Производные 4-циано-4-деформилсордарина

























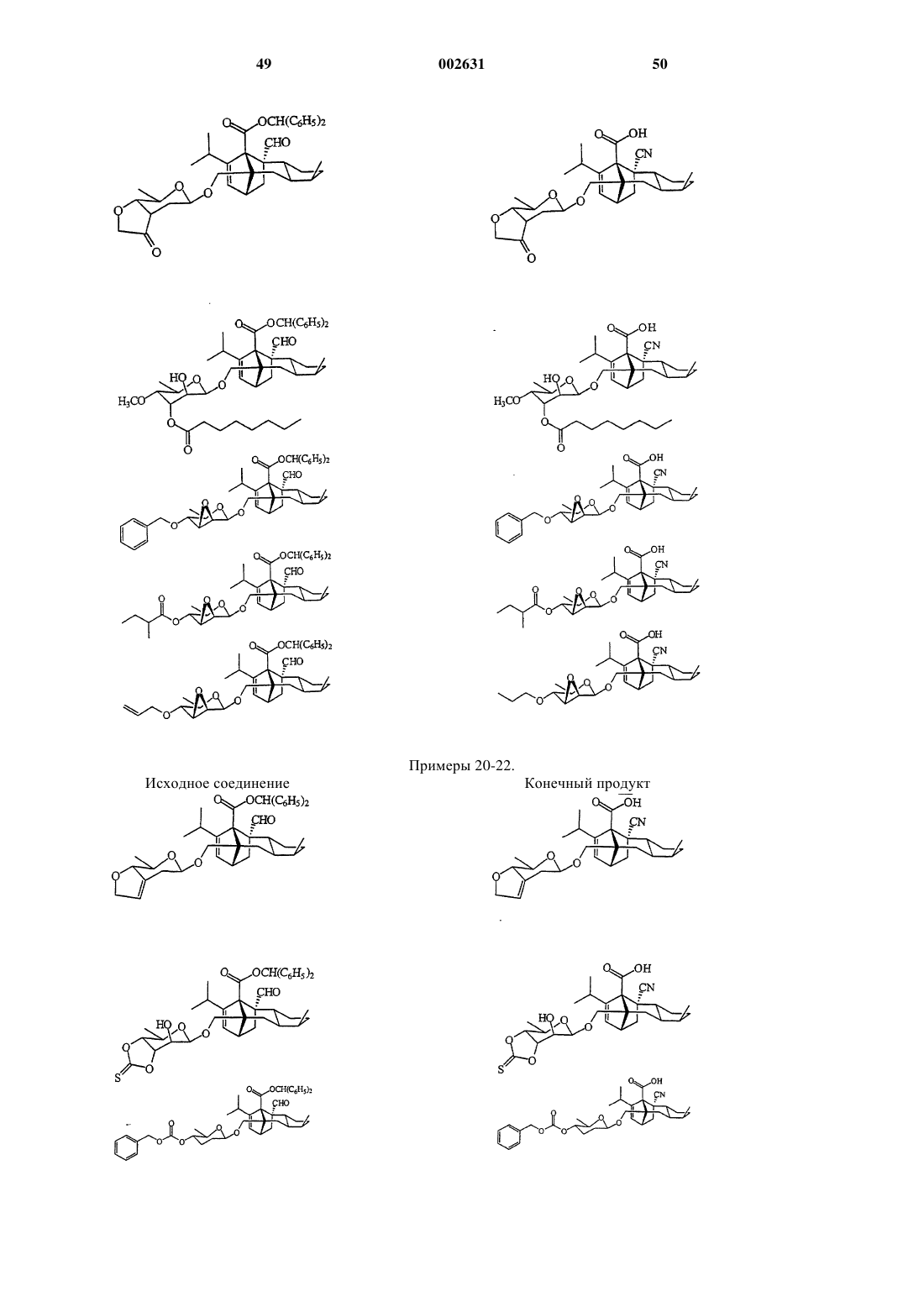

Формула / Реферат
1. Соединение, отвечающее формуле I

в которой Z представляет собой тетрагидропираногруппу, выбранную из

а также соли и сольваты (например, гидраты) или метаболически лабильные его производные, где Ra представляет собой С(O)СН3 или СН3;
R1 представляет собой водород, галоген, гидроксил, C1-4алкокси или ацилокси;
каждый из радикалов R2 и R3 независимо друг от друга представляет собой водород, C1-6алкил или С1-4алкоксиС1-4алкил, или
R2 и R3 вместе с атомом углерода, к которому они присоединены, представляют собой С=O, C=S или С3-8циклоалкил;
R4 представляет собой водород или CH2R7 (где R7 представляет собой водород, гидроксил, С1-4алкокси или группу OCOR8, в которой R8 представляет собой С1-4алкил или арил);
каждый из радикалов R5 и R6 независимо друг от друга представляет собой водород, C1-6алкил или С1-4алкоксиС1-4алкил, или
R5 и R6 вместе с атомом углерода, к которому они присоединены, представляют собой С=O, C=S или С3-8циклоалкил;
n равно нулю или 1;
каждый из Х и Y независимо друг от друга представляют собой кислород, серу или CR9R10 (где каждый из радикалов R9 и R10 независимо друг от друга представляет собой водород, C1-6алкил, С1-4алкокси или С1-4алкоксиС1-4алкил; или R9 и R10 совместно с соединенным с ними атомом углерода представляют собой С=O, C=S или С3-8циклоалкил или C=CHR11, где R11 представляет собой водород или С1-4алкил); или, когда Х и Y представляют собой кислород, а n равно нулю, -Y-CR2R3 или -X-CR2R3- соответственно могут также представлять собой -N=CR3- или -NR12-CR2R3- (где CR2 и CR3 представляют собой С=O, а R12 представляет собой С1-4алкил или ацильную группу COR13, в которой R13 представляет собой C1-6алкил), или, когда Y представляет собой кислород, а n равно нулю, Х может представлять собой группу CR11 (в котором R11 имеет указанные выше значения), которая присоединена к пирановому кольцу двойной связью;
R15 представляет собой водород, галоген, азидо, C1-6алкил, гидрокси, C1-6алкокси (необязательно замещенную 1 или 2 гидроксигруппами или их кеталями, либо 1 или 2 C1-3алкоксигруппами), арилС1-4алкокси, С3-6алкенилокси, группу OCOR18 (в которой R18 представляет собой арилС1-4алкокси или C1-10-алкильную группу, необязательно содержащую одну или две двойных связи) или C1-6алкоксикарбонилС1-4алкокси, а R16 представляет собой водород, или R15 и R16 совместно с соединенным с ними углеродным атомом могут представлять собой С=O или С=СН2;
R17 представляет собой CH2R19, где R19 представляет собой водород, гидроксил, C1-4алкокси или группу OCOR20, в которой R20 представляет собой С1-4алкил;
W представляет собой кислород, серу или СН2;
а волнистая линия в группе (а) указывает на необязательное присутствие дополнительной связи;
R1a представляет собой водород, галоген, гидроксил или С1-4алкокси;
R2a представляет собой водород, галоген, гидроксил, C1-10 алкокси, C1-10алкилтио, С1-6алкоксиС1-4алкокси, арилС1-6алкилокси, арилС3-6алкенилокси, азидо, NR5aCOR5a (где каждый из радикалов R5a независимо друг от друга представляет собой водород или C1-6алкил), OR6a (где R6a представляет собой циклическую эфирную группу, содержащую 4-8 атомов, соединенных с кислородным атомом через углеродный атом кольца, соседний с кольцевым кислородным атомом) или группу YaС(=O)-Xa-R7a, в которой Ya представляет собой кислород, серу или NH, Xa представляет собой связь, кислородный атом или фрагмент NR8a, в котором R8a представляет собой водород или C1-6алкил, а R7 представляет собой C1-10алкил, необязательно содержащий одну или две двойные связи, арил, арилС1-4алкил, арилС2-4алкенил, галоС1-6алкил или С1-6алкоксиС1-4алкил), a R3a представляет собой водород, или
R2a и R3a совместно с соединенным с ними углеродным атомом представляют собой С=O или C=NOR9a (где R9a представляет собой C1-6алкил); а
R4a представляет собой гидроксил, C1-6алкокси или OC(=O)R7a (где R7a имеет указанные выше значения).
2. Соединение по п.1, отвечающее следующей структурной формуле

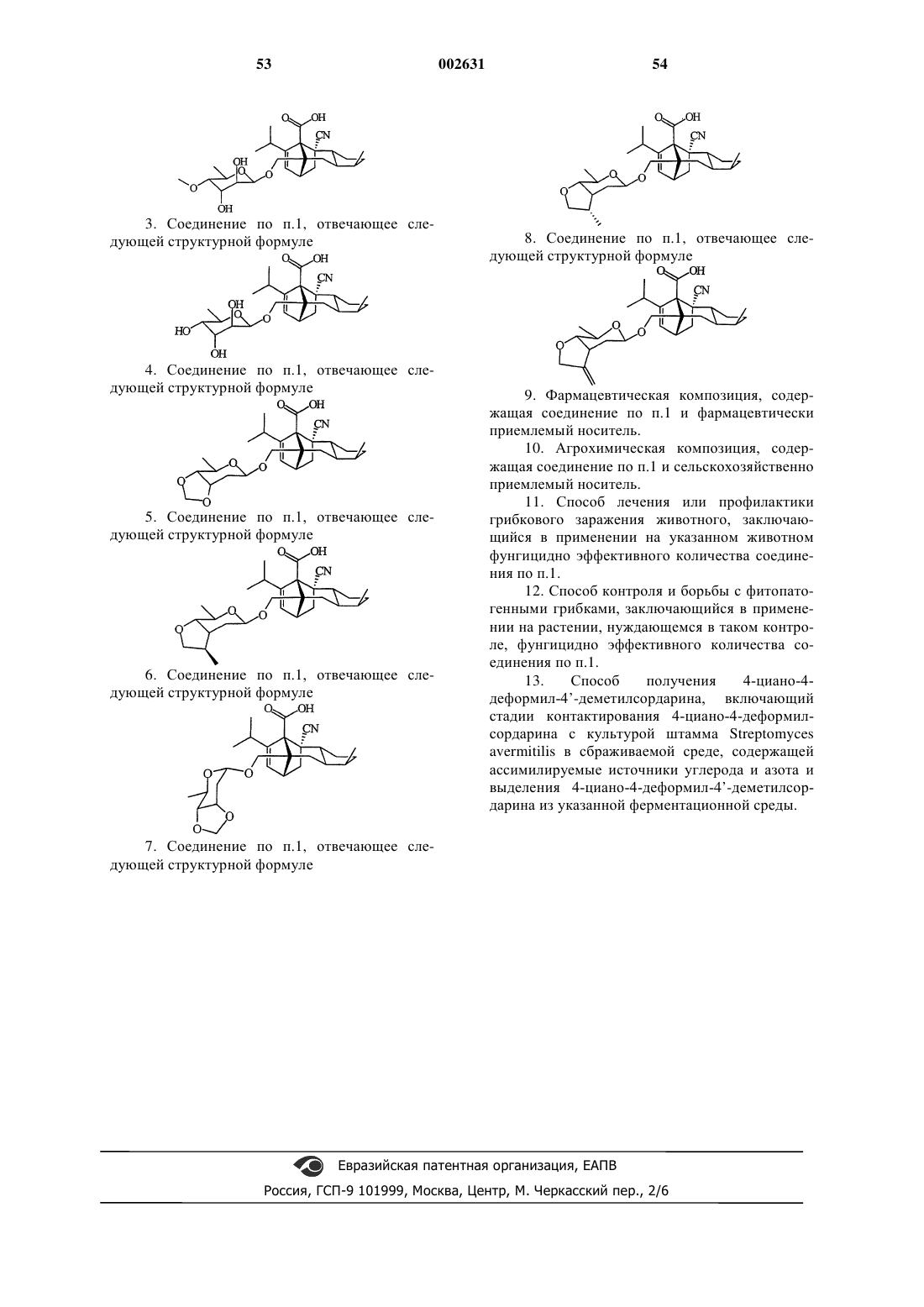
3. Соединение по п.1, отвечающее следующей структурной формуле

