материал, который выступает в качестве носителя, наполнителя либо среды для активного ингредиента. Таким образом, композиции могут быть в форме таблеток,пилюль, порошков, лепешек, саше, каше, эликсиров, суспензий, эмульсий, растворов, сиропов,аэрозолей (в виде твердого вещества либо в жидкой среде), мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильных расфасованых порошков. К числу примеров приемлемых носителей,наполнителей и разбавителей относятся лактоза,декстроза, сахароза, сорбит, маннит, крахмалы,аравийская камедь, фосфат кальция, альгинаты,трагакант, желатина, силикат кальция, микрокристаллическая целлюлоза, поливинилпирролидон, целлюлоза, жидкий сироп, метилцеллюлоза, метил и пропилгидроксибензоаты, тальк,стеарат магния и минеральное масло. В состав лекарственных форм дополнительно могут входить смазывающие вещества, увлажняющие средства, эмульгаторы и суспендирующие вещества, консерванты, подслащивающие вещества либо ароматизаторы. Композиции, соответствующие настоящему изобретению, могут составляться таким образом, чтобы обеспечивать ускоренное, замедленное либо пролонгированное выделение активного ингредиента после введения пациенту. Упомянутые композиции, в 50 предпочтительном варианте, изготовляются в виде стандартной лекарственной формы, причем в состав каждой стандартной дозы входит приблизительно от 1 мг до приблизительно 500 мг, в более предпочтительном варианте приблизительно от 5 мг до приблизительно 300 мг активного ингредиента. Следует понимать, однако, что введенная терапевтическая доза будет определяться врачом, исходя из соответствующих обстоятельств, в том числе состояния, подлежащего излечению, выбора соединения, предназначенного для введения, и избранного пути введения. Таким образом, указанные ранее диапазоны дозировок никоим образом не предназначены для ограничения объема настоящего изобретения. Термин "стандартная лекарственная форма" обозначает физически раздельные формы, пригодные в качестве стандартных(унифицированных) доз для людей и других млекопитающих, причем в состав каждой формы входит предопределенное количество активного вещества, рассчитанное на достижение необходимого терапевтического эффекта, в сочетании с соответствующим фармацевтическим носителем. Наряду с вышеупомянутыми лекарственными формами, соединения, соответствующие настоящему изобретению, могут использоваться для местного применения. Лекарственными формами, предназначенными для местного применения, являются мази, кремы и гели. Мази, как правило, готовятся с использованием (1) маслянистой основы, т.е. основы, в состав которой входят нелетучие масла либо углеводороды,например белый вазелин либо минеральное масло, или (2) абсорбционной базы, т.е. базы, в состав которой входит безводное вещество либо вещества, которые могут поглощать воду, например безводный ланолин. Обычно после образования основы, маслянистой либо абсорбционной, упомянутый активный ингредиент (соединение) добавляется в количестве, обеспечивающем получение необходимой концентрации. Кремы представляют собой эмульсии масла в воде. Они включают масляную фазу (внутреннюю фазу), в состав которой, как правило,входят нелетучие масла, углеводороды и т.п.,например воски, вазелин, минеральное масло и т.п., и водную фазу (непрерывную фазу), в состав которой входит вода и любые водорастворимые вещества, например соли, получаемые добавлением кислот либо оснований. Обе упомянутые фазы стабилизируются с помощью эмульгатора,например,поверхностноактивного вещества, например, лаурилсульфата натрия; гидрофильных коллоидов, например аравийских коллоидных глин, вигума и т.