4. Соединение по п.1, отвечающее следующей структурной формуле

5. Соединение по п.1, отвечающее следующей структурной формуле

6. Соединение по п.1, отвечающее следующей структурной формуле

7. Соединение по п.1, отвечающее следующей структурной формуле

8. Соединение по п.1, отвечающее следующей структурной формуле

9. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически приемлемый носитель.
10. Агрохимическая композиция, содержащая соединение по п.1 и сельскохозяйственно приемлемый носитель.
11. Способ лечения или профилактики грибкового заражения животного, заключающийся в применении на указанном животном фунгицидно эффективного количества соединения по п.1.
12. Способ контроля и борьбы с фитопатогенными грибками, заключающийся в применении на растении, нуждающемся в таком контроле, фунгицидно эффективного количества соединения по п.1.
13. Способ получения 4-циано-4-деформил-4'-деметилcордарина, включающий стадии контактирования 4-циано-4-деформилсордарина с культурой штамма Streptomyces avermitilis в сбраживаемой среде, содержащей ассимилируемые источники углерода и азота и выделения 4-циано-4-деформил-4'-деметилсордарина из указанной ферментационной среды.
Текст
1 Настоящее изобретение относится к производным 4-циано-4-деформилсордарина, являющимся мощными фунгицидными агентами с широким спектром активности и повышенной устойчивостью, способам их получения, фармацевтическим и сельскохозяйственным композициям, содержащим эти соединения, а также к способам борьбы с грибковыми инфекциями людей, животных и растительных материалов с применением таких соединений. Сордарин представляет собой противогрибковый антибиотик, выделенный из плесениSordaria areneosa (см. патент Великобритании 1162027 и статью в Helvetica Chimica Acta, 1971,51:119-20). Имеются также сообщения о других соединениях со структурой сордарина, обладающих противогрибковой активностью. В японском патенте Kokai J62040292 раскрывается соединение зофимарин, выделенное из видаSCH57404 сообщается в J.Antibiotics, 1995,48:1171-1172. Полусинтетические производные сордарина раскрыты в заявках РСТ WO 96/14326 и WO 96/14327. Настоящее изобретение относится к соединению общей формулы (I) а также соли и сольваты (например, гидраты) или метаболически лабильные производные таких соединений,где Ra представляет собой С(O)СН 3 или СН 3;R1 представляет собой водород, галоген,гидроксил, C1-4 алкокси или ацилокси; Каждый из R2 и R3, независимо друг от друга, представляет собой водород, C1-6 алкил или С 1-4 алкоксиС 1-4 алкил, илиR2 и R3 совместно с атомом углерода, к которому присоединены эти радикалы, представляют собой С=O, C=S или С 3-8-циклоалкил;R4 представляет собой водород или CH2R7 2 каждый из R5 и R6, независимо друг от друга, представляет собой водород, C1-6 алкил или С 1-4 алкоксиС 1-4 алкил, илиR5 и R6 совместно с соединенным с ними атомом углерода представляют собой C=O, C=S или С 3-8 циклоалкил;n равно нулю или 1; каждый из Х и Y, независимо друг от друга, представляет собой кислород, серу или группу CR9R10 (где R9 и R10, независимо друг от друга, представляют собой водород, C1-6 алкил, С 1-4 алкокси или С 1-4 алкоксиС 1-4 алкил; или R9 и R10 совместно с соединенным с ними атомом углерода представляют собой С=O, C=S, С 3-8 циклоалкил или C=CHR11, где R11 представляет собой водород или С 1-4 алкил); а в том случае, когда Х или Y представляют собой кислород, а n равно 0, группы -Y-CR2R3 или -X-CR2R3- могут представлять собой, соответственно, -N=CR3- или-NR12-CR2R3- (где CR2 и R3 представляют собой С=O, а R12 представляет собой С 1-4 алкил или ацильную группу COR13, в которой R13 представляет собой C1-6 алкил), или в том случае когда Y представляет собой кислород, а n равно нулю, Х может представлять собой группу CR11(в которой R11 имеет указанные выше значения),которая присоединена посредством двойной связи к пирановому кольцу;R15 представляет собой водород, галоген,азидо, C1-6 алкил, гидрокси, C1-6 алкокси (необязательно замещенную 1 или 2 гидроксигруппами, или их кеталями, либо 1 или 2 C1-3 алкокси группами), арилС 1-4 алкокси, С 3-6 алкенилокси,группу OCOR18 (в которой R18 представляет собой арилС 1-4 алкокси или C1-10 алкильную группу,необязательно содержащую одну или две двойных связи) или С 1-6 алкоксикарбонилС 1-4 алкокси,a R16 представляет собой водород, либо R15 и R16 совместно с соединенным с ними атомом углерода могут представлять С=O или С=СН 2;R17 представляет собой группу CH2R19, в которой R19 представляет собой водород, гидроксил, С 1-4 алкокси или группу OCOR20, в которойW представляет собой кислород, серу или СН 2; причем ломаная линия в группе (а) указывает на необязательное присутствие дополнительной связи;R2a представляет собой водород, галоген,гидроксил, C1-10 алкокси, C1-10 алкилтио, С 1-6 алкоксиС 1-4 алкокси, арилС 1-6 алкилокси, арилС 3-6 алкенилокси, азидо, NR5aCOR5a (где каждый R5a,независимо друг от друга, представляет собой водород или C1-6 алкил), OR6a (где R6a представляет собой фрагмент простого циклического эфира, содержащий 4-8 атомов, соединенный с кислородным атомом через углеродный атом кольца, соседний с кислородным атомом кольца) или группу YaC(=O)-Xa-R7a, в которой Ya 3 представляет собой кислород, серу или NH, Xa представляет собой связь, атом кислорода или фрагмент NR8a, в котором R8a представляет собой водород или C1-6 алкил, a R7a представляет собой C1-10 алкил, необязательно содержащий одну или две двойных связи, арил, арил С 1-4 алкил, арилС 2-4 алкенил, галоC1-6 алкил, или С 1-6 алкоксиС 1-4 алкил, a R3a представляет собой водород, илиR4a представляет собой гидроксил, С 1-6 алкокси или OC(=O)R7a (где R7a имеет указанные выше значения). Одно из технических решений настоящего изобретения предусматривает соединения формулы I, в которой Другим объектом настоящего изобретения является фармацевтическая композиция, содержащая фунгицидно эффективное количество соединения формулы I и фармацевтически применимый носитель. Предусматривается также фармацевтическая композиция, которую готовят 4 объединением соединения формулы I и фармацевтически применимого носителя. Объектом изобретения является также сельскохозяйственная композиция, включающая фунгицидно эффективное количество соединения формулы I и его сельскохозяйственно применимый носитель. Предусматривается также сельскохозяйственная композиция, которую получают смешиванием соединения формулы I с сельскохозяйственно применимым носителем. Объектом изобретения является и способ лечения грибковой инфекции на животном(включая людей), заключающийся в применении на животном, нуждающемся в такой обработке, фунгицидно эффективного количества соединения формулы I. Изобретение относится также к способу борьбы с фитопатогенными грибками растений,заключающийся в применении на указанном растении фунгицидно эффективного количества соединения формулы I. В тексте заявки, если не оговорено особо,используются следующие определения. Термин "контроль" или "борьба" относится к профилактическому применению (т.е. направленному на защиту от инфекции) и лечебному применению (т.е. направленному на уничтожение очага инфекции). Термин "растения" относится к живым растениям полностью или к их частям, листве,цветам, семенам, плодам, и другим материалам,являющимся производными растений. Этот термин включает также корни растений, при применении активного ингредиента на почве. Термин "композиция", подразумевающий сельскохозяйственную или агрохимическую композицию, относится к продукту содержащему активный ингредиент(ы) и инертный ингредиент(ы), входящий в состав носителя, а также любому продукту, который непосредственно или косвенно образуется в результате соединения, комплексообразования или агрегации любых двух или более ингредиентов, или диссоциации одного или более ингредиентов, или в результате других типов реакций, или взаимодействий одного или более ингредиентов. В соответствии с этим, композиции согласно изобретению включают любую композицию, полученную смешиванием соединения настоящего изобретения с сельскохозяйственно применимым носителем. Термин "алкил", относящийся к группе,или части группы, обозначает такой алкильный фрагмент нормального или изостроения, как метил, этил, н-пропил, н-бутил, изопропил,втор-бутил, трет-бутил, н-гексил и н-октил. Термин "арил", относящийся к группе или ее части, обозначает фенил или гетероарил, каждый из которых может быть необязательно замещен одной или тремя группами, независимо выбранными из галогена, гидроксила, C1-6 алкила, C1-6 алкокси или С 1-4 алкоксикарбонила. 5 Гетероарильная группа может представлять собой 5- или 6-членное гетероароматическое кольцо, содержащее один или более гетероатомов, выбранных из азота, кислорода и серы. Подходящими примерами гетероарильных групп могут служить пиридил, фурил, тиенил и пирролил. Термины "галоген" или "гало" обозначают фтор, хлор, бром или иод. В том случае, когда R1 представляет собой ацилоксигруппу, она может представлять собой,например, группу OCOR13, в которой R13 имеет указанные выше значения. Подходящие соли соединения формулы I включают такие соли неорганических оснований, как соль щелочного металла (например,соли натрия и калия), аммониевые соли и соли органических оснований. Подходящие соли органических оснований включают соли такого амина, как триалкиламин (например, триэтиламин), диалкиламин (например, дициклогексиламин), необязательно замещенный бензиламин (например, фенилбензиламин или пбромбензиламин), этаноламин, диэтаноламин,N-метилглюкозамин, N-метилпиперидин, пиридин и замещенный пиридин (например, коллидин, лутидин, 4-диметиламинопиридин) и три(гидроксиметил)метиламин, а также соли аминокислот (например, соли лизина или аргинина). Метаболически лабильные производные соединений формулы I представляют собой соединения, которые превращаются в объекте обработки (будь это животное, растение (включая листву, цветки, плоды, семена и другие части или растительную продукцию) или почва) в соединения формулы I. Примерами таких производных могут служить традиционные, метаболически лабильные сложные эфиры, образующиеся из карбоновых кислот молекулы. Получение соединений. Соединения формулы I могут быть получены из сордарина и его производных, сордарицина, и других соединений сордаринового типа, причем все перечисленные соединения описаны в литературе. Сордарин представляет собой [1R-(1,3,4,4,7,7 а,8 а)]8 а-[(6-деокси-4-O-метил-D-альтропиранозилокси)метил]-4-формил 4,4 а,5,6,7,7 а,8,8 а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3 а(1 Н)-карбоновую кислоту, отвечающую формуле II Сордарин может быть получен культивированием Sodaria araneosa NRRL 3196 (также депонировано АТСС, как АТСС 36386), согласно методике, описанной в патенте Великобри 002631 6 тании 1162027, или в WO 96/14326. Кроме этого, сордарин может быть выделен из продуктов ферментации Rosellinia subiculata и неидентифицированного грибка АТСС 74387 по описанной ниже методике. Зофимарин может быть получен из ферментационного бульона Zofiela marina SANK 21274 (АТСС 34456) в соответствии с описанным в японском патенте Kokai 62040292. Соединение ВЕ 31405 (I, где А отвечает формуле(f), a Ra представляет собой метил) продуцируется грибком, идентифицированным, как Shering culture number SCF1082A. Исходные материалы для производных сордарина (I, где Z представляет собой (а) или(b описаны в заявке РСТ WO 96/14326; а исходные материалы для производных сордарина(I, где Z представляет собой (с описаны в заявке РСТ WO 96/14327. Сордарицин представляет собой [1R-(1,3 а,4,4 а,7 а,7,8 а)]-4-формил-8 а-(гидроксиметил)-4,4 а,5,6,7,7 а,8,8 а-октагидро-7-метил-3(1-метилэтил)-1,4-метано-s-индацен-3 а(1 Н)карбоновую кислоту, отвечающую формуле VI Сордарицин может быть получен из сардарина в результате обработки концентрированной хлористо-водородной кислотой. Как указывается в WO 96/14326, сордарицин получают также путем ферментации мутанта, производного Sordaria araneosa NRRL 3196 и биотрансформации сордарина с использованием вида Coryneform. Как отмечалось выше, были найдены два других организма, продуцирующих сордарин. Одним из грибковых штаммов, используемых для получения сордарина, является неидентифицированный стерильный грибок GB 3109,который выделяли из внутренних корневых тканей мангрового дерева, Conocarpus erectusRincon, Peninsula de Osa, Provincia de Puntarenas,Costa Rica, и идентифицировали, как MF6232 в культурной коллекции MerckCo., Inc.,Rahway, New Jersey. Эта культура депонирована 27 августа 1996 г. в постоянной коллекции при коллекции американских типовых культур,12301 Parklawn Drive, Rockville, Maryland 20852,США, в соответствии с Будапештским договором по Международному признанию депонирования микроорганизмов в целях патентования,под номером АТСС 74387. 7 Грибок выращивали в разнообразных микологических средах, при различных режимах освещения, на стерильных листьях и фильтровальной бумаге, однако, во всех случаях не удалось получить и идентифицировать репродуктивные структуры. Колонии грибка в культуре на агаре имели следующую морфологию: Колонии в агаре на отваре овсяной муки(Difco) при 23 С, 12-часовом световом периоде растут умеренно быстро, достигая за 14 дней размера в 85-90 мм, имеют сдавленную переднюю зону, ровные, со скрытым окаймлением, с выраженными радиальными полосками, содержащие в центре влажный сдавленный мицелий,становящийся шелковистым с радиально расходящимися, стелющимися пучками гиф или мицелилальными тяжами, просвечивающий до бледно-розового цвета, близкого по оттенку кPale Ochraceous Salmon (названия цветовых оттенков из Ridgway, R.1912. Color Standards andNomenclature, Washington, D.C.), Light Ochraceous Salmon, розовато-серому Avellaneous, Cinnamon-Drab, или с белым верхним воздушным мицелием, изменяющим оттенок на бледный розовато-серый, эксудаты отсутствуют, издают слабый аромат. При 37 С роста в агаре на отваре овсяной муки не наблюдается. Колонии в агаре на соке V8 (Stevens, R.B. 1981. Mycology Guidebook. University of Washington Press, Seattle, стр. 665) при 23 С, 12 часовом световом периоде растут медленно,достигая за 14 дней 37-42 мм, погружены по краям, в основном со сдавленным мицелием, со скудным хлопьевидноопушенным воздушным мицелием во внешней трети, окаймленные, просвечивающие до бледно-розового оттенка, близкого к цвету колоний в агаре на отваре овсяной муки, меняющие цвет при обратном просвечивании на бледный красновато-коричневый,близкие к Wood Brown, Fawn цвету. Колонии в агаре на отваре кукурузной муки (Difco), при 12-часовом световом периоде с температурой 25 С растут медленно, достигая за 14 дней 33-34 мм, погружены по краям, характеризуются отсутствием воздушных гиф,окаймлены, прозрачны. Мицелий состоит из высоко разветвленных, стекловидных гид с простыми перегородками. Второй грибковый штамм (GB3719), используемый для получения сордарина, представляет собой штамм Rosellina subiculata (Ascomycotina, Xylariaceae), обозначенный, какMF6239 в коллекции культур MerckCo., Inc.,Rahway, New Jersey. Эта культура депонирована 27 августа 1996 г. в постоянной коллекции приAmerican Type Culture Collection, 12301 Parklawn Drive, Rockville, Maryland 20852, США в соответствии с Будапештским договором по Международному признанию депонирования 8 микроорганизмов в целях патентования с присвоением номера АТСС 74386. Аскомы Rosellina subiculata (GB3719) были обнаружены на лишенной коры твердой древесине на берегу Navesink River, Monmouth Co.,New Jersey. В лабораторных условиях верхушки нескольких аском удаляли лезвием стерилизованного микротома, после чего с помощью булавки из сердцевины удаляли спороносный слой, парафизы и аскоспоры и делали посев на агар с солодо-дрожжевым экстрактом. Аскоспоры инкубировали в течение ночи до их прорастания и переносили в пробирки с солододрожжевым агаром с целью инициирования чистых колоний. Морфология Rosellina subiculata (GB3719) в основном соответствует литературным данным (J.B. EllisВ.М. Everhart. 1892. The Northzones. Sydowia 44:169-281). Следующие ключевые особенности, позволили идентифицировать грибок, как Rosellina subiculata строматные аскомы встречаются в отдельных экземплярах,однако, они агрегируют или сливаются в мелкие грозди на основании мицелия ошкуренной древесины; стромы имеют полусферическую форму, покрыты сосочками, являются гладкими,блестящими, темными, основание представляет собой тонкий мицелиальный слой бледнопалевого цвета, иногда он проявляется, как светлоокрашенное обесцвечивание древесины рядом со стромами; спороносный слой имеет цилиндрическую форму с крахмальной верхней пробкой; коричневато-серые аскоспоры имеют,в основном, форму эллипса со слегка почковидным характером, они гладкие без отростков или ложных стеблей, с прямой, вентральной зародышевой щелью, 10-125-6 мкм. В культуре на агаре колонии грибков обладают следующей морфологией. Колонии в агаре на отваре овсяной муки при 12-часовом световом периоде умеренно быстро растут при 23 С, достигая размера в 73-75 мм за 14 дней, характеризуются тем, что передняя зона сдавлена, является гладкой и латентно окаймленной, с белым мицелием в области внутренней трети, имеющим структуру от бархатистой до хлопьевидноопушенной, с влажным сдавленным мицелием в области внешних двух третей, на просвет они окрашены в оттенки белого или бледно-розового, бледно-винограднорозовые, Light Vinaceous Cinnamon с обратной стороны, эксудаты отсутствуют, издают слабый аромат. В овсяномучном агаре, при 37 С роста не наблюдается. Колонии в агаре с соком V8 медленно растут при 23 С и 12-часовом световом периоде,достигая за 14 дней 25-35 мм, они погружены по краям, большая часть мицелия, в основном,сдавлена, имеется хлопьевидноопушенный воз 9 душный мицелий в области внутренней трети,окаймлены, на просвет они бледно-сероваторозовые, Vinaceous Cinnamon, с обратной стороны окрашены в коричный, Orange-Cinnamon,Cinnamon или красновато-коричневый, Russet,Flawn Color, издают аромат. Колонии в агаре на отваре кукурузной муки медленно растут при 25 С и 12-часовом световом периоде, достигая за 14 дней 29-34 мм,погружены по краям, не имеют воздушных гиф,не окаймлены, прозрачны или имеют скудный белый мицелий в точке прививки, бесцветны с обратной стороны. При первом выращивании в культуре в августе 1993 г. штамм продуцировал скудные конидиофоры и конидии Geniculosporium анаморфа, аналогичные тем, что были описаны Petrini в 1993 г. Однако спорообразования больше не наблюдалось, по всей вероятности, из-за длительного хранения и повторных пересадок. По крайней мере, в одном случае, через 5 недель выращивания в агаре на отваре овсяной муки формировалось несколько зрелых перетеций со спороносным слоем и аскоспорами, которые были идентичны, наблюдающимся в природных условиях. Аскоспоры прорастали в течение ночи при инкубировании на агаре с солодовым дрожжевым экстрактом при комнатной температуре. Мицелий состоял из высоко разветвленных, разделенных простыми перегородками,стекловидных гиф. Сордарин получают культивированием штамма Rosellina subiculata или неидентифицированного грибка MF6232 (АТСС 74387), способного вырабатывать указанное соединение на обычной твердой среде, или в общепринятой водной среде. Микроорганизм выращивают в питательной среде, содержащей известные источники питания аналогичных грибков, т.е. ассимилируемые источники углерода и азота,плюс необязательные неорганические соли и другие известные факторы роста. Общепринятые методы, применяемые для культивирования других аналогичных грибков, могут использоваться и в настоящем изобретении. Питательная среда должна содержать соответствующий источник ассимилируемого углерода, например, рибозу, глюкозу, сахарозу,целлобиозу или фруктозу. В качестве источника азота могут применяться хлористый аммоний,сульфат аммония, мочевина, нитрат аммония,нитрат натрия и т.п., которые используются, как таковые, или совместно с такими источниками органического азота, как пептон, экстракт рыбной муки, дрожжевой экстракт, жидкость для замачивания зерна, соевый порошок, мука хлопкового семени и т.п. Если необходимо, могут добавляться питательные неорганические соли, являющиеся источниками натрия, калия,кальция, аммония, фосфата, сульфата, хлорида,бромида, карбоната, цинка, магния, марганца,кобальта, железа и т.