п. При образовании эмульсии, активный ингредиент 51 Гели включают основу, выбираемую из числа маслянистых основ, воды либо эмульсионно-суспензионных основ. К упомянутой основе добавляется желирующий компонент, образующий в упомянутой основе матрицу, повышающую вязкость последней. Примерами желирующих компонентов являются гидроксипропилцеллюлоза, полимеры акриловой кислоты и т.п. Активный ингредиент (соединения),как правило, добавляют к лекарственной форме в необходимой концентрации в момент, предшествующий добавлению желирующего компонента. Количество соединения, включаемое в состав лекарственной формы, предназначенной для местного применения, не является критическим; концентрация должна быть в пределах,достаточных для обеспечения готовности к нанесению упомянутой лекарственной формы на участок пораженной ткани в таком объеме, которое обеспечит доставку необходимого количества соединения. Традиционное количество лекарственной формы для местного применения, предназначенное для нанесения на пораженную ткань,будет зависеть от величины пораженной ткани и концентрации соединения в упомянутой лекарственной форме. Как правило, упомянутая лекарственная форма будет наноситься на пораженную ткань в количестве, обеспечивающем приблизительно от 1 мкг до приблизительно 500 мкг соединения на см 2 пораженной ткани. В предпочтительном варианте нанесенное количество соединения будет колебаться в пределах приблизительно от 30 до приблизительно 300 мкг/см 2, в более предпочтительном варианте приблизительно от 50 до приблизительно 200 мкг/см 2 и в наиболее предпочтительном варианте приблизительно от 60 до приблизительно 100 мкг/см 2. Представленные далее примеры лекарственных форм имеют чисто иллюстративное предназначение и ни в коей мере не предназначены для ограничения объема настоящего изобретения. Лекарственная форма 1. Твердые желатиновые капсулы изготовляют с использованием следующих ингредиентов: Количество(мг/капсулу) Активный агент 250 Сухой крахмал 200 Стеарат магния 10 Всего 460 мг Вышеупомянутые ингредиенты смешиваются и засыпаются в твердые желатиновые капсулы в количестве 460 мг. Лекарственная форма 2. 52 Для изготовления таблеток используют указанные далее ингредиенты: Количество(мг/капсулу) Активный агент 250 Микрокристаллическая целлюлоза 400 Диоксид кремния,фумигованный 10 Стеариновая кислота 5 Итого 665 мг Упомянутые компоненты смешивают и прессуют до образования таблеток, масса каждой из которых составляет 665 мг. Лекарственная форма 3. Готовят аэрозольный раствор, в состав которого входят следующие компоненты: Количество(хлордифторметан) 70,00 Итого 100,00 Упомянутый активный ингредиент смешивают с этанолом. Полученную смесь добавляют к порции пропеллента 22, охлаждают до -30 С и переносят в устройство для наполнения. После этого необходимое количество упомянутой смеси переносится в контейнер из нержавеющей стали и разбавляется остаточным количеством пропеллента. После этого контейнер снаряжается клапанным устройством. Лекарственная форма 4. Таблетки, каждая из которых включает 60 мг активного ингредиента, изготавливают следующим образом: Количество(мг/капсулу) Активный агент 60 мг Крахмал 45 мг Микрокристаллическая целлюлоза 35 мг Поливинилпирролидон (в виде 10% раствора в воде) 4 мг Натрийкарбоксиметилированный крахмал 4,5 мг Стеарат магния 0,5 мг Тальк 1 мг Итого 150 мг Активный ингредиент, крахмал и целлюлозу пропускают через сито 45 (0,353 мм) и тщательно перемешивают. Раствор поливинилпирролидона смешивают с образовавшимися порошками, которые после этого пропускают через сито 14 (1,41 мм). Полученные подобным образом гранулы высушивают при 50 С и пропускают через сито 18 (1,00 мм). Натрий 53 карбоксиметилированный крахмал, стеарат магния и тальк, предварительно пропущенные через сито 60 (0,248 мм) добавляют к гранулам,которые после перемешивания прессуют на таблеточной машине до образования