д. 10 Продуцирование сордарина может осуществляться при любой температуре, способствующей удовлетворительному росту продуцирующего микроорганизма, например, при 2030 С. Обычно оптимальное продуцирование целевого соединения достигается во встряхиваемых колбах после периода инкубации 7-21 день. Аэрация во встряхиваемых колбах достигается путем перемешивания, например, путем встряхивания в ротационном шейкере. Если ферментацию следует проводить в бродильном чане, то желательно получать вегетативный посевной материал в питательной среде путем инокуляции бульонной культуры из культуры на скошенном агаре, лиофилизированной культуре или замороженной культуре микроорганизма. После получения активного инокулята указанным способом, его асептически переносят в среду бродильного чана. Продукция целевого соединения в бродильном чане обычно достигает оптимального значения через 7-21 день инкубации. Взбалтывание среды в бродильном чане обеспечивается перемешиванием, а аэрация путем ввода воздуха или кислорода в перемешиваемую смесь. За получением целевого соединения можно следить с использованием методов хроматографии или спектроскопии, либо с помощью традиционного биологического анализа. Сордарин легко выделяется из ферментационного бульона путем экстракции всего бульона таким органическим растворителем, как метилэтил кетон. Целевое соединение может быть очищено с использованием таких хорошо известных стандартных методов, как гельфильтрационная хроматография, тонкослойная хроматография, жидкостная хроматография высокого разрешения, выпаривание, осаждение и/или кристаллизация, или их комбинаций. С другой стороны, весь бульон или его органический экстракт может быть подвергнут сушке распылением или вымораживанием, с последующей очисткой описанными выше методами. Соединения настоящего изобретения(формула I) могут быть получены описанными ниже способами. Указанные условия являются репрезентативными и они не ограничивают сферу изобретения. Как показано на схеме 1, соединения формулы I, в которой Z представляет собой группу(а) или (b), могут быть получены из исходных веществ, описанных в заявке РСТ WO 96/14326,или из исходных веществ, описанных в заявке РСТ WO 96/14327 для соединений формулы I, в которой Z представляет собой группу (с). Исходное вещество в виде карбоновой кислоты дереватизируют с помощью подходящей защитной группы (например, бензила или пметоксибензила) и в результате обработки гидрохлоридом гидроксиламина в спиртовом растворителе, содержащем пиридин, получают альдоксим. Полученный альдоксим трансфор 11 мируют в соединение с нитрильной группой с помощью подходящего дегидратирующего агента (например, внутренней соли гидроксида(метоксикарбонилсульфамоил)триэтиламмония или цианурхлорида) и удаляют защитную группу с образованием соединения формулы (I). Схема 1R'ОН представляет собой низший алкиловый спиртовый растворитель;PG представляет собой карбоновую кислотную защитную группу;Z имеет указанные выше значения или представляет собой подходящим образом защищенное производное. Соединение формулы I(d) может быть получено способом, проиллюстрированным на схеме 2. Сордарин надлежащим образом защищают и полученный в результате этого альдегид реагирует с гидрохлоридом гидроксиламина в среде спиртового растворителя, в присутствии пиридина. Полученный в результате альдоксим дегидрируют с помощью такого реагента, как внутренняя соль гидроксида (метоксикарбонилсульфамоил)триэтиламмония или цианурхлорид, с образованием соединения с нитрильной группой (IV). В результате удаления защитной группы (PG) получают соединение формулы На схеме 3 продемонстрировано микробиологическое деметилирование 4'-мeтoкcигруппы соединения I(d) с получением соедине 002631 12 ния (V). Такое деметилирование осуществляется путем контактирования соединения формулыI(d) с культурой штамма Streptomyces avermitilis в ферментационной среде, содержащей ассимилируемые источники углерода и азота; и выделения соединения (V) из ферментационной среды. Подходящие штаммы Streptomyces avermitilis включают штамм МА 4848, депонированный в American Type Collection, Rockvile, MD под номером АТСС 31272. Соединение (V) может использоваться в синтезе соединений формулы С другой стороны, как показано на схеме 4, сордарицин (VI) может использоваться в качестве исходного материала для синтеза соединений формулы (I). Дериватизация карбоновой кислоты с помощью подходящей защитной группы, сопровождающаяся защитной первичной гидроксильной группы, позволяет синтезировать нитрилагликоновое соединение (VII) по реакции с гидрохлоридом гидроксиламина с последующим дегидратированием и удалением гидроксизащитной группы. В результате присоединения сахара или модифицированного сахарного субстрата методами, известными специалистам в этой области, получают соединения формулы (I). Схема 4 Промышленная применимость. Соединения формулы I представляют собой противогрибковые агенты, применяемые в качестве медикаментов для лечения людей и животных, а также средств защиты культурных растений. Соединения формулы I представляют собой очень активные фунгициды, используемые в борьбе с грибковыми инфекциями животных, 13 включая людей. Так, например, они могут использоваться для обработки грибковых инфекций, вызываемых такими микроорганизмами,как виды Candida (например, Candida albicans,Candida glabrata, (Torulopsis glabrata), CandidaBlastomyces dermatitidis). Они могут также использоваться для борьбы с другими видами грибковых инфекций, вызываемых видами(например, Trichophyton mentographytes, Trichophyton rubrum, Microsporum canis или Epidermophyton floccosum), или со слизистыми инфекциями, вызываемыми Candida albicans. Соединения формулы I могут также использоваться для борьбы с другими инфекциями, вызываемыми разновидностями такого нитевидного грибка, как Geotrichum (например,Geotrichum clavatum), Trichosporon (например,Trichosporon beigelii), Blastoschizomyces (например, Blastoschizomyces capitatus), Sporothris (например, Sporothris schenskii), Scedosporium (например, Scedosporium apiosperum), CladosporiumIn vitro оценку противогрибковой активности соединений изобретения осуществляли в жидкой или твердой среде, с помощью противогрибкового метода двухкратного серийного разбавления, предназначенного для определения минимальной ингибиторной концентрации(МИК) противогрибкового агента, который ингибирует развитие роста через 24-48 ч инкубации при 35 С. На практике агаровые пластинки или панели для микроразведения бульона исследовали на предмет наличия или отсутствия роста грибков и отмечали соответствующие значения МИК. Визуализации конечных точек способствовало применение витального окрашивания Alamar Blue.In vivo оценку соединений формулы I можно осуществлять сериями дозировочных уровней путем применения (например, подкожного, орального, внутрибрюшинного или внутривенного) на мышах инокулированных внутривенно штаммом Candida spp. Почки испытуемых животных могут быть удалены и количественно охарактеризованы на присутствие жизнеспособного Candida spp., а снижение инфицирования может быть определено сравнением с необработанными контрольными животными. В зависимости от уровня противогрибковой активности, соединения формулы I применяются для лечения и/или профилактики большого числа грибковых инфекций людей и животных. Такие инфекции включают такие поверхностные, кожные, подкожные и системные 14 микотические инфекции, как инфекции дыхательных путей,инфекции желудочнокишечного тракта, сердечно-сосудистые инфекции, инфекции мочевых путей, инфекции ЦНС,кандидоз и хронический мукокандидоз (например, кандидозный стоматит и вульвовагинит), а также кожные инфекции, вызываемые грибками, кожный кандидоз и кандидоз слизистых оболочек, грибковые болезни кожи, включая дерматомикоз и дерматофитию, эпидермофития стопы, паронихия, разноцветный лишай, эритразма, интертриго, грибковая опрелость, кандида вульвит, кандида баланит, и наружный отит. Эти соединения могут также применяться в качестве профилактических агентов для предотвращения системных и местных грибковых инфекций. Применение в качестве профилактических агентов может, например, быть частью схемы селективной деконтаминации кишечника при профилактике заражения у пациентов иммунологической группы риска (например, пациентов с симптомами СПИДа, пациентов, перенесших онкотерапию или трансплантацию). Предотвращение избыточного грибкового роста в ходе лечения антибиотиками также может оказаться желательным при некоторых болезненных синдромах или ятрогенных состояниях. Соединения формулы (I) также находят применение в качестве противогрибковых агентов для широкого спектра культурных растений и обладают эффективностью в отношении широкого спектра фитопатогенных грибков, особенно тех, что принадлежат к классу, состоящему из дейтеромицет (например, Botrytis spp.,Septoria spp., Pyricularia spp., Stagnospora spp.,Helminthosporium spp., Fusarium spp., Cercosporaspp. и Alternaria spp.); базидиомицет (например,Puccinia spp. и Hemileia); аскомицет (например,Venturia spp., Podospharera spp., Erysiphe spp.,Monilinia spp. и Uncinula spp.); и оомицет (например, Phytophthora spp., Pemospora spp., Bremia spp., Pythium spp. и Plasmopara spp.). В приведенном перечне представлены примеры фитопатогенных грибков, в отношении к которым названные соединения проявляют свою активность, и эти примеры никоим образом не ограничивают сферу изобретения. Эти соединения обладают очень полезными лечебными и профилактическими фунгицидными свойствами в отношении защиты растений, и могут применяться для ингибирования или уничтожения микроорганизмов, существующих на растениях или частях (плодах, цветках, листьях, стеблях,клубнях или корнях) ценных растений различных культур, при этом вырастающие позже части таких растений также предохраняются от воздействия указанных микроорганизмов. Они могут также использоваться в качестве удобрения при обработке материала для размножения растений, особенно семян (плоды, клубни, зерна) и растительных черенков (например, риса) с 15 целью обеспечения защиты от грибковых инфекций и фитопатогенных грибков, находящихся в почве. Отличительной особенностью соединений формулы (I) является тот факт, что они очень хорошо переносятся растениями и благоприятны для окружающей среды. Сельскохозяйственная оценка соединений формулы I может осуществляться с использованием следующих тестов. 1. Действие против Erysiphe graminus на пшенице.a). Через 1 неделю культивации, пшеничные растения опрыскивали до стекания распылительной смесью (200 млн.д. системы активный ингредиент/20% ацетона/0,25% TritonX155). Через 2 ч обработанные растения инфицировали аскоспорами, стряхиваемыми с растений-инокулятов. Воздействие грибков оценивали после инкубации в течение 8 дней при 22 С и 50% относительной влажности с целью определения защиты, обеспечиваемой данным соединением.b). Через 1 неделю после культивации,пшеничные растения инфицировали акоспорами, стряхиваемыми с растений-инокулятов. Через 24 ч пшеничные растения опрыскивали распылительной смесью (200 млн.д. активного ингредиента/20% ацетона/0,25% Triton X155). Грибковое воздействие оценивали после инкубации в течение 8 дней при 22 С и 50% относительной влажности с целью определения степени лечебной активности, обеспечиваемой испытуемым соединением. с). Через 1 неделю после культивации,пшеничные растения инфицировали аскоспорами, стряхиваемыми с растений-инокулятов. Через 24 ч почву, на которой росли пшеничные растения, смачивали дренажной смесью (200 млн.д. активного ингредиента/20% ацетона/ 0,25% Triton X155). Грибковое воздействие оценивали после инкубации в течение 8 дней при 22 С и 50% относительной влажности с целью определения степени лечебного действия, обеспечиваемого данным соединением. 2. Действие против Puccinia recondita на пшенице.a). Через 1 неделю культивации пшеничные растения опрыскивали до стекания жидкости распылительной смесью (200 млн.д. активного ингредиента/20% ацетона/0,25% TritonX155). Через 2 ч обработанные растения инфицировали спорами. Грибковое воздействие оценивали после инкубации в течение 1 дня при 95100% относительной влажности и температуре 20 С и последующих 7 дней при 25 С и 50% относительной влажности с целью определения защитного действия испытуемого соединения.b). Через 1 неделю культивации пшеничные растения инфицировали суспензией спор. Через 24 ч инфицированные растения опрыскивали до стекания жидкости распылительной смесью (200 млн.д. активного ингредиента/20% 16 ацетона/0,25% Triton X155). Грибковое воздействие оценивали после инкубации в течение 1 дня при 20 С и относительной влажности 95100% и 7 последующих дней при 25 С и 50% относительной влажности с целью определения степени лечебной активности, обеспечиваемой испытуемым соединением.c). Через 1 неделю культивации пшеничные растения инфицировали суспензией спор. Через 24 ч почву, на которой росли пшеничные растения, смачивали дренажной смесью (200 млн.ч. активного ингредиента/20% ацетона/ 0,25% Triton X155). Грибковое воздействие оценивали после инкубации в течение 1 дня при 20 С и относительной влажности 95-100% и последующих 7 дней при 25 С и относительной влажности 50% с целью определения степени лечебного действия испытуемого соединения. В зависимости от спектра активности, соединения согласно изобретению могут применяться для защиты или лечения ценных сельскохозяйственных растений, зараженных фитопатогенными грибками. Следующие ниже виды растений подходят для применения соединений,согласно изобретению: зерновые культуры (например, пшеница, рожь, овес, ячмень, рис, сорго и родственные культуры); свекла (сахарная свекла и кормовая свекла); семечковые, плодовые и ягодные культуры (например, яблони,груши, сливы, персики, миндаль, вишни, земляника, малина, ежевика); бобовые растения (например, бобы, горох, чечевица и соя); масляничные растения (например, рапс, горчица, мак,маслины, подсолнечник, кокос, касторовые масляные растения, какао-бобы и арахис); тыквенные (огурцы, тыква, дыня); волокнистые растения (например, хлопок, лен, конопля и джут); цитрусовые (например, апельсины, лимоны,мандарины и грэйпфруты); овощи (например,салат, кабачки, шпинат, морковь, спаржа, перец,лук, томаты и картофель); лавровые (авокадо,коричное дерево и камфарное дерево); или такие растения, как маис, табак, орехи, кофе, сахарный тростник, чай, виноград, хмель, бананы и природные каучуковые растения, а также декоративные растения (цветы, кустарники, широколистные деревья и такие вечнозеленые растения, как хвойные). Следует отметить, что указанные выше виды растений не являются ограничительным списком в отношении спектра активности указанных соединений. Соединения формулы I особенно полезны в борьбе со следующими видами болезней растений:Alternaria фруктов и овощей. Соединения формулы I могут также использоваться для защиты материалов (например, предохранения лесоматериалов от воздействия Paecilomyces variotii). Фармацевтические композиции. Хотя в терапевтической практике соединения согласно изобретению могут применяться в неочищенном виде, предпочтительно, чтобы они были активными ингредиентами фармацевтических композиций. Таким образом, настоящее изобретение предусматривает фармацевтическую композицию, содержащую соединение формулы I, или его фармацевтически применимую соль совместно с одним или более фармацевтически применимыми носителями и, необязательно, другие терапевтические и/или профилактические ингредиенты. Носитель должен быть "применимым" в том смысле, что он совместим с другими ингредиентами рецептуры и не оказывает вредного влияния на его реципиента. Композиции согласно изобретению включают рецептуры в форме, специально сформированной для перорального, трансбукального,парентерального, имплантантного, ректального,местного, офтальмического или мочеполового применения, или в форме, подходящей для применения путем ингаляции или вдувания. Таблетки и капсулы для перорального применения могут содержать такие общепринятые наполнители, как связующие агенты, например, патоку, аравийскую камедь, желатин,сорбит, трагант, крахмальную слизь или поливинилпирролидон; наполнители, например, лактозу, сахар, микрокристаллическую целлюлозу,кукурузный крахмал, фосфат кальция или сорбит; смазывающие вещества, например, стеарат магния, стеариновую кислоту, тальк, полиэтиленгликоль, или оксид кремния; дезинтегранты, например, картофельный крахмал или гликолят натриевой формы крахмала, либо натрий кросскармелозу; или смачивающие агенты,например, лаурилсульфат натрия. На таблетки,включающие жевательные, диспергируемые и вспенивающиеся таблетки, может быть нанесено покрытие с помощью способов, хорошо известных в этой области. Пероральные жидкие препараты могут изготовляться в виде жидких или масляных суспензий, растворов, эмульсий,сиропов или эликсиров, или могут представлять собой сухой продукт, предназначенный для разбавления водой или другими подходящими жидкими носителями перед применением. Эти жидкие препараты могут содержать такие об 002631 18 щепринятые присадки, как суспендирующие агенты, например, сироп, на основе сорбита,метилцеллюлозу, глюкоза/сахарный сироп, желатин, гидроксиэтиллцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия, или гидрированные съедобные жиры; эмульгирующие агенты, например, лецитин, моноолеат сорбита или аравийскую камедь; неводные наполнители (которые могут включать съедобные жиры), например, миндальное масло, фракционированное масло кокосового ореха, жирные сложные эфиры, пропиленгликоль или этиловый спирт; а также предохраняющие агенты,например, метил или пропил-н-гидроксибензоаты или сорбиновую кислоту. Композиции, предназначенные для трансбукального применения, могут иметь форму таблеток или лепешек, сформированных общепринятыми способами. Композиции согласно изобретению могут быть изготовлены для парентерального применения путем инъекции или непрерывного вливания. Рецептуры для инъекции могут быть приготовлены в виде единичной стандартной дозы, находящейся в ампулах, или в упаковке для многократных дозировок, в которые добавляют консервант. Композиции могут использоваться в виде суспензий, растворов или эмульсий в масляных или водных наполнителях, и могут содержать формирующие агенты, например, суспендирующие, стабилизирующие и/или диспергирующие агенты. С другой стороны,активный ингредиент может использоваться в виде порошка, предназначенного для растворения перед применением в подходящем носителе, например, в стерильной, апирогенной воде. Композиции согласно изобретению, предназначенные для ингаляции, обычно применяют в виде аэрозольных спреев из герметичных блоков с использованием подходящего распределяющего средства, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа, или из распылителя. В случае применения герметичных аэрозольных устройств, единичная доза может быть определена с помощью клапана, распределяющего отмеренное количество. С другой стороны, композиции согласно изобретению, применяемые ингаляцией, могут иметь форму сухой порошкообразной композиции, например, порошкообразной смеси активного соединения и такой подходящей порошковой основы, как лактоза или крахмал, либодля этой цели может использоваться модифцированная физическая форма самого лекарственного вещества. Порошкообразная композиция может быть представлена в форме единичной дозы, содержащейся, например, в капсулах, обоймах из желатины, или в прозрачных упаковках,из которых можно применять порошок с помощью ингалятора или инсуффлятора. 