таблеток,масса каждой из которых составляет 150 мг. Лекарственная форма 5. Капсулы, каждая из которых содержит 80 мг медикамента, изготовляют следующим образом: Количество(мг/капсулу) Активный агент 80 мг Крахмал 59 мг Микрокристаллическая целлюлоза 59 мг Стеарат магния 2 мг Итого 200 мг ной кислоты, корригент и краситель разводят некоторым количеством воды и добавляют с перемешиванием. После этого добавляют воду в количестве, достаточном для получения необходимого объема. Лекарственная форма 8. Лекарственная форма для внутривенного введения может быть получена следующим образом: Количество(мг/капсулу) Активный агент 250 мг Изотонический физраствор 1000 мг Упомянутый активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают,пропускают через сито 45 и засыпают в твердые желатиновые капсулы в количестве 200 мг. Лекарственная форма 6. Суппозитории, каждый из которых содержит 225 мг активного ингредиента, могут быть изготовлены следующим образом: Количество(мг/капсулу) Активный агент 225 мг Глицериды насыщенных жирных кислот 2000 мг Итого 2225 мг Упомянутый активный ингредиент пропускают через сито 60 (0,248 мм) и суспендируют в глицеридах насыщенных жирных кислот, предварительно расплавленных с использованием минимального необходимого нагрева. После этого смесь вливают в суппозиторную форму номинальной емкостью 2 г и выдерживают до охлаждения. Лекарственная форма 7. Суспензии, 5 мл доза каждой из которых включает 50 мг медикамента, изготовляются следующим образом: Количество(мг/капсулу) Активный агент 50 мг Натрийкарбоксиметилированная целлюлоза 50 мг Сироп 1,25 мг Раствор бензойной кислоты 0,10 мг Корригент Сколько угодно Краситель Сколько угодно Дистиллированная вода до 5 мл Упомянутый медикамент пропускают через сито 45 и смешивают с натрийкарбоксиметилированной целлюлозой и сиропом до образования однородной пасты. Раствор бензой Упомянутый раствор вышеупомянутых ингредиентов вводится внутривенно со скоростью 1 мл/мин субъекту, нуждающемуся в подобном лечении.X - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;R5 и R6 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) или вместе образуют двухвалентную группу формулы -CR7R8-, где R7 и R8 независимо водород, C1-C4 алкил либо галоген(C1C4 алкил), либо R7 и R8, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6 членное кольцо; при условии, что, как минимум, один из Y,S, Т или W - галоген либо галогензамещенная группа, либо оба Т и W - метилен. 2. Соединение по п.1, где, как минимум,один из Y, S, Т или W - фтор либо фторзамещенная группа. 3. Соединение формулыW - этилен, факультативно замещенный галогеном, либо C1-C4 алкил; Х - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;R5 и R6 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) или вместе образуют двухвалентную группу формулы -CR7R8-, где R7 и R8 независимо водород, C1-C4 алкил либо галоген(C1 001450C4 алкил), либо R7 и R8, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6 членное кольцо при условии, что, как минимум, один из Y,S, Т или W - галоген либо галогензамещенная группа. 4. Соединение по п.3, где, как минимум,один из Y, S, Т или W - фтор либо фторзамещенная группа. 5. Соединение по п.3, где W - фторзамещенный этилен. 6. Соединение по п.3, где Т - фторзамещенный этилен. 7. Соединение по п.3, где Т - фторзамещенный триметилен. 8. Соединение по п.3, где R1 и Y - водород,X - кислород. 9. Соединение по п.5, где R1 и Y - водород,X - кислород,R3 и R4 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил), либо R3 и R4, взятые вместе с атомом N, с которым они связаны, образуют 5- либо 6-членное кольцо. 12. Соединение по п.9, где R1 - водород. 13. Соединение по п.9, где галогеновым заместителем J является фтор. 14. Соединение формулы где n и m - независимо 1 либо 2; Х - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;R5 и R6 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) или вместе образуют двухвалентную группу формулы -CR7R8-, где R7 и R8 - независимо водород, C1-C4 алкил либо галоген(C1C4 алкил), либо R7 и R8, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6 членное кольцо при условии, что, как минимум, один из Y,S, Т или W - галоген либо галогензамещенная группа, либо оба Т и W - метилен. 15. Фармацевтическая композиция,включающая соединение формулы где n и m - независимо 1 либо 2; Х - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;R5 и R6 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) или вместе образуют двухвалентную группу формулы -CR7R8-,где R7 и R8 - независимо водород, C1C4 алкил либо галоген(C1-C4 алкил), либо R7 и R8,взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6-членное кольцо,при условии, что, как минимум, один из Y,S, Т или W - галоген либо галогензамещенная группа, либо оба Т и W - метилен; и фармацевтически приемлемый наполнитель, носитель либо разбавитель. 16. Фармацевтическая композиция по п.15,где 17. Фармацевтическая композиция по п.16,где SR1 - водород. 18. Фармацевтическая композиция по п.17,где, как минимум, один из Y, S, Т или W - фтор либо фторзамещенная группа. 19. Фармацевтическая композиция по п.15,где Т и W - метилен, а галогеновым заместителем является фтор. 20. Способ лечения млекопитающего,имеющего заболевание либо состояние, связанное с аномальной активностью протеинкиназы С, причем упомянутый способ включает введение упомянутому млекопитающему фармацевтически эффективного количества соединения формулыR5 и R6 - независимо водород, C1-C4 алкил,галоген(C1-C4 алкил), C1-C4 алканоил, галоген(C1C4 алканоил) или вместе образуют двухвалентную группу формулы -CR7R8-, где R7 и R8 - независимо водород, C1-C4 алкил либо галоген(C1C4 алкил), либо R7 и R8, взятые вместе с атомом С, с которым они связаны, образуют 5- либо 6 членное кольцо,при условии, что, как минимум, один из Y,S, Т или W - галоген либо галогензамещенная группа, либо оба Т и W - метилен; и фармацевтически приемлемый наполнитель, носитель либо разбавитель. 21. Способ по п.20, где J22. Способ по п.21, где SX - кислород; и R1 - водород. 23. Способ по п.21, где, как минимум, один из Y, S, Т или W - фтор либо фторзамещенная группа. 24. Способ по п.20, где Т и W - метилен, а галогеновым заместителем является фтор. 25. Способ по п.20, где W - фторзамещенный этилен. 26. Способ по п.20, где Т - фторзамещенный этилен. 27. Способ по п.20, где Т - фторзамещенный триметилен. где n и m - независимо 1 либо 2; Х - кислород, сера либо связь между атомами углерода, соединенными мостиковой связью, обеспечиваемой X;
МПК / Метки
МПК: A61P 9/10, C07D 498/22, A61K 47/16
Метки: протеинкиназы, галогензамещенные, ингибиторы
Код ссылки
<a href="https://eas.patents.su/30-1450-galogenzameshhennye-ingibitory-proteinkinazy-s.html" rel="bookmark" title="База патентов Евразийского Союза">Галогензамещенные ингибиторы протеинкиназы с.</a>
Ингибиторы протеинкиназы с.

Номер патента: 598
Опубликовано: 29.12.1999
Авторы: Макдональд Джон Х., Джироусек Майкл Р., Рито Кристофер Дж., Хит Уильям Ф.
МПК: C07D 471/22, A61K 47/22
Метки: ингибиторы, протеинкиназы
Формула / Реферат:
1. Соединение формулы где W является О, -S- или NH, R1 представляет собой независимо водород, галоген, С1-С4-алкил, гидрокси, С1-С4-алкокси, галогеналкил, нитро, -NH(С1-С4-алкил), N(С1-С4-алкил)2 или -NНСО(С1-С4-алкил), R2 является водородом, СН3СО-, NН2 или гидроксигруппой, Z является -(СН2)р- или (СН2)р-O-(СН2)p-, R6 является -NH(CF3) или -N(CF3)(СН3), m независимо является 0, 1, 2 или 3, р независимо является 0, 1 или 2 или...