19 Композиции могут использоваться в виде суппозитория, например, содержащего традиционную суппозиторную основу, или маточного кольца, например, содержащего общепринятую для этой цели основу. Композиции согласно изобретению могут также формироваться для местного применения в виде мазей, кремов, гелей, лосьонов, шампуней, порошков (включая распылительные порошки), маточных колец, тампонов, спреев, дезинфицирующих растворов, аэрозолей, капель(например, глазных, ушных и носовых капель) или растворов для вливаний. Мази и кремы могут формироваться с помощью водной или масляной основы, с добавлением подходящего загустителя и/или гелеобразующего агента. Мази,предназначенные для глазного применения, могут выпускаться в стерильном исполнении с использованием стерильных компонентов. Препараты для вливаний могут формироваться для ветеринарного применения в маслах, содержащих органические растворители, необязательно в присутствии формирующих агентов, например, стабилизирующих и солюбилизирующих агентов. Маточные кольца и тампоны для вагинального введения могут изготавливаться с использованием традиционных методов и, если необходимо, могут содержать вспенивающийся носитель. Такие композиции могут дополнительно содержать другие активные ингредиенты, например, кортикостероиды, антибиотики или противопаразитарные средства. Жидкие препараты для внутриназальной доставки могут применяться в виде растворов или суспензий и могут содержать такие традиционные эксципиенты, как тонизирующие агенты, например, хлористый натрий, декстроза или маннит; предохраняющие агенты, например,хлористый бензалконий,тиомерсал,фенилэтиловый спирт; а также другие формирующие агенты, например, суспендирующий агент,буферный агент, стабилизатор, диспергатор и отдушки. Трансдермальное применение может осуществляться путем конструирования подходящей системы, которая усиливает абсорбцию активного соединения через кожу и которая обычно состоит из базовой рецептуры вложенной внутрь клейкой повязки, имеющей пленку с задней стороны, мембраны и прокладки для выделений. Эти системы могут включать такие усилители абсорбции, как спирты, или их действие связано со стимуляцией лекарственного электрофореза. Композиция согласно изобретению может также формироваться в виде препарата типа депо. Такие рецептуры пролонгированного действия могут применяться путем имплантации(например, подкожной или внутримышечной) или путем внутримышечной инъекции. Так, например, соединение изобретения может формироваться в композицию с подходящими поли 002631 20 мерными или гидрофобными материалами (например, в виде эмульсии в применимом масле) или ионообменными смолами или использоваться в виде частично растворимых производных, например, в виде частично растворимых солей. В том случае, когда композиции содержат единичные дозировки, каждая стандартная доза предпочтительно содержит 0,001-1000 мг, лучше всего 0,01-400 мг, активного ингредиента при пероральном применении соединения изобретения. Дневная дозировка, используемая для лечения взрослого человека, предпочтительно составляет 0,001-5000 мг активного ингредиента, наиболее предпочтительно 0,01-2000 мг, и может применяться в виде 1-4 дневных доз в зависимости от типа применения, состояния пациента и природы заболевания. Соединение согласно изобретению может применяться путем внутривенного вливания с использованием, например, 50 мг/день активного ингредиента. Длительность лечения будет определяться скоростью ответной реакции, а не произвольным числом дней. Соединения согласно изобретению также могут применяться в комбинации с другими терапевтическими агентами, и, таким образом,объектом изобретения является также комбинация, включающая соединение изобретения совместно с другим терапевтически активным агентом. Так, например, соединения согласно изобретению могут применяться в комбинации с одним или более противогрибковыми агентами,например, с полиеновым производным (амфотерицин В, нистатин, липидная рецептура амфотерицина В), производным азола (флюконазол,интраконазол, кетоконазол, миконазол, клотримазол, ZD-08070, UK-109496, SCH 56592), 5 фтороцитозином, таким производным пневмокандина или эхинокандина, как килофунгин,LY-303366, L-733560, L-743872 или другим соединением, обладающим активностью в отношении клеточной стенки, например, никомицином Z, и/или одним или более таких иммуномодулирующих агентов, как интерферон, например, (IFN-), интерлейкин, например (IL-1, IL-2,IL-3, и IL-8), факторами стимуляции колоний[(G)-CSF, (M)-CSF и (GM)-CSF] и дефензинами. Особенно подходящими соединениями для совместного применения с соединениями изобретения являются интраконазол, флуцитозин,флуконазол или амфотерицин В. В том случае, когда соединения настоящего изобретения применяются в комбинации с другим противогрибковым агентом, соединения изобретения и другой противогрибковый агент могут применяться в рекомендованной максимальной клинической дозировке или с более низкими дозировками. Указанные выше комбинации могут быть представлены в виде фармацевтической рецеп 21 туры и, таким образом, эти фармацевтические рецептуры, включающие указанную выше комбинацию, совместно с фармацевтически применимым носителем составляют еще один аспект настоящего изобретения. Индивидуальные компоненты таких комбинаций могут применяться последовательно или одновременно, в виде отдельных или совместных фармацевтических рецептур. В том случае, когда соединение согласно изобретению используется в комбинации со вторым терапевтическим агентом для лечения одного и того же состояния, дозировка каждого соединения может отличаться от той, что применяется в том случае, когда каждое соединение используется по отдельности. Соответствующие дозировки могут быть легко установлены специалистом в данной области. Сельскохозяйственные композиции. Соединения формулы I могут применяться в немодифицированной форме или предпочтительно совместно с адьювантами, традиционно используемыми в области составления сельскохозяйственных рецептур, которые, главным образом, известны как эмульгируемые концентраты, наносимые пасты, растворы для непосредственного распыления или разбавления, разбавленный раствор, суспензии (включая суспензии с высоким процентным содержанием воды, масла или других веществ), дисперсии, масляные дисперсии, агенты для посева вразброс, смачиваемые порошки, растворимые порошки, дусты,гранулы и инкапсулированные препараты. Такие рецептуры готовят известным способом,например, путем гомогенного смешивания и/или измельчения активных ингредиентов с разбавителями, например, с растворителями,твердыми носителями и, когда необходимо, с поверхностно-активными соединениями. Порошки, дусты и агенты для посева вразброс могут быть приготовлены смешиванием или измельчением активных ингредиентов с твердым носителем. Гранулы, например гранулы с покрытием, импрегнированные или гомогенные гранулы, могут быть получены путем связывания активных ингредиентов с твердыми носителями. Подходящие растворители представляют собой ароматические углеводороды, предпочтительно фракции, содержащие 8-12 углеродных атомов, например ксилольные смеси или замещенные нафталины, такие хлорированные ароматические углеводороды, как хлорбензолы,такие фталаты, как дибутил или диоктилфталат, такие алифатические углеводороды, как циклогексан или парафины, спирты и гликоли, а также их простые и сложные эфиры, например,этанол, этиленгликоль, монометиловый или моноэтиловый эфир этиленгликоля, такие кетоны,как циклогексанон, такие амины, как этаноламин, такие сильно полярные растворители, как 22 или диметилформамид, а также растительные масла или эпоксидированные растительные масла, например, эпоксидированное масло кокосового ореха или соевое масло; а также вода. Примерами поверхностно-активных веществ могут служить соли ароматических сульфокислот с катионами, представляющими собой щелочные и щелочно-земельные металлы, а также аммоний, например соли лигнинсульфокислоты, фенолсульфокислоты, нафталинсульфокислоты и дибутилнафталинсульфокислоты,а также соли жирных кислот, алкил и алкиларилсульфонаты, а также алкилсульфаты лаурилового эфира и жирного спирта, соли сульфированных гексадеканолов, гептадеканолов и октадеканолов, соли простых эфиров жирного спирта и гликолей, продукты конденсации сульфонированного нафталина и нафталиновых производных с формальдегидом, продукты конденсации нафталина или нафталинсульфокислот с фенолом и формальдегидом, простые эфиры полиоксиэтилен октилфенола, этоксилированный изооктилфенол, этоксилированный октилфенол и этоксилированный нонилфенол, простые эфиры алкилфенола и полигликоля, трибутилфенилполигликолевый эфир, алкиларилполиэфирные спирты, изотридециловый спирт,конденсаты жирного спирта и этиленоксида,этоксилированное касторовое масло, полиоксиэтиленалкиловые эфиры, этоксилированный полиоксипропилен, ацеталь лаурилового спирта и полигликолевого эфира, сложные эфиры сорбита, лигнинсульфитная барда, и метилцеллюлоза. Примерами твердых носителей могут служить такие минеральные материалы, как кремниевая кислота, силикагели, силикаты, тальк,каолин, аттапульгитные глины, известняк, известь, мел, железистая известковая глина, лсс,глина, доломит, диатомовая земля, смешанный сульфат оксида алюминия и кальция, сульфат магния, оксид магния, грунтовые пластики, такие удобрения, как сульфат аммония, фосфат аммония, нитрат аммония и мочевины, а также такие растительные продукты, как зерновая мука, корневая мука, древесная мука и мука из скорлупы орехов, целлюлозные порошки и т.п. Соединения формулы I могут смешиваться и применяться совместно с другими активными ингредиентами, например, с гербицидами, инсектицидами, бактерицидами, нематоцидами,моллюскицидами, регуляторами роста, питательными микроэлементами и удобрениями. В качестве других ингредиентов могут также использоваться один или более фунгицидов, принадлежащих к следующим классам фунгицидов,но не ограничивающихся ими: карбоксамиды,бензимидазолы, триазолы, гидроксипиридины,дикарбоксамиды, фениламиды, тиадиазолы,карбаматы, цианооксимы, производные коричной кислоты, морфолины, имидазолы, Вметоксиакрилаты и пиридины/пиримидины. 23 Кроме этого, такие дополнительные активные ингредиенты могут использоваться в качестве смесей из нескольких препаратов, если желательно, совместно с адьювантами, промотирующими другие применения, которые обычно используются в области составления рецептур. Подходящие для этой цели носители и адьюванты могут представлять собой твердые и жидкие вещества, соответствующие тем, что обычно применяются в рецептурной технологии (например, природные или регенерированные минеральные вещества, растворители, диспергенты и смачивающие агенты). Следующий ниже перечень фунгицидов с которыми могут комбинироваться соединения формулы I, приведен в целях иллюстрации возможных комбинаций и не является ограничительным. Примерами фунгицидов, которые используются совместно с соединениями формулы I,могут служить сера, дитиокарбаматы и их производные, например диметилдитиокарбамат трехвалентного железа, диметилдитиокарбамат цинка, этиленбисдитиокарбамат цинка, этиленбисдитиокарбамат марганца, смешанный этилендиаминбисдитиокарбамат марганца и цинка,дисульфиды тетраметилтиурам, аммиачный комплекс N,N'-этиленбисдитиокарбамат цинка,аммиачный комплексDL-N-(2,6-диметилфенил)-N-(фенилацетаил) аланат, 5-метил-5-винил-3-(3,5-дихлорфенил)2,4-диоксо-1,3-оксазолидин,3-(3,5-дихлорфенил)-5-метил-5-метоксиметил-1,3-оксазолидин 2,4-дион, 3-(3,5-дихлорфенил)-1-изопропилкарбамилгидантоин,N-(3,5-дихлорфенил)-1,2 диметилциклопропан-1,2-дикарбоксимид,2 циано-[N-(этиламинокарбонил)-2-метоксимино] ацетамид,1-[2-(2,4-дихлорфенил)пентил]-1 Н 1,2,4-триазол, 2,4-дифтор-(1 Н-1,2,4-триазол-1 илметил)бензгидриловый спирт, N-(3-хлор-2,6 динитро-4-трифторметилфенил)-5-трифторметил-3-хлор-2-аминопиридин и 1-[(бис-(4 фторфенил)метилсилил)метил]-1 Н-1,2,4-триазол. Относительно природы композиций следует отметить, что способ применения, например,распрыскивание, распыление, опыление, разбрасывание, нанесение, подкормка и выливание выбираются в соответствии с предполагаемыми целями применения и существующими обстоятельствами. Одним из способов применения активного ингредиента или агрохимической композиции, содержащей, по крайней мере, одно из указанных соединений является применение на растениях (например, некорневое питание растений). Однако активный ингредиент может также проникать в растение из почвы через корни (т.е. почвенная подкормка). Это может быть применение жидкости на почве (полив) или применение в гранулированной форме. Активный ингредиент может также применяться на таком материале размножения растений, как семена (плоды, клубни или зерна) или черенках растений, причем такое применение может осуществляться в жидкой форме (путем нанесения покрытия) или в твердой форме(протравливание). Семена, например, могут протравливаться перед посевом. Соединения настоящего изобретения могут также применяться на зернах путем их пропитки жидкой рецептурой или их покрытия твердыми составами. Композиция может также применяться в месте посадки при посеве размножающего материала, например, в семенной борозде в ходе посева. Предпочтительные нормы внесения обычно составляют от 10 г до 50 кг активного ингредиента (а.и.) на гектар, предпочтительно 10 г - 2 кг а.и./га, наиболее предпочтительно 100-600 г а.и./га. Активные ингредиенты заявленных соединений, как правило, используются в виде композиции и могут применяться на растении или на частях растения одновременно или последовательно с дополнительными активными ингредиентами. Такими дополнительными активными ингредиентами могут служить удобрения, дополнительные микроэлементы или другие соединения, влияющие на рост растений. Однако они могут также представлять собой селективные гербициды, инсектициды, бактерициды, нематоциды, инсектициды и моллюскициды, а также другие фунгициды. Получение исходного материала Ферментационное получение сордарина. Следующие культурные среды использовали при ферментации Rosellinia subiculata(ATCC 74386) и АТСС 74387 для получения сордарина. Посевная среда 1. Компонент Дрожжевой экстракт Солодовый экстракт Глюкоза Среду готовили с использованием дистиллированной воды, рН устанавливали равным 7,0 перед стерилизацией и среду распределяли 50 мл порциями в Эрленмейровских колбах без дросселя, емкостью 250 мл. Использовали хлопковые перекрытия. Стерилизацию проводили в течение 20 мин при 121 С. Посевная среда 2. Компонент Жидкость для замачивания пшена(высушенная) Томатная паста Овсяная мука Глюкоза Раствор микроэлементов Микроэлементы готовили в 6N HCl. Среду готовили с использованием дистиллированной воды, перед стерилизацией рН устанавливали равным 6,8 и среду распределяли 50 мл порциями в 250 мл Эрленмейровских колбах без дросселей. Применяли хлопковые прокладки. Стерилизацию проводили в течение 20 мин при 121 С. Твердая продуцирующая среда 1. 1. Твердая часть. 675 см 3 вермикулита загружали в 2 литровый вращающийся флакон. Флакон закрывали каучуковой пробкой; выдерживали в течение 60 мин в автоклаве и сушили в течение 30 мин. 2. Жидкая часть. В сосуд емкостью 500 мл помещали 220 мл следующей смеси: Компонент Глюкоза Глицерин Дрожжевой экстракт Среду готовили с использованием дистиллированной воды, устанавливая перед стерилизацией рН 7,0. Глюкозу обрабатывали в автоклаве отдельно. Полученную смесь распределяли по сосудам емкостью 500 мл и обрабатывали в автоклаве в течение 15 мин при 121 С. Жидкая продукционная среда 1. Компонент Глицерин Глюкоза Томатная паста Среду готовили с использованием дистиллированной воды, устанавливая перед стерилизацией рН 7,0. Полученную среду распределяли порциями по 50 мл в Эрленмейровских колбах без дросселей емкостью 250 мл. Колбы закрывали хлопковыми прокладками и обрабатывали в автоклаве в течение 20 мин при 121 С. Твердая продукционная среда 2. 1. Твердая часть. 675 см 3 вермикулита помещали в 2 литровый вращающийся флакон. Сосуд закрывали каучуковой пробкой; обрабатывали в автоклаве в течение 60 мин и сушили в течение 30 мин. 2. Жидкая часть. В 500 мл сосуд загружали 220 мл следующей смеси: Компонент Сахароза Глюкоза Глицерин Лимонная кислота Среду готовили с помощью дистиллированной воды, устанавливая перед стерилизацией рН 7,0. Ее распределяли порциями по 220 мл в колбах емкостью 500 мл и обрабатывали в течение 15 мин в автоклаве при 121 С. Жидкая продукционная среда 2. Состав соответствовал жидкой части твердой продукционной среды 1. Среду готовили с использованием дистиллированной воды, устанавливая рН равным 7,0 перед стерилизацией. Глюкозу обрабатывали в автоклаве отдельно. Полученную среду распределяли порциями по 50 мл в Эрленмейеровских колбах без дросселей емкостью 250 мл. Колбы закрывали хлопковыми прокладками и в течение 15 мин обрабатывали в автоклаве при 121 С.Rossellina subiculata (MF6239, АТСС 74386) 1. Культура. Часть скошенного агара, содержащего культуру, асептически переносили в посевную среду 1 (50 мл в 250-миллилитровой колбе без дросселя). Этот материал инкубировали во вращающемся шейкере с ходом в 2 дюйма (5,1 см) при скорости вращения 220 об./мин в течение 5 дней при 25 С и относительной влажности (rh) 85% с получением биомассы. Части такой биомассы переносили в стерильные ампулы, содержащие глицерин, и замораживали (в качестве замороженного вегетативного мицелия(FVM. Концентрация глицерина в ампулах составляла 10-15% и их хранили при -75 С. Вторичный FVM получали из первичного FVM путем переноса 1,0 мл оттаявшего первичногоFVM в посевную среду 2, инкубации в течение 7 дней при 25 С и скорости вращения 220 об./мин, после чего материал замораживали, как описано выше. 2. Посевной материал. Замороженную ампулу (FVM) с MF6239 оттаивали до комнатной температуры и использовали для инокуляции посевных культур в количестве 1,0 мл на 50 мл посевной культуры 2. Полученный материал выращивали во вращающемся шейкере (скорость вращения 220 об./мин) в течение 7 дней при 25 С и относительной влажности 85%. 3. Продуцирование. На твердой продукционной среде. Аликвоту (10-12 мл) посевного материала помещали в 220 мл жидкой части твердой продукционной среды 1. Колбу интенсивно вращали с целью диспергирования биомассы. Содержимое переливали в 2 л сосуд для роллерной культуры,содержащий 675 см 3 крупнокускового вермикулита. Содержимое роллерного сосуда встряхивали/смешивали для обеспечения гомогенной инокуляции и заполнения. Вращающиеся сосуды инкубировали в горизонтальном положении,вращали со скоростью примерно 4 об./мин на роллерном устройстве Wheaton в течение 17 дней при 22 С и относительной влажности 70% с получением вторичного метаболита в ферментационной среде. В жидкой продукционной среде. Посевные культуры инокулировали описанным выше способом. Аликвоту посевного материала (1,5 мл) использовали для инокуляции каждой продукционной колбы, содержащей 50 мл жидкой продукционной среды 1 в расчете на 250 мл колбу. Колбы инкубировали на вращающемся шейкере(220 об./мин) в течение 7-21 дней при 25 С и относительной влажности 50-85%. Получение сордарина ферментацией MF6232 29 среду 1 (50 мл/250 мл колбы без дросселя). Полученный материал инкубировали во вращающемся шейкере с ходом 2 дюйма (5,1 см) при скорости вращения 220 об./мин, в течение 3 дней при 25 С и относительной влажности 85% с получением биомассы. Части такой биомассы переносили в стерильные ампулы, содержащие глицерин, и замораживали (в виде FVM). Их хранили при -75 С и конечной концентрации глицерина 10-15%. Вторичные FVM получали из первичных FVM путем переноса 1,0 мл оттаявшего первичного FVM в посевную культуру 2(имеющую состав, указанный ниже), инкубации в течение 7 дней при 25 С и скорости вращения 220 об./мин и последующего замораживания по описанному выше способу. 2. Посевной материал. Замороженную ампулу (FVM) с MF6232 оттаивали до комнатной температуры и использовали для инокуляции посевных культур в количестве 1,0 мл на 50 мл посевной культуры 2. Полученный материал выращивали во вращающемся шейкере (220 об./мин) в течение 7 дней при 25 С и относительной влажности 85%. 3. Продуцирование. На твердой продукционной среде. Аликвоту (10-12 мл) посевного материала помещали в 220 мл твердой продукционной среды 2. Полученный материал интенсивно вращали с целью диспергирования биомассы. Содержимое переливали в 2 л сосуд для роллер-культуры, который содержал 675 см 3 крупнокускового вермикулита. Содержимое вращающегося сосуда подвергали встряхиванию/смешиванию для обеспечения гомогенной инокуляции и заполнения. Вращающиеся сосуды инкубировали в горизонтальном положении, вращали со скоростью примерно 4 об./мин на вращающемся устройстве Wheaton в течение 21 дня при 22 С и относительной влажности 70% с получением вторичного метаболита в ферментационной среде. В жидкой продукционной среде. Семенные культуры инокулировали описанным выше способом. Аликвоту семенного материала (1,5 мл) использовали для инокуляции каждой продукционной колбы, содержащей 50 мл жидкой продукционной среды 2/250 мл колбу. Колбы инкубировали во вращающемся шейкере (220 об./мин) в течение 7-21 дня при 25 С и относительной влажности 50-85%. Крупномасштабное продуцирование сордарина MF6232 (АТСС 74387) Жидкую часть твердой продукционной среды 1 использовали как в посевном, так и в продукционном ферментере. После стерилизации в среду посевного ферментера добавляли 30 г/л церелозы, тогда как для продукционного ферментера это количество составляло 150 г/л. Посевные ферментеры инокулировали 2 л культуры, которую выращивали во встряхиваемых колбах. В этих ферментерах проводили культивацию при 25 С в течение 30 ч до значения ско 002631 рости поглощения кислорода 3 ммоль/лч. Через 30 ч 25 л ферментационной посевной культуры переносили в продукционный ферментер. Через 50 ч скорость роста в продукционном ферментере достигала 8-10 ммоль/лч,уменьшаясь к концу культивации до значения 57 ммоль/лч. Количество растворенного кислорода регулировали увеличением скорости перемешивания. рН бульона не регулировали и обычно оно снижалось до значения 5,3 через 200 ч. Процесс проводили при 25 С. Через 280 ч культивации ферментацию прерывали и начинали подготовку к сбору урожая. С помощью гидроксида натрия устанавливали рН равным примерно 12 и в течение 20 ч проводили старение смеси при температуре ферментации. Перед переносом в барабан для дальнейшей обработки, рН среды снова устанавливали равным 6,0 с помощью серной кислоты. Выделение сордарина Метод выделения 1. В результате экстракции ферментационной среды культуры MF6232 (АТСС 74387) метилэтилкетоном из всего бульона получали 64 мл материала, который концентрировали досуха в вакууме (365 мг). Полученный материал растворяли в смеси, состоящей из 2 частей метанола и 98 частей хлористого метилена, и смесь доводили до конечного объема 4,6 мл. Часть полученного раствора объемом в 4,3 мл (341 мг) очищали методом тонкослойной хроматографии на колонке с 60 мл силикагеля 60 (0,040-0,0630 мм,230-400 меш, E.Merck), уравновешенной 2% метанола в хлористом метилене. Колонку элюировали ступенчатыми градиентами 2, 5, 10 и 30% метанола в хлористом метилене, каждый из которых имел объем 240 мл, и далее 120 мл метанола. В каждой системе растворителей отбирали шестнадцать фракций объемом 15 мл каждая. Обогащенные продуктом фракции 39-56 подвергали биологическому анализу. Группу неочищенных фракций концентрировали в вакууме до сухого состояния (103,1 мг). 34,4 мг такого образца дополнительно очищали методом ЖХВР (Zorbax Rx-C8, 5 мкм,колонка размером 9,4 мм 250 мм, элюирование с помощью подвижной фазы, состоящей из 20% ацетона в 80% водном растворе 0,1 М КН 2 РO4, при рН 6,9, установленным с помощью концентрированной Н 3 РO4, объемная скорость 4 мл/мин при 40 С, детекция с антенным диодом). Отбирали фракции объемом 4 мл. Обогащенные продуктом фракции 16-20 сливали и концентрировали в вакууме до объема в 25% от исходного. Концентрат дважды экстрагировали одинаковыми объемами этилацетата и этилацетатные слои промывали равным объемом рассола, сушили над безводным Na2SO4 и концентрировали в вакууме с получением 3,7 мг сордарина. Метод выделения 2. Материал, полученный экстракцией продукта периодической -004Y ферментации культуры MF6232 (АТСС 74387), в количестве 980 мл от всего бульона, концентрировали досуха в вакууме (4,9 г). Этот материал растворяли в 1 части метанола и 9 частях хлористого метилена до конечного объема 21,5 мл. Порцию объемом 21 мл (4,8 г) обрабатывали на хроматографической колонке с 500 мл силикагеля 60 (фракция 0,040-0,0630 мм, 230-400 меш, E.Merck), уравновешенной 2% метанола в хлористом метилене. Колонку элюировали со скоростью 25 мл/мин ступенчатым градиентом 2 и 5% раствора метанола в хлористом метилене, начиная с объема в 1 л для каждой концентрации, после чего применяли 2 л 15% метанола. Элюирование колонки завершали пропусканием 1 л 30% и 100% метанола. Отбирали фракции объемом в 25 мл. Обогащенные целевым продуктом фракции 75-85 и 111-121 определяли биологическим анализом и согласно данным обращеннофазовой ЖХВР в кислых условиях эти фракции содержали соединение формулы I. Пулы сырых фракций 75-85 и 111-121 по отдельности концентрировали досуха в вакууме(69,3 мг и 95,3 мг соответственно). Две 34 мг порции пула 75-85 дополнительно очищали идентичными методами ЖХВР (Zorbax Rx-C8, 7 мкм, 21,2250 мм, элюирование подвижной фазой, состоящей из 40% ацетона/60% H2O, в присутствии 0,1% Н 3 РO4, объемная скорость 20 мл/мин при 25 С, длина волны 220 нм). Отбирали фракции объемом в 1 мл. Обогащенные продуктом фракции 27-31 из обоих опытов сливали и концентрировали в вакууме до объема в 40% от исходного. Полученный концентрат экстрагировали равным объемом этилацетата и промывали равным объемом рассола, сушили над безводным Na2SO4 и концентрировали в вакууме с получением 27 мг сордарина. 46 мг порции пула 111-121 также дополнительно очищали методом ЖХВР при указанных выше условиях. Фракции 25-28 из обоих опытов объединяли и обрабатывали, как описано выше, с дополнительным образованием 17 мг сордарина. Интермедиат 1. [1R-(1,3 а,4,4 а,4 а,7,7a,8a)]-Бензил-4-формил-8 а-(гидроксиметил)-4,4a,5,6,7,7 а,8,8 а-октагидро-7-метил-3-(1 метилэтил)-1,4-метано-s-индацен-3 а(1 Н)-карбоксилат (бензиловый эфир сордарицина). туре в течение 1 дня. После разбавления водой и обработки водной фазы (СН 2 Сl2), органическую фракцию сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученную смесь растворяли в 2 мл ДМФ, к которому добавляли 0,1 мл бромистого бензила с последующим добавлением твердого NаНСО 3. Смесь перемешивали при комнатной температуре в течение ночи и затем концентрировали в вакууме. К смеси добавляли хлороформ и проводили фильтрацию для удаления NаНСО 3. Фильтрат концентрировали в вакууме и очищали методом препаративнои тонкослойной хроматографии (ПТСХ) с получением 1,0 мг бензилового эфира сордарицина. Спектр 1H ЯМР (СDСl3):0,51 (3H, д,J=6,9), 0,82 (3 Н, д, J=6,6), 0,91 (1 Н, м), 1,0 (3 Н,д, J=6,6), 1,18 (1H, д, J=12,6), 1,50-2,00 (9 Н, м),2,24 (1 Н, м), 2,51 (1 Н, м), 3,48 (1H, д, J=11,0),3,87 (1H, д, J=11,0), 5,11 (1 Н, д, J=11,7), 5.31 Сордарин (2 мг) растворяли в 1 мл ацетона. Добавляли концентрированную НСl (0,2 мл). Смесь перемешивали при комнатной темпера Следовали методике получения бензилового эфира сордарицина, за исключением того, что вместо бромистого бензила использовали бро Повторяли методику получения бензилового эфира сордарицина, но вместо бромистого бензила использовали хлористый 4-метоксибензил. Спектр 1 Н ЯМР (СDСl3):0,51 (3 Н, д,J=6,9), 0,82 (3 Н, д, J=6,9), 1,00 (3 Н, д, J=6,9),0,90-2,00 (11H, м), 2,23 (1 Н, м), 2,49 (1H, т,J=3,8), 3,79 (3 Н, с), 4,61 (2 Н, с), 5,05 (1H, д,J=11,7), 5,26 (1H, д, J=11,7), 6,03 (1H, д, J=3,2),6,88 (2 Н, д, J=8,7), 7,28 (2 Н, д, J=8,7), 9,60 (1 Н,с). Интермедиат 3. [1R-(1,3a,4,4a,4 а,7,7 а,8 а)]-Аллил 4-формил-8 а-(гидроксиметил)4,4 а,5,6,7,7 а,8,8 а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3 а(1H)-карбоксилат 33 мистый аллил. Таким способом получали целевое соединение. Интермедиат 4. [1R-(1,3 а,4,4 а,4 а,7,7 а,8 а)]-4-формил-8 а-(гидроксиметил)-4,4 а,5,6,7,7 а,8,8 а-октагидро-7-метил-3-(1-метилэтил)1,4-метано-s-индацен-3 а(1H)-карбоновая кислота (сордарицин). К раствору бензилового эфира сордарицина (0,6 мг) в МеОН добавляли катализаторPearlman. Полученную смесь перемешивали в атмосфере водорода (под давлением из баллона) в течение 15 мин. После фильтрации через хлопковое волокно и концентрирования в вакууме получали 0,4 мг сордарицина. Спектр 1H ЯМР (CDCl3):0,82 (3 Н, д,J=6,8), 0,98 (3 Н, д, J=6,6), 1,01 (3 Н, д, J=6,9),1,23 (1 Н, м), 1,25 (1 Н, д, J=12,6), 1,58-2,10 (9 Н,м), 2,34 (1 Н, м), 2,41 (1H, т, J=3,6), 3,45 (1H, д,J=11,0), 4,14 (1H, д, J= 11,0), 6,05 (1H, д, J=3,0),9,75 (1H,c). Интермедиат 5. [1R-(1,3 а,4,4 а,4a,7,7a,8a)]-Бензил-4-циано-8 а-(гидроксиметил)4,4 а,5,6,7,7 а,8,8 а-октагидро-7-метил-3-(1 метилэтил)-1,4-метано-s-индацен-3 а(1H)-карбоксилат. Бензиловый эфир сордарицина (161,2 мг) растворяли в 6 мл N,N-диметилформамида и добавляли хлористый п-метоксибензил (1 мл) с последующим добавлением избытка гидрида натрия (50 мг 60% дисперсии в минеральном масле). Полученную смесь разбавляли эфиром и тщательно промывали водой. Эфирный слой сушили над безводным сульфатом натрия и летучие вещества удаляли в вакууме. Остаток очищали хроматографией на силикагеле с получением 192,5 мг (93%) п-метоксибензилового эфира. Полученный выше эфир (150 мг) растворяли в 5 мл сухого этанола и добавляли 3 мл сухого пиридина. Добавляли гидрохлорид гидроксиламина (96 мг) и полученную смесь нагревали до 70 С в течение 3 ч. Реакционную смесь охлаждали и концентрировали в вакууме. Остаток растворяли в эфире, промывали водой и сушили над безводным сульфатом натрия. Остаток, полученный после удаления эфира в вакууме, очищали методом препаративной ТСХ с получением 143,4 мг альдоксима (93%). Оксим (143 мг) растворяли в 5 мл толуола,к которому добавляли избыток внутренней соли(метоксикарбонилсульфамоил) триэтиламмония (700 мг). Полученную смесь перемешивали в течение 2 ч при 70 С. После концентрирования в вакууме остаток очищали методом препаративной ТСХ с получением 116,6 мг нитрила (84%). Нитрил с предыдущей стадии (67,5 мг) растворяли в 5 мл дихлорметана, к которому добавляли DDQ (43 мг) и 0,5 мл воды. Полученную смесь перемешивали в течение 2 ч при комнатной температуре. После обработки водой и очистки методом препаративной ТСХ, получали 47,6 мг (91%) целевого соединения. Следующие ниже примеры представлены для более полной иллюстрации изобретения и они никоим образом не ограничивают область изобретения. Пример 1. Часть А. Соединение (1 эквивалент), отвечающее приведенной выше формуле, в которойR=-CH(C6H5), a X представляет собой -СНО,получение которого описано в WO 96/14326,растворяли в этаноле и добавляли равный объем пиридина, после чего добавляли гидрохлорид гидроксиламина (10 эквивалентов). Реакционную смесь перемешивали в атмосфере азота примерно в течение 20 мин или до заметной глубины реакции, о чем судили с помощью аналитической ТСХ. Полученную смесь концентрировали при пониженном давлении и распределяли в смеси воды и дихлорметана. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали методом препаративной ТСХ (гексан:этилацетат) с получением продукта. Часть В. Продукт из части А (1 эквивалент) растворяли в толуоле и добавляли сольBurgess (5 эквивалентов). Полученную смесь перемешивали в атмосфере азота при 60 С в течение 1 ч или до того, как прореагирует значительная часть исходного материала. Реакционную смесь охлаждали и летучие вещества удаляли в вакууме. В результате очистки методом препаративной ТСХ (гексан:этилацетат) получали продукт. Часть С. Продукт из части В растворяли в 2% растворе трифторуксусной кислоты в дихлорметане (вес./вес.) при 0 С и затем перемешивали в течение 4 ч или до значительной глубины протекания реакции. Летучие вещества удаляли при пониженном давлении и продукт 35 реакции очищали методом препаративной ТСХ с получением целевого соединения. Пример 2. [1R-(1,3a,4,4a,4a,7,7a,8a)]-8 а-[(6-деокси-4-O-метилD-алтропиранозилокси)метил]-4-циано-4,4 а,5,6,7,7 а,8,8 аоктагидро-7-метил-3-(1-метилэтил)-1,4-метаноs-индацен-3 а(1 Н)-карбоновая кислота (4-циано 4-деформилсордарин). Сордарин (50 мг) растворяли в 3 мл N,Nдиметилформамида и добавляли 0,3 мл бромистого бензила с последующим добавлением 200 мг гидрида натрия (в виде 60% дисперсии в минеральном масле). Полученную смесь перемешивали в течение ночи при комнатной температуре. После водной обработки (диэтиловый эфир) и очистки методом препаративной ТСХ,получали бензиловый эфир 2',3'-ди-O-бензилсордарина. Раствор бензилового эфира 2',3'-ди-Oбензилсордарина (1 эквивалент) с предыдущей стадии получали в системе этанол/пиридин(2:1). Добавляли избыток гидрохлорида гидроксиламина и смесь в течение 2 ч при перемешивании нагревали до 70 С. Полученную смесь концентрировали в вакууме и проводили жидкостную обработку (дихлорметан). В результате очистки методом препаративной ТСХ получали бензиловый эфир 2',3'-ди-O-бензил-4-альдоксимсордарина. К толуольному раствору бензилового эфира 2',3'-ди-O-бензил-4-альдоксимсордарина с предыдущей стадии добавляли избыток внутренней соли гидроксида (метоксикарбонилсульфамоил)триэтиламмония (реагент Burgess). Полученную смесь перемешивали в атмосфере азота в течение 2 ч при 70 С. После концентрирования в вакууме и очистки методом препаративной ТСХ получали бензиловый эфир 2',3'-диО-бензил-4-циано-4-деформилсордарина. Готовили метанольный раствор бензилового эфира с предыдущей стадии. Добавляли гидроксид палладия на угле (катализатор Pearlman) и реакционный сосуд продували газообразным водородом. Полученную смесь интенсивно перемешивали под давлением водорода 1 атм в течение 15 мин. Смесь фильтровали и раствор концентрировали в вакууме с получением целевого соединения. Масс-спектр (Сl): m/z = 507,5 (M+NH4)+. Пример 3. [1R-(1,3a,4,4a,4a,7,7a,8 а)]-8 а-[(6-деоксиD-алтропиранозилокси) метил]-4-циано-4,4 а,5,6,7,7 а,8,8 а-октагидро-7 метил-3-(1-метилэтил)-1,4-метано-s-индацен 3 а(1 Н)-карбоновая кислота (4-цианo-4-дeфopмил-4'-дeмeтилcopдаpин). 1 мл замороженного мицелия Streptomycesavermitilis МА 4848 (АТСС 31272) инокулировали в каждую из 8 Эрленмейеровских колб с дросселями, содержащих 4 мл среды BaSa [в одном литре: 20 г дрожжевого экстракта (Difco),20 г Hycase (бессолевая форма, Sheffield), 20 г дестрозы, 2 г KNO3, 10 мл микроэлементов (указаны ниже), pН 7,0, обработка в автоклаве в течение 20 мин]. Колбы инкубировали в течение 24 ч при 21 С и скорости вращения 220 об./мин. Значение рН составляло 6,7-6,8. Мицелии исследовали под микроскопом на присутствие немицелиальных примесей. Микроэлементы Сорок мг целевого соединения примера 2(4-циано-4-деформилсордарин) растворяли в 0,40 мл 80% этанола и по 50 мкл такой смеси добавляли в каждую колбу. Полученную смесь инкубировали в течение 18 ч при 27 С. Согласно данным, полученным методом ЖХВР, имела место исчерпывающая конверсия. Бульон (200 мл) разбавляли равным объемом метанола (200 мл) для экстракции и твердые вещества удаляли центрифугированием. Супернатант концентрировали в вакууме с целью удаления основного количества метанола и в оставшемся раствореNaOH. Этот раствор дважды экстрагировали дихлорметаном (200 мл). рН водного слоя устанавливали равным 2,5 с помощью разбавленной серной кислоты и проводили двукратную экстракцию дихлорметаном. Объединенные дихлорметановые слои промывали водой, рассолом и сушили над безводным сульфатом натрия. Сульфат натрия удаляли фильтрацией, а дихлорметан отгоняли в вакууме с получением твердого вещества (53 мг). Остаток очищали методом препаративной обращеннофазовой ЖХВР на Phenomenex Primesphere C8 (5 ,9,4250 мм). Подвижную фазу, представляющую собой смесь ацетонитрил:вода (34:46), содержащую 0,1% фосфорной кислоты, применяли с объемной скоростью 3,5 мл/мин при 40 С. Целевое соединение элюировали через 14,4 мин. Обогащенные фракции объединяли и ацетонитрил удаляли в потоке N2. Оставшийся водный раствор экстрагировали дихлорметаном, как описано выше, с получением 7,2 мг целевого соединения. Часть А. Дигитоксозу (0,500 г, 3,37 ммоль) помещали в колбу и подвергали азеотропной перегонке с бензолом. Материал растворяли в 5 мл сухого пиридина и добавляли 5 мл уксусного ангидрида. Реакционную смесь перемешивали в течение 18 ч. Согласно анализу, проведенному методом тонкослойной хроматографии (силикагель, этилацетат/гексаны 1:2), зафиксировано полное исчезновение дигитоксозы. Спектр 1H ЯМР (СDСl3):1,11 (д, 3 Н),1,96-2,16 (м, 2 Н), 2,02 (с, 3 Н), 2,11 (с, 6 Н), 4,06(м, 1 Н), 4,61 (дд, 1 Н), 5,49 (широкий квартет,1 Н), 6,02 (дд, 1 Н). Масс-спектр: 215,1 (M-C2H3O2)+. Часть 2. Перацетилдигитоксозу, полученную по методике, описанной выше в части А,(9,31 г, 33,9 ммоль) добавляли к 150 мл воды и в полученную смесь добавляли 50 мл ледяной уксусной кислоты. Реакционную смесь перемешивали при окружающей температуре в течение 3 дней. Растворитель удаляли в вакууме, а сырой остаток очищали методом препаративной тонкослойной хроматографии(94%) бледно-желтого сиропообразного продукта. Полученный продукт, 3,4-диацетоксидигитоксоза, существует в виде смеси аномеров. Часть С. 3,4-Диацетоксидигитоксозу (1,23 г, 5,3 ммоль) помещали в колбу и в атмосфере азота добавляли 25 мл безводного дихлорметана. Затем добавляли карбонат цезия (0,35 г, 1,1 ммоль), после чего добавляли трихлорацетонитрил (7,2 г, 50 ммоль). Реакционную смесь перемешивали примерно в течение одного часа,фильтровали и летучие вещества удаляли в вакууме. Полученный в результате сырой трихлорацетимидат (2,00 г) использовали в дальнейших реакциях без дополнительной очистки. Спектр 1H ЯМР (СDСl3):1,34 (д, 3 Н),2,03 (с, 3 Н), 2,07 (с, 3 Н), 2,11 (м, 1 Н), 2,29 (ддд,1 Н), 4,17 (м, 1 Н), 4,80 (дд, 1 Н), 5,52 (м, 1 Н), 6,20 38 Часть D. Интермедиат 1 (1,6 г, 3,8 ммоль) подвергали азеотропной перегонке с бензолом и высушенное соединение растворяли в 10 мл дихлорметана. Добавляли безводный бромистый цинк (0,340 г, 1,5 ммоль) и полученную смесь охлаждали до 0 С. Раствор продукта из части С (2,8 г, 7,5 ммоль) в 5 мл дихлорметана добавляли с помощью шприцевого насоса в течение одного часа. Реакционную смесь переливали в водный раствор кислого карбоната натрия и экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Сырой материал очищали методом препаративной тонкослойной хроматографии(силикагель, этилацетат/гексан 1:4) с получением 2,09 г (86%) целевого продукта в виде бесцветного сиропообразного вещества. Продукт представлял собой смесьиизомеров, в соотношении 2:3. Масс-спектр: 654,2 (М+ NH4). Часть Е. Продукт из части D (2,09 г, 33 ммоль) растворяли в 50 мл метанола и добавляли карбонат калия (0,20 г). Реакционную смесь перемешивали в течение 2 ч при температуре окружающего воздуха. Карбонат калия удаляли фильтрацией, а летучие вещества отгоняли в вакууме. Сырой продукт очищали методом тонкослойной хроматографии (силикагель, вначале элюирование смесью этилацетат/гексаны 1:2,затем смесью этилацетат/гексаны 1:1) с получением 0,84 г (49%) целевого деацетилированного(д, 1 Н), 7.35 (м, 5 Н), 9.69 (с, 1 Н). Масс-спектр: 423,3 (М+Н-дигитоксоза). Часть F. -аномер из части Е (35.6 мг) растворяли в 3 мл дибромметана и добавляли 3 мл 50% водного раствора гидроксида натрия, после чего добавляли бромистый тетрабутиламмоний(4.2 мг, 0.013 ммоль). Полученную смесь интенсивно перемешивали в течение 18 ч. Реакционную смесь экстрагировали дихлорметаном и органическую фазу сушили над безводным сульфатом натрия, фильтровали и летучие вещества удаляли в вакууме. Остаток очищали методом препаративной ТСХ (9:1, гек 39 сан/этилацетат) с получением 16,4 мг (45%) белого твердого вещества. Спектр парциального 1H ЯМР (CDCl3):0.52 (д, 3 Н), 0.90 (д, 3 Н), 0.9 (м), 1.00 (д, 3 Н),1.04 (м), 1.13 (д, 3 Н), 2.71 (т, 1 Н), 3.38 (м, 1 Н),3.60 и 3.89 (АВ квартет, 2 Н), 3.64 (м, 1 Н), 4.13(м, 1 Н), 4.48 (дд, 1 Н), 4.85 (с, 1 Н), 5.14 (с, 1 Н),5.18 (АВ квартет, 2 Н), 6.01 (д, 1 Н), 7.36 (м, 5 Н),9.71 (с, 1 Н). Часть G. Продукт из части F (9,7 мг, 0,017 ммоль) растворяли в 1 мл пиридина и добавляли 1 мл этанола, после чего добавляли гидрохлорид гидроксиламина (12 мг, 0,17 ммоль). Полученную смесь нагревали до 70 С и перемешивали в течение 1 ч. Реакционную смесь охлаждали и растворитель удаляли в вакууме. Остаток распределяли в смеси воды с дихлорметаном и водный слой дополнительно экстрагировали дихлорметаном. Объединенные органические фазы сушили над безводным сульфатом натрия,фильтровали и летучие вещества удаляли в вакууме. В результате очистки остатка методом препаративной ТСХ (4:1/гексан:этилацетат) получали 8,6 мг (87%) продукта в виде твердого вещества. Спектр парциального 1H ЯМР (СDСl3):0.52 (д, 3 Н), 0.81 (д, 3 Н), 0.9 (м), 0.98 (д, 3 Н),1.01 (м), 1.23 (д, 3 Н), 1.34 (д, 1 Н), 2.62 (т, 1 Н),3.36 (м, 1 Н), 3.60 (д, 1 Н), 3.64 (дд, 1 Н), 3.87 (д,1 Н), 4.13 (м, 1 Н), 4.49 (дд, 1 Н), 4.85 (с, 1 Н), 5.15(м, 5 Н), 7.46 (широкий с, 1 Н), 7.81 (с, 1 Н). Часть Н. Продукт из части G (8.6 мг, 0,015 ммоль) растворяли в 2 мл толуола и добавляли реагент Burgess (18 мг, 0,074 ммоль). Реакционную смесь перемешивали в атмосфере сухого азота при 60 С в течение примерно одного часа. Добавляли дополнительное количество реагентаBurgess и полученную смесь