Ингибитор протеинкиназы с.

Номер патента: 967
Опубликовано: 28.08.2000
Авторы: Фарид Наджи А., Инджел Гари Л., Ричардсон Лори А., Джираусек Майкл Р., Фол Маргарет М., Виннероски Леонард Л.
МПК: A61K 31/40, A61P 5/48, C07D 413/14...
Метки: протеинкиназы, ингибитор
Формула / Реферат:
1. Соль формулы и ее сольваты. 2. Соль по п.1 формулы и ее сольваты. 3. Соль по п.1 или 2, которая по существу является кристаллической. 4. Соль по п.3, которая представляет собой моногидрат метансульфоната (S)-13-[(диметиламино)метил]-10,11,14,15-тетрагидро-4,9:16,21-диметено-1Н,13Н-дибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3(2Н)-диона. 5. Соль по любому одному из пп.1-4, отличающаяся тем, что имеет общее...
Фармацевтическая композиция, содержащая ингибиторы протонного насоса

Номер патента: 87
Опубликовано: 25.06.1998
Авторы: Дэйв Кошик Дж., Уилльямс Джеймс Б.
МПК: A61K 31/44
Метки: ингибиторы, композиция, протонного, содержащая, насоса, фармацевтическая
Формула / Реферат:
1. Фармацевтическая композиция для перорального введения, которая включает ингибитор протонного насоса, загуститель, подщелачивающий агент и гидрофобный масляный жидкий носитель. 2. Композиция по п.1, в которой указанный ингибитор протонного насоса представляет собой омепразол. 3. Композиция по п.1, в которой указанный загуститель представляет собой гидрогенизированное касторовое масло. 4. Композиция по п.1, в которой указанный гидрофобный...
Аминосилановые соли и силаноамиды карбоновых кислот как ингибиторы коррозии

Номер патента: 794
Опубликовано: 24.04.2000
Авторы: Крамер Андреас, Фрей Маркус, Брайг Адальберт
МПК: C07F 7/18, C09D 5/08, C08F 8/42...
Метки: соли, ингибиторы, карбоновых, аминосилановые, кислот, коррозии, силаноамиды
Формула / Реферат:
1. Композиция для покрытия, содержащая: a) органическое пленкообразующее вещество, и b) в качестве ингибитора коррозии a) по крайней мере, одну соль и/или b) по крайней мере, один амид, полученные из i)полиакриловой кислоты, сополимера акриловой кислоты и малеиновой кислоты, или карбоновой кислоты формулы I где R1 представляет водород, С1-С25алкил, С2-С25алкил, цепь которого прерывается кислородом или серой; С2-С24алкенил,...
Тгф-содержащие сульфонамидные ингибиторы аспартил-протеазы

Номер патента: 1221
Опубликовано: 25.12.2000
Автор: Тунг Роджер Д.
МПК: A61K 31/164, A61P 31/18
Метки: ингибиторы, аспартил-протеазы, тгф-содержащие, сульфонамидные
Формула / Реферат:
1. Фармацевтическая композиция, содержащая а) фармацевтически приемлемое количество ингибитора протеазы - соединения формулы I где каждый R1, независимо, выбран из группы, включающей -С(О)-, -S(O)2-, -С(O)-С(O)-, -О-С(О)-, -O-S(O)2, -NR2-S(O)2-, -NR2-С(O)- и -NR2-C(O)-С((О)-; каждый Het, независимо, выбран из группы, включающей С3-С7 карбоцикл; C6-С10 арил; фенил, конденсированный с гетероциклом; и гетероцикл; где любой член указанного Het...
Предыдущий патент: Устройство для индикации электрической неисправности в устройстве коммутации, в частности, в дифференциальном выключателе.
Следующий патент: Волокна из сильноразветвленного полиамида.
Случайный патент: Предшественник катализатора для полимеризации олефинов, способ его получения, катализатор полимеризации олефинов и способ полимеризации олефинов