перемешивали еще в течение 20 мин. Реакционную смесь охлаждали и летучие вещества удаляли в вакууме. В результате очистки методом препаративной ТСХ (4:1/гексан:этилацетат) получали 7,3 мг(м, 1 Н), 4.48 (дд, 1 Н), 4.85 (с, 1H), 5.14 (с, 1H),5.18 (АВ квартет, 2 Н), 6.12 (м, 1H), 7.34 (м, 3 Н),7.46 (м, 2 Н). Часть I. Продукт, полученный в разделе,часть Н (7,3 мг, 0,013 ммоль), растворяли в 2 мл метанола и добавляли катализатор Pearlman (2 мг). Реакционный сосуд продували водородом и содержимое перемешивали в атмосфере водорода в течение 2 ч. Полученную смесь фильтровали через слой целита и летучие вещества удаляли в вакууме с получением 6,1 мг (100%) целевого соединения в виде белого твердого вещества. Спектр парциального 1H ЯМР (СDСl3):(0,84 г, 1,5 ммоль) растворяли в 100 мл толуола. В атмосфере азота добавляли оксид дибутилолова (0,568 г, 2,3 ммоль) и полученную смесь нагревали с обратным холодильником и при перемешивании в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли бромистый аллил (0,544 г, 4,5 ммоль), после чего добавляли фтористый тетрабутиламмоний (2,3 мл, 1,0 М раствор в ТГФ, 2,3 ммоль). Реакционную смесь нагревали до 50 С. Через 1,5 дня смесь охлаждали и летучие вещества удаляли в вакууме. В результате очистки методом тонкослойной хроматографии (ступенчатый градиент: 9:1 4:102:1/гексаы:этилацетат) получали 0,454 г (51%) продукта, активного в УФ-области. Спектр парциального 1H ЯМР (CDCl3):0.51 (д, 3 Н), 0.81 (д, 3 Н), 0.9 (м), 1.00 (д, 3 Н),1.05 (м), 1.22 (д, 3 Н), 2.21 (м, 1 Н), 2.72 (т, 1 Н),3.01 (дд, 1 Н), 3.60 (д, 1 Н), 3.68 (м, 1 Н), 3.87 (д,1 Н), 3.98 (м, 1 Н), 4.10 (м, 1 Н), 4.18 (м, 1 Н), 4.59(с, 1 Н). Часть В. Продукт из части А (288 мг, 0,49 ммоль), трифенилфосфин (511 мг, 1,95 ммоль) и имидазол (133 мг, 1,95 ммоль) помещали в колбу и растворяли в 40 мл свежеперегнанного тетрагидрофурана. Добавляли твердый иод (371 мг,1,46 ммоль) и полученную смесь перемешивали в атмосфере азота при комнатной температуре в течение 1,5 ч. Добавляли 1N хлористоводородную кислоту и полученную смесь экстрагировали этилацетатом. Органическую фазу промывали водой, раствором тиосульфата натрия и затем рассолом. Затем органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали методом препаративной ТСХ(АВ квартет, 2 Н), 4.01 (м, 1 Н), 4.38 (дд, 1 Н), 41 5.18 (АВ квартет, 2 Н), 5.20 (с, 1 Н), 5.30 (д, 1 Н),5.96 (м, 1 Н), 6.00 (д, 1 Н), 7.37 (м, 5 Н), 9.69 (с,1 Н). Масс-спектр: 725,0 (M+Na). Часть С. К 8 мл толуола добавляли гидрид три-н-бутилолова (0,267 мл, 0,993 ммоль). Полученный раствор нагревали с обратным холодильником в течение 2 ч. Продукт со стадии,описанной в части В (211 мг, 0,301 ммоль), растворяли в 10 мл сухого толуола и в течение 2 ч с помощью шприцевого насоса добавляли в орашающий раствор гидрида три-н-бутилолова. Добавляли дополнительный эквивалент гидрида три-н-бутилолова и полученную смесь нагревали с обратным холодильником еще в течение 30 мин, после чего методом аналитической ТСХ было установлено полное расходование исходного материала. Полученную смесь охлаждали и летучие вещества удаляли при пониженном давлении. В результате очистки методом препаративной ТСХ получали желтое масло. Полученный материал дополнительно очищали методом препаративной ЖХВР (Zorbax RxC18, 5% воды/95% ацетонитрил, =220 нм) с получением двух продуктов, 23,0 мг (13%) -изомера и изомера.(д, 1 Н), 3.65 (т, 1 Н), 3.77 (д, 1 Н), 3.82 (т, 1 Н),4.64 (м, 1 Н), 5.18 (с, 2 Н), 5.99 (м, 1 Н), 7.32 (м,5 Н), 9.69 (с, 1 Н). Часть D. -изомер, полученный по методике части С (5,6 мг, 0,0097 ммоль), растворяли в 0,5 мл этанола и добавляли 0,5 мл пиридина,после чего добавляли гидрохлорид гидроксиламина (6,7 мг, 0,097 ммоль). Реакционную смесь перемешивали в атмосфере азота в течение примерно 20 мин. Полученную смесь концентрировали при пониженном давлении и распределяли в смеси воды с дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали методом препаративной ТСХ (4:1/гексан:этилацетат) с получением 6,6 мг (количественный выход) продукта,Спектр парциального 1 Н ЯМР (СDСl3):0.51 (д, 3 Н), 0.81 (д, 3 Н), 0.98 (д, 3 Н), 1.02 (д,3 Н), 1.20 (д, 3 Н), 2.23 (м, 1 Н), 2.63 (т, 1 Н), 3.29 42 Часть Е. Продукт со стадии, описанной в части D (6,6 мг, 0,011 ммоль) растворяли в 1 мл толуола и добавляли соль Burgess (13,3 мг, 0,055 ммоль). Полученную смесь перемешивали в атмосфере азота при 60 С в течение часа. Реакционную смесь охлаждали и летучие вещества удаляли в вакууме. В результате очистки методом препаративной ТСХ (9:1/гексан:этилацетат) получали 5,1 мг (81%) продукта. Спектр парциального 1H ЯМР (CDCl3):0.39 (д, 3 Н), 0.86 (д, 3 Н), 1.01 (д, 3 Н), 1.15 (д,3 Н), 1.20 (д, 3 Н), 2.27 (дд, 1 Н), 2.63 (м, 1 Н), 2.73(т, 1 Н), 3.29 (м, 1 Н), 3.58 (д, 1 Н), 3.65 (т, 1 Н),3.83 (д, 1 Н), 3.99 (т, 1 Н), 4.41 (дд, 1 Н), 5.27 (АВ квартет, 2 Н), 6.13 (м, 1 Н), 7.33 (м, 3 Н), 7.46 (м,2 Н). Часть F. Продукт со стадии, описанной в части Е (5,1 мг, 0,0087 ммоль), растворяли в 1 мл метанола и добавляли примерно 1 мг гидроксида палладия на угле (катализатор Pearlman). Реакционный сосуд продували газообразным водородом и полученную смесь в течение 30 мин интенсивно перемешивали при давлении водорода в 1 атм. Катализатор удаляли фильтрацией через слой целита и фильтрат концентрировали при пониженном давлении с получением 3,8 мг (88%) целевого соединения в виде белого твердого вещества. Спектр парциального 1H ЯМР (СDСl3):0.77 (д, 3 Н), 1,01 (д, 3 Н), 1.04 (д, 3 Н), 1.02 (д,3 Н), 1.17 (д, 3 Н), 1.22 (д, 3 Н), 3.67 (т, 1 Н), 3.98 Часть А. -Аномер полученный на стадии,описанной в части Е примера 4 (18,3 мг, 0,033 ммоль), растворяли в 1,5 мл дибромметана. К полученному раствору добавляли 1,5 мл 50% водного раствора гидроксида натрия, после чего добавляли бромистый тетрабутиламмоний (2,1 мг, 0,006 ммоль). Полученную смесь интенсивно перемешивали в течение 24 ч, добавляли еще 6 мг бромистого тетрабутиламмония и смесь перемешивали в течение дополнительных 24 ч. Реакцию тушили добавлением 9,4 мл 2N хлористо-водородной кислоты и полученную смесь распределяли в смеси воды с дихлорметаном. Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Полученный в результате остаток очищали методом препаративной ТСХ (4:1/гексаны:этилацетат) с получением 3,1 мг (17%) продукта.(м, 1 Н), 4.94 (с, 1 Н), 5.13 (с, 1 Н), 5.18 (АВ квартет, 2 Н), 5.99 (д, 1 Н), 7.35 (м, 5 Н), 9.69 (с, 1 Н). Часть В. Продукт со стадии, описанной в части А (3,1 мг, 0,0055 ммоль), растворяли в 1 мл метанола и добавляли примерно 1 мг гидроксида палладия на угле (катализатор Pearlman). Реакционный сосуд продували газообразным водородом и содержимое перемешивали в атмосфере водорода в течение 40 мин. Катализатор удаляли фильтрацией через целит. В результате концентрации фильтрата при пониженном давлении получали 3,1 мг (количественный выход) целевого соединения. Спектр парциального 1 Н ЯМР (CDCl3):0.80 (д, 3 Н), 0.95 (д, 3 Н), 1.01 (д, 3 Н), 1.27 (д,3 Н), 2.39 (м), 2.78 (т, 1 Н), 3.76 (м, 4 Н), 4.06 (м,1 Н), 4.71 (м, 1 Н), 4.87 (с, 1 Н), 5.13 (с, 1 Н), 6.02 Часть А. -Изомер примера 5, часть С (1 эквивалент) растворяли в этаноле и добавляли равный объем пиридина после чего добавляли гидрохлорид гидроксиламина (10 эквивалентов). Реакционную смесь перемешивали в атмосфере азота в течение примерно 20 мин или до достаточной глубины протекания реакции, которую устанавливали методом аналитической ТСХ. Смесь концентрировали при пониженном давлении и распределяли в смеси воды с дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали методом препаративной ТСХ (гексан:этилацетат) с получением продукта. Часть В. Продукт, полученный в части А(1 эквивалент), растворяли в толуоле и добавляли соль Burgess. Смесь перемешивали в атмосфере азота при 60 С в течение 1 ч или до вступления в реакцию существенной части исходного материала. Реакционную смесь охлаждали и летучие вещества удаляли в вакууме. В результате очистки методом препаративной ТСХ (гексан:этилацетат) получали целевой продукт. Часть С. Продукт, полученный в части В,растворяли в метаноле и добавляли каталитическое количество гидроксида палладия на угле(катализатор Pearlman). Реакционный сосуд продували газообразным водородом и смесь интенсивно перемешивали под давлением водорода в 1 атм в течение примерно 30 мин или до тех пор, пока не прореагирует весь исходный 44 материал. Катализатор удаляли фильтрацией через слой целита и фильтрат концентрировали при пониженном давлении с получением целевого продукта. Пример 8. Часть А. Интермедиат 5 (16,3 мг, 0,039 ммоль) подвергали трехкратной азеотропной дистилляции с бензолом и высушенное соединение совместно с безводным бромистым цинком (2,0 мг, 0,0078 ммоль) загружали в колбу в атмосфере азота. Добавляли сухой дихлорметан(1,0 мл) и полученную смесь охлаждали до 0 С. 3,4-Диацетилдигитоксоза-1-трихлорацетимидат(полученный по методике, описанной в части С примера 4) (29,2 мг, 0,078 ммоль) растворяли в 0,075 мл сухого дихлорметана и добавляли в реакционную смесь в течение 1 ч. Реакционной смеси давали нагреваться до окружающей температуры в течение ночи. Полученную смесь переливали в насыщенный водный раствор бикарбоната натрия и экстрагировали дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и летучие вещества удаляли в вакууме. Остаток очищали методом препаративной ТСХ (обработка смесью гексан:этилацетат, вначале в соотношении 9:1 и затем 4:1) с получением 17,0 мг (69%) смеси - и -изомеров в соотношении около 1:2. Часть В. Продукт, полученный в части А,растворяли в метаноле и добавляли гидроксид палладия на угле (катализатор Pearlman). Реакционный сосуд продували газообразным водородом и тщательно перемешивали в течение 30 мин или до полного расходования исходного материала. Катализатор отфильтровывали и в результате удаления из фильтрата летучих веществ получали целевое соединение. Масс-спектр: 561,4 (M+NH4). Пример 9. Часть А. Соединение (1 эквивалент), отвечающее приведенной выше формуле, в которойR = -СН(С 6 Н 5)2, а Х = -СНО, получение которого описано в WO 96/14326, растворяли в этаноле и добавляли равный объем пиридина, после чего добавляли гидрохлорид гидроксиламина (10 эквивалентов). Реакционную смесь перемешивали в атмосфере азота в течение примерно 20 мин или до существенной глубины протекания 45 реакции, о чем судили по данным аналитической ТСХ. Полученную смесь концентрировали при пониженном давлении и распределяли в смеси воды с дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия,фильтровали и концентрировали в вакууме. Остаток очищали методом препаративной ТСХ(гексан:этилацетат) с получением продукта. Часть В. Продукт, полученный на стадии,описанной в части А (1 эквивалент), растворяли в толуоле и добавляли соль Burgess (5 эквивалентов). Полученную смесь перемешивали в атмосфере азота при 60 С в течение 1 ч или до вступления в реакцию значительной части исходного материала. Реакционную смесь охлаждали и летучие вещества удаляли в вакууме. В результате очистки методом препаративной ТСХ (гексан:этилацетат) получали целевой продукт. Часть С. Продукт со стадии, описанной в части В, растворяли в метаноле и добавляли каталитическое количество гидроксида палладия на угле (катализатор Pearlman). Реакционный сосуд продували газообразным водородом и смесь интенсивно перемешивали под давлением водорода в 1 атм в течение 30 мин или до полного прореагирования исходного материала. Катализатор удаляли фильтрацией через слой целита и фильтрат концентрировали при пониженном давлении с получением целевого соединения. Пример 10. Часть А. Соединение (1 эквивалент), отвечающее приведенной выше формуле, в которойR = -CH(C6H5)2, a X = -CHO, получение которого описано в WO 96/14326, растворяли в этаноле и добавляли равный объем пиридина с последующим добавлением гидрохлорида гидроксиламина (10 эквивалентов). Реакционную смесь перемешивали в атмосфере азота примерно в течение 20 мин или до достаточной глубины протекания реакции, о чем судили по данным аналитической ТСХ. Полученную смесь концентрировали при пониженном давлении и распределяли в смеси воды с дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали методом препаративной ТСХ (гексан:этилацетат) с получением целевого продукта. Часть В. Продукт со стадии, описанной в части А (1 эквивалент) , растворяли в толуоле и добавляли соль Burgess (5 эквивалентов). Полученную смесь перемешивали в атмосфере азота при 60 С в течение 1 ч или до вступления в ре 002631 46 акцию значительной части исходного материала. Реакционную смесь охлаждали и летучие вещества удаляли в вакууме. В результате очистки методом препаративной ТСХ (гексан:этилацетат) получали продукт. Часть С. Продукт со стадии, описанной в части В, растворяли в 2% растворе трифторуксусной кислоты в дихлорметане (вес./вес.) при 0 С и перемешивали в течение 4 ч или до значительной глубины протекания реакции. Летучие вещества удаляли при пониженном давлении и продукт реакции очищали методом препаративной ТСХ с получением целевого соединения. Пример 11.-Н, Х = -СНО) растворяли в сухом ДМФ, содержащем 5% (об./об.) бромистого пметоксибензила. Добавляли избыточное количество твердого NаНСО 3 и полученную смесь перемешивали при комнатной температуре в течение примерно 18 ч или до расходования значительной части зофимарина. Полученную смесь концентрировали в вакууме и добавляли хлороформ. Смесь фильтровали с целью удаленияNаНСО 3, фильтрат концентрировали в вакууме и очищали методом препаративной тонкослойной хроматографии (ПТСХ) с получением продукта, отвечающего приведенной выше формуле, в которой R = -СН 2 С 6 Н 4-п-ОСН 3, а Х =-СНО. Часть В. Продукт со стадии, описанной в части А (1 эквивалент), растворяли в этаноле и добавляли равный объем пиридина с последующим добавлением гидрохлорида гидроксиламина (10 эквивалентов). Реакционную смесь перемешивали в атмосфере азота, примерно, в течение 20 мин или до существенной глубины протекания реакции, о чем судили по данным аналитической ТСХ. Полученную смесь концентрировали при пониженном давлении и распределяли в смеси воды с дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали методом препаративной ТСХ (гексан:этилацетат) с получением продукта. Часть С. Продукт со стадии, описанной в части В (1 эквивалент), растворяли в толуоле и добавляли соль Burgess (5 эквивалентов). Полученную смесь перемешивали в атмосфере азота при 60 С в течение 1 ч или до вступления в реакцию значительной части исходного материала. Реакционную смесь охлаждали и летучие вещества удаляли в вакууме. В результате очи 47 стки методом препаративной ТСХ (гексан:этилацетат) получали продукт. Часть D. Продукт со стадии, описанной в части С, растворяли в 2% растворе дифторуксусной кислоты в дихлорметане (вес./вес.) при 0 С и перемешивали в течение 4 ч или до существенной глубины протекания реакции. Летучие Исходное соединение 48 вещества удаляли при пониженном давлении и продукт реакции очищали методом ПТСХ с получением целевого соединения. Примеры 12-19. Способом, аналогичным описанному в примере 9, получали следующие соединения. Конечный продукт а также соли и сольваты (например, гидраты) или метаболически лабильные его производные,где Ra представляет собой С(O)СН 3 или СН 3;R1 представляет собой водород, галоген,гидроксил, C1-4 алкокси или ацилокси; каждый из радикалов R2 и R3 независимо друг от друга представляет собой водород,C1-6 алкил или С 1-4 алкоксиС 1-4 алкил, илиR4 представляет собой водород или CH2R7(где R7 представляет собой водород, гидроксил,С 1-4 алкокси или группу OCOR8, в которой R8 представляет собой С 1-4 алкил или арил); каждый из радикалов R5 и R6 независимо друг от друга представляет собой водород,C1-6 алкил или С 1-4 алкоксиС 1-4 алкил, илиn равно нулю или 1; каждый из Х и Y независимо друг от друга представляют собой кислород, серу или CR9R10(где каждый из радикалов R9 и R10 независимо друг от друга представляет собой водород, C1-6 алкил, С 1-4 алкокси или С 1-4 алкоксиС 1-4 алкил; или R9 и R10 совместно с соединенным с ними атомом углерода представляют собой С=O, C=S или С 3-8 циклоалкил или C=CHR11, где R11 представляет собой водород или С 1-4 алкил); или, когда Х и Y представляют собой кислород, а n равно нулю, -Y-CR2R3 или -X-CR2R3- соответственно могут также представлять собой -N=CR3 или -NR12-CR2R3- (где CR2 и CR3 представляют собой С=O, а R12 представляет собой С 1-4 алкил 52 или ацильную группу COR13, в которой R13 представляет собой C1-6 алкил), или, когда Y представляет собой кислород, а n равно нулю, Х может представлять собой группу CR11 (в котором R11 имеет указанные выше значения), которая присоединена к пирановому кольцу двойной связью;R15 представляет собой водород, галоген,азидо, C1-6 алкил, гидрокси, C1-6 алкокси (необязательно замещенную 1 или 2 гидроксигруппами или их кеталями, либо 1 или 2 C1-3 алкоксигруппами), арилС 1-4 алкокси, С 3-6 алкенилокси,группу OCOR18 (в которой R18 представляет собой арилС 1-4 алкокси или C1-10-алкильную группу, необязательно содержащую одну или две двойных связи) или C1-6 алкоксикарбонил С 1-4 алкокси, а R16 представляет собой водород, илиR15 и R16 совместно с соединенным с ними углеродным атомом могут представлять собой С=O или С=СН 2;R17 представляет собой CH2R19, где R19 представляет собой водород, гидроксил, C1-4 алкокси или группу OCOR20, в которой R20 представляет собой С 1-4 алкил;W представляет собой кислород, серу или СН 2; а волнистая линия в группе (а) указывает на необязательное присутствие дополнительной связи;R2a представляет собой водород, галоген,гидроксил, C1-10 алкокси, C1-10 алкилтио, С 1-6 алкоксиС 1-4 алкокси, арилС 1-6 алкилокси, арилС 3-6 алкенилокси, азидо, NR5aCOR5a (где каждый из радикалов R5a независимо друг от друга представляет собой водород или C1-6 алкил), OR6a(где R6a представляет собой циклическую эфирную группу, содержащую 4-8 атомов, соединенных с кислородным атомом через углеродный атом кольца, соседний с кольцевым кислородным атомом) или группу YaС(=O)-Xa-R7a, в которой Ya представляет собой кислород, серу илиNH, Xa представляет собой связь, кислородный атом или фрагмент NR8a, в котором R8a представляет собой водород или C1-6 алкил, а R7 представляет собой C1-10 алкил, необязательно содержащий одну или две двойные связи, арил,арилС 1-4 алкил, арилС 2-4 алкенил, галоС 1-6 алкил или С 1-6 алкоксиС 1-4 алкил, a R3a представляет собой водород, илиR2a и R3a совместно с соединенным с ними углеродным атомом представляют собой С=O или C=NOR9a (где R9a представляет собой C1-6 алкил); аR4a представляет собой гидроксил, C1-6 алкокси или OC(=O)R7a (где R7a имеет указанные выше значения). 2. Соединение по п.1, отвечающее следующей структурной формуле 3. Соединение по п.1, отвечающее следующей структурной формуле 4. Соединение по п.1, отвечающее следующей структурной формуле 5. Соединение по п.1, отвечающее следующей структурной формуле 6. Соединение по п.1, отвечающее следующей структурной формуле 8. Соединение по п.1, отвечающее следующей структурной формуле 9. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически приемлемый носитель. 10. Агрохимическая композиция, содержащая соединение по п.1 и сельскохозяйственно приемлемый носитель. 11. Способ лечения или профилактики грибкового заражения животного, заключающийся в применении на указанном животном фунгицидно эффективного количества соединения по п.1. 12. Способ контроля и борьбы с фитопатогенными грибками, заключающийся в применении на растении, нуждающемся в таком контроле, фунгицидно эффективного количества соединения по п.1. 13. Способ получения 4-циано-4 деформил-4-деметилcордарина, включающий стадии контактирования 4-циано-4-деформилсордарина с культурой штамма Streptomycesavermitilis в сбраживаемой среде, содержащей ассимилируемые источники углерода и азота и выделения 4-циано-4-деформил-4-деметилсордарина из указанной ферментационной среды. 7. Соединение по п.1, отвечающее следующей структурной формуле
МПК / Метки
МПК: A61K 31/34, C07D 307/77
Метки: производные, 4-циано-4-деформилсордарина
Код ссылки
<a href="https://eas.patents.su/28-2631-proizvodnye-4-ciano-4-deformilsordarina.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 4-циано-4-деформилсордарина</a>
Новые производные 5-амино-3-циано-4-этилсульфинил-1-фенилпиразола и их применение в качестве пестицидов

Номер патента: 973
Опубликовано: 28.08.2000
Авторы: Вю Тай-Тех, Хаас Чарльз Ли, Пилато Майкл Томас
МПК: C07D 233/44, A01N 43/56
Метки: 5-амино-3-циано-4-этилсульфинил-1-фенилпиразола, применение, производные, качестве, новые, пестицидов
Формула / Реферат:
1. Соединение формулы (I) где Х - хлор или бром; Y - трифторметил или трифторметокси и R1 - водород, метил или этил. 2. Соединение по п.1, в котором Х - хлор и Y - трифторметил. 3. Соединение по п.1 или 2, в котором R1 - водород или метил. 4. Соединение по пп.1, 2 или 3, которое представляет собой: 5-амино-3-циано-1-(2,6-дихлор-4-трифторметилфенил)-4-этилсульфинилпиразол; ...
Производные 5н, 10н-имидазо{1,2-а}индено {1,2-е} пиразин-4-она, способы их получения, содержащее их лекарственное средство и производные инданона в качестве промежуточных продуктов

Номер патента: 218
Опубликовано: 24.12.1998
Авторы: Немесе Патрик, Барро Мишель, Жимоне Патрик, Арди Жан-Клод, Манфр Франко, Дамур Доминик, Женевуа-Борелла Ариель, Рибей Ив, Алу Жан-Клод, Миньяни Серж, Одьо Франсуа
МПК: A61K 31/495, C07F 9/6561, C07D 487/04...
Метки: 10н-имидазо{1,2-а}индено, продуктов, пиразин-4-она, получения, способы, содержащее, инданона, лекарственное, качестве, 1,2-е, производные, промежуточных, средство
Формула / Реферат:
1. Производные 5Н,10H-имидазо[1,2-а] индено[1,2-е]пиразин-4-она формулы (I) в которой R означает атом водорода или карбоксил, алкоксикарбонил, -CO-NR4R5, -РО3H2 или -СН2ОН; R1 означает радикалы -aлк-NH2, -алк-NН-СО-R3, -алк-СООR4, -aлк-CO-NR5R6 или -CO-NH-R7; R3 означает алкил, фенил, фенилалкил, циклоалкил или -NR6R8; R4 означает атом водорода или алкильный радикал; R5 означает атом водорода, алкил, фенил, циклоалкил или...
Производные пиридилкарбамоилиндолинов в качестве антагонистов 5-нт2с-рецепторов

Номер патента: 1780
Опубликовано: 27.08.2001
Авторы: Бромидж Стивен Марк, Форбес Ян Томсон
МПК: A61P 25/28, A61K 31/4439, C07D 401/14...
Метки: 5-нт2с-рецепторов, пиридилкарбамоилиндолинов, антагонистов, качестве, производные
Формула / Реферат:
1. Соединение формулы (I) или его соль где Х обозначает СН или N; R1 обозначает водород или C1-6-алкил; R2 и R3 обозначают независимо C1-6-алкил или трифторметил. 2. Соединение по п.1, в котором Х обозначает СН. 3. Соединение по п.1 или 2, в котором R1 обозначает метил. 4. Соединение по любому из пп.1-3, в котором R2 обозначает СF3. 5. Соединение по любому из пп.1-4, в котором R3 обозначает C1-6-алкил. 6. Соединение по любому из пп.1-5, в...
Производные гуанина

Номер патента: 364
Опубликовано: 24.06.1999
Авторы: Конвей Грегори Элан, Лейк Филипп Джордж, Варлэшкин Питер Грегори, Партин Джейн Мьюз, Грабб Уильям Бейн(третий), Картер Бэрри Хауард, Скиннер Дэйвид Майкл, Уотрап Дэйвид Джеймс, Уинник Ричард Огастас
МПК: C07D 473/00, A61K 31/52
Метки: гуанина, производные
Формула / Реферат:
1. Гидрохлорид валацикловира в безводной, по существу, кристаллической форме, имеющей следующие значения расстояний d между кристаллографическими плоскостями (в ангстремах): 10,20 + 0,08; 8,10 + 0,06; 7,27 + 0,06; 6,08 + 0,05; 5,83 + 0,03; 5,37 + 0,02; 5,23 + 0,02; 4,89 + 0,02; 4,42 + 0,02; 4,06 + 0,02; 3,71 + 0,02; 3,39 + 0,02; 3,32 + 0,02; 2,91 + 0,02; 2,77 + 0,02. 2. Кристаллический гидрохлорид валацикловира по п.1, отличающийся тем, что...
Производные 8-замещенного-1,3,8-триазаспиро [4.5]декан-4-она.

Номер патента: 1357
Опубликовано: 26.02.2001
Авторы: Адам Гео, Монсма Фредерик, Галлей Гуидо, Чесура Андреа, Женк Франсуа, Рёвер Стефан, Вихманн Йюрген
МПК: C07D 471/10, A61K 31/435, A61P 3/04...
Метки: 8-замещенного-1,3,8-триазаспиро, 4.5]декан-4-она, производные
Формула / Реферат:
1. Соединения общей формулы где R1 и R2 каждый независимо друг от друга обозначает водород, C1-C6алкил, C1-C6алкокси или галоген; R3 обозначает фенил, необязательно замещенный C1-C6алкилом, CF3, C1-C6 алкоксигруппой или галогеном; и R4 обозначает водород, C1-C6алкил, C1-C6 алкенил, -С(О)- C1-C6алкил, -С(О)-фенил, C1-C6 алкил-С(О)-фенил, C1-C6алкилен-С(О)О- C1-C6 алкил, C1-C6алкантриилди-С(О)О- C1-C6алкил, гидрокси- C1-C